

Abstract
Background & Aims:
Eosinophilic esophagitis (EoE) can progress to fibrostenosis by unclear mechanisms. Herein, we investigated gene dysregulation in fibrostenotic EoE, its association with clinical parameters and specific pathways, and the functional consequences.
Methods:
Esophageal biopsies from subjects with EoE were collected across 11 Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR) sites (n = 311) and two independent replication cohorts (n = 83). Inclusion criteria for fibrostenotic EoE were endoscopic rings, stricture, and/or a history of dilation. Endoscopic, histologic, and molecular features were assessed by the EoE endoscopic reference score (EREFS), EoE Histology Scoring System (HSS), EoE Diagnostic Panel (EDP), and RNA sequencing. Esophageal endothelial TSPAN12 expression and functional effects on barrier integrity and gene expression were analyzed in vitro.
Results:
TSPAN12 was the gene most correlated with fibrostenosis (r = −0.40, P < .001). TSPAN12 was lower in fibrostenotic EoE and correlated with EREFS, EDP, and HSS (r = 0.34–0.47, P < .001). Lower TSPAN12 associated with smaller esophageal diameter (r = 0.44, P = .03), increased lamina propria fibrosis (r = −0.41, P < .001), and genes enriched in cell cycle–related pathways. IL-13 reduced TSPAN12 expression in endothelial cells. Conversely, anti-IL-13 therapy increased TSPAN12 expression. TSPAN12 gene silencing increased endothelial cell permeability and dysregulated genes associated with extracellular matrix (ECM) pathways. Endothelial cell–fibroblast crosstalk induced ECM changes relevant to esophageal remodeling.
Conclusions:
Patients with fibrostenotic EoE express decreased levels of endothelial TSPAN12. We propose that IL-13 decreases TSPAN12, likely contributing to the chronicity of EoE by promoting tissue remodeling through fibroblast-endothelial cell crosstalk.
Keywords: endothelium, eosinophil, eosinophilic esophagitis, fibrosis, transcriptome
Graphical Abstract
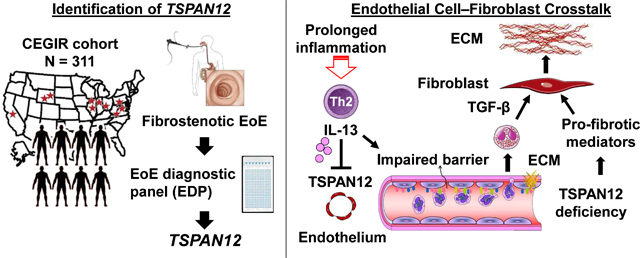
Lay summary
We deciphered the role of TSPAN12 in fibrostenotic EoE by transcriptomic analysis across a multi-site cohort, its association with clinical parameters and specific pathways, and the functional consequences in vitro.
INTRODUCTION
Eosinophilic esophagitis (EoE) is an immune/antigen-mediated, progressive fibrostenotic disease characterized by marked esophageal specific eosinophilia and esophageal dysfunction.1, 2 Evidence is accumulating that the disease starts with eosinophil-rich, type 2–mediated inflammation that depends on epithelial barrier impairment, accompanying basal cell hyperplasia, and loss of epithelial cell differentiation. Activity of this chronic disease typically waxes and wanes in response to therapy (e.g., dietary modifications, topical steroids) with fibrotic features developing over time.3 It is unclear which patients will go on to develop fibrostenosis. Only some patients with untreated disease and ongoing long-term inflammation develop fibrostenotic changes over many years,4 whereas others develop fibrostenosis at a young age.5, 6 The fibrostenotic endoscopic phenotype associates with a distinct EoE endotype based on esophageal gene expression.7, 8 Understanding the key features that differentiate EoE phenotypes will potentially facilitate personalized care and earlier interventions.
The molecular cause of fibrogenesis in EoE is not well understood. Previous studies suggest that uncontrolled type 2–associated eosinophilic esophageal inflammation leads to esophageal rigidity in children and adults through subepithelial tissue remodeling that includes basal zone hyperplasia (BZH), fibrosis, angiogenesis, and smooth muscle hyperplasia with hypertrophy.9, 10 Although eosinophils densely infiltrate the esophagus in EoE, their complex interactions with non-immune structural cells likely dictate the histologic and clinical remodeling outcomes. Among tissue structural cells, epithelial cells are an important source of a wide variety of immune mediators, including eotaxin-3, thymic stromal lymphopoietin (TSLP), and calpain-14.1, 11 Cumulative evidence suggests that EoE pathogenesis is mediated by an IL-13–stimulated, epithelial derived transcriptome that is largely reversible with corticosteroid treatment.12 Given that the EoE fibrostenotic phenotype is often steroid resistant,13 tissue cells other than epithelial cells may modulate EoE-related key molecules in a corticosteroid-insensitive manner. For example, eosinophil- and mast cell–derived TGF-β induces fibroblast activation, promotes fibroblast-eosinophil tethering,14 and promotes epithelial mesenchymal transition in EoE, thereby increasing tissue remodeling via the synthesis and deposition of extracellular matrix in subepithelial layers.15 In addition, eosinophil expression of vascular endothelial growth factor likely supports increased angiogenic responses of vascular endothelium with vascular cell adhesion molecule-1 activation by IL-13, contributing to the increased eosinophil trafficking and esophageal thickening.16 However, the molecular pathogenesis of the EoE fibrostenotic phenotype remains largely undefined.
Herein, we sought insight into the molecular pathogenesis of the EoE fibrostenotic phenotype by comparing fibrostenotic and non-fibrostenotic disease by analysis of EoE transcript signatures with the EoE diagnostic panel (EDP),17 a set of 94 esophageal mRNAs dysregulated in EoE. Children and adults with a fibrostenotic or non-fibrostenotic EoE phenotype across multiple sites associated with the Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR) and a replication cohort were examined.18 We identified a previously unappreciated mechanism involving loss of esophageal tetraspanin 12 (TSPAN12) and deciphered its molecular control and likely contribution to the disease process and the implications for therapeutic strategies.
METHODS
Study design and subjects
This study was conducted within CEGIR,18 a national collaborative network of 16 academic centers caring for adults and children with eosinophilic gastrointestinal disorders (EGID). The CEGIR clinical study Outcome Measures in Eosinophilic Gastrointestinal disorders Across the ages (OMEGA, ClinicalTrials.gov Identifier: NCT02523118) is a longitudinal cohort study aimed at understanding EGID natural history during routine clinical care. For the discovery cohort, children and adults with EoE (age ≥3 years) were enrolled in a CEGIR-associated, multicenter, prospective, observational study. For the validation cohorts, we employed two separate cohorts with the same disease definition: 1) Cincinnati Children’s Hospital Medical Center (CCHMC) cohort; children and adults with EoE presenting for standard care were enrolled in a biologically independent cohort at CCHMC and 2) Six Food vs One Food Eosinophilic Esophagitis Elimination Diet (SOFEED) Followed by Swallowed Glucocorticoid Trial (ClinicalTrials.gov Identifier: NCT02778867) cohort; the latter included adults having active EoE without treatment (e.g., steroids, diet restriction) at baseline in the SOFEED trial. The institutional review boards of the participating institutions approved this study via a central institutional review board at CCHMC.
Definitions of fibrostenotic endoscopic phenotype
Clinical features were defined on the basis of previous reports,7, 19 as summarized in Supplementary Table 1. Inclusion criteria for fibrostenotic endoscopic phenotype (termed fibrostenotic EoE) were endoscopic rings, stricture, and/or a history of dilation, regardless of other endoscopic and histologic findings. Subjects who did not meet the above inclusion criteria were categorized as non-fibrostenotic phenotype (termed non-fibrostenotic EoE).
Endoscopic and histologic features
Findings by esophagogastroduodenoscopy were prospectively recorded and assessed by the EoE endoscopic reference score (EREFS) as previously described.20 The EREFS total score was calculated as the sum of the EREFS scores for the distal and proximal/mid esophagus. Fibrostenotic signs were calculated as the sum of the total scores of rings and stricture.21 Estimating stricture diameter at the distal and proximal esophagus was done by inspection on retroflection.22 Esophageal biopsy specimens were analyzed using described approaches, including quantifying peak esophageal eosinophil counts and the EoE Histology Scoring System (HSS).23
Molecular evaluation
Molecular profiles of patients’ esophageal biopsies and human esophageal microvascular endothelial cells (HEsMEC) (IL-13–stimulated and untreated) were assessed by EDP with normalization to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH).17 In addition to EDP datasets,17, 24, 25 both bulk (GSE58640)26 and single-cell RNA sequencing (GSE126250)27 data were used for esophageal biopsy specimens of patients with EoE or control subjects. Genes were tested for differential expression (EoE vs. controls) using DESeq2,28 yielding a P value for each gene [EGIDExpress (https://egidexpress.research.cchmc.org/data/)]. Gene ontology enrichment analysis was performed with the ToppGene suite.29
Cell culture and treatment
For cell culture and stimulation, HEsMEC were maintained with endothelial cell medium per manufacturer recommendations (ScienCell Research Laboratories, Carlsbad, CA). Recombinant human TNF-α, TGF-β, IL-4, and IL-13 were purchased from PeproTech (Rocky Hill, NJ). Fluticasone propionate (FP) was purchased from SIGMA (St. Louis, MO). Cells were treated with the indicated cytokines and concentrations. Other esophageal cells (epithelial [EPC2] and fibroblast [FEF3] cells; a kind gift from Dr. Anil Rustgi and Dr. Hiroshi Nakagawa [Columbia University]) were cultured as described previously.30, 31
In vitro assays
For TSPAN12 gene silencing in HEsMEC, lentiviral transduction was performed, as described previously.32 Lentiviral shRNA vectors against TSPAN12 (MISSION shRNA, Sigma-Aldrich, TRCN0000159397 [shTSPAN12_1] and TRCN0000379849 [shTSPAN12_2]) and no known mammalian genes as a control (shCtrl) were used to produce lentiviral particles. For barrier function, transendothelial electrical resistance (TEER) and fluorescein isothiocyanate (FITC)-dextran flux measurements were assessed, as previously described.32 Whole-transcriptome analysis was performed in endothelial cells (TSPAN12 silencing vs. control) as described previously.31 qPCR, western blotting, and immunofluorescence analyses were performed as described previously.32 For media-swap experiments, endothelial cell supernatants were collected from confluent HEsMECs (shCtrl, shTSPAN12) and used to stimulate confluent FEF3 for 24 hours. Fresh media with or without TGF-β were also used as controls.
