

Abstract
Neuroinflammation plays an important role in neurodegenerative diseases, such as Parkinson’s disease (PD) and Alzheimer’s disease. HACE1 (HECT domain and Ankyrin repeat Containing E3 ubiquitin-protein ligase 1) is a tumor suppressor. Recent evidence suggests that HACE1 may be involved in oxidative stress responses. Due to the critical role of ROS in neuroinflammation, we speculated that HACE1 might participate in neuroinflammation and related neurodegenerative diseases, such as PD. In this study, we investigated the role of HACE1 in neuroinflammation of PD models. We showed that HACE1 knockdown exacerbated LPS-induced neuroinflammation in BV2 microglial cells in vitro through suppressing ubiquitination and degradation of activated Rac1, an NADPH oxidase subunit. Furthermore, we showed that HACE1 exerted vital neuronal protection through increasing Rac1 activity and stability in LPS-treated SH-SY5Y cells, as HACE1 knockdown leading to lower tolerance to LPS challenge. In MPTP-induced acute PD mouse model, HACE1 knockdown exacerbated motor deficits by activating Rac1. Finally, mutant α-synuclein (A53T)-overexpressing mice, a chronic PD mouse model, exhibited age-dependent reduction of HACE1 levels in the midbrain and striatum, implicating that HACE1 participated in PD pathological progression. This study for the first time demonstrates that HACE1 is a negative regulator of neuroinflammation and involved in the PD pathogenesis by regulating Rac1 activity. The data support HACE1 as a potential target for PD and other neurodegenerative diseases.
Keywords: HACE1, Rac1 activity, neuroinflammation, LPS, BV2 microglial cells, MPTP-induced acute PD mouse model, α-synuclein transgenic mice, Parkinson’s disease
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease, characterized by a selective loss of dopaminergic neurons and Lewy body (LB) formation [1]. Though the etiology of PD remains elusive, several hypotheses have been proposed, including genetic and age factors, neuroinflammation, oxidative stress, mitochondrial dysfunction, and so on. Overwhelming evidence indicated that microglia activation occurs in the surrounding of the injury neurons in the substantia nigra (SN), which implicate that neuroinflammation participates in PD etiology [2, 3]. Central nervous system (CNS) inflammatory responses, mediated by activated microglia and astrocytes contribute to PD progressive neuronal degeneration [4, 5]. Microglia, the resident innate immune cell in the CNS, act as the line against inflammation. Astrocytes, the most abundant cell type in the CNS, appear to be more passive than microglia against inflammation, and secrete neurotrophic factors to support neurons [6, 7]. Evidence indicate that the persistent activation of microglia results in neuronal damage or even death [8]. Therefore, the inhibition of microglia activation might be an effective therapeutic strategy for PD future treatment.
Therefore, we focused on neuroinflammation to find new targets for PD treatment. HACE1 (HECT domain and Ankyrin repeat Containing E3 ubiquitin-protein ligase 1), a tumor suppressor, was first identified in human Wilms’ tumors. It is reported to be epigenetically inactivated in several other tumors [9–11]. Recently, it was reported that HACE1 knockdown in tumors leads to increased reactive oxygen species (ROS) level, resulting in excessive production of ROS by Rac1-dependent nicotinamide adenine dinucleotide phosphate (NADPH) oxidases. Moreover, HACE1 could promote the activation of nuclear factor erythroid 2-related factor 2, suggesting that HACE1 might be involved in oxidative stress responses. However, the role of HACE1 in neuroinflammation has not been elucidated. Due to the critical role of ROS in neuroinflammation, we speculated that HACE1 might participate in neuroinflammation and related neurodegenerative diseases, such as PD.
In this study, we attempted to investigate whether HACE1 knockdown confers hypersensitivity to LPS through targeting Rac1 in vitro study. We discovered that HACE1 regulates neuroinflammation by ubiquitylating and degrading Rac1 upon its translocation to the plasma membrane. Moreover, we demonstrate that HACE1 knockdown promotes microglia activation following exposure to LPS, which accelerated tyrosine hydroxylase (TH) neuronal death in vitro. We also confirmed that HACE1 knockdown exacerbates the motor behavioral dysfunction of MPTP-induced PD mice. Furthermore, we found that HACE1 expression decreases in the brain of α-synuclein A53T transgenic PD mice compared to wild type (WT) mice. Finally, this study represents a scientific report that demonstrates the negative regulation of HACE1 in neuroinflammation, and highlights its neuroprotective role in PD.
Materials and methods
Cell cultures
Murine BV2 microglial cells, human HEK 293T cells and human neuroblastoma SH-SY5Y cells were purchased from the culture center at the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College. The cells were grown in 90% Dulbecco’s Modified Eagle’s Medium (Solarbio, Beijing, China) and 10% fetal bovine serum (Gibco, New York, USA). All the cells were kept in the 5% CO2 incubator at 37 °C.
RNA interference
The negative control and HACE1 siRNA (sense: 5’-GCGGAUGUCAACAUUUGUATT-3’; anti-sense: 5’-UACAAAUGUUGACAUCCGCTT-3’) were supplied by Invitrogen (Dublin, Ireland). Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen, Dublin, Ireland) was used to transfect the siRNA (final concentration: 25 nM) and its control in BV2 cells following the manufacturer’s instructions.
