

Summary
N-glycans are displayed on cell surface proteins, and can engage in direct binding interactions with membrane-bound and secreted glycan-binding proteins (GBPs). Biochemical identification and characterization of glycan-mediated interactions is often made difficult by low binding affinities. Here we describe the metabolic introduction of a diazirine photocrosslinker onto GlcNAc residues of N-linked glycoproteins on cell surfaces. We characterize sites at which diazirine-modified GlcNAc is incorporated, as well as modest perturbations to glycan structure. We show that diazirine-modified GlcNAc can be used to covalently crosslink two extracellular GBPs, galectin-1 and cholera toxin subunit B (CTB), to cell surface N-linked glycoproteins. The extent of crosslinking correlates with display of the preferred glycan ligands for the GBPs. Additionally, covalently crosslinked complexes could be isolated, and protein components of crosslinked N-linked glycoproteins were identified by proteomics analysis. This method may be useful in the discovery and characterization of binding interactions that depend on N-glycans.
Graphical Abstract

Wu et al. report that a photocrosslinking analog of GlcNAc can be incorporated into cellular N-glycans and used to covalently crosslink glycan-protein interactions in their native cellular context. This method can be used to identify novel glycan-protein interactions, and to evaluate the extent of glycan-protein binding occurring under diverse conditions.
INTRODUCTION
Cell surfaces are covered with diverse glycosylated molecules. The identity, abundance, and distribution of cell surface glycans provide an extracellular report on cell type and physiological state. This cell surface glycan communique is detected and interpreted by glycan-binding proteins (GBPs) that recognize specific glycan features. (Varki, 2017) For example, galectins are a family of animal GBPs that share a conserved carbohydrate recognition domain (CRD) and preferentially bind β-galactosides, recognizing both animal and microbial glycans. (Kamili et al., 2016; Vasta, 2020) Binding of galectins to cell surfaces activates intracellular signaling pathways, but galectins are also found inside of cells where they act directly on signaling components. (Johannes et al., 2018) Acting in diverse ways, galectins play immunomodulatory roles and to contribute to metastatic phenotypes. (Girotti et al., 2020; Thiemann and Baum, 2016) Human cell surface glycans are also recognized by microbial pathogens through cell surface and secreted GBPs. (Cummings et al., 2015) For instance, Vibrio cholerae produces a secreted toxin that causes the severe diarrhea that characterizes the disease cholera. (Holmgren, 1981) In the initial step of host cell intoxication, cholera toxin subunit B (CTB) binds glycan receptors on the surface of intestinal epithelial cells. (Wernick et al., 2010) CTB binding partners include the ganglioside GM1, which CTB binds with high affinity, as well as lower affinity fucosylated glycoconjugate ligands that are abundant on intestinal epithelial cell surfaces. (Cervin et al., 2018; Holmgren et al., 1975)
Historically, affinity purification has been a powerful approach for identifying protein binding partners but, in the case of GBPs, affinity purification has limitations. First, GBPs commonly exhibit moderate to low single-site binding affinities for ligands, with micromolar and sometimes even millimolar Kds. (Nizet et al., 2015) Low affinity binding interactions dissociate rapidly and can be difficult to capture. Second, affinity purification requires lysis of cells and destruction of the cellular architecture, which can result in scrambling of binding partners. Spurious interactions may occur between molecules that would normally reside in separate compartments. Conversely, molecules that normally interact when concentrated within a specific organelle can dissociate when organellar structure is disrupted. Thus, alternate approaches have been pursued for identifying true GBP ligands.
Photoactivatable crosslinking probes (photocrosslinkers) are tools used to capture binding interactions in normal cellular contexts. (Mishra et al., 2020; Wu and Kohler, 2019) Photocrosslinkers can be appended to cellular biomolecules. UV irradiation converts the crosslinker to a highly reactive species that covalently reacts with nearby molecules. Crosslinked complexes can be isolated and characterized by various approaches, including immunoblot and mass spectrometry. We and others have appended photocrosslinkers to monosaccharides and used metabolic glycan engineering (MGE) to incorporate photocrosslinking sugars into cellular glycoconjugates. (Feng et al., 2013; Han et al., 2005; Luchansky et al., 2004; Tanaka and Kohler, 2008; Yu et al., 2012) Such strategies have been used to capture and identify glycoconjugate ligands of GBPs.
We reported a method to produce a diazirine-functionalized UDP-GlcNAc donor (UDP-GlcNDAz) and showed that the nucleocytoplasmic O-GlcNAc transferase (OGT) can accept this modified donor, resulting in production of diazirine-functionalized O-GlcNAc.(Yu et al., 2012) We speculated that UDP-GlcNDAz might also be a substrate for Golgi-resident glycosyltransferases, resulting in production of diazirine-modified cell surface glycans. Evaluating whether GlcNDAz is incorporated into cell surface glycans requires an analytic approach that provides information about sites of diazirine modification as well as overall glycan structures. First, because normal metabolic pathways interconvert sugar analogs, the possibility exists for the photocrosslinkers to be displayed in unexpected locations. Indeed, in other MGE applications, interconversion of azide-modified monosaccharides has been observed and exploited. (Boyce et al., 2011) A second consideration arises regarding sites of unnatural sugar incorporation. For example, typical N-glycans contain multiple GlcNAc residues added by different GlcNAc transferases: two GlcNAc residues in the core, GlcNAc residues initiating each mature antenna, and possibly bisecting GlcNAc and/or additional GlcNAc residues within poly-N-acetylactosamine (polyLacNAc) chains attached to antennae. The relative ability of different glycosyltransferases to add photocrosslinking sugars and the resulting positions at which unnatural photocrosslinking sugars are incorporated will affect crosslinking events. Finally, incorporation of unnatural sugars has the potential to perturb glycan biosynthesis in ways that could affect glycan recognition and function. For instance, production of UDP-GlcNDAz has the potential to disrupt regulation of the hexosamine biosynthetic pathway. Also, incorporation of unnatural sugars could prevent addition of subsequent sugars, resulting in truncated glycan structures. Together, these concerns highlight the need for comprehensive analysis of unnatural sugar metabolism and incorporation for reliable use of this chemical biology tool.
Here we report that diazirine-modified GlcNAc (GlcNDAz) is incorporated into N-glycans of human cells grown in culture. The method builds on our prior demonstration that cells can be engineered to produce the diazirine-modified donor, UDP-GlcNDAz. (Yu et al., 2012) We find that, like OGT, at least one Golgi-resident GlcNAc-transferase, MGAT2, can use UDP-GlcNDAz as a substrate, while some Golgi-resident GlcNAc-transferases do not tolerate the diazirine. Glycoproteomics analysis demonstrates production of mature complex N-glycans containing GlcNDAz and no other diazirine-modified sugars. However, lectin binding analysis does reveal modest perturbations to N-glycan structure. Using metabolically-incorporated GlcNDAz, we show crosslinking of two GBPs – galectin-1 and CTB – to glycoprotein binding partners. Crosslinking intensity reflects the affinity of binding interactions. Also, galectin-1 crosslinked complexes were purified and analyzed by mass spectrometry-based proteomics analysis to provide a list of candidate galectin-1 binding partners. We confirmed the crosslinking of galectin-1 to several glycoproteins including LY75, which, to our knowledge, has not been described previously as a galectin-1 ligand. We find that the crosslinking method provides context-specific information about binding interactions: crosslinking of extracellularly-added galectin-1 to LAMP1 is only detected when the cells are lysed allowing compartmental mixing to occur. Thus, GlcNDAz crosslinking has potential application for discovery of cellular binding interactions that depend on N-linked glycosylation, and characterization of cellular conditions under which these interactions occur.
RESULTS
Strategy for metabolic production of photocrosslinking N-glycans
Previously, we reported that HeLa (Yu et al., 2012) and K562 (Rodriguez et al., 2015) cells stably expressing the F383G mutant of UAP1 and treated with a cell-permeable, diazirine-modified analog of GlcNAc-1-phosphate (Ac3GlcNDAz-1P(AcSATE)2) produce diazirine-modified UDP-GlcNAc (UDP-GlcNDAz; Fig. 1). We found that O-GlcNAc transferase (OGT) accepts UDP-GlcNDAz in place of UDP-GlcNAc, enabling production of O-GlcNDAzylated proteins with diazirine attached to O-GlcNAc. We demonstrated use of this metabolically-incorporated crosslinker to covalently crosslink O-GlcNAcylated proteins to nearby proteins in living cells.
Figure 1. Strategy for metabolic production of photocrosslinking N-glycans.

N-glycans are synthesized in the ER and Golgi through the sequential actions of enzymes, including multiple GlcNAc-transferases. N-linked glycoproteins are displayed on the cell surface, where glycans are recognized by extracellular GBPs. Individual binding interactions are typically low-affinity, although high avidity binding can be achieved through multivalency. To introduce diazirine, cells expressing UAP1(F383G) are cultured with Ac3GlcNDAz-1P(AcSATE)2. Ac3GlcNDAz-1P(AcSATE)2 is deprotected, potentially by intracellular esterases, and transformed to UDP-GlcNDAz. In this work, we asked whether UDP-GlcNDAz enters the Golgi and is a substrate for Golgi GlcNAc-transferases. Cell surface N-linked glycoproteins displaying the diazirine modification can be activated by UV light, resulting in covalent crosslinking to extracellular GBPs. See also Fig. S1.
With a robust method for cellular production of UDP-GlcNDAz in hand, in this work we asked whether GlcNDAz was also incorporated into N-glycans. We performed experiments in T84 cells, a colorectal cancer cell line capable of producing complex type N-glycans, including multi-antennary, fucosylated, and sialylated structures. (Holst et al., 2016) T84 cells stably expressing UAP1(F383G) were prepared by lentiviral infection.(Rodriguez et al., 2015; Yu et al., 2012) T84 UAP1(F383G) cells cultured with Ac3GlcNDAz-1P(AcSATE)2 produced UDP-GlcNDAz efficiently, with UDP-GlcNDAz levels exceeding UDP-GlcNAc levels after 8 h (Fig. S1).
GlcNDAz is incorporated into N-glycans
To incorporate GlcNDAz into N-glycans, UDP-GlcNDAz must enter the Golgi and be accepted by a Golgi-resident GlcNAc-transferase (Fig. 1). We assayed nine Golgi-resident GlcNAc-transferases for GlcNDAz-transferase activity. Each GlcNAc-transferase was incubated with an appropriate acceptor substrate (see Methods) and UDP-GlcNDAz. Comparison reactions were performed with UDP-GlcNAc. Activity was quantified by measuring release of UDP. MGAT2 showed modest activity with UDP-GlcNDAz (activity with UDP-GlcNDAz 19 % of that observed with UDP-GlcNAc) and minimal activity was observed for MGAT5 (activity with UDP-GlcNDAz 4 % of that observed with UDP-GlcNAc). No activity with UDP-GlcNDAz was detected for other GlcNAc-transferases examined (Fig. 2A and Fig. S2). MGAT2 and MGAT5 are involved in establishment of β1–2- and β1–6-antennae from α1–6-linked mannose in complex N-glycan biosynthesis. Several GlcNAc-transferases involved in biosynthesis of polyLacNAc, a terminal structure found on multiple types of glycans, and O-mannose glycans were also evaluated. While none of these enzymes appreciably transferred GlcNDAz, other functionally redundant GlcNAc-transferases were not available. Therefore, it remains possible that GlcNDAz may also be incorporated into cell surface O-linked glycans or polyLacNAc.
Figure 2. GlcNDAz is incorporated into N-glycans.
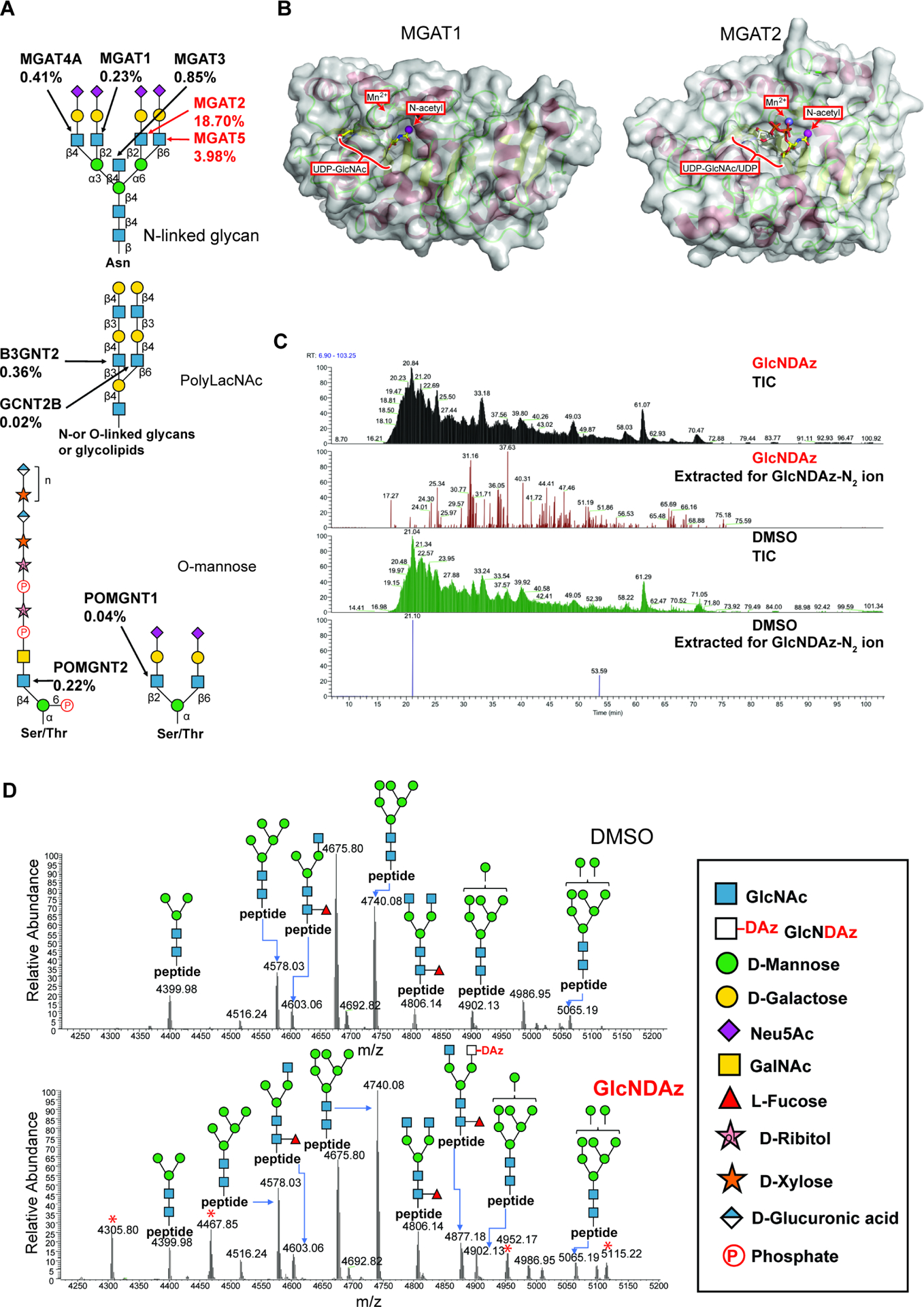
(A) Biosynthetic roles of GlcNAc-transferases were examined using recombinant Golgi GlcNAc-transferases (Fig. S2). Percentages represent relative activity of GlcNAc transferases in using UDP-GlcNDAz as a substrate compared to UDP-GlcNAc. Percentages are averaged from two trials. (B) Modeled structures of human MGAT1 and MGAT2 (PDB 5VCM) (Kadirvelraj et al., 2018) in complex with UDP-GlcNAc. The human MGAT1 structure was modeled based on the structure of the rabbit MGAT1:UDP-GlcNAc:Mn2+ (PDB 1FOA). (Unligil et al., 2000) A magenta sphere marks the methyl group to which the diazirine is attached in GlcNDAz. (C) Glycopeptides from T84 UAP1(F383G) cells treated with Ac3GlcNDAz-1P(AcSATE)2 or DMSO were analyzed by mass spectrometry, and ion chromatograms were extracted for the GlcNDAz – N2 ion. Chromatograms are from single trial. (D) Comparison of glycoforms detected on peptide 38VVRPDSELGERPPEDNQSFQYDHEAFLGK66 from human reticulocalbin-1 from T84 UAP1(F383G) cells treated with Ac3GlcNDAz-1P(AcSATE)2 or vehicle. Red asterisks label peaks (m/z 4305.80, 4467.85, 4952.17, 5115.22) assigned to other co-eluting glycopeptides. Spectrum is from single trial. See also Fig. S2, Fig. S3, Fig. S4, and Data S1.
