

ABSTRACT
Hepatitis B virus (HBV) utilizes host DNA repair mechanisms to convert viral relaxed circular DNA (rcDNA) into a persistent viral genome, the covalently closed circular DNA (cccDNA). To identify host factors involved in cccDNA formation, we developed an unbiased approach to discover proteins involved in cccDNA formation by precipitating nuclear rcDNA from induced HepAD38 cells and identifying the coprecipitated proteins by mass spectrometry. DNA damage binding protein 1 (DDB1) surfaced as a hit, coinciding with our previously reported short hairpin RNA (shRNA) screen in which shRNA-DDB1 in HepDES19 cells reduced cccDNA production. DDB1 binding to nuclear rcDNA was confirmed in HepAD38 cells via ChIP-qPCR. DDB1 and DNA damage binding protein 2 (DDB2) form the UV-DDB complex, and the latter senses DNA damage to initiate the global genome nucleotide excision repair (GG-NER) pathway. To investigate the role of the DDB complex in cccDNA formation, DDB2 was knocked out in HepAD38 and HepG2-NTCP cells. In both knockout cell lines, cccDNA formation was stunted significantly, and in HepG2-NTCP-DDB2 knockout cells, downstream indicators of cccDNA such as HBV RNA, HBcAg, and HBeAg were similarly reduced. Knockdown of DDB2 in HBV-infected HepG2-NTCP cells and primary human hepatocytes (PHH) also resulted in cccDNA reduction. Transcomplementation of wild-type DDB2 in HepG2-NTCP-DDB2 knockout cells rescued cccDNA formation and its downstream indicators. However, ectopic expression of DDB2 mutants deficient in DNA binding, DDB1 binding, or ubiquitination failed to rescue cccDNA formation. Our study thus suggests an integral role of UV-DDB, specifically DDB2, in the formation of HBV cccDNA.
IMPORTANCE Serving as a key viral factor for chronic hepatitis B virus (HBV) infection, HBV covalently closed circular DNA (cccDNA) is formed in the cell nucleus from viral relaxed circular DNA (rcDNA) by hijacking host DNA repair machinery. Previous studies have identified several host DNA repair factors involved in cccDNA formation through hypothesis-driven research with some help from RNA interference (RNAi) screening and/or biochemistry approaches. To enrich the landscape of tools for discovering host factors responsible for rcDNA-to-cccDNA conversion, we developed an rcDNA immunoprecipitation paired mass spectrometry assay, which allowed us to pull down nuclear rcDNA in its transitional state to cccDNA and observe the associated host factors. From this assay, we discovered a novel relationship between the UV-DDB complex and cccDNA formation, providing a proof of concept for a more direct discovery of novel HBV DNA-host interactions that can be exploited to develop new cccDNA-targeting antivirals.
KEYWORDS: DNA repair, HBV, UV-DDB, cccDNA
INTRODUCTION
The hepatitis B virus (HBV) is a hepatotropic pathogen that causes acute and chronic hepatitis B in humans, representing a major etiologic agent for cirrhosis and liver cancer (1). The infectious HBV particle contains a partially double-stranded relaxed circular DNA (rcDNA) genome as genetic material (2). To initiate infection, the enveloped HBV virion binds to a hepatocyte-specific receptor sodium taurocholate cotransporting polypeptide (NTCP), which triggers virus entry through the endocytosis pathway (3–6). After de-envelopment, the viral capsid delivers rcDNA into the nucleus, where the rcDNA is converted into a persistent infection form, covalently closed circular DNA (cccDNA), which is chromatinized and serves as the transcription template for viral mRNAs (2, 7). The HBV pregenomic RNA (pgRNA) is packaged into a cytoplasmic viral capsid, inside which the viral polymerase reverse transcribes pgRNA into rcDNA (8, 9). The mature rcDNA can be either secreted as virion DNA or recycled into the nucleus to replenish the cccDNA pool (2, 10–12). Therefore, both the de novo HBV infection and intracellular rcDNA recycling contribute to cccDNA formation, thus maintaining a stable pool of cccDNA in patients with chronic hepatitis B despite antiviral treatment (11).
HBV cccDNA formation is a mandatory step for establishing viral infection due to the “incomplete” and modified nature of rcDNA, which is incapable of transcribing intact viral mRNAs or replicating itself directly. There are five key structural aberrations in rcDNA that must be repaired during cccDNA formation: (i) the plus (positive-sense) strand is incomplete, thus leaving a variable portion of the minus (negative-sense) strand open; (ii) a capped RNA primer, a remnant of pgRNA during reverse transcription, remains connected to the 5′ end of the plus strand; (iii) the viral polymerase is covalently attached to the 5′ end of the minus-strand DNA; (iv) there is a redundant and overlapping portion of the minus-strand DNA termini, creating a flap of DNA hanging from the structure; (v) due to the viral polymerase and terminal redundant sequences, the minus strand is not a closed circular strand but is instead held together by the complementary plus strand spanning the cohesive end region; similarly, the plus strand is not closed due to the incomplete strand and the vestigial RNA primer on the 5′ end, giving rise to the peculiar structure of rcDNA. In a broad sense, cccDNA formation is a dynamic process that involves rcDNA nuclear import, uncoating, and repair, which require both viral and host factors and machineries. While the HBV capsid serves as a vehicle for delivering rcDNA into the nucleus, viral polymerase activity and other nonstructural viral proteins, including precore and HBx, are dispensable in cccDNA formation (13–17). It is generally acknowledged that HBV relies on host DNA replication and repair factors for converting rcDNA to cccDNA in the nucleus, and there have been recent advancements in our understanding of this process with successful identification of several cccDNA formation intermediates and cellular DNA repair factors involved in cccDNA biosynthesis (reviewed in reference 18) (Fig. 1).
FIG 1.

Proposed molecular pathway of HBV cccDNA formation. The plus (+) and minus (−) strands of rcDNA are labeled, and direct repeat 1 (DR1) and DR2 are indicated by rectangles. The terminal modifications of rcDNA are indicated as follows: the dotted line portion of the plus-strand DNA indicates strand incompletion, the capped RNA primer at the 5′ end of (+) strand is shown as a curved line, HBV Pol covalently attached to the 5′ end of the minus strand is illustrated as a filled oval, and the terminal redundancy of the minus strand is labeled with an “r.” The bona fide cccDNA ought to be converted from rcDNA through a series of DNA repair processes that fix the peculiarities on the termini of rcDNA as indicated. The identified DP-rcDNA and CM-rcDNA are putative transitional intermediates during rcDNA-to-cccDNA conversion. The lines drawn between the listed DNA species correspond to the certainty (solid lines) and uncertainty (dotted lines) of their direct relationships, and the cellular DNA repair factors that have been implicated in each DNA transitions are indicated. See the text for more details.
The deproteinated rcDNA (DP-rcDNA; also known as protein-free rcDNA), which has lost the covalently bound viral polymerase, has been discovered as a probable precursor to cccDNA (19–21). DP-rcDNA exists in both the cytoplasm and nucleus, and it appears first in the cytoplasm upon HBV infection (20–22). A recent in-depth analysis of cytoplasmic DP-rcDNA revealed that it lacks the entire viral polymerase at the 5′ end of the negative strand as well as the RNA primer on the 5′ end of the positive strand (23), suggesting that an initial rcDNA repair step(s) takes place in the cytoplasm while the viral capsid is being shuttled to the nucleus. Another potential precursor to cccDNA was discovered to be a closed minus-strand rcDNA (CM-rcDNA), which possesses a covalently closed minus strand but an unligated plus strand (24). Further study suggested that CM-rcDNA is a derivative of DP-rcDNA during rcDNA-to-cccDNA conversion (25).
Along with these potential precursors being discovered, several host factors associated with DNA replication and repair have been implicated to be directly involved in the formation of cccDNA in cell cultures through hypothesis-driven and/or RNA interference (RNAi) and inhibitor screening approaches, including (i) cellular tyrosyl-DNA phosphodiesterase 2 (TDP2), which has been shown to biochemically unlink HBV polymerase from nascent single-strand DNA (ssDNA) or mature rcDNA in vitro (26–29), although inhibition or depletion of TDP2 had no or little effect on DP-rcDNA or cccDNA production in cells (23, 29, 30); (ii) the cell cycle checkpoint protein ataxia-telangiectasia-mutated-and-Rad3-related kinase (ATR) and its major downstream effector checkpoint kinase 1 (CHK1), which protect the 5′ end of DP-rcDNA minus strand from being extensively truncated during CM-rcDNA and cccDNA formation (25); (iii) the flap endonuclease 1 (FEN-1), which may be involved in removal of the 5′ terminal redundancy of the rcDNA minus strand (31); (iv) cellular DNA polymerases (Pol) κ and λ and B-family polymerases α and δ, which are essential to cccDNA formation via de novo infection and intracellular rcDNA recycling, respectively (16, 32); (v) topoisomerase 1 and 2, which have been indicated to play a primary role in the formation of CM-rcDNA and cccDNA, respectively (33); and (vi) cellular DNA ligase 1 and 3, which exert overlapping functions in cccDNA formation, likely by ligating both DNA strands of rcDNA (34). In addition, recent studies with the assembled cell-free cccDNA formation assay confirmed that previously reported host factors involved in DNA lagging strand synthesis, including Pol δ, FEN-1, and LIG1, were necessary for the strand-specific repair of rcDNA to cccDNA and also implicated for the first time that proliferating cell nuclear antigen (PCNA) and replication factor C (RFC), additional core components of DNA lagging-strand synthesis apparatus, are essential to cccDNA formation, within the context of an in vitro assay (35, 36).
Based on the aforementioned viral DNA intermediates and host DNA repair factors involved in cccDNA formation, a preliminary molecular pathway of rcDNA-to-cccDNA conversion can be proposed, though the detailed mechanisms remain largely obscure in terms of the temporospatial distribution of DNA repair factors and the precise bioreactions on each specific viral DNA substrate (Fig. 1). In order to achieve a coherent understanding of cccDNA formation, searching for additional cccDNA intermediates and their associated host factors is a task of top priority. To address this research need, we sought to develop a targeted and unbiased assay to identify host factors associated with nuclear HBV rcDNA by a proteomic approach and revealed a critical role of DNA damage binding proteins in cccDNA formation.
RESULTS
CoIP-MS identified novel interactions between host proteins and nuclear HBV rcDNA.
