

Abstract
Interactions between human leukocyte antigen (HLA) molecules on target cells and the inhibitory killer cell immunoglobulin-like receptors (KIRs) and heterodimeric inhibitory receptor CD94-NKG2A on human natural killer (NK) cells shape and program various response capacities. A functionally orthologous system exists in mice, consisting of major histocompatibility complex (MHC) molecules on target cells and the inhibitory Ly49 and CD94-NKG2A receptors on NK cells. Here, we found that the abundance of Src homology 2 domain–containing phosphatase-1 (SHP-1) in NK cells was established by interactions between MHCs and NK cell inhibitory receptors, although phenotypically identical NK cell populations still showed substantial variability in endogenous SHP-1 abundance and NK cell response potential. Human and mouse NK cell populations with high responsiveness had low SHP-1 abundance, and a reduction in SHP-1 abundance in NK cells enhanced their responsiveness. Computational modeling of NK cell activation by membrane-proximal signaling events identified SHP-1 as a negative amplitude regulator, which was validated by single-cell analysis of human NK cell responsiveness. The amount of mRNA and protein varied among responsive NK cells despite their similar chromatin accessibility to that of unresponsive cells, suggesting dynamic regulation of SHP-1 abundance. Low intracellular SHP-1 abundance was a biomarker of responsive NK cells. Together, these data suggest that enhancing NK cell function through the acute loss of SHP-1 abundance or activity may enhance the tumoricidal capacity of NK cells.
Introduction
Natural killer (NK) cells provide rapid immune surveillance against virally infected cells and tumor cells, while maintaining tolerance to healthy tissues. Most NK cells express inhibitory receptors, such as the killer cell immunoglobulin-like receptors (KIRs) in humans, the Ly49 receptors in mice, and the heterodimeric inhibitory receptor CD94-NKG2A (hereafter referred to as NKG2A) in both humans and mice, all of which recognize major histocompatibility complex (MHC) class I molecules. Inhibitory receptor–expressing NK cells are efficiently inhibited by cognate MHC molecules expressed on host cells as a mechanism of self-tolerance. The same NK cells, however, attack MHClow/neg targets, such as virally infected cells and tumor cells, upon simultaneous triggering by activating receptors (1). This recognition of pathologic loss of self-MHC class I on targets and the subsequent stimulation of effector activity has been described as the “missing self” response (2).
NK cells expressing NKG2A receptors or self-MHC-specific inhibitory KIR/Ly49 receptors are better effectors against MHClow/neg targets than are NK cells lacking these receptors or NK cells expressing receptors for non-self-MHC class I antigens (3, 4). Historically referred to as “licensing” (3) or “disarming” (4), the poorly understood process by which an NK cell becomes tolerized to cells bearing self-MHC class I while simultaneously being endowed with a higher capacity to kill cells lacking self-MHC class I is now generally referred to as “NK cell education,” an active process that lends itself to fine-tuning, referred to as the “rheostat model” (5). How inhibitory signaling promotes and controls NK cell responsiveness is unclear, but it requires interactions between inhibitory KIR/Ly49 or NKG2A receptors on the NK cell and MHC class I molecules on neighboring cells and on the NK cell itself (6,7,8).
Self-MHC–specific inhibitory KIR/Ly49-expressing and NKG2A-expressing NK cells, or so-called “educated NK cells,” are, in general, better effector cells than are “uneducated NK cells” lacking self-MHC–specific receptors, and demonstrate a greater likelihood of target cell conjugation (9), activation (3), and killing of MHCneg targets (3, 4). Educated human NK cells express more DNAM-1 (10) and granzyme B (11), and exhibit higher baseline glycolysis (12, 13). In B6 mice, educated NK cells also express more DNAM-1, but less T cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domains (TIGIT) (14) and Ly49C (15), and exhibit increased basal mammalian target of rapamycin (mTOR) activity (16) and PI3Kδ abundance (17) compared to uneducated NK cells. Furthermore, distinct cytoskeletal distributions and dynamics of activating and inhibitory receptors characterize educated NK cells (18, 19). The loss of genes encoding beta-2 microglobulin (B2M) (3, 20), Ly49 (21), ITIM (22), and SHP-1 (23) proteins in mice eliminates NK cell education, resulting in universal NK cell hyporesponsiveness. Whereas demonstrating that ablation of these molecules underscores their essential roles in NK cell education, no studies have addressed which signaling molecule establishes and adjusts the NK cell response in real time and whether there is a single molecular determinant that digitally controls the all-or-none response at the level of the individual NK cell.
The phosphatase Src homology 2 domain–containing phosphatase-1 (SHP-1) is the protein product of PTPN6 and is expressed predominantly in hematopoietic cells of all lineages and at all stages of maturation (24). In T cells, SHP-1 is a negative regulator of T cell receptor (TCR)–mediated signaling, counterbalancing the activation of protein tyrosine kinases and modulating the magnitude and kinetics of signaling downstream of the TCR (25). Modeling of single-cell responses upon T cell activation reveals that despite stochastic variability in protein abundance among clonal populations, the cell is sensitive to small changes in the amount of SHP-1 (26). In T cells, the coreceptor CD8 acts to adjust the response threshold, whereas SHP-1 functions as a negative amplitude regulator of cell responsiveness (27), where regulators of threshold and amplitude are defined by their respective effects on parameters characterizing the dose-response to stimuli: namely, EC50 (the concentration of ligand required to achieve a 50% response) and amplitude (the maximal response of the cell as the ligand dose approaches infinity) (27). In human NK cells, SHP-1 directly associates with KIR, whose inhibitory function is dependent on its tyrosine phosphorylation and the recruitment and activation of SHP-1 (28). In mice deficient in SHP-1 (23) and in mice transgenic for a catalytically inactive form of SHP-1 (29), NK cells are universally hyporesponsive to tumor cells and MHC class I–deficient target cells. Together, these studies point to a requirement for SHP-1 for categorical NK cell responsiveness, but without establishing exactly what role the protein plays.
We found variability in the abundance of SHP-1 among NK cells, based on inhibitory receptor abundance and MHC environment. In both humans and mice, SHP-1 amounts were reduced at the level of transcript and protein in educated NK cells compared to uneducated NK cells and were modulated by inhibitory receptor density and MHC ligand concentration. The abundance of SHP-1 rapidly responded to changes in environmental MHC, and an acute reduction in SHP-1 increased NK cell responsiveness, indicating its underlying role in the elastic tuning of the NK cell response. Computational modeling of human NK cell responses identified SHP-1 as a negative amplitude regulator, with the KIR-HLA interaction broadly establishing the threshold of the response. Together, these findings establish that the abundance of SHP-1 determines the responsiveness of an individual NK cell. Furthermore, the amount of SHP-1 was more accurate than that of self-MHC-specific receptor as a marker of responsive NK cells, making it a suitable target for the therapeutic enhancement of NK cell activity.
Results
Educated human NK cells exhibit reduced amounts of PTPN6 transcripts and SHP-1 protein
In humans, KIR2DL1, KIR2DL2/3, KIR3DL1, and NKG2A are the inhibitory receptors that contribute to NK cell education. To identify the transcriptional differences between educated and uneducated NK cells, we performed transcriptome profiling of NK cells sorted for single-positive (sp) expression of KIR2DL3, KIR2DL1, and NKG2A, from an individual homozygous for KIR haplotype-A and homozygous for HLA-C1 (fig. S1, A and B). The inhibitory receptor–rich KIR haplotype-A in an HLA-C1 KIR ligand environment designates NK cells that are positive for self-HLA–specific KIR2DL3 and NKG2A as being educated, and cells solely expressing the nonself-HLA–specific KIR2DL1 as being uneducated (30). We found that NK cells that were single-positive for self-MHC–specific receptors express reduced amounts of PTPN6 transcripts when compared with NK cells single-positive for nonself-MHC–specific receptors, linking SHP-1 abundance to education status (fig. S1C).
We validated the link with SHP-1 abundance between educated and uneducated NK cell populations from the same individual by flow cytometric staining of intracellular protein. In experiments with polyclonal and monoclonal antibodies against SHP-1, we confirmed that KIR2DL3sp and NKG2Asp NK cells had less SHP-1 protein compared to KIR2DL1sp and KIR−NKG2A− NK cells (Fig. 1A). In addition to HLA-C1, the donor also exhibited the KIR ligand HLA-Bw4, thereby denoting NK cells expressing the cognate receptor KIR3DL1 as being educated and therefore predicted to be more responsive. Consistent with the findings for the HLA-C1–educated KIR2DL3sp NK cells, the HLA-Bw4–educated KIR3DL1sp NK cells had less SHP-1 when compared to the uneducated KIR2DL1sp and KIR−NKG2A− cells from the same individual (Fig. 1A).
Fig. 1. Low SHP-1 abundance in human NK cells correlates with high responsiveness.
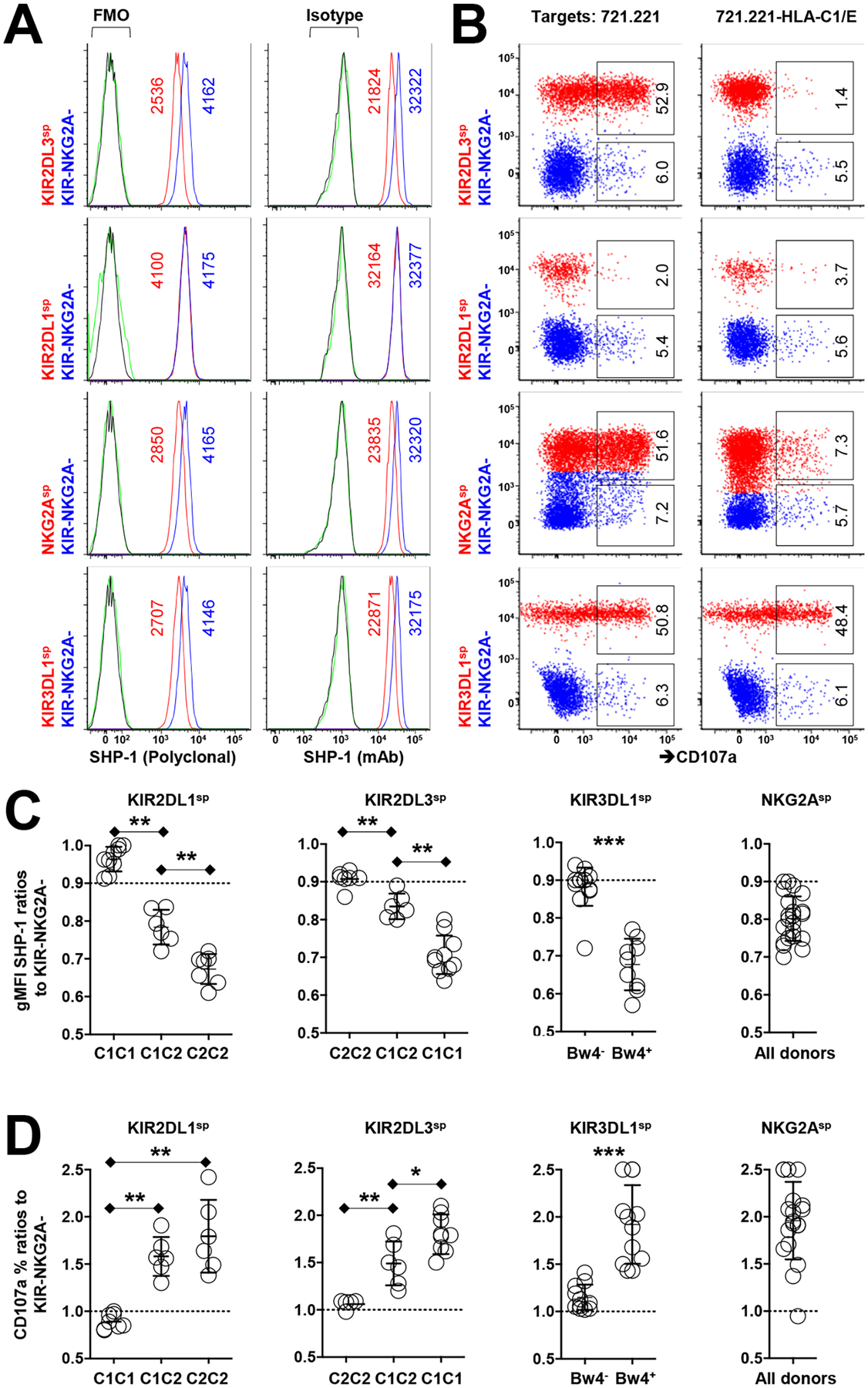
(A) Flow cytometric analysis of relative SHP-1 abundance among the indicated human NK cell subsets (stained with the polyclonal antibody C19 or the monoclonal antibody Y476) from an HLA-C1+C2−Bw4+ donor. gMFI values of SHP-1 are indicated in the plots. Staining controls are indicated in green for KIR+ or NKG2A+ cells and in black for KIR−NKG2A− cells. FMO, fluorescence minus one. (B) Analysis of the degranulation of the indicated NK cell subsets in response to treatment with 721.221 cells or HLA-C1/E-expressing 721.221 cells. Red populations are KIR-expressing or NKG2A-expressing cells. The indicated percentages of positive cells in the dot plots were determined as a percentage of the KIR+ and NKG2A+ single-positive (sp) cells or the KIR−NKG2A− NK cells. (C) Ratio of relative SHP-1 abundance (as determined with the Y476 antibody) among cells single-positive for KIR or NKG2A from 20 KIR haplotype-A homozygous donors. (D) Ratio of the degranulation activity of the indicated NK cell subsets to K562 cells from the indicated HLA genotypes. Data are representative of three (A and B) independent experiments. Data are means ± SD in (C) and (D), n = 20 human donors. *P < 0.05; **P < 0.01; ***P < 0.001; Friedman test and post hoc pairwise Wilcoxon test for multi-group comparison were used. Each symbol represents an individual human donor.
