

Abstract
Emerging evidence shows that chronic inflammation mediated by toll-like receptors (TLRs) contributes to diabetic nephropathy. Myeloid differentiation primary-response protein-88 (MyD88) is an essential adapter protein of all TLRs except TLR3 in innate immunity. It is unclear whether MyD88 could be a therapeutic target for diabetic nephropathy. Here, we used a new small-molecule MyD88 inhibitor, LM8, to examine the pharmacological inhibition of MyD88 in protecting kidneys from inflammatory injury in diabetes. We showed that MyD88 was significantly activated in the kidney of STZ-induced type 1 diabetic mice in tubular epithelial cells as well as in high glucose-treated rat tubular epithelial cells NRK-52E. In cultured tubular epithelial cells, we show that LM8 (2.5−10 μM) or MyD88 siRNA attenuated high-concentration glucose-induced inflammatory and fibrogenic responses through inhibition of MyD88-TLR4 interaction and downstream NF-κB activation. Treatment with LM8 (5, 10 mg/kg, i.g.) significantly reduced renal inflammation and fibrosis and preserved renal function in both type 1 and type 2 diabetic mice. These renoprotective effects were associated with reduced MyD88-TLR4 complex formation, suppressed NF-κB signaling, and prevention of inflammatory factor expression. Collectively, our results show that hyperglycemia activates MyD88 signaling cascade to induce renal inflammation, fibrosis, and dysfunction. Pharmacological inhibition of MyD88 may be a therapeutic approach to mitigate diabetic nephropathy and the inhibitor LM8 could be a potential candidate for such therapy.
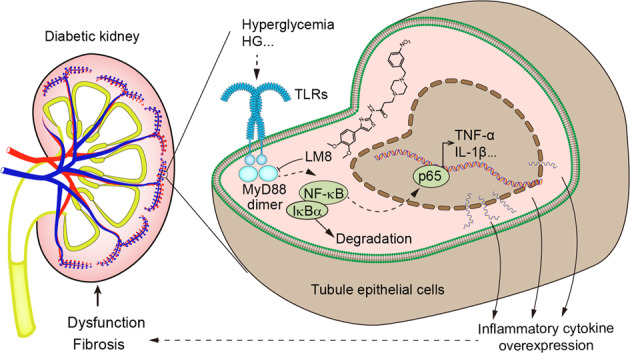
Keywords: MyD88, NF-κB, renal inflammation, renal fibrosis, diabetic nephropathy, renal tubular epithelial cells
Introduction
Diabetes is the leading cause of end-stage renal failure, and therefore all-cause mortality in type 1 and type 2 diabetic patients [1–3]. The key risk factors associated with the development of diabetic kidney disease (DKD) include poor glycemic control, hypertension, and dyslipidemia [4]. The disease progresses to microalbuminuria and a significantly lowered glomerular filtration rate [5]. Kidney biopsy specimens from advanced cases show mesangial hypercellularity, thickening of the basement membranes and interstitial fibrosis, arteriolar hyalinosis, and tubular atrophy. Due to the immense health and economic burden, discovering new therapeutic interventions is urgently needed to treat DKD.
Hyperglycemia-induced chronic inflammation appears to be a central pathological process in diabetic nephropathy [6]. Renal inflammation leads to renal fibrosis, which occurs in all forms of chronic kidney diseases. Emerging evidence shows that chronic sterile inflammation contributes to diabetic nephropathy [7] and further supports a putative role of toll-like receptors (TLRs), especially TLR2 and TLR4, in diabetic nephropathy via mediating renal inflammatory genesis [8]. Although no clinical studies have examined the effect of TLR antagonists in diabetic nephropathy, increasing experimental data suggest that inhibition of TLR signaling, particularly that of TLR2 and/or TLR4, may confer renal protection [7].
Pro-inflammatory signaling initiated by TLR2 and/or TLR4 activation is mediated by unique interaction between Toll/interleukin-1 receptor (TIR)-domain-containing cytosolic adapters including TLR2/4 and myeloid differentiation primary-response protein-88 (MyD88) [9, 10]. MyD88 is an essential adapter protein used by TLRs-mediated inflammatory response. Activation of MyD88 culminates in the activation of nuclear factor-κB (NF-κB) and the production of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin (IL)-1β [11]. Deficiencies in MyD88 by RNA interference knockdown or MyD88 gene knockout show a protective effect on inflammatory responses induced by endotoxin [12]. Considering that MyD88 is an important adapter protein in TLRs’ pro-inflammatory signaling pathway, we speculate that MyD88 may be as a potential target for the treatment of diabetic nephropathy. However, it is unclear if pharmacological inhibition of MyD88 can protect kidneys from inflammatory injuries in diabetes, partly due to the absence of small-molecule MyD88 inhibitors.
Recently, we designed and discovered a series of new small-molecule MyD88 inhibitors, which bind to the TIR domain of MyD88 and interfere with the dimerization of MyD88 and MyD88-TLR4 complex formation [13]. One of these compounds, LM8 (named originally as compound 15c), significantly reduced lipopolysaccharide-induced inflammatory cytokines in macrophages [13] and showed good chemical property and druggability for further investigations. Based on these studies, we hypothesized that LM8 may potentially attenuate DKD via inhibiting MyD88. Our results show that MyD88 activity is increased in mouse tubular epithelial cells and mediates high-concentration glucose-induced pro-inflammatory and pro-fibrotic responses. LM8 treatment inhibited high glucose (HG)-induced inflammatory and fibrotic changes in renal epithelial cells and prevented renal inflammation and fibrosis in diabetic mice. These results show that pharmacological inhibition of MyD88 may potentially curb diabetic nephropathy.
Materials and methods
Reagents
MyD88 inhibitor LM8 was synthesized as described by us previously [13] (identified originally as compound 15c). LM8 has a purity of 98.9% and was dissolved in dimethyl sulfoxide (DMSO) for in vitro studies or in 1% sodium carboxyl methyl cellulose (CMC-Na, cat no: 419338, Sigma, St. Louis, MO, USA) for in vivo studies. Biotinylated LM8 (Bio-LM8) was also synthesized and structurally characterized with a purity of 98.1%. Antibodies against inhibitor of NF-κB α subunit (IκBα, cat no: sc-1643), Tissue Growth Factor-β (TGF-β, cat no: sc-130348), Collagen IV (Col-4, cat no: sc-59814), aquaporin-1 (AQP-1, cat no: sc-25287), glyceraldehyde-3-phosphate dehydrogenase (cat no: sc-365062), TLR4 (cat no: sc-293072), and anti-rabbit horseradish peroxidase-conjugated secondary antibody (cat no: sc-2357) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against TNF-α (cat no: ab183218), MyD88 (cat no: ab219413), anti-mouse horseradish peroxidase-conjugated secondary antibody (cat no: ab47827) and TRITC- and Alexa Fluor488-conjugated secondary antibodies (cat no: ab150077, cat no: ab7065) were obtained from Abcam (Cambridge, MA, USA). Wilms tumor-1 (WT1) antibody (cat no: NBP2-67587) was from Novus Biologicals (Littleton, CO, USA). Myeloid differentiation factor 2 (MD2, cat no: PA5-20058) antibody was from Invitrogen (Carlsbad, CA, USA). Antibodies against Flag tags (cat no: F2555) and HA tags (cat no: 05-904) were obtained from Sigma (St. Louis, MO, USA). Mouse ELISA kits for IL-1β (cat no: 88-7013-88) and TNF-α (cat no: 88-7324-77) were obtained from eBioscience (San Diego, CA, USA). Urine micro albumin (cat no: E038-1-1), blood urea nitrogen (BUN, cat no: C013-2-1), and creatinine assay (cat no: C011-2-1) kits were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Renal epithelial cell culture and transfection
Rat tubular epithelial cell line NRK-52E (cat no: GNR 8) was obtained from the Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China). NRK-52E cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (cat no: 10567022, Gibco, Eggenstein, Germany) containing 5.5 mM D-glucose, 5% fetal bovine serum, 100 U/mL of penicillin, and 100 mg/mL of streptomycin in 5% CO2 cell culture incubator at 37 °C. Cells were challenged with DMEM medium containing 33 mM of glucose for the HG groups. In LM8-treated cellular experiments, the vehicle DMSO (final concentration, 1%) was used for control.