To accelerate the search for the DNA repair factors involved in cccDNA formation, we developed a new screen to uncover novel interactions between rcDNA and the host DNA repair machinery (Fig. 2). A biotinylated DNA oligonucleotide complementary to the single-stranded region of HBV rcDNA was synthesized and used as bait to pull down nuclear rcDNA together with the binding proteins. To this end, HBV replication was induced in HepAD38 for 6 days in tetracycline (Tet)-free medium, a time point at which nuclear DP-rcDNA has emerged without detectable cccDNA by Southern blotting (20), which is ideal to capture nuclear rcDNA during early transition to mid-transition to cccDNA. The uninduced HepAD38 cells would not produce rcDNA and would therefore serve as a control for nonspecific or integrated HBV DNA binding of the biotinylated oligonucleotide or streptavidin-coated magnetic beads. The nuclear extract was prepared from induced and uninduced cells separately and incubated with the biotinylated oligonucleotide “bait”; then, oligonucleotide-rcDNA-protein complexes were subsequently precipitated with streptavidin-covered paramagnetic beads. It is noted that the nuclear extraction kit used in this assay does not extract chromatin DNA, thus avoiding the potential coprecipitation of HBV transgenes by oligonucleotide bait. On-bead trypsin digestion was then performed, and the digested peptides were analyzed and identified through liquid chromatography paired with mass spectrometry (LC-MS). The UniProt database of human proteins was used to match the peptide fingerprints with their probable protein sources. When the coimmunoprecipitation-mass spectrometry (CoIP-MS) lists from noninduced and induced HepAD38 cells (see Table S1 in the supplemental material) were compared, while many of the proteins were nonspecific to the rcDNA-positive cells, as revealed by the noninduced control, or are not the known DNA repair factors, a few proteins of interest (POI) caught our attention (Fig. 2). Since the MS assay used in this study was qualitative but not quantitative, we thus focused on the protein hits unique to induced samples. Consistent with previous reports, several known proteins or components of pathways involved in cccDNA formation were reidentified in our assay, including topoisomerases, Pol δ, RFC subunits, and ATR pathway-related proteins (25, 33, 35). The potential novel nuclear rcDNA-binding DNA repair factors identified from the assay are the Fanconi anemia group proteins and DNA damage binding protein 1 (DDB1). DDB1 is the large subunit of the UV-DDB complex, a heterodimer comprising DDB1 and a small DDB2 subunit (37), and DDB1 surfaced as a hit in a search for cccDNA formation participants in a previous short hairpin RNA (shRNA) screen (34). Therefore, we prioritized DDB1 for further investigation in this study.
FIG 2.

Nuclear HBV rcDNA CoIP-MS workflow. HepAD38 cells were cultured in Tet-free medium for 6 days to induce HBV pgRNA transcription and DNA replication. Some of the rcDNA, including DP-rcDNA, is transported into the nucleus, where it undergoes a rate-limiting DNA repair to form cccDNA. The uninduced HepAD38 cells served as a control. The nuclear fractions are isolated and incubated with the biotinylated oligonucleotides (rcDNA bait), complementary to the open (minus) strand of rcDNA. Streptavidin-coated magnetic beads were then used to precipitate the oligonucleotide-bound rcDNA and proteins associated with the complex. An on-bead trypsin digestion was performed, and the resulting peptides were analyzed by liquid chromatography-mass spectrometry (LC-MS). See Materials and Methods for technical details. The representative proteins of interest (POI) with known DNA repair functions and their subunits are listed.
UV-DDB complex binds to nuclear rcDNA.
To validate the binding of DDB1 to nuclear rcDNA, we performed a chromatin immunoprecipitation (ChIP) assay in HepAD38 cells induced in Tet-free medium for 6 days, which gave ample time for rcDNA to be accumulated in the nucleus but was before cccDNA became unambiguously detectable. Since the PCR primers used in this assay do not discriminate between rcDNA and cccDNA, the 6-day induction time point allowed a temporal separation between rcDNA and cccDNA populations. Uninduced HepAD38 cells were set as a negative control. The cells were collected, and ChIP coupled with quantitative PCR (ChIP-qPCR) targeting each of the two components of the UV-DDB complex, DDB1 and DDB2, was performed (Fig. 3). Nonimmune serum IgG and anti-HBc served as background and positive controls, respectively. The ChIP-qPCR results showed specific precipitation of rcDNA along with DDB1, DDB2, and HBc, suggesting that the DDB complex does in fact bind to nuclear rcDNA. This also confirmed the validity of the novel rcDNA CoIP-MS assay in its potential ability to identify novel host-HBV DNA interactions. The strong ChIP signal exhibited by anti-HBc indicated a tight association of HBc with nuclear rcDNA, which may exist predominantly in nucleocapsid format after nuclear import, followed by slow rcDNA uncoating and cccDNA formation.
FIG 3.
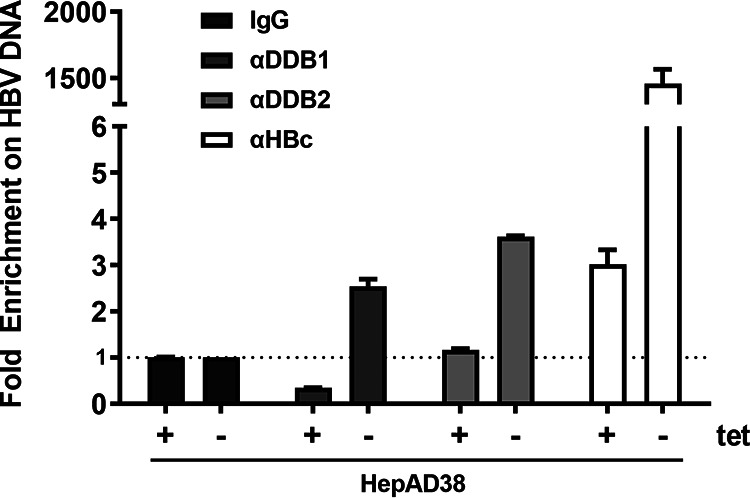
Association of UV-DDB with nuclear rcDNA. HepAD38 cells were induced (Tet-) for 6 days to induce nuclear rcDNA accumulation. A ChIP assay was then conducted with nonimmune serum IgG or antibodies against DDB1, DDB2, and HBV core protein (HBc), followed by qPCR of total HBV DNA. The same rcDNA ChIP-qPCR assay performed on uninduced HepAD38 cells (Tet+) served as a cell background control for the integrated HBV transgene. The ChIP-qPCR results were expressed as fold enrichment over the IgG controls (mean and SD; n = 3).
DDB2 knockout in HepAD38 cells reduces HBV cccDNA formation.
CRISPR RNAs (crRNAs) targeting DDB1 and DDB2 were designed and created. We proceeded to transduce HepAD38 cells with the lentiCRISPR virus encoding DDB1 or DDB2 crRNA. However, DDB1 crRNA failed to produce clones, and we noted that cells treated with the DDB1-crRNA lentivirus suffered extensive cell death prior to puromycin selection. This could suggest that the complete loss of DDB1 may be a lethal mutation to cells, given the importance of DDB1 in host genome stability and cell cycle progression (37, 38). Alternatively, the DDB2 crRNA targeting the third exon in its open reading frame (ORF) successfully knocked out the gene in HepAD38 cells (Fig. 4A). The successful knockout of DDB2 was confirmed through sequencing and Western blotting (Fig. 4A and B). DDB2 is the DNA binding subunit of the UV-DDB complex, and by knocking it out, the UV-DDB’s DNA binding and downstream DNA repair signaling pathway would be completely disrupted (37, 39). The HepAD38 DDB2 knockout (KO) cells were induced for 14 days in Tet-free medium to maximize time allotted to cccDNA formation. In comparison to control KO cells, HBV core DNA replicative intermediates, including rcDNA and DP-rcDNA (the precursors for cccDNA), were slightly decreased in the DDB2 KO cells, accompanied by a more profound reduction of cccDNA, as revealed by Southern blotting. Furthermore, a cccDNA-specific qPCR normalized to total HBV Hirt DNA and cellular mitochondrial DNA confirmed a similar reduction in cccDNA corresponding to the Southern blot results. These results suggest a substantial role for DDB2 in the formation of HBV cccDNA through the rcDNA recycling pathway.
FIG 4.

DDB2 KO in HepAD38 cells reduced cccDNA production. (A) A crRNA (underlined sequence) was designed to target the third exon of the DDB2 gene. The knockout of DDB2 gene in HepAD38 DDB2 KO cells was confirmed by indel-PCR sequencing, which revealed an ORF-disrupting deletion around the protospacer-adjacent motif (PAM) site. The HepAD38 control KO cell line made with scramble crRNA contains wild-type DDB2 sequence. (B) The HepAD38 scramble KO and DDB2 KO cells were induced for 12 days. The expression levels of DDB2 protein were analyzed by Western blotting, with β-actin as a loading control. The cytoplasmic HBV core DNA and total HBV Hirt DNA were analyzed by Southern blotting. The Hirt DNA samples were heated at 85°C for 5 min and subsequently digested with EcoRI before gel loading. Relative levels of HBV rcDNA and cccDNA hybridization signal in each sample are expressed as the percentage of the control (ctrl) and indicated underneath the blots. The Southern blots represent one of two trials. HBV cccDNA was also quantified by using a cccDNA-specific qPCR assay; the cccDNA copy numbers were normalized to total HBV Hirt DNA and mitochondrial DNA and plotted relative to the control (fold change of 1) (mean and SD; n = 3). ***, P < 0.001.
Next, we examined the effect of DDB2 KO on production of CM-rcDNA, a potential intermediate during rcDNA-to-cccDNA conversion. As shown in Fig. 5, after normalizing the input amount of HBV core DNA to an equal amount of rcDNA between control and DDB2 KO HepAD38 cells, the proportional exonuclease I and III (ExoI/III)-treated Hirt DNA samples exhibited a significant reduction of both CM-rcDNA and cccDNA in DDB2 KO cells. The results indicate that the nuclear rcDNA-binding DDB2 may embark on cccDNA formation at a step prior to CM-rcDNA formation.
FIG 5.