Transfection of HACE1 cDNA
The HACE1 cDNA fragment was synthesized by Invitrogen. The pIRES2-HACE1 plasmids were transiently transfected into 293T cells using Lipofectamine 3000 (Invitrogen, Dublin, Ireland) according to the manufacturer’s instructions.
Measurement of inflammatory cytokines
BV2 cells were seeded at a density of 2 × 105 cells/well in a 6-well plate. After 24 h, the cells were treated with HACE1 siRNA and its negative control for 24 h, and then stimulated with 1.0 μg/mL LPS for 6 h. The release of PGE2 and TNF-α was measured by ELISA kits (Enzo Biochem, New York, USA) following the manufacturer’s instructions.
Measurement of ROS production
ROS production was assayed with 2’,7’-dichlorofluorescein diacetate (DCFH-DA) using a fluorescence microplate reader. BV2 cells were seeded in a 24-well plate (2 × 104/well), transfected with siRNA and Lipofectamine RNAiMAX reagent for 24 h. ROS production was induced by LPS for 3 h. Following this steps, DCFH-DA (20 μM) was added for 30 min and the fluorescence was analyzed.
Western blot
RIPA buffer was applied to lyse the samples (Sangon Biotech, Shanghai, China). A membrane and cytosol protein extraction kit (Sangon Biotech, Shanghai, China) was used to extract the cytoplasm and plasma membrane proteins (Applygen, Beijing, China). The proteins were separated by a 12% SDS-polyacrylamide gel and transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were incubated for 12–16 h at 4 °C with the following primary antibodies: rabbit anti-p47phox antibody (1:1000, Cat#BS3261, Bioss, Beijing, China), rabbit anti-p67phox antibody (1:500, sc-15342, Santa Cruz Biotechnology, Dallas, USA), mouse anti-gp91phox antibody (1:500, 611414, BD Transduction Laboratories, San Jose, USA), rabbit anti-p22phox antibody (1:500, sc-20781, Santa Cruz Biotechnology, Dallas, USA), mouse anti-Rac1 antibody (1:1000, Cat#ab129758, Abcam, USA), rabbit anti-β-actin antibody (1:10000, Cat#AC026, ABclonal, Wuhan, China), rabbit anti-JAK2 antibody (1:500, sc-294, Santa Cruz Biotechnology, Dallas, USA), rabbit anti-p-STAT1 antibody (1:1000, Cat#9167, Cell Signaling Technology, Danvers, USA), rabbit anti-STAT1 antibody (1:1000, Cat#ab2415, Abcam, Cambridge, UK), rabbit anti-iNOS antibody (1:1000, Cat#ab15323, Abcam, Cambridge, UK), rabbit anti-IGF-IR antibody (1:1000, Cat#9750, Cell Signaling Technology, USA), rabbit anti-HACE1 antibody (1:1000, Cat#A9593, ABclonal, Wuhan, China), rabbit anti-COX-2 antibody (1:1000, Cat#ab23672, Abcam, Cambridge, UK), rabbit anti-TH antibody (1:1000, Cat#A12756, ABclonal, Wuhan, China) and horseradish peroxidase-conjugated secondary antibodies: anti-mouse IgG (1:2000, Cat#AS071, ABclonal, Wuhan, China) and anti-rabbit IgG (1:2000, Cat#AS014, Abclonal, Wuhan, China) for 2 h. Chemiluminescence was detected with Super ECL Detection Reagent kits (Yeasen, Shanghai, China) and Gel-pro analyzer 3.2 was used to analyze the intensity of band.
Immunoprecipitated experiments
Briefly, BV2 cells were seeded at a density of 8 × 105 cells in 10-cm plates (diameter). Lysis buffer was added to lyse the samples and the proteins’ concentration was adjusted to 2 mg/mL. HACE1 antibody (10 μL) and agarose beads (10 μL) were added to the samples (500 μL), and incubated overnight at 4 °C. The samples were separated by SDS-PAGE, and the level of Rac1 was analyzed by Gel-pro analyzer 3.2.
Measurement of Rac1 activity
For the measurement of Rac1 GTPase activity in BV2 cells, the G-LISA Rac1 colorimetric-based kit (Cytoskeleton, Denver, USA) was used. BV2 cells were incubated with siRNA for 24 h in a 10-cm plate (diameter). LPS (1 μg/mL) was added to the plate for 30 min to activate Rac1. The cells were lysed in lysis buffer for 20 min and centrifuged to obtain soluble cell extracts. Proteins’ concentration was adjusted to 2 mg/mL and 50 μL of proteins (on ice) was incubated in the G-LISA plate for 30 min. The amount of active Rac1 was analyzed by a microplate reader at 490 nm (Bio-Tek uQuant, Hercules, USA).
Rac1 pull-down assay
The Cell Biolabs’s Rac1 Activation Assay Kit (Cytoskeleton, Denver, USA) was applied. The samples were lysed and centrifuged to obtain soluble cell extracts. Proteins’ concentration was corrected to 1 mg/mL and the extracts were incubated with GST-PBD (p21-binding domain of PAK) and rotated at 4 °C for 2 h. The Rac1-GTP was separated by SDS-PAGE and analyzed by Gel-pro analyzer 3.2.