Silencing of MyD88 in NRK-52E was performed by transfecting cells with siRNA using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). siRNA sequences were obtained from Gene Pharma Co. Ltd. (Shanghai, China). Specific siRNA sequences were 5′-GGAGAUGAUCCGGCAACUATT-3′ and 5′-UAGUUGCCGGAUCAUCUCCTT-3′ for rat MyD88, and 5′-UUCUCCGAACGUGUCACGUTT-3′ and 5′-ACGUGACACGUUCGGAGAATT-3′ for negative control. In total, 5 × 104 cells/well were plated in six-wells and then cultured for 24 h in 5% CO2 cell culture incubator at 37 °C. Target siRNA or non-targeting control siRNA were transfected at a final concentration of 50 nM. Culture medium was replaced with fresh growth medium after 6 h. The effect of knockdown was determined by Western blotting. HA- and FLAG-MyD88 plasmids were constructed and sequenced by Shanghai GeneChem. Co. Ltd. (Shanghai, China). NRK-52E cells were co-transfected with HA- and FLAG-MyD88 plasmids using Lipofectamine 3000 (cat no: L3000008, Invitrogen, Carlsbad, CA, USA).
Animal experiments
All animal care and experimental procedures were approved by the Wenzhou Medical University Animal Policy and Welfare Committee, and all animals were given humane care according to the National Institutes of Health (USA) guidelines. Male C57BL/6 mice weighing 18–20 g were obtained from Animal Center of Wenzhou Medical University. Male db/db mice (BKSdb/db; stock # T002407, n = 21), and male littermate db/m (BKSdb/m, n = 7) mice were purchased from GemPharmatech Co., Ltd. (Nanjing, China). Male mice were widely used in both T1DM and T2DM models [14]. Animals were housed at a constant room temperature with a 12:12 h light–dark cycle and were fed with water and a standard rodent diet (Cat. #MD12031, MediScience Diets Co. Ltd, Yangzhou, China,). The animals were acclimatized to the laboratory for at least 2 weeks before initiating the studies. Experimenters were blinded to all animal genotype and treatment. Treatment groups were assigned in random model.
STZ-induced type 1 diabetic model: 7-week-old male C57BL/6 mice (n = 30) were used to induce type 1 diabetes by intraperitoneal injection of STZ (cat no: S0130, Sigma, 50 mg/kg in citrate buffer, pH 4.5) for five consecutive days, while eight 7-week-old male C57BL/6 mice, as the control group, received the same volume of citrate buffer. The blood glucose level was monitored using glucometer after 4 h of fasting. One week after STZ injection, 24 mice with fasting-blood glucose >16.6 mM were selected and considered as diabetic. The diabetic nephropathy will be developed in another 18 weeks after STZ-induced hyperglycemia. The control mice (n = 8) received the same volume of citrate buffer. All mice were fed with water and standard rodent diet. At 11th week after diabetes was onset, 24 diabetic animals were randomly divided into three groups: T1DM (n = 8), LM8 (5 mg·kg−1 per day)-treated T1DM (T1DM + LM8-5, n = 8), and LM8 (10 mg·kg−1 per day)-treated T1DM (T1DM + LM8-10, n = 8). In the LM8-treated groups, LM8 (5 or 10 mg/kg) was administered as oral gavage once every 2 days for another 8 weeks (from 11th to 19th week), respectively. The T1DM group and age-matched control group (n = 8) received 1% CMC-Na solution alone according to the same schedule. No mice died during the whole experiment. Bodyweight and blood glucose levels were recorded weekly.
db/db type 2 diabetic model: 21 7-week-old male db/db and seven littermates db/m mice was used. The experimental groups were as follows: non-diabetic controls (db/m, n = 7), T2D (db/db, n = 7), 5 mg·kg−1 per day LM8-administered T2D mice (db/db + LM8-5), 10 mg·kg−1 per day LM8-administered T2D mice (db/db + LM8-10). In LM8-administed T2D mice, LM8 (5 or 10 mg/kg) was administered by oral gavage every 2 days for 8 weeks, while the db/db group and db/m group received the same volume of diluent (1% CMC-Na solution) in the same schedule. No mice died during the experiment. Bodyweight and blood glucose levels were recorded weekly.
At 6 h before sacrifice, the spontaneous urine was collected using the previously described method [15]. At the end of treatment (T1DM at 19th weeks and T2DM at 16th weeks, respectively), mice were killed under sodium pentobarbital anesthesia (i.p. injection of 0.2 mL sodium pentobarbital at 100 mg/mL). Blood and renal tissues were collected at the time of sacrifice. The body weight and right kidney weight were recorded. Serum creatinine, urinary albumin, and urinary creatinine were detected using commercial kits to evaluate the nephropathy profile (Nanjing Jiancheng, Nanjing, China).
Kidney tissue staining analysis
The kidney tissues from all mice were fixed in 4% paraformaldehyde for paraffin embedding and histopathological analysis, or were snap-frozen in liquid nitrogen for gene and protein expression analysis. The fixed kidney tissues were sectioned at 5 µm thickness. The sections were stained with Haematoxylin and Eosin for histopathological analysis. The slides were examined under light microscope (×400 amplification; Nikon, Japan). Paraffin sections (5 µm) of the kidney tissues were stained using Masson’s three-color staining kit or sirius red staining kit (Beyotime Biotech, Nantong, China), respectively, according to the manufacturer’s instructions. The slides were examined under light microscope (×400 amplification; Nikon, Japan).
For immunofluorescent double-staining, frozen sections (5 μm) of renal tissues were washed three times with PBS at room temperature and each wash was carried out for 5 min. Slides were blocked using 5% BSA for 30 min and then incubated overnight at 4 °C with both MyD88 antibody (1:200) and WT1 (1:200) or AQP-1 (1:200) antibody, respectively. Slides were then correspondingly incubated with two kinds of secondary antibody (TRITC and Alexa Fluor488 labeled antibody, 1:500) at 37 °C for 1 h, and washed by PBS for four times. DAPI was used to stain nucleus for 10 min and all stained sections were viewed by fluorescence microscopy (Nikon, Japan).
