

ABSTRACT
Alphaviruses and flaviviruses have class II fusion glycoproteins that are essential for virion assembly and infectivity. Importantly, the tip of domain II is structurally conserved between the alphavirus and flavivirus fusion proteins, yet whether these structural similarities between virus families translate to functional similarities is unclear. Using in vivo evolution of Zika virus (ZIKV), we identified several novel emerging variants, including an envelope glycoprotein variant in β-strand c (V114M) of domain II. We have previously shown that the analogous β-strand c and the ij loop, located in the tip of domain II of the alphavirus E1 glycoprotein, are important for infectivity. This led us to hypothesize that flavivirus E β-strand c also contributes to flavivirus infection. We generated this ZIKV glycoprotein variant and found that while it had little impact on infection in mosquitoes, it reduced replication in human cells and mice and increased virus sensitivity to ammonium chloride, as seen for alphaviruses. In light of these results and given our alphavirus ij loop studies, we mutated a conserved alanine at the tip of the flavivirus ij loop to valine to test its effect on ZIKV infectivity. Interestingly, this mutation inhibited infectious virion production of ZIKV and yellow fever virus, but not West Nile virus. Together, these studies show that shared domains of the alphavirus and flavivirus class II fusion glycoproteins harbor structurally analogous residues that are functionally important and contribute to virus infection in vivo.
IMPORTANCE Arboviruses are a significant global public health threat, yet there are no antivirals targeting these viruses. This problem is in part due to our lack of knowledge of the molecular mechanisms involved in the arbovirus life cycle. In particular, virus entry and assembly are essential processes in the virus life cycle and steps that can be targeted for the development of antiviral therapies. Therefore, understanding common, fundamental mechanisms used by different arboviruses for entry and assembly is essential. In this study, we show that flavivirus and alphavirus residues located in structurally conserved and analogous regions of the class II fusion proteins contribute to common mechanisms of entry, dissemination, and infectious-virion production. These studies highlight how class II fusion proteins function and provide novel targets for development of antivirals.
KEYWORDS: alphavirus, assembly, flavivirus, fusion protein, glycoprotein
INTRODUCTION
Arthropod-borne viruses (arboviruses) are a diverse group of pathogens that can cause explosive epidemics and devasting disease (1–3). Arboviruses can be transmitted by a wide variety of arthropod vectors, including mosquitoes, ticks, and sandflies (4). Importantly, vectors such as Aedes mosquitoes can transmit several pathogenic arboviruses, such as dengue virus, Zika virus (ZIKV), and chikungunya virus (CHIKV), suggesting that there may be common mechanisms of infectivity among specific arboviruses. Currently there are limited vaccines and no specific antiviral therapies targeting these viral threats, highlighting the need to better understand at the molecular level how these viruses replicate and are transmitted. In particular, identifying common mechanisms shared among arboviruses and genetically distant arbovirus families could help determine broad-spectrum characteristics needed for effective antivirals.
A feature shared by all arboviruses is their need to be transmitted from an insect vector to the human host, yet we understand little of the molecular mechanisms involved in arbovirus transmission and infectivity. To study what contributes to arbovirus transmission, we can look at West Nile virus (WNV) (5, 6), ZIKV (7), Venezuelan equine encephalitis virus (8, 9), and CHIKV (10). Natural epidemics of these viruses have identified key residues in the viral glycoproteins that promote virus transmission and infectivity. These findings along with years of extensive experimentation have defined the viral glycoproteins as critical factors for virus assembly, attachment and entry, transmission, and pathogenesis (11–17).
Alphaviruses and flaviviruses encode class II fusion proteins that are required for pH-dependent entry and fusion (18, 19). In a previous study (22), we used the in vivo evolution of CHIKV during mosquito-to-mammal transmission to identify and study key determinants for alphavirus infectivity and pathogenesis. In that study, we identified an adaptive variant (V80I) in β-strand c of the E1 glycoprotein domain II which contributes to CHIKV transmission in mosquitos and pathogenesis in mice (20). In subsequent studies, we further characterized this residue and found that V80 functions together with residue 226 on the ij loop of the tip of domain II to impact pH- and cholesterol-dependent entry (21). Importantly, the tip of domain II of the class II fusion glycoprotein is structurally conserved between alphaviruses and flaviviruses. This observation suggests that while these viruses are genetically unrelated, structural conservation in their glycoproteins could translate to functional similarities between virus families.
In this study, we used in vivo evolution of ZIKV to identify factors involved in flavivirus replication and infectivity. We identified novel variants in NS2A, NS3, and β-strand c of domain II of the ZIKV envelope protein. Given the structural similarities between domain II of the alphavirus and flavivirus fusion glycoprotein, we hypothesized that β-strand c and the ij loop may have analogous functions in both virus families. Here, we found that a highly conserved valine in the ZIKV β-strand c attenuated virus replication in A549 cells and mice and impacted pH-dependent entry. Moreover, we found that analogous residues in the tip of the envelope ij loop were important for infectious-virion production in ZIKV and yellow fever virus but not in WNV. Together, these data provide functional evidence that alphaviruses and flaviviruses use similar mechanisms and structural domains for infectivity and infectious-virion production. These studies further our understanding of arbovirus biology as well as opening new avenues for the development of antivirals targeting multiple virus families.
RESULTS
Identification of emerging ZIKV variants in the envelope, NS2A, and NS3 during vector-borne transmission to type I interferon-deficient mice.
In a previous study to understand how transmission route and organ microenvironment impacts ZIKV evolution, we dissected the in vivo evolution of the prototype ZIKV strain (MR766) in type I interferon-deficient (Ifnar1−/−) mice (22). To do this, we infected Ifnar1−/− mice either by needle inoculation in the footpad or by mosquito bite from ZIKV-infected Aedes aegypti mosquitoes, harvested organs at 7 days postinfection, and analyzed ZIKV variants by deep sequencing. Using the data from this study, we analyzed the synonymous and nonsynonymous minority variants from mice after mosquito transmission (Fig. 1). We identified unique variants in two mice (mouse A and mouse B) that had the same point mutations in multiple organs (Fig. 1A and B, boxes). In mouse A, there were two synonymous changes (C1690T [8.2 to 12.9%] and T3931C [9.4 to 17.0%]). In mouse B, there were three nonsynonymous changes (E: G1316A and V114M [8.9 to 9.2%]; NS2A: A3638G, I33V [13.1 to 16.8%]; and NS3: G4788A, R59K [19.4 to 22.7%]). When we mapped these changes onto the protein structures, we found that the EV114M mutation is located in the tip of domain II, which coincides with a highly conserved glycerophospholipid binding pocket (23) (Fig. 1C), and the R59K mutation in NS3 maps near a cellular 14-3-3 binding site (24) (Fig. 1D). Finally, the I33V mutation in NS2A is located in a putative transmembrane alpha-helix predicted to be in the endoplasmic reticulum (ER) lumen (13) (Fig. 1E).
FIG 1.

Identification of emerging ZIKV minority variants after vector-borne transmission. ZIKV MR766 vector-transmitted synonymous (A) and nonsynonymous (B) minor variants (<50%) present in individual mice (n = 3). Boxes indicate variants present in multiple organs of individual mice. (C) Structure of the tip of ZIKV envelope protein (PDB ID 5JHM) with the V114M variant in red. (D) Structure of the ZIKV NS3 protease (PDB ID 5GJ4) with the R59K mutation in red and the 14-3-3 RLDP binding motif in black. (E) Schematic representation of the ZIKV NS2A protein. The I33V mutation is depicted in red.
ZIKV E, NS3, and NS2A variants have no significant advantage in Ifnar1−/− mice and are attenuated in A549 cells.
