

Summary
Despite the great diversity of vertebrate limb proportion and our deep understanding of the genetic mechanisms that drive skeletal elongation, little is known about how individual bones reach different lengths in any species. Here, we directly compare the transcriptomes of homologous growth cartilages of the mouse (Mus musculus) and bipedal jerboa (Jaculus jaculus), the latter of which has ‘mouse-like’ arms but extremely long metatarsals of the feet. Intersecting gene expression differences in metatarsals and forearms of the two species revealed that about 10% of orthologous genes are associated with the disproportionately rapid elongation of neonatal jerboa feet. These include genes and enriched pathways not previously associated with endochondral elongation as well as those that might diversify skeletal proportion in addition to their known requirements for bone growth throughout the skeleton. We also identified transcription regulators that might act as ‘nodes’ for sweeping differences in genome expression between species. Among these, Shox2, which is necessary for proximal limb elongation, has gained expression in jerboa metatarsals where it has not been detected in other vertebrates. We show that Shox2 is sufficient to increase mouse distal limb length, and a nearby putative cis-regulatory region is preferentially accessible in jerboa metatarsals. In addition to mechanisms that might directly promote growth, we found evidence that jerboa foot elongation may occur in part by de-repressing latent growth potential. The genes and pathways that we identified here provide a framework to understand the modular genetic control of skeletal growth and the remarkable malleability of vertebrate limb proportion.
eTOC Blurb
How did the jerboa get its long feet? Using an interspecies transcriptome approach, Saxena et al. identify genes associated with the disproportionate elongation of jerboa metatarsals compared to mouse. Altogether, they provide a framework to understand the modular genetic control of skeletal growth and the striking malleability of limb proportion.
Introduction
Diversification of limb skeletal proportion from the most primitive four-legged ancestor allowed animals to fly high above land, to travel quickly across its surface, and to arduously burrow beneath. Even in an individual person, skeletal proportion changes from infancy to adulthood such that growth of the arms and legs surpasses the head and torso by a phenomenon known as ‘allometry’. Despite differences in the initial size and subsequent rate and duration of growth of individual bones, loss-of-function mutations in many genes affect growth of all long bones1–3. We therefore do not yet know what subset of genes is responsible for differential growth, let alone how their expression is regulated to allow for modular development and evolution of the forelimb, hindlimb, and of more than a dozen long bones within each limb.
Quantitative trait locus (QTL) analyses and genome-wide association studies (GWAS) can identify genes associated with the variance of limb proportion within populations4–7. However, the variance in length of a particular bone does not compare to the scale of length difference among different bones. Bone lengths in a mouse or human, for example, span an order of magnitude from the short toe bones to the long tibia, but here it is difficult to distinguish genes that might be responsible for differential growth from their association with positional identity (e.g., the roles of different Hox genes to specify the foot and lower leg). An alternative approach would be to directly compare homologous skeletal elements in two or more species with dramatically different proportions. However, large phylogenetic distances can make it difficult to identify phenotypically relevant orthologous gene expression differences among extensive expression level and sequence divergence.
Here, we addressed these challenges using the uniquely suitable lesser Egyptian jerboa, Jaculus jaculus, and the laboratory mouse, Mus musculus, which diverged from a common ancestor ~50 million years ago8. Although the jerboa has since acquired extraordinarily elongated hindlimbs with disproportionately long feet, it retained more ‘mouse-like’ forelimbs that are a valuable control for gene expression divergence in these two species that is unrelated to the evolution of skeletal proportion9. We took advantage of these morphological similarities and differences, and the relatively close phylogenetic relationship between these two rodents, to directly compare the transcriptomes of homologous growth cartilages (metatarsals of the hindfeet and radius/ulna of the forearm). We then intersected these data to identify disproportionate gene expression differences that are associated with disproportionately accelerated metatarsal elongation in the jerboa.
Although a majority of the growth cartilage transcriptome has diverged since the last common ancestor of mice and jerboas, we found that most gene expression has changed to the same extent in both metatarsals and radius/ulna. Expression of only about 10% of genes has evolved in metatarsals independent of radius/ulna. In addition to genes that are expected to directly increase the rate of limb bone elongation (e.g., Shox2), we found differentially expressed genes (e.g., Crabp1, Gdf10, and Mab21L2) that suggest latent growth potential in mouse metatarsals may be de-repressed in jerboa metatarsals. Our data demonstrate the power of an inter-species transcriptomic approach to directly compare similar and extremely divergent structures in order to identify expression differences with relevance to a genetically complex phenotype in the vast background of gene expression divergence arising over macro-evolutionary timescales.
Results and Discussion
Interspecies gene expression differences between metatarsals and between radius/ulna
Establishment of skeletal proportion, whether over developmental or evolutionary time, is a complex and dynamic process influenced by differences in the initial size of embryonic cartilage rudiments that are amplified by allometric differences in the relative rates of elongation and by the timing of senescence whereby growth slows and ultimately stops10–12. At birth, the third metatarsal of jerboa measures already about 55% longer than mouse (3.1 mm versus 2.0 mm) while femur lengths are more similar. In order to achieve the extremely exaggerated proportion of adult jerboa hindlimbs, however, these initial differences are amplified postnatally with jerboa metatarsals elongating about twice as fast as mouse in the first week after birth (Figure 1). Soon following formation of the secondary ossification center, and thus a true ‘growth plate’ by postnatal day 7 (P7), mouse metatarsal elongation begins to slow as femur elongation continues. By contrast, a rapid rate of jerboa hindlimb elongation continues, including metatarsals, with secondary ossification of the metatarsals delayed to about P2113.
Figure 1 |. Jerboa metatarsals elongate disproportionately faster than mouse during neonatal development.
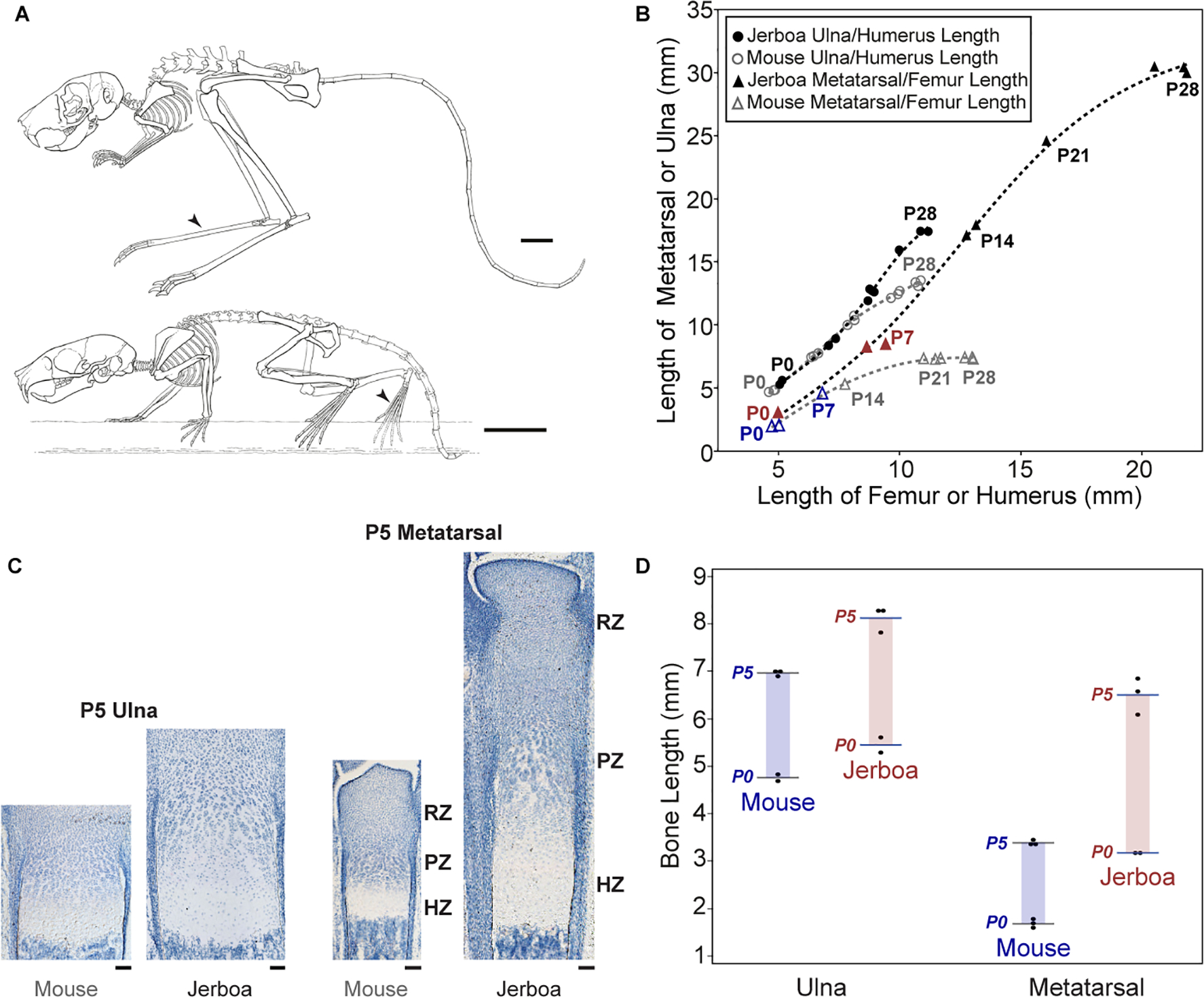
(A) Postural reconstructions of adult jerboa and mouse skeletons adapted from Moore, et al. 2015 (arrowheads point to metatarsals, scalebars = 10 mm) (B) Postnatal (P0 to P28) growth trajectory of metatarsal or ulna (y-axis) relative to femur or humerus (x-axis), respectively. P0 and P7 hindlimb measurements are emphasized in blue for mouse and in red for jerboa (C) Histological sections of P5 distal ulna and metatarsal growth cartilages of mouse and jerboa. Resting (RZ), proliferative (PZ), and hypertrophic (HZ) zones in mouse and jerboa metatarsal are indicated to the right of each growth cartilage (scalebars = 100 μm). (D) Colored bars highlight increase in ulna and metatarsal whole bone lengths from P0 (n=3 mouse and 2 jerboas) to P5 (n=3 each). Individual (dots) and average (line) measurements are shown for each element at P0 and P5. The third (middle) jerboa metatarsal elongates at a rate 2.1-times faster than mouse while the ulna grows 1.2-times faster than mouse.
To identify genes associated with the disproportionately accelerated rate of jerboa metatarsal elongation, we focused here on mouse and jerboa postnatal day 5 (P5). This represents a timepoint soon after the metatarsal growth rate difference increases perinatally and before secondary ossification and senescence alter the composition of mouse metatarsals14. We chose the distal radius/ulna for comparison of growth cartilages that elongate at a more similar rate in the forelimbs of the two species, because metacarpals were too small to dissect with sufficient tissue purity and RNA quality. We performed RNA-Seq on mRNA isolated from the distal metatarsal (MT) and distal radius/ulna (RU) and included both the growth cartilage and surrounding perichondrial membrane, since crosstalk between the two tissues is critical to control endochondral skeletal elongation15.
In order to directly quantify gene expression differences between homologous skeletal elements of the jerboa and mouse, we first annotated a set of 17,464 orthologous genes comprised of at least one exon with no frameshift, nonsense, or splice site mutation using CESAR16 (STAR Methods) and the M. musculus (mm10) and J. jaculus (JacJac1.0) genome assemblies (NCBI). This 1:1 orthologous gene set represents 92.5% of predicted protein-coding genes in the jerboa and 79.8% of annotated mouse genes, with the difference likely due to a higher completeness of the mouse versus jerboa genome assemblies and annotations.
For each of the four sample sets, we sequenced five biological replicates and mapped reads to their respective genome using annotations from our 1:1 orthologous gene set. We performed differential expression analysis using DESeq217 with an additional custom normalization for gene length differences between species (STAR Methods). We then applied a principal component (PC) analysis and found that all samples are primarily separated by species (PC1) and secondarily by growth cartilage type (PC2) (Figure 2A). To reduce the potential effects of technical variation18,19 and to reserve samples for validation of reproducibility below, we selected for our primary analysis three of the five samples from each dataset that clustered most closely.
Figure 2 |. Gene expression profiling identifies 10% of orthologous genes that are associated with disproportionate jerboa metatarsal elongation.