Real-time quantitative PCR
Total RNA from NRK-52E cells and kidney tissues were extracted using TRIzol (cat no: 15596018, Invitrogen, Carlsbad, CA, USA). Reverse transcription and quantitative PCR (RT-qPCR) were performed using M-MLV Platinum RT-qPCR Kit (cat no: 28025013, Invitrogen, Carlsbad, CA, USA). Real-time qPCR was carried out using the Eppendorf Real plex 4 instruments (Eppendorf, Hamburg, Germany). Primers for genes including TNF-α, IL-1β, Col-4, TGF-β, and β-actin were synthesized and obtained from Invitrogen. The primer sequences used are provided in Supplementary Table S1. The relative amount of each gene was detected and normalized to β-actin, using the calculation and comparison methods described in our previous paper [15].
Western blotting and immunoprecipitation
Lysates were prepared from cells and kidney tissues. Protein levels were determined by the Bradford Assay (cat no: 5000201, Bio-Rad, Hercules, CA, USA). Proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electro-transferred to a polyvinylidene fluoride membrane (cat no: 1620177, Bio-Rad, Hercules, CA, USA). Each membrane was preincubated for 1.5 h at room temperature in blocking buffer (5% non-fat milk in Tris-buffered saline containing 0.05% Tween 20, TBS-T). Membranes were then incubated with specific primary antibodies. Immunoreactive bands were detected by incubating with secondary antibodies conjugated with horseradish peroxidase for 1–2 h at room temperature and visualized with enhanced chemiluminescence reagent (Bio-Rad, Hercules, CA, USA).
For immunoprecipitation, cell and tissue extracts were prepared and incubated with anti-TLR4 or anti-HA antibody at 4 °C overnight. Proteins were immunoprecipitated with protein A/G-sepharose beads at room temperature for 1 h. Samples were used for the detection of MyD88 or MD2 as co-precipitated proteins of TLR4, or Flag epitope for MyD88 dimerization. The density of the immunoreactive band was analyzed using ImageJ analysis software version 1.38e and normalized to their respective controls.
Biotin-based pulldown assay
Bio-LM8 was synthesized and structurally characterized with a purity of 98.1%. Bio-LM8 at 1 μM was added to 200 μL pre-blocked streptavidin-agarose beads and incubated at room temperature for 1 h. LM8 and Biotin alone were used as controls. The lysates from mouse kidney tissues were incubated with the beads preloaded with Bio-LM8 streptavidin-agarose at room temperature for 4 h. Finally, 50 μL 1 × SDS loading buffer was added to precipitates the beads and elutes proteins, which then were analyzed using SDS-PAGE immunobloting. Total lysates were used as input control.
Surface plasmon resonance analysis
The binding affinity of LM8 to recombinant MyD88 was determined using ProteOnXPR36 Protein Interaction Array system (Bio-Rad, Hercules, CA, USA) with a ProteOn GLH sensor chip (cat no: 1765013, Bio-Rad, Hercules, CA, USA) using ProteOn Amine Coupling Kit (cat no: 1762410, Bio-Rad, Hercules, CA, USA) as described previously [13]. Briefly, after activated using EDC (40 mM) and NHS (10 mM), the recombinant MyD88 protein dissolved in acetate acid buffer was immobilized on the sensor. Then the LM8 compound at different doses was dissolved in running buffer containing 5% DMSO. Compounds at different concentrations were injected simultaneously at a flow rate of 30 μL/min for 120 s of association phase, followed with 120 s of dissociation phase at 25 °C. Data were analyzed using ProteOn manager software. The KD value was calculated using global fitting of the kinetic data from various concentrations by 1:1 Langmuir binding model.
Statistical analysis
All experiments were randomized and blinded. In all in vitro experiments, data represented at least three independent experiments and expressed as means ± SEM. Statistical analysis was performed with GraphPad Prism 6.0 software (San Diego, CA, USA). One-way ANOVA followed by Dunnett’s post hoc test was used when comparing more than two groups of data and one-way ANOVA, non-parametric Kruskal–Wallis test, followed by Dunn’s post hoc test when comparing multiple independent groups. P values of <0.05 was considered to be statistically significant. Posttests were run only if F achieved P < 0.05 and there was no significant variance in homogeneity.
Results
Hyperglycemia increases MyD88 activity in renal tubular epithelial cells
We firstly examined the expression and activation of MyD88 in diabetic mouse kidney. As shown in Fig. 1a, however, only slight increase in MyD88 protein level was observed in STZ-induced type 1 diabetic mice, compared to the control mice. As an adapter protein of TLRs, MyD88 activation means that MyD88 is recruited by TLRs and forms dimers to transduce signals. To determine whether MyD88 activity is upregulated, we immunoprecipitated kidney lysates with TLR4 antibody and detected associated MyD88 proteins. Our results showed that diabetes increased MyD88-TLR4 complex formation, indicative of increased MyD88 activity (Fig. 1b). Thus, our data indicated that diabetes activated MyD88, rather than increased MyD88 expression in mouse kidneys. Immunofluorescence staining of kidney tissues showed that MyD88 was primarily localized to AQP-1-positive tubular epithelial cells (Fig. 1c). Minimal co-labeling was seen in cells positive for WT1, a marker of podocytes. Our results suggest that renal tubular epithelial cells are the primary source of increased MyD88 in kidney tissues.
Fig. 1. MyD88 is activated in renal tubular epithelial cells in diabetes.

a Diabetes was induced in mice by streptozotocin. Kidney tissues were harvested at 16 weeks following the onset of diabetes. Representative blots show levels of MyD88 in kidney samples. GAPDH was used as loading control. Densitometric quantification was performed by ImageJ (mean ± SEM; n = 5 per group; ns not significant). b Immunoblotting showing MyD88 and TLR4 complex formation in kidney samples from diabetic mice. Samples were immunoprecipitated using TLR4 antibody and MyD88 was detected. Lower panel showing densitometric quantification (mean ± SEM; n = 5; *P < 0.05 compared to non-diabetic control mice). c Immunofluorescence double-staining of mouse kidney tissues for MyD88 (green), aquaporin-1 (AQP-1; red) and Wilms tumor-1 (WT1; red). Tissues were counterstained with DAPI (blue). Representative images shown (n = 5; scale bar = 200 µm). d Rat tubular epithelial cells NRK-52E were exposed to 33 mM glucose (HG) for indicated time periods ranging from 0 to 60 min. Lysates were immunoprecipitated using TLR4 antibody and probed for MyD88. Representative blots were shown (n = 3 independent experiments). e NRK-52E cells were transfected with Flag-MyD88 and HA-MyD88 plasmids. Cells were then exposed to HG for 0–30 min. Lysates were immunoprecipitated with HA antibody and probed for Flag epitope. Representative blots are shown (n = 3 independent experiments).
