


Keywords: cancer-associated weight loss, mitochondria, muscle strength, physical activity, skeletal muscle
Abstract
Reductions in skeletal muscle mass and function are often reported in patients with cancer-associated weight loss and are associated with reduced quality of life, impaired treatment tolerance, and increased mortality. Although cellular changes, including altered mitochondrial function, have been reported in animals, such changes have been incompletely characterized in humans with cancer. Whole body and skeletal muscle physical function, skeletal muscle mitochondrial function, and whole body protein turnover were assessed in eight patients with cancer-associated weight loss (10.1 ± 4.2% body weight over 6–12 mo) and 19 age-, sex-, and body mass index (BMI)-matched healthy controls to characterize skeletal muscle changes at the whole body, muscle, and cellular level. Potential pathways involved in cancer-induced alterations in metabolism and mitochondrial function were explored by interrogating skeletal muscle and plasma metabolomes. Despite similar lean mass compared with control participants, patients with cancer exhibited reduced habitual physical activity (57% fewer daily steps), cardiorespiratory fitness [22% lower V̇o2peak (mL/kg/min)] and leg strength (35% lower isokinetic knee extensor strength), and greater leg neuromuscular fatigue (36% greater decline in knee extensor torque). Concomitant with these functional declines, patients with cancer had lower mitochondrial oxidative capacity [25% lower State 3 O2 flux (pmol/s/mg tissue)] and ATP production [23% lower State 3 ATP production (pmol/s/mg tissue)] and alterations in phospholipid metabolite profiles indicative of mitochondrial abnormalities. Whole body protein turnover was unchanged. These findings demonstrate mitochondrial abnormalities concomitant with whole body and skeletal muscle functional derangements associated with human cancer, supporting future work studying the role of mitochondria in the muscle deficits associated with cancer.
NEW & NOTEWORTHY To our knowledge, this is the first study to suggest that skeletal muscle mitochondrial deficits are associated with cancer-associated weight loss in humans. Mitochondrial deficits were concurrent with reductions in whole body and skeletal muscle functional capacity. Whether mitochondrial deficits are causal or secondary to cancer-associated weight loss and functional deficits remains to be determined, but this study supports further exploration of mitochondria as a driver of cancer-associated losses in muscle mass and function.
INTRODUCTION
Skeletal muscle is often dramatically affected by cancer. Between 50% and 80% of patients with cancer suffer from muscle wasting and loss of muscle functional capacity, with or without concomitant losses in adipose tissue (1). This loss of muscle mass, which cannot be reversed with nutritional support, contributes to patient morbidity and is associated with impaired responses to chemotherapy, increased postsurgical complications, elevated susceptibility to infection, and increased mortality (1). Skeletal muscle metabolic derangements, including hypermetabolism, attenuated muscle protein synthesis, and heightened muscle protein breakdown, have also been associated with cancer (2, 3). In addition to the loss of muscle mass and the metabolic derangements, patients with cancer also commonly exhibit physical impairments and muscular functional deficits, including muscular fatigue and reduced muscle strength, that are associated with significant reductions in quality of life, can limit antineoplastic therapy, and are predictive of reduced responses to chemotherapy and survival (4).
Although functional declines and changes at the whole muscle level have been reported, changes at the cellular and subcellular levels have been infrequently examined. Mitochondrial derangements that sometimes precede muscle wasting have been observed in rodent models of cancer-associated muscle wasting (3, 5, 6). As highly dynamic organelles that play a significant role in energy production, regulation of metabolism, redox homeostasis, cell death, and cell survival, mitochondria may lie at the nexus of the muscular deficits associated with cancer. Although mitochondrial morphological changes and alterations in the expression of genes associated with mitochondrial quality control have been reported in the skeletal muscle of human patients with cancer (7–9), no studies have systematically examined mitochondrial function in human subjects with cancer.
Although deficits in muscle mass and function have profound consequences for patients with cancer, mechanistic insights into the causes of these muscular declines in patients with cancer are unknown. To generate insights into cancer-associated muscle derangements at the whole body, muscle, and cellular level, assessments of physical function, whole body protein turnover, and skeletal muscle mitochondrial function were performed in a cross-sectional representative sample of patients with cancer exhibiting cancer-associated weight loss. Nontargeted metabolomics was performed to identify potential novel pathways involved in cancer-induced alterations in metabolism and mitochondrial function. Compared with healthy age-, sex-, and body mass index (BMI)-matched control participants, patients with cancer exhibited functional impairments including reduced whole body oxygen consumption, increased neuromuscular fatigue, and reduced knee extensor strength. Concomitantly, patients with cancer exhibited decreased mitochondrial function; metabolomic profiling identified significant changes in the phospholipid profiles of both the plasma and skeletal muscle of patients with cancer.
METHODS
Participants and Study Design
This was a cross-sectional comparison of patients with cancer who were experiencing weight loss and healthy age-, sex-, and BMI-matched control participants. All patients with cancer had received a diagnosis of metastatic cancer or localized tumors and exhibited a loss of ≥5% body weight in the 6–12 mo before study participation with no other evident cause of weight loss (Table 1). Cancer type, status, and extant treatment are reported in Table 1. For patients undergoing chemotherapy treatment, study procedures were not performed in the week following infusion. The patients with cancer were matched with healthy control participants based on age (±5 yr), body mass index (±5 kg/m2), and sex at a patient:control ratio of 1:2–3. All participants were 21 yr of age or older, and for both patients with cancer and healthy controls, exclusion criteria included: BMI ≥ 45 kg/m2; impaired fasting glucose (≥126 mg/dL); renal failure (serum creatinine > 1.5 mg/dL in males, >1.3 mg/dL in females); chronic active liver disease [bilirubin >17 mmol/L, aspartate aminotransferase (AST) >144 IU/L, or alanine transferase (ALT) >165 IU/L]; active coronary artery disease or history of unstable macrovascular disease; oral warfarin group medications or history of blood clotting disorders; platelet count < 100,000/µL; untreated or uncontrolled thyroid disorders [thryoid stimulating hormone (TSH) < 0.5 mIU/L or > 10 mIU/L]; pregnancy or breastfeeding; alcohol or substance abuse; infection; communicable disease; or debilitating chronic disease assessed by the study team. Participants with contraindications to MRI or exercise or with any other disease or condition that would preclude participation or increase study risks were also excluded. Following a screening visit to determine eligibility, study participants underwent physical function assessment, activity monitoring, muscle biopsies, and assessment of whole body protein turnover. All study procedures were approved by the Mayo Foundation Institutional Review Board and conformed to the principles of the Declaration of Helsinki. The study was registered under Clinical Trial Number NCT03012139 on January 6, 2017, before enrollment of the first participant in May of 2017. Written informed consent was obtained from all participants before study inclusion.
Table 1.
Patient characteristics
| Patient | Cancer Type | Cancer Status | Extant Cancer Therapy at Time of Study | Weight Loss*, % Body Weight |
|---|---|---|---|---|
| 1 | Breast and ovarian cancer | Metastatic | None | −8.7% |
| 2 | Colon cancer | Metastatic | None | −8.3% |
| 3 | Uterine carcinosarcoma | Metastatic | Doxil, carboplatin; study participation 21 days postinfusion | −7.3% |
| 4 | Appendiceal cancer | Metastatic | None | −11.8% |
| 5 | Colon cancer | Metastatic | 5-Fluorouracil, oxaliplatin, leucovorin; study participation 12 days postinfusion | −8.5% |
| 6 | Uterine cancer | No evidence of disease | Carboplatin, paclitaxel; study participation 19 days postinfusion | −6.5% |
| 7 | Pancreatic cancer | Metastatic | Gemcitabine, Abraxane; study participation 7 days postinfusion | −19.7% |
| 8 | Esophageal cancer | Metastatic | 5-Fluorouracil, oxaliplatin, leucovorin; study participation 30 days postinfusion | −10.1% |
Percent of body weight lost in the 6–12 mo prior to study participation.
Screening
Potential participants reported to the Mayo Clinic Clinical Research and Trials Unit (CRTU). After obtaining consent, participant height, weight, blood pressure, and heart rate were recorded, participant medical history was reviewed, and a blood sample was obtained for measurement of complete erythrocyte and leukocyte counts, serum creatinine, bilirubin, AST, ALT, activated partial thromboplastin time, prothrombin time, glucose, and TSH. Blood was also obtained for the measurement of inflammatory marker concentrations [tumor necrosis factor α (TNFα), C-reactive protein (CRP), and interleukin (IL)-6], and anabolic and mineral-regulatory hormone concentrations [human growth hormone (HGH), insulin-like growth factor-1 (IGF1), testosterone, parathyroid hormone (PTH), fibroblast growth factor-23 (FGF23), and total 25-hydroxyvitamin D]. Analyses were performed by the Mayo Clinic Central Clinical and Immunochemical Core Laboratories. Body composition was measured by dual-energy X-ray absorptiometry (DEXA; GE Lunar iDXA, GE Healthcare, Chicago, IL). Skeletal muscle index (SMI) was calculated as the appendicular lean mass, obtained by DEXA, normalized to height square. In addition, participants completed an online food frequency questionnaire (VioFFQ, VIOCARE, Inc., Princeton, NJ) to estimate daily macro and micronutrient consumption (10).
Free-Living Physical Activity
Participant physical activity was assessed in the patient’s free-living environment using triaxial accelerometers (ActiGraph GT3X+, Pensacola, FL) worn on the waist, thigh, and bilateral ankles for four consecutive days (2 weekdays, 2 weekend days) during waking hours, as previously described (11). A minimum “wear time” of 8 h was required to be considered a valid day for data analysis. Accelerometer data were collected at 50 Hz, and raw signals were processed using MATLAB (Version 7.11.0, MathWorks, MA) and analyzed using custom algorithms for measuring posture, movement, step counts, and cadence (12, 13). Upright, dynamic activity was determined over 1-s epochs from the waist and thigh accelerometer signals (12). Step counts and heel strike frequency (cadence) were assessed from the ankle accelerometers during all upright dynamic activity periods using a peak detection algorithm with adaptive acceleration and timing thresholds (12).