To confirm that SHP-1low cells are more responsive than SHP-1high cells, we evaluated the activity of each NK cell population against the HLA-negative 721.221 target cell line. We found that SHP-1low NK cells responded robustly against 721.221 target cells, as measured by CD107a mobilization, in comparison to SHP-1high NK cells (Fig. 1B). That low amounts of SHP-1 were found among cell populations expressing self-HLA–specific KIRs and that a higher response was associated with low amounts of SHP-1 suggests that SHP-1 abundance determines NK education and the likelihood of a cell to respond to an NK cell–sensitive target.
We measured the amounts of SHP-1 in NK cell populations from an additional 20 different KIR haplotype-A homozygous donors, using the uneducated KIR−NKG2A− NK cells from each donor as an internal control. Normalizing the geometric mean fluorescence intensity (gMFI) of SHP-1 in the cell population of interest to the gMFI of SHP-1 in autologous KIR−NKG2A− NK cells, we demonstrated that SHP-1 amounts were lower in NK cells with surface expression of inhibitory KIR2DL1, KIR2DL3, or KIR3DL1, once the respective cognate HLA ligand was present in the same individual. HLA-E, the ligand for NKG2A, was present universally in all donors, leading to reduced SHP-1 abundance in NKG2A+ cells compared to that in their KIR−NKG2A− counterparts (Fig. 1C). In all cases, an inverse correlation was observed between SHP-1 abundance and the degree of responsiveness of each NK cell population to the HLA-class I negative target cell K562 (Fig. 1D). Consistent with previous studies that demonstrated that ligand copy number titrates NK cell responsiveness (31, 32), KIR2DL3sp and KIR2DL1sp NK cells from donors with two copies of HLA-C1 or HLA-C2, respectively, had less SHP-1 compared to those from donors with only one copy of each ligand. In all comparisons, the greatest amounts of SHP-1 were in the uneducated, hyporesponsive cells from donors lacking the cognate ligand (Fig. 1C). To attempt to understand how SHP-1 abundance was regulated in NK cells, we assessed the chromatin accessibility of PTPN6 from the same human NK cell populations by ATACseq (fig. S1D). We did not observe a difference in chromatin accessibility between the NK cell populations, suggesting that SHP-1 abundance was regulated in a dynamic fashion at the transcriptional level and not due to changes in chromatin availability at PTPN6.
Modeling NK cell responsiveness
In a standard degranulation assay to HLA class I-negative target cells, not all phenotypically identical NK cells expressing inhibitory receptors for self-MHC responded, despite having similar exposure and target cell contacts (Fig. 1B). This suggests a variability in the activation thresholds among NK cells that are traditionally defined by phenotype to be educated. Although being SHP-1low appeared to be a general phenotypic marker for the more responsive, self-MHC inhibitory receptor-bearing NK cells, whether and how SHP-1 regulates single NK cell responsiveness is not clear. In T cell activation, endogenous variation in the abundances of signaling proteins affects antigen responsiveness, where SHP-1 abundance acts as a digital regulator, switching a cell between responsive and unresponsive states in real-time during signaling events (26) and as a negative amplitude regulator (27). Together, these mechanisms ensure that variability in cell-to-cell SHP-1 protein abundance enables a population of cells to make a graded response from decisions that are all-or-none at a single-cell level (27).
Hypothesizing that SHP-1 behaves similarly in NK cells, we applied a quantitative assessment of phosphorylated S6 ribosomal protein (pS6) in individual primary NK cells after short-term stimulation with anti-CD16 antibody (fig. S2A). As a readout of mTORC1 signaling in NK cell activation (33), the abundance of pS6 in human NK cells provides a positivity gradient with increasing intensity of stimulation, a critical feature for the quantitative approach of this study. CD16, a potent activating receptor for NK cells, is found on most CD56dim NK cells, regardless of their education state, thereby providing the opportunity to titrate NK cell activity through CD16 stimulation. CD56bright NK cells were used as a control population because they lack CD16 and cannot be activated in this experimental setting (fig. S2B).
KIR3DL1+ NK cells from HLA-Bw4+ donors are considered to be educated, with a higher likelihood of cytotoxic and cytokine responses to target cells. Among the KIR3DL1+ NK cells, however, there was a range of SHP-1 abundance, where cells expressing lower amounts of SHP-1 demonstrated a higher frequency of pS6 response when activated by antibody stimulation of CD16 (Fig. 2A). This suggests that variation in SHP-1 amount, even among cells whose receptor abundance predicts that they should be similarly educated, controls the NK cell response. The same inverse correlation between SHP-1 abundance and responsiveness was observed when the NK cells were stimulated with cellular targets (Fig. 2B).
Fig. 2. Modeling NK cell responsiveness based on SHP-1 abundance.
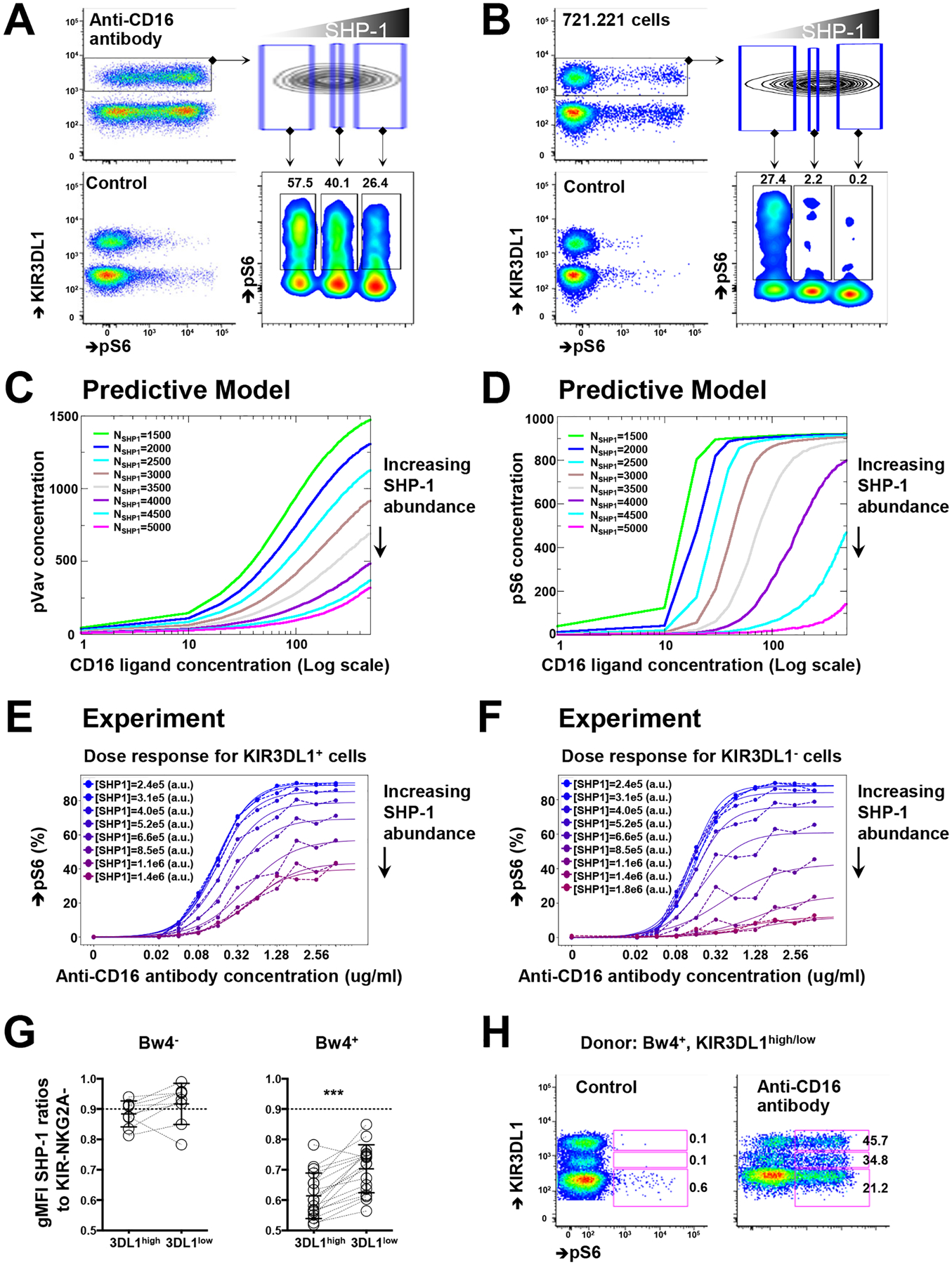
(A) Purified NK cells were treated with the crosslinking anti-CD16 antibody for 5 min and then fixed to detect the relative abundance of pS6 based on the amounts of SHP-1. Unstimulated NK cells served as the control. Plots are representative of six independent experiments. (B) Freshly purified NK cells were incubated with 721.221 cells for 15 min and then fixed to determine the relative amounts of pS6 and SHP-1. Plots are representative of three independent experiments. (C and D) Simulations of the amounts of pVav (C) and pS6 (D) in 4000 cells after CD16 stimulation with increasing concentrations of human IgG. Colored lines indicate equal amounts of SHP-1 molecules in the simulation box (see also Materials and Methods). (E and F) Single-cell analysis of KIR3DL1+ (E) and KIR3DL1− (F) NK cell responses to the indicated concentrations of anti-CD16 antibody for different amounts of SHP-1. Plots are representative of two independent experiments. (G) SHP-1 abundance among KIR3DL1high and KIR3DL1low NK cells from HLA-Bw4− and HLA-BW4+ donors. NK cells from the same donors are paired by lines. (H) pS6 abundance among KIR3DL1high, KIR3DL1low, and KIR3DL1− NK cells after treatment with a crosslinking anti-CD16 antibody. The indicated percentages of positive cells in the dot plots were determined as percentages of the KIR3DL1high, KIR3DL1low, and KIR3DL1− NK cells. Plots are representative of two independent experiments from four donors in total. Data are means ± SD in (G); n = 9 for HLA-Bw4–negative donors and n = 18 for HAL-Bw4–positive donors. ***P < 0.001; nonparametric Wilcoxon signed rank sum test was used. Each symbol represents an individual human donor.