We exposed NRK-52E renal epithelial cells in culture to high levels of glucose (HG, 33 mM) and examined MyD88 activation. HG exposure of NRK-52E cells increased MyD88 association with TLR4 (Fig. 1d). This complex formation was quite rapid and was seen maximally at 15 min post-HG exposure. Consistent with these data, increased interaction between TLR4 and TLR4 co-receptor MD2 [16] was noted in NRK-52E cells exposed to HG at 5 min (Supplementary Fig. S1a). The MD2-TLR4 and MyD88-TLR4 interactions were not seen with 33 mM mannitol, excluding the involvement of osmotic stress (Supplementary Fig. S1b). We then co-transfected NRK-52E cells with Flag- and HA-MyD88 to determine whether HG increases MyD88 dimerization. As expected, HG induced MyD88 dimerization at 15 min with a further increase at 30 min (Fig. 1e). These results show that diabetes in mice and HG exposure of renal epithelial cells induces MyD88 activation.
MyD88 deficiency attenuates high glucose-induced inflammatory and fibrotic responses in renal epithelial cells
To determine the functional significance of increased MyD88 activity in renal epithelial cells, we silenced the expression of MyD88 (Fig. 2a) and then exposed the NRK-52E cells to HG. Our results show that HG decreases the levels of IκBα, indicating increased NF-κB activity, in control cells but not in cells transfected with MyD88 siRNA (Fig. 2b). These results linked the increased MyD88 activity in cells challenged with HG to downstream signaling through NF-κB. Induction of over-expression of downstream pro-inflammatory cytokines, TNF-α and IL-1β, which have been found to be significantly upregulated in diabetic renal tissues [17, 18], was also examined in HG-challenged NRK-52E cells (Supplementary Fig. S2a, b). Mannitol at the same concentration was used to eliminate the effect of high osmotic pressure. As shown in Fig. 2c, d, HG-induced TNF-α/IL-1β expression was significantly supressed in NRK-52E cells following knockdown of MyD88. As TLR4 has been shown to induce renal fibrogenesis by the modulation of inflammatory cytokines [19], we examined the expression of extracellular matrix protein Col-4 and fibrogenic TGF-β. Both mRNA (Fig. 2e, f) and protein (Fig. 2g) levels of Col-4 and TGF-β were induced by HG. No such induction was noted in cells transfected with MyD88 siRNA. These results suggest that HG activates MyD88 in renal epithelial cells to induce a pro-inflammatory and -fibrotic phenotype.
Fig. 2. MyD88 knockdown prevents HG-induced inflammatory and fibrotic responses in NRK-52E cells.

a NRK-52E cells were transfected with siRNA targeting MyD88 (siMyD88) or negative control siRNA (NC). MyD88 protein levels were detected by immunoblotting. GAPDH was used as loading control (Ctrl non-transfected control cells). b NRK-52E cells transfected with MyD88 siRNA or NC were exposed to 33 mM glucose (HG) for 1 h. Lysates were probed for IκBα levels. GAPDH was used as loading control. Representative blots and densitometric quantification were shown (siMyD88 = MyD88 siRNA transfected cells). c, d NRK-52E cells transfected with MyD88 siRNA were exposed to HG for 8 h. Levels of TNF-α and IL-1β transcripts were measured by real-time qPCR assay. β-Actin was used for normalization. e, f NRK-52E cells with MyD88 knockdown were exposed to HG for 8 h. TGF-β and Col-4 transcript levels were measured by real-time qPCR assay. β-Actin was used for normalization. g NRK-52E cells with MyD88 siRNA were exposed to HG for 24 h. Protein levels of Col-4 and TGF-β were measured by Western blot assay. GAPDH was used as loading control. Densitometric quantification was performed by ImageJ. Mean ± SEM; n = 3 independent experiments; *P < 0.05 compared to NC; #P < 0.05 compared to siMyD88.
LM8 prevents TLR4-MyD88 interaction and supresses HG-induced inflammatory responses in renal epithelial cells
Our next objective was to determine the benefits of pharmacological inhibition of MyD88 by a small-molecule inhibitor LM8 (Fig. 3a). Firstly, we confirmed the binding ability of LM8 with recombinant MyD88 protein using surface plasmon resonance assay as described previously [13]. As shown in Fig. 3b, LM8 exhibited a high binding affinity to MyD88 protein in a dose-dependent manner with a KD value of 15.6 μM. We then examined the interaction of LM8 with MyD88 in kidney tissues using Bio-LM8 (Fig. 3c) and pulldown assays. Fig. 3d showed the successful binding of LM8 with MyD88 protein in mouse kidney tissues. Next, we treated NRK-52E cells with 5 µM LM8 and then exposed the cells to HG. We used LM8 at the dose of 5 µM based on our previous studying showing significant reduction in TNF-α expression in primary macrophages in response to lipopolysaccharide [13]. In addition, LM8 below 40 μM showed no cytotoxicity in NRK-52E cells (Supplementary Fig. S3a). Here, we showed that LM8 treatment of NRK-52E cells reduced HG-induced TLR4-MyD88 complex formation (Fig. 3e) and MyD88 dimerization (Fig. 3f). However, no changes were noted in upstream MD2-TLR4 interaction upon treatment of cells with LM8 (Supplementary Fig. S3b). These results suggested that LM8 specifically prevented MyD88 activation in renal cells. Analysis of downstream mechanisms showed that all three doses of LM8 significantly inhibited HG-induced NF-κB activity as assessed through IκBα levels (Fig. 3g), and expression of TNF-α and IL-1β mRNA (Fig. 3h, i). Similarly, we noted dose-dependent suppression of HG-induced Col-4 and TGF-β at both mRNA and protein levels (Fig. 3j–l). These results mimicked MyD88 silencing. These studies show that LM8 is effective in preventing MyD88 activation and pro-inflammatory and fibrogenic signaling in renal epithelial cells in response to HG.
Fig. 3. LM8 prevents HG-induced MyD88 activation and inflammatory responses in NRK-52E cells.

a Chemical structure of MyD88 inhibitor LM8. b SPR analysis of LM8 to recombinant MyD88 protein. c Chemical structure of Bio-LM8. d Biotin-based pulldown assay shows that LM8 could interact with MyD88 in renal tissues. Lysates from mouse kidney tissues were incubated with the streptavidin-agarose beads preloaded with Bio-LM8. Biotin (Bio) and LM8 were used as controls. Eluent from agarose beads was then examined for MyD88 using Western blot assay. Total renal lysates were used as an input control. e NRK-52E cells were pretreated with 5 µM LM8 for 1 h before exposure to 33 mM glucose (HG) for 15 min. DMSO was used as vehicle control (Ctrl). Lysates were immunoprecipitated with TLR4 antibody and probed for MyD88. Representative blots were shown. f NRK-52E cells transfected with Flag- and HA-MyD88 were pretreated with 5 µM LM8 for 1 h and then exposed to HG for 30 min. Lysates were immunoprecipitated using HA antibody and probed for the Flag epitope. Representative blots showing Flag-MyD88 and HA-MyD88 complex. Representative blots were shown. g NRK-52E cells were pretreated with LM8 at increasing concentrations for 1 h and then exposed to HG for 1 h. Levels of IκBα were determined by Western blot assay. GAPDH was used as loading control. Lower panel shows densitometric quantification. h, i NRK-52E cells were pretreated with LM8 at 2.5, 5.0, or 10.0 µM for 1 h and then exposed to HG for 8 h. Levels of TNF-α and IL-1β transcripts were measured by real-time qPCR. β-Actin was used for normalization. j, k NRK-52E cells were pretreated with LM8 at 2.5, 5.0, or 10.0 µM for 1 h prior to an 8 h HG challenge. TGF-β and Col-4 transcript levels were measured by real-time qPCR. β-Actin was used for normalization. l NRK-52E cells were pretreated with LM8 at 2.5, 5.0, or 10.0 µM for 1 h and then exposed to HG for 24 h. Protein levels of Col-4 and TGF-β were measured by Western blotting. GAPDH was used as loading control. Densitometric quantification was performed by ImageJ. Mean ± SEM; n = 3 independent experiments; *P < 0.05 compared to Ctrl; #P < 0.05 compared to HG.
