

Abstract
Improving CAR-T cell therapy for solid tumors requires a better understanding of CAR design and cellular composition. Here, we compared second-generation (BBζ and 28ζ) with third-generation (28BBζ) carbonic anhydrase IX (CAIX)-targeted CAR constructs and investigated the antitumor effect of CAR-T cells with different CD4/CD8 proportions in vitro and in vivo. The results demonstrated that BBζ exhibited superior efficacy compared with 28ζ and 28BBζ CAR-T cells in a clear-cell renal cell carcinoma (ccRCC) skrc-59 cell bearing NSG-SGM3 mouse model. The mice treated with a single dose of BBζ CD4/CD8 mixture (CAR4/8) showed complete tumor remission and remained tumor-free 72 days after CAR-T cells infusion. In the other CAR-T and control groups, tumor-infiltrating T cells were recovered and profiled. We found that BBζ CAR8 cells upregulated expression of major histocompatibility complex (MHC) class II and cytotoxicity-associated genes, while downregulating inhibitory immune checkpoint receptor genes and diminishing differentiation of regulatory T cells (Treg cells), leading to excellent therapeutic efficacy in vivo. Increased memory phenotype, elevated tumor infiltration, and decreased exhaustion genes were observed in the CD4/8 untransduced T (UNT) cells compared with CD8 alone, indicating that CD4/8 would be the favored cellular composition for CAR-T cell therapy with long-term persistence. In summary, these findings support that BBζ CAR4/8 cells are a highly potent, clinically translatable cell therapy for ccRCC.
Keywords: chimeric antigen receptor T, CAR-T, clear cell renal cell carcinoma, ccRCC, carbonic anhydrase IX, CAIX, ccRCC orthotopic NSG-SGM3 mouse model, 4-1BB (CD137), single-cell RNA sequencing, scRNA-seq
Graphical abstract
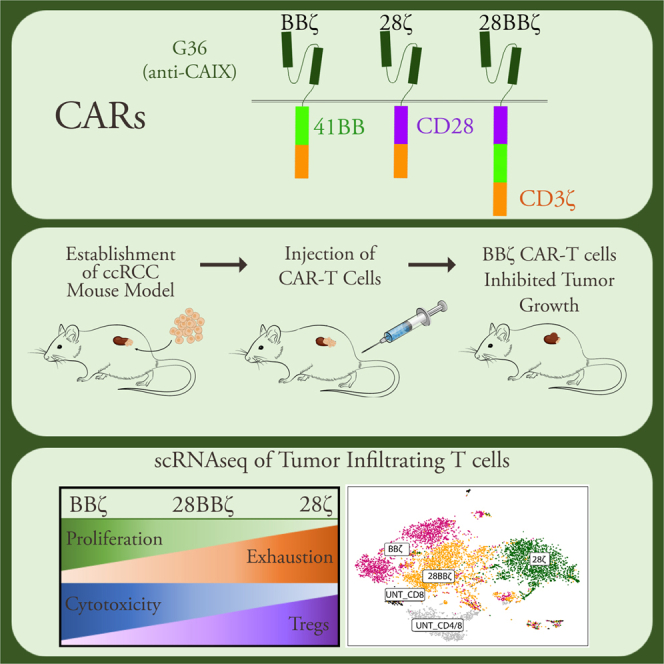
CAIX-targeted BBζ CAR8 showed superior efficacy against ccRCC in vivo compared with 28ζ and 28BBζ CAR8, and scRNA-seq revealed enhanced function, diminished exhaustion, and decreased Treg cell differentiation. BBζ CAR4/8 has even more superior efficacy and long-term immune surveillance and is a highly potent, clinically translatable cell therapy for ccRCC.
Introduction
CAR-T cells are a new type of “living drug,” in which the CAR contains a single-chain variable antibody fragment (scFv) linked to an intracellular signaling block that includes the CD3ζ activation domain (first generation), CD3ζ with CD28 or 4-1BB (second generation), or CD3ζ with both costimulatory domains (third generation).1 CAR-T cells have proven to be a powerful, clinically translatable immunotherapy for hematologic malignancies.2, 3, 4 Based on the promising clinical trial data, in 2017, the US Food and Drug Administration (FDA) approved the first CAR-T cell therapy, tisagenlecleucel (Kymriah), for treatment of certain children and young adults with a form of acute lymphoblastic leukemia (ALL)5,6 and the second CAR-T cell therapy, axicabtagene ciloleucel (Yescarta), for patients with large-B-cell lymphomas who had received at least two prior treatment regiments.7,8 So far, there are five CAR-T products approved by the FDA, four CD19-targeted CAR-T cells9,10 and one targeting B-cell maturation antigen (BCMA).11
The focus of CAR designs has shifted to incorporation of intracellular signaling domains from T cell costimulatory molecules to overcome problems with inefficient effector function and anergic status of antitumor CAR-T cells. The most frequently used domains are derived from the CD28 family (CD28 and ICOS) or the tumor necrosis factor receptor (TNFR) family (4-1BB, CD27, and OX40).12 Incorporation of CD28-signaling domain into CARs generates CAR-T cells with superior antitumor activities, including tumor-induced proliferation, cytotoxicity, cytokine secretion, clonal expansion, and tumor regression. CAR-T cells that utilize the 4-1BB signaling domain have slower onset of antitumor activity but have the advantage of providing durable activity in vivo due to resistance of anergy induction.13,14 In recent clinical trials, CD28-encoded CARs targeting CD19 have shown promising results in chronic lymphocytic leukemia (CLL), indolent B-cell lymphoma, and ALL.3,15 Similarly, complete remission and long-term survival of effector memory T cells have been observed in patients with advanced chemotherapy-resistant CLL who had received adoptive immunotherapy using the 4-1BB CAR targeting CD19, illustrating successful translation of such a therapeutic approach.16
Wang et al.17 reported that CD4 CAR-T cells were considered to be a highly potent and clinically important T cell subset for effective CAR therapy, which was validated in their orthotopic glioblastoma (GBM) mouse model by using GBM-associated antigen interleukin-13 receptor a2 (IL-13Ra2)-targeted CAR-T cells with different compositions of CD4 and CD8 subsets. The Fry team has shown that CD4 CAR-T cells alone can be cytolytic and equally potent as CD8 CAR-T cells at eradicating leukemia in vivo and may be superior due to better persistence and reduced susceptibility toward exhaustion, thereby preventing relapse.18
The results from the Riddell and Maloney teams suggested that infusion of CD8 CAR-T cells combined with CD4 CAR-T cells might provide optimal antitumor efficacy.19,20 Sheih et al.21 studied clonal kinetics and transcriptional programs that regulate the fate of anti-CD19 CD8 CAR-T cells isolated from the infusion product and from blood of patients treated with CD19-targeted CAR-T cell immunotherapy via T cell receptor β (TCRB) sequencing, integration site analysis, and single-cell RNA sequencing (scRNA-seq). The results demonstrated that expanded clones after infusion mainly originate from infused clusters with higher expression of cytotoxicity and proliferation genes.
Long et al.22 and Boroughs et al.23 performed transcriptional analysis on ex vivo cultured CD19-targeted CAR-T cells with different costimulatory domains via transcriptome sequencing, which revealed the transcriptional signatures of CARs resulting from different costimulatory domains. However, the transcriptome of solid-tumor-infiltrating CAR-T cells has not been well studied but may uncover novel correlations between functional activity and transcriptional signatures of CAR-T cells. Here, we evaluated the killing capacity of BBζ, 28ζ, and 28BBζ CAIX-targeted CAR-T cells with different cellular composition in a ccRCC orthotopic NSG-SGM3 mouse model. Our results showed that anti-CAIX BBζ CAR-T cells with a CD4/CD8 ratio of 2:1, CAR4/8, resulted in complete tumor regression and maintained immune surveillance. For other CAR-T and control groups, profiling transcriptional signatures of tumor-infiltrating CAR-T cells revealed that 4-1BB signaling led to elevated expression of pro-inflammatory genes and downregulated expression of negative immune modulator genes, as well as less differentiation of BBζ CAR8 into CD25+FOXP3+ CD8 Treg cells.