Cardiorespiratory Fitness
Participant peak whole body oxygen consumption (V̇o2peak) was measured during a maximal graded treadmill test. Following 1 min of quiet standing, treadmill speed and/or incline were increased every 3 min in accordance with the Bruce Protocol (14) or the Modified Bruce Protocol (15), depending on participant physical abilities. Inspired and expired gases were measured continuously by breath-by-breath indirect calorimetry, and the individual V̇o2peak was defined as the highest average V̇o2 over an interval lasting at least 15 s. Oxygen saturation and heart rhythm and rate were continuously measured by pulse oximetry and 12-lead electrocardiography, respectively. Blood pressure was measured during each exercise stage and participants’ rating of perceived exertion was assessed in the last 30 s of each exercise stage using the Borg 6–20 scale (16). All tests were monitored by a physician or PhD physiologist trained in clinical exercise testing, and tests were terminated if the participants reached volitional exhaustion, developed ECG changes indicative of cardiac ischemia, or at the discretion of the supervising study team member.
Muscle Strength and Endurance Assessment
Handgrip strength of the dominant hand was measured using a hand dynamometer (NK Biotechnical Corp, Minneapolis, MN), as previously described (17). Maximal grip strength was determined from the highest value of three maximal repetitions (30 s rest between attempts). Participant knee extensor and flexor isokinetic strength, isometric strength, and neuromuscular fatigue were assessed using an isokinetic dynamometer (HUMAC NORM, Computer Sports Medicine Inc., Stoughton, MA). Participants sat upright in the dynamometer chair with the hip joint flexed to 70° and the knee joint axis aligned with the isokinetic device center of rotation. The leg was secured with Velcro straps to the knee attachment pad. Stabilizing straps around the waist and trunk restrained trunk and hip movement during testing. Following familiarization and a warmup, participants performed three maximum voluntary isometric contractions (MVICs) of 5 s with 30 s rest between each attempt. The highest recorded contraction was considered maximum isometric strength. Participants then completed three maximum voluntary isokinetic contractions at 60°/s and maximum isokinetic strength was defined as the highest torque obtained. Strength data were normalized to participant height and weight and expressed as % body weight (grip strength) and % body weight × height (isometric and isokinetic strength). Neuromuscular fatigue was assessed as previously described (18). Participants performed 30 isokinetic concentric contractions at 180°/s, and the percent decline in torque {[(Initial Torque − Final Torque)/Initial Torque] × 100} was calculated, with higher values reflecting poorer neuromuscular endurance.
Stable Isotope Tracer Administration
Stable isotope tracers (Cambridge Isotopes Inc., Cambridge, MA) were used for the assessment of whole body protein turnover. Following an overnight fast, participants reported to the Mayo Clinic CRTU. A peripheral venous catheter was placed and priming boluses of [13C6]phenylalanine [1 mg/kg fat free mass (FFM)], [13C6]tyrosine (0.5 mg/kg FFM), and [15N]tyrosine (0.5 mg/kg FFM) were administered at 0715. Immediately following the priming doses, a continuous infusion of [13C6]phenylalanine (1 mg/kg FFM/h), and [15N]tyrosine (0.5 mg/kg FFM/h) was initiated and maintained for 6 h.
Blood and Muscle Collection
A retrograde intravenous catheter was placed in the hand opposite the infusion line, and the hand was placed in a plexiglass box maintained at 55°C for collection of arterialized venous blood samples. Blood samples were collected before isotope bolus administration for metabolomics analysis and at 12 time points during tracer infusion for the measurement of isotopic enrichments (IEs). A percutaneous muscle biopsy of the vastus lateralis was performed at 1015. Muscle biopsies were performed under sterile conditions and local anesthesia (2% lidocaine) using a modified Bergstrom needle, as previously described (19). Approximately 100 mg of tissue was isolated for mitochondrial functional assessments, and the remainder was snap-frozen in liquid nitrogen and stored at −80°C for metabolomics analysis.
Whole Body Protein Turnover
Plasma was isolated from blood samples obtained before isotope bolus injections and at 12 time points during the continuous infusion. Plasma enrichments of [13C6]phenylalanine, [13C6]tyrosine, and [15N]tyrosine were measured by gas chromatography/mass spectrometry (GCMS) as previously described (20). Briefly, phenylalanine and tyrosine were isolated from plasma by ion-exchange chromatography and dried. Dried samples were derivatized with N-methyl-N(t-butyldimethylsilyl)-trifluoroacetamide plus 1% t-butyl-dimethylchlorosilane in acetonitrile and the derivatives were analyzed on an Agilent MSD5977A GCMS under electron impact conditions. Single-ion monitoring was carried out at m/z 342/336 for phenylalanine and at m/z 472/467/466 for tyrosine. Isotopic enrichment was calculated against a calibration curve constructed with phenylalanine, [13C6]phenylalanine, tyrosine, [13C6]tyrosine, and [15N]tyrosine standards.
Average IEs of [13C6]phenylalanine, [13C6]tyrosine, and [15N]tyrosine in plasma after achieving isotopic steady state were used to calculate whole body amino acid flux rates using the single-pool steady-state model (21). The whole body rate of appearance of [13C6]phenylalanine (Ra), representing protein breakdown was calculated from the equation:
where I is the infusion rate, Ei is the mean percent enrichment (MPE) of the isotope in the infusate, and EA is the MPE in the plasma. Because all measurements were performed during isotopic steady state, Ra is assumed to be equal to the rate of disappearance (Rd).
The oxidation rate of phenylalanine (QPT) was estimated by the conversion rate of phenylalanine to tyrosine using the equation:
where RaTyr and RaPhe are the rates of appearance of [15N]tyrosine and [13C6]phenylalanine, respectively, EATyr and EAPhe are the plasma enrichments of [13C6]tyrosine and [13C6]phenylalanine, respectively, and IPhe is the infusion rate of [13C6]phenylalanine.
The oxidation rate of phenylamine was then subtracted from the rate of disappearance of phenylalanine to estimate whole body protein synthesis (QPS):
Whole body protein balance was estimated by subtracting the RaPhe (protein breakdown) from QPS.
Ex Vivo Mitochondrial Functional Assessment
Mitochondria were isolated by differential centrifugation from ∼100 mg of fresh muscle tissue for assessment of mitochondrial respiration, ATP production, and reactive oxygen species (ROS) generation, as previously described (22, 23). Isolated mitochondria were resuspended at a volume of 4.2 µL buffer per milligram of wet tissue weight. Mitochondrial oxidative capacity was assessed by high-resolution respirometry (Oxygraph, Oroboros Instruments, Innsbruck, Austria) using 90 µL of mitochondrial suspension per chamber and a stepwise protocol that isolated electron flow through the respiratory chain complexes and assessed oxygen flux (JO2) during respiratory states 1–4 in the presence of glutamate, malate, and succinate (GMS) as previously described (23–25). The average oxygen flux rates were calculated for each state and corrected for background oxygen flux using Datlab software (Oroboros Instruments). The respiratory control ratio (RCR) was calculated from the ratio of state 3 to state 4 respiration and served as an indicator of mitochondrial proton leak (23, 26). The ratio of ADP to oxygen (ADP:O), an indicator of mitochondrial phosphorylation efficiency, was determined by measuring the amount of oxygen consumed in response to a nonsaturating pulse of ADP (15 µmol/L) (27). Mitochondrial ROS production (H2O2 emission) was measured by fluorometric monitoring of the oxidation of Amplex Red in conjunction with the measurement of mitochondrial respiration using the Oxygraph O2K-Fluorescence LED2-Module (22). To evaluate excessive ROS generation beyond what is produced during normal respiration, H2O2 flux rates were normalized to oxygen consumption at the corresponding respiratory state. Mitochondrial ATP production was assessed fluorometrically (Fluorolog 3; Horiba Jobin Yvon, Piscataway, NJ) as previously described (28, 29). Briefly, supplementation with hexokinase, glucose-6-phosphate dehydrogenase, and NADP+ during mitochondrial respiration catalyzed the conversion of ATP to NADPH, and the autofluorescence of NADPH was continuously monitored during respiratory states 1–4 as a surrogate indicator of ATP production. The rate of ATP production was calculated using MATLAB (MathWorks). All measurements were performed in duplicate, and JO2 and ATP production were normalized to tissue wet weight and to mitochondrial protein content (average measured protein content: 2.66 ± 0.63 µg/µL), measured using the Pierce 660-nm protein assay.
In Vivo Mitochondrial Functional Assessment
In vivo skeletal muscle maximal ATP production was measured by phosphorous magnetic resonance spectroscopy (31P-MRS) and the phosphocreatine (PCr) recovery kinetics following brief muscle activity, as previously described (30, 31). Briefly, participants lay supine on the bed of a 3.0 Tesla GE Signa MRI scanner with the right leg positioned in a custom knee extension apparatus capable of measuring isometric force. A transmit/receive phosphorous radiofrequency coil was placed over the vastus lateralis muscle. The magnet was shimmed on the muscle phosphorous signal and two phosphorus acquisitions in resting muscle were obtained, one fully relaxed spectrum (TR = 16 s) and one partially saturated spectrum using a short repetition time (TR = 2 s). Correction factors for partial saturation at the 2 s TR were determined for each participant for inorganic phosphate (Pi), PCr, and ATP. A single phosphorus acquisition (TR = 2 s) consisting of 60 s of rest, 30 s of maximal effort sustained isometric knee extension, and 10 min of recovery was obtained. 31P data were processed using jMRUI (32). The free induction decays (FIDs) from the resting TR = 2 and TR = 16 s acquisitions were averaged and those averages analyzed; the individual FIDs from the exercise TR = 2 acquisition were analyzed separately. All data were apodized using a 5 Hz Lorentzian, followed by conversion to the frequency domain by Fourier transform. The PCr peak was shifted in frequency space such that all spectra had PCr centered at exactly 0.0 ppm. AMARES was then used with a custom basis set to quantify the averaged resting spectra and the individual exercise spectra. Phosphorous metabolites concentrations were calculated assuming that [PCr] + [Cr] = 42.5 mM and resting [ATP] = 8.2 mM and that Δ[Pi] is equivalent to Δ[Cr] during rest, exercise, and recovery. Intramuscular pH was calculated based on the chemical shift (σ) of Pi relative to PCr in parts per million:
For each participant, a rate constant (kPCr) from the kinetics of PCr recovery after the 30-s muscle contraction was determined by fitting a single exponential curve:
where t is time, PCrex is [PCr] at the end of exercise, . The kPCr reflects the rate of oxidative phosphorylation.
