

SUMMARY
The epitranscriptome has emerged as a new fundamental layer of control of gene expression. Nevertheless, the determination of the transcriptome-wide occupancy and function of RNA modifications remains challenging. Here we have developed Rho-seq, an integrated pipeline detecting a range of modifications through differential modification-dependent rhodamine labeling. Using Rho-seq, we confirm that the reduction of uridine to dihydrouridine (D) by the Dus reductase enzymes targets tRNAs in E. coli and fission yeast. We find that the D modification is also present on fission yeast mRNAs, particularly those encoding cytoskeleton-related proteins, which is supported by large-scale proteome analyses and ribosome profiling. We show that the α-tubulin encoding mRNA nda2 undergoes Dus3-dependent dihydrouridylation, which affects its translation. The absence of the modification on nda2 mRNA strongly impacts meiotic chromosome segregation, resulting in low gamete viability. Applying Rho-seq to human cells revealed that tubulin mRNA dihydrouridylation is evolutionarily conserved.
In brief
Finet et al. report Rho-seq, a new method to determine the transcriptome occupancy of a range of RNA modifications. Rho-seq reveals that the dihydrouridine tRNA modification expands to a subset of fission yeast mRNAs. The modification of the tubulin mRNA is essential for proper meiotic chromosome segregation and gamete viability.
Graphical abstract
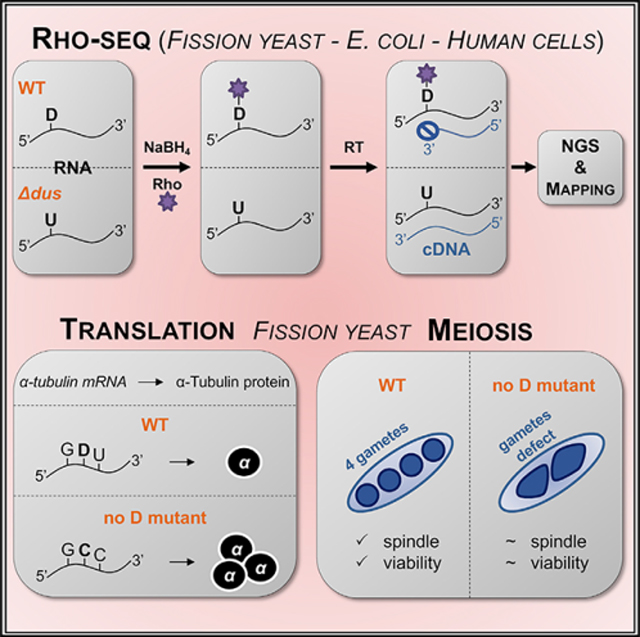
INTRODUCTION
Dihydrouridine (D) results from reduction of uridine, making it a fully saturated pyrimidine ring with no remaining double bonds, and is the second most abundant modified nucleoside found in tRNAs (Machnicka et al., 2014), predominantly installed within the eponym loop (D-loop). The conversion of uridine to D is catalyzed by Dus enzymes (dihydrouridine synthetases) using NADPH as a cofactor (Xing et al., 2002). In budding yeast, there are four Dus enzymes, each displaying distinct specificities (Xing et al., 2004), with each enzyme reducing either one or two specific uridines in tRNAs (Figure S1A). Most bacteria have only three Dus enzymes retaining this exquisite specificity (Kasprzak et al., 2012). A unique feature of D is its ability to remove the aromaticity of the ring. The incorporation of this nonplanar molecule therefore has the potential to interfere with base stacking within an RNA molecule with consequences on base-pairing and folding.
The flexibility introduced by D is correlated with cell adaptation to the environment, with psychrophilic and thermophilic organisms having either more or less D, respectively, per tRNA (Dalluge et al., 1997; Edmonds et al., 1991). From a biomedical standpoint, cancer cells have higher D levels than non-cancerous cells, which correlates with Dus2 overexpression and suppression of tumor growth after its depletion (Kato et al., 2005).
D associates with plant and mammalian histone-bound RNAs (Huang and Bonner, 1965; Shih and Bonner, 1969), suggesting that its occurrence may not be restricted to tRNAs, which remains to be established. Here we have developed Rho-seq, a method to determine the transcriptome-wide distribution of D. Our analyses confirmed the abundant presence of D on E. coli tRNAs and revealed that D is present on a subset of fission yeast mRNAs. Detailed analysis of one modified site on the nda2 tubulin encoding mRNA shows that it controls its translation efficiency (TE) and is essential for meiotic spindle dynamics and gamete viability.
RESULTS
We have reported that the long non-coding RNA rse1, which regulates gametogenesis in fission yeast, potentially interacts with the tRNA D-synthase Dus2 (Cassart et al., 2012; Fauquenoy et al., 2018). In addition, the dus2 and dus3 deletion strains were recovered in a genome-wide screen for gametogenesis defects in fission yeast (Dudin et al., 2017). To further investigate this connection, we generated a strain lacking all four Dus enzymes. D was undetectable in that strain (Figure S1B), which displayed normal growth at 32°C but a severe thermosensitivity at 37°C (Figure S1C). Furthermore, the dus3-deleted strain indeed displayed a decreased ability to complete gametogenesis (Figure S1D). Such specific phenotypes appeared hardly compatible with a general tRNA modification defect and supported the possibility that the transcriptome-wide distribution of D may extend beyond tRNAs. This led us to develop a global profiling method of D, hereafter referred to as “Rho-seq.”
Rhodamine labeling of D results in specific reverse transcription (RT) arrest
Contrary to uridine, the ring of D is subject to reductive cleavage by sodium borohydride (NaBH4) treatment, resulting in the formation of a ureido-group (NH2CONH) linked to an alcohol (Cerutti and Miller, 1967). Upon addition of a fluorophore bearing a primary amino group (NH2 dye, e.g., rhodamine [Rho]) in acidic conditions, a nucleophilic substitution results in the covalent binding of the fluorophore to the ureido group (Kaur et al., 2011). We set up rhodamine labeling and defined the R+ condition (NaBH4 + Rho) and a control condition, R− (KOH + Rho). A dot blot assay indicated that a specific labeling of RNA occurred in the R+ condition but not in the R− condition (Figure 1A). When RNA from a strain deleted of the four Dus-encoding genes was used (hereafter referred to as Δ4), a marked decrease of the labeling was observed. The remaining signal indicates that other RNA modifications were targeted by the protocol (see Discussion). tRNAs were a major target of D-specific labeling, as expected (Figure 1B).
Figure 1. Single-nucleotide detection of dihydrouridine (D) by rhodamine labeling.