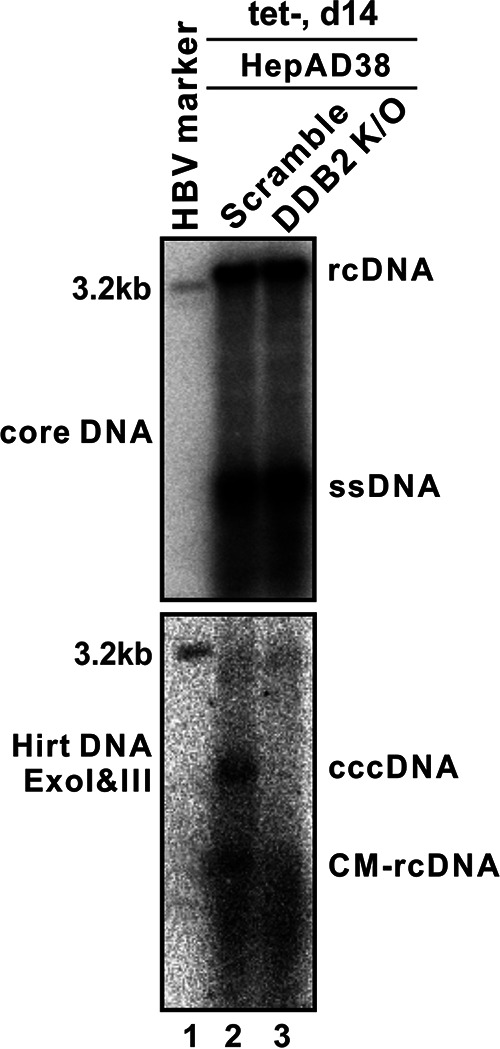
DDB2 KO reduced CM-rcDNA production in HepAD38 Cells. The HepAD38 scramble KO and DDB2 KO cells were induced for 14 days. HBV core DNA was analyzed by Southern blotting (top). Hirt DNA was extracted and treated with ExoI/III, and the remaining cccDNA and the minus-strand circular-DNA moiety of CM-rcDNA were detected by Southern blotting (bottom). The results are representative of three separate trials.
DDB2 siRNA KD in in vitro HBV infection systems reduces de novo cccDNA formation.
Having demonstrated the role of DDB2 in cccDNA formation via rcDNA recycling pathway in HBV stably transfected HepAD38 cells, we set out to investigate its role in de novo HBV cccDNA formation following initial infection. To this end, we used HepG2-NTCP cells as well as primary human hepatocytes (PHH), both of which are permissive to HBV infection and first-round cccDNA formation (40–42). As shown in Fig. 6, DDB2 small interfering RNA (siRNA) efficiently knocked down DDB2 expression in HepG2-NTCP and PHH cells. HBV cccDNA was observed to be largely reduced through Southern blotting and/or qPCR analysis, compared to control siRNA knockdown (KD). DP-rcDNA was essentially undetected in either the control or DDB2 KD cells, which is consistent with previous reports that the rcDNA recycling pathway in the HepG2-NTCP-based HBV infection system is much less efficient than that of HBV stable cell lines (16, 22, 40, 43). Though the underlying mechanism remains unknown, this phenotype makes HepG2-NTCP cells a suitable system for specifically studying the de novo cccDNA formation from the incoming virus. Thus, the reduction of cccDNA mediated by DDB2 transient knockdown in HepG2-NTCP and PHH cells suggests that DDB2 plays an important role in de novo HBV infection and cccDNA formation.
FIG 6.

DDB2 KD reduced cccDNA formation in HBV-infected cells. (A) HepG2-NTCP cells were transfected with control siRNA or DDB2-specific siRNA for 32 h, followed by HBV infection at an MOI of 500 for 6 days. The DDB2 KD was assessed by Western blotting (top). HBV cccDNA was analyzed by Southern blotting after heat denaturation and EcoRI linearization (middle), and the relative level of cccDNA hybridization signal in each sample is expressed as the percentage of the control (ctrl). The cccDNA copy numbers were quantified by qPCR, normalized to mitochondrial DNA, and expressed relative to the control (mean and SD; n = 3) (bottom). (B) PHH cells were transfected with control siRNA or DDB2-specific siRNA for 48 h, followed by HBV infection at an MOI of 500 for 4 days. The levels of DDB2 protein and HBV cccDNA were analyzed by Western blotting and qPCR, respectively, as described for panel A. **, P < 0.01; ***, P < 0.001.
DDB2 KO in HepG2-NTCP cells reduces cccDNA and its downstream indicators upon HBV infection.
To further investigate the dependence on DDB2 by de novo HBV cccDNA formation, we decided to knock out DDB2 expression in HepG2-NTCP cells. The same crRNA designed and used to knock out DDB2 in HepAD38 cells was used in HepG2-NTCP cells. A successful knockout clone was selected through sequencing and Western blotting (Fig. 7A and B). Compared to HBV infection in the scramble control KO cells, the infection of DDB2 KO cells exhibited markedly reduced levels of cccDNA and multiple downstream indicators of cccDNA, such as intracellular HBV RNA and HBcAg (Fig. 7B to D).
FIG 7.

DDB2 KO reduced cccDNA and downstream indicators in HBV-infected HepG2-NTCP cells. (A) The DDB2 crRNA-mediated deletion mutation (dashed line) of the DDB2 gene in HepG2-NTCP DDB2 KO cells was detected by indel PCR sequencing and aligned with the wild-type sequence from HepG2-NTCP scramble control KO cells. (B) The control and DDB2 KO cells were infected by HBV (MOI, 500) for 6 days. The expression levels of DDB2 protein were analyzed by Western blotting, with β-actin as the loading control; the intracellular total HBV RNA was detected by Northern blotting, with rRNA (28S and 18S) as the loading control. (C) cccDNA copy numbers were quantified by qPCR, and normalized to mitochondrial DNA, and expressed relative to the control (mean and SD; n = 3). (D) HepG2-NTCP scramble KO and DDB2 KO cells were infected by HBV or HDV at an MOI of 500 for 6 days. The intracellular HBcAg (top) and HDδAg (bottom) were detected by immunofluorescence. Cell nuclei were counterstained with DAPI. Each image is representative of five microscopic fields; the percent antigen-positive cells is indicated. ***, P < 0.001.
To ensure that DDB2 specifically played a role in de novo cccDNA formation but not the upstream step(s) in viral entry, hepatitis D virus (HDV) infections were carried out in HepG2-NTCP DDB2 KO cells. HDV, a satellite RNA virus to HBV, can propagate only in HBV-infected cells, as it coats itself with the HBV envelope proteins and therefore invades hepatocytes via NTCP identically to HBV but possesses a distinct postentry replication cycle (3, 4). Therefore, if DDB2 was involved in HBV entry but not cccDNA formation, it would also inhibit HDV infection. However, HDV-infected DDB2 KO cells had levels of HDδAg immunofluorescence signals similar to those of HDV-infected scramble KO cells (Fig. 7D), indicating that DDB2 does not play a role in HBV/HDV entry but is rather a critical factor in de novo HBV cccDNA formation.
It is worth noting that DDB2 KO did not completely block cccDNA formation in HBV stable cell lines or an infection model (Fig. 4 and 7), indicating that a redundancy mechanism(s) exists to maintain partial capacity of cccDNA formation or compensate for the loss of DDB2.
Ectopic DDB2 expression rescues cccDNA formation in HepG2-NTCP DDB2 KO cells.
After demonstrating that a loss of DDB2 led to a reduction in cccDNA, we wanted to strategically confirm that the cccDNA reduction was specifically due to DDB2 depletion. We transfected HepG2-NTCP DDB2 KO cells with a wild-type (WT) DDB2 expression plasmid to rescue DDB2 expression, followed by HBV infection. As shown in Fig. 8, Western blotting confirmed that DDB2 expression was restored in DDB2 KO cells transfected by WT DDB2; while the expression of endogenous DDB1 was unchanged in the absence or presence of DDB2 (Fig. 8A, blots), the levels of cccDNA and HBeAg were restored in the DDB2 KO cells upon DDB2 transcomplementation (Fig. 8A, graphs). HBcAg immunofluorescence also showed a rebound of HBcAg expression in the HepG2-NTCP DDB2 KO cells upon restoration of DDB2 expression (Fig. 8B). Collectively, the above results further demonstrated a critical role of DDB2 in cccDNA formation.
FIG 8.

Ectopic DDB2 expression rescued HBV infection in HepG2-NTCP DDB2 KO cells. HepG2-NTCP scramble KO cells and DDB2 KO cells were transfected with control vector or plasmid Flag-DDB2 as indicated for 32 h, followed by HBV infection at an MOI of 500 for 6 days. (A) DDB1 and DDB2 protein expression was detected by Western blotting using antibodies against endogenous DDB1 and DDB2, respectively. The intracellular HBV cccDNA and supernatant HBeAg were analyzed by qPCR and ELISA, respectively, and expressed relative to the control (mean and SD; n = 3). (B) Intracellular HBcAg was detected by immunofluorescence. Cell nuclei were counterstained with DAPI. Each image is representative of five microscopic fields; the percent HBcAg-positive cells is indicated. ***, P < 0.001.
The DNA- and DDB1-binding activity and ubiquitination of DDB2 play a role in establishing HBV infection.
To investigate what functional domains of DDB2 may be involved in the interaction between UV-DDB and HBV cccDNA formation, we employed several DDB2 mutant forms (Fig. 9A), including (i) two naturally occurring mutations, K244E and R273H, involved in the rare genetic disorder xeroderma pigmentosum (type E) (44, 45), which result in structural instability within the protein, leading to their inability to tightly bind damaged DNA (46–48); (ii) the WD motif (amino acids [aa] 238 to 278) and C-terminal-deletion DDB2 mutants (WDΔ, 1–320, and 1–380) that have lost their DDB1-binding ability, leaving DDB2 unable to form the DDB heterodimeric complex (49, 50); (iii) an N7KR ubiquitination mutant which had its seven N-terminal lysine residues replaced with arginine and an Ndel mutant with an N-terminal deletion removing the entire region containing the seven N-terminal lysines (51). The ubiquitination of DDB2 by DDB1-CUL4A E3 ligase is essential for it to hand off the damaged DNA to its downstream effectors for the initiation of the nucleotide excision repair (NER) pathway (48, 52, 53). We transfected the HepG2-NTCP DDB2 KO cells with the above-described mutant DDB2 expression plasmids and confirmed their expression by Western blotting (Fig. 9B). The HepG2-NTCP scramble control KO cells and DDB2 KO cells transfected with WT and mutant DDB2 were infected with HBV for 6 days, and the infection was assessed by HBc immunofluorescence. As observed before, WT DDB2 rescued widespread HBc expression in the DDB2 KO cells; however, the DDB2 mutants all failed to rescue HBV infection in DDB2 KO cells (Fig. 9C). Therefore, the results indicate that, for optimal cccDNA formation, DDB2 must be part of the UV-DDB complex that binds directly to a precursor to cccDNA and signals its downstream effector(s) via its N-terminal ubiquitination.
FIG 9.