In silico modeling of murine NK cell activation, which is based on the signaling pathway involving membrane-proximal, receptor-ligand interactions, predicts that NK cell activation is digital in nature (34). This model also suggests that stochastic fluctuations in the numbers of signaling molecules, which is the intrinsic random nature of biochemical reactions, determine NK cell activity at the single-cell level. Using such a computational approach, we tested the role of SHP-1 abundance in human NK cell responsiveness by simulating CD16 activation. The mathematical model enables the user to titrate the amount of input ligands (immunoglobulin G, IgG) and to vary the amounts of signaling molecules to measure the abundances of pS6 and phosphorylated Vav (pVav), markers of the NK cell response based on known membrane-proximal, receptor-ligand interactions (fig. 3A) (34). We found that as the total concentration of SHP-1 increased, the amounts of pVav and pS6 decreased directly and rapidly, indicating that SHP-1 is a negative amplitude regulator of human NK cell activation (Fig. 2, C and D). The mathematical model also predicts that the quantitative nature of digital activation does not change with variation in the number of activating ligands (fig. 3B) (34); instead, increased SHP-1 amounts correlated with a decreased amplitude of response without affecting the minimal number of ligands able to stimulate S6 phosphorylation in 50% of the cells (EC50). In other words, the EC50 value reflects the probability of assembling a successful ligand-receptor pair, whereas the amplitude reflects the probability of translating receptor engagement into S6 phosphorylation.
Fig. 3. Functional and transcriptional analyses of responsive murine NK cells.
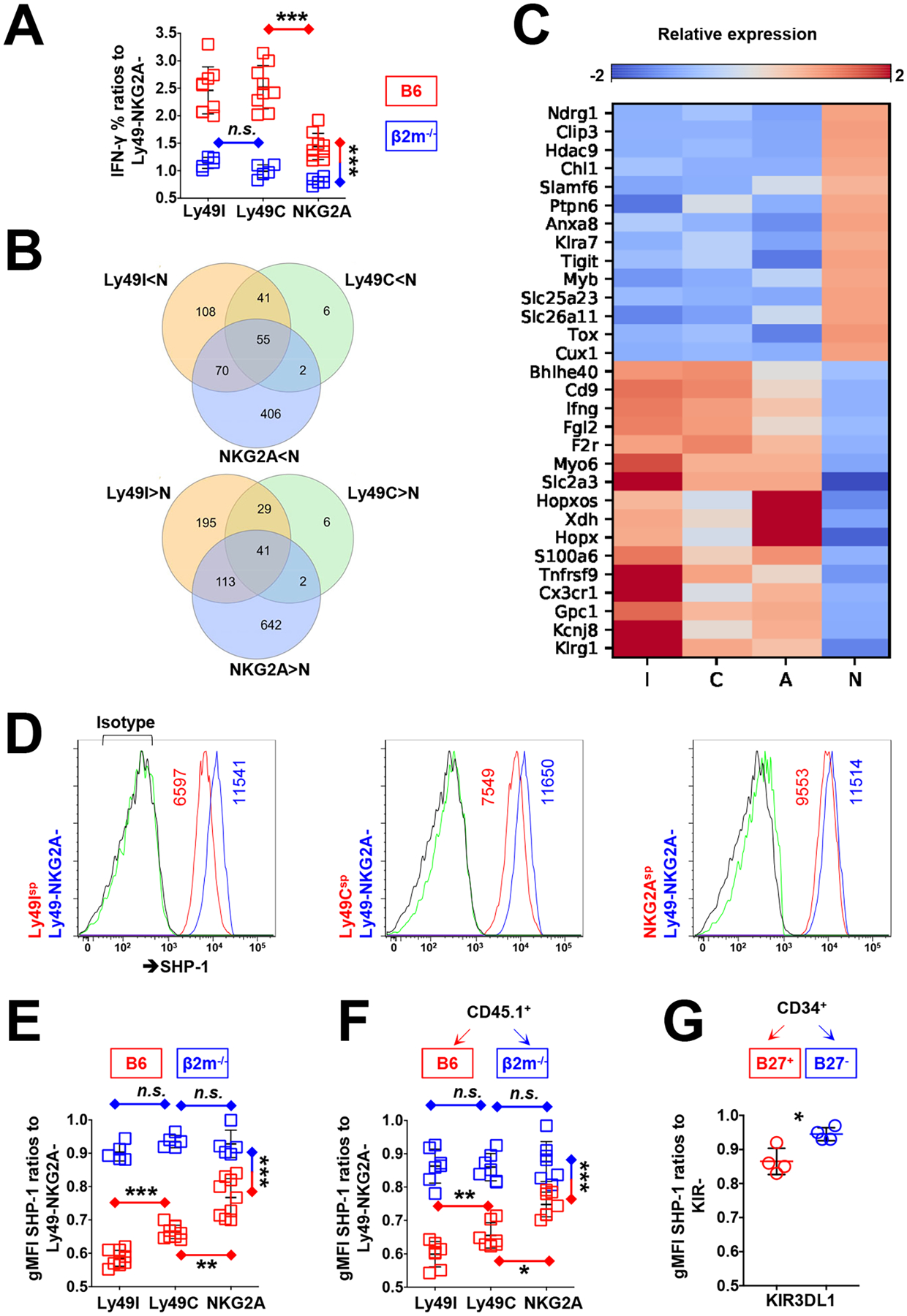
(A) Analysis of the percentages of IFN-γ+ among Ly49I, Ly49C, and NKG2A single-positive NK cells from B6 and β2m−/− mice after treatment with a crosslinking anti-NK1.1 antibody. Results are pooled from two independent experiments. (B) Overlap of differentially expressed genes among Ly49Isp, Ly49Csp, and NKG2Asp NK cells vs. Ly49−NKG2A− (N) NK cells that were sorted from B6 mice. (C) Representative results of differentially expressed genes between nonresponsive and responsive NK cells: Ly49I single-positive (I), Ly49C single-positive (C), and NKG2A single-positive (A). (D) Flow cytometric analysis of SHP-1 abundance among educated and uneducated NK cell populations from B6 mice. gMFI values of SHP-1 are indicated in the plots. Isotype control antibodies are indicated in green (for Ly49+ or NKG2A+ cells) and black lines (for Ly49−NKG2A− cells). (E) Analysis of SHP-1 abundance among the indicated NK cell populations from B6 and β2m−/− mice. Results are pooled from two independent experiments. (F) Analysis of SHP-1 abundance among CD45.1+ NK cells 10 days after their transfer to irradiated B6 or β2m−/− recipient mice. Results are pooled from two independent experiments. (G) Analysis of SHP-1 abundance among KIR3DL1+ NK cells 8 weeks after the transplantation of CD34+ human umbilical cord blood cells into irradiated HLA-B27+ or HLA-B27− NRG mice. Data are representative of two human donors, with cells from each donor used to implant four NRG mice. Data are means ± SD in (A), (E), (F), and (G), n=14, 14, 14, and 8 mice. *P < 0.05; **P < 0.01; ***P < 0.001; Friedman test and post hoc pairwise Wilcoxon test for multi-group comparison (A, E, and F), and Mann-Whitney U test (G) were used. Each symbol represents an individual mouse.
To determine whether this model predicted the responsiveness of primary human NK cells, we demonstrated that the frequency of pS6+ NK cells was titrated by applying differing concentrations of anti-CD16 antibody to the NK cells (fig. S3C). We compared the KIR3DL1+ population with the KIR3DL1− NK cell population within the same Bw4+ individuals, noting that the KIR3DL1− population consisted of a mixture of uneducated NK cells lacking self-HLA–specific receptors and NK cells educated by self-HLA–specific, but non-KIR3DL1 inhibitory receptors. Reflecting its inclusion of uneducated NK cells, the KIR3DL1− NK cell subset has a lower frequency of pS6+ NK cells compared to the KIR3DL1+ NK cell population from the same individual (Fig. 2, E and F). Among the two NK cell populations, the amount of SHP-1 did not affect the EC50 value, but KIR3DL1+ NK cells exhibited an enhanced amplification of responsiveness (fig. S3D), consistent with the in silico simulation.
Allelic polymorphism at the KIR3DL1 locus determines the cell surface density of KIR3DL1, resulting in persons who bear two different KIR3DL1 alleles having NK cell populations with high or low KIR3DL1 surface abundance (35, 36, and 37). Due to predominant monoallelic expression (32, 38), an individual NK cell typically expresses one KIR3DL1 allele, so that NK cells expressing one or the other allele can easily be discriminated from each other based on flow cytometric measurement of cell surface receptor density. Thus, whereas both KIR3DL1high and KIR3DL1low NK cells are considered to be educated in HLA-Bw4+ donors, their response capacities differ based on KIR allotype, its abundance, and its affinity for the HLA-Bw4 allotype (39, 40). We previously showed that KIR3DL1high NK cells are more responsive than KIR3DL1low NK cells in individuals with HLA-Bw4 alleles characterized by an isoleucine at amino acid position 80, for which high-expressing KIR3DL1 allotypes display high-affinity binding (35, 39). By measuring SHP-1 abundance in KIR3DL1high and KIR3DL1low NK cells from the same Bw4-I80 donors, we found that the more responsive KIR3DL1high NK cells had less SHP-1 protein than did the KIR3DL1low NK cells (Fig. 2G). In contrast, autologous KIR3DL1high and KIR3DL1low NK cells from donors in the absence of the cognate ligand Bw4 express similarly high amounts of SHP-1. Furthermore, in HLA-Bw4+ donors, KIR3DL1negative, KIR3DL1low, and KIR3DL1high NK cells showed increasing frequencies of pS6 positivity after anti-CD16 crosslinking (Fig. 2H). Therefore, similar to KIR ligand copy number, KIR receptor density and ligand affinity are positively correlated with NK responsiveness (35) and inversely correlated with SHP-1 abundance, confirming that receptor and ligand combine to set threshold for the cellular response through SHP-1 in a manner directly related to receptor-ligand avidity.
MHC class I and inhibitory receptor interactions shape the NK cell transcriptome
In B6 mice, NK cells expressing one or more of the inhibitory receptors (Ly49C, Ly49I, and NKG2A) that bind to H-2b MHC class I molecules are considered to be educated and responsive NK cell subsets, whereas NK cells lacking all three receptors are uneducated and hyporesponsive (4). In β2m−/− mice, all NK cells are hyporesponsive because of the lack of MHC class I expression (3, 20). We evaluated in B6 mice the responsiveness of NK cell populations that were single-positive for each inhibitory receptor to NK1.1 triggering, normalizing the frequency of IFN-γ+ cells among each population to the autologous Ly49−NKG2A− NK population. We demonstrated that the Ly49Isp and Ly49Csp cells were the most responsive (Fig. 3A). NKG2Asp NK cells, although less responsive than Ly49I/C+ cells, were still more responsive than Ly49−NKG2A− NK cells (Fig. 3A). This suggests that NKG2A+ cells were educated in B6 mice, but to a lesser extent compared with Ly49C+ or Ly49I+ NK cells. In contrast, all of the NK cell subsets were hyporesponsive in β2m−/− mice (Fig. 3A).
We performed global transcriptional profiling of four populations from B6 mice by RNA-seq analysis, sorting splenic NK cells into Ly49Isp, Ly49Csp, NKG2Asp, and Ly49−NKG2A− populations (Fig. S4). To identify the transcriptional signatures of responsive NK cells, we overlapped the differentially expressed genes between the three responsive NK cell subsets and Ly49−NKG2A− NK cells (Fig. 3B). In all three responsive NK cell subsets, we found 41 genes whose expression was greater and 55 genes whose expression was less relative to those in the hyporesponsive Ly49−NKG2A− NK cells (Fig. 3B). Among the genes whose expression was greater in responsive NK cells (Fig. 3C), Kcnj8, Gpc1, S100a6, Hopx, Xdh, Myo6, F2r, Fgl2, and Bhlhe40 have been described as NK cell signature genes (41), although their exact roles in NK cell biology are unknown. Some differentially expressed genes, including Chl1, Klra7, Clip3, and Slamf6, were reported in a previous study comparing the hyporesponsive, uneducated Ly49G2+Ly49I− population and the responsive, educated Ly49G2−Ly49I+ NK cell population in C57BL/6 mice (42). Previously described to be regulated by MHC class I interaction with self-MHC–specific Ly49 molecules and SHP-1 signaling, transcription of Klrg1 was confirmed to be increased in this dataset (43).
In addition, we found a different set of genes that were preferentially expressed in hyporesponsive NK cells. These included genes encoding NK cell receptors, such as Klra7 (Ly49G2), Slamf6 (Ly108), and Tigit (TIGIT), as well as genes encoding solute carrier family molecules, adhesion molecules, and transcription factors Myb, Tox, and Cux1. These findings are consistent with those of a previous study, which suggested that NK cells developed in an MHC-class I–negative environment, and were therefore predicted to be uneducated and hyporesponsive, express more cell surface Ly49G2 (44). Most pertinent to our studies is the finding that Ptpn6 (SHP-1) was more expressed among the uneducated NK cell population lacking inhibitory receptors for self-MHC class I and less expressed among the educated NK cell populations expressing inhibitory receptors for self-MHC class I.