Statistical analysis
Data are presented as n (%) or median (interquartile range [IQR]) unless otherwise stated. Missing data were excluded from all formal statistical analyses. Non-parametric correlation analysis was performed using Spearman’s rank correlation coefficient followed by Bonferroni adjustment. For 2-group comparisons, we used t-test or Mann-Whitney U test. For ≥3-group comparisons, we used one-way analysis of variance (ANOVA) followed by a Dunnett’s multiple-comparison test or the Kruskal-Wallis test followed by a Dunn multiple-comparison test, unless otherwise stated. A Benjamini–Hochberg correction was applied for multiple testing to control the false-discovery rate (FDR). A significant P value was defined as P < .05.
For detailed information, see the Supplementary Material and Methods.
RESULTS
Study Population
Gene expression profiles from 394 esophageal biopsy samples were analyzed (n = 311 discovery cohort and 83 replication cohorts [n = 62 CCHMC, n = 21 SOFEED]). Demographic characteristics are detailed in Table 1 and Supplementary Table 2–7 and briefly summarized here.
Table 1.
Demographic and clinical characteristics of study subjects*
| CEGIR cohort (Discovery) | CCHMC cohort (Validation)# | SOFEED cohort (Validation) | Combined EoE cohorts | |||
|---|---|---|---|---|---|---|
|
| ||||||
| N = 311 | N = 62 | N = 21 | Non-fibrostenotic | Fibrostenotic | P value | |
| Fibrostenotic EoE | 151 (48.6%) | 16 (25.8%) | 17 (81%) | 210 (53.3%) | 184 (46.7%) | |
| Demographics | ||||||
| Age at biopsy (y) | 25.5 (14.4–41.4) | 9.4 (5.3–17.3) | 33.1 (30.5–42.8) | 13.8 (8.8–21.2) | 37.1 (28.1–46.8) | < 0.00001 |
| Age at Diagnosis (y) | 19.0 (7.0–36.0) | 6.0 (3.0–13.0) | NA | 6.8 (2.9–13.7) | 31.1 (20.8–41.0) | < 0.00001 |
| Age at First Endoscopy (y) | 16.2 (6.0–33.9) | NA | NA | 7.0 (2.9–15.0) | 30.0 (19.0–38.1) | < 0.00001 |
| Age at First Symptoms (y) | 10.0 (2.0–24.0) | 0.8 (0–5.0) | NA | 3.0 (0.6–9.0) | 16.1 (9.0–30.0) | < 0.00001 |
| Disease duration (y) | 9.8 (6.4–15.2) | 4.9 (2.7–8.0) | NA | 7.6 (5.4–11.4) | 12.4 (5.9–20.1) | < 0.00001 |
| Diagnostic delay (y) | 2.0 (0.8–8.7) | 2.8 (1.6–5.2) | NA | 1.3 (0.5–3.3) | 6.3 (2.0–15.2) | < 0.00001 |
| Gender (Male) | 204 (65.6%) | 52 (83.9%) | 9 (42.9%) | 154 (73.3%) | 111 (60.3%) | 0.00712 |
| Race (White) | 288 (92.6%) | 58 (93.5%) | 20 (95.2%) | 191 (91.0%) | 175 (95.1%) | 0.11980 |
| Fibrostenotic EoE | 151 (48.6%) | 16 (25.8%) | 17 (81%) | |||
| History of dilation | 96 (30.9%) | 10 (16.1%) | NA | 0 (0%) | 106 (57.6%) | < 0.00001 |
| Tissue eosinophil counts | ||||||
| Peak (eos/HPF) | 20 (2–44) | 64 (37–109) | 68 (24–95) | 24 (4–56) | 32 (11–65) | 0.07284 |
| Range (min.–max.) | (0–252) | (16–269) | (15–181) | (0–269) | (0–252) | |
| Disease parameters | ||||||
| EDP score | 259 (97–353) | 150 (67–284) | 152 (73–257) | 268 (113–363) | 173 (58–309) | 0.00023 |
| HSS total score | 0.6 (0.3–0.9) | 0.8 (0.6–0.9) | 0.9 (0.7–1.1) | 0.5 (0.3–0.8) | 0.8 (0.5–1.0) | 0.00005 |
| EREFS total score | 3 (1–6) | 2 (1–2) | 8 (6–10) | 1 (0–3) | 6 (3–9) | < 0.00001 |
| Treatment at biopsy | ||||||
| Ongoing diet therapy | 149 (47.9%) | 35 (56.5%) | 0 (0%) | 122 (58.1%) | 62 (33.7%) | < 0.00001 |
| Proton pump inhibitor | 138 (44.4%) | 43 (69.4%) | 0 (0%) | 83 (39.5%) | 71 (38.6%) | 0.91762 |
| Swallowed topical steroids | 151 (48.6%) | 16 (25.8%) | 0 (0%) | 81 (38.6%) | 71 (38.6%) | 1.00000 |
| Oral systemic steroids | 1 (0.3%) | 1 (1.6%) | 0 (0%) | 32 (15.2%) | 12 (6.5%) | 0.00637 |
Data are n (%) or median (interquartile range [IQR]) unless otherwise stated.
Simplified endoscopic severity score (ESS) was used. CEGIR, Consortium of Eosinophilic Gastrointestinal Disease Researchers; CCHMC, Cincinnati Children’s Hospital Medical Center; SOFEED, Six Food vs. One Food Eosinophilic Esophagitis Diet Study; EDP, EoE Diagnostic Panel; EoE, eosinophilic esophagitis; HSS, EoE Histology Scoring System; EREFS, EoE endoscopic reference score; eos/HPF, eosinophils per high-power microscopic field; max., maximum; min., minimum; NA, not assessed.
For all subjects (n = 394), the age was 2.3–72.7 years, with 155 children (median 10.5 years [IQR 7.5–14.3]) and 239 adults (median 36.2 years [IQR 26.4–46.0]). Approximately 70% were men, and >90% self-identified as White. The peak esophageal eosinophil counts ranged from 0–269 eosinophils/HPF and had no significant difference among CEGIR sites (P = .57). Over half of subjects (n = 270, 69%; discovery cohort 187/311, 60%; CCHMC/SOFEED validation cohort 83/83, 100%) had active EoE (≥15 eosinophils/HPF). Similar to peak eosinophils, the HSS and EDP showed consistency and had similar values across CEGIR sites (P = .31 and P = .80, respectively). At biopsy, 284 subjects (72%) were taking one or more medication (39% proton pump inhibitor [PPI], 39% swallowed topical steroids, 11% oral systemic steroids). About half of subjects (n = 184, 47%) were being treated for EoE with diet therapy. Subjects in the SOFEED cohort were not receiving medication nor dietary therapy for EoE at the time of biopsy.
About half of subjects (n = 184, 47%; discovery cohort 151/311, 49%; CCHMC/SOFEED validation cohort 33/83, 40%) were identified as having fibrostenotic EoE (175 defined by rings, stricture, and/or a history of dilation; 9 defined solely by a history of dilation). Among the 184 subjects with fibrostenotic EoE, 106 (58%; 96 adults and 10 children) had a history of dilation. In fibrostenotic EoE, endoscopic abnormalities were common, including 58% with edema, 40% with rings, 33% with exudates, 57% with furrows, and 18% with stricture.
Compared to non-fibrostenotic EoE, fibrostenotic EoE was more prevalent in adults (P < .001) and females (P = .007) and less prevalent in subjects on elimination diet therapy (P < .001) or oral systemic steroids (P = .006). Prevalence of fibrostenotic EoE increased with longer duration of disease and diagnostic delay (P < .001 for each). Disease parameters (EDP, HSS, EREFS) had more severe scores in fibrostenotic EoE. Conversely, there were no significant differences in peak eosinophil counts nor treatment modality (PPIs and topical steroids) between fibrostenotic and non-fibrostenotic EoE. However, fibrostenotic EoE and non-fibrostenotic EoE showed variability when further stratified (Supplementary Table 3–7).