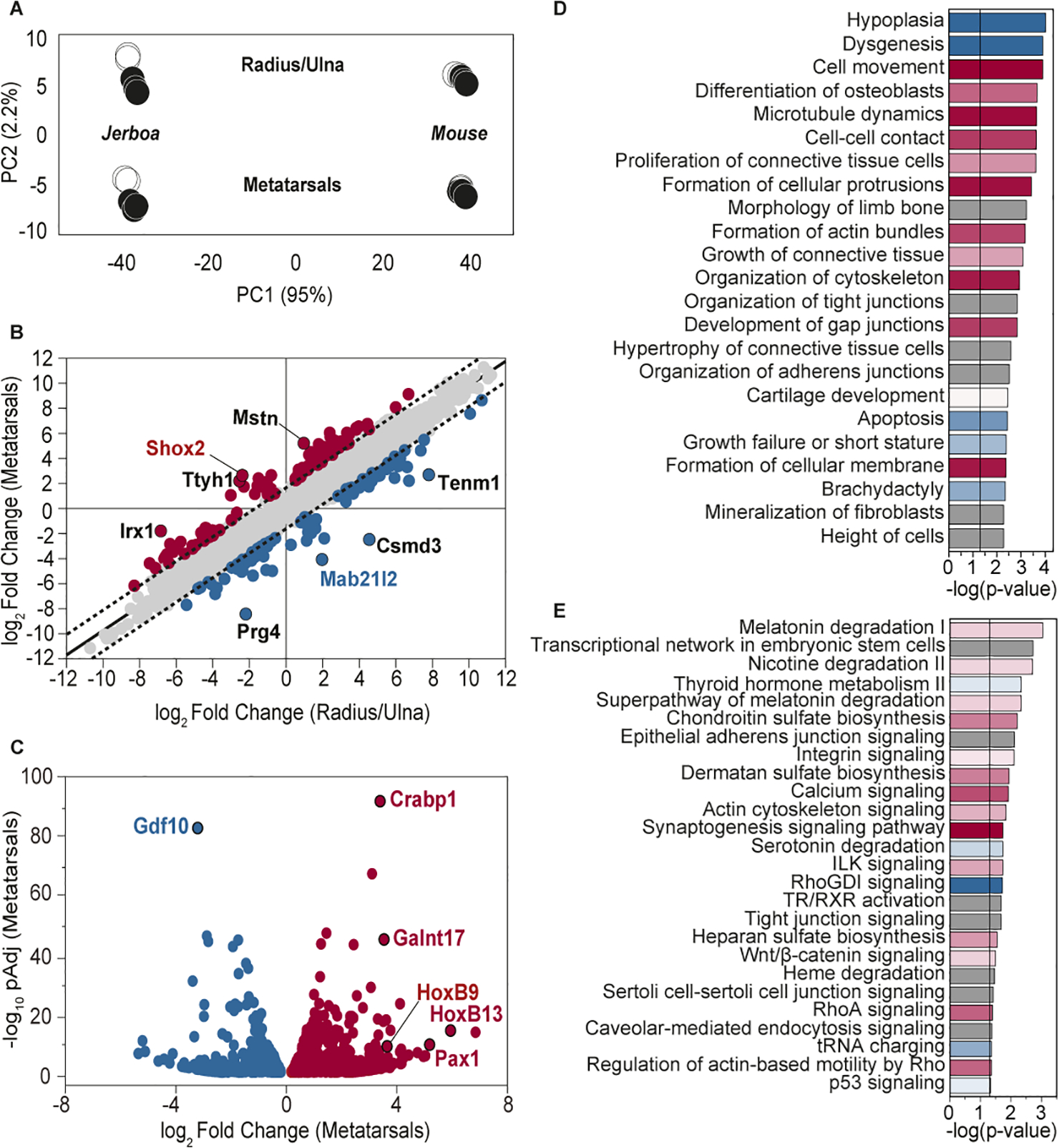
(A) Principal component analysis (PCA) shows that PC1 (species) explains 95% of the variance and PC2 (tissue-type) explains 2.2% of the variance (n=5 each). Black circles denote the three samples used in primary analyses. (B) 8,734 genes are differentially expressed in jerboa compared to mouse both in metatarsals (MT, y-axis) and in radius/ulna (RU, x-axis). Of these, 8,493 are equivalently differentially expressed between species in the MT and RU (grey points) and lie within the 99% confidence interval (dashed line) of the linear fit (solid black line, slope=0.977). 241 genes outside of the confidence interval are non-equivalently differentially expressed in the MT and RU. Eight genes with least-correlated expression differences in MT and RU are labeled. (C) 1,514 genes are significantly differentially expressed between jerboa and mouse MT and not RU. Genes that are expressed higher in jerboa than in mouse MT are denoted red and those that are lower are denoted blue in (B) and (C). (D) A selection of cellular functions, organismal and tissue developmental processes, and developmental disorders of interest to cartilage biology and enriched (padj<0.05) among all 1,755 genes that are associated with disproportionate jerboa MT elongation. (E) Canonical signaling pathways that are significantly enriched among these genes. In (D) and (E), activation of a function or a pathway is indicated in red and inhibition in blue. Grey bars indicate unknown activation status. Bar colour intensity indicates confidence of predicted activation status (z-score). Black vertical lines in (D) and (E) indicate p-value threshold (<0.05). Numerical values for selected annotations in (D) and for all enriched canonical pathways (E) are in Table S6. See also Figure S1 and S2 and Table S1–S4.
Given ~50 million years of evolutionary divergence since the common ancestral expression state, we were not surprised to find that most genes are differentially expressed between jerboa and mouse metatarsals and/or radius/ulna [58.6% and 57.6% of transcripts, respectively, with an adjusted p-value (padj) <0.05] (Tables S1–S2). Of genes differentially expressed in metatarsals, however, a majority (~83%) are equivalently differentially expressed between species in both skeletal elements (Figure 2B; m=0.977; b=0.025; R2=0.930; Table S1). Gene expression in these two growth cartilages of the limb skeleton is therefore highly coordinated, even after extensive evolutionary divergence since the last common ancestor and regardless of exaggerated jerboa hindlimb elongation. This suggests that many gene regulatory mechanisms are common to multiple cartilages, and mutations that altered DNA binding sites or expression of transcription regulators perturbed target gene expression coordinately in metatarsals and radius/ulna. These results illustrate the value of directly comparing divergent and more conserved structures of the same tissue composition, because genes that differ equivalently in both locations cannot explain the disproportionate rate of jerboa metatarsal elongation.
Exclusion of these genes revealed a much smaller set (about 10% of all orthologous genes) that likely contribute to the evolution of skeletal proportion, since expression differences between the two species do not correlate in these two skeletal elements. These include 241 genes with non-equivalent expression differences in both the metatarsals and radius/ulna that could contribute to disproportionate growth rate differences between species at both locations (Figure 2B and Table S1). An additional 1,514 genes differ significantly in expression between the metatarsals of the two species but not between the radius/ulna (Figure 2C and Table S2). These include genes that are not detected in the radius/ulna of either species as well as genes with expression that seems constrained in these forearm elements and divergent in metatarsals (Figure S1). Thus, in total, we identified 1,755 genes that are associated with the disproportionate acceleration of jerboa metatarsal elongation, which provide the foundation for a mechanistic understanding of the modular nature of skeletal proportion.
Traditionally, differential microarray and RNA-Seq analyses have been validated by real-time quantitative reverse transcription PCR (qRT-PCR) of a subset of genes20,21. However, a direct comparison of individual gene expression between species is challenging due to the potential for differences in primer annealing affinities and differences in the expression of reference transcripts used for normalization in each species22. We therefore employed two approaches to verify differential expression, in addition to performing qRT-PCR on select genes (Figure S2F–G).
We first repeated DESeq2 analyses using the two independently collected biological replicates for each growth cartilage that were not included in the primary analysis (Tables S3–S4). Despite the variance between these and the primary samples, there is a strong correlation among the genes that are associated with the independent acceleration of jerboa metatarsal growth, including all genes we discuss in detail below (p=1.7E-106, Fisher’s exact test; Figure S2C). We also assessed the expression of selected transcripts in growth cartilages using RNAScope in situ hybridization, as detailed below. RNAScope is a sensitive technique that can detect single RNA molecules23, and it has been used to validate next-generation expression profiling results in a variety of biological contexts24–27.
Concordant genetic control of the evolution and development of skeletal proportion
It is not known if there are similarities between the genetic mechanisms that diversify limb skeletal proportion over many millions of years and those that establish skeletal proportion during development of an individual and/or that vary proportion in a population. To answer this question, we tested whether there is significant overlap between the genes we identified as associated with the evolution of skeletal proportion and three other datasets that are associated with the development of limb proportion in laboratory rodents or the variance of body proportion in a human population. Although the available datasets represent a narrow slice of phylogeny, such correlations might reveal a common subset of genes in the cartilage growth ‘toolkit’ that are recurrently utilized to establish growth rate differences in the vertebrate limb.
First, an analysis of mouse and rat growth plate RNA-Seq identified differentially expressed genes that are associated with the slowing of tibia growth rate as the growth plate matures in juvenile animals of both species12. Second, the same study also identified differentially expressed genes associated with growth rate differences by location in the rapidly elongating proximal tibia growth plate of young animals versus the slowly elongating distal phalanx of the same individuals. Third, though representing only an indirect measure of skeletal proportion within limbs, the human sitting height ratio (SHR) GWAS identified SNPs associated at a population level with leg length as a proportion of total body length6.
We first identified genes within each of these datasets that were assigned the same gene name in our 1:1 jerboa and mouse orthologous reference set (Table S5). Using our orthologous reference set as background for comparison to genes associated with the evolutionary acceleration of jerboa metatarsal elongation, we found significant correlation with genes associated with tibia maturation as well as genes that are differentially expressed by location (p=5.4E-03 and 3.6E-06, respectively; Fisher’s exact test) but no correlation with SHR-associated genes (p=0.41). We then intersected the three correlating datasets and found thirteen genes that are associated with the evolution of skeletal proportion, slowed growth rate during maturation, and growth rate differences by location (Table 1). Six of these have mouse mutant phenotypes that include bone length alterations, skeletal malformations, bone mineral deficiencies, and/or body size abnormalities that could be a direct result of skeletal growth perturbation. Together, these data suggest that a similar subset of growth mechanisms determine skeletal proportion during development and diversify adult skeletal proportion during evolution.
Table 1:
Genes that are associated with the development and evolution of limb skeletal proportion.
| Gene Symbol | Gene Name | Known Skeletal/Body Size Phenotypes |
|---|---|---|
| Alpl | alkaline phosphatase, liver/bone/kidney | Abnormal long bone metaphysis morphology28
Decreased long bone epiphyseal plate size28 Decreased bone mineral density29 Increased bone resorption30 Decreased body size31 |
| B2m | beta-2 microglobulin | None reported |
| Baiap2l2 | BAI1-associated protein 2-like 2 | None reported |
| Enpp2 | ectonucleotide pyrophosphatase/phosphodiesterase 2 | None reported |
| Ibsp | integrin binding sialoprotein | Abnormal trabecular bone morphology32
Decreased bone mineral density32 Increased bone resorption33 Decreased body size32 Short femur32 |
| Lmcd1 | LIM and cysteine-rich domains 1 | None reported |
| Mcoln3 | mucolipin 3 | Decreased body size34 |
| Myh10 | myosin, heavy polypeptide 10, non-muscle | Abnormal body size35 |
| Nlgn3 | neuroligin 3 | None reported |
| Npr3* | natriuretic peptide receptor 3 | Increased width of hypertrophic chondrocyte zone36
Delayed endochondral bone ossification37 Increased length of long bones36,38 Elongated metatarsal bones38 Increased body length36,38,39 |
| Nprl3 | nitrogen permease regulator-like 3 | None reported |
| Tbx18 | T-box18 | Abnormal skeleton morphology40,41
Decreased body length41,42 |
| Ttll9 | tubulin tyrosine ligase-like family, member 9 | None reported |
Asterisk designates Npr3, which also appears in the human SHR GWAS. See also Table S5.
Mechanistic insights from enrichment analyses
Although the knowledge base of gene function is biased by an historical focus on a minority of all known genes43, pathway and network enrichment analyses can provide valuable information about biological functions, signaling pathways, and transcription networks that might causally explain a phenotype. Among all 1,755 differentially expressed genes that are associated with the disproportionate acceleration of jerboa metatarsal elongation, Ingenuity Pathway Analysis (IPA)44 revealed significant enrichment of annotations with relevance to cartilage biology. These include signatures for cytoskeletal organization, proliferation and hypertrophy of connective tissue, suppression of apoptosis, and developmental processes that, when perturbed, result in hypoplasia, growth failure, and short stature (Figure 2D, Table S6).
Among all significantly enriched canonical signaling pathways, the most significant suggests activation of melatonin degradation; melatonin appears to be sufficient to promote growth plate chondrogenesis45,46. We also find genes that promote cell-cell (adherens and tight junction signaling) or cell-extracellular matrix (ECM) contacts (integrin and integrin-linked kinase signaling) and an overall increase in the biosynthesis of ECM proteins (heparan, chondroitin, and dermatan sulfates) (Figure 2E). Integrins and integrin-linked kinases (ILK) regulate chondrocyte proliferation, and mice lacking these genes in growth cartilages develop chondroplasia47,48, consistent with the enrichment of cell-ECM signaling in our dataset. Our IPA analysis also revealed an increase in Rho family GTPase signaling and suppression of its inhibitor, RhoGDI (Figure 2E, Table S6). Members of the Rho family of GTPases regulate chondrocyte proliferation and size of the hypertrophic chondrocyte zone in mouse growth cartilages, and disruption of this signaling shortens limb bones49. Finally, we see enrichment of the WNT/β-catenin network (Figure 3A, Table S6) and evidence of activation of this signaling pathway (Figure 2E, Table S6), which is a key regulator of skeletal growth50.
Figure 3 |. Identification of hypertrophy pathways and transcription networks associated with disproportionate jerboa metatarsal elongation.
