

Visual Abstract
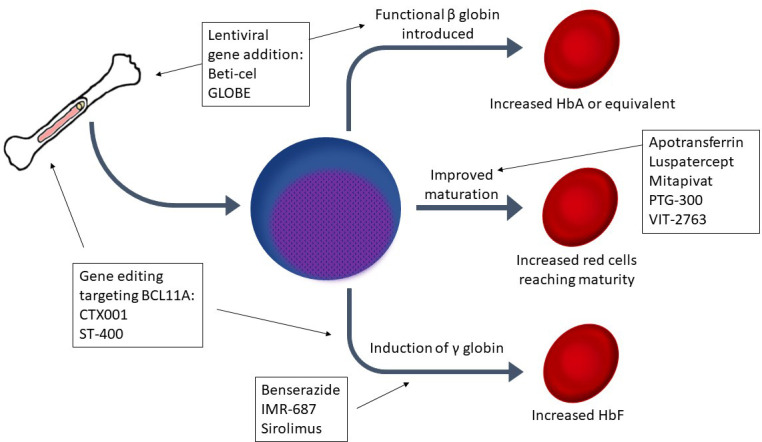
Abstract
After years of reliance on transfusion alone to address anemia and suppress ineffective erythropoiesis in β-thalassemia, many new therapies are now in development. Luspatercept, a transforming growth factor–β inhibitor, has demonstrated efficacy in reducing ineffective erythropoiesis, improving anemia, and possibly reducing iron loading. However, many patients do not respond to luspatercept, so additional therapeutics are needed. Several medications in development aim to induce hemoglobin F (HbF): sirolimus, benserazide, and IMR-687 (a phosphodiesterase 9 inhibitor). Another group of agents seeks to ameliorate ineffective erythropoiesis and improve anemia by targeting abnormal iron metabolism in thalassemia: apotransferrin, VIT-2763 (a ferroportin inhibitor), PTG-300 (a hepcidin mimetic), and an erythroferrone antibody in early development. Mitapivat, a pyruvate kinase activator, represents a unique mechanism to mitigate ineffective erythropoiesis. Genetically modified autologous hematopoietic stem cell transplantation offers the potential for lifelong transfusion independence. Through a gene addition approach, lentiviral vectors have been used to introduce a β-globin gene into autologous hematopoietic stem cells. One such product, betibeglogene autotemcel (beti-cel), has reached phase 3 trials with promising results. In addition, 2 gene editing techniques (CRISPR-Cas9 and zinc-finger nucleases) are under investigation as a means to silence BCL11A to induce HbF with agents designated CTX001 and ST-400, respectively. Results from the many clinical trials for these agents will yield results in the next few years, which may end the era of relying on transfusion alone as the mainstay of thalassemia therapy.
Learning Objectives
Understand the strengths and limitations of luspatercept for the treatment of TDT and NTDT
Understand the potential benefits and toxicity of genetically modified autologous HSCT for TDT
Describe therapies under development for the treatment of TDT and NTDT
After many years without novel disease-modifying therapeutics, numerous agents are now in development for β-thalassemia. We review therapies that have been recently approved or are in development for transfusion-dependent thalassemia (TDT) and non-transfusion-dependent thalassemia (NTDT) β-thalassemia using 4 patient cases (Table 1).
Table 1.
Current limitations of thalassemia care
| Limitation | Potential solutions |
|---|---|
| Regular transfusion appointments | Reduce transfusion requirement Stem cell transplantation |
| Iron overload | Reduce transfusion requirement Block intestinal iron absorption Escalate chelation |
| Iron chelator toxicity | Reduce transfusion requirement Block intestinal iron absorption Additional chelation options |
| Lack of allogeneic stem cell donor | Genetically modified autologous transplantation Nontransplant therapies that preclude desirability of transplant |
CLINICAL CASE 1: USE OF LUSPATERCEPT
A 54-year-old woman with congestive heart failure, paroxysmal atrial fibrillation on warfarin, and pulmonary hypertension due to β-thalassemia intermedia has a hemoglobin ranging from 7.5 to 8.5 g/dL without packed red blood cell (RBC) transfusion. Transfusion to maintain hemoglobin nadir of approximately 9.5 g/dL is indicated because of end-organ damage. However, she has been unable to tolerate the transfusion volumes, as attempts at transfusion frequently trigger a heart failure exacerbation and acute worsening of her pulmonary pressures. She is started on luspatercept, which leads to a rise in hemoglobin to 9.2 to 10.1 g/dL with drops below this range only in the setting of acute illness.