TSPAN12 is the most dysregulated gene in fibrostenotic EoE
To identify genes that contributing to fibrostenotic EoE, we evaluated the association between esophageal fibrostenotic signs and individual expression levels of EDP genes in the discovery cohort (Figure 1A). Using fibrostenotic signs (total scores of rings and stricture) to represent the overall value of fibrostenosis,21 we determined that the most correlated gene was TSPAN12, having substantial correlation (r = −0.40, P < .001). TSPAN12 is a member of the transmembrane 4 superfamily (tetraspanin family)33 and regulates cell development, activation, growth, and motility.34 Although TSPAN12 has been shown to be down-regulated in the EoE transcriptome,17, 26 its relationship with fibrostenotic EoE was hitherto unidentified. To better understand TSPAN12 expression in fibrostenotic EoE, we stratified the subjects by disease activity (active, inactive), age (adults, children), and gender (male, female) (Supplementary Table 3–6). Notably, TSPAN12 was significantly lower in fibrostenotic than non-fibrostenotic EoE regardless of age group or gender (Figure 1B–E). Notably, peak eosinophil counts did not differ between fibrostenotic and non-fibrostenotic EoE (Figure 1B–E). The differences in TSPAN12 between fibrostenotic and non-fibrostenotic EoE were independent of not only peak eosinophil levels, but also disease duration per regression analysis (P < .001). To further verify the lower TSPAN12 expression in fibrostenotic EoE, we employed 2 separate cohorts, the CCHMC cohort (children and adults with active EoE from a single center) and SOFEED cohort (adults with active, untreated EoE at biopsy from multiple centers). Our findings were consistent with those in the discovery cohort; TSPAN12 was significantly lower in fibrostenotic EoE than non-fibrostenotic EoE (Supplementary Figure 1).
Figure 1. TSPAN12 most highly dysregulated gene for fibrostenotic EoE.

A, Negative log10 FDR P value of the Spearman correlation between the fibrostenotic score (total score of rings and stricture) and a diagnostic subset of genes from the EoE transcriptome (EDP). Red indicates a positive correlation and blue indicates a negative correlation. The dashed line indicates an FDR P value of 0.05. B-E, Peak esophageal eosinophil counts and TSPAN12 expression are plotted by groups for EoE disease activity (active, inactive), age (adults, children), gender (male, female), and EoE phenotype (NF, non-fibrostenotic [blue]; F, fibrostenotic [red]) for the Discovery cohort (CEGIR, active and inactive EoE). Data are mean ± SEM; markers represent individual subjects. NS, *P < .05, and **P < .01 using the Mann-Whitney U test. CEGIR, Consortium of Eosinophilic Gastrointestinal Disease Researchers; EoE, eosinophilic esophagitis; EDP, EoE Diagnostic Panel; FDR, false-discovery rate; GWAS, genome-wide association study; NS, not significant; HPF, high-power microscopic field.
TSPAN12 correlates with endoscopic, histologic, molecular and fibrostenotic features of EoE
To define the relationships among TSPAN12 and various clinical, endoscopic, and histologic features, we evaluated the associations between TSPAN12 expression and disease parameters (peak eosinophil count, EREFS, EDP, HSS) in the discovery cohort. Using total scores, which represent the overall values of each parameter, we found correlations between TSPAN12 expression and the diagnostic parameters: peak eosinophil counts (current diagnostic standard, r = −0.26, P < .001), endoscopic score (EREFS [total scores of edema, rings, exudate, furrows, and stricture], r = −0.47, P < .001), EDP score (EDP, r = 0.39, P < .001), and histologic score (HSS, r = −0.34, P < .001) (Figure 2A). The level of TSPAN12 directly correlated with esophageal diameter (both distal and proximal) in adults (Figure 2B). Furthermore, we examined the relationship of TSPAN12 expression level and the individual histologic components in EoE (i.e., HSS). Notably, TSPAN12 expression inversely correlated with basal zone hyperplasia (BZH) and lamina propria fibrosis (LPF) domains (grade and stage) (BZH, grade: r = −0.31, P < .01, stage: r = −0.31, P < .01; LPF, grade: r = −0.41, P < .01, stage: r = −0.37, P < .01) (Figure 2C, red bar).
Figure 2. TSPAN12 associates with EoE diagnostic parameters and fibrostenotic features.
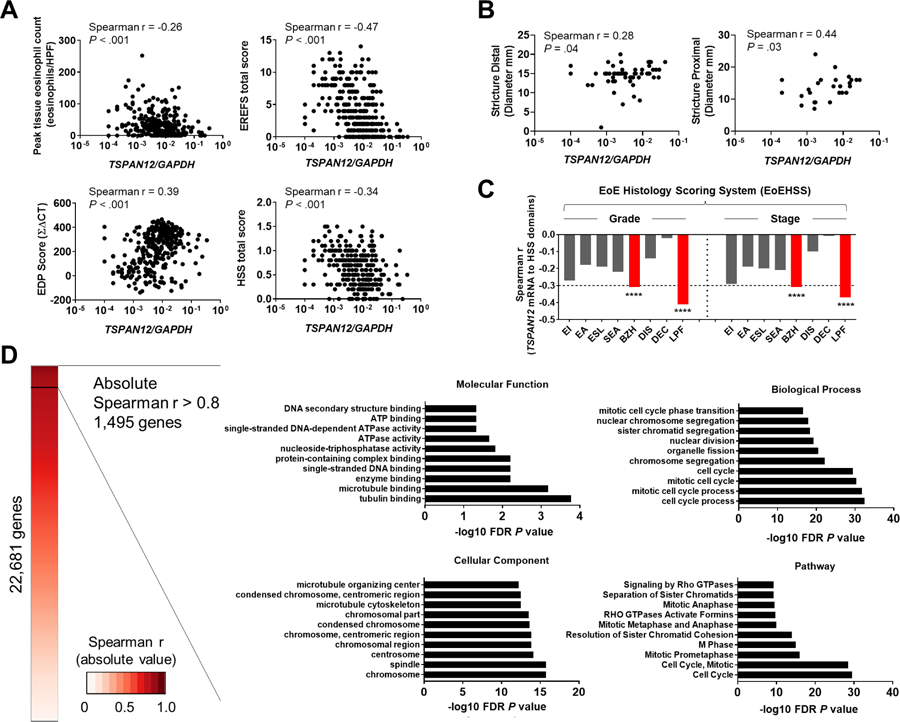
A, Associations between TSPAN12 expression and diagnostic parameters of gene expression, endoscopic, and histologic platforms. TSPAN12 expression levels correlate with peak esophageal eosinophils per HPF (upper left), total score from EREFS (upper right), EDP (lower left), and HSS (lower right). Markers represent individual subjects. B, Associations between TSPAN12 expression and stricture diameter (left, distal; right, proximal). Markers represent individual subjects. C, Associations (Spearman r values) between TSPAN12 expression and HSS domains (EI, eosinophilic inflammation; EA, eosinophilic abscess; ESL, eosinophilic surface layering; SEA, surface epithelial alteration; BZH, basal zone hyperplasia; DIS, dilated intercellular spaces; DEC, dyskeratotic epithelial cells; LPF, lamina propria fibrosis) for grade and stage are shown. ****P < .001. D, Functional enrichment analysis of the genes that strongly correlated with TSPAN12 (1,495 genes). Heatmap based on absolute Spearman r values between TSPAN12 and genes assessed by means of RNA sequencing.26 Shown are the 10 most significant terms by functional enrichment analysis in the following categories: Molecular Function (upper left), Biological Processes (upper right), Cellular Component (lower left), and Pathway (lower right). The x-axes represent the negative log10 FDR P value. EoE, eosinophilic esophagitis; EDP, EoE Diagnostic Panel; EREFS, EoE reference score; HSS, EoE Histology Scoring System; HPF, high-power microscopic field; FDR, false-discovery rate.
To determine the significant and relevant molecular functions associated with TSPAN12 at the whole-transcriptome level, we utilized RNA-sequencing data from esophageal biopsies.26 Of the 22,681 genes analyzed, 1,495 genes strongly correlated with TSPAN12 (absolute Spearman r > 0.8, P < .001) (Figure 2D). Functional enrichment gene ontology (GO) analysis of these 1,495 genes identified the enrichment of cell cycle–related terms (e.g., tubulin binding, cell cycle process, chromosome) in molecular functions, biological processes, and cellular component analyses (P < .001; Figure 2D). Moreover, pathway analysis on these 1,495 genes revealed significant enrichment of cell cycle pathways (Figure 2D). Interestingly, in addition to enrichment pathways, we observed that TSPAN12 was inversely correlated with extracellular matrix (ECM)-related genes (GO:0031012, Supplementary Figure 2), including collagens and metalloproteinases (MMPs) that are elevated in the lamina propria of esophageal biopsies and correlated with fibrotic severity.35
Esophageal TSPAN12 is primarily expressed in endothelial cells
On the basis of single-cell RNA sequencing (Supplementary Table 8),27 TSPAN12 was preferentially expressed in the vascular endothelium in esophageal tissue and significantly enriched in endothelium compared to other cell populations (adjusted P value < 0.001) (Figure 3A–B). Immunofluorescence staining and qPCR analysis for TSPAN12 among esophageal cells (epithelial cells, fibroblasts, endothelial cells) demonstrated that esophageal TSPAN12 is primarily expressed in endothelial cells at the mRNA and protein levels (Figure 3C–D). Notably, active EoE had lower TSPAN12 expression than did controls (Figure 3B). These results show that TSPAN12 was substantially downregulated in the inflamed esophageal endothelium in patients with active EoE.