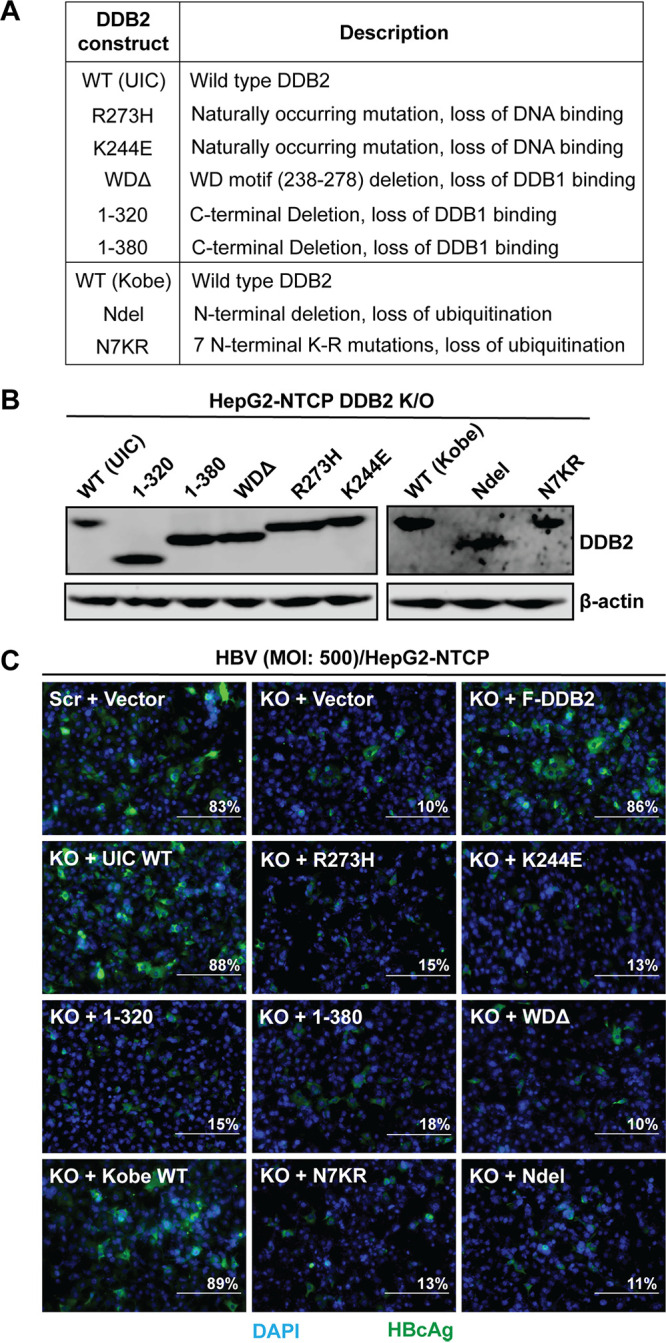
DDB2 loss-of-function mutants were unable to rescue HBV infection in HepG2-NTCP DDB2 KO cells. (A) DDB2 mutant plasmids, with brief descriptions of mutations, and the corresponding wild-type (WT) DDB2 expression plasmids. (B) Each of the plasmids listed in panel A was transfected into HepG2-NTCP DDB2 KO cells for 32 h, followed by HBV infection (MOI, 500) for 6 days. The expression of WT and mutant DDB2 proteins was assessed by Western blotting using antibodies against the T7 tag (left) and the HA tag (right), respectively. β-Actin served as a loading control. (C) Intracellular HBcAg was detected by immunofluorescence. DAPI was used to stain nuclei. HepG2-NTCP scramble KO cells transfected with control vector and HepG2-NTCP DDB2 KO cells transfected with control vector and Flag-DDB2 (F-DDB2) were infected with HBV as described above and served as references for the rescue experiment (top). Each image is representative of five microscopic fields; the percent HBcAg-positive cells is indicated. Bars, 200 μm.
DISCUSSION
The mechanism of cccDNA formation remains a major knowledge gap in our understanding of the HBV life cycle. It is generally acknowledged that HBV rcDNA hijacks host DNA repair machinery to repair it to form cccDNA. To date, most if not all the known host factors involved in cccDNA formation have been discovered by hypothesis testing and/or phenotypic screening (18), but targeted identification of rcDNA-interacting host DNA repair factors has not been reported. For this reason, we sought out an unbiased exploratory assay to discover novel interactions between host factors and cccDNA formation (Fig. 2). With our nuclear rcDNA CoIP-MS assay, we were able to confirm previously reported interactions, such as those with ATR pathway proteins, RFC subunits, multiple host polymerase subunits, and topoisomerases, which validated the assay’s feasibility and practicality.
A limitation of our CoIP-MS assay is the limited region of rcDNA sequence for designing biotinylated DNA oligonucleotide to target and pull down nuclear rcDNA complexes. The oligonucleotide must target the single-stranded region of rcDNA, where a gap of approximately several hundred nucleotides is left between the variable 3′ end of plus-strand DNA and the DR2 region (Fig. 1). Therefore, we designed a 52-nucleotide (nt) oligonucleotide with the immediate upstream sequence of DR2 (nt 1540 to 1591) to maximize the coverage of nuclear rcDNA molecules (Fig. 2). However, we previously found that a portion of cytoplasmic DP-rcDNA population contained the 3′ end of the plus strand beyond nt 1540 or even into the DR2 motif (23), which, if present in the nucleus, would not be efficiently annealed to the oligonucleotide bait. To a lesser extent, the oligonucleotide could possibly competitively bind to these rcDNA species with complete or almost complete but unligated plus strands. However, the hypothetical rcDNA intermediates with a complete and ligated plus strand (36) (if any), the cccDNA, and the integrated HBV transgene would not be able to be pulled down by the rcDNA oligonucleotide bait. On the other hand, the nuclear HBV double-stranded linear DNA (dslDNA) or deproteinated dslDNA, a minor HBV genome by-product resulting from the failed second template switch during rcDNA synthesis (18, 54), could be precipitated by the oligonucleotide bait, granted that the plus strand is incomplete and the minus strand complementary to the oligonucleotide is readily available for binding.
Though dslDNA can be a precursor to cccDNA, the dslDNA-derived cccDNA is commonly observed with extensive indels (19, 55). Studies investigating dsl-DNA-derived cccDNA have discovered accumulating evidence that the nonhomologous end joining (NHEJ) DNA repair mechanism is responsible for the conversion of dslDNA to cccDNA, including the involvement of NHEJ sensor Ku70/Ku80 complex and the NHEJ-specific DNA ligase LIG4 (34, 56). However, Ku70/80 and LIG4 were not detected in the CoIP-MS assay (Table S1), perhaps due to the low abundance of nuclear dslDNA input and/or the sensitivity of the assay. Nonetheless, the small population of incomplete nuclear dslDNA, if any, is not expected to create serious complications in our results. Another potential concern is that there is a possibility that the induced HBV transgene could have single-strand DNA regions exposed for oligonucleotide binding, but the DNA repair factor(s) may not be associated with the integrated HBV DNA if no DNA damage is generated. Furthermore, the nuclear extraction kit used in this study does not extract chromosome DNA. On the other hand, there may be a small number of cccDNA molecules present at 6 days postinduction in this assay, and the cccDNA being actively transcribed may also be targeted by the oligonucleotide bait; however, if cccDNA is precipitated, it would likely be in insignificant amounts, and it also does not interfere with the principle of the assay, as host DNA repair factors could possibly still be bound to the newly formed cccDNA.
With all these considerations in mind, the designed oligonucleotide bait should be able to precipitate a majority of nonencapsidated rcDNA populations within the nucleus, including DP-rcDNA and CM-rcDNA, which contain incomplete and unligated plus strands (19, 20, 23, 24). If we accept these populations to be intermediates to cccDNA formation, as has been implied, then our assay should be able to observe host factors involved in cccDNA formation at least after rcDNA nuclear uncoating and before plus-strand completion and ligation.
The rcDNA CoIP-MS assay delivered a long list of proteins; we specifically focused on the DNA repair proteins that were unique to induced HepAD38 cells. A new nuclear rcDNA-interacting hit that stood out to us was the DDB1 protein, a subunit in the UV-DDB complex and major factor in the global genome NER (GG-NER) pathway (37). Interestingly, DDB1 was also a hit in our previous shRNA screen for host DNA repair genes involved in cccDNA formation (34). UV-DDB is composed of DDB1 and DDB2, with DDB2 being the DNA scanning and binding subunit of the complex (37). In line with this, the ChIP-qPCR assay showed that both subunits of the UV-DDB complex are associated with the nuclear HBV rcDNA (Fig. 3). However, DDB2 was not present in the rcDNA CoIP-MS assay; the reason for this was unclear, but it could perhaps be due to the lack of suitable DDB2-specific proteolytic peptides for mass spectrometry detection. To further investigate the role of UV-DDB in cccDNA formation, we attempted a knockout of DDB1 and DDB2 in HepAD38 cells. The DDB2 knockout was successful and confirmed with sequencing and Western blotting (Fig. 4). However, we failed to establish viable DDB1 knockout cells likely due to DDB1 being essential to cell viability. In line with this, previous studies have demonstrated that the loss of DDB1 leads to cell apoptosis (57–59). Furthermore, in the rare genetic disorder xeroderma pigmentosum (XP), which leaves carriers with extreme sensitivity to UV light-mediated DNA damage, human DDB2 has several natural mutations which lead to this genetic disorder (47), while, to the best of our knowledge, there are no DDB1 mutations described leading to XP, leading us to believe that further efforts to knock out DDB1 would be futile. DDB2 is the DNA-binding and -sensing subunit of the UV-DDB complex (39); therefore, we continued with our focus on assessing the effect of DDB2 on cccDNA formation. The HepAD38 DDB2 KO cells showed a marked decrease in HBV cccDNA formation exclusively mediated by the intracellular rcDNA recycling pathway (Fig. 4).
Furthermore, DDB2 KO resulted in a concurrent reduction of CM-rcDNA (Fig. 5), a putative intermediate precursor for cccDNA formation (24), indicating that DDB2 plays a role in cccDNA formation prior to the covalent circularization of the minus strand of nuclear rcDNA. It is plausible that the binding of DDB2 (or the UV-DDB complex) to nuclear rcDNA recruits downstream factors to repair the minus strand of rcDNA first. It has been reported that topoisomerase 1 and the ATR-CHK1 DNA damage response (DDR) pathway are involved in CM-rcDNA formation (25, 33); thus, there may be a potential interplay among UV-DDB and these factors in CM-rcDNA synthesis. In line with this notion, DDB2 has been shown to recruit ATR kinase to the UV DNA damage sites for initiating DDR (60). Furthermore, as mentioned above, whether the nuclear HBV dslDNA can be sensed by UV-DDB or ATR-CHK1 awaits further investigation.