Luspatercept is a recombinant fusion protein that blocks transforming growth factor–β (TGF-β) superfamily ligands and promotes late-stage erythroid maturation, resulting in effective reduction in transfusion requirements in some patients with TDT.1 In a randomized, placebo-controlled trial, 21.4% of patients met the primary end point of at least a 33% reduction in transfusion volume during weeks 13 through 24 compared with 4.5% receiving placebo (P < .001). This equated to a least mean square difference in transfusion burden of 1.35 fewer RBC transfusions in weeks 13 through 24 in the TDT population. Follow-up data show transfusion reduction continued at 48 weeks and up to 4.8 years into treatment.2,3 Common side effects included headache, myalgia, and bone pain. Although rare, 8 of 223 (3.6%) patients in the luspatercept arm had thromboembolism compared with 1 of 109 patients in the placebo arm (Table 2).1
Table 2.
Luspatercept considerations1-6
| Strengths | Limitations | |
|---|---|---|
| Subcutaneous administration | Does not require intravenous access | Requires infusion center visit every 3 weeks |
| Reduction of transfusion in TDT | 21.4% had >33% reduction 7.6% had >50% reduction |
Most patients without significant reduction Majority still have visits every 3 weeks |
| Increase in hemoglobin in NTDT | 52.1% had 1.5-g/dL increase | Many patients without significant increase Requires increased appointment burden Not yet approved by regulatory agencies |
| Risk of thromboembolism | Rates low: 8 in 223 (3.6%) in trial | Patients with thalassemia already at increased risk of thromboembolism, especially if prior splenectomy |
| Impact on iron loading | Preliminary data showed lower ferritin and LIC in some patients, including some who did not meet primary end point | Additional data needed to verify findings |
In the NTDT population, data from a phase 1/2 nonrandomized trial showed an increase in hemoglobin of at least 1.5 g/dL in 58% of patients receiving higher dose levels.4 A randomized, placebo-controlled trial for luspatercept in patients with NTDT has finished accrual, and preliminary results showed hemoglobin of at least 1.5 g/dL in 52.1% in the luspatercept arm compared with 0% in the placebo arm and no thromboembolic events in either arm (NCT03342404).5
Case 1 demonstrates several strengths and limitations of luspatercept. This medication is most likely to be useful in patients for whom a moderate increase in hemoglobin or a moderate reduction in transfusion volume would be impactful. This may be the case because of difficulty with volume status, as was the case in this patient; for patients with alloimmunization for whom obtaining RBC units is more difficult; or for patients whose quality of life would improve by spending less time at transfusion appointments. The risk of thromboembolism should be considered when initiating this medication; although low in clinical trials, the incidence of thrombosis may compound other risk factors, such as prior splenectomy, and this may affect the risk-benefit ratio of initiating luspatercept. In case 1, the patient was already on lifelong anticoagulation because of atrial fibrillation, so this risk was mitigated. Finally, published data primarily address the TDT population; forthcoming results will help clarify how best to tailor the use of luspatercept for patients with NTDT.
Further follow-up will be required to determine if luspatercept may improve iron overload, although preliminary results are promising.6 Early findings that ferritin and liver iron concentration (LIC) improved in both those who met the primary end point and those who did not suggest that reduced iron loading from the gut is likely playing a role, rather than just reduced transfusion burden. If this bears out, it would represent a significant additional benefit, particularly for individuals in poor iron balance. Although luspatercept represents an exciting new therapeutic option and is the first novel approved therapy in many years, caution is merited in setting expectations, as most patients will not be able to forgo transfusion, and increasing the interval between transfusions is often not practically feasible or appealing since visits every 3 weeks are required for this subcutaneous medication. As such, a great unmet need remains.
