

Abstract
PURPOSE
In neuroblastoma (NB), the ALK receptor tyrosine kinase can be constitutively activated through activating point mutations or genomic amplification. We studied ALK genetic alterations in high-risk (HR) patients on the HR-NBL1/SIOPEN trial to determine their frequency, correlation with clinical parameters, and prognostic impact.
MATERIALS AND METHODS
Diagnostic tumor samples were available from 1,092 HR-NBL1/SIOPEN patients to determine ALK amplification status (n = 330), ALK mutational profile (n = 191), or both (n = 571).
RESULTS
Genomic ALK amplification (ALKa) was detected in 4.5% of cases (41 out of 901), all except one with MYCN amplification (MNA). ALKa was associated with a significantly poorer overall survival (OS) (5-year OS: ALKa [n = 41] 28% [95% CI, 15 to 42]; no-ALKa [n = 860] 51% [95% CI, 47 to 54], [P < .001]), particularly in cases with metastatic disease. ALK mutations (ALKm) were detected at a clonal level (> 20% mutated allele fraction) in 10% of cases (76 out of 762) and at a subclonal level (mutated allele fraction 0.1%-20%) in 3.9% of patients (30 out of 762), with a strong correlation between the presence of ALKm and MNA (P < .001). Among 571 cases with known ALKa and ALKm status, a statistically significant difference in OS was observed between cases with ALKa or clonal ALKm versus subclonal ALKm or no ALK alterations (5-year OS: ALKa [n = 19], 26% [95% CI, 10 to 47], clonal ALKm [n = 65] 33% [95% CI, 21 to 44], subclonal ALKm (n = 22) 48% [95% CI, 26 to 67], and no alteration [n = 465], 51% [95% CI, 46 to 55], respectively; P = .001). Importantly, in a multivariate model, involvement of more than one metastatic compartment (hazard ratio [HR], 2.87; P < .001), ALKa (HR, 2.38; P = .004), and clonal ALKm (HR, 1.77; P = .001) were independent predictors of poor outcome.
CONCLUSION
Genetic alterations of ALK (clonal mutations and amplifications) in HR-NB are independent predictors of poorer survival. These data provide a rationale for integration of ALK inhibitors in upfront treatment of HR-NB with ALK alterations.
INTRODUCTION
Neuroblastoma (NB), the most frequent solid, extracranial malignancy in children, exhibits wide clinical and genetic heterogeneity. High-risk neuroblastoma (HR-NB), defined as metastatic disease over the age of 12 months or MYCN-amplified (MNA) disease at any age, remains associated with long-term survival rates of only 50%.1 Current treatment approaches consist of intensive induction chemotherapy, surgical resection of the primary tumor, consolidation with high-dose chemotherapy (HDC), and autologous stem-cell rescue, and for minimal residual disease, isotretinoin in combination with human or mouse chimeric anti-GD2 antibody, ch14.18.2-8
CONTEXT
Key Objective
High risk neuroblastoma (HR-NB) is one of the most difficult childhood cancers to cure. This study examined whether the presence of an ALK alteration (amplification or mutation) was associated with a poor prognosis in a large patient series treated on the prospective European high-risk neuroblastoma trial (HR-NBL1).
Knowledge Generated
We found that ALK amplification or clonal mutation was associated with inferior prognosis in patients with HR-NB and both are independent prognostic variables on multivariate analysis. To our knowledge, this is the first study to report the highly prognostic significance of ALK amplification in HR-NB.
Relevance
As ALK can be targeted therapeutically, this study convincingly argues for the introduction of ALK inhibitors for upfront management of patients with HR-NB with ALK aberrations. Importantly, the prognostic significance of ALK alterations included a subgroup of trial patients treated with the current standard of care for HR-NB including anti-GD2 immunotherapy.
In NB, several recurrent genetic alterations have been described. MNA is a strong biomarker associated with rapid tumor growth.9 Other copy-number alterations occur over more extensive chromosome regions, with segmental chromosome alterations being associated with a poor outcome.10 Recurrent mutations have been described in the RAS-MAPK pathway, chromatin remodeling genes (ATRX and ARID1A), and TERT rearrangements.11-14
Activating anaplastic lymphoma kinase (ALK) mutations are the most frequent mutations in NB, occurring in both familial and sporadic cases, with somatically acquired ALK mutations (ALKm) observed in 6%-12% of sporadic NBs in all risk groups.15-18
These ALK activating mutations are localized most frequently within the kinase domain at hotspots identified at the F1174, R1275, and F1245 positions, with mutations occurring both at clonal (> 20% mutated allele fraction [MAF]) or subclonal levels (< 20% MAF).19-23
ALK can also be activated by genomic focal amplification, described in 1%-2% of NBs, almost exclusively with MNA,17,24 or, more rarely, following structural rearrangements.25 Genetic alterations of ALK are associated with poorer survival in the overall NB population.24,26 However, their prognostic role in HR-NB has been less well studied.10,17,24 Altogether, ALK alterations are an important molecular target, given the role of ALK as a driver oncogene in NB and its actionability with small molecule therapies.27-29
To determine the frequency of ALK alterations (mutations and amplifications), their correlation with clinical characteristics, and their prognostic impact in HR-NB, we analyzed a large series of 1,092 diagnostic NB samples from patients on the HR-NBL1/SIOPEN trial.
MATERIALS AND METHODS
Patients and Samples
Patients were treated within the HR-NBL1/SIOPEN Protocol (ClinicalTrials.gov: NCT01704716, EudraCT: 2006-001489-17; Protocol [online only]), an international, randomized, multiarm, open-label, phase III trial.2-5,30,31 Patients with International Neuroblastoma Staging System stages 2, 3, 4, or 4S with MNA, or International Neuroblastoma Staging System stage 4 without MNA ≥ 12 months of age at diagnosis were eligible for the trial up to 20 years of age. Within the trial, several randomized treatment arms were conducted over different periods (Appendix Fig A1, online only). Induction random assignments included the following: R0—random assignment of prophylactic granulocyte colony-stimulating factor during rapid COJEC induction31; R3—comparison of two induction regimens, rapid-COJEC versus modified N7.32 HDC was evaluated in the R1 random assignment: busulfan or melphalan versus carboplatin or etoposide or melphalan.3 Anti-GD2 immunotherapy random assignments during maintenance phase were explored in R2 (2009-2013) and R4 (2014-2017), both comparing dinutuximab beta with oral isotretinoin to dinutuximab beta and subcutaneous interleukin-2 with oral isotretinoin, but with altered schedules.5,30 In the interim, dinutuximab beta with oral isotretinoin was the recommended standard.
Patients were enrolled on the HR-NBL1/SIOPEN trial after approval by national regulatory authorities and by national, and institutional, ethical committees or review boards in participating countries. Parents or guardians and patients according to age provided written informed consent for treatment, data collection, and analysis.
The ALK analysis cohort consisted of patients for whom a contributive tumor sample obtained at diagnosis was available in a SIOPEN reference laboratory33 for additional molecular analysis with available follow-up data (Fig 1).
FIG 1.
