

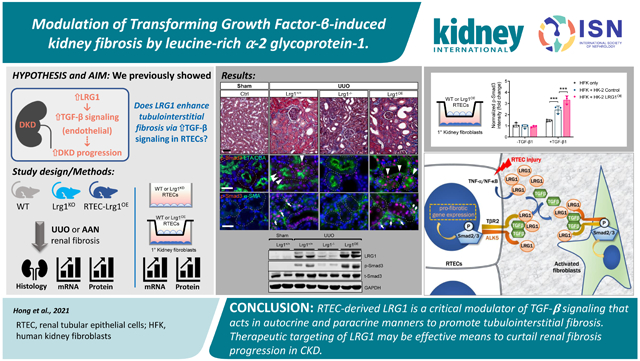
Abstract
Kidney fibrosis is considered the final convergent pathway for progressive chronic kidney diseases, but there is still a paucity of success in clinical application for effective therapy. We recently demonstrated that the expression of secreted leucine-rich α-2 glycoprotein-1 (LRG1) is associated with worsened kidney outcomes in patients with type 2 diabetes and that LRG1 enhances endothelial transforming growth factor-β signaling to promote diabetic kidney disease progression. While the increased expression of LRG1 was most prominent in the glomerular endothelial cells in diabetic kidneys, its increase was also observed in the tubulointerstitial compartment. Here, we explored the potential role of LRG1 in kidney epithelial cells and TGF-β-mediated tubulointerstitial fibrosis independent of diabetes. LRG1 expression was induced by tumor necrosis factor-α in cultured kidney epithelial cells and potentiated TGF-β/Smad3 signal transduction. Global Lrg1 loss in mice led to marked attenuation of tubulointerstitial fibrosis in models of unilateral ureteral obstruction and aristolochic acid fibrosis associated with concomitant decreases in Smad3 phosphorylation in tubule epithelial cells. In mice with kidney epithelial cell-specific LRG1 overexpression, while no significant phenotypes were observed at baseline, marked exacerbation of tubulointerstitial fibrosis was observed in the obstructed kidneys. This was associated with enhanced Smad3 phosphorylation in both kidney epithelial cells and α-smooth muscle actin-positive interstitial cells. Co-culture of kidney epithelial cells with primary kidney fibroblasts confirmed the potentiation of TGF-β-1mediated Smad3 activation in kidney fibroblasts through epithelial-derived LRG1. Thus, our results indicate that enhanced LRG1 expression induced epithelial injury is an amplifier of TGF-β signaling in autocrine and paracrine manners promoting tubulointerstitial fibrosis. Hence, therapeutic targeting of LRG1 may be an effective means to curtail kidney fibrosis progression in chronic kidney disease.
Keywords: renal fibrosis, TGF-β, Smad3, renal tubular epithelial cells
Graphical Abstract
INTRODUCTION
Although much has been learned of the molecular mechanisms underlying renal fibrogenesis, there is still a paucity of success in translating this knowledge to clinical application 1, 2, and the need for a better understanding of molecular mechanisms remains for effective therapeutic targeting for renal fibrosis. The central role of transforming growth factor- β (TGF-β) in the initiation and progression of renal fibrosis is well established 3, 4, which exerts pleiotropic effects through the activation of canonical Smad-dependent and noncanonical Smad-independent pathways. In particular, the blockade of TGF-β/Smad3 pathway has been demonstrated to significantly attenuate fibrosis in animal models of kidney disease 5, 6. Nevertheless, given the multifaceted effects of TGF-β signaling that include the regulation of inflammation and vascular development, a complete blockade of TGF-β may not be an ideal therapeutic approach.
The potency and the specificity of TGF-β signaling are also regulated by cell surface co-receptors and secreted molecules that interact with TGF-β and cognate receptors 7 to amplify or dampen the signaling elicited in a cell-specific manner. For example, endoglin, which is preferentially expressed on endothelial cells, acts as an accessory protein for endothelial TGF-β signaling via ALK1-Smad1/5 signaling 8-10. In this context, TGF-β signaling is further amplified by leucine-rich alpha-2 glycoprotein 1 (LRG1), a secreted glycoprotein that was shown to facilitate the recruitment of TGF-β with both endoglin and TGF-β receptors 11. On the other hand, BMP and activin membrane-bound inhibitor (BAMBI) acts as a decoy receptor to antagonize TGF-β signaling in a context-dependent manner 12. Therefore, therapeutic targeting of modifiers of TGF-β signaling may be a viable approach in dampening excessive and uncontrolled TGF-β signaling in kidney disease without complete abrogation of the necessary homeostatic function of TGF-β. In support of this notion, we recently demonstrated that the genetic loss of TGF-β signaling modifiers can have profound effects on the progression of DKD in two independent studies: 1) We showed that BAMBI expression is reduced in DKD and that the genetic ablation of BAMBI enhanced TGF-β signaling in glomerular cells to heighten podocyte loss and endothelial cell activation to accelerate DKD progression 13, 14; and 2) conversely, we also showed that the expression of LRG1 is significantly enhanced in endothelial cells in DKD15, 16 and that its loss curtailed endothelial activation and injury and DKD progression in mice. These results suggested that the manipulation of TGF-β signaling modifiers, such as LRG1 and BAMBI, may be a feasible approach to attenuate TGF-β-mediated fibrosis progression in vivo. Because our previous study indicated that the increased LRG1 expression in human and mouse diabetic kidneys was not limited to the glomerular endothelium, but observed also in the tubulointerstitial compartment 16, and LRG1 was shown to interact with ALK5 type 1 receptor (TGFBR1) in addition to endothelial-restricted ALK1 receptor (ACVRL1) 11, we posited that LRG1 may influence the canonical TGF-β/Smad3 signaling in renal tubular epithelial cells (RTECs) to promote renal fibrosis. Therefore, in this study, we examined the effects of global loss and RTEC-specific overexpression of LRG1 in the setting of tubular injury and its influence on renal fibrosis progression.
METHODS
Study Approval
All mouse procedures were performed according to the protocol approved by the Institutional Animal Care and Use Committee at Icahn School of Medicine at Mount Sinai (IACUC-2018-0033).
Mouse Models
Mice were housed in a specific pathogen-free facility with free access to chow and water and a 12-hour day-to-night cycle.
