

Abstract
Currently, medicine lacks the ability to reprogram selected immune cells so they possess all the functions which, from a clinical standpoint, physicians might wish them to have. To solve this problem, scientists have been marrying concepts from materials science, immunology, and genetic engineering to develop novel nanotherapeutics that directly genetically reprogram immune cells inside the body. These products could address key limitations of existing ex vivo-engineered cell immunotherapies and substantially enhance patient access and outcomes. This review highlights the latest advances in this rapidly emerging biotech field and discusses challenges in translating these preclinical studies into successful clinical nanomedicines.
Keywords: In vivo cell reprogramming, Nonviral gene therapy, T-cell therapy, Macrophage, Dendritic cell, Nanotechnology, CAR T-cell
1. Introduction
Transplantation of genetically engineered immune cells has become a powerful tool in medicine. In particular, the rise of Chimeric Antigen Receptor (CAR) T-cell immunotherapy, currently with three Food and Drug Administration (FDA)-approved agents (Kymriah™, Yescarta™, and Tecartus™), was a major advance in the treatment of cancer. While the potential of other types of immune cell therapy has been known for a while, only in recent years has the field really begun to blossom. Beyond T lymphocytes, invariant natural killer T cells(1), γδ T cells(2), natural killer cells(3), macrophages(4), and even B cells(5) have all been added to the repertoire of genetically engineered cell therapies against a wide spectrum of diseases. The accelerated pace of these developments has undoubtedly benefited from multiple innovative technologies, such as the CRISPR-Cas9 gene editing system, which allows researchers to perform virtually any desired gene modification in vitro. Furthermore, alternatives to conventional viral vectors have become widely available to the gene therapy community. In vitro-transcribed messenger RNA (IVT mRNA), in particular, is now established as an extremely versatile class of ex vivo gene therapy reagent for transient expression(6). Notably, IVT mRNA of uniformly high quality can be produced in large amounts under good manufacturing practices (GMP). For more durable transgene expression, novel closed-linear DNA constructs, referred to as closed-ended (ce) DNA or doggybone™ DNA, have advanced into clinical cell-engineering applications(7). These constructs have a unique ability to translocate from the cytoplasm to the nucleus without the use of a viral capsid. Once inside the nucleus, they form stable, non-integrating episomes that produce high levels of long-term gene expression(8). Additionally, the recent commercialization of high-throughput electroporation devices has advanced many adoptive cell therapy programs, as they enable clinical investigators to transfect billions of cultured immune cells while maintaining cellular health and function(9).
While there are now more therapies with genetically engineered immune cells on offer than ever before, there are also obstacles preventing them from becoming widely used (Figure 1, left panel). Despite the remarkable increase in the development of cell and gene therapies over the past couple of years, manufacturing technology for these therapies is still largely at the first-generation stage, which makes scaling up challenging if not impossible(10). In autologous cell therapies (meaning the cells are derived from the patient, modified, and administered back to the patient) the units of operation are not scalable. The process can be summed up as ‘one patient, one batch,’ which limits batch volume and precludes economies of scale. Unlike autologous cell therapies, ‘off-the-shelf’ allogeneic cells from non-related donors could, in theory, be expanded in high numbers prior to treatment and made quickly available to patients. However, allogeneic cells possess foreign immunological identities that can lead to histo-incompatibility such as graft-versus-host disease and cell rejection(11). A solution might be to immunologically conceal allogeneic cells from the host immune cells by adding complex genome editing and cell purification steps to the manufacturing protocols (12), but these procedures would not only delay production time and increase costs (also factoring in costs for gene-editing intellectual property), they would also compromise the viability of the lymphocytes and substantially reduce their yield. Another problem common to all bio-based therapeutics is that any product sourced from a live cell is inherently more variable than a conventional pharmaceutical product, resulting in unavoidable heterogeneity and inter-batch variability(13). With price points sky high and laborious manufacturing processes, these bespoke adoptive cell therapies are out of reach for most patients.
Figure 1: In vivo cell reprogramming platforms are built for widescale adoption and expanded patient access.
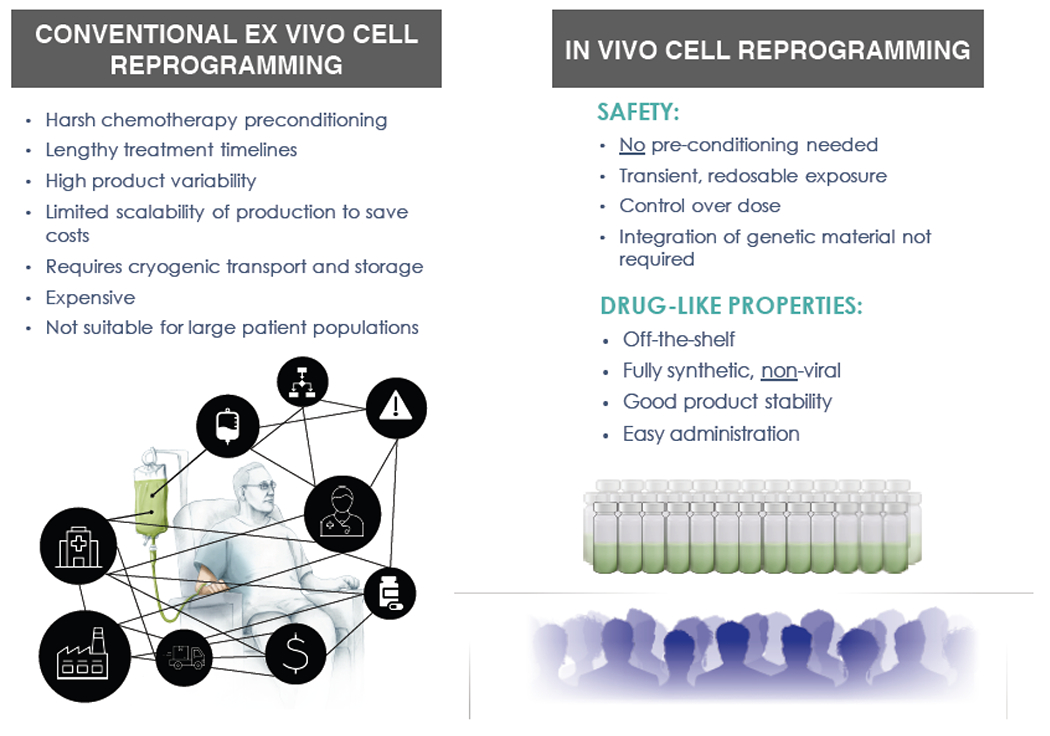
Listed are key advantages of in vivo cell reprogramming platforms based on injectable nanomedicines compared to conventional ex vivo engineered cell therapies.
In situ programming reagents could provide cell therapy options for patients and compete with frontline therapeutics such as small molecule drugs or monoclonal antibodies as they can be produced in bulk quantities just like conventional pharmaceuticals (Figure 1, right panel). These reagents could be centrally manufactured and distributed for administration at the point of care, thus significantly broadening patient access to gene therapeutics. Furthermore, preconditioning of the recipient is not necessary because the cells targeted to be programmed by the transgene-bearing reagent are the existing populations of circulating immune cells. During this reprogramming, immune cells never exit their physiological environment and are not exposed to supra-physiological levels of cytokines, as happens during ex vivo propagation. Moreover, unlike in vitro expansion, a process that can functionally exhaust cells before they are reintroduced (14), immune cells programmed in situ are already at their job site.
Synthetic nanoparticles are ideal reagents for cell-selective in vivo gene therapies. They are engineered in a modular fashion, which makes it easy to switch out individual modules and incorporate custom modalities, which is not possible for viral vectors because of their biological constraints. Depending on the desired characteristics, nanoparticles can be fabricated with organic materials, such as protein, polymer or lipid, or with inorganic substances (e.g., gold or silica). Also, the cargo encoding the therapeutic transgene can be customized, and ranges from lVT mRNA for transient in situ gene expression to constructs that deliver a transgene into the nucleus for persistent gene expression, such as linear, ceDNA (8), or minicircle vectors (15). Similarly, the full range of immune cell binding proteins or synthetic antibody mimetics can be considered when designing targeted nonviral gene delivery systems. Examples include, Designed Ankyrin Repeat Proteins (DARPins) (16), Affimers (17) and Aptamers (18). Several continuous flow microfluidics platforms designed for scalable manufacturing of nanoparticles under GMP conditions are now available (19; 20). These instruments enable scale-independent synthesis of nanoparticles, from milligram to gram amounts in a single day (21). Thus, nanomedicines could be readily fabricated on a large scale and in a stable form, would be easy to distribute as lyophilized reagents, would be inexpensive to administer, and could be delivered to sizeable patient populations in outpatient settings.
In this review, we discuss established nanotechnology platforms that are used to genetically modify immune cells in the body and do not require the removal and reinfusion of a patient’s cells. Our focus is on methods for specific alteration of gene expression in monocytes/macrophages, dendritic cells, and T cells (Figure 2, Figure 3). We cover the entire spectrum of nanoparticle-based nucleic acid delivery, including plasmid DNA, IVT mRNA, siRNA, and microRNA (summarized in Table 1). Vaccines using nanodelivery systems to transfect antigen-presenting cells in situ with nucleic acid encoding vaccine antigens are not discussed, as this topic has been extensively covered in previous reviews(22–24). Rather, we provide a detailed overview of nucleic acid nanotherapeutics that can re-educate/retool a patient’s immune cells and enhance their natural capacity to fight specific diseases.