We compared structures of MGAT1 and MGAT2 in their binding sites for UDP-GlcNAc donor. Because structures of human MGAT1 and human MGAT2 with UDP-GlcNAc are not available, models were built using available structural data (Fig. 2B). (Kadirvelraj et al., 2018; Unligil et al., 2000) For MGAT1, the N-acetyl group of UDP-GlcNAc is buried with limited space to accommodate the diazirine, consistent with its inability to use UDP-GlcNDAz. In contrast, MGAT2 has a more surface exposed donor binding pocket consistent with the modest activity with UDP-GlcNDAz we observed. Recent structural data for MGAT5 indicate that this enzyme also binds UDP-GlcNAc with the N-acetyl group exposed to solvent. (Darby et al., 2020)
With the knowledge that UDP-GlcNDAz was produced and that some Golgi-resident GlcNAc-transferases could accept it, we used glycoproteomics mass spectrometry to determine whether GlcNDAz was incorporated into N-glycans. We employed a multi-stage enrichment strategy.(Shajahan et al., 2020) Briefly, membrane proteins from T84 UAP1(F383G) cells cultured with Ac3GlcNDAz-1P(AcSATE)2 were enriched through two-step ultracentrifugation by differential dissolution of proteins.(Nielsen et al., 2005) The membrane protein pellet was dissolved in urea lysis buffer with reducing agent, alkylated and precipitated. Precipitated proteins were proteolyzed by trypsin, enriched by zwitterionic hydrophilic interaction liquid chromatography (ZIC-HILIC) and analyzed by LC-MS/MS with higher-energy collisional dissociation (HCD) and product-triggered collision-induced dissociation (CID). When the total ion chromatogram (TIC) was extracted for the oxonium ions corresponding to GlcNDAz – N2, a number of peaks were detected, which were absent in a sample from cells cultured without Ac3GlcNDAz-1P(AcSATE)2 (Fig. 2C). Because previous studies showed that azide-functionalized GlcNAc (GlcNAz) can be epimerized to ManNAz by GlcNAc 2-epimerase,(Luchansky et al. 2003) resulting in the azide group being incorporated into sialic acid,(Shajahan et al. 2020) we asked whether GlcNDAz was converted to diazirine-modified sialic acid (SiaDAz) in a similar manner. Extraction for the SiaDAz – N2 ion did not support the presence of diazirine-modified sialic acid (Fig. S3A). MS/MS analysis was performed to gain insight into position(s) at which GlcNDAz was incorporated. MS2 fragmentation by HCD and CID on N-glycopeptides with GlcNDAz modification confirmed that GlcNDAz is on terminal arms of the glycans but not in the glycan core. The presence of peaks corresponding to peptide with GlcNAc fragments (Y1 fragment) and lack of peaks corresponding to peptide with GlcNDAz fragments in HCD spectra indicates that the location of GlcNDAz is not at the glycan core. Moreover, CID fragmentation showed neutral loss of GlcNDAz fragments from glycan termini (Data S1). GlcNDAz was detected in biantennary and triantennary complex N-glycans of multiple proteins (Fig. 3), and the extent of GlcNDAz incorporation varied among glycoforms examined (Fig. S4). Also, the ratio of Hex:HexNAc found in some detected glycans implied that incorporated GlcNDAz could be further extended with Gal (Fig. 3).
Figure 3. GlcNDAz-containing glycopeptides.

Glycopeptides extracted from T84 UAP1(F383G) cells treated with Ac3GlcNDAz-1P(AcSATE)2 were identified by mass spectrometry. Possible glycan structures are proposed based on glycan composition, MS2 analysis, and biosynthesis pathways. We make the assumption that GlcNDAz is most likely to be found attached to α1–6Man in the N-glycan core, but data do not exclude other possible antennary arrangements. Data are from single trial.
GlcNDAz engineering results in small perturbations to N-glycan structure
We considered whether GlcNDAz engineering perturbed N-glycan structures. Glycoproteomics analysis of an N-linked glycopeptide 38VVRPDSELGERPPEDNQSFQYDHEAFLGK66 from human reticulocalbin-1 revealed GlcNDAz incorporation on a biantennary glycoform. Comparison of data from control cells and Ac3GlcNDAz-1P(AcSATE)2 treated cells revealed only modest differences in the glycosylation pattern except for the presence of GlcNDAz-modified glycoforms in the treated case (Fig. 2D). Thus, these data do not show evidence for dramatic alterations in N-glycan structure due to GlcNDAz engineering.
We also measured the effects of GlcNDAz incorporation on binding of lectins that recognize various glycan structures (Fig. 4A). (Gao et al., 2019) T84 UAP1(F383G) cells were cultured with Ac3GlcNDAz-1P(AcSATE)2 or vehicle control. First, cells were fixed with paraformaldehyde and binding of lectins to cell surface glycoconjugates was measured by enzyme-linked lectin binding (ELLA), using multiple lectin concentrations (Fig. S5). Statistical comparison was performed at non-saturating lectin concentrations (Fig. 4B). One GlcNAc-recognizing lectin (succinylated wheat germ agglutinin, WGA) (Monsigny et al., 1980) showed no Ac3GlcNDAz-1P(AcSATE)2-dependent changes in lectin binding. However, Ac3GlcNDAz-1P(AcSATE)2 treatment led to an increase in binding of another GlcNAc-recognizing lectin, Griffonia (Bandeiraea) simplicifolia lectin II (GSL II) that preferentially recognizes highly branched N-glycans. (Nakamura-Tsuruta et al., 2006) Binding of Maackia amurensis lectin II (MAL II), which recognizes α2–3-linked sialic acid, (Geisler and Jarvis, 2011) was unaffected by Ac3GlcNDAz-1P(AcSATE)2 treatment, but a small increase in binding of Sambucus nigra lectin (SNA), which recognizes α2–6-linked sialic acid was observed. Binding of phytohemagglutinin-L (PHA-L), which recognizes β1–6 branches on N-glycans, of CTB, which recognizes multiple structures including fucosylated glycans, (Prudden et al., 2017) and of Datura stramonium lectin (DSL), which recognizes both polyLacNAc and β1–4 GlcNAc on α1–3 mannose,(Cummings and Kornfeld, 1984) were also unaffected by Ac3GlcNDAz-1P(AcSATE)2 treatment. A lectin that recognizes polyLacNAc and β-linked galactose (human galectin-1), (Merkle and Cummings, 1987) (Nielsen et al., 2018; Stowell et al., 2008a) and a lectin that recognizes β1–4-linked galactose (Erythrina cristagalli lectin, ECA) also showed no change of binding with Ac3GlcNDAz-1P(AcSATE)2 treatment, although one polyLacNAc recognizing lectin (Lycopersicon esculentum lectin, LEL) showed an insignificant decrease in binding to cells cultured with Ac3GlcNDAz-1P(AcSATE)2. Next, we performed lectin blotting of lysates from cells treated with Ac3GlcNDAz-1P(AcSATE)2 or vehicle control (Fig. 4C). Lectin blot analysis provides information about both intracellular and extracellular glycoproteins. Lectin blots confirmed the Ac3GlcNDAz-1P(AcSATE)2-dependent increase in GSL II binding, although the GSL II lectin blot could also reflect recognition of O-GlcNAc glycans.(Ma and Hart, 2014) Also, a dramatic Ac3GlcNDAz-1P(AcSATE)2-dependent reduction in PHA-L binding was observed, mostly confined to a single band. Ac3GlcNDAz-1P(AcSATE)2-dependent increase in mobility of lectin-reactive species was observed for some lectins, notably DSL and LEL.
Figure 4. Effects of GlcNDAz incorporation on N-glycan structure.
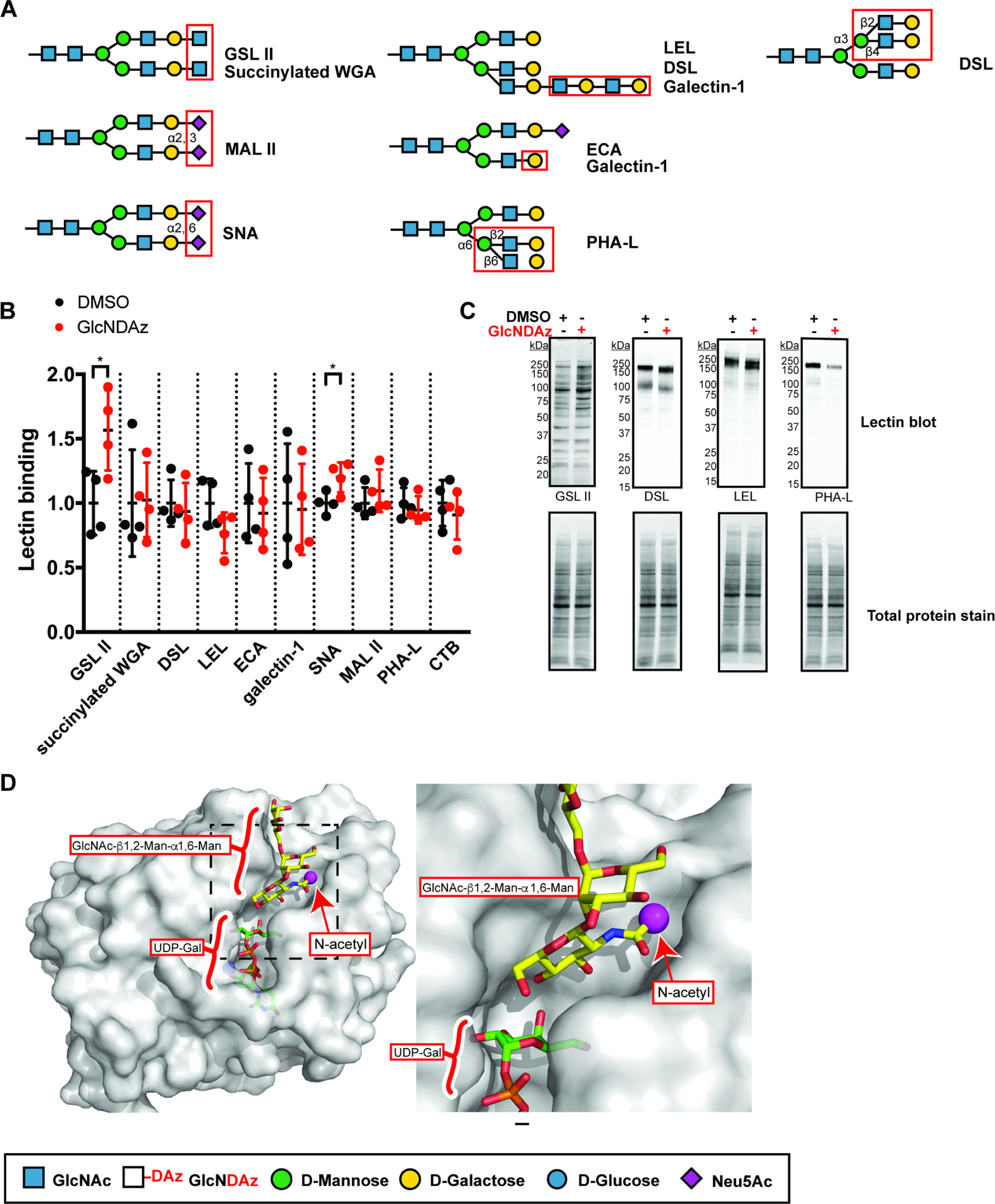
(A) Specificity of lectins used in (B) and (C). (B) Lectin binding to T84 UAP1(F383G) cells treated with Ac3GlcNDAz-1P(AcSATE)2 at non-saturating concentrations (Fig. S3) and normalized to vehicle-treated cells. Lectin concentration used: GSL II (1 μg/ml), succinylated WGA (0.1 μg/ml), DSL (1 μg/ml), LEL (1 μg/ml), ECA (0.1 μg/ml), galectin-1 (1 μg/ml), SNA (3 μg/ml), MAL II (3 μg/ml), PHA-L (3 μg/ml), CTB (3 μg/ml). Data represent mean ±SD from four biological replicates, each of which is averaged from three technical replicates. (C) Lectin blot of total lysates from T84 UAP1(F383G) cells treated with Ac3GlcNDAz-1P(AcSATE)2 or vehicle. Blots are representative of biological triplicate. (D) Structure of human B4GALT1 (overlay of PDB 2AEC (Ramasamy et al., 2005) and PDB 1TVY (Ramakrishnan et al., 2006)) in complex with UDP-Gal and trisaccharide acceptor containing terminal GlcNAc is shown with zoom-in for the area indicated by the dotted box. A magenta sphere marks the methyl group to which the diazirine is attached in GlcNDAz. See also Fig. S5.
Taken together, GlcNAc-transferase assays, glycoproteomics analysis, and lectin binding data demonstrate that GlcNDAz can be metabolically incorporated into antennae of complex N-glycans. While diazirine-containing glycans are similar to those found in non-engineered cells, the process does result in some perturbations to N-glycan structure. Notably, increased GSL II binding was detected by both ELLA and lectin blot, suggesting that Gal may be added less efficiently to GlcNDAz than to GlcNAc residues. Similarly, the increased mobility of DSL and LEL reactive bands in the lectin blots suggests that GlcNDAz incorporation may interfere with antennae extension. Also, decreased PHA-L binding observed in the lectin blot but not the ELLA assay could reflect decreased N-glycan branching, particularly on intracellular glycoproteins. Nonetheless, glycoproteomics analysis demonstrates that Gal addition to GlcNDAz can occur, and mature GlcNDAz-containing glycans were detected, implying that GlcNDAz does not always act as a chain terminator. This observation is consistent with structural analysis of B4GALT1, a galactosyltransferase that is ubiquitously expressed and a likely candidate for catalyzing addition of Gal to GlcNDAz.(Hennet, 2002) Inspection of B4GALT1 crystal structures reveals that the N-acetyl group of GlcNAc on trisaccharide acceptors is localized in a relatively exposed binding pocket (Fig. 4D).(Ramakrishnan et al., 2012; Ramasamy et al., 2005) In sum, GlcNDAz incorporation yields N-glycans that are similar but not identical to those produced by normal cells.