LM8 treatment prevents inflammatory responses and attenuates DKD in STZ-induced T1DM mice
We then examined the pharmacological effects of LM8 in mouse models with DKD. We treated STZ-induced diabetic mice with LM8 for 8 weeks before sacrifice. Two doses of LM8 (5 and 10 mg/kg) were selected according to our previous studies using LM8 and another structurally similar MyD88 inhibitor LM9 in mouse model with obesity-related cardiomyopathy [20, 21]. In addition, LM8 at 10 mg/kg should be very safe since our toxicity study showed that administration of LM8 at 100 mg/kg for 30 days failed to cause any toxic phenotypes in normal mice. No changes to body weights or plasma glucose levels were noted between untreated diabetic mice and diabetic mice treated with LM8 (Fig. 4a, b). Immunoprecipitate assay using kidney tissue lysates showed that LM8 administration significantly reduced TLR4-MyD88 interaction in diabetic mice (Fig. 4c). Associated with this reduced MyD88 activation by LM8, we observed increased IκBα levels indicating dampened NF-κB activity (Fig. 4d). LM8 treatment of diabetic mice also prevented the induction of TNF-α and IL-1β at both mRNA (Fig. 4e, f) and protein levels (Fig. 4g, h) in kidney tissues. Treatment of mice with LM8 produced results indistinguishable from non-diabetic control mice. Together, these results show that LM8 suppresses chronic inflammatory signaling in diabetic kidney.
Fig. 4. LM8 prevents MyD88 activation and inflammatory responses in STZ-induced diabetic mice.

Animal experiments were performed as described in “Methods” section. Body weights (a) and plasma glucose levels (b) were recorded weekly. Figures show that LM8 treatment does not affect body weight gain or glucose levels in diabetic mice. Data are shown in mean ± SEM. c The representative blots showed the co-precipitation analysis for TLR4-MyD88 complex in kidney samples. Lysates were immunoprecipitated using TLR4 antibody and probed for MyD88. Densitometric quantification is shown in the lower panel. d The representative blots showed IκBα protein levels in mouse kidneys detected by Western blot assay. GAPDH was used as loading control. Densitometric quantification is shown in the lower panel. e, f mRNA levels of TNF-α and IL-1β in the kidney tissues were examined by real-time qPCR assay. β-Actin was used for normalization. g, h TNF-α and IL-1β protein levels in the kidney tissue lysates as detected by ELISA. Ctrl untreated control mice, T1DM STZ-induced diabetic mice, LM8-5 5 mg/kg LM8 treatment, LM8-10 10 mg/kg LM8 treatment; mean ± SEM; n = 8 per group; *P < 0.05 compared to Ctrl; #P < 0.05 compared to T1DM.
We next examined the renal protection of LM8 in type 1 diabetic mice. Increased kidney to body weight ratios were seen in diabetic mice compared to non-diabetic control mice (Fig. 5a). However, this increase was not evident in diabetic mice treated with LM8. Further evidence of potential renoprotective activity of LM8 came from analysis of kidney function biomarkers. Increased serum creatine, BUN, albumin to creatinine ratio levels in diabetic mice were prevented in mice treated with LM8 (Fig. 5b–d). Both doses of LM8 were effective in normalizing the changes seen in diabetic mice. Routine histological analysis through H&E-stained sections showed atypical tubular epithelia, glomerulosclerosis, glomerular vascular tufts, and interstitial expansion in diabetic mice (Fig. 5e). Treatment of diabetic mice with LM8 normalized this diabetes-associated tubulo-glomerular histopathology. Sirius Red staining and Masson’s Trichrome staining showed increased fibrosis and collagen deposition in the interstitial compartment of kidney tissues from diabetic mice, while LM8 reduced the levels of collagen deposition in the kidneys of diabetic mice (Fig. 5e and Supplementary Fig. S4). Immunoblotting and qPCR analysis of Col-4 and TGF-β showed increased levels in kidney tissues from diabetic mice, while LM8 treatment at both 5 and 10 mg/kg effectively prevented the increase in the levels of Col-4 and TGF-β (Fig. 5f–h). These results showed that LM8 potentially preserved kidney function in diabetic mice and prevented matrix expansion. These data demonstrate that LM8 protects kidneys from type 1 diabetes via inhibiting MyD88-mediated inflammation.
Fig. 5. LM8 attenuates renal dysfunction and fibrosis in STZ-induced diabetic mice.

Animal experiments were performed as described in “Methods” section. a Kidney to body weight ratios of mice were recorded. Levels of b serum creatine, c blood urea nitrogen (BUN), and d urine albumin to creatinine ratio were examined using commercial kits. e Representative images for hematoxylin and eosin (H&E; upper), Sirius Red (middle), and Masson’s Trichrome (lower) staining of kidney tissues from mice (scale bar = 20 µm). f, g The mRNA levels of Col-4 and TGF-β in the kidney tissues as determined by real-time qPCR. β-Actin was used for normalization. h Protein levels of Col-4 and TGF-β in kidney tissues of mice were determined by Western blot assay. GAPDH was used as loading control. Densitometric quantification is shown in the lower panel. Ctrl untreated control mice, T1DM STZ-induced diabetic mice, LM8-5 5 mg/kg LM8 treatment, LM8-10 10 mg/kg LM8 treatment; mean ± SEM; n = 8 per group; *P < 0.05 compared to Ctrl; #P < 0.05 compared to T1DM.
