

ABSTRACT
Although a broad range of viruses cause myocarditis, the mechanisms that underlie viral myocarditis are poorly understood. Here, we report that the M2 gene is a determinant of reovirus myocarditis. The M2 gene encodes outer capsid protein μ1, which mediates host membrane penetration during reovirus entry. We infected newborn C57BL/6 mice with reovirus strain type 1 Lang (T1L) or a reassortant reovirus in which the M2 gene from strain type 3 Dearing (T3D) was substituted into the T1L genetic background (T1L/T3DM2). T1L was nonlethal in wild-type mice, whereas more than 90% of mice succumbed to T1L/T3DM2 infection. T1L/T3DM2 produced higher viral loads than T1L at the site of inoculation. In secondary organs, T1L/T3DM2 was detected with more rapid kinetics and reached higher peak titers than T1L. We found that hearts from T1L/T3DM2-infected mice were grossly abnormal, with large lesions indicative of substantial inflammatory infiltrate. Lesions in T1L/T3DM2-infected mice contained necrotic cardiomyocytes with pyknotic debris, as well as extensive lymphocyte and histiocyte infiltration. In contrast, T1L induced the formation of small purulent lesions in a small subset of animals, consistent with T1L being mildly myocarditic. Finally, more activated caspase-3-positive cells were observed in hearts from animals infected with T1L/T3DM2 than T1L. Together, our findings indicate that substitution of the T3D M2 allele into an otherwise T1L genetic background is sufficient to change a nonlethal infection into a lethal infection. Our results further indicate that T3D M2 enhances T1L replication and dissemination in vivo, which potentiates the capacity of reovirus to cause myocarditis.
IMPORTANCE Reovirus is a nonenveloped virus with a segmented double-stranded RNA genome that serves as a model for studying viral myocarditis. The mechanisms by which reovirus drives myocarditis development are not fully elucidated. We found that substituting the M2 gene from strain type 3 Dearing (T3D) into an otherwise type 1 Lang (T1L) genetic background (T1L/T3DM2) was sufficient to convert the nonlethal T1L strain into a lethal infection in neonatal C57BL/6 mice. T1L/T3DM2 disseminated more efficiently and reached higher maximum titers than T1L in all organs tested, including the heart. T1L is mildly myocarditic and induced small areas of cardiac inflammation in a subset of mice. In contrast, hearts from mice infected with T1L/T3DM2 contained extensive cardiac inflammatory infiltration and more activated caspase-3-positive cells, which is indicative of apoptosis. Together, our findings identify the reovirus M2 gene as a new determinant of reovirus-induced myocarditis.
KEYWORDS: dissemination, inflammation, myocarditis, pathogenesis, reovirus
INTRODUCTION
Numerous viruses, such as cytomegalovirus (CMV), coxsackievirus B3 (CVB3), parvovirus B19, and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), can cause myocarditis, which is characterized by inflammation of the heart muscle that can lead to cardiac damage, electrophysiological abnormalities, heart failure, or sudden death (1). Viral myocarditis is driven by a combination of virus-induced cytopathic effects in cardiac cells and the host inflammatory response to infection (2–5). Although a significant public health risk, many open questions remain with respect to the viral factors and host mechanisms that underlie viral myocarditis.
Mammalian orthoreovirus (reovirus) is a nonenveloped, double-stranded RNA (dsRNA) virus with a segmented genome that serves as a model for studying viral myocarditis (6). Some, but not all, reovirus strains cause myocarditis in neonatal mice (7, 8). The segmented nature of the reovirus genome has enabled the identification of specific genes that associate with myocarditis induction. Such studies have identified proteins that make up the viral inner shell (core) as determinants of myocarditis (7, 9, 10). However, amino acid changes in core proteins of myocarditic and nonmyocarditic strains that define differences in heart disease remain unidentified. Interestingly, most myocarditic strains are reassortants. Clone 8B, a highly myocarditic reovirus strain, was generated via reassortment between the mildly myocarditic reovirus strain type 1 Lang (T1L) and the nonmyocarditic strain type 3 Dearing (T3D) (11). Clone 8B contains 8 gene segments from T1L in combination with the S1 and M2 genes from T3D (11). The reovirus S1 gene encodes viral attachment protein σ1 and nonstructural protein σ1s, while M2 encodes outer capsid protein μ1 (6). T1L/T3Dμ1σ3 contains 8 gene segments from T1L with the M2 and S4 genes from T3D (12). The S4 gene encodes outer capsid protein σ3, which complexes with μ1 on the reovirus virion (6). The KC19 strain, which contains 7 genes from T1L and the S1, S3 (encoding nonstructural protein σNS), and M2 genes from T3D also has higher myocarditic potential than T1L (7). Common among clone 8B, T1L/T3Dμ1σ3, and KC19 is that they contain the M2 gene from T3D. This observation raises the possibility that the T3D-derived M2 gene impacts the myocarditic potential of reovirus strains. However, this idea has not been formally tested.