GlcNDAz is used to covalently capture glycan-mediated binding interactions
Having established production of mature GlcNDAz-containing glycans, we tested whether metabolically incorporated diazirine could be used to photochemically crosslink known glycan recognition events. We selected two GBPs – galectin-1 and CTB – for which information about glycan specificity has been reported. (Li et al., 2019; Prudden et al., 2017; Stowell et al., 2008a) T84 UAP1(F383G) cells were cultured with Ac3GlcNDAz-1P(AcSATE)2, then the GBP was added extracellularly and allowed to bind to cell surface glycoconjugates at 4 °C to prevent endocytosis (Fig. 5A). Cells were irradiated to activate the crosslinker, after which they were immediately lysed. Lysates were analyzed by immunoblot using antibodies against GBPs. Both GBPs have low molecular weights (~15 kDa for galectin-1 and ~12 kDa for CTB) and are not retained in low percentage gels. However, when GBPs are crosslinked to N-linked glycoproteins, their apparent molecular weight increases and crosslinked species can be detected (Fig. 5B). For both galectin-1 (Fig. 5C) and CTB (Fig, 5D), no crosslinked species were observed when UV was not applied and only minimal crosslinking was detected when Ac3GlcNDAz-1P(AcSATE)2 was omitted, while robust crosslinking was detected when Ac3GlcNDAz-1P(AcSATE)2 was included and UV was applied. Background crosslinking observed in the absence of the crosslinker might be due to radical species formed by UV irradiation of aromatic amino acid residues. (Russ et al., 2005)
Figure 5. GlcNDAz can be used to covalently capture glycan-mediated binding interactions.

(A) Scheme of crosslinking experiment. (B) Scheme of detecting crosslinking complexes by immunoblot analysis of total cell lysates. Crosslinking of human galectin-1 (~15 kDa) and CTB (~14 kDa) to larger cell surface glycoproteins results in a shift of galectin-1 and CTB to apparent higher molecular weight. (C) Crosslinking of galectin-1 to glycoproteins from T84 UAP1(F383G) cells. (D) Crosslinking of cholera toxin to glycoproteins from T84 UAP1(F383G) cells. (E) Crosslinking of galectin-1 with pre-treatment with 40 μg/ml ECA lectin. (F) Crosslinking of galectin-1 in presence of 100 mM of mono- or disaccharides, or with pretreatment of cells with 1 μg/ml of kifunensine for 72 h (G) Crosslinking of galectin-1 to glycoproteins with pre-treatment by StcE. (H) Crosslinking of CTB with cells cultured with 2F-fucose for 72 h, pretreatment with 40 μg/ml of lectin AAL, or in presence of 100 mM of l-fucose, but not d-fucose. All blots are representative of biological triplicate.
We interrogated the nature of the galectin-1-glycoprotein crosslinked species. Galectin-1 preferentially binds Galβ1–4GlcNAc (N-acetylactosamine, LacNAc) and inclusion of the ECA lectin, which also recognizes LacNAc, resulted in reduced crosslinking (Fig. 5E). Galectin-1 binding to LacNAc can be competitively inhibited by Galβ1–4Glc (lactose, Lac), so we also tested whether different disaccharides and monosaccharides interfered with crosslinking. (Lantéri et al., 2003; Shimura et al., 2002) Crosslinking was eliminated by inclusion of 100 mM Lac, but not by Glcα1–6Glc (maltose), Glcβ1–4Glc (cellobiose), or Glcα1–2Fruβ (sucrose) (Fig. 5F). Similarly, the terminal sugar of LacNAc, d-Gal, inhibited galectin-1-glycoprotein crosslinking, but l-Gal, d-Glc, and d-GlcNAc did not. Also, culturing cells with kifunensine, an inhibitor of N-glycan maturation, (Elbein et al., 1990) reduced crosslinking, indicating that at least some glycans recognized by galectin-1 are displayed on N-linked glycoproteins (Fig. 5G). However, pre-treatment of cells with the protease StcE, which cleaves adjacent to GalNAc-type O-linked glycosylation sites, (Malaker et al., 2019) also reduced crosslinking. This result suggests that some crosslinking may occur through recognition of O-linked glycans. In sum, the galectin-1 crosslinking results are consistent with previous studies demonstrating that galectin-1 binds to cells through both N-linked and O-linked glycans.(Nielsen et al., 2018; Seelenmeyer et al., 2003)
We performed similar analysis for CTB (Fig. 5H). CTB recognizes multiple glycans including the canonical receptor ganglioside GM1 as well as fucosylated glycoconjugates, which mediate a large fraction of CTB binding to T84 cells.(Wands et al., 2015) As predicted, culturing cells with a metabolic inhibitor of fucosylation, 2F-fucose,(Rillahan et al., 2012) reduced crosslinking. Inclusion of l-Fuc, but not d-Fuc, in the binding buffer reduced crosslinking. Finally, inclusion of Aleuria aurantia lectin (AAL), which competes with CTB for binding to fucosylated glycoproteins, (Wands et al., 2015) resulted in reduced crosslinking.
Together, these results demonstrate that metabolically-incorporated GlcNDAz can be used to covalently crosslink known GBP-glycoprotein binding interactions, and that the extent of crosslinking observed depends on the presence and accessibility of preferred glycan ligands for the GBP.
GlcNDAz-enabled proteomics facilitates identification of galectin-1 binding partners
Covalent crosslinking offers the capacity to isolate and identify glycoprotein binding partners of GBPs. To do so, we cultured T84 UAP1(F383G) cells with Ac3GlcNDAz-1P(AcSATE)2 or vehicle, then added biotinylated galectin-1 extracellularly and UV irradiated. Crosslinked complexes were purified using streptavidin beads. Purified material was separated into three regions of an SDS polyacrylamide gel, digested with trypsin, and analyzed by LC-MS/MS. Protein abundance was evaluated and compared to control samples in which Ac3GlcNDAz-1P(AcSATE)2, UV irradiation and biotinylated galectin-1 were omitted (Table S1). Proteins were considered hits if they were enriched more than 5-fold in the experimental sample, if the abundance was greater than 100,000, and with a minimum number of peptide spectrum matches (PSMs) of 2. Proteins that were identified but not quantified and potential contaminants were excluded. The high molecular weight region of the gel yielded 86 hits, the medial molecular weight region yielded 73 hits, and the low molecular weight region yielded 30 hits (Fig. 6A). A total of 103 protein hits were identified (Table S1). More than half of hits were identified in both biological replicates (Fig. 6B). Candidate galectin-1 binding partners were further analyzed with David Informatics Resource 6.8. (Huang et al., 2009b; 2009a) Among the 102 recognized hits (Table S1), the majority are known to be glycosylated: 70 proteins were annotated glycoproteins based on UniProt keywords and 67 proteins were annotated to have N-linked glycosylation sites based on UniProt. We also used the GlyGen Bioinformatics Resource, which catalogs experimentally-demonstrated glycosylation sites.(York et al. 2020) Narrowing analysis to the 46 hits detected in the experimental sample but not in the control sample (i.e. those displaying infinite enrichment) in at least one region of the gel, we found that GlyGen cites experimental evidence of N-glycans for forty hits while additional literature reports evidence for N-linked glycosylation for at least three additional hits.(Kariya et al., 2008; Nittis et al., 2004: Samanta et al., 2011) Thus, from the most highly enriched subset of 46 proteins, 93% are known N-glycoproteins. We conclude that GlcNDAz crosslinking and enrichment is effective at identifying glycoproteins whose characteristics are consistent with galectin-1 binding.
Figure 6. GlcNDAz-enabled proteomics facilitates identification of candidate galectin-1 binding partners.
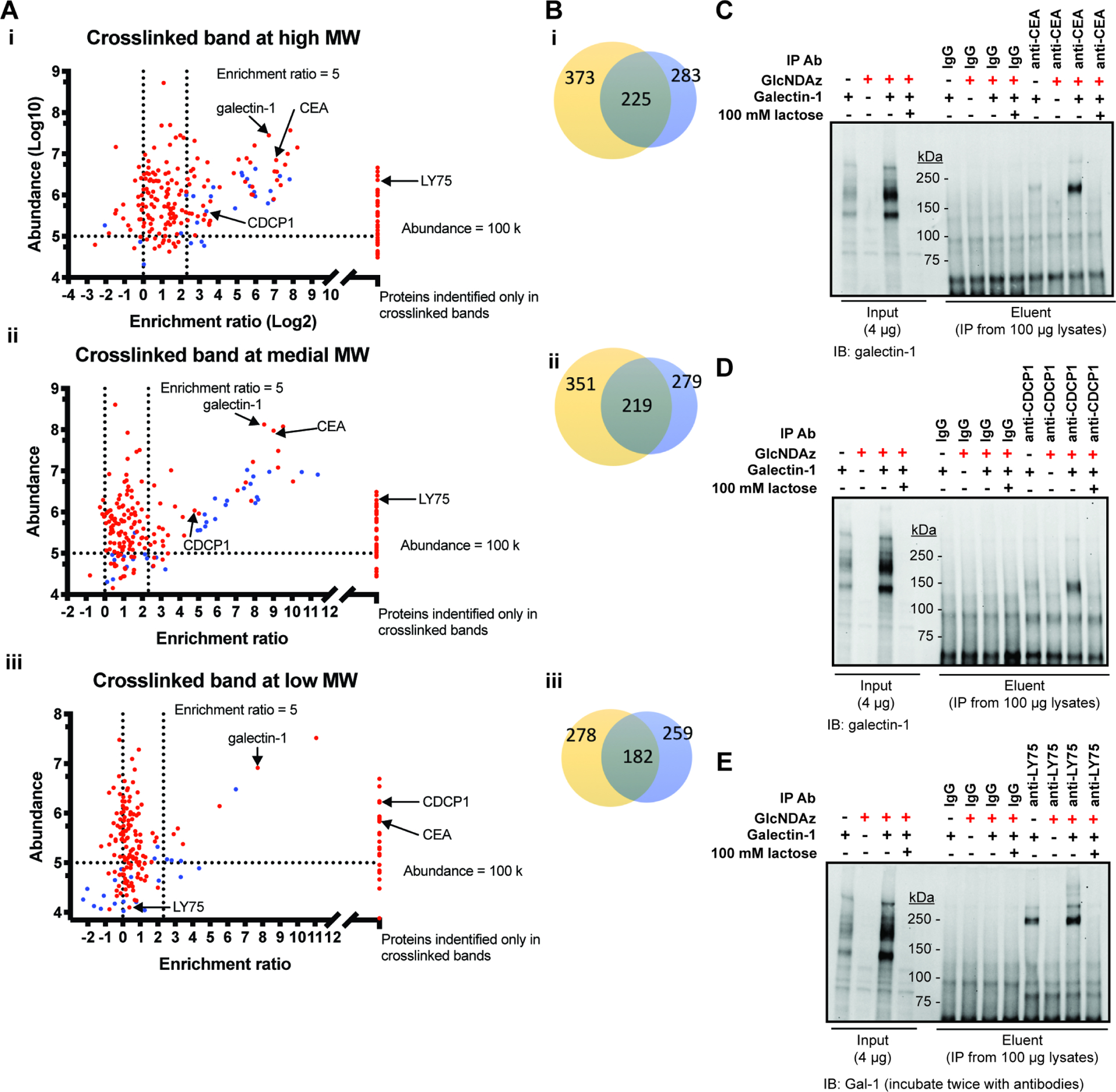
(A) Scatter-plots of proteins from T84 UAP1(F383G) cells identified in the three major crosslinked bands in two biological replicates. Abundance and fold-enrichment represent the average of two biological replicates. Proteins detected in crosslinked but not control samples are shown on the right of each plot. Proteins detected in crosslinked samples in two replicates but detected in control samples in only one replicate are represented with blue symbols. For these proteins, fold-enrichment from the trial in which proteins were identified in both crosslinked and control samples was used in the plot. (B) Venn diagram of proteins identified from each region in two replicates. (C) – (E) CEA (C), CDCP1 (D) and LY75 (E) were enriched by immunoprecipitation with antiCEA, anti-CDCP1 and anti-LY75 antibodies and crosslinked complexes were detected with anti-galectin-1 antibody by immunoblot. Confirmation of successful immunoprecipitation is presented in Fig. S6. See also Table S1. Figures (C)–(E) are representative of biological duplicate using 100 μg/ml galectin-1 for crosslinking. In a third trial conducted with 30 μg/ml galectin-1, similar results albeit with fainter crosslinked bands were obtained.
We evaluated whether previously reported galectin-1 binding partners were identified. Carcinoembryonic antigen (CEA), a galectin-1 ligand identified in an early study from colon carcinoma cell line KM12, was among the top hits from all three crosslinked regions. (Ohannesian et al., 1994) Galectin-1 ligands reported from other cell lines, such as CDCP1, laminin, integrin β1 and CD44, (Camby et al., 2006; Elola et al., 2005; Ito and Ralph, 2012; Law et al., 2016) (Moiseeva et al., 2003) were found among the hits (Table S1). Crosslinking of CEA, CDCP1 and LY75 to galectin-1 was further confirmed by immunoprecipitation (IP) (Fig. 6C–E, Fig. S6) and immunoblot, where a band(s) migrating near or slightly above the molecular weight of the putative binding partner can be detected by anti-galectin-1 antibody. This result demonstrates direct crosslinking of galectin-1 to each of these binding partners.
LAMP1 and LAMP2, identified as galectin-1 ligands from KM12 cells and other cell lines, (Do et al., 1990; Obermann et al., 2017; Ohannesian et al., 1994) were not enriched in the proteomics analysis. We also failed to detect crosslinked complexes between LAMP1 and galectin-1 in IP-enriched samples (Fig. 7A), even though LAMP1 is readily detected in these cells (Fig. S7A and B). We speculated that our inability to detect galectin-1 crosslinking to LAMP1 could be because the extracellularly-added galectin-1 was unable to contact LAMP1 localized to the lysosome or other intracellular compartments. Indeed, galectin-1 displays a different crosslinking pattern when crosslinking was performed in lysates rather than intact cells (Fig. 7B, input lanes). Further, when crosslinking was performed in lysates, crosslinked complexes between LAMP1 and galectin-1 were enriched by LAMP1 immunoprecipitation and detected by galectin-1 immunoblot (Fig. 7B). In contrast, CEA, which was highly enriched in proteomics analysis, was crosslinked to galectin-1 both in lysates and in intact cells (Fig. S7C and D).
Figure 7. GlcNDAz-mediated crosslinking of LAMP1 to galectin-1 occurs in cell lysates but not intact cells.