Results
Generation of anti-CAIX second-generation and third-generation CAR-T cells
Previously, we constructed a first-generation CD3ζ24 and a second-generation 28ζ CAR25 targeting CAIX. CAIX is highly expressed in ccRCC and hence used as a therapeutic target for patients with ccRCC.26,27 To enhance the therapeutic effect of anti-CAIX CAR-T cells and translate the cellular therapy to solid tumor treatment, we built a second-generation CAR with an anti-CAIX G36 scFv followed by a 4-1BB costimulatory domain and CD3ζ activation signal (BBζ) and a third-generation CAR G36-CD28-4-1BB (28BBζ) with both CD28 and 4-1BB modules. All of these CAR constructs have an IRES domain after the CAR moiety to express the ZsGreen fluorophore. We excluded the first-generation CARs because of their well-known poor persistence in vivo and compared two second-generation (28ζ and BBζ) CARs with the third-generation CAR (28BBζ) in vivo (Figure 1A). CD4 and CD8 T cells were isolated from healthy-donor peripheral blood mononuclear cells (PBMCs) and transduced separately using the same multiplicity of infection (MOI). CAR-T cells were purified by flow sorting for in vitro and in vivo evaluation.
Figure 1.

Evaluation of anti-CAIX BBζ CAR-T cells in vitro
(A) Designs of anti-CAIX BBζ, 28ζ, and 28BBζ CAR-T cells. The same anti-CAIX G36 scFv was used as CAR moiety followed by CD8 hinge, costimulatory domain (s), and activation cassette CD3ζ. ZsGreen was introduced after IRES. (B) Cytotoxicity of anti-CAIX BBζ CAR-T cells with different CD4:CD8 composition ratios. From left to right, the cytotoxicity of CD4, CD4:CD8 = 2:1, CD4:CD8 = 1:1, CD4:CD8 = 1:2, and CD8 anti-CAIX BBζ CAR-T against CAIX-expressing skrc-59 cells is shown. P values are defined by unpaired two-tailed t-tests ( ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and ∗∗∗∗p < 0.0001). (C) Heatmap of log2 fold change cytokine and chemokine profiles in supernatant. After 48 h co-culture of skrc-59 tumor cells and CAR-T cells with different CD4:CD8 composition ratios, the media were assessed for cytokine and chemokine release. The fold change was defined by the value of experimental group divided by the one of UNT negative control. All values of fold change are labeled in the heatmap.
Assessment of anti-CAIX BBζ with different CD4/8 ratios in vitro
We utilized anti-CAIX G36 BBζ CAR-T cells to optimize the CD4:CD8 cellular ratio to be used in the in vivo experiment. We observed that CD4 T cells in general had a higher transduction efficiency compared with CD8 T cells among three CAR constructs, 28ζ, BBζ, and 28BBζ (Figure S1). The antitumor efficacy of BBζ CAR-T cells with pure CD4, CD4:CD8 ratio of 2:1, CD4:CD8 ratio of 1:1, CD4:CD8 ratio of 1:2, and pure CD8, was evaluated using image cytometry on mCardinal-expressing CAIX+ ccRCC patient-derived cell line skrc-59 as described previously.28,29
With effector to target cell (E:T) ratio of 2:1, CD8-only CAR-T cells exhibited the strongest killing capacity and pure CD4 CAR-T cells showed modest cytotoxicity in a 48 h in vitro co-incubation (Figure 1B). With increasing proportions of CD8 in the CD4/8 CAR-T cell mixture, killing of CAIX+ skrc-59 cells was significantly elevated, demonstrating the positive correlation of the CD8 proportion in the CAR-T cell mixture with CAR-T cytotoxicity. We analyzed the pattern of cytokine release at 48 h via Luminex (Figure 1C). A greater than 8-fold change in the BBζ CAR-T cell cultures compared with the untransduced T cells (UNT) was seen for IL-2, granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon γ (IFN-γ), TNF, and MIP-1a, while lesser fold changes were seen with other cytokines. We also demonstrated that the CD4 CAR-T cells exhibited higher levels of cytokine release compared with CD8, and cytokine release was positively correlated with the proportion of CD4. To balance cytolytic CD8 T cell killing and cytokine-induced killing, CD8 only (CAR8) and a CD4:CD8 ratio of 2:1 (CAR4/8) BBζ CAR-T cells were selected to be evaluated in vivo.
Anti-CAIX BBζ CAR8 and CAR4/8 showed superior efficacy in ccRCC orthotopic mouse model
An orthotopic ccRCC NSG-SGM3 mouse model, bearing luciferized skrc-59 CAIX+ cells under the left kidney capsule, was established and used to compare two second-generation (28ζ and BBζ) with a third-generation CAR (28BBζ) and investigate the impact of cellular composition on antitumor efficacy (Figure 2A). Ten groups were tested in this model, including CD8 only of 28ζ, BBζ, 28BBζ, and UNT and CD4/8 of BBζ and UNT at different doses (Figure S2). One week after implantation, tumor engraftment was confirmed by bioluminescence imaging (BLI) and followed by randomized grouping (n = 5 per group). One million, three million, or ten million human CAR-T or UNT cells were injected through the tail vein (day 0). Tumor growth was monitored by BLI weekly and magnetic resonance imaging (MRI) biweekly. CAR-T cell expansion and persistence were evaluated by assessing peripheral blood via flow cytometry weekly.
Figure 2.

Anti-CAIX BBζ CAR4/8 T cells had superior activity in ccRCC orthotopic NSG-SGM3 mouse model
(A) Timeline of the experiment. A week after tumor engraftment, BLI was performed to confirm tumor growth followed by CAR-T cell injection (day 0). BLI and bleeding were performed weekly to monitor tumor growth and CAR-T persistence. MRI was performed biweekly to monitor tumor growth and metastasis. (B) Tumor growth curve of the mice treated with 3 million of anti-CAIX BBζ (pink curve), 28ζ (green curve), and 28BBζ (orange curve) CAR-T cells and UNT (black curve) cells. n.s., not significant. P values are defined by ANOVA ( ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and ∗∗∗∗p < 0.0001). (C) Tumor weight of anti-CAIX BBζ CAR-T cells (pink circle) compared with 28ζ (green circle) and 28BBζ (orange circle) CAR-T cells and UNT (black curve). P values are defined by unpaired two-tailed t-tests ( ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and ∗∗∗∗p < 0.0001). (D) Tumor growth curve of the mice treated with 3 million of anti-CAIX BBζ CAR8 (pink curve), BBζ CAR4/8 (blue curve), UNT CD8 (black curve), and UNT CD4/8 (gray curve) cells. P values are defined by ANOVA ( ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and ∗∗∗∗p < 0.0001). (E) Tumor weight of CAR8 (pink curve) and CAR4/8 (blue curve) of BBζ CAR-T cells compared with the CD8 (black curve) and CD4/8 (gray curve) of UNT cells. P values are defined by unpaired two-tailed t-tests ( ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and ∗∗∗∗p < 0.0001). Note that the endpoint of BBζ CAR4/8 group is day 72 instead of day 28. (F) BLI images of mice treated with 3 million of BBζ, 28ζ, 28BBζ, BBζ CAR4/8, UNT CD4/8, and UNT CD8 cells are shown here. (G) Day 0 (D0), D14, and D28 of MRI on BBζ-, 28ζ-, 28BBζ-, BBζ-CAR4/8-, UNT-CD4/8-, and UNT-CD8-treated mice. Red arrow indicates the tumor site. All data with error bars are presented as mean ± SD.
These imaging studies over the course of 4 weeks showed that, in the CAR8 treatment groups, only BBζ CAR-T cells resulted in significant antitumor activity, whereas mice treated with 28ζ and 28BBζ CAR-T cells showed no inhibition of tumor growth compared with UNT CD8 control cells at a dosage of 3 million cells (Figures 2B and 2C). Decreasing the dose of BBζ CAR-T cells to 1 million cells, BBζ lost efficacy, while increasing the dose of 28BBζ CAR-T cells to 10 million cells, 28BBζ showed significant antitumor activity (Figure S2), demonstrating the dose-dependent therapeutic effect of CAR-T cells. In addition, the exhaustion marker PD-1 was profiled and demonstrated that peripheral circulating BBζ CAR-T cells had significantly lower PD-1 compared with 28ζ and 28BBζ CAR-T cells, which may contribute to the superior antitumor activity of BBζ CAR-T cells (Figure 3B).
Figure 3.

Flow cytometry analysis of peripheral circulating and tumor-infiltrating CAR-T cells
(A) T cell count in peripheral blood. Peripheral blood was analyzed weekly via flow cytometry. T cell count per 100 μL was quantified in BBζ CAR8 (pink), 28ζ CAR8 (green), 28BB CAR8 (orange), and BBζ CAR4/8 (blue) CAR-T cells and CD8 (black) and CD4/8 (gray) UNT cells. (B) Exhaustion of peripheral circulating T cells. The percentage of PD-1 high-expressing T cells was quantified in BBζ CAR8 (pink), 28ζ CAR8 (green), 28BB CAR8 (orange), and CD8 (black) UNT cells on day 7. P values are defined by unpaired two-tailed t-tests ( ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and ∗∗∗∗p < 0.0001). (C) Tumor-infiltrating T cell percentage. At the endpoint of the animal study, tumor-infiltrating human CD45+ lymphocytes were isolated. T cell infiltration of BBζ (pink), 28ζ (green), 28BBζ (orange), and BBζ CD4/8 (blue) CAR-T cells and CD8 (black) and CD4/8 (gray) UNT cells in tumors was quantified. The percentage was defined by human CD45 population divided by total viable cells. (D) Percent CD4 and CD8 TILs. (E) CAR+ percentage. (F and G) Exhaustion marker expression (PD-1 high; F) and the memory T cell subset (CD45RO+CD45RA−; G) of CAR4/8 BBζ CAR-T cells on day 72 were analyzed. ZsGreen signal was used to define CAR+ population. P values are defined by unpaired two-tailed t-tests ( ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and ∗∗∗∗p < 0.0001).