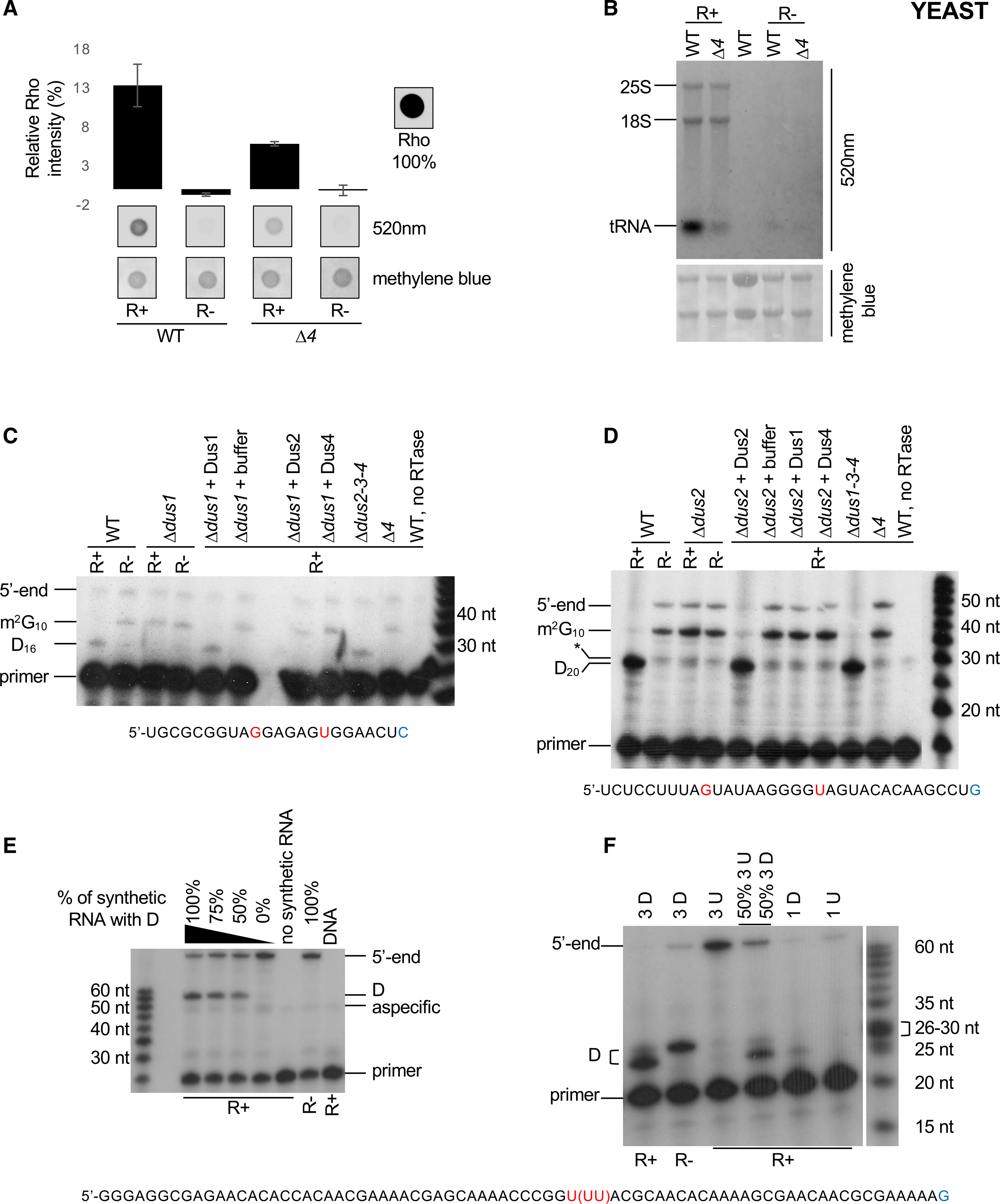
(A) Comparative Rho (rhodamine) detection at 520 nm on WT (wild-type) and Δ4 (Δdus1–2-3–4) total RNAs in R+ and mock-treated (R−) samples. Intensity of a Rho drop is scaled at 100%, a drop of water is considered as the background, and methylene blue staining serves as loading control. n = 3 biological replicates; error bars represent SEM.
(B) Fluorescent northern blotting of WT and Δ4 total RNAs in R+ and mock-treated samples. Untreated total RNA from WT strain is shown in lane 3. Representative result from a biological triplicate; methylene blue staining serves as loading control.
(C) Primer extension assays specific to tRNAiMet (Dus1-dependent D16) were performed on total RNA from WT, single dus (Δdus1), triple dus (Δdus2–3-4), or quadruple dus (Δ4) mutant strains, R+ or R− treated as indicated. Before the assay, in vitro dihydrouridylation reactions were performed on total RNA with GST-fusion S. cerevisiae proteins (+ Dus1, 2, or 4) or buffer. G10 and U16 are highlighted in red, and the last nucleotide of the primer is in blue. m2G10 indicates a labeling-independent RT stop.
(D) Primer extension assays specific to tRNAAspGUC (Dus2-dependent D20) performed on total RNA from WT, single dus (Δdus2), triple dus (Δdus1–3-4), or quadruple dus (Δ4) mutant strains, R+ or R− treated as indicated. Before the assay, in vitro dihydrouridination reactions were performed on total RNA with GST-fusion S. cerevisiae proteins (+ Dus1, 2, or 4) or buffer. G10 and U20 are highlighted in red, and the last nucleotide bound by the 3′ end of the primer id in blue. m2G10 indicates a labeling-independent RT stop, and the asterisk indicates a nonspecific band.
(E) Primer extension assay was performed on an in vitro-transcribed RNA containing 1 U/D mixed with S. pombe total RNA. A decreasing ratio of the dihydrouridinated RNA was added before R+ or R− labeling reactions. DNA is the PCR product used for in vitro transcription. The unique U43 is highlighted in red, and the last nucleotide bound by the 3′ end of the primer is in blue.
(F) Primer extension assay was performed on an in vitro-transcribed RNA mixed with S. pombe total RNA before R+ or R− labeling reactions. 3 D indicates an in vitro RNA containing three consecutive D residues at position 25, 26, and 27, respectively. 3 U indicates an in vitro-transcribed RNA where the corresponding positions are uridine. 1 D and 1 U are the same synthetic RNAs used in (E). The triplet U43−45 is highlighted in red, and the last nucleotide bound by the 3′ end of the primer is in blue.
The presence of D barely affects RT (Motorin et al., 2007). We hypothesized that the addition of the bulky rhodamine moiety onto the D residue might block the progression of the polymerase during RT, leading to a specific “drop-off.”
This was tested by primer extension, where a robust R+ dependent arrest was indeed observed at predicted Dus1 (D16) and Dus2 (D20) positions. Confirming the high specificity of the reaction, the RT arrest was absent when RNA from the corresponding dus1 or dus2 mutants was used, and it was restored by an in vitro treatment using the recombinant respective Dus enzymes (Figures 1C and 1D). An in vitro-transcribed synthetic RNA harboring a single U/D confirmed that the presence of D was required to generate an arrest one nucleotide downstream of the modified position in the R+ condition (Figure 1E), while a D triplet resulted in a main yet not unique arrest downstream of the third position. In that context, the D triplet was sufficient to stall the RT reaction even in the R− condition (Figure 1F).
Based on these data, we implemented Rho-seq to profile D transcriptome wide. The method relies on the labeling of D with rhodamine (R+) and a systematic comparison with the R− “mock” treatment. As a control for specificity of D labeling (Figure 1A), we used a strain lacking the four Dus enzymes (Δ4).
Global profiling of D using Rho-seq
The method is aimed at mapping specifically the positions at which the RT “drops off” from the RNA substrate during cDNA synthesis in a rhodamine-labeling- and Dus-dependent fashion. A multifactorial workflow highlights transcriptomic positions where RT is more likely to stop in a rhodamine- and Dus-dependent manner (Figures S2A and S2B). The test condition refers to the wild-type (WT) R+; the control conditions correspond to the WT R−, Δ4 R+ and Δ4 R− (Figure S3A). A “D-ratio” is defined as the proportion of reads attesting RT termination with respect to all reads overlapping the position of interest. To further control for specificity, we used an in vitro synthesized RNA as “spike in” control (Figure 1E). The single D present in this RNA generates a single RT stop precisely one nucleotide downstream of the modified position in the R+ condition (D-ratio 17.5%), but not in the R− condition (D-ratio 1%) or when a U instead of a D is present (D-ratio 0.1%) (Figures S2C–S2F, and S3). Linear regression on D-ratios from mixed populations of U and D spike-ins revealed a high correlation (R2 = 0.94) between the RT stop site D-ratios and the percentage of D (from 0% to 100%) containing spike-in in R+ but not in R− conditions (Figures S3B and S3C).