Reovirus infection is initiated by binding of viral attachment protein σ1 to cell surface receptors, followed by internalization of the particle by receptor-mediated endocytosis (13, 14). In the endosome, heterohexameric complexes of μ1 and σ3 that form the viral outer capsid are cleaved by the activity of endosome-resident cathepsin proteases to facilitate virus entry (15–17). The σ3 protein is degraded, and μ1 is cleaved to enable conformational changes in μ1 required for endosomal membrane penetration and release of viral cores into the cytoplasm for transcription and subsequent replication (6, 15–17). In addition to its role in entry, the μ1 protein also contributes to induction of apoptosis in reovirus-infected cells (6, 18, 19). T3 and T1 reoviruses differ in many aspects of virus replication and host response. Among them, several phenotypes are associated with T3-derived M2. A single-gene reassortant containing the M2 gene from T3D strain in an otherwise T1L genetic background (T1L/T3DM2) has increased attachment to host cells compared to T1L (20). Enhanced binding of T1L/T3DM2 correlated with greater infectivity and cell death induction in cultured cells. These observations suggest that the reovirus M2 gene, and by extension the μ1 protein, can influence reovirus tropism and disease in vivo. The presence of T3D M2 also allows more efficient conformational changes that occur during cell entry. Consequently, membrane penetration and core delivery are also more efficient for T3D M2-containing viruses (21–23). The higher apoptotic potential of T3 strains in comparison to T1 strains also maps to the μ1-encoding M2 gene segment (18, 19). The precise function of μ1 in apoptosis induction is unclear, since seemingly contradictory data exist to support roles for both incoming μ1 from capsids and newly synthesized μ1 produced during virus replication (18, 24–26). Nonetheless, mutations in the ϕ domain of T3D μ1 diminish the apoptotic capacity of reovirus in vivo which is associated with attenuated virulence (27).
Here, we assessed the function of M2 in reovirus pathogenesis. We found that in contrast to T1L, T1L/T3DM2 was lethal in neonatal mice. T1L/T3DM2 replicated to higher titers than T1L in all organs tested. Hearts from mice infected with T1L/T3DM2 displayed signs of overt myocarditis, including extensive purulent lesions with marked inflammatory infiltrate. In contrast, hearts from T1L-infected animals appeared largely normal or had small lesions, which is consistent with T1L being mildly myocarditic. Together, these findings indicate that the T3D M2 gene is a determinant of reovirus-induced myocarditis.
RESULTS
T1L/T3DM2 is more virulent than T1L in neonatal mice.
To determine whether the T3D M2 allele modulates reovirus virulence, we assessed survival of neonatal C57BL/6 mice infected orally with T1L or T1L/T3DM2 viruses (Fig. 1A). Consistent with previous findings (6), all mice inoculated with T1L survived. In contrast, 90% of mice succumbed to infection with T1L/T3DM2. Similar results were obtained with intracranial inoculation (Fig. 1B). Following T1L infection, 11 of 12 (91.7%) mice survived, whereas 1 of 14 (7.1%) mice survived T1L/T3DM2 infection. These data indicate that replacing the T1L M2 gene with the T3D M2 allele converts the nonlethal T1L strain into a lethal pathogen in neonatal mice. Further, because infection with T1L/T3DM2 was lethal following both peroral and intracranial inoculation, our work indicates that the effect of T3D M2 on reovirus virulence is independent of the inoculation route.
FIG 1.

T1L/T3DM2 is more virulent than T1L in neonatal mice. (A) Neonatal C57BL/6 mice were inoculated orally with 104 PFU of T1L or T1L/T3DM2 viruses. Mice were monitored for 25 days. (B) Neonatal C57BL/6 mice were inoculated intracranially with 100 PFU of T1L or T1L/T3DM2 viruses. Mice were monitored for 20 days. ****, P < 0.0001 (determined by the log-rank test, in comparison to T1L).
T1L/T3DM2 replicates and disseminates more efficiently than T1L in vivo.
We next measured viral burdens following oral inoculation with T1L or T1L/T3DM2 viruses (Fig. 2). At days 1 and 2 postinfection, T1L/T3DM2 produced higher viral titers in the intestine than T1L. However, T1L and T1L/T3DM2 intestinal viral titers equilibrated by day 4. In secondary organs (liver, heart, brain, and spleen), T1L/T3DM2 produced higher viral loads than T1L at most time points and achieved higher peak viral titers than T1L in each organ tested. The largest difference in peak viral titers between T1L/T3DM2 and T1L was observed in the heart, where T1L/T3DM2 viral loads were approximately 50-fold higher than T1L at 8 days postinfection.
FIG 2.

T1L/T3DM2 disseminates more efficiently than T1L following oral inoculation. Neonatal C57BL/6 mice were inoculated orally with 104 PFU of T1L or T1L/T3DM2 viruses. At days 1, 2, 4, and 8, mice were euthanized and the indicated organs were resected. Viral titers in each organ were determined by plaque assay. Error bars represent standard deviations (SD). *, P < 0.05; **, P < 0.01; ***, P < 0.001 (determined by the Mann-Whitney test).
We also assessed virulence following intracranial (i.c.) inoculation. Similar to oral inoculation, T1 reoviruses also disseminate systemically following i.c. inoculation (28, 29). As we observed following oral inoculation, T1L/T3DM2 produced higher viral titers at earlier time points and had higher peak viral loads than T1L in each organ when virus was introduced i.c. (Fig. 3). Viral titers in the heart were higher following i.c. inoculation than with oral inoculation at day 4 (Fig. 2 versus Fig. 3). Reovirus may disseminate more rapidly after i.c. inoculation than following oral inoculation, possibly due to the virus needing to overcome fewer physiological barriers when introduced into the brain than the intestine. Again, the largest difference in peak viral titers between T1L/T3DM2 and T1L was found in the heart. T1L/T3DM2 viral loads were 10-fold higher than those of T1L at 8 days postinfection. Together, these data indicate that the M2 allele influences reovirus replication and dissemination in vivo.