According to previous studies and this study, the relative proportions of nuclear DP-rcDNA (or PF-rcDNA), CM-rcDNA, and cccDNA are approximately 10 (or more):1:1 in HepAD38 and other inducible HBV stable cell lines (Fig. 4 and 5) (19, 20, 24, 32, 61). Serving as a putative precursor for cccDNA, DP-rcDNA greatly outnumbers CM-rcDNA and cccDNA, but the reason for such an inefficient conversion of DP-rcDNA to CM-rcDNA/cccDNA via the rcDNA recycling pathway is largely unclear. The rate-limiting step(s) is possibly nuclear rcDNA uncoating and/or DNA repair, leaving a significant fraction of DP-rcDNA behind as laggards or even dead ends. As shown in Fig. 3, a large amount of nuclear rcDNA remain associated with HBc, indicating an inefficient rcDNA uncoating in the nucleus. Furthermore, although the cytoplasmic DP-rcDNA exhibits almost homogenous characteristics in terms of DNA terminal sequences and modifications (23), it remains possible that a large portion of DP-rcDNA is further modified upon delivery into the nucleus, making it incapable of cccDNA formation. Thus, further characterizing the nuclear DP-rcDNA molecules in detail would help to address these issues. On the other hand, although the cccDNA formation efficiency appears to be low in in vitro HBV infection systems despite the high virus inoculum used, there is a much lower accumulation of DP-rcDNA compared to that in HBV stable cell lines. In this regard, growing evidence has indicated that certain different regulations of DP-rcDNA and cccDNA formation exist between the intracellular rcDNA recycling and de novo HBV infection (16, 18, 32, 62).
We continued to explore the role of DDB2 in the HBV infection model cell line HepG2-NTCP by knocking down DDB2 with siRNA. This resulted in a greater than 50% reduction in cccDNA formation (Fig. 6A). To further validate the physiological relevance of these findings, we wanted to confirm these results in PHH. The DDB2 KD in PHH cells revealed a similar impact on cccDNA formation (Fig. 6B). CRISPR-based DDB2 KO in HepG2-NTCP cells similarly resulted in a roughly 50% reduction in cccDNA levels following HBV infection (Fig. 7). Furthermore, the reduced cccDNA phenotype in DDB2 KO cells could then be rescued by DDB2 transcomplementation (Fig. 8), suggesting a direct role of DDB2 in cccDNA formation during de novo HBV infection.
Though DDB2 is involved in cccDNA formation through both de novo infection and rcDNA recycling route, a complete loss of cccDNA was not achieved under DDB2 KO conditions, indicating that a compensatory mechanism(s) is in place for cccDNA biosynthesis in the absence of DDB2. In line with this notion, the redundant effects of host DNA repair factors on cccDNA formation have been reported in previous studies, including topoisomerases (33), DNA polymerases (16, 32), and ligases (34). These phenomena indicate that HBV has evolved to take advantage of the host redundant DNA repair systems for maintaining the viral replication cycle.
It appears that the manipulation of DDB2 expression level had a slightly higher impact on viral transcription and antigen expression than cccDNA formation in HBV infection setting (Fig. 7 and 8), which indicated that DDB2 might have an additional effect on cccDNA transcription. In this regard, a well-studied function of DDB1, the binding partner of DDB2 in UV-DDB complex, in the HBV life cycle is its interaction with HBx protein to recruit the CUL4 E3 ubiquitin ligase to ubiquitinate SMC5/6 complex for proteasomal degradation, a process to protect the cccDNA minichromosome from SMC5/6-mediated epigenetic silencing (63–65). However, HBx-independent stimulation of HBV transcription by DDB1 has also been reported (66). Furthermore, it has been reported that DDB2 induces nuclear accumulation of HBx independently of binding to DDB1 (49). Considering that HBx is not required for cccDNA formation but is indispensable for cccDNA transcription (15) and that the loss of DDB2 does not affect the steady-state level of DDB1 (Fig. 8), the potential effect of DDB2 on cccDNA transcription in the context of UV-DDB complex and in the presence or absence of HBx awaits investigation in future studies.
The DDB1 and DDB2 duet uses DDB2 as the DNA damage binding subunit, and DDB1 forms a ubiquitin E3 ligase complex with CUL4A, which in turn ubiquitinates DDB2 and xeroderma pigmentosum complementation group C protein (XPC) to kick off the GG-NER pathway (67, 68). In this scenario, DDB2 also serves as a DDB1- and CUL4-associated-factor (DCAF) like HBx. Indeed, the DDB1-CUL4 complex pairs with dozens of known host and viral DCAFs to ubiquitinate specific protein substrates to regulate a variety of cellular functions (69, 70). In order to determine the domains/functions of DDB2 necessary for its role in HBV cccDNA formation, we assessed the mutant forms of DDB2 for their ability to rescue the DDB2 KO phenotype in HBV infection (Fig. 9). The well-characterized DDB2 mutants contained aberrations inhibiting complexing with DDB1, DNA damage binding, and ubiquitination for downstream signaling/handoff to XPC (Fig. 9A) (44–51). All the mutant DDB2 constructs failed to rescue HBV infection in DDB2 KO cells (Fig. 9C), indicating that DDB2 must be in complex with DDB1 to scan nuclear HBV rcDNA, identify it as damaged, and be ubiquitinated to hand off the rcDNA to the next factor(s) in the cccDNA formation pathway. These observations demonstrate a significant and intimate role played by the UV-DDB complex, or more specifically DDB2, in the crucial life cycle step of HBV cccDNA formation. DDB2 likely plays an early role in sensing the DP-rcDNA or other unidentified rcDNA species upon nuclear entry and signaling for host DNA repair factors to repair rcDNA to cccDNA.
While we have identified DDB2 as an early effector in cccDNA formation, this was concluded mostly from DDB2’s function as a DNA damage-detecting factor and the early-nuclear-rcDNA-selective bias of our assay. In this regard, there is, thus far, a lack of direct evidence for establishing a timeline/chronological order of host factor binding to nuclear rcDNA (12, 18). In this study, one question that remains to be elucidated is the specific binding site of DDB2 along the nuclear rcDNA. Mechanistically, the UV-DDB complex employs DDB2 to continually scan the genome for DNA damage and establishes high-affinity DNA interactions once damage is detected, such as UV-induced cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts (71). DDB2 also exhibits a remarkable substrate diversity to recognize a variety of helix-distorting “blocking” DNA lesions (72–74). Thus, we hypothesize that DDB2 within the UV-DDB complex randomly finds HBV rcDNA upon nuclear import and repeatedly loosely binds to it before finding a specific structure along rcDNA, likely the terminal redundancy of the minus strand and its resulting flap formation or the gap/nick on plus strand DNA, where it will tightly bind and recruit presumably the GG-NER pathway or another unidentified pathway to begin forming CM-rcDNA and then cccDNA (Fig. 10). While this updated model of cccDNA formation awaits further development, it is noteworthy that some late factors in the NER pathway, like PCNA, RFC, polymerases (Pol δ and Pol κ), and ligases (LIG1 and LIG3), have also been implicated in cccDNA formation (16, 31, 32, 34, 35, 68, 75). Therefore, it is of interest to further assess the potential involvement of other GG-NER midstream components in cccDNA formation, such as XPC, transcription factor II H (TFIIH), XPG, etc. Moreover, it will also be interesting to investigate the prevalence and pathophysiology of HBV infection in XP patients.
FIG 10.

Model of UV-DDB-mediated cccDNA formation. The cytoplasmic HBV rcDNA undergoes deproteination to generate DP-rcDNA; the latter is transported into the nucleus and identified as damaged DNA by UV-DDB and/or ATR-CHK1 complexes. While there are potential single-strand break (ssb) sites on DP-rcDNA for ATR to bind to, it is uncertain what structure triggers DDB2 to tightly bind to rcDNA. It is possible that DDB2 and ATR could detect the nick and/or terminal redundancy on minus-strand DNA individually or cooperatively, as both are involved in CM-rcDNA formation. DDB2, in conjunction with DDB1, is ubiquitinated by the CUL4A ubiquitin ligase, facilitating its handoff to a yet-to-be-defined DNA damage response pathway(s). The pathway then assembles DNA repair factors to facilitate the repair of the remaining terminal aberrations of rcDNA into cccDNA. Interestingly, six of the previously reported proteins implicated in cccDNA formation are involved in the late stages of the GG-NER pathway, which is commonly activated by the UV-DDB complex upon substrate anchoring.
In summary, by making use of a newly developed HBV rcDNA CoIP-MS assay, this study has identified UV-DDB as a novel nuclear rcDNA binding factor and established adequate evidence to implicate DDB2, as part of the UV-DDB complex, in the formation of HBV cccDNA, providing new insights into the molecular mechanism of cccDNA formation and potential antiviral targets for treatment of hepatitis B.
MATERIALS AND METHODS
Cell lines.
HepAD38 cells, a tetracycline-inducible HBV stable cell line, were maintained in DMEM/F12 medium (Corning) and supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin, and 1 μg/ml tetracycline (Tet) (20, 76). To induce HBV pgRNA transcription, DNA replication, and cccDNA formation in confluent HepAD38 cells, Tet was withdrawn from the culture medium and cells were cultured up to 14 days. HepG2-NTCP cells, a cell line permissive to HBV infection, were cultured in the same medium as HepAD38 cells without Tet (40). Primary human hepatocytes were purchased from BioIVT (Liverpool 5-donor human cryoplateable hepatocytes, mixed gender; BioIVT, X008052-P, lot SML). PHH were cultured in hepatocyte maintenance medium (HMM; Lonza, CC-3197) supplemented with HMM SingleQuot (Lonza, CC-4192) according to the manufacturer’s instructions. All cell lines were cultured under standard conditions of 37°C and 5% CO2.
Nuclear rcDNA pulldown and mass spectrometry.