Untargeted Metabolomic Analysis
Metabolomic profiles of snap-frozen pulverized vastus lateralis and plasma samples were assessed in triplicate by LC-quadrupole time-of-flight (QTOF)-MS by the Mayo Clinic Metabolomics Core Laboratory. Pulverized vastus lateralis samples (∼20 mg) were suspended in PBS and homogenized via sonication. Plasma samples and tissue homogenates were deproteinized with 1:1 acetonitrile:methanol after the addition of 13C6-phenylalanine (3 µL at 250 ng/µL) as an internal standard, vortexed, and centrifuged at 18,000 g. The supernatants were divided into two aliquots and dried for analysis on a quadrupole time-of-flight mass spectrometer (Agilent Technologies 6550 QTOF) coupled with an ultra-high-pressure liquid chromatograph (1290 Infinity UHPLC Agilent Technologies). LC-QTOF-MS was performed in positive and negative electrospray ionization modes using polar (HILIC) and nonpolar (C18) ultra-performance liquid chromatography separation within the mass to charge (m/z) ratio range of 100–1,200. A total of four runs per sample were performed to achieve maximum metabolite coverage. A quality control sample comprising a subset of samples from the study was injected at multiple intervals throughout each run. Raw data files were converted to compound exchange file format using Masshunter DA reprocessor software (Agilent). Mass Profiler Professional (Agilent) was used for data alignment and to convert each metabolite feature (m/z × intensity × time) into a matrix of detected peaks for compound identification. Accurate mass and a detection window of 7 ppm or less were used for metabolite identification against the Metlin database. Mass accuracy of the QTOF method was <5 ppm with retention time precision greater than 0.2% such that a 1.2 times fold change can be detected with a precision of 4%. The 13C6 phenylalanine internal standard was used to check for sample recovery and was not included in the normalization of the data.
A metabolite peak intensity threshold of ≥5,000 was required in at least 75% of samples to be eligible for downstream analyses to filter low-intensity hits and was applied to all modes. For each mode, metabolite peak intensities for unique compounds were normalized by sum, log2 transformed, and mean-centered using MetaboAnalyst 4.0 (33). To identify differences in the relative abundance of individual metabolites between patients with cancer and control participants, the MetaboAnalyst 4.0 Statistical Analysis feature was used to perform fold-change analyses and unpaired t tests for each identified metabolite. P values were false discovery rate (FDR)-adjusted to account for multiple comparisons. Significance was defined as an FDR > 0.05 and an absolute log2fold change > 0.58 (corresponding to a fold change >1.5). Analyses from each mode were combined and duplicate metabolites were removed.
Statistics
Data for each group were summarized as median (range). To compare groups while accounting for matching and potential skew related to the small sample size, outcomes for each variable were ranked, independent of group, and generalized estimating equations were performed to compare ranks between the matched study groups. Each variable was compared in a separate model. Significance was set a priori at P < 0.05.
RESULTS
Participant Characteristics
Eight individuals (7 females/1 male) with cancer and with a loss of ≥5% body weight in the preceding 6- to 12-mo period and 19 healthy age-, sex-, and BMI-matched control participants (17 females/2 males) were identified by the study team and participated in the study (Table 2). By design, body composition measured by DEXA did not differ between the patients with cancer and the healthy controls (Table 2). All participants were non-Hispanic white individuals. In addition, self-reported macronutrient and micronutrient intake assessed using the VioCare Food Frequency questionnaire was similar between patients with cancer and controls, except for cobalamin B12, the consumption of which was lower in patients with cancer (Supplemental Table S1; see https://doi.org/10.6084/m9.figshare.16869201).
Table 2.
Participant demographics, body composition, blood chemistry, and inflammatory marker concentrations
| CancerMedian, Rangen = 8 | ControlMedian, Rangen = 19 | β,95% CI | P Value | |
|---|---|---|---|---|
| Demographics and body composition | ||||
| Age, yr | 66.5 (62, 77) | 67 (57, 75) | 0.36 (−1.84, 2.56) | 0.7516 |
| Height, cm | 160.25 (156.2, 180.2) | 161.2 (146.1, 180.3) | −0.71 (−4.8, 3.38) | 0.7333 |
| Weight, kg | 72.15 (40.8, 100.4) | 69.5 (45.3, 85.5) | 1.07 (−5.4, 7.53) | 0.7465 |
| BMI, kg/m2 | 28.65 (15.8, 38.3) | 25.5 (19, 33.9) | 1.24 (−4.71, 7.2) | 0.6822 |
| SMI, kg/m2 | 6.76 (4.54, 7.81) | 6.64 (5, 8.25) | −1.07 (−5.32, 3.18) | 0.6231 |
| Fat free mass, kg | 41.52 (30.39, 56.66) | 39.28 (31.06, 60.77) | 0.71 (−4.32, 5.75) | 0.7821 |
| Fat mass, kg | 27.41 (5.91, 53.09) | 26.76 (10.29, 42.95) | 0.36 (−5.61, 6.32) | 0.9071 |
| Body fat, % | 39.4 (9.4, 54.5) | 41.3 (20.1, 52.7) | 0.18 (−5.13, 5.48) | 0.9477 |
| BMD T-score | −0.65 (−3.9, 1) | −0.2 (−1.7, 2.9) | −2.22 (−8, 3.56) | 0.4517 |
| Blood chemistry | ||||
| Hemoglobin, g/dL | 11.1 (10.4, 13.3) | 13.1 (11.3, 14.6) | −11.28 (−15.39, −7.17) | <0.0001 |
| Bilirubin, mg/dL | 0.3 (0.2, 0.9) | 0.5 (0.3, 1.4) | −6.48 (−12.24, −0.72) | 0.0273 |
| Creatinine, mg/dL | 0.8 (0.6, 1) | 0.86 (0.7, 1.04) | −4.09 (−11.39, 3.22) | 0.2731 |
| ALT, U/L | 21 (12, 34) | 18 (11, 39) | 2.84 (−4, 9.69) | 0.4152 |
| AST, U/L | 28.5 (19, 50) | 23 (15, 43) | 3.38 (−3.47, 10.22) | 0.3339 |
| TSH, mIU/L | 1.8 (0.5, 6.2) | 2.8 (1.1, 6.9) | −2.75 (−9.39, 3.88) | 0.4158 |
| HGH, ng/mL | 0.92 (0.41, 3.26) | 0.32 (0.03, 6.32) | 6.57 (0.69, 12.45) | 0.0284 |
| IGF1, ng/mL | 104.7 (29.3, 145) | 89.4 (48.5, 144) | 0.71 (−8.02, 9.44) | 0.8732 |
| Testosterone, ng/dL | 16.5 (6.5, 153) | 15 (3.9, 988.1) | 3.46 (−1.29, 8.22) | 0.1533 |
| PTH, pg/mL | 52.6 (22.2, 242) | 45.7 (25.1, 64.7) | 1.69 (−5.42, 8.79) | 0.6416 |
| FGF23, pg/mL | 49.75 (15.44, 108.74) | 43.5 (32.95, 120.82) | −0.71 (−8.63, 7.21) | 0.8603 |
| Total 25OH vitamin D, ng/mL | 22.5 (4.3, 60) | 32 (7, 55) | −2.22 (−9.22, 4.78) | 0.5341 |
| WBC, ×109/L | 4.5 (1.5, 11) | 4.9 (3.4, 7.6) | −1.51 (−8.15, 5.13) | 0.6557 |
| Platelets, ×109/L | 228.5 (127, 434) | 243 (68, 354) | −0.62 (−8.26, 7.02) | 0.8733 |
| APT time, s | 28 (26, 37) | 28 (26, 33) | 0.18 (−6.49, 6.85) | 0.9584 |
| Prothrombin time, s | 11.5 (10.6, 12.4) | 11.3 (9.7, 12.4) | 1.51 (−2.05, 5.07) | 0.4053 |
| Fasting glucose, mg/dL | 95 (85, 115) | 95 (74, 109) | 3.29 (−3.63, 10.21) | 0.3520 |
| Inflammatory markers | ||||
| TNFα, pg/mL | 1.5 (0.84, 2.3) | 1 (0.81, 1.9) | 6.93 (1.8, 12.06) | 0.0244 |
| C-reactive protein, mg/dL | 0.5 (0.04, 3.32) | 0.16 (0.03, 1.64) | 7.02 (1.46, 12.57) | 0.0398 |
| IL-6, pg/mL | 8.7 (1.3, 37) | 2 (0.4, 80) | 8.26 (3.9, 12.62) | 0.0006 |
Matched generalized estimating equations were performed on ranked variables to determine significant differences between cancer and control, and the β coefficients and P values of the regression are presented. Bold P values represent significant differences between patients with cancer and healthy controls; n, number of subjects. ALT, alanine transferase; APT, activated partial thromboplastin; AST, aspartate aminotransferase; BMD, bone mineral density; BMI, body mass index; FGF23, fibroblast growth factor 23; HGH, human growth hormone; IGF1, insulin-like growth factor 1; IL-6, interleukin-6; PTH, parathyroid hormone; SMI, skeletal muscle mass index; TNFα, tumor necrosis factor-α; TSH, thyroid-stimulating hormone; WBC, white blood count.
Hemoglobin concentrations were significantly lower in the patients with cancer (Table 2). B12 intake was decreased but no macrocytosis was observed. Concentrations of serum bilirubin were also significantly lower in the patients with cancer but remained within normal ranges. Compared with the healthy control participants, patients with cancer had significantly higher HGH concentrations (Table 2). In addition, compared with the healthy controls, patients with cancer had significantly higher concentrations of TNFα, CRP, and IL-6 (Table 2).