Distribution of D in fission yeast tRNAs
Rho-seq identified 228 modified positions (Table S1) on 141 fission yeast tRNAs (Candiracci et al., 2019). Strikingly, 98.7% of the identified D-sites were located within the D-loop, with the most prevalent positions being D16 and D20 (Figure 2A), in agreement with previous work in budding yeast (Xing et al., 2004; Figure S1A). When lowering the stringency of the pre-filtering analysis by removing the requirement of the RT stop −1 position being a T in the genome sequence, an additional 36 new sites were discovered. Among these, 95% had a T at the RT stop itself, suggesting that the RT terminated at the modified position rather than one nucleotide downstream and that the stringent parameters may generate some false-negative signals. Four randomly chosen positions were confirmed by primer extension assays (Figure 2B).
Figure 2. Rho-seq detects D on yeast tRNAs.
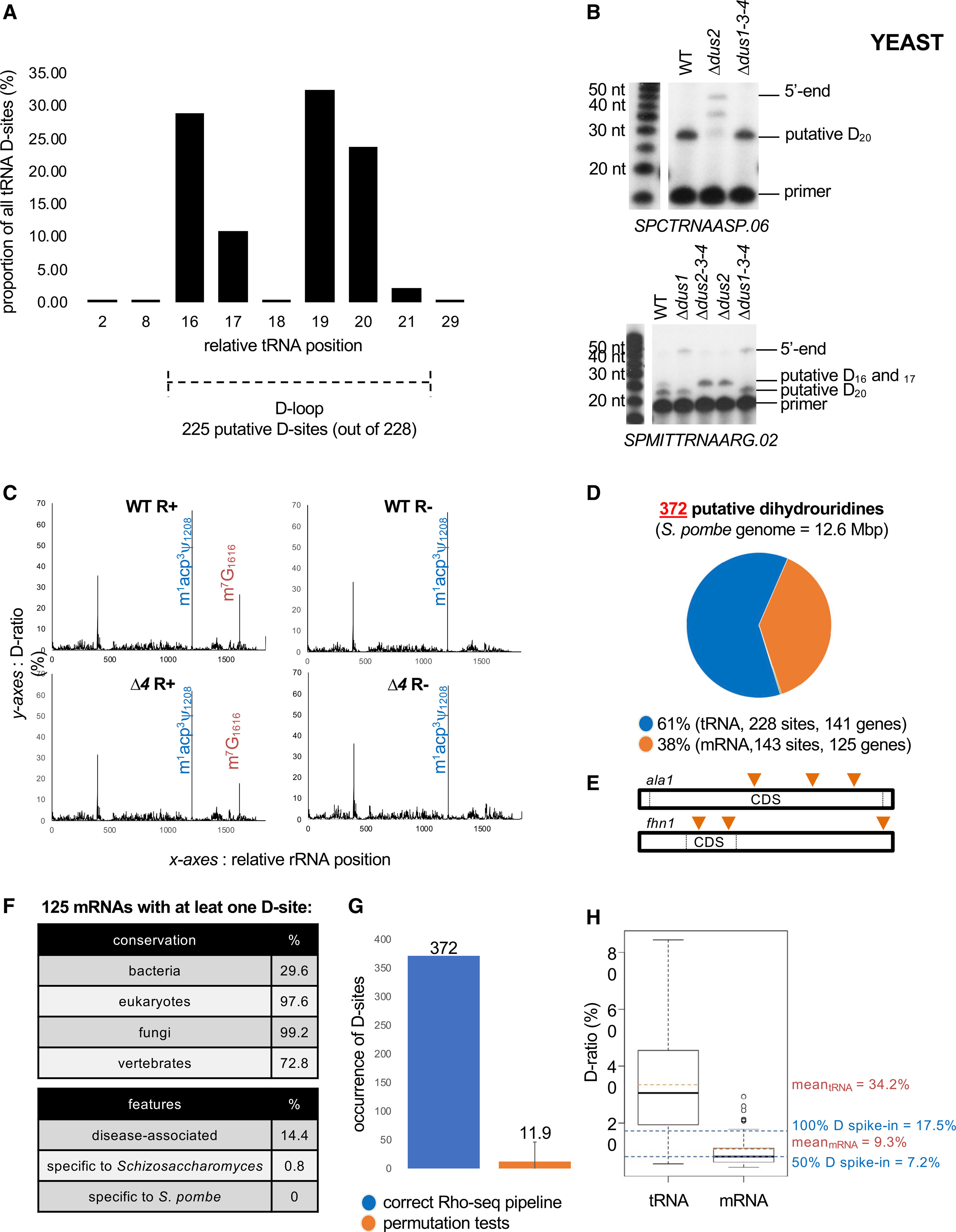
(A) Distribution of the 228 tRNA D-sites according to their relative position on tRNA.
(B) Upper panel: primer extension assays specific to tRNAAspGUC (containing a putative Dus2-dependent D20 based on Rho-seq) performed on R+ total RNA from WT, single dus (Δdus2), and triple dus (Δdus1–3-4) mutant strains. Bottom panel: primer extension assays specific to tRNAArgUCG (putative Dus1-dependent D16–17 and putative Dus2-dependent D20 based on Rho-seq) performed on R+ total RNA from WT, single dus (Δdus1, Δdus2), and triple dus (Δdus1–3-4, Δdus2–3-4) mutant strains.
(C) D-ratio profiles along the entire 18S rRNA in R+ and R− conditions in WT or Δd4 strains. The m1acp3y modification (blue) naturally blocks a RT reaction. The m7G modification (orange) is sensitive to R labeling and therefore blocks the RTase in R+ conditions independently of the strain. Representative example for SPRRNA.43 is shown.
(D) Distribution of 372 detected D-sites in fission yeast within tRNAs and mRNAs.
(E) Putatively dihydrouridylated mRNAs have up to three distinct D-sites on their sequence. ala1 and fhn1 are the two mRNAs dihydrouridylated at three different positions (orange triangles).
(F) General features of the 125 protein-coding genes whose RNA products are modified.
(G) Effect of the assignment of test and control conditions in the Rho-seq analysis pipeline. Sixteen permutation tests were performed in which the properly committed information was randomly mixed. Error bars represent max and min numbers of detected D-sites upon permutation.
(H) Boxplot representation of WT R+ D-ratio distribution for tRNAs (228 values) and mRNAs (143 values) and corresponding means highlighted by orange dashed lines. Blue dashed lines indicate the D-ratios of R+ 100% D spike-in and 50% D spike-in.
No D was detected on rRNA, which is in line with the fact that this modification was never reported to be present on eukaryotic rRNAs. The profile of the 18S-rRNA constitutes an internal control showing that Rho-seq is insensitive to RT termination caused by non-D modifications, particularly m1acp3ψ1208 and m7G1616. The m1acp3ψ1208 peak was invariable across all conditions (WT R+, WT R−, Δ4 R+, and Δ4 R−), while the m7G1616 was detected in a rhodamine-dependent but, importantly, Dus-independent manner (Figures 2C and S4).
Collectively, these data indicate that Rho-seq accurately predicts the position of D and discriminates against non-D modifications. Additional modifications sensitive to the chemistry used (rhodamine derivatization) might be detectable using the method, providing that a mutant of the corresponding synthetase is used as control (see Discussion).
D is present within yeast mRNAs
Almost 40% (143 sites) of all 372 detected D-sites were not on tRNAs (Figure 2D) but were spread across 125 protein-coding genes and 1 lncRNA (Table S1). Among the 125 D-containing protein-coding genes, 87% have a unique putative D on their mRNA, whereas only two genes (fhn1 and ala1) carry at least three distinct D-sites (Figure 2E); 91 (73%) are conserved in vertebrates, whereas less than 1% is specific to the Schizosaccharomyces genus, suggesting that dihydrouridylation occurs on mRNAs with conserved functions (Figure 2F). Several mRNAs encoding cytoskeleton-related proteins were identified as containing D, including the nda2 and nda3 mRNAs encoding α- and β-tubulins and 4 of the 8 subunits of the CCT (chaperonin-containing tailless complex polypeptide 1) chaperonin complex, which is required for the folding of newly synthesized tubulin and actin proteins (Vallin and Grantham, 2019). The eng1 mRNA that encodes an endo-1,3-β-glucanase required for actin filaments to form an aster-like structure during mating was also recovered (Table S1).