Given that these ZIKV variants were identified in Ifnar1−/− mice, we tested whether this variant had a replication advantage in Ifnar1−/− mice. We introduced each variant individually into an infectious clone of ZIKV strain MR766, infected Ifnar1−/− mice with each virus, and monitored survival and weight loss. All four viruses caused 100% lethality, with no significant difference in the average time to death (Fig. 2A). Interestingly, we found that although mice infected with each variant succumbed to infection at the same time as those with wild-type (WT) ZIKV, the mice infected with the NS2AI33V and NS3R59K variants initially gained weight before undergoing rapid weight loss and death (Fig. 2B, green and purple triangles). These data suggest that NS2A and NS3 may play critical roles in ZIKV pathogenesis later in infection.
FIG 2.
ZIKV variants impact weight loss and virus replication in Ifnar1−/− mice. (A) Survival of 5- to 6-week-old Ifnar1−/− mice infected with 103 FFU of wild-type (WT) (n = 4), EV114M (n = 5), NS2AI33V (n = 6), or NS3R59K (n = 6) ZIKV. No significant difference between groups. (B) Weight loss of mice from panel A. Data are censored after the first mouse in each group died. ***, P < 0.001; *, P < 0.05 (two-way analysis of variance [ANOVA]). Viral loads in serum (C) and spleen (D) from Ifnar1−/− mice infected with WT ZIKV or variants. n = 8 for each virus. *, P < 0.05 (one-way ANOVA, Kruskal-Wallis test).
To investigate differences in viral replication between variants, we infected Ifnar1−/− mice with each variant and quantified infectious virus in the serum at 2 and 4 days postinfection and in the spleens at 4 days postinfection (Fig. 2C and D). We found no significant advantage of any of these variants in Ifnar1−/− mice, and if anything, there was a reduction in replication consistent with the weight loss results. Finally, to investigate the replication of each ZIKV variant in cell culture, we performed multistep growth curves in human A549, monkey Vero, and Aedes albopictus C6/36 cells. We found that ZIKV EV114M and NS2AI33V were statistically significantly attenuated in A549 cells compared to WT virus (Fig. 3A) yet showed no differences in replication in Vero or C6/36 cells (Fig. 3B and C). Together, these data indicate that these ZIKV variants show no significant replication advantage in Ifnar1−/− mice yet are attenuated in IFN-competent human cells, highlighting a role for these residues in ZIKV replication.
FIG 3.

Replication of ZIKV envelope and NS2A variants is attenuated in A549 cells. Multistep growth curves of WT ZIKV and variants in A549 (A), Vero (B), and C6/36 cells (C). Each cell line was infected with each virus at an MOI of 0.1, and virus titers in the supernatant were quantified by focus-forming assay. Dotted lines indicate the limit of detection. Data are from 3 independent experiments. **, P < 0.01 (two-way ANOVA).
ZIKV E residue V114 is structurally analogous to alphavirus E1 residue V80 and is sensitive to low concentrations of ammonium chloride.
We previously identified the CHIKV E1 glycoprotein residue V80 in β-strand c as important for CHIKV pH-dependent entry (21). Interestingly, the ZIKV envelope residue V114 falls into a structurally analogous site in β-strand c of the ZIKV E protein (Fig. 4A), and similar to alphaviruses (21), a valine at flavivirus residue 114 is highly conserved (Fig. 4B). Given the similarities between ZIKV EV114 and CHIKV E1V80, we hypothesized that ZIKV EV114 may also contribute to ZIKV pH-dependent entry. To test this hypothesis, we first determined that the ZIKV EV114M variant did not lead to any major differences in viral protein accumulation or processing. To address this, we transfected 293T cells with infectious clones encoding WT ZIKV, a replication-dead ZIKV harboring a mutation (GNN) in the polymerase, or the EV114M variant. We harvested cells at 72 h posttransfection and evaluated the accumulation of ZIKV proteins by Western blotting. While we found a slight reduction in all proteins in the EV114M variant, possibly due to the replication defect in human cells, there were no major differences in protein processing with the EV114M variant (Fig. 4C). Next, we asked whether the ZIKV EV114M variant was sensitive to the lysosomotropic agent ammonium chloride, which deacidifies the endosome and blocks pH-dependent entry. We infected Vero cells with each virus in the presence of increasing concentrations of ammonium chloride. Interestingly, we found that ZIKV EV114M replication was most sensitive to low concentrations of ammonium chloride yet showed a consistent, although not statistically significant, reduction at higher concentrations (Fig. 4D). Together, these data indicate that flavivirus glycoprotein β-strand c contributes to pH-dependent entry, as seen for alphaviruses.
FIG 4.

ZIKV V114M is structurally analogous to chikungunya virus E1 V80 and sensitive to ammonium chloride inhibition. (A) ZIKV MR766 envelope (PDB ID 5JHM) and CHIKV E1 (PDB ID 3N42). The fusion loop, ij loop, and bc loop are labeled, and the V114 and V80 variants are in red. (B) Flavivirus envelope protein sequence alignment around residue V114. (C) 293T cells were transfected with a ZIKV WT or ZIKV EV114M infectious clone. Cells were harvested at 72 h postinfection (hpi) and lysed in Laemmli buffer, and ZIKV proteins were analyzed by SDS-PAGE and immunoblotting. The blot is representative of at least three independent transfections. (D). Vero cells were pretreated for 1 h with increasing concentrations of NH4Cl and infected at an MOI of 0.1. Supernatant was collected 36 hpi, and viral titers were quantified by plaque assay. Data are means and standard errors of the means (SEM). There were 3 independent experiments with internal technical triplicates. Student's t test was used.
ZIKV EV114M is attenuated in wild-type neonatal mice with no impact in replication in A. aegypti mosquitoes.
We observed that CHIKV E1V80 contributes to dissemination in wild-type mice and in A. aegypti mosquitoes (21). Therefore, we hypothesized that ZIKV EV114M and ZIKV envelope β-strand c may also play essential roles in dissemination in mice and mosquitoes. To study ZIKV replication in WT immunocompetent mice, we first infected 6-week-old C57BL/6 mice, and as expected, neither virus replicated in these mice (data not shown). As an alternative approach, we used 4- and 7-day-old C57BL/6 neonatal mouse models. We hypothesized that the 4-day-old mice, which are more susceptible to ZIKV, would mimic Ifnar1−/− mice, while the 7-day-old mice would allow us to study ZIKV infection in a model that is closer to adult mice. We infected each neonatal model with WT ZIKV or the ZIKV EV114M variant. We found that ZIKV EV114M was attenuated in its ability to replicate and disseminate in both 4- and 7-day-old mice (Fig. 5A and B). This attenuation of ZIKV EV114M was dependent on the age of the mice, as infection of the ZIKV EV114M variant was not detectable in the brains of several 7-day-old mice, suggesting that the virus may be cleared faster than WT ZIKV or that there is a subset of 7-day-old mice that are specifically not susceptible to infection with the ZIKV EV114M variant.
FIG 5.

Replication of WT and E V114M ZIKV in neonatal mice and A. aegypti mosquitoes. Four-day-old (A) and 7-day-old (B) C57BL/6J mice were infected subcutaneously with 104 PFU of each virus. Virus titers were quantified in each organ at 7 days postinfection. Data are means and SEM from two independent infections. For four-day-old mice, n = 11 (WT) and 7 (EV114M). For seven-day-old mice, n = 7 (WT) and 8 (EV114M). The Mann-Whitney test was used. (C to F) A. aegypti mosquitoes were infected with 106 PFU of each virus, and viral titers were determined in the bodies (C and E) and legs and wings (D and F) at 14 days postinfection. Data are means and SEM from two independent infections. WT, n = 39; EV114M, n = 36. There was no significant difference between groups (Mann-Whitney compare-ranks test).