Lrg1 mouse models:
Lrg1−/− mouse strain in C57BL/6J background was obtained from the Knock-Out Mouse Project (KOMP) Repository (www.komp.org; #VG10067). Genotyping was performed as described previously 16. For inducible LRG1 transgenic mice, the full-length mLrg1 ORF (NM_029796.2) with C-terminal V5 tag was subcloned into pTRE-tight vector (Takara Bio, Mountain View, CA). The sequence and orientation of the TRE-Tight-Lrg1-V5 insert were confirmed by restriction endonuclease and DNA sequencing. The DNA fragment encoding TRE-Tight-Lrg1-V5 was used for microinjection to generate the TRE-LRG1 transgenic mice in the FVB/NJ background. TRE-LRG1 mice were crossed with Pax8-rtTA mice (in FVB/NJ) to generate Pax8-rtTA;TRE-mLrg1 mice. Tubular overexpression of LRG1 was validated by quantitative PCR and western blot analysis of kidney cortices after 3 weeks of doxycycline-supplemented chow feeding (625mg/kg, Envigo). For the induction of LRG1 overexpression for fibrosis models, mice were given doxycycline-supplemented chow 1 week before UUO surgery or AAN injection and for the experimental duration. Phenotypes of Pax8-rtTA;TRE-mLrg1 mice without doxycycline induction and control littermates expressing either Pax8-rtTA or TRE-mLrg1 transgene with doxycycline induction were indistinguishable from normal mouse kidneys and used interchangeably as wildtype (WT) controls.
UUO model:
UUO model was established by ligation of the left ureter as described previously 17. The contralateral sham-operated kidneys were used as controls. All mice were euthanized at 10 days post-surgery.
AAN model:
AAN was established in mice at 7-8 weeks of age with an intraperitoneal injection of aristolochic acid I (Sigma-Aldrich, 3 mg/kg in PBS) once every two days for 3 weeks, followed by 3 weeks of recovery, as described previously 18. PBS-injected mice were used as controls, and all mice were euthanized after 6 weeks post-injection for analysis.
BUN measurement
Blood urea nitrogen (BUN) levels were measured using a commercially available kit according to the manufacturer's instruction (Bioassay Systems, Hayward, CA).
Kidney histology
Kidney samples were fixed in 10% formalin, embedded in paraffin, and sectioned to 4-μm thickness. Renal fibrosis was evaluated histologically by picrosirius red (#ab150681, Abcam, Danvers, MA) and Masson’s trichrome (#87019, Thermo Scientific, Waltham, MA) staining on paraffin slides of mouse kidney tissues according to the manufacturer’s instructions. Ten or more non-overlapping fields were randomly selected and imaged per mouse. Renal fibrosis was quantified using ImageJ software (National Institutes of Health, Bethesda, MD), and expressed as an average percentage of fibrotic area relative to the total area (positively stained areas of glomeruli, Bowman's capsules, or arterial vessels were excluded from the analysis).
Statistical analysis
Data are expressed as mean±SD. For comparisons of means between two groups, two-tailed, unpaired t-tests were performed. For comparison of means between three or more groups, 1-way or 2-way ANOVA with Tukey’s post hoc test was applied. Statistical significance was achieved when P<0.05. Prism software (GraphPad, La Jolla, CA) was used for statistical analysis.
Additional detailed methods are included in the Supplemental Methods.
RESULTS
LRG1 expression is increased in RTECs in tubular injury and CKD
We previously showed that LRG1 expression is increased in glomerular and tubulointerstitial compartments of mouse and human diabetic kidneys 16. A recent study also reported the enhanced LRG1 expression in injured proximal and distal tubules of mice with albumin overload-induced kidney damage 19. Therefore, we further examined the expression of LRG1 in human kidneys in CKD using a publicly available RNA sequencing (RNAseq) dataset from Nephroseq (nephroseq.org). Indeed, the expression of LRG1 was increased five-fold in LRG1 mRNA expression in the micro-dissected tubulointerstitial compartments from renal biopsy samples of DN versus those from healthy living donors (HLD) (Fig. 1A). Similarly, greater than six-fold increase was observed in tubulointerstitial LRG1 expression in focal segmental glomerulosclerosis (Fig. 1B), and a three-fold increase in lupus nephritis in comparison to HLD (Fig. 1C).
Figure 1. LRG1 expression is increased in tubulointerstitium in CKD and models of renal fibrosis.

(A-C) Median-centered Log2 hLRG1 mRNA expression in the microdissected tubulointerstitial specimen from renal biopsy samples of healthy living donors (HLD) versus diabetic nephropathy (DN), minimal change disease (MCD), lupus nephritis (LN), or focal segmental glomerulosclerosis (FSGS) from Nephroseq database (nephroseq.org; ENSG00000171236). **P<0.01 between indicated groups. (D) RNAseq analysis of mLrg1 mRNA expression in control and Tg26 kidney cortex samples at 4, 8, and 12 weeks of age (n=3 mice per group per time point). RPKM, reads per kilobase per million mapped reads. (E) Quantitative PCR analysis of mLrg1 mRNA expression in kidney cortices of sham control or UUO kidneys at days 3 and 10 post-surgery. (F) Representative images of in situ RNA hybridization in sham and UUO kidneys at 3 days post-surgery (D3) and in control and aristolochic-acid nephropathy (AAN) using Lrg1 (blue) and Krt18 (red) probes. Scale bars, 20μm (top panels) and 50μm (bottom panels). Quantitative scoring of Lrg1 mRNA signal is shown on the right (fold change relative to control; n=30 fields per group, consisting of 3 mice and 10 fields per mouse evaluated). *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 between indicated groups by unpaired, two-tailed t-test.
We next examined the LRG1 expression in mouse models of HIV-associated nephropathy (HIVAN), Tg26, which develops severe renal injury with progressive fibrosis development 20. RNAseq of serial kidney biopsies performed at 4, 8, and 12 weeks of age in control and Tg26 mice showed increasing Lrg1 transcript that correlated with the kidney disease progression (Fig. 1D). Similarly, quantitative PCR analysis of kidney cortices of mice with unilateral ureteral obstruction (UUO) showed a progressive increase in Lrg1 mRNA expression when examined at 3- or 10-days post-surgery in comparison to sham-surgery controls (Fig. 1E). To localize the increased LRG1 in the tubulointerstitial compartment in tubular injury settings, we performed in situ mRNA hybridization analysis on previously collected mouse kidneys after 3 days post-UUO surgery, as well as those with aristolochic acid-induced nephropathy (AAN)18. A scrambled probe was used as a negative control (data not shown for space limitation), and the probe for Keratin-18, upregulated in injured tubules 21, was used as a positive control. Lrg1 expression was relatively low in most kidney cells in normal kidneys, but it significantly increased in UUO and AAN kidneys (Fig. 1F-G). Similar to Krt18, the enhanced Lrg1 expression was detected largely in tubular cells, with some detection in the interstitial area (Fig. 1F).
