

Abstract
Background:
Dyskeratosis congenita (DC) is a rare disease and is a heterogenous disorder, with its inheritance patterns as autosomal dominant, autosomal recessive, and X-linked recessive. This disorder occurs due to faulty maintenance of telomeres in stem cells. This congenital condition is diagnosed with three symptoms: oral leukoplakia, nail dystrophy, and abnormal skin pigmentation. However, because it has a wide range of symptoms, it may have phenotypes similar to other diseases. For this reason, it is necessary to use methods of measuring the Telomere Length (TL) and determining the shortness of the telomere in these patients so that it can be distinguished from other diseases. Today, the Next Generation Sequencing technique accurately detects mutations in the target genes.
Aim:
This work aims to review and summarize how each of the DC genes is involved in TL, and how to diagnose and differentiate the disease using clinical signs and methods to measure TL. It also offers treatments for DC patients, such as Hematopoietic Stem Cell Transplantation and Androgen therapy.
Relevance for Patients:
In DC patients, the genes involved in telomere homeostasis are mutated. Because these patients may have an overlapping phenotype with other diseases, it is best to perform whole-exome sequencing after genetics counseling to find the relevant mutation. As DC is a multi-systemic disease, we need to monitor patients frequently through annual lung function tests, ultrasounds, gynecological examinations, and skin examinations.
Keywords: dyskeratosis congenita, shelterin, telomerase, diagnosis, treatment
1. Introduction
Vertebrate telomeres are a hexanucleotide complex of TTAGGG repeats, with the range between 10 to 15 kb in humans and 25 to 50 kb at the ends of mouse chromosomes [1]. Telomeres are specialist nucleoprotein structures at the extremities of chromosomes that protect the natural chromosomal end against the signaling and DNA repair activities produced at the end created by the DNA cleavage [2]. Telomeres are due to incomplete amplification of linear DNA molecules by DNA polymerases shrink with each cell division, which is called the telomere replication problem [3]. The longest telomere length (TL) is at the beginning of birth that gradually diminishes with age. Because of the end proliferation problem, telomeres are shortened at each cell division, with the human cells proliferation repeating regions of telomeres with each cell division at the 3′ chromosome end and their telomeres gradually shortened [4]. The mechanism to counteract the problem of ultimate replication is reduced to 200–50 bp. Eventually, telomeres reach an important threshold, leading to an irreversible stop in growth, a phenomenon known as cell senescence. Short telomeres activate a control point depending on p53 leading to apoptosis or senescence. Telomere shortening as a major molecular factor plays an important function in the aging phenomenon. Thus, telomeres act as “clocks” that determine the lifetime of the cell surface. The emergence of telomeres can cause short-lived telomeres that bring about cell aging or cellular crisis, including apoptosis, genomic instability, and shortening cell lifespan [5]. The question remains as to how the ends of the conserved chromosomes can lead to the discovery of telomeres and telomerase. Research in this area has led to the identification of new age-related diseases during the past few years with the identification of a group of telomeric mediated syndromes [6]. Shortened telomeres can be extended by telomerase, a ribonucleoprotein complex, consisting of telomerase RNA component (TERC) and telomerase reverse transcription (TERT) and shelterin [7].
1.1. Telomerase
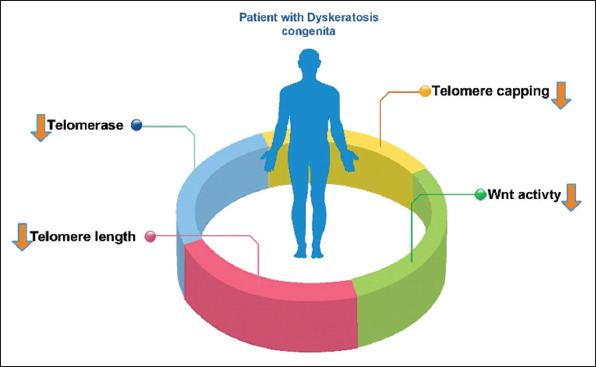
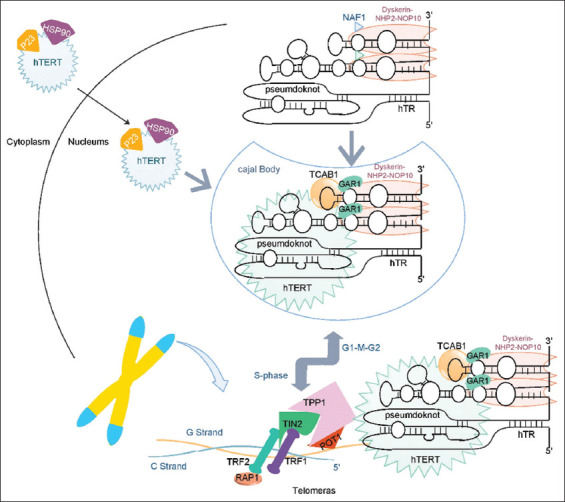
The telomere maintenance requires the aggregation of the protein components, RNA from telomerase, and telomerase maturation and stability factors. Telomerase compensates for telomere aberration by adding TTAGGG repeats to the end of chromosomes in normally expressed cells, such as pluripotent stem cells. Telomerase is composed of a TERT and a TERC, which serve as a model for the de novo addition of telomere repetitions. Telomerase, which can produce new telomeres, solves the problems of ultimate proliferation [8]. Telomerase biogenesis requires the assembling of TERT and TERC in a stable complex that can function in telomeres. TERT after synthesis into the cytoplasm with HSP 90 and P23, which penetrates the nucleus by its nuclear location signal. On the other hand, telomerase RNA (TR) synthesis in the nucleus binds to the NOP10, NHP2, GAR1, and dyskerin complexes (TERC complex). TERT and TERC are then assembled to form the final telomerase. The telomerase enters the Cajal Body (CB) through the CAB box in TR. In the S phase of the cellular cycle, the telomerase is connected to the TPP1 in the Shelterin complex through the TEN domain in TERT and enters the telomere. In other cell cycle phases, it is separated from the telomere [9]. A specific reverse transcriptase can help to extend the 3′ end of the chromosomes from the telomerase complex. Disruptions in telomerase activity can damage the chromosome and cause the cell to become cancerous. Telomere dysfunction is also associated with other conditions including dyskeratosis congenita (DC) (Figure 1), Hoyeraal-Hreidarsson (HH) syndrome, and Idiopathic Pulmonary Fibrosis (IPF) [10].
Figure 1. An overview of what happened to dyskeratosis congenita patients.
1.2. Shelterin
The telomerase enzyme protects the TL of DNA. The shelterin complex accumulates along the telomere, holds the telomere, and regulates telomere protection and regulation of TL and formation of T-loop [11]. Shelterin comprises of six proteins that include Telomeric Repeat binding Factor1 (TRF1), Telomeric Repeat binding Factor2 (TRF2), TRF1-Interacting Nuclear protein2 (TIN2/TINF2), ACD/TPP1, TERF2 Interacting Protein (TERF2IP/RAP1), and Protection of Telomer1 (POT1), which prevent telomeres from binding to each other. Shelterin detects the TTAGGG repeat through TRAF1,2 (which connects to the double-strand part of the telomere) and POT1 (which connects to the single-strand part of the telomere D-loop). One of the functions of Shelterin is to regulate telomerase activity. TPP1 is an interaction with the telomerase. Other factors, such as CB and tankyrase1 and 2, also positively affect telomerase activity. By increasing the TTAGGG repeat and the length of ssDNA, POT1 binds to these repeats which leads to a negative regulator of telomerase (Figure 2). Another function of the shelterin is the prevention of the response of DNA damage response. These signals, generated when DNA is damaged, increase Non-Homologous End Joining (NHEJ) in the cell [12]. In Mouse models, it has been shown that the absence of TRF2 and RAD1 can cause more NHEJ. Therefore, shelterin plays a role in the inhibition of NHEJ [13].