FIG 3.

T1L/T3DM2 disseminates more efficiently than T1L following intracranial inoculation. Neonatal C57BL/6 mice were inoculated intracranially with 100 PFU of T1L or T1L/T3DM2 viruses. At days 1, 2, 4, and 8, mice were euthanized and the indicated organs were resected. Viral titers in each organ were determined by plaque assay. Error bars represent SD. *, P < 0.05; ***, P < 0.001 (determined by the Mann-Whitney test).
Systemic dissemination is required for T3D M2-mediated lethality.
All reovirus serotypes require junctional adhesion molecule A (JAM-A) for systemic dissemination (1, 3, 4). To determine whether systemic spread is required for the virulence differences between T1L and T1L/T3DM2, we measured survival of neonatal JAM-A−/− mice infected with T1L or T1L/T3DM2 viruses (Fig. 4). Following oral inoculation, none of the JAM-A−/− mice succumbed to T1L and 10 of 11 (>90%) mice inoculated with T1L/T3DM2 survived (Fig. 4A). T1L/T3DM2 produced higher viral titers in the intestine than T1L at days 1 and 2 postinfection, but T1L/T3DM2 and T1L viral loads were equal by day 8 (Fig. 4A). Intestinal viral loads in JAM-A−/− mice were comparable to those observed in wild-type mice (Fig. 2 versus Fig. 4A). This observation is consistent with published results demonstrating that JAM-A is dispensable for reovirus replication in the intestine (30). Low levels of virus were found in the heart and brain from a small number of JAM-A−/− mice, but the majority of animals had no detectable virus. Although most JAM-A−/− animals had measurable T1L and T1L/T3DM2 titers in the liver and spleen, viral loads produced in all organs from JAM-A−/− mice were substantially lower than in wild-type mice (Fig. 2 versus Fig. 4A).
FIG 4.

Systemic dissemination is required for T1L/T3DM2 lethality. Neonatal JAM-A−/− mice were inoculated (A) orally with 104 PFU or (B) intracranially with 100 PFU of T1L or T1L/T3DM2 viruses. For survival experiments, mice were monitored for 25 days. For titer experiments, at days 1, 2, 4, and 8, mice were euthanized, and the indicated organs were resected. Viral titers in each organ were determined by plaque assay. Error bars represent SD. *, P < 0.05; **, P < 0.01 (determined by the Mann-Whitney test).
Similar results were obtained following intracranial inoculation. All JAM-A−/− mice infected with T1L or T1L/T3DM2 survived (Fig. 4B). These data indicate that JAM-A expression is required for T1L/T3DM2 lethality in vivo. T1L and T1L/T3DM2 replicated comparably in the brain (Fig. 4B), which is consistent with JAM-A being dispensable for reovirus replication in the central nervous system (CNS) (30). In secondary organs, T1L and T1L/T3DM2 viral titers were below the level of detection at 1 and 2 days postinfection, but some virus was detected in the heart, liver, and spleen at days 4 and 8 (Fig. 4B). However, the viral titers produced in JAM-A−/− mice were substantially lower than the viral loads measured in wild-type animals (Fig. 3 versus Fig. 4B). T1L and T1L/T3DM2 viral titers in the intestine increased over time, and T1L/T3DM2 produced higher intestinal viral loads than T1L at day 8 (Fig. 4B). The dramatic increase in intestinal titers at day 8 may be due to T1L and T1L/T3DM2 establishing viremia in JAM-A−/− mice following i.c. inoculation, albeit at lower levels than in wild-type mice. Once virus reaches the intestine, it can replicate independently of JAM-A to reach peak titers comparable to those observed following oral inoculation (30). Taken together, these data suggest that systemic dissemination is required for the enhanced virulence of T1L/T3DM2 compared to T1L.
T1L/T3DM2 is more myocarditic than T1L.
Regardless of the inoculation route, T1L/T3DM2 was substantially more virulent than T1L in wild-type mice (Fig. 1). During necropsy, we observed that hearts from T1L infected mice were largely normal, with a few mice having small cardiac lesions (Fig. 5A and B). This result is consistent with published reports that T1L is mildly myocarditic (7, 31, 32). In contrast, hearts from T1L/T3DM2-infected mice were grossly abnormal. In some animals, the entire heart appeared opaque, which is indicative of severe myocarditis. These data indicate that substitution with reovirus T3D M2 gene enhances the capacity of T1L to cause cardiac pathology. In JAM-A−/− mice, hearts from T1L- and T1L/T3DM2-infected animals lacked any signs of inflammation (data not shown). This finding indicates that reovirus dissemination to the heart is required for the development of cardiac lesions upon reovirus infection. It also is possible that JAM-A functions as a reovirus receptor for cardiac cells. Thus, in addition to decreased dissemination in JAM-A-deficient mice, any virus reaching the heart may not be able to infect cardiac cells and cause heart pathology.
FIG 5.