CLINICAL CASE 2: AUTOLOGOUS GENETIC THERAPIES
A 17-year-old boy with TDT on a stable transfusion regimen and in good iron balance wishes to consider curative intent therapy. He is considering his goals for adulthood, including attending college that is not near a large medical center, and is interested in avoiding frequent medical and transfusion appointments. He has no available donor for allogeneic stem cell transplantation. He opts to enroll in a clinical trial of genetically modified autologous hematopoietic stem cell transplantation (HSCT) using a lentivirus vector.
Patients may desire a curative intent therapy given the long-term impact on morbidity, mortality, and quality of life associated with chronic transfusion therapy. Until recently, the only curative option was allogeneic HSCT. Allogeneic HSCT has provided a cure to many patients with TDT, with the best outcomes reported for matched sibling donors and recipients who underwent transplantation in teenage years.7 However, the risk of acute and chronic complications and the lack of available donors have limited its use.
Ex vivo modification of CD34+ hematopoietic stem cells using different techniques has opened the possibility of autologous HSCT as a curative therapy, thereby eliminating the need for a donor as well as the risks specific to allogeneic HSCT, such as graft-versus-host disease. In this context, genetically modified autologous HSCT is an area of active research with significant advancements in the past few years.
One ex vivo gene therapy, betibeglogene autotemcel (beti-cel; formerly LentiGlobin), uses a lentiviral vector to add a modified β-globin gene into CD34+ cells for infusion by autologous HSCT after myeloablative conditioning. In phase 1/2 trials, beti-cel treatment led to transfusion independence (TI) in 12 of 13 non-β0/β0 genotype and 3 of 9 β0/β0 genotype patients with TDT, or 68% overall.8 The remaining patients had a reduction in transfusion volume. Toxicity was primarily related to busulfan used for HSCT conditioning. The original beti-cel trial established the potential for genetically modified autologous HSCT, although most patients with the β0/β0 genotype did not achieve TI. Preliminary data from 2 phase 3 trials of beti-cel for β0/β0 (NCT03207009) and non-β0/β0 (NCT02906202) patients have demonstrated TI at >12 months of follow-up in 88% of 34 evaluable patients, including 24 of 27 non-β0/β0 and 6 of 7 β0/β0 patients, with no additional toxicities noted.9 Preliminary reports of long-term follow up of 44 patients with >2 years of follow-up after beti-cel treatment (23-76 months) showed a durable hemoglobin response, reduction in iron burden, and favorable safety profile.10 Initial evaluation of 27 pediatric patients treated with beti-cel, 16 of whom were under 12 years old, showed a comparable TI rate of 84.6% (Table 3).11
Table 3.
Gene therapy trials for β-thalassemia
| Therapy | Gene modification technique | Genetic mechanism | Phase of development | Also in development for sickle cell disease? | Comments |
|---|---|---|---|---|---|
| Betibeglogene autotemcel (beti-cel; formerly BB305)8-11,17,18 | Lentiviral vector | Insertion of modified β-globin | Phase 3 trials | Yes | Reopened after temporary halt due to AML cases in sickle cell patients |
| GLOBE12 | Lentiviral vector | Insertion of human β-globin | Phase 1/2 trial completed | No | Intrabone administration |
| CTX00114,15 | CRISPR-Cas9 | Targets BCL11A enhancer → HbF induction | Phase 1/2 trial | Yes | |
| ST-40016 | ZFN | Targets BCL11A enhancer → HbF induction | Phase 1/2 trial | No |
Another phase 1/2 trial uses a lentiviral vector (the GLOBE vector) to add a β-globin gene to autologous hematopoietic stem cells, with intrabone administration of the transduced cells. Three of 4 evaluable pediatric patients achieved TI, whereas 3 adult patients had a reduced transfusion burden without achieving TI.12 At present, a subsequent trial is not registered.