Physical Activity and Strength Assessment
Patients with cancer exhibited significant decrements in physical fitness and activity despite similar appendicular lean mass compared with healthy control participants (Fig. 1A). Whole body cardiorespiratory fitness normalized to both total body mass and lean mass were lower in patients with cancer (Fig. 1B), as were habitual physical activity levels. In addition to taking fewer steps per day, patients with cancer had significantly slower walking cadence (Fig. 1C). Although grip strength and knee flexor strength were largely preserved in patients with cancer (Fig. 1, D–F), isometric and isokinetic knee extensor strength were lower in patients with cancer (Fig. 1, E and F), and there was a trend for reduced isokinetic knee flexor strength. Furthermore, the patients with cancer exhibited greater peripheral fatigue of the knee extensors compared with healthy control participants, as evidenced by the greater decline in peak torque levels generated over 30 s of isokinetic concentric contraction exercises (Fig. 1G). These data demonstrate significant functional deficits in the antigravity muscles used for upright posture, particularly the knee extensors, and the associated self-motivated movement in the free-living environment by patients with cancer, independent of muscle mass.
Figure 1.

Functional assessment of patients with cancer. A: appendicular lean mass was assessed by dual-energy X-ray absorptiometry in patients with cancer (n = 8) and healthy age-, body mass index-, and sex-matched controls (n = 19). B: whole body cardiorespiratory fitness in patients with cancer (n = 7) and healthy controls (n = 16) was determined by indirect calorimetry during an incremental treadmill test. C: the average number of steps per day and the average walking pace (cadence) for patients with cancer (n = 8) and healthy control (n = 19) participants was evaluated during free-living conditions using hip, waist, and ankle-worn accelerometers. D: grip strength was assessed using a hand-held dynamometer and normalized to body weight (cancer, n = 7; control, n = 16). Isometric (cancer, n = 7; control, n = 16) (E) and isokinetic (cancer, n = 7; control, n = 14) (F) strength of the knee extensors and flexors were determined using an isokinetic dynamometer and normalized to height and weight. G: the percent decline in knee extensor and flexor strength was assessed using an isokinetic dynamometer as a measure of neuromuscular fatigue (cancer, n = 6; control, n = 12). For each outcome, data were ranked, independent of group, and matched generalized estimating equations were performed on ranked variables to determine significant differences between patients with cancer and healthy age-, sex-, and body mass index-matched control participants. Each variable was compared in a separate model. Blue data points indicate male participants. Box plots extend from the 25th to the 75th percentile, and center line represents the median value. Whiskers extend from the minimum to the maximum. *Significant (P < 0.05) difference between groups; n, number of subjects.
Mitochondrial Function
Oxygen consumption and ATP production in respiratory states 2, 3 through complexes I and II, and respiratory state 3 through complex II alone, were significantly lower per milligram of skeletal muscle tissue in biopsy samples of patients with cancer compared with the healthy control participants (Fig. 2, A and B). When respiration was normalized to mitochondrial protein content, respiration in states 2 and 3 remained lower in patients with cancer, but was only significantly lower during state 3 through complex II (Fig. 2C). ATP production also remained significantly lower in patients with cancer during state 2 and state 3 through complex II when normalized to mitochondrial protein content, but was not lower during state 3 respiration through both complex I and II (Fig. 2D). In line with these ex vivo measurements, skeletal muscle oxidative capacity measured in vivo was lower in patients with cancer compared with controls (Fig. 2F, P = 0.053). Respiration resulting from proton leak across the mitochondrial membrane (state 4) and uncoupled respiration through CII [measured with the addition of the protonophore carbonylcyanide-4-(trifluoromethoxy)-phenylhydrazone (FCCP) in the presence of rotenone] were also higher in healthy controls (Fig. 2, A and C). Correspondingly, the RCR was significantly higher in patients with cancer, indicating greater mitochondrial efficiency, though no differences in the ADP:O ratio, a measure of phosphorylation efficiency, were observed (Fig. 2E). Despite the observed increased efficiency measured by RCR, isolated mitochondria from patients with cancer exhibited increased ROS production, as determined by the emission of H2O2 during respiratory states 2 and 4 and normalized to oxygen consumption at the corresponding respiratory state (Fig. 2G).
Figure 2.

In vivo and in vitro measurement of mitochondrial function in patients with cancer. A–D: mitochondrial respiration (cancer, n = 7; control, n = 17) was measured by high-resolution respirometry and ATP production (cancer, n = 7; control, n = 15) was measured by fluorometry in mitochondria isolated from skeletal muscle biopsies of the vastus lateralis from patients with cancer and healthy age-, sex-, and body mass index-matched control participants. Respiration (A and C) and ATP production (B and D) were normalized to wet tissue weight (A and B) or to mitochondrial protein content (C and D). E: mitochondrial integrity and coupling efficiency (RCR) and phosphorylation efficiency (ADP:O) were assessed by high-resolution respirometry. F: phosphocreatine (PCr) recovery kinetics following a single bout of muscle activity were measured by phosphorous magnetic resonance spectroscopy (31P-MRS) to measure oxidative capacity in vivo (cancer, n = 5; control, n = 11). G: mitochondrial reactive oxygen species production by isolated mitochondria was measured fluorometrically as H2O2 emissions during respiratory states 2 and 4. For each outcome, data were ranked, independent of group, and matched generalized estimating equations were performed on ranked variables to determine significant differences between patients with cancer and healthy age-, sex-, and body mass index-matched control participants. Each variable was compared in a separate model. Blue data points indicate male participants. Respiration, ATP production data, and ROS generation are presented as median and interquartile range. Box plots extend from the 25th to the 75th percentile, and center line represents the median value. Whiskers extend from the minimum to the maximum. *Significant (P < 0.05) difference between groups; n, number of subjects. CI, complex I; CII, complex II; FCCP, carbonylcyanide-4-(trifluoromethoxy)-phenylhydrazone; JATP, ATP production rate; JO2, oxygen flux; RCR, respiratory control ratio; ROS, reactive oxygen species.
Whole Body Protein Turnover
Whole body protein turnover was assessed by measuring the isotopic enrichment of amino acids in blood during a primed, continuous infusion of stable isotope-labeled amino acids (21, 34). After achieving steady-state tracer enrichments (Supplemental Fig. S1; see https://doi.org/10.6084/m9.figshare.16869233), whole body protein synthesis, protein degradation, and protein balance were modeled from the plasma enrichment of phenylalanine and tyrosine isotopes. Compared with the healthy controls, patients with cancer did not exhibit significant alterations in whole body protein turnover (Fig. 3).
Figure 3.

Whole body protein turnover in patients with cancer. After achieving steady state isotopic enrichment of phenylalanine and tyrosine stable isotope amino acid tracers, plasma phenylalanine flux was calculated as an indicator of whole body rates of protein synthesis, protein breakdown, and protein balance (cancer, n = 7; control, n = 15). For each outcome (synthesis, balance, and breakdown), data were ranked, independent of group, and matched generalized estimating equations were performed on ranked variables to determine significant differences between patients with cancer and healthy age-, sex-, and body mass index-matched control participants. Each variable was compared in a separate model. Blue data points indicate male participants. Whole body protein kinetics are presented as means and SD; n, number of subjects. Phe, phenylalanine.
Plasma and Skeletal Muscle Metabolite Profiles
To characterize alterations in the metabolite profiles of plasma and skeletal muscle of patients with cancer that may serve as indicators or potential drivers of impaired mitochondrial function and cancer-associated muscle atrophy, nontargeted profiling of the plasma and skeletal muscle metabolomes were performed. Of the 876 unique identified metabolites in plasma, the relative abundance of 93 metabolites significantly (FDR < 0.05; fold change > 1.5) differed between patients with cancer and healthy control participants (abundance of 74 metabolites lower, 19 higher in patients with cancer, Supplemental Data Set 1; see https://doi.org/10.6084/m9.figshare.17124290). In skeletal muscle, 1,147 unique metabolites were identified, and the relative abundance of 56 significantly differed between patients with cancer and healthy controls (21 lower, 36 higher in patients with cancer, Supplemental Data Set 2; see https://doi.org/10.6084/m9.figshare.17124308).
In the plasma, key membrane phospholipid metabolites were lower in patients with cancer (Fig. 4A). The relative abundance of phosphatidylcholines (PC) and phosphatidylethanolamines (PE) and their precursors (phosphocholines, phosphoethanolamines) and breakdown products (lysoPC, lysoPE) were lower in the plasma of patients with cancer compared with the healthy control participants. Similarly, the relative abundance of other membrane phospholipids, including phosphatidylserines (PS) and phosphatidylinositols (PI), tended to be lower in the plasma of patients with cancer. In addition to serving as the most abundant phospholipids in the plasma membranes of mammalian cells, PC and PE are implicated in mitochondrial and metabolic health (35, 36). Within the skeletal muscle, PC and PE concentrations and the PC:PE ratio are associated with insulin sensitivity, mitochondrial area, and the transcription of genes associated with oxidative phosphorylation and the mammalian target of rapamycin (mTOR) (35, 37). Metabolomic profiling of the skeletal muscle of patients with cancer revealed overall reductions in multiple phospholipids, including PC and PE species (Fig. 4B).
Figure 4.