Figure 2: Creating living drugs with injectable synthetic nanoparticles.
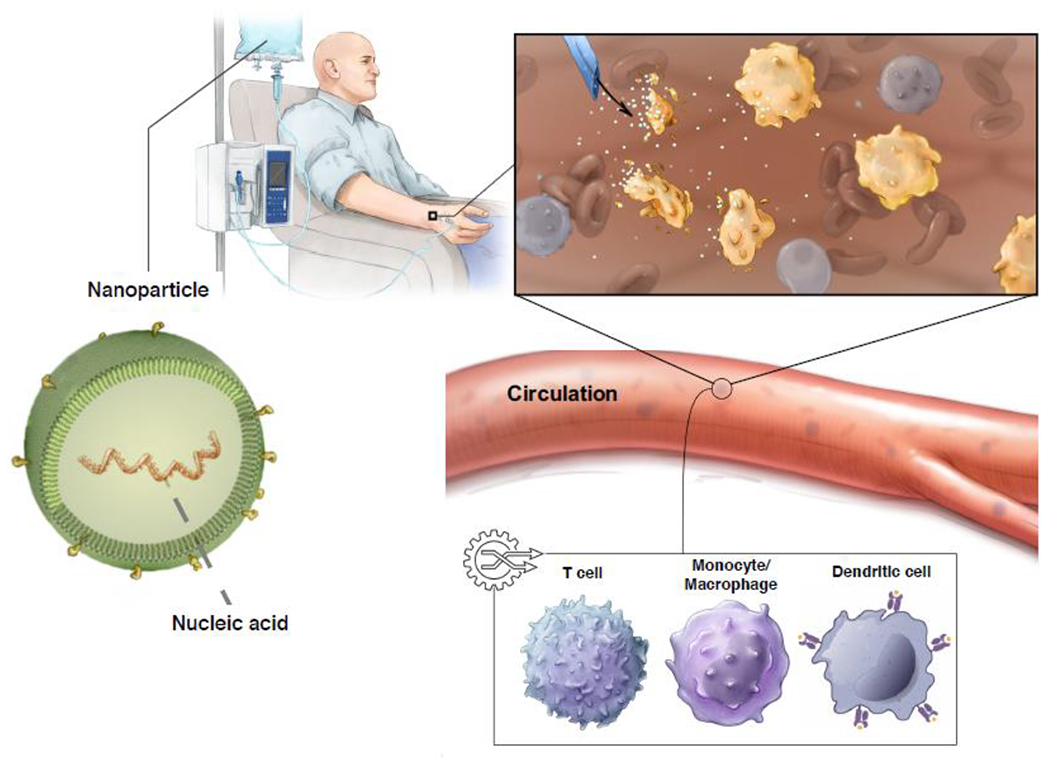
Schematic diagram illustrating genetic reprogramming of immune cells in situ using genes carried by polymeric nanoparticles. These particles are coated with ligands that target them to therapeutically desired immune cell subsets, so once infused into the patient’s circulation they can transfer the genes they carry into the target cells.
Figure 3: T-cell reprogramming and beyond.
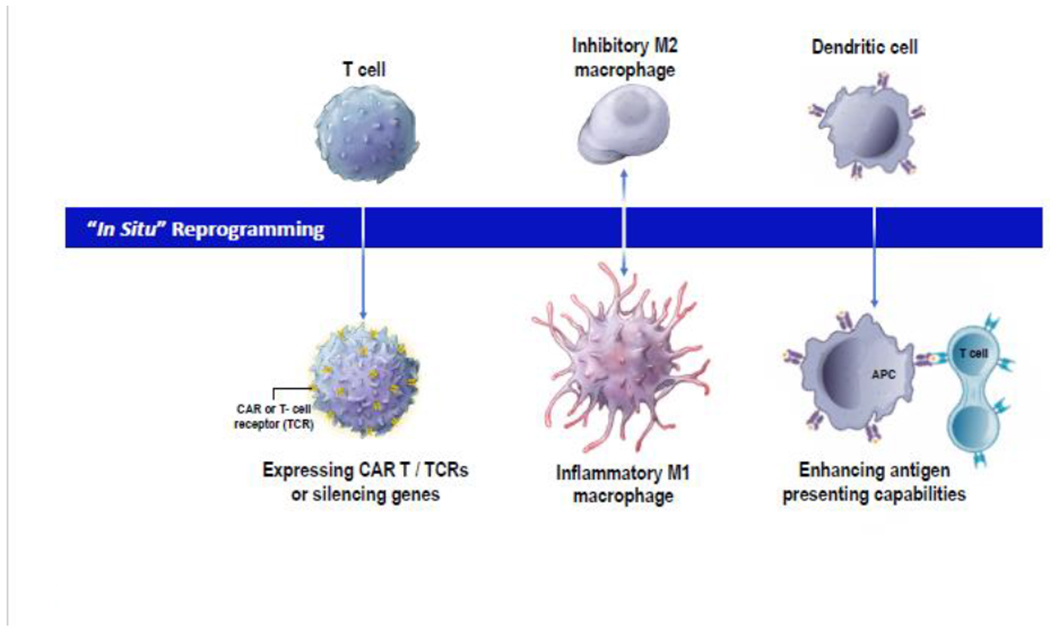
This schematic summarizes potential therapeutic approaches which are developing nanotherapeutics to directly reprogram immune cells inside the body.
Table 1:
Summary of literature reviewed.
| Target cell | Nanoparticle cargo | Nanocarrier | Active Targeting | Therapy | Ref. |
|---|---|---|---|---|---|
| Macrophage | Plasmid DNA (encoding IL-10) | Alginate nanoparticles | Tuftsin peptide | Rheumatoid arthritis | Jain et al |
| Macrophage | Plasmid DNA (encoding IL-4 or IL-10) | Hyaluronic acid-poly(ethylene imine) nanoparticles | Hyaluronic acid binds CD44 | Immunosuppression | Tran et al |
| Macrophage | Plasmid DNA (encoding CRISPR/Cas9 under the control of the CD68 promoter) + sgRNA targeting Ntn1 | Cationic lipid-assisted PEG-b-PLGA nanoparticles (CLAN) | None | High-fat diet induced Type 2 diabetes | Luo et al |
| Macrophage | IVT-mRNA (encoding IL-10) | Lipid nanoparticles | Anti-Ly6c antibody (conjugated using the ASSET linker strategy) | Inflammatory bowel disease | Veiga et al |
| Macrophage | IVT-mRNA (encoding IRF5 and IKKβ) | Poly (β-amino ester) PbAE core with a poly-glutamic acid (PGA) envelope | Di-mannose (which binds CD206) | Immunosuppressive M2 → Anti-tumor M1 | Zhang et al |
| Macrophage | IVT-mRNA (encoding CRISPR/Cas9) + gRNA targeting NLRP3 | Cationic lipid-assisted PEG-b-PLGA nanoparticles (CLAN) | None | LPS-induced Septic shock, Peritonitis, High-fat diet induced Type 2 diabetes | Xu et al |
| Macrophage | siRNA (targeting STAT3 and HIF-1α) | Pegylated lipid nanoparticles (CL4H6, chol, DSG-PEG 2000) | None | Immunosuppressive M2 → Anti-tumor M1 | Shobaki et al |
| Macrophage | siRNA (targeting IRF8) | Lipid nanoparticles | Anti-Ly6c antibody (conjugated using the ASSET linker strategy) | Colitis | Veiga et al |
| Macrophage | siRNA (targeting TNF-α) | Lipid nanoparticles | Anti-Ly6c antibody (conjugated using the ASSET linker strategy) | Colitis | Kedmi et al |
| Macrophage | siRNA (targeting TNF-α) | Hyaluronic acid nanoparticles | Hyaluronic acid binds CD44 | LPS-induced peritonitis | Kosovrasti et al |
| Macrophage | siRNA (targeting CaMKIIɣ) | Poly(lactic-co-glycolic) acid (PLGA) polymer core coated with polyethylene glycol (lipid-PEG) | S2P peptide, which recognizes the macrophage receptor stabilin-2 | Atherosclerosis | Tao et al |
| Macrophage | miRNA (miR-155) | Self-crosslinked redox-responsive nanoparticles based on galactose-functionalized n-butylamine-poly(L-lysine)-b-poly(L-cysteine) polypeptides (GLC) coated with DCA-grafted sheddable PEG-PLL (sPEG) copolymers | Galactose, which binds lectin receptors on macrophages | Immunosuppressive M2 → Anti-tumor M1 | Liu et al |
| Macrophage | miRNA (miR-125b) | Hyaluronic acid-poly(ethylene imine) nanoparticles | Hyaluronic acid binds CD44 | Immunosuppressive M2 → Anti-tumor M1 (ovarian cancer) | Parayath et al |
| Macrophage | miRNA (miR-223) | Hyaluronic acid-poly(ethylene imine) nanoparticles | Hyaluronic acid binds CD44 | Immunsuppression (Inflammatory M1 → Anti-inflammatory M2) | Tran et al |
| Macrophage | miRNA (miR-21) | Hyaluronan-sulfate (HAS) nanoparticles | None | Myocardial infarction (promotes angiogenesis, reduces hypertrophy, fibrosis and cell apoptosis) | Bejerano |
| Macrophage | miRNA (miR-33) | Pegylated Chitosan nanoparticles | None | Regulation of cholesterol efflux from lipid-laden macrophages in atherosclerotic plaques | Nguyen et al |
| Dendritic cell | Plasmid DNA (encoding CRISPR/Cas9) + gRNA targeting CD80, CD86, and CD40 + autoimmune-diabetes-relevant peptide | Cationic lipid-assisted PFG-b-PLGA nanoparticles (CLAN) | None | Generating tolerogenic dendritic cells and autoantigen-specific regulatory T cells to treat autoimmune type 1 diabetes | Luo et al |
| Dendritic cell | IVT-mRNA (encoding CRISPR/Cas9) + gRNA targeting CD40 | Cationic lipid-assisted PFG-b-PLGA nanoparticles (CLAN) | None | Generating tolerogenic dendritic cells and regulatory T cells to relieve graft rejection | Zhang et al |
| Dendritic cell | siRNA (targeting CD40, CD80, and CD86) | Pegylated lipid nanoparticles | scFv specific tor DEC205 | Immunosuppression | Katakowski et al |
| Dendritic cell | siRNA (targeting IDO) | Cationic lipid-assisted PEG-b-PLGA nanoparticles (CLAN) | None | Improved therapeutic response in orthtopic pancreatic tumors (co-administered with immunogenic chemotherapy) | Huang et al |
| Dendritic cell | siRNA (targeting STAT3) + vaccine antigen + Poly I:C adjuvant | Poly(ethylene glycol)-b-poly (L-lysine)-b-poly (L-leucine) (PEG-PLL-PLLeu) polypeptide micelles | None | Inducing anti-tumor immunity | Luo et al |
| Dendritic cell | siRNA (targeting SOCS1) + vaccine antigen | Poly(lactic-co-glycolic) acid (PLGA) nanoparticles | None | Inducing anti-tumor immunity | Heo et al |
| Dendritic cell | siRNA (targeting SNAP-23) | Poly(lactic-co-glycolic) acid (PLGA)/Polyethylenimine (PEI) nanoparticles release from a biodegradable film consisting of poly vinyl alcohol (PVA) and λ-carrageenan | Anti-HLA-DR antibody | Prevention of HIV infection when administered intravaginally | Gu et al |
| T cell | Plasmid DNA (encoding the 194-1BBz CAR and the hyperactive iPB7 transposase) | Poly (β-amino ester) PbAE core with a poly-glutamic acid (PGA) envelope | Anti-CD3 f(ab′)2 | In situ programming of leukemia-specific T cells | Smith et al |
| T cell | IVT-mRNA (encoding the 19-28z CAR, or the ROR1-28z CAR, or the HBcore 18-27 specific TCR) | Poly (β-amino ester) PbAE core with a poly-glutamic acid (PGA) envelope | Anti-CD8 or anti-CD8 antibody (deglycosylated Fc) | Transient in situ programming of leukemia-specific, prostate tumor-specific, or HBV antigen-specific T cells. | Parayath et al |
| T cell | siRNA (targeting CCR5) | Multilamellar vesicle liposomes composed of soybean PC (phosphatidylcholine), DPPE (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine), and Chol (cholesterol) | Anti-LFA-1 antibody | Prevention of HIV infection | Kim et al |
| T cell | siRNA (targeting CD4 or TNPO3) | Cationic Poly(amidoamine) (PAMAM) dendrimer nanoparticles | None | HIV-1 inhibition | Zhou et al |
| T cell | siRNA (targeting Cyclin D1) | Multilamellar vesicle liposomes composed of 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) | anti-β7 integrin antibodies | Colitis | Peer et al |