Figure 3. IL-13 decreases TSPAN12 expression in endothelium.
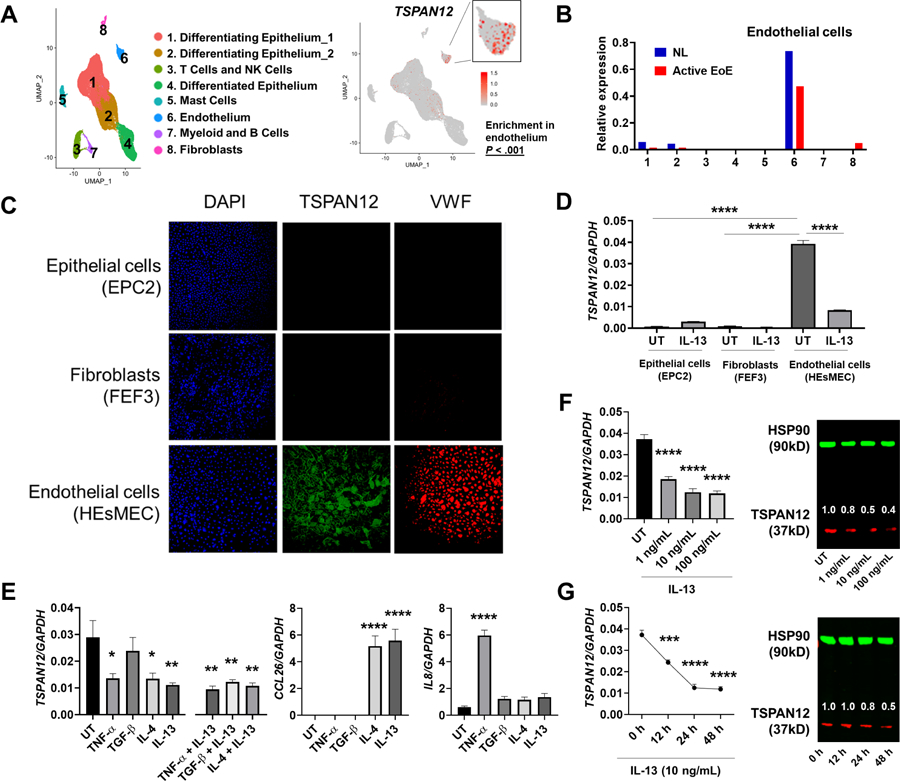
A, TSPAN12 expression in endothelial cells. UMAP plot displaying single cells, colored by shared nearest neighbor clusters and cell types from a single-cell RNA-sequencing analysis of esophageal biopsies (left). The featured plot demonstrates TSPAN12 expression, with each dot representing a single cell (right). B, Relative expression of TSPAN12 by single-cell RNA sequencing; the cluster numbers match those shown in the UMAP plot of panel A. C, Representative immunofluorescence images of TSPAN12 (green) in the esophageal cells. Nuclei was stained by DAPI (blue), and VWF was stained as a specific marker for endothelium (red). D, The TSPAN12 expression levels among the esophageal cells stimulated with or without IL-13 were assessed by qPCR and normalized to GAPDH. E, Effects of EoE-related cytokine stimulation. HEsMEC were treated with the indicated cytokines for 24 h and evaluated by qPCR for TSPAN12, CCL26, and IL8 mRNA. F-G, Dose dependency and time course of IL-13–induced TSPAN12 decrease. HEsMECs were treated with IL-13 (for the indicated dose and time) and evaluated by using qPCR (left) and western blot (right). Numbers above TSPAN12 band represent the signal intensity normalized by HSP90 and relative to the untreated (UT) set as 1. Data (E-G) are the means ± SEM of three independent experiments performed in duplicate. *P < .05, **P < .01, and ****P < .0001 compared with UT using the one-way ANOVA test followed by a Dunnett’s multiple-comparison test. UMAP, uniform manifold approximation and projection; EoE, eosinophilic esophagitis, NL, normal controls; HEsMEC, human esophageal microvascular endothelial cells; UT, untreated; DAPI, 4ʹ,6-diamidino-2-phenylindole; VWF, von Willebrand factor.
IL-13 regulates TSPAN12 loss and induces EoE-like changes in endothelial cells in vitro
To determine which factors may be contributing to the decreased TSPAN12 expression in EoE, we stimulated primary endothelial cells (HEsMEC) with the EoE-relevant cytokines IL-4, IL-13, TNF-α, and TGF-β. We found that IL-4, IL-13, and TNF-α (but not TGF-β) significantly decreased TSPAN12 expression in vitro; however, no synergistic effects were observed with co-stimulation (Figure 3E). As a positive control, we found CCL26 induction by IL-4 and IL-13 and IL8 induction by TNF-α, consistent with previous reports.36 Of the cytokines tested, IL-13 induced the most robust response and in a dose- and time-dependent manner at mRNA and protein levels (Figure 3F–G). In addition to these in vitro data, TSPAN12 had an inverse correlation with IL13 (r = −0.86, P < .001) in esophageal biopsies.26
To gain insight into the potential role of the endothelium in EoE, particularly in the context of IL-13, we analyzed IL-13–treated HEsMEC by assessing the 94 EDP genes. We identified 42 differentially expressed genes (DEGs) in IL-13–treated HEsMEC versus control cells (Figure 4A). Interestingly, the gene most overexpressed in IL-13–treated HEsMEC was CCL26, which is known as the gene most overexpressed in IL-13–stimulated epithelial cells.12 We compared the differential gene signature from these endothelial cells with that of the inflamed esophageal tissue of patients with active EoE (76 dysregulated genes in EDP).17 Notably, among 31 genes shared with esophageal mucosa, 24 genes—CCL26, PMCH, TNFAIP6, SUSD2, CFB, CD200R1, POSTN, CXCL6, CPA3, RTP4, CITED2, GRPEL2, CLDN10, EML1, HILPDA, TSPAN12, CDA, MT1M, ACTG2, ACPP, FLG, SLC16A6, GYS2, and UPK1A—were regulated in similar manners in both IL-13–stimulated endothelial cells and esophageal specimens of patients with EoE (Figure 4B, Supplementary Table 9–10). Stimulating endothelial cells with IL-13 partially reproduced key molecules of the EoE transcriptome, indicating that this cell type likely contributes to the abnormal response seen in endoscopic biopsy specimens.
Figure 4. IL-13 regulates loss of TSPAN12 and induces EoE-like changes in endothelial cells.
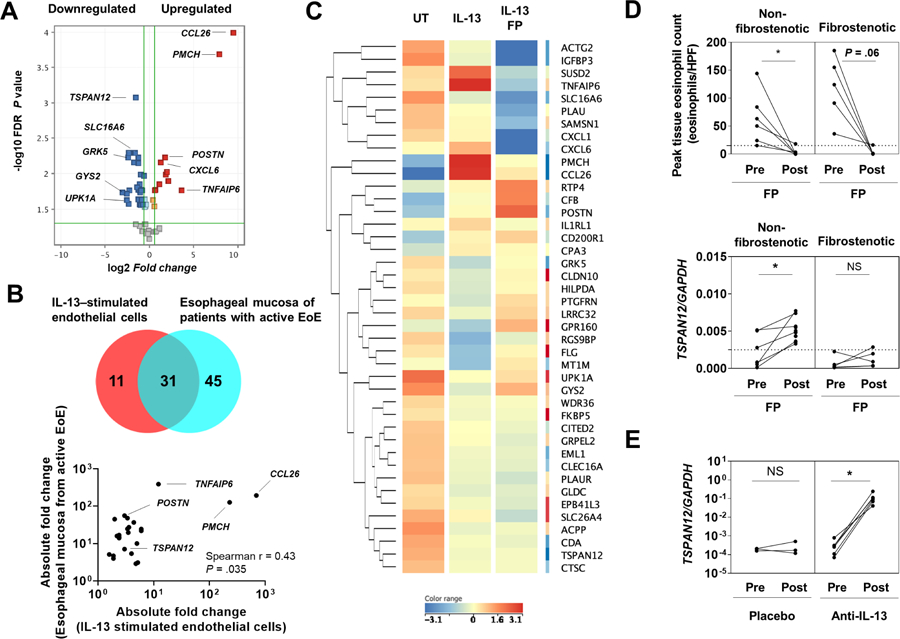
A, Volcano plot of the 42 EDP genes differentially expressed between the IL-13–stimulated endothelial cells and untreated cells. Top 5 of upregulated and downregulated genes were labeled. B, Venn diagram analysis of shared differentially expressed genes in EDP between IL-13–stimulated endothelial cells (HEsMEC) and esophageal mucosa in EoE.17 Spearman correlation comparing absolute fold change values for the overlapping genes regulated in similar manners in both IL-13–stimulated endothelial cells and the inflamed esophageal tissue of patients with active EoE. C, Heat diagram of 31 differentially expressed genes as determined by EDP analysis (A). Each column indicates endothelial cells that were untreated (left), stimulated with IL-13 (100 ng/mL) for 24 h (middle), or treated with IL-13 (100 ng/mL) and fluticasone propionate (FP) (100 nM) for 24 h (right). Hierarchical clustering was used to analyze data and generate heat diagrams (red, upregulated; blue, downregulated). D, Peak esophageal eosinophil counts and and TSPAN12 expression were assessed in patients with EoE by EoE phenotype treated with FP, as described.24 “Pre” corresponds to the beginning of the study, and “Post” corresponds to day 85 of the study. *P < .05, t test. E, Relative expression level of TSPAN12 was assessed in patients with EoE who were either treated with anti–IL-13 antibody or placebo, as described in previous study.25 “Pre” corresponds to the study beginning, and “Post” corresponds to study day 85. *P < .05, t test. EoE, eosinophilic esophagitis; EDP, EoE Diagnostic Panel; HEsMEC, human esophageal microvascular endothelial cells; UT, untreated; TEER, transendothelial electrical resistance; FITC, fluorescein isothiocyanate; NS, not significant; HPF, high-power microscopic field.