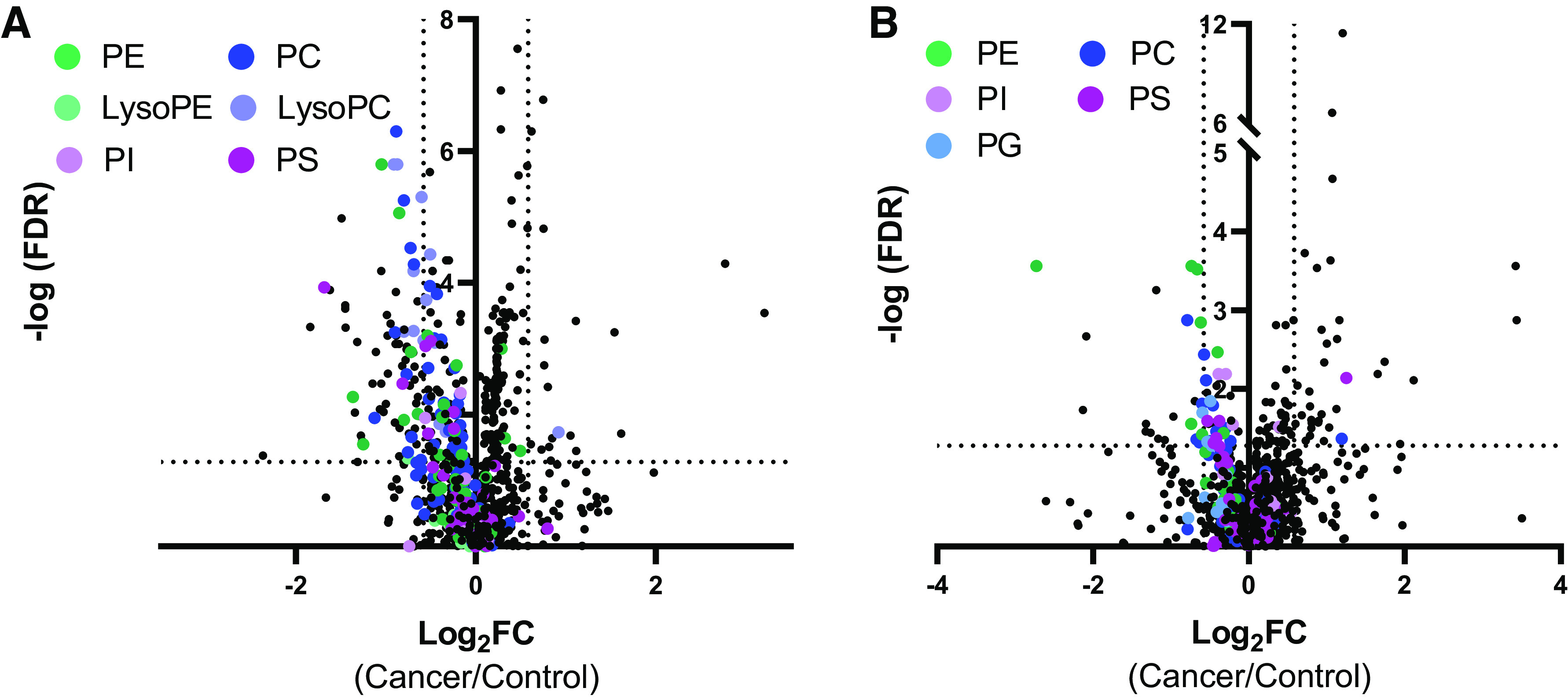
Plasma and skeletal muscle metabolomic profiles of patients with cancer. Untargeted metabolomic profiles of the plasma (cancer, n = 8; control, n = 19) (A) and skeletal muscle (cancer, n = 6; control, n = 8) (B) were assessed by LC- QTOF-MS. To identify differences in the relative abundance of individual metabolites between patients with cancer and control participants, fold-change analyses and unpaired t tests were performed for each identified metabolite using the MetaboAnalyst 4.0 Statistical Analysis Tool. P values were false discovery rate (FDR)-adjusted to account for multiple comparisons, and significant differences were defined as false discovery rates > 0.05 (represented by the dashed horizontal line), and an absolute log2fold change (cancer/control) > 0.58 (represented by the vertical dashed lines); n, number of subjects. All unique identified metabolites are presented and specific phospholipid species are represented by the colors denoted in the figure. FC, fold change; FDR, false discovery rate; LC-QTOF-MS, liquid chromatography-quadrupole time of flight-mass spectrometry; PC, phosphatidylcholine/phosphocholine; PE, phosphatidylethanolamine/phosphoethanolamine; PG, phosphatidylglycerol/phosphoglycerol; PI: phosphatidylinositol/phosphoinositol; PS, phosphatidylserine/phosphoserine.
DISCUSSION
In the present study, cancer-associated muscle derangements were investigated in a group of patients with cancer-associated weight loss. Despite similar muscle mass compared with healthy control participants, patients with cancer exhibited significant functional deficits including decreased muscle strength and endurance, reduced cardiorespiratory fitness, and lower activity levels. Concomitant with these functional declines, patients with cancer demonstrated reduced mitochondrial function and altered skeletal muscle membrane phospholipid metabolite profiles.
The functional deficits associated with cancer significantly impair quality of life, reducing the ability to perform activities of daily living and maintain independence (4, 38). Despite having similar levels of appendicular lean mass compared with the healthy age-, sex-, and BMI-matched participants, the patients with cancer exhibited numerous functional deficits. Lower limb isometric and isokinetic strength was particularly impacted in patients with cancer. The significant functional deficits in the knee extensors and in muscle fatigability in patients with cancer, independent of muscle mass, are in agreement with previous reports of reduced knee extensor torque and power output during walking in patients with cancer (9). Compared with the knee extensors, knee flexor function and knee flexion isometric strength, in particular, were relatively preserved in patients with cancer. During aging, the loss of knee extensor strength has been shown to precede detectable losses in knee flexor strength (39). However, due to the large antagonist activity of the quadriceps during knee flexion, the error associated with isometric knee flexor strength testing is significantly higher than that of knee extensor strength testing (40). Therefore, it is possible that protocols for testing knee flexor strength are not sensitive enough to detect subtle losses in knee flexor function. Reduced leg extensor strength and isokinetic strength have important clinical consequences, significantly contributing to impaired mobility and increased fall risk (41). The causes of the observed reductions in strength in patients with cancer compared with healthy controls are likely multifactorial. Malnutrition was found to be an independent contributor to losses in muscle strength and function in patients with cancer (42). In aging, losses in muscle strength precede losses in muscle mass (43), and both inflammation (43) and impairments in skeletal muscle proteostasis (44) are proposed to contribute to these functional declines. The patients with cancer exhibited signs of systemic inflammation that is inherently linked with cancer (45) and that has been associated with functional deficits in skeletal muscle (38, 46) and proposed to play a mechanistic role in the etiology of cancer-associated muscle loss (47, 48). In addition, the accumulation of misfolded proteins driven by oxidative stress and the dysregulation of the ubiquitin-proteasomal and autophagy-lysosomal systems contributes to skeletal muscle functional decline in aging (44). Disruption of these proteostatic systems has also been reported in cancer animal models and patients (3, 49) and may inhibit sarcomeric and mitochondrial protein quality control, impairing skeletal muscle function. Future work will be required to explore inflammation and impaired proteostasis as potential mechanisms for the loss in muscle function in patients with cancer.
Whole body cardiorespiratory fitness, habitual activity levels, and walking speed were also lower in patients with cancer, and as with the losses in muscle strength, the causes of these decrements are likely complex and multifaceted. The fatigue that the vast majority of patients with cancer experience (50) undoubtedly contributes to the reductions in self-motivated free-living movement and whole body cardiorespiratory fitness. Skeletal muscle impairments may also contribute to the whole body functional declines as increased muscular fatigability and decreased dynamic, isokinetic strength may limit activity and cardiorespiratory fitness. The patients with cancer also had significantly lower hemoglobin concentrations than the healthy control participants, and anemia is a common feature of cancer (51, 52) that is associated with both fatigue and reduced cardiorespiratory fitness (53, 54). These reductions in cardiorespiratory fitness have important clinical consequences, as low V̇o2peak is a significant, independent predictor of cancer-associated mortality (55).
In addition to the observed functional deficits, patients with cancer also exhibited reduced mitochondrial respiration and ATP production and increased mitochondrial ROS generation observed using both in vivo and ex vivo methods. State 3, maximal ADP-stimulated mitochondrial respiration and ATP production measured ex vivo were significantly lower in patients with cancer, particularly when respiration was normalized to wet tissue weight. Our observation that the losses in respiratory capacity were somewhat attenuated when respiration was normalized to mitochondrial protein content suggests that the reductions in mitochondrial function in patients with cancer may partially be the result of a reduced number of mitochondria. These results are in agreement with previous reports of decreased mitochondrial density in patients with cancer (9). However, respiration did remain lower in patients with cancer when normalized to mitochondrial protein content, indicating an intrinsic dysfunction at the level of the organelle beyond the decline in mitochondrial abundance. ROS generation was also higher in patients with cancer. Notably, in addition to detecting the production of H2O2, Amplex Red also detects lipid hydroperoxides, though at a much lower intensity (56, 57). In some conditions, such as denervation, the contribution of lipid hydroperoxides to the hydroperoxide production by mitochondria is substantial (56, 57). In future experiments, catalase scavenging of H2O2 could be measured to determine the relative contributions of H2O2 and lipid peroxidases on the Ample Red signal. Interestingly, the RCR, a measure of mitochondrial efficiency, was actually higher in patients with cancer. Theoretically, increased mitochondrial efficiency indicates that less substrate would be required to produce ATP (58). Although this may be an adaptive mitochondrial alteration to preserve energy during weight loss, increased mitochondrial efficiency has also been proposed to result in increased intramuscular triglyceride deposition and contribute to insulin resistance (58), and cancer-associated muscle wasting is also often accompanied by insulin resistance (59).
Additional work is required to elucidate the causes of these intrinsic mitochondrial alterations. Mitochondria are dynamic organelles that undergo processes of fusion and fission, proteolysis, and mitophagy to maintain the health and integrity of the mitochondrial network (60, 61). Evidence of alterations in these mitochondrial processes have been reported in models of cancer-associated muscle wasting and likely contribute to the observed decrements in mitochondrial function (3, 60). Importantly, multiple participants were undergoing chemotherapy regimens at the time of study participation, which may also contribute to the observed mitochondrial abnormalities and functional deficits. Chemotherapeutics are commonly associated with significant fatigue and have been shown to reduce mitochondrial number and increase ROS production in cultured myotubes (62) and to alter the expression of proteins associated with mitochondrial fusion and fission in mice (60). Therefore, future work will be required to distinguish the independent effects of cancer and chemotherapy on skeletal muscle and mitochondrial functional capacity. The mitochondrial derangements may also be a consequence of the reduced whole body and skeletal muscle physical function observed in patients with cancer. Physical activity is a potent stimulator of mitochondrial biogenesis, and therefore the reduced physical activity and skeletal muscle function may contribute to the observed reductions in mitochondrial function.