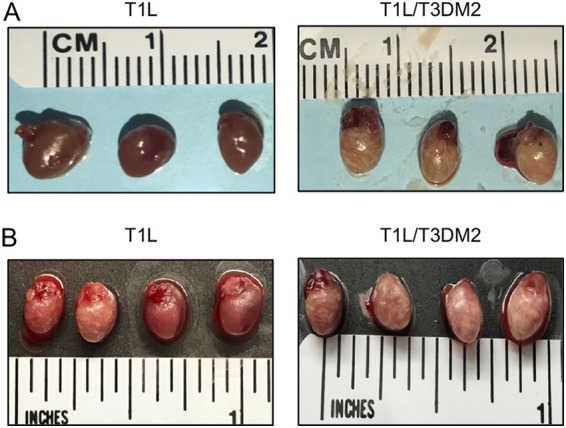
T1L/T3DM2 causes more cardiac pathology than T1L. Neonatal C57BL/6 mice were inoculated (A) orally with 104 PFU or (B) intracranially with 100 PFU of T1L or T1L/T3DM2. At 8 days, mice were euthanized and hearts were resected and imaged.
We next performed histological analysis of hearts from wild-type mice infected with T1L or T1L/T3DM2 (Fig. 6A). In contrast to mock-infected hearts, T1L-infected hearts had small patches of cardiomyocyte necrosis admixed with pyknotic nuclear debris and histiocytes. T1L lesions typically contained two or three cardiomyocytes exhibiting myolysis, which is an indicator of destruction of cardiac muscle tissue. In addition, scant lymphocyte and histiocyte infiltration were observed in the epicardium. These data are consistent with previous observations of T1L inducing mild myocarditis in neonatal wild-type mice. On the other hand, hearts from mice infected with T1L/T3DM2 displayed more frequent cardiomyocyte damage and had increased lymphocytic and histiocytic infiltration in the epicardium compared to T1L-infected hearts (Fig. 6A). We quantified the number of inflammatory lesions (Fig. 6B), lesion size (Fig. 6C), and total percentage of the heart with infiltrating immune cells (Fig. 6D) in hematoxylin-and-eosin (H&E)-stained sections of hearts from mock-, T1L-, and T1L/T3DM2-infected mice. There was no difference in the number of cardiac lesions between T1L- and T1L/T3DM2-infected hearts. This finding suggests that T1L and T1L/T3DM2 initiate infection of the heart at similar rates. Cardiac lesions from mice infected with T1L/T3DM2 were larger, and the overall level of cardiac immune cell infiltration was significantly higher, than in T1L-infected mice (Fig. 6C). Hearts from mice infected with T1L/T3DM2 showed markedly more viral antigen staining than those with T1L (Fig. 6B). This observation is consistent with the higher peak titers produced by T1L/T3DM2 than T1L in the heart at day 8 (Fig. 2 and 3). It is noteworthy that regions of viral antigen staining in T1L- and T1L/T3DM2-infected hearts overlapped myocardial lesions (Fig. 6B). This finding indicates that reovirus replication in cardiomyocytes is associated with lesion formation (11). Lesions from T1L/T3DM2-infected hearts also contained cells that stained positive for activated caspase-3, which is a marker of apoptosis (Fig. 6C). Together, these data indicate that T1L- and T1L/T3DM2-induced cardiac lesions contain reovirus-infected cells, infiltrating immune cells, and apoptotic cells, with T1L/T3DM2 causing more extensive lesion formation than T1L.
FIG 6.

T1L/T3DM2 is more myocarditic than T1L. Neonatal C57BL/6 mice were mock infected with PBS or inoculated orally with 104 PFU of T1L or T1L/T3DM2 viruses. At 8 days, the mice were euthanized, and the hearts were resected, paraffin embedded, and sectioned. Consecutive sections were stained with (A) H&E, (E) polyclonal reovirus antiserum, or (F) cleaved-caspase-3 antibodies. Sections were imaged at ×10 magnification (top) or 20× (bottom). Using H&E-stained sections, (B) cardiac lesion number, (C) lesion size (in arbitrary units [AU]), and (D) total area of inflammation (expressed as percentage of the total heart) were quantified (5 mice per group). Error bars indicate SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant (determined by Student's t test).
DISCUSSION
In this study, we found that the M2 gene is a determinant of reovirus myocarditis. Reovirus strain T1L is nonlethal in neonatal mice, although it induces mild heart inflammation in a subset of animals. However, replacement of the T1L M2 gene with the T3D M2 allele (T1L/T3DM2) was sufficient to confer a lethal phenotype that is characterized by marked inflammation in nearly 100% of infected animals. T1L/T3DM2 replicated to significantly higher titers at the initial site of inoculation (Fig. 2 and 3), disseminated to secondary organs with more rapid kinetics, and produced higher peak organ titers than T1L (Fig. 2 and 3). T1L/T3DM2 binds and infects L929 cells more efficiently than T1L, leading to a higher percentage of cells becoming infected and ultimately production of higher progeny yields than T1L (20). It is possible that T1L/T3DM2 initially infects more cells than T1L, leading to higher progeny yields at the primary site of replication that disseminate systemically and infect target organs more efficiently, including the heart. The greatest difference in peak viral titers between T1L and T1L/T3DM2 was observed in the heart, where T1L/T3DM2 viral progeny were approximately 50-fold and 10-fold higher than T1L following oral and intracranial inoculation, respectively (Fig. 2 and 3). Previous studies indicate that reovirus replication levels in the heart do not correlate with myocarditis induction (10). It is possible that delivery of T1L/T3DM2 virus to the heart more rapidly than T1L allows T1L/T3DM2 to reach a threshold that contributes to the myocarditic capacity of T1L/T3DM2.