A 52-mer oligonucleotide complementary to the HBV minus-strand DNA sequence (nt 1540 to 1591; GenBank no. U95551) was synthesized by Integrated DNA Technologies (IDT), and a biotin tag was ligated to the 5′ end of the oligonucleotide by means of a triethylene glycol spacer (TEG), giving rise to the rcDNA oligonucleotide bait (Table 1), which was further purified by high-performance liquid chromatography (HPLC). HepAD38 cells were cultured in T225 flasks with Tet-free medium to induce HBV replication for 6 days with daily medium change. HepAD38 cells cultured in the presence of Tet for 6 days were used as a negative control. The cells were collected, and the nuclear fractions were isolated using a nuclear extraction kit (Cayman Chemical, 10009277). The obtained 300 μl of nuclear fractions was then incubated with 1.25 nmol of biotinylated rcDNA oligonucleotide bait at 4°C for 16 h under constant gentle rotation. Next, 50 μl of streptavidin-coated MyOneC1 Dynabeads (Thermo Fisher) was added to the reaction and incubated for 4 h at 4°C under constant slow rotation. The paramagnetic beads were then pulled out of solution using a permanent neodymium magnet. The beads were washed in wash buffer containing 5 mM Tris-HCl (pH 7.5), 0.5 mM EDTA, and 1 M NaCl 3 times. The beads were then spun down on a tabletop centrifuge, and excess wash buffer was carefully removed with a micropipette. The rcDNA-bound proteins were reduced, alkylated, and trypsin digested directly on beads, and the resulting peptides were identified with liquid chromatography interfaced with a hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Q Exactive HF Orbitrap liquid chromatography-tandem mass spectrometry [LC-MS/MS] system) at the Purdue University Proteomics Facility. A database search against the UniProt human database (August 2017) was conducted using MaxQuant to identify the probable source proteins of the peptides. A hit list was compiled comparing oligonucleotide pulldown samples from induced (Tet−) versus uninduced (Tet+) conditions to differentiate between potential rcDNA-specific and -nonspecific bound proteins.
TABLE 1.
Oligonucleotides used in this study
| Name | Direction and type | Sequence (5′→3′)a |
|---|---|---|
| rcDNA oligonucleotide bait | Biotin-TEG-ACGCGGACTCCCCGTCTGTGCCTTCTCATCTGCCGGACCGTGTGCACTTCGC | |
| HBV total DNA | Forward primer | CCGTCTGTGCCTTCTCATCTG |
| Reverse primer | AGTCCAAGAGTYCTCTTATGYAAGACCTT | |
| Probe | FAM-CCGTGTGCACTTCGCTTCACCTCTGC-TAMRA | |
| HBV cccDNA | Forward primer | ATGGAGACCACCGTGAACGCCC |
| Reverse primer | TCCCGATACAGAGCTGAGGCGG | |
| Probe | FAM-TTCAAGCCTCCAAGCTGTGCCTTGGGTGGC-TAMRA | |
| Mitochondrial DNA | Forward primer | CCCTCTCGGCCCTCCTAATAACCT |
| Reverse primer | GCCTTCTCGTATAACATCGCGTCA | |
| DDB2 crRNAb | Sense oligonucleotide | CACCGGGAGGGCTACATCCTTGGCG |
| Antisense oligonucleotide | AAACCGCCAAGGATGTAGCCCTCCC | |
| DDB2 indel PCR | Forward primer | TCATTTCTCTCTGTGGCAGGGG |
| Reverse primer | GAACTGCTCACCCCTTTGATGA |
FAM, 6-carboxyfluorescein; TAMRA, 6-carboxytetramethylrhodamine.
The sticky ends for cloning are in italics and underlined.
Chromatin immunoprecipitation.
HepAD38 cells were seeded and grown to confluence in T75 flasks. Once they were confluent, the medium was switched to Tet-free medium for 6 days to induce rcDNA synthesis. After 6 days of induction, the cells were fixed with 1% formaldehyde, and the chromatin immunoprecipitation assay was conducted using the ChIP-IT Express kit with protein G magnetic beads (Active Motif, 53008) according to the manufacturer’s manual, with modifications. Briefly, chromatin was sheared using an EpiShear ultrasound sonicator (Active Motif), with delivery of 50 to 250 J, depending on the cell line, for obtaining chromatin fragments with DNA lengths of around 500 to 1,500 bp. The sheared chromatin was precipitated with 5 μg of a nonimmune serum (NIS) IgG control (Sigma, I8765) or specific antibodies against DDB1 (Abcam, ab97522), DDB2 (Rockland, 100-401-A10) or HBcAg (Dako, B0586). After the removal of formaldehyde cross-links, immunoprecipitated chromatin was deproteinized with 10 μg/ml proteinase K. The input control and immunoprecipitated DNA fractions were purified with a QIAquick PCR purification kit and served as templates for qPCR amplification in a LightCycler 96 system (Roche) using a FastStart essential DNA probe master kit (Roche) with HBV total DNA primers and probes as previously described (34). Occupancy of the specific protein on rcDNA was expressed as fold enrichment above NIS using −ΔΔCq methods.
Plasmids, siRNA, and cell transfection.
The N-terminally Flag-tagged wild-type human DDB2 expression plasmid Flag-DDB2 was purchased from Sino Biological (HG14460-NF). The T7-tagged wild-type DDB2 plasmid WT (UIC) and plasmids expressing DDB2 naturally occurring mutants (K244E and R273H) and WD deletions (ΔWD, 1–320, and 1–380) were provided by Pradip Raychaudhuri (University of Illinois Chicago) (49, 50). The HA-tagged wild-type DDB2 plasmid WT (Kobe) and plasmids expressing an N-terminal deletion (Ndel) and seven N-terminal lysine-to-arginine mutants (N7KR), were provided by Kaoru Sugasawa (Kobe University) (51). The HDV genome replication-competent plasmid pSVL(D3) was provided by Jinhong Chang (Baruch S. Blumberg Institute) (77). Plasmid pLMS, expressing the HBV large (L), middle (M), and small (S) envelope proteins, was provided by Youhua Xie (Fudan University) (78). Plasmid DNA transfections of cells were conducted with Lipofectamine 3000 according to the manufacturer’s directions (Invitrogen). DDB2 siRNA was purchased from Santa Cruz (sc-37799), and cell transfections were performed with Lipofectamine RNAiMAX reagent (Invitrogen).
DDB2 knockout cell line establishment.
DDB2 knockout cell lines were generated through CRISPR-mediated genome editing of DDB2 gene loci. The CRISPR RNA (crRNA) targeting the human DDB2 gene was designed using tools available at http://www.e-crisp.org/E-CRISP. The synthetic scramble and DDB2 crRNA oligonucleotide pairs were annealed and cloned into BsmBI-digested lentiCRISPRv2 control vector (Addgene no. 52961; gift from Feng Zhang). Lentivirus preparation and titration were performed as previously described (34). Lentiviral transduction of HepAD38 or HepG2-NTCP cells and puromycin selection were conducted to generate scramble control and DDB2 stable knockout cell lines. The DDB2 knockout phenotype was confirmed by Western blotting and indel sequencing assay. The corresponding coding sequence for the epitope of the DDB2 antibody has no overlap with the gene targeting sites of the designed crRNA. For indel sequencing analysis of DDB2 genes, total genomic DNA from the control and DDB2 knockout cells was extracted using a DNeasy blood and tissue kit (Qiagen) according to the manufacturer’s protocol. The genomic sequence region covering the crRNA target site was amplified by PCR using the indel detection primers (Table 1) and cloned into T vector pMD19 (Clontech) for Sanger sequencing. The DDB2 DNA sequences from control and knockout cells were aligned to determine the CRISPR-induced mutations.
HBV and HDV infection in vitro.
The infectious HBV inoculum was prepared from the supernatant of the HBV stable cell line HepDE19 as described previously (40). To prepare the HDV inoculum, plasmids pSVL(D3) and pLMS were cotransfected into Huh7 cells, the virus particles were harvested from the supernatant, and virus titer was determined by qPCR according to a previous publication (79). HepG2-NTCP or PHH cells were seeded at a density of 1 × 106 cells/well of a 6-well plate (2 days before infection [D−2]). The next day (D−1), the cells were preincubated for 24 h with HMM (TaKaRa). After 24 h (D0), the medium was replaced with an infection medium prepared with HMM, 4% (wt/wt) polyethylene glycol 8000 (PEG-8000), and HBV at a multiplicity of infection (MOI) of 500 per well. After 16 h inoculation with virus (D1), the medium was replaced with fresh HMM. From day 2 (D2) onward until collection, the medium was changed daily with primary hepatocyte maintenance medium (PMM) (17). HDV infections were performed in an identical manner except that the HDV virus stock was used in place of HBV. When siRNA knockdown was included in the infection experiment, the siRNA transfection was conducted 1 day after cell seeding for the indicated time, followed by the HBV infection workflow described above.
Immunofluorescence.
The HBV- or HDV-infected HepG2-NTCP cells were fixed with 4% paraformaldehyde for 20 min and then washed three times with cold phosphate-buffered saline (PBS) for 5 min each time. The cells were then permeabilized with 0.5% Triton X-100 for 1 h at room temperature and washed again three times with cold PBS. The cells were then incubated with immunofluorescence assay (IFA) blocking buffer for 1 h at room temperature. The cells were stained with rabbit anti-HBcAg (Dako, B0586) or anti-HDδAg (a gift from Severin Gudima) (80) for 16 h at 4°C under constant gentle agitation. The cells were then washed three times with cold PBS and blocked again for 30 min, followed by incubation with Alexa-Fluor 488 dye-conjugated goat anti-rabbit secondary antibody (Thermo Fisher), and the nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole) for 60 min at room temperature. The cells were washed 3 more times and left in PBS. An EVOS M5000 microscope was used to take images of the stained cells under 20× magnification. ImageJ was used for counting cells on the images (81).
ELISA.
HBeAg in the supernatant of HBV-infected HepG2-NTCP cells was detected by an enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s instructions (CUSABIO, CSB-E13557h).
Western blot assay.
Laemmli buffer was used to collect whole-cell lysates, which were run on a 12% SDS-PAGE gel and then transferred onto Immobilon-FL polyvinylidene difluoride (PVDF) membranes (Millipore). The membrane was blocked with Western Breeze blocking buffer (Thermo Fisher) and probed with antibodies against β-actin (Santa Cruz, sc-47778), DDB2 (Santa Cruz, sc-81246), DDB1 (Santa Cruz, sc-137142), Flag tag (Sigma, F3165), hemagglutinin (HA) tag (Sigma, H3663), and T7 tag (Cell Signaling, 13246T).
HBV nucleic acid analyses.
HBV total RNA and core DNA were extracted and subjected to Northern and Southern blotting as previously described (82). HBV cccDNA was extracted by a modified Hirt DNA extraction method (83). For cccDNA Southern blot assay, the Hirt DNA sample was heat denatured at 85°C for 5 min and then digested by EcoRI before gel loading, in order to denature the DP-rcDNA into ssDNA and subsequently linearize cccDNA into double-stranded DNA for improved electrophoretic separation and hybridization (61). To detect CM-rcDNA, the Hirt DNA was treated with 3′→5′ ExoI/III and subjected to Southern blotting assay as previously described (24). A phosphorimager screen was used to record hybridization signals and was scanned using a Typhoon FLA-7000 (GE Healthcare). Hybridization signals were quantified with QuantityOne software (Bio-Rad). The quantitative PCR analysis of cccDNA was conducted with heat-denatured and plasmid-safe ATP-dependent DNase (PSAD)-treated Hirt DNA samples and normalized to mitochondrial DNA according to our previous publication (34).