The reduced mitochondrial respiration and ATP production and increased mitochondrial ROS generation observed using both in vivo and in vitro methods in patients with cancer may represent both a cause and a consequence of the functional declines observed. The increased fatigability and the loss in dynamic strength may be indicative of the observed reduced oxidative and ATP production capacity of the mitochondria in patients with cancer. Indeed, decreased mitochondrial content and biogenesis are associated with a more fatigable muscle phenotype (5, 38) and mitochondrial derangements have been proposed to contribute to impaired skeletal muscle function in the ApcMin/+ mouse model of cancer-associated muscle wasting (63). In addition to contributing to reduced skeletal muscle function, impaired mitochondrial function may also contribute to the observed reduced habitual physical activity, slower walking speed, and reduced overall cardiorespiratory capacity observed in the patients with cancer. In older adults, lower skeletal muscle mitochondrial capacity is associated with both reduced walking speed and V̇o2peak (64). Furthermore, fatigue is often considered a hallmark of mitochondrial disease, and associations between mitochondrial dysfunction and fatigue in multiple disease states have also been reported (65). Indeed, it is likely that reduced physical activity leads to impaired mitochondrial function, which in turn serves as an additional barrier to performing physical activity, creating a vicious cycle that serves to reduce muscle function and physical capacity (66). Physical deconditioning is also considered a contributor to cancer-associated weight and muscle loss, and promoting physical activity, in conjunction with adequate nutrition, may partially relieve the “cachexia burden” (67). However, the reversible contributors to cancer-associated weight loss (physical inactivity, malnutrition) appear to be only minor factors in the etiology of the syndrome (67), highlighting the need to identify other causes of cancer-associated muscle derangements.
As with mitochondrial dysfunction, alterations in protein turnover are also postulated to contribute to cancer-associated muscle derangements. Both increased protein breakdown (2) and reduced protein synthesis (2, 3, 68) have been reported to contribute to cancer-associated muscle loss. Despite these reports and in-line with previous work (68), whole body protein synthesis and breakdown rates were similar between healthy volunteers and patients with cancer in the present study. Importantly, whole body protein kinetics include both muscle and nonmuscle (including the tumor) compartments that may undergo divergent responses to the disease (68). Future work will be required to assess muscle-specific protein synthesis and breakdown in patients with cancer.
To identify potential novel pathways involved in cancer-induced alterations in muscle and mitochondrial function, nontargeted metabolomic profiling of the skeletal muscle was performed. Interestingly, within the skeletal muscle, an overall reduction in phospholipid species in patients with cancer, and in PE species, in particular, was observed. The phospholipid composition of the mitochondria has important implications for numerous mitochondrial functions, including respiration, programmed cell death, mitochondrial fusion, and autophagy (69–71). Inner mitochondrial membranes are enriched with PE, which contributes to the highly folded morphology of the membrane that is essential for oxidative capacity (35, 70). Mitochondrial-specific reductions in PE concentrations result in impaired cell growth and survival, reduced oxygen consumption and ATP production, and aberrant mitochondrial morphology (36, 71). Therefore, observed reductions in PE species and alterations in the phospholipid composition of skeletal muscle of patients with cancer may have important implications and consequences for mitochondrial function. Indeed, concomitant mitochondrial dysfunction and alterations in mitochondrial phospholipid composition have been reported in rodent models of cancer-associated muscle wasting (69). In addition to their potential roles in mitochondrial function, skeletal muscle phospholipids are associated with insulin sensitivity and metabolic health (35, 37). In addition to reduced skeletal muscle functional capacity, cancer-associated muscle wasting is also often accompanied by insulin resistance (59). PC and PE are the most abundant phospholipids in cellular membranes, and total skeletal muscle PC and PE levels are positively associated with insulin sensitivity (35, 37). The relative abundance of each phospholipid also has implications for metabolic health, as individuals with obesity or type 2 diabetes have lower skeletal muscle levels of PC and PE, and an elevated PC:PE ratio (35, 37). In addition to its associations with insulin sensitivity, the PC:PE ratio has been implicated in glucose metabolism and skeletal muscle contractile function (37, 72). As the metabolomics platform used in the present study was nonquantitative and provided only relative abundance and not concentrations of metabolites, an accurate PC:PE ratio could not be calculated, but the observed alterations in phospholipid composition in patients with cancer may have implications for skeletal muscle mitochondrial function and metabolism and warrant further investigation.
To identify additional mechanistic indicators and potential biomarkers of the losses in muscle mass and function associated with cancer, nontargeted plasma metabolomic profiling was also performed. Within the plasma of the patients with cancer, overall reductions in PC and PE levels, and reductions in the breakdown products (lysoPE, lysoPC) of these phospholipids were observed. Reduced overall phospholipid concentrations and LysoPC concentrations have previously been observed in the plasma of patients with cancer, and LysoPC concentrations were negatively associated with both inflammation and degree of weight loss (73, 74). Phospholipid metabolism is an important contributor to whole body energy metabolism, and alterations in the relative concentrations of phospholipids in multiple tissues appear to contribute to various metabolic disorders (71). Therefore, the observed reductions in phospholipids may be indicative of mechanistic causes of cancer-associated muscle wasting.
The limitations of the present study include the small number of patients with cancer, as well as the unequal distribution of sex within the participants. In addition to the small sample size, for some outcomes, data were not obtained for all participants. For multiple outcomes, missing data were related to disease severity (e.g., muscle biopsies were not able to be obtained in some patients due to small muscle size), and therefore, the results of the present study may underestimate the effects of cancer. For other outcomes, data were missing at random. Importantly, data from control participants were only included in each analysis if data were available for the matched patient with cancer. In addition, to maximize the potential to observe skeletal muscle changes associated with cancer, only patients who had experienced significant weight loss (≥5% body weight within the previous 12 mo) were included in the study, and the observed functional deficits and mitochondrial changes may represent late-stage skeletal muscle abnormalities associated with muscle loss.
In summary, cancer is associated with significant skeletal muscle functional deficits, alterations in mitochondrial function, and changes in the concentrations of plasma and tissue phospholipids that modulate mitochondrial activity. To our knowledge, the present study demonstrates for the first time mitochondrial deficits in human patients with cancer concomitant with functional derangements. It is uncertain whether the changes in mitochondrial function are drivers of muscle weakness, or whether the changes in mitochondrial function occur as a result of muscle weakness that may occur due to factors that have yet to be identified. These human data provide compelling evidence for future study of mitochondrial abnormalities and alterations in membrane phospholipid profiles for future investigations and therapeutics.
DATA AVAILABILITY
The data generated in this study are available upon request from the corresponding author.
SUPPLEMENTAL DATA
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.16869201.
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.16869233.
Supplemental Data Set 1: https://doi.org/10.6084/m9.figshare.17124290.
Supplemental Data Set 2: https://doi.org/10.6084/m9.figshare.17124308.
GRANTS
The project was funded by an award from the Fred B. and Katherine C. Andersen Foundation (to I. R. Lanza and R. Kumar) and R01CA195473 (to A. Jatoi). H. E. Kunz was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases for the Musculoskeletal Research Training Program Grant T32AR056950.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.E.K., J.D.P., K.R.K., A.J., C.R.H., K.J.G., I.R.L., and R.K. conceived and designed research; H.E.K., J.D.P., K.R.K., C.R.H., K.J.G., I.R.L., and R.K. performed experiments; H.E.K., J.D.P., K.R.K., A.J., C.R.H., K.J.G., I.R.L., and R.K. analyzed data; H.E.K., J.D.P., K.R.K., A.J., C.R.H., K.J.G., I.R.L., and R.K. interpreted results of experiments; H.E.K. prepared figures; H.E.K. drafted manuscript; H.E.K., J.D.P., K.R.K., A.J., C.R.H., K.J.G., I.R.L., and R.K. edited and revised manuscript; H.E.K., J.D.P., K.R.K., A.J., C.R.H., K.J.G., I.R.L., and R.K. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Bobbie Soderberg for biopsy support; Zachary Ryan, Taylor Berent, and Kathie Bernhardt for assistance with data collection; and Christine Huyber for study coordination. Dr. Felicity Enders and Jason Viehman in the Mayo Clinic Division of Biomedical Statistics and Informatics provided statistical support. Work was also supported by the staff at the Mayo Clinic Clinical Research and Trials Unit and Metabolomics Core. Graphical Abstract created with BioRender.com.
Present addresses: C. R. Hart, Air Force Research Laboratory, 711th Human Performance Wing, Wright Patterson Air Force Base, Dayton, Ohio; K. J. Gries, Exercise and Sports Science Department, College of Health Professions, Marian University, Indianapolis, Indiana.