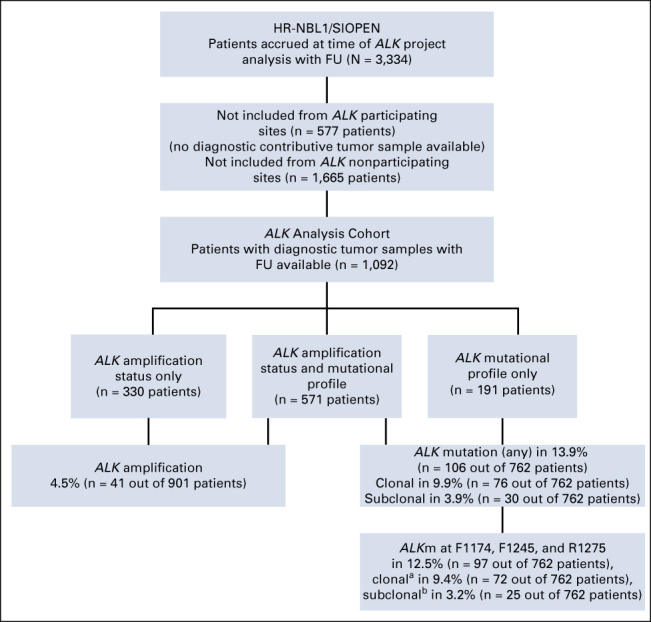
Flow diagram of patient inclusion. A total of 3,334 patients with HR-NB were enrolled in the HR-NBL1 trial from 188 centers. Among these, 2,350 patients were not included in this study, either because no contributive tumor material was available, or because these was no FU data, or both. Thus, 1,092 patients from 132 centers were included in this study. aClonal level: > 20% MAF. bSubclonal level: MAF 0.1%-20%. FU, follow-up; HR-NB, high-risk neuroblastoma; MAF, mutated allele fraction.
MYCN status and tumor genomic copy-number profiles were determined in SIOPEN reference laboratories as described previously.10,33-36 Samples were required to contain at least 20% tumor cells on pathologic examination.
The ALK amplification (ALKa) status was evaluated using either fluorescence in situ hybridization and/or multiplex ligation polymerase chain reaction–dependent amplification, array comparative genomic hybridization (aCGH), and/or array single-nucleotide polymorphism according to established guidelines.10,33,34,37 ALK gene amplification was defined as more than fourfold increase of ALK signals in relation to numbers of chromosome 2 by fluorescence in situ hybridization, or as more than 10 copies of the gene estimated by multiplex ligation–dependent amplification, aCGH, or array single-nucleotide polymorphism.
The ALK mutational (ALKm) status was determined by Sanger sequencing, next-generation sequencing (NGS) techniques (coverage > 80×), targeted deep sequencing (TDS), or a combination of the latter techniques, covering the ALK regions of interest (exon 23: chr2:29443647-29443776; exon 24: chr2:29436830-29436935; exon 25: chr2:29432603-29432704; UCSC Genome Browser Home,38 hg19) containing the ALK mutational hotspots F1174 (exon 23), F1245 (exon 24), and R1275 (exon 25).20,22
MAF ≥ 20% were defined as clonal events and MAF < 20% as subclonal events, as reported previously.20,22 No correction for tumor cell content was undertaken when reporting MAF. Mutations identified by Sanger sequencing were considered clonal. All detected mutations were validated by a second independent experiment: for clonal events, TDS data were validated by Sanger sequencing, and for subclonal events, NGS or TDS was validated in an independent second experiment.
Standard bioinformatics were used to detect mutations in NGS experiments as previously reported. Mutations in TDS experiments were determined as described previously.20,22 In brief, to highlight mutations, in each NB sample, the frequencies of each base at each position of the analyzed regions were compared with those observed in all other samples and controls. This approach enabled the identification of mutations with a statistically significant increase in percentage of a variant base, compared with background noise.
Statistical Analysis
Event-free survival (EFS) was calculated from diagnosis to the first relapse, progressive disease, secondary malignancy, or death from any cause, or until last patient contact. Overall survival (OS) was calculated from diagnosis to death from any cause, or until the last patient contact. EFS and OS were estimated using the Kaplan-Meier method and compared using the logrank test, and if indicated with pseudo-value regression for 5-year OS.39-41 EFS and OS are presented as 5-year point estimates together with 95% CIs using log-log transformation.41 To adjust for established risk-factors (age at diagnosis, stage, number of metastatic compartments, and MYCN amplification), a Cox proportional hazards regression model was used.
Correlations between patient and disease characteristics and ALK genetic alterations were explored using chi-square tests.
To allow for sufficient follow-up time, only patients enrolled until December 31, 2019, were considered. The data cutoff for the final analysis was October 3, 2020. We calculated median follow-up using the inverse Kaplan-Meier estimate. Statistical analysis was performed using SAS (version 9.4).
RESULTS
Of 3,334 patients enrolled on the HR-NBL1/SIOPEN trial between November 24, 2002, and December 31, 2019, 1,092 patients were included in the ALK analysis cohort (Fig 1; Appendix Table A1, online only). Patients were accrued from 132 SIOPEN member institutions or hospitals in 19 countries (Appendix Table A2, online only). Among these 1,092 patients, 81% (889 out of 1,092) were > 18 months of age at diagnosis, 47% (521 out of 1,092) showed MNA, and 88% (966 out of 1,092) had stage 4 disease, with no statistically significant difference in EFS or OS between the ALK analysis cohort and the overall HR-NBL1 cohort (Appendix Fig A2, online only).42 The median follow-up period was 6.8 years (0.1-17.4 years).
ALK Alterations
Within the ALK cohort, the ALKm status was analyzed in 762 patients, the ALKa status in 901 cases, with both ALKm and ALKa studied in 571 patients (Fig 1, Table 1).
TABLE 1.
Characteristics of Patients According to the ALK Amplification or ALK Mutation Status

ALK alterations were detected in 146 out of 1,092 patients with ALKa occurring in 4.5% (41 out of 901 cases) and ALKm in 13.9% (106 out of 762 cases). Only one case showed ALKa and a concomitant ALK R1275Q mutation with an MAF of 93%, suggesting that the mutated allele is contained in the amplicon (Appendix Fig A3, online only).
ALK Amplification and Correlation With Risk Factors
High-level genomic amplification of the ALK gene was found in 4.5% (41 out of 901) of cases (Fig 2A, Table 1). All but one also had MNA. ALKa significantly correlated with MNA (P < .001), non–stage 4 disease (P < .001), and age at diagnosis < 18 months (P = .005). No correlation between the presence of ALKa and response at the end of induction treatment was observed.
FIG 2.