ACKNOWLEDGMENTS
We thank Jinhong Chang (Blumberg Institute), Severin Gudima (University of Kansas), Pradip Raychaudhuri (University of Illinois), Kaoru Sugasawa (Kobe University), and Youhua Xie (Fudan University) for providing reagents for this study and Bennett Van Houten (University of Pittsburgh) for helpful discussions on the study.
This study was supported by the U.S. National Institutes of Health (NIH) grants R01AI110762, R01AI134818, and R01AI150255 (to H.G.). A.L.M. was partly supported by NIH training grant T32AI060519.
Footnotes
Supplemental material is available online only.
Contributor Information
Haitao Guo, Email: guoh4@upmc.edu.
J.-H. James Ou, University of Southern California.
REFERENCES
- 1.Revill PA, Chisari FV, Block JM, Dandri M, Gehring AJ, Guo H, Hu J, Kramvis A, Lampertico P, Janssen HLA, Levrero M, Li W, Liang TJ, Lim SG, Lu F, Penicaud MC, Tavis JE, Thimme R, Zoulim F, Members of the ICE-HBV Working Gouprs, ICE-HBV Stakeholders GroupChairs, ICE-HBV Senior Advisors. 2019. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol Hepatol 4:545–558. 10.1016/S2468-1253(19)30119-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seeger C, Mason WS. 2015. Molecular biology of hepatitis B virus infection. Virology 479–480:672–686. 10.1016/j.virol.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 1:e00049. 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Falth M, Stindt J, Koniger C, Nassal M, Kubitz R, Sultmann H, Urban S. 2014. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 146:1070–1083. 10.1053/j.gastro.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 5.Huang HC, Chen CC, Chang WC, Tao MH, Huang C. 2012. Entry of hepatitis B virus into immortalized human primary hepatocytes by clathrin-dependent endocytosis. J Virol 86:9443–9453. 10.1128/JVI.00873-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwamoto M, Saso W, Nishioka K, Ohashi H, Sugiyama R, Ryo A, Ohki M, Yun JH, Park SY, Ohshima T, Suzuki R, Aizaki H, Muramatsu M, Matano T, Iwami S, Sureau C, Wakita T, Watashi K. 2020. The machinery for endocytosis of epidermal growth factor receptor coordinates the transport of incoming hepatitis B virus to the endosomal network. J Biol Chem 295:800–807. 10.1074/jbc.AC119.010366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Block TM, Guo H, Guo JT. 2007. Molecular virology of hepatitis B virus for clinicians. Clin Liver Dis 11:685–706, vii. 10.1016/j.cld.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nassal M. 2008. Hepatitis B viruses: reverse transcription a different way. Virus Res 134:235–249. 10.1016/j.virusres.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 9.Hu J, Seeger C. 2015. Hepadnavirus genome replication and persistence. Cold Spring Harb Perspect Med 5:a021386. 10.1101/cshperspect.a021386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nassal M. 2015. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64:1972–1984. 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- 11.Guo JT, Guo H. 2015. Metabolism and function of hepatitis B virus cccDNA: implications for the development of cccDNA-targeting antiviral therapeutics. Antiviral Res 122:91–100. 10.1016/j.antiviral.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xia Y, Guo H. 2020. Hepatitis B virus cccDNA: formation, regulation and therapeutic potential. Antiviral Res 180:104824. 10.1016/j.antiviral.2020.104824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kock J, Schlicht HJ. 1993. Analysis of the earliest steps of hepadnavirus replication: genome repair after infectious entry into hepatocytes does not depend on viral polymerase activity. J Virol 67:4867–4874. 10.1128/JVI.67.8.4867-4874.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moraleda G, Saputelli J, Aldrich CE, Averett D, Condreay L, Mason WS. 1997. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J Virol 71:9392–9399. 10.1128/JVI.71.12.9392-9399.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lucifora J, Arzberger S, Durantel D, Belloni L, Strubin M, Levrero M, Zoulim F, Hantz O, Protzer U. 2011. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol 55:996–1003. 10.1016/j.jhep.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Qi Y, Gao Z, Xu G, Peng B, Liu C, Yan H, Yao Q, Sun G, Liu Y, Tang D, Song Z, He W, Sun Y, Guo JT, Li W. 2016. DNA polymerase kappa is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog 12:e1005893. 10.1371/journal.ppat.1005893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitra B, Wang J, Kim ES, Mao R, Dong M, Liu Y, Zhang J, Guo H. 2019. Hepatitis B virus precore protein p22 inhibits alpha interferon signaling by blocking STAT nuclear translocation. J Virol 93:e00196-19. 10.1128/JVI.00196-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchetti AL, Guo H. 2020. New insights on molecular mechanism of hepatitis B virus covalently closed circular DNA formation. Cells 9:2430. 10.3390/cells9112430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao W, Hu J. 2007. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol 81:6164–6174. 10.1128/JVI.02721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo H, Mao R, Block TM, Guo JT. 2010. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol 84:387–396. 10.1128/JVI.01921-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dezhbord M, Lee S, Kim W, Seong BL, Ryu WS. 2019. Characterization of the molecular events of covalently closed circular DNA synthesis in de novo hepatitis B virus infection of human hepatoma cells. Antiviral Res 163:11–18. 10.1016/j.antiviral.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Cai D, Yan R, Xu JZ, Zhang H, Shen S, Mitra B, Marchetti A, Kim ES, Guo H. 2020. Characterization of the termini of cytoplasmic hepatitis B virus deproteinated relaxed circular DNA. J Virol 95:e00922-20. 10.1128/JVI.00922-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo J, Cui X, Gao L, Hu J. 2017. Identification of an intermediate in hepatitis B virus covalently closed circular (CCC) DNA formation and sensitive and selective CCC DNA detection. J Virol 91:e000539-17. 10.1128/JVI.00539-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo J, Luckenbaugh L, Hu H, Yan Z, Gao L, Hu J. 2020. Involvement of host ATR-CHK1 pathway in hepatitis B virus covalently closed circular DNA formation. mBio 11:e03523-19. 10.1128/mBio.03423-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koniger C, Wingert I, Marsmann M, Rosler C, Beck J, Nassal M. 2014. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc Natl Acad Sci USA 111:E4244–E4253. 10.1073/pnas.1409986111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones SA, Boregowda R, Spratt TE, Hu J. 2012. In vitro epsilon RNA-dependent protein priming activity of human hepatitis B virus polymerase. J Virol 86:5134–5150. 10.1128/JVI.07137-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones SA, Hu J. 2013. Protein-primed terminal transferase activity of hepatitis B virus polymerase. J Virol 87:2563–2576. 10.1128/JVI.02786-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui X, McAllister R, Boregowda R, Sohn JA, Ledesma FC, Caldecott KW, Seeger C, Hu J. 2015. Does tyrosyl DNA phosphodiesterase-2 play a role in hepatitis B virus genome repair? PLoS One 10:e0128401. 10.1371/journal.pone.0128401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winer BY, Huang TS, Pludwinski E, Heller B, Wojcik F, Lipkowitz GE, Parekh A, Cho C, Shrirao A, Muir TW, Novik E, Ploss A. 2017. Long-term hepatitis B infection in a scalable hepatic co-culture system. Nat Commun 8:125. 10.1038/s41467-017-00200-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kitamura K, Que L, Shimadu M, Koura M, Ishihara Y, Wakae K, Nakamura T, Watashi K, Wakita T, Muramatsu M. 2018. Flap endonuclease 1 is involved in cccDNA formation in the hepatitis B virus. PLoS Pathog 14:e1007124. 10.1371/journal.ppat.1007124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang L, Sheraz M, McGrane M, Chang J, Guo JT. 2019. DNA polymerase alpha is essential for intracellular amplification of hepatitis B virus covalently closed circular DNA. PLoS Pathog 15:e1007742. 10.1371/journal.ppat.1007742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheraz M, Cheng J, Tang L, Chang J, Guo JT. 2019. Cellular DNA topoisomerases are required for the synthesis of hepatitis B virus covalently closed circular DNA. J Virol 93:e02230-18. 10.1128/JVI.02230-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Long Q, Yan R, Hu J, Cai D, Mitra B, Kim ES, Marchetti A, Zhang H, Wang S, Liu Y, Huang A, Guo H. 2017. The role of host DNA ligases in hepadnavirus covalently closed circular DNA formation. PLoS Pathog 13:e1006784. 10.1371/journal.ppat.1006784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei L, Ploss A. 2020. Core components of DNA lagging strand synthesis machinery are essential for hepatitis B virus cccDNA formation. Nat Microbiol 5:715–726. 10.1038/s41564-020-0678-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei L, Ploss A. 2021. Hepatitis B virus cccDNA is formed through distinct repair processes of each strand. Nat Commun 12:1591. 10.1038/s41467-021-21850-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beecher M, Kumar N, Jang S, Rapic-Otrin V, Van Houten B. 2020. Expanding molecular roles of UV-DDB: shining light on genome stability and cancer. DNA Repair (Amst)) 94:102860. 10.1016/j.dnarep.2020.102860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iovine B, Iannella ML, Bevilacqua MA. 2011. Damage-specific DNA binding protein 1 (DDB1): a protein with a wide range of functions. Int J Biochem Cell Biol 43:1664–1667. 10.1016/j.biocel.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Jang S, Kumar N, Beckwitt EC, Kong M, Fouquerel E, Rapic-Otrin V, Prasad R, Watkins SC, Khuu C, Majumdar C, David SS, Wilson SH, Bruchez MP, Opresko PL, Van Houten B. 2019. Damage sensor role of UV-DDB during base excision repair. Nat Struct Mol Biol 26:695–703. 10.1038/s41594-019-0261-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan R, Zhang Y, Cai D, Liu Y, Cuconati A, Guo H. 2015. Spinoculation enhances HBV infection in NTCP-reconstituted hepatocytes. PLoS One 10:e0129889. 10.1371/journal.pone.0129889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu J, Lin YY, Chen PJ, Watashi K, Wakita T. 2019. Cell and animal models for studying hepatitis B virus infection and drug development. Gastroenterology 156:338–354. 10.1053/j.gastro.2018.06.