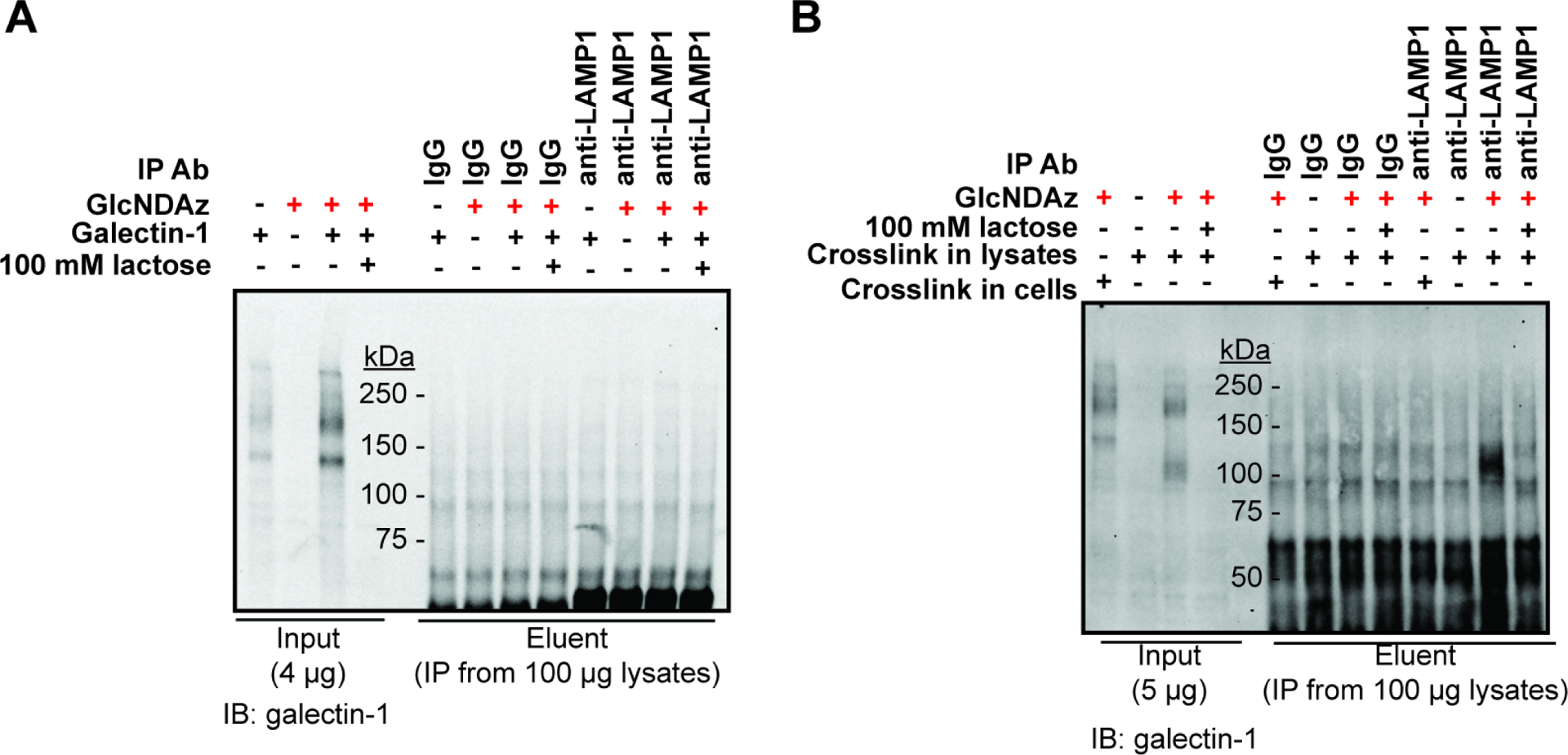
(A) T84 UAP1(F383G) cells were cultured with 100 μM of Ac3GlcNDAz-1P(AcSATE)2 or vehicle. Galectin-1 was added to a final concentration of 100 μg/mL, then samples were UV irradiated. Cells were lysed, and immunopurified for LAMP1. Immunoblot analysis was performed with a galectin-1 antibody. Data are representative of biological duplicate using 100 μg/ml galectin-1 for crosslinking. In a third trial conducted with 30 μg/ml galectin-1, similar results were obtained. (B) T84 UAP1(F383G) cells were cultured with 100 μM of Ac3GlcNDAz-1P(AcSATE)2 or vehicle. Cells were lysed, then 30 μg/mL of galectin-1 was added and UV irradiation applied. Immunopurification was performed with a LAMP1 antibody. Immunoblot analysis was performed with a galectin-1 antibody. Data are representative of biological triplicate. See also Fig. S7.
Discussion
MGE is a powerful method to introduce unnatural functional groups into cellular glycans.(Du et al., 2009) The relaxed substrate tolerance of metabolic enzymes is critical for MGE, but can also allow metabolic interconversions that yield unnatural sugar incorporation at unexpected or undesired locations. For example, MGE conducted with GlcNAz has revealed metabolic conversion to azide-modified N-acetylneuraminic acid (SiaNAz).(Saxon et al., 2002; Shajahan et al., 2020) Here we used MGE to introduce diazirine on GlcNAc residues of N-glycans, which were characterized by mass spectrometry-based glycoproteomics. Diazirine was found attached to antennary GlcNAc, and no SiaDAz was detected. One possible explanation for different outcomes for azide-modified versus diazirine-modified GlcNAc is the size of the unnatural functional groups. Indeed, RENBP, which interconverts GlcNAc and ManNAc, epimerizes azide-modified ManNAc 345 times more rapidly than a ManNAc analog with a levulinoyl modification that is about the same size as the diazirine modification employed here.(Luchansky et al., 2003) Alternately, our strategy of delivering a GlcNAc-1-P analog and overexpressing a UAP1 mutant with relaxed specificity (Yu et al., 2012) may direct more of the unnatural sugar flux through UDP-GlcNAc analogs, rather than toward ManNAc and sialic acid analogs. Also, we found that the Golgi-resident GlcNAc-transferase MGAT2 uses the UDP-GlcNDAz although with reduced efficiency as compared to the natural substrate. This preference is similar that of OGT, which displayed a 12-fold difference in catalytic efficiency between UDP-GlcNAc and UDP-GlcNDAz.(Rodriguez, et al., 2015) We propose that GlcNDAz is most likely to be found attached to α1–6Man in the N-glycan core; isolation of GlcNDAz modified N-glycans or N-glycopeptides and analysis by NMR would be required to demonstrate GlcNDAz position(s) more definitively. While the efficiency of GlcNDAz incorporation was sufficient to support crosslinking, further improvements may be possible through targeted mutations to MGAT2 or other enzymes.
Binding of galectin-1 and CTB to T84 cell surfaces was unaffected by Ac3GlcNDAz-1P(AcSATE)2 treatment, suggesting that the MGE did not eliminate production of preferred glycan ligands for these GBPs. Nonetheless, knowledge of even modest perturbations to glycan structures is essential for reliable use of this tool. While mass spectrometry-based glycoproteomics demonstrated that mature GlcNDAz-containing N-glycans were produced, lectin binding experiments revealed some alterations in glycan structures. One possible explanation is that B4GALTs may inefficiently extend GlcNDAz-containing glycans, resulting in fewer mature terminal structures and shorter polyLacNAc chains. Also, the MGE approach results in a high concentration of UDP-GlcNDAz. This metabolic perturbation could affect the production of GlcNAc/GlcNDAz-containing glycans. Elevated UDP-GlcNAc also has the potential to lead to increased sialylation through activation of GNE, (Hinderlich et al., 2015) consistent with the small increase in SNA binding.
GlcNDAz crosslinking can offer insight into the preferred glycan ligands for a GBP. For example, the intensity of CTB-glycoprotein crosslinking was reduced when preferred fucosylated ligands were absent. Also, glycoproteins that display the preferred ligands for a GBP can be identified by covalent crosslinking, followed by purification and proteomics analysis. Known galectin-1 binding partners, such as CEA, (Ohannesian et al., 1994) CDCP1, (Law et al., 2016) and integrin β4, (Moiseeva et al., 2003) were identified through such a workflow, which also revealed additional potential galectin-1 partners. The common occurrence of N-linked glycoproteins among the most highly enriched hits implies a high specificity for identification of genuine galectin-1 binding partners but further improvements may be possible. These might include the use of PNGase F to specifically elute crosslinked partners with N-glycans or the use of lactose-pretreated cells as a comparator in the proteomics analysis. We confirmed crosslinking to galectin-1 for multiple proteins from the proteomics hit list, including one, LY75, that we believe was not previously identified as a galectin-1 binding partner. In this method, the appearance of a crosslinked complex is indicative of a direct interaction between the crosslinking partners. The represents an advantage over traditional immunopurification approaches in which a third party may mediate an observed binding interaction.
Another factor affecting GlcNDAz crosslinking is the incorporation efficiency of GlcNDAz at different glycan positions. GlcNDAz may not be incorporated into the preferred glycan structures of some GBPs. For example, glycoproteomic analysis did not detect GlcNDAz incorporation into the Man3GlcNAc2 core of N-glycans, which is recognized by some E3 ligases.(Yoshida et al., 2019) Thus, our data confirm the incorporation of GlcNDAz into N-glycans and also set limits on use of this tool. However, the decreased galectin-1 crosslinking to T84 UAP1(F383G) cells treated with StcE also suggests the possible incorporation of GlcNDAz into O-GalNAc glycans, which we plan to explore in future work. Expanding the positions of GlcNDAz incorporation by structure-based engineering of GlcNAc-transferases, such as MGAT1 and POMGNT1, may also facilitate broader use.
Proteomics analysis of galectin-1 crosslinking partners offers some indication of the types of biological insights that may be obtained with this method. Typically, discovery of GBP binding partners is performed by affinity purification from cell lysates, conditions that disrupt the normal cellular architecture and may allow interactions among proteins that normally reside in distinct compartments. Indeed, an early study employing immobilized galectin-1 and KM12 cell extracts identified carcinoembryonic antigen (CEA), LAMP1, and LAMP2 as galectin-1 binding partners. (Ohannesian et al., 1994) The ability of LAMP1 and LAMP2 to interact with galectin-1 was confirmed in lysates from other cell types.(Do et al., 1990; Obermann et al., 2017) However, our proteomics analysis of the binding partners of extracellularly added galectin-1 identified the CEA-displaying protein CEACAM5, but not LAMP1. Because glycoproteomic analysis confirmed the incorporation of GlcNDAz into glycans on multiple asparagine residues of LAMP1, we speculated that the absence of LAMP1 and LAMP2 in the proteomics analysis arose from the fact that the crosslinking was performed in intact cells where extracellularly added galectin-1 did not contact lysosomal proteins. Indeed, when cells were lysed to allow mixing of intracellular and extracellular components prior to UV irradiation, we observed crosslinking of LAMP1 to galectin-1. Thus, the ability to report on cellular context is an advantage of the crosslinking method. We plan to examine whether cytoplasmic galectin-1 is capable of crosslinking to lysosomal proteins such as LAMP1 and LAMP2, and whether the extent of these interactions changes under conditions that induce lysosomal damage pathways, which expose lysosomal glycoproteins to cytoplasmic GBPs and recruit multiple galectins, including galectin-1, to damaged lysosomes. (Aits et al., 2015; Jia et al., 2018; 2020)
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jennifer J. Kohler (jennifer.kohler@utsouthwestern.edu).
Materials availability
This study did not generate new unique reagents. Cell lines and limited amounts of Ac3GlcNDAz-1P(AcSATE)2 and UDP-GlcNDAz are available through the lead contact. The pSin4 UAP1(F383G) plasmid is available from Addgene using ID 169891. GlcNAc-transferase expression constructs are available from dnasu.org using the indicated clone IDs.
Data and code availability
Raw data of proteomics analysis were deposited to MassIVE with the identifier MSV000087640. Crosslinked bands from the first biological replicate from high, medial, and low molecular weight have the sample names f.MSV000087640/raw/LUM1_661503.raw, f.MSV000087640/raw/LUM1_661504.raw and f.MSV000087640/raw/LUM1_661505.raw, respectively. Control bands from the first biological replicate from high, medial, and low molecular weight have the sample names f.MSV000087640/raw/LUM1_661500.raw, f.MSV000087640/raw/LUM1_661501.raw, and f.MSV000087640/raw/LUM1_661502.raw, respectively. Crosslinked bands from the second biological replicate from high, medial, and low molecular weight have the sample names f.MSV000087640/raw/LUM1_671792.raw, f.MSV000087640/raw/LUM1_671790.raw and f.MSV000087640/raw/LUM1_671788.raw, respectively. Control bands from the second biological replicate from high, medial, and low molecular weight have the sample names f.MSV000087640/raw/LUM1_671793.raw, f.MSV000087640/raw/LUM1_671791.raw and f.MSV000087640/raw/LUM1_671789.raw, respectively.
The glycoproteomics data have been deposited at GlycoPost (Watanabe et al., 2021) with the dataset identifier GPST000201.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Male T84 cells stably overexpressing UAP1(F383G) were generated by infection with pSIN4 UAP1(F383G) lentivirus. (Rodriguez et al., 2015; Yu et al., 2012) Male T84 UAP1(F383G) cells (Yu et al., 2012) were cultured in DMEM/F12 medium (Thermo Fisher Scientific, Cat. No. 11330032) containing 5 % fetal bovine serum (Thermo Fisher Scientific, Cat. No. 16000044), 100 μg/mL streptomycin and 100 units/mL penicillin (Thermo Fisher Scientific, Cat. No. 15140122). Cells were free of mycoplasma contamination as confirmed by MycoAlert Detection Kit (Lonza, Basel, Switzerland; Cat# LT07–118). Cell line has not been authenticated.
METHOD DETAILS
Chemicals, antibodies, lectins, and other protein reagents
Dimethyl sulfoxide (DMSO) was purchased from Fischer Scientific (Cat. No. BP231–100). Ac3GlcNDAz-1P(AcSATE)2 and UDP-GlcNDAz disodium salt were synthesized by Wuxi AppTec (Tianjin). Stock concentration of Ac3GlcNDAz-1P(AcSATE)2 is 50 mM in DMSO. Lactose (Cat. No. 61339–25g), cellobiose (Cat. No. 22150–10G), d-galactose (Cat. No. G0750–100G), N-acetylglucosamine (Cat. No. A3286–25G), d-glucose (Cat. No. G7021–100G), l-galactose (Cat. No. G7134–100MG), d-fucose (Cat. No. F8150–1G) and l-fucose (Cat. No. F2252–5G) were purchased from Sigma. Maltose (Cat. No. 190237) was purchased from MP Biomedicals. Sucrose was purchased from Fisher Scientific (Cat. No. BP220–1). Kifunensine (Cat. No. K1140–1MG) was purchased from Sigma and stock concentration is 1 mg/mL in Dulbecco’s phosphate-buffered saline (DPBS). 2F-peracetyl-fucose (Cat. No. 344827–10MG) was purchased from Millipore and the stock concentration is 200 mM in DMSO. All stocks are stored at −20 °C unless otherwise specified.
Antibodies were from the following sources: galectin-1 (clone D608T, #12936) (1:1000 for WB), LAMP1 (clone D2D11, #9091) (1:1000 for WB, 1:100 for IP), CDCP1 (clone D1W9N, #13794) (1:1000 for WB, 1:200 for IP), CDCP1(#4115) (1:1000 for WB) were purchased from Cell Signaling Technologies; cholera toxin B subunit (ab34992) (1:5000 for WB) and LY75 (ab124897) (1:2000 for WB, 1:50 for IP) were purchased from Abcam; α-tubulin (T6199) (1:2000 – 1:5000 for WB) was purchased from Sigma; CEA (clone 1106, MA5–14675) (1:2000 for WB, 1:100 for IP) was purchased from Thermo Fisher Scientific; galectin-1 (AF1152) (0.1 mg/mL stock, 1:500 dilution for WB used in comparison of crosslinking in intact cells and in lysates) was purchased from R&D Systems. IRDye® 680 RD conjugated goat anti mouse IgG (H+L) (926–68070), IRDye® 800CW conjugated goat anti rabbit IgG (H+L) (926–32211), IRDye® 800CW conjugated goat anti mouse IgG (H+L) (926–32210) and IRDye® 800CW conjugated donkey anti goat IgG (H+L) (925–32214) were purchased from LI-COR and used as 1:10000 dilution for WB.
Biotin-labeled lectins were purchased from Vector Laboratories: GSL II (B-1215), succinylated WGA (B-1025S), DSL (B-1185), LEL (B-1175), ECA (B-1145), SNA (B-1305), MAL II (B-1265), PHA-L (B-1115), ConA (B-1005). Unlabeled AAL (L-1390) was purchased from Vector Laboratories.
Recombinant galectin-1 and biotin-labeled galectin-1 were generated by Dr. Connie Arthur and Dr. Sean Stowell at Emory University.[ref] StcE was a gift from Drs. Stacy Malaker and Carolyn Bertozzi at Stanford University. (Malaker et al., 2019) Cholera toxin B subunit (C9903) was purchased from Sigma. Biotin-conjugated cholera toxin B subunit (C-34779) was purchased from Thermo Fisher Scientific. IRDye® 800CW conjugated streptavidin (926–32230) was purchased from LI-COR. Normal rabbit IgG (NI01–100UG) and normal mouse IgG (NI03–100UG) were purchased from Millipore Sigma.