In contrast to the gene addition approach, another strategy for autologous genetic therapies for β-hemoglobinopathies is to decrease BCL11A expression in order to induce expression of γ-globin and hemoglobin F (HbF). The first trial targeting BCL11A uses a lentiviral vector, BCH-BB694, to introduce short hairpin RNA to decrease BCL11A expression.13 Six patients with sickle cell disease who received BCH-BB694 demonstrated safety and feasibility and had substantial induction of HbF. This vector has not been evaluated in patients with TDT. Trials are currently open for patients with thalassemia, in whom a gene editing technique, either CRISPR-Cas9 or zinc-finger nuclease (ZFN), is employed to disrupt an erythroid enhancer of BCL11A. CTX001 is a CRISPR-Cas9–modified autologous HSCT product being investigated in TDT as well as sickle cell disease (NCT03655678). Preliminary results from the first 10 patients treated with CTX001 for TDT showed substantial HbF induction, broad distribution of HbF, and achievement of TI in all patients.14,15 Toxicity was related to busulfan conditioning. A phase 1/2 study of ST-400, an autologous HSCT product in which the BCL11A enhancer is disrupted by a ZFN, is currently under way (NCT03432364), but only very preliminary data on the first 2 patients have been reported.16
Case 2 demonstrates the clinical demand for curative intent therapy and its potential impact on quality of life as well as health. Although genetically modified autologous HSCT may offer a more feasible and less toxic option for curative intent therapy compared with allogeneic HSCT, limitations are likely to remain. HSCT of any kind is a costly therapy and not likely to be available in low-resource settings. In addition to monetary costs, undergoing autologous HSCT requires a significant time commitment and interruption of other life events. The potential for reduced fertility or infertility due to exposure to myeloablative alkylator conditioning is an important consideration for many patients. Finally, 2 cases of acute myeloid leukemia (AML) in patients with sickle cell disease treated with beti-cel have raised concerns.17,18 Evaluation suggests that neither case was caused by insertional mutagenesis. To date, no patients with thalassemia have developed myelodysplastic syndrome or AML after genetically modified autologous HSCT, but additional data will be necessary before final conclusions may be drawn about what factors influence malignancy risk.
CLINICAL CASE 3: TARGETING ABNORMAL IRON METABOLISM
A 34-year-old man with NTDT is concerned about ongoing iron overload. His hemoglobin averages 8.4 g/dL, and he receives transfusion a few times per year. His LIC is estimated at 8 mg per gram of dry weight on magnetic resonance imaging. In addition to intensifying his iron chelation, he inquires whether there are any therapies that may raise his hemoglobin or reduce his iron absorption. He is open to participation in clinical trials.
Currently, there are no established therapies to address this patient's anemia. However, several medications in development may improve anemia in patients with NTDT that also target iron metabolism. Apotransferrin has been shown to upregulate hepcidin and downregulate transferrin receptor 1.19,20 This leads to decreased iron absorption from the gut,19 as well as potentially less cardiac iron loading.21 The correction of pathologic iron metabolism led to more effective erythropoiesis in mouse models.20 These preclinical findings have spurred a phase 2 trial of intravenous apotransferrin every 2 weeks in patients with β-thalassemia intermedia that seeks to raise hemoglobin and reduce transfusion requirements (NCT03993613).
Similarly, a ferroportin inhibitor (VIT-2763) has begun a phase 2 trial (NCT04364269) after demonstrating improvement in anemia and dysregulated iron metabolism in a mouse model of thalassemia.22 Like the apotransferrin trial, the VIT-2753 is focused on correction of anemia but includes secondary outcomes measures of iron overloading. Notably, VIT-2753 is administered orally. In considering treatment for patients with NTDT, this is particularly impactful for quality of life, as many of these patients do not otherwise have recurring infusion center appointments (Table 4).
Table 4.