Ubiquitination assay
The BV2 cells were transfected with siRNA for 24 h. Before cells’ harvest, the proteasome inhibitor MG132 (10 μM) was added while Rac1 activation was achieved by LPS for 2 h. All samples were incubated with high-binding affinity resin (Ubiquitinated Protein Enrichment kit, Millipore, Massachusetts, USA) and rotated for 2 h at 4 °C. The resin was then washed four times, and the proteins were separated by SDS-PAGE and immunoblotted for Rac1.
Measurement of the viability of SH-SY5Y cells co-cultured with BV2 cells
The BV2 cells were seeded on top of the Trans-well inserts (Corning Life Sciences, Tewksburry, USA). SiRNA was added into the plates and the inserts containing BV2 cells that were stimulated with LPS, were transferred onto plates containing SH-SY5Y cells for an additional 24 h co-culture. The MTT assay was used to measure the viability of SH-SY5Y cells.
Immunofluorescent staining
The cells were fixed in 4% paraformaldehyde for 15 min and permeabilized with 0.1% Triton X-100 for 5 min. After blocking, the cells were incubated with primary antibodies (Rac1, 1:200; TH, 1:50), and then with secondary antibodies: Rhodamine goat anti-rabbit IgG (1:200, Cat#AS040, ABclonal, Wuhan, China) and FITC goat anti-mouse IgG (1:200, Cat#AS001, Abclonal, Wuhan, China) for 2 h. Following these steps, the cells were counterstained with 4’,6-diamidino-2-phenylindole (DAPI, 2 μg/mL), to reveal the nuclei, and observed by a Carl Zeiss microscope (Jena, Munich, Germany).
Histochemistry staining
Brain sections were incubated with primary antibodies (Iba1, 1:200; TH, 1:2000) and then incubated with an HRP-goat anti-rabbit IgG (H+L) secondary antibody. The stained sections were observed using a light microscope and analyzed using Image-Pro Plus 6.0.
Experimental animals and treatments
Male C57 mice (22–25 g) were supplied by HuaFuKang biotechnology (Beijing, China). C57BL-6J mice overexpressing mutant α-synuclein (A53T) (Lifespan: 16–20 months) and WT mice were constructed and purchased from the Animal Center of the Chinese Academy of Medical Sciences. The mice were maintained in 24 °C environment (12/12 h light/dark) and given ad libitum access to food and drinking water. All experiments were performed in accordance with the guidelines of the Beijing Municipal Ethics Committee for the care and use of laboratory animals. The male C57 mice were anesthetized by 2.5% isoflurane and the lentiviruses HBLV-NC and HBLV-HACE1-shRNA were injected into the SN region (AP: −3.3 mm, ML:+1.2 mm, DV: −4.0 mm from bregma). Seven days later, MPTP (Sigma, Darmstadt, Germany) (30 mg/kg) was intraperitoneally injected into the mice for 7 consecutive days.
Rotarod test
The rotarod test was applied to assess the mice motor skills. The mice were positioned on the rotarod and tested on the revolving rod for up to 120 s at a speed of 35 r/min. The rotarod automatically recorded the time when the mice first fell off the rotarod.
Statistical analysis
The data were expressed as the mean ± SD (n = 3 or 4) or mean ± SEM (n = 15). Results were analyzed by t-test or one-way analysis of variance (ANOVA) followed by Tukey test when the differences between two groups or three and more groups were compared, respectively. A value of P < 0.05 was considered to be statistically different.
Results
HACE1 knockdown aggravated neuroinflammation in BV2 cells
To investigate the role of HACE1 in neuroinflammation, a loss-of-function approach was employed using a specific siHACE1. Following the transfection of siHACE1and its negative control (NC) into the BV2 cells, HACE1 expression was significantly downregulated compared to the NC (Fig. 1a). ROS and key pro-inflammatory cytokines (TNF-α and PGE2) released from BV2 cells were also determined, and the results suggested that siHACE1 treatment exaggerates the production of LPS-induced inflammatory mediators (Fig. 1b–d, Supplementary Fig. S1a, b). Major enzymes, iNOS and COX-2, contribute to inflammation [12, 13]. LPS increased the expression of iNOS and COX-2, and HACE1 knockdown further intensified these effects. It is well-known that stress and pro-inflammatory stimuli rapidly activate the JAK2/STAT1 pathway, which subsequently induces the gene transcription of inflammatory cytokines and chemokines [14, 15]. As shown in Fig. 1e–i, HACE1 knockdown significantly aggravated the LPS-induced-JAK2/STAT1 pathway activation, which might have contributed to the amplification of inflammatory responses. Collectively, HACE1 knockdown led to exaggerated microglial inflammatory responses, which indicates that HACE1 might be a negative regulator of neuroinflammation.
Fig. 1. HACE1 knockdown in BV2 cells exacerbated neuroinflammation.

a The expression of HACE1 following siRNA transfection was detected by Western blot. b The release of TNF-α production was measured by ELISA kit. c The release of PGE2 production was measured by ELISA kit. d The level of ROS was assayed using a fluorescence microplate reader. e–i The levels of expression of iNOS, COX-2, JAK2, p-STAT1 and STAT1 were analyzed by Western blot. The data was presented as mean ± SD, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001.