Glycopeptide analysis of GlcNDAz incorporation into N-glycans
Cell culture.
For analysis of GlcNDAz incorporation into N-glycans by mass spectrometry, 2.25 × 106 of T84 UAP1(F383G) cells were plated with 20 mL of complete medium in nine 15-cm tissue culture plates. After 24 h and 48 h, 40 μL of Ac3GlcNDAz-1P(AcSATE)2 stock or DMSO was added to each plate. About 72 hr after cells were plated, cells were lifted from the plates by incubation with 1 mM of ethylenediaminetetraacetic acid (EDTA) in DPBS at 37 °C and quenched with cell culture medium. Cells were collected by centrifugation and washed with DPBS. Cell pellets were frozen in liquid nitrogen and stored at −80 °C.
Enrichment of membrane proteins from the cells and protease digestion.
For the membrane proteins enrichment, the T84 cells (30 × 106) were pelleted (W, whole cell lysate was prepared from an aliquot) at 1000 × g for 5 min and homogenized in high salt buffer (1.0 mL; 2.0 M NaCl, 5.0 mM EDTA, pH 7.4) by passing through 26-gauge syringe for 10 times. The homogenate was then probe sonicated for 5 min using 10 sec intervals pulsed program and ultracentrifuged at 90000 × g for 15 min at 4 °C. The supernatant (S1) was separated and the pellet (P1) was re-suspended in 1.0 mL sodium carbonate buffer (0.1 M Na2CO3, 1.0 mM EDTA, pH 11.3) and kept for 30 min on ice. The suspension was subsequently ultracentrifuged at 90000 × g for 90 min at 4 °C. The supernatant (S2) and the pellet (P2) was separated and the pellet containing membrane fraction was dissolved in 1.0 mL urea lysis buffer (8.0 M urea, 1.0 M NaCl, 4 % CHAPS, 100 mM dithiothreitol (DTT), 200 mM Tris-HCl, pH 8.0) and kept at 50 °C for 45 min for denaturation. Subsequently, 55 mg of iodoacetamide was added and incubated at room temperature in dark for 45 min. The proteins were precipitated by sequential addition of chloroform (1.5 mL), methanol (4.5 mL) and water (5.0 mL) and the precipitated proteins were separated by centrifugation at 3000 × g for 5 min. The proteins were washed once with 500 μL of methanol, centrifuged, and resuspended in 50 mM ammonium bicarbonate buffer (pH 8.0).The proteins were digested by adding a cocktail of 25 μg each of trypsin-LysC protease and incubated at 37 °C for 24 h. MS compatible protease inhibitor cocktail was added to all buffers prior to addition.
Enrichment of glycopeptides from the protease digest.
The glycopeptides were enriched by ZIC-HILIC method using ProteoExtract® Glycopeptide Enrichment Kit (Millipore, 72103–3) with some modification from manufacturer’s protocol, as reported previously. (Shajahan et al., 2020) About 100 μg of peptides (estimated by nanodrop spectrophotometer) in 30 μL were mixed with 150 μL of binding buffer. 150 μL of ZIC-HILIC resin was centrifuged to remove the solvents, the samples were added to it and vortexed for 20 min at room temperature. The samples with resin were centrifuged, supernatants were discarded, the resin was resuspended in 450 μL washing buffer and incubated at room temperature for 10 min. The washing procedure was repeated two more times. The enriched glycopeptides were eluted by adding 225 μL of elution buffer and subsequently by 225 μL of 0.1 % formic acid, incubating for 5 min at room temperature in each case. The eluted fractions were combined, evaporated to dryness and the enriched glycopeptides were dissolved in 25 μL of 0.1 % formic acid for the subsequent LC-MS/MS analysis.
Data acquisition of protein digest samples using nano-LC-MS/MS.
The glycopeptides were analyzed on an Orbitrap Fusion Tribrid mass spectrometer coupled with a Dionex Ultimate 3000 LC-MS/MS system. Acclaim PepMap® nano-LC columns (Thermo Scientific; Cat No. 164568) of 150 mm length with 75 μm internal diameter (id), filled with 3 μm, 100 °A C18 material (reverse phase) were used for chromatographic separation. Solvent A consisted of 0.1% formic acid (Fluka) in LC-MS grade water (Sigma Aldrich), and solvent B consisted of 0.1% formic acid, 80 % acetonitrile (Sigma Aldrich) and 20 % LC-MS grade water. Samples were run for 180 min at 300 nL/min with gradients as follows, 180 min LC method: 5–30 % solvent B within 90 min; 30–60 % solvent B within 60 min; 60–99 % solvent B within 15 min; 99–80 % solvent B within 5 min; 80–10 % solvent B within 5 min and 10–5 % solvent B within 5 min. All instrument methods for the mass spectrometer were set up in data dependent acquisition mode with an automatic gain control (AGC) target value of 5 × 105 for precursor scans. After the precursor ion scan at 120,000 resolution in Orbitrap analyzer, intense precursors were selected for subsequent fragmentation using HCD and CID (product-triggered based on the presence of glycan oxonium ions in the HCD) within 3 sec at a normalized collision energy of 28 and 35, respectively. For internal mass calibration 445.120025 ion was used as lock mass with a target lock mass abundance of 0 %. Charge state screening and dynamic exclusion was enabled (exclusion size list 100, exclusion duration 30 s), and precursors with unknown charge state or a charge state of +1 were excluded. The fragment ions were analyzed in the Orbitrap analyzer at 15000 resolution.
Identification of GlcNDAz modified glycopeptides.
The identification of the LC-MS/MS glycopeptide spectra file was performed using the commercial software Byonic™ 2.3 (https://www.proteinmetrics.com/products/byonic/). In order to improve the detection of GlcNDAz modified N-glycopeptides, the LC-MS/MS data files were processed as described previously. (Shajahan et al., 2020) Briefly, an in-house software SpectraPicker (freely available at our source code repository https://github.com/ReneRanzinger/SpectraFiltering/) that allows extracting MS scans based on user defined key ions in the MS2 data is used to extract spectra with oxonium ions of GlcNDAz-N2 (M+H − 244.12, +/− 5ppm accuracy). The spectra with an intensity value of more than 10 % of the highest MS2 intensity was saved into a separate filtered data file in mzXML format. Further, Byonic™ software-based search was conducted on the filtered spectra file for the annotation of the GlcNDAz bearing glycoproteins using a custom created glycan database with GlcNDAz modified N-glycans. The UniProtKB human reviewed N-glycoproteome proteome data set (download date 14.10.2018) was used for the search with oxidation of methionine and carbamidomethylation of cysteine as variable modifications. A precursor ion tolerance of 5 ppm and fragment ion tolerance of 15 ppm was set for the search with up to two missed cleavage for the target enzyme trypsin/Lys-C.
In vitro GlcNAc transferase activity assay
Protein expression constructs encoding the catalytic domains of MGAT1, MGAT2, MGAT3, MGAT4A, MGAT5, POMGNT1, POMGNT2, B3GNT2, GCNT2B were generated by PCR from a Mammalian Gene Collection clone followed by Gateway recombination into the pDONR221 vector (Moremen et al., 2018). The PCR amplification extended the truncated protein coding regions by inclusion of flanking Gateway att1 recombination sites as well as an extension of the NH2- terminus of the coding region with a TEV protease recognition site as previously described (Moremen et al., 2018). The fusion protein constructs encode an NH2- terminal signal sequence, 8xHis tag, AviTag, “superfolder” GFP, the TEV protease recognition site, and the corresponding truncated protein coding region behind a CMV promoter. Recombinant enzyme expression was accomplished by transient transfection of HEK293 cells (FreeStyle 293-F cells, Life Technologies, Grand Island, NY) as previously described (Moremen et al., 2018). Briefly, HEK293 cells were maintained in serum free Freestyle 293 expression medium (Life Technologies). Transfections were initiated at cell densities of 2.5 × 106 cells/ml by addition of 4 μg/mL of the respective expression plasmid and 9 μg/mL polyethylenimine (linear 25 kDa PEI, Polysciences, Inc., Warrington, PA) to the suspension culture. The cultures were diluted 1:1 with culture medium containing 4.4 mM valproic acid (2.2 mM final) 24 h after transfection, and protein production was continued for a further 5 days at 37°C.
Culture medium was harvested on day 6 and clarified by sequential centrifugation at 1,500 rpm for 10 min, 2,500 rpm for 15 min, 4,000 rpm for 20 min and then passed through a 0.8 μM filter (Pall Corporation). The conditioned culture medium was loaded on a Ni2+-NTA Superflow (Qiagen) column equilibrated with 20 mM HEPES, 300 mM NaCl, 20 mM imidazole, pH 7.4, washed with column buffer, and eluted successively with column buffers containing stepwise increasing imidazole concentrations (40–300 mM). The protein preparations were further purified on a Superdex 75 column (GE Healthcare) preconditioned with a buffer containing 20 mM HEPES, 300 mM NaCl and the final purified protein was concentrated by ultrafiltration to 1 mg/ml.
Enzyme activity was determined using the UDP-Glo™ Glycosyltransferase Assay (Promega) that determined UDP concentration formed as a by-product of the GlcNAc transferase reaction. Assays were performed according to the manufacturer’s instructions using reactions (5 μL) that consisted of a universal buffer containing 100 mM each of MES, MOPS, TRIS, pH 7.0, 0.5 mM donor (UDP-GlcNDAz and UDP-GlcNAc), 0.5 mM acceptor, 5 mg/ml BSA, 5 mM MnCl2 (for MGAT1, MGAT2, MAGT3, MGAT4A, POMGNT1, B3GNT2), and purified enzymes (1–500 ng). Acceptor structures for the respective enzymes were: Man3GlcNAc2-Asn (MGAT1), GlcNAcMan3GlcNAc2-Asn (MGAT2), GlcNAc2Man3GlcNAc2-Asn (MGAT3, MGAT4A, MGAT5) (all kind gifts of Geert-Jan Boons, University of Georgia), O-Man glycopeptide 317 (POMGNT1 and POMGNT2) (kind gift of Lance Wells, University of Georgia), lacto-N-neotetraose (B3GNT2 and GCNT2B) (Carbosynth). Assays were initiated with the addition of enzyme and carried out for 1 h at 37°C. Reactions were stopped by mixing with an equal volume of UDP Detection Reagent (5 μL) in white polystyrene, low-volume, 384-well assay plates (Corning) and incubated for 60 min at room temperature. After incubation, luminescence measurements were performed using a GloMax Multi Detection System plate reader (Promega). Luminescence values were compared to a standard curve for quantification of UDP produced.
Lectin binding
Cell culture.
To evaluate the global cellular glycome changes resulting from GlcNDAz incorporation, 500,000 T84 UAP1(F383G) cells were plated with 5 mL of complete medium in a 6-cm tissue culture plate. At 24 h and 48 h after cells were plated, 10 μL of Ac3GlcNDAz-1P(AcSATE)2 stock or DMSO was added to each plate. About 72 h after cells were plated, cells were rinsed with DPBS and scraped into 500 μL of RIPA buffer (50 mM Tris-HCl, pH=8.0, 150 mM NaCl, 0.1% (w/v) SDS, 0.5% (w/v) sodium deoxycholate and 1% (w/v) IGEPAL CA-630) with protease inhibitor cocktail (Sigma, Cat. No. 11836170001). Cells were lysed by vortexing followed by incubation on ice for 30 min. Lysates were centrifuged at 20,817 g at 4 °C for 10 min. Concentrations of proteins were determined using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Cat. No. 23225) with BSA as standard. Supernatant was frozen in liquid nitrogen and stored at −80 °C.
Lectin binding.
For lectin blots, samples were prepared by dilution of lysates with water and addition of 4 x SDS loading dye (200 mM Tris-HCl pH 6.8, 8 % (w/v) SDS, 0.08 % (w/v) bromophenol blue, 40 % (v/v) glycerol, and 40 mM DTT). Samples were denatured at 90 °C for 5 min and 10 μg of lysates were loaded on each lane and separated by SDS-PAGE using a 12 % TGX FastCast gel (Bio-Rad, Cat. No. 1610175). Proteins were transferred to a Immobilon-FL PVDF membrane (Millipore, Cat. No. IPFL00010 or IPFL00005) by wet transfer with 87 mA of current at 4 °C overnight. After the transfer, the membrane was rinsed with ultrapure water and stained with a Revert™ 700 Total Protein Stain Kits (LI-COR, Cat. No. 926–11016 or 926–11010). Total protein stain was imaged with a ChemiDoc MP Imaging system (Bio-Rad, Hercules, CA). Membrane was destained with the Revert™ 700 Total Protein Stain Kits, rinsed with ultrapure water, and equilibrated with TBS (10 mM Tris, pH 8.0, 150 mM NaCl) for about 2 min at room temperature. Blots were blocked with Carbo-free Blocking Solution (Vector Laboratories, SP-5040) diluted in TBS and then probed with 10 μg/ml of biotin-labeled lectins or galectin-1, or 1 μg/mL of biotin-labeled cholera toxin B subunit diluted in Carbo-free Blocking Solution in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.1% Tween 20) at 4 °C overnight. Blots were washed with TBST and probed with IRDye® 800CW conjugated streptavidin at 1:4000 dilution in Carbo-free Blocking Solution in TBST with 0.02 % SDS for 1 h at room temperature. Blots were washed with TBST and imaged on a ChemiDoc MP Imaging system.
On-Cell ELLA.
To each well in a 96-well stripwell plate (Costar, 9102), 25,000 T84 UAP1(F383G) cells were added in 200 μL of complete medium. At 24 h and 48 h after cells were plated, medium was replaced with complete medium containing 100 μM of Ac3GlcNDAz-1P(AcSATE)2 or equal volume of DMSO. About 72 h after cells were plated, cells were rinsed three times with DPBS and fixed with 4 % paraformaldehyde (PFA, Electron Microscopy Sciences, Cat. No. 15713) for 30 min at room temperature. Cells were then washed with DPBS. DPBS supplemented with 2 % (w/v) BSA and 0.1 % (w/v) lysine were added to cells and incubated for 1 h at room temperature to quench unreacted PFA and block for nonspecific binding. Cells were then incubated with biotin-labeled lectins diluted in 2 % BSA in DPBS overnight at 4 °C and washed three times with DPBS. Cells were then incubated with 1:4000 dilution of streptavidin-HRP (500 U/ml, Roche, Cat. No. 11089153001) in 2 % BSA in DPBS for 1 h at room temperature and washed three times with DPBS. To evaluate the activity of HRP, HRP solution was prepared by dissolving o-phenylenediamine dihydrate (OPD) (Sigma, Cat. No. P1526–10G) in HRP buffer (2.56 g citric acid and 3.65 g anhydrous dibasic sodium phosphate in 500 mL of water) to a final concentration of (w/v) 0.1 % and 30 % (w/w) H2O2 was added at a 1:1000 dilution. Reaction was started by addition of 100 μL of HRP solution to each well and quenched with 100 μL of 1 M H2SO4. Absorbance at 490 nm was determined with a Synergy™ 2 Multi-Mode Microplate Reader (BioTek, Winooski, VT) and corrected with absorbance at 650 nm. Cells were then washed and incubated with BCA reagents for 2 h at 37 °C. Absorbance at 562 nm was determined, corrected with the average absorbance of blank 96-well plate and used to normalize the readout of HRP activity to total protein concentration.
For each biological replicate, three technical replicates were included. The average of lectin binding from three technical replicates was normalized to the highest value in DMSO-treated cells at the following lectin concentrations: 30 μg/ml of lectins for GSL II, SNA, MAL II and CTB; 1 μg/ml of lectins for succinylated WGA and ECA; 3 μg/ml of lectins for DSL and LEL; 10 μg/ml of lectins for PHA-L and galectin-1. The normalized average lectin binding from four biological replicates were used for statistical analysis using Student’s t-test and plotting.