Permutation tests in which all conditions were randomly mixed in silico before the Rho-seq analysis resulted, on average, in the identification of fewer than 12 D-sites, which supports the robustness of the Rho-seq method (Figure 2G). On average, the D-ratio on D identified on mRNA (9.3%) was lower than on tRNA (34.2%) (Figure 2H), which suggests that tRNA-specific features may exacerbate the D-ratio. Indeed, the D-ratio from the spike-in RNA linearly ranged from 7.2% when 50% of the molecules are modified to 17.5% when the entire population is modified, suggesting that despite an apparent low D-ratio, the ratio of modified versus unmodified molecules for a given mRNA should be more than 50% on average.
We observed a sharp and significant enrichment (Fisher’s exact test, p < 10−9) of D on coding sequences (CDSs) that harbor 90% of the detected D-sites, while only 62% of U are present within CDSs, the rest covering UTRs (Figure S5A). D was mostly found within the context of a U-tract in the CDSs (Figure S5B). Among the 124 D-sites found on CDSs, the most represented codon coded for leucine (Figure S5C). Among the six different possible leucine codons, three contain two uridines (CUU, UUA, and UUG), but D was unevenly distributed among them (Figure S5D). Moreover, only 33% of D-sites found on CDSs were localized on a codon containing a single U, whereas the naive probability of finding D on a single U-containing codon at the transcriptomic level is of 42%, indicating that the D formation occurs preferentially on codons with two or three uridines (Fisher’s exact test, p = 0.046). Finally, no D-site was detected on ten different single U-containing codons, including STOP codons, which represents a statistically significant depletion (Fisher’s exact test, p = 8 × 10−5) as those codons represent 8% of the coding uridines and account for 8% of the possibilities to get a D-site on the yeast coding transcriptome. The non-random distribution of D across the transcriptome—and more specifically on the coding region of conserved genes—argues in favor of a biologically relevant function of D.
To gain further insight on the specificity of the four Dus enzymes, we repeated Rho-seq in the single Δdus1, Δdus2, Δdus3, and Δdus4 mutants using the same analytical pipeline. For each detected D-site, the RT stop signal in the single Dus mutant was incorporated in the binomial model, and Dus dependency was statistically tested (Figure S3A). At the tRNA level, 251 D-sites were detected, of which 96% were located on the D-loop between positions 16 and 22 (Figures 3A and 3B) as expected. We could unambiguously assign 87, 113, 9, and 28 D-sites to Dus1, Dus2, Dus3, and Dus4, respectively. The single-nucleotide distribution of the D-sites matched with the known substrate specificities of the Dus enzymes (Figure 3C). Intriguingly, the dus dependency of 14 sites could not be statistically determined (Figures 3B and S5E), which suggests that the presence of a D-site at a given position may be required for the subsequent modification of another. Alternatively, several Dus enzymes may target the same site in rare cases.
Figure 3. Fission yeast protein-coding genes products are dihydrouridinated.
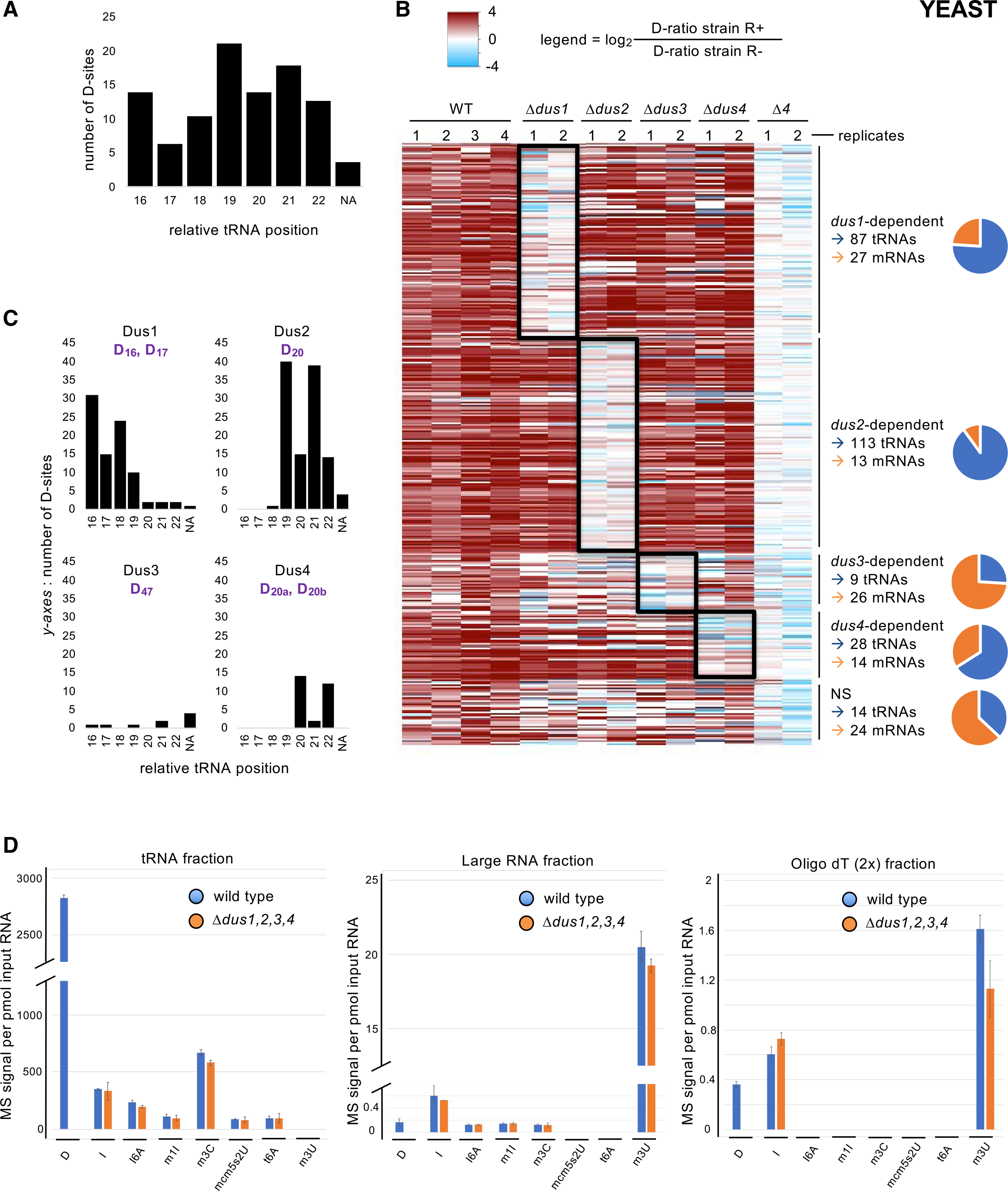
(A) Distribution of the 251 tRNA D-sites according to their relative position on tRNA. NA, not assigned (for positions that were out of the D-loop).
(B) Heatmap showing all detected D-sites sorted by comparison of their respective D-ratios in test (R+) and control (R−) conditions. Dark red hue highlights more RT terminations in the test condition (D-ratio R+ > D-ratio R−). The columns depict all replicates from the indicated strains. All sites were clustered according to their dus1, dus2, dus3, or dus4 dependency, as shown to the right of the map. If no dependency was revealed by the statistical analysis, the sites were clustered in the NS (non-significant) category. The ratio of tRNA (blue) and mRNA (orange) is shown on the graphic. A total of 237 tRNA and 80 mRNA D-sites show Dus specificity.