Previous reports have shown that LRG1 expression is increased in various tissues at sites of inflammation 22-25 and that its expression can be induced by inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-β (IL-1β) 11, 26. Querying of putative transcription factors that regulate LRG1 expression using ChIP Enrichment Analysis (ChEA) and ENCODE (accessed from Harmonizomes database 27) showed four overlapping transcription factors between the two datasets (Fig. 2A), one of which was NF-κB p65 subunit (Rel A). Chromatin immunoprecipitation (ChIP) assay confirmed the binding of NF-κB p65 to LRG1 promoter in cultured primary mouse RTECs stimulated with TNF-α (Fig. 2B). Indeed, TNF-α treatment led to ~3-fold increase in LRG1 mRNA and protein expression in both primary mouse RTECs and conditionally immortalized human RTECs, HPT-1b cells 28, 29 (Fig. 2C-D). Therefore, these results suggest that while the basal expression of LRG1 is relatively low in normal kidneys, its expression is induced in injured kidney cells, in part through inflammatory cytokines, such as TNF-α, in disease settings.
Figure 2. TNF-α upregulates LRG1 expression in RTECs in vitro.

(A) Venn diagram of transcription factors predicted to bind to LRG1 promoter region using ENCODE and CHEA tools (obtained from https://maayanlab.cloud/Harmonizome/gene/LRG1). CEBPB, ESR1, RELA1, and SPI1 were common between 84 TFs predicted by ENCODE and 15 TFs predicted by CHEA. (B) Chromatin-bound proteins were immunoprecipitated with control immunoglobin (IgG) or anti-p65 antibody in primary mouse RTECs after TNF-α (10ng/mL for 24 hours) stimulation. Co-precipitating DNA fragments were analyzed by quantitative PCR, and results are shown as normalized values to total input (n=3 samples). ****p<0.0001 between indicated groups by 2-way ANOVA with Tukey’s post hoc test. (C) Quantitative PCR analysis of LRG1 mRNA in differentiated HPT-1b cells (hLRG1) and primary mouse RTECs (mLrg1) with or without TNF-α stimulation. **p<0.01 between indicated groups by unpaired, two-tailed t-test. (D) Representative western blot of LRG1 protein in primary mouse RTECs after TNF-α stimulation. Densitometric analysis of LRG1 protein normalized to β-actin is shown on the right as fold change relative to control. **p<0.01 between indicated groups by unpaired, two-tailed t-test.
LRG1 expression potentiates TGF-β/Smad3 signaling in RTECs
Various studies have previously demonstrated that LRG1 amplifies TGF-β signaling in various cell types and tissue contexts 11, 30, 31. In the context of diabetic kidney disease, we showed that increased LRG1 expression significantly augments the glomerular endothelial cell activation and proliferation via TGF-β-induced ALK1-Smad1/5 activation 16. However, ALK1 (or ACVRL1) expression is largely restricted to endothelial cells32, 33, in contrast to the ubiquitously expressed ALK5 (or TGFBR1) that typically activates Smad2/3 signaling. Therefore, we posited that the increased LRG1 expression in RTECs, which predominantly expresses ALK5, would lead to amplified TGF-β/Smad3 signaling. To test this in vitro, we utilized primary mouse RTECs with stable expression of short hairpin (shRNA) sequence against Lrg1 (shLrg1) 16, which showed significantly reduced levels of secreted LRG1 in the supernatant and total cell LRG1 protein compared to control or RTECs expressing scrambled shRNA sequence (shScr) (Fig. 3A-C). Treatment of cells with TGF-β1 (5ng/mL) led to rapid phosphorylation of Smad3 in shLrg1 and shScr RTECs, but its level was significantly attenuated in shLrg1 cells in comparison to shScr controls (Fig. 3B-C). As anticipated, the phosphorylation status of Smad1/5 remained unchanged irrespective of TGF-β1 stimulation in either shScr or shLrg1 RTECs (Fig. 3B-C). Consistent with these findings, reduced expression of extracellular matrix and profibrotic genes were observed in shLrg1 RTECs in comparison to the shScr RTECs in presence of TGF-β (Fig. 3D).
Figure 3. LRG1 potentiates the TGF-β/Smad3-signaling in RTECs in vitro.

Immortalized human proximal tubular HK-2 cells were stably transduced with lentivirus expressing shRNA against LRG1 (shLRG1) or scrambled control shRNA (shScr). (A) The concentration of secreted LRG1 protein in supernatants of cultured HK-2 cells by ELISA. ****p<0.0001 between indicated groups by 1-way ANOVA with Tukey’s post hoc test (n=6 in each group). (B) shScr and shLrg1 cells were treated with TGF-β1 (5ng/mL), and lysates were examined at 10 or 30 minutes post-treatment with antibodies as indicated (n=3 experiments; representative blots are shown). (C) Quantification of western blot analysis in (B) for LRG1 (normalized to β-actin), p-Smad3 and p-Smad1/5 (normalized to respective total Smad proteins). ****p<0.0001 when compared between indicated groups by 1-way ANOVA with Tukey’s post hoc test. (D) Control and shRNA knockdown cells were treated with or without TGF-β (5ng/mL) for 24 hours. Quantitative PCR analysis of TGF-β-responsive genes as relative fold change to control cells. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 when compared between indicated groups by 2-way ANOVA with Tukey’s post hoc test.
LRG1 loss attenuates UUO- and AA-induced renal fibrosis
We next examined the effects of LRG1 deficiency in the renal fibrosis progression after unilateral ureteral obstruction (UUO) using Lrg1−/− mice. Lrg1−/− mice are viable and without gross defects or kidney function deficits 11, 16. Histological analysis showed significant tubular dilatation and atrophy, and interstitial inflammation in the obstructed kidneys of both Lrg1+/+ and Lrg1−/− mice in comparison to the sham-operated contralateral kidneys (Fig. 4A, top panel). However, these changes were much milder Lrg1−/− mice in comparison to Lrg1+/+ mice (Fig. 4A, top panel). The fibrosis evaluation by Masson’s trichrome (MTC) and picrosirius red (PSR) staining, and collagen 1 (COL1A1) immunostaining confirmed a marked reduction in extracellular matrix accumulation in the obstructed kidneys of Lrg1−/− mice (Fig. 4A-B). Consistent with dampened TGF-β signaling in Lrg1−/− UUO kidneys, we also observed a reduction in mRNA expression of collagen 1, fibronectin, and α-SMA (Fig. 4C). Moreover, as TGF-β signaling results in its autoinduction 34-36, consistent with dampened TGF-β signaling in Lrg1−/− UUO kidneys, a reduced expression of Tgfb1 was also detected in Lrg1−/− UUO kidneys in comparison to Lrg1+/+ UUO kidneys (Fig. 4C).