Crosslinking of galectin-1 and CTB to glycoproteins
To crosslink galectin-1 and CTB to glycoproteins, 150,000 T84 UAP1(F383G) cells were plated in 1 mL of complete media in each well in 12-well plates. To each well, 2 μL of Ac3GlcNDAz-1P(AcSATE)2 stock or DMSO was added at 24 h and 48 h after cells were plated. At about 72 h after cells were plated, cells were rinsed with DPBS and changed to 500 μL of serum-free media. Galectin-1 or CTB was added to cells at a final concentration of 10 μg/mL, or 4 μg/mL, respectively. Cells were incubated with galectin-1 or CTB for 45 min at 4 °C in dark and irradiated with a UVP Blak-Ray Lamp (Model XX-20BLB, VWR, Cat. No. 21474–676) for 45 min on an ice-water bath. Control cells were incubated with galectin-1 or CTB for 90 min at 4 °C in dark. After UV irradiation, cells were scraped into 80 μL of RIPA buffer with protease inhibitor cocktail. Cells were lysed and protein concentrations were determined as described for the lectin blots.
For immunoblots, 30 μg of lysates were loaded on each lane and separated by SDS-PAGE using a 7.5% TGX FastCast Gel (BioRad, 1610171). Proteins were transferred to Immobilon-FL PVDF membrane with 87 mA of current at 4 °C overnight and stained for total protein as described for lectin blots. After destaining, blots were rinsed with ultrapure water, equilibrated with TBS and blocked with a Intercept™ (TBS) blocking buffer (LI-COR, 927–66003) for 1 h at room temperature. Blots were then incubated with anti-galectin-1 or anti-CTB and anti-α-tubulin antibodies diluted in a Intercept™ T20 (TBS) antibody diluent (LI-COR, 927–66003) at 4 °C overnight. Blots were washed with TBST and incubated with IRDye® 800CW conjugated goat anti rabbit IgG (H+L) and IRDye® 680 RD conjugated goat anti mouse IgG (H+L) diluted in Intercept™ T20 (TBS) antibodies diluent containing 0.01 % SDS for 1 h at room temperature. Blots were then washed with TBST and imaged with a ChemiDoc MP Imaging system.
For experiments in which inhibitors of glycosylation were used, 200 μM of 2F-peracetyl-fucose or 1 μg/mL of kifunensine was included in the cell culture media when cells were plated. Cells were treated with Ac3GlcNDAz-1P(AcSATE)2 stock or DMSO, crosslinked to galectin-1 or CTB and analyzed by immunoblot as described above.
To evaluate inhibition of crosslinking by free sugars, lactose, maltose, sucrose, cellobiose, d-galactose, d-glucose, l-galactose, d-GlcNAc, d-fucose and l-fucose were dissolved in serum-free media to a final concentration of 100 mM and filtered through a 0.2 μm Nalgene syringe filter (Thermo Fisher Scientific, Cat. No. 720-1320). T84 UAP1(F383G) cells were cultured as described above. Serum-free media (500 μL) with free sugars was added to each well. Galectin-1 or CTB was then immediately added and UV irradiation was performed. Cells were lysed and analyzed by immunoblot as described above.
To evaluate inhibition of crosslinking by lectins, T84 UAP1(F383G) cells were cultured as described above and changed to 500 μL of serum-free media. Biotin-labeled lectins were added to cells to a final concentration of 40 μg/mL immediately before the addition of galectin-1 or CTB. UV irradiation was performed and lysates were analyzed by immunoblot as described above.
To remove O-glycans on mucin or mucin-like proteins with StcE, cells were washed with DPBS and incubated with 5 μg/mL of StcE in serum-free media at 37 °C for 2 h before adding galectin-1 and performing the crosslinking experiment.
Proteomic analysis of galectin-1 crosslinked complexes
Cell culture and crosslinking.
To prepare crosslinked samples for proteomic analysis, 2 × 106 T84 UAP1(F383G) cells were plated in 10 mL of media in a 10 cm tissue culture plate. At 24 h and 48 h after cells were plated, 20 μL of Ac3GlcNDAz-1P(AcSATE)2 stock or DMSO was added to each plate. About 72 h after cells were plated, cells were rinsed with DPBS and 3 mL of serum-free media was added to each plate. Biotin-labeled galectin-1 was added to serum-free media to a final concentration of 100 μg/mL. Cells treated with Ac3GlcNDAz-1P(AcSATE)2 were incubated with galectin-1 for 45 min at 4 °C and UV irradiated for 45 min on ice. Cells treated with DMSO were kept at 4 °C in dark for 90 min without galectin-1. Cells were then washed twice with 5 mL of 100 mM lactose in cold DPBS followed by 10 mL of DPBS to remove uncrosslinked galectin-1. After washing, cells were scraped into 1 mL of RIPA buffer with protease inhibitor cocktail, vortexed, and incubated on ice for 30 min. Lysates were centrifuged at 20,817 g for 10 min at 4 °C. Concentration of supernatant was determined with a Pierce BCA Protein Assay Kit. Lysates were frozen in liquid nitrogen and stored at −80 °C.
Immunoprecipitation and silver stain.
To purify the crosslinked complexes, 1043 μg of lysates were diluted with RIPA buffer containing protease inhibitor cocktail to 1 mL and added to 10 mg of Dynabeads™ M-280 streptavidin (Thermo Fisher Scientific, Cat. No. 11206D), which had been pre-equilibrated with RIPA buffer. Lysates were incubated with beads at 4 °C overnight with end-over-end rotation. Beads were washed 5 times with 1 mL of RIPA buffer and 5 times with 1 mL of high salt RIPA buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 0.1 % (w/v) SDS, 0.5 % (w/v) sodium deoxycholate and 1 % (w/v) IGEPAL CA-630). After washing, beads were transferred to a fresh tube and eluted by incubating beads with 100 μL of 1 x SDS dye with biotin (50 mM Tris-HCl, pH 6.8, 2 % (w/v) SDS, 0.02 % (w/v) bromophenol blue, 10 % (v/v) glycerol, 10 mM DTT and 20 mM biotin) at 90 °C for 5 min. Eluent from each sample was loaded in 2 lanes of a 7.5% precast gel (Bio-Rad, Cat. No. 4561024) for separation by SDS-PAGE. After separation, gels were stained with a mass compatible silver staining kit (Thermo Fisher Scientific, Cat. No. 24600).
In-gel digestion and mass spectrometry.
Gel bands were digested overnight with trypsin (Pierce) following destaining, reduction with DTT and alkylation with iodoacetamide (Sigma–Aldrich). The samples then underwent solid-phase extraction cleanup with an Oasis HLB plate (Waters) and the resulting samples were injected onto an Orbitrap Fusion Lumos mass spectrometer coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system. Samples were injected onto a 75 um i.d., 75-cm long EasySpray column (Thermo) and eluted with a gradient from 0–28% buffer B over 90 min. Buffer A contained 2% (v/v) ACN and 0.1% formic acid in water, and buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. The mass spectrometer operated in positive ion mode with a source voltage of 1.5–2.2 kV and an ion transfer tube temperature of 275 °C. MS scans were acquired at 120,000 resolution in the Orbitrap and up to 10 MS/MS spectra were obtained in the ion trap for each full spectrum acquired using higher-energy collisional dissociation (HCD) for ions with charges 2–7. Dynamic exclusion was set for 20 s after an ion was selected for fragmentation.
Raw MS data files were analyzed using Proteome Discoverer v2.2 (Thermo), with peptide identification performed using Sequest HT searching against the human protein database from UniProt. Fragment and precursor tolerances of 10 ppm and 0.6 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification, with oxidation of Met set as a variable modification. The false-discovery rate (FDR) cutoff was 1% for all peptides.
Immunoprecipitation
Cell culture and crosslinking.
To prepare crosslinked samples for immunoprecipitation, T84 UAP1(F383G) cells were treated, crosslinked and harvested as sample preparation for proteomics, except that 100 μg/mL of unlabeled galectin-1 was used during crosslinking, and that 100 mM lactose was added to one sample right before the addition of galectin-1 as a negative control.
In-lysate crosslinking.
To compare the crosslinking of LAMP1 and CEA to galectin-1 in intact cells and in lysates, T84 UAP1(F383G) cells were plated and treated as for proteomics. For crosslinking in lysates without lactose, cells were scraped into 1 mL of RIPA buffer with a protease inhibitor cocktail, vortexed and incubated on ice for 30 min. Lysates were then centrifuged at 20,817 g for 10 min at 4 °C and 900 μL of the supernatant from each sample was transferred to one well in a 6-well plate. For crosslinking in lysates with the presence of lactose, cells were lysed as before, except that RIPA buffer with 100 mM of lactose and protease inhibitor cocktail was used to lyse the cells. Unlabeled galectin-1 was added to intact T84 cells in serum-free media or to cell lysates to a final concentration of 30 μg/mL, incubated for 45 min at 4 °C in dark and UV-irradiated for 45 min on an ice-water bath. Intact T84 cells were then lysed as before. Crosslinked lysates were directly transferred to a fresh 1.5 mL tube, incubated on ice for 30 min and centrifuged at 20,817 g for 10 min at 4 °C. Protein concentration was determined with the BCA kit using RIPA buffer or RIPA buffer with 100 mM lactose as blank.
Immunoprecipitation and Western blot.
To enrich for selected proteins, 140 μg of lysates were diluted to 1 μg/mL in RIPA buffer with a protease inhibitor cocktail and incubated with LAMP1, CEA, CDCP1 and LY75 antibody or the same amount of mouse or rabbit IgG control overnight in dark at 4 °C with end-over-end rotation. For 140 μg of lysates, 28 μL of Dynabeads™ Protein G (Thermo Fisher Scientific, Cat. No. 10004D) pre-equilibrated with RIPA buffer were added and incubated with lysates at room temperature with end-over-end rotation for 1 hour. Beads were washed 4 times with 200 μL of RIPA buffer and eluted with 28 μL of 1 x SDS loading dye (50 mM Tris-HCl, pH 6.8, 2 % (w/v) SDS, 0.02 % (w/v) bromophenol blue, 10 % (v/v) glycerol and 10 mM DTT) at 90 °C for 5 min. For IP control (IP and probe for the same protein), 4 μL of the eluent was loaded and separated on a 7.5% SDS PAGE. For IP experiment (IP for potential galectin-1 binding partner and probe for galectin-1), 20 μL of the eluent was loaded and separated on a 7.5% SDS PAGE. Proteins were then transferred to PVDF-FL membrane and membrane was blocked same as for detection of galectin-1 crosslinking. After blocking, membrane was probed with primary antibody overnight at 4 °C, washed with TBST and incubated with IRDye conjugated secondary antibody for 1 hour at room temperature. Blots were imaged with a ChemiDoc MP Imaging system, after which the region below around 50 kDa was removed. To enhance the signal to noise ratio, blots were then probed again with primary antibody overnight at 4 °C, washed with TBST and incubated with IRDye conjugated secondary antibody for 1 hour at room temperature, and imaged again with ChemiDoc MP Imaging system.
High performance anion exchange chromatography (HPAEC) analysis of UDP-GlcNDAz
To assess production of UDP-GlcNDAz, 2 × 106 T84 UAP1(F383G) cells were plated in a 6-cm tissue culture plate and allowed to adhere to plates overnight. Cells were treated with DMSO or Ac3GlcNDAz-1P(AcSATE)2 at a final concentration of 100 μM for 8 h. Cells were lifted from plates by TrypLE (Thermo Fisher Scientific, Cat. No. 12605–010), quenched with complete medium and washed with DPBS. Cells were frozen in liquid nitrogen and stored at −80 °C.
To extract the nucleotide sugars, cells were resuspended in 1 ml of ice-cold 50 % acetonitrile, vortexed, and incubated on ice for 10 min. Debris was removed by centrifugation at 20,000 g at 4 °C for 10 min. Supernatant was then frozen in liquid nitrogen and dried with a SAVANT SC210A SpeedVac Concentrator (Thermo Fisher Scientific).
Before analysis by HPAEC, extract was resuspended in 100 μL of 40 mM sodium phosphate buffer (pH 7.4) and centrifuged at 20,817 g at 4 °C for 1 min. The supernatant was filtered through a 3K Amicon Ultra centrifugal filter unit at 4 °C (Millipore, Cat. No. UFC5003BK), and 20 μL of each sample was injected for analysis by HPAEC using a CarboPac PA1 column (Thermo Fisher Scientific, Cat. No. 035391) with a guard column (Thermo Fisher Scientific, Cat. No. 0343096). Samples were eluted with buffer A (1 mM NaOH) and buffer B (1 M NaOAc, 1 mM NaOH) at a flow rate of 1 mL/min using either of the following gradient of buffer B in buffer A: T 0 min = 5 %, T 40 min = 30 %, T 45 min = 50 %, T 60 min = 55 %, T 65 min = 100 %, T 75 min = 100 %, T 80 min = 5 %, T 90 min = 5 %; or T 0 min = 20 %, T 10 min = 45 %, T 25 min = 45 %, T 35 min = 55 %, T 40 min = 100 %, T 50 min = 100 %, T 55 min = 20 %, T 65 min = 20 %. Elution of nucleotide sugars were monitored by UV absorption at the wavelength of 260 nm. UDP-GlcNDAz and UDP-GlcNAc (Promega, Cat. No. V7071) were used as standards.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of lectin binding using ELLA was performed using two-tailed unpaired t test at concentrations indicated in the figure legend. A significant difference is defined when p<0.05. ELLA data are from four biological replicates, each of which is averaged from three technical replicates, and error bars represent ±SD. Details of data acquisition can be found in the methods details. All Western blots and immunoprecipitation data are representative of three biological replicates, except for immunoprecipitation experiment to confirm proteomics hits, where data are representative of two biological replicates using 100 ug/mL galectin-1 and similar results have been obtained using 30 ug/mL galectin-1 from one trial. Proteomics results are averaged from two biological replicates. Glycoproteomics data are from single trial due to discontinuation of the ZIC-HILIC enrichment kit.
Supplementary Material
Supplementary Data 1. Representative MS2 spectra of glycopeptides containing GlcNDAz, Related to Figure 2. See attached word file. Related to main Figure 2.