(C) Distribution of the 87 Dus1-dependent, 113 Dus2-dependent, 9 Dus3-dependent, and 28 Dus4-dependent tRNA D-sites according to their relative position on tRNA. NA, not assigned (for positions that were out of the D-loop). The expected target sites are shown in purple.
(D) LC-MS/MS analyses of the D content of several classes of RNA. Left panel: a tRNA-enriched fraction was isolated from the indicated strains by size-exclusion chromatography (SEC), hydrolysed into single ribonucleosides, and analyzed by LC-MS. Middle panel: a corresponding “large RNA” fraction from the same SEC was similarly analyzed. Right panel: an mRNA-enriched fraction from the indicated strains was obtained by two poly-T affinity purifications in tandem and analyzed as above. Each column represents the averaged value ± SEM (n = 3 biological replicates).
In parallel, 104 D-sites were also detected on 92 mRNAs, 90% of which were located within the coding region, similarly to the previous dataset. The dus dependency of these D-sites revealed that all four Dus enzymes potentially modify mRNAs, while nearly 75% of the Dus3-dependent sites were found in mRNAs, including nda2, cct3, and cct1.
To strengthen the discovery of the presence of D in mRNA, we used RNA mass spectrometry (MS) as an orthogonal approach that does not rely on the Rho-seq chemistry. We used size-exclusion chromatography to isolate a tRNA and a “large” RNA fraction from total RNA prepared from the Δ4 strain and its isogenic WT counterpart. After hydrolysis of RNA into single nucleotide, liquid chromatography-mass spectrometry (LC-MS) analyses were performed, and a set of modified nucleosides were quantified, including D, inosine (I), N6-isopentenyladenosine (i6A), 1-methylinosine (m1I), 3-methylcytidine (m3C), 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U), N6-threonylcarbamoyladenosine (t6A), and 3-methyluridine (m3U). D, i6A, m1I, m3C, mcm5s2U, and t6A are well-characterized tRNA modifications, while m3U is a rRNA marker (Sharma et al., 2014) and I is found on both tRNA and mRNA (Presnyak et al., 2015; Srinivasan et al., 2021). In the absence of mRNA-associated m6A in fission yeast, I is the best mRNA marker at our disposal. Analysis of the tRNA fraction corroborated previous findings, with detection of all marks except m3U and dus-dependent D being most prominent (Figure 3D). A reverse picture was obtained in the “large” RNA fraction, with m3U being dominant while both the anticodon mcm5s2 and the tRNA position-37-associated t6A were under the detection limit. D and I were also detected, and traces of I6A, m1I, and m3C were present. In parallel, we also analyzed RNA fractions resulting from two consecutive purifications of total RNA on oligo dT columns aiming at enriching for mRNAs. While m3U, most likely resulting from persistent rRNA, was the most prominent, D and I were detected in a similar range in this poly-A fraction (Figure 3D). Compared to the large RNA fraction, there was a 2-fold increase (MS signals from 0.16 to 0.36 per pmol of input RNA) in the D level in the oligo dT (2x) fraction, while the tRNA-specific marks (i6A, m1I, m3C, mcm5s2U, and t6A) were undetectable. The specific D signal enrichment is evidence that D exists in mRNA, even if part of the signal could be from a small amount of contaminating tRNA.
Taken together, these data suggest that a low amount of D is specifically found in mRNA in fission yeast, supporting the Rho-seq data.
Dihydrouridylation is limited to tRNAs in E. coli
Rho-seq is amenable to systematic D mapping to any organism, providing that a mutant lacking the modification is available as normalization control. We performed Rho-seq on fastgrowing E. coli cells to assess whether mRNA may also be dihydrouridylated in that species. The previously generated triple dus mutant (ΔdusA-B-C or Δ3) (Bishop et al., 2002) lacked detectable D (Figure 4A). Dot blot and northern blotting analyses of R+- and R–-treated RNAs confirmed the conclusions drawn in yeast, i.e., an overall decrease of Rho labeling in the Δ3 strain (Figures 4B and 4C). Rho-seq detected 106 modified positions onto tRNA positions. Manual curation (using tRNAmodviz and GtRNAdb) revealed 63 distinct modified nucleotides on 35 different tRNA species (Table S1). Fifty-three D-sites were previously reported (representing 72% of all known dihydrouridylated tRNA positions in E. coli). The remaining 10 are new D spread over six tRNA species (Figure 4D). In total, the 63 D-sites are distributed along the D-loop following the exact same pattern as the 74 known D (Figure 4E). Together, these data confirm the power and reliability of Rho-seq to detect D at the single-nucleotide level.
Figure 4. Implementation of Rho-seq on the E. coli transcriptome.
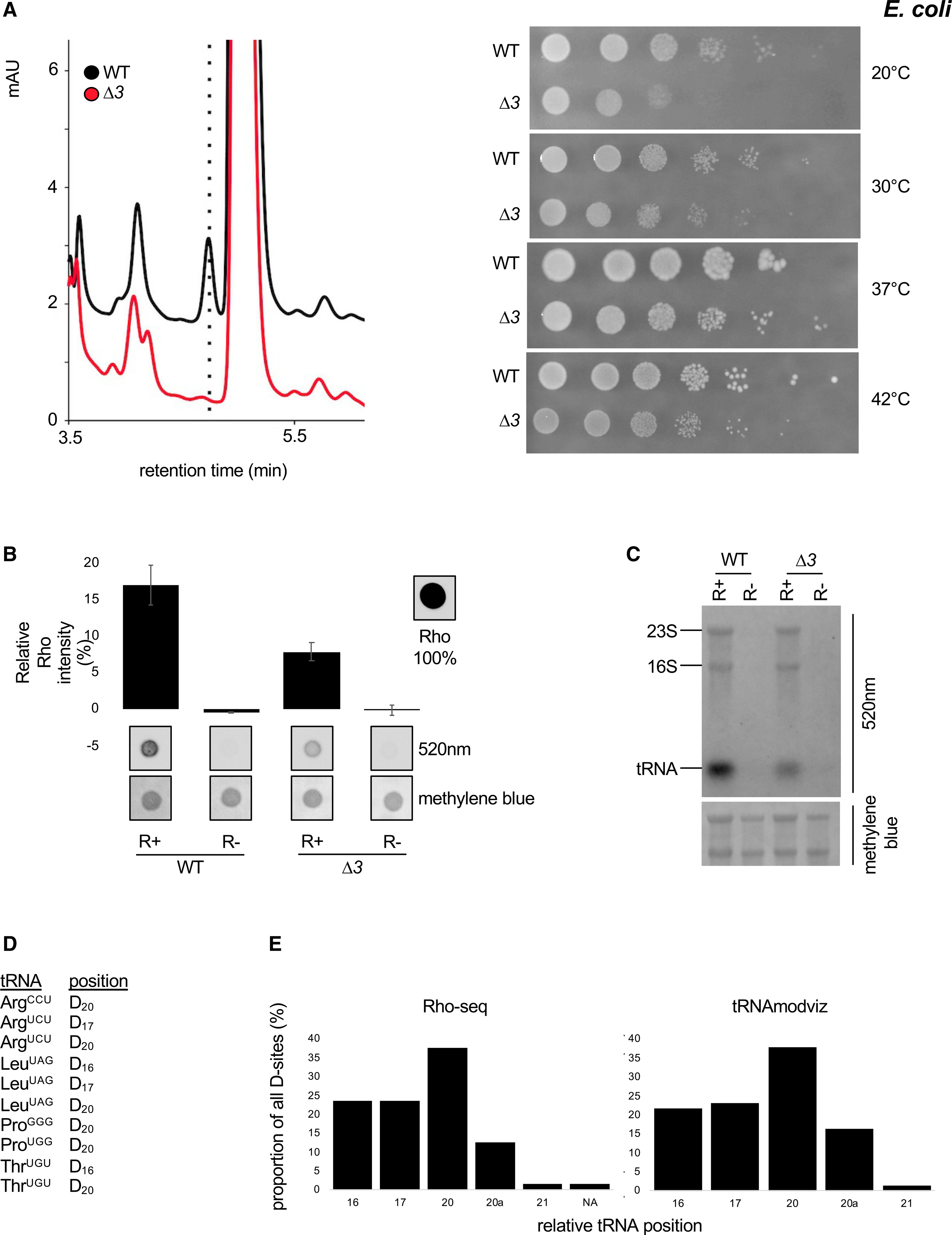
(A) Left: HPLC analysis of D content of RNA from a WT E. coli strain and a strain deleted for all dus gene family members. Right: chromatogram of pure D at the indicated concentrations. Right: growth assay at the indicated temperatures of a WT E. coli strain and a strain deleted for all dus gene family members.