Genetic alterations of ALK in patients with HR-NB. (A) Copy-number profile of case 536. Genomic coamplification of MYCN and ALK is observed on chromosome 2, encompassing the regions between position 15,440,477 and 16,822,999 and between 29,113,790 and 30,309,749 bp (human genome assembly hg19; UCSC Genome Browser Home38). (B) Frequency distribution of mutated ALK alleles at the studied chromosome regions, encompassing the AA positions F1174, L1190, L1196, R1245, D1270, G1272, M1273, A1274, R1275, and Y1278 detected, in 762 samples. ALK mutations involved the common mutational hotspots (F1174, F1245, and R1275) in 12.5% (97 out of 772) of cases, at a clonal level (MAF 20%-93%) in 72 cases, and at a subclonal level (MAF < 20%) in 25 cases. At the F1174 hotspot (chr2: 29,443,695-29,443,697), alterations were observed in 44 cases: 42 cases harbored a mutation leading to the AA change F1174L, one case with F1174I, and one case with F1174S, with MAFs ranging from 0.12% to 78%. At the R1275 hotspot (chr2: 29,432,849-29,430,139), mutations were detected in 43 cases: 38 cases harbored a mutation leading to the AA change R1275Q and five cases with R1275L, with the MAFs ranging from 0.2% to 93%. Ten cases showed ALK mutations at the F1245 hotspot (chr2: 29,436,858-29,436,860) within exon 24. Three samples showed the F1245L mutation, three cases carried the F1245C mutation, three showed the F1245I mutation, and one showed mutation F1245V mutation (Fig 1 and Appendix Table A1). Other ALK mutations were detected at residues I1170, L1190 (two cases), L1196, D1270, G1272, M1273, A1274, and Y1278 within the explored regions, leading to a nonsynonymous AA change with a predicted functional impact. All these mutations were clonal (MAF > 20%) except for M1273I (MAF 0.2%) and I1170 (MAF 2.8%). AA, amino acid; aCGH, array comparative genomic hybridization; bp, base pair; HR-NB, high-risk neuroblastoma; MAF, mutated allele fraction; UCSC, University of California, Santa Cruz.
A statistically significant poorer 5-year OS was observed in patients whose tumors harbored ALKa (5-year OS: ALKa 28% [95% CI, 15 to 42] v non-ALKa 51% [95% CI, 47 to 54]; P < .0001; Fig 3A, Table 2) with a stronger prognostic effect in patients with stage 4 or 4S MNA.
FIG 3.

Survival in the ALK analysis cohort. (A) OS according to ALK amplification status in 901 patients: presence of ALK amplification (n = 41), 5-year OS 28% (95% CI, 15 to 42) versus absence of ALK amplification (n = 860), 5-year OS 51% (95% CI, 47 to 54); P < .0001. (B) OS according to ALK mutation status in 762 patients: presence of an ALK mutation (n = 106), 5-year OS 41% (95% CI, 31 to 51) versus absence of an ALK mutation (n = 656), 5-year OS 49% (95% CI, 45 to 53); P = NS. (C) OS according to ALK clonal or subclonal mutation status in 762 patients: no mutation (n = 656), 5-year OS 49% (95% CI, 45 to 53); clonal mutations (n = 76), 5-year OS 34% (95% CI, 23 to 45); and subclonal mutations (n = 30), 5-year OS 59% (95% CI, 39 to 74), respectively; P = .018. (D) OS according to the presence of any ALK alterations in 611 patients with known ALK amplification and ALK mutation status: presence of an ALK alteration (n = 146), 5-year OS 37% (95% CI, 29 to 45); versus absence of ALK alterations (n = 465), 5-year OS 51% (95% CI, 46 to 55); P = .005. (E) OS according to the type of ALK alteration in the cohort of 571 patients with known ALK amplification and ALK mutation status: no alteration (n = 465), 5-year OS 51% (95% CI, 46 to 55); clonal mutations (n = 65), 5-year OS 33% (95% CI, 21 to 44); subclonal mutations (n = 12), 5-year OS 48% (95% CI, 26 to 67); and ALK amplification (n = 19), 5-year OS 26% (95% CI, 10 to 47), respectively; P = .001. (F) OS according to ALK alterations (ALK amplification or clonal ALK mutation) in patients who received immunotherapy (n = 141): To evaluate the impact of ALK alterations (ALK amplification or clonal ALK mutation) in patients who received dinutuximab beta, OS was calculated from the start of dinutuximab beta treatment and evaluated using the same approaches as described in the Materials and Methods section. ALK alteration (ALK amplification or clonal ALK mutation, n = 29, 5-year OS 48% [95% CI, 28 to 65]) versus no ALK alteration (n = 112) 67% (95% CI, 56 to 75); P = .034. Patient details: Appendix Table A3. HR, hazard ratio; NS, not significant; OS, overall survival; ref, reference.
TABLE 2.
EFS and OS According to ALK Alterations

ALK Mutation and Correlation With Risk Factors
ALK mutational status was studied in 762 cases by Sanger sequencing (n = 163), by NGS techniques (n = 13), or by TDS (n = 650, including 64 by TDS and Sanger). The biologic data for 52 cases have been reported previously.22
Among these, 13.9% (106 out of 762) showed at least one ALKm within the explored ALK regions of interest, with 10% (76 out of 762) harboring mutations at a clonal level (MAF > 20%) and 3.9% (30 out of 762) at a subclonal level (MAF ≤ 20%): nine cases—MAF 0.1% to < 1%, 10 cases MAF 1% to < 5%, two cases MAF 5% to < 10%, and nine cases MAF 10% to < 20% (Figs 1 and 2B; Table 1).
Concordance between results analyzed by two different techniques was observed in 64 cases with clonal ALKm (TDS and Sanger). Subclonal ALKm were validated by a second independent TDS experiment, with an excellent correlation of MAF between the two experiments (R2 = 0.9924; P < .0001) (Appendix Fig A4, online only).
ALKm involved the common mutational hotspots (F1174, F1245, and R1275) in 12.5% (97 out of 762) of cases, comprising 91% (97 out of 106) of all detected ALKm (Fig 2B).
Interestingly, three cases harbored two or more distinct mutations. In the first case, both F1174L and F1245L mutations were observed (MAF 2% and 0.8%, respectively). The second case showed three subclonal mutations F1174L, R1275Q, and R1275L (MAF 2.9%, 8.9%, and 2.9%, respectively). A third case harbored a mutation at the F1174 and R1275 hotspots (MAF 27% and 1.3%, respectively).
There were no statistically significant correlations between ALKm and stage, age at diagnosis, or localization of the primary tumor (adrenal, abdominal, or other) (Table 1). However, a significant correlation was observed between the presence of an ALKm and MNA (P < .001), with an enrichment of ALKm F1174 in MNA tumors (P = .0005). This was also observed when analyzing only stage 4 tumors. No correlation between ALKm and response at the end of induction treatment was observed.
No statistically significant difference in outcome was observed between patients harboring any ALKm versus none (Fig 3B, Table 2). However, when distinguishing clonal and subclonal mutations, a poorer OS was observed only in patients with clonal ALKm, as opposed to subclonal or no mutations (5-year OS, clonal ALKm 34% [95% CI, 23 to 45], subclonal ALKm 59% [95% CI, 39 to 74], and no ALKm 49% [95% CI, 45 to 53]; P = .018) (Fig 3C, Table 2).
Patients with metastatic disease (stage 4 or 4S MNA) and a clonal ALKm showed a trend toward poorer OS. However, in patients with localized disease, the presence of ALKm did not confer poorer survival (Table 2).