Figure 4. Genetic ablation of LRG1 attenuates UUO-induced renal fibrosis.

UUO or sham surgery was performed in Lrg1+/+ and Lrg1−/− mice (n=8 mice in each group). All mice were euthanized 10 days after surgery. (A) Representative images of periodic acid Schiff (PAS)-, picrosirius red (PSR)-, and Masson’s trichrome (MTC)-staining and collagen 1 (COL1A1) immunostaining of mouse kidneys. Scale bars, 50μm. (B) Quantification of the average percentage of PSR and MTC-stained area and COL1 staining intensity (in arbitrary units) are shown per mouse (n=5 sham, 8 UUO kidneys with 10 or more fields per kidney analyzed). (C) Quantitative PCR analysis of TGF-β-induced gene expression of collagen 1 (Col1a1), α-SMA (Acta2), fibronectin (Fn1), and Tgfb1 is shown as a relative fold change to sham-operated Lrg1+/+ control (n=5 mice per group). *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 when compared between indicated groups by 2-way ANOVA with Tukey’s post hoc test.
To corroborate the above findings and to examine the effect of LRG1 loss in renal function, we utilized a CKD model of AAN, in which RTECs are the primary target of aristolochic acid-induced toxicity 18, 37, 38. Lrg1+/+ and Lrg1−/− mice were subjected to AA or PBS injections for three weeks and allowed to recover for three additional weeks. After 6 weeks of first exposure to AA, significant tubular damage and inflammatory cell infiltration were evident in kidneys of both groups in comparison to control mice (Fig. 5A); but these were all significantly attenuated in Lrg1−/− versus Lrg1+/+ AAN kidneys. Similarly, renal fibrosis, as assessed by Masson’s trichrome, picrosirius staining, and by collagen 1 immunostaining showed significant attenuation in Lrg1−/− mice than in Lrg1+/+ mice (Fig. 5A-B). Consistent with these histologic changes, a diminution of blood urea nitrogen (BUN) levels was observed in Lrg1−/− AAN mice in comparison to Lrg1+/+ AAN mice (Fig. 5C), and expression of Col1a1, Fn1, Acta2, and Tgfb1 were all reduced in Lrg1−/− AAN kidneys (Fig. 5D). These results corroborate the finding that LRG1 deficiency mitigates tubular injury and renal fibrosis to improve renal function in vivo.
Figure 5. Genetic ablation of LRG1 attenuates renal fibrosis and improves kidney function in mice with AAN.

Aristolochic acid-nephropathy (AAN) was induced in Lrg1+/+ and Lrg1−/− mice (n=5 control, n=6 AAN mice per group). All mice were euthanized 6 weeks post AA or vehicle injection. (A) Representative images of periodic acid Schiff (PAS)-, picrosirius red (PSR)-, and Masson's trichrome (MTC)-staining and collagen 1 (COL1A1) immunostaining of mouse kidneys. Scale bars, 50μm. (B) Quantification of the average percentage of PSR and MTC-stained area and COL1 staining intensity (in arbitrary units) are shown per mouse (n=5 sham, 8 UUO kidneys with 10 or more fields per kidney analyzed). (C) Blood urea nitrogen (BUN) levels in mice. (D) Quantitative PCR analysis of TGF-β-induced gene expression of collagen 1 (Col1a1), α-SMA (Acta2), fibronectin (Fn1), and Tgfb1 is shown as a relative fold change to sham-operated Lrg1+/+ control (n=5 mice per group). **p<0.01, ***p<0.001 and ****p<0.0001 when compared between indicated groups by 2-way ANOVA with Tukey’s post hoc test.
The RTEC-specific LRG1 overexpression exacerbates UUO-induced injury and renal fibrosis
Given that LRG1 expression is increased in RTECs in kidney injury and disease, we next explored how RTEC-specific increase in LRG1 would influence TGF-β signaling and renal fibrosis in vivo. We first generated a mouse model with a tetracycline-inducible LRG1 expression specifically in the RTECs (Pax8-rtTA;TRE-mLrg1) in the FVB/NJ background, as described in Methods. As shown in Supp. Figure 1A-B, doxycycline induction led to an average of 5-fold increase in LRG1 expression in the kidneys of transgenic mice (referred to as LRG1OE) in comparison to mice without induction. Phenotypes of Pax8-rtTA;TRE-mLrg1 mice without doxycycline induction and control littermates expressing either Pax8-rtTA or TRE-mLrg1 transgene with doxycycline induction were indistinguishable from normal mouse kidneys (data not shown); therefore either group of mice was used as controls (referred to as WT). The RTEC overexpression of LRG1 did not affect the overall kidney architecture (data not shown) or function when examined up to 8 weeks of Dox induction (Supp. Figure 1C). UUO was performed in WT and Lrg1OE mice, and renal fibrosis was examined after 10 days post-surgery. Renal fibrosis assessment by Masson’s trichrome and picrosirius red staining, and collagen 1 immunostaining similarly showed markedly enhanced matrix deposition in the obstructed kidneys of Lrg1OE mice in comparison to those of WT mice (Fig. 6A-B). Consistently, TGF-β-induced fibrosis gene expression was further augmented in the Lrg1OE UUO kidneys in comparison to the WT UUO kidneys (Fig. 6C). Thus, together with the above data in Lrg1−/− mice, these results indicate that LRG1 is a key regulator of renal fibrosis progression in vivo.
Figure 6. Tubular overexpression of LRG1 exacerbates UUO-induced renal fibrosis.

UUO or sham surgery was performed in control (WT) and Lrg1OE mice (n=6 mice in each group). All mice were euthanized 10 days after surgery. (A) Representative images of periodic acid Schiff (PAS)-, picrosirius red (PSR)-, and Masson's trichrome (MTC)-staining and collagen 1 (COL1A1) immunostaining of mouse kidneys. Scale bars, 50μm. (B) Quantification of the average percentage of PSR and MTC-stained area and COL1 staining intensity (in arbitrary units) are shown per mouse (n=5 sham, 8 UUO kidneys with 10 or more fields per kidney analyzed). (C) Quantitative PCR analysis of TGF-β-induced gene expression of collagen 1 (Col1a1), α-SMA (Acta2), fibronectin (Fn1), and Tgfb1 is shown as a relative fold change to sham-operated Lrg1+/+ control (n=5 mice per group). **p<0.01, ***p<0.001 and ****p<0.0001 when compared between indicated groups by 2-way ANOVA with Tukey’s post hoc test.