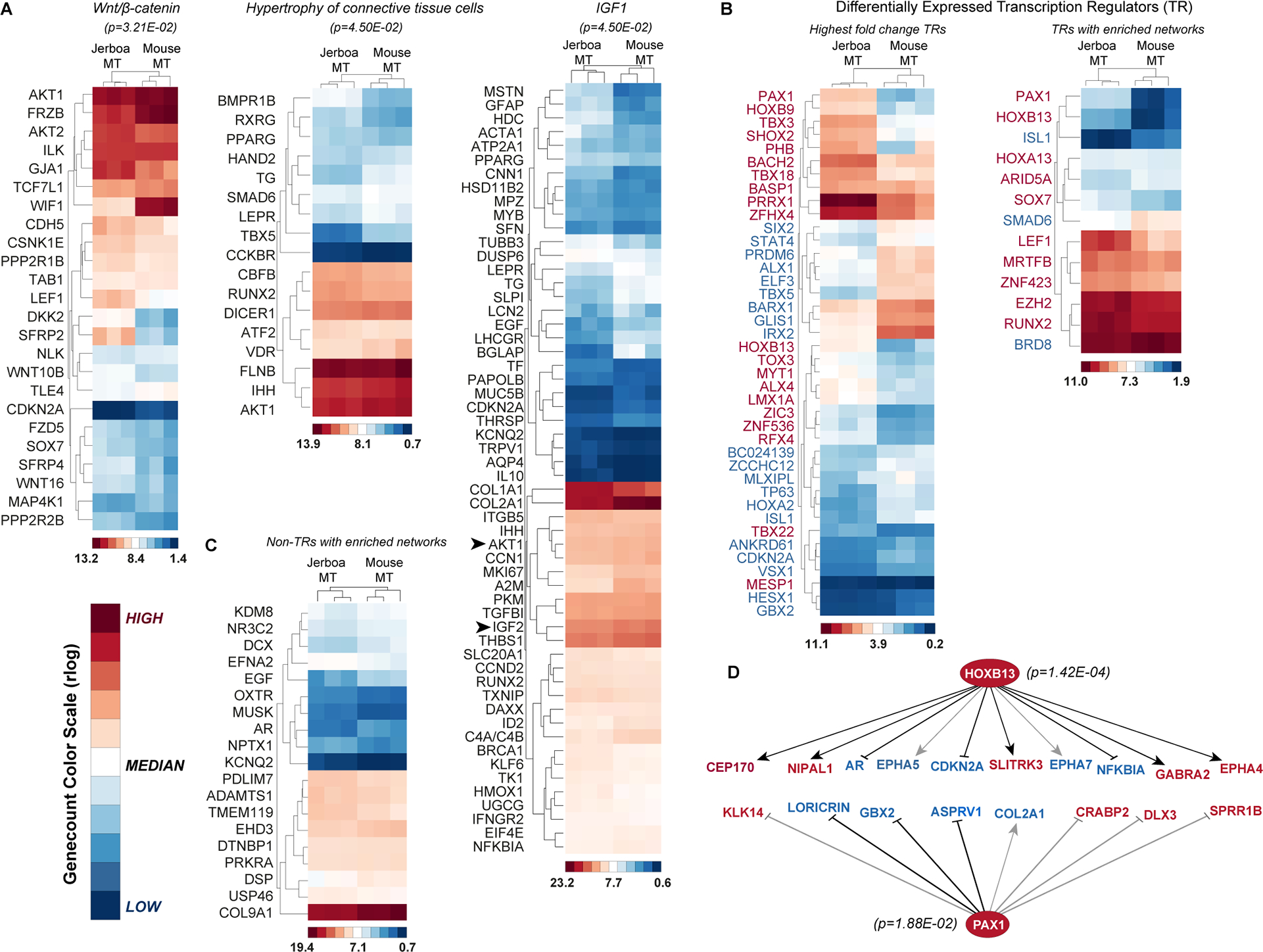
(A-C) Regularized log-transformed gene count heatmaps for genes that are disproportionately differentially expressed between jerboa (n=3) and mouse (n=3) MTs. Red, white, and blue colors indicate high, median, and low r-log genecount values, respectively. Color scales that indicate the genecount range within each heatmap are shown with internally assigned color values. (A) Genes in the enriched Wnt/β-catenin signaling network, those enriched within a hypertrophy of connective tissue network, and genes enriched in the IGF-1 signaling network (p-values <0.05). (B) The 40 transcription regulators that are most differentially expressed in MT of the two species. Thirteen differentially expressed transcription regulators that are each in networks with putative target genes that are enriched in our dataset. (C) Differentially expressed non-transcriptional regulators with putative networks enriched in our dataset. (D) Downstream networks of HoxB13 and Pax1 transcription regulators. Grey lines indicate that the direction of gene expression change is the opposite of what has been demonstrated in other contexts. Red and blue gene names in (B,D) indicate genes with higher or lower expression, respectively, in jerboa metatarsals compared to mouse. See also Table S6 and S7.
We previously showed that insulin-like growth factor 1 (IGF-1) is necessary in mice to establish the hypertrophic chondrocyte size differences that contribute to differential skeletal growth rate in many mammals, including jerboas14. Here, we see enrichment of genes associated with ‘hypertrophy of connective tissue’ among 1,755 genes that are associated with disproportionate acceleration of jerboa metatarsal elongation (Figures 2D and 3A). Although IGF-1 itself is not differentially expressed between jerboa and mouse metatarsals, we find significant enrichment of its network in this dataset (Figure 3A, Table S6). Additionally, IGF-2 and a major downstream effector of IGF-signaling, AKT1, are differentially expressed (Figure 3A, arrowheads). A complete list of signaling pathways and a selection of biological functions that are enriched in our dataset can be found in Table S6.
Finally, we focused on transcription regulators that may act as ‘nodes’ of evolutionary divergence due to their ability to have sweeping trans-regulatory effects on gene expression networks (Figure 3B). These include developmental transcription factors with expression in jerboa metatarsals that has not been reported in the distal mouse hindlimb (e.g. HoxB9, HoxB13, Pax1, and Shox2) and higher expression of Prrx1, which has been associated with the elongation of bat wing digits51 (Figure 3B and Figure S4). The expression patterns of HoxB9, HoxB13, Pax1, and Prrx1 in jerboa distal hindlimb raise the possibility that exaggerated elongation of the metatarsals may be influenced by surrounding perichondrium and connective tissues. Alternatively, these novel or expanded expression domains in the jerboa foot may underlie other unknown connective tissue differences between the species.
Thirteen of the 32 differentially expressed genes with enriched networks are transcription regulators (Figure 3B,C and Table S7). At least two of these, Pax1 (62.7-fold higher, padj=6.2E-16) and HoxB13 (37-fold higher, padj=3.1E-11), are detected in jerboa metatarsals despite no reported expression in distal mouse limbs (Figure 3B,D and Figure S4). In the developing mouse embryo, Pax1 expression is restricted to the proximal-most limb where it is required for elongation of the acromion, a protrusion of the scapula that articulates with the clavicle, but Pax1 knockout mice have no other limb abnormality52. Similarly, although HoxB13 is expressed during development and regeneration in amphibian limbs53, its expression is not detected in mouse limbs by our analyses or in prior studies54,55. Expression of these two transcription factors in jerboa metatarsals and enrichment of their targets within our dataset might therefore represent recruitment of new transcription networks to regulate disproportionate bone growth in jerboas. As analysis tools and whole genome annotations of all gene functions improve in the future, it will be important to revisit these data to gain new insight into pathways and networks that may control the development and evolution of skeletal proportion.
Evidence for both de-repression and acceleration of jerboa metatarsal elongation
Complementary to whole transcriptome analyses of biological functions and genetic networks, individual genes among the most significantly and highly differentially expressed reveal insights into complex genetic mechanisms of differential growth. The disproportionately exaggerated length of the foot in jerboa compared to mouse may result from a difference in molecular mechanisms that directly accelerate growth rate (e.g., more growth factor expression in jerboa metatarsals compared to mouse metatarsals). It is also possible that there are mechanisms that repress the rate of mouse metatarsal elongation, and that these ‘brakes’ may have been released in jerboa metatarsals. Here, we highlight a number of differentially expressed genes that suggest tuning both repression and acceleration may explain growth rate differences that establish and vary adult skeletal proportion.
The two most significantly differentially expressed genes among the 1,514 genes that differ between jerboa and mouse metatarsals and not between radius/ulna provide support for the hypothesis that differential growth may be achieved in part by differences in growth repression (Figure 2C). Cellular retinoic acid binding protein 1 (Crabp1) is expressed 10.6-fold higher in jerboa metatarsals (padj=1.5E-92). Crabp1 has been used as a marker of perichondrium56–58, and its expression indeed appears higher in the perichondrium of jerboa metatarsals compared to mouse by mRNA in situ hybridization (Figure 4A,B). Growth and differentiation factor 10 (Gdf10/Bmp3b), an inhibitory transforming growth factor beta (TGFβ) ligand, is expressed 9.3-fold lower in jerboa metatarsals than in mouse (padj=1.3E-83). Gdf10 mRNA is localized to the proliferative zone of both growth cartilages in each species, with strong expression in mouse metatarsals, consistent with our RNA-Seq results (Figure 4C,D and Figure S3A,B and G,H).
Figure 4 |. Spatial pattern of Crabp1, Gdf10, and Mab21L2 expression in jerboa and mouse metatarsal growth cartilages.
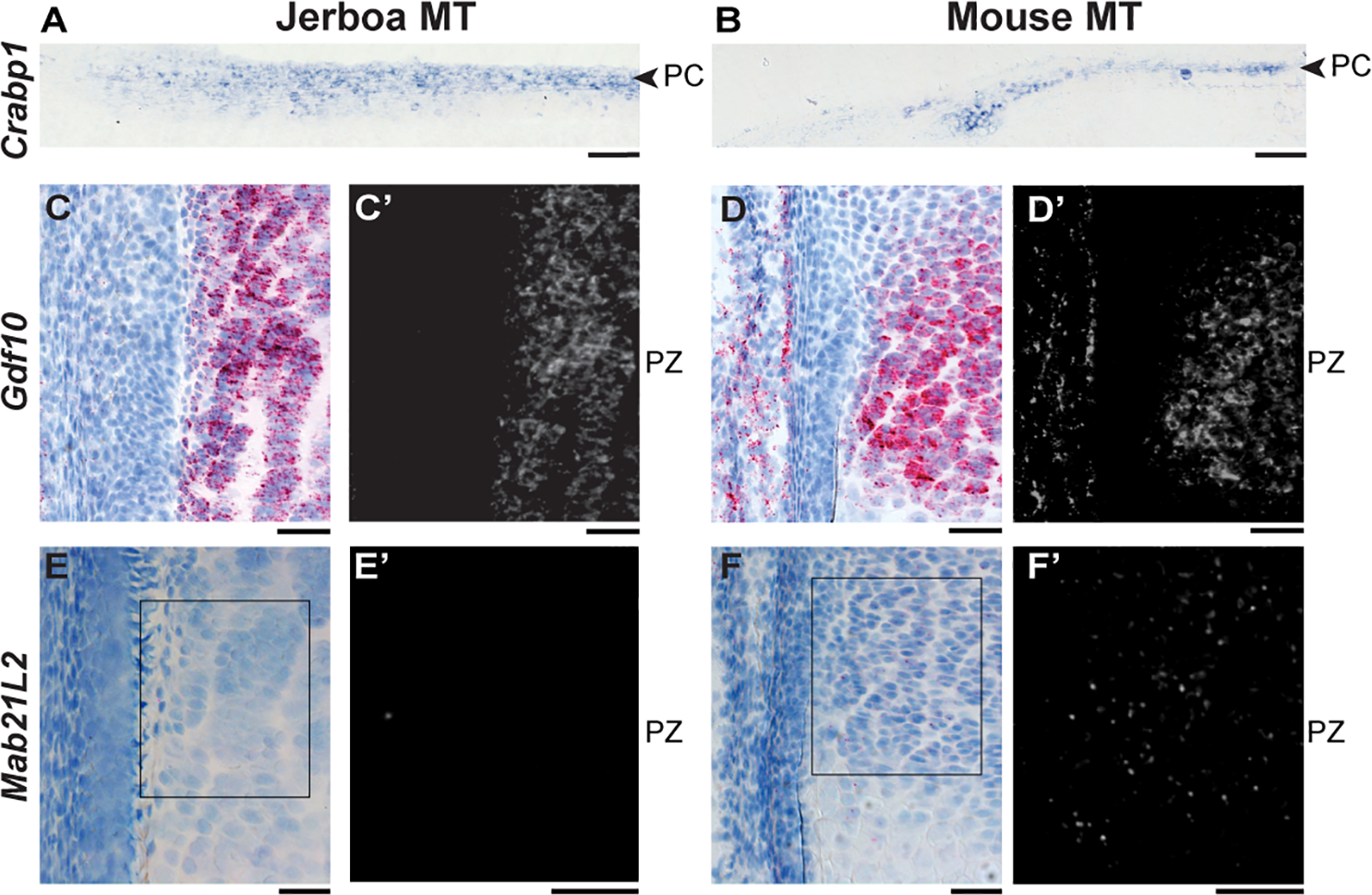
Expression patterns in distal growth cartilages of P5 jerboa (left) and mouse (right). (A-B) Crabp1 colorimetric (blue) in situ hybridization in perichondrium (PC), (C-H) RNAScope colorimetric and (C’-H’) fluorescence detection of (C-D) Gdf10, and (E-F) Mab21L2 in proliferative zones (PZ). (C-F), Hematoxylin-stained nuclei (blue) and Fast Red in situ signal (red). Fluorescent Fast Red in situ signal is shown in (C’-F’). Scale bars, A-B = 100 μm, C-F = 50 μm. See also Figures S3–S5.
The functions of Crabp1 and Gdf10 during skeletal development remain unknown in part because no abnormal phenotype was detected in knockout mice59,60, though precise analyses of bone lengths were not reported. However, several pieces of evidence from other studies intersect to suggest that Crabp1 and Gdf10 could participate in a mechanism to de-repress distal jerboa hindlimb elongation. In cancer cells, Crabp1 inhibits the anti-proliferative effects of retinoic acid (RA) by sequestering RA in the cytoplasm61–63. Exogenous RA, a potent teratogen, inhibits skeletal elongation in juvenile rats, and pharmacological inhibition of RA signaling accelerates rat metatarsal elongation in vitro, suggesting that endogenous RA suppresses metatarsal growth64. Intriguingly, Gdf10 is one of two genes upregulated by RA or an RA Receptor-γ agonist in mouse limb culture65. Together, these data suggest that endogenous RA signaling may repress growth in mouse metatarsals, perhaps in part by increasing Gdf10 expression. If true, such a repressive effect could be alleviated by higher Crabp1 expression in jerboa metatarsals to sequester RA and decrease Gdf10 expression.
Among the 241 genes with non-equivalent expression differences in both forearms and in metatarsals, Mab21L2 is one of the genes with the greatest deviation from the linear regression of correlated expression differences in both growth cartilages [16.8-fold lower expression in jerboa metatarsals (padj=1.8E-26) and 3.9-fold higher expression in the radius/ulna (padj=3.9E-16) compared to mice]. RNAScope in situ hybridization confirmed expression in resting zone and proliferative chondrocytes as well as the perichondrium of mouse metatarsals, whereas Mab21L2 expression was not detected in jerboa metatarsals (Figure 4E,F). Mab21L2 inhibits the effects of BMP4 overexpression in Xenopus laevis embryos, and it immunoprecipitates with Smad1 transcription factors in vivo and with Smad1/Smad4 complexes in vitro66. BMP signaling promotes bone growth, which is evident from loss-of-function mutations in BMP receptors that cause severe chondrodysplasia in mice67,68 and from overexpression of Bmp2 and Bmp4 in chicken embryo limbs that each increase the size of skeletal elements69. Consistent with the hypothesis that Mab21L2 inhibition of BMP signaling is less in jerboa than in mouse metatarsals, we see significant enrichment in our dataset of BMP2, BMP4, SMAD1, and SMAD4 networks that predict activation of SMAD1 and SMAD4 in jerboa metatarsals (Figure S5 and Table S7).