LM8 treatment prevents inflammatory responses and attenuates DKD in db/db T2DM mice
To further validate the renoprotective effects of LM8, we extended our investigation of LM8 to T2DM mice. Six-weeks old db/db diabetic and db/m control mice were treated with 5 or 10 mg/kg of LM8 for 8 weeks. Similar to the T1DM mice, LM8 treatment to db/db mice showed no effects on body weight change (Fig. 6a) and blood glucose levels (Fig. 6b). As shown in Fig. 6c, type 2 diabetes also increased MyD88-TLR4 complex level in kidney tissues compared to that in db/m group, while LM8 treatment inhibited MyD88 activation in db/db mouse kidney. Similarly, LM8 administration inhibited IκBα degradation (Fig. 6d) and TNF-α and IL-1β production (Fig. 6e–h) in db/db mouse kidneys. Subsequently, diabetes-increased KW/BW ratio, serum creatine, BUN, and albumin to creatinine ratio in db/db mice were significantly inhibited by LM8 treatment (Fig. 7a–d). H&E staining analysis showed that LM8 attenuated pathological changes in db/db mouse kidney (Fig. 7e). Sirius red and Masson’s trichrome staining showed that LM8 treatment also significantly reduced renal fibrosis in db/db mice (Fig. 7e and Supplementary Fig. S5). LM8-mediated prevention of renal fibrosis was further confirmed by examination of the levels of Col-4 and TGF-β in the kidneys of db/db mice (Fig. 7f–h). However, LM8 did not show good dose-dependent manner in db/db mouse model, which be associated with the pharmacokinetic/pharmacodynamic parameters of LM8. Overall, LM8 showed similar protections against DKD by inhibiting MyD88-mediated inflammation in T2D mouse models.
Fig. 6. LM8 prevents MyD88 activation and inflammatory responses in db/db mice.

Animal experiments were performed as described in “Methods” section. Body weights (a) and plasma glucose levels (b) were recorded weekly. Data are shown in mean ± SD. c The representative blots showed the co-precipitation analysis for TLR4-MyD88 complex in kidney samples. Lysates were immunoprecipitated using TLR4 antibody and probed for MyD88. Densitometric quantification is shown in the lower panel. d The representative blots showed IκBα protein levels in mouse kidneys detected by Western blot assay, with GAPDH as loading control. Densitometric quantification is shown in the lower panel. e, f mRNA levels of TNF-α and IL-1β in the kidney tissues were examined by real-time qPCR assay. β-Actin was used for normalization. g, h TNF-α and IL-1β protein levels in the kidney tissue lysates as detected by ELISA. db/m Control db/m group, db/db db/db T2DM mice, LM8-5 5 mg/kg LM8 treatment, LM8-10 10 mg/kg LM8 treatment; mean ± SEM; n = 7 per group; *P < 0.05 compared to db/m; #P < 0.05 compared to db/db.
Fig. 7. LM8 attenuates renal dysfunction fibrosis in db/db mice.

Animal experiments were performed as described in “Methods” section. a Kidney to body weight ratios of mice were recorded. Levels of b serum creatine, c blood urea nitrogen (BUN), and d urine albumin to creatinine ratio were examined using commercial kits. e Representative images for hematoxylin and eosin (H&E; upper), Sirius Red (middle), and Masson’s Trichrome (lower) staining of kidney tissues from mice (scale bar = 20 µm). f, g The mRNA levels of Col-4 and TGF-β in the kidney tissues as determined by real-time qPCR. β-Actin was used for normalization. h Protein levels of Col-4 and TGF-β in kidney tissues of mice were determined by Western blot assay. GAPDH was used as loading control. Densitometric quantification is shown in the lower panel. db/m Control db/m group, db/db db/db T2DM mice, LM8-5 5 mg/kg LM8 treatment, LM8-10 10 mg/kg LM8 treatment; mean ± SEM; n = 7 per group; *P < 0.05 compared to db/m; #P < 0.05 compared to db/db.
Discussion
Our results show that MyD88 activity, rather than its expression, is upregulated in the kidneys of diabetic mice. MyD88 expression was found to be localized to renal tubular epithelial cells. Exposure of cultured renal epithelial cells to HG increased MyD88 activity, its association with TLR, activation of NF-κB, and induction of downstream pro-inflammatory cytokines and fibrotic factors. Treatment of renal epithelial cells with LM8 or MyD88 siRNA prevented HG-induced MyD88 activation and inflammatory and fibrotic changes. In vivo, treatment with LM8 prevented kidney dysfunction, expansion of extracellular matrix production, and expression of inflammatory factors in both type 1 and type 2 diabetic mice.
Initially, DKD was viewed as a nonimmune disease, however, emerging evidence suggests that inflammatory mechanisms play a central role in disease pathogenesis and progression [22]. Deficiencies in renal function in diabetic patients are positively associated with tubulointerstitial inflammation [23]. One signaling pathway which may orchestrate an inflammatory context in kidneys of diabetic patients is the TLR4 pathway. TLR4 levels have also been shown to be increased in tubular epithelial cells in kidneys of type 1 [24] and type 2 [6] diabetic patients. Experimental models of diabetic complications also show increased TLR4 in kidney tissues [25–27]. Furthermore, TLR4 deficiency reduces diabetes-induced inflammation [27] and renal fibrosis [27, 28]. Similarly, increasing evidence suggest that TLR2 also plays an important role to mediate inflammatory and fibrotic responses in DKD [7]. These studies clearly highlight the importance of TLR2 and TLR4 in DKD. However, we know that TLR4 may signal through multiple nodes and deciphering which signaling arms are involved in diabetes-induced inflammation and fibrosis is important to develop and test interventions. Upon activation, both TLR2 and TLR4 trigger two major downstream signaling pathways, the MyD88-dependent and TRIF-dependent (TIR domain-containing adapter inducing IFN-β) pathways. The MyD88-dependent pathway stimulates NF-κB activation and the expression of pro-inflammatory cytokines, while TRIF-dependent pathway leads to the expression of interferon (IFN)-inducible genes. It is still unknown which pathway (MyD88 or TRIF) contributes more to diabetes-associated renal injury.
A series of compounds that target TLR4 have been investigated for the treatment of sepsis but have failed in preclinical study or in clinical trials [29]. The therapeutic options existing so far to target TLR4 are quite limited. Therefore, MyD88 may emerge as a favorable candidate target for the treatment of inflammatory diseases, since MyD88 mediates the inflammatory signaling transduction of all TLRs except TLR3. However, it is unclear if pharmacological small-molecule inhibitor of MyD88 is effective for the treatment of inflammatory DKD. Lack of good small-molecular inhibitors of MyD88 may be a main reason. Our group designed and developed some effective MyD88 inhibitors, which effectively suppress LPS-induced inflammatory responses in macrophages [13]. LM8 is one of most active compounds. Here, we confirmed that LM8 remarkably suppressed TLR4-MyD88 interaction, MyD88 dimerization, and downstream pro-inflammatory signaling activation in HG-stimulated cells. LM8 also showed selectivity since it failed to affect the upstream MD2/TLR4 complex formation in cells. Importantly, we show that inhibiting MyD88 activity by LM8 is able to prevent activation of NF-κB and downstream signaling responsible for inflammatory cytokines and matrix expansion. This is the first time to show that pharmacological inhibition of MyD88 is able to treat DKD, suggesting that MyD88 could be a good target for the therapy and drug development of diabetic nephropathy. TLRs and MyD88 have a wide range of effect on organs other than kidney. We showed that MyD88 in renal tubular epithelial cells mediated HG-induced inflammation and fibrosis. To completely exclude the effects of MyD88 in other tissues, however, the renal cell-specific MyD88 knockout mice should be used. Therefore, extra-renal mechanism may also contribute to the renal protection of MyD88 inhibition.