Overall Prognostic Impact of ALK Genetic Alterations
To determine the overall prognostic impact of ALK genetic alterations, we focused on the subgroup of 571 patients with both known ALKa and ALKm status. In this subgroup of patients, a statistically significant poorer OS was observed in patients whose tumors harbored any ALK alteration (5-year OS, any alteration 37% [95% CI, 29 to 45] v no alteration 51% [95% CI, 46 to 55]; P = .005; Fig 3D). ALKa or clonal ALKm were associated with a poorer outcome (5-year OS, ALKa 26% [95% CI, 10 to 47], clonal ALKm 33% [95% CI, 21 to 44], subclonal ALKm 48% [95% CI, 26 to 67], and no ALK alteration 51% [95% CI, 46 to 55]; P = .001; Fig 3E, Table 2).
Among the subgroup of patients with known ALK status, we sought to determine the prognostic impact of ALK alterations according to the different treatment arms of HR-NBL1. Indeed, in the HR-NBL01/SIOPEN trial, the introduction of busulfan and melphalan as standard for HDC, and anti-GD2 maintenance therapy as a new standard since 2010, has led to significantly improved survival (Appendix Fig A5F, online only).3-5 Importantly, when considering patients treated according to the SIOPEN standard with busulfan and melphalan HDC and maintenance immunotherapy, the presence of an ALK alteration (ALKa or clonal ALKm) remained associated with a poorer 5-year OS of 48% (95% CI, 28 to 65), versus no ALK alteration 67% (95% CI, 56 to 75); P = .03 (Fig 3F, Appendix Table A3, online only), with a trend also observed when taking into account all ALKm (clonal and subclonal, P = .059).
Based on univariate risk factor exploration of the whole ALK analysis cohort (Appendix Fig A5), we developed a Cox model for multivariate analysis including clinical and biologic parameters previously shown to be of prognostic impact (n = 571 patients). Involvement of two or more metastatic compartments (OS: hazard ratio [HR], 2.87 [95% CI, 1.73 to 4.78]; P = .001) and the presence of ALKa (OS: HR, 2.38 [95% CI, 1.32 to 4.27]; P = .004) and clonal ALKm (OS: HR, 1.77 [95% CI, 1.25 to 2.49]; P = .001) were of independent prognostic significance, whereas MNA and age were not (Table 3).
TABLE 3.
Multivariate Analysis in 571 Patients With a Known ALK Amplification and ALK Mutation Status

DISCUSSION
In HR-NB, the identification of prognostic biomarkers is crucial for the development of new treatment approaches. Recent studies have shown that MNA is not associated with poorer outcome among the overall cohort of patients with HR-NB, but the presence of genomic amplifications other than MYCN might constitute a poor outcome biomarker.43 We now show in this large ALK analysis cohort that the presence of ALKa or clonal ALKm resulted in significantly worse outcome.
Given the oncogenic driver role of ALK activation, and the prognostic impact of ALKa or clonal ALKm, the introduction of frontline ALK-targeted treatment is now strongly supported by the current study. Although early phase clinical trials of first- and second-generation ALK inhibitors showed modest efficacy of the first-generation inhibitor crizotinib in NB with F1174 hotspot mutations being resistant,44 third-generation ALK inhibitors such as lorlatinib exhibit improved efficacy alone and when combined with chemotherapy.28,44-46 Crizotinib is currently being administered with chemotherapy in a phase III upfront trial for patients with HR-NB with ALK alterations (ClinicalTrials.gov: NCT03126916).
Improvements in HR-NB patient survival have been achieved with intensification of HDC and immunotherapy with dinutuximab (ch14.18/Sp02 and ch14.18/CH0),3-5,7 and our results highlight the potential of ALK inhibition as an attractive upfront precision-medicine strategy in patients with ALK alterations to further improve survival. Importantly, in patients reaching the maintenance treatment phase with dinutuximab beta in the HR-NBL1/SIOPEN trial, the presence of an ALK alteration was still associated with poorer survival, thus strongly suggesting that integration of ALK-targeted therapy is warranted throughout all treatment phases of modern-era HR-NB therapy.
ALKa was observed in 4% of NB cases, accounting for approximately 1 out of 3 of ALK-activated NB cases. To date, co-occurrence of ALK hotspot mutations and genomic amplification has rarely been reported in NB.17 In this extensive cohort of patients, one case harboring both ALKa and an R1275 ALKm was identified. This indicates that these alterations are not fully mutually exclusive, although co-occurrence is extremely rare.
ALKm were found in 13.9% of cases at the studied exonic regions harboring known ALK mutational hotspots.17,24 This is higher than previously reported frequencies of ALKm in HR-NB of approximately 10%, most likely as previous reports using Sanger sequencing or standard-resolution NGS approaches.24,26 Sanger sensitivity is limited to the detection of MAF > 15%-20%, but in NB, ALK mutations with lower MAFs have been reported.14,19-21
Ultradeep sequencing used in this analysis has a sensitivity limit of MAF of 0.1%.19,20 This approaches the theoretical limit of detection based on the genomic DNA input of 50 ng for one experiment, equivalent to 5,000 diploid genomes.
This study demonstrates that use of higher-resolution techniques enables a higher detection rate of ALKm. The MAF distribution indicated a majority of clonal events (76 out of 106 cases). Importantly, clonal ALKm were associated with poorer outcome and were of independent prognostic significance, but subclonal events were not. Subclonal events, defined in this study by MAF < 20%, comprised 28% (30 out of 106) of all ALKm, with a very low MAF (< 5%) observed in 19 cases.
However, when considering ALKm, the OS remains poor in all patient subgroups (5-year OS < 62%). Furthermore, although of different prognostic impact in this study, the biomarker (ALK mutation) might not be of distinct predictive impact, and even in patients with subclonal ALK mutations, ALK inhibitor treatment might be effective in the targeted cell population. Thus, future upfront trials should consider ALK-targeted treatment based on clinically applicable reliable detection limits (for instance MAF 5% for NGS techniques) rather than the MAF defining prognostic subgroups.
As tumor samples harbored at least 20% tumor cells by pathologic examination, with additional confirmation provided by a dynamic aCGH or SNPa profile in the majority of cases, the observed low MAF is likely to correspond to intratumoral heterogeneity. In NB, intratumor heterogeneity has been reported for MNA and segmental chromosome alterations.47-49 The coexistence of ALK nonmutated and mutated cells within a single tumor suggests that these different subclones might coexist in an advantageous equilibrium, which might crucially affect the dynamics of cancer progression.50,51 Correlation with pathologic findings, single-cell RNA or DNA experiments, and in situ approaches will elucidate how ALK-mutated cells are distributed throughout an NB. A higher frequency of ALKm at NB relapse has been demonstrated, suggesting clonal evolution of a minor ALK-mutated subclone to a dominant ALK mutated clone at relapse, but these cases might not represent clinically unfavorable cases initially.23,52,53 Further studies focusing on serial blood samples for ctDNA studies will further elucidate clonal evolution, also under targeted therapy.54
In HR-NB, mutations in the p53 or RAS-MAPK pathways, including ALK, together with telomere maintenance caused by induction of telomerase or ALT (alternative lengthening of telomere) are thought to increase tumor aggressiveness, resulting in even poorer survival among patients with HR-NB.55,56 As MYCN leads to upregulation of TERT expression, MNA associated with any ALK alteration might lead to inferior outcome. Cases with ALKa show both ALK pathway activation and activation of telomere maintenance through MNA, with a suggested additive effect of these genetic events. The very poor survival of ALKa patients is concordant with this observation. However, survival of patients whose tumors harbored ALKm and MNA was not different from those without MNA, suggesting that ALKm cases constitute a more heterogeneous group with regards to the mechanistic tumor classification.55
ALKa and ALK clonal mutation were both independent predictors of poor outcome in our multivariate Cox model. Notably, the end-of-induction response rate was not associated with ALK genetic alterations, suggesting that ALK-altered tumor cells are unlikely to be primarily chemotherapy resistant.