| T cell | siRNA (targeting adenosine A2a receptor) | PEG-chitosan-lactate (PCL) nanoparticles | None | Improve anti-tumor T cell response of dendritic cell-based cancer vaccine | Masjedi et al |
2. Monocytes/macrophages
2.1. Rationale
Monocytes and macrophages are now recognized as attractive therapeutic targets for virtually all major immune-related diseases, including cancer, inflammation, autoimmunity, and transplant rejection(25). In fact, the enormous plasticity of macrophages, which includes their potentiality to be ‘reprogrammed’ between immune-suppressive (pro-tumoral) M2-like and inflammatory (anti-tumor) M1-like phenotypes, is key to their hopeful application in therapeutic strategies. While immunomodulatory small molecule compounds, recombinant cytokines or antibodies are being developed as macrophage-polarizing agents, they require high systemic drug exposures and thus increase the risk of off-target adverse effects. For example, interleukin-12 (IL-12), interferon gamma (IFNγ), Toll-like receptor (TLR−) agonists, and CD40 agonists have all been reported to induce repolarization of tumor-associated macrophages (TAMs) (26–28). However, these immunomodulatory agents can also activate a broad range of other cell types, which means they are associated with dose-limiting adverse effects and systemic toxicities (29–31). Likewise, several small molecule drugs have been developed that focus on blocking the localization of TAM-precursor cells to tumors by targeting pathways involved in cell recruitment or expansion (e.g., inhibitors of CSF-1/CSF-1R (32; 33) or CCL2 (34)). Unfortunately, these approaches do not specifically promote macrophage antitumor activities and require repeated systemic exposure to large doses of the drugs. Furthermore, clinical trials of these pharmaceuticals produced low responses unless they were combined with cyto-reductive therapies or checkpoint inhibitors (34; 35). Another complication of the clinical use of CSF-1R inhibitors is they cause systemic depletion of normal monocytic cells, which results in high toxicity if patients are treated for prolonged periods (36). More recently, macrophage-based cell therapy products have entered clinical testing for anticancer activity or tolerance induction(37; 38). However, the elaborate and expensive protocols currently required to manufacture engineered macrophages ex vivo put this approach beyond the reach of many patients who might benefit.
Harnessing synthetic nanoparticles as reagents for selective in situ reprogramming of monocytes/macrophages for therapeutic purposes comes with notable advantages. For example, macrophages are inherently phagocytic cells that are extremely effective at clearing nanomaterials, and they are present in high numbers, thus ensuring high transfection rates with only moderate off-target gene transfer. Moreover, they can quickly and directionally migrate to pathological sites, such as tumor lesions or inflamed tissue. This native homing ability favors them as vehicles for the focused delivery and expression of therapeutic transgenes at the disease site. In many cases, however, direct targeting and genetic engineering of tissue resident disease-causing macrophages is not a realistic goal. In particular, tumor resident macrophages are difficult to access by systemically administered nanotherapeutics since blood and lymphatic vessels are greatly compressed within the tumor bulk(39). A large percentage of the injected nanoparticle dose is therefore taken up by circulating monocytes before they extravasate into tissue and differentiate into macrophages. This strategy is appealing because of the relative abundance of monocytes in the peripheral blood as well as their inherent ability to take up nanoparticles. As a result, the fate of monocytes can be switched by genetic reprogramming before they are recruited into tissue and contribute to disease progression.
2.2. Delivering plasmid DNA
Unlike other mammalian cells, macrophages are difficult to transfect with plasmid DNA, not only because they have evolved to recognize foreign nucleic acids and to initiate an immune response to these molecules, but mostly because they are almost non-proliferating cells(40). Thus, timely arrival at the nucleus during mitotic envelope breakdown is challenging for plasmid DNA. To balance inefficient gene transfer, DNA-containing nanomedicines need to be administered at high and repeated doses to achieve a therapeutic benefit. This raises safety concerns about nanoparticle immunogenicity and infusion reactions. Jain and colleagues encapsulated plasmid DNA encoding the immune-suppressive IL-10 cytokine into alginate nanoparticles and modified the surface of the nanocarriers with tuftsin peptide to achieve active macrophage targeting(41). Using a model of adjuvant-induced arthritis, they demonstrated that, following intraperitoneal administration, nanoparticles were effectively internalized by peritoneal macrophages and subsequently localized into the inflamed paws of arthritic rats. Here, transfected macrophages were successfully reprogrammed from M1-like to M2-like, which led to the downregulation of proinflammatory cytokines and ultimately prevented the progression of inflammation and joint damage. In a parallel study, the same group demonstrated that administration of hyaluronic acid-poly(ethyleneimine) (HA-PEI) nanoparticles loaded with plasmids expressing IL-4 or IL-10 suppressed local inflammation induced by lipopolysaccharide(42). HA-based nanocarriers are very attractive macrophage delivery systems because the glycoprotein CD44, which is strongly expressed on macrophages and tumor cells, binds to the extracellular domain of HA. This inherent macrophage targeting of HA-based formulations is appealing as it bypasses the need for surface-functionalization of the nanocarriers, which usually complicates scale-up manufacturing and quality control, and adds cost. One advantage of plasmid DNA is that transgenes can be expressed under the control of a tissue-specific promoter, thus limiting gene expression only to therapeutically desired cell types and avoiding off-target effects. Luo et al., for instance, have developed CRISPR/Cas9 vectors using a plasmid delivery system under the control of the CD68 promoter, which drives gene expression specifically in monocytes and macrophages(43). A single guide RNA (sgRNA) sequence targeting netrin-1 (Ntn1), a gene that promotes macrophage accumulation in adipose tissue, was included in the plasmid. The authors used a Cationic Lipid N,N-bis(2hydroxyethyl)e-N-methyl-N-(2-cholesteryoxycarbonyl-aminoethyl) ammonium bromide (BHEM-Chol)-Assisted polyethylene glycol (PEG)-b-poly(lactic-co-glycolic) acid (PLGA) Nanoparticle (CLAN) system to encapsulate the large plasmid. Following systemic administration, the Ntn1 gene was selectively disrupted in macrophages and their precursor monocytes, which reduced the expression of Ntn1 and subsequently improved Type-2 diabetes signs.