Lower Mab21L2 expression in jerboa metatarsals is exciting in light of the fact that its expression is also lower in tissues associated with the elongated digits of the fetal bat wing compared to the short first wing digit, bat hindlimb digits, and all mouse digits70. Although a specific role for Mab21L2 in growth cartilage elongation remains unknown, as the gene is also required for mouse embryo viability71, two presumed gain-of-function missense mutations that appear to stabilize the human MAB21L2 protein each cause shortening of the proximal limb skeleton in addition to eye malformations72. By retroviral misexpression in the developing chicken wing skeleton, we demonstrate a small but significant inhibition of bone elongation; in limbs that over-express Mab21L2 the humerus is 1.9% shorter on average, in contrast to the symmetric limbs of infected control embryos that overexpress eGFP (p=0.0221, Wilcoxon/Mann-Whitney nonparametric test, and Figure S5C). Together, these data suggest that Mab21L2 inhibits skeletal elongation, and decreasing its expression may have contributed to the convergent evolution of an elongated distal skeleton in the jerboa hindlimb and bat forelimb. That Mab21L2 appears to be a relatively weak BMP inhibitor might reflect the fact that there are multiple intracellular effectors of BMP signaling that can compensate for one another73,74 contrasting the potent inhibition by Noggin75,76 or Gremlin77,78, which each block ligand-receptor interactions. The relatively weak inhibitory effect might make Mab21L2 amenable to processes of natural selection that can act through subtle multigenic changes over time.
The Shox2 transcription factor is a key example of genetic control that is likely to accelerate jerboa metatarsal elongation. Shox2 is expressed 9.1-fold higher in jerboa metatarsals compared to mouse (padj=2.2E-16) and 4.9-fold lower in jerboa radius/ulna (padj=1.3E-50; Figure 2B). Loss-of-function mutation of the Short stature homeobox transcription factor, Shox, causes short stature in a variety of human genetic disorders, including Turner syndrome79,80 and Langer syndrome81. Although Shox is not present in rodent genomes, its paralogue, Shox2, is necessary for elongation of the proximal mouse limb skeleton. It is not, however, required for growth of the hands and feet where its expression has never been detected in multiple vertebrate species82–85. We observed no Shox2 expression in mouse metatarsals (Figure 5B and Figure S2F) or the metacarpals of either species (Figure S3I,J), as expected, but Shox2 appears in the proliferative zone and perichondrium of jerboa metatarsals (Figure 5A) as in the radius/ulna of both species (Figure S3C,D). Quantitative RT-PCR shows the level of expression in jerboa metatarsals is comparable to expression in jerboa radius/ulna (Figure S2G).
Figure 5 |. Derived Shox2 expression in jerboa metatarsals may have contributed to increased distal limb elongation.
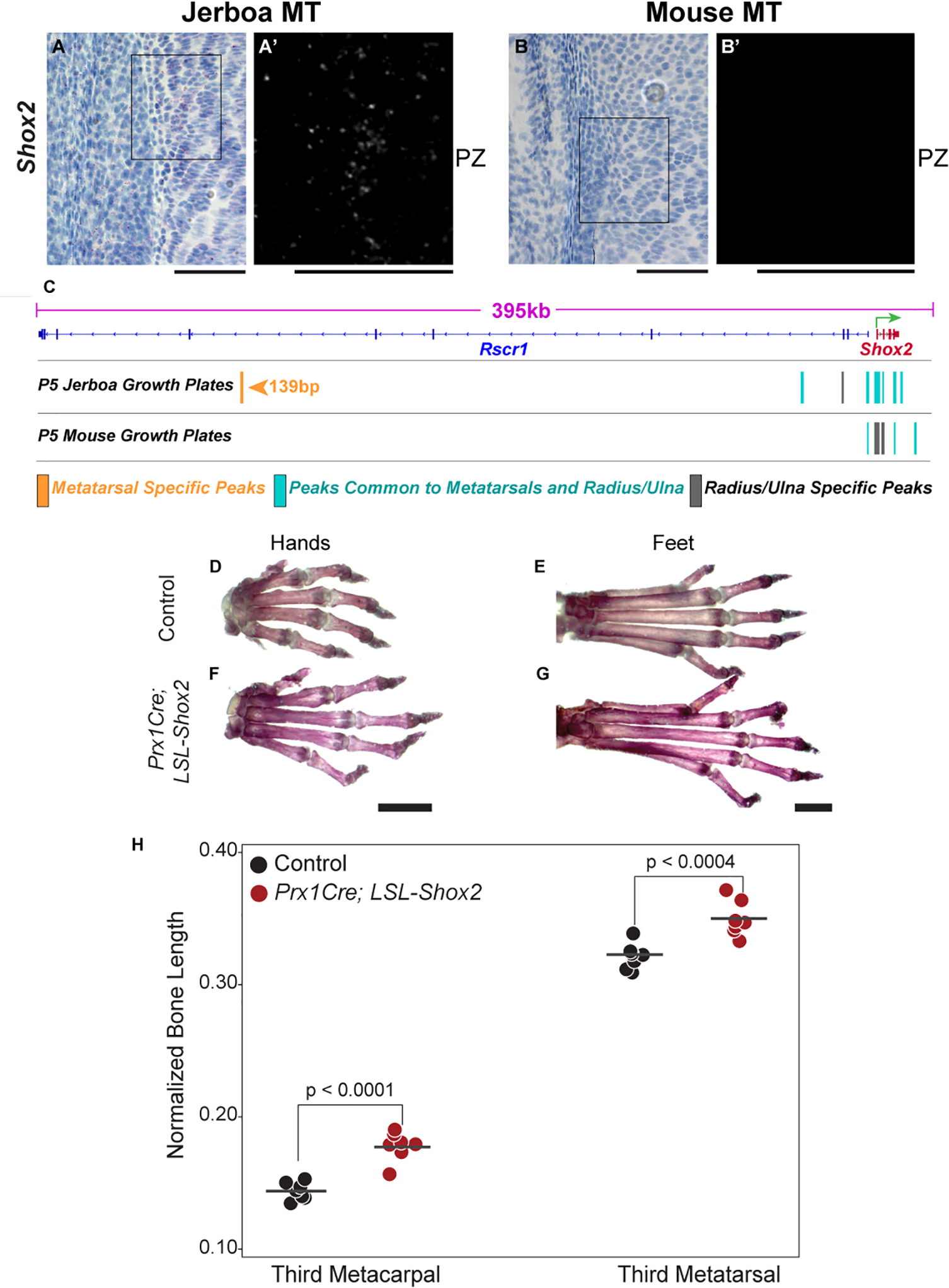
(A-B) Shox2 is expressed in the proliferative zone (PZ) of jerboa metatarsals (MT) where it is not present in mouse. (C) Open chromatin regions in P5 jerboa and mouse growth cartilage near the Shox2 transcription start site (TSS, green arrow). A 139 bp region of open chromatin, preferentially accessible in jerboa metatarsals (orange), lies ~285 kb upstream of the Shox2 TSS in an Rsrc1 intron at a position orthologous to mouse chr3:67266489–67267716. (D,F) Metacarpal and (E,G) metatarsal lengths are increased in Prx1Cre;LSL-Shox2 (Shox2-overexpressing) mice at eight weeks of age compared to control littermates. In (H), measurements are normalized to the skull length of each individual, which was not affected by Shox2-overexpression (Figure S6D). p-values are derived from a paired t-test between matched-sex littermates (n=5 females and 2 males of each genotype). Shox2-overexpressing metacarpals are 23.6% and metatarsals are 9.1% longer than controls. Scale bars, (A-B) = 50 μm, (D-G) = 2 mm. See also Figure S6.
Although viral misexpression of the chicken Shox ortholog is sufficient to increase wing digit length85, genetic misexpression of Shox2 in mice has a mild shortening effect on proximal limb elements86 (Figure S6C). In order to test the hypothesis that exogenous Shox2 is sufficient to increase the length of distal skeletal elements in rodents, we measured metacarpals and metatarsals of mice in which Shox2 was genetically overexpressed during limb development for a previous study86–88. The study demonstrated 2- to 2.5-fold increased Shox2 expression in Prrx1-Cre;Rosa-LSL-Shox2 embryonic limb buds compared to controls86. While not reported at later stages, exogenous Shox2 expression is expected to persist postnatally in descendants of Prrx1-expressing cells that include chondrocytes and perichondrium87. Though similar to controls at birth (Figure S6B), we observed that distal limb skeletal elements of Prrx1-Cre;Rosa-LSL-Shox2 mice grew more by eight weeks of age. The same absolute increase in growth added 23.6% to the length of short metacarpals and 9.1% to longer metatarsals compared to littermate controls (Figure 5D–H).
To identify cis-regulatory changes that might be responsible for modular gain of Shox2 expression in jerboa feet, which has not been observed in other species, we performed comparative ATAC-Seq89 using cartilage cells from P5 metatarsals and radius/ulna of jerboa and mouse. We analyzed open chromatin regions within ~500 kb of the jerboa Shox2 transcription start site and found a 139 bp region that is preferentially accessible in metatarsals (Figures 5C and S7). The jerboa metatarsal peak lies upstream of Shox2 in an intron of the Rsrc1 gene, which itself is equivalently differentially expressed between species in both metatarsals and radius/ulna (gray interval in Figure 2B). Mouse Rsrc1 intronic regions are known to harbor enhancers90, at least one of which (m741/hs741) contributes to Shox2 expression and femur elongation90,91.
Interestingly, the genomic region of the 139 bp jerboa metatarsal peak aligns to the mouse genome, but the orthologous mouse genomic region exhibits no ATAC-Seq peak of accessibility in either mouse metatarsals or radius/ulna, showing differential chromatin accessibility in the two species. Surprisingly, we also found that the 139 bp jerboa metatarsal peak corresponds to 1228 bp of mouse genomic sequence that is present in an alignment of 40 placental mammal genomes and not for its full length in jerboa. We confirmed by PCR and Sanger sequencing that the 139 bp jerboa metatarsal peak joins 122 bp upstream and 17 bp downstream of an approximate 1 kb deletion in the jerboa genome (Figure 6). Future work will endeavor to dissect this presumed cis-regulatory region to determine whether it acts in the jerboa as a novel gain of regulatory control or loss of a repressive element controlling Shox2 expression. Although incompleteness of the current draft jerboa genome precludes a thorough genome-wide analysis at this time, we also identified an open chromatin region preferentially accessible in jerboa metatarsals among these growth cartilages that is near HoxB13, which has also gained derived expression in jerboa metatarsals (Figure S7).
Figure 6 |. Jerboa Rsrc1 metatarsal ‘unique’ peak shows deletion of a genomic region that is retained across placental mammals.
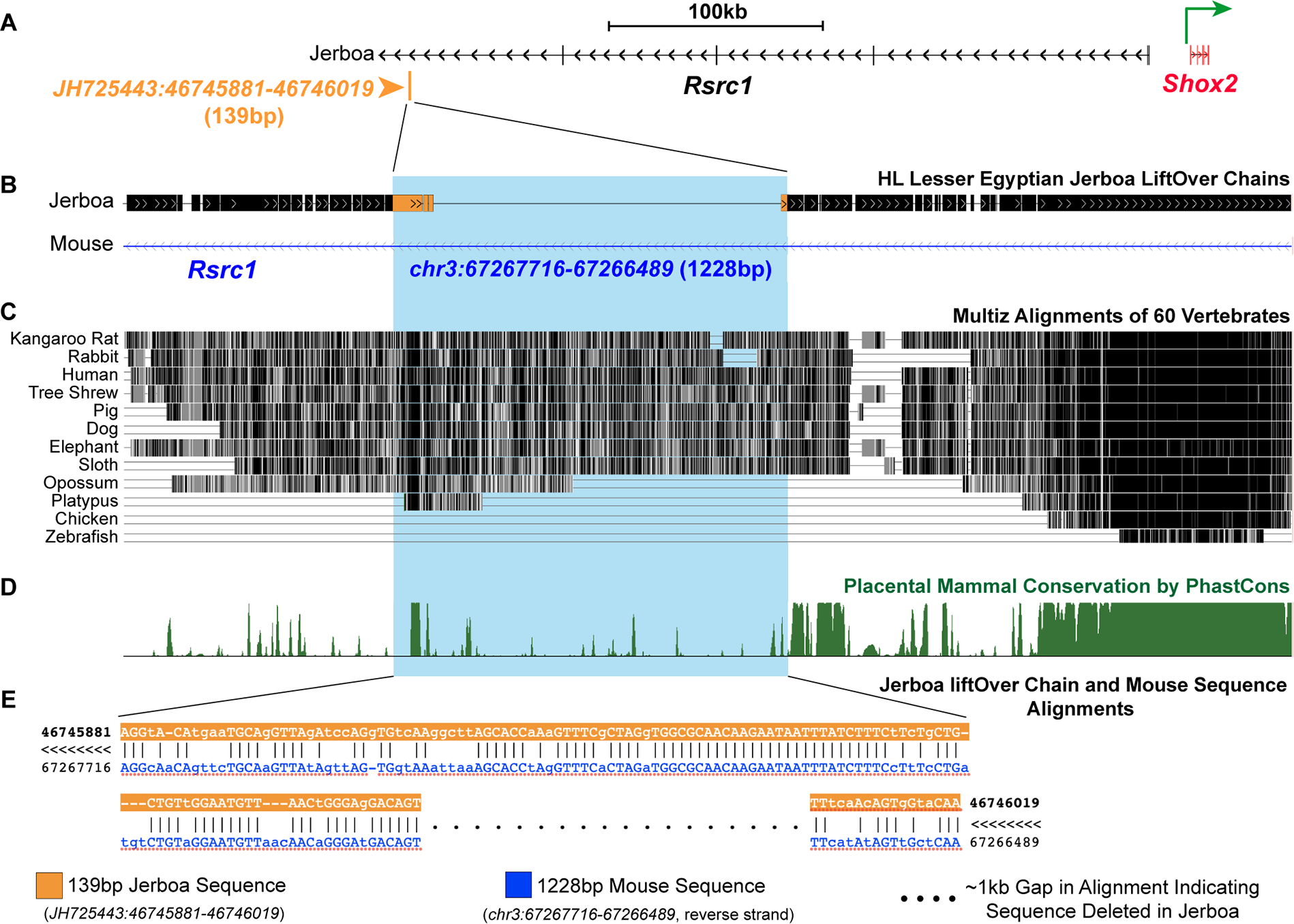
(A) The 139 bp Rsrc1 open chromatin peak that is preferentially accessible in jerboa metatarsals partially maps to a longer 1228 bp orthologous region in mouse Rsrc1 intron (B, blue highlight). Jerboa LiftOver chains show ~1 kb gap in this alignment to the mouse Rsrc1 intron. (c) This gap indicates deletion of sequence in the jerboa genome that is broadly present in an alignment of placental mammals with overall low sequence conservation (D). (E) Alignment of the 139 bp jerboa sequence to the 1228 bp mouse sequence. Dotted line indicates a 1089 bp mouse sequence without orthology in the jerboa genome. Sanger sequencing of a larger region that spans the 139 bp jerboa metatarsal peak confirms the presumed deletion at this site (JH725443:46745881:46746019). Multiple sequence alignment and conservation for this region in C-D were extracted from UCSC genome browser. See also Figure S7.