Apoptosis is a key pathogenic mechanism that contributes to reovirus myocarditis (6, 33). Clone 8B induces caspase-3 activation within the myocardium of infected hearts, enhancing myocardial injury (34). Moreover, myocarditis development is dramatically reduced in caspase-3-deficient mice and mice treated with caspase inhibitors. (34–36). Reoviruses display serotype-specific differences in apoptosis induction, with T3 reoviruses inducing substantially more apoptosis than T1 strains (18). A determinant of reovirus apoptosis is the M2 gene (6, 18, 27). We found that hearts from mice infected with T1L/T3DM2 had more prominent activated caspase-3 staining than T1L-infected hearts (Fig. 6). Therefore, we hypothesize that the T3D M2 allele enhances apoptosis in infected cells, which promotes myocardial injury and inflammation, which is the hallmark of myocarditis.
The inflammatory response also may contribute to the enhanced myocarditic capacity of the T1L/T3DM2 virus. The adaptive immune response is dispensable for reovirus-induced myocarditis (6, 37). However, innate immune cells may contribute to the heart inflammation and cell death observed following infection with the T1L/T3DM2 virus. Similar to what we observed for T1L/T3DM2, clone 8B induced significant mononuclear cardiac infiltration (11). The myocarditic reovirus strain T1L/T3Dμ1σ3, which contains the M2 and S4 genes from T3D, elicits substantial proinflammatory cytokine production in the heart that promotes cardiomyocyte damage (12).
T1L is mildly myocarditic, whereas T3D is nonmyocarditic (6, 7, 31). However, reassortant viruses generated from cells coinfected with T1L and T3D have the capacity to cause severe myocarditis (11, 38). A number of highly myocarditic reassortants contain the T3D M2 gene. Clone 8B contains the S1 and M2 genes from T3D, while clone KC19 contains the T3D S1, M2, and S3 genes (7, 11). Clone EW60 is genetically similar to the T1L/T3DM2 virus used in this study, as it is a single-gene reassortant containing the T3D M2 gene in a T1L genetic background (31). Clone E3, a single-gene reassortant with the T1L M2 gene in an otherwise T3D genetic background, is nonmyocarditic in neonatal mice (8). Together, these findings suggest that reovirus myocarditic capacity can be modulated by the M2 gene. Here, we used T1L/T3DM2 produced using reverse genetics to clarify and characterize how M2 contributes to reovirus myocarditis. However, the M2 gene is not the sole determinant of reovirus myocarditis. The M1 gene, which encodes inner core protein μ2, which functions as a cofactor for the reovirus RNA-dependent RNA polymerase, also is a determinant of reovirus myocarditis (6, 8, 10, 32). M1 enhances reovirus myocarditis by potentiating replication in cardiac cells and modulating the sensitivity to type-1 interferon (8, 39). In addition, distinct T3D clones have evolved as the virus is passaged by different laboratories, and these clones can differ phenotypically (40–42). Comparing the myocarditic capacity of M2 genes from different T3D clones may provide insight into how M2 causes myocarditis. There may not be a clear-cut mechanism underlying how T1L/T3DM2 enhances myocarditis. It is possible that enhanced dissemination to and replication in the heart, coupled with amplified apoptotic capacity, combine to increase the level of cardiac damage induced by T1L/T3DM2. In turn, the recruitment of immune cells to fight the infection may further exacerbate cardiac damage by producing proinflammatory cytokines and directly or indirectly killing cells.
Other models of virus-induced myocarditis share similarities with reovirus (43–46). SARS-CoV-2 replication in the myocardium is hypothesized to drive the pathological changes observed in the heart tissue (47). CVB3 and murine CMV myocarditis require viral replication and apoptosis induction (43–46). Studies of how reovirus causes pathology in cardiac muscle cells can provide valuable information about how the heart combats viral infection.
MATERIALS AND METHODS
Cells.
Spinner-adapted murine L929 fibroblasts were maintained in Joklik’s modified minimum essential medium (JMEM; Sigma) supplemented to contain 5% heat-inactivated fetal bovine serum (FBS; Invitrogen), 2 mM l-glutamine (Invitrogen), 100 U/mL penicillin–100 μg/mL streptomycin (Invitrogen), and 25 μg/mL amphotericin B (Sigma).
Viruses.
Recombinant reoviruses (T1L and T1L/T3DM2) were generated using plasmid-based reverse genetics (48, 49). Reovirus virions were purified from second- or third-passage L929 cell lysates infected with twice-plaque-purified reovirus (50). Reovirus particles were Vertrel (TMC Industries) extracted from infected-cell lysates, layered onto 1.2- to 1.4-g/cm3 CsCl gradients, and centrifuged at 107,240 × g for 18 h or 144,302 × g for 5 h. Virions were collected and dialyzed against dialysis buffer (150 mM NaCl, 15 mM MgCl, and 10 mM Tris-HCl [pH 7.4]). Viral titers were determined by plaque assay using L929 fibroblasts (51). To confirm the viral gene segments, viral dsRNA was purified from virions and separated by SDS-PAGE. Gene segments were visualized by ethidium bromide staining (48).
Mouse experiments.