2.3. Delivering in vitro-transcribed (IVT) mRNA
IVT mRNA has emerged as a new drug class for delivering genetic information directly into cells (44). These synthetic medicines can be engineered to induce the transient expression of selected proteins because they structurally resemble natural mRNA. In contrast to plasmids, which need to be situated in the nucleus to be transcribed into mRNA, IVT mRNA is immediately translated in the cytoplasm. This not only circumvents the risk of genomic integration, but substantially increases transfection efficiency and ensures that the cells are reprogrammed quickly and reliably before the disease becomes intractable. The relatively short half-life results in transient and more controlled expression of the encoded protein. Moreover, IVT mRNA can be produced in a cell-free environment by in vitro transcription, thereby obviating the need for microbes or cultured cells for production, and avoiding the quality and safety issues that accompany such production. This permits simple downstream purification and rapid and cost-effective manufacturing. However, efficient and safe in vivo delivery of IVT mRNA requires nanocarriers that bind and condense the mRNA, protect it from degradation by omnipresent RNases, and facilitate cellular uptake and endosomal escape into the cytosol without interfering with the cellular translation machinery. Peer’s group at Tel Aviv University developed a self-assembled modular platform for targeted nucleic acid delivery named ASSET (Anchored Secondary scFv Enabling Targeting), which coats lipid-based nanoparticles (LNPs) with monoclonal antibodies(45). Recently, Veiga et al. utilized the ASSET platform to deliver IVT mRNA encoding the anti-inflammatory protein IL-10 into Ly6c+ inflammatory leukocytes(46). Using a mouse colitis model, they demonstrated selective and efficient mRNA-based IL-10 expression in diseased tissue by disease-related Ly6c+ inflammatory leukocytes. By targeting expression of IL-10, they were able to achieve therapeutic concentrations of this cytokine in the colon, resulting in significant reductions in colitis-related pathological signs and the severity of intestinal inflammation. Our group explored the use IVT mRNA formulated into an injectable nanotherapeutic to genetically reprogram TAMs into antitumor macrophages without disrupting immune homeostasis or causing systemic toxicity(47). We developed a targeted mRNA delivery system that can introduce robust gene expression in the targeted cells by taking advantage of electrostatic interactions between cationic poly(β-amino ester) (PbAE) polymers and anionic mRNA To target the nanoparticles to TAMs as well as further stabilize the mRNA-PbAE complexes they contain, we engineered Di-mannose moieties onto their surface using polyglutamic acid (PGA) as a linker. The nanoparticles were manufactured by using a simple two-step, charge-driven self-assembly process. First, the synthetic mRNA was complexed with a positively-charged PbAE polymer, which condenses the mRNA into nano-sized complexes. This step was followed by the addition of PGA functionalized with Di-mannose, which shields the positive charge of the PbAE-mRNA particles and confers macrophage-targeting. We chose to express the M1-polarizing transcription factor Interferon regulatory factor 5 (IRF5) and its activating kinase IKKβ to reprogram TAMs. By applying in vivo test systems that faithfully model advanced-stage ovarian cancer and glioblastoma, we established that serial administration of IRF5/IKKβ-encoding nanoparticles (via an intraperitoneal route for ovarian cancer, and injected intravenously to treat glioblastoma) reverses the immunosuppressive, tumor-supporting state of TAMs and reprograms them to a phenotype that induces antitumor immunity and promotes tumor regression. IVT mRNA has also become a powerful tool for in situ genome editing. Xu and colleagues encapsulated mRNA encoding mCas9 and gRNA targeting the NLR family pyrin domain containing 3 (NLRP3) gene into a CLAN for delivery into mice(48). The NLRP3 protein is predominantly expressed in macrophages as a cytosolic sensor that triggers inflammation. The study showed that repeated intravenous dosing of CLANmCas9/gNLRP3 could ablate NLRP3 in transfected macrophages and inhibit inflammasome activation. Using this therapy, the authors demonstrated amelioration of a range of both acute (septic shock, peritonitis) and chronic inflammatory diseases (Type-2 diabetes), highlighting the therapeutic potential of CRISPR-mediated in situ editing of monocytes/macrophages.
2.4. Delivering small interfering RNA (siRNA)
Another exciting RNA modality recently added to the drug discovery toolbox is siRNA, which can be applied to target novel pathways not amenable to more traditional drug discovery approaches(49). It took two decades from the initial discovery of siRNA for the first treatment using this technology to get past regulatory scrutiny, and the field faced numerous obstacles and setbacks along the way. To date, only three siRNA drugs, ONPATTRO™, GIVLAARI™ and OXLUMO™, have been cleared by the United States FDA, all for the treatment of inherited rare liver diseases(50). A technical challenge with this approach in terms of immunological disease is how to deliver siRNA to specific immune cells in the body at high enough concentrations to exert a therapeutic effect. siRNA molecules are too large to cross cell membranes but small enough to be freely cleared by glomeruli, as molecules with a size smaller than 8 nm are easily filtered into the urine(51). Hence, once unformulated siRNAs leave the bloodstream, they will end up in the bladder and be excreted within a few minutes to half an hour, which prevents them from accumulating in the targeted tissues or cells. To overcome this barrier, LNPs have been explored extensively as a promising carrier for siRNA delivery into immune cells. For example, Shobaki et al. prepared LPNs composed of the pH-sensitive cationic lipid CL4H6, in addition to cholesterol and PEGylated-lipids, and achieved high (>90%) siRNA encapsulation(52). Following intravenous infusion, these particles — even without targeting ligands on their surface — were efficiently taken up by CD45+, CD11b+, and F4/80+ TAMs in a human xenograft mouse model. The authors demonstrated that delivery of siRNA against the signal transducer and activator of transcription 3 (STAT3) and hypoxia inducible factor 1 α (HIF-1α) genes into TAMs reversed their pro-tumorous functions, such as angiogenesis and tumor cell activation/invasiveness, which ultimately triggered a significant antitumor therapeutic response. Also, macrophage-targeted LNPs, such as the Ly6c-specific ASSET platform we described above for the delivery of IVT mRNA into inflammatory myeloid cells, have been explored as siRNA carriers. Through this strategy, Veiga and colleagues demonstrated that in vivo silencing of the Interferon regulatory factor 8 (IRF8) gene, a master regulator of pro-inflammatory signals in myeloid cells, can effectively treat inflammatory bowel disease in mice(53). A comparable therapeutic effect was achieved when using the same nanoplatform to silence Tumor Necrosis Factor alpha (TNF-α) in macrophages, highlighting the versatility of the ASSET platform for antibody-directed delivery of RNA therapeutics into hard-to-transfect immune cells(45). In parallel to LNPs, investigators have optimized polymeric nanoparticles for delivery of siRNA into macrophages. CD44-targeting HA-PEI nanoparticles, for instance, have been successfully used to silence TNF-α in situ, specifically in macrophages(54). A more elaborate polymeric nanoparticle formulation to deliver siRNA into macrophages was developed by Tao and colleagues (55). In their study, atherosclerosis-promoting genes in plaque macrophages were silenced with siRNA nanoparticles to promote plaque stability. To enable long-term circulation and macrophage targeting, the nanoparticles were composed of PLGA polymer and lipid-PEG. In addition, a peptide called S2P, which recognizes the macrophage receptor stabilin-2, was anchored to the lipid PEG layer on the surface. Using a mouse model of atherosclerosis, this group demonstrated that treatment with siRNA nanoparticles targeting Ca2+/calmodulin-dependent protein kinase II subunit γ (CaMKIIγ), a molecule expressed in lesional macrophages, resulted in strengthening of the fibrous cap, thus improving plaque stability. Overall, this study provides a promising macrophage-targeted siRNA delivery platform for atherosclerosis therapy, which is a fairly recent, and frankly somewhat risky, treatment strategy for cardiovascular disease. One challenge all siRNA nanodrugs need to overcome when treating inflammatory disease is potential off-target effects. Many studies have reported innate immune stimulation by siRNA or the siRNA delivery vehicle, which might be a desired adjuvant effect for cancer immunotherapy, but could exacerbate autoimmune disease(56; 57). Various features of the siRNA structure and sequence have contributed to the immune stimulation effect. To minimize RNA-associated immune stimulation in clinical applications, known immunostimulatory motifs, such as 5’-UGUGU-3’, and 5’-GUCCUUCAA-3’ should be avoided without reducing the potency of the siRNA construct(58). Furthermore, base modifications can reduce immune activation, and the addition of modified nucleotides into siRNA suppresses unwanted immunostimulation(59). Lastly, the nanovector itself should be rationally designed to avoid any potentially immunogenic components.