When CAR8 was compared with CAR4/8, remarkably on day 28, the mice treated with BBζ CAR4/8 CAR-T cells did not show any tumor signals on BLI (Figure 2D). All mice from this group were carried for an additional 44 days (total 72 days post-treatment). At the endpoint, day 72 for BBζ CAR4/8 group and day 28 for the other five groups, the tumors were collected and weighted (Figure 2E). The BBζ-CAR4/8-treated mice remained tumor free, and as already observed in the tumor growth curve, the BBζ CAR-T cell treatment displayed enhanced efficacy compared with the other CAR constructs (Figures 2C and 2E). Consistent with a greater therapeutic effect of BBζ CAR-T cell treatment, BBζ-treated tumors showed the least Ki67 compared with 28ζ and 28BBζ in immunohistochemistry (IHC) staining (Figure S4).
MRI was performed biweekly to confirm orthotopic tumor implantation and also detect tumor metastases. As shown in Figure 2G, metastatic lesions (red arrows) were observed in the abdominal cavity in all treatment groups except for CAR8 and CAR4/8 BBζ T cell groups.
CAR4/8 cells exhibited enhanced long-term immune surveillance compared with CAR8
We compared BBζ CAR8 and CAR4/8 in vivo; CAR4/8-treated mice (5/5) had no tumors even 72 days after the initial single-dose administration, while two mice (2/5) in the CAR8 group developed small tumors 4 weeks after CAR-T cell administration (Figures 2E and S3). However, the difference in tumor inhibition between CAR8 and CAR4/8 T cells within 28 days was not significant (Figure 2D). Limited proliferation and persistence of the CAR8 cells in peripheral blood of all constructs were observed compared with CAR4/8 cells, which were abundant after 72 days and provided long-term immune surveillance in the treated mice after tumor disappearance (Figure 3A). BBζ CAR8 and CAR4/8 cells exhibited effective tumor control and remarkable tumor regression that can be seen even on day 7 (Figure 2F).
On day 72, we profiled circulating CAR4/8 cells in peripheral blood and observed that the CD4:CD8 ratio had changed from the initial 2:1 to about 1:1 (Figure 3D). There was no significant difference in CAR expression (Figure 3E) or in exhaustion marker expression or memory phenotype between CD4 and CD8 CAR-T cells (Figures 3F and 3G). Due to the potent antitumor activity of BBζ CAR4/8 cells, there were no tumors in BBζ-CAR4/8-treated group. Thus, tumor-infiltrating BBζ CAR4/8 could not be profiled via scRNA-seq.
Flow and scRNA-seq analysis of tumor-infiltrating T cells
To investigate tumor-infiltrating T cells, tumor tissues collected from mice were dissociated into single-cell suspensions. Tumor-infiltrating lymphocytes (TILs) from the groups of BBζ CAR8, 28ζ CAR8, 28BBζ CAR8, CD4/8 UNT, and CD8 UNT at the dosage of 3 million cells were sorted and characterized. BBζ CAR-T cells showed the highest infiltration among the different constructs with circa 0.6% human CD45-positive cells of total viable cells. 28ζ CAR8, 28BBζ CAR8, CD4/8 UNT, and CD8 UNT groups had progressively lower levels of tumor infiltration, indicating that BBζ CAR-T cells have the most favorable trafficking among these three CAR constructs. CD4/8 UNT cells exhibited higher tumor penetration compared with CD8 UNT (Figure 3C).
The sorted TILs were labeled with hashtag antibodies and pooled together to study their transcriptional profile via 10X Genomics scRNA-seq. A total of 14,446 single human T cell transcriptomes were obtained. Following quality control, 2,100 BBζ, 2,280 28ζ, 2,569 28BBζ, 254 CD8 UNT, and 1,256 CD4/8 UNT cells were assigned to barcodes (demultiplexing) by independent components analysis (ICA) and were visualized with t-distributed stochastic neighbor embedding (t-SNE) plots in Figure 4A. TILs from the three CAR-T conditions grouped in the upper part of the plot and were visually distinct from the two UNT cell conditions that grouped in the lower part of the plot. The TILs grouped into 19 clusters (Figures S5A and S5B) using Louvain community detection to partition a graph in which cells were weighted by the number of shared nearest neighbors. Clusters included tissue-resident memory CD8 T cells (cluster 10), which were enriched in BBζ, PDGFA+ tissue-resident CD8 Treg cells (cluster 12) enriched in 28ζ, and dysfunctional CD8 T cells (cluster 3) enriched in 28BBζ. In addition, CD40L+ CD4 naive T cells (cluster 11) and CXCL13-producing follicular helper CD4 T cells (cluster 15) were enriched in CD4/8 UNT (Figure S5C).
Figure 4.

scRNA-seq analysis of tumor-infiltrating T cells
(A) t-SNE plot of tumor-infiltrating BBζ CAR8 (pink), 28ζ CAR8 (green), and 28BBζ CAR8 (orange) cells and CD8 UNT (black) and CD4/8 UNT (gray) cells. CD4 and CD8 gene distribution is shown in the lower t-SNE plots. (B) CD8 regulatory T cell (Treg cell) subset analysis of the five treatments by determining FOXP3 and IL-2RA (CD25) gene expression. FOXP3−IL-2RA− (gray), FOXP3−IL-2RA+ (brown), FOXP3+IL-2RA− (orange), and FOXP3+IL-2RA+ (yellow) distribution is shown in the t-SNE plot. (C and D) Quantification of the percentage (C) and cell count (D) of each population.
Transcriptomic signatures of tumor-infiltrating CAR8 cells
Analysis of differentially expressed genes (DEGs) revealed differences in activation-, function-, and exhaustion-associated gene expression among the three CAR8 groups (Figure 5A; Table S1). BBζ CAR8 cells showed high expression of major histocompatibility complex class II (MHC II) genes, including histocompatibility leukocyte antigen (HLA)-DPA1 and HLA-DRB1 (Figures 5C and S6), similar to the in vitro cultured 4-1BB CAR-T cells with and without stimulation reported before.23 The elevated MHC class II expression may facilitate epitope spreading via enhanced antigen presentation.
Figure 5.

Transcriptional signatures of tumor-infiltrating BBζ CAR8, 28ζ CAR8, and 28BBζ CAR8 cells
(A) Heatmap of top 20 differential expression genes from each group in the comparison of BBζ (pink), 28ζ (green), and 28BBζ (orange) CAR-T cells. (B) Gene expression of XCL1, XCL2, KLRB1, IL-2RA, CTLA-4, LAG-3, TNFRSF18 (GITR), BATF, and PD-1 in BBζ (pink), 28ζ (green), and 28BBζ (orange) CAR-T cells. (C) HLA class II gene expression in BBζ, 28ζ, and 28BBζ CAR-T cells. CD74, HLA-DRA, HLA-DRB5, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DMA, HLA-DPA1, and HLA-DPB1 are shown in the heatmap after removing seven low-expression genes HLA-DQB1-AS1, HLA-DQA2, HLA-DQB2, HLA-DOB, HLA-DMB, HLA-DOA, and CIITA (Figure S6). (D) Violin plot of mean Z score expression of MHC class II gene expression in BBζ CAR8 (pink), 28ζ CAR8 (green), and 28BB CAR8 (orange). A Kruskal-Wallis rank-sum test was applied.
Among the three CAR8 groups, BBζ CAR-T cells expressed highest levels of cell-mediated-cytotoxicity-associated genes, such as the chemoattractants XCL1 and XCL2 and CD161 (KLRB1), which upregulate IFN-γ secretion30 and stimulate innate response,31 and also had the lowest expression levels of activation-associated gene (IL-2RA), inhibitory immune checkpoint receptors (CTLA-4, LAG-3, and GITR), and AP-1/ATF superfamily transcription factor BATF (Figure 5B). These results illustrated that 4-1BB signaling is able to promote CD8 T cell cytotoxicity and persistence, as well as reduce exhaustion in vivo.