Novel medications for β-thalassemia
| Medication | Mechanism of action | Route and frequency of administration | Phase of development | Current target patient population | In development for sickle cell disease? | Comments |
|---|---|---|---|---|---|---|
| Luspatercept1,5 | TGF-β inhibitor | Subcutaneous, every 3 weeks | EMA and FDA approved | NTDT, TDT | No | Full data for NTDT pending; in use for MDS |
| Sotatercept36,37 | TGF-β inhibitor | Subcutaneous, every 3 weeks | Development halted | NTDT, TDT | No | In development for pulmonary hypertension |
| Sirolimus25-28 | mTOR inhibitor; HbF induction | Oral, daily | Phase 2 trials | TDT | No | In use for other disorders |
| Benserazide31,32 | HbF induction | Oral, daily | Phase 1 trial | NTDT | Yes | In use for Parkinson disease |
| IMR-68729,30 | PDE-9 inhibitor; HbF induction | Oral, daily | Phase 2 trial | NTDT, TDT | Yes | |
| Apotransferrin19-21 | Hepcidin upregulation | Intravenous, every 2 weeks | Phase 2 trial | NTDT | No | |
| VIT-276322 | Ferroportin inhibitor | Oral, one or twice daily | Phase 2 trial | NTDT | Yes | |
| PTG-30023 | Hepcidin mimetic | Subcutaneous, once weekly | Phase 2 trials | NTDT, TDT | No | In development for hemochromatosis and polycythemia vera |
| Mitapivat33-35 | Pyruvate kinase activator | Oral, twice daily | Phase 3 trials | NTDT, TDT | Yes | In development for pyruvate kinase deficiency |
| Ruxolitinib38 | JAK 1/2 inhibitor | Oral, twice daily | Development halted | TDT | No | Spleen size reduction; no change in transfusion |
EMA, European Medicines Agency; FDA, Food and Drug Administration; JAK, Janus-associated kinase; MDS, myelodysplastic syndrome; mTOR, mechanistic target of rapamycin.
Hepcidin itself has also been a target for drug development, because increasing hepcidin has been shown to both reduce iron absorption and ameliorate ineffective erythropoiesis.23 A hepcidin mimetic, PTG-300, has completed accrual for a phase 2 study, including both NTDT and TDT arms (NCT03802201). A recent mouse study of an erythroferrone antibody showing improved anemia as well as iron homeostasis provides another potential therapeutic target in the iron regulation pathway.24
Case 3 emphasizes the large unmet need of therapies for NTDT and the appeal of a therapy that may also address nontransfusional hemosiderosis in these patients. These trials not only highlight the role of altered iron metabolism in ineffective erythropoiesis but also provide several promising therapeutic targets for ameliorating anemia.
CLINICAL CASE 4: FETAL HEMOGLOBIN INDUCTION AND OTHER MECHANISMS
A 21-year-old woman with TDT and in good iron balance recently tried luspatercept with only a minimal reduction in transfusion burden. She inquires if there are other therapies that might reduce her transfusion requirement. She is not interested in allogeneic or genetically modified autologous HSCT, as she is worried about the upfront risk and disruption in her life, because she is doing quite well overall on her current transfusion and chelation regimen.
Several agents currently in clinical trials could provide good options for this patient. Sirolimus, the mechanistic target of rapamycin inhibitor, is being investigated as a method of increasing HbF expression. Sirolimus appears to upregulate the expression of HbF in erythroid cell cultures derived from patients with β-thalassemia, as well as sickle cell patients, and may increase clearance of α-globin in RBC precursors.25-28 The latter finding may have the potential to reduce ineffective erythropoiesis separate from HbF induction. In this context, 2 ongoing phase 2 trials of sirolimus in patients with TDT (NCT03877809, NCT04247750) will track markers of ineffective erythropoiesis in addition to the primary end point of HbF induction.
Another pathway for HbF induction may be phosphodiesterase 9 inhibition with IMR-687.29,30 A phase 2 trial of IMR-687 for both patients with TDT and NTDT is currently under way (NCT04411082). Benserazide, which is currently used in combination with levodopa for the treatment of Parkinson disease, showed promising induction of HbF, although the study was not of thalassemia models,31 but it did not induce HbF in patients on this medication for Parkinson disease.32 A phase 1 trial of benserazide in patients with NTDT is ongoing (NCT04432623).
Mitapivat (formerly AG-348), an allosteric activator RBC-specific pyruvate kinase, represents a distinct and novel mechanism. RBC-specific pyruvate kinase activation increases adenosine triphosphate synthesis, as well as reduces the production of reactive oxygen species and concentration of 2,3-diphosphoglycerate.33 Preliminary results of a phase 2 trial of mitapivat in α and β NTDT showed a response, defined as an increase in hemoglobin of at least 1 g/dL in 16 of 20 patients, including all 5 patients with α- thalassemia.34 Phase 3 studies of mitapivat in TDT (NCT04770779) and NTDT (NCT04770753) patients are under way.35 Like the phase 2 trial, these trials include patients with α-thalassemia, a patient population that is underserved but outside the scope of this review.