LRG1 heightens tubulointerstitial TGF-β/Smad3 signaling in the obstructed kidneys
As LRG1 is a secreted glycoprotein, we further examined the autocrine and potential paracrine effects of LRG1 in UUO kidneys. Since our above in vitro data indicated that LRG1 potentiated TGF-β-induced Smad3 activation in RTECs (Fig. 2), we first examined the extent of autocrine effects of RTEC- overexpression of LRG1 in obstructed kidneys. While phosphorylated Smad3 (p-Smad3) was hardly detectable in the control kidneys of all groups, its increase was prominent in the tubulointerstitial compartment in the UUO kidneys of Lrg1+/+, Lrg1−/−, and Lrg1OE mice, particularly in RTECs (Fig. 7A-B), such as those labeled with Lotus tetragonolobus agglutinin for proximal tubules and Dolichos biflorus agglutinin for distal collecting duct cells. However, p-Smad3 expression was much more pronounced in the Lrg1OE UUO kidneys in comparison to Lrg1+/+ UUO kidneys; conversely, p-Smad3 expression was markedly reduced in Lrg1−/− UUO kidneys when compared to the other UUO kidneys (Fig. 7A-B). We further confirmed these findings by western blot analysis of kidney cortices (Fig. 7C-D). Notably, the extent of p-Smad3 expression in Lrg1+/+, Lrg1−/−, and Lrg1OE kidneys corresponded with the expression levels of tubular injury markers, KIM-1 and NGAL (Fig. 8A-B). While KIM-1 and NGAL mRNA and proteins were upregulated in obstructed kidneys of all groups, the highest expression was found in Lrg1OE UUO kidneys, and lowest in Lrg1−/− UUO kidneys. Moreover, a similar pattern of reduced KIM-1 and NGAL was found in the Lrg1−/− AAN kidneys (Supp. Fig. 2A-B). These results are consistent with the enhanced tubular damage and fibrosis associated with increased LRG1 expression locally produced in injured kidneys (Supp. Fig. 3A), as circulating LRG1 levels remained unchanged in UUO or AAN mice (Supp. Fig. 3B), unlike in the diabetic settings 16. We also did not detect a significant change in the Smad1/5 phosphorylation in the kidneys of control and experimental mice (Supp. Fig. 4).
Figure 7. LRG1 enhances the TGFβ-mediated Smad3-activation in the RTECs of UUO kidneys.

(A) Representative images of phospho-Smad3 (p-Smad3) immunostaining on formalin-fixed paraffin-embedded sections and p-Smad3 immunofluorescence (red) co-stained with tubular markers (proximal tubular marker, Lotus tetragonolobus agglutinin (LTA) and collecting duct marker, Dolichos biflorus agglutinin (DBA), both in green) on frozen kidney sections of Lrg1+/+, Lrg1−/−, and Lrg1OE UUO mice. Scale bars, 50μm. Arrowheads show examples of p-Smad3+ tubular cells. (B) Quantification of immunohistochemical p-Smad3 intensity (in arbitrary units). (C) Representative western blot image of LRG1, phosphorylated and total Smad3 in kidneys of Lrg1+/+, Lrg1−/−, and Lrg1OE mice (n=6 mice per group). (D) Densitometric analysis of normalized pSmad3 intensity as a relative fold change to sham control. **p<0.01 and ****p<0.0001 when compared between indicated groups by 2-way ANOVA with Tukey’s post hoc test.
Figure 8: Extent of tubular injury is associated with LRG1 expression in UUO kidneys.

(A) Quantitative PCR analysis of Havcr1 (KIM-1) and Lcn2 (NGAL) expression shown as fold change to WT for individual mice. ***P<0.001 and ****P<0.0001 between indicated groups by 2-way ANOVA with Tukey’s post hoc test. (C) Representative images of KIM-1 and NGAL immunostaining in UUO kidneys. Scale bar, 50μm.
In addition to enhanced matrix accumulation, leukocyte infiltration is another prominent feature in UUO- and AA-induced fibrotic kidneys 39-41. Although the expression of both F4/80+ macrophages/dendritic cells and CD3+ T cells were significantly increased in UUO and AAN kidneys, we did not detect a significant difference in their expression between the diseased kidneys of Lrg1+/+, Lrg1−/−, and Lrg1OE mice (Supp. Fig. 5A, 5C). Similarly, as obstructed kidneys tend to have significant rarefaction of peritubular capillaries 42, no apparent difference was found among the obstructed kidneys of Lrg1+/+, Lrg1−/−, and Lrg1OE mice by CD34 immunostaining 43, 44 (Supp. Fig. 5B). We also did not observe a significant change between AAN kidneys of Lrg1+/+ and Lrg1−/− mice (Supp. Fig. 5B). As these fibrosis models involve an acute tubular injury that leads to significant peritubular capillary rarefaction, a detailed mechanism of RTEC-derived LRG1’s influence on peritubular capillary endothelial cells in promoting renal fibrosis may be better examined in other CKD settings, such as diabetic kidney disease.
RTEC-derived LRG1 enhances TGF-β/Smad3 signaling in kidney fibroblasts
While the increased Smad3 phosphorylation of RTECs was pronounced in the obstructed kidneys of Lrg1+/+ and Lrg1OE mice, it was not limited to the RTECs, but observed also in the interstitial cells (Fig. 7A). Interestingly, the majority of p-Smad3+ interstitial cells co-expressed α-SMA (Fig. 9A), and the alteration in the expression of α-SMA was associated with levels of LRG1 expression between obstructed kidneys of Lrg1+/+, Lrg1−/−, and Lrg1OE mice (Fig. 9A-B). These results suggested that the increased RTEC-derived LRG1 in obstructed kidneys enhances TGF-β signaling in an autocrine and paracrine manner in the injured kidneys.
Figure 9. RTEC-derived LRG1 enhances the TGF-β-mediated Smad3-activation in the interstitial fibroblasts of UUO kidneys.