To investigate how ZIKV EV114M impacts replication in mosquitoes, we infected A. aegypti mosquitoes with WT ZIKV or the ZIKV EV114M variant and quantified viral titers in the bodies and in the legs and wings at 14 days postinfection (Fig. 5C to F). We found no major difference in the percentage of infected mosquitoes (Fig. 5C) or the percentage of mosquitoes that had virus in the legs and wings (disseminated virus) (Fig. 5D). Interestingly, while we did not find a statistically significant increase in viral titers with the ZIKV EV114M variant, the titers were higher in multiple mosquitoes with no clear replication disadvantage (Fig. 5E and F). Together, these data suggest that the flavivirus envelope β-strand c contributes to replication in neonatal mice with no negative impact on replication in mosquitoes.
A conserved alanine at the tip of the flavivirus envelope protein ij loop is important for ZIKV infectious particle production and RNA accumulation.
CHIKV has undergone several natural adaptation events that have shaped its infectivity and transmission (25, 26). One of the most significant was the emergence of a single amino acid substitution in the E1 glycoprotein (A226V) during an epidemic on La Reunion Island (10). This variant and residue 226, located in the ij loop of the alphavirus E1 glycoprotein, has been shown to increase replication and transmission in A. albopictus mosquitoes (27, 28) and is important for cholesterol-dependent entry (29, 30). Interestingly, flaviviruses contain a highly conserved alanine at the tip of the ij loop similar to CHIKV (Fig. 6A). Given the structural similarities between the flavivirus and alphavirus class II fusion proteins, we hypothesized that the ZIKV alanine would be important for virus infectivity. We mutated this alanine (A250) to a valine in both the MR766 and Brazilian strains of ZIKV and found that this single mutation completely blocked the production of infectious ZIKV particles in 293T cells as well as in BHK-21 and Vero cells (Fig. 6B and data not shown). In line with this observation, we found undetectable levels of ZIKV EA250V RNA in the supernatant compared to our wild-type ZIKV and ZIKV EV114M controls (Fig. 6C). One potential explanation for this phenotype is that ZIKV EA250V inhibits RNA replication or accumulation and subsequent protein production, as is the case for the NS5 GNN mutant (Fig. 6). To address whether ZIKV EA250V impacted replication and/or protein production, we transfected 293T cells with each ZIKV plasmid, harvested cells at 72 h posttransfection, and analyzed intracellular viral RNA levels by qPCR (Fig. 6D) and protein accumulation by Western blotting (Fig. 6E). Interestingly, we found a dramatic reduction in ZIKV RNA accumulation with the ZIKV EA250V mutant compared to wild-type virus and the ZIKV EV114M mutant. However, while RNA levels for ZIKV EA250V were barely above those for the NS5 GNN mutant, we did detect the majority of ZIKV proteins in transfected cells at higher levels than for the NS5 GNN mutant, albeit at lower levels than wild-type ZIKV and the ZIKV EV114M variant due to the lack of spread of the ZIKV EA250V variant (Fig. 6E). Interestingly, the envelope protein did not accumulate in the ZIKV EA250V variant, as demonstrated by the lack of detection by an anti-E polyclonal antibody, suggesting that the virus replicated at low levels yet may not have produced infectious particles due to a defect in envelope accumulation. These results indicate that the ZIKV ij loop is critical for infectious particle production potentially through the stability of the envelope glycoprotein and/or the impact of the E protein on RNA replication or accumulation.
FIG 6.
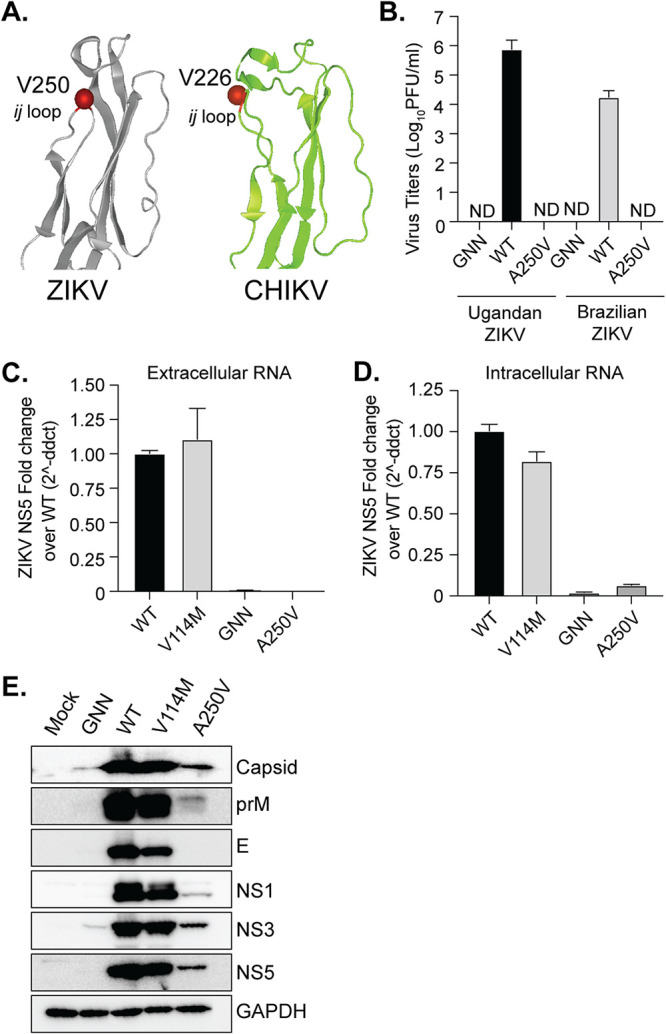
A single point mutation in the flavivirus E ij loop inhibits ZIKV infectious particle production and impacts envelope protein accumulation. (A) ZIKV MR766 envelope (PDB ID 5JHM) and CHIKV E1 (PDB ID 3N42). The ij loop and ZIKV A250 and CHIKV A226 residues are in red. (B) 293T cells were transfected with WT ZIKV or ZIKV EA250V infectious clone plasmids, and virus supernatants were collected at 48 hpi. Virus titers were quantified by plaque assay. n = 2 with internal technical duplicates. Extracellular (C) and intracellular (D) ZIKV RNA was quantified by RT-qPCR at 72 h posttransfection. RNA is normalized to WT ZIKV. There were three independent experiments with internal technical duplicates. (E) 293T cells were transfected with WT ZIKV or ZIKV EA250V plasmids. Cells were harvested at 72 h posttransfection and lysed in Laemmli buffer, and ZIKV proteins were analyzed by SDS-PAGE and immunoblotting. The immunoblot is representative of at least 3 independent transfections.
Conserved alanine at the tip of flavivirus ij loop blocks YFV but not WNV infectious-particle production.
The alanine at the tip of the flavivirus ij loop is highly conserved among flaviviruses and sits in the middle of a histidine and lysine which have been shown to be important for virion assembly (31) (Fig. 7A and B). Given this conservation, we hypothesized that the ij loop could also be critical for infectious particle production of other flaviviruses. To test this hypothesis, we mutated the alanine at the tip of the E protein ij loop to a valine in the WNV and yellow fever virus (YFV), transfected Vero cells with in vitro-transcribed RNA, and quantified infectious virions in the supernatant 48 h later by plaque assay. We found that the A-to-V mutation in WNV did not affect infectious particle production, yet the same mutation in YFV significantly impaired infectious-virus production (Fig. 7C and D). Importantly, when we sequenced the infectious virus from WNV, the A247V mutation was maintained and genetically stable. Finally, given these results with YFV in mammalian cells, we then transfected C6/36 mosquito cells with YFV wild-type or EA239V RNA and harvested virus at 72 and 96 h posttransfection (Fig. 7E). At 72 h postinfection, we found no infectious particles in the supernatant of YFV EA239V-transfected cells, suggesting that the lack of infectious particle production is not host specific. Interestingly, we did start to see viral particles at 96 h posttransfection, which may have been revertants. These results indicate that the ij loop plays flavivirus-specific roles in virion assembly and that the single alanine at the tip of the ij loop contributes to infectious-particle production. Future work will be essential to understand how flaviviruses and alphaviruses coordinate infections in insects and mammals and to elucidate the differences in virus entry between flaviviruses.