Despite the effect of 4-1BB on CD8 T cells, it has been reported that 4-1BB agonist can stimulate pro-inflammatory cytokine production from CD4 effector T cells and modulate suppressive Treg cells.32 Consequently, we studied the distribution of Treg cells among the three CAR8 groups, revealing that the presence of 4-1BB significantly inhibited the differentiation of CD8 Treg cells (Figures 4B and 4C), which were defined by overexpression of FOXP3 and IL-2RA (CD25).33,34
In contrast, tumor-infiltrating 28ζ CAR-T cells showed the highest proportion of CD8 Treg cells with 78.3% cells expressing either FOXP3 or IL-2RA and a terminal exhausted T cell phenotype (PD-1, LAG-3, and CTLA-4; Figure 5B). 28BBζ CAR-T cells showed similar CD8 Treg cell percentage compared with BBζ; however, 28BBζ displayed poorer therapeutic effects in the ccRCC orthotopic mouse model than BBζ at the same dosage of 3 million cells. Using Reactome pathway analysis, we identified a few deactivated pathways in 28BBζ compared with BBζ CAR-T cells, including formyl peptide receptor, IL-6, IL-15, and leptin pathways, indicating that the CD28 module attenuated trafficking and cytotoxicity of 28BBζ CAR-T cells (Figure S7; Tables S4 and S5).
Transcriptomic signatures of tumor-infiltrating UNT cells
While neither UNT-CD8 or CD4/8 treatments were able to halt tumor growth in mice, we were interested in determining any contribution of CD4 cells to the overall transcriptional profile of the TILs that could provide additional information on the superiority of BBζ CAR4/8 over BBζ CAR8 T cells in curing the mice of their tumors. Here, we compared tumor-infiltrating CD8 only and the CD4/8 mixture of UNT cells (Figure 6A; Table S2). Increased MHC II gene (HLA-DRB1) and memory T cell gene (KLF6 and IL-7R), as well as decreased activation marker (GZMB) and exhaustion marker (NR4A2) gene expression were observed in CD4/8 UNT cells. Interestingly, we also found the upregulation of JUN in the CD4/8 mixture (Figure 6B). Its gene product c-Jun has been engineered in CAR-T cells, as c-Jun overexpression was linked to long-term proliferation capacity,35 which may result in the high amount of circulating BBζ CAR4/8 cells that were observed 72 days after the single-dose administration.
Figure 6.

Transcriptional signatures of tumor-infiltrating CD8 UNT and CD4/8 UNT cells
(A) Heatmap of top 50 differential expression genes in the comparison between CD8 UNT (black) and CD4/8 (gray) UNT cells. (B) Gene expression of HLA-DRB1, KLF6, IL-7R, GZMB, NR4A2, and JUN in CD8 (black) and CD4/8 (gray) UNT cells. (C) Heatmap of top 72 differential expression genes in the comparison between CD8 T cells in CD8 UNT (black) and the ones in CD4/8 (gray) UNT cells.
We also performed DEGs analysis to compare CD8 T cells in the CD8-only group with those in the CD4/8 mixture group (Figure 6C; Table S3). We were able to recover 147 CD8 T cells from UNT CD8 group and 40 from UNT CD4/8 group. Increased MHC II gene (HLA-DRB1 and HLA-DQA1) and memory T cell gene (IL-7R), as well as decreased activation marker (GZMA and GZMB) gene expression were observed in CD8 cells recovered from UNT CD4/8 group. Interestingly, we also observed 13 CD4 CD8 co-expressing T cells in UNT CD4/8 group, which have been reported to produce enhanced Th2 cytokines.36
These results suggest that CD4 T cells contributed to the complete tumor elimination by BBζ CAR4/8 through improving antigen presentation, enhancing proliferative potential, increasing functional capacity, diminishing terminal differentiation, and elevating T cell persistence.
Discussion
To optimize CAR-T cell therapy for ccRCC, we investigated different costimulatory signaling domains in the same CAIX-targeted CAR and interrogated the cellular composition of the CAR-T product in vivo using a ccRCC orthotopic xenograft mouse model. We showed that anti-CAIX BBζ CAR8 cells exhibited superior efficacy compared with 28ζ and 28BBζ CAR8 cells. We further demonstrated the even more superior efficacy, persistence, and long-term immune surveillance of the BBζ CAR4/8 cells. We generated a transcriptional atlas of tumor-infiltrating BBζ, 28ζ, and 28BBζ CAR8 cells, as well as CD8 UNT and CD4/8 UNT cells to understand the therapeutic outcome mediated by the intracellular costimulatory domains and the CD4:CD8 ratio of the CAR-T product. We demonstrated that BBζ CAR-T cells upregulated MHC II genes and cytotoxic-associated genes, downregulated exhaustion markers, and significantly diminished CD8 Treg cells, whereas CD4 T cells helped CD8 T cells maintain the favored memory phenotype in vivo. These results not only showed the exceptional therapeutic efficacy of BBζ CAR8 and CAR4/8 against ccRCC in vivo but also uncovered favorable transcriptional programs associated with the 4-1BB costimulation cassette and cellular composition.
Costimulatory domains in general enhance T cell proliferation, cytokine secretion, cytotoxic function, memory formation, and survival.37 The immunoglobulin (Ig) superfamily member CD28 is considered the prototypical T cell costimulatory receptor and competes with its co-inhibitory receptor counterpart CTLA4 for binding to the B7 molecules CD80 and CD86 on antigen-presenting cells (APCs).38 In comparison, 4-1BB (TNFRSF9 and CD137), a TNFR superfamily member, is expressed on a subset of resting CD8 T cells and is upregulated on both CD4 and CD8 T cells following activation.39 CD28 is critical for the initial activation of naive T cells, while 4-1BB is more important in previously activated T cells. It has been reported that, in animal studies, CD28-costimulated CAR-T cells exhibit improved early expansion and cytotoxic activity compared with their 4-1BB-costimulated counterparts in vivo, while 4-1BB-costimulated CAR-T cells exhibit better long-term persistence.6,7,14,40, 41, 42 In our ccRCC orthotopic mouse model, BBζ exhibited excellent therapeutic effects and persistence compared with 28ζ and 28BBζ CAR-T cells.
The third-generation CAR-T cells were reported to have superior efficacy that benefits from combining the tumoricidal capacity of CD28-based CARs with the persistence generated by 4-1BB-based CARs in CD19-targeted therapy both in a xenograft mouse model and in the clinic.14,43, 44, 45 We did observe the induction of strong intrinsic activation of the type I IFN pathway in 28BBζ CAR-T cells (cluster 13) as reported before14 as well as cluster 14, which has upregulated CD38 TIGIT expression (Figure S5).46 28ζ had significantly more CD8 Treg cells, while 28BBζ and BBζ had few CD8 Treg cells (Figures 4B and 4C); however, 28BBζ did not have superior efficacy or persistence in vivo. We speculate from the pathway analysis on several reasons why 28BBζ CAR-T cells may have decreased therapeutic efficacy compared with BBζ (Figure S7). First, the downregulation of formyl peptide chemotaxis receptors may lead to less chemotactic recruitment of CAR-T cells to the tumor site, which was seen in the lower number of tumor-infiltrating 28BBζ CAR-T cells compared with BBζ (Figure 3C). Second, 28BBζ CAR-T cells had less activated IL-6 and IL-15 pathways, indicating attenuated cytotoxicity against tumor cells compared with BBζ CAR-T cells. Finally, downregulating leptin pathway could inhibit the specific activation of 28BBζ effector CAR-T cells.47
In solid-tumor CAR-T cell therapies, different tumor types may be susceptible to distinct costimulatory domains. For example, in an anti-B7-H3 CAR study in three animal models, including non-small cell lung cancer (NSCLC) and osteosarcoma, the authors showed that the CD28 second-generation CAR-T cells have superior antitumor activity in comparison with those with a 4-1BB costimulatory domain.48 The molecular signatures that are associated with these differential outcomes by solid-tumor-infiltrating CAR cells have not been elucidated to date but can be delineated at the single-cell transcriptional level. We profiled CAR-T cells isolated from tumor tissues and illustrated the transcriptional signatures of BBζ, 28ζ, and 28BBζ CAR-T cells as well as CD8 and CD4/8 UNT cells. Interestingly, the BBζ signatures are consistent with those of the in vitro cultured CD19 BBζ and EGFR BBζ CAR-T cells,23 including a higher MHC II gene expression and an enriched memory phenotype compared with 28ζ CAR-T cells.