Conclusion
The limb skeleton is highly modular with dozens of individual long bones elongating at different rates during development, and each rate has been individually tuned to diversify skeletal proportion across species. Although this study does not encompass the full complexity of allometric skeletal growth and its evolution, which also includes size differences during embryonic mesenchymal condensation and differential senescence to terminate growth, we provide valuable insight to understand mechanisms that establish disproportionate growth rate.
Our innovative approach directly compared gene expression in homologous growth cartilages of two closely related rodents that manifest strikingly different limb proportions. Though some genes unrelated to the evolution of skeletal proportion certainly remain in our dataset, including cartilages that grow at a similar rate allowed us to remove thousands of genes that are equivalently differentially expressed in both cartilages and to focus on those genes whose differential expression associates with disproportionate elongation of jerboa metatarsals. We identified biological processes, pathways, and individual genes that provide insight into how growth modularity is genetically encoded. Significant overlap between genes associated with the development and evolution of skeletal proportion suggests that some of the same mechanisms that establish proportion in an individual may also diversify proportion across species. All together, these are an ensemble of genes whose modular cis-regulatory control within the skeleton might constrain growth in some bones while tuning the growth of others to bring about the extraordinary malleability of limb form and function.
STAR METHODS
RESOURCE AVAILABILITY:
Lead Contact and Materials Availability
Further information and requests for resources generated in this study should be directed to and will be fulfilled by the lead contact, Dr. Kimberly L. Cooper (kcooper@ucsd.edu).
Data and code availability
The RNA-Seq and ATAC-Seq illumina reads generated in this study have been submitted as fastq files to the Zenodo repository. These datasets are publicly accessible at https://doi.org/10.5281/zenodo.5123254 and https://doi.org/10.5281/zenodo.5120588.
All original code and additional tables with STAR gene counts, rlog transformed gene counts, and DESeq2 differential expression results for all the 17,464 mouse and jerboa orthologs are publicly available on the Zenodo repository. These datasets can be accessed at https://doi.org/10.5281/zenodo.5123384.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS:
Jerboas were housed and bred as previously described92. CD-1 mice and fertilized chicken eggs were obtained from Charles River Laboratories (MA, USA). Mice that were hemizygous for Rosa26CAG-loxSTOPlox-Shox2 and Prx1-Cre were generated for the purposes of a previous study86. Controls for this cross were siblings that were hemizygous for only one or neither of these transgenes. All animal care and use protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California San Diego or the Life and Environmental Sciences Animal Care Committee of the University of Calgary.
METHOD DETAILS:
Skeletal Preparations and Measurements:
Freshly dissected chicken wings were fixed while rocking at room temperature in 100% ethanol for 24 hours followed by 100% acetone for 24 hours. Specimens were then stained for 3–5 days with alcian blue/alizarin red staining solution (one volume each of 0.3% alcian blue-8GX in 70% ethanol, 0.1% alizarin red-S in 95% ethanol, and glacial acetic acid plus 17 volumes of 70% ethanol). After staining, specimens were rinsed with de-ionised water and destained over several days in a graded series of 1% KOH followed by 20%, 50%, and 80% glycerol diluted with 1% KOH until stain was sufficiently cleared from the soft tissues. Specimens were stored and imaged in 100% glycerol at room temperature.
Eight-week-old mouse skeletons (Figure 5D–H, Figure S6C–D) were stained with Alizarin red as follows. Mice were skinned and eviscerated, and internal organs were removed. Mice were fixed in 100% ethanol for 4 days followed by acetone for 3 days rocking at room temperature. The specimens were then incubated in 2% KOH for 5 days to dissolve tissue. The skeletons were subsequently stained with 0.005% Alizarin red (Sigma-Aldrich A5533) in 2% KOH for 3 days until the bones appeared red. Following staining, the skeletons were de-stained with 20% glycerol in 1% KOH to remove excess stain followed by 100% glycerol. All chicken and mouse specimens were blinded prior to measuring skeletal elements using a digital caliper. The length of each skeletal element in each specimen is represented as the average of three separate blinded measurements to improve precision and to reduce measurement error, which was quantified as less than 0.5% of total length. Mouse limb bone lengths were normalized either to skull (Figures 5H and S6C) or lumbar vertebrae lengths (Figure S6B) prior to analyses.
RNA Sequencing and Analysis:
Distal metatarsal (MT) and radius/ulna (RU) growth cartilages with intact perichondrium were dissected in ice cold 1X phosphate-buffered saline (PBS) from freshly euthanized mice and jerboas at postnatal day five (P5). Compared to the forelimb RU elements, the P5 metacarpals (MC) are extremely small and therefore difficult to dissect with the same degree of tissue purity and high-quality RNA as metatarsals and radius/ulna. Dissected growth cartilages were treated for three minutes at room temperature in a 25% dilution of Proteinase-K (Qiagen) in 1X PBS to remove muscle and connective tissue. Samples were equilibrated in RNAlater solution (Qiagen) overnight at 4°C and stored at −80°C until ready for RNA extraction. To extract RNA, growth cartilages were pulverized in a liquid nitrogen cooled chamber. Pulverized samples were further homogenized with QIAshredder columns (Qiagen). Homogenate was treated with Proteinase-K (Qiagen) at 55°C for 10 min. RNA was extracted using the RNeasy Micro kit (Qiagen) following the manufacturer’s protocol that included an on-column DNase treatment step. cDNA libraries were prepared using Illumina TruSeq® stranded mRNA library preparation kit following the manufacturer’s protocol. We multiplexed libraries and sequenced single-end 50-bp reads (SR50) on an Illumina HiSeq-4000 platform at the Institute for Genomic Medicine (IGM) at UC San Diego. Low quality reads and residual adaptor sequences were trimmed using Trimmomatic93 (version 0.35).
Using the Coding Exon-Structure Aware Realigner (CESAR)16 we aligned exons from jerboa (JacJac1.0, NCBI) and mouse (mm10, NCBI) genomes to generate a custom 1:1 orthologous transcript annotation set with no paralogs. To this end, we first generated a whole genome alignment between the mouse (reference) and jerboa (query) genome assemblies94. Briefly, we computed local alignments using lastz95 (alignment parameters K = 2400, L = 3000, Y = 9400, H = 2000, default scoring matrix), ‘chained’ these local alignments using axtChain96 linearGap=loose, otherwise default parameters), used RepeatFiller97 (default parameters) to incorporate previously-undetected alignments between repetitive sequences and used chainCleaner98 (default parameters) to improve alignment specificity. Chains were converted to alignment nets using a modified version of chainNet96 that computes real scores of partial nets98. To filter out exonic alignments that represent potential paralogs or intact exons from processed pseudogenes, we removed all such alignments using the procedure described in Sharma et al99. CESAR was then applied to the filtered genome alignment to annotate mouse exons (Ensembl version 87, longest isoform per gene) in the jerboa genome. Exonic regions that appear only once in the resulting jerboa annotation file were retained. Exons corresponding to a particular mouse transcript were combined together to create the orthologous jerboa gene structure. Lastly, those jerboa genes where all the constitutive exons do not come from the same scaffold and strand were discarded. The resulting orthologous reference gene set contains 17,464 transcripts present in both genomes with at least one exon free of frame-shift, non-sense, and splice site mutations. CESAR related computational code and tools used for generating 1:1 custom jerboa-mouse orthologous annotations can be accessed at https://github.com/hillerlab/CESAR and https://github.com/hillerlab/CESAR2.0/tree/master/tools
We mapped MT and RU RNASeq reads to jerboa or mouse genomes using these 1:1 orthology GTF annotations and computed gene counts for each library with the STAR aligner100. We were able to uniquely map 78.3–88.4% of mouse and 88.2–92.9% of jerboa SR50 reads to their respective genomes with this approach. STAR gene counts were used to perform differential expression analysis between jerboa and mouse metatarsals and radius/ulna with DESeq2, a robust approach that employs a negative binomial generalized linear model to identify differentially expressed genes17. Since orthologous gene/transcript lengths could vary between jerboa and mouse genomes, we implemented an additional length normalization step in the DESeq2 pipeline to avoid false comparative quantifications resulting from species-specific gene/transcript length variation. To do this, we first calculated gene lengths in the number of base pairs in non-overlapping exons of each gene from our 1:1 orthologous GTF annotations. We then created a matrix of these lengths for each gene in each sample and input these into the DESeq2 ‘DESeqDataSet’ object so that they are included in the normalization for downstream analysis. We have provided the code for this length normalization and the supporting R library for this analysis in our primary data archive at Zenodo, associated with this paper. Principal component analysis (PCA) was performed with default DESeq2 settings to identify variance components associated with our MT and RU comparisons (n=5 in each species). In DESeq2, differential expression analysis was performed using the Wald test, and the DESeq function performed log2 fold-change shrinkage by default. We considered all differentially expressed genes with an adjusted p-value <0.05 to be statistically significant in our analyses. Hierarchical clustering in Figure 3, Figure S1 and Figure S5 was performed using DESeq2 rlog-transformed gene counts to stabilize variance. Gene counts for all 17,464 transcripts were considered for the rlog transformation.
ATAC Sequencing and Associated Analysis:
Distal metatarsal and radius/ulna growth cartilages were dissected from P5 mice (n=2) and jerboas (n=3). Samples were transferred to 1.5 ml microfuge tubes containing 200 μl of Dulbecco’s Modified Eagle Medium (DMEM) (high glucose)-Glutamax (Gibco) with 10% fetal bovine serum (Sigma-Aldrich), 2% C. histolyticum collagenase type II (Gibco) and 0.2% bovine testes hyaluronidase (Sigma-Aldrich). Samples were digested in this solution for 2–3 hours at 37°C in a CO2 tissue culture incubator without shaking or agitation. After digestion, samples were filtered through a 100 μm mesh, and single cell suspension was collected in a fresh microfuge tube. Samples were centrifuged at 400 g at 4°C for 5 min, and supernatant was discarded. The cartilage cell pellets were resuspended and washed in 500 μl sterile, ice-cold 1x PBS. Samples were centrifuged again at 400g/4°C for 5 min. After discarding the supernatants, the cell pellets were resuspended in 100 μl sterile, ice-cold 1x PBS. Next, cells were counted in trypan blue solution, and only those samples that had 10% or less dead cells were subjected to ATAC-Seq protocol.
50,000 cells from each biological replicate, resuspended in sterile 1x PBS, were subjected to a previously described ATAC-Seq protocol89,101 with minor modifications102. The transposed DNA was purified using a DNA Clean & Concentrator kit (Zymo Research) and eluted in 10 μl of pre-warmed, molecular biology grade water (Sigma). Sequencing libraries were prepared following a previously described protocol102 using primers described in Buenrostro JD et al; 2015101. Mouse and jerboa samples were sequenced in independent lanes on Illumina NextSeq500 and NovaSeq6000 platforms.
Mouse and jerboa sequence read quality were checked with FastQC. Reads sequenced across runs were pooled for each sample, and adapters were removed with NGMerge103. Mouse reads were aligned to the mouse reference mm10 genome assembly with Bowtie2 v2.3.26 using default parameters for paired-end alignment104. Jerboa reads were aligned to the jerboa draft reference JacJac1.0 genome assembly with Bowtie2 v2.3.26 using local read alignment mode to maximize the alignment score. The mouse and jerboa aligned reads were filtered for duplicates using Picard’s MarkDuplicates, and mitochondrial reads were also removed. BAM files were subsequently used for peak calling using MACS2 software (version 2.1.1.2), with the following flags for ‘callpeak’: --BAMPE –nolambda105. For mouse and jerboa samples, peaks reproducible across biological replicates were screened using a stringent IDR threshold of <0.01, as defined by the IDR statistical test (version 2.0.3)106. Briefly, the IDR method looks for overlaps in called peaks across pairs of replicate samples by comparing ranked peak lists (using MACS2 q-value) to define a reproducibility score curve. All paired ranks were assigned a pointwise score based on this curve, subsequently sorted, and all peaks falling below an ‘irreproducible discovery rate’ (IDR) threshold of 0.01 were taken as the final set of reproducible peaks.