Figure 2. Telomere assembly and recruitment. TERT, after being synthesized in the cytoplasm enters the nucleus with the chaperones. TR is made in the nucleus but is not expressed because it is a non-coding RNA. TR has different domains to which NOP10, NHP2, Dyskerin, and GAR1 proteins bind and form the TERC complex. TERT and TERC are then entered into CB by TCAB1. In CB, telomerase is formed and matures. In the S phase of the cell cycle for telomere lengthening, telomerase is recruited to telomere by TPP1 Shelterin complex (the shelterin complex consists of TPP1, POT1, TRF1, TRF2, TIN2, Rap1 proteins). Telomerase, along with the shelterin, adds TTAGGG repetitions to the telomere. Once the TL is sufficient, the negative feedback generated by the POT1 disables the telomerase and re-enters the CB.
1.3. DC
DC is a rare illness with a prevalence of one in one million. A total of 400 families worldwide are reported to have been affected by the DC [14], but this complex disorder accounts for about 1% of all telomere syndromes. This congenital condition can be inherited as autosomal recessive (AR), autosomal dominant (AD), and X-Link recessive (XLR). This is a premature aging syndrome with short telomeres. Telomere dysfunction can be caused by telomere loss, such as telomerase deficiency or loss, leading to inappropriate activation of DNA repair pathways [6]. The decrease in TL generally shows its symptoms in the skin, blood, and nails because these tissues are highly renewable. This renewability depends on the stem cells in these tissues. In stem cells, telomerase activity is high (TLs are almost constant). Therefore, any defect in telomerase or other factors that affect TL will damage stem cells and impair tissue renewability [15]. DC is a disorder of several systems in its classical form with triple skin abnormalities, nails and leukoplakia of the mouth (white spots on the inside of the mouth) or the presence of one of the features of the triplet in combination with BM failure and two other related findings [16] (Figure 3). The main reason for death among these patients is BMF, although PF, cancer, and hepatic cirrhosis are secondary causes of death [17]. In genetic terms, DC is almost equally heterogenous. Mutations directly involved in DC have been identified in the DKC1, TERC, TERT, NOP10, NHP2, PARN, RTEL1, TPP1, TCAB1, CTC1, POT1, STN1, and TINF2 genes [18] (Table 1).
Figure 3. DC is diagnosed by triad significant symptoms of oral leukoplakia, dysplastic nail, reticular pigmentation. Having two major signs or one major sign and two or more minor signs (written in the figure) can diagnose DC. Because DC causes a wide range of symptoms, it needs careful diagnosis because it overlaps with the symptoms of other diseases.
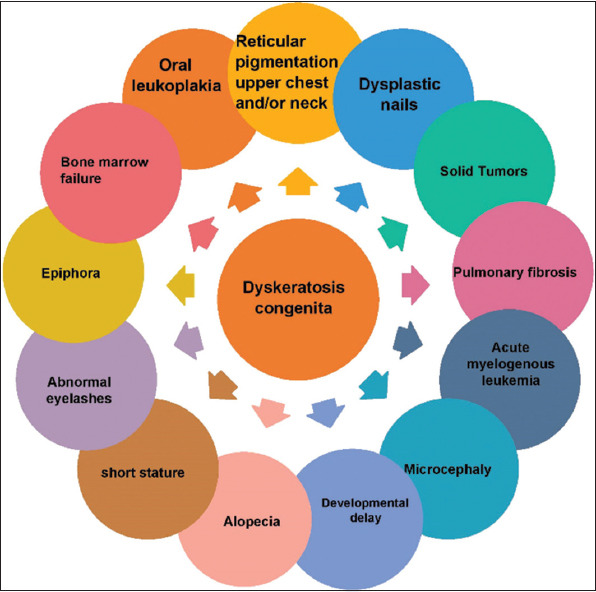
Table 1. Dyskeratosis congenita genes and related disorders (OMIM).
| GENE/OMIM NUMBER | Protein function | Disorders/Inheritance |
|---|---|---|
| TERT (187270) | Reverse transcriptase in telomerase | DC (AD, AR) Leukemia-acute myeloid (AD) Pulmonary fibrosis and/or bone marrow failure (AD) |
| TERC (602322) | RNA template in telomerase for addition telomeric repeat | Aplastic anemia (AD) Pulmonary fibrosis idiopathic (AD) DC (AD) |
| DKC1 (300126) | Component of telomerase | DC (X-linked) |
| NOP10/NOLA3 (606471) | Component of telomerase | DC (AR) |
| NHP2/NOLA2 (606470) | Component of telomerase | DC (AR) |
| TINF2/TIN2 (604319) | Component of shelterin | DC (AD) Revesz syndrome (AD) |
| TPP1/ACD (609377) | Component of shelterin | DC (AD, AR) |
| TCAB1 (612661) | Recruitment telomerase to Cajal body | DC (AR) |
| RTEL1 (608833) | Prevent the formation of the second structure in G-overhang and T-loop unwinding | DC (AD, AR) Pulmonary fibrosis and/or bone marrow failure (AD) |
| PARN (604212) | Degrades poly (A) tail in 3′ end TR | DC (AR) Pulmonary fibrosis and/or bone marrow failure (AD) |
| STN1 (613128) | Component of the CST complex | DC (AR) Cerebroretinal microangiopathy with calcifications and cysts 2 (AR) |
| CTC1 (613129) | Component of the CST complex | DC (AR) Cerebroretinal microangiopathy with calcifications and cysts (AR) |
| POT1 (606478) | Component of shelterin | DC |
2. DC Genes
2.1. TERT
TERT (also called TP2, hTRT, and DKCA2) is an abbreviated form of TERT. It is situated at 5p15.33 with 16 exons. TERT is expressed in Testis, lymph node, bone marrow, brain, skin, and 22 other tissues [19]. TERT forms the catalytic part of the telomerase, which in replication, adds TTAGGG repeats to the telomere end of each chromosome [20]. TERT has four functional areas: 1- telomerase N -terminal (TEN) domain. 2- TR -binding domain (TRBD). 3- Reverse transcriptase (RT) domain. 4- C -terminal extension (CTE) [21]. RT plays a major role in TERT. TERT binds to TR through TRBD and RT. These two areas are bound to the CR4/CR5 and pseudoknot domains in TR, respectively [22]. The TEN domain is involved in telomerase recruitment to telomeres and CTE is part of the reverse transcriptase complex [9]. In cancerous cells and stem cells, telomerase is highly active. These cells increase telomerase activity by augmenting TERT expression [23]. Higher TERT activity in cancer cells can be due to epigenetic changes, mutations in the promoter, and the impact on signaling pathways in TERT [24]. However, a decrease in telomerase activity that shortens TL due to a mutation in TERT causes DC disease. The AD mutation in TERT causes DC classic [25], but the AR mutation causes a more severe form called HH syndrome. In addition to the three symptoms of DC, intrauterine growth delay, cerebellar hypoplasia, gradual failure of the bone marrow in microcephaly, and immunodeficiency are seen in HH syndrome [26]. More severe symptoms of HH syndrome surface because the TL in these patients is much shorter than that in DC patients [27].
2.2. TERC
TERC (also called hTR, TRC3, and DKCA1) is an abbreviated form of TERC. It is located in 3q26.2 with one exon. Telomerase is a ribonucleoprotein polymerase that retains the ends of the telomere by adding TTAGGG to the telomere. The enzyme is composed of a reverse transcriptase protein component and an RNA component coded by this gene, which serves as a telomere repetition model. Deregulation of expression telomerase in somatic cells may be involved in oncogenesis [28]. TR has five important areas: 1- A template area for telomere synthesis. 2- pseudoknot domain. 3- CR4/CR5 domain. 4- H/ACA domain. 5- CR7 domain [29]. H/ACA interacts with NOP10, GAR1, NHP2, and dyskerin. H/ACA has two hairpin areas bound by the H box and ACA areas. In the H/ACA domain, there is an area called the CAB Box, which is bound to the TCAB1 and assists with telomerase localization in the CB [9]. For two reasons, the accumulation of TR in CB is implicated in the telomere lengthening, first in CB, telomerase maturation, and second in telomerase recruitment to telomeres [30].