FIG 7.

The alanine at the tip of flavivirus envelope inhibits infectious particle production of yellow fever virus but not West Nile virus. (A) Flavivirus envelope ij loop protein sequence alignment. The orange box indicates the conserved alanine at the tip of the ij loop. (B) Domain II of the WNV (PDB ID 2HG0) and YFV (PDB ID 6IW4) envelope protein. The ij loop and YFV A239 and WNV A247 residues are in red. (C and D) Vero cells were transfected with WT or variant WNV and YFV in vitro-transcribed RNA, and virus-containing supernatant was collected at 48 hpi. Virus titers were quantified by plaque assay. There were 3 independent experiments. (E) C6/36 cells were transfected with WT or variant YFV RNA, and virus-containing supernatant was collected at 72 and 96 hpi. Virus titers were quantified by plaque assay. There were 3 independent experiments.
DISCUSSION
Arboviruses are a diverse group of human pathogens belonging to multiple virus families. There are no antiviral therapies targeting these viruses. This problem is in part due to our incomplete understanding of the common, fundamental molecular mechanisms different arboviruses use for infectivity and spread. To identify and study the fundamental mechanisms of arbovirus biology, we use samples collected from natural infections and in vivo lab experiments. In this study, we observed the emergence of ZIKV variants during vector-borne transmission where three variants of interest were found in the E (V114M, ∼10%), NS2A (I33V, ∼15%), and NS3 (R59K, ∼20%) proteins. The envelope variant is located at the tip of the class II fusion protein, near the fusion loop and the highly conserved glycerophospholipid-binding pocket (23). The NS2A protein variant is located in a predicted alpha-helix near the ER membrane, and the NS3 protease variant is close to an RLDP motif that was recently shown to be important for interactions between NS3 and cellular 14-3-3 proteins to antagonize the innate immune response (24). Given that we originally found these variants in Ifnar1−/− mice, we hypothesized that they would play significant roles in ZIKV infection in mice. However, when in separate experiments we introduced these variants into Ifnar1−/− mice, they had no significant replication advantage in the organs we investigated. These data and the fact that these variants were not found in mice infected via needle inoculation (22) suggest that these variants may have been selected for not in mice but rather in A. aegypti mosquitoes. This observation is particularly interesting as the alphavirus glycoproteins have been highlighted many times in nature and in the lab as a key evolutionary determinant for alphavirus infectivity and transmission in mosquitoes (20, 25, 29). This study provides evidence that this may also be the case for flaviviruses. Importantly, future studies regarding the role of NS2A and NS3 in mosquitoes and mice will be important to better understand the molecular mechanisms of the flavivirus life cycle in vivo.
The ZIKV envelope has been shown to significantly contribute to virulence (7, 32–34). The ZIKV envelope residue V114 is located in β-strand c of glycoprotein domain II. Because β-strand c of CHIKV plays an important role in virus dissemination and pathogenesis in mice and transmission in mosquitoes, we hypothesize this would also be the case for flaviviruses. Interestingly, we found that the ZIKV EV114M variant was attenuated in multiple mouse models and in A549 cells, confirming that E residue V114 and β-strand c are important for flavivirus infection. Given this attenuation in mammals, we thought that the ZIKV EV114M would have a significant replication advantage in mosquitoes, yet while we did see an increase in replication of the ZIKV EV114M variant in A. aegypti mosquitoes, this was not statistically significant. One potential explanation for this could be that the EV114M mutation alone is not enough to enhance replication, and perhaps the combination of E, NS2A, and NS3 variants will drive enhanced replication in mosquitoes and/or mice. Indeed, mutations in ZIKV NS3 have been implicated in ZIKV infection in mice (35). Moreover, as we looked at only one late time point, it will be interesting to explore the temporal replication of these variants in mosquitoes and mice to understand where the variants act during the viral life cycle. This is particularly important for the NS2A and NS3 variants in mice, as mice infected with these variants gained weight in the beginning of the infection and rapidly succumbed to the infection, suggesting that NS2A and NS3 may play a role late in the infection. Additionally, domain II, which contains β-strand c, occupies different orientations in the postfusion trimer between alphaviruses and flaviviruses. While the distal ends of E domain II are more tightly arranged in the flavivirus postfusion trimer, the analogous region of alphavirus E1 exhibits a more outward splayed arrangement. These differing orientations occupied by the fusogenic glycoproteins may contribute to the phenotypes we observe between the two virus families.
In addition to the envelope domain II β-strand c, we also observed structural similarities between the alphavirus and flavivirus glycoprotein ij loop. In CHIKV, a mutation from alanine to valine at position 226, in the tip of the ij loop, was responsible for a significant outbreak that led to increased replication in A. albopictus mosquitoes and to the emergence of the Indian Ocean lineage of CHIKV (10, 27). However, when we made this same A-to-V mutation in ZIKV, there was complete inhibition of infectious particle production. This result suggests that this residue of the ij loop may play a role in ZIKV particle assembly or that assembled particles have compromised fusogenic function, as has been observed for other ij loop variants (36–38). This is plausible, as previous work with Japanese encephalitis virus (JEV) has shown that mutation of a conserved histidine and lysine in the ij loop impacts JEV assembly (31). In that study, the authors had hypothesized that these conserved charged His and Lys residues in the envelope ij loop interact with negatively charged residues in prM to promote assembly. The crystal structure of prM-E of dengue virus further reveals complementary electrostatic patches on the two proteins facing each other in this region (39). Additional studies demonstrated that mutating H249 and H288 abolishes fusion in YFV and mutating H246 in WNV yields no infectious particles (36, 37). These critical histidine residues of the ij loop are conserved in both flaviviruses and alphaviruses. When histidine 230 of the Semliki Forest virus (SFV) ij loop is mutated to an alanine, this is detrimental to membrane fusion (38). It is possible that the valine mutation adjacent to these other critical residues in the ij loop also destabilizes interactions between E and prM, leading to reduction in infectious particle production. In a deep mutational scanning study of ZIKV E, residue A250 was strongly selected, along with the adjacent histidine (H249), supporting their functional importance (40). Further, the tip of domain II functions in sustaining contacting regions within the E-E intradimer interface, crucial for dimerization and formation of the glycoprotein lattice of antiparallel homodimers (41).
However, the above observations alone do not directly explain the specific reduction in the accumulation of the cellular EA250V protein or viral RNA. This observation that a mutation in the envelope glycoprotein can impact RNA replication is intriguing, as it is generally thought that the structural proteins are dispensable for RNA replication. While RNA levels are low for the ZIKV EA250V variant, they are still above those of the GNN mutant. More important, when we passage ZIKV EA250V cells, the virus reverts to wild type and infectious particles are generated. This result indicates that RNA replication functions at low levels. This raises the question of how the envelope protein impacts RNA levels. One potential explanation may be that the envelope protein, or the RNA encoding the protein, is important for RNA stability. Therefore, the RNA is degraded after replication, leading to less overall RNA, less protein, and no packaged particles. Future studies will be important to understand how the ij loop contributes to ZIKV E stability and how the ij loop impacts ZIKV RNA accumulation and assembly.