Our results showed that multiple MHC class II genes increased in BBζ compared with 28ζ and 28BBζ (Figure 5C), including HLA-DR, which is recognized as a marker of T cell activation.49, 50, 51, 52, 53 We hypothesize that activated human CAR-T cells promote a T cell response through modulating MHC class II expression. However, when we investigated the antigen-presenting function of MHC class II molecules on BBζ CAR8 cells, we did not observe a corresponding upregulation of CD80 or CD86 (data not shown). The antigen-presenting function could be further validated in a humanized mouse model with endogenous tumor microenvironment (TME) via T cell repertoire analysis.
In contemporary trials, patients are typically administrated with CD4/CD8 mixed CAR-T cell products, including Yescarta and Kymriah with a fixed CD4/8 ratio of 1 and Tecartus (brexucabtagene autoleucel) with a median CD4/8 ratio of 0.7.5,7,9 In this study, we observed the more rapid expansion of CD8 BBζ CAR-T cells in the CD4/8 mixture in vivo, resulting in the CD4/8 ratio changing from 2:1 to about 1:1. Our study demonstrated that it is optimal to utilize CD4 and CD8 T cell mixture to generate CAR-T cells, as CD4 secreted higher levels of cytokines in vitro and CAR4/8 cells have superior efficacy and persistence in vivo compared with CAR8. However, no specific cytokine release pattern, such as CD4-dependent Th1/Tc1 or Th2/Tc2, was found in vitro.
There are several limitations in this study. First, we only profiled the transcriptome of tumor-infiltrating CAR-T cells at the endpoint. However, multiple time points should be included to monitor the dynamic change of CAR-T signatures. It would be ideal to compare pre-infusion CAR-T cells with tumor-resident and circulating CAR-T cells to understand their dynamic activation and differentiation. Second, the ccRCC orthotopic mouse model was built in NSG-SGM3 mouse, which has diminished innate immunity, in particular lower macrophage function and NK activity.54, 55, 56 This model may not provide a suitable TME to study the crosstalk between CAR-T cells and endogenous immune cells.57 We look forward to translating cell therapy evaluation to a humanized NSG-SGM3 mouse model. Third, we only compared conventional CD8 CAR-T cells with a CD4/8 2:1 mixture in vivo. However, CD4 alone should be evaluated, as cytolytic CD4 CAR-T cells have been shown to be potent.17,18 In the future, in addition to scRNA-seq, we plan to incorporate cellular indexing of transcriptomes and epitopes (CITE)-seq and other methods to verify proteomic expression of the important biomarkers identified from transcriptional studies.
Taken together, by using skrc-59 cells and the ccRCC-NSG-SGM3 mouse model, we evaluated anti-CAIX CAR-T cells with different costimulatory domains (BBζ, 28ζ, and 28BBζ) and studied their therapeutic effects and the influence of their CD4/CD8 cellular composition. The results revealed unique as well as some overlapping transcriptional signatures of tumor-infiltrating BBζ, 28ζ, and 28BBζ CAR-T cells as well as CD8 UNT and CD4/8 UNT cells. We found that CAIX-targeted, second-generation BBζ CAR-T cells with a CD4/8 ratio of 2:1 exhibited outstanding long-term tumor regression and inhibition with one single dose of 3 million cells. This resulted from enhanced cytotoxic capacity, an enriched memory phenotype, downregulation of terminal exhaustion markers, diminished CD8 Treg cell differentiation, and increased persistence leading to immune surveillance. We believe that anti-CAIX BBζ CAR4/8 CAR-T cells have the potential to be translated to clinic for treatment of ccRCC.
Materials and methods
Cell lines and culture
Human ccRCC cell line skrc-59 (obtained from Dr. Gerd Ritter, Memorial Sloan-Kettering Cancer Center, New York, USA) was engineered to express high levels of human CAIX (skrc-59 CAIX+)25 and mCardinal fluorophore. These cells were grown in RPMI-1640 medium (Gibco) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (Gibco) at 37°C with 5% CO2.
Production of lentivirus particles
For lentivirus production, polyethylenimine (PEI), DNA of the helper plasmids vesicular stomatitis virus G protein (VSV-G), TAT, GAG, and REV (10 μg per 15 cm dish of 293T cells) and 20 μg of the respective CAR DNA was added to Opti-MEM medium (Gibco). This mixture was incubated for 20 min at room temperature (RT) and was afterward added drop by drop to a 15-cm dish of LentiX-293T cells (Clontech Laboratories). After 48 h of incubation, the supernatant was collected, debris was removed, and lentiviral concentrator (Clontech) was added in a 1:3 (v/v) ratio. This mixture was incubated overnight at 4°C; the next day, the tubes were centrifuged for 45 min at 1,500 g and the supernatant was discarded. The pelleted lentivirus was resuspended in RPMI-1640 medium and stored at −80°C.
Generation of CAR-T cells
Human PBMCs were either purchased commercially (STEMCELL Technologies) or separated from apheresis leukoreduction collars obtained from the blood bank of the Brigham and Women's Hospital under a Dana-Farber Cancer Institute (DFCI)-approved IRB protocol no. 14–343 using Ficoll-Paque-PLUS (GE Healthcare). CD4 and CD8 T cells were isolated by using CD8 MicroBeads and CD4 MicroBeads (Miltenyi Biotec) accordingly from PBMCs. T cells were cultured by using RPMI-1640 medium supplemented with 10% FBS, IL-21 (30 ng/mL; Miltenyi Biotec) and activated by T cell TransAct (Miltenyi Biotec). CAR-T cells were generated by using lentiviral transduction (MOI 20) with 10 μg/mL diethylaminoethyl (DEAE). The CAR-positive T cells were sorted by SONY sorter MA900 (Sony Biotechnology). After sorting, the T cells were cultured in RPMI-1640 medium supplemented with 10% FBS, IL-7 (5 ng/mL; Miltenyi Biotec), IL-15 (2.5 ng/mL; Miltenyi Biotec), and gentamicin (50 μg/mL; Gibco).
Celigo in vitro killing assay
Approximately 3,000 skrc-59 tumor cells (target cells) were seeded in a 96-well plate (Greiner). After 12 h of incubation, the plate was scanned and analyzed in bright-field and far-red channel for mCardinal as the 0-h time point. CAR-T cells were added and co-incubated with the target cells. Additional control wells were prepared with target cells only (negative control) and target cells with 1% Triton X (positive control). Subsequently, the plate was scanned and analyzed at the 48 h time point with the equation .
Multiplexed cytokine profiling
The supernatant collected from in vitro cultures after 48 h of incubation of CAR-T cells with target cells was used for cytokine profiling using Luminex human magnetic 30-plex kit (Life Technologies) on a Luminex MAGPIX system (Life Technologies). Conditioned media concentration levels (pg/mL) of each protein were derived from five-parameter, curve-fitting models.
Flow cytometry
All samples were analyzed with an LSR Fortessa (BD Bioscience), and data were analyzed using FlowJo software (FlowJo). T cell phenotype was evaluated via Zombie Yellow Fixable Viability Kit (BioLegend), human CD45 (clone HI30; BioLegend), mouse CD45 (clone 30F11; BioLegend), CD3 (clone OKT3; BioLegend), CD4 (clone A161A1; BioLegend), CD8 (clone SK1; BioLegend), PD-1 (clone MIH4; BD Bioscience), CD45RO (clone UCHL1; BioLegend), and CD45RA (HI100; BioLegend).
In vivo orthotopic ccRCC model
All mice were housed and treated in ethical compliance with DFCI IACUC apporved protocols. A total of 5 × 104 skrc-59 CAIX+ luciferase+ cells were resuspended in 10 μL of RPMI-1640 medium and diluted 1:1 in Matrigel (Corning Life Sciences). This cell mixture was injected under the left kidney capsule of NSG-SGM3 mice (Jackson Laboratory). One week after, tumor engraftment was confirmed with BLI and different doses of CAR-T cells or UNT cells were injected through the tail vein of the mice (day 0; n = 5 mice per group). The tumor BLI was determined 7, 14, 21, and 28 days post-CAR-T cell injection. MRI was performed on day 0, day 14, and day 28. The mice were euthanized by CO2 inhalation, final blood was drawn, and organs and tumors were harvested for downstream analysis.
Bioluminescence imaging (BLI)
Tumor growth was monitored weekly using the IVIS Spectrum In Vivo Imaging System (PerkinElmer). Briefly, mice were injected subcutaneously with 75 mg/kg D-luciferin potassium salt (Promega E1605) in sterile PBS and anesthetized with 2% isoflurane in medical air. Serial bioluminescence images were acquired using the automated exposure setup. The peak bioluminescence signal intensity within selected regions of interest (ROIs) was quantified using the Living Image Software (PerkinElmer) and expressed as photon flux (p/s/cm2/sr). Representative planar bioluminescence images were displayed with indicated adjusted minimal and maximal thresholds.