2.3. DKC1
DKC1 (also called CBF5 and DKCX) is a shortened form of dyskerin pseudouridine synthase 1. It is located in Xq28, and it has 14 exons. DKC1 is expressed in bone marrow, lymph node, testis, skin, and 23 other tissues [31]. DKC plays a role in TR stability [9]. By binding to the H/ACA Domain, DKC prevents excessive cleavage at the TR 3′ end. Mutations in DKC reduce TERC [32]. One of the post-translational changes that occur in DKC is sumoylation, which occurs in specific DKC lysines. Reportedly, sumoylation plays a role in regulating DKC stability. Defects in sumoylation reduce DKC stability and decrease TR, shortening telomere and causing the DC [33]. Defects in DKC also disrupt the rRNA and biogenesis of the ribosome because DKC interacts with the H/ACA domain in snoRNA to form a complex that plays a role in altering certain uridines in the rRNA. DKC in this complex activates the enzyme pseudouridine synthase [34].
2.4. NOP10
NOP10 (also called DKCB1 and NOLA3(is located in 15q14 and has two exons. NOP10 is expressed in bone marrow, spleen, appendix, skin, and 23 other tissues. This gene is part of the family of H/ACA snoRNPs (small nuclear ribonucleoprotein). SnoRNPs contribute to various aspects of the treatment and modify the rRNA [35]. NOP10 plays a role in telomerase stability. Mutations in NOP10 are associated with a decrease in TERC [36].
2.5. NHP2
NHP2 (also called DKCB2 and NOLA2) is located in 5q35.3 and has four exons. It is expressed in Esophagus, brain, bone marrow, skin, and 23 other tissues. This gene belongs to the H/ACA snoRNPs (small nuclear ribonucleoprotein) gene family [37]. Mutations in NHP2 reduce TR and subsequently lower telomerase activity. NHP2, together with NOP10, GAR1, and DKC interact with snoRNA and form the snoRNP complex. This complex is involved in pseudouridylation and cleavage rRNA. Therefore, NHP2 deficiency is associated with pre-rRNA maturation defects and ribosome biogenesis. In 2020, Benyelles and et al reported the first case of HH syndrome with mutations in both NHP2 alleles [38].
2.6. TINF2
TINF2 (also called include TIN2 and DKCA3) is an abbreviated form of TERF1 interacting nuclear factor 2. It is located in 14q12 and has six exons. It is expressed in the Lymph node, adrenal, bone marrow, skin, and 23 other tissues [39]. TINF2 is one of the six proteins that make up the shelterin [34]. It also binds from C-terminal to TRF1 and from N-terminal to TRF2 and TPP1 (binding to TPP1 causes the TIN2, with indirect binding to POT1). These binds establish communication between the double-strand and single-strand telomere areas, which has a pivotal role in the stability of the Shelterin complex [40,41]. Furthermore, TINF2 binds to TRF1 in the cytoplasm to prevent ubiquitination by E3-ligase Fbx4 and its degradation. TINF2 is ubiquitinated and destroyed by E3-ligase siah2 after entering the nucleus. In general, TINF2 affects the dynamics of the Shelterin and Telomere complexes to match the telomere with the cell cycle [42]. The mutation in TINF2 disrupts the stability of the shelterin and recruitment telomerase complex [43]. Mutations in TINF2 have been seen in patients with Revesz syndrome, a severe form of DC. In addition to the triad symptoms of DC, pancytopenia and cerebellar hypoplasia can be seen in Revesz syndrome [44]. TINF2 is also involved in metabolism. In this way, in its N-terminal, it has targeting sequences for mitochondria. In mitochondria, it undergoes post-translational changes. Studies have shown that when TINF2 expression decreases with RNAi, oxidative phosphorylation is reduced and glycolysis is weakened in cancer cells [45].
2.7. TPP1
TPP1 (Also called ACD, PTOP, and TINT1) is located in 16q22.1 and has 12 exons. TPP1 is expressed in Testis, ovary, skin, bone marrow, and 23 other tissues. This gene codes for a protein associated with telomere function. This protein assembles and stabilizes this complex and facilitates telomerase access [46]. TPP1 binds from the C-terminal to the TIN2, from its central domain to the POT1 [47], and from the OB (oligosaccharide/oligonucleotide -binding) domain to the TERT in the N-terminal. In the OB domain, there is an area called TELpatch (TPP1 glutamate(E)- leucine(L)- rich patch) which is involved in telomerase activity [48]. Heterozygous mutations in TELpatch have been seen in people with HH syndrome [49]. Growth signals, which the cell responds to changes in the environment, play a role in protecting the telomere. One of the pathways involved in signal response is the PI3-K/AKT. This pathway protects telomere and prevents cellular senescence. This pathway adjusts the telomere by the impact on TPP1. Through the OB domain, TPP1 forms a homodimer. By binding to the TPP1, AKT increases homodimerization. This dimerization is essential for telomerase performance and increasing TL as regulated by AKT. During nutritional stress, AKT activity is inhibited, which disrupts the formation of the homodimer, TPP1-POT1 heterodimer, and the shelterin complex. In general, disturbance in the AKT pathway disrupts the function of the TPP1 and causes damage to the telomere [50]. Heterodimer TPP1-POT1 is involved in binding the primer (the same 3′ end) to the template strand in the TERT, which reduces the primer detachment and increases the RAP (Repeat addition processivity). On the other hand, TPP1-POT1 causes slipped DNA strand on the TR template strand, which causes repetitions to be made on DNA in a row [51]. In general, TPP1 recruits telomerase into the telomere, telomerase activity, and the negative feedback of the TL through the POT1 [52].
2.8. TCAB1
TCAB1 (also called DKCB3 and WRAP53) is an abbreviated form of Telomerase CB protein 1. It is located in 17p13.1 and has 13 exons. TCAB1 is expressed in the Lymph node, testis, bone marrow, skin, and 23 other tissues [53]. Telomerase is found in most cellular cycles in CB. Its placement in CB is determined by the interaction between CAB Box in hTR and TCAB1 in CB [54]. In the CB, telomerase matures. Thus, TCAB1 is involved in telomerase maturation and telomerase recruitment to telomere [55]. TACB1 is involved in repairing HR and NHEJ. In response to DNA damage, ATM phosphorylates TCAB1 in S64. TCAB1 forms a complex before being phosphorylated by MDC1 and RNF8. After TCAB1 is phosphorylated, the complex binds to H2AX through TCAB1 and phosphorylates it. Phosphoryl TCAB1 also causes 53BP1 to be applied to the lesion site. In general, TCAB1 initiates repair pathways in response to DNA damage [56].
2.9. RTEL1
RTEL1 (also called NHL and DKCA4) is an abbreviated regulator of telomere elongation helicase 1. It is located in 20q13.33 and has 35 exons. RTEL1 is expressed in Testis, appendix, skin, bone marrow, and 23 other tissues [57]. RTEL1, with its helicase activity, prevents the formation of the second structure in the G -overhang so that POT1 can bind to G -overhang. Since POT1 is involved in telomerase activity, then disruption of RTEL1 reduces POT1 binding and subsequently reduces telomerase activity and telomere shortening [58]. RTEL1 is involved in T-loop unwinding. TRF2 is involved in the recruitment of RTEL1 to telomere by binding to the C4C4 motif in RTEL1 in the S phase of the cell cycle. After the RTEL1 is placed in the telomere, the RTEL1 -PCNA causes the T-loop to disassemble so that there is no barrier at the end of the telomere for the replication fork. In the event of an RTEL1 dysfunction, the replication fork stops near the T-loop. Cleavage by nucleases shortens the telomere. Therefore, RTEL1 is involved in replication, telomerase activity, and telomere protection [59].