Case 4 is a reminder that even in well-managed patients, there is significant room for improvement in care. All 4 patients presented highlight the nuanced needs of patients with thalassemia and the importance of tailoring therapeutic choices to the individual as new treatments emerge. Although the medications and interventions reviewed here are grounded in strong preclinical research on thalassemia, the results of these trials are needed to understand which agents will be clinically impactful and which agents will not be fruitful for patient care. If early studies are borne out, it is likely that both patients with TDT and NTDT will have several options beyond transfusion and iron chelation in the coming years.
Conflict-of-interest disclosure
Arielle L. Langer: no competing financial interests to declare.
Erica B. Esrick: steering committee (consulting) for bluebird bio and research funding to institution from Celgene (site for luspatercept trial).
Off-label drug use
Arielle L. Langer: nothing to disclose. The majority of the drugs discussed are not yet approved.
Erica B. Esrick: nothing to disclose. The majority of the drugs discussed are not yet approved.
References
- 1.Cappellini MD, Viprakasit V, Taher AT, et al; BELIEVE Investigators. A phase 3 trial of luspatercept in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2020;382(13):1219-1231. doi: 10.1056/NEJMoa1910182. [DOI] [PubMed] [Google Scholar]
- 2.Taher AT, Porter JB, Hermine O, et al.. Fewer red blood cell transfusion units and visits across baseline transfusion burden levels in patients with beta-thalassemia treated with luspatercept in the phase 3 believe trial. Paper presented at the 26th Congress of the European Hematology Association (EHA), June 9-17, 2021. Abstract 325024. [Google Scholar]
- 3.Piga A, Tartaglione I, Gamberini R, et al.. Long-term efficacy and safety outcomes in the phase 2 study of luspatercept in Β-thalassemia. Paper presented at the 26th Congress of the European Hematology Association (EHA), June 9-17, 2021. Abstract EP1304. [Google Scholar]
- 4.Piga A, Perrotta S, Gamberini MR, et al.. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with β-thalassemia. Blood. 2019;133(12):1279-1289. doi: 10.1182/blood-2018-10-879247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taher AT, Cappellini MD, Kattamis A, et al.. The beyond study: results of a phase 2, double-blind, randomized, placebo-controlled multicenter study of luspatercept in adult patients with non-tranfusion dependent beta-thalassemia. Paper presented at the 26th Congress of the European Hematology Association (EHA), June 9-17, 2021. Abstract S101. [Google Scholar]
- 6.Hermine O, Cappellini MD, Taher AT, et al.. Longitudinal effect of luspatercept treatment on iron overload and iron chelation therapy (ICT) in adult patients (Pts) with β-thalassemia in the believe trial. Blood. 2020;136(suppl 1): 47-48. doi: 10.1182/blood-2020-136517. [DOI] [Google Scholar]
- 7.Baronciani D, Angelucci E, Potschger U, et al.. Hemopoietic stem cell transplantation in thalassemia: a report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000-2010. Bone Marrow Transplant. 2016;51(4):536-541. doi: 10.1038/bmt.2015.293. [DOI] [PubMed] [Google Scholar]
- 8.Thompson AA, Walters MC, Kwiatkowski J, et al.. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018;378(16):1479-1493. doi: 10.1056/NEJMoa1705342. [DOI] [PubMed] [Google Scholar]
- 9.Locatelli F, Kwiatkowski JL, Walters MC, et al.. Betibeglogene autotemcel in patients with transfusion-dependent β-thalassemia: updated results from HGB-207 (NORTHSTAR-2) and HGB-212 (NORTHSTAR-3). Paper presented at the 26th Congress of the European Hematology Association (EHA), June 9-17, 2021. Abstract S266. [Google Scholar]
- 10.Yannaki E, Locatelli F, Kwiatkowski JL, et al.. Betibeglogene autotemcel gene therapy for the treatment of transfusion-dependent β-thalassemia: updated long-term efficacy and safety results. Paper presented at the 26th Congress of the European Hematology Association (EHA), June 9-17, 2021. Abstract S267. [Google Scholar]