Currently, topical corticosteroids are a first-line therapy and one of the most effective therapies for EoE;37 however, endothelial cells are known to not respond as robustly as epithelial cells to steroids.36 Thus, we investigated whether fluticasone propionate (FP) was able to directly modulate the differential gene signature in IL-13–treated HEsMEC. Of 42 DEGs in IL-13–treated HEsMEC, FP affected 25 genes (reversed 5, partially reversed 11, worsened 9) (Figure 4C). For controls, we observed that FP decreased PMCH induction and increased POSTN induction by IL-13 (Supplementary Figure 3), consistent with previous reports.36, 38 Contrary to its general clinical effectiveness for EoE, FP did not prevent TSPAN12 downregulation in endothelial cells (Supplementary Figure 3). In addition to in vitro data, we further assessed whether swallowed FP treatment improved TSPAN12 expression by analyzing a separate set of patients with EoE (Supplementary Table 8).24 Notably, we found that swallowed FP treatment did not normalize TSPAN12 expression in patients with fibrostenotic EoE, yet it did in non-fibrostenotic EoE (Figure 4D).
Anti–IL-13 treatment of human EoE restores esophageal TSPAN12 expression
Early studies have shown a benefit of anti–IL-13 therapy in EoE.25, 39, 40 We hypothesized that anti–IL-13 treatment of patients with EoE may increase esophageal TSPAN12 expression. By analyzing a separate set of patients with EoE (Supplementary Table 8),25 we found that anti–IL-13 antibody (QAX576) treatment, but not placebo treatment, increased TSPAN12 expression in esophageal biopsies in EoE (P = .03) (Figure 4E). Collectively, these results provide evidence that IL-13 regulates TSPAN12 loss in endothelial cells in patients with EoE.
Endothelial TSPAN12 deficiency promotes tissue remodeling via endothelial cell–fibroblast crosstalk
To investigate the potential role of TSPAN12 in mediating esophageal endothelial dysfunction, we gene-silenced TSPAN12 in HEsMEC. HEsMEC that were transduced with two independent TSPAN12 shRNAs exhibited significant reduction in TSPAN12 expression compared with those transduced with non-silencing control shRNA (shCtrl) (Figure 5A). TSPAN12-deficient cells had decreased endothelial integrity and increased permeability compared to that of control cells (Figure 5B–C). Notably, IL-13 (but not TGF-β) treatment further impaired barrier function in TSPAN12-deficient cells.
Figure 5. TSPAN12 deficiency affects endothelial functions.
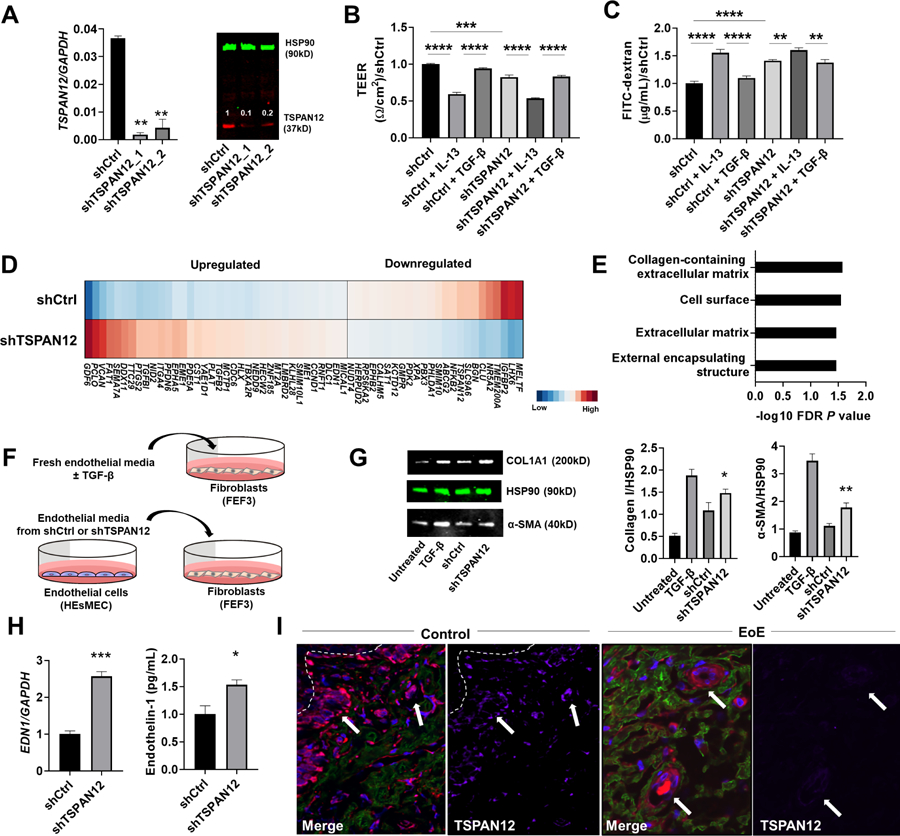
A, Representative qPCR (left) and western blot (right) of TSPAN12 after TSPAN12 downregulation by shRNA in HEsMEC. Numbers above TSPAN12 band represent the signal intensity normalized by HSP90 and relative to the control (shCtrl) set as 1. B-C, The effect of TSPAN12 gene silencing on the endothelial functions of barrier integrity (TEER [B] and FITC-dextran flux measurements [C]). Data were normalized to the control (shCtrl) set as 1. D, Heat diagram representing RNA-sequencing analysis of the TSPAN12-deficient cells and control cells. Differentially expressed genes (DEGs) were identified by filtering on TPM>1, moderated t-test with FDR P < .05. E, Gene ontology analysis of DEGs in the TSPAN12-deficient cells. F, Schematic of media swap experiments. G, Representative western blots demonstrating levels of collagen I and α-SMA in esophageal fibroblasts of media swap experiments. H, EDN1 (endothelin-1) expression in HEsMECs (shCtrl, shTSPAN12). qPCR (left) and ELISA of supernatants (right). I, Representative immunofluorescence staining of esophageal biopsy sections from normal control individuals and patients with fibrostenotic EoE; TSPAN12 (magenta), collagen (cyan), and VWF (red) with DAPI–stained nuclei (blue). White arrows indicate blood vessels, and white broken line indicates basal membrane. For A-C, G, and H, data are the means ± SEM of three independent experiments performed in duplicate. *P < .05, **P < .01, ***P < .001, and ****P < .0001 using the Mann-Whitney U test (H), one-way ANOVA test followed by a Sidak’s multiple comparisons test (B-C), or compared with shCtrl using one-way ANOVA test followed by a Tukey’s multiple comparisons test (A, G). DAPI, 4ʹ,6-diamidino-2-phenylindole; EoE, eosinophilic esophagitis; EDP, EoE Diagnostic Panel; HEsMEC, human esophageal microvascular endothelial cells; TEER, transendothelial electrical resistance; FITC, fluorescein isothiocyanate; FDR, false-discovery rate; shCtrl, shRNA non-silencing control; shTSPAN12, shRNA silencing TSPAN12; VWF, von Willebrand factor.
Next, we investigated the potential molecular mechanism of how TSPAN12 deficiency affects endothelial responses associated with fibrostenotic remodeling in EoE. By RNA sequencing, there were 60 unique DEGs in TSPAN12-deficient endothelial cells compared to control cells (Figure 5D, Supplementary Table 11), including downregulated TSPAN12, serving as an internal positive control. Gene Ontology analysis of these 60 DEGs found functional terms enriched in pathways involved in ECM (Figure 5E, Supplementary Table 12).
Crosstalk between different cell types can induce a profibrotic milieu,30 even in the absence of inflammatory cells. To test whether TSPAN12 deficiency in HEsMEC affects tissue remodeling via interaction with fibroblasts, we performed a media-swap experiment (Figure 5F). We cultured esophageal fibroblasts (FEF3) with media from TSPAN12-deficient or control esophageal endothelial cells (HEsMEC) and found that the fibroblasts stimulated with the media from TSPAN12-deficient HEsMEC showed increased ECM protein expression (Collagen I, α-SMA) (Figure 5G). We used TSPAN12-deficient cells to identify potential pro-fibrotic mediators; by RNA sequencing, TSPAN12-deficient HEsMEC showed up-regulation of pro-fibrotic genes, including endothelin-1 (EDN1).41 We confirmed these findings by qPCR and increased levels of endothelin-1 protein in media of TSPAN12-deficient HEsMEC compared with control cells (Figure 5H).