(B) Comparative Rho (rhodamine) detection at 520 nm on WT (wild-type) and Δ3 (ΔdusA-B-C) total RNAs in R+ and mock-treated (R−) samples. Intensity of a Rho drop is scaled at 100%, a drop of water is considered as the background, and methylene blue staining serves as loading control. n = 3 biological replicates; error bars represent SEM.
(C) Fluorescent northern blotting of WT and Δ3 total RNAs in R+ and mock-treated samples. Representative result from a biological triplicate; methylene blue staining serves as loading control.
(D) Ten previously unknown positions (tRNAmodviz) on six E. coli tRNA species are found by Rho-seq to be dihydrouridinated.
(E) Left panel: distribution of the 63 unique tRNA D-sites identified by Rho-seq. NA, not assigned due to sequence incompatibility (D22 on tRNASerGGA). Right panel: distribution of the 74 unique tRNA D-sites as reported by the tRNAmodviz database.
In contrast to the fission yeast Rho-seq, no D-site was detected on bacterial mRNA. The rRNA 23S-U2449 position was reported to be a D in E. coli (Popova and Williamson, 2014). RT termination signals were observed one nucleotide downstream of this position in both WT R+ and Δ3 R+, but not in mock-treated samples (Figure S6A), as confirmed with a primer extension assay (Figure S6B). This result suggests that D is present in rRNA in E. coli, but it is surprisingly deposited independently of the DusA,B,C enzymes. The possibility that this modification is not a D is unlikely based on previous work (Popova and Williamson, 2014) but cannot be excluded (see Discussion).
The presence of D within an mRNA decreases translation in vitro and in vivo
To test possible effects of D on protein expression, we set up an in vitro 35S methionine-based translation assay relying on wheat germ extracts and T7-generated mRNA containing U or D at specific positions. mRNA containing D at the initiation (ADG) and STOP (DAA) codons showed an average 10% decrease of protein production compared to the corresponding U-containing mRNA. An ACG codon that was reported to substitute for AUG as initiation codon (Kearse and Wilusz, 2017) completely abolished protein expression (Figure 5A). We next added either a cysteine UGC codon, an aspartic acid GAU codon, or a leucine CUG codon within the CDS. The UGC codon was not recovered as modified in Rho-seq, while the GAU codon was the most abundant modified codon containing a single U. The CUG codon was recovered at lower frequency (Figure S5B). While the presence of the cysteine DGC codon did not affect the level of translation, the presence of a single GAD aspartic acid codon or a CDG leucine codon decreased protein expression compared to the corresponding U-containing mRNA (Figures 5A, S8A, and S8B). These results support that the codon-dependent modification of U to D has the potential to modulate translation.
Figure 5. mRNA-associated D decreases the abundance of some encoded protein in vitro and in vivo.
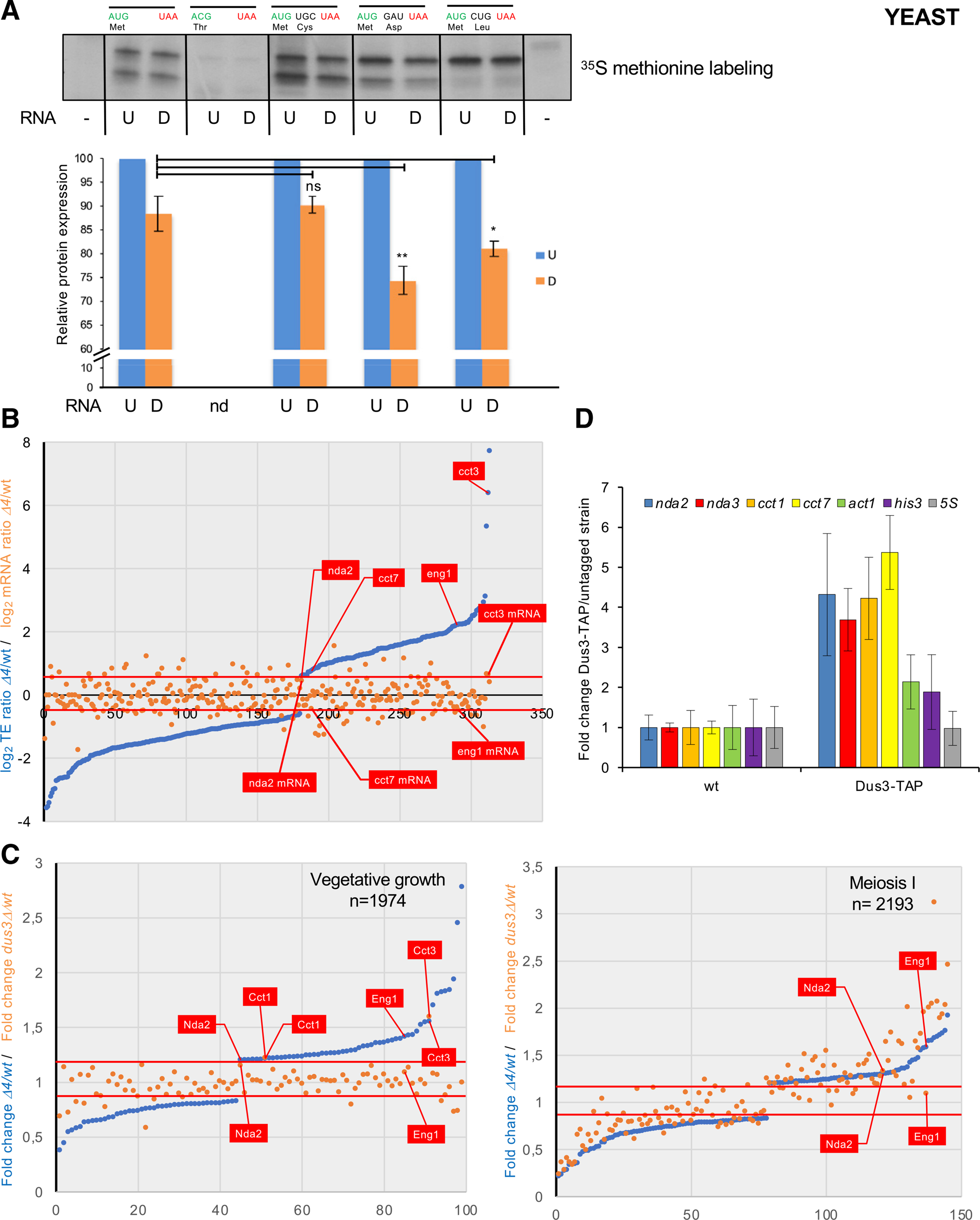
(A) 35S methionine in vitro translation assay using in vitro-synthetized mRNAs with indicated codon specificity. The signal obtained from the uridine-containing RNA (U) was set as 1. Error bars represent standard error from three assays.