Small-animal magnetic resonance imaging (MRI)
MRI experiments were performed on a Bruker BioSpec 7T/30 cm USR horizontal bore Superconducting Magnet System (Bruker) equipped with the B-GA12S2 gradient and integrated with up to second-order RT shim system, which provides a maximum gradient amplitude of 440 mT/m and slew rate of 3440 T/m/s. The Bruker-made 35-mm ID volume radiofrequency (RF) coil was used for both RF excitation and receiving. The Bruker AutoPac with laser positioning was used for accurate definition of the ROI. Animals were anesthetized with 1.5% isoflurane mixed in medical air at a flow rate of 2 L/min. Body temperature was maintained at 37°C using a warm-air fan. A pressure transducer for respiratory gating was placed on the abdomen. Animal respiration and temperature were monitored and regulated by the SAII (Sa Instruments) monitoring and gating system model 1025T. Bruker Paravision 6.0.1 was used for MRI data acquisition. Bruker Paravision 6.0.1 was used for MRI data acquisition. T2 weighted images of the kidneys were obtained engaging a fast spin echo (rapid acquisition with refocusing echoes [RARE]) with fat-suppression sequence. The following parameters were used: repetition time (TR) = 2,306 ms; time to echo (TE) = 27 ms; rare factor = 8; number of averages = 8; total acquisition time 5:30 min; field of view (FOV) = 35 × 24 mm2; matrix size = 256 × 192; spatial resolution = 136 × 125 μm2; slice thickness = 0.7 mm; and number of slices = 24.
Tumor dissociation and tumor-infiltrating CAR-T cell isolation
Renal tumor samples were dissociated into single cells by a semi-automated combined mechanical and enzymatic process. The tumor tissue was cut into pieces of 2 to 3 mm in size and transferred to C Tubes (Miltenyi Biotec) containing a mix of enzymes H, R, and A (Tumor Dissociation Kit, human; Miltenyi Biotec). Mechanical dissociation was accomplished by performing a program (37C_h_TDK_1) on the gentleMACS Octo Dissociator with Heaters (Miltenyi Biotec). Single-cell suspensions generated from ccRCC tumor samples were fluorescence-activated cell sorting (FACS) sorted on a SONY MA900 sorter (SONY) for viable human T cells (Zombie−mCD45−hCD45+CD3+; BioLegend).
RNA-seq 10X Genomics library preparation, hash tag, and single-cell 3′ sequencing
scRNA-seq experiments were performed by the Brigham and Women's Hospital Single Cell Genomics Core. Sorted viable human T cells were stained with a distinct barcoded antibody (Cell-Hashing antibody, TotalSeq-A; BioLegend), as previously described.58 Next, 7,500 cells from each condition were resuspended in 0.4% BSA in PBS at a concentration of 1,000 cells per μL, pooled together, and then loaded onto a single lane (Chromium chip B, 10X Genomics) followed by encapsulation in a lipid droplet (Single Cell 3′ kit V3, 10X Genomics) followed by cDNA and library generation according to the manufacturer's protocol. mRNA library was sequenced to an average of 50,000 reads per cell and hashtag-oligos (HTOs) (Cell Hashing antibodies) library sequenced to an average of 5,000 reads per cell, both using Illumina Novaseq. scRNA-seq reads of 14,446 cells were processed with Cell Ranger v3.1,59 where read quantification was performed using the STAR aligner60 against the GRCh38 transcriptome. There were 33,908 mean reads per cell, 21,577 genes were detected, and there were 4,855 median unique molecular identifier (UMI) counts per cell. Single-cell data analysis was performed using R version 4.0.3, and code is available from https://github.com/aedin.
Demixing of HTO tags
Centering and log ratio transformation of the HTO tags (features) and centered log-ratio (CLR) transformation normalization of features performed using the NormalizeData in the Seurat R package (version 3.9.9.9). To assign the sample of origin for each cell, HTODemux with a positive quantile of 0.99 was applied to identify doublets (n = 2,654) and negative (n = 1,037) and assign cells to singlet HTO tags (n = 10,755). Raw, unprocessed 14,446 HTO counts indicated that one hashtag (#7) had higher background raw counts compared with other HTO tags. The threshold used by HTODemux was greater for this hashtag than the other hashtag, reflecting its high background. This was not resolved by normalization or by increasing the HTODemux settings. Independent component analysis (using the R package fastICA) of the HTO data indicated that HTO tags that were called negative by HTODemux clustered with the cells assigned to this hashtag #7 (n = 191). By curating these data, this HTO group was expanded to 436 putative cells. Clustering of cells to this hashtag was confirmed with mRNA clustering. Remaining cells with doublet or negative HTO tags were excluded. In the analysis of CD8 T cells in both UNT CD8 and UNT CD4/8 groups, we called CD8A counts >0 as CD8+ cells. There were 147 and 40 cells in each UNT group, in which there were 13 CD4+ CD8A+ cells from the 40 cells in UNT CD4/8 group.
Removal of low-quality cells and normalization
The Bioconductor package, scuttle,61 was used to extract cell-level metrics and to exclude low-quality cells: low log-total count or number of detected features or high percentage of mitochondrial genes to retain 8, 459 cells.
Dimension reduction and cluster analysis
Correspondence analysis was performed using the Corral Bioconductor package,62 which visually had greater discrimination of groups (treatments and control) when compared with principal-component or independent-component analysis. Figures show t-SNE clustering of Corral reduced data with a seed of 945. t-SNE clustering was stable over a range of perplexity values 10, 100, and 500 (data not shown). To define cell clusters, a shared nearest neighbor graph63 was constructed in which 50 nearest neighbors were considered during graph construction (k = 50), and Louvain clustering with Jaccard distance was used to define cell clusters.
MHC class II gene analysis
The expression of 16 MHC class II genes was examined (HLA-DRA, HLA-DRB5, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DQB1-AS1, HLA-DQA2, HLA-DQB2, HLA-DOB, HLA-DMB, HLA-DMA, HLA-DOA, HLA-DPA1, HLA-DPB1, CIITA, and CD74); however, seven genes were not expressed (HLA-DQB1-AS1, HLA-DQA2, HLA-DQB2, HLA-DOB, HLA-DMB, HLA-DOA, and CIITA), which was defined as expressed with a count of 5 or more in 10 or more cells (Figure S6). The group gene expression was compared using a pairwise Wilcoxon signed rank test, and a Kruskal-Wallis rank-sum test was applied to compute the overall statistic. Violin plots were generated using the dittoSeq64 function dittoPlotVarsAcrossGroups.
Differential expression gene analysis
DEG analysis was performed using a Poisson generalized linear model using the function FindMarkers in Seurat.65 The FindMarkers min.pct parameter, which speeds up the function by testing fewer genes when the data have high dimension, was reduced from its default value of 0.1 to 0.05 in analyses where there were few cells (UNT analysis). P values were corrected for multiple testing based on Bonferroni correction using all genes in the matrix, and DEGs were ranked by P value. In DE analysis of UNT CD8 and CD4/CD8, genes were filtered to the top 10 and 40 DEGs (respectively), which had an absolute fold change of >0.7 and adjusted p < 0.05.
Pathway analysis
Pathway in analysis of Reactome gene sets was performed using the Bioconductor package AUCell,66 which generates an “area under the curve” (AUC) score for each gene set that estimates the proportion of genes in the gene set that are highly expressed in each cell. Reactome pathways in the Bioconductor package “graphite” (version 1.38.0) were subset to the 2,390 Homo sapiens reactome pathways. AUCell “scored” each pathway in 6,949 cells, which included the BBζ (n = 2,100), 28ζ (n = 2,280), and 28BBζ (n = 2,569). Default parameters for AUCell ranking were used, and the AUC scores represent the fraction of genes, within the top 1,677 genes in the ranking, that are included in the signature. The default aucMaxRank parameter of 5% of the total number of genes (n = 33,538) was used as the threshold to calculate the AUC rankings. A pairwise Wilcoxon rank-sum test (scran:pairwiseWilcox) was used to test which Reactome pathways were differentially regulated between the three groups (BBζ, 28ζ, and 28BBζ). Pathways were filtered using both scran:pairwiseWilcox P values and the scran:pairwiseWilcox test statistic (which is called AUC and represents the probability of sampling a value in the first group greater than a random value from the second group). Figures report the AUCell AUC scores, not the scran:pairwiseWilcox test statistic.
Accession codes
All generated scRNA-seq data are available at the Gene Expression Omnibus (GEO) with accession code GEO: GSE190964.