In summary, our data contribute to the rationale for future clinical trials introducing ALK-targeted treatment in the frontline setting together with chemotherapy and immunotherapy, and the distinct prognostic impact of different ALK alterations (ALKa and ALKm) needs to be considered.
ACKNOWLEDGMENT
The authors would like to thank the following Biobanks for providing samples: In Italy, the BIT-Gaslini Biobank, IRCCS Istituto Giannina Gaslini, Via G. Gaslini 5, Genova. In Spain, the Clinic Hospital INCLIVA-Valencia NB Tissue Bank (ISCIII, Reference: B0000339). They also thank the Children's Cancer & Leukemia Group (CCLG) Tissue Bank for access to DNA samples (CCLG 2015 BS 04), and contributing CCLG Centers, including members of the Experimental Cancer Medicine Centers Pediatric network.
Appendix
FIG A1.

Treatment flowchart of the HR-NBL1 Protocol (ClinicalTrials.gov: NCT01704716, EudraCT: 2006-001489-17) over the whole period. aInfants and children with a body weight below 12 kg will be dosed at 0.67 mg/kg/d. In infants weighing ≤ 5 kg, a further 1/3 dose reduction is advised. AUC, area under the curve; BUMEL, busulfan and melphalan; CAV, cyclophosphamide plus doxorubicin or vincristine; CEM, carboplatin, etoposide, and melphalan; CH14.18/CHO, human-mouse chimeric monoclonal anti-disialoganglioside GD2 antibody ch14.18 produced in Chinese hamster ovary (CHO) cells; COJEC, chemotherapy schedule COJEC defined below; GFR, glomerular filtration rate; IL-2, interleukin-2; IV, intravenous; P or E, cisplatin or etoposide; R1, randomization 1; R2, randomization 2; R3, randomization 3; R4, randomization 4; RT, radiotherapy; SCR, stringent complete response; TP, time period; TVD, topotecan-vincristine-doxorubicin.
FIG A2.

Comparison of patients in the ALK analysis cohort and patients not in the ALK analysis cohort. (A and B) EFS and OS of the ALK analysis cohort and patients not in the ALK cohort. (A) No statistically significant difference in EFS and (B) OS was observed between patients included in the ALK analysis cohort (n = 1,092, from 132 centers; red line), patients not included in this study from the same centers (n = 1,665, blue line) and patients not included in this study from centers not participating in this study (n = 577, green line) (5-year EFS: 40% [95% CI, 37 to 43] v 37% [95% CI, 35 to 40] v 33% [95% CI, 29 to 37]; 5-year OS: 49% [95% CI, 46 to 53] v 48% [95% CI, 46 to 51] v 44% [95% CI, 40 to 59]; P = NS). (C) Recruitment, by year (x-axis), in the ALK analysis cohort (% of patients: y-axis; absolute numbers: in the blue bars). The % and number of patients not included in the ALK analysis cohort from centers participating, and from nonparticipating centers, are indicated in orange and gray, respectively. EFS, event-free survival; NS, not significant; OS, overall survival.
FIG A3.

Double event of ALK amplification and ALK mutation detected in one case (case 15). The SNP array shows an amplified region in chromosome 2 encompassing the ALK gene. Sanger sequencing profile shows R1275Q mutation (MAF = 93.3%) in the same case. HD, high definition; MAF, mutated allele fraction; SNP, single-nucleotide polymorphism.
FIG A4.
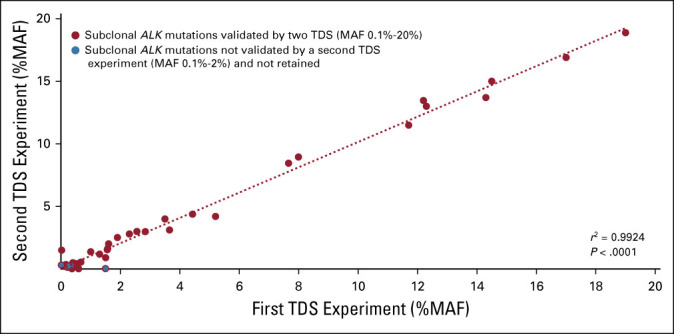
MAF of subclonal ALK mutations detected by TDS and confirmed by a second independent TDS experiment. Red spots representing the MAF for each ALK mutation are plotted on the x-axis (first TDS experiment) and y-axis (second TDS experiment), with a strong correlation between the two independent experiments (r2 = 0.9924, P < .0001). Blue spots represent subclonal ALK mutations with a very low MAF (< 0.1%) not confirmed in an independent experiment and not retained in the analysis (n = 6). MAF, mutated allele fraction; TDS, targeted deep sequencing.
FIG A5.

Survival in the ALK analysis cohort (n = 1,092 patients) according to known prognostic factors. (A) EFS and OS in the ALK analysis cohort population (n = 1,092 patients). Five-year EFS (blue line) 40% (95% CI, 37 to 43); 5-year OS (red line) 49% (95% CI, 46 to 53). (B) OS according to age. Five-year OS in patients < 1 year of age at diagnosis (red line) 50% (95% CI, 37 to 61); in patients 1-1.5 years of age at diagnosis (blue line) 58% (95% CI, 49 to 66); in patients 1.5-5 years of age at diagnosis (green line) 50% (95% CI, 46 to 53); and in patients > 5 years of age at diagnosis (purple line) 43% (95% CI, 35 to 50); P = NS (pseudo-value regression). (C) OS according to number of involved MCs. Five-year OS in patients with localized disease (red line) 67% (95% CI, 58 to 75), in patients with involvement of one MC (blue line) 65% (95% CI, 55 to 73), two MCs (green line) 52% (95% CI, 46 to 58), or over two MCs (purple line) 41% (95% CI, 36 to 46); P < .001. (D) OS according to stage. Five-year OS in patients with localized disease (red line) 67% (95% CI, 58 to 75), in patients with stage 4 disease (blue line) 47% (95% CI, 44 to 50), or stage 4s disease (green line) 54% (95% CI, 25 to 76); P < .001. (E) OS according to MYCN amplification in stage 4 disease. Five-year OS in patients with MNA (blue line) 46% (95% CI, 41 to 51), in patients without MNA (red line) 48% (95% CI, 44 to 53), NS (pseudo-value regression). (F) OS according to treatment period, before (< March 2010) or after (> March 2010) the definition of HDC by BUMEL and immunotherapy maintenance as standard treatment. A significant improvement survival because of BUMEL and GD2 standard therapy is observed. Five-year OS in patients having been treated before March 2010 (red line) 46% (95% CI, 41 to 51) versus after March 2010 (blue line) 51% (95% CI, 47 to 56); P = .039.3-5 BUMEL, busulfan and melphalan; cHR, crude hazard ratio; EFS, event-free survival; HDC, high-dose chemotherapy; HR, hazard ratio; MC, metastatic compartment; MNA, MYCN-amplified; NS, not significant; OS, overall survival; ref, reference.