Environmental MHC class I resets SHP-1 amounts in mouse and human NK cells
We measured the amount of SHP-1 protein in different murine NK cell subsets expressing inhibitory self-MHC–specific receptors normalized to that in autologous NK cells lacking all receptors. We found that Ly49Isp and Ly49Csp cells had the lowest amounts of SHP-1, NKG2Asp NK cells had an increased amount of SHP-1, but still at a ratio significantly <1 compared to that in NK cells lacking all receptors (Fig. 3, D and E). In contrast, in β2m−/− mice, in which all NK cell populations were hyporesponsive due to the lack of MHC class I, the SHP-1 ratios were equivalent between receptor-bearing populations and virtually equivalent to 1 with the “uneducated” NK cell population lacking all receptors (Fig. 3E).
Murine NK cell transfer studies have demonstrated that environmental MHC class I can reshape NK cell responsiveness (45, 46), whereupon responsive NK cells developed in an MHC class I–sufficient environment become hyporesponsive one week after their adoptive transfer to an MHC-I–deficient host (45). Reciprocally, hyporesponsive NK cells from MHC class I–deficient mice gain effector function after their transfer to an MHC class I–sufficient host (46). To test whether environmental MHC class I resets SHP-1 abundance in NK cells, we transferred splenic NK cells from CD45.1+ B6 mice into irradiated CD45.2+ wild-type or β2m−/− mice. Ten days after transfer, we evaluated SHP-1 abundance and NK cell responsiveness after NK1.1 crosslinking of NK cell populations predicted to be educated and uneducated based on the expression of self-MHC–specific receptors. Consistent with previous findings, Ly49Isp, Ly49Csp, and NKG2Asp CD45.1+ NK cells transferred to MHC-sufficient CD45.2+ WT recipients exhibited high responsiveness to NK1.1 stimulation, whereas the same NK populations lost responsiveness after transfer to the β2m−/− recipients, most substantially in the previously highly responsive Ly49I+ and Ly49C+ NK cells (fig. S5). Correspondingly, the same Ly49I+ and Ly49C+ NK cells transferred to the β2m−/− recipients were found to switch from low to high amounts of SHP-1 after transfer (Fig. 3F).
We previously demonstrated that environmental HLA class I molecules can also adjust human NK cell reactivity (8). KIR3DL1+ NK cells developed in HLA-B27 (HLA-Bw4+) transgenic (Tg) nonobese diabetic (NOD).Cg-Rag1tm1MomIl2rgtm1Wjl/SzJ (NRG) mice display enhanced missing-self activity (8). To test whether human NK cells reset their SHP-1 amounts in the NRG mouse model, human umbilical cord blood CD34+ cells were transplanted into irradiated WT or B27 Tg+ mice, and SHP-1 amounts were evaluated after 8 weeks. KIR3DL1+ NK cells that developed in HLA-B27 Tg animals expressed less SHP-1 than KIR3DL1+ NK cells that developed in HLA-negative recipients (Fig. 3G). Thus, both murine and human NK cells changed their SHP-1 abundance according to changes in environmental MHC class I molecules.
An acute reduction in SHP-1 abundance enhances NK cell responsiveness
Our results correlating SHP-1 abundance with inhibitory receptors, MHC class I ligand, and the capacity of NK cells to respond suggested that inhibitory receptors and MHC class I interactions set the threshold for responsiveness through dynamic modulation of SHP-1 abundance. Previous attempts to ablate SHP-1 among NK cells and directly manipulate responsiveness did not demonstrate this point, however, because germline deletion of Ptpn6 results in complete loss of responsiveness among all NK cells (23). We therefore reasoned that a minimal presence of SHP-1 protein among NK cells, similarly to MHC class I and inhibitory receptor, is necessary to establish a capacity for responsiveness and that active adjustments in SHP-1 abundance then sets the amplitude of the response. An intervention to reduce SHP-1 protein abundance, therefore, should result in an increased NK cell response. To achieve an acute diminution of SHP-1 abundance in NK cells without complete loss of the protein, we generated an inducible, NK cell–specific SHP-1 heterozygous-deficient mouse strain. Ptpn6fl/fl mice were bred with NKp46-CreERT2 Tg mice carrying the Rosa26-td-Tomato allele (47) to generate NKp46-CreERT2+/−Ptpn6fl/+tdTomatofl/− mice. In this mouse model, tdTomato marks Cre recombinase-edited NK cells (Fig. 4A) and enables phenotypic and functional comparison between tdTomato+ and tdTomato− NK cells from the same host treated with tamoxifen (fig. S6).
Fig. 4. NK cells have enhanced activity when SHP-1 is reduced.
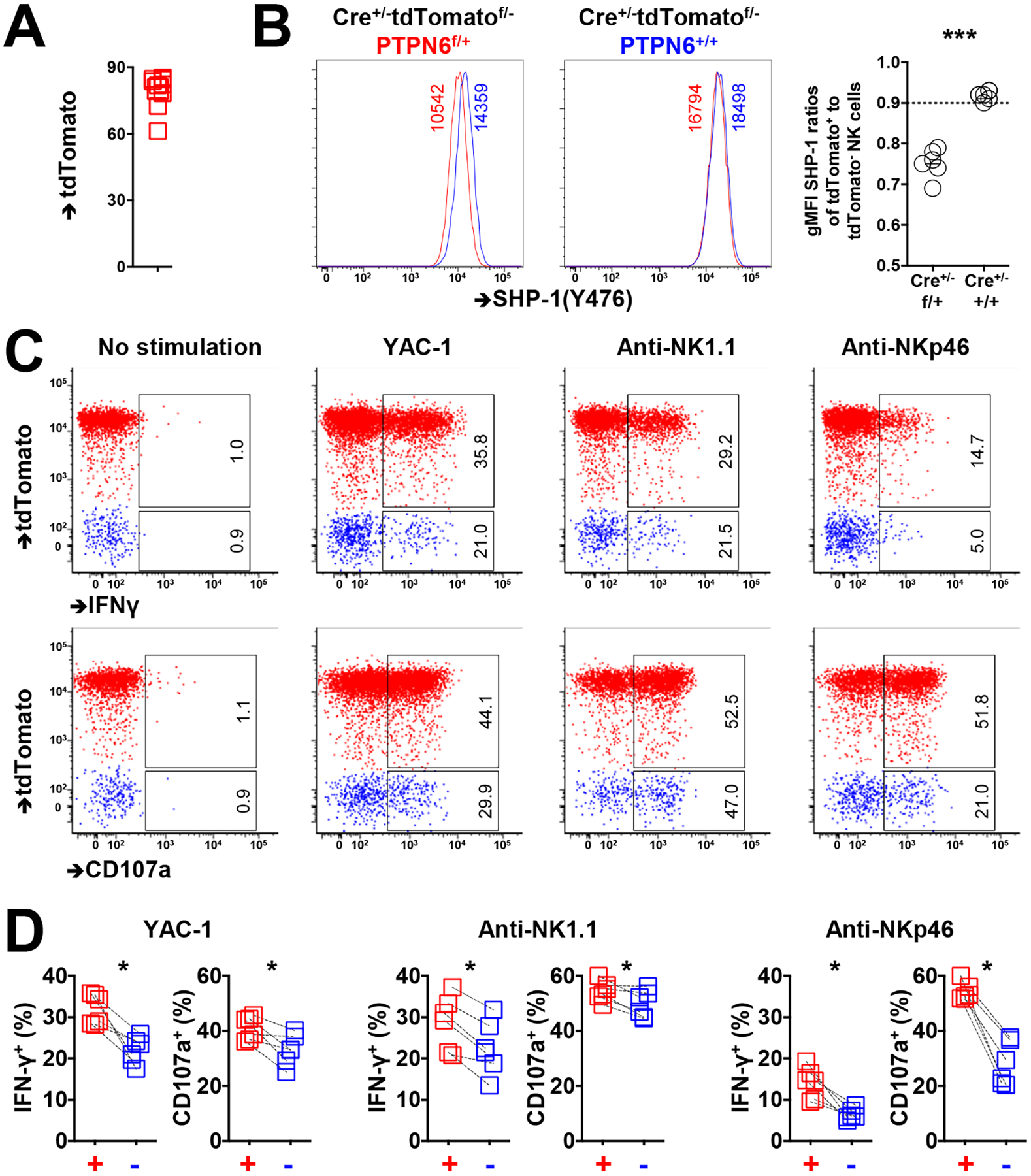
(A) TdTomato abundance among NKp46+ splenic NK cells from CreERT2+/−Ptpn6fl/+tdtomatofl/− mice 3 days after tamoxifen administration. (B) SHP-1 abundance among tdTomato+ and tdTomato− NK cells from CreERT2+/−Ptpn6fl/+tdtomatofl/− and CreERT2+/−Ptpn6+/+tdtomatofl/− mice 3 days after tamoxifen administration. (C and D) IFN-γ production and CD107a activity among tdTomato+ and tdTomato− NK cells in response to YAC-1 cells or treatment with crosslinking anti-NK1.1 or anti-NKp46 antibodies. Paired symbols indicate populations from the same mouse. Results are representative of three independent experiments. The indicated percentages of positive cells in the dot plots were determined as percentages of the tdTomato+ and tdTomato− NK cells. Data are means ± SD in (B), n = 11 mice. *P < 0.05; ***P < 0.001; Mann-Whitney U test (B), and Nonparametric Wilcoxon signed rank sum test (D) were used. Each symbol represents an individual mouse.
We confirmed that tdTomato+ NK cells had measurably less SHP-1 protein at day 3 after tamoxifen administration compared to tdTomato− NK cells from the same host of CreERT2+/−Ptpn6fl/+tdTomatofl/− mice (Fig. 4B). Similarly to human and wild-type murine NK cells, differences in SHP-1 protein abundance were measurable but small, with SHP-1 amounts in the tdTomato+ cells averaging 75% of those in tdTomato− cells. We then evaluated differences in responsiveness between the induced tdTomato+ NK cells (SHP-1low) and the tdTomato− cells against the NK cell–sensitive YAC-1 target cell line and upon activation with the plate-bound antibodies anti-NK1.1 and anti-NKp46. In response to all of these stimuli, tdTomato+ NK cells demonstrated significantly enhanced IFN-γ production and CD107a mobilization compared to tdTomato− NK cells (Fig. 4, C and D). Our results indicate that an acutely induced reduction in SHP-1 abundance rapidly enhanced NK cell responsiveness, suggesting a direct role for SHP-1 in determining the responsiveness of NK cells.
Previous studies investigating complete germline loss of Ptpn6 in NK cells reported universal hypo-responsiveness, suggesting the importance of SHP-1 in NK cell development to NK cell function (23). To further test whether induction of complete loss of SHP-1 among developed NK cells would result in even greater response, we generated NKp46-CreERT2+/−Ptpn6fl/fl mice. After confirming loss of SHP-1 among NK cells, but not among T cells, after tamoxifen administration (Fig. 5A), we used mixed bone marrow chimeric mice to evaluate the phenotype and functionality of NK cells with induced loss of SHP-1 (Fig. 5B). Compared to wild-type (SHP-1pos) NK cells developed in the same environment, the SHP-1neg NK cell population exhibited similar frequencies of inhibitory receptor expression (Fig. 5C), suggesting that loss of SHP-1 did not lead to skewing toward a particular NK cell subpopulation. Furthermore, SHP-1neg NK cells showed similar activity to that of SHP-1pos NK cells when stimulated with the YAC-1 target cell line or plate-bound anti-NK1.1 and anti-NKp46 antibodies (Fig. 5D). Because the inhibitory Ly49+ population frequencies were similar between SHP-1neg and SHP-1pos cell populations, lack of induced hypo- or hyperresponsiveness upon loss of SHP-1 expression suggests a lack of effect across all NK cell populations.
Fig. 5. SHP-1–deficient NK cells show enhanced activity upon crosslinking of activating and inhibitory receptors.