HACE1 regulated neuroinflammation through Rac1-NADPH oxidase
As HACE1 knockdown-induced exaggerated neuroinflammation, we set out to investigate the mechanisms by which HACE1 regulates microglia neuroinflammation. It has been reported that NADPH oxidase is a major generator of ROS release in microglia [16, 17]. Consequently, we demonstrated whether HACE1 knockdown influences the expression levels of NADPH oxidase subunits, including cytosolic components (p47phox, p67phox and Rac1) and membrane subunits (gp91phox and p22phox), in the LPS-treated BV2 cells. The results showed an equivalent level for each subunit except for Rac1, which was significantly increased in siHACE1-treated BV2 cells compared to the control (Fig. 2a). Previous studies reported that NADPH oxidase activation depends on its cytosolic subunits translocation into the membranes [18]. Therefore, we sought to determine whether HACE1 could influence the activation of NADPH oxidase by measuring the translocation of Rac1 in microglia. To investigate the interaction between HACE1 and Rac1, immune-precipitated experiments were conducted and the results showed that there is indeed an interaction between HACE1 and Rac1 in BV2 cells (Fig. 2b). The results showed that HACE1 knockdown exacerbates LPS-induced translocation of Rac1 from the cytoplasm to the plasma membrane, suggesting that the elevated neuroinflammation in the HACE1-deficient cells might originate from the Rac1-dependent NADPH oxidase pathway (Fig. 2c, d). Rac1, a small GTPase, has been reported to locate in the cytoplasm and plasma membrane, forming an inactive GDP-bound state and an active GTP-bound state, respectively [19]. Pull-down and GLISA assays were used to measure the activity of Rac1. The results showed that HACE1 knockdown induces an increase in Rac1-GTP level (Fig. 2e, f) following LPS stimulation for 30 min. Together, these data indicate that HACE1 knockdown enhances LPS-induced activation of Rac1-NADPH oxidase in microglia, resulting in magnified neuroinflammation.
Fig. 2. HACE1 regulated neuroinflammation through the Rac1-NADPH oxidase pathway.

a Levels of NADPH oxidase subunits were determined using Western blot assay. b IP images of HACE1 and Rac1 expression in BV2 cells. c Rac1 translocation was determined using Western blot assay. d Immunofluorescence and statistical analysis of Rac1 in BV2 cells. e The G-LISA assay was applied to measure Rac1 activity. f The Rac1 pull-down assay was used to assess Rac1 activity. The results were presented as mean ± SD, n = 3. The scale bar represents 25 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
HACE1 promoted the ubiquitination and degradation of Rac1-GTP
Overwhelming evidences indicated that HACE1 exerts its roles through binding to Rac1-GTP and then catalyzing its poly-ubiquitination and degradation [20, 21]. To investigate whether HACE1 affects the degradation of endogenous Rac1 in the process of neuroinflammation, Rac1 level was detected in the presence of cycloheximide (CHX), an inhibitor of protein translation, for indicated times (0, 2, 4 and 8 h). The results revealed that the level of Rac1 was obviously reduced following CHX treatment of NC cells for 8 h. However, no significant change was observed when HACE1 was knocked down. All the results suggested that HACE1 might promote the degradation of Rac1 (Fig. 3a). To further assess Rac1 level following BV2 cells exposure to CHX for 8 h, cytoplasm and plasma membrane were extracted. The results revealed that HACE1-induced degradation of Rac1 was mainly observed in the membranes of NC cells, while HACE1 knockdown induced no change on Rac1 level of expression in the membranes, indicating that HACE1 might promote the degradation of activated Rac1 (Fig. 3b). To confirm that Rac1-GTP decrease was due to its poly-ubiquitination, ubiquitin precipitation experiments were performed. The results showed that HACE1 knockdown reduces Rac1 poly-ubiquitination compared with NC cells (Fig. 3c). The results reveal that HACE1 regulates neuroinflammation through Rac1 poly-ubiquitination and degradation.
Fig. 3. HACE1 promoted the ubiquitination and degradation of Rac1- GTP.

a HACE1 siRNA treated BV2 cells were exposed to CHX (10 μg/mL) for the indicated times and analyzed by Western blot assay. b HACE1 siRNA treated BV2 cells were exposed to CHX (8 h), and the cytoplasm and plasma membranes were extracted and analyzed by Western blot assay. c BV2 cells were transfected with HACE1 siRNA and separated on 12% SDS-PAGE and detected by Western blot assay. d 293T cells were transfected with HACE1-cDNA or empty vector, and Rac1 expression in the cytoplasm and plasma membrane were detected by Western blot. e 293T cells were treated with CHX for the indicated times and analyzed by Western blot assay. f 293T cells transfected with HACE1-cDNA were treated with CHX for 8 h, and Rac1 in the cytoplasm and plasma membrane were detected with an anti-Rac1 antibody by Western blot. g 293T cells transfected with HACE1-cDNA, and analyzed by Western blot assay. Results were expressed as mean ± SD, n = 3. *P < 0.05, **P < 0.01.
To further confirm these results, we examined the degradation of endogenous Rac1 in 293T cells. These cells were transfected with HACE1 cDNA, and the cytoplasmic and membranous levels of Rac1 were detected. The results suggested that HACE1 overexpression significantly decreases Rac1 level in the membrane, revealing that HACE1 tagged Rac1-GTP (Fig. 3d). Meanwhile, the degradation of CHX-exposed Rac1 for indicated times (0, 2, 4 and 8 h), was significantly elevated in HACE1 overexpressing cells (Fig. 3e). Consistent with the above studies, HACE1-induced degradation and poly-ubiquitination of Rac1 were mainly observed in the plasma membranes (Fig. 3f, h). Altogether, the results further confirm that HACE1 tags Rac1 in the plasma membranes for ubiquitination and proteasomal degradation.