The implications of our results are broad and may possibly impact other kidney diseases as well. TLR4 and TLR2 have been shown to promote renal injury in renal ischemia-reperfusion injury [30, 31], endotoxin-induced acute kidney injury [32], and acute allograft rejection [33]. The common hallmarks of these kidney diseases include inflammation and fibrosis. The fibrosis is the final outcome. It is difficult to separate inflammatory and fibrotic responses because renal fibrosis is almost always preceded by the infiltration of inflammatory cells including lymphocytes and monocytes. Infiltrating inflammatory cells generate factors that damage tissues such as reactive oxygen species, fibrogenic cytokines and growth factors [34, 35]. These factors, acting on kidney fibroblasts, mesangial cells, endothelial cells, and tubular epithelial cells cause interstitial accumulation of extracellular matrix proteins [36, 37]. Unfortunately, these changes elicit a self-propagating axis. During progressive/sustained renal fibrosis, increased matrix production and turnover may generate endogenous TLR ligands to continue the inflammatory-fibrotic assault. In addition, cellular damage-associated proteins such as heat shock proteins [38, 39] and HMGB1 may also act as ligands of TLR4 [40]. Therefore, agents blocking TLR4/MyD88 signaling related to the pleotropic transcription factor NF-κB may be proved to be invaluable.
There are a few unanswered questions in our study which warrant future investigations. First, among the TLRs, which ones are involved in DKD and what is their contribution? We know that MyD88 is a common adapter protein for all TLRs except TLR3 [41]. MyD88 knockout mice show a lack of inflammatory cytokine induction in response to TLR4 ligands [42] and TLR2 ligands [43], and a blunted response through TLR5/7/9 [44–46]. It is possible that some of these TLRs were also activated in our experimental model systems and treatment with LM8 inhibited or blunted their respective signaling. On the positive side, if the notion is correct, LM8 may offer broad-spectrum anti-inflammatory activity through inhibiting the common adapter protein. On the cautionary side, however, are studies showing that MyD88 knockout mice are susceptible to a large variety of bacterial pathogens and parasites[47, 48]. Therefore, further validations on the activity and safety of LM8, in experimental DKD models, are needed. The possibility of pharmacological inhibition of MyD88 represents an exciting research area for the treatment of inflammatory diseases.
Conclusion
In summary, we have shown diabetes increases the interaction between MyD88 and TLR to initiate inflammatory injury in the kidneys. A new small-molecule LM8, through targeting MyD88 activation, markedly alleviated the impaired kidney function by reducing renal inflammation in both type 1 and type 2 diabetic mice. Also, similar results were obtained in renal epithelial cells challenged with high levels of glucose in culture. These findings indicate that MyD88 may be a new therapeutic target for diabetes-associated kidney disease, and evaluate the potential of LM8 for the development as the therapeutics for diabetic nephropathy patients.
Supplementary information
Acknowledgements
We thank Dr. Zia A. Khan (Department of Pathology and Laboratory Medicine, Western University, London, ON, Canada) for language editing of this manuscript. This work was supported by the National Key Research Project (2017YFA0506000 to GL), the National Natural Science Foundation of China (81770825 to GL, 820007938 to WL, 81900737 to XH, 81872918 to YLZ, 81803600 to JCQ) and Zhejiang Key Research Project (2021C03044 and 2018C03068 to GL).
Author contributions
GL, WL, and JCQ contributed to the literature search and study design. QYZ, SJX, JCQ, LBY, and PQC carried out the experiments. QYZ, SJX, and WL contributed to data collection and analysis. GL, WL, and YW participated in the drafting of the article. YW, XH, and YLZ revised the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Qiu-yan Zhang, Su-jing Xu, Jian-chang Qian.
Contributor Information
Wu Luo, Email: wuluo@wmu.edu.cn.
Guang Liang, Email: wzmcliangguang@163.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00766-6.
References
- 1.Fineberg D, Jandeleit-Dahm KA, Cooper ME. Diabetic nephropathy: diagnosis and treatment. Nat Rev Endocrinol. 2013;9:713–23. doi: 10.1038/nrendo.2013.184. [DOI] [PubMed] [Google Scholar]
- 2.Groop PH, Thomas MC, Moran JL, Waden J, Thorn LM, Makinen VP, et al. The presence and severity of chronic kidney disease predicts all-cause mortality in type 1 diabetes. Diabetes. 2009;58:1651–8. doi: 10.2337/db08-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Afkarian M, Sachs MC, Kestenbaum B, Hirsch IB, Tuttle KR, Himmelfarb J, et al. Kidney disease and increased mortality risk in type 2 diabetes. J Am Soc Nephrol. 2013;24:302–8. doi: 10.1681/ASN.2012070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim A. Diabetic nephropathy—complications and treatment. Int J Nephrol Renov. 2014;7:361–81. doi: 10.2147/IJNRD.S40172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bermejo S, Pascual J, Soler MJ. The large spectrum of renal disease in diabetic patients. Clin Kidney J. 2017;10:255–6. doi: 10.1093/ckj/sfw137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin M, Yiu WH, Wu HJ, Chan LY, Leung JC, Au WS, et al. Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J Am Soc Nephrol. 2012;23:86–102. doi: 10.1681/ASN.2010111210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wada J, Makino H. Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol. 2016;12:13–26. doi: 10.1038/nrneph.2015.175. [DOI] [PubMed] [Google Scholar]
- 8.Yiu W, Lin M, Tang S. Toll-like receptor activation: from renal inflammation to fibrosis. Kidney Int Suppl. 2014;4:20–5. doi: 10.1038/kisup.2014.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–5. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 10.Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science. 1997;278:1612–5. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 11.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 12.Noulin N, Quesniaux VF, Schnyder-Candrian S, Schnyder B, Maillet I, Robert T, et al. Both hemopoietic and resident cells are required for MyD88-dependent pulmonary inflammatory response to inhaled endotoxin. J Immunol. 2005;175:6861–9. doi: 10.4049/jimmunol.175.10.6861. [DOI] [PubMed] [Google Scholar]