Finally, we found that the alanine at the tip of the flavivirus ij loop behaves in a flavivirus-specific manner in that a mutation of alanine to valine in ZIKV and YFV blocked infectious-virus production, yet there was no such effect with WNV. In comparing the crystal structures of the ZIKV, YFV, and WNV ij loops (Fig. 7), we noted that the alanine is oriented in a different direction in WNV, and perhaps this change in structure is responsible for this phenotype. Overall the ZIKV envelope protein is more phylogenetically similar to WNV (42), but ZIKV and YFV are both transmitted by Aedes mosquitoes, and WNV is transmitted by Culex species. (43). There could be many other genetic differences between the envelopes of ZIKV and WNV that account for these differences in infectious-particle production. Importantly, residue 114 is a valine in ZIKV and an aspartic acid in WNV, already suggesting differences in E β-strand c function. This may help shed light on how flaviviruses such as ZIKV and WNV infect different mosquito species and hosts such as birds.
Together, these studies identified novel flavivirus variants that impact flavivirus infection. We have shown that structurally similar domains of the flavivirus and alphavirus envelope glycoproteins contribute to similar functions in virus infection. These common mechanisms of infection could potentially provide the determinants needed for the development of broad-spectrum antivirals. More importantly, these studies lead to new insight into the fundamental biology of flaviviruses. Future studies to dissect the molecular details of the flavivirus and alphavirus envelope proteins are critical to understand how these viruses function and cause disease.
MATERIALS AND METHODS
Cell lines.
293T cells (ATCC CRL3216) and human foreskin fibroblasts (HFF1; ATCC SCRC-1041) were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 1% penicillin-streptomycin (P/S), 1% nonessential amino acids, and 10% heat-inactivated fetal bovine serum (FBS, Atlanta Biologicals) at 37°C with 5% CO2. Stapleford lab Vero cells (ATCC CCL-81) were maintained in DMEM supplemented with 1% P/S and 10% heat-inactivated newborn calf serum (Gibco) at 37°C with 5% CO2. Lazear lab Vero and A549 cells were maintained in DMEM containing 5% heat-inactivated FBS and l-glutamine at 37°C with 5% CO2. C6/36 cells were maintained in DMEM containing 6% FBS, nonessential amino acids (NEAA), and P/S at 28°C with 5% CO2. All cells were verified to be mycoplasma free.
Viruses.
The ZIKV (Ugandan 1947; MR766) wild-type and replication-deficient (NS5 GNN mutant) plasmid infectious clones were a gift from Matthew Evans at the Icahn School of Medicine at Mt. Sinai (44). The ZIKV Brazilian strain infectious clone was a gift from Alexander Pletnev at the National Institutes of Health (45). The yellow fever virus (vaccine strain 17-D) infectious clone was a gift from Julie Pfeiffer at the University of Texas Southwestern (46). The West Nile virus infectious clone was a gift from Gregory Ebel at Colorado State University (47). The ZIKV, WNV, and YFV envelope variants were generated by site-directed mutagenesis using Phusion high-fidelity polymerase (Thermo) and the primers in Table 1. All plasmids were sequenced in full to confirm that no second-site mutations were introduced.
TABLE 1.
Primers used for this study
| Virus, variant, and primer directiona | Sequence |
|---|---|
| MR766 ZIKV EV114M, For | GGCAAAGGGAGCTTGATGACATGTGCCAAG |
| MR766 ZIKV EV114M, Rev | CTTGGCACATGTCATCAAGCTCCCTTTGCC |
| MR766 ZIKV NS2AI21V, For | GAGAATGACCACAAAGGTCATCATGAGCACATC |
| MR766 ZIKV NS2AI21V, Rev | GATGTGCTCATGATGACCTTTGTGGTCATTCTC |
| MR766 ZIKV NS3R42K, For | CAAAAGGAGCCGCACTGAAGAGCGGTGAGGGAAGAC |
| MR766 ZIKV NS3R42K, Rev | GTCTTCCCTCACCGCTCTTCAGTGCGGCTCCTTTTG |
| MR766 ZIKV EA250V, For | CAAGGATGCCCACGTCAAGAGGCAAACCG |
| MR766 ZIKV EA250V, Rev | CGGTTTGCCTCTTGACGTGGGCATCCTTG |
| Brazilian ZIKV EA250V, For | GTTCAAGGACGCACATGTCAAAAGGCAAACTGTCG |
| Brazilian ZIKV EA250V, Rev | CGACAGTTTGCCTTTTGACATGTGCGTCCTTGAAC |
| YFV EA239V, For | GAATTTGAACCTCCGCATGTCGCCACTATCAGAGTAC |
| YFV EA239V, Rev | GTACTCTGATAGTGGCGACATGCGGAGGTTCAAATTC |
| WNV EA247V, For | GTTTGAGGAACCACACGTCACGAAGCAGTCTGTG |
| WNV EA247V, Rev | CACAGACTGCTTCGTGACGTGTGGTTCCTCAAAC |
For, forward; Rev, reverse.
The wild-type and variant Ugandan and Brazilian ZIKV were produced by transfecting 293T cells with 0.5 μg of each plasmid with the Lipofectamine 2000 transfection reagent (Invitrogen). Virus-containing supernatants were harvested 48 h posttransfection, centrifuged at 1,200 × g for 5 min, aliquoted and stored at −80°C. To generate a working virus stock, Vero cells were infected with transfection virus stocks and virus-containing supernatants were harvested 48 h postinfection, centrifuged at 1,200 × g for 5 min, aliquoted, and stored at −80°C. Viral titers were determined by plaque assay on Vero cells, as described below.
Wild-type and variant YFV and WNV were generated by the transfection of in vitro-transcribed RNA into Vero cells. In brief, YFV and WNV infectious clones were linearized overnight with XhoI and XbaI, respectively. Each plasmid was purified by phenol-chloroform extraction and ethanol precipitation, resuspended in nuclease-free water. Linearized YFV and WNV plasmids were used for in vitro transcription using the mMessage mMachine SP6 and T7 kits, respectively, following the manufacturer’s instructions. In vitro-transcribed RNAs were purified by phenol-chloroform extraction and ethanol precipitation, diluted to 1 mg/mL, aliquoted and stored at −80°C. Vero cells (200,000 cells/well) were transfected with 5 μg of YFV and WNV in vitro-transcribed RNA with Lipofectamine 2000 following the manufacturer’s instructions and incubated at 37°C for 48 h. Following incubation, virus titers were quantified by plaque assay as described below. All work with WNV was performed under biosafety level 3 conditions. For C6/36 transfections, C6/36 cells were trypsinized and washed twice in phosphate-buffered saline (PBS). Cells were resuspended in PBS at a concentration of 107/mL and electroporated with 10 μg of in vitro-transcribed RNA using the following conditions: 250 V, 25 Ω, and 550 μF. Following electroporation, cells were placed back in the medium and incubated at 28°C for 72 or 96 h. Virus titers were quantified by plaque assay as described below.
Deep sequencing analysis.
Sequencing data were published previously and can be found in the Sequence Read Archive (SRA) under BioProject ID PRJNA589089 (22). Sequencing reads were processed and aligned as described previously (22). Minority variants were identified using an in-house variant caller (https://github.com/GhedinLab/ZIKV_Analysis). Coverage and minority variant calls were checked to ensure that overlapping regions were identical in their nucleotide composition before merging the fastq files and then realigning the 3 amplicons to the reference file at once. Minority variants were called again on the merged alignment files, and the amino acid position was added using the positions indicated on the MR766 NCBI site (KX830961.1). Minority variants were called if coverage at the given nucleotide was at or above 500×, and the frequency of the variant was above 1%, and the variant was present in both forward and reverse reads and had a quality score above 25.