HACE1 protected SH‐SY5Y cells from LPS‐mediated neurotoxicity
Neuroinflammation is identified as an important participant in the pathogenesis of PD. Microglial cells serve as innate immune cells in the brain, and their activation under diverse conditions, leads to the development of neuroinflammation. Excessive microglial activation damages the surrounding healthy neural tissues, and the secreted factors by the dead or dying neurons exacerbate microglia activation, causing progressive loss of neurons [22, 23]. Based on the above observations that HACE1 is involved in neuroinflammation, we evaluated the effect of HACE1 in neuroprotection. BV2 cells were treated with HACE1 siRNA prior to LPS stimulation, and the SH-SY5Y cell viability following co-culture with LPS-treated BV2 cells for 24 h was examined using the MTT assay. An increased SH-SY5Y death was observed following co-culture with LPS-treated BV2 cells, indicating that inflammatory cytokines secreted by BV2 cells induced neuro-cytotoxicity (Fig. 4a). HACE1 knockdown led to an increased SH-SY5Y cells death, suggesting that HACE1 might have a vital role in neuroprotection. To further uncover the role of HACE1 in neurons, the dopaminergic maker, TH was analyzed by Western blot and immunofluorescence. The expression of TH was decreased following co-culture with LPS-treated BV2 cells, and the TH level was further decreased in SH-SY5Y cells following HACE1 knockdown (Fig. 4b, c). These findings demonstrate that HACE1 might have an important role in neuroprotection through counteracting neuroinflammation.
Fig. 4. HACE1 protected SH‐SY5Y cells from LPS‐mediated neurotoxicity.

a The viability was analyzed after co-culture with HACE1-deficient BV2 cells with LPS stimulation for 24 h. b Expression of TH in SH-SY5Y cells was detected by Western blot. c TH level in SH-SY5Y cells co-cultured with BV2 cells was detected by immunofluorescence. Results were presented as mean ± SD, n = 3. The scale bar represents 25 μm. *P < 0.05, **P < 0.01, ***P < 0.001.
HACE1 knockdown exacerbated behavior impairments in the MPTP-induced mouse model
To verify whether HACE1 exerts a vital effect on neurons in vivo, lentiviral HBLV-HACE1-shRNA was microinjected into the left side of mice SN region, and the mice were challenged with MPTP one week later (Fig. 5a). As a control group, mice were also microinjected with the lentiviral HBLV-NC. MPTP-treated mice had a worse motion ability compared with control mice, indicating the successful establishment of the PD mouse model (Fig. 5b). Among the MPTP-treated PD mice, the HACE1 knockdown mice spent less time on the rotating rod (Fig. 5b), suggesting that HACE1 knockdown exacerbates the motor behavioral dysfunction of MPTP-induced PD mice. We also found that the expression of HACE1 is significantly decreased (Fig. S2). Immunostaining results showed that HACE1 knockdown led to greater TH positive neuronal death compared with MPTP-induced PD mice (Fig. 5c). Similarly, Western blot results also showed a decreased TH level and increased RAC1 level following HACE1 knockdown and after MPTP administration (Fig. 5d). These results suggested that HACE1 knockdown accelerates neuronal loss in vivo. It is known that MPTP administration can damage neurons and induce microglia activation in vivo. As shown in Fig. 5e, MPTP injection induced the overactivation of microglia in SN. Interestingly, we also found that HACE1 knockdown induces microglia activation in the SN (Fig. 5e). The results implied that HACE1 knockdown induced microglia overactivation accompanying MPTP killed TH positive neurons in vivo. Altogether, the data indicated HACE1 knockdown accelerated neuronal degeneration by revoking microglia overactivation in the MPTP mouse model.
Fig. 5. HACE1 knockdown exacerbated behavior impairments in the MPTP-induced mouse model.

a Experimental procedures of MPTP-induced PD model and lentiviruses’ injection. b Time staying on the rod. c Immunohistochemistry of TH (1: NC, 2: HACE1-shRNA, 3: NC+MPTP, 4: HACE1-shRNA+MPTP). d TH expression in the striatum was assessed by Western blot. e Immunohistochemistry of Iba1 (1: NC, 2: HACE1-shRNA, 3: NC+MPTP, 4: HACE1-shRNA+MPTP). The data were expressed as the mean ± SD (n = 3) or mean ± SEM (n = 15), *P < 0.05, **P < 0.01, ***P < 0.001
HACE1 is reduced in the brain of A53T transgenic mice
Since the MPTP model is an acute PD model, it might be more reasonable to measure HACE1 change in a chronic PD mouse model, such as α-synuclein transgenic mouse model. The C57BL-6J overexpressing mutant α-synuclein (A53T) mouse model is one of the most comprehensive and well-established PD model, which mimics mutations identified in familial forms of PD. It is proposed that neuroinflammation is influenced by aging, therefore, we further monitored HACE1 expression in 6-month-old and 12-month-old A53T transgenic mice, respectively. Results showed that HACE1 expression changed with the increase in age. Notably, HACE1 level was decreased in A53T transgenic mice compared with WT mice at age of 12 months, however, there was no change at the age of 6 months. Furthermore, the decreased HACE1 level correlated with an increased Rac1. We also found that Rac1 level increased in A53T mice compared with WT mice at age of 6-month-old until 12-month-old (Fig. 6a). Meanwhile, 12-month-old A53T transgenic mice exhibited greater microglia activation and DA neuronal loss compared to the 6-month-old A53T transgenic mice (Fig. 6b–e). Together, the findings suggest that a low HACE1 level might contribute to the increase of Rac1 expression in PD, resulting in the activation of neuroinflammatory responses and a selective vulnerability of neurons to α-synuclein-induced toxicity.