Supplementary Table 1. List of candidate galectin-1 binding proteins identified by proteomics analysis of crosslinked bands, Related to Figure 6. See attached excel file.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-galectin-1 | Cell Signaling Technology (Danvers, MA, USA) | Cat# 12936; RRID: AB_2798065 |
| Goat polyclonal anti-galectin-1 | R&D Systems (Minneapolis, MN, USA) | Cat# AF1152; RRID: AB_2136626 |
| Rabbit monoclonal anti-LAMP1 | Cell Signaling Technology | Cat# 9091; RRID: AB_2687579 |
| Rabbit monoclonal anti-CDCP1 | Cell Signaling Technology | Cat# 13794; RRID: AB_2798314 |
| Rabbit polyclonal anti-CDCP1 | Cell Signaling Technology | Cat# 4115; RRID: AB_2078818 |
| Rabbit monoclonal anti-LY75 | Abcam (Cambridge, UK) | Cat# ab124897; RRID: AB_10976058 |
| Rabbit polyclonal anti-cholera toxin B subunit | Abcam | Cat# ab34992; RRID: AB_726859 |
| Mouse monoclonal anti-α-tubulin | Sigma (St. Louis, MO, USA) | Cat# T6199; RRID: AB_477583 |
| Mouse monoclonal anti-CEA | Thermo Fisher Scientific (Waltham, MA, USA) | Cat# MA5-14675; RRID: AB_10985171 |
| IRDye® 680 RD goat anti-mouse IgG (H+L) | LI-COR Biosciences (Lincoln, USA) | Cat# 926-68070; RRID: AB_10956588 |
| IRDye® 800 CW goat anti-mouse IgG (H+L) | LI-COR Biosciences | Cat# 926-32210; RRID: AB_621842 |
| IRDye® 800 CW goat anti-rabbit IgG (H+L) | LI-COR Biosciences | Cat# 926-32211; RRID: AB_621843 |
| IRDye® 800 CW donkey anti-goat IgG (H+L) | LI-COR Biosciences | Cat# 925-32214; RRID: AB_2687553 |
| Chemicals, peptides, and recombinant proteins | ||
| Ac3GlcNDAz-1P(AcSATE)2 | (Yu et al., 2012) Wuxi AppTec (Tianjin) |
N/A |
| UDP-GlcNDAz | (Yu et al., 2012) Wuxi AppTec (Tianjin) |
N/A |
| UDP-GlcNAc | Promega (Madison, WI, USA) | Cat# V707A |
| Dimethyl sulfoxide | Fisher Scientific (Hampton, NH, USA) | Cat# BP231-100 |
| Lactose | Sigma | Cat# 61339-25G |
| Cellobiose | Sigma | Cat# 22150-10G |
| d-galactose | Sigma | Cat# G0750-100G |
| l-galactose | Sigma | Cat# G7134-100MG |
| d-fucose | Sigma | Cat# F8150-1G |
| l-fucose | Sigma | Cat# F2252-5G |
| d-glucose | Sigma | Cat# G7021-100G |
| N-acetylglucosamine | Sigma | Cat# A3286-25G |
| Maltose | MP Biomedicals (Santa Ana, CA, USA) | Cat# 190237 |
| Sucrose | Fisher Scientific | Cat# BP220-1 |
| Kifunensine | Sigma | Cat# K1140-1MG |
| 2F-peracetyl-fucose | Millipore (Burlington, MA, USA) | Cat# 344827-10MG |
| Biotinylated Griffonia (Bandeiraea) Simplicifolia Lectin II | Vector Laboratories (Burlingame, CA, USA) | Cat# B-1215 |
| Biotinylated Succinylated Wheat Germ Agglutinin | Vector Laboratories | Cat# B-1025S |
| Biotinylated Datura Stramonium Lectin | Vector Laboratories | Cat# B-1185 |
| Biotinylated Lycopersicon Esculentum (Tomato) Lectin | Vector Laboratories | Cat# B-1175 |
| Biotinylated Erythrina Cristagalli Lectin | Vector Laboratories | Cat# B-1145 |
| Biotinylated Sambucus Nigra Lectin | Vector Laboratories | Cat# B-1305 |
| Biotinylated Maackia Amurensis Lectin II | Vector Laboratories | Cat# B-1265 |
| Biotinylated Phaseolus Vulgaris Leucoagglutinin | Vector Laboratories | Cat# B-1115 |
| Aleuria Aurantia Lectin | Vector Laboratories | Cat# L-1390 |
| Cholera toxin B subunit | Sigma | Cat# C9903 |
| Recombinant cholera toxin B subunit, biotinylated | Thermo Fisher Scientific | Cat# C-34779 |
| Recombinant human galectin-1 | (Wu et al., 2021) | N/A |
| Recombinant human galectin-1, biotinylated | (Stowell et al., 2008a, Stowell et al., 2008b) | N/A |
| IRDye® 800 CW streptavidin | Li-COR Biosciences | Cat# 926-32230 |
| Streptavidin-POD conjugate | Sigma | Cat# 11089153001 |
| O-phenylenediamine dihydrate | Sigma | Cat# P1526-10G |
| Normal rabbit IgG | Millipore | Cat# NI01-100UG |
| Normal mouse IgG | Millipore | Cat# NI03-100UG |
| Carbo-free Blocking Solution | Vector Laboratories | Cat# SP-5040 |
| Intercept™ (TBS) Blocking Buffer and Diluent Kit | LI-COR Biosciences | Cat# 927-66003 |
| Dynabeads™ M-280 streptavidin | Thermo Fisher Scientific | Cat# 11206D |
| Dynabeads™Protein G | Thermo Fisher Scientific | Cat# 10004D |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Sigma | Cat# 11836170001 |
| 20% Paraformaldehyde (formaldehyde) aqueous solution | Electron Microscopy Sciences (Hatfield, PA, USA) | Cat# 15713 |
| StcE | (Malaker et al., 2019) | N/A |
| Man3GlcNAc2-Asn | (Kadirvelraj et al., 2020) | N/A |
| GlcNAcMan3GlcNAc2-Asn | (Kadirvelraj et al., 2020) | N/A |
| GlcNAc2Man3GlcNAc2-Asn | (Kadirvelraj et al., 2020) | N/A |
| O-Man glycopeptide 317 | (Halmo et al., 2017) | N/A |
| Lacto-N-neotetraose | Carbosynth (Compton, UK) | Cat# OL05765 |
| Critical commercial assays | ||
| ProteoExtract® Glycopeptide Enrichment Kit | Millipore | Cat# 72103-3 |
| UDP-Glo™ Glycosyltransferase Assay | Promega | Cat# V6961 |
| Revert™ 700 Total Protein Stain Kit | LI-COR Biosciences | Cat# 926-11016 or 926-11010 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Pierce™ Silver Stain for Mass Spectrometry | Thermo Fisher Scientific | Cat# 24600 |
| Deposited data | ||
| Glycoproteomics raw data and results | This paper | GlycoPOST: GPST000201 |
| Proteomics raw data | This paper | MassIVE: MSV000087640 |
| Experimental models: Cell lines | ||
| Male T84 UAP1(F383G) cells | (Yu et al., 2012) | N/A |
| Recombinant DNA | ||
| MGAT1-PGEn2 | (Moremen et al., 2018) dnasu.org |
DNASU clone ID: HsCD00413193 |
| MGAT2-PGEn2 | (Moremen et al., 2018) dnasu.org |
DNASU clone ID: HsCD00413203 |
| MGAT3-PGEn2 | (Moremen et al., 2018) dnasu.org |
DNASU clone ID: HsCD00413114 |
| MGAT4A-PGEn2 | (Moremen et al., 2018) dnasu.org |
DNASU clone ID: HsCD00413158 |
| MGAT5-PGEn2 | (Moremen et al., 2018) dnasu.org |
DNASU clone ID: HsCD00522293 |
| POMGNT1-PGEn2 | (Moremen et al., 2018) dnasu.org |
DNASU clone ID: HsCD00413191 |
| POMGNT2-PGEn2 | (Moremen et al., 2018) dnasu.org |
DNASU clone ID: HsCD00413126 |
| B3GNT2-PGEn2 | (Moremen et al., 2018) dnasu.org |
DNASU clone ID: HsCD00413085 |
| GCNT2B-PGEn2 | (Moremen et al., 2018) dnasu.org |
DNASU clone ID: HsCD00413202 |
| Software and algorithms | ||
| MATLAB R2019a | MathWorks | https://www.mathworks.com/products/matlab.html |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Byonic™ 2.3 | Protein Metrics | https://proteinmetrics.com/byos/ |
| SpectrsPicker | Dr. Rene Ranzinger | https://github.com/ReneRanzinger/SpectraFiltering/ |
| Proteome Discover v2.2 | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
Significance.
N-linked glycoproteins are key components of many extracellular recognition events. Their N-glycans are recognized by a variety of glycan-binding proteins. These glycan recognition events regulate diverse cellular processes, including cell-cell interactions, signal transduction, and host-microbe recognition. However, glycan recognition events are often low-affinity and transient, characteristics that pose challenges for identifying binding partners through traditional affinity purification approaches. Here we describe a metabolic labeling method to incorporate the diazirine photocrosslinking functional group onto GlcNAc residues of N-glycans. Importantly, the method results in only modest perturbations to N-glycan structures. We demonstrate that the metabolically incorporated diazirine can be activated by UV light, yielding covalent crosslinking between N-linked glycoproteins and extracellular glycan-binding proteins. The resulting crosslinked complexes can be analyzed by biochemical methods including immunoblot, immunoprecipitation, and proteomics mass spectrometry analysis. Pharmacological agents that modulate glycan structure as well as protein and glycan reagents that competitively interfere with glycan binding can be used in conjunction with immunoblot analysis of crosslinked complexes, revealing information about glycan features that are required for specific glycan recognition events. Protein components of crosslinked complexes can be identified by proteomics mass spectrometry analysis of the crosslinked complexes. Finally, information about the cellular context of glycan-recognition events can be discerned by evaluating the conditions under which crosslinking is observed. These results support the utility of cell surface photocrosslinking sugars in identifying and defining glycan-dependent binding interactions within their native context.
Highlights.
A photocrosslinking analog of GlcNAc can be incorporated into cellular N-glycans.
Incorporation of photocrosslinking GlcNAc modestly perturbs N-glycan structures.
Interactions between N-glycans and glycan binding proteins are covalently captured.
In-cell crosslinking gives context-specific information about glycan binding events.
Acknowledgments
This project was supported by NIH R21DK112733 and U01CA242115 to JJK, S10OD018530 to PA, P41GM103390 and R01GM130915 to KWM, and Welch Foundation (I-1686) to JJK. HW was supported by the Sara and Frank McKnight Fund for Research in Biochemistry. We acknowledge support from the UT Southwestern Proteomics Core Facility, directed by Dr. Andrew Lemoff and thank Dr. Daniela Carroll and Atossa Ghorashi for comments on the manuscript.
Declaration of Interests
KWM acknowledges ownership interest and roles as President and CEO of Glyco Expression Technologies, Inc., a biotechnology spinout commercializing recombinant glycosyltransferases, and may conceivably profit from the results described herein. JJK is member of the Advisory Board for Cell Chemical Biology. Other authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aits S, Kricker J, Liu B, Ellegaard A-M, Hämälistö S, Tvingsholm S, Corcelle-Termeau E, Høgh S, Farkas T, Holm Jonassen A, et al. (2015). Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 11, 1408–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce M, Carrico IS, Ganguli AS, Yu S-H, Hangauer MJ, Hubbard SC, Kohler JJ, and Bertozzi CR (2011). Metabolic cross-talk allows labeling of O-linked beta-N-acetylglucosamine-modified proteins via the N-acetylgalactosamine salvage pathway. Proc. Natl. Acad. Sci. U.S.A 108, 3141–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camby I, Le Mercier M, Lefranc F, and Kiss R (2006). Galectin-1: a small protein with major functions. Glycobiology 16, 137R–157R. [DOI] [PubMed] [Google Scholar]
- Cervin J, Wands AM, Casselbrant A, Wu H, Krishnamurthy S, Cvjetkovic A, Estelius J, Dedic B, Sethi A, Wallom K-L, et al. (2018). GM1 ganglioside-independent intoxication by Cholera toxin. PLoS Pathog. 14, e1006862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings RD, and Kornfeld S (1984). The distribution of repeating [Gal beta 1,4GlcNAc beta 1,3] sequences in asparagine-linked oligosaccharides of the mouse lymphoma cell lines BW5147 and PHAR 2.1. J Biol Chem 259, 6253–6260. [PubMed] [Google Scholar]
- Darby JF, Gilio AK, Piniello B, Roth C, Blagova E, Hubbard RE, Rovira C, Davies GD and Wu L (2020) Substrate Engagement and Catalytic Mechanisms of N-Acetylglucosaminyltransferase V. ACS Catalysis 10, 8590–8596 [Google Scholar]
- Do KY, Smith DF, and Cummings RD (1990). LAMP-1 in CHO cells is a primary carrier of poly-N-acetyllactosamine chains and is bound preferentially by a mammalian S-type lectin. Biochem. Biophys. Res. Commun 173, 1123–1128. [DOI] [PubMed] [Google Scholar]
- Du J, Meledeo MA, Wang Z, Khanna HS, Paruchuri VDP, and Yarema KJ (2009). Metabolic glycoengineering: sialic acid and beyond. Glycobiology 19, 1382–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbein AD, Tropea JE, Mitchell M, and Kaushal GP (1990). Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J Biol Chem 265, 15599–15605. [PubMed] [Google Scholar]
- Elola MT, Chiesa ME, Alberti AF, Mordoh J, and Fink NE (2005). Galectin-1 receptors in different cell types. J. Biomed. Sci 12, 13–29. [DOI] [PubMed] [Google Scholar]
- Feng L, Hong S, Rong J, You Q, Dai P, Huang R, Tan Y, Hong W, Xie C, Zhao J, et al. (2013). Bifunctional unnatural sialic acids for dual metabolic labeling of cell-surface sialylated glycans. J. Am. Chem. Soc 135, 9244–9247. [DOI] [PubMed] [Google Scholar]
- Gao C, Hanes MS, Byrd-Leotis LA, Wei M, Jia N, Kardish RJ, McKitrick TR, Steinhauer DA, and Cummings RD (2019). Unique Binding Specificities of Proteins toward Isomeric Asparagine-Linked Glycans. Cell Chem Biol 26, 535–547.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler C, and Jarvis DL (2011). Effective glycoanalysis with Maackia amurensis lectins requires a clear understanding of their binding specificities. Glycobiology 21, 988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti MR, Salatino M, Dalotto-Moreno T, and Rabinovich GA (2020). Sweetening the hallmarks of cancer: Galectins as multifunctional mediators of tumor progression. J. Exp. Med 217, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Collins BE, Bengtson P, and Paulson JC (2005). Homomultimeric complexes of CD22 in B cells revealed by protein-glycan cross-linking. Nat. Chem. Biol 1, 93–97. [DOI] [PubMed] [Google Scholar]
- Hennet T (2002). The galactosyltransferase family. Cell. Mol. Life Sci 59, 1081–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinderlich S, Weidemann W, Yardeni T, Horstkorte R, and Huizing M (2015). UDP-GlcNAc 2-Epimerase/ManNAc Kinase (GNE): A Master Regulator of Sialic Acid Synthesis. Top Curr Chem 366, 97–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren J (1981). Actions of cholera toxin and the prevention and treatment of cholera. Nature 292, 413–417. [DOI] [PubMed] [Google Scholar]
- Holmgren J, Lönnroth I, Månsson J, and Svennerholm L (1975). Interaction of cholera toxin and membrane GM1 ganglioside of small intestine. Proc. Natl. Acad. Sci. U.S.a 72, 2520–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst S, Deuss AJM, van Pelt GW, van Vliet SJ, Garcia-Vallejo JJ, Koeleman CAM, Deelder AM, Mesker WE, Tollenaar RA, Rombouts Y, et al. (2016). N-glycosylation Profiling of Colorectal Cancer Cell Lines Reveals Association of Fucosylation with Differentiation and Caudal Type Homebox 1 (CDX1)/Villin mRNA Expression. Mol. Cell Proteomics 15, 124–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009a). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009b). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, and Ralph SJ (2012). Inhibiting galectin-1 reduces murine lung metastasis with increased CD4(+) and CD8 (+) T cells and reduced cancer cell adherence. Clin. Exp. Metastasis 29, 561–572. [DOI] [PubMed] [Google Scholar]
- Jia J, Abudu YP, Claude-Taupin A, Gu Y, Kumar S, Choi SW, Peters R, Mudd MH, Allers L, Salemi M, et al. (2018). Galectins Control mTOR in Response to Endomembrane Damage. Mol. Cell 70, 120–135.e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Claude-Taupin A, Gu Y, Choi SW, Peters R, Bissa B, Mudd MH, Allers L, Pallikkuth S, Lidke KA, et al. (2020). Galectin-3 Coordinates a Cellular System for Lysosomal Repair and Removal. Dev. Cell 52, 69–87.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes L, Jacob R, and Leffler H (2018). Galectins at a glance. J. Cell. Sci 131, jcs208884. [DOI] [PubMed] [Google Scholar]