- 11.Kulozik AE, Thuret I, Kwiatkowski JL, et al.. Interim results of betibeglogene autotemcel gene therapy in pediatric patients with transfusion-dependent Β-thalassemia (TDT) treated in the phase 3 NORTHSTAR-2 and NORTHSTAR-3 studies. Paper presented at the 26th Congress of the European Hematology Association (EHA), June 9-17, 2021. Abstract EP1301. [Google Scholar]
- 12.Marktel S, Scaramuzza S, Cicalese MP, et al.. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat Med. 2019;25(2):234-241. doi: 10.1038/s41591-018-0301-6. [DOI] [PubMed] [Google Scholar]
- 13.Esrick EB, Lehmann LE, Biffi A, et al.. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N Engl J Med. 2021;384(3):205-215. doi: 10.1056/NEJMoa2029392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frangoul H, Altshuler D, Cappellini MD, et al.. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med. 2021;384(3):252-260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 15.Locatelli F, Ailinca-Luchian S, Bobruff Y, et al.. CTX001 for transfusion-dependent β-thalassemia: safety and efficacy results from the ongoing CLIMB THAL-111 study of autologous CRISPR-CAS9-modified CD34+ hematopoietic stem and progenitor cells. Paper presented at the 26th Congress of the European Hematology Association (EHA), June 9-17, 2021. Abstract EP733. [Google Scholar]
- 16.Smith AR, Schiller GJ, Vercellotti GM, et al.. Preliminary results of a phase 1/2 clinical study of zinc finger nuclease-mediated editing of BCL11A in autologous hematopoietic stem cells for transfusion-dependent beta thalassemia. Blood. 2019;134(suppl 1):3544-3544. doi: 10.1182/blood-2019-125743. [DOI] [Google Scholar]
- 17.Hsieh MM, Bonner M, Pierciey FJ, et al.. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020;4(9):2058-2063. doi: 10.1182/bloodadvances.2019001330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.bluebird bio Provides updated findings from reported case of acute myeloid leukemia (AML) in LentiGlobin for Sickle Cell Disease (SCD) Gene Therapy Program—bluebird bio, Inc. Accessed 6 July 2021. https://investor.bluebirdbio.com/news-releases/news-release-details/bluebird-bio-provides-updated-findings-reported-case-acute
- 19.Gelderman MP, Baek JH, Yalamanoglu A, et al.. Reversal of hemochromatosis by apotransferrin in non-transfused and transfused Hbbth3/+ (heterozygous B1/B2 globin gene deletion) mice. Haematologica. 2015;100(5):611-622. doi: 10.3324/haematol.2014.117325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Choesang T, Bao W, et al.. Decreasing TfR1 expression reverses anemia and hepcidin suppression in β-thalassemic mice. Blood. 2017;129(11):1514-1526. doi: 10.1182/blood-2016-09-742387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garbowski MW, Evans P, Vlachodimitropoulou E, Hider R, Porter JB. Residual erythropoiesis protects against myocardial hemosiderosis in transfusion-dependent thalassemia by lowering labile plasma iron via transient generation of apotransferrin. Haematologica. 2017;102(10):1640-1649. doi: 10.3324/haematol.2017.170605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manolova V, Nyffenegger N, Flace A, et al.. Oral ferroportin inhibitor ameliorates ineffective erythropoiesis in a model of β-thalassemia. J Clin Invest. 2019;130(1):491-506. doi: 10.1172/JCI129382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt PJ, Fleming MD. Modulation of hepcidin as therapy for primary and secondary iron overload disorders: preclinical models and approaches. Hematol Oncol Clin North Am. 2014;28(2):387-401. doi: 10.1016/j.hoc.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arezes J, Foy N, McHugh K, et al.. Antibodies against the erythroferrone N-terminal domain prevent hepcidin suppression and ameliorate murine thalassemia. Blood. 2020;135(8):547-557. doi: 10.1182/blood.2019003140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fibach E, Bianchi N, Borgatti M, et al.. Effects of rapamycin on accumulation of alpha-, beta- and gamma-globin mRNAs in erythroid precursor cells from beta-thalassaemia patients. Eur J Haematol. 2006;77(5):437-441. doi: 10.1111/j.1600-0609.2006.00731.x. [DOI] [PubMed] [Google Scholar]