(A) Representative images of phospho-Smad3 (p-Smad3) immunostaining on formalin-fixed paraffin-embedded kidney sections and p-Smad3 immunofluorescence (red) co-stained with α-smooth muscle actin (α-SMA, green) on frozen kidney sections of Lrg1+/+, Lrg1−/−, and Lrg1OE UUO mice. Scale bars, 50μm. Arrowheads show examples of p-Smad3+, α-SMA+ cells. (B) Quantification of immunohistochemical α-SMA intensity (in arbitrary units). ****p<0.0001 when compared between indicated groups by 2-way ANOVA with Tukey’s post hoc test. (C) Quantitative PCR analysis of LRG1 mRNA expression in human umbilical vein endothelial cells (HUVEC), human proximal tubular cells (HK-2), and primary human kidney fibroblasts (HKF) as a fold change relative to expression in HUVECs. **p<0.01 and ****p<0.0001 when compared between indicated groups by 1-way ANOVA with Tukey’s post hoc test. (D) Concentration of secreted LRG1 protein in supernatants of cultured cells by ELISA. (E) Concentration of secreted LRG1 protein in supernatants of HK-2 cells stably transduced with control lentivector (control) or LRG1 overexpressing lentivector (LRG1OE) by ELISA. (F-I) Control and LRG1OE HK-2 cells grown on transwell inserts were co-cultured with HKF cells. (G) Western blot analysis of Smad3 phosphorylation in HKF lysates after 30 minutes of TGF-β1 stimulation (5ng/mL). Representative data of three independent experiments are shown. (H) Quantification of western blot analysis for p-Smad3 (normalized to t-Smad3). (I) Quantitative PCR analysis of fibrogenic genes in HFK cells co-cultured with HK2 cells. Gene expression is shown as relative fold change to control cells without TGF-β1 stimulation. **p<0.01, ***p<0.001, and ****p<0.0001 when compared between indicated groups by 2-way ANOVA with Tukey’s post hoc test.
To confirm these findings, we carried out an in vitro co-culture experiment using the human proximal tubular cell line, HK-2 cells, and primary human kidney fibroblasts (HKF). We first checked for the level of LRG1 transcript and protein levels in the cultured HK-2 and HKF cells relative to the human umbilical vein endothelial cells (HUVECs) that had been used in previous reports to explore LRG1 function in vitro 11, 45, 46. Quantitative PCR analysis showed a modestly increased LRG1 transcript in HK-2 cells in comparison to HUVECs, but little expression in HKF cells (Fig. 9C). The level of secreted LRG1 protein in the supernatant of the cultured cells also mirrored what we observed with the transcript levels in HUVECs, HK-2 and HKF cells (7.3±1.5, 10.3±2.7, and 0.43±0.5 ng/mL, respectively; Fig. 9D). The comparison of secreted LRG1 in wildtype HK-2 cells (Control) or LRG1 overexpressing HK-2 cells (LRG1OE) showed about a 4-fold increase in LRG1 in LRG1OE cells (36.1±7.6 vs. 9.3±2.7ng/mL, respectively; Fig, 9E). HKF cells without or with co-cultured with either control HK-2 or LRG1OE HK-2 cells were stimulated with TGF-β1 (5ng/mL), which led to enhanced phosphorylation of Smad3 in HKF cells co-cultured with LRG1OE HK-2 in comparison to HKF cultured alone or co-cultured with HK-2 control cells (Fig. 9G-H). Similarly, TGF-β-induced gene expression was further augmented in HKFs co-cultured with LRG1OE HK-2 in comparison to those with control HK-2 cells (Fig. 9I). These further corroborate the above in vivo finding that RTEC-derived LRG1 can augment the TGF-β/Smad3 signaling cascade in RTECs, as wells as the neighboring fibroblasts in a paracrine manner.
DISCUSSION
In our previous report, we demonstrated that LRG1 acts as a novel angiogenic factor that amplifies TGF-β signaling in glomerular endothelial cells through the activation of ALK1-Smad1/5 pathway to promote DKD progression 16. Whether LRG1 likewise would influence TGF-β signaling in other kidney cells in vivo, such as RTECs, had not been determined. Our present study demonstrates the role of LRG1 as a key modulator of TGF-β/Smad3 signal transduction to influence tubulointerstitial fibrosis progression in two models of renal fibrosis: the loss of LRG1 led to a significant diminution of renal fibrosis, whereas its tubular overexpression conversely exacerbated the process. Thus, it remains to be determined whether LRG1 could be a better therapeutic target than the main components of TGF-β signaling (i.e. TGF-β ligand or receptors) against renal fibrosis progression. However, the following aspects of LRG1 function may be more advantageous as a drug target: 1) since LRG1 acts as a key modifier of TGF-β signaling, the inhibition of LRG1 would not completely suppress TGF-β pathway, such that basal level of TGF-β pathway that is required to maintain homeostatic function may be unaffected. In support of this, while the genetic ablation of TGF-β signaling components is lethal in mice (e.g. TGF- β1, ALK1, ALK5, and endoglin), LRG1 ablation in mice does not result in major physiological deficit 11, and we also did not observe any deficits in renal function in Lrg1-null mice when examined up to 9 months of age; 2) the overexpression of LRG1 did not affect TGF-β/Smad3 signaling in absence of TGF-β in cultured RTECs in vitro or kidneys prior to injury in vivo. These results strongly suggest that LRG1 acts to accelerate disease progression through the potentiation of TGF-β signaling axis; 3) our previous and current data indicate that pan-LRG1 inhibition may have a dual benefit to attenuate the glomerular endothelial injury and tubulointerstitial fibrosis in a chronic kidney disease setting, such as DKD; and 4) as a small, secreted molecule that acts on the cell surface in complex with TGF-β signaling components, the pharmacological inhibition of LRG1 may be more feasible than membrane-bound kinases or intracellular proteins.
LRG1 has been shown to increase in plasma and urine of patients with a variety of cancer malignancies 47-50 and inflammatory diseases 24, 51-53. We and others also found that LRG1 is associated with renal outcomes in type 2 diabetic patients 16, 54. As a mechanism of increased LRG1 expression, we found that LRG1 expression is upregulated in response to high glucose conditions in endothelial cells 15, and our present study indicates that its expression in tubular cells is stimulated in response to TNF-α. Thus, it is likely that, in addition to hyperglycemia, the inflammatory cytokines may further upregulate LRG1 in kidney disease settings. Recently, increased urinary LRG1 was reported for IgA nephropathy 55, steroid-resistant nephrotic syndrome 56, and in the mouse model of albumin overload-induced renal injury 19. Since our study demonstrates a salient role of LRG1 in tubulointerstitial fibrosis, it would be important to assess the efficacy of urinary LRG1 detection as a prognostic marker of tubulointerstitial fibrosis progression in future studies. We also demonstrated that RTEC-derived LRG1 acts in an autocrine and paracrine manner to enhance TGF-β signaling in RTECs and myofibroblasts. Whether RTEC-derived LRG1 can act in a paracrine manner to affect other renal cell types (i.e. endothelial cells) in disease settings remains to be investigated.
In conclusion, together with our previous work 16, the findings in this study further underscores the complexities of TGF-β signaling regulation in kidney disease and suggest that the inhibition of LRG1 may confer dual benefit in kidney disease in that it would be anti-angiogenic to attenuate uncontrolled endothelial activation and proliferation in such settings as DKD, and anti-fibrotic in mediating the progression of renal fibrosis.
Supplementary Material
Translational Statement.