- Kadirvelraj R, Yang J-Y, Sanders JH, Liu L, Ramiah A, Prabhakar PK, Boons G-J, Wood ZA, and Moremen KW (2018). Human N-acetylglucosaminyltransferase II substrate recognition uses a modular architecture that includes a convergent exosite. Proc. Natl. Acad. Sci. U.S.A 115, 4637–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadirvelraj R, Yang JY, Kim HW, Sanders JH, Moremen KW & Wood ZA 2020. Comparison of human poly-N-acetyl-lactosamine synthase structure with GT-A fold glycosyltransferases supports a modular assembly of catalytic subsites. J Biol Chem, 296, 100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamili NA, Arthur CM, Gerner-Smidt C, Tafesse E, Blenda A, Dias-Baruffi M, and Stowell SR (2016). Key regulators of galectin-glycan interactions. Proteomics 16, 3111–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya Y, Kato R, Itoh S, Fukuda T, Shibukawa Y, Sanzen N, Sekiguchi K, Wada Y, Kawasaki N, Gu J (2008) N-Glycosylation of laminin-332 regulates its biological functions. A novel function of the bisecting GlcNAc. J Biol Chem. 283, 33036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halmo SM, Singh D, Patel S, Wang S, Edlin M, Boons GJ, Moremen KW, Live D & Wells L (2017) Protein O-Linked mannose beta-1,4-N-acetylglucosaminyl-transferase 2 (POMGNT2) is a gatekeeper enzyme for functional glycosylation of alpha-dystroglycan. J Biol Chem, 292, 2101–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantéri M, Giordanengo V, Hiraoka N, Fuzibet J-G, Auberger P, Fukuda M, Baum LG, and Lefebvre J-C (2003). Altered T cell surface glycosylation in HIV-1 infection results in increased susceptibility to galectin-1-induced cell death. Glycobiology 13, 909–918. [DOI] [PubMed] [Google Scholar]
- Law ME, Ferreira RB, Davis BJ, Higgins PJ, Kim J-S, Castellano RK, Chen S, Luesch H, and Law BK (2016). CUB domain-containing protein 1 and the epidermal growth factor receptor cooperate to induce cell detachment. Breast Cancer Res. 18, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Guan W, Zhang G, Wu Z, Yu H, Chen X, and Wang PG (2019). Microarray analyses of closely related glycoforms reveal different accessibilities of glycan determinants on N-glycan branches. Glycobiology 30, 3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchansky SJ, Goon S, and Bertozzi CR (2004). Expanding the diversity of unnatural cell-surface sialic acids. Chembiochem 5, 371–374. [DOI] [PubMed] [Google Scholar]
- Luchansky SJ, Yarema KJ, Takahashi S, and Bertozzi CR (2003). GlcNAc 2-epimerase can serve a catabolic role in sialic acid metabolism. J Biol Chem 278, 8035–8042. [DOI] [PubMed] [Google Scholar]
- Ma J, and Hart GW (2014). O-GlcNAc profiling: from proteins to proteomes. Clin Proteomics 11, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaker SA, Pedram K, Ferracane MJ, Bensing BA, Krishnan V, Pett C, Yu J, Woods EC, Kramer JR, Westerlind U, et al. (2019). The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc. Natl. Acad. Sci. U.S.A 116, 7278–7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkle RK, and Cummings RD (1987). Relationship of the terminal sequences to the length of poly-N-acetyllactosamine chains in asparagine-linked oligosaccharides from the mouse lymphoma cell line BW5147. Immobilized tomato lectin interacts with high affinity with glycopeptides containing long poly-N-acetyllactosamine chains. J Biol Chem 262, 8179–8189. [PubMed] [Google Scholar]
- Mishra PK, Yoo C-M, Hong E, and Rhee HW (2020). Photo-crosslinking: An emerging chemical tool for investigating molecular networks in live cells. Chembiochem 21, 924–932. [DOI] [PubMed] [Google Scholar]
- Moiseeva EP, Williams B, Goodall AH, and Samani NJ (2003). Galectin-1 interacts with beta-1 subunit of integrin. Biochem. Biophys. Res. Commun 310, 1010–1016. [DOI] [PubMed] [Google Scholar]
- Monsigny M, Roche AC, Sene C, Maget-Dana R, and Delmotte F (1980). Sugar-lectin interactions: how does wheat-germ agglutinin bind sialoglycoconjugates? Eur. J. Biochem 104, 147–153. [DOI] [PubMed] [Google Scholar]
- Moremen KW, Ramiah A, Stuart M, Steel J, Meng L, Forouhar F, Moniz HA, Gahlay G, Gao Z, Chapla D, Wang S, Yang J-Y, Prabahkar PK, Johnson R, dela Rosa M, Geisler C, Nairn AV, Wu S-C, Tong L, Gilbert HJ, LaBaer J, and Jarvis DL (2018) Expression system for structural and functional studies of human glycosylation enzymes. Nature Chem. Biol 14, 156–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura-Tsuruta S, Kominami J, Kamei M, Koyama Y, Suzuki T, Isemura M, and Hirabayashi J (2006). Comparative analysis by frontal affinity chromatography of oligosaccharide specificity of GlcNAc-binding lectins, Griffonia simplicifolia lectin-II (GSL-II) and Boletopsis leucomelas lectin (BLL). J. Biochem 140, 285–291. [DOI] [PubMed] [Google Scholar]
- Nielsen MI, Stegmayr J, Grant OC, Yang Z, Nilsson UJ, Boos I, Carlsson MC, Woods RJ, Unverzagt C, Leffler H, et al. (2018). Galectin binding to cells and glycoproteins with genetically modified glycosylation reveals galectin-glycan specificities in a natural context. J Biol Chem 293, 20249–20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen PA, Olsen JV, Podtelejnikov AV, Andersen JR, Mann M, and Wisniewski JR (2005). Proteomic mapping of brain plasma membrane proteins. Mol. Cell Proteomics 4, 402–408. [DOI] [PubMed] [Google Scholar]
- Nittis T, Gitlin JD (2004) Role of copper in the proteosome-mediated degradation of the multicopper oxidase hephaestin. J Biol Chem. 279, 25696. [DOI] [PubMed] [Google Scholar]
- Nizet V, Varki A and Aebi M (2015). Microbial Lectins: Hemagglutinins, Adhesins, and Toxins., In Essentials of Glycobiology, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, eds. (Cold Spring Harbor Laboratory Press; ) [PubMed] [Google Scholar]
- Obermann J, Priglinger CS, Merl-Pham J, Geerlof A, Priglinger S, Götz M, and Hauck SM (2017). Proteome-wide Identification of Glycosylation-dependent Interactors of Galectin-1 and Galectin-3 on Mesenchymal Retinal Pigment Epithelial (RPE) Cells. Mol. Cell Proteomics 16, 1528–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohannesian DW, Lotan D, and Lotan R (1994). Concomitant increases in galectin-1 and its glycoconjugate ligands (carcinoembryonic antigen, lamp-1, and lamp-2) in cultured human colon carcinoma cells by sodium butyrate. Cancer Res. 54, 5992–6000. [PubMed] [Google Scholar]
- Prudden AR, Liu L, Capicciotti CJ, Wolfert MA, Wang S, Gao Z, Meng L, Moremen KW, and Boons G-J (2017). Synthesis of asymmetrical multiantennary human milk oligosaccharides. Proc. Natl. Acad. Sci. U.S.a 114, 6954–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan B, Boeggeman E, and Qasba PK (2012). Binding of N-acetylglucosamine (GlcNAc) β1–6-branched oligosaccharide acceptors to β4-galactosyltransferase I reveals a new ligand binding mode. J Biol Chem 287, 28666–28674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan B, Ramasamy V, and Qasba PK (2006). Structural snapshots of beta-1,4-galactosyltransferase-I along the kinetic pathway. J. Mol. Biol 357, 1619–1633. [DOI] [PubMed] [Google Scholar]
- Ramasamy V, Ramakrishnan B, Boeggeman E, Ratner DM, Seeberger PH, and Qasba PK (2005). Oligosaccharide preferences of beta1,4-galactosyltransferase-I: crystal structures of Met340His mutant of human beta1,4-galactosyltransferase-I with a pentasaccharide and trisaccharides of the N-glycan moiety. J. Mol. Biol 353, 53–67. [DOI] [PubMed] [Google Scholar]
- Rillahan CD, Antonopoulos A, Lefort CT, Sonon R, Azadi P, Ley K, Dell A, Haslam SM, and Paulson JC (2012). Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Biol 8, 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AC, Yu S-H, Li B, Zegzouti H, and Kohler JJ (2015). Enhanced transfer of a photocrosslinking N-acetylglucosamine (GlcNAc) analog by an O-GlcNAc transferase mutant with converted substrate specificity. J Biol Chem 290, 22638–22648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ M, Lou D, and Kohler H (2005). Photo-activated affinity-site cross-linking of antibodies using tryptophan containing peptides. J. Immunol. Methods 304, 100–106. [DOI] [PubMed] [Google Scholar]
- Samanta S, Dutta D, Ghoshal A, Mukhopadhyay S, Saha B, Sundar S, Jarmalavicius S, Forgber M, Mandal C, Walden P, Mandal C (2011) Glycosylation of erythrocyte spectrin and its modification in visceral leishmaniasis. PLoS One. 6, e28169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon E, Luchansky SJ, Hang HC, Yu C, Lee SC, and Bertozzi CR (2002). Investigating cellular metabolism of synthetic azidosugars with the Staudinger ligation. J. Am. Chem. Soc 124, 14893–14902. [DOI] [PubMed] [Google Scholar]
- Seelenmeyer C, Wegehingel S, Lechner J, and Nickel W (2003). The cancer antigen CA125 represents a novel counter receptor for galectin-1. J. Cell. Sci 116, 1305–1318. [DOI] [PubMed] [Google Scholar]
- Shajahan A, Supekar NT, Wu H, Wands AM, Bhat G, Kalimurthy A, Matsubara M, Ranzinger R, Kohler JJ, and Azadi P (2020). Mass spectrometric method for the unambiguous profiling of cellular dynamic glycosylation. ACS Chem. Biol 15, 2692–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura K, Arata Y, Uchiyama N, Hirabayashi J, and Kasai K-I (2002). Determination of the affinity constants of recombinant human galectin-1 and −3 for simple saccharides by capillary affinophoresis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci 768, 199–210. [DOI] [PubMed] [Google Scholar]
- Stowell SR, Arthur CM, Mehta P, Slanina KA, Blixt O, Leffler H, Smith DF, and Cummings RD (2008a). Galectin-1, −2, and −3 exhibit differential recognition of sialylated glycans and blood group antigens. J Biol Chem 283, 10109–10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell SR, Qian Y, Karmakar S, Koyama NS, Dias-Baruffi M, Leffler H, McEver RP & Cummings RD (2008b) Differential roles of galectin-1 and galectin-3 in regulating leukocyte viability and cytokine secretion. J Immunol, 180, 3091–102. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, and Kohler JJ (2008). Photoactivatable crosslinking sugars for capturing glycoprotein interactions. J. Am. Chem. Soc 130, 3278–3279. [DOI] [PubMed] [Google Scholar]
- Thiemann S, and Baum LG (2016). Galectins and Immune Responses-Just How Do They Do Those Things They Do? Annu. Rev. Immunol 34, 243–264. [DOI] [PubMed] [Google Scholar]
- Unligil UM, Zhou S, Yuwaraj S, Sarkar M, Schachter H, and Rini JM (2000). X-ray crystal structure of rabbit N-acetylglucosaminyltransferase I: catalytic mechanism and a new protein superfamily. Embo J. 19, 5269–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A (2017). Biological roles of glycans. Glycobiology 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasta GR (2020). Galectins in Host-Pathogen Interactions: Structural, Functional and Evolutionary Aspects. Adv. Exp. Med. Biol 1204, 169–196. [DOI] [PubMed] [Google Scholar]
- Wands AM, Fujita A, McCombs JE, Cervin J, Dedic B, Rodriguez AC, Nischan N, Bond MR, Mettlen M, Trudgian DC, et al. (2015). Fucosylation and protein glycosylation create functional receptors for cholera toxin. Elife 4, e09545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Aoki-Kinoshita KF, Ishihama Y, Okuda S (2021). GlycoPOST realizes FAIR principles for glycomics mass spectrometry data. Nucleic Acids Research 49, D1523–D1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernick NLB, Chinnapen DJ-F, Cho JA, and Lencer WI (2010). Cholera toxin: an intracellular journey into the cytosol by way of the endoplasmic reticulum. Toxins (Basel) 2, 310–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, and Kohler J (2019). Photocrosslinking probes for capture of carbohydrate interactions. Curr Opin Chem Biol 53, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SC, Paul A, Ho A, Patel KR, Allen JWL, Verkerke H, Arthur CM & Stowell SR (2021) Generation and use of recombinant galectins. Curr Protoc, 1, e63. [DOI] [PubMed] [Google Scholar]
- York WS, Mazumder R, Ranzinger R, Edwards N, Kahsay R, Aoki-Kinoshita KF, Campbell MP, Cummings RD, Feizi T, Martin M, Natale DA, Packer NH, Woods RJ, Agarwal G, Arpinar S, Bhat S, Blake J, Castro LJG, Fochtman B, Gildersleeve J, Goldman R, Holmes X, Jain V, Kulkarni S, Mahadik R, Mehta A, Mousavi R, Nakarakommula S, Navelkar R, Pattabiraman N, Pierce MJ, Ross K, Vasudev P, Vora J, Williamson T, Zhang W (2020) GlyGen: Computational and Informatics Resources for Glycoscience. Glycobiology. 30, 72–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Mizushima T, and Tanaka K (2019). Sugar-Recognizing Ubiquitin Ligases: Action Mechanisms and Physiology. Front Physiol 10, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S-H, Boyce M, Wands AM, Bond MR, Bertozzi CR, and Kohler JJ (2012). Metabolic labeling enables selective photocrosslinking of O-GlcNAc-modified proteins to their binding partners. Proc. Natl. Acad Sci. U.S.a 109, 4834–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data 1. Representative MS2 spectra of glycopeptides containing GlcNDAz, Related to Figure 2. See attached word file. Related to main Figure 2.
Supplementary Table 1. List of candidate galectin-1 binding proteins identified by proteomics analysis of crosslinked bands, Related to Figure 6. See attached excel file.
Data Availability Statement
Raw data of proteomics analysis were deposited to MassIVE with the identifier MSV000087640. Crosslinked bands from the first biological replicate from high, medial, and low molecular weight have the sample names f.MSV000087640/raw/LUM1_661503.raw, f.MSV000087640/raw/LUM1_661504.raw and f.MSV000087640/raw/LUM1_661505.raw, respectively. Control bands from the first biological replicate from high, medial, and low molecular weight have the sample names f.MSV000087640/raw/LUM1_661500.raw, f.MSV000087640/raw/LUM1_661501.raw, and f.MSV000087640/raw/LUM1_661502.raw, respectively. Crosslinked bands from the second biological replicate from high, medial, and low molecular weight have the sample names f.MSV000087640/raw/LUM1_671792.raw, f.MSV000087640/raw/LUM1_671790.raw and f.MSV000087640/raw/LUM1_671788.raw, respectively. Control bands from the second biological replicate from high, medial, and low molecular weight have the sample names f.MSV000087640/raw/LUM1_671793.raw, f.MSV000087640/raw/LUM1_671791.raw and f.MSV000087640/raw/LUM1_671789.raw, respectively.
The glycoproteomics data have been deposited at GlycoPost (Watanabe et al., 2021) with the dataset identifier GPST000201.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request