- 26.Mischiati C, Sereni A, Lampronti I, et al.. Rapamycin-mediated induction of gamma-globin mRNA accumulation in human erythroid cells. Br J Haematol. 2004;126(4):612-621. doi: 10.1111/j.1365-2141.2004.05083.x. [DOI] [PubMed] [Google Scholar]
- 27.Pecoraro A, Troia A, Calzolari R, et al.. Efficacy of rapamycin as inducer of Hb F in primary erythroid cultures from sickle cell disease and β-thalassemia patients. Hemoglobin. 2015;39(4):225-229. doi: 10.3109/03630269.2015.1036882. [DOI] [PubMed] [Google Scholar]
- 28.Lechauve C, Keith J, Khandros E, et al.. The autophagy-activating kinase ULK1 mediates clearance of free α-globin in β-thalassemia. Sci Transl Med. 2019;11(506):eaav4881. doi: 10.1126/scitranslmed.aav4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Almeida CB, Traina F, Lanaro C, et al.. High expression of the cGMP-specific phosphodiesterase, PDE9A, in sickle cell disease (SCD) and the effects of its inhibition in erythroid cells and SCD neutrophils. Br J Haematol. 2008;142(5):836-844. doi: 10.1111/j.1365-2141.2008.07264.x. [DOI] [PubMed] [Google Scholar]
- 30.McArthur JG, Svenstrup N, Chen C, et al.. A novel, highly potent and selective phosphodiesterase-9 inhibitor for the treatment of sickle cell disease. Haematologica. 2020;105(3):623-631. doi: 10.3324/haematol.2018.213462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pace BS, Perrine S, Li B, et al.. Benserazide racemate and enantiomers induce fetal globin gene expression in vivo: studies to guide clinical development for beta thalassemia and sickle cell disease. Blood Cells Mol Dis. 2021;89:102561. doi: 10.1016/j.bcmd.2021.102561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santos MEHP, Olops L, Vendrame F, et al.. Benserazide as a potential novel fetal hemoglobin inducer: an observational study in non-carriers of hemoglobin disorders. Blood Cells Mol Dis. 2021;87:102511. doi: 10.1016/j.bcmd.2020.102511. [DOI] [PubMed] [Google Scholar]
- 33.Kung C, Hixon J, Kosinski PA, et al.. AG-348 enhances pyruvate kinase activity in red blood cells from patients with pyruvate kinase deficiency. Blood. 2017;130(11):1347-1356. doi: 10.1182/blood-2016-11-753525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuo KHM, Layton DM, Lal A, et al.. Results from a phase 2, open-label, multicenter study of the oral pyruvate kinase activatorr mitapivat in adults with non–transfusion-dependent alpha- or beta-thalassemia. Paper presented at the 26th Congress of the European Hematology Association (EHA), June 9-17, 2021. Abstract S267. [Google Scholar]
- 35.Kuo KHM, Layton DM, Al-Samkari H, et al.. ENERGIZE and ENERGIZE-T: two phase 3, randomized, double-blind, placebo-controlled studies of mitapivat in adults with non–transfusion-dependent or transfusion-dependent alpha- or beta-thalassemia. Paper presented at the 26th Congress of the European Hematology Association (EHA), June 9-17, 2021. Abstract PB1805. [Google Scholar]
- 36.Cappellini MD, Porter J, Origa R, et al.. Sotatercept, a novel transforming growth factor β ligand trap, improves anemia in β-thalassemia: a phase II, open-label, dose-finding study. Haematologica. 2019;104(3):477-484. doi: 10.3324/haematol.2018.198887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humbert M, McLaughlin V, Gibbs JSR, et al; PULSAR Trial Investigators. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med. 2021;384(13):1204-1215. doi: 10.1056/NEJMoa2024277. [DOI] [PubMed] [Google Scholar]
- 38.Taher AT, Karakas Z, Cassinerio E, et al.. Efficacy and safety of ruxolitinib in regularly transfused patients with thalassemia: results from a phase 2a study. Blood. 2018;131(2):263-265. doi: 10.1182/blood-2017-06-790121. [DOI] [PMC free article] [PubMed] [Google Scholar]