To confirm the link between esophageal fibrosis and endothelial function in patients with EoE, we analyzed EoE and control esophageal biopsy specimens by immunofluorescence staining (Supplementary Table 8). Esophageal endothelial cells were located at the reduced levels of TSPAN12 and in close proximity to fibrotic areas in active EoE (Figure 5I, Supplementary Figure 4). These collective findings suggest that loss of endothelial TSPAN12 contributes to fibrostenotic EoE via endothelial cell–fibroblast crosstalk.
DISCUSSION
Herein, we deciphered the role of TSPAN12 in fibrostenotic EoE by transcriptomic analysis across a multi-site cohort associated with CEGIR using a combination of standardized histologic, endoscopic, and clinical platforms and examined its function in endothelial cells in the context of EoE. TSPAN12 was the most dysregulated gene in fibrostenotic EoE and showed significantly lower expression regardless of age group or gender. The decreased TSPAN12 in EoE in remission underscores the potential of TSPAN12 to contribute to the fibrostenotic process despite resolved eosinophilia, suggesting that molecular dysregulation of TSPAN12 could promote disease chronicity. TSPAN12 expression correlated with cardinal EoE features, especially EREFS and multiple structural components as assessed by endoscopy and histology, and enrichment of cell cycle pathways. Notably, TSPAN12 was uniquely expressed in endothelial cells, and stimulating endothelial cells with IL-13 reduced TSPAN12 expression and partially reproduced key components of the EoE transcriptome, indicating a previously underrecognized role of the endothelium in disease pathogenesis. Anti–IL-13 therapy of EoE increased TSPAN12 expression, adding clinical significance of this and related anti–type 2 therapies.25, 39, 40 Finally, TSPAN12 gene silencing promoted endothelial dysfunction characterized by increased endothelial permeability and ECM-related gene expression; these changes, especially the increase in permeability and profibrotic mediators (e.g., endothelin-1) are likely contributory to tissue remodeling on the basis of prior studies.42 In support of our findings, crosstalk between TSPAN12-deficient endothelial cells and fibroblasts induced ECM changes relevant to esophageal remodeling. Taken together, we propose that endothelial cell TSPAN12 loss is induced by IL-13 and contributes to EoE by promoting tissue remodeling, likely contributing to the chronicity of EoE, particularly related to fibrostenosis.
There has been recent interest in investigating whether unique molecular profiles are involved in the fibrostenosis-related EoE disease group on the basis of a subtype approach (e.g., phenotype, endotype).19, 43 Our findings dovetail with previous work reporting that three transcript clusters associate with distinct endotypes—EoE endotype 1–3 (EoEe1–3)—despite similar eosinophil levels.43 Notably, EoEe3 was associated with a narrow-caliber esophagus and enriched for structural genes that lose expression, including TSPAN12. Although the molecular mechanism to the gene level was uncertain, it is consistent with our current findings. Moreover, herein, TSPAN12 expression was associated with esophageal diameter, alterations of structural features in histology, and cell cycle pathways that signify the potential role of endothelial proliferation during EoE-associated angiogenesis. Significant correlations of decreased TSPAN12 with smaller proximal and distal esophageal diameters, which reflect fibrostenotic severity, support direct involvement of TSPAN12 in esophageal remodeling; these findings are the first molecular correlate of esophageal diameter. Notably, TSPAN12 expression also exhibited strongest association with the LPF and BZH HSS domains, corroborating recent work supporting a substantial role of the lamina propria and basal epithelium, particularly related to tissue remodeling.44 In EoE, TSPAN12 inversely correlated with MMPs (e.g., MMP-2, MMP-14), which were upregulated in active EoE biopsies with LPF.35 These findings suggest that TSPAN12 has a strong, potential mechanistic link with fibrostenotic EoE.
Our data suggest a novel paradigm wherein disease-associated endothelial changes may influence the fibrostenotic disease process and chronicity in the EoE inflammatory milieu. Our data identify IL-13 as a potent regulator of TSPAN12 expression in human endothelial cells. Previous work demonstrated a pronounced effect of IL-13 on global gene expression in primary esophageal epithelial cells and, in particular, genes involved in epithelial differentiation.12 Interestingly, we found TSPAN12 to be uniquely expressed in endothelial cells. TSPAN family members participate in diverse cellular processes, including signaling platforms by forming TSPAN-enriched microdomains in plasma membranes, but have been associated with several pathologic conditions (e.g., cancer, retinal dystrophies, viral infections, mental retardation), though not previously with the esophagus nor allergic disease.45–48 The IL-13–TSPN12 axis in endothelial cells identified herein and IL-13 being significantly increased in the peripheral blood and esophageal mucosa of patients with active EoE49 indicate that endothelial TSPAN12 expression is negatively regulated by type 2 cytokines during allergic inflammation.
Due to their anatomical location, vascular endothelial cells have a critical “gatekeeper” role in the inflammatory process through their ability to recruit circulating immune cells into tissues.36 Endothelial dysfunction suggests a mechanism whereby immune cells easily penetrate the endothelial barrier and migrate into the tissue, thereby amplifying allergic inflammation. Our findings underscore the capacity of endothelial cells to produce key inflammatory molecules that participate in EoE and other type 2 immune responses (e.g., CCL26, POSTN, TARC, TSLP, IL33),36, 50 especially those related to tissue fibrosis in EoE. Consistent with previous reports that TSPAN12 is required for endothelial barrier function and our observed decrease in endothelial barrier function in TSPAN12-deficient cells,51, 52 endothelial dysfunction is a potential consequence of lost TSPAN12 expression.
Furthermore, we found that the loss of endothelial TSPAN12 contributed to the fibrostenotic phenotype via endothelial cell–fibroblast crosstalk. We found that TSPAN12-deficient endothelial cells have dysregulated expression of ECM-related genes involved in tissue remodeling. This is relevant because ECM production and maintenance is an essential aspect of endothelial cell function, with several ECM proteins being up-regulated in fibrotic processes. Also, we demonstrated the ability of media from TSPAN12-deficient HEsMEC to up-regulate ECM production from esophageal fibroblasts, likely by an HEsMEC-derived pro-fibrotic mediator (e.g., endothelin-1). For instance, endothelin-1, is a potent vasoconstrictor polypeptide produced and secreted by endothelial cells.53 Besides its vascular effects, numerous studies have described a variety of endothelin-1 profibrogenic activities, including stimulating the synthesis of collagens and α-smooth muscle actin.41, 54 Furthermore, various human fibrotic diseases across organs have been shown to display increased production of endothelin-1.41, 42 Although it remains to be studied how TSPAN12 deficiency affected endothelin-1 production in our study, TGF-β has been reported as a potential mediator of TSPAN12-deficient effects on gene expression.55 Indeed, TGF-β is known to induce endothelin-1.56 In support of our findings, transcripts encoding proteins involved in ECM organization, remodeling of collagen, and endothelin family (EDN2) have been reported to be significantly elevated in the retina of TSPAN12-deficient mice; the esophagus has not been studied.52 Importantly, observations in vitro were further reinforced by esophageal biopsy staining that showed these ECM proteins were present in close proximity to the vessels in vivo. These findings suggest the TSPAN12-dependent molecular changes in endothelium may contribute to the pathogenesis of fibrostenosis and warrant further study.
Our study has strengths and limitations. First, analysis of 398 samples from multiple sites across the USA increased the generalizability of the results, especially across ages in children and adults. Second, the cardinal findings were validated in independent cohorts. Third, participants were assessed by several approaches, allowing us to examine associations between gene expression and endoscopic and histologic parameters. Fourth, we studied the mechanistic link between endothelial TSPAN12 expression and fibrostenotic EoE. In terms of limitations, first, most of the analyses for gene expression were restricted to the 94 EDP DEGs. Unbiased, genome-wide transcriptome approaches would likely reveal additional genes and pathways of interest. Second, the study definition for fibrostenosis balanced feasibility and accuracy, warranting future analyses to define fibrostenotic EoE with further accurate evaluations (e.g., esophageal wall compliance data for all subjects). Third, esophageal biopsies inconsistently sample the subepithelial space; this heterogeneity could affect the results. Although the analysis focusing on the subjects having subepithelial spaces also showed that TSPAN12 was significantly lower in fibrostenotic EoE than non-fibrostenotic EoE, this was still an assumption because biopsies for histology and those for molecular analysis were different. Finally, the data were limited by the cross-sectional approach, highlighting the importance of additional replication, particularly in prospective studies.
In conclusion, we provide multiple lines of evidence that TSPAN12 contributes to fibrostenosis in EoE. Patients with fibrostenotic EoE express decreased levels of endothelial TSPAN12. We propose that TSPAN12 loss is induced by IL-13 and contributes to EoE by promoting increased endothelial permeability and tissue remodeling via interaction with esophageal fibroblasts, likely contributing to the chronicity of EoE. Mechanistically, we propose that steroid-resistant IL-13–induced TSPAN12 loss modulates endothelial dysfunction and gene expression, contributing to EoE tissue remodeling. We substantiate IL-13’s role in this process by demonstrating that anti–IL-13 therapy in patients restores TSAPN12 expression. Our data provide new insights regarding the pathogenesis of fibrostenotic EoE, drawing attention to the roles of TSPAN12 and endothelial cells.
Supplementary Material
What You Need to Know.