Fig. 6. HACE1 is reduced in the brain of A53T transgenic mice.

a HACE1 and Rac1 levels in midbrain and striatum detected by Western blot. b, c Immunohistochemistry of TH. d Immunohistochemistry of Iba1. e Iba1 level in the SN was measured by Western blot. Results were presented as mean ± SD, n = 3 or 4. *P < 0.05, **P< 0.01, ***P< 0.001.
Discussion
In the present study, we demonstrate, for the first time, that HACE1 plays a crucial role in microglia neuroinflammation, mediated by Rac1 ubiquitination and degradation. We found that HACE1 knockdown induces microglia activation and accelerates neuronal death in vitro and in vivo, indicating that HACE1 plays an important effect on neuroprotection. Moreover, HACE1 expression level in the brain of A53T transgenic mice decreased as age increased. This study indicates that HACE1 is a negative regulator of neuroinflammation, and further confirms the neuroprotective role of HACE1 in PD.
Considering the overwhelming evidence that indicates the involvement of neuroinflammation in various neurodegenerative diseases, therapeutic strategies that regulate this process might become an effective disease-modifying therapies [24, 25]. The tumor suppressor HACE1 is involved in protecting against ROS-induced tumors [9, 26, 27]. Since ROS is an important mediator of neuroinflammation [28, 29], we assumed that HACE1 might be participating in neuroinflammation and related diseases, such as AD and PD. Based on this, BV2 cells were used to study HACE1 role in neuroinflammation. In the current study, we found that HACE1 knockdown in BV2 cells results in a magnified neuroinflammation. A further mechanistic study revealed that following LPS stimulation, HACE1 knockdown accelerates the activation of JAK2-STAT1 pathway and induces a higher expression of inflammatory protein and release of various pro-inflammatory cytokines. These data revealed that HACE1 might be involved in neuroinflammation as a negative regulator.
As a result of these findings, it is interesting to investigate the mechanism how HACE1 regulates neuroinflammation. It is well known that NADPH oxidase is solely dedicated to ROS production [30, 31]. In this study, we demonstrated that HACE1 knockdown solely induces the increase of Rac1 subunit in BV2 microglia, and further investigated that how HACE1 influences Rac1. HACE1 tags activated Rac1 for proteasomal degradation [32, 33]. Rac1 has been reported to form an inactive GDP-bound state and an active GTP-bound state in different conditions [19, 34]. In the present study, HACE1 knockdown in BV2 cells leads to Rac1-GTP increased expression level and stability, indicating the essential role of endogenous HACE1 in promoting Rac1 ubiquitination and degradation in vitro. Furthermore, we confirmed that HACE1 overexpression promotes Rac1 ubiquitination and degradation in the plasma membrane of 293T cells. Taken together, our data suggested that HACE1 induces Rac1 ubiquitination and its degradation, thereby regulating neuroinflammation.
Evidence has indicated that continuously activated microglia releasing pro-inflammatory factors leads to the exacerbation of neuronal injury and loss [35, 36]. Therefore, a co-culture system of SH-SY5Y and BV2 cells [37] was used to assess whether HACE1 was involved in neuroprotection. The results suggested that HACE1 could protect SH-SY5Y cells from cytotoxicity induced by LPS-treated BV2 cells. The findings suggested that HACE1 exerts a neuroprotective effect against inflammation‐induced neurotoxicity. Therefore, it might be of interest to know whether HACE1 plays a neuroprotective role in vivo.
HACE1-shRNA lentivirus was microinjected in the left side of the SN region to examine the role of HACE1 in vivo. Among the MPTP-treated PD mice, the HACE1 knockdown mice had a worse performance score than the PD mice. Moreover, the findings revealed that the knockdown of HACE1 exacerbated neuronal death in MPTP-induced PD mouse model. In line with the in vitro findings, HACE1 knockdown induced excessive activation of microglia. The results revealed that HACE1 suppression accelerated MPTP-induced DA neuronal death through activating microglia. Taken together, the findings indicated that HACE1 knockdown in vivo accelerates DA neuronal death by suppression of excessive activation of microglia.
Since the MPTP model is an acute model of neuronal death [38], we investigated HACE1 long-term dynamics in a chronic PD mouse model. In the present study, we chose the A53T transgenic model (6-month-old and 12-month-old A53T transgenic mice) to investigate the effect of HACE1 in the brain. In the midbrain and striatum, Rac1 was obviously elevated in the 6-month-old A53T mice compared to WT mice, however, there was no change in HACE1 expression. Interestingly, HACE1 expression was remarkably reduced in the 12-month-old A53T mice compared to WT mice. This reduction in HACE1 expression was accompanied with an elevated Rac1 expression. These results gave us a hint that genetic or environmental insults might reduce HACE1 expression, magnify neuroinflammation, and thus promote brains likelihood to be affected by insults during PD. Therefore, it might be meaningful to uncover whether HACE1 overexpression facilitates the resolution of inflammation and protects neurons.