For a direct comparison between mouse and jerboa ATAC-seq data, jerboa IDR-called peaks were assigned to orthologous mouse mm10 genome coordinates using UCSC’s liftOver utility (minMatch = 0.1). The jacJac1 to mm10 liftOver chains were computed from the alignment net using the procedure described above but with jerboa as the reference and the netChainSubset utility96. Only those jerboa genome peaks that ‘lifted-over’ and hence have an orthologous region present in the mouse mm10 genome were further analyzed.
Endpoint PCR and Quantitative Real Time-PCR:
RNA was extracted from growth cartilage as described above. cDNA libraries were prepared using a QuantiTect® reverse transcription kit (Qiagen) following the manufacturer’s protocol that included an additional genomic DNA elimination step before cDNA synthesis. Primers spanning exon-exon junctions that prevent amplification from genomic DNA were designed in-silico using NCBI Primer-BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast). Note: Mab21L2 is a single exon gene and primers for Mab21L2 in both species bind within this exon. Quantitative Real Time-PCR (qRT-PCR) was performed on a CFX96™ real-time system with a C1000™ thermal cycler (Bio-Rad) using SsoAdvanced™ universal SYBR green supermix (Bio-Rad) following manufacturer’s protocol. Shox2 and Mab21L2 expression levels in each species were calculated using the 2−ΔCt method and were normalised to their respective Sdha expression levels. HoxB13 endpoint PCRs were performed on a C1000™ touch thermal cycler (Bio-Rad) for 35 cycles using 2x MyTaq™ red mix (Bioline). Sdha amplification was used as a positive control for each of the cDNA libraries. Absence of ~400bp HoxB13 amplicons in mouse cartilage cDNA libraries was confirmed with two independent primer pairs. A list of PCR primers used in this study are provided in the Key Resource Table associated with this manuscript.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AMV-3C2 Monoclonal Antibody (supernatant) | Developmental Studies Hybridoma Bank | AMV-3C2 |
| Bacterial and virus strains | ||
| RCASBP(A)-deltaF1’ | Dr. Stephen Hughes’ Laboratory, NCI-NIH, Bethesda | Replication competent RCAS virus |
| RCASBP(A)-deltaF1’::GFP | Dr. Stephen Hughes’ Laboratory, NCI-NIH, Bethesda | Replication competent RCAS virus expressing GFP |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| OCT compound | Sakura Inc. | Ref# 4582 |
| DMEM (High Glucose) Media | ATCC | Cat# 30–2002 |
| DMEM (GlutaMAX, High Glucose, HEPES) Media | Gibco/Thermo Fisher | Cat# 10564011 |
| Collagenase Type II | Gibco | Ref# 17101–015 |
| Hyaluronidase Type IV-S | Sigma Life Sciences | Cat# H4272 |
| Proteinase K | Qiagen | Cat# 19131 |
| Hematoxylin Gill-1 | Ricca Chemical Company | Cat# 3535–32 |
| Ammonium Hydroxide Solution | Sigma Aldrich | Cat# 221228 |
| Ampure XP magnetic beads | Beckman Coulter | Ref# A63880 |
| Mag-Bind Rxn Pure Plus magnetic beads | Omega Bio-Tek | Cat# 1386 |
| MyTaq Red PCR Mix | Bioline/Meriadian BioScience | Cat# BIO-25044 |
| Superscript II Reverse Transcriptase | Invitrogen | Cat# 18064014 |
| Sso Advanced SYBR Green Supermix | BioRad | Cat# 1725271 |
| NEBNext High-Fidelity PCR MasterMix | New England Biolabs | Cat# M0541 |
| Q5 High-Fidelity Mastermix | New England Biolabs | Cat# M0492 |
| Critical commercial assays | ||
| Jerboa Shox2 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 578821 |
| Jerboa Mab21L2 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 723661 |
| Jerboa Gdf10 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 578801 |
| Jerboa Pax1 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 578851 |
| Jerboa HoxB13 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 578841 |
| Jerboa HoxB9 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 1072041 |
| Jerboa Prrx1 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 1072031 |
| Mouse Galnt17 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 579031 |
| Mouse Shox2 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 579051 |
| Mouse Mab21L2 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 456901 |
| Mouse Gdf10 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 525161 |
| Mouse Pax1 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 579091 |
| Mouse HoxB13 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 579101 |
| Mouse HoxB9 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 452821 |
| Mouse Prrx1 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 485231 |
| Mouse Galnt17 in situ Probe | Advanced Cell Diagnostics Inc. | Part ID 578861 |
| Protease Plus Solution | Advanced Cell Diagnostics Inc. | Part ID 322331 |
| Protease III Solution | Advanced Cell Diagnostics Inc. | Part ID 322337 |
| RED Chromogenic in situ staining and Detection Kits |
Advanced Cell Diagnostics Inc. | Part ID 322350 Part ID 323971 |
| TruSeq Stranded RNA Library Preparation Kit | Illumina Inc. | RS-122-2101 |
| Nextera DNA Sample Preparation Kit | Illumina Inc. | FC-121-1030 |
| Deposited data | ||
| RNA-Seq fastq illumina sequencing reads for Mouse and Jerboa cartilage samples | This paper | Zenodo |
| ATAC-Seq fastq illumina sequencing reads for Mouse and Jerboa cartilage samples | This paper | Zenodo |
| DESeq2 R-Script and associated library used for differential expression analysis | This paper | Zenodo |
| RNA in situ hybridization for Galnt17 in mouse and jerboa growth cartilages. | This paper | Zenodo |
| DESeq2 MA-plot for Jerboa and Mouse metatarsal comparison | This paper | Zenodo |
| Data tables with STAR gene counts and DESeq2 differential expression results for all of the 17,464 mouse and jerboa orthologous. | This paper | Zenodo |
| Experimental models: Cell lines | ||
| DF-1 Chicken Fibroblast Cells | Dr. Clifford J. Tabin’s Laboratory, Harvard University | N/A |
| Experimental models: Organisms/strains | ||
| Mus musculus (CD-1 Strain) | Charles River Labs | N/A |
| Jaculus jaculus | Dr. Kim Cooper’s Lab | N/A |
| Gallus gallus (SPF eggs; White Leghorn Strain) | Charles River Labs | N/A |
| Oligonucleotides | ||
| Fwd (5’-3’): AGAGCTCCGCAGAAAGCGAC Rev (5’-3’): GCGTCTTCAGCACCGAGAGG | This Study | Mouse Mab21L2, qRT-PCR primers |
| Fwd (5’-3’): ACCGCGACGTGGTCAAGATG Rev (5’-3’): CAGGGGATATGGGGCATGGG | This Study | Jerboa Mab21L2, qRT-PCR primers |
| Fwd (5’-3’): ACGGAGGTGTCCCCTGAACT Rev (5’-3’): CGCCTCTGCTTGATTTTGGT | Cobb & Duboule108 | Mouse Shox2, qRT-PCR primers |
| Fwd (5’-3’): AAAGGTGTTCTCATAGGGGCTGCT Rev (5’-3’):CGTCACGTTGCAATGACTATCCTGC | This Study | Jerboa Shox2, qRT-PCR primers |
|
Fwd1 (5’-3’): GTTTGCAGAGCCCAGTGTCC. Rev1 (5’-3’): GATAGCCCCGAGGAACAGTC Fwd2 (5’-3’): CAACGCTGATGCCAACTGTC. Rev2 (5’-3’): GACACTGGGCTCTGCAAAC |
This Study | Mouse HoxB13, PCR primers |
| Fwd (5’-3’): CAGGAGGGGGTCGGAATCTA Rev (5’-3’): GCACGCTCGACTCTGCAAAT | This Study | Jerboa HoxB13, PCR primers |
| Fwd (5’-3’): GGAACACTCCAAAAACAGACCT Rev (5’-3’): CCACCACTGGGTATTGAGTAG | This Study | Mouse Sdha, qRT-PCR primers |
| Fwd (5’-3’): ACTGGAGGTGGCATTTCTAC Rev (5’-3’): TTTTCTAGCTCGACCACAGATG | This Study | Jerboa Sdha, qRT-PCR primers |
| Recombinant DNA | ||
| Software and algorithms | ||
| JMP | SAS Institute Inc. | N/A |
| DESEQ2 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | N/A |
| STAR | https://github.com/alexdobin/STAR | N/A |
| Ingenuity Pathway Analysis | Qiagen | N/A |
| CESAR | https://github.com/hillerlab/CESAR | N/A |
| liftOver | https://genome.ucsc.edu/cgi-bin/hgLiftOver | N/A |
| Other | ||
Growth Cartilage Section in situs:
Metatarsal (MT) and radius/ulna (RU) growth cartilages from P5 animals were fixed in 4% paraformaldehyde (PFA) in 1X PBS for 24 hours at 4°C. Samples were allowed to equilibrate in 20% sucrose-PBS for 2 days at 4°C. Cartilages were mounted in Tissue-Tek OCT (Sakura) or SCEM compound (SECTION-LAB Co. Ltd., Japan) and cryo-sectioned at 20–30 μm thickness. Species-specific digoxigenin (DIG) labeled riboprobes were generated by in vitro transcription of Crabp1 fragments cloned in pGEMT-Easy (Promega) vector. Colorimetric in situs were performed on neonatal limb sections following a standard protocol as previously described107. For RNAScope23 hybridization (Advanced Cell Diagnostics, Inc.), custom designed ZZ probes were used to detect Shox2, Mab21L2, Gdf10, Pax1, HoxB9 HoxB13, and Prrx1 transcripts in P5 growth cartilages of jerboas and mice. Hybridization was performed following manufacturer’s protocol for RED detection kits cat # 322360 and cat # 323910. ACD Protease Plus or Protease-III solutions were used for tissue permeabilization prior to probe hybridization. Probe hybridization was detected with Fast Red chromogenic dye. Tissue sections were counterstained with 50% Gill1 hematoxylin solution and were rinsed in ammonium hydroxide solution to achieve blue nuclear staining. The red chromogenic in situ staining was assessed under visible (DIC) and UV (550 nm) light on an Olympus Model BX61 compound microscope with a 20x N.A. 0.75 objective, and a Nikon Eclipse Ti2 microscope with a 40x N.A. 0.9 objective.
GFP and Mab21L2 RCAS Injections in Chickens:
RCAS vectors that improve the ease of cloning cargo genes, RCASBP(A)-F1 and RCASBP(A)-F1::GFP, were kind gifts from Drs. Andrea L Ferris and Stephen H Hughes (National Cancer Institute, NIH, MD, USA). The 1080 bp jerboa Mab21L2 (XM_004655859, NCBI) CDS region was synthesised as a gene-block (Integrated DNA Technologies, Inc.) and cloned between ClaI and MluI restriction sites of RCASBP(A)-F1. DF-1 chicken fibroblast cells (kind gift of Prof. Cliff Tabin, Harvard University, USA) were used to produce virus. Virus production, concentration and pressurized injections into 3–3.5 day old (Hamilton-Hamburg stages 19–22) chicken forelimb buds were performed as previously described108. Embryos were collected 13–14 days after injection. A subset of forelimbs from each injection cohort were sectioned and stained with AMV-3C2 antibody (1:50, Developmental Studies Hybridoma Bank) to confirm widespread RCAS viral infection only in the injected limb.
4. QUANTIFICATION AND STATISTICAL ANALYSIS:
Test for Significance of Overlap:
Fisher’s exact test was performed using the GeneOverlap package109 (version 1.20.0) in R to test the statistical significance of overlap between DESeq2 differentially expressed genes in mouse and jerboa for the three primary samples and for the two independently analyzed samples (Figure S4B–C).
We also used Fisher’s exact test to evaluate the significance of overlap between gene lists from multiple differential growth datasets in our meta-analysis (Table 1). For these comparisons, we first identified genes within each of these datasets that were assigned the same gene name in our 1:1 jerboa and mouse orthologous reference set. Of these, there are 287 differentially expressed genes associated with growth rate slowing during tibia maturation, 493 genes that are associated with growth rate differences by location, and 99 unique genes assigned to SNPs with nominal significance in the human sitting height ratio GWAS (Table S5). Overlapping genes in Table 1 were then searched against the Mouse Genome Informatics database (Jackson Laboratory) to obtain a curation of known phenotypes. These phenotypes were then confirmed by reading the primary citations.
Ingenuity Pathway Analysis (IPA, Qiagen):
IPA was performed using log2 fold change values of 1,755 genes that are disproportionately differentially expressed between jerboa and mouse metatarsals. We used a ‘user defined reference set’ of 15,947 genes that were detected in mouse and/or jerboa metatarsal growth cartilages (basemean count >0 in the DESeq2 differential expression analysis). A Fisher’s exact test was used in IPA to determine significantly enriched signaling pathways, cellular & developmental processes, and to predict upstream regulatory networks in the Ingenuity Knowledge Base restricted to experimentally validated interactions. IPA platform was also used to compute z-score to determine activation and inhibition states shown in Figure 2D–E and Figure S5. Enriched Disease and Development terms in Figure 2D are selected from hundreds of significantly enriched annotations based on a priori interest in cartilage biology.
JMP®, Versions 14 and 15 (SAS Institute Inc., Cary, NC, 1989–2019) were used to generate all graphs and to perform the Wilcoxon test and paired t-tests to assess significance of differences.
Supplementary Material
Table S1: Genes with non-equivalent differences in Metatarsals and Radius/Ulna comparisons (padj<0.05 for MT); n=3, Related to Figure 2.
Table S2: Genes with significant expression differences ONLY in Metatarsal comparisons (padj<0.05 for MT); n=3, Related to Figure 2.
Table S3: Genes with non-equivalent differences in Metatarsals and Radius/Ulna comparisons (padj<0.05 for MT); n=2, Related to Figure 2.
Table S4: Genes with significant expression differences ONLY in Metatarsal comparisons (padj<0.05 for MT); n=2, Related to Figure 2.
Table S5: A list of gene IDs used for GeneOverlap analysis, Related to Table 1.