2.5. Delivering microRNA (miRNA)
MicroRNAs (miRNAs) are small RNAs that do not code for proteins, but function by controlling the expression of other genes(60). Numerous studies have described the influence of miRNAs on the survival and functioning of various immune cell types. In particular, the essential role of miRNAs in the differentiation of dendritic cells and macrophages via toll-like receptors is well characterized. For example, miR-127, miR-155 and miR-125b have been shown to promote M1 polarization of macrophages, while miR-124, miR-223, and miR-132 induce M2 polarization by targeting various transcription factors(61). However, despite this preclinical promise, the efficacy of miRNA therapeutics is limited by poor targeting ability, short circulation time and off-target effects of naked miRNA-based agents. To overcome these barriers, nanoparticles have been explored for their capability to shield miRNA from the external environment, thereby reducing inactivation or degradation and enhancing the circulation time and accumulation at the target cell. Liu and colleagues, for instance, reported a redox/pH dual-responsive polypeptide nanovehicle consisting of self-crosslinked galactose-functionalized polypeptides (GLC) coated with sheddable PEG-poly(L-lysine) (sPEG) copolymers(62). Injection of miR-155-loaded sPEG/GLC elevated miR-155 expression in TAMs and effectively repolarized them to antitumor M1 macrophages, which increased tumor infiltration by T cells and natural killer cells and consequently triggered tumor regression. In another study, Parayath et al. encapsulated miR-125b with HA-PEI to target CD44+ macrophages(63). Intraperitoneal injection of HA-PEI/miR-125b selectively elevated miR-125b in peritoneal macrophages over 100-fold and repolarized them from a suppressive M2-like phenotype to an antitumor M1 phenotype. More interestingly, activated peritoneal macrophages effectively migrated into mouse lung cancer and significantly elevated lung M1 macrophages, suggesting a potential to boost cancer immunotherapy. The therapeutic use of miRNA nanoparticles to dampen aberrant macrophage activation in inflammatory disease was demonstrated by Tran et al(64). By using HA-PEI nanocarriers to overexpress miR-223 in peritoneal macrophages prior to lipopolysaccharide administration, the authors were able to reduce systemic inflammation, as evidenced by a significant decrease in the levels of the pro-inflammatory cytokines TNF-α, IL-1β and IL-6.
Based on the rapidly emerging appreciation of the role of cardiac macrophages in heart disease, Cohen’s group investigated whether targeting macrophages in the infarct zone by using nanoparticle-delivered miRNA could switch their phenotype from a pro-inflammatory to an anti-inflammatory state, thereby promoting cardiac healing(65). Using a mouse model of myocardial infarction, her team demonstrated that, following intravenous infusion, miR-21 particles targeted macrophages in the infarct zone and changed their phenotype, resulting in reduced cell apoptosis, fibrosis and hypertrophy. Other studies have focused on preventing rather than treating cardiac infarction and atherosclerosis with miRNA Nguyen and colleagues formulated chitosan nanoparticles loaded with miR-206 or miR-223 for systemic administration with the goal to stimulate cholesterol efflux from macrophages that populate the atherosclerotic plaques(66). Mice treated with these nanoparticles showed reduced vascular lipid accumulation and atherosclerosis as well as increased cholesterol excretion by the liver and intestine. These highlighted studies provide just a glimpse of the potential of miRNA therapeutics to modulate macrophages in the context of several diseases. As our understanding of the role of miRNAs in human disease continues to expand, miRNA nanotherapeutics are likely to gain more attention in the clinical immunotherapy landscape.
3. Dendritic cells
3.1. Rationale
Dendritic cells (DCs) are powerful modulators of immune responses at the interface between the innate and adaptive immune systems; therefore, they have been extensively scrutinized for immunotherapy applications(67). DCs are an ideal antigen delivery vehicle and thus are frequently used in patient trials, but clinical responses have been largely disappointing(68). To rationally design more effective DC vaccines, various ex vivo gene modifications have been tried either to boost antigen presentation, provide sufficient T-cell stimulatory signals, or improve migration to lymphoid tissues(69). However, the process used to generate DCs ex vivo is expensive, labor-intensive, and often difficult to standardize and scale up. Direct in situ genetic manipulation of DCs is therefore appealing, all the more so because in this approach the cells never exit their physiological environment.
3.2. Delivering plasmid DNA or IVT mRNA
Luo and coworkers prepared CLANs to simultaneously encapsulate an autoimmune diabetes-relevant peptide, a CRISPR-Cas9 plasmid (pCas9), and three guide RNAs targeting the costimulatory molecules CD80, CD86 and CD40(70). Using autoimmune Type-1 diabetes as a disease model, this team demonstrated that nanoparticle infusions generate tolerogenic DCs in situ, which triggered the expansion of autoreactive regulatory T cells (Tregs) and prevented diabetes development. In a separate study, this group further extended their in situ DC genome editing approach to treat organ rejection(71). Instead of plasmid DNA encoding Cas9, they used IVT mRNA to express the Cas enzyme and co-encapsulated it with gRNA targeting CD40. After intravenous injection into an acute mouse skin transplant model, these nanoparticles effectively delivered mCas9/gCD40 into DCs and disrupted CD40, which significantly inhibited T cell activation and prolonged graft survival.
3.3. Delivering small interfering RNA (siRNA)
Silencing the expression of costimulatory molecules to inhibit immune responses has also been accomplished with siRNA Katakowski and colleagues, for instance, showed significant knockdown of CD40, CD80 and CD86 following injection of LNPs containing siRNA specific for these molecules (72). The authors took advantage of single chain antibodies against the DC marker DEC205 to guide the therapy. DEC205 is a C-type lectin expressed at high levels on CD8(+) DCs, which are uniquely able to cross-prime host T cells. Their results demonstrate the ability of targeted siRNA-loaded LNPs to inhibit powerful immune responses such as mixed lymphocyte reactions. In the cancer immunotherapy field, siRNA-mediated silencing of counter-regulators of DC activation, such as Indoleamine 2,3-dioxygenase-1 (IDO1), STAT3 or Suppressor Of Cytokine Signaling 1 (SOCS1), is a promising strategy. Traditionally, these targets were inhibited by small molecule drugs which have been widely applied in several clinical trials(73–75). However, since these signaling pathways are not selective for DCs, but are important in almost every organ and tissue in the body, off-target effects are common. This motivated the development of an siRNA-based nanomedicine. Huang and colleagues delivered IDO siRNA into DCs using a CLAN vehicle(76). When infused into mice bearing established colorectal carcinoma, this treatment achieved substantial antitumor effects by promoting DC maturation, increasing tumor-infiltrating T lymphocytes and decreasing the number of Tregs. In a study by Luo et al., co-delivery of STAT3 siRNA with poly I:C adjuvant and vaccine antigen substantially improved the therapeutic potential of a cancer nanovaccine(77). Heo and coworkers reported that a PLGA polymeric nanoparticle which combined the delivery of tumor antigens and SOCS siRNA to DCs induced an enhanced antitumor immune response(78). A novel intravaginal film platform for targeted delivery of siRNA-loaded nanoparticles to DCs was reported by Gu and colleagues(79). They produced siRNA-loaded, anti-human leukocyte antigen-DR (HLA-DR) antibody-functionalized PEG-PLGA-based nanoparticles, with a ~230 nm mean diameter, and associated it to polyvinyl alcohol/λ-carrageenan-based films using solvent-casting. The team demonstrated that, after placement on the vaginal mucosa, siRNA nanoparticles were slowly released from the film to penetrate epithelial cells and specifically target siRNA into HLA-DR+ immune cells (HIV target cells). This approach could therefore be developed as a promising platform for preventing HIV infection within the female genital tract.
4. T cells
4.1. Rationale
The adoption of T cells as a mainstream therapy is still some years away, as the biotechnology industry currently lacks off-the-shelf injectable reagents that (i) do not require complex manufacturing and (ii) can be easily re-dosed for as long as medically necessary. Instead, the technology is now limited to allogeneic platforms manufactured from healthy donors(80). However, this immunological tailoring of allogeneic T cells by adding genome editing and cell purification steps complicates the manufacturing protocol, which not only delays production and increases costs (including costs for gene-editing intellectual property), but also reduces the viability of the lymphocytes and their yield (81). Allogeneic T cells have clear advantages over autologous therapies, particularly in patients with advanced malignancy who have an extensive treatment history, and where collection of T cells sufficient for the cell-manufacturing cycle may be difficult. Nonetheless, sourcing T cells from healthy donors instead of patients will not solve the overall challenge of providing large numbers of patients with a mainstream T-cell therapy that is affordable, effective and safe.