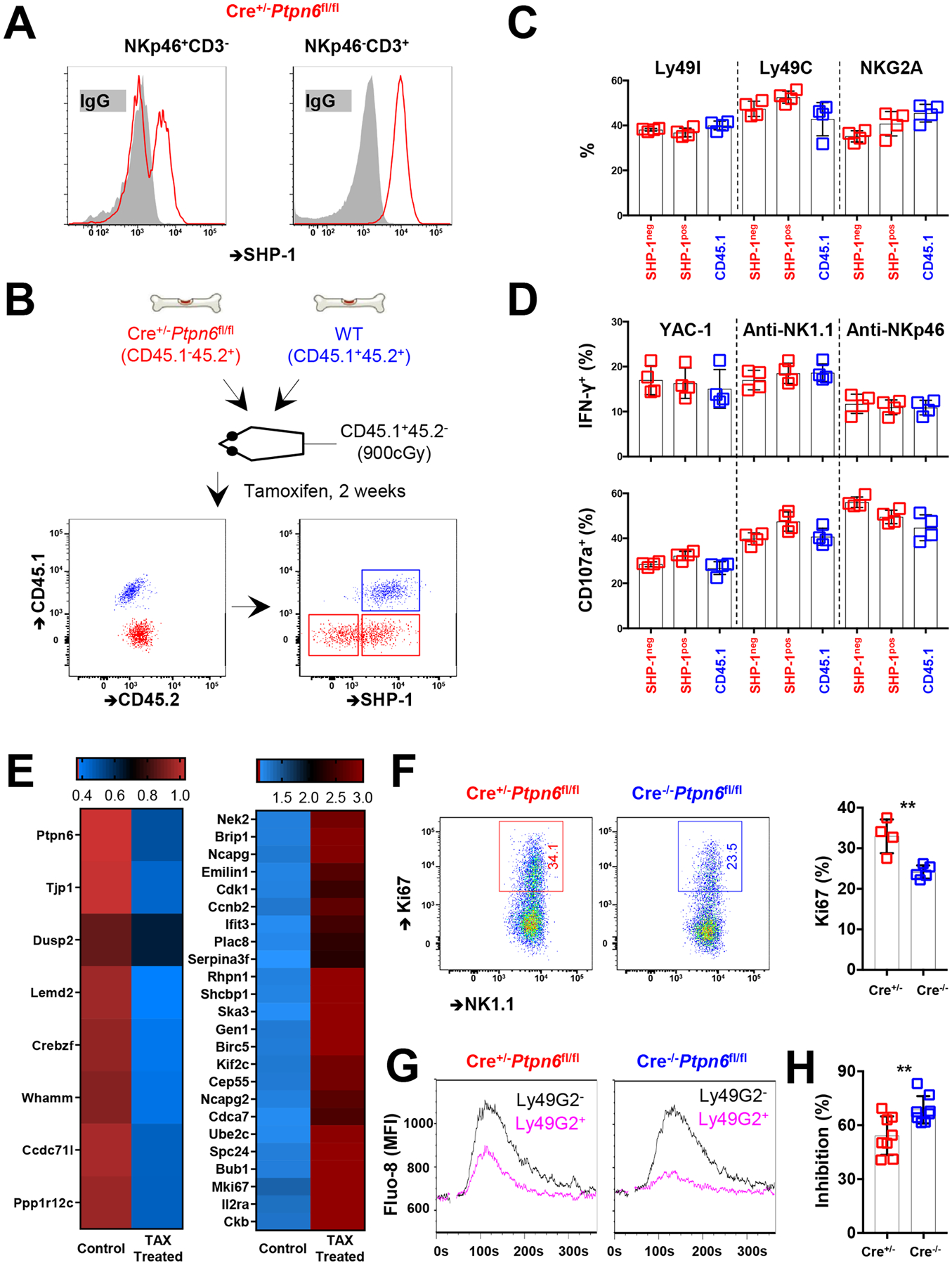
(A) SHP-1 abundance among NKp46+CD3− and NKp46−CD3+ splenic lymphocytes from CreERT2+/−Ptpn6fl/fl mice 14 days after tamoxifen administration. (B) Experimental design. Mixed bone marrow chimeric mice were established with both WT (CD45.1+CD45.2+) and CreERT2+/−Ptpn6fl/fl (CD45.1−CD45.2+) bone marrow at an equal ratio. SHP-1 abundance among splenic NK cells (NKp46+NK1.1+CD3−) was evaluated 2 weeks after the administration of tamoxifen to the chimeric animals. (C and D) Ly49I, Ly49C, and NKG2A cell surface expression (C) and IFN-γ production and CD107a activity (D) in response to co-culture with YAC-1 cells or treatment with crosslinking anti-NK1.1 or anit-NKp46 antibodies among CD45.1−SHP-1+, CD45.1−SHP-1−, and CD45.1+ NK cells. Results are pooled from two independent experiments. (E) CD45.1−CD45.2+ NK cells were sorted from chimeric animals that did or did not receive tamoxifen for 2 weeks. Differentially expressed genes are determined by RNAseq analysis. (F) Representative and accumulated Ki67 expression among splenic NK cells from CreERT2+/−Ptpn6fl/fl and CreERT2−/−Ptpn6fl/fl mice with tamoxifen treatment. Results are pooled from two independent experiments. (G) Representative kinetics of Fluo-8 fluorescence (Ca2+ flux) after the treatment of NK cells with antibodies against NKp46 and Ly49G2. Splenic NK cells were purified from CreERT2+/−Ptpn6fl/fl and CreERT2+/−Ptpn6fl/fl mice 14 days after tamoxifen administration. Ly49G2+ NK cells are indicated in pink, and Ly49G2− NK cells are indicated in black. (H) Ly49G2-mediated inhibition among NK cells assessed by area under curve (AUC) values of the kinetics of Ca2+ flux as shown in (G). Results are pooled from two independent experiments. (E to H) NK cells from Cre+ animals were a mixture of SHP-1–positive and –negative cells. Data are means ± SD in (C), (D), (F), and (H), n = 4, 4, 4, and 8 mice. **P < 0.01; Mann-Whitney U test (F and I) was used. Each symbol represents an individual mouse.
We further compared the responsiveness of SHP-1neg and SHP-1pos NK cells from NKp46-CreERT2+/−Ptpn6fl/fl mice at sequential times after treatment with one dose of tamoxifen. NKp46-CreERT2−/−Ptpn6fl/fl littermates were applied as controls. SHP-1neg NK cells were found with a mean frequency of 43.8% (range: 24.5 to 64.0%) from NKp46-CreERT2+/−Ptpn6fl/fl mice at two weeks after tamoxifen administration (fig. S7A). Again, compared to SHP-1pos cells and cells from Cre−/− littermates, SHP-1neg NK cells did not have significant differences in their response, even 6 weeks after tamoxifen administration (fig. S7, B and C). Together with the findings from the mice with heterozygous loss of Ptpn6, these results suggest that NK cells require SHP-1 protein to establish and adjust responsiveness, but that when SHP-1 is lost after the NK cell has developed, the response threshold of the NK cell becomes fixed and is no longer adjustable.
To study transcriptional differences among NK cells after acute ablation of SHP-1, we globally profiled differentially expressed genes among NK cells from chimeric mice with or without tamoxifen treatment. NKp46-CreERT2+/−Ptpn6fl/fl mice do not express the td-Tomato allele to mark SHP-1–negative NK cells. We therefore analyzed samples containing NK cells cytometrically negative for SHP-1 for comparison (fig. S8). Consistent with the response data, we did not observe a difference among genes that contribute to NK cell effector function, inhibition, and responsiveness. Instead, we found that the expression of Dusp2, Whamm, and Ppp1r12c was reduced in NK cells with induced loss of SHP-1 (Fig. 5E). Dusp2, which encodes dual specificity protein phosphatase 2, inhibits mitogenic signaling, and the gene products of Whamm and Ppp1r12c have been implicated in cytoskeleton formation and assembly (48, 49). Cell cycle–related genes, including Nek2, Ncapg, Ncapg2, Cdk1, Ccnd2, Ska3, Birc5, Cdca7, Mki67, and Ckb were preferentially expressed among NK cells with induced loss of SHP-1, with no evidence of compensation by the expression of genes encoding other NK cell–related phosphatases, such as SHP-2 (Fig. 5E) (50). We found that NK cell proliferation was indeed enhanced upon ablation of SHP-1, as evidenced by increased Ki67 expression among NK cells from NKp46-CreERT2+/−Ptpn6fl/fl mice in comparison to that in NK cells from NKp46-CreERT2−/−Ptpn6fl/fl mice after treatment with tamoxifen (Fig. 5F). These results suggest that acute SHP-1 ablation affects NK cells transcriptionally and promotes NK cell proliferation.
Finally, SHP-1 is mediator of inhibitory signaling, leading us to hypothesize that induced SHP-1neg NK cells were less likely be inhibited upon stimulation. This was confirmed by comparing the amplitude of Ca2+ flux in NK cells after NKp46 triggering and crosslinking of the inhibitory receptor Ly49G2. A decrease in Ly49G2-mediated inhibition was observed among NK cells from NKp46-CreERT2+/−Ptpn6fl/fl mice when compared to that in NK cells from NKp46-CreERT2−/−Ptpn6fl/fl mice (Fig. 5, G and H). These findings indicate that ablation of SHP-1 in mature NK cells dampens inhibitory receptor–mediated inhibition. Together, these findings are consistent with a model in which SHP-1 is necessary to establish the ability of an NK cell to respond and for titrating the amplitude of the response. Upon complete loss of SHP-1 in a mature NK cell, however, the threshold to respond becomes fixed and is not lost. While the same individual NK cell retains the capacity to respond, it also becomes less sensitive to receptor-mediated inhibition.
Discussion
Central to the role of the NK cell in tumor surveillance and pathogen control is its ability to mount an immune response while maintaining self-tolerance. A diverse spectrum of NK cells is endowed with varied capacities for an effector response through a process that is generally referred to as “education” and recognized to be governed by self-inhibitory receptor interactions with MHC class I proteins. Through RNA-seq analysis and intracellular staining, we have found that SHP-1 is a measurable protein marker that distinguishes responsive NK cells from hyporesponsive NK cells in humans and in mice more accurately than does self-MHC–specific receptor expression. SHP-1 therefore is a protein marker to identify populations previously only functionally defined in mice and humans.
NK cells constantly scan cells in their environment, but it is not known how they distinguish between healthy and diseased cells with high accuracy and speed in real time. An essential role of SHP-1 in this process was underscored through in silico mathematical modeling and through computational analysis of primary NK cell responsiveness, both of which demonstrated a nonlinear dependency between input and response. We demonstrated that SHP-1 was a digital regulator of NK cell responsiveness, determining the response state of a given cell. When SHP-1 abundance exceeded a critical amount, as in uneducated NK cells, the EC50 for the signaling network became infinite and NK cells became resistant to activation, concurrently leading to the general inability to tune NK cell activity among uneducated NK cells. In contrast, subpopulations with varied amounts of SHP-1 in educated NK cells exhibited different degrees of S6 phosphorylation, while the responsive fractions had the same EC50 values. These findings suggest that SHP-1 plays a second key role as a negative amplitude regulator in educated NK cell responsiveness, whereby small fluctuations in SHP-1 protein abundance tune NK cell reactivity. We further found that an activation threshold was established through SHP-1 by self-MHC-class I–specific KIR and NKG2A in a dose-dependent manner for both receptors and ligands (Fig. 6). In further support of its active role in controlling dynamic response, an acute reduction, but not complete abrogation, of SHP-1 abundance in NK cells augments their reactivity.
Fig. 6. The relationship between SHP-1 abundance and NK cell responsiveness.