Table S6: Canonical pathways and disease function terms from Ingenuity Pathway Analysis, Related to Figure 2 and Figure 3.
Table S7: Networks discovered with Ingenuity Pathway Analysis, Related to Figure 3.
Highlights.
About 10% of genes are associated with disproportionate growth of jerboa feet.
Pathway and network analyses reveal known and novel growth mechanisms.
Putative mechanisms might directly accelerate and also de-repress foot growth.
Derived Shox2 expression associates with a novel putative jerboa regulatory sequence.
Acknowledgements
We are grateful to Dr. Wayne Pfeiffer and Dr. Mahidhar Tatineni at the San Diego Super Computing Cluster (SDSC) for computational assistance and to Dr. David Traver for microinjector usage. We are grateful to Dr. Fan Wang (Duke University) for supplying the Rosa26 mice. We thank Dr. Stephen Hughes and Andrea Ferris (NCI-NIH) for RCAS vectors. We also thank Dr. C. Tabin, Dr. J. Posakony, Dr. J. Monda and A. Weitzel for comments on the manuscript. This work used the Extreme Science and Engineering Discovery Environment (XSEDE) at SDSC, which is supported by National Science Foundation grant number ACI-1548562. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health SIG grant (#S10 OD026929). We are grateful to Drs Daphne Bindels and Eric Griffis at UC San Diego Nikon imaging center and Dr. Kristen Jepsen at the UC San Diego IGM Genomics Center for their help and guidance. This work was also supported by Natural Sciences and Engineering Research Council grant RGPIN/355731-2013 to JC and by a Searle Scholar Award from the Kinship Foundation, a Pew Biomedical Scholar Award from the Pew Charitable Trusts, a Packard Fellowship in Science and Engineering from the David and Lucile Packard Foundation, and by the National Institutes of Health under award number R01AR075415 awarded to KLC.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Inclusion and Diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper self-identifies as living with a disability. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kronenberg HM (2003). Developmental regulation of the growth plate. Nature 423, 332–336. [DOI] [PubMed] [Google Scholar]
- 2.Kozhemyakina E, Lassar AB, and Zelzer E (2015). A pathway to bone: signaling molecules and transcription factors involved in chondrocyte development and maturation. Development 142, 817–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krakow D, and Rimoin DL (2010). The skeletal dysplasias. Genet. Med 12, 327–341. [DOI] [PubMed] [Google Scholar]
- 4.Christians JK, Bingham VK, Oliver FK, Heath TT, and Keightley PD (2003). Characterization of a QTL affecting skeletal size in mice. Mamm. Genome 14, 175–183. [DOI] [PubMed] [Google Scholar]
- 5.Parker HG, VonHoldt BM, Quignon P, Margulies EH, Shao S, Mosher DS, Spady TC, Elkahloun A, Cargill M, Jones PG, et al. (2009). An Expressed Fgf4 Retrogene Is Associated with Breed-Defining Chondrodysplasia in Domestic Dogs. Science 325, 995–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan Y, Salem RM, Hsu Y-HH, McMahon G, Pers TH, Vedantam S, Esko T, Guo MH, Lim ET, Franke L, et al. (2015). Genome-wide Analysis of Body Proportion Classifies Height-Associated Variants by Mechanism of Action and Implicates Genes Important for Skeletal Development. Am. J. Hum. Genet 96, 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castro JP, Yancoskie MN, Marchini M, Belohlavy S, Hiramatsu L, Kučka M, Beluch WH, Naumann R, Skuplik I, Cobb J, et al. (2019). An integrative genomic analysis of the Longshanks selection experiment for longer limbs in mice. eLife 8, e42014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar S, Stecher G, Suleski M, and Hedges SB (2017). TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol 34, 1812–1819. [DOI] [PubMed] [Google Scholar]
- 9.Moore TY, Organ CL, Edwards SV, Biewener AA, Tabin CJ, Jenkins FA Jr., and Cooper KL (2015). Multiple Phylogenetically Distinct Events Shaped the Evolution of Limb Skeletal Morphologies Associated with Bipedalism in the Jerboas. Curr. Biol 25, 2785–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grüneberg H (1963). The pathology of development;a study of inherited skeletal disorders in animals. (New York, Wiley; ). [Google Scholar]
- 11.Wilsman NJ, Farnum CE, Leiferman EM, Fry M, and Barreto C (1996). Differential growth by growth plates as a function of multiple parameters of chondrocytic kinetics. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 14, 927–936. [DOI] [PubMed] [Google Scholar]
- 12.Lui JC, Jee YH, Garrison P, Iben JR, Yue S, Ad M, Nguyen Q, Kikani B, Wakabayashi Y, and Baron J (2018). Differential aging of growth plate cartilage underlies differences in bone length and thus helps determine skeletal proportions. PLOS Biol. 16, e2005263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie M, Gol’din P, Herdina AN, Estefa J, Medvedeva EV, Li L, Newton PT, Kotova S, Shavkuta B, Saxena A, et al. (2020). Secondary ossification center induces and protects growth plate structure. eLife 9, e55212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper KL, Oh S, Sung Y, Dasari RR, Kirschner MW, and Tabin CJ (2013). Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 495, 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kronenberg HM (2007). The Role of the Perichondrium in Fetal Bone Development. Ann. N. Y. Acad. Sci 1116, 59–64. [DOI] [PubMed] [Google Scholar]
- 16.Sharma V, Elghafari A, and Hiller M (2016). Coding exon-structure aware realigner (CESAR) utilizes genome alignments for accurate comparative gene annotation. Nucleic Acids Res. 44, e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, et al. (2016). A survey of best practices for RNA-seq data analysis. Genome Biol. 17, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marioni JC, Mason CE, Mane SM, Stephens M, and Gilad Y (2008). RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 18, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morey JS, Ryan JC, and Van Dolah FM (2006). Microarray validation: factors influencing correlation between oligonucleotide microarrays and real-time PCR. Biol. Proced Online 8, 175–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffith M, Griffith OL, Mwenifumbo J, Goya R, Morrissy AS, Morin RD, Corbett R, Tang MJ, Hou Y-C, Pugh TJ, et al. (2010). Alternative expression analysis by RNA sequencing. Nat. Methods 7, 843–847. [DOI] [PubMed] [Google Scholar]
- 22.Nolan T, Hands RE, and Bustin SA (2006). Quantification of mRNA using real-time RT-PCR. Nat. Protoc 1, 1559–1582. [DOI] [PubMed] [Google Scholar]
- 23.Wang F, Flanagan J, Su N, Wang L-C, Bui S, Nielson A, Wu X, Vo H-T, Ma X-J, and Luo Y (2012). RNAscope: A Novel in Situ RNA Analysis Platform for Formalin-Fixed, Paraffin-Embedded Tissues. J. Mol. Diagn 14, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lescroart F, Wang X, Lin X, Swedlund B, Gargouri S, Sànchez-Dànes A, Moignard V, Dubois C, Paulissen C, Kinston S, et al. (2018). Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 359, 1177–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin A, Li C, Xing Z, Hu Q, Liang K, Han L, Wang C, Hawke DH, Wang S, Zhang Y, et al. (2016). The LINK-A lncRNA activates normoxic HIF1α signalling in triple-negative breast cancer. Nat. Cell Biol 18, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silberstein L, Goncalves KA, Kharchenko PV, Turcotte R, Kfoury Y, Mercier F, Baryawno N, Severe N, Bachand J, Spencer JA, et al. (2016). Proximity-Based Differential Single-Cell Analysis of the Niche to Identify Stem/Progenitor Cell Regulators. Cell Stem Cell 19, 530–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cimino PJ, Zhao G, Wang D, Sehn JK, Lewis JS, and Duncavage EJ (2014). Detection of viral pathogens in high grade gliomas from unmapped next-generation sequencing data. Exp. Mol. Pathol 96, 310–315. [DOI] [PubMed] [Google Scholar]
- 28.Narisawa S, Fröhlander N, and Millán JL (1997). Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev. Dyn 208, 432–446. [DOI] [PubMed] [Google Scholar]
- 29.Harmey D, Johnson KA, Zelken J, Camacho NP, Hoylaerts MF, Noda M, Terkeltaub R, and Millán JL (2006). Elevated Skeletal Osteopontin Levels Contribute to the Hypophosphatasia Phenotype in Akp2−/− Mice. J. Bone Miner. Res 21, 1377–1386. [DOI] [PubMed] [Google Scholar]
- 30.Foster BL, Sheen CR, Hatch NE, Liu J, Cory E, Narisawa S, Kiffer-Moreira T, Sah RL, Whyte MP, Somerman MJ, et al. (2015). Periodontal Defects in the A116T Knock-in Murine Model of Odontohypophosphatasia. J. Dent. Res 94, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waymire KG, Mahuren JD, Jaje JM, Guilarte TR, Coburn SP, and MacGregor GR (1995). Mice lacking tissue non–specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B–6. Nat. Genet 11, 45–51. [DOI] [PubMed] [Google Scholar]
- 32.Malaval L, Wade-Guéye NM, Boudiffa M, Fei J, Zirngibl R, Chen F, Laroche N, Roux J-P, Burt-Pichat B, Duboeuf F, et al. (2008). Bone sialoprotein plays a functional role in bone formation and osteoclastogenesis. J. Exp. Med 205, 1145–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foster BL, Ao M, Willoughby C, Soenjaya Y, Holm E, Lukashova L, Tran AB, Wimer HF, Zerfas PM, Nociti FH, et al. (2015). Mineralization defects in cementum and craniofacial bone from loss of bone sialoprotein. Bone 78, 150–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lane PW (1972). Two New Mutations in Linkage Group XVI of the House MouseFlaky tail and varitint-waddler-J. J. Hered 63, 135–140. [DOI] [PubMed] [Google Scholar]
- 35.Tullio AN, Accili D, Ferrans VJ, Yu Z-X, Takeda K, Grinberg A, Westphal H, Preston YA, and Adelstein RS (1997). Nonmuscle myosin II-B is required for normal development of the mouse heart. Proc. Natl. Acad. Sci 94, 12407–12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esapa CT, Piret SE, Nesbit MA, Loh NY, Thomas G, Croucher PI, Brown MA, Brown SDM, Cox RD, and Thakker RV (2016). Mice with an N-Ethyl-N-Nitrosourea (ENU) Induced Tyr209Asn Mutation in Natriuretic Peptide Receptor 3 (NPR3) Provide a Model for Kyphosis Associated with Activation of the MAPK Signaling Pathway. PLOS ONE 11, e0167916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaubert J, Jaubert F, Martin N, Washburn LL, Lee BK, Eicher EM, and Guénet J-L (1999). Three new allelic mouse mutations that cause skeletal overgrowth involve the natriuretic peptide receptor C gene (Npr3). Proc. Natl. Acad. Sci 96, 10278–10283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsukawa N, Grzesik WJ, Takahashi N, Pandey KN, Pang S, Yamauchi M, and Smithies O (1999). The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Proc. Natl. Acad. Sci 96, 7403–7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dauphinee SM, Eva MM, Yuki KE, Herman M, Vidal SM, and Malo D (2013). Characterization of Two ENU-Induced Mutations Affecting Mouse Skeletal Morphology. G3 Genes Genomes Genet. 3, 1753–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bussen M, Petry M, Schuster-Gossler K, Leitges M, Gossler A, and Kispert A (2004). The T-box transcription factor Tbx18 maintains the separation of anterior and posterior somite compartments. Genes Dev. 18, 1209–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grisanti L, Clavel C, Cai X, Rezza A, Tsai S-Y, Sennett R, Mumau M, Cai C-L, and Rendl M (2013). Tbx18 Targets Dermal Condensates for Labeling, Isolation, and Gene Ablation during Embryonic Hair Follicle Formation. J. Invest. Dermatol 133, 344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu S-P, Dong X-R, Regan JN, Su C, and Majesky MW (2013). Tbx18 regulates development of the epicardium and coronary vessels. Dev. Biol 383, 307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stoeger T, Gerlach M, Morimoto RI, and Amaral LAN (2018). Large-scale investigation of the reasons why potentially important genes are ignored. PLOS Biol. 16, e2006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krämer A, Green J, Pollard J, and Tugendreich S (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu S, Kuwahara M, Uchida Y, Kondo S, Hayashi D, Shimomura Y, Takagaki A, Nishida T, Maruyama Y, Ikegame M, et al. (2019). Circadian production of melatonin in cartilage modifies rhythmic gene expression. J. Endocrinol 241, 161–173. [DOI] [PubMed] [Google Scholar]
- 46.Gao W, Lin M, Liang A, Zhang L, Chen C, Liang G, Xu C, Peng Y, Chen C, Huang D, et al. (2014). Melatonin enhances chondrogenic differentiation of human mesenchymal stem cells. J. Pineal Res 56, 62–70. [DOI] [PubMed] [Google Scholar]
- 47.Aszodi A, Hunziker EB, Brakebusch C, and Fässler R (2003). β1 integrins regulate chondrocyte rotation, G1 progression, and cytokinesis. Genes Dev. 17, 2465–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terpstra L, Prud’homme J, Arabian A, Takeda S, Karsenty G, Dedhar S, and St-Arnaud R (2003). Reduced chondrocyte proliferation and chondrodysplasia in mice lacking the integrin-linked kinase in chondrocytes. J. Cell Biol 162, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki W, Yamada A, Aizawa R, Suzuki D, Kassai H, Harada T, Nakayama M, Nagahama R, Maki K, Takeda S, et al. (2015). Cdc42 Is Critical for Cartilage Development During Endochondral Ossification. Endocrinology 156, 314–322. [DOI] [PubMed] [Google Scholar]
- 50.Teufel S, and Hartmann C (2019). Wnt-signaling in skeletal development. In Vertebrate Skeletal Development, Olsen BR, ed. (Elsevier Academic Press Inc; ), pp. 235–+. [DOI] [PubMed] [Google Scholar]