4.1. Delivering plasmid DNA
Our group designed injectable DNA nanocarriers that choreograph robust Chimeric Antigen Receptor (CAR) production in host T cells(82). To create a reagent that can genetically modify primary T lymphocytes (which are notoriously refractory to non-viral transfection methods) simply by contact, we bioengineered polymeric nanocarriers with four functional components: (i) surface-anchored anti-CD3e f(ab’)2 fragments that selectively bind the nanocarriers to T cells and initiate rapid receptor-induced endocytosis to internalize them; (ii) a negatively charged PGA coating to minimize off-target binding by reducing the net surface charge of the nanocarriers; (iii) a PBAE polymer-based carrier matrix that condenses and protects nucleic acid from enzymatic degradation; and (iv) DNA plasmids encoding the CAR and a transposase gene to enable stable integration of the CAR DNA into the host genome. Using a mouse version of a CD19 CAR, we compared this nanoparticle system to CAR-T cells generated ex vivo, using a protocol that mimics clinical trials, in mice with established leukemia. We found that nanoparticles could program T cells in quantities that were sufficient to bring about tumor regression at similar efficacies as conventional infusions of T cells transduced ex vivo with CAR-encoding viral vectors. Notably, nanoparticle-reprogrammed T cells continued to produce these receptors for weeks, allowing them to act as a ’living drug’ that increases in number and serially destroys tumor cells. The CD3 molecule we targeted in these experiments is one of many options to selectively shuttle transgenes into lymphocytes. Our group also tested nanocarriers targeted with anti-CD4 or anti-CD8 antibodies and achieved comparable gene transfer efficiencies. Thus, it is probably possible to selectively modify only defined T cell subsets, such as antigen-experienced lymphocytes, using activation markers (e.g., CD25, 4-1BB, OX40, or CD40L).
4.2. Delivering IVT mRNA
In a follow-up study, we explored the use of IVT mRNA to quickly and specifically program antigen-recognizing capabilities into circulating T cells as a strategy to treat cancer and infectious disease(83). Using clinically relevant preclinical models of leukemia, prostate cancer and hepatitis-induced hepatocellular carcinoma, we demonstrated that repeated infusions of rationally designed mRNA nanocarriers can selectively deliver CAR- or TCR-genes into host T cells and program them in quantities sufficient to bring about disease regression with efficacies similar to adoptive methods.
4.3. Delivering siRNA
Systemic delivery of siRNA into T lymphocytes has primarily been explored as a treatment for HIV-1 infection. In particular, cellular receptors that mediate entry of HIV into T cells, such as CCR5 or CD4, have been downregulated in CD4+ T cells as one of the main hosts and reservoirs of HIV. The Shankar group at Texas Tech University, for instance, described in 2010 a T-cell binding nanoreagent built from neutral phospholipids and targeted to the Lymphocyte Function-associated Antigen-1 (LFA-1) integrin(84). They showed that a single in vivo administration of these nanocarriers loaded with siRNA against CCR5, selectively silenced CCR5 expression in T cells and macrophages in humanized mice for up to 10 days and protected these animals from HIV infection. Similar protection was reported by Zhou and colleagues, who infused humanized mice with cationic poly(amidoamine) (PAMAM) dendrimer nanoparticles that delivered siRNA silencing CD4 or Transportin-3 (TNPO3), which is a cellular factor involved in facilitating cytoplasmic-nuclear trafficking of the HIV-1 preintegration complex(85). Peer and coworkers demonstrated in 2008 that delivery of siRNA to leukocytes could be used to treat autoimmune disease(86). The siRNA target in that study was Cyclin D1, a cell cycle regulator that is aberrantly upregulated in immune cells in an inflammatory environment. Antibodies to β7 integrin were used to target siRNA-loaded lipid nanoparticles to specific leukocyte subsets involved in gut inflammation. Systemic administration of this nanomedicine silenced Cyclin D1 in leukocytes and reversed experimentally induced colitis in mice. A somewhat unconventional approach to improve the efficacy of a cancer vaccine using a T-cell targeted siRNA nanoparticle was reported by Masjedi and coworkers(87). Instead of genetically enhancing antigen processing and presentation in DCs, this group developed PEG-chitosan-lactate (PCL) nanoparticles that silence adenosine A2A receptor (A2aR) expression in T cells. Immunosuppressive pathways mediated through A2aR are known to inhibit T lymphocytes in hypoxic, inflamed and cancerous microenvironments, and A2aR small molecule antagonists are an emerging class of agents to treat cancer. In that study, a DC cancer vaccine and A2aR siRNA-loaded nanoparticles were co-administered into mice bearing established 4T1 breast tumors. Although the siRNA particles were not targeted to T cells, they effectively downregulated A2aR expression in tumor infiltrating lymphocytes, which markedly improved their expansion and therapeutic potential in response to the DC cancer vaccine.
5. Conclusions and future directions
Currently, efforts to apply nonviral in vivo immunotherapy platforms to translational research are proceeding apace. The driving force behind this development is the pressing need to dramatically simplify the logistics and expand the reach of the curative power of cell-based immunotherapy. All the reprogramming nanoreagents highlighted in this review have the potential to leapfrog current ex vivo cell engineering approaches as scalable, cost-effective, and minimally disruptive medicines, that could be delivered to sizeable patient populations in outpatient settings. Despite this promise, nanotechnology is far from being a panacea. Modified viral vectors, specifically designed to bind and genetically retool therapeutically relevant immune cells in vivo, are being developed by various biotech startups as medicines that can be directly administered into the patient. In parallel, hybrid delivery systems such as polymer-coated viral vectors with defined tropism for specific immune cell subtypes are entering clinical testing as injectable agents. The nascent field of in vivo genetic engineering of immune cells will therefore be extremely competitive and fast-paced.
While viral reprogramming reagents will face inherent bottlenecks, such as complex scale-up production, innate immune responses against the viral particle (88), inactivation by human serum complement(89), and the risk of uncontrollable vector-based insertional mutagenesis(90), synthetic nanomedicines also come with challenges. In contrast to therapeutics that are already established in the clinic, such as small molecules or antibodies, immune cell nanoparticles are multi-component three-dimensional constructs that require a reproducible manufacturing process to reliably achieve the intended physicochemical characteristics, biological behaviors, and pharmacological profiles. The safety and efficacy of such nanodrugs can be influenced by minor variations in many parameters and need to be carefully monitored, particularly in the context of targeting to unintended sites and potential toxicities. Furthermore, nanomedicines require additional developmental and regulatory considerations compared with conventional medicines. Only a few manufacturing facilities with the requisite degree of expertise are currently operational in the United States. Also, the gene transfer efficiency of nonviral systems is usually less than that of viral systems (91), which means that patients will likely need repeated dosing to program immune cells in quantities that are sufficient to bring about the desired therapeutic benefit. Viral particles possess innate machinery to overcome cellular barriers (cellular uptake, endosomal escape, nuclear entry and nucleic acid release). Overcoming the same barriers with rationally designed nucleic acid nanoformulations requires great effort and is often challenging. On the plus side, with this new treatment modality the patient could be easily re-dosed for as long as medically necessary. In contrast to integrating viral vectors, that require suicide genes as safety switches in case of unpredictable immediate or persistent toxicities, physicians administering nanoparticles gain the ability to control pharmacokinetic properties of the therapy and to periodically in situ reprogram fresh populations of therapeutic host immune cells—thus bypassing not only cell exhaustion and dysfunction, but also long-term toxicities.
In conclusion, nanotherapeutics that genetically modify immune cells within the body to improve their therapeutic action are poised to change the way we treat disease. This approach makes it possible to eliminate all stages of genetic modification carried out ex vivo, as required by currently marketed products, and bring life-changing therapies to a large patient population, including in rural settings where access to sophisticated healthcare systems is limited. To translate the scientific potential of this potentially disruptive technology into breakthrough treatments, it is now imperative to bring together scientists, entrepreneurs and leading pharmaceutical companies. The young field of nanotechnology is still causing a great deal of confusion among senior level decision-makers at venture capital firms and drug companies. Also, building a productive research team with in-depth cross-disciplinary expertise, spanning immunotherapy, nanotechnology, and genetic engineering takes more time than recruiting narrowly focused talent. Nonetheless, we are optimistic that by joining efforts among leaders in the nanotechnology field and the immunotherapy industry, and by merging resources, inventions and know-how, nanomedicines for in vivo immune cell reprogramming will evolve into a new biotechnology arena with broad applications.
Acknowledgments
This work was supported with funds provided by the National Cancer Institute (CA207407), a 2018 Emerging Leader Award from the Mark Foundation for Cancer Research, and a 2018 Investigator Award from the Alliance for Cancer Gene Therapy. M.T.S. was also supported by an Allen Distinguished Investigator Award, a Paul G. Allen Frontiers Group advised grant of the Paul G. Allen Family Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
M.T.S. is a consultant of Sanofi.