REFERENCES
- 1.von Haehling S, Anker SD. Prevalence, incidence and clinical impact of cachexia: facts and numbers-update 2014. J Cachexia Sarcopenia Muscle 5: 261–263, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown JL, Lee DE, Rosa-Caldwell ME, Brown LA, Perry RA, Haynie WS, Huseman K, Sataranatarajan K, Van Remmen H, Washington TA, Wiggs MP, Greene NP. Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J Cachexia Sarcopenia Muscle 9: 987–1002, 2018. [Erratum in J Cachexia Sarcopenia Muscle 10: 712, 2019]. doi: 10.1002/jcsm.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kunz HE, Dorschner JM, Berent TE, Meyer T, Wang X, Jatoi A, Kumar R, Lanza IR. Methylarginine metabolites are associated with attenuated muscle protein synthesis in cancer-associated muscle wasting. J Biol Chem 295: 17441–17459, 2020. doi: 10.1074/jbc.RA120.014884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, Jatoi A, Kalantar-Zadeh K, Lochs H, Mantovani G, Marks D, Mitch WE, Muscaritoli M, Najand A, Ponikowski P, Rossi Fanelli F, Schambelan M, Schols A, Schuster M, Thomas D, Wolfe R, Anker SD. Cachexia: a new definition. Clin Nutr 27: 793–799, 2008. doi: 10.1016/j.clnu.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 5.Brown JL, Rosa-Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA, Haynie WS, Hardee JP, Carson JA, Wiggs MP, Washington TA, Greene NP. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J Cachexia Sarcopenia Muscle 8: 926–938, 2017. doi: 10.1002/jcsm.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fontes-Oliveira CC, Busquets S, Toledo M, Penna F, Paz Aylwin M, Sirisi S, Silva AP, Orpi M, Garcia A, Sette A, Ines Genovese M, Olivan M, Lopez-Soriano FJ, Argiles JM. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim Biophys Acta 1830: 2770–2778, 2013. doi: 10.1016/j.bbagen.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Marzetti E, Lorenzi M, Landi F, Picca A, Rosa F, Tanganelli F, Galli M, Doglietto GB, Pacelli F, Cesari M, Bernabei R, Calvani R, Bossola M. Altered mitochondrial quality control signaling in muscle of old gastric cancer patients with cachexia. Exp Gerontol 87: 92–99, 2017. doi: 10.1016/j.exger.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 8.de Castro GS, Simoes E, Lima J, Ortiz-Silva M, Festuccia WT, Tokeshi F, Alcantara PS, Otoch JP, Coletti D, Seelaender M. Human cachexia induces changes in mitochondria, autophagy and apoptosis in the skeletal muscle. Cancers (Basel) 11: 1264, 2019. doi: 10.3390/cancers11091264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toth MJ, Miller MS, Callahan DM, Sweeny AP, Nunez I, Grunberg SM, Der-Torossian H, Couch ME, Dittus K. Molecular mechanisms underlying skeletal muscle weakness in human cancer: reduced myosin-actin cross-bridge formation and kinetics. J Appl Physiol (1985) 114: 858–868, 2013. doi: 10.1152/japplphysiol.01474.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patterson RE, Kristal AR, Tinker LF, Carter RA, Bolton MP, Agurs-Collins T. Measurement characteristics of the Women's Health Initiative food frequency questionnaire. Ann Epidemiol 9: 178–187, 1999. doi: 10.1016/s1047-2797(98)00055-6. [DOI] [PubMed] [Google Scholar]
- 11.Kaufman KR, Bernhardt KA, Symms K. Functional assessment and satisfaction of transfemoral amputees with low mobility (FASTK2): a clinical trial of microprocessor-controlled vs. non-microprocessor-controlled knees. Clin Biomech (Bristol, Avon) 58: 116–122, 2018. doi: 10.1016/j.clinbiomech.2018.07.012. [DOI] [PubMed] [Google Scholar]
- 12.Fortune E, Lugade V, Morrow M, Kaufman K. Using tri-axial accelerometers to measure human movement. II. Step counts at a wide range of gait velocities. Med Eng Phys 36: 659–669, 2014. doi: 10.1016/j.medengphy.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lugade V, Fortune E, Morrow M, Kaufman K. Validity of using tri-axial accelerometers to measure human movement. I. Posture and movement detection. Med Eng Phys 36: 169–176, 2014. doi: 10.1016/j.medengphy.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruce RA, Kusumi F, Hosmer D. Maximal oxygen intake and nomographic assessment of functional aerobic impairment in cardiovascular disease. Am Heart J 85: 546–562, 1973. doi: 10.1016/0002-8703(73)90502-4. [DOI] [PubMed] [Google Scholar]
- 15.McInnis KJ, Balady GJ. Comparison of submaximal exercise responses using the Bruce vs. modified Bruce protocols. Med Sci Sports Exerc 26: 103–107, 1994. [PubMed] [Google Scholar]
- 16.Borg GA. Perceived exertion: a note on “history” and methods. Med Sci Sports 5: 90–93, 1973. [PubMed] [Google Scholar]
- 17.Peolsson A, Hedlund R, Oberg B. Intra- and inter-tester reliability and reference values for hand strength. J Rehabil Med 33: 36–41, 2001. doi: 10.1080/165019701300006524. [DOI] [PubMed] [Google Scholar]
- 18.Katsiaras A, Newman AB, Kriska A, Brach J, Krishnaswami S, Feingold E, Kritchevsky SB, Li R, Harris TB, Schwartz A, Goodpaster BH. Skeletal muscle fatigue, strength, and quality in the elderly: the Health ABC Study. J Appl Physiol (1985) 99: 210–216, 2005. doi: 10.1152/japplphysiol.01276.2004. [DOI] [PubMed] [Google Scholar]
- 19.Lalia AZ, Dasari S, Johnson ML, Robinson MM, Konopka AR, Distelmaier K, Port JD, Glavin MT, Esponda RR, Nair KS, Lanza IR. Predictors of whole-body insulin sensitivity across ages and adiposity in adult humans. J Clin Endocrinol Metab 101: 626–634, 2016. doi: 10.1210/jc.2015-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Short KR, Meek SE, Moller N, Ekberg K, Nair KS. Whole body protein kinetics using Phe and Tyr tracers: an evaluation of the accuracy of approximated flux values. Am J Physiol Endocrinol Physiol 276: E1194–E1200, 1999. doi: 10.1152/ajpendo.1999.276.6.E1194. [DOI] [PubMed] [Google Scholar]
- 21.Rooyackers O, Kouchek-Zadeh R, Tjader I, Norberg A, Klaude M, Wernerman J. Whole body protein turnover in critically ill patients with multiple organ failure. Clin Nutr 34: 95–100, 2015. doi: 10.1016/j.clnu.2014.01.020. [DOI] [PubMed] [Google Scholar]
- 22.Krumschnabel G, Fontana-Ayoub M, Sumbalova Z, Heidler J, Gauper K, Fasching M, Gnaiger E. Simultaneous high-resolution measurement of mitochondrial respiration and hydrogen peroxide production. Methods Mol Biol 1264: 245–261, 2015. doi: 10.1007/978-1-4939-2257-4_22. [DOI] [PubMed] [Google Scholar]
- 23.Lanza IR, Nair KS. Functional assessment of isolated mitochondria in vitro. Methods Enzymol 457: 349–372, 2009. doi: 10.1016/S0076-6879(09)05020-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kunz HE, Dasari S, Lanza IR. EPA and DHA elicit distinct transcriptional responses to high-fat feeding in skeletal muscle and liver. Am J Physiol Endocrinol Metab 317: E460–E472, 2019. doi: 10.1152/ajpendo.00083.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanza IR, Blachnio-Zabielska A, Johnson ML, Schimke JM, Jakaitis DR, Lebrasseur NK, Jensen MD, Sreekumaran Nair K, Zabielski P. Influence of fish oil on skeletal muscle mitochondrial energetics and lipid metabolites during high-fat diet. Am J Physiol Endocrinol Metab 304: E1391–E1403, 2013. [Erratum in Am J Physiol Endocrinol Metab 305: E1048, 2013]. doi: 10.1152/ajpendo.00584.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perry CG, Kane DA, Lanza IR, Neufer PD. Methods for assessing mitochondrial function in diabetes. Diabetes 62: 1041–1053, 2013. doi: 10.2337/db12-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konopka AR, Asante A, Lanza IR, Robinson MM, Johnson ML, Dalla Man C, Cobelli C, Amols MH, Irving BA, Nair KS. Defects in mitochondrial efficiency and H2O2 emissions in obese women are restored to a lean phenotype with aerobic exercise training. Diabetes 64: 2104–2115, 2015. doi: 10.2337/db14-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gouspillou G, Rouland R, Calmettes G, Deschodt-Arsac V, Franconi JM, Bourdel-Marchasson I, Diolez P. Accurate determination of the oxidative phosphorylation affinity for ADP in isolated mitochondria. PLoS One 6: e20709, 2011. doi: 10.1371/journal.pone.0020709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lark DS, Torres MJ, Lin CT, Ryan TE, Anderson EJ, Neufer PD. Direct real-time quantification of mitochondrial oxidative phosphorylation efficiency in permeabilized skeletal muscle myofibers. Am J Physiol Cell Physiol 311: C239–C245, 2016. doi: 10.1152/ajpcell.00124.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanza IR, Bhagra S, Nair KS, Port JD. Measurement of human skeletal muscle oxidative capacity by 31P-MR spectroscopy: a cross-validation with in vitro measurements. J Magn Reson Imaging 34: 1143–1150, 2011. doi: 10.1002/jmri.22733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lanza IR, Nair KS. Mitochondrial metabolic function assessed in vivo and in vitro. Curr Opin Clin Nutr Metab Care 13: 511–517, 2010. doi: 10.1097/MCO.0b013e32833cc93d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naressi A, Couturier C, Castang I, de Beer R, Graveron-Demilly D. Java-based graphical user interface for MRUI, a software package for quantitation of in vivo/medical magnetic resonance spectroscopy signals. Comput Biol Med 31: 269–286, 2001. doi: 10.1016/s0010-4825(01)00006-3. [DOI] [PubMed] [Google Scholar]
- 33.Chong J, Wishart DS, Xia J. Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr Protoc Bioinformatics 68: e86, 2019. doi: 10.1002/cpbi.86. [DOI] [PubMed] [Google Scholar]