2.10. PARN
PARN (also called DAN and DKCB6) is an abbreviated poly(A)-specific ribonuclease. It is located in 16p13.12 and has 27 exons. PARN is expressed in the thyroid, testis, bone marrow, skin, and 23 other tissues [60]. Poly A tail mRNA affects stability. In any case, the stability of mRNAs decreases over time and decomposes, which is essential. Several factors are involved in the breakdown of mRNA, one of which is PARN, a deadenylase. PARN has a domain with nuclease activity and two domains bound to RNA called R3H and RRM [61]. TR is an RNA that needs to be processed. It is necessary for TR to deadenylation in the 3′ end by PARN. If the PARN is disrupted and lacks deadenylation, several adenyls attach to the 3′ end of the TR, making the TR susceptible to destruction by exonucleases such as Exosc10. In general, mutations in PARN reduce the amount of TR and impair telomerase and telomere [62].
2.11. STN1
STN1 is located in 10q24.33 and has 10 exons. STN1 is expressed in the testis, lung, esophagus, skin, and 23 other tissues [63]. The CST complex (CTC1- STN1- TEN1) binds single-stranded DNA with high affinity in a sequence-independent manner, while isolated subunits bind DNA with low affinity by themselves [64]. Initially, the CST complex has been proposed to protect telomeres from DNA degradation. However, the CST complex is involved in several aspects of telomere replication [65]. The CST complex inhibits telomerase and is involved in TL homeostasis; it is proposed to bind to newly telomerase-synthesized 3’ overhangs and terminates telomerase action implicating the association with the ACD: POT1 complex, thus interfering with its telomerase stimulation activity [66]. The CST complex is also proposed to be involved in fill-in synthesis of the telomeric C-strand, probably implicating recruitment and activation of DNA polymerase alpha [67].
2.12. POT1
POT1 is an abbreviation of protection of telomeres 1. It is located in 7q31.33 and has 23 exons. POT1 is a member of the telomere family and encodes a nuclear protein involved in telomere maintenance [68]. Specifically, this protein functions as a member of a multi-protein complex that binds to the TTAGGG repeats of telomeres, regulating TL and protecting chromosome ends from illegitimate recombination, catastrophic chromosome instability, and abnormal chromosome segregation [69].
2.13. CTC1
CTC1 gene is an abbreviated form of CST telomere replication complex component. It is located in 17p13.1 and has 23 exons. This gene is expressed in the spleen, lymph node, and 25 other tissues [70]. CTC1 encodes a component of the CST complex. Essentially, it protects telomeres from degradation [71]. The component of the CST complex acts as a specialized replication factor promoting DNA replication under conditions of replications stress or natural replication barriers such as telomere duplex. The complex is involved in TL homeostasis [72].
3. Diagnosis
3.1. Overlapping phenotype and whole-exome sequencing
To diagnose DC patients, the three main symptoms, including nail dystrophy, oral leukoplakia, and abnormal skin pigmentation, or the association of one or two of the main symptoms with other symptoms are used as demonstrated in the figure. Some people have been shown to have delayed symptoms in adulthood, a condition known as cryptic DC [6]. Diagnosing cryptic DC patients is a challenging issue because they have different symptoms. As physicians expect early symptoms for hereditary diseases, diagnosing cryptic DC may not be easily performed, but its symptoms appear in adulthood [73]. Furthermore, DC is hard to accurately diagnose because it includes a wide range of symptoms and overlapping phenotypes with other diseases. For example, in a survey of 12 families of patients with DC-like symptoms, they were initially diagnosed with DC. However, by Whole Exome Sequencing (WES), mutant genes, none of which were associated with DC but related to different syndromes. Error in diagnosis due to nail dystrophy, abnormal skin pigmentation, bone marrow failure in 60% of these patients has been reported. The genes in which the mutation was found were related to the following syndromes: 1- Poikiloderma with Neutropenia, in which patients have symptoms of short stature, nail abnormalities, and suspected infection; 2- Ectodermal dysplasia/short stature syndrome, in which patients have symptoms of short stature, nail dystrophy, and abnormal pigmentation; and 3- Dubowitz syndrome, where patients have symptoms of microcephaly, eczema, short stature, and mental retardation. The phenotype of these syndromes overlaps with the DC phenotype, which might cause misdiagnosis [74]. Besides, the mucocutaneous symptoms appear late, so the presence of cryptic DC and overlapping phenotype make the diagnosis even more difficult. Also, early treatment for DC patients is vital. Given the above, we need a method to quickly and accurately detect mutations. Today, the WES technique detects Telomere Biology Disorders (TBD) such as DC [75]. WES helps diagnose heterogeneous diseases and diseases that are difficult to diagnose on phenotypic symptoms. WES also helps physicians diagnose the disease quickly to provide the best treatment for the patient and provide more accurate genetic counseling [74].
3.2. TL measurement methods
In DC, various methods are used to measure TL such as quantitative -PCR, Terminal Restriction Fragment (TRF) analysis, Q -FISH, Single TL Analysis, Telomere Shortest Length Assay, Monochrome Multiplex -qPCR (MM-qPCR), and flow -FISH [76,77]. Peripheral Blood Leukocytes (PBL) are used to measure TL. All of the above methods have advantages and disadvantages. In most studies, two or more methods have been compared with each other in the case of DC patients. For example, two flow-FISH and MM-qPCR were used to measure TL in 105 samples in one study. People with very short TL performed the WES to identify the mutation accurately. In general, with the results of this study, flow-FISH is more accurate to measure TL, but MM-qPCR is more suitable for study on a large cohort. Flow-FISH is a method routinely used to measure TL. One of its advantages is high accuracy and sensitivity to TL changes, a low rate of false-positive results, and reproducibility. Yet, the disadvantages include the difficulty of analysis, the need for a suitable cell population, and the need for a fresh sample [76,78]. High-throughput Q -FISH is an accurate method of checking short telomeres (<3 kbp). After taking a blood sample, the PBL is purified and stored in liquid nitrogen in this method. To perform the test after thawing, the cells are counted and fixed on a plate. Fluorescent probes (CCCTAA) are then bound to the telomere repeats with high affinity. Photographs are taken using digital microscopes and evaluated by the photo analysis software. Flow-FISH, like Q-FISH, uses fluorescent probes analyzed by flow cytometry [78,79]. Q -PCR is less expensive than other methods, has higher sensitivity, and can be performed with a small amount of DNA sample in this technique. Furthermore, the size of the TL estimating is based on the Threshold Cycle (CT). The sample CT depends on the number of repetitions of the telomeres. The q -PCR result is expressed as a Telomeric repeats/Single copy gene (T/S) ratio. In fact, it is difficult to optimize this method, so standards are set for all stages to make the test more reliable [76]. TRF measures the average TL in a cell population. This method is the oldest method for measuring TL and is also the gold standard [79]. TRF has six steps:
1- DNA extraction from the sample, which can be used by phenol-chloroform or salt precipitation
2- Examining DNA integrity to ensure a good DNA sample. Run the sample together in the Gel electrophoresis. The sample that forms a long smear in the gel is not a good sample and its DNA has been decomposed.
3- DNA cleavages by restriction endonuclease enzymes, usually HinfI and RsaI. The area of repeated telomere repetitions does not require cleavage.
4- The cleavage DNA to be run on the Agarose gel.
5- Probes that have CCCTAA sequence and complement the repetitive telomere sequence to be hybridized with the telomere sequence after being labeled with digoxigenin (DIG). These probes are identified by chemiluminescence and Anti-DIG -AP.
6- Data analysis to be performed after exposure to the X-ray.
One of the disadvantages of this method is that because the short telomeres have low signal intensity in hybridization, it might not accurately detect short telomeres [80].
4. Treatment
To diagnose and manage the complications in DC patients, we need to monitor patients frequently through annual lung function tests, ultrasounds, gynecological examinations, and skin examinations [81]. As DC is a multi-systemic disease, parts of the body need to be under constant supervision [82]. Since no specific treatment can be assigned to DC, the most commonly used treatment is supportive therapy with blood transfusion. Diagnosis of genetic causes of DC has an important role in managing blood problems [83].