C57BL/6 mice were obtained from Jackson Laboratory. C57BL/6-JAM-A−/− mice were obtained from the Dermody Laboratory at the University of Pittsburgh Medical Center. Animal husbandry and housing were performed following the guidelines of the Division of Laboratory Animal Medicine (DLAM) at the University of Arkansas for Medical Sciences (UAMS). Neonatal mice (3 to 5 days old) were infected orally with 104 PFU or intracranially with 100 PFU of T1L or T1L/T3DM2. For survival experiments, infected mice were monitored for 20 or 25 days and sacrificed if moribund. Brain, heart, spleen, liver, and intestines were resected at the times indicated in the figure legends and homogenized, and viral titers were determined by plaque assay on L929 cells. Resected hearts were imaged at the times indicated in the figure legends using a hand-held camera.
Histology and immunohistochemistry.
Neonatal C57BL/6 mice (3 to 5 days old) were mock-infected (phosphate-buffered saline [PBS]) or infected orally with 104 PFU of T1L or T1L/T3DM2. At 8 days postinfection, mice were euthanized and the hearts were resected. Vertical longitudinal cuts of the infected hearts were prepared and fixed in methanol Carnoy’s solution for 24 h followed by 70% ethanol until embedding. The fixed hearts were submitted to the UAMS Experimental Pathology Core Laboratory for paraffin embedding and sectioning (4-μm thickness). Cardiac sections were stained with H&E and a pathologist who was blind to the identities of the inocula examined them under a light microscope. ImageJ software was used to count the number of cardiac lesions, defined as distinct area of immune infiltration. For each cardiac section (×10 magnification), the total heart surface area was determined using the HSB (hue saturation and brightness) threshold color tool and measured in arbitrary units (pixels). Using the Magic Wand tool in ImageJ, each area of immune cell aggregate was selected as a cardiac lesion, and the size was measured in arbitrary units (pixels). The total area of inflammation was calculated as a percentage of the total heart section. Results were collected from 5 hearts per infection. Consecutive sections were processed for immunohistochemical staining with reovirus-specific rabbit polyclonal antiserum (1:1,000) or activated caspase-3 (Abcam; 1:100). Images were taken on an EVOS FL Auto 2 (Invitrogen) using the 10× and 20× objectives (133× and 266× total magnification, respectively).
Statistics.
Statistical analysis was performed using Prism software (GraphPad Software Inc.). Differences in survival were determined by the log-rank test. Differences in mean viral titers were determined using the Mann-Whitney nonparametric t test. Differences in cardiac lesion number, size, and total area of inflammation were evaluated using an unpaired Student's t test. P values less than 0.05 were considerate statistically significant.
ACKNOWLEDGMENTS
We thank the Dermody Lab (University of Pittsburgh Medical Center) for providing the JAM-A−/− mice. We are grateful to Jennifer James at the UAMS Experimental Pathology Core for assistance with histological sample preparations.
This research was supported by Public Health Award R01 AI118801 (K.W.B.) and R01 AI110637 (P.D.). Additional support was provided by the Center for Microbial Pathogenesis and Host Inflammatory Response (P20 GM103625).
The authors declare no conflict of interest.
Contributor Information
Karl W. Boehme, Email: kwboehme@uams.edu.
Susana López, Instituto de Biotecnologia/UNAM.
REFERENCES
- 1.Fung G, Luo H, Qiu Y, Yang D, McManus B. 2016. Myocarditis. Circ Res 118:496–514. 10.1161/CIRCRESAHA.115.306573. [DOI] [PubMed] [Google Scholar]
- 2.Opavsky MA, Penninger J, Aitken K, Wen WH, Dawood F, Mak T, Liu P. 1999. Susceptibility to myocarditis is dependent on the response of alphabeta T lymphocytes to coxsackieviral infection. Circ Res 85:551–558. 10.1161/01.res.85.6.551. [DOI] [PubMed] [Google Scholar]
- 3.Bock CT, Klingel K, Kandolf R. 2010. Human parvovirus B19-associated myocarditis. N Engl J Med 362:1248–1249. 10.1056/NEJMc0911362. [DOI] [PubMed] [Google Scholar]
- 4.Duechting A, TschöPe C, Kaiser H, Lamkemeyer T, Tanaka N, Aberle S, Lang F, Torresi J, Kandolf R, Bock C-T. 2008. Human parvovirus B19 NS1 protein modulates inflammatory signaling by activation of STAT3/PIAS3 in human endothelial cells. J Virol 82:7942–7952. 10.1128/JVI.00891-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez-Bermejo JA, Kang S, Rockwood SJ, Simoneau CR, Joy DA, Silva AC, Ramadoss GN, Flanigan WR, Fozouni P, Li H, Chen PY, Nakamura K, Whitman JD, Hanson PJ, McManus BM, Ott M, Conklin BR, McDevitt TC. 2021. SARS-CoV-2 infection of human iPSC-derived cardiac cells reflects cytopathic features in hearts of patients with COVID-19. Sci Transl Med 13:eabf7872. 10.1126/scitranslmed.abf7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dermody TS, Parker JSL, Sherry B. 2013. Orthoreovirus, p 1304–1346. In Knipe DM, Howley PM (ed), Fields virology, 6th ed, vol 2. Lippincott, Williams, & Wilkins, Philadelphia, PA. [Google Scholar]