- 13.Chen L, Chen H, Chen P, Zhang W, Wu C, Sun C, et al. Development of 2-amino-4-phenylthiazole analogues to disrupt myeloid differentiation factor 88 and prevent inflammatory responses in acute lung injury. Eur J Med Chem. 2019;161:22–38. doi: 10.1016/j.ejmech.2018.09.068. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Luo W, Han J, Khan ZA, Fang Q, Jin Y, et al. MD2 activation by direct AGE interaction drives inflammatory diabetic cardiomyopathy. Nat Commun. 2020;11:2148. doi: 10.1038/s41467-020-15978-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Fang Q, Jin Y, Liu Z, Zou C, Yu W, et al. Blockade of myeloid differentiation 2 attenuates diabetic nephropathy by reducing activation of the renin-angiotensin system in mouse kidneys. Br J Pharmacol. 2019;176:2642–57. doi: 10.1111/bph.14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–72. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 17.Barutta F, Bruno G, Grimaldi S, Gruden G. Inflammation in diabetic nephropathy: moving toward clinical biomarkers and targets for treatment. Endocrine. 2015;48:730–42. doi: 10.1007/s12020-014-0437-1. [DOI] [PubMed] [Google Scholar]
- 18.Lee SH, Lee TW, Ihm CG, Kim MJ, Woo JT, Chung JH. Genetics of diabetic nephropathy in type 2 DM: candidate gene analysis for the pathogenic role of inflammation. Nephrology (Carlton) 2005;10:S32–36. doi: 10.1111/j.1440-1797.2005.00454.x. [DOI] [PubMed] [Google Scholar]
- 19.Campbell MT, Hile KL, Zhang H, Asanuma H, Vanderbrink BA, Rink RR, et al. Toll-like receptor 4: a novel signaling pathway during renal fibrogenesis. J Surg Res. 2011;168:e61–9. doi: 10.1016/j.jss.2009.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng XY, Sun CC, Liu Q, Lu XY, Fu LL, Liang G, et al. Compound LM9, a novel MyD88 inhibitor, efficiently mitigates inflammatory responses and fibrosis in obesity-induced cardiomyopathy. Acta Pharmacol Sin. 2020;41:1093–101. doi: 10.1038/s41401-020-0410-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu H, Jia W, Tang Y, Zhang W, Qi J, Yan J, et al. Inhibition of MyD88 by LM8 attenuates obesity-induced cardiac injury. J Cardiovasc Pharmacol. 2020;76:63–70. doi: 10.1097/FJC.0000000000000846. [DOI] [PubMed] [Google Scholar]
- 22.Navarro-Gonzalez JF, Mora-Fernandez C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol. 2008;19:433–42. doi: 10.1681/ASN.2007091048. [DOI] [PubMed] [Google Scholar]
- 23.Taft JL, Nolan CJ, Yeung SP, Hewitson TD, Martin FI. Clinical and histological correlations of decline in renal function in diabetic patients with proteinuria. Diabetes. 1994;43:1046–51. doi: 10.2337/diab.43.8.1046. [DOI] [PubMed] [Google Scholar]
- 24.Leemans JC, Butter LM, Pulskens WP, Teske GJ, Claessen N, van der Poll T, et al. The role of Toll-like receptor 2 in inflammation and fibrosis during progressive renal injury. PLoS One. 2009;4:e5704. doi: 10.1371/journal.pone.0005704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuwabara T, Mori K, Mukoyama M, Kasahara M, Yokoi H, Saito Y, et al. Exacerbation of diabetic nephropathy by hyperlipidaemia is mediated by Toll-like receptor 4 in mice. Diabetologia. 2012;55:2256–66. doi: 10.1007/s00125-012-2578-1. [DOI] [PubMed] [Google Scholar]
- 26.Liu P, Li F, Qiu M, He L. Expression and cellular distribution of TLR4, MyD88, and NF-kappaB in diabetic renal tubulointerstitial fibrosis, in vitro and in vivo. Diabetes Res Clin Pr. 2014;105:206–16. doi: 10.1016/j.diabres.2014.04.020. [DOI] [PubMed] [Google Scholar]
- 27.Jialal I, Major AM, Devaraj S. Global Toll-like receptor 4 knockout results in decreased renal inflammation, fibrosis and podocytopathy. J Diabetes Complications. 2014;28:755–61. doi: 10.1016/j.jdiacomp.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 28.Ma J, Chadban SJ, Zhao CY, Chen X, Kwan T, Panchapakesan U, et al. TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PLoS One. 2014;9:e97985. doi: 10.1371/journal.pone.0097985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peri F, Calabrese V. Toll-like receptor 4 (TLR4) modulation by synthetic and natural compounds: an update. J Med Chem. 2014;57:3612–22. doi: 10.1021/jm401006s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–59. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J, John R, Richardson JA, Shelton JM, Zhou XJ, Wang Y, et al. Toll-like receptor 4 regulates early endothelial activation during ischemic acute kidney injury. Kidney Int. 2011;79:288–99. doi: 10.1038/ki.2010.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cunningham PN, Wang Y, Guo R, He G, Quigg RJ. Role of Toll-like receptor 4 in endotoxin-induced acute renal failure. J Immunol. 2004;172:2629–35. doi: 10.4049/jimmunol.172.4.2629. [DOI] [PubMed] [Google Scholar]
- 33.Ducloux D, Deschamps M, Yannaraki M, Ferrand C, Bamoulid J, Saas P, et al. Relevance of Toll-like receptor-4 polymorphisms in renal transplantation. Kidney Int. 2005;67:2454–61. doi: 10.1111/j.1523-1755.2005.00354.x. [DOI] [PubMed] [Google Scholar]
- 34.Vielhauer V, Kulkarni O, Reichel CA, Anders HJ. Targeting the recruitment of monocytes and macrophages in renal disease. Semin Nephrol. 2010;30:318–33. doi: 10.1016/j.semnephrol.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 35.Vernon MA, Mylonas KJ, Hughes J. Macrophages and renal fibrosis. Semin Nephrol. 2010;30:302–17. doi: 10.1016/j.semnephrol.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 36.Farris AB, Colvin RB. Renal interstitial fibrosis: mechanisms and evaluation. Curr Opin Nephrol Hypertens. 2012;21:289–300. doi: 10.1097/MNH.0b013e3283521cfa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–96. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–61. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 39.Vabulas RM, Braedel S, Hilf N, Singh-Jasuja H, Herter S, Ahmad-Nejad P, et al. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem. 2002;277:20847–53. doi: 10.1074/jbc.M200425200. [DOI] [PubMed] [Google Scholar]
- 40.Dikmen M, Canturk Z, Ozturk Y, Tunali Y. Investigation of the apoptotic effect of curcumin in human leukemia HL-60 cells by using flow cytometry. Cancer Biother Radiopharm. 2010;25:7497–55. doi: 10.1089/cbr.2010.0822. [DOI] [PubMed] [Google Scholar]
- 41.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–50. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 42.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–22. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 43.Takeuchi O, Takeda K, Hoshino K, Adachi O, Ogawa T, Akira S. Cellular responses to bacterial cell wall components are mediated through MyD88-dependent signaling cascades. Int Immunol. 2000;12:113–7. doi: 10.1093/intimm/12.1.113. [DOI] [PubMed] [Google Scholar]
- 44.Hacker H, Vabulas RM, Takeuchi O, Hoshino K, Akira S, Wagner H. Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker 88 and tumor necrosis factor receptor-associated factor (TRAF)6. J Exp Med. 2000;192:595–600. doi: 10.1084/jem.192.4.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schnare M, Holt AC, Takeda K, Akira S, Medzhitov R. Recognition of CpG DNA is mediated by signaling pathways dependent on the adaptor protein MyD88. Curr Biol. 2000;10:1139–42. doi: 10.1016/s0960-9822(00)00700-4. [DOI] [PubMed] [Google Scholar]
- 46.Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 47.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–6. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 48.Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, et al. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol. 2002;168:5997–6001. doi: 10.4049/jimmunol.168.12.5997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.