- 34.Lalia AZ, Dasari S, Robinson MM, Abid H, Morse DM, Klaus KA, Lanza IR. Influence of omega-3 fatty acids on skeletal muscle protein metabolism and mitochondrial bioenergetics in older adults. Aging (Albany NY) 9: 1096–1129, 2017. doi: 10.18632/aging.101210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S, Norheim F, Gulseth HL, Langleite TM, Aker A, Gundersen TE, Holen T, Birkeland KI, Drevon CA. Skeletal muscle phosphatidylcholine and phosphatidylethanolamine respond to exercise and influence insulin sensitivity in men. Sci Rep 8: 6531, 2018. [Erratum in Sci Rep 8: 7885, 2018]. doi: 10.1038/s41598-018-24976-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tasseva G, Bai HD, Davidescu M, Haromy A, Michelakis E, Vance JE. Phosphatidylethanolamine deficiency in mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J Biol Chem 288: 4158–4173, 2013. doi: 10.1074/jbc.M112.434183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newsom SA, Brozinick JT, Kiseljak-Vassiliades K, Strauss AN, Bacon SD, Kerege AA, Bui HH, Sanders P, Siddall P, Wei T, Thomas M, Kuo MS, Nemkov T, D'Alessandro A, Hansen KC, Perreault L, Bergman BC. Skeletal muscle phosphatidylcholine and phosphatidylethanolamine are related to insulin sensitivity and respond to acute exercise in humans. J Appl Physiol (1985) 120: 1355–1363, 2016. doi: 10.1152/japplphysiol.00664.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.VanderVeen BN, Hardee JP, Fix DK, Carson JA. Skeletal muscle function during the progression of cancer cachexia in the male Apc(Min/+) mouse. J Appl Physiol (1985) 124: 684–695, 2018. doi: 10.1152/japplphysiol.00897.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee H, Kim IG, Sung C, Jeon TB, Cho K, Ha YC, Park KS, Yoo JI, Kang GH, Kim SJ, Kim JS. Exercise training increases skeletal muscle strength independent of hypertrophy in older adults aged 75 years and older. Geriatr Gerontol Int 19: 265–270, 2019. doi: 10.1111/ggi.13597. [DOI] [PubMed] [Google Scholar]
- 40.Krishnan C, Williams GN. Error associated with antagonist muscle activity in isometric knee strength testing. Eur J Appl Physiol 109: 527–536, 2010. doi: 10.1007/s00421-010-1391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lord SR, Ward JA, Williams P, Anstey KJ. Physiological factors associated with falls in older community-dwelling women. J Am Geriatr Soc 42: 1110–1117, 1994. doi: 10.1111/j.1532-5415.1994.tb06218.x. [DOI] [PubMed] [Google Scholar]
- 42.Norman K, Stobaus N, Smoliner C, Zocher D, Scheufele R, Valentini L, Lochs H, Pirlich M. Determinants of hand grip strength, knee extension strength and functional status in cancer patients. Clin Nutr 29: 586–591, 2010. doi: 10.1016/j.clnu.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 43.Goodpaster BH, Park SW, Harris TB, Kritchevsky SB, Nevitt M, Schwartz AV, Simonsick EM, Tylavsky FA, Visser M, Newman AB. The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol A Biol Sci Med Sci 61: 1059–1064, 2006. doi: 10.1093/gerona/61.10.1059. [DOI] [PubMed] [Google Scholar]
- 44.Fernando R, Drescher C, Nowotny K, Grune T, Castro JP. Impaired proteostasis during skeletal muscle aging. Free Radic Biol Med 132: 58–66, 2019. doi: 10.1016/j.freeradbiomed.2018.08.037. [DOI] [PubMed] [Google Scholar]
- 45.Chechlinska M, Kowalewska M, Nowak R. Systemic inflammation as a confounding factor in cancer biomarker discovery and validation. Nat Rev Cancer 10: 2–3, 2010. doi: 10.1038/nrc2782. [DOI] [PubMed] [Google Scholar]
- 46.VanderVeen BN, Fix DK, Montalvo RN, Counts BR, Smuder AJ, Murphy EA, Koh HJ, Carson JA. The regulation of skeletal muscle fatigability and mitochondrial function by chronically elevated interleukin-6. Exp Physiol 104: 385–397, 2019. doi: 10.1113/EP087429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 16: 153–166, 2012. doi: 10.1016/j.cmet.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 48.Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev 89: 381–410, 2009. doi: 10.1152/physrev.00016.2008. [DOI] [PubMed] [Google Scholar]
- 49.Tardif N, Klaude M, Lundell L, Thorell A, Rooyackers O. Autophagic-lysosomal pathway is the main proteolytic system modified in the skeletal muscle of esophageal cancer patients. Am J Clin Nutr 98: 1485–1492, 2013. doi: 10.3945/ajcn.113.063859. [DOI] [PubMed] [Google Scholar]
- 50.Wagner LI, Cella D. Fatigue and cancer: causes, prevalence and treatment approaches. Br J Cancer 91: 822–828, 2004. doi: 10.1038/sj.bjc.6602012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dicato M, Plawny L, Diederich M. Anemia in cancer. Ann Oncol 21 Suppl 7: vii167–vii172, 2010. doi: 10.1093/annonc/mdq284. [DOI] [PubMed] [Google Scholar]
- 52.Dodson S, Baracos VE, Jatoi A, Evans WJ, Cella D, Dalton JT, Steiner MS. Muscle wasting in cancer cachexia: clinical implications, diagnosis, and emerging treatment strategies. Annu Rev Med 62: 265–279, 2011. doi: 10.1146/annurev-med-061509-131248. [DOI] [PubMed] [Google Scholar]
- 53.Gregg SG, Willis WT, Brooks GA. Interactive effects of anemia and muscle oxidative capacity on exercise endurance. J Appl Physiol (1985) 67: 765–770, 1989. doi: 10.1152/jappl.1989.67.2.765. [DOI] [PubMed] [Google Scholar]
- 54.Sobrero A, Puglisi F, Guglielmi A, Belvedere O, Aprile G, Ramello M, Grossi F. Fatigue: a main component of anemia symptomatology. Semin Oncol 28: 15–18, 2001. doi: 10.1016/s0093-7754(01)90207-6. [DOI] [PubMed] [Google Scholar]
- 55.Groarke JD, Payne DL, Claggett B, Mehra MR, Gong J, Caron J, Mahmood SS, Hainer J, Neilan TG, Partridge AH, Di Carli M, Jones LW, Nohria A. Association of post-diagnosis cardiorespiratory fitness with cause-specific mortality in cancer. Eur Heart J Qual Care Clin Outcomes 6: 315–322, 2020. doi: 10.1093/ehjqcco/qcaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A, Van Remmen H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol 293: R1159–R1168, 2007. doi: 10.1152/ajpregu.00767.2006. [DOI] [PubMed] [Google Scholar]
- 57.Pharaoh G, Brown JL, Sataranatarajan K, Kneis P, Bian J, Ranjit R, Hadad N, Georgescu C, Rabinovitch P, Ran Q, Wren JD, Freeman W, Kinter M, Richardson A, Van Remmen H. Targeting cPLA2 derived lipid hydroperoxides as a potential intervention for sarcopenia. Sci Rep 10: 13968, 2020. doi: 10.1038/s41598-020-70792-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iossa S, Mollica MP, Lionetti L, Crescenzo R, Tasso R, Liverini G. A possible link between skeletal muscle mitochondrial efficiency and age-induced insulin resistance. Diabetes 53: 2861–2866, 2004. doi: 10.2337/diabetes.53.11.2861. [DOI] [PubMed] [Google Scholar]
- 59.Honors MA, Kinzig KP. The role of insulin resistance in the development of muscle wasting during cancer cachexia. J Cachexia Sarcopenia Muscle 3: 5–11, 2012. doi: 10.1007/s13539-011-0051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barreto R, Mandili G, Witzmann FA, Novelli F, Zimmers TA, Bonetto A. Cancer and chemotherapy contribute to muscle loss by activating common signaling pathways. Front Physiol 7: 472, 2016. doi: 10.3389/fphys.2016.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Romanello V, Sandri M. The connection between the dynamic remodeling of the mitochondrial network and the regulation of muscle mass. Cell Mol Life Sci 78: 1305–1328, 2021. doi: 10.1007/s00018-020-03662-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guigni BA, Callahan DM, Tourville TW, Miller MS, Fiske B, Voigt T, Korwin-Mihavics B, Anathy V, Dittus K, Toth MJ. Skeletal muscle atrophy and dysfunction in breast cancer patients: role for chemotherapy-derived oxidant stress. Am J Physiol Cell Physiol 315: C744–C756, 2018. doi: 10.1152/ajpcell.00002.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.White JP, Puppa MJ, Sato S, Gao S, Price RL, Baynes JW, Kostek MC, Matesic LE, Carson JA. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet Muscle 2: 14, 2012. doi: 10.1186/2044-5040-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Coen PM, Jubrias SA, Distefano G, Amati F, Mackey DC, Glynn NW, Manini TM, Wohlgemuth SE, Leeuwenburgh C, Cummings SR, Newman AB, Ferrucci L, Toledo FG, Shankland E, Conley KE, Goodpaster BH. Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults. J Gerontol A Biol Sci Med Sci 68: 447–455, 2013. doi: 10.1093/gerona/gls196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Filler K, Lyon D, Bennett J, McCain N, Elswick R, Lukkahatai N, Saligan LN. Association of mitochondrial dysfunction and fatigue: a review of the literature. BBA Clin 1: 12–23, 2014. doi: 10.1016/j.bbacli.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP, Nair KS. Endurance exercise as a countermeasure for aging. Diabetes 57: 2933–2942, 2008. [Erratum in Diabetes 61: 2653, 2012]. doi: 10.2337/db08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 10: 90–99, 2013. doi: 10.1038/nrclinonc.2012.209. [DOI] [PubMed] [Google Scholar]
- 68.Dworzak F, Ferrari P, Gavazzi C, Maiorana C, Bozzetti F. Effects of cachexia due to cancer on whole body and skeletal muscle protein turnover. Cancer 82: 42–48, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 69.Antunes D, Padrao AI, Maciel E, Santinha D, Oliveira P, Vitorino R, Moreira-Goncalves D, Colaco B, Pires MJ, Nunes C, Santos LL, Amado F, Duarte JA, Domingues MR, Ferreira R. Molecular insights into mitochondrial dysfunction in cancer-related muscle wasting. Biochim Biophys Acta 1841: 896–905, 2014. doi: 10.1016/j.bbalip.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 70.Heden TD, Neufer PD, Funai K. Looking beyond structure: membrane phospholipids of skeletal muscle mitochondria. Trends Endocrinol Metab 27: 553–562, 2016. doi: 10.1016/j.tem.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van der Veen JN, Kennelly JP, Wan S, Vance JE, Vance DE, Jacobs RL. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr 1859: 1558–1572, 2017. doi: 10.1016/j.bbamem.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 72.Funai K, Lodhi IJ, Spears LD, Yin L, Song H, Klein S, Semenkovich CF. Skeletal muscle phospholipid metabolism regulates insulin sensitivity and contractile function. Diabetes 65: 358–370, 2016. doi: 10.2337/db15-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kriat M, Vion-Dury J, Confort-Gouny S, Favre R, Viout P, Sciaky M, Sari H, Cozzone PJ. Analysis of plasma lipids by NMR spectroscopy: application to modifications induced by malignant tumors. J Lipid Res 34: 1009–1019, 1993. [PubMed] [Google Scholar]
- 74.Taylor LA, Arends J, Hodina AK, Unger C, Massing U. Plasma lyso-phosphatidylcholine concentration is decreased in cancer patients with weight loss and activated inflammatory status. Lipids Health Dis 6: 17, 2007. doi: 10.1186/1476-511X-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.16869201.
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.16869233.
Supplemental Data Set 1: https://doi.org/10.6084/m9.figshare.17124290.
Supplemental Data Set 2: https://doi.org/10.6084/m9.figshare.17124308.
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.