Immunohistochemistry staining and quantitation
Tissues were fixed in 10% formalin (Sigma-Aldrich) for 24 h and replaced by 70% ethanol (Decon Laboratories) and then processed and embedded into blocks. Immunohistochemical staining was performed on 4-μm sections of the tissue slides on an automated Leica Bond Rx system, with antibody granzyme B (EPR20129-217, rabbit monoclonal; Abcam) at 1:250 for 60 min, PD-L1 (E1L3N, rabbit monoclonal; Cell Signaling Technology) at 1:200 for 60 min, and Ki67 (SP6, rabbit monoclonal; Biocare Medical) at 1:100 for 60 min. The sections were then treated according to the streptavidin-biotin-peroxidase complex method (Bond Polymer Refine Detection Kit; Leica Microsystems) with diaminobenzidine (DAB) as a chromogen and counterstained with hematoxylin.
Whole slide images were acquired from stained slides using a Vectra 3.0 Automated Quantitative Pathology Imaging System (Akoya Biosciences) and analyzed using Halo Image Analysis platform (Indica Labs).
Statistical analysis
The statistical significance of the data was evaluated using ANOVA or Student's t test in GraphPad Prism 9. p < 0.05 was considered significant. Student's t test: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; and ∗∗∗∗p < 0.0001.
Acknowledgments
This work was partially supported by the Kidney Cancer Research Alliance (KCCure) 2017 Research Grant Award (W.A.M.), National Foundation for Cancer Research fellowship (W.A.M.), and NCI P50CA101942 (G.J.F.). This work was also supported by the Assistant Secretary of Defense for Health Affairs endorsed by the Department of Defense, through the FY18, KCRP, Idea Development Award (W81XWH-18-1-0568), FY21 Translational Research Partnership Award (W81XWH-21-1-0442) and FY21 Idea Development Award (W81XWH-21-1-0482) to W.A.M. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. In addition, this work was supported by the Wong Family Award to Y.W.
Author contributions
Y.W. and W.A.M. designed the study. Y.W., A.B., M.G., D.M.B., Z.Z., and M.L.O. performed experiments. Y.W., A.B., M.G., A.C.C., S.K., C.R., D.M.B., Q.-D.N., M.L.O., and Y.H. analyzed data. Y.W., A.B., G.J.F., and W.A.M. wrote the manuscript. All authors edited and approved the manuscript.
Declaration of interests
W.A.M. has patents in the PD-1/PDL1 field. G.J.F. has patents and pending royalties on the PD-1/PD-L1 pathway from Roche, Merck MSD, Bristol-Myers-Squibb, Merck KGA, Boehringer-Ingelheim, AstraZeneca, Dako, Leica, Mayo Clinic, Eli Lilly, and Novartis. G.J.F. has served on advisory boards for Roche, Bristol-Myers-Squibb, Xios, Origimed, Triursus, iTeos, NextPoint, IgM, Jubilant, Trillium, GV20, and Geode. G.J.F. has equity in Nextpoint, Triursus, Xios, iTeos, IgM, GV20, IOME, and Geode. The other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.12.019.
Supplemental information
References
- 1.Lee D.W., Barrett D.M., Mackall C., Orentas R., Grupp S.A. The future is now: chimeric antigen receptors as new targeted therapies for childhood cancer. Clin. Cancer Res. 2012;18:2780–2790. doi: 10.1158/1078-0432.Ccr-11-1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Srivastava S., Riddell S.R. Engineering CAR-T cells: design concepts. Trends Immunol. 2015;36:494–502. doi: 10.1016/j.it.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F., et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu Y., Tan Y., Ou R., Zhong Q., Zheng L., Du Y., Zhang Q., Huang J. Anti-CD19 chimeric antigen receptor-modified T cells for B-cell malignancies: a systematic review of efficacy and safety in clinical trials. Eur. J. Haematol. 2016;96:389–396. doi: 10.1111/ejh.12602. [DOI] [PubMed] [Google Scholar]
- 5.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D., et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 7.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y., et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Locke F.L., Ghobadi A., Jacobson C.A., Miklos D.B., Lekakis L.J., Oluwole O.O., Lin Y., Braunschweig I., Hill B.T., Timmerman J.M., et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019;20:31–42. doi: 10.1016/s1470-2045(18)30864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang M., Munoz J., Goy A., Locke F.L., Jacobson C.A., Hill B.T., Timmerman J.M., Holmes H., Jaglowski S., Flinn I.W., et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2020;382:1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abramson J.S., Palomba M.L., Gordon L.I., Lunning M.A., Wang M., Arnason J., Mehta A., Purev E., Maloney D.G., Andreadis C., et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839–852. doi: 10.1016/s0140-6736(20)31366-0. [DOI] [PubMed] [Google Scholar]
- 11.FDA FDA approves first cell-based gene therapy for adult patients with multiple myeloma. 2021. https://www.fda.gov/news-events/press-announcements/fda-approves-first-cell-based-gene-therapy-adult-patients-multiple-myeloma
- 12.Lindner S.E., Johnson S.M., Brown C.E., Wang L.D. Chimeric antigen receptor signaling: functional consequences and design implications. Sci. Adv. 2020;6:eaaz3223. doi: 10.1126/sciadv.aaz3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Stegen S.J., Hamieh M., Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015;14:499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Z., Condomines M., van der Stegen S.J.C., Perna F., Kloss C.C., Gunset G., Plotkin J., Sadelain M. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28:415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pegram H.J., Park J.H., Brentjens R.J. CD28z CARs and armored CARs. Cancer J. 2014;20:127–133. doi: 10.1097/ppo.0000000000000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalos M., Levine B.L., Porter D.L., Katz S., Grupp S.A., Bagg A., June C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D., Aguilar B., Starr R., Alizadeh D., Brito A., Sarkissian A., Ostberg J.R., Forman S.J., Brown C.E. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight. 2018;3 doi: 10.1172/jci.insight.99048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y., Lin T., Jacoby E., Qin H., Gardner E.G., Chien C.D., Lee D.W., III, Fry T. J. CD4 CAR T cells mediate CD8-like cytotoxic anti-leukemic effects resulting in leukemic clearance and are less susceptible to attenuation by endogenous TCR activation than CD8 CAR T cells. Blood. 2015;126:100. doi: 10.1182/blood.V126.23.100.100. [DOI] [Google Scholar]
- 19.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M., et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/jci85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terakura S., Yamamoto T.N., Gardner R.A., Turtle C.J., Jensen M.C., Riddell S.R. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood. 2012;119:72–82. doi: 10.1182/blood-2011-07-366419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheih A., Voillet V., Hanafi L.A., DeBerg H.A., Yajima M., Hawkins R., Gersuk V., Riddell S.R., Maloney D.G., Wohlfahrt M.E., et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 2020;11:219. doi: 10.1038/s41467-019-13880-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long A.H., Haso W.M., Shern J.F., Wanhainen K.M., Murgai M., Ingaramo M., Smith J.P., Walker A.J., Kohler M.E., Venkateshwara V.R., et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boroughs A.C., Larson R.C., Marjanovic N.D., Gosik K., Castano A.P., Porter C.B.M., Lorrey S.J., Ashenberg O., Jerby L., Hofree M., et al. A distinct transcriptional program in human CAR T cells bearing the 4-1BB signaling domain revealed by scRNA-seq. Mol. Ther. 2020;28:2577–2592. doi: 10.1016/j.ymthe.2020.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lo A.S., Xu C., Murakami A., Marasco W.A. Regression of established renal cell carcinoma in nude mice using lentivirus-transduced human T cells expressing a human anti-CAIX chimeric antigen receptor. Mol. Ther. Oncolytics. 2014;1:14003. doi: 10.1038/mto.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suarez E.R., Chang de K., Sun J., Sui J., Freeman G.J., Signoretti S., Zhu Q., Marasco W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7:34341–34355. doi: 10.18632/oncotarget.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen C., Kaelin W.G., Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin. Cancer Biol. 2013;23:18–25. doi: 10.1016/j.semcancer.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruf M., Mittmann C., Nowicka A.M., Hartmann A., Hermanns T., Poyet C., van den Broek M., Sulser T., Moch H., Schraml P. pVHL/HIF-regulated CD70 expression is associated with infiltration of CD27+ lymphocytes and increased serum levels of soluble CD27 in clear cell renal cell carcinoma. Clin. Cancer Res. 2015;21:889–898. doi: 10.1158/1078-0432.Ccr-14-1425. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y., Chan L.L., Grimaud M., Fayed A., Zhu Q., Marasco W.A. High-throughput image cytometry detection method for CAR-T transduction, cell proliferation, and cytotoxicity assays. Cytometry A. 2020 doi: 10.1002/cyto.a.24267. [DOI] [PubMed] [Google Scholar]