(B) Blue dots: 337 mRNAs sorted by translation efficiency fold change (Δ4/WT) with thresholds of a minimum 1.5-fold change and adjusted p value < 0.01 indicated by the two red lines. The orange dots show the fold change of the level (Δ4/WT) of the corresponding set of mRNAs. Translation efficiencies are normalized on mRNA level. The indicated nda2, cct7, eng1, and cct3 mRNAs were identified by Rho-seq.
(C) TMT-based quantitative proteomic analysis of the WT, Δ4, and dus3Δ strains during vegetative growth and meiosis.
Left panel, blue dots: 99 proteins sorted by mitotic growth expression fold change (Δ4/WT) with thresholds of a minimum 1.2-fold change and adjusted p value < 0.01 indicated by the two red lines. 1,974 proteins were detected in total by TMT-based analysis. The orange dots show the mitotic growth expression fold change (dus3Δ/WT) of the same set of proteins. The Nda2, Cct1, Eng1, and Cct3 proteins whose mRNAs were identified by Rho-seq are indicated.
Right panel, blue dots: 145 proteins sorted by meiotic growth expression fold change (Δ4/WT, Δ4/wild-type) with thresholds of a minimum 1.2-fold change and adjusted p value < 0.01 indicated by the two red lines. 2,193 proteins were detected in total by TMT-based analysis. The orange dots show the meiotic growth expression fold change (dus3Δ/WT) of the same set of proteins. The Nda2 and Eng1 proteins whose mRNAs were identified by Rho-seq are indicated.
(D) RNA immunoprecipitation (RIP) of Dus3-TAP of the untagged control. The fold change (Dus3-TAP/WT) of the indicated mRNAs is shown. Each column represents the averaged value ± SEM (n = 3 biological replicates).
To expand these findings in vivo, we used ribosome profiling (Ribo-seq) to compare the WT and Δ4 strains. Ribosome-protected fragments were analyzed after normalization by the number of RNA-seq reads to calculate relative TEs transcriptome-wide. A set of about 300 mRNAs were identified with thresholds of a minimum 1.5-fold change and adjusted p value < 0.01 (Figure 5B; Table S2). These data indicate that the complete absence of D does not globally impact TEs, at least in the tested condition. Examination of individual genes revealed that nda2, cct3, cct7, and eng1 that were isolated in Rho-seq were translationally upregulated when D was absent (Figures 5B and S8C). Considering that the modified uridine represents only a subset of the population for a given codon, we did not expect strong effects on the relative codon occupancy of all codons in the Δ4 compared to the WT. Nevertheless, we noticed that when considering the 11 codons that have the highest occurrence of D (based on Rho-seq; Figure S5B), all but one displayed reduced occupancy within the ribosomal A site in the mutant when D is absent, which constitutes a significant enrichment (Fisher’s exact test, p value = 9 × 10−4, estimated odds ratio = 17.5; Figure S8D). This suggests these codons are translated faster when the Dus enzymes are absent.
To further analyze how the absence of D impacts protein expression level, we performed a large-scale proteome analysis by tandem mass tag (TMT)-based quantitative proteomic analysis. Considering that Rho-seq on single dus mutants revealed that a set of cytoskeleton-related mRNAs is targeted by Dus3 and that remodeling of the cytoskeleton is critical for both mitosis and meiosis, we analyzed the Δ4 and dus3 mutant strains during both vegetative growth and at a meiotic time point corresponding roughly to meiosis I (MI) (Figure S8E) for differential expression with thresholds of a minimum 1.2-fold change and p value < 0.01.
During mitotic growth, about 100 proteins were found differentially expressed in the Δ4 strain, including Nda2, Cct1, Cct3, and Eng1, all of which were overexpressed (Figure 5C; Table S3). Remarkably, the dependence on Dus3 (based on Rho-seq in single mutants) of Nda2, Cct1, and Cct3 (but not Eng1) was confirmed, as these proteins were also upregulated in the single dus3 mutant. Both Nda2 and Eng1 were also overexpressed during MI (Figure 5C; Table S3). Contrary to vegetative growth, the deletion of dus3 appeared to have a much more prominent effect during meiosis, with about 35% of the proteins affected both in the Δ4 and the dus3 mutant (Figure 5C).
The overlap between the different sets of mRNAs/proteins identified by Rho-seq, Ribo-seq, and TMT-based proteomics ranged from 8% to 25%. This is often the case, as different technologies do not necessarily capture the same features of a given biological process. Nevertheless, the subset of targets we are focusing on behaved consistently, as summarized in Figure S9A.
Among the mRNA targets, nda2 has a uridine at position 1133 that Rho-seq identified as a D. This corresponds to the middle position of a valine codon, GUU. We therefore used CRISPR to mutate this codon to GCC (nda2noD mutant), which encodes alanine. Indeed, all the possible valine codons harbor a U at the middle position, and the valine-to-alanine replacement is conservative, as both amino acids are aliphatic with a hydrophobic side chain. The level of α-tubulin Nda2 was increased in the nda2noD mutant (Figures S9B, S9F, and S9G). Deleting dus3 alone was sufficient to mimic this effect, confirming that it may directly contribute to the modification of the nda2 mRNA (Figures S9C, S9F, and S9G). We noticed that nda3, which encodes β-tubulin, was also recovered in Rho-seq, but the protein was not detected by TMT-based proteomics. We used CRISPR to mutate the target serine UCU codon to ACC (nda3noD mutant), which encodes threonine. The level of β-tubulin was increased in the nda3noD mutant (Figures S9D, S9F, and S9G), and deleting dus3 alone was also sufficient to reproduce this effect (Figures S9E, S9F, and S9G). Deleting the other three Dus enzymes (Dus1, Dus2, and Dus4) did not affect the level of Nda2 (Figures S9C, S9F, and S9G).
Taken together, these data show that, both in vivo and in vitro, the presence of D rather than U in specific mRNAs alters the level of the encoded protein.
If Dus3 indeed directly modifies specific mRNAs, we made the assumption that the resulting transient interaction may be captured by RNA immunoprecipitation (RIP). Indeed, the nda2, nda3, cct1, and cct7 mRNAs that were identified by Rho-seq were immunoprecipitated with Dus3-TAP with a 4- to 5-fold enrichment to the untagged control, while the act1, his3, and 5S RNAs, though notoriously abundant, were not (Figure 5D). These data support the conclusion that Dus3 specifically binds some mRNA in vivo.
Modification of the nda2 mRNA is required for meiotic chromosome segregation
Neither the nda2noD, the nda3noD, nor the Δ4 mutants display any growth defect on complete medium at 32°C, but they are sensitive to the tubulin depolymerizing drug TBZ (Figure S7A), which suggested defects in the microtubule cytoskeleton. Intriguingly, the double nda2noD nda3noD mutant was less sensitive than the single ones. It is known that α- and β-tubulin monomers associate with strong binding constants to form hetero-dimers at a stable 5 μM concentration in vivo (Adachi et al., 1986; Loiodice et al., 2019). Therefore, a possible explanation for the lower sensitivity of the double mutant is that defects resulting from the elevated level of one monomer are compensated for by a similar overexpression of the second monomer.
Considering that Dus3 is required for gametogenesis (Figure S1) and that some tubulin mutants are specifically defective for meiosis (Paluh et al., 2004), we next measured the level of Nda2 in the WT and the nda2noD mutant during meiosis (Figures S7B and S7C). This revealed an overexpression of the protein when the dihydrouridylation is abolished.