Plaque assay and focus-forming assay.
Viral titers were determined by plaque assays or focus-forming assays (FFA) on Vero cells. For plaque assays, virus was subjected to 10-fold serial dilutions in DMEM and added to a monolayer of Vero cells for 1 h at 37°C. Following incubation, a 0.8% agarose overlay was added, and cells were incubated for 5 days at 37°C. Five days postinfection, cells were fixed with 4% formalin, the agarose overlay was removed, and plaques were visualized by staining with crystal violet (10% crystal violet and 20% ethanol). Viral titers were determined on the highest dilution virus could be counted.
For FFA, duplicates of serial 10-fold dilutions of virus in viral growth medium (DMEM containing 2% FBS and 20 mM HEPES) were applied to Vero cells in 96-well plates and incubated for 1 h at 37°C. Following virus adsorption, the monolayers were overlaid with 1% methylcellulose in minimum essential Eagle medium (MEM). Infected-cell foci were detected 42 to 46 h after infection. Following fixation with 2% paraformaldehyde (PFA) for 1 h at room temperature, plates were incubated with 500 ng/mL of flavivirus-cross-reactive mouse monoclonal antibody (MAb) E60 (48) for 2 h at room temperature or overnight at 4°C. After incubation at room temperature for 2 h with a 1:5,000 dilution of horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Sigma), foci were detected by addition of TrueBlue substrate (KPL). Foci were quantified with an ImmunoSpot analyzer (Cellular Technology, Ltd. [CTL]).
Virus growth curves.
For multistep growth analysis, cells were infected at a multiplicity of infection (MOI) of 0.1 and incubated at 37°C or 28°C with 5% CO2 for 1 h. Then, inoculum was aspirated, cells were washed with PBS, and medium was replenished. Samples of infected cell culture supernatant were collected at 4, 24, 48, and 72 h postinfection and stored at −80°C for virus titration. Virus quantification was performed by FFA on Vero cells as described above.
Immunoblotting.
293T cells (500,000 cells/well in a 6-well plate) were transfected with 1.2 μg of each plasmid as described above. At predetermined time points, cells were harvested, washed twice with PBS, and resuspended in 2× Laemmli buffer containing 10% β-mercaptoethanol. Samples were boiled for 10 min and centrifuged for 5 min at 12,000 × g to clarify debris, and proteins were separated by SDS-PAGE. Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane, and membranes were blocked with 5% nonfat milk in Tris-buffered saline (TBS; 50 mM Tris [pH 7.5], 150 mM NaCl) containing 0.1% Tween 20 (TBS-T). Membranes were incubated with anti-ZIKV capsid (GeneTex; catalog no. GTX133317), prM (GeneTex; GTX133305), envelope (GeneTex; catalog no. GTX133314), NS1 (GeneTex; catalog no. GTX133307), NS3 and NS5 (gifts from Andres Merits at the University of Tartu, Estonia), and anti-GAPDH (GA1R; Thermo Fisher; catalog no. MA5-15738) antibodies, washed with TBS-T, and incubated with an HRP-conjugated secondary antibody. After incubation with secondary antibody, membranes were washed extensively, developed with the SuperSignal West Pico chemiluminescent substrate (Pierce), and imaged on the Bio-Rad ChemiDoc imager. Images were analyzed and processed using ImageLab (version 6.0.0).
ZIKV RNA replication assay.
293T cells (250,000 cells/well in a 12-well plate) were transfected with 1.2 μg of each plasmid as described above. At 72 h posttransfection, cells were harvested and washed in PBS. RNA was purified using TRIzol (Fisher-Scientific) following the manufacturer’s instructions from both cells and supernatant. RNA was treated with Turbo DNase (Thermo) for 2 h at 37°C and repurified by phenol-chloroform extraction and ethanol precipitation. Viral genomes relative to cellular GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were quantified in both fractions using SYBR green qPCR and the following primers: GAPDH, 5′-GAAGGTCGGAGTCAACGGATTT-3′ and 5′-GAATTTGCCATGGGTGGAAT-3′, and ZIKV NS5, 5′-AGATGACTGCGTTGTGAAGC-3′ and 5′-GAGCAGAACGGGACTTCTTC-3′.
Mosquito infections and harvests.
A. aegypti mosquitoes (Poza Rica, Mexico; F30-35) were a gift from Gregory Ebel at Colorado State University (49). Mosquitoes were reared and maintained in the NYU School of Medicine ABSL3 facility at 28°C and 70% humidity with a 12-h–12-h diurnal light cycle. The day before infection, female mosquitoes were sorted and starved overnight. The day of infection, mosquitoes were exposed to an infectious bloodmeal containing freshly washed sheep blood, 5 mM ATP, and 107 PFU/mL virus. After approximately 30 min, mosquitoes were cold-anesthetized, and engorged female mosquitoes were sorted into new cups (21, 50). Engorged mosquitoes were incubated at 28°C with 70% humidity for 14 days and fed ad libitum with 10% sucrose. Following incubation, mosquitoes were cold-anesthetized and the legs and wings removed. Mosquito bodies, legs, and wings were put into 2-mL round-bottom tubes containing 250 mL of PBS and a steel ball (Qiagen). Samples were homogenized using a TissueLyser (Qiagen) with 30 shakes/s for 2 min and centrifuged at 8,000 × g for 5 min to remove debris, and viral titers were quantified by plaque assay.
Mouse infections.
Four- and 7-day-old WT C57BL/6J mouse experiments were completed in the NYU ABSL3 facility and performed with the approval of the NYU School of Medicine Institutional Animal Care and Use Committee (IACUC) (protocol IA16-01783). Four- and 7-day-old male and female C57BL/6J mice were infected subcutaneously with 104 PFU of WT ZIKV or the ZIKV EV114M variant. Mice were euthanized by decapitation 7 days postinfection (dpi), organs were harvested and homogenized as described above, and ZIKV titers were quantified by plaque assay.
Husbandry and infections in type I interferon receptor knockout (Ifnar1−/−) mice were performed with the approval of the University of North Carolina at Chapel Hill IACUC (protocol 19-185). Five- to 6-week-old male Ifnar1−/− mice on a C57BL/6 background were used. Mice were inoculated with 103 FFU of ZIKV in a volume of 50 μL by a subcutaneous route in the footpad. Survival and weight loss were monitored for 14 days. Animals that lost ≥30% of their starting weight or that exhibited severe disease signs were euthanized by isoflurane overdose. Weights are reported as percentages of weights on the day of infection, and group means were censored once one animal in a group died. To measure viral loads, ZIKV-infected mice were euthanized at 4 dpi as described above and perfused with 20 mL of PBS. Spleens were harvested and homogenized with zirconia beads (BioSpec) in a MagNA Lyser instrument (Roche Life Science) in 1 mL of PBS. Blood was collected at 2 dpi by submandibular bleed with a 5 mm Goldenrod lancet and by cardiac puncture at 4 dpi. Blood was collected in serum separator tubes (BD), and serum was separated by centrifugation at 8,000 rpm for 5 min. Tissues and serum from infected animals were stored at −80°C until titration by focus-forming assay.
Virus protein accession numbers.
GenBank accession numbers used for flavivirus protein alignments were as follows: Zika MR766 (KX830960.1), ZIKV Puerto Rican (KX377337), ZIKV Brazilian (KX280026.1), ZIKV French Polynesia (KJ776791.2), ZIKV Cambodian (KU955593.1), DENV1 (BCG29749), DENV2 (BCG29762), DENV3 (AVF19960), DENV4 (BCG29769), YFV (NP_740305), and WNV (MN849176).