TABLE A1.
Clinical Characteristics of 1,092 Patients Included in the ALK Analysis Cohort

TABLE A2.
Number of Patients Included in the ALK Analysis Cohort by Country and Center
TABLE A3.
Clinical Characteristics of 35 Patients Treated by Immunotherapy Whose Tumors Harbored ALK Genetic Alterations

Walentyna Balwierz
Honoraria: Shire, Gilead Sciences, Novartis, Amgen
Consulting or Advisory Role: Amgen, Novartis, Roche, Takeda
Travel, Accommodations, Expenses: Jazz Pharmaceuticals, Shire, Roche, Servier
Martin Elliott
Consulting or Advisory Role: Bayer
Dominique Valteau-Couanet
Consulting or Advisory Role: EUSA Pharma
Research Funding: Orphelia Pharma
Patents, Royalties, Other Intellectual Property: Royalties from Apeiron to SIOPEN
Travel, Accommodations, Expenses: EUSA Pharma, Jazz Pharmaceuticals
Deborah A. Tweddle
Honoraria: Eusa Pharma
Travel, Accommodations, Expenses: EUSA Pharma
Ruth Ladenstein
Honoraria: Apeiron Biologics, Boehringer Ingelheim, EUSA Pharma
Consulting or Advisory Role: Apeiron Biologics, Boehringer Ingelheim, EUSA Pharma
Research Funding: Apeiron Biologics, EUSA Pharma
Patents, Royalties, Other Intellectual Property: Apeiron Biologics, EUSA Pharma
Expert Testimony: Apeiron Biologics, EUSA Pharma
Travel, Accommodations, Expenses: Apeiron Biologics, EUSA Pharma
Gudrun Schleiermacher
Honoraria: BMS
Research Funding: Bristol Myers Squibb, Pfizer, MSDavenir, Roche
Travel, Accommodations, Expenses: Roche
No other potential conflicts of interest were reported.
SUPPORT
Supported by the Annenberg Foundation and the Association Hubert Gouin Enfance et Cancer, France. This study was also funded by the Fédération Enfants Cancers Santé, Les Bagouz à Manon, Les amis de Claire. Funding was also obtained from SiRIC/INCa (Grant INCa-DGOS-4654) and PHRC IC2007-09 grant.
High-throughput sequencing was performed by the ICGex NGS platform of the Institut Curie supported by the grants ANR-10-EQPX-03 (Equipex) and ANR-10-INBS-09-08 (France Génomique Consortium) from the Agence Nationale de la Recherche (Investissements d’Avenir program), by the Canceropole Ile-de-France, and by the SiRIC-Curie program - SiRIC Grant INCa-DGOS-4654.
In the United Kingdom, this work was supported by Neuroblastoma UK, Cancer Research UK, Department of Health, Families against Neuroblastoma, Solving Kids' Cancer, and Action Medical Research/Great Ormond Street Hospital Charity. The CCLG Tissue Bank is funded by Cancer Research UK and CCLG.
The funding of the European Commission 5th Frame Work Grant (SIOPEN-R-NET EC Grant No. QLRI-CT-2002-01768, www.siopen-r-net.org) supporting the HR-NBl1/SIOPEN trial is disclosed as funding source in the author statement. Pierre Fabre Médicament providing Busilvex (Paris, France), APEIRON (Vienna, Austria) providing dinutuximab beta (ch14.18/CHO) and the St Anna Kinderkrebsforschung GmbH (Vienna, Austria). The St Anna Kinderkrebsforschung was the academic sponsor of the HR-NBL1/SIOPEN trial providing resources for the remote trial data base and central trial management.
Recloning and production of the ch14.18 monoclonal antibody was done at Polymun, Vienna, Austria, and was enabled by a SIOPEN fundraising effort in 2001. APEIRON provided additional product at a later stage. The authors express their gratitude and appreciation to SIOPEN investigators, treating physicians, clinical research and care teams, and most importantly to patients and families facing high-risk neuroblastoma for their committed participation in the trial. The European Commission, Pierre Fabre Médicament, and Apeiron had no involvement in the conduct of the research and preparation of the article.
In addition, this work was supported as follows: Belgium: vzw Kinderkankerfonds and Kom op tegen Kanker. Czech Republic: MH CZ—DRO, University Hospital Motol, Prague, Czech Republic; Israel: Hayim Association—for Children with Cancer in Israel, Ramat Gan. Italy: Fondazione Italiana per la Lotta al Neuroblastoma O.N.L.U.S. c/o Istituto G. Gaslini, Genova, Associazione Bianca Garavaglia O.N.L.U.S., Busto Arsizio. Spain: Grant FIS EC10/303, Asociación Pablo Ugarte, Cancercare Xavia, Sumemos Muchas Manos, Heath Institute Carlos III (ISCIII) and FEDER (European Regional Development Fund): Grants PI17/01558 and CIBERONC-CB16/12/00484. NEN Association (Nico contra el cáncer infantil 2017-PVR00157). Switzerland: Oncosuisse, Bern; Swiss Cancer League, Bern; Fond'action contre le Cancer, Lausanne; FORCE Fondation Recherche sur le Cancer de l’Enfant, Ecublens.
CLINICAL TRIAL INFORMATION
NCT01704716 (HR-NBL1/SIOPEN)
A.B. and U.P. contributed equally to this work and are to be considered as joint first authors. D.A.T., R.L., and G.S. contributed equally to this work and are to be considered joint senior authors.