BACKGROUND AND CONTEXT:
Although eosinophilic esophagitis (EoE) is a chronic, antigen-mediated immunologic disease that can progress to fibrostenosis, the molecular pathogenesis of fibrostenotic EoE is not well understood.
NEW FINDINGS:
Patients with fibrostenotic EoE express decreased levels of endothelial TSPAN12. Mechanistically, IL-13 induces loss of TSPAN12 in endothelial cells, and this loss modulates endothelial dysfunction and gene expression leading to remodeling.
LIMITATIONS:
This study is limited by the restricted number of genes within the EoE Diagnostic Panel (EDP) and cross-sectional approach, highlighting the importance of additional replication.
IMPACT:
Endothelial TSPAN12 contributes to fibrostenotic EoE and is the first molecular correlate of esophageal diameter. These findings provide new insight into previously underrecognized roles of the endothelium in disease pathogenesis. Anti–IL-13 therapy may improve fibrostenotic EoE through normalizing TSPAN12 levels.
Acknowledgments
The authors would like to thank all patients who participated in the study. The authors are also grateful to their colleagues and clinical support staff for procuring biopsies and clinical data.
Grant Support
This study was supported by CEGIR (U54 AI117804), which is part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS), and is co-funded by National Institute of Allergy and Infectious Diseases (NIAID), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), and NCATS. CEGIR is also supported by patient advocacy groups including the American Partnership for Eosinophilic Disorders (APFED), Campaign Urging Research for Eosinophilic Disease (CURED), and Eosinophilic Family Coalition (EFC). As a member of the RDCRN, CEGIR is also supported by its Data Management and Coordinating Center (DMCC) (U2CTR002818).
List of Abbreviations
- ANOVA
one-way analysis of variance
- APFED
American Partnership for Eosinophilic Disorders
- AD
atopic dermatitis
- BZH
basal zone hyperplasia
- CCED
Cincinnati Center for Eosinophilic Disorders
- CCHMC
Cincinnati Children’s Hospital Medical Center
- CEGIR
Consortium of Eosinophilic Gastrointestinal Disease Researchers
- CURED
Campaign Urging Research for Eosinophilic Disease
- DEC
dyskeratotic epithelial cells
- DIS
dilated intercellular spaces
- DMCC
Data Management and Coordinating Center
- EA
eosinophilic abscess
- ECM
extracellular matrix
- EDP
EoE diagnostic panel
- EFC
Eosinophilic Family Coalition
- EGID
eosinophilic gastrointestinal disorders
- EI
eosinophilic inflammation
- EoE
eosinophilic esophagitis
- EREFS
EoE Endoscopic Reference Score
- ESL
eosinophilic surface layering
- DEG
differentially expressed genes
- FDR
false-discovery rate
- FITC
fluorescein isothiocyanate
- FP
fluticasone propionate
- GO
gene ontology
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GI
gastrointestinal
- HPF
high-power microscopic field
- HSS EoE
Histology Scoring System
- HEsMEC
human esophageal microvascular endothelial cells
- IQR
interquartile range
- LPF
lamina propria fibrosis
- MCH
melanin-concentrating hormone
- MMP
matrix metalloproteinase
- OMEGA
Outcomes Measures in Eosinophilic Gastrointestinal disorders Across the ages
- ORDR
Office of Rare Diseases Research
- PPI
proton pump inhibitor
- qPCR
quantitative real-time PCR
- RDCRN
Rare Diseases Clinical Research Network
- SEA
surface epithelial alteration
- SOFEED
Six Food vs. One Food Eosinophilic Esophagitis Elimination Diet
- TEER
transendothelial electrical resistance
- TSLP
thymic stromal lymphopoietin
- TSPAN
tetraspanin
Footnotes
The list of participants is provided in this article’s Supplementary Material
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
M.E.R. is a consultant for Pulm One, Spoon Guru, Allakos, ClostraBio, Serpin Pharm, Celgene, Shire, Astra Zeneca, GlaxoSmithKline, Allakos, Adare, Regeneron, and Novartis and has an equity interest in the first five, as well as royalties from reslizumab (Teva Pharmaceuticals) and Up-To-Date. M.E.R. is an inventor of patents, owned by Cincinnati Children’s. G.W.F. has received research support from Lucid, Allakos, Regeneron, Takeda/Shire, and Adare and is a consultant for Adare, Allakos, Lucid and Takeda/Shire. I.H. is a consultant for Adare, Allakos, Arena, AstraZeneca, Boston Scientific, Eli Lilly, EsoCap, Gossamer Bio, Parexel, Receptos/Celegene/BMS, Regeneron, and Shire/Takeda; has received research funding from Adare, Allakos, Meritage, Receptos/Celgene/BMS, Regeneron, and Shire/Takeda. M.H.C. is a consultant for Allakos, Arena, Astra Zeneca, Calypso, GSK, Meritage/Shire/Takeda, Robarts/Alimentiv, Regeneron, Receptos/Celgene/BMS, and Esocap and has received research funding from Meritage/Shire/Takeda, Regeneron, Receptos, and Astra Zeneca. S.K.G. is a consultant for Abbott, Adare, Allakos, Gossamer Bio, MedScape, QOL, Receptos/Celgene, UpToDate, and Viaskin and receives research support from Shire. V.A.M. is a consultant for Shire and has received research funding from Shire. N.G. is a consultant for Allakos. E.S.D. is a consultant for Abbott, Adare, Aimmune, Allakos, Amgen, Arena, AstraZeneca, Biorasi, Calypso, Eli Lilly, EsoCap, Gossamer Bio, GlaxoSmithKline, Parexel, Receptos/Celegene/BMS, Regeneron, Robarts, Salix, and Shire/Takeda; has received research funding from Adare, Allakos, GlaxoSmithKline, Meritage, Miraca, Nutricia, Receptos/Celgene/BMS, Regeneron, and Shire/Takeda; and has received educational grants from Allakos, Banner, and Holoclara. S.S.A. is a consultant for Regeneron, AImmune Therapeutics, DBV, and AstraZeneca and is an inventor of oral viscous budesonide, patented by UCSD and licensed by Shire/Takeda. J.M.S. is a consultant for Regeneron and DBV Technology, and his research is supported by the NIH, Everbody Eats (EATS) foundation, AImmune Therapeutics, Food Allergy Research & Education (FARE), and DBV Technology. I.H. is a consultant for Regeneron, Receptos, Shire, Allakos, and Adare and has received research funding from Regeneron, Receptos, Shire, and Adare. G.T.F. is a consultant for Shire and a co-founder of EnteroTrack. J.B.W. is a consultant for Allakos and Regeneron. J.L. is a consultant for Adare, Eli Lilly, AbbVie, Genzyme and Shire/Takeda and has received research funding from Adare, Allakos, Celgene, Regeneron, AstraZeneca, and Shire/Takeda. A.K.R.S.’ co-authorship of this publication does not necessarily constitute endorsement by the National Institute of Allergy and Infectious Diseases, the National Institutes of Health, or any other agency of the United States government. All other authors declare that they have no competing interests.
Writing Assistance
Shawna Hottinger provided editorial assistance as a medical writer funded by Cincinnati Children’s Hospital Medical Center.