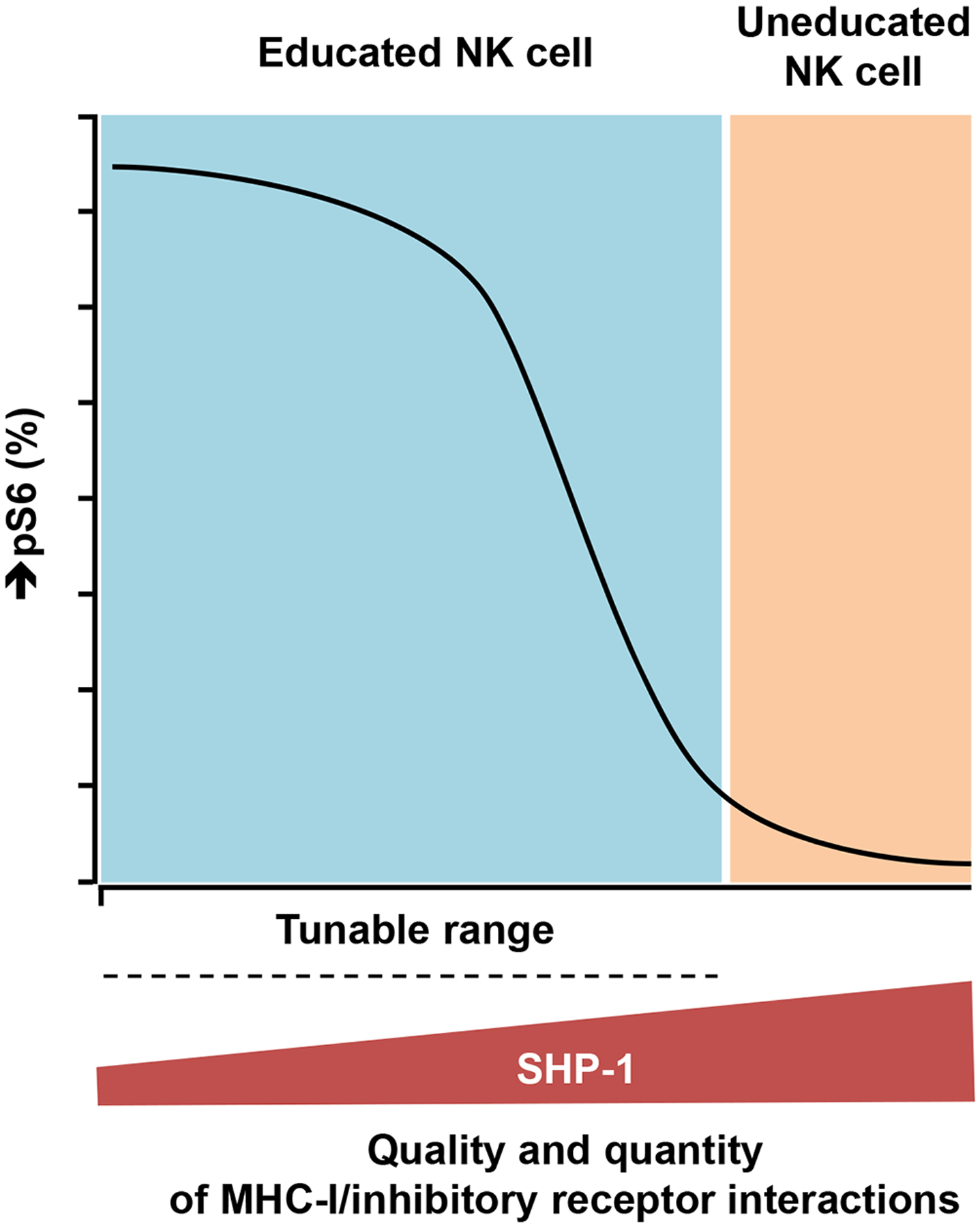
SHP-1 is a digital regulator of NK cell responsiveness, determining the response state of a given cell. When the abundance of SHP-1 exceeds a critical threshold, as in uneducated NK cells, the EC50 value for the signaling network becomes infinite and NK cells become resistant to activation, concurrently leading to the general inability to tune NK cell activity among uneducated NK cells. SHP-1 is a negative amplitude regulator in NK cell responsiveness. Subpopulations among educated NK cells with varying amounts of SHP-1 have different Pmax values (pS6), despite having consistent EC50 values. The relative amounts of SHP-1 among the NK cells were established by self-MHC-class I–specific KIR and NKG2A in a dose-dependent manner for both receptors and ligands.
Educated NK cells exhibit distinct cytoskeletal distributions (18) and dynamics (19) of activating and inhibitory receptors at rest, whereby activating receptors are confined in signaling-favorable nanodomains instead of the meshwork linked to the actin cytoskeleton. In contrast, uneducated cells have both activating and inhibitory receptors confined to the actin meshwork on the plasma membrane (18). A faster NKp46 diffusion rate with less time within each microdomain in educated NK cells suggests that actin cytoskeleton–dependent activating receptor dynamics contribute to NK cell responsiveness (19) and are therefore likely vital to an active education process. SHP-1 directly binds to β-actin upon inhibitory receptor engagement in human NK cells, leading to control of the conformational state and catalytic activity of SHP-1 by actin dynamics upon inhibitory signaling (51). SHP-1 also forms complexes with γ-tubulin (52) and myosin-9 (53) in immune cells, suggesting SHP-1 may reciprocally influence cytoskeleton-related cell biology, whereupon complete loss of SHP-1 leads to increased cytoskeletal assembly and cell cycling. Together, these findings suggest that NK cell education is a dynamic process, linking NK cell function to receptor-ligand distribution, the actin cytoskeleton, and the abundance of SHP-1 protein.
Germline deletion of the gene encoding the activating receptor NKG2D or its ligand RAE-1 leads to the generation of hyperresponsive NK cells in mice, suggesting that NKG2D signaling desensitizes the NK cell response globally in response to various stimuli (54). Moreover, a loss-of-function mutation in the gene encoding the activating receptor NKp46 (55) but not full deletion of NKp46 (56) results in the generation of hyperresponsive NK cells. These observations support the disarming NK cell education model in which baseline activation signaling desensitizes NK cells at rest (57). Furthermore, NKG2D-deficient NK cells in mice show enhanced activity only through NKp46 triggering, leading to better control of tumor and viral infection compared to that by wild-type NK cells (58). Similar to what we observed in NK cells, low SHP-1 expression at the level of transcript and protein has also been observed among other hyperresponsive innate immune cells. Invariant NKT (iNKT) cells made hyperresponsive by the deletion of CD1d have less SHP-1 protein compared to wild-type iNKT cells (59). It is not clear, however, whether SHP-1 mediates activation-induced NK cell desensitization or how expression of the gene encoding SHP-1 is regulated among innate cells.
A complete absence of SHP-1 during NK cell development results in universal nonresponsiveness (23), indicating that despite its inhibitor role, SHP-1 must be present at a minimal amount to enable NK cells to respond. The exact mechanism underlying how SHP-1 contributes to NK cell responsiveness during development remains unclear. The disarming hypothesis proposes that tonic activation during development leads the NK cell to an anergic and hyporesponsive state, unless inhibitory signaling triggered by the binding of MHC class I to inhibitory receptors interrupts activation and preserves response capacity (54, 57). Fulfilling the role of a negative regulator, SHP-1 associates with inhibitory NK cell receptors and blocks NK cell activation through the dephosphorylation of Vav-1, the adaptor protein LAT, and phospholipase C-γ (60, 61). Interestingly, the responsiveness of SHP-1-negative NK cells is preserved in SHP-1-defective, motheaten-viable (me-v) mice in a transplantation setting (3) and upon induced deletion among mature NK cells in our study. These seemingly discordant findings may be reconciled if SHP-1, at any level during development, is necessary for establishing the capacity to respond; upstream molecular interactions between inhibitory receptor and ligand can then adjust SHP-1 abundance, leading to tuning of the response capacity, as has been proposed in the “rheostat model” (5). Lack of engagement of MHC class I and inhibitory NK receptors, whether during development or due to the transfer of mature cells into an MHC class I-deficient environment (45, 46), leads to an increase in SHP-1 abundance and a decrease in NK cell responsiveness, consistent with a process of desensitization or disarming in the setting of persistent NK cell activation unchecked by inhibitory signaling (55, 57). Together, these data suggest that SHP-1 has several roles in the NK cell response: enabling the capacity for a response, controlling the likelihood of a response, and adjusting the response according to environmental MHC class I.
As a critical signaling molecule and regulator of responsiveness in NK cells, SHP-1 may be a reasonable immunotherapy target in disease states. After normal NK cell development and the acquisition of response potential, a reduction in SHP-1 abundance enhances effector function, an outcome that could be achieved by treatment with a SHP-1 inhibitor (58). We showed that complete ablation of SHP-1 decreased SHP-1-mediated NK cell inhibition while protecting NK cell responsiveness, advantageous qualities that were replicated by CRISPR-mediated editing of PTPN6 in an NK cell line (62). Enhanced NK cell proliferation upon loss of SHP-1 provides potential additional therapeutic benefit. Adoptive NK cell therapies are emerging as an important tool in the treatment of malignancies (63, 64), with the goal of achieving tumor killing through missing-self activation. Because the amount of SHP-1 is reset upon transfer of NK cells to an environment lacking the MHC class I molecules that were present in the donor, the long-term prospect of retaining high NK cell responsiveness is unlikely without additional manipulation. Reduction of SHP-1 activity or abundance in NK cells, either with an inhibitor or through the use of SHP-1−/− cells, may enhance the effects of this promising cellular therapy.
Materials and Methods
Human samples
Buffy coats and umbilical cord blood samples were collected from volunteer blood donors at the New York Blood Center. These samples were obtained anonymously for research purposes; therefore, the Memorial Sloan Kettering Cancer Center (MSKCC) Institutional Review Board waived the need for additional research consent. Peripheral blood was additionally collected from healthy donors at MSKCC after approval by the MSKCC Institutional Review Board, and donors provided informed written consent. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll purification, aliquoted, and stored in liquid nitrogen before experimentation. High-resolution HLA typing (Histogenetics) and KIR genotyping were performed as previously described (35).
Mice
C57BL/6J (B6), B6.129P2-B2mtm1Unc/J (β2m−/−), B6.SJL-PtprcaPepcb/BoyJ (B6 CD45.1), B6.129P2-Ptpn6tm1Rsky/J (Ptpn6fl/+), and NOD.Cg-Rag1tm1MomIl2rgtm1Wjl/SzJ (NRG) mice were obtained from The Jackson Laboratory. NOD.Cg-Rag1tm1MomIL2rgtmWjl Tg (B2M, HLA-B*27–05)56–3 (B27 Tg+) mice were described previously (39). NKp46-CreERT2 Tg mice carrying the Rosa26-td-Tomato allele (40) were kindly provided by Dr. L. Lanier (University of California), courtesy of Dr. J. Sun (MSKCC). Where indicated in the figure legends, mice were administered 4 mg of tamoxifen (Sigma) by oral gavage in 200 μl of corn oil (Sigma). All of the mice used in this study were housed and bred under specific pathogen-free conditions at Memorial Sloan Kettering Cancer Center and handled in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC).
Cells
Human NK cells were enriched by negative selection from PBMCs (STEMCELL). CD34+ cells were purified by positive selection (Miltenyi) from umbilical cord blood. Murine NK cells were enriched by negative selection (Miltenyi) from splenocytes for RNAseq analysis. Murine NK cells were enriched from spleens by CD4, CD8, CD19, and Ter-119 depletion for phenotypic and functional studies in some experiments. The cell lines 721.221, K562, YAC-1, Ba/F3, and all primary NK cells were cultured in RPMI 1640 medium containing 10% fetal calf serum (FCS) supplemented with penicillin and streptomycin (MSKCC). 721.221-C1 cells were provided by Dr. Peter Parham (Stanford University).
Antibodies and flow cytometry analysis
Antibodies used for flow cytometry are listed in table S1. The Ly49C antibody 4LO3311 was kindly provided by Drs. S. Lemieux and W. Yokoyama (Washington University School of Medicine). Murine NKG2A was stained with the antibody 20d5, which detects the same NK population as that detected with clone 16a11 in B6 mice (65). For intracellular SHP-1 staining, cells were fixed and permeabilized with a staining kit (GAS004, Thermo Fisher Scientific) before staining. Murine cells were preincubated for 10 min on ice with 2.4G2 to block FcγRII and FcγIII before staining. Flow cytometry and cell sorting were performed on the Fortessa (BD Biosciences) and Aria II cytometers (BD Biosciences), respectively. The data were analyzed with FlowJo software (Tree Star).