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schulze-Bergkamen H, Untergasser A, Dax A, Vogel H, Buchler P, Klar E, Lehnert T, Friess H, Buchler MW, Kirschfink M, Stremmel W, Krammer PH, Muller M, Protzer U. 2003. Primary human hepatocytes–a valuable tool for investigation of apoptosis and hepatitis B virus infection. J Hepatol 38:736–744. 10.1016/s0168-8278(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 43.Ko C, Chakraborty A, Chou WM, Hasreiter J, Wettengel JM, Stadler D, Bester R, Asen T, Zhang K, Wisskirchen K, McKeating JA, Ryu WS, Protzer U. 2018. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J Hepatol 69:1231–1241. 10.1016/j.jhep.2018.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Payne A, Chu G. 1994. Xeroderma pigmentosum group E binding factor recognizes a broad spectrum of DNA damage. Mutation Res 310:89–102. 10.1016/0027-5107(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 45.Tang J, Chu G. 2002. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair (Amst) 1:601–616. 10.1016/S1568-7864(02)00052-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takedachi A, Saijo M, Tanaka K. 2010. DDB2 complex-mediated ubiquitylation around DNA damage is oppositely regulated by XPC and Ku and contributes to the recruitment of XPA. Mol Cell Biol 30:2708–2723. 10.1128/MCB.01460-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feltes BC, Pedebos C, Bonatto D, Verli H. 2018. Dynamics of DDB2-DDB1 complex under different naturally-occurring mutants in Xeroderma Pigmentosum disease. Biochim Biophys Acta Gen Subj 1862:2579–2589. 10.1016/j.bbagen.2018.08.007. [DOI] [PubMed] [Google Scholar]
- 48.Ghodke H, Wang H, Hsieh CL, Woldemeskel S, Watkins SC, Rapić-Otrin V, Van Houten B. 2014. Single-molecule analysis reveals human UV-damaged DNA-binding protein (UV-DDB) dimerizes on DNA via multiple kinetic intermediates. Proc Natl Acad Sci USA 111:E1862–E1871. 10.1073/pnas.1323856111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nag A, Datta A, Yoo K, Bhattacharyya D, Chakrabortty A, Wang X, Slagle BL, Costa RH, Raychaudhuri P. 2001. DDB2 induces nuclear accumulation of the hepatitis B virus X protein independently of binding to DDB1. J Virol 75:10383–10392. 10.1128/JVI.75.21.10383-10392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shiyanov P, Hayes SA, Donepudi M, Nichols AF, Linn S, Slagle BL, Raychaudhuri P. 1999. The naturally occurring mutants of DDB are impaired in stimulating nuclear import of the p125 subunit and E2F1-activated transcription. Mol Cell Biol 19:4935–4943. 10.1128/MCB.19.7.4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsumoto S, Fischer ES, Yasuda T, Dohmae N, Iwai S, Mori T, Nishi R, Yoshino K-i, Sakai W, Hanaoka F, Thomä NH, Sugasawa K. 2015. Functional regulation of the DNA damage-recognition factor DDB2 by ubiquitination and interaction with xeroderma pigmentosum group C protein. Nucleic Acids Res 43:1700–1713. 10.1093/nar/gkv038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sugasawa K. 2009. UV-DDB: a molecular machine linking DNA repair with ubiquitination. DNA Repair (Amst) 8:969–972. 10.1016/j.dnarep.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 53.Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Tanaka K, Hanaoka F. 2005. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 121:387–400. 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 54.Seeger C, Mason WS. 2000. Hepatitis B virus biology. Microbiol Mol Biol Rev 64:51–68. 10.1128/MMBR.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang W, Summers J. 1995. Illegitimate replication of linear hepadnavirus DNA through nonhomologous recombination. J Virol 69:4029–4036. 10.1128/JVI.69.7.4029-4036.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo H, Xu C, Zhou T, Block TM, Guo JT. 2012. Characterization of the host factors required for hepadnavirus covalently closed circular (ccc) DNA formation. PLoS One 7:e43270. 10.1371/journal.pone.0043270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cang Y, Zhang J, Nicholas SA, Kim AL, Zhou P, Goff SP. 2007. DDB1 is essential for genomic stability in developing epidermis. Proc Natl Acad Sci USA 104:2733–2737. 10.1073/pnas.0611311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu Z, Holzschuh J, Driever W. 2015. Loss of DDB1 leads to transcriptional p53 pathway activation in proliferating cells, cell cycle deregulation, and apoptosis in zebrafish embryos. PLoS One 10:e0134299. 10.1371/journal.pone.0134299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cang Y, Zhang J, Nicholas SA, Bastien J, Li B, Zhou P, Goff SP. 2006. Deletion of DDB1 in mouse brain and lens leads to p53-dependent elimination of proliferating cells. Cell 127:929–940. 10.1016/j.cell.2006.09.045. [DOI] [PubMed] [Google Scholar]
- 60.Ray A, Milum K, Battu A, Wani G, Wani AA. 2013. NER initiation factors, DDB2 and XPC, regulate UV radiation response by recruiting ATR and ATM kinases to DNA damage sites. DNA Repair (Amst)) 12:273–283. 10.1016/j.dnarep.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cai D, Wang X, Yan R, Mao R, Liu Y, Ji C, Cuconati A, Guo H. 2016. Establishment of an inducible HBV stable cell line that expresses cccDNA-dependent epitope-tagged HBeAg for screening of cccDNA modulators. Antiviral Res 132:26–37. 10.1016/j.antiviral.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hu J, Tang L, Cheng J, Zhou T, Li Y, Chang J, Zhao Q, Guo JT. 2021. Hepatitis B virus nucleocapsid uncoating: biological consequences and regulation by cellular nucleases. Emerg Microbes Infect 10:852–864. 10.1080/22221751.2021.1919034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hodgson AJ, Hyser JM, Keasler VV, Cang Y, Slagle BL. 2012. Hepatitis B virus regulatory HBx protein binding to DDB1 is required but is not sufficient for maximal HBV replication. Virology 426:73–82. 10.1016/j.virol.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Decorsiere A, Mueller H, van Breugel PC, Abdul F, Gerossier L, Beran RK, Livingston CM, Niu C, Fletcher SP, Hantz O, Strubin M. 2016. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531:386–389. 10.1038/nature17170. [DOI] [PubMed] [Google Scholar]
- 65.Murphy CM, Xu Y, Li F, Nio K, Reszka-Blanco N, Li X, Wu Y, Yu Y, Xiong Y, Su L. 2016. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep 16:2846–2854. 10.1016/j.celrep.2016.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim W, Lee S, Son Y, Ko C, Ryu WS. 2016. DDB1 stimulates viral transcription of hepatitis B virus via HBx-independent mechanisms. J Virol 90:9644–9653. 10.1128/JVI.00977-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li J, Wang QE, Zhu Q, El-Mahdy MA, Wani G, Praetorius-Ibba M, Wani AA. 2006. DNA damage binding protein component DDB1 participates in nucleotide excision repair through DDB2 DNA-binding and cullin 4A ubiquitin ligase activity. Cancer Res 66:8590–8597. 10.1158/0008-5472.CAN-06-1115. [DOI] [PubMed] [Google Scholar]
- 68.Kumar N, Raja S, Van Houten B. 2020. The involvement of nucleotide excision repair proteins in the removal of oxidative DNA damage. Nucleic Acids Res 48:11227–11243. 10.1093/nar/gkaa777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee J, Zhou P. 2007. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol Cell 26:775–780. 10.1016/j.molcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 70.Li T, Robert EI, van Breugel PC, Strubin M, Zheng N. 2010. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat Struct Mol Biol 17:105–111. 10.1038/nsmb.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yeh JI, Levine AS, Du S, Chinte U, Ghodke H, Wang H, Shi H, Hsieh CL, Conway JF, Van Houten B, Rapic-Otrin V. 2012. Damaged DNA induced UV-damaged DNA-binding protein (UV-DDB) dimerization and its roles in chromatinized DNA repair. Proc Natl Acad Sci USA 109:E2737–46. 10.1073/pnas.1110067109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Van Houten B. 1990. Nucleotide excision repair in Escherichia coli. Microbiol Rev 54:18–51. 10.1128/mr.54.1.18-51.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gomez-Pinto I, Cubero E, Kalko SG, Monaco V, van der Marel G, van Boom JH, Orozco M, Gonzalez C. 2004. Effect of bulky lesions on DNA: solution structure of a DNA duplex containing a cholesterol adduct. J Biol Chem 279:24552–24560. 10.1074/jbc.M311751200. [DOI] [PubMed] [Google Scholar]
- 74.Jones KL, Zhang L, Seldeen KL, Gong F. 2010. Detection of bulky DNA lesions: DDB2 at the interface of chromatin and DNA repair in eukaryotes. IUBMB Life 62:803–811. 10.1002/iub.391. [DOI] [PubMed] [Google Scholar]
- 75.Iyama T, Wilson DM. 3rd, 2013. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 12:620–636. 10.1016/j.dnarep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41:1715–1720. 10.1128/AAC.41.8.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chao M, Hsieh SY, Taylor J. 1990. Role of two forms of hepatitis delta virus antigen: evidence for a mechanism of self-limiting genome replication. J Virol 64:5066–5069. 10.1128/JVI.64.10.5066-5069.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hong R, Bai W, Zhai J, Liu W, Li X, Zhang J, Cui X, Zhao X, Ye X, Deng Q, Tiollais P, Wen Y, Liu J, Xie Y. 2013. Novel recombinant hepatitis B virus vectors efficiently deliver protein and RNA encoding genes into primary hepatocytes. J Virol 87:6615–6624. 10.1128/JVI.03328-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gudima S, He Y, Meier A, Chang J, Chen R, Jarnik M, Nicolas E, Bruss V, Taylor J. 2007. Assembly of hepatitis delta virus: particle characterization, including the ability to infect primary human hepatocytes. J Virol 81:3608–3617. 10.1128/JVI.02277-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ryu WS, Bayer M, Taylor J. 1992. Assembly of hepatitis delta virus particles. J Virol 66:2310–2315. 10.1128/JVI.66.4.2310-2315.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu Y, Nie H, Mao R, Mitra B, Cai D, Yan R, Guo JT, Block TM, Mechti N, Guo H. 2017. Interferon-inducible ribonuclease ISG20 inhibits hepatitis B virus replication through directly binding to the epsilon stem-loop structure of viral RNA. PLoS Pathog 13:e1006296. 10.1371/journal.ppat.1006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cai D, Nie H, Yan R, Guo JT, Block TM, Guo H. 2013. A Southern blot assay for detection of hepatitis B virus covalently closed circular DNA from cell cultures. Methods Mol Biol 1030:151–161. 10.1007/978-1-62703-484-5_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1<br>. Download JVI.01360-21-s0001.xlsx, XLSX file, 14.8 MB (14.8MB, xlsx)