REFERENCES
- 1.O’Shea KM, Aceves SS, Dellon ES, et al. Pathophysiology of Eosinophilic Esophagitis. Gastroenterology 2018;154:333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kavitt RT, Hirano I, Vaezi MF. Diagnosis and Treatment of Eosinophilic Esophagitis in Adults. Am J Med 2016;129:924–34. [DOI] [PubMed] [Google Scholar]
- 3.Dellon ES, Hirano I. Epidemiology and Natural History of Eosinophilic Esophagitis. Gastroenterology 2018;154:319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schoepfer AM, Safroneeva E, Bussmann C, et al. Delay in diagnosis of eosinophilic esophagitis increases risk for stricture formation in a time-dependent manner. Gastroenterology 2013;145:1230–6. [DOI] [PubMed] [Google Scholar]
- 5.Straumann A, Aceves SS, Blanchard C, et al. Pediatric and adult eosinophilic esophagitis: similarities and differences. Allergy 2012;67:477–90. [DOI] [PubMed] [Google Scholar]
- 6.Bolton SM, Kagalwalla AF, Wechsler JB. Eosinophilic Esophagitis in Children: Endoscopic Findings at Diagnosis and Post-intervention. Curr Gastroenterol Rep 2018;20:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dellon ES, Kim HP, Sperry SL, et al. A phenotypic analysis shows that eosinophilic esophagitis is a progressive fibrostenotic disease. Gastrointest Endosc 2014;79:577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shoda T, Wen T, Aceves SS, et al. Eosinophilic oesophagitis endotype classification by molecular, clinical, and histopathological analyses: a cross-sectional study. Lancet Gastroenterol Hepatol 2018;3:477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nhu QM, Aceves SS. Tissue Remodeling in Chronic Eosinophilic Esophageal Inflammation: Parallels in Asthma and Therapeutic Perspectives. Front Med (Lausanne) 2017;4:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rieder F, Nonevski I, Ma J, et al. T-helper 2 cytokines, transforming growth factor β1, and eosinophil products induce fibrogenesis and alter muscle motility in patients with eosinophilic esophagitis. Gastroenterology 2014;146:1266–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arias Á, Lucendo AJ. Molecular basis and cellular mechanisms of eosinophilic esophagitis for the clinical practice. Expert Rev Gastroenterol Hepatol 2019;13:99–117. [DOI] [PubMed] [Google Scholar]
- 12.Blanchard C, Mingler MK, Vicario M, et al. IL-13 involvement in eosinophilic esophagitis: transcriptome analysis and reversibility with glucocorticoids. J Allergy Clin Immunol 2007;120:1292–300. [DOI] [PubMed] [Google Scholar]
- 13.Eluri S, Runge TM, Cotton CC, et al. The extremely narrow-caliber esophagus is a treatment-resistant subphenotype of eosinophilic esophagitis. Gastrointest Endosc 2016;83:1142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manresa MC, Chiang AWT, Kurten RC, et al. Increased Production of LIGHT by T Cells in Eosinophilic Esophagitis Promotes Differentiation of Esophageal Fibroblasts Toward an Inflammatory Phenotype. Gastroenterology 2020;159:1778–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muir AB, Wang JX, Nakagawa H. Epithelial-stromal crosstalk and fibrosis in eosinophilic esophagitis. J Gastroenterol 2019;54:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aceves SS, Ackerman SJ. Relationships Between Eosinophilic Inflammation, Tissue Remodeling, and Fibrosis in Eosinophilic Esophagitis. Immunol Allergy Clin North Am 2009;29:197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen T, Stucke EM, Grotjan TM, et al. Molecular diagnosis of eosinophilic esophagitis by gene expression profiling. Gastroenterology 2013;145:1289–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta SK, Falk GW, Aceves SS, et al. Consortium of Eosinophilic Gastrointestinal Disease Researchers: Advancing the Field of Eosinophilic GI Disorders Through Collaboration. Gastroenterology 2019;156:838–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dellon ES, Selitsky SR, Genta RM, et al. Gene expression-phenotype associations in adults with eosinophilic esophagitis. Dig Liver Dis 2018;50:804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirano I, Moy N, Heckman MG, et al. Endoscopic assessment of the oesophageal features of eosinophilic oesophagitis: validation of a novel classification and grading system. Gut 2013;62:489–95. [DOI] [PubMed] [Google Scholar]
- 21.Straumann A, Lucendo AJ, Miehlke S, et al. Budesonide Orodispersible Tablets Maintain Remission in a Randomized, Placebo-Controlled Trial of Patients With Eosinophilic Esophagitis. Gastroenterology 2020;159:1672–1685.e5. [DOI] [PubMed] [Google Scholar]
- 22.Hirano I How to Approach a Patient With Eosinophilic Esophagitis. Gastroenterology 2018;155:601–606. [DOI] [PubMed] [Google Scholar]
- 23.Collins MH, Martin LJ, Alexander ES, et al. Newly developed and validated eosinophilic esophagitis histology scoring system and evidence that it outperforms peak eosinophil count for disease diagnosis and monitoring. Dis Esophagus 2017;30:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butz BK, Wen T, Gleich GJ, et al. Efficacy, dose reduction, and resistance to high-dose fluticasone in patients with eosinophilic esophagitis. Gastroenterology 2014;147:324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothenberg ME, Wen T, Greenberg A, et al. Intravenous anti-IL-13 mAb QAX576 for the treatment of eosinophilic esophagitis. J Allergy Clin Immunol 2015;135:500–7. [DOI] [PubMed] [Google Scholar]
- 26.Sherrill JD, Kiran KC, Blanchard C, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun 2014;15:361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wen T, Aronow BJ, Rochman Y, et al. Single-cell RNA sequencing identifies inflammatory tissue T cells in eosinophilic esophagitis. J Clin Invest 2019;129:2014–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J, Bardes EE, Aronow BJ, et al. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009;37:W305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kasagi Y, Dods K, Wang JX, et al. Fibrostenotic eosinophilic esophagitis might reflect epithelial lysyl oxidase induction by fibroblast-derived TNF-alpha. J Allergy Clin Immunol 2019;144:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rochman M, Xie YM, Mack L, et al. Broad transcriptional response of the human esophageal epithelium to proton pump inhibitors. J Allergy Clin Immunol 2021;147:1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rochman M, Travers J, Abonia JP, et al. Synaptopodin is upregulated by IL-13 in eosinophilic esophagitis and regulates esophageal epithelial cell motility and barrier integrity. JCI Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Serru V, Dessen P, Boucheix C, et al. Sequence and expression of seven new tetraspans. Biochim Biophys Acta 2000;1478:159–63. [DOI] [PubMed] [Google Scholar]
- 34.Junge HJ, Yang S, Burton JB, et al. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell 2009;139:299–311. [DOI] [PubMed] [Google Scholar]
- 35.Beppu L, Yang T, Luk M, et al. MMPs-2 and −14 Are Elevated in Eosinophilic Esophagitis and Reduced Following Topical Corticosteroid Therapy. J Pediatr Gastroenterol Nutr 2015;61:194–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shoda T, Futamura K, Orihara K, et al. Recent advances in understanding the roles of vascular endothelial cells in allergic inflammation. Allergol Int 2016;65:21–9. [DOI] [PubMed] [Google Scholar]
- 37.Gonsalves NP, Aceves SS. Diagnosis and treatment of eosinophilic esophagitis. J Allergy Clin Immunol 2020;145:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shoda T, Futamura K, Kobayashi F, et al. Cell type-dependent effects of corticosteroid on periostin production by primary human tissue cells. Allergy 2013;68:1467–70. [DOI] [PubMed] [Google Scholar]
- 39.Hirano I, Collins MH, Assouline-Dayan Y, et al. RPC4046, a Monoclonal Antibody Against IL13, Reduces Histologic and Endoscopic Activity in Patients With Eosinophilic Esophagitis. Gastroenterology 2019;156:592–603. [DOI] [PubMed] [Google Scholar]
- 40.Hirano I, Dellon ES, Hamilton JD, et al. Efficacy of Dupilumab in a Phase 2 Randomized Trial of Adults With Active Eosinophilic Esophagitis. Gastroenterology 2020;158:111–122. [DOI] [PubMed] [Google Scholar]
- 41.Rodríguez-Pascual F, Busnadiego O, González-Santamaría J. The profibrotic role of endothelin-1: is the door still open for the treatment of fibrotic diseases? Life Sci 2014;118:156–64. [DOI] [PubMed] [Google Scholar]
- 42.Fagan KA, McMurtry IF, Rodman DM. Role of endothelin-1 in lung disease. Respir Res 2001;2:90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shoda T, Wen T, Aceves SS, et al. Eosinophilic oesophagitis endotype classification by molecular, clinical, and histopathological analyses: a cross-sectional study. Lancet Gastroenterol Hepatol 2018;3:477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vanoni S, Zeng C, Marella S, et al. Identification of anoctamin 1 (ANO1) as a key driver of esophageal epithelial proliferation in eosinophilic esophagitis. J Allergy Clin Immunol 2020;145:239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Termini CM, Gillette JM. Tetraspanins Function as Regulators of Cellular Signaling. Front Cell Dev Biol 2017;5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poulter JA, Ali M, Gilmour DF, et al. Mutations in TSPAN12 cause autosomal-dominant familial exudative vitreoretinopathy. Am J Hum Genet 2010;86:248–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye X, Wang Y, Nathans J. The Norrin/Frizzled4 signaling pathway in retinal vascular development and disease. Trends Mol Med 2010;16:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hemler ME. Tetraspanin proteins promote multiple cancer stages. Nat Rev Cancer 2014;14:49–60. [DOI] [PubMed] [Google Scholar]
- 49.Blanchard C, Stucke EM, Rodriguez-Jimenez B, et al. A striking local esophageal cytokine expression profile in eosinophilic esophagitis. J Allergy Clin Immunol 2011;127:208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emi-Sugie M, Shoda T, Futamura K, et al. Robust production of IL-33 and TSLP by lung endothelial cells in response to low-dose dsRNA stimulation. J Allergy Clin Immunol 2020;146:1449–1452. [DOI] [PubMed] [Google Scholar]
- 51.Bucher F, Zhang D, Aguilar E, et al. Antibody-Mediated Inhibition of Tspan12 Ameliorates Vasoproliferative Retinopathy Through Suppression of beta-Catenin Signaling. Circulation 2017;136:180–195. [DOI] [PubMed] [Google Scholar]
- 52.Zhang C, Lai MB, Pedler MG, et al. Endothelial Cell-Specific Inactivation of TSPAN12 (Tetraspanin 12) Reveals Pathological Consequences of Barrier Defects in an Otherwise Intact Vasculature. Arterioscler Thromb Vasc Biol 2018;38:2691–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barton M, Yanagisawa M. Endothelin: 20 years from discovery to therapy. Can J Physiol Pharmacol 2008;86:485–98. [DOI] [PubMed] [Google Scholar]
- 54.Villaschi S, Nicosia RF. Paracrine interactions between fibroblasts and endothelial cells in a serum-free coculture model. Modulation of angiogenesis and collagen gel contraction. Lab Invest 1994;71:291–9. [PubMed] [Google Scholar]
- 55.Knoblich K, Wang HX, Sharma C, et al. Tetraspanin TSPAN12 regulates tumor growth and metastasis and inhibits beta-catenin degradation. Cell Mol Life Sci 2014;71:1305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prud’homme GJ. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab Invest 2007;87:1077–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.