Protein structures.
The E glycoprotein structures of ZIKV (PDB ID 5JHM), WNV (PDB ID 2HG0), and YFV (PDB ID 6IW4) and the CHIKV E1 glycoprotein structure (PDB ID 3N42) were visualized using Protein 3D (DNASTAR).
Statistical analysis.
GraphPad Prism (version 8.0.2) was used to analyze all data and perform statistical analyses. All in vitro experiments were completed in triplicate with internal duplicates or triplicates. Mosquito and mouse experiments were completed in two independent infections. A P value of <0.05 is considered significant.
ACKNOWLEDGMENTS
We thank all members of the Stapleford lab for critical comments on the experiments and manuscript. We thank our collaborators Julie Pfeiffer, Matthew Evans, Alexander Pletnev, Andres Merits, and Gregory Ebel for essential reagents.
This work was supported by a start-up package from NYU Grossman School of Medicine (K.A.S.), Public Health Service Institutional Research Training Award T32 AI007180 (M.V.R. and K.E.E.J.), an American Heart Association Postdoctoral Fellowship (19-A0-00-1003686), and R01 AI39512 (H.M.L. and N.C.). This work was supported in part by the Division of Intramural Research (DIR) and the NIAID/NIH (E.G.).
Contributor Information
Kenneth A. Stapleford, Email: Kenneth.stapleford@nyulangone.org.
Colin R. Parrish, Cornell University
REFERENCES
- 1.Coffey LL, Forrester N, Tsetsarkin K, Vasilakis N, Weaver SC. 2013. Factors shaping the adaptive landscape for arboviruses: implications for the emergence of disease. Future Microbiol 8:155–176. 10.2217/fmb.12.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierson TC, Diamond MS. 2020. The continued threat of emerging flaviviruses. Nat Microbiol 5:796–812. 10.1038/s41564-020-0714-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weaver SC, Charlier C, Vasilakis N, Lecuit M. 2018. Zika, chikungunya, and other emerging vector-borne viral diseases. Annu Rev Med 69:395–408. 10.1146/annurev-med-050715-105122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kramer LD. 2016. Complexity of virus-vector interactions. Curr Opin Virol 21:81–86. 10.1016/j.coviro.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brault AC, Huang CY, Langevin SA, Kinney RM, Bowen RA, Ramey WN, Panella NA, Holmes EC, Powers AM, Miller BR. 2007. A single positively selected West Nile viral mutation confers increased virogenesis in American crows. Nat Genet 39:1162–1166. 10.1038/ng2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ebel GD, Carricaburu J, Young D, Bernard KA, Kramer LD. 2004. Genetic and phenotypic variation of West Nile virus in New York, 2000-2003. Am J Trop Med Hyg 71:493–500. 10.4269/ajtmh.2004.71.493. [DOI] [PubMed] [Google Scholar]
- 7.Shan C, Xia H, Haller SL, Azar SR, Liu Y, Liu J, Muruato AE, Chen R, Rossi SL, Wakamiya M, Vasilakis N, Pei R, Fontes-Garfias CR, Singh SK, Xie X, Weaver SC, Shi PY. 2020. A Zika virus envelope mutation preceding the 2015 epidemic enhances virulence and fitness for transmission. Proc Natl Acad Sci USA 117:20190–20197. 10.1073/pnas.2005722117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brault AC, Powers AM, Ortiz D, Estrada-Franco JG, Navarro-Lopez R, Weaver SC. 2004. Venezuelan equine encephalitis emergence: enhanced vector infection from a single amino acid substitution in the envelope glycoprotein. Proc Natl Acad Sci USA 101:11344–11349. 10.1073/pnas.0402905101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brault AC, Powers AM, Holmes EC, Woelk CH, Weaver SC. 2002. Positively charged amino acid substitutions in the e2 envelope glycoprotein are associated with the emergence of venezuelan equine encephalitis virus. J Virol 76:1718–1730. 10.1128/jvi.76.4.1718-1730.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schuffenecker I, Iteman I, Michault A, Murri S, Frangeul L, Vaney MC, Lavenir R, Pardigon N, Reynes JM, Pettinelli F, Biscornet L, Diancourt L, Michel S, Duquerroy S, Guigon G, Frenkiel MP, Brehin AC, Cubito N, Despres P, Kunst F, Rey FA, Zeller H, Brisse S. 2006. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med 3:e263. 10.1371/journal.pmed.0030263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jose J, Snyder JE, Kuhn RJ. 2009. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol 4:837–856. 10.2217/fmb.09.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicholls CMR, Sevvana M, Kuhn RJ. 2020. Structure-guided paradigm shifts in flavivirus assembly and maturation mechanisms. Adv Virus Res 108:33–83. 10.1016/bs.aivir.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnard TR, Abram QH, Lin QF, Wang AB, Sagan SM. 2021. Molecular determinants of flavivirus virion assembly. Trends Biochem Sci 46:378–390. 10.1016/j.tibs.2020.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Carro SD, Cherry S. 2020. Beyond the surface: endocytosis of mosquito-borne flaviviruses. Viruses 13:13. 10.3390/v13010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown RS, Wan JJ, Kielian M. 2018. The alphavirus exit pathway: what we know and what we wish we knew. Viruses 10:89. 10.3390/v10020089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Button JM, Qazi SA, Wang JC, Mukhopadhyay S. 2020. Revisiting an old friend: new findings in alphavirus structure and assembly. Curr Opin Virol 45:25–33. 10.1016/j.coviro.2020.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holmes AC, Basore K, Fremont DH, Diamond MS. 2020. A molecular understanding of alphavirus entry. PLoS Pathog 16:e1008876. 10.1371/journal.ppat.1008876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaney MC, Rey FA. 2011. Class II enveloped viruses. Cell Microbiol 13:1451–1459. 10.1111/j.1462-5822.2011.01653.x. [DOI] [PubMed] [Google Scholar]
- 19.Kielian M. 2006. Class II virus membrane fusion proteins. Virology 344:38–47. 10.1016/j.virol.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 20.Stapleford KA, Coffey LL, Lay S, Bordería AV, Duong V, Isakov O, Rozen-Gagnon K, Arias-Goeta C, Blanc H, Beaucourt S, Haliloğlu T, Schmitt C, Bonne I, Ben-Tal N, Shomron N, Failloux A-B, Buchy P, Vignuzzi M. 2014. Emergence and transmission of arbovirus evolutionary intermediates with epidemic potential. Cell Host Microbe 15:706–716. 10.1016/j.chom.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 21.Noval MG, Rodriguez-Rodriguez BA, Rangel MV, Stapleford KA. 2019. Evolution-driven attenuation of alphaviruses highlights key glycoprotein determinants regulating viral infectivity and dissemination. Cell Rep 28:460–471.E5. 10.1016/j.celrep.2019.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson KEE, Noval MG, Rangel MV, De Jesus E, Geber A, Schuster S, Cadwell K, Ghedin E, Stapleford KA. 2020. Mapping the evolutionary landscape of Zika virus infection in immunocompromised mice. Virus Evol 6:veaa092. 10.1093/ve/veaa092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guardado-Calvo P, Atkovska K, Jeffers SA, Grau N, Backovic M, Perez-Vargas J, de Boer SM, Tortorici MA, Pehau-Arnaudet G, Lepault J, England P, Rottier PJ, Bosch BJ, Hub JS, Rey FA. 2017. A glycerophospholipid-specific pocket in the RVFV class II fusion protein drives target membrane insertion. Science 358:663–667. 10.1126/science.aal2712. [DOI] [PubMed] [Google Scholar]