In vivo NK cell development and adoptive transfer
For the humanized mouse model, NRG wild-type and HLA-B27 Tg mice were irradiated one day before receiving 1 × 105 human CD34+ cells, which were confirmed to be >95% pure by FACS. Ba/F3-IL15-IL15R stable transfectants were irradiated (100 cGy) and intravenously (i.v.) injected twice weekly beginning 3 weeks after transplantation, as previously described (8). Human NK cells were harvested from mouse spleens at 8 weeks after bone marrow reconstitution and were defined as mCD45−hCD45+CD3−CD14−CD19−CD122+CD16+ among live cells. For murine NK cell transfer experiments, CD45.1+ splenocytes were injected i.v. into recipients (B6 or β2m−/− mice) that had received 600 cGy of irradiation 5 hours earlier, as previously described (46). Each recipient received 50 × 106 donor splenocytes. NK cell phenotype and responsiveness were tested 10 days later.
In vitro NK cell assays
Human PBMCs were co-cultured with target cells for 5 hours in the presence of anti-CD107a antibody to detect degranulation. Frozen PBMCs were cultured overnight in RPMI 1640 medium containing 10% FCS and 200 IU/ml IL-2 before undergoing functional analysis. To evaluate the functions of murine NK cells, flat-bottomed, high protein-binding plates (Corning) were coated with anti-NK1.1(10 μg/ml) or anti-NKp46 (5 μg/ml) antibody overnight at 4°C. NK cells from the spleen were stimulated for 5 hours in the presence of Monensin (BD Biosciences), Brefeldin A (Sigma), and 1,000 U/ml recombinant human IL-2 (Roche) before intracellular IFN-γ was detected as previously described (46). The Ca2+ flux assay was performed and quantified as previously optimized (66). Splenic NK cells were freshly enriched with a negative selection kit (Miltenyi) and labeled with the Ca2+ indicator Fluo-8 (Abcam) in assay buffer. Purified NK cells were then labeled with color-conjugated, rat anti-mouse NKp46 and rat anti-mouse Ly49G2. The cells were then washed and kept on ice until acquisition by flow cytometry. To record Ca2+ flux, the cells were pre-warmed at 37°C in a water bath for 10 min after which fluorescence signal was acquired for 30 s as baseline. After adding crosslinking donkey anti-rat IgG (Jackson Immunoresearch), acquisition was continued for 360 s. To determine the area under the curve of each Ca2+ flux plot, kinetics analysis was performed with the Flowjo algorithm (BD Biosciences).
Anti-CD16 antibody stimulation and flow cytometric analysis of NK cell responsiveness
Freshly purified human NK cells were stimulated with different amounts of pre-warmed biotin-conjugated anti-CD16 antibody and crosslinked with avidin (Thermo Fisher Scientific) for 5 min at 37°C. The NK cells were fixed immediately with 1.6% paraformaldehyde for 10 min at room temperature with excessive washes. After fixation, the cells were stained for surface markers and then were permeabilized with Perm Buffer III (BD Biosciences) for 30 min on ice for intracellular SHP-1 detection. For KIR3DL1high/low/negative NK cell activation studies, cells were stimulated with mouse anti-human CD16 antibody (3G8) and crosslinked with goat anti-mouse IgG, F(ab’)2 (115-006-006) at a concentration of 13 μg/ml. KIR2DL1-, KIR2DL2/L3-, and NKG2A-positive NK cells were excluded from the analysis with biotin-conjugated antibodies against KIR2DL1/KIR2DL2/L3/NKG2A and Pacific Blue-conjugated streptavidin (Thermo Fisher Scientific).
Computational modeling of NK cell signaling
The signaling proteins and receptors in the computational model were assumed to be well-mixed in a simulation box of volume V = 2μm × 2μm × 0.02μm. The simulation box represents a 4 μm2 area in the NK cell plasma membrane and a 0.02-μm thick cytosolic region beneath the plasma membrane. The signaling reactions were simulated through the standard Gillespie algorithm (67), which includes intrinsic stochastic fluctuations in the biochemical signaling reactions. Simulations were performed with the software package Stochastic Simulator Compiler (MIT) (67). SHP-1 concentrations were varied in the simulations from 500 to 8000 molecules in the simulation box V. The reactions, reaction rates, and the initial protein abundances were set as previously described (34). We used binding (kon = 6500M−1s−1) and unbinding (koff = 0.0047s−1) rates for CD16 and its cognate ligand based on reported values for the interaction of soluble CD16A with human IgG (68). The binding and unbinding rates for mouse NKp46 and its ligand heparin were set to kon=0.0072 (μM)−1s−1 and koff=0.0029 (69). The average values of pVav and pS6 were calculated at t = 10 min after stimulation and the averages were performed over 4000 single cells in silico.
RNAseq and ATACseq analyses
RNA was isolated from sorted cell populations with TRIzol (Invitrogen), which was followed by SMARTer amplification. Paired-end Illumina next-generation sequencing was performed for RNAseq (70). DNA was prepared from sorted cells for ATACseq. Libraries were sequenced on a HiSeq 2500 in High Output mode and a HiSeq4000 in a 50bp/50bp paired end run, using the TruSeq SBS Kit v4 or HiSeq 3000/4000 SBS Kit (Illumina) as previously described (70).
Statistical analysis
The nonparametric Wilcoxon signed rank sum test was used to compare paired ratios from the same donor. The Mann-Whitney U test was used to compare ratios between two groups. The Friedman test and post hoc pairwise Wilcoxon test were performed for multi-group comparisons. Results were considered to be significant at the two-sided P level of 0.05. *P < 0.05; **P < 0.01; ***P < 0.001.
Supplementary Material
Acknowledgments:
We thank Dr. Jean-Benoît Le Luduec for flow cytometry assistance. We thank Ms. Cindy Zhang for mouse colony management. We thank Dr. L. Lanier for providing NKp46-CreERT2 Tg mice carrying the Rosa26-td-Tomato allele. We thank Drs. S. Lemieux and W. Yokoyama for providing Ly49C antibody.
Funding:
Z.W., S.P., and K.C.H. were supported by NIH P01 CA23766, NIH/NIAID U01 AI25651, and R01 AI125651. C.M.L. was supported by the Cancer Research Institute as a Cancer Research Institute - Carson Family Fellow and by the Center for Experimental Immuno-Oncology and Comedy Vs. Cancer at MSKCC. J.C.S. was supported by the Ludwig Center for Cancer Immunotherapy, the American Cancer Society, the Burroughs Wellcome Fund, and the NIH (AI100874, AI130043, and AI155558). J.D. was supported by NIH R01 AI143740 and R01 AI146581. G.A.-B. was supported by the intramural research program of the National Cancer Institute, USA. Z.W, S.P., K.C.H, J.C.S., C.M.L, Y.Z., and S.S. were supported by NIH/NCI Cancer Center Support Grant P30 CA008748.
Footnotes
Data and materials availability:
RNA-seq data are accessible through the NCBI Sequence Read Archive database with accession code PRJNA723694. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Lanier LL, NK cell recognition. Annu Rev Immunol 23, 225–274 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Karre K, Ljunggren HG, Piontek G, Kiessling R, Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319, 675–678 (1986). [DOI] [PubMed] [Google Scholar]
- 3.Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang L, French AR, Sunwoo JB, Lemieux S, Hansen TH, Yokoyama WM, Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 436, 709–713 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Fernandez NC, Treiner E, Vance RE, Jamieson AM, Lemieux S, Raulet DH, A subset of natural killer cells achieves self-tolerance without expressing inhibitory receptors specific for self-MHC molecules. Blood 105, 4416–4423 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brodin P, Karre K, Hoglund P, NK cell education: not an on-off switch but a tunable rheostat. Trends Immunol 30, 143–149 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Doucey MA, Scarpellino L, Zimmer J, Guillaume P, Luescher IF, Bron C, Held W, Cis association of Ly49A with MHC class I restricts natural killer cell inhibition. Nat Immunol 5, 328–336 (2004) [DOI] [PubMed] [Google Scholar]
- 7.Bessoles S, Angelov GS, Back J, Leclercq G, Vivier E, Held W, Education of murine NK cells requires both cis and trans recognition of MHC class I molecules. J Immunol 191, 5044–5051 (2013) [DOI] [PubMed] [Google Scholar]
- 8.Boudreau JE, Liu XR, Zhao Z, Zhang A, Shultz LD, Greiner DL, Dupont B, Hsu KC, Cell-Extrinsic MHC Class I Molecule Engagement Augments Human NK Cell Education Programmed by Cell-Intrinsic MHC Class I. Immunity 45, 280–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas LM, Peterson ME, Long EO, Cutting edge: NK cell licensing modulates adhesion to target cells. J Immunol 191, 3981–3985 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enqvist M, Ask EH, Forslund E, Carlsten M, Abrahamsen G, Beziat V, Andersson S, Schaffer M, Spurkland A, Bryceson Y, Onfelt B, Malmberg KJ, Coordinated expression of DNAM-1 and LFA-1 in educated NK cells. J Immunol 194, 4518–4527 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Goodridge JP, Jacobs B, Saetersmoen ML, Clement D, Hammer Q, Clancy T, Skarpen E, Brech A, Landskron J, Grimm C, Pfefferle A, Meza-Zepeda L, Lorenz S, Wiiger MT, Louch WE, Ask EH, Liu LL, Oei VYS, Kjallquist U, Linnarsson S, Patel S, Tasken K, Stenmark H, Malmberg KJ, Remodeling of secretory lysosomes during education tunes functional potential in NK cells. Nat Commun 10, 514 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schafer JR, Salzillo TC, Chakravarti N, Kararoudi MN, Trikha P, Foltz JA, Wang R, Li S, Lee DA, Education-dependent activation of glycolysis promotes the cytolytic potency of licensed human natural killer cells. J Allergy Clin Immunol 143, 346–358 e346 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Pfeifer C, Highton AJ, Peine S, Sauter J, Schmidt AH, Bunders MJ, Altfeld M, Korner C, Natural Killer Cell Education Is Associated With a Distinct Glycolytic Profile. Front Immunol 9, 3020 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagner AK, Kadri N, Snall J, Brodin P, Gilfillan S, Colonna M, Bernhardt G, Hoglund P, Karre K, Chambers BJ, Expression of CD226 is associated to but not required for NK cell education. Nat Commun 8, 15627 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersson M, Freland S, Johansson MH, Wallin R, Sandberg JK, Chambers BJ, Christensson B, Lendahl U, Lemieux S, Salcedo M, Ljunggren HG, MHC class I mosaic mice reveal insights into control of Ly49C inhibitory receptor expression in NK cells. J Immunol 161, 6475–6479 (1998). [PubMed] [Google Scholar]
- 16.Marcais A, Marotel M, Degouve S, Koenig A, Fauteux-Daniel S, Drouillard A, Schlums H, Viel S, Besson L, Allatif O, Blery M, Vivier E, Bryceson Y, Thaunat O, Walzer T, High mTOR activity is a hallmark of reactive natural killer cells and amplifies early signaling through activating receptors. Elife 6, (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi L, Li K, Guo Y, Banerjee A, Wang Q, Lorenz UM, Parlak M, Sullivan LC, Onyema OO, Arefanian S, Stelow EB, Brautigan DL, Bullock TNJ, Brown MG, Krupnick AS, Modulation of NKG2D, NKp46, and Ly49C/I facilitates natural killer cell-mediated control of lung cancer. Proc Natl Acad Sci U S A 115, 11808–11813 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guia S, Jaeger BN, Piatek S, Mailfert S, Trombik T, Fenis A, Chevrier N, Walzer T, Kerdiles YM, Marguet D, Vivier E, Ugolini S, Confinement of activating receptors at the plasma membrane controls natural killer cell tolerance. Sci Signal 4, ra21 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Staaf E, Hedde PN, Bagawath Singh S, Piguet J, Gratton E, Johansson S, Educated natural killer cells show dynamic movement of the activating receptor NKp46 and confinement of the inhibitory receptor Ly49A. Sci Signal 11, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao NS, Bix M, Zijlstra M, Jaenisch R, Raulet D, MHC class I deficiency: susceptibility to natural killer (NK) cells and impaired NK activity. Science 253, 199–202 (1991). [DOI] [PubMed] [Google Scholar]