4.1. Immunosuppressive Therapy (IST)
IST with cyclosporine and anti-thymocyte globulin was used to treat severe Aplastic Anemia. DC patients treated with IST developed a transient response [83]. Researches have shown that BMF and immunodeficiency cause between 60% and 70% of premature deaths in DC patients. DC patients are not treated with IST [84]. Therefore, it cannot be considered a treatment for DC, but some evidence suggests an improvement in adult patients with mutations in TERT or TERC [85].
4.2. Growth factors
Granulocyte-Macrophage Colony- Stimulating Factor (GM-CSF) and erythropoietin did not show therapeutic success by affecting platelet count, but it has been suggested that combination therapy may be successful for neutropenia and anemia. Furthermore, it could be used as a Therapeutic approach for DC patients without a suitable donor [86]. Hematopoietic growth factors are sometimes used for BMF patients, but it is not advisable to use them with androgen [87].
4.3. Androgen therapy
According to studies, androgens can cause hematological responses in patients with BMF syndrome, such as DC and FA. In these, Danazol, Fluoxymesterone, and Oxymetholone can be mentioned as androgens [88,89], accounting for 70% response without transfusion dependence. The mechanism that stimulates hematopoiesis by androgens is unclear. Androgens can act by increasing telomerase activity [90] and stimulating erythropoietic stem cells, and to a lesser extent, myeloid progenitor cells stimulator in the bone marrow [91]. Recent studies have shown the effect of androgens on erythropoietin receptors (not erythropoietin itself) and receiving hematological responses [92]. Treatment with androgens has several side effects. For example, Oxymetholone can cause dyslipidemia and virilization [88]. Danazol is another synthetic androgen candidate for treating DC for its known side effects and good efficacy [83]. The estimated time for androgens can be between 1 and 3 months. Patients receiving androgens should avoid growth factors such as granulocyte-stimulating factor because of the risk of developing splenic peliosis and vascular Rupture [88].
4.4. Hematopoietic stem cell transplantation (HSCT)
Allogeneic HSCT is the only therapy that works for BMF in DC patients. In this regard, for a successful transplant, an HLA donor is needed. In the absence of a related donor, an unrelated donor can be used for transplantation. Therefore, donors should be evaluated for TL or mutations [82]. In a study for HCT, using the flow FISH method, a donor with a long telomere leukocyte was selected. They found that a donor with a long telomere increased the chances of survival and reduced the incidence of malignancies in recipients. It has also been shown that increasing each Kb of leukocyte telomere in the donor causes a reduction of 23% in the risk of death in the recipients [93]. HSCT can be associated with toxicity and high rates of premature mortality in patients. This toxicity can be due to the use of high doses of chemo-radiotherapy and chemotherapy [94]. Issues related to transplants include transplant failure, Graft versus host disease, organ toxicity such as liver cirrhosis, and pulmonary fibrosis [95], which probably causes the immunosuppressive condition, busulfan, high dose cyclophosphamide and increased use of Fludarabine [96]. Fludarabine is a type of reduced-intensity packaging diet used in HSCT with the safety suppressor effect of T-Cells in DC [97]. The use of non-myeloablative allogeneic HSCT is promising due to lower toxicity and greater success in transplantation [82]. Reduced-intensity conditioning (RIC) HSCT is a treatment to enhance the survival chance of patients with low side effects. Although there is no evidence of RIC support in DC patients, it requires less toxicity [98]. Recent reports have shown that RIC can reduce the likelihood of lung diseases without a significant effect on survival. However, evidence suggests better survival with complications in some cases [99]. A new study in patients with DC demonstrates that mesenchymal stem cells derived from the bone marrow can construct peripheral hematopoietic and may effectively manage DC patients with hematopoietic stem cell deficiency [100]. HSCTs cannot correct genetic defects such as TERT or TERC germ cell mutations related to aplastic anemia and lung fibrosis, so pulmonary complications after HSCT may be accelerated [82]. These patients are also highly vulnerable to malignancy after treatment, so contact with factors that trigger DNA damage should be minimized [101].
4.5. New treatment for mutation in PARN
TR plays a crucial role in maintaining telomeres. TR is affected by post-translational changes to maturity. One of the factors involved in the maturation of TR is PARN, which causes the deadenylation to the 3′ end of the TR. On the other hand, PAPD5 is poly (A) polymerase, and by adding adenine to the 3′ end, it causes TR to be targeted and decomposed by RNA exome, which in turn shortens telomere in stem cells. It was found in one study that PARN gene mutated cells without deadenylation in TR can be used as PAPD5 inhibitors such as BCCH001 and RG7834, which can inactivate PAPD5 to prevent the addition of poly(A) to TR and its degradation, in order to enhance telomerase activity [102].
5. Conclusion
TL homeostasis is essential for cell development in highly renewable tissues. Defects in TL homeostasis are seen in cancer and BMFs such as DC. TL is maintained by the two main components of telomerase and shelterin. Telomerase, which includes TERC and TERT, has a vital role in the TL. TERC as a template and TERT with their enzymatic activity add TTAGGG repetitions to the telomere. Mutations that increase and decrease telomerase activities are seen in cancer and DC, respectively. As DC has a wide range of phenotypes, caution needs to be practiced for an overlapping phenotype with other diseases. However, TL measurement methods can be used to distinguish it from other disorders. Today, the Next Generation Sequencing technique helps accurate diagnosis of the disease. This study addressed the role of each of the DC genes and the diagnosis of DC using clinical phenotype and TL measurement methods and treatments for DC patients.
Acknowledgments
The authors would like to express gratitude to the Department of Medical Genetics faculty, School of Advanced Technologies in Medicine, Golestan University of Medical Sciences.
Footnotes
Publisher’s note
Whioce Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Conflict of Interests
The authors declare no conflict of interests.
References
- [1].Martínez P, Blasco MA. Telomere-driven Diseases and Telomere-targeting Therapies. J Cell Biol. 2017;216:875–87. doi: 10.1083/jcb.201610111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Oganesian L, Karlseder J. Telomeric Armor:The Layers of End Protection. J Cell Sci. 2009;122:4013–25. doi: 10.1242/jcs.050567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Maestroni L, Matmati S, Coulon S. Solving the Telomere Replication Problem. Genes. 2017;8:55. doi: 10.3390/genes8020055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jafri MA, Ansari SA, Alqahtani MH, Shay JW. Roles of Telomeres and Telomerase in Cancer, and Advances in Telomerase-targeted Therapies. Genome Med. 2015;8:69. doi: 10.1186/s13073-016-0324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wai LK. Telomeres, Telomerase, and Tumorigenesis a Review. Med Gen Med. 2004;6:19. [PMC free article] [PubMed] [Google Scholar]
- [6].Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361:2353–65. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fiorini E, Santoni A, Colla S. Dysfunctional Telomeres and Hematological Disorders. Differentiation. 2018;100:1–11. doi: 10.1016/j.diff.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Huang Y, Liang P, Liu D, Huang J, Songyang Z. Telomere Regulation in Pluripotent Stem Cells. Protein Cell. 2014;5:194–202. doi: 10.1007/s13238-014-0028-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schmidt JC, Cech TR. Human Telomerase:Biogenesis, Trafficking, Recruitment, and Activation. Genes Dev. 2015;29:1095–105. doi: 10.1101/gad.263863.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Blasco MA. Telomeres and Human Disease:Ageing, Cancer and Beyond. Nat Rev Genet. 2005;6:611–22. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- [11].Diotti R, Loayza D. Shelterin Complex and Associated Factors at Human Telomeres. Nucleus. 2011;2:119–35. doi: 10.4161/nucl.2.2.15135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Denchi EL, De Lange T. Protection of Telomeres through Independent Control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–71. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- [13].Konishi A, De Lange T. Cell Cycle Control of Telomere Protection and NHEJ Revealed by a TS Mutation in the DNA-binding Domain of TRF2. Genes Dev. 2008;22:1221–30. doi: 10.1101/gad.1634008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dyskeratosis Congenita Orphanet. 2021. [Last accessed on 2021 Dec]. Available from: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=1775 .