References
- 1.Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, et al. 2020. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat Med 26:1686–90 [DOI] [PubMed] [Google Scholar]
- 2.Rozenbaum M, Meir A, Aharony Y, Itzhaki O, Schachter J, et al. 2020. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front Immunol 11:1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. 2020. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 59:102975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, et al. 2020. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol 38:947–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moffett HF, Harms CK, Fitzpatrick KS, Tooley MR, Boonyaratanakornkit J, Taylor JJ. 2019. B cells engineered to express pathogen-specific antibodies protect against infection. Sci Immunol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kariko K 2019. In vitro-Transcribed mRNA Therapeutics: Out of the Shadows and Into the Spotlight. Mol Ther 27:691–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bishop DC, Caproni L, Gowrishankar K, Legiewicz M, Karbowniczek K, et al. 2020. CAR T Cell Generation by piggyBac Transposition from Linear Doggybone DNA Vectors Requires Transposon DNA-Flanking Regions. Mol Ther Methods Clin Dev 17:359–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, Dimitriadis EK, Yang Y, Li J, Yuan Z, et al. 2013. Production and characterization of novel recombinant adeno-associated virus replicative-form genomes: a eukaryotic source of DNA for gene transfer. PLoS One 8:e69879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hung CF, Xu X, Li L, Ma Y, Jin Q, et al. 2018. Development of Anti-Human Mesothelin-Targeted Chimeric Antigen Receptor Messenger RNA-Transfected Peripheral Blood Lymphocytes for Ovarian Cancer Therapy. Hum Gene Ther 29:614–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moutsatsou P, Ochs J, Schmitt RH, Hewitt CJ, Hanga MP. 2019. Automation in cell and gene therapy manufacturing: from past to future. Biotechnol Lett 41:1245–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.2018. The Quest for Off-the-Shelf CAR T Cells. Cancer Discov 8:787–8 [DOI] [PubMed] [Google Scholar]
- 12.Alzubi J, Lock D, Rhiel M, Schmitz S, Wild S, et al. 2021. Automated generation of gene-edited CAR T cells at clinical scale. Mol Ther Methods Clin Dev 20:379–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subklewe M 2021. COUNTERPOINT BiTEs better than CAR T cells. Blood Adv 5:607–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tumeh PC, Koya RC, Chodon T, Graham NA, Graeber TG, et al. 2010. The impact of ex vivo clinical grade activation protocols on human T-cell phenotype and function for the generation of genetically modified cells for adoptive cell transfer therapy. J Immunother 33:759–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, et al. 2017. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 31:186–94 [DOI] [PubMed] [Google Scholar]
- 16.Schweizer A, Rusert P, Berlinger L, Ruprecht CR, Mann A, et al. 2008. CD4-specific designed ankyrin repeat proteins are novel potent HIV entry inhibitors with unique characteristics. PLoS Pathog 4:e1000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang AA, Tiede C, Hughes DJ, McPherson MJ, Tomlinson DC. 2017. Isolation of isoform-specific binding proteins (Affimers) by phage display using negative selection. Sci Signal 10. [DOI] [PubMed] [Google Scholar]
- 18.Kacherovsky N, Cardle II, Cheng EL, Yu JL, Baldwin ML, et al. 2019. Traceless aptamer-mediated isolation of CD8(+) T cells for chimeric antigen receptor T-cell therapy. Nat Biomed Eng 3:783–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Operti MC, Fecher D, van Dinther EAW, Grimm S, Jaber R, et al. 2018. A comparative assessment of continuous production techniques to generate sub-micron size PLGA particles. Int J Pharm 550:140–8 [DOI] [PubMed] [Google Scholar]
- 20.Leaver T. Nanoparticles - a Revolution in the Development of Drug Delivery Vehicles. Drug Development & Delivery [Google Scholar]
- 21.Petschacher CEA, Besenhard M, Wagner J, Barthelmes J, Bernkop-Schnuerch A, Khinast JG. Zimmer A. 2013. Thinking continuously: a microreactor for the production and scale-up of biodegradable, self-assembled nanoparticles. Polymer Chemistry 4:2342–52 [Google Scholar]
- 22.Zhang C, Maruggi G, Shan H, Li J. 2019. Advances in mRNA Vaccines for Infectious Diseases. Front Immunol 10:594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pardi N, Hogan MJ, Porter FW, Weissman D. 2018. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov 17:261–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho W, Gao M, Li F, Li Z, Zhang XQ, Xu X. 2021. Next-Generation Vaccines: Nanoparticle-Mediated DNA and mRNA Delivery. Adv Healthc Mater:e2001812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poltavets AS, Vishnyakova PA, Elchaninov AV, Sukhikh GT, Fatkhudinov TK. 2020. Macrophage Modification Strategies for Efficient Cell Therapy. Cells 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. 2004. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–86 [DOI] [PubMed] [Google Scholar]
- 27.Muller E, Christopoulos PF, Halder S, Lunde A, Beraki K, et al. 2017. Toll-Like Receptor Ligands and Interferon-gamma Synergize for Induction of Antitumor M1 Macrophages. Frontiers in Immunology 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiehagen KR, Girgis NM, Yamada DH, Smith AA, Chan SR, et al. 2017. Combination of CD40 Agonism and CSF-1R Blockade Reconditions Tumor-Associated Macrophages and Drives Potent Antitumor Immunity. Cancer Immunol Res 5:1109–21 [DOI] [PubMed] [Google Scholar]
- 29.Sangro B, Mazzolini G, Ruiz J, Herraiz M, Quiroga J, et al. 2004. Phase I trial of intratumoral injection of an adenovirus encoding interleukin-12 for advanced digestive tumors. J Clin Oncol 22:1389–97 [DOI] [PubMed] [Google Scholar]
- 30.Vonderheide RH, Burg JM, Mick R, Trosko JA, Li DG, et al. 2013. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pockros PJ, Guyader D, Patton H, Tong MJ, Wright T, et al. 2007. Oral resiquimod in chronic HCV infection: Safety and efficacy in 2 placebo-controlled, double-blind phase IIa studies. J Hepatol 47:174–82 [DOI] [PubMed] [Google Scholar]
- 32.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, et al. 2013. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19:1264–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, et al. 2015. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N Engl J Med 373:428–37 [DOI] [PubMed] [Google Scholar]
- 34.Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, et al. 2016. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol 17:651–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, et al. 2016. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol 18:557–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sauter KA, Pridans C, Sehgal A, Tsai YT, Bradford BM, et al. 2014. Pleiotropic effects of extended blockade of CSF1R signaling in adult mice. J Leukocyte Biol 96:265–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mukhopadhyay M 2020. Macrophages enter CAR immunotherapy. Nat Methods 17:561. [DOI] [PubMed] [Google Scholar]
- 38.Anderson NR, Minutolo NG, Gill S, Klichinsky M. 2021. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res 81:1201–8 [DOI] [PubMed] [Google Scholar]
- 39.Jain RK, Martin JD, Stylianopoulos T. 2014. The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng 16:321–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, Edwards JP, Mosser DM. 2009. The expression of exogenous genes in macrophages: obstacles and opportunities. Methods Mol Biol 531:123–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jain S, Tran TH, Amiji M. 2015. Macrophage repolarization with targeted alginate nanoparticles containing IL-10 plasmid DNA for the treatment of experimental arthritis. Biomaterials 61:162–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tran TH, Rastogi R, Shelke J, Amiji MM. 2015. Modulation of Macrophage Functional Polarity towards Anti-Inflammatory Phenotype with Plasmid DNA Delivery in CD44 Targeting Hyaluronic Acid Nanoparticles. Sci Rep 5:16632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo YL, Xu CF, Li HJ, Cao ZT, Liu J, et al. 2018. Macrophage-Specific in Vivo Gene Editing Using Cationic Lipid-Assisted Polymeric Nanoparticles. ACS Nano 12:994–1005 [DOI] [PubMed] [Google Scholar]
- 44.Kwon H, Kim M, Seo Y, Moon YS, Lee HJ, et al. 2018. Emergence of synthetic mRNA: In vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials 156:172–93 [DOI] [PubMed] [Google Scholar]
- 45.Kedmi R, Veiga N, Ramishetti S, Goldsmith M, Rosenblum D, et al. 2018. A modular platform for targeted RNAi therapeutics. Nat Nanotechnol 13:214–9 [DOI] [PubMed] [Google Scholar]