- 21.Belanger S, Tu MM, Rahim MM, Mahmoud AB, Patel R, Tai LH, Troke AD, Wilhelm BT, Landry JR, Zhu Q, Tung KS, Raulet DH, Makrigiannis AP, Impaired natural killer cell self-education and “missing-self” responses in Ly49-deficient mice. Blood 120, 592–602 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bern MD, Beckman DL, Ebihara T, Taffner SM, Poursine-Laurent J, White JM, Yokoyama WM, Immunoreceptor tyrosine-based inhibitory motif-dependent functions of an MHC class I-specific NK cell receptor. Proc Natl Acad Sci U S A 114, E8440–E8447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Viant C, Fenis A, Chicanne G, Payrastre B, Ugolini S, Vivier E, SHP-1-mediated inhibitory signals promote responsiveness and anti-tumour functions of natural killer cells. Nat Commun 5, 5108 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Lorenz U, SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev 228, 342–359 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altan-Bonnet G, Germain RN, Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol 3, e356 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feinerman O, Veiga J, Dorfman JR, Germain RN, Altan-Bonnet G, Variability and robustness in T cell activation from regulated heterogeneity in protein levels. Science 321, 1081–1084 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cotari JW, Voisinne G, Altan-Bonnet G, Diversity training for signal transduction: leveraging cell-to-cell variability to dissect cellular signaling, differentiation and death. Curr Opin Biotechnol 24, 760–766 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burshtyn DN, Scharenberg AM, Wagtmann N, Rajagopalan S, Berrada K, Yi T, Kinet JP, Long EO, Recruitment of tyrosine phosphatase HCP by the killer cell inhibitor receptor. Immunity 4, 77–85 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowin-Kropf B, Kunz B, Beermann F, Held W, Impaired natural killing of MHC class I-deficient targets by NK cells expressing a catalytically inactive form of SHP-1. J Immunol 165, 1314–1321 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Yu J, Heller G, Chewning J, Kim S, Yokoyama WM, Hsu KC, Hierarchy of the human natural killer cell response is determined by class and quantity of inhibitory receptors for self-HLA-B and HLA-C ligands. J Immunol 179, 5977–5989 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Charoudeh HN, Schmied L, Gonzalez A, Terszowski G, Czaja K, Schmitter K, Infanti L, Buser A, Stern M, Quantity of HLA-C surface expression and licensing of KIR2DL+ natural killer cells. Immunogenetics 64, 739–745 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Le Luduec JB, Boudreau JE, Freiberg JC, Hsu KC, Novel Approach to Cell Surface Discrimination Between KIR2DL1 Subtypes and KIR2DS1 Identifies Hierarchies in NK Repertoire, Education, and Tolerance. Front Immunol 10, 734 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mao Y, van Hoef V, Zhang X, Wennerberg E, Lorent J, Witt K, Masvidal L, Liang S, Murray S, Larsson O, Kiessling R, Lundqvist A, IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood 128, 1475–1489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Das J, Activation or tolerance of natural killer cells is modulated by ligand quality in a nonmonotonic manner. Biophys J 99, 2028–2037 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boudreau JE, Mulrooney TJ, Le Luduec JB, Barker E, Hsu KC, KIR3DL1 and HLA-B Density and Binding Calibrate NK Education and Response to HIV. J Immunol 196, 3398–3410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yawata M, Yawata N, Draghi M, Little AM, Partheniou F, Parham P, Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J Exp Med 203, 633–645 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gardiner CM, Guethlein LA, Shilling HG, Pando M, Carr WH, Rajalingam R, Vilches C, Parham P, Different NK cell surface phenotypes defined by the DX9 antibody are due to KIR3DL1 gene polymorphism. J Immunol 166, 2992–3001 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Beziat V, Traherne JA, Liu LL, Jayaraman J, Enqvist M, Larsson S, Trowsdale J, Malmberg KJ, Influence of KIR gene copy number on natural killer cell education. Blood 121, 4703–4707 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boudreau JE, Giglio F, Gooley TA, Stevenson PA, Le Luduec JB, Shaffer BC, Rajalingam R, Hou L, Hurley CK, Noreen H, Reed EF, Yu N, Vierra-Green C, Haagenson M, Malkki M, Petersdorf EW, Spellman S, Hsu KC, KIR3DL1/ HL A-B Subtypes Govern Acute Myelogenous Leukemia Relapse After Hematopoietic Cell Transplantation. J Clin Oncol 35, 2268–2278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saunders PM, Pymm P, Pietra G, Hughes VA, Hitchen C, O’Connor GM, Loiacono F, Widjaja J, Price DA, Falco M, Mingari MC, Moretta L, McVicar DW, Rossjohn J, Brooks AG, Vivian JP, Killer cell immunoglobulin-like receptor 3DL1 polymorphism defines distinct hierarchies of HLA class I recognition. J Exp Med 213, 791–807 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bezman NA, Kim CC, Sun JC, Min-Oo G, Hendricks DW, Kamimura Y, Best JA, Goldrath AW, Lanier LL, Molecular definition of the identity and activation of natural killer cells. Nat Immunol 13, 1000–1009 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCullen MV, Li H, Cam M, Sen SK, McVicar DW, Anderson SK, Analysis of Ly49 gene transcripts in mature NK cells supports a role for the Pro1 element in gene activation, not gene expression. Genes Immun 17, 349–357 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Corral L, Hanke T, Vance RE, Cado D, Raulet DH, NK cell expression of the killer cell lectin-like receptor G1 (KLRG1), the mouse homolog of MAFA, is modulated by MHC class I molecules. Eur J Immunol 30, 920–930 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Salcedo M, Diehl AD, Olsson-Alheim MY, Sundback J, Van Kaer L, Karre K, Ljunggren HG, Altered expression of Ly49 inhibitory receptors on natural killer cells from MHC class I-deficient mice. J Immunol 158, 3174–3180 (1997); [PubMed] [Google Scholar]
- 45.Joncker NT, Shifrin N, Delebecque F, Raulet DH, Mature natural killer cells reset their responsiveness when exposed to an altered MHC environment. J Exp Med 207, 2065–2072 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elliott JM, Wahle JA, Yokoyama WM, MHC class I-deficient natural killer cells acquire a licensed phenotype after transfer into an MHC class I-sufficient environment. J Exp Med 207, 2073–2079 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nabekura T, Lanier LL, Tracking the fate of antigen-specific versus cytokine-activated natural killer cells after cytomegalovirus infection. J Exp Med 213, 2745–2758 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen QT, Hsiue PP, Sindelar CV, Welch MD, Campellone KG, Wang HW, Structural insights into WHAMM-mediated cytoskeletal coordination during membrane remodeling. J Cell Biol 199, 111–124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan I, Ng CH, Lim L, Leung T, Phosphorylation of a novel myosin binding subunit of protein phosphatase 1 reveals a conserved mechanism in the regulation of actin cytoskeleton. J Biol Chem 276, 21209–21216 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Niogret C, Miah SMS, Rota G, Fonta NP, Wang H, Held W, Birchmeier W, Sexl V, Yang W, Vivier E, Ho PC, Brossay L, Guarda G, Shp-2 is critical for ERK and metabolic engagement downstream of IL-15 receptor in NK cells. Nat Commun 10, 1444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matalon O, Ben-Shmuel A, Kivelevitz J, Sabag B, Fried S, Joseph N, Noy E, Biber G, Barda-Saad M, Actin retrograde flow controls natural killer cell response by regulating the conformation state of SHP-1. EMBO J 37, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klebanovych A, Sladkova V, Sulimenko T, Vosecka V, Capek M, Draberova E, Draber P, Sulimenko V, Regulation of Microtubule Nucleation in Mouse Bone Marrow-Derived Mast Cells by Protein Tyrosine Phosphatase SHP-1. Cells 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Speir M, Nowell CJ, Chen AA, O’Donnell JA, Shamie IS, Lakin PR, D’Cruz AA, Braun RO, Babon JJ, Lewis RS, Bliss-Moreau M, Shlomovitz I, Wang S, Cengia LH, Stoica AI, Hakem R, Kelliher MA, O’Reilly LA, Patsiouras H, Lawlor KE, Weller E, Lewis NE, Roberts AW, Gerlic M, Croker BA, Ptpn6 inhibits caspase-8- and Ripk3/Mlkl-dependent inflammation. Nat Immunol 21, 54–64 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thompson TW, Kim AB, Li PJ, Wang J, Jackson BT, Huang KTH, Zhang L, Raulet DH, Endothelial cells express NKG2D ligands and desensitize antitumor NK responses. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Narni-Mancinelli E, Jaeger BN, Bernat C, Fenis A, Kung S, De Gassart A, Mahmood S, Gut M, Heath SC, Estelle J, Bertosio E, Vely F, Gastinel LN, Beutler B, Malissen B, Malissen M, Gut IG, Vivier E, Ugolini S, Tuning of natural killer cell reactivity by NKp46 and Helios calibrates T cell responses. Science 335, 344–348 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Gazit R, Gruda R, Elboim M, Arnon TI, Katz G, Achdout H, Hanna J, Qimron U, Landau G, Greenbaum E, Zakay-Rones Z, Porgador A, Mandelboim O, Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol 7, 517–523 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Raulet DH, Vance RE, Self-tolerance of natural killer cells. Nat Rev Immunol 6, 520–531 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Jelencic V, Sestan M, Kavazovic I, Lenartic M, Marinovic S, Holmes TD, Prchal-Murphy M, Lisnic B, Sexl V, Bryceson YT, Wensveen FM, Polic B, NK cell receptor NKG2D sets activation threshold for the NCR1 receptor early in NK cell development. Nat Immunol 19, 1083–1092 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Napolitano A, Pittoni P, Beaudoin L, Lehuen A, Voehringer D, MacDonald HR, Dellabona P, Casorati G, Functional education of invariant NKT cells by dendritic cell tuning of SHP-1. J Immunol 190, 3299–3308 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, Long EO, Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol Cell Biol 23, 6291–6299 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matalon O, Fried S, Ben-Shmuel A, Pauker MH, Joseph N, Keizer D, Piterburg M, Barda-Saad M, Dephosphorylation of the adaptor LAT and phospholipase C-gamma by SHP-1 inhibits natural killer cell cytotoxicity. Sci Signal 9, ra54 (2016) [DOI] [PubMed] [Google Scholar]
- 62.Ben-Shmuel A, Biber G, Sabag B, Barda-Saad M, Modulation of the intracellular inhibitory checkpoint SHP-1 enhances the antitumor activity of engineered NK cells. Cell Mol Immunol, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, Leong JW, Abdel-Latif S, Schneider SE, Willey S, Neal CC, Yu L, Oh ST, Lee YS, Mulder A, Claas F, Cooper MA, Fehniger TA, Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med 8, 357ra123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, Nassif Kerbauy L, Overman B, Thall P, Kaplan M, Nandivada V, Kaur I, Nunez Cortes A, Cao K, Daher M, Hosing C, Cohen EN, Kebriaei P, Mehta R, Neelapu S, Nieto Y, Wang M, Wierda W, Keating M, Champlin R, Shpall EJ, Rezvani K, Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med 382, 545–553 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vance RE, Jamieson AM, Cado D, Raulet DH, Implications of CD94 deficiency and monoallelic NKG2A expression for natural killer cell development and repertoire formation. Proc Natl Acad Sci U S A 99, 868–873 (2002); [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ganesan S, Hoglund P, Inhibitory Receptor Crosslinking Quantitatively Dampens Calcium Flux Induced by Activating Receptor Triggering in NK Cells. Front Immunol 9, 3173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lis M, Artyomov MN, Devadas S, Chakraborty AK, Efficient stochastic simulation of reaction-diffusion processes via direct compilation. Bioinformatics 25, 2289–2291 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li P, Jiang N, Nagarajan S, Wohlhueter R, Selvaraj P, Zhu C, Affinity and kinetic analysis of Fcgamma receptor IIIa (CD16a) binding to IgG ligands. J Biol Chem 282, 6210–6221 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Zilka A, Landau G, Hershkovitz O, Bloushtain N, Bar-Ilan A, Benchetrit F, Fima E, van Kuppevelt TH, Gallagher JT, Elgavish S, Porgador A, Characterization of the heparin/heparan sulfate binding site of the natural cytotoxicity receptor NKp46. Biochemistry 44, 14477–14485 (2005). [DOI] [PubMed] [Google Scholar]
- 70.Lau CM, Adams NM, Geary CD, Weizman OE, Rapp M, Pritykin Y, Leslie CS, Sun JC, Epigenetic control of innate and adaptive immune memory. Nat Immunol 19, 963–972 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data are accessible through the NCBI Sequence Read Archive database with accession code PRJNA723694. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.