- [15].Gu BW, Bessler M, Mason PJ. A Pathogenic Dyskerin Mutation Impairs Proliferation and Activates a DNA Damage Response Independent of Telomere Length in Mice. Proc Natl Acad Sci U S A. 2008;105:10173–8. doi: 10.1073/pnas.0803559105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Atkinson JC, Harvey KE, Domingo DL, Trujillo MI, Guadagnini JP, Gollins S, et al. Oral and Dental Phenotype of Dyskeratosis Congenita. Oral Dis. 2008;14:419–27. doi: 10.1111/j.1601-0825.2007.01394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ballew BJ, Savage SA. Updates on the Biology and Management of Dyskeratosis Congenita and Related Telomere Biology Disorders. Expert Rev Hematol. 2013;6:327–37. doi: 10.1586/ehm.13.23. [DOI] [PubMed] [Google Scholar]
- [18].Bergstrand S, Böhm S, Malmgren H, Norberg A, Sundin M, Nordgren A, et al. Biallelic Mutations in WRAP53 Result in Dysfunctional Telomeres, Cajal Bodies and DNA Repair, thereby Causing Hoyeraal-Hreidarsson Syndrome. Cell Death Dis. 2020;11:238. doi: 10.1038/s41419-020-2421-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].National Center for Biotechnology Information:TERT Telomerase Reverse Transcriptase [Homo sapiens (human)] 2020 [PubMed] [Google Scholar]
- [20].Gilson E, Géli V. How Telomeres are Replicated. Nat Rev Mol Cell Biol. 2007;8:825–38. doi: 10.1038/nrm2259. [DOI] [PubMed] [Google Scholar]
- [21].Blackburn EH, Collins K. Telomerase:An RNP Enzyme Synthesizes DNA. Cold Spring Harb Perspect Biol. 2011;3:a003558. doi: 10.1101/cshperspect.a003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lai CK, Mitchell JR, Collins K. RNA Binding Domain of Telomerase Reverse Transcriptase. Mol Cell Biol. 2001;21:990–1000. doi: 10.1128/MCB.21.4.990-1000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li Y, Tergaonkar V. Noncanonical Functions of Telomerase:Implications in Telomerase-targeted Cancer Therapies. Cancer Res. 2014;74:1639–44. doi: 10.1158/0008-5472.CAN-13-3568. [DOI] [PubMed] [Google Scholar]
- [24].Leão R, Apolónio JD, Lee D, Figueiredo A, Tabori U, Castelo-Branco P. Mechanisms of Human Telomerase Reverse Transcriptase (hTERT) Regulation:Clinical Impacts in Cancer. J Biomed Sci. 2018;25:22. doi: 10.1186/s12929-018-0422-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lamm N, Ordan E, Shponkin R, Richler C, Aker M, Tzfati Y. Diminished Telomeric 3'Overhangs are Associated with Telomere Dysfunction in Hoyeraal-Hreidarsson Syndrome. PLoS One. 2009;4:e5666. doi: 10.1371/journal.pone.0005666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Glousker G, Touzot F, Revy P, Tzfati Y, Savage SA. Unraveling the Pathogenesis of Hoyeraal-Hreidarsson Syndrome, a Complex Telomere Biology Disorder. Br J Haematol. 2015;170:457–71. doi: 10.1111/bjh.13442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere Length is Associated with Disease Severity and Declines with Age in Dyskeratosis Congenita. Haematologica. 2012;97:353–9. doi: 10.3324/haematol.2011.055269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].National Center for Biotechnology Information:TERC Telomerase RNA Component [Homo sapiens (human)] 2020 [Google Scholar]
- [29].Theimer CA, Feigon J. Structure and Function of Telomerase RNA. Curr Opin Struct Biol. 2006;16:307–18. doi: 10.1016/j.sbi.2006.05.005. [DOI] [PubMed] [Google Scholar]
- [30].Cristofari G, Adolf E, Reichenbach P, Sikora K, Terns RM, Terns MP, et al. Human Telomerase RNA Accumulation in Cajal Bodies Facilitates Telomerase Recruitment to Telomeres and Telomere Elongation. Mol Cell. 2007;27:882–9. doi: 10.1016/j.molcel.2007.07.020. [DOI] [PubMed] [Google Scholar]
- [31].National Center for Biotechnology Information:DKC1 Dyskerin Pseudouridine Synthase [Homo sapiens (human)] 2020 [Google Scholar]
- [32].Mochizuki Y, He J, Kulkarni S, Bessler M, Mason PJ. Mouse Dyskerin Mutations Affect Accumulation of Telomerase RNA and Small Nucleolar RNA, Telomerase Activity, and Ribosomal RNA Processing. Proc Natl Acad Sci U S A. 2004;101:10756–61. doi: 10.1073/pnas.0402560101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Brault ME, Lauzon C, Autexier C. Dyskeratosis Congenita Mutations in Dyskerin SUMOylation Consensus Sites Lead to Impaired Telomerase RNA Accumulation and Telomere Defects. Hum Mol Genet. 2013;22:3498–507. doi: 10.1093/hmg/ddt204. [DOI] [PubMed] [Google Scholar]
- [34].Mason PJ, Bessler M. The Genetics of Dyskeratosis Congenita. Cancer Genet. 2011;204:635–45. doi: 10.1016/j.cancergen.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].National Center for Biotechnology Information:NOP10 Ribonucleoprotein [Homo sapiens (human)] 2020 [Google Scholar]
- [36].Vulliamy T, Beswick R, Kirwan M, Marrone A, Digweed M, Walne A, et al. Mutations in the Telomerase Component NHP2 Cause the Premature Ageing Syndrome Dyskeratosis Congenita. Proc Natl Acad Sci U S A. 2008;105:8073–8. doi: 10.1073/pnas.0800042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].National Center for Biotechnology Information:NHP2 Ribonucleoprotein [Homo sapiens (human)] 2020 [Google Scholar]
- [38].Benyelles M, O'Donohue MF, Kermasson L, Lainey E, Borie R, Lagresle-Peyrou C, et al. NHP2 Deficiency Impairs rRNA Biogenesis and Causes Pulmonary Fibrosis and Høyeraal-Hreidarsson Syndrome. Hum Mol Genet. 2020;29:907–22. doi: 10.1093/hmg/ddaa011. [DOI] [PubMed] [Google Scholar]
- [39].National Center for Biotechnology Information:TINF2 TERF1 Interacting Nuclear Factor [Homo sapiens (human)] 2020 [Google Scholar]
- [40].Bianchi A, Shore D. How Telomerase Reaches its End:Mechanism of Telomerase Regulation by the Telomeric Complex. Mol Cell. 2008;31:153–65. doi: 10.1016/j.molcel.2008.06.013. [DOI] [PubMed] [Google Scholar]
- [41].Ye JZ, Donigian JR, Van Overbeek M, Loayza D, Luo Y, Krutchinsky AN, et al. TIN2 binds TRF1 and TRF2 Simultaneously and Stabilizes the TRF2 Complex on Telomeres. J Biol Chem. 2004;279:47264–71. doi: 10.1074/jbc.M409047200. [DOI] [PubMed] [Google Scholar]
- [42].Bhanot M, Smith S. TIN2 Stability is Regulated by the E3 Ligase Siah2. Mol Cell Biol. 2012;32:376–84. doi: 10.1128/MCB.06227-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pereboeva L, Hubbard M, Goldman FD, Westin ER. Robust DNA Damage Response and Elevated Reactive Oxygen Species in TINF2-Mutated Dyskeratosis Congenita Cells. PLoS One. 2016;11:e0148793. doi: 10.1371/journal.pone.0148793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 Mutations Result in Very Short Telomeres:Analysis of a Large Cohort of Patients with Dyskeratosis Congenita and Related Bone Marrow Failure Syndromes. Blood. 2008;112:3594–600. doi: 10.1182/blood-2008-05-153445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen LY, Zhang Y, Zhang Q, Li H, Luo Z, Fang H, et al. Mitochondrial Localization of Telomeric Protein TIN2 Links Telomere Regulation to Metabolic Control. Mol Cell. 2012;47:839–50. doi: 10.1016/j.molcel.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].National Center for Biotechnology Information:ACD ACD Shelterin Complex Subunit and Telomerase Recruitment Factor [Homo sapiens (human)] 2020 [Google Scholar]