In conclusion, HACE1, a tumor suppressor, is participating neuroinflammatory response through regulating Rac1 activity (Fig. 7). The results supported that HACE1 might be a potential target for PD and other neurodegenerative diseases.
Fig. 7. Schematic diagram of HACE1 role in regulating the neuroinflammatory responses of BV2 microglial cells.
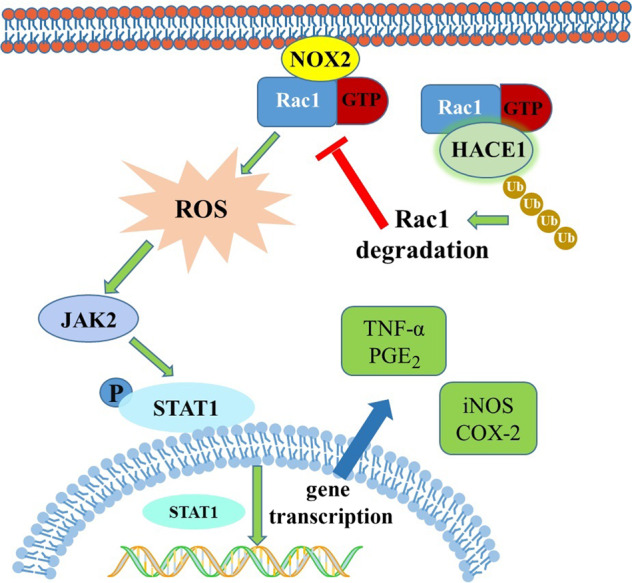
HACE1 targets Rac1 for proteasomal degradation, thus regulating neuroinflammation. In the absence of HACE1, the activity of Rac1 significantly increases, leading to a higher level of ROS and the activation of the JAK2-STAT1 signaling pathway.
Supplementary information
Acknowledgements
The study was supported by grants from Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (No. 2016-I2M-3-011), Chinese Academy of Medical Sciences Fundamental Research Funds for the Central Universities (No. 2018RC350002), National Natural Science Foundation of China (No. 81630097, 81773718, 21772235).
Author contributions
CXZ and DZ designed the study and drafted the manuscript. CXZ, LW, HYY, JMS carried out behavioral tests, immunohistochemistry and Western blot assay. HL, ZHZ, CJ, FYY and FYL participated in the Western blot assay. XQB revised the manuscript.
Competing interests
The authors declare no competing interests.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00778-2.
References
- 1.Samii A, Nutt JG, Ransom BR. Parkinson’s disease. Lancet. 2004;363:1783–93. doi: 10.1016/S0140-6736(04)16305-8. [DOI] [PubMed] [Google Scholar]
- 2.Ha D, Stone DK, Mosley RL, Gendelman HE. Immunization strategies for Parkinson’s disease. Parkinsonism Relat D. 2012;18:S218–S221. doi: 10.1016/S1353-8020(11)70067-0. [DOI] [PubMed] [Google Scholar]
- 3.Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–97. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 4.Damier P, Hirsch EC, Zhang P, Agid Y, Javoy-Agid F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience. 1993;52:1–6. doi: 10.1016/0306-4522(93)90175-F. [DOI] [PubMed] [Google Scholar]
- 5.Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, et al. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol. 2005;57:168–75. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- 6.Linnerbauer M, Wheeler MA, Quintana FJ. Astrocyte crosstalk in CNS inflammation. Neuron. 2020;108:608–22. doi: 10.1016/j.neuron.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang QQ, Zhou JW. Neuroinflammation in the central nervous system: symphony of glial cells. Glia. 2019;67:1017–35. doi: 10.1002/glia.23571. [DOI] [PubMed] [Google Scholar]
- 8.Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003;106:518–26. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
- 9.Cetinbas N, Daugaard M, Mullen AR, Hajee S, Rotblat B, Lopez A, et al. Loss of the tumor suppressor Hace1 leads to ROS-dependent glutamine addiction. Oncogene. 2015;34:4005–10. doi: 10.1038/onc.2014.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daugaard M, Nitsch R, Razaghi B, McDonald L, Jarrar A, Torrino S, et al. Hace1 controls ROS generation of vertebrate Rac1-dependent NADPH oxidase complexes. Nat Commun. 2013;4:2180. doi: 10.1038/ncomms3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Razaghi B, Steele SL, Prykhozhij SV, Stoyek MR, Hill JA, Cooper MD, et al. Hace1 influences zebrafish cardiac development via ROS-dependent mechanisms. Dev Dyn. 2018;247:289–303. doi: 10.1002/dvdy.24600. [DOI] [PubMed] [Google Scholar]
- 12.Chiurchiù V, Maccarrone M. Chronic inflammatory disorders and their redox control: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2011;15:2605–41. doi: 10.1089/ars.2010.3547. [DOI] [PubMed] [Google Scholar]
- 13.Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol. 2012;84:581–90. doi: 10.1016/j.bcp.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 14.Yan ZQ, Gibson SA, Buckley JA, Qin HW, Benveniste EN. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin Immunol. 2018;189:4–13. doi: 10.1016/j.clim.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin HW, Buckley JA, Li XR, Liu YD, Fox TH, Meares GP, et al. Inhibition of the JAK/STAT pathway protects against α-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J Neurosci. 2016;36:5144–59. doi: 10.1523/JNEUROSCI.4658-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yauger YJ, Bermudez S, Moritz KE, Glaser E, Stoica B, Byrnes KR. Iron accentuated reactive oxygen species release by NADPH oxidase in activated microglia contributes to oxidative stress in vitro. J Neuroinflammation. 2019;16:41. doi: 10.1186/s12974-019-1430-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin LY, Liu YX, Wang TG, Wei SJ, Block ML, Wilson B, et al. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–21. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- 18.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 19.Bos JL, Rehmann H, Wittinghofe A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–77. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 20.Goka ET, Lippman ME. Loss of the E3 ubiquitin ligase HACE1 results in enhanced Rac1 signaling contributing to breast cancer progression. Oncogene. 2015;34:5395–405. doi: 10.1038/onc.2014.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mettouchi A, Lemichez E. Ubiquitylation of active Rac1 by the E3 ubiquitin-ligase HACE1. Small GTPases. 2012;3:102–6. doi: 10.4161/sgtp.19221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hurley LL, Tizabi Y. Neuroinflammation, neurodegeneration, and depression. Neurotox Res. 2013;23:131–44. doi: 10.1007/s12640-012-9348-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kempuraj D, Thangavel R, Selvakumar GP, Zaheer S, Ahmed ME, Raikwar SP, et al. Brain and peripheral atypical inflammatory mediators potentiate neuroinflammation and neurodegeneration. Front Cell Neurosci. 2017;11:216. doi: 10.3389/fncel.2017.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perry VH, Teeling J. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol. 2013;35:601–12. doi: 10.1007/s00281-013-0382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schain M, Kreisl WC. Neuroinflammation in neurodegenerative disorders-a review. Curr Neurol Neurosci Rep. 2017;17:25. doi: 10.1007/s11910-017-0733-2. [DOI] [PubMed] [Google Scholar]
- 26.Cetinbas N, Daugaard M, Mullen AR, Hajee S, Rotblat B, Lopez A, et al. Loss of the tumor suppressor Hace1 leads to ROS-dependent glutamine addiction. Oncogene. 2015;34:4005–10. doi: 10.1038/onc.2014.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar B, Roy A, Asha K, Walia NS, Ansari MA, Chandran B. HACE1, an E3 ubiquitin protein ligase, mitigates Kaposi’s Sarcoma-associated herpesvirus infection-induced oxidative stress by promoting Nrf2 activity. J Virol. 2019;93:e01812–01818. doi: 10.1128/JVI.01812-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milton VJ, Sweeney ST. Oxidative stress in synapse development and function. Dev Neurobiol. 2012;72:100–10. doi: 10.1002/dneu.20957. [DOI] [PubMed] [Google Scholar]
- 29.Bischoff LJM, Kuijper IA, Schimming JP, Wolters L, Braak BT, Langenberg JP, et al. A systematic analysis of Nrf2 pathway activation dynamics during repeated xenobiotic exposure. Arch Toxicol. 2019;93:435–51. doi: 10.1007/s00204-018-2353-2. [DOI] [PubMed] [Google Scholar]
- 30.Lam GY, Huang J, Brumell JH. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin Immunopathol. 2010;32:415–30. doi: 10.1007/s00281-010-0221-0. [DOI] [PubMed] [Google Scholar]
- 31.Fernandez-Marcos PJ, Nóbrega-Pereira S. NADPH: new oxygen for the ROS theory of aging. Oncotarget. 2016;7:50814–5. doi: 10.18632/oncotarget.10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang LY, Chen X, Sharma P, Moon M, Sheftel AD, Dawood F, et al. HACE1-dependent protein degradation provides cardiac protection in response to haemodynamic stress. Nat Commun. 2014;5:3430. doi: 10.1038/ncomms4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Acosta MI, Urbach S, Doye A, Ng YW, Boudeau J, Mettouchi A, et al. Group-I PAKs-mediated phosphorylation of HACE1 at serine 385 regulates its oligomerization state and Rac1 ubiquitination. Sci Rep. 2018;8:1410. doi: 10.1038/s41598-018-19471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rathinam R, Berrier A, Alahari SK. Role of Rho GTPases and their regulators in cancer progression. Front Biosci. 2011;16:2561–71. doi: 10.2741/3872. [DOI] [PubMed] [Google Scholar]
- 35.Kempuraj D, Thangavel R, Natteru PA, Selvakumar GP, Saeed D, Zahoor H, et al. Neuroinflammation induces neurodegeneration. J Neurol Neurosurg Spine. 2016;1:1003. [PMC free article] [PubMed] [Google Scholar]
- 36.Tansey MG, Goldberg MS. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–8. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lawrimore CJ, Coleman LG, Zou J, Crews FT. Ethanol induction of innate immune signals across BV2 microglia and SH-SY5Y neuroblastoma involves induction of IL-4 and IL-13. Brain Sci. 2019;9:228. doi: 10.3390/brainsci9090228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park J, Lim CS, Seo H, Park CA, Zhuo M, Kaang BK, et al. Pain perception in acute model mice of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Mol Pain. 2015;11:28. doi: 10.1186/s12990-015-0026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.