AUTHOR CONTRIBUTIONS
Conception and design: Angela Bellini, Ulrike Pötschger, Tommy Martinsson, Louis Chesler, Dominique Valteau-Couanet, Deborah A. Tweddle, Ruth Ladenstein, Gudrun Schleiermacher
Financial support: Deborah A. Tweddle, Gudrun Schleiermacher
Administrative support: Louis Chesler, Olivier Delattre, Gudrun Schleiermacher
Provision of study materials or patients: Angela Bellini, Peter F. Ambros, Nathalie Auger, Klaus Beiske, David R. Betts, Katleen de Preter, Nathalie Clément, Valérie Combaret, Jaime Font de Mora, Irene Jiménez, Marta Jeison, Tommy Martinsson, Katia Mazzocco, Martina Morini, Annick Mühlethaler-Mottet, Rosa Noguera, Gaelle Pierron, Sabine Taschner-Mandl, Nadine Van Roy, Louis Chesler, Victoria Castel, Martin Elliott, Per Kogner, Geneviève Laureys, Josef Malis, Maja Popovic-Beck, Shifra Ash, Olivier Delattre, Dominique Valteau-Couanet, Deborah A. Tweddle, Ruth Ladenstein, Gudrun Schleiermacher
Collection and assembly of data: Angela Bellini, Ulrike Pötschger, Eve Lapouble, Sylvain Baulande, Nathalie Auger, Klaus Beiske, Marie Bernkopf, Jaydutt Bhalshankar, Nick Bown, Katleen de Preter, Nathalie Clément, Valérie Combaret, Jaime Font de Mora, Sally L. George, Irene Jiménez, Marta Jeison, Tommy Martinsson, Katia Mazzocco, Martina Morini, Annick Mühlethaler-Mottet, Rosa Noguera, Gaelle Pierron, Maria Rossing, Sabine Taschner-Mandl, Nadine Van Roy, Ales Vicha, Louis Chesler, Walentyna Balwierz, Victoria Castel, Martin Elliott, Per Kogner, Geneviève Laureys, Josef Malis, Maja Popovic-Beck, Shifra Ash, Olivier Delattre, Dominique Valteau-Couanet, Deborah A. Tweddle, Ruth Ladenstein, Gudrun Schleiermacher
Data analysis and interpretation: Angela Bellini, Ulrike Pötschger, Virginie Bernard, Peter F. Ambros, Nathalie Auger, Marie Bernkopf, David R. Betts, Jaime Font de Mora, Barbara Marques, Tommy Martinsson, Sabine Taschner-Mandl, Nadine Van Roy, Per Kogner, Geneviève Laureys, Roberto Luksch, Deborah A. Tweddle, Ruth Ladenstein, Gudrun Schleiermacher
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Frequency and Prognostic Impact of ALK Amplifications and Mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1)
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Walentyna Balwierz
Honoraria: Shire, Gilead Sciences, Novartis, Amgen
Consulting or Advisory Role: Amgen, Novartis, Roche, Takeda
Travel, Accommodations, Expenses: Jazz Pharmaceuticals, Shire, Roche, Servier
Martin Elliott
Consulting or Advisory Role: Bayer
Dominique Valteau-Couanet
Consulting or Advisory Role: EUSA Pharma
Research Funding: Orphelia Pharma
Patents, Royalties, Other Intellectual Property: Royalties from Apeiron to SIOPEN
Travel, Accommodations, Expenses: EUSA Pharma, Jazz Pharmaceuticals
Deborah A. Tweddle
Honoraria: Eusa Pharma
Travel, Accommodations, Expenses: EUSA Pharma
Ruth Ladenstein
Honoraria: Apeiron Biologics, Boehringer Ingelheim, EUSA Pharma
Consulting or Advisory Role: Apeiron Biologics, Boehringer Ingelheim, EUSA Pharma
Research Funding: Apeiron Biologics, EUSA Pharma
Patents, Royalties, Other Intellectual Property: Apeiron Biologics, EUSA Pharma
Expert Testimony: Apeiron Biologics, EUSA Pharma
Travel, Accommodations, Expenses: Apeiron Biologics, EUSA Pharma
Gudrun Schleiermacher
Honoraria: BMS
Research Funding: Bristol Myers Squibb, Pfizer, MSDavenir, Roche
Travel, Accommodations, Expenses: Roche
No other potential conflicts of interest were reported.
REFERENCES
- 1.Matthay KK, Maris JM, Schleiermacher G, et al. : Neuroblastoma. Nat Rev Dis Primers 2:16078, 2016 [DOI] [PubMed] [Google Scholar]
- 2.Holmes K, Potschger U, Pearson ADJ, et al. : Influence of surgical excision on the survival of patients with stage 4 high-risk neuroblastoma: A report from the HR-NBL1/SIOPEN study. J Clin Oncol 38:2902-2915, 2020 [DOI] [PubMed] [Google Scholar]
- 3.Ladenstein R, Potschger U, Pearson ADJ, et al. : Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): An international, randomised, multi-arm, open-label, phase 3 trial. Lancet Oncol 18:500-514, 2017 [DOI] [PubMed] [Google Scholar]
- 4.Ladenstein R, Potschger U, Valteau-Couanet D, et al. : Investigation of the role of dinutuximab beta-based immunotherapy in the SIOPEN high-risk neuroblastoma 1 trial (HR-NBL1). Cancers (Basel) 12:309, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ladenstein R, Potschger U, Valteau-Couanet D, et al. : Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): A multicentre, randomised, phase 3 trial. Lancet Oncol 19:1617-1629, 2018 [DOI] [PubMed] [Google Scholar]
- 6.Ozkaynak MF, Gilman AL, London WB, et al. : A comprehensive safety trial of chimeric antibody 14.18 with GM-CSF, IL-2, and isotretinoin in high-risk neuroblastoma patients following myeloablative therapy: Children's Oncology Group study ANBL0931. Front Immunol 9:1355, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park JR, Kreissman SG, London WB, et al. : Effect of tandem autologous stem cell transplant vs single transplant on event-free survival in patients with high-risk neuroblastoma: A randomized clinical trial. JAMA 322:746-755, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinto N, Naranjo A, Hibbitts E, et al. : Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112:66-79, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seeger RC, Brodeur GM, Sather H, et al. : Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med 313:1111-1116, 1985 [DOI] [PubMed] [Google Scholar]
- 10.Janoueix-Lerosey I, Schleiermacher G, Michels E, et al. : Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol 27:1026-1033, 2009 [DOI] [PubMed] [Google Scholar]
- 11.Peifer M, Hertwig F, Roels F, et al. : Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526:700-704, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molenaar JJ, Koster J, Zwijnenburg DA, et al. : Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483:589-593, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Pugh TJ, Morozova O, Attiyeh EF, et al. : The genetic landscape of high-risk neuroblastoma. Nat Genet 45:279-284, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sausen M, Leary RJ, Jones S, et al. : Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet 45:12-17, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Takita J, Choi YL, et al. : Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455:971-974, 2008 [DOI] [PubMed] [Google Scholar]
- 16.George RE, Sanda T, Hanna M, et al. : Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455:975-978, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janoueix-Lerosey I, Lequin D, Brugieres L, et al. : Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455:967-970, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Mosse YP, Laudenslager M, Longo L, et al. : Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455:930-935, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bellini A, Bessoltane-Bentahar N, Bhalshankar J, et al. : Study of chromatin remodeling genes implicates SMARCA4 as a putative player in oncogenesis in neuroblastoma. Int J Cancer 145:2781-2791, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Javanmardi N, Fransson S, Djos A, et al. : Low frequency ALK hotspots mutations in neuroblastoma tumours detected by ultra-deep sequencing: Implications for ALK inhibitor treatment. Sci Rep 9:2199, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Combaret V, Iacono I, Bellini A, et al. : Detection of tumor ALK status in neuroblastoma patients using peripheral blood. Cancer Med 4:540-550, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bellini A, Bernard V, Leroy Q, et al. : Deep sequencing reveals occurrence of subclonal ALK mutations in neuroblastoma at diagnosis. Clin Cancer Res 21:4913-4921, 2015 [DOI] [PubMed] [Google Scholar]