- [47].Liu D, Safari A, O'Connor MS, Chan DW, Laegeler A. PTOP Interacts with POT1 and Regulates its Localization to Telomeres. Nat Cell Biol. 2004;6:673–80. doi: 10.1038/ncb1142. [DOI] [PubMed] [Google Scholar]
- [48].Bisht K, Smith EM, Tesmer VM, Nandakumar J. Structural and Functional Consequences of a Disease Mutation in the Telomere Protein TPP1. Proc Natl Acad Sci U S A. 2016;113:13021–6. doi: 10.1073/pnas.1605685113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kocak H, Ballew BJ, Bisht K, Eggebeen R, Hicks BD, O'Neil A, et al. Hoyeraal-Hreidarsson Syndrome Caused by a Germline Mutation in the TEL Patch of the Telomere Protein TPP1. Genes Dev. 2014;28:2090–102. doi: 10.1101/gad.248567.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Han X, Liu D, Zhang Y, Li Y, Lu W, Chen J, et al. Akt Regulates TPP1 Homodimerization and Telomere Protection. Aging Cell. 2013;12:1091–9. doi: 10.1111/acel.12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Latrick CM, Cech TR. POT1-TPP1 Enhances Telomerase Processivity by Slowing Primer Dissociation and Aiding Translocation. EMBO J. 2010;29:924–33. doi: 10.1038/emboj.2009.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sexton AN, Regalado SG, Lai CS, Cost GJ, O'Neil CM, Urnov FD, et al. Genetic and Molecular Identification of Three Human TPP1 Functions in Telomerase Action:Recruitment, Activation, and Homeostasis Set Point Regulation. Genes Dev. 2014;28:1885–99. doi: 10.1101/gad.246819.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].National Center for Biotechnology Information:WRAP53 WD Repeat Containing Antisense to TP53 [Homo sapiens (human)] 2020 [Google Scholar]
- [54].Venteicher AS, Artandi SE. TCAB1:Driving Telomerase to Cajal Bodies. Cell Cycle. 2009;8:1329–31. doi: 10.4161/cc.8.9.8288. [DOI] [PubMed] [Google Scholar]
- [55].Stern JL, Zyner KG, Pickett HA, Cohen SB, Bryan TM. Telomerase Recruitment Requires Both TCAB1 and Cajal Bodies Independently. Mol Cell Biol. 2012;32:2384–95. doi: 10.1128/MCB.00379-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Coucoravas C, Dhanjal S, Henriksson S, Böhm S, Farnebo M. Phosphorylation of the Cajal Body Protein WRAP53βby ATM Promotes its Involvement in the DNA Damage Response. RNA Biol. 2017;14:804–13. doi: 10.1080/15476286.2016.1243647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].National Center for Biotechnology Information:RTEL1 Regulator of Telomere Elongation Helicase 1 [Homo sapiens (human)] 2020 [Google Scholar]
- [58].Porreca RM, Glousker G, Awad A, Fernandez MI, Gibaud A, Naucke C, et al. Human RTEL1 Stabilizes Long G-overhangs Allowing Telomerase-dependent Over-extension. Nucleic Acids Res. 2018;46:4533–45. doi: 10.1093/nar/gky173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sarek G, Vannier JB, Panier S, Petrini JH, Boulton SJ. TRF2 Recruits RTEL1 to Telomeres in S Phase to Promote T-Loop Unwinding. Mol Cell. 2015;57:622–35. doi: 10.1016/j.molcel.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].National Center for Biotechnology Information:PARN Poly (A)-specific Ribonuclease [Homo sapiens (human)] 2020 [Google Scholar]
- [61].Balatsos NA, Maragozidis P, Anastasakis D, Stathopoulos C. Modulation of Poly(A)-specific Ribonuclease (PARN):Current Knowledge and Perspectives. Curr Med Chem. 2012;19:4838–49. doi: 10.2174/092986712803341539. [DOI] [PubMed] [Google Scholar]
- [62].Shukla S, Parker R. PARN Modulates Y RNA Stability and its 3'-End Formation. Mol Cell Biol. 2017;37:e00264–17. doi: 10.1128/MCB.00264-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].National Center for Biotechnology Information:STN1 Subunit of CST Complex [Homo sapiens (human)] 2020 [Google Scholar]
- [64].Lue NF, Zhou R, Chico L, Mao N, Steinberg-Neifach O, Ha T. The Telomere Capping Complex CST Has an Unusual Stoichiometry, Makes Multipartite Interaction with G-Tails, and Unfolds Higher-Order G-Tail Structures. PLoS Genet. 2013;9:e1003145. doi: 10.1371/journal.pgen.1003145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Huang C, Dai X, Chai W. Human Stn1 Protects Telomere Integrity by Promoting Efficient Lagging-strand Synthesis at Telomeres and Mediating C-strand Fill-in. Cell Res. 2012;22:1681–95. doi: 10.1038/cr.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].STN1 Function. 2021. [Last accessed on 2021 Dec]. Available from: https://www.nextprot.org/entry/NX_Q9H668 .
- [67].Feng X, Hsu SJ, Kasbek C, Chaiken M, Price CM. CTC1-mediated C-strand Fill-in is an Essential Step in Telomere Length Maintenance. Nucleic Acids Res. 2017;45:4281–93. doi: 10.1093/nar/gkx125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].National Center for Biotechnology Information:Protection of Telomeres [Homo sapiens (human)] 2020 [Google Scholar]
- [69].Mir SM, Tehrani SS, Goodarzi G, Jamalpoor Z, Asadi J, Khelghati N, et al. Shelterin Complex at Telomeres:Implications in Ageing. Clin Interv Aging. 2020;15:827–39. doi: 10.2147/CIA.S256425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].National Center for Biotechnology Information:CST Telomere Replication Complex Component [Homo sapiens (human)] 2020 [Google Scholar]
- [71].Lim CJ, Cech TR. Shaping Human Telomeres:From Shelterin and CST Complexes to Telomeric Chromatin Organization. Nat Rev Mol Cell Biol. 2021;22:283–98. doi: 10.1038/s41580-021-00328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Stewart JA, Wang Y, Ackerson SM, Schuck PL. Emerging Roles of CST in Maintaining Genome Stability and Human Disease. Front Biosci (Landmark Ed) 2018;23:1564–86. doi: 10.2741/4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kirwan M, Dokal I. Dyskeratosis Congenita:A Genetic Disorder of Many Faces. Clin Genet. 2008;73:103–12. doi: 10.1111/j.1399-0004.2007.00923.x. [DOI] [PubMed] [Google Scholar]
- [74].Dorgaleleh S, Naghipoor K, Hajimohammadi Z, Oladnabi M. Molecular Basis of Ectodermal Dysplasia:A Comprehensive Review of the Literature. Egypt J Dermatol Venereol. 2021;41:55–66. [Google Scholar]
- [75].Trotta L, Norberg A, Taskinen M, Béziat V, Degerman S, Wartiovaara-Kautto U, Välimaa H, et al. Diagnostics of Rare Disorders:Whole-exome Sequencing Deciphering Locus Heterogeneity in Telomere Biology Disorders. Orphanet J Rare Dis. 2018;13:139. doi: 10.1186/s13023-018-0864-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ferreira MS, Kirschner M, Halfmeyer I, Estrada N, Xicoy B, Isfort S, et al. Comparison of Flow-FISH and MM-qPCR Telomere Length Assessment Techniques for the Screening of Telomeropathies. Ann N Y Acad Sci. 2020;1466:93–103. doi: 10.1111/nyas.14248. [DOI] [PubMed] [Google Scholar]