- 7.Sherry B, Blum MA. 1994. Multiple viral core proteins are determinants of reovirus-induced acute myocarditis. J Virol 68:8461–8465. 10.1128/JVI.68.12.8461-8465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sherry B, Torres J, Blum MA. 1998. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol 72:1314–1323. 10.1128/JVI.72.2.1314-1323.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matoba Y, Sherry B, Fields BN, Smith TW. 1991. Identification of the viral genes responsible for growth of strains of reovirus in cultured mouse heart cells. J Clin Invest 87:1628–1633. 10.1172/JCI115177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherry B, Fields BN. 1989. The reovirus M1 gene, encoding a viral core protein, is associated with the myocarditic phenotype of a reovirus variant. J Virol 63:4850–4856. 10.1128/JVI.63.11.4850-4856.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sherry B, Schoen FJ, Wenske E, Fields BN. 1989. Derivation and characterization of an efficiently myocarditic reovirus variant. J Virol 63:4840–4849. 10.1128/JVI.63.11.4840-4849.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doyle JD, Danthi P, Kendall EA, Ooms LS, Wetzel JD, Dermody TS. 2012. Molecular determinants of proteolytic disassembly of the reovirus outer capsid. J Biol Chem 287:8029–8038. 10.1074/jbc.M111.334854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barton ES, Chappell JD, Connolly JL, Forrest JC, Dermody TS. 2001. Reovirus receptors and apoptosis. Virology 290:173–180. 10.1006/viro.2001.1160. [DOI] [PubMed] [Google Scholar]
- 14.Campbell JA, Schelling P, Wetzel JD, Johnson EM, Forrest JC, Wilson GA, Aurrand-Lions M, Imhof BA, Stehle T, Dermody TS. 2005. Junctional adhesion molecule A serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J Virol 79:7967–7978. 10.1128/JVI.79.13.7967-7978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liemann S, Chandran K, Baker TS, Nibert ML, Harrison SC. 2002. Structure of the reovirus membrane-penetration protein, Mu1, in a complex with is protector protein σ3. Cell 108:283–295. 10.1016/S0092-8674(02)00612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Snyder AJ, Danthi P. 2018. Cleavage of the C-terminal fragment of reovirus mu1 is required for optimal infectivity. J Virol 92:e01848-17. 10.1128/JVI.01848-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snyder AJ, Danthi P. 2018. Infectious subviral particle to membrane penetration active particle (ISVP-to-ISVP*) conversion assay for mammalian orthoreovirus. Bio Protoc 8:e2700. 10.21769/BioProtoc.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tyler KL, Squier MK, Brown AL, Pike B, Willis D, Oberhaus SM, Dermody TS, Cohen JJ. 1996. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes. J Virol 70:7984–7991. 10.1128/JVI.70.11.7984-7991.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oberhaus SM, Dermody TS, Tyler KL. 1998. Apoptosis and the cytopathic effects of reovirus. Curr Top Microbiol Immunol 233:23–49. 10.1007/978-3-642-72095-6_2. [DOI] [PubMed] [Google Scholar]
- 20.Thete D, Snyder AJ, Mainou BA, Danthi P. 2016. Reovirus mu1 protein affects infectivity by altering virus-receptor interactions. J Virol 90:10951–10962. 10.1128/JVI.01843-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lucia-Jandris P, Hooper JW, Fields BN. 1993. Reovirus M2 gene is associated with chromium release from mouse L cells. J Virol 67:5339–5345. 10.1128/JVI.67.9.5339-5345.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandran K, Walker SB, Chen Y, Contreras CM, Schiff LA, Baker TS, Nibert ML. 1999. In vitro recoating of reovirus cores with baculovirus-expressed outer-capsid proteins mu1 and sigma3. J Virol 73:3941–3950. 10.1128/JVI.73.5.3941-3950.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarkar P, Danthi P. 2010. Determinants of strain-specific differences in efficiency of reovirus entry. J Virol 84:12723–12732. 10.1128/JVI.01385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Danthi P, Coffey CM, Parker JS, Abel TW, Dermody TS. 2008. Independent regulation of reovirus membrane penetration and apoptosis by the mu1 phi domain. PLoS Pathog 4:e1000248. 10.1371/journal.ppat.1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Danthi P, Hansberger MW, Campbell JA, Forrest JC, Dermody TS. 2006. JAM-A-independent, antibody-mediated uptake of reovirus into cells leads to apoptosis. J Virol 80:1261–1270. 10.1128/JVI.80.3.1261-1270.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roebke KE, Danthi P. 2019. Cell entry-independent role for the reovirus mu1 protein in regulating necroptosis and the accumulation of viral gene products. J Virol 93:e00199-19. 10.1128/JVI.00199-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oberhaus SM, Smith RL, Clayton GH, Dermody TS, Tyler KL. 1997. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J Virol 71:2100–2106. 10.1128/JVI.71.3.2100-2106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boehme KW, Guglielmi KM, Dermody TS. 2009. Reovirus nonstructural protein sigma1s is required for establishment of viremia and systemic dissemination. Proc Natl Acad Sci USA 106:19986–19991. 10.1073/pnas.0907412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phillips MB, Dina Zita M, Howells MA, Weinkopff T, Boehme KW. 2021. Lymphatic type 1 interferon responses are critical for control of systemic reovirus dissemination. J Virol 95:e02167-20. 