- 51.Cretekos CJ, Wang Y, Green ED, Program NCS, Martin JF, Rasweiler JJ, and Behringer RR (2008). Regulatory divergence modifies limb length between mammals. Genes Dev. 22, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aubin J, Lemieux M, Moreau J, Lapointe J, and Jeannotte L (2002). Cooperation of Hoxa5 and Pax1 genes during formation of the pectoral girdle. Dev. Biol 244, 96–113. [DOI] [PubMed] [Google Scholar]
- 53.Carlson MRJ, Komine Y, Bryant SV, and Gardiner DM (2001). Expression of Hoxb13 and Hoxc10 in Developing and Regenerating Axolotl Limbs and Tails. Dev. Biol 229, 396–406. [DOI] [PubMed] [Google Scholar]
- 54.Zeltser L, Desplan C, and Heintz N (1996). Hoxb-13: a new Hox gene in a distant region of the HOXB cluster maintains colinearity. Development 122, 2475–2484. [DOI] [PubMed] [Google Scholar]
- 55.Economides KD, Zeltser L, and Capecchi MR (2003). Hoxb13 mutations cause overgrowth of caudal spinal cordand tail vertebrae. Dev. Biol 256, 317–330. [DOI] [PubMed] [Google Scholar]
- 56.Bandyopadhyay A, Kubilus JK, Crochiere ML, Linsenmayer TF, and Tabin CJ (2008). Identification of unique molecular subdomains in the perichondrium and periosteum and their role in regulating gene expression in the underlying chondrocytes. Dev. Biol 321, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Villavicencio-Lorini P, Kuss P, Friedrich J, Haupt J, Farooq M, Türkmen S, Duboule D, Hecht J, and Mundlos S (2010). Homeobox genes d11–d13 and a13 control mouse autopod cortical bone and joint formation. J. Clin. Invest 120, 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuss P, Kraft K, Stumm J, Ibrahim D, Vallecillo-Garcia P, Mundlos S, and Stricker S (2014). Regulation of cell polarity in the cartilage growth plate and perichondrium of metacarpal elements by HOXD13 and WNT5A. Dev. Biol 385, 83–93. [DOI] [PubMed] [Google Scholar]
- 59.de Bruijn DRH, Oerlemans F, Hendriks W, Baats E, Ploemacher R, Wieringa B, and van Kessel AG (1995). Normal development, growth and reproduction in cellular retinoic acid binding protein-I (CRABPI) null mutant mice. Differentiation 58, 141–148. [DOI] [PubMed] [Google Scholar]
- 60.Zhao R, Lawler AM, and Lee S-J (1999). Characterization of GDF-10 Expression Patterns and Null Mice. Dev. Biol 212, 68–79. [DOI] [PubMed] [Google Scholar]
- 61.Dong D, Ruuska SE, Levinthal DJ, and Noy N (1999). Distinct Roles for Cellular Retinoic Acid-binding Proteins I and II in Regulating Signaling by Retinoic Acid. J. Biol. Chem 274, 23695–23698. [DOI] [PubMed] [Google Scholar]
- 62.Boylan JF, and Gudas LJ (1991). Overexpression of the cellular retinoic acid binding protein-I (CRABP-I) results in a reduction in differentiation-specific gene expression in F9 teratocarcinoma cells. J. Cell Biol 112, 965–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu R-Z, Garcia E, Glubrecht DD, Poon HY, Mackey JR, and Godbout R (2015). CRABP1 is associated with a poor prognosis in breast cancer: adding to the complexity of breast cancer cell response to retinoic acid. Mol. Cancer 14, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Luca F, Uyeda JA, Mericq V, Mancilla EE, Yanovski JA, Barnes KM, Zile MH, and Baron J (2000). Retinoic Acid Is a Potent Regulator of Growth Plate Chondrogenesis. Endocrinology 141, 346–353. [DOI] [PubMed] [Google Scholar]
- 65.Galdones E, and Hales BF (2008). Retinoic Acid Receptor Gamma-Induced Misregulation of Chondrogenesis in the Murine Limb Bud In Vitro. Toxicol. Sci 106, 223–232. [DOI] [PubMed] [Google Scholar]
- 66.Baldessari D, Badaloni A, Longhi R, Zappavigna V, and Consalez GG (2004). MAB21L2, a vertebrate member of the Male-abnormal 21 family, modulates BMP signaling and interacts with SMAD1. BMC Cell Biol. 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yi SE, Daluiski A, Pederson R, Rosen V, and Lyons KM (2000). The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development 127, 621–630. [DOI] [PubMed] [Google Scholar]
- 68.Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, and Lyons KM (2005). Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc. Natl. Acad. Sci. U. S. A 102, 5062–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duprez D, de H, Bell EJ, Richardson MK, Archer CW, Wolpert L, Brickell PM, and Francis-West PH (1996). Overexpression of BMP-2 and BMP-4 alters the size and shape of developing skeletal elements in the chick limb. Mech. Dev 57, 145–157. [DOI] [PubMed] [Google Scholar]
- 70.Dai M, Wang Y, Fang L, Irwin DM, Zhu T, Zhang J, Zhang S, and Wang Z (2014). Differential Expression of Meis2, Mab21l2 and Tbx3 during Limb Development Associated with Diversification of Limb Morphology in Mammals. PLOS ONE 9, e106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamada R, Mizutani-Koseki Y, Koseki H, and Takahashi N (2004). Requirement for Mab21l2 during development of murine retina and ventral body wall. Dev. Biol 274, 295–307. [DOI] [PubMed] [Google Scholar]
- 72.Rainger J, Pehlivan D, Johansson S, Bengani H, Sanchez-Pulido L, Williamson KA, Ture M, Barker H, Rosendahl K, Spranger J, et al. (2014). Monoallelic and biallelic mutations in MAB21L2 cause a spectrum of major eye malformations. Am. J. Hum. Genet 94, 915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi Y, and Massagué J (2003). Mechanisms of TGF-β Signaling from Cell Membrane to the Nucleus. Cell 113, 685–700. [DOI] [PubMed] [Google Scholar]
- 74.Zhang J, Tan X, Li W, Wang Y, Wang J, Cheng X, and Yang X (2005). Smad4 is required for the normal organization of the cartilage growth plate. Dev. Biol 284, 311–322. [DOI] [PubMed] [Google Scholar]
- 75.Capdevila J, and Johnson RL (1998). Endogenous and ectopic expression of noggin suggests a conserved mechanism for regulation of BMP function during limb and somite patterning. Dev. Biol 197, 205–217. [DOI] [PubMed] [Google Scholar]
- 76.Pizette S, and Niswander L (2000). BMPs Are Required at Two Steps of Limb Chondrogenesis: Formation of Prechondrogenic Condensations and Their Differentiation into Chondrocytes. Dev. Biol 219, 237–249. [DOI] [PubMed] [Google Scholar]
- 77.Capdevila J, Tsukui T, Esteban CR, Zappavigna V, and Belmonte JCI (1999). Control of Vertebrate Limb Outgrowth by the Proximal Factor Meis2 and Distal Antagonism of BMPs by Gremlin. Mol. Cell 4, 839–849. [DOI] [PubMed] [Google Scholar]
- 78.Merino R, Rodriguez-Leon J, Macias D, Ganan Y, Economides AN, and Hurle JM (1999). The BMP antagonist Gremlin regulates outgrowth, chondrogenesis and programmed cell death in the developing limb. Development 126, 5515–5522. [DOI] [PubMed] [Google Scholar]
- 79.Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A, Muroya K, Binder G, Kirsch S, Winkelmann M, et al. (1997). Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat. Genet 16, 54–63. [DOI] [PubMed] [Google Scholar]
- 80.Ellison JW, Wardak Z, Young MF, Gehron Robey P, Laig-Webster M, and Chiong W (1997). PHOG, a Candidate Gene for Involvement in the Short Stature of Turner Syndrome. Hum. Mol. Genet 6, 1341–1347. [DOI] [PubMed] [Google Scholar]
- 81.Zinn AR, Wei F, Zhang L, Elder FF, Scott CI, Marttila P, and Ross JL (2002). Complete SHOX deficiency causes Langer mesomelic dysplasia. Am. J. Med. Genet 110, 158–163. [DOI] [PubMed] [Google Scholar]
- 82.Cobb J, Dierich A, Huss-Garcia Y, and Duboule D (2006). A mouse model for human short-stature syndromes identifies Shox2 as an upstream regulator of Runx2 during long-bone development. Proc. Natl. Acad. Sci 103, 4511–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu L, Liu H, Yan M, Yang J, Long F, Muneoka K, and Chen Y (2007). Shox2 is required for chondrocyte proliferation and maturation in proximal limb skeleton. Dev. Biol 306, 549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bobick BE, and Cobb J (2012). Shox2 regulates progression through chondrogenesis in the mouse proximal limb. J. Cell Sci 125, 6071–6083. [DOI] [PubMed] [Google Scholar]
- 85.Tiecke E, Bangs F, Blaschke R, Farrell ER, Rappold G, and Tickle C (2006). Expression of the short stature homeobox gene Shox is restricted by proximal and distal signals in chick limb buds and affects the length of skeletal elements. Dev. Biol 298, 585–596. [DOI] [PubMed] [Google Scholar]
- 86.Neufeld SJ, Wang F, and Cobb J (2014). Genetic Interactions Between Shox2 and Hox Genes During the Regional Growth and Development of the Mouse Limb. Genetics 198, 1117–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, and Tabin CJ (2002). Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. genesis 33, 77–80. [DOI] [PubMed] [Google Scholar]
- 88.Scott A, Hasegawa H, Sakurai K, Yaron A, Cobb J, and Wang F (2011). Transcription Factor Short Stature Homeobox 2 Is Required for Proper Development of Tropomyosin-Related Kinase B-Expressing Mechanosensory Neurons. J. Neurosci 31, 6741–6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Osterwalder M, Barozzi I, Tissières V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, Lee EA, Zhu Y, Plajzer-Frick I, Pickle CS, et al. (2018). Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ye W, Song Y, Huang Z, Osterwalder M, Ljubojevic A, Xu J, Bobick B, Abassah-Oppong S, Ruan N, Shamby R, et al. (2016). A unique stylopod patterning mechanism by Shox2 - controlled osteogenesis. Development 143, 2548–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jordan B, Vercammen P, and Cooper KL (2011). Husbandry and Breeding of the Lesser Egyptian Jerboa, Jaculus jaculus. Cold Spring Harb. Protoc 2011, pdb.prot066712. [DOI] [PubMed] [Google Scholar]
- 93.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharma V, and Hiller M (2017). Increased alignment sensitivity improves the usage of genome alignments for comparative gene annotation. Nucleic Acids Res. 45, 8369–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Harris RS (2007). Improved pairwise alignment of genomic DNA. Ph.D. Thesis. (The Pennsylvania State University). [Google Scholar]
- 96.Kent WJ, Baertsch R, Hinrichs A, Miller W, and Haussler D (2003). Evolution’s cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci 100, 11484–11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Osipova E, Hecker N, and Hiller M (2019). RepeatFiller newly identifies megabases of aligning repetitive sequences and improves annotations of conserved non-exonic elements. GigaScience 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Suarez HG, Langer BE, Ladde P, and Hiller M (2017). chainCleaner improves genome alignment specificity and sensitivity. Bioinformatics 33, 1596–1603. [DOI] [PubMed] [Google Scholar]
- 99.Sharma V, Hecker N, Roscito JG, Foerster L, Langer BE, and Hiller M (2018). A genomics approach reveals insights into the importance of gene losses for mammalian adaptations. Nat. Commun 9, 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinforma. Oxf. Engl 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Guo M, Liu Z, Willen J, Shaw CP, Richard D, Jagoda E, Doxey AC, Hirschhorn J, and Capellini TD (2017). Epigenetic profiling of growth plate chondrocytes sheds insight into regulatory genetic variation influencing height. eLife 6, e29329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gaspar JM (2018). NGmerge: merging paired-end reads via novel empirically-derived models of sequencing errors. BMC Bioinformatics 19, 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li Q, Brown JB, Huang H, and Bickel PJ (2011). Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat 5, 1752–1779. [Google Scholar]
- 107.McGlinn E, and Mansfield JH (2011). Detection of gene expression in mouse embryos and tissue sections. Methods Mol. Biol. Clifton NJ 770, 259–292. [DOI] [PubMed] [Google Scholar]
- 108.Logan M, and Tabin C (1998). Targeted gene misexpression in chick limb buds using avian replication-competent retroviruses. Methods San Diego Calif 14, 407–420. [DOI] [PubMed] [Google Scholar]
- 109.Shen L (2019). GeneOverlap: Test and visualize gene overlaps. R package version 1.20.0, http://shenlab-sinai.github.io/shenlab-sinai/. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Genes with non-equivalent differences in Metatarsals and Radius/Ulna comparisons (padj<0.05 for MT); n=3, Related to Figure 2.
Table S2: Genes with significant expression differences ONLY in Metatarsal comparisons (padj<0.05 for MT); n=3, Related to Figure 2.
Table S3: Genes with non-equivalent differences in Metatarsals and Radius/Ulna comparisons (padj<0.05 for MT); n=2, Related to Figure 2.
Table S4: Genes with significant expression differences ONLY in Metatarsal comparisons (padj<0.05 for MT); n=2, Related to Figure 2.
Table S5: A list of gene IDs used for GeneOverlap analysis, Related to Table 1.
Table S6: Canonical pathways and disease function terms from Ingenuity Pathway Analysis, Related to Figure 2 and Figure 3.
Table S7: Networks discovered with Ingenuity Pathway Analysis, Related to Figure 3.
Data Availability Statement
The RNA-Seq and ATAC-Seq illumina reads generated in this study have been submitted as fastq files to the Zenodo repository. These datasets are publicly accessible at https://doi.org/10.5281/zenodo.5123254 and https://doi.org/10.5281/zenodo.5120588.
All original code and additional tables with STAR gene counts, rlog transformed gene counts, and DESeq2 differential expression results for all the 17,464 mouse and jerboa orthologs are publicly available on the Zenodo repository. These datasets can be accessed at https://doi.org/10.5281/zenodo.5123384.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.