Although much has been learned of the molecular mechanisms underlying renal fibrogenesis, there is still a paucity of success in translating this knowledge to clinical application, and the need remains for effective therapeutic targeting for renal fibrosis. Our current study attributes the role of LRG1 as a key amplifier of tubulointerstitial TGF-β/Smad3 signal transduction that promotes renal fibrosis in CKD. As such, full inhibition of LRG1 may effectively reduce TGF-β signaling, without complete blockade of its multifaceted functions in vivo. Therefore, targeting LRG1 may be more advantageous as a therapeutic approach than targeting TGF-β or TGF-β receptors against renal fibrosis progression.
ACKNOWLEDGEMENTS
QH is supported by the National Natural Science Foundation of China (No.81870491, 82070741) and National Key Research and Development Project (No.2018YFE0126600); JCH is supported by NIH/NIDDK R01DK109683, R01DK122980, R01DK129467, P01DK56492, and VA Merit Award I01BX000345; KL is supported by NIH/NIDDK R01DK117913-01 and R01DK129467.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES: The authors have declared that no conflict of interest exists.
SUPPLEMENTARY MATERIAL:
Supplementary information is available on Kidney International's website.
Suppl. Figures 1-5; Supp. Methods and References.
REFERENCES
- 1.Boor P, Sebekova K, Ostendorf T, et al. Treatment targets in renal fibrosis. Nephrol Dial Transplant 2007; 22: 3391–3407. [DOI] [PubMed] [Google Scholar]
- 2.Boor P, Ostendorf T, Floege J. Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol 2010; 6: 643–656. [DOI] [PubMed] [Google Scholar]
- 3.Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol 2016; 12: 325–338. [DOI] [PubMed] [Google Scholar]
- 4.Sureshbabu A, Muhsin SA, Choi ME. TGF-beta signaling in the kidney: profibrotic and protective effects. American journal of physiology Renal physiology 2016; 310: F596–F606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato M, Muragaki Y, Saika S, et al. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest 2003; 112: 1486–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Xia Y, Lin X, et al. Smad3 signaling activates bone marrow-derived fibroblasts in renal fibrosis. Lab Invest 2014; 94: 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heldin CH, Moustakas A. Signaling Receptors for TGF-beta Family Members. Cold Spring Harb Perspect Biol 2016; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. The Journal of biological chemistry 1999; 274: 584–594. [DOI] [PubMed] [Google Scholar]
- 9.Guerrero-Esteo M, Sanchez-Elsner T, Letamendia A, et al. Extracellular and cytoplasmic domains of endoglin interact with the transforming growth factor-beta receptors I and II. The Journal of biological chemistry 2002; 277: 29197–29209. [DOI] [PubMed] [Google Scholar]
- 10.Pomeraniec L, Hector-Greene M, Ehrlich M, et al. Regulation of TGF-beta receptor hetero-oligomerization and signaling by endoglin. Mol Biol Cell 2015; 26: 3117–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Abraham S, McKenzie JAG, et al. LRG1 promotes angiogenesis by modulating endothelial TGF-beta signalling. Nature 2013; 499: 306–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Onichtchouk D, Chen YG, Dosch R, et al. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 1999; 401: 480–485. [DOI] [PubMed] [Google Scholar]
- 13.Fan Y, Li X, Xiao W, et al. BAMBI elimination enhances alternative TGF-beta signaling and glomerular dysfunction in diabetic mice. Diabetes 2015; 64: 2220–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai H, Chen A, Cai H, et al. Podocyte and endothelial-specific elimination of BAMBI identifies differential transforming growth factor-beta pathways contributing to diabetic glomerulopathy. Kidney Int 2020; 98: 601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu J, Wei C, Zhang W, et al. Gene expression profiles of glomerular endothelial cells support their role in the glomerulopathy of diabetic mice. Kidney Int 2018; 94: 326–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong Q, Zhang L, Fu J, et al. LRG1 Promotes Diabetic Kidney Disease Progression by Enhancing TGF-beta-Induced Angiogenesis. J Am Soc Nephrol 2019; 30: 546–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao W, E J, Bao L, et al. Tubular HIPK2 is a key contributor to renal fibrosis. JCI Insight 2020; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan Y, Xiao W, Lee K, et al. Inhibition of Reticulon-1A-Mediated Endoplasmic Reticulum Stress in Early AKI Attenuates Renal Fibrosis Development. J Am Soc Nephrol 2017; 28: 2007–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee H, Fujimoto M, Ohkawara T, et al. Leucine rich alpha-2 glycoprotein is a potential urinary biomarker for renal tubular injury. Biochemical and biophysical research communications 2018; 498: 1045–1051. [DOI] [PubMed] [Google Scholar]
- 20.Fan Y, Wei C, Xiao W, et al. Temporal profile of the renal transcriptome of HIV-1 transgenic mice during disease progression. PLoS One 2014; 9: e93019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Djudjaj S, Papasotiriou M, Bulow RD, et al. Keratins are novel markers of renal epithelial cell injury. Kidney Int 2016; 89: 792–808. [DOI] [PubMed] [Google Scholar]
- 22.Fujimoto M, Serada S, Suzuki K, et al. Leucine-rich alpha2 -glycoprotein as a potential biomarker for joint inflammation during anti-interleukin-6 biologic therapy in rheumatoid arthritis. Arthritis & rheumatology 2015; 67: 2056–2060. [DOI] [PubMed] [Google Scholar]
- 23.Honda H, Fujimoto M, Miyamoto S, et al. Sputum Leucine-Rich Alpha-2 Glycoprotein as a Marker of Airway Inflammation in Asthma. PLoS One 2016; 11: e0162672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serada S, Fujimoto M, Ogata A, et al. iTRAQ-based proteomic identification of leucine-rich alpha-2 glycoprotein as a novel inflammatory biomarker in autoimmune diseases. Ann Rheum Dis 2010; 69: 770–774. [DOI] [PubMed] [Google Scholar]
- 25.Serada S, Fujimoto M, Terabe F, et al. Serum leucine-rich alpha-2 glycoprotein is a disease activity biomarker in ulcerative colitis. Inflamm Bowel Dis 2012; 18: 2169–2179. [DOI] [PubMed] [Google Scholar]