10.1128/JVI.02167-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antar AA, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59–71. 10.1016/j.chom.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherry B, Baty CJ, Blum MA. 1996. Reovirus-induced acute myocarditis in mice correlates with viral RNA synthesis rather than generation of infectious virus in cardiac myocytes. J Virol 70:6709–6715. 10.1128/JVI.70.10.6709-6715.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Irvin SC, Zurney J, Ooms LS, Chappell JD, Dermody TS, Sherry B. 2012. A single-amino-acid polymorphism in reovirus protein mu2 determines repression of interferon signaling and modulates myocarditis. J Virol 86:2302–2311. 10.1128/JVI.06236-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeBiasi RL, Robinson BA, Leser JS, Brown RD, Long CS, Clarke P. 2010. Critical role for death-receptor mediated apoptotic signaling in viral myocarditis. J Card Fail 16:901–910. 10.1016/j.cardfail.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeBiasi RL, Robinson BA, Sherry B, Bouchard R, Brown RD, Rizeq M, Long C, Tyler KL. 2004. Caspase inhibition protects against reovirus-induced myocardial injury in vitro and in vivo. J Virol 78:11040–11050. 10.1128/JVI.78.20.11040-11050.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeBiasi RL, Edelstein CL, Sherry B, Tyler KL. 2001. Calpain inhibition protects against virus-induced apoptotic myocardial injury. J Virol 75:351–361. 10.1128/JVI.75.1.351-361.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim M, Hansen KK, Davis L, van Marle G, Gill MJ, Fox JD, Hollenberg MD, Rancourt DE, Lee PW, Yun CO, Johnston RN. 2010. Z-FA-FMK as a novel potent inhibitor of reovirus pathogenesis and oncolysis in vivo. Antivir Ther 15:897–905. 10.3851/IMP1646. [DOI] [PubMed] [Google Scholar]
- 37.Sherry B, Li XY, Tyler KL, Cullen JM, Virgin HW IV.. 1993. Lymphocytes protect against and are not required for reovirus-induced myocarditis. J Virol 67:6119–6124. 10.1128/JVI.67.10.6119-6124.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wenske EA, Chanock SJ, Krata L, Fields BN. 1985. Genetic reassortment of mammalian reoviruses in mice. J Virol 56:613–616. 10.1128/JVI.56.2.613-616.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stewart MJ, Smoak K, Blum MA, Sherry B. 2005. Basal and reovirus-induced beta interferon (IFN-beta) and IFN-beta-stimulated gene expression are cell type specific in the cardiac protective response. J Virol 79:2979–2987. 10.1128/JVI.79.5.2979-2987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohamed A, Clements DR, Gujar SA, Lee PW, Smiley JR, Shmulevitz M. 2020. Single amino acid differences between closely related reovirus T3D lab strains alter oncolytic potency in vitro and in vivo. J Virol 94:e01688-19. 10.1128/JVI.01688-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mohamed A, Konda P, Eaton HE, Gujar S, Smiley JR, Shmulevitz M. 2020. Closely related reovirus lab strains induce opposite expression of RIG-I/IFN-dependent versus -independent host genes, via mechanisms of slow replication versus polymorphisms in dsRNA binding sigma3 respectively. PLoS Pathog 16:e1008803. 10.1371/journal.ppat.1008803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mohamed A, Smiley JR, Shmulevitz M. 2020. Polymorphisms in the most oncolytic reovirus strain confer enhanced cell attachment, transcription, and single-step replication kinetics. J Virol 94:e01937-19. 10.1128/JVI.01937-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lenzo JC, Fairweather D, Cull V, Shellam GR, James Lawson CM. 2002. Characterisation of murine cytomegalovirus myocarditis: cellular infiltration of the heart and virus persistence. J Mol Cell Cardiol 34:629–640. 10.1006/jmcc.2002.2003. [DOI] [PubMed] [Google Scholar]
- 44.Henke A, Launhardt H, Klement K, Stelzner A, Zell R, Munder T. 2000. Apoptosis in coxsackievirus B3-caused diseases: interaction between the capsid protein VP2 and the proapoptotic protein siva. J Virol 74:4284–4290. 10.1128/jvi.74.9.4284-4290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. 1997. Apoptosis in the failing human heart. N Engl J Med 336:1131–1141. 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 46.Colston JT, Chandrasekar B, Freeman GL. 1998. Expression of apoptosis-related proteins in experimental coxsackievirus myocarditis. Cardiovasc Res 38:158–168. 10.1016/S0008-6363(97)00323-4. [DOI] [PubMed] [Google Scholar]
- 47.Zeng JH, Liu YX, Yuan J, Wang FX, Wu WB, Li JX, Wang LF, Gao H, Wang Y, Dong CF, Li YJ, Xie XJ, Feng C, Liu L. 2020. First case of COVID-19 complicated with fulminant myocarditis: a case report and insights. Infection 48:773–777. 10.1007/s15010-020-01424-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi T, Antar AA, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157. 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. 2010. An improved reverse genetics system for mammalian orthoreoviruses. Virology 398:194–200. 10.1016/j.virol.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol 62:246–256. 10.1128/JVI.62.1.246-256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tyler KL, Bronson RT, Byers KB, Fields B. 1985. Molecular basis of viral neurotropism: experimental reovirus infection. Neurology 35:88–92. 10.1212/wnl.35.1.88. [DOI] [PubMed] [Google Scholar]