- 29.Xu C., Lo A., Yammanuru A., Tallarico A.S.C., Brady K., Murakami A., Barteneva N., Zhu Q., Marasco W.A. Unique biological properties of catalytic domain directed human anti-CAIX antibodies discovered through phage-display technology. PLoS One. 2010;5:e9625. doi: 10.1371/journal.pone.0009625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang P., Zhou Y., Liu Z., Zhang P. Interaction between ANXA1 and GATA-3 in immunosuppression of CD4(+) T cells. Mediators Inflamm. 2016:1701059. doi: 10.1155/2016/1701059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fergusson J.R., Smith K.E., Fleming V.M., Rajoriya N., Newell E.W., Simmons R., Marchi E., Björkander S., Kang Y.H., Swadling L., et al. CD161 defines a transcriptional and functional phenotype across distinct human T cell lineages. Cell Rep. 2014;9:1075–1088. doi: 10.1016/j.celrep.2014.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartkowiak T., Curran M.A. 4-1BB agonists: multi-potent potentiators of tumor immunity. Front. Oncol. 2015:117. doi: 10.3389/fonc.2015.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mishra S., Srinivasan S., Ma C., Zhang N. CD8(+) regulatory T cell - a mystery to Be revealed. Front. Immunol. 2021;12:708874. doi: 10.3389/fimmu.2021.708874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Churlaud G., Pitoiset F., Jebbawi F., Lorenzon R., Bellier B., Rosenzwajg M., Klatzmann D. Human and mouse CD8(+)CD25(+)FOXP3(+) regulatory T cells at steady state and during interleukin-2 therapy. Front. Immunol. 2015;6:171. doi: 10.3389/fimmu.2015.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lynn R.C., Weber E.W., Sotillo E., Gennert D., Xu P., Good Z., Anbunathan H., Lattin J., Jones R., Tieu V., et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576:293–300. doi: 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bohner P., Chevalier M.F., Cesson V., Rodrigues-Dias S.-C., Dartiguenave F., Burruni R., Tawadros T., Valerio M., Lucca I., Nardelli-Haefliger D., et al. Double positive CD4+CD8+ T cells are enriched in urological cancers and favor T helper-2 polarization. Front. Immunol. 2019;10 doi: 10.3389/fimmu.2019.00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen L., Flies D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esensten J.H., Helou Y.A., Chopra G., Weiss A., Bluestone J.A. CD28 costimulation: from mechanism to therapy. Immunity. 2016;44:973–988. doi: 10.1016/j.immuni.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H., Snyder K.M., Suhoski M.M., Maus M.V., Kapoor V., June C.H., Mackall C.L. 4-1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J. Immunol. 2007;179:4910–4918. doi: 10.4049/jimmunol.179.7.4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu P., Lu X.-A., Zhang X., Xiong M., Zhang J., Zhou X., Qi F., Yang J., He T. Which is better in CD19 CAR-T treatment of r/r B-ALL, CD28 or 4-1BB? A parallel trial under the same manufacturing process. J. Clin. Oncol. 2018;36:3041. doi: 10.1200/JCO.2018.36.15_suppl.3041. [DOI] [Google Scholar]
- 41.Ying Z., He T., Wang X., Zheng W., Lin N., Tu M., Xie Y., Ping L., Zhang C., Liu W., et al. Parallel comparison of 4-1BB or CD28 Co-stimulated CD19-targeted CAR-T cells for B cell non-hodgkin's lymphoma. Mol. Ther. Oncolytics. 2019;15:60–68. doi: 10.1016/j.omto.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Majzner R.G., Mackall C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019;25:1341–1355. doi: 10.1038/s41591-019-0564-6. [DOI] [PubMed] [Google Scholar]
- 43.Ramos C.A., Rouce R., Robertson C.S., Reyna A., Narala N., Vyas G., Mehta B., Zhang H., Dakhova O., Carrum G., et al. In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B cell non-hodgkin's lymphomas. Mol. Ther. 2018;26:2727–2737. doi: 10.1016/j.ymthe.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee D.W., Santomasso B.D., Locke F.L., Ghobadi A., Turtle C.J., Brudno J.N., Maus M.V., Park J.H., Mead E., Pavletic S., et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol. Blood Marrow Transpl. 2019;25:625–638. doi: 10.1016/j.bbmt.2018.12.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weinkove R., George P., Dasyam N., McLellan A.D. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin. Transl. Immunol. 2019;8:e1049. doi: 10.1002/cti2.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Philip M., Fairchild L., Sun L., Horste E.L., Camara S., Shakiba M., Scott A.C., Viale A., Lauer P., Merghoub T., et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545:452–456. doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saucillo D.C., Gerriets V.A., Sheng J., Rathmell J.C., Maciver N.J. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J. Immunol. 2014;192:136–144. doi: 10.4049/jimmunol.1301158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nguyen P., Okeke E., Clay M., Haydar D., Justice J., O'Reilly C., Pruett-Miller S., Papizan J., Moore J., Zhou S., et al. Route of 41BB/41BBL costimulation determines effector function of B7-H3-CAR.CD28ζ T cells. Mol. Ther. Oncolytics. 2020;18:202–214. doi: 10.1016/j.omto.2020.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saraiva D.P., Jacinto A., Borralho P., Braga S., Cabral M.G. HLA-DR in cytotoxic T lymphocytes predicts breast cancer patients' response to neoadjuvant chemotherapy. Front. Immunol. 2018;9:2605. doi: 10.3389/fimmu.2018.02605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Viallard J.F., Blanco P., André M., Etienne G., Liferman F., Neau D., Vidal E., Moreau J.F., Pellegrin J.L. CD8+HLA-DR+ T lymphocytes are increased in common variable immunodeficiency patients with impaired memory B-cell differentiation. Clin. Immunol. 2006;119:51–58. doi: 10.1016/j.clim.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 51.Baecher-Allan C., Wolf E., Hafler D.A. MHC class II expression identifies functionally distinct human regulatory T cells. J. Immunol. 2006;176:4622–4631. doi: 10.4049/jimmunol.176.8.4622. [DOI] [PubMed] [Google Scholar]
- 52.Viallard J.F., Bloch-Michel C., Neau-Cransac M., Taupin J.L., Garrigue S., Miossec V., Mercie P., Pellegrin J.L., Moreau J.F. HLA-DR expression on lymphocyte subsets as a marker of disease activity in patients with systemic lupus erythematosus. Clin. Exp. Immunol. 2001;125:485–491. doi: 10.1046/j.1365-2249.2001.01623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sáez-Cirión A., Lacabaratz C., Lambotte O., Versmisse P., Urrutia A., Boufassa F., Barré-Sinoussi F., Delfraissy J.F., Sinet M., Pancino G., et al. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc. Natl. Acad. Sci. U S A. 2007;104:6776–6781. doi: 10.1073/pnas.0611244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wunderlich M., Chou F.S., Link K.A., Mizukawa B., Perry R.L., Carroll M., Mulloy J.C. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia. 2010;24:1785–1788. doi: 10.1038/leu.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ito M., Hiramatsu H., Kobayashi K., Suzue K., Kawahata M., Hioki K., Ueyama Y., Koyanagi Y., Sugamura K., Tsuji K., et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100:3175–3182. doi: 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- 56.Shultz L.D., Lyons B.L., Burzenski L.M., Gott B., Chen X., Chaleff S., Kotb M., Gillies S.D., King M., Mangada J., et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 57.Boulch M., Cazaux M., Loe-Mie Y., Thibaut R., Corre B., Lemaître F., Grandjean C.L., Garcia Z., Bousso P. A cross-talk between CAR T cell subsets and the tumor microenvironment is essential for sustained cytotoxic activity. Sci. Immunol. 2021;6 doi: 10.1126/sciimmunol.abd4344. [DOI] [PubMed] [Google Scholar]
- 58.Stoeckius M., Zheng S., Houck-Loomis B., Hao S., Yeung B.Z., Mauck W.M., 3rd, Smibert P., Satija R. Cell hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018;19:224. doi: 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng G.X., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCarthy D.J., Campbell K.R., Lun A.T.L., Wills Q.F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 2017;33:1179–1186. doi: 10.1093/bioinformatics/btw777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hsu L.L., Culhane A.C. corral: single-cell RNA-seq dimension reduction, batch integration, and visualization with correspondence analysis. bioRxiv. 2021 doi: 10.1101/2021.11.24.469874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu C., Su Z. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics. 2015;31:1974–1980. doi: 10.1093/bioinformatics/btv088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bunis D.G., Andrews J., Fragiadakis G.K., Burt T.D., Sirota M. dittoSeq: universal user-friendly single-cell and bulk RNA sequencing visualization toolkit. Bioinformatics. 2020;36:5535–5536. doi: 10.1093/bioinformatics/btaa1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.