- 26.Shirai R, Hirano F, Ohkura N, et al. Up-regulation of the expression of leucine-rich alpha(2)-glycoprotein in hepatocytes by the mediators of acute-phase response. Biochemical and biophysical research communications 2009; 382: 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rouillard AD, Gundersen GW, Fernandez NF, et al. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford) 2016; 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen P, Chen BK, Mosoian A, et al. Virological synapses allow HIV-1 uptake and gene expression in renal tubular epithelial cells. J Am Soc Nephrol 2011; 22: 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ross MJ, Wosnitzer MS, Ross MD, et al. Role of ubiquitin-like protein FAT10 in epithelial apoptosis in renal disease. J Am Soc Nephrol 2006; 17: 996–1004. [DOI] [PubMed] [Google Scholar]
- 30.Honda H, Fujimoto M, Serada S, et al. Leucine-rich alpha-2 glycoprotein promotes lung fibrosis by modulating TGF-beta signaling in fibroblasts. Physiol Rep 2017; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Urushima H, Fujimoto M, Mishima T, et al. Leucine-rich alpha 2 glycoprotein promotes Th17 differentiation and collagen-induced arthritis in mice through enhancement of TGF-beta-Smad2 signaling in naive helper T cells. Arthritis Res Ther 2017; 19: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lebrin F, Deckers M, Bertolino P, et al. TGF-beta receptor function in the endothelium. Cardiovasc Res 2005; 65: 599–608. [DOI] [PubMed] [Google Scholar]
- 33.Seki T, Yun J, Oh SP. Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circulation research 2003; 93: 682–689. [DOI] [PubMed] [Google Scholar]
- 34.Kim SJ, Angel P, Lafyatis R, et al. Autoinduction of transforming growth factor beta 1 is mediated by the AP-1 complex. Molecular and cellular biology 1990; 10: 1492–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dockrell ME, Phanish MK, Hendry BM. Tgf-beta auto-induction and connective tissue growth factor expression in human renal tubule epithelial cells requires N-ras. Nephron Exp Nephrol 2009; 112: e71–79. [DOI] [PubMed] [Google Scholar]
- 36.Ding Y, Kim S, Lee SY, et al. Autophagy regulates TGF-beta expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol 2014; 25: 2835–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Debelle FD, Nortier JL, De Prez EG, et al. Aristolochic acids induce chronic renal failure with interstitial fibrosis in salt-depleted rats. J Am Soc Nephrol 2002; 13: 431–436. [DOI] [PubMed] [Google Scholar]
- 38.Yang L, Su T, Li XM, et al. Aristolochic acid nephropathy: variation in presentation and prognosis. Nephrol Dial Transplant 2012; 27: 292–298. [DOI] [PubMed] [Google Scholar]
- 39.Anders HJ, Vielhauer V, Frink M, et al. A chemokine receptor CCR-1 antagonist reduces renal fibrosis after unilateral ureter ligation. J Clin Invest 2002; 109: 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eis V, Luckow B, Vielhauer V, et al. Chemokine receptor CCR1 but not CCR5 mediates leukocyte recruitment and subsequent renal fibrosis after unilateral ureteral obstruction. J Am Soc Nephrol 2004; 15: 337–347. [DOI] [PubMed] [Google Scholar]
- 41.Pozdzik AA, Salmon IJ, Husson CP, et al. Patterns of interstitial inflammation during the evolution of renal injury in experimental aristolochic acid nephropathy. Nephrol Dial Transplant 2008; 23: 2480–2491. [DOI] [PubMed] [Google Scholar]
- 42.Ohashi R, Shimizu A, Masuda Y, et al. Peritubular capillary regression during the progression of experimental obstructive nephropathy. J Am Soc Nephrol 2002; 13: 1795–1805. [DOI] [PubMed] [Google Scholar]
- 43.Ligresti G, Nagao RJ, Xue J, et al. A Novel Three-Dimensional Human Peritubular Microvascular System. J Am Soc Nephrol 2016; 27: 2370–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masum MA, Ichii O, Elewa YHA, et al. Local CD34-positive capillaries decrease in mouse models of kidney disease associating with the severity of glomerular and tubulointerstitial lesions. BMC Nephrol 2017; 18: 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Z, Zeng C, Nong Q, et al. Exosomal Leucine-Rich-Alpha2-Glycoprotein 1 Derived from Non-Small-Cell Lung Cancer Cells Promotes Angiogenesis via TGF-beta Signal Pathway. Mol Ther Oncolytics 2019; 14: 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Xu J, Zhang X, et al. TNF-alpha-induced LRG1 promotes angiogenesis and mesenchymal stem cell migration in the subchondral bone during osteoarthritis. Cell Death Dis 2017; 8: e2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, Zhang Y, Qiu F, et al. Proteomic identification of exosomal LRG1: a potential urinary biomarker for detecting NSCLC. Electrophoresis 2011; 32: 1976–1983. [DOI] [PubMed] [Google Scholar]
- 48.Andersen JD, Boylan KL, Jemmerson R, et al. Leucine-rich alpha-2-glycoprotein-1 is upregulated in sera and tumors of ovarian cancer patients. J Ovarian Res 2010; 3: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linden M, Lind SB, Mayrhofer C, et al. Proteomic analysis of urinary biomarker candidates for nonmuscle invasive bladder cancer. Proteomics 2012; 12: 135–144. [DOI] [PubMed] [Google Scholar]
- 50.Furukawa K, Kawamoto K, Eguchi H, et al. Clinicopathological Significance of Leucine-Rich alpha2-Glycoprotein-1 in Sera of Patients With Pancreatic Cancer. Pancreas 2015; 44: 93–98. [DOI] [PubMed] [Google Scholar]
- 51.Ha YJ, Kang EJ, Lee SW, et al. Usefulness of serum leucine-rich alpha-2 glycoprotein as a disease activity biomarker in patients with rheumatoid arthritis. J Korean Med Sci 2014; 29: 1199–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kentsis A, Ahmed S, Kurek K, et al. Detection and diagnostic value of urine leucine-rich alpha-2-glycoprotein in children with suspected acute appendicitis. Ann Emerg Med 2012; 60: 78–83 e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shinzaki S, Matsuoka K, Iijima H, et al. Leucine-rich Alpha-2 Glycoprotein is a Serum Biomarker of Mucosal Healing in Ulcerative Colitis. J Crohns Colitis 2017; 11: 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu JJ, Pek SLT, Ang K, et al. Plasma Leucine-Rich alpha-2-Glycoprotein 1 Predicts Rapid eGFR Decline and Albuminuria Progression in Type 2 Diabetes Mellitus. J Clin Endocrinol Metab 2017; 102: 3683–3691. [DOI] [PubMed] [Google Scholar]
- 55.Kalantari S, Rutishauser D, Samavat S, et al. Urinary prognostic biomarkers and classification of IgA nephropathy by high resolution mass spectrometry coupled with liquid chromatography. PLoS One 2013; 8: e80830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suresh CP, Saha A, Kaur M, et al. Differentially expressed urinary biomarkers in children with idiopathic nephrotic syndrome. Clin Exp Nephrol 2016; 20: 273–283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.