- 24.Riedl W, Acharya D, Lee JH, Liu G, Serman T, Chiang C, Chan YK, Diamond MS, Gack MU. 2019. Zika virus NS3 mimics a cellular 14–3-3-binding motif to antagonize RIG-I- and MDA5-mediated innate immunity. Cell Host Microbe 26:493–503.E6. 10.1016/j.chom.2019.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsetsarkin KA, Chen R, Sherman MB, Weaver SC. 2011. Chikungunya virus: evolution and genetic determinants of emergence. Curr Opin Virol 1:310–317. 10.1016/j.coviro.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsetsarkin KA, Chen R, Leal G, Forrester N, Higgs S, Huang J, Weaver SC. 2011. Chikungunya virus emergence is constrained in Asia by lineage-specific adaptive landscapes. Proc Natl Acad Sci USA 108:7872–7877. 10.1073/pnas.1018344108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S. 2007. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog 3:e201. 10.1371/journal.ppat.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arias-Goeta C, Moutailler S, Mousson L, Zouache K, Thiberge JM, Caro V, Rougeon F, Failloux AB. 2014. Chikungunya virus adaptation to a mosquito vector correlates with only few point mutations in the viral envelope glycoprotein. Infect Genet Evol 24:116–126. 10.1016/j.meegid.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 29.Tsetsarkin KA, McGee CE, Higgs S. 2011. Chikungunya virus adaptation to Aedes albopictus mosquitoes does not correlate with acquisition of cholesterol dependence or decreased pH threshold for fusion reaction. Virol J 8:376. 10.1186/1743-422X-8-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vashishtha M, Phalen T, Marquardt MT, Ryu JS, Ng AC, Kielian M. 1998. A single point mutation controls the cholesterol dependence of Semliki Forest virus entry and exit. J Cell Biol 140:91–99. 10.1083/jcb.140.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu H, Liu Y, Wang S, Zhang Y, Zu X, Zhou Z, Zhang B, Xiao G. 2015. Structure-based mutational analysis of several sites in the E protein: implications for understanding the entry mechanism of Japanese encephalitis virus. J Virol 89:5668–5686. 10.1128/JVI.00293-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carbaugh DL, Lazear HM. 2020. Flavivirus envelope protein glycosylation: impacts on viral infection and pathogenesis. J Virol 94:e00104-20. 10.1128/JVI.00104-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duggal NK, McDonald EM, Weger-Lucarelli J, Hawks SA, Ritter JM, Romo H, Ebel GD, Brault AC. 2019. Mutations present in a low-passage Zika virus isolate result in attenuated pathogenesis in mice. Virology 530:19–26. 10.1016/j.virol.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carbaugh DL, Baric RS, Lazear HM. 2019. Envelope protein glycosylation mediates Zika virus pathogenesis. J Virol 93:e00113-19. 10.1128/JVI.00113-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collette NM, Lao VHI, Weilhammer DR, Zingg B, Cohen SD, Hwang M, Coffey LL, Grady SL, Zemla AT, Borucki MK. 2020. Single amino acid mutations affect Zika virus replication in vitro and virulence in vivo. Viruses 12:1295. 10.3390/v12111295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson S, Poddar S, Lin T-Y, Pierson TC. 2009. Protonation of individual histidine residues is not required for the pH-dependent entry of West Nile virus: evaluation of the “histidine switch” hypothesis. J Virol 83:12631–12635. 10.1128/JVI.01072-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fritz R, Stiasny K, Heinz FX. 2008. Identification of specific histidines as pH sensors in flavivirus membrane fusion. J Cell Biol 183:353–361. 10.1083/jcb.200806081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chanel-Vos C, Kielian M. 2004. A conserved histidine in the ij loop of the Semliki Forest virus E1 protein plays an important role in membrane fusion. J Virol 78:13543–13552. 10.1128/JVI.78.24.13543-13552.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li L, Lok SM, Yu IM, Zhang Y, Kuhn RJ, Chen J, Rossmann MG. 2008. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science 319:1830–1834. 10.1126/science.1153263. [DOI] [PubMed] [Google Scholar]
- 40.Sourisseau M, Lawrence DJP, Schwarz MC, Storrs CH, Veit EC, Bloom JD, Evans MJ, Pfeiffer JK. 2019. Deep mutational scanning comprehensively maps how Zika envelope protein mutations affect viral growth and antibody escape. J Virol 93:e01291-19. 10.1128/JVI.01291-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luca VC, AbiMansour J, Nelson CA, Fremont DH. 2012. Crystal structure of the Japanese encephalitis virus envelope protein. J Virol 86:2337–2346. 10.1128/JVI.06072-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barba-Spaeth G, Dejnirattisai W, Rouvinski A, Vaney MC, Medits I, Sharma A, Simon-Loriere E, Sakuntabhai A, Cao-Lormeau VM, Haouz A, England P, Stiasny K, Mongkolsapaya J, Heinz FX, Screaton GR, Rey FA. 2016. Structural basis of potent Zika-dengue virus antibody cross-neutralization. Nature 536:48–53. 10.1038/nature18938. [DOI] [PubMed] [Google Scholar]
- 43.Cook S, Holmes EC. 2006. A multigene analysis of the phylogenetic relationships among the flaviviruses (family: Flaviviridae) and the evolution of vector transmission. Arch Virol 151:309–325. 10.1007/s00705-005-0626-6. [DOI] [PubMed] [Google Scholar]
- 44.Schwarz MC, Sourisseau M, Espino MM, Gray ES, Chambers MT, Tortorella D, Evans MJ. 2016. Rescue of the 1947 Zika virus prototype strain with a cytomegalovirus promoter-driven cDNA clone. mSphere 1:e00246-16. 10.1128/mSphere.00246-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsetsarkin KA, Kenney H, Chen R, Liu G, Manukyan H, Whitehead SS, Laassri M, Chumakov K, Pletnev AG. 2016. A full-length infectious cDNA clone of Zika virus from the 2015 epidemic in Brazil as a genetic platform for studies of virus-host interactions and vaccine development. mBio 7:e01114-16. 10.1128/mBio.01114-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Erickson AK, Pfeiffer JK. 2013. Dynamic viral dissemination in mice infected with yellow fever virus strain 17D. J Virol 87:12392–12397. 10.1128/JVI.02149-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grubaugh ND, Fauver JR, Ruckert C, Weger-Lucarelli J, Garcia-Luna S, Murrieta RA, Gendernalik A, Smith DR, Brackney DE, Ebel GD. 2017. Mosquitoes transmit unique West Nile virus populations during each feeding episode. Cell Rep 19:709–718. 10.1016/j.celrep.2017.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oliphant T, Nybakken GE, Engle M, Xu Q, Nelson CA, Sukupolvi-Petty S, Marri A, Lachmi BE, Olshevsky U, Fremont DH, Pierson TC, Diamond MS. 2006. Antibody recognition and neutralization determinants on domains I and II of West Nile Virus envelope protein. J Virol 80:12149–12159. 10.1128/JVI.01732-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruckert C, Weger-Lucarelli J, Garcia-Luna SM, Young MC, Byas AD, Murrieta RA, Fauver JR, Ebel GD. 2017. Impact of simultaneous exposure to arboviruses on infection and transmission by Aedes aegypti mosquitoes. Nat Commun 8:15412. 10.1038/ncomms15412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaczmarek ME, Herzog NL, Noval MG, Zuzworsky J, Shah Z, Bajwa WI, Stapleford KA. 2020. Distinct New York City Aedes albopictus mosquito populations display differences in salivary gland protein D7 diversity and chikungunya virus replication. Viruses 12:698. 10.3390/v12070698. [DOI] [PMC free article] [PubMed] [Google Scholar]