- 46.Veiga N, Goldsmith M, Granot Y, Rosenblum D, Dammes N, et al. 2018. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat Commun 9:4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, et al. 2019. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat Commun 10:3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu C, Lu Z, Luo Y, Liu Y, Cao Z, et al. 2018. Targeting of NLRP3 inflammasome with gene editing for the amelioration of inflammatory diseases. Nat Commun 9:4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu B, Zhong L, Weng Y, Peng L, Huang Y, et al. 2020. Therapeutic siRNA: state of the art. Signal Transduct Target Ther 5:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang MM, Bahal R, Rasmussen TP, Manautou JE, Zhong XB. 2021. The growth of siRNA-based therapeutics: Updated clinical studies. Biochem Pharmacol:114432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang Y, Cheng Q, Ji JL, Zheng S, Du L, et al. 2016. Pharmacokinetic Behaviors of Intravenously Administered siRNA in Glandular Tissues. Theranostics 6:1528–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shobaki N, Sato Y, Suzuki Y, Okabe N, Harashima H 2020. Manipulating the function of tumor-associated macrophages by siRNA-loaded lipid nanoparticles for cancer immunotherapy. J Control Release 325:235–48 [DOI] [PubMed] [Google Scholar]
- 53.Veiga N, Goldsmith M, Diesendruck Y, Ramishetti S, Rosenblum D, et al. 2019. Leukocyte-specific siRNA delivery revealing IRF8 as a potential anti-inflammatory target. J Control Release 313:33–41 [DOI] [PubMed] [Google Scholar]
- 54.Kosovrasti VY, Nechev LV, Amiji MM. 2016. Peritoneal Macrophage-Specific TNF-alpha Gene Silencing in LPS-Induced Acute Inflammation Model Using CD44 Targeting Hyaluronic Acid Nanoparticles. Mol Pharm 13:3404–16 [DOI] [PubMed] [Google Scholar]
- 55.Tao W, Yurdagul A Jr., Kong N, Li W, Wang X, et al. 2020. siRNA nanoparticles targeting CaMKIIgamma in lesional macrophages improve atherosclerotic plaque stability in mice. Sci Transl Med 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marques JT, Williams BR. 2005. Activation of the mammalian immune system by siRNAs. Nat Biotechnol 23:1399–405 [DOI] [PubMed] [Google Scholar]
- 57.Sioud M 2007. RNA interference and innate immunity. Adv Drug Deliv Rev 59:153–63 [DOI] [PubMed] [Google Scholar]
- 58.Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. 2005. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol 23:457–62 [DOI] [PubMed] [Google Scholar]
- 59.Morrissey DV, Lockridge JA, Shaw L, Blanchard K, Jensen K, et al. 2005. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat Biotechnol 23:1002–7 [DOI] [PubMed] [Google Scholar]
- 60.Forterre A, Komuro H, Aminova S, Harada M. 2020. A Comprehensive Review of Cancer MicroRNA Therapeutic Delivery Strategies. Cancers (Basel) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Curtale G, Rubino M, Locati M. 2019. MicroRNAs as Molecular Switches in Macrophage Activation. Front Immunol 10:799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu L, Yi H, He H, Pan H, Cai L, Ma Y. 2017. Tumor associated macrophage-targeted microRNA delivery with dual-responsive polypeptide nanovectors for anti-cancer therapy. Biomaterials 134:166–79 [DOI] [PubMed] [Google Scholar]
- 63.Parayath NN, Parikh A, Amiji MM. 2018. Repolarization of Tumor-Associated Macrophages in a Genetically Engineered Nonsmall Cell Lung Cancer Model by Intraperitoneal Administration of Hyaluronic Acid-Based Nanoparticles Encapsulating MicroRNA-125b. Nano Lett 18:3571–9 [DOI] [PubMed] [Google Scholar]
- 64.Tran TH, Krishnan S, Amiji MM. 2016. MicroRNA-223 Induced Repolarization of Peritoneal Macrophages Using CD44 Targeting Hyaluronic Acid Nanoparticles for Anti-Inflammatory Effects. PLoS One 11:e0152024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bejerano T, Etzion S, Elyagon S, Etzion Y, Cohen S. 2018. Nanoparticle Delivery of miRNA-21 Mimic to Cardiac Macrophages Improves Myocardial Remodeling after Myocardial Infarction. Nano Lett 18:5885–91 [DOI] [PubMed] [Google Scholar]
- 66.Nguyen MA, Wyatt H, Susser L, Geoffrion M, Rasheed A, et al. 2019. Delivery of MicroRNAs by Chitosan Nanoparticles to Functionally Alter Macrophage Cholesterol Efflux in Vitro and in Vivo. ACS Nano 13:6491–505 [DOI] [PubMed] [Google Scholar]
- 67.Perez CR, De Palma M. 2019. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun 10:5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saxena M, Bhardwaj N. 2018. Re-Emergence of Dendritic Cell Vaccines for Cancer Treatment. Trends Cancer 4:119–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cornel AM, van Til NP, Boelens JJ, Nierkens S. 2018. Strategies to Genetically Modulate Dendritic Cells to Potentiate Anti-Tumor Responses in Hematologic Malignancies. Frontiers in Immunology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luo YL, Liang LF, Gan YJ, Liu J, Zhang Y, et al. 2020. An All-in-One Nanomedicine Consisting of CRISPR-Cas9 and an Autoantigen Peptide for Restoring Specific Immune Tolerance. Acs Appl Mater Inter 12:48259–71 [DOI] [PubMed] [Google Scholar]
- 71.Zhang Y, Shen S, Zhao G, Xu CF, Zhang HB, et al. 2019. In situ repurposing of dendritic cells with CRISPR/Cas9-based nanomedicine to induce transplant tolerance. Biomaterials 217. [DOI] [PubMed] [Google Scholar]
- 72.Katakowski JA, Mukherjee G, Wilner SE, Maier KE, Harrison MT, et al. 2016. Delivery of siRNAs to Dendritic Cells Using DEC205-Targeted Lipid Nanoparticles to Inhibit Immune Responses. Molecular Therapy 24:146–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Van den Eynde BJ, van Baren N, Baurain JF. 2020. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annu Rev Canc Biol 4:241–56 [Google Scholar]
- 74.Zou SL, Tong QY, Liu BW, Huang W, Tian Y, Fu XH. 2020. Targeting STAT3 in Cancer Immunotherapy. Mol Cancer 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sharma J, Larkin J. 2019. Therapeutic Implication of SOCS1 Modulation in the Treatment of Autoimmunity and Cancer. Front Pharmacol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang H, Jiang CT, Shen S, Liu A, Gang YJ, et al. 2019. Nanoenabled Reversal of IDO1-Mediated Immunosuppression Synergizes with Immunogenic Chemotherapy for Improved Cancer Therapy. Nano Letters 19:5356–65 [DOI] [PubMed] [Google Scholar]
- 77.Luo ZC, Wang C, Yi HQ, Li P, Pan H, et al. 2015. Nanovaccine loaded with poly I:C and STAT3 siRNA robustly elicits anti-tumor immune responses through modulating tumor-associated dendritic cells in vivo. Biomaterials 38:50–60 [DOI] [PubMed] [Google Scholar]
- 78.Heo MB, Cho MY, Lim YT. 2014. Polymer nanoparticles for enhanced immune response: Combined delivery of tumor antigen and small interference RNA for immunosuppressive gene to dendritic cells. Acta Biomaterialia 10:2169–76 [DOI] [PubMed] [Google Scholar]
- 79.Gu JJ, Yang SD, Ho EA 2015. Biodegradable Film for the Targeted Delivery of siRNA-Loaded Nanoparticles to Vaginal Immune Cells. Mol Pharmaceut 12:2889–903 [DOI] [PubMed] [Google Scholar]
- 80.Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 2020. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nature Reviews Drug Discovery 19:185–99 [DOI] [PubMed] [Google Scholar]
- 81.Moffett HF, Coon ME, Radtke S, Stephan SB, McKnight L, et al. 2017. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat Commun 8:389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji WH, et al. 2017. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nature Nanotechnology 12:813–+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Parayath NN, Stephan SB, Koehne AL, Nelson PS, Stephan MT. 2020. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nature Communications 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim SS, Peer D, Kumar P, Subramanya S, Wu HQ, et al. 2010. RNAi-mediated CCR5 Silencing by LFA-1-targeted Nanoparticles Prevents HIV Infection in BLT Mice. Molecular Therapy 18:370–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou JH, Neff CP, Liu XX, Zhang J, Li HT, et al. 2011. Systemic Administration of Combinatorial dsiRNAs via Nanoparticles Efficiently Suppresses HIV-1 Infection in Humanized Mice. Molecular Therapy 19:2228–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Peer D, Park EJ, Morishita Y, Carman CV, Shimaoka M. 2008. Systemic leukocyte-directed siRNA delivery revealing cyclin D1 as an anti-inflammatory target. Science 319:627–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Masjedi A, Ahmadi A, Ghani S, Malakotikhah F, Nabi Afjadi M, et al. 2020. Silencing adenosine A2a receptor enhances dendritic cell-based cancer immunotherapy. Nanomedicine 29:102240. [DOI] [PubMed] [Google Scholar]
- 88.Breckpot K, Escors D, Arce F, Lopes L, Karwacz K, et al. 2010. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J Virol 84:5627–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.DePolo NJ, Reed JD, Sheridan PL, Townsend K, Sauter SL, et al. 2000. VSV-G pseudotyped lentiviral vector particles produced in human cells are inactivated by human serum. Mol Ther 2:218–22 [DOI] [PubMed] [Google Scholar]
- 90.White M, Whittaker R, Gandara C, Stoll EA. 2017. A Guide to Approaching Regulatory Considerations for Lentiviral-Mediated Gene Therapies. Hum Gene Ther Methods 28:163–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Patil S, Gao YG, Lin X, Li Y, Dang K, et al. 2019. The Development of Functional Non-Viral Vectors for Gene Delivery. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]