- [77].Morinha F, Magalhães P, Blanco G. Standard Guidelines for the Publication of Telomere qPCR Results in Evolutionary Ecology. Mol Ecol Resour. 2020;20:635–48. doi: 10.1111/1755-0998.13152. [DOI] [PubMed] [Google Scholar]
- [78].De Pedro N, Díez M, García I, García J, Otero L, Fernández L, et al. Analytical Validation of Telomere Analysis Technology®for the High-Throughput Analysis of Multiple Telomere-Associated Variables. Biol Proc Online. 2020;22:2. doi: 10.1186/s12575-019-0115-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Aubert G, Hills M, Lansdorp PM. Telomere Length Measurement-caveats and a Critical Assessment of the Available Technologies and Tools. Mutat Res. 2012;730:59–67. doi: 10.1016/j.mrfmmm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kimura M, Stone RC, Hunt SC, Skurnick J, Lu X, Cao X, et al. Measurement of Telomere Length by the Southern Blot Analysis of Terminal Restriction Fragment Lengths. Nat Protoc. 2010;5:1596–607. doi: 10.1038/nprot.2010.124. [DOI] [PubMed] [Google Scholar]
- [81].Savage SA, Alter BP. Dyskeratosis Congenita. Hematol Oncol Clin North Am. 2009;23:215–31. doi: 10.1016/j.hoc.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].García MS, Teruya-Feldstein J. The Diagnosis and Treatment of Dyskeratosis Congenita:A Review. J Blood Med. 2014;5:157–67. doi: 10.2147/JBM.S47437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Agarwal S. Evaluation and Management of Hematopoietic Failure in Dyskeratosis Congenita. Hematol Oncol Clin North Am. 2018;32:669–85. doi: 10.1016/j.hoc.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Gadalla SM, Sales-Bonfim C, Carreras J, Alter BP, Antin JH, Ayas M, et al. Outcomes of Allogeneic Hematopoietic Cell Transplant in Patients with Dyskeratosis Congenita. Biol Blood Marrow Transplant. 2013;19:1238–43. doi: 10.1016/j.bbmt.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Comoli P, Basso S, Huanga GC. Intensive Immunosuppression Therapy for Aplastic Anemia Associated with Dyskeratosis Congenita:Report of a Case. Int J Hematol. 2005;82:35–7. doi: 10.1532/IJH97.A10416. [DOI] [PubMed] [Google Scholar]
- [86].Erduran E, Hacisalihoglu S, Ozoran Y. Treatment of Dyskeratosis Congenita with Granulocyte-macrophage Colony-stimulating Factor and Erythropoietin. J Pediatr Hematol Oncol. 2003;25:333–5. doi: 10.1097/00043426-200304000-00015. [DOI] [PubMed] [Google Scholar]
- [87].Giri N, Pitel PA, Green D, Alter BP. Splenic Peliosis and Rupture in Patients with Dyskeratosis Congenita on Androgens and Granulocyte Colony-stimulating Factor. Br J Haematol. 2007;138:815–7. doi: 10.1111/j.1365-2141.2007.06718.x. [DOI] [PubMed] [Google Scholar]
- [88].Khincha PP, Wentzensen IM, Giri N, Alter BP, Savage SA. Response to Androgen Therapy in Patients with Dyskeratosis Congenita. Br J Haematol. 2014;165:349–57. doi: 10.1111/bjh.12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Islam A, Rafiq S, Kirwan M, Walne A, Cavenagh J, Vulliamy T, et al. Haematological Recovery in Dyskeratosis Congenita Patients Treated with Danazol. Br J Haematol. 2013;162:854–6. doi: 10.1111/bjh.12432. [DOI] [PubMed] [Google Scholar]
- [90].Nobili B, Rossi G, De Stefano P, Zecca M, Giorgiani G, Perrotta S, et al. Successful Umbilical Cord Blood Transplantation in a Child with Dyskeratosis Congenita after a Fludarabine-based Reduced-intensity Conditioning Regimen. Br J Haematol. 2002;119:573–4. doi: 10.1046/j.1365-2141.2002.03835_2.x. [DOI] [PubMed] [Google Scholar]
- [91].Shahidi NT. A Review of the Chemistry, Biological Action, and Clinical Applications of Anabolic-androgenic Steroids. Clin Ther. 2001;23:1355–90. doi: 10.1016/s0149-2918(01)80114-4. [DOI] [PubMed] [Google Scholar]
- [92].Maggio M, Snyder PJ, Ceda GP, Milaneschi Y, Luci M, Cattabiani C, et al. Is the Haematopoietic Effect of Testosterone Mediated by Erythropoietin?The Results of a Clinical Trial in Older Men. Andrology. 2013;1:24–8. doi: 10.1111/j.2047-2927.2012.00009.x. [DOI] [PubMed] [Google Scholar]
- [93].Gadalla SM, Aubert G, Wang T, Haagenson M, Spellman SR, Wang L, et al. Donor Telomere Length and Causes of Death after Unrelated Hematopoietic Cell Transplantation in Patients with Marrow Failure. Blood. 2018;131:2393–8. doi: 10.1182/blood-2017-10-812735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Dokal I. Dyskeratosis Congenita. Hematol Am Soc Hematol Educ Program. 2011;2011:480–6. doi: 10.1182/asheducation-2011.1.480. [DOI] [PubMed] [Google Scholar]
- [95].Nelson AS, Marsh RA, Myers KC, Davies SM, Jodele S, O'Brien TA, et al. A Reduced-Intensity Conditioning Regimen for Patients with Dyskeratosis Congenita Undergoing Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2016;22:884–8. doi: 10.1016/j.bbmt.2016.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Al-Rahawan MM, Giri N, Alter BP. Intensive Immunosuppression Therapy for Aplastic Anemia Associated with Dyskeratosis Congenita. Int J Hematol. 2006;83:275–6. doi: 10.1532/IJH97.06030. [DOI] [PubMed] [Google Scholar]
- [97].Kharfan-Dabaja MA, Otrock ZK, Bacigalupo A, Mahfouz RA, Geara F, Bazarbachi A. A Reduced Intensity Conditioning Regimen of Fludarabine Cyclophosphamide, Antithymocyte Globulin, Plus 2 Gy TBI Facilitates Successful Hematopoietic Cell Engraftment in an Adult with Dyskeratosis Congenita. Bone Marrow Transplant. 2012;47:1254–5. doi: 10.1038/bmt.2011.257. [DOI] [PubMed] [Google Scholar]
- [98].Barbaro P, Vedi A. Survival after Hematopoietic Stem Cell Transplant in Patients with Dyskeratosis Congenita:Systematic Review of the Literature. Biol Blood Marrow Transplant. 2016;22:1152–8. doi: 10.1016/j.bbmt.2016.03.001. [DOI] [PubMed] [Google Scholar]
- [99].De la Fuente J, Dokal I. Dyskeratosis Congenita:Advances in the Understanding of the Telomerase Defect and the Role of Stem Cell Transplantation. Pediatr Transplant. 2007;11:584–94. doi: 10.1111/j.1399-3046.2007.00721.x. [DOI] [PubMed] [Google Scholar]
- [100].Balakumaran A, Mishra PJ, Pawelczyk E, Yoshizawa S, Sworder BJ, Cherman N, et al. Bone Marrow Skeletal Stem/Progenitor Cell Defects in Dyskeratosis Congenita and Telomere Biology Disorders. Blood. 2015;125:793–802. doi: 10.1182/blood-2014-06-566810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Bessler M, Wilson DB, Mason PJ. Dyskeratosis Congenita. FEBS Lett. 2010;584:3831–8. doi: 10.1016/j.febslet.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Nagpal N, Wang J, Zeng J, Lo E, Moon DH, Luk K, et al. Small-Molecule PAPD5 Inhibitors Restore Telomerase Activity in Patient Stem Cells. Cell Stem Cell. 2020;26:896–909. doi: 10.1016/j.stem.2020.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]