- 23.Eleveld TF, Oldridge DA, Bernard V, et al. : Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 47:864-871, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bresler SC, Weiser DA, Huwe PJ, et al. : ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 26:682-694, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fransson S, Hansson M, Ruuth K, et al. : Intragenic anaplastic lymphoma kinase (ALK) rearrangements: Translocations as a novel mechanism of ALK activation in neuroblastoma tumors. Genes Chromosomes Cancer 54:99-109, 2015 [DOI] [PubMed] [Google Scholar]
- 26.De Brouwer S, De Preter K, Kumps C, et al. : Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res 16:4353-4362, 2010 [DOI] [PubMed] [Google Scholar]
- 27.Friboulet L, Li N, Katayama R, et al. : The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov 4:662-673, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guan J, Tucker ER, Wan H, et al. : The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis Model Mech 9:941-952, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Solomon BJ, Besse B, Bauer TM, et al. : Lorlatinib in patients with ALK-positive non-small-cell lung cancer: Results from a global phase 2 study. Lancet Oncol 19:1654-1667, 2018 [DOI] [PubMed] [Google Scholar]
- 30.Ladenstein R, Potschger U, Siabalis D, et al. : Dose finding study for the use of subcutaneous recombinant interleukin-2 to augment natural killer cell numbers in an outpatient setting for stage 4 neuroblastoma after megatherapy and autologous stem-cell reinfusion. J Clin Oncol 29:441-448, 2010 [DOI] [PubMed] [Google Scholar]
- 31.Ladenstein R, Valteau-Couanet D, Brock P, et al. : Randomized trial of prophylactic granulocyte colony-stimulating factor during rapid COJEC induction in pediatric patients with high-risk neuroblastoma: The European HR-NBL1/SIOPEN study. J Clin Oncol 28:3516-3524, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Garaventa A, Poetschger U, Valteau-Couanet D, et al. : Randomized trial of two induction therapy regimens for high-risk neuroblastoma: HR-NBL1.5 International Society of Pediatric Oncology European Neuroblastoma Group study. J Clin Oncol 39:2552-2563, 2021 [DOI] [PubMed] [Google Scholar]
- 33.Ambros PF, Ambros IM, Brodeur GM, et al. : International consensus for neuroblastoma molecular diagnostics: Report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br J Cancer 100:1471-1482, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ambros IM, Brunner B, Aigner G, et al. : A multilocus technique for risk evaluation of patients with neuroblastoma. Clin Cancer Res 17:792-804, 2011 [DOI] [PubMed] [Google Scholar]
- 35.Schleiermacher G, Michon J, Ribeiro A, et al. : Segmental chromosomal alterations lead to a higher risk of relapse in infants with MYCN-non-amplified localised unresectable/disseminated neuroblastoma (a SIOPEN collaborative study). Br J Cancer 105:1940-1948, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schleiermacher G, Mosseri V, London WB, et al. : Segmental chromosomal alterations have prognostic impact in neuroblastoma: A report from the INRG project. Br J Cancer 107:1418-1422, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ambros IM, Brunner C, Abbasi R, et al. : Ultra-high density SNParray in neuroblastoma molecular diagnostics. Front Oncol 4:202, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.UCSC Genome Browser Home : https://genome.ucsc.edu/index.html
- 39.Kaplan E, Meier P: Nonparametric estimation from incomplete observations. J Am Stat Assoc 53:457-481, 1958 [Google Scholar]
- 40.Peto R, Pike MC, Armitage P, et al. : Design and analysis of randomized clinical trials requiring prolonged observation of each patient. II. Analysis and examples. Br J Cancer 35:1-39, 1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andersen PK, Perme MP: Pseudo-observations in survival analysis. Stat Methods Med Res 19:71-99, 2011 [DOI] [PubMed] [Google Scholar]
- 42.Morgenstern DA, Potschger U, Moreno L, et al. : Risk stratification of high-risk metastatic neuroblastoma: A report from the HR-NBL-1/SIOPEN study. Pediatr Blood Cancer 65:e27363, 2018 [DOI] [PubMed] [Google Scholar]
- 43.Depuydt P, Boeva V, Hocking TD, et al. : Genomic amplifications and distal 6q loss: Novel markers for poor survival in high-risk neuroblastoma patients. J Natl Cancer Inst 110:1084-1093, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mosse YP, Lim MS, Voss SD, et al. : Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A Children's Oncology Group phase 1 consortium study. Lancet Oncol 14:472-480, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guan J, Fransson S, Siaw JT, et al. : Clinical response of the novel activating ALK-I1171T mutation in neuroblastoma to the ALK inhibitor ceritinib. Cold Spring Harb Mol Case Stud 4:a002550, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krytska K, Ryles HT, Sano R, et al. : Crizotinib synergizes with chemotherapy in preclinical models of neuroblastoma. Clin Cancer Res 22:948-960, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bogen D, Brunner C, Walder D, et al. : The genetic tumor background is an important determinant for heterogeneous MYCN-amplified neuroblastoma. Int J Cancer 139:153-163, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marrano P, Irwin MS, Thorner PS: Heterogeneity of MYCN amplification in neuroblastoma at diagnosis, treatment, relapse, and metastasis. Genes Chromosomes Cancer 56:28-41, 2017 [DOI] [PubMed] [Google Scholar]
- 49.Berbegall AP, Bogen D, Potschger U, et al. : Heterogeneous MYCN amplification in neuroblastoma: A SIOP Europe Neuroblastoma study. Br J Cancer 118:1502-1512, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turajlic S, Sottoriva A, Graham T, et al. : Resolving genetic heterogeneity in cancer. Nat Rev Genet 20:404-416, 2019 [DOI] [PubMed] [Google Scholar]
- 51.Williams JB, Li S, Higgs EF, et al. : Tumor heterogeneity and clonal cooperation influence the immune selection of IFN-gamma-signaling mutant cancer cells. Nat Commun 11:602, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Padovan-Merhar OM, Raman P, Ostrovnaya I, et al. : Enrichment of targetable mutations in the relapsed neuroblastoma genome. PLoS Genet 12:e1006501, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schleiermacher G, Javanmardi N, Bernard V, et al. : Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol 32:2727-2734, 2014 [DOI] [PubMed] [Google Scholar]
- 54.Chicard M, Colmet-Daage L, Clement N, et al. : Whole-exome sequencing of cell-free DNA reveals temporo-spatial heterogeneity and identifies treatment-resistant clones in neuroblastoma. Clin Cancer Res 24:939-949, 2018 [DOI] [PubMed] [Google Scholar]
- 55.Ackermann S, Cartolano M, Hero B, et al. : A mechanistic classification of clinical phenotypes in neuroblastoma. Science 362:1165-1170, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koneru B, Lopez G, Farooqi A, et al. : Telomere maintenance mechanisms define clinical outcome in high-risk neuroblastoma. Cancer Res 80:2663-2675, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]