Next, we analyzed the effect of the Δ4 mutant on chromosome segregation in mitosis using Atb2 (the second α-tubulin-encoding gene in fission yeast) linked to the ENVY fluorescent protein. Maximum spindle length and mitosis duration were not significantly different compared to WT cells (Figures S10A and S10B). Similarly, the dus3Δ strain did not show any prominent mitotic defects beside a short, yet significant, delay in metaphase (Figure S10C).
We next focused on the dus3Δ and the nda2noD mutants and analyzed their effect on meiosis. Both mutants displayed meiosis defects with a reduction in gamete numbers and viability more marked in the nda2noD mutant (Figures 6A, 6B, and 6C). To further analyze these defects, we performed live-cell imaging at short time (1’) intervals (Figure 6D). When dus3Δ diploids were induced to enter meiosis, the time needed in prophase for spindle assembly in MI was increased, while a marked increase in the duration of metaphase was noted in both MI and MII (Figures 6D–6H).
Figure 6. The deletion of dus3 or the absence of D on the nda2 mRNA results in meiotic spindle defects and low gamete viability.
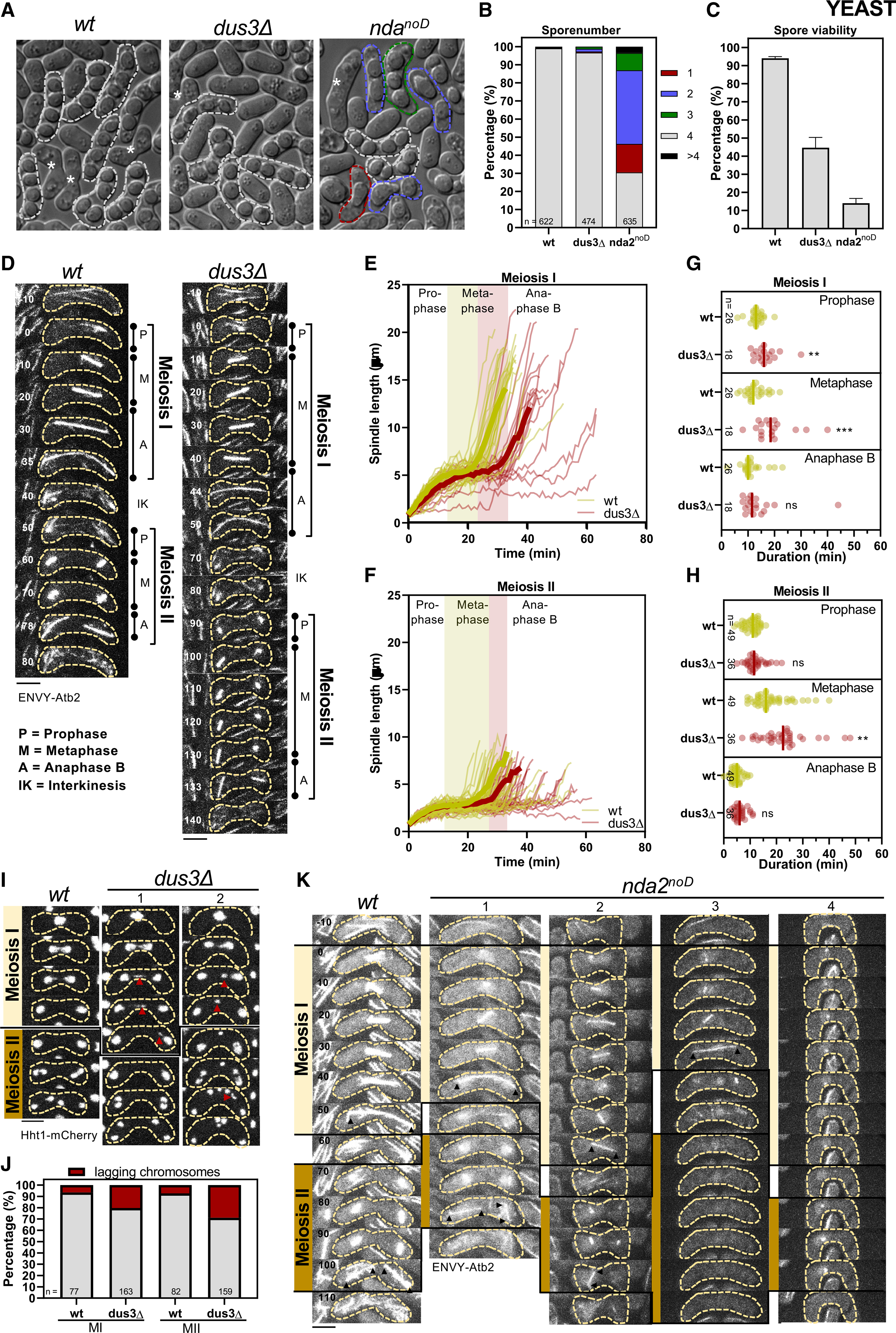
(A) Brightfield images of h90 WT, nda2noD, and dus3Δ mutants after 48 h on malt extract (ME) plates. Zygotes without spores (gametes) are marked by an asterisk, the dashed white line marks zygotes with 4 spores, and dashed green, blue, and red lines mark zygotes with aberrant spore numbers.
(B) Stacked bar plot of the spore number per zygote of h90 WT, nda2noD, and dus3Δ mutants.
(C) Gamete viability was assessed from the ability of gametes to form colonies in the indicated strains. Error bars represent the standard error from three biological replicates.
(D) Time-lapse images of WT and the dus3Δ cells expressing ENVY-Atb2 (α-tubulin). Scale bar, 5 μm.
(E and F) Comparative plot of spindle length dynamics of WT (n = 26 spindles) and the dus3Δ cells (n = 18 spindles). Bold curves correspond to mean spindle dynamics. Green bar illustrates the metaphase duration of WT cells and the red bar the additional time needed for metaphase in the dus3Δ cell.
(G and H) Dot plot comparison of the duration of the meiotic phases for the WT and dus3Δ mutant. p values were calculated by Mann-Whitney U test.
(I) Time-lapse images of WT and dus3Δ cells expressing Hht1-RFP. Lagging chromosomes are marked by the red arrowheads. Time interval corresponds to 10 min. Scale bar, 5 μm.
(J) Stacked bar plot of the number of WT and dus3Δ cells with lagging chromosomes during meiosis I and meiosis II.
(K) Time-lapse images of WT and nda2noD mutant expressing ENVY-Atb2. Arrowheads illustrate spindle poles of final MI or MII spindles. Scale bar, 5 μm. Representative pictures are shown: zygotes forming normal MI and MII spindles (1), zygotes forming an MI spindle but only one MII spindle (2), zygotes forming an MI spindle but no MII spindle (3), zygotes forming no proper MI or MII spindle (4).
We next scrutinized if these meiotic spindle defects resulted in defective chromosome segregation using histone H3 (Hht1) fused to mCherry. During both MI and MII, we observed an increase of lagging chromosomes (Figures 6I and 6J). These lagging chromosomes could eventually be correctly separated during anaphase B, or they remained unseparated and formed an additional DNA mass (Figure 6I), likely explaining the low gamete viability of the dus3Δ strain.
While similarly testing the effect of the nda2noD mutation on meiosis, we noticed that the intensity of the ENVY-Atb2 reporter and the number of microtubule bundles was strongly diminished in the nda2noD mutant strain (Figure 6K). We therefore could not perform time-lapse images of meiosis with short time intervals. Nevertheless, following the morphology of the spindles during the whole meiotic cycle with a longer (10’) interval revealed that the nda2noD mutant displayed various meiotic defects (Figure 6K), including only one spindle formed during MII, no MII spindle formed, and neither MI nor MII spindle formed (Figure 6K).
Taken together, these data show that the deletion of dus3, or the mutation of the nda2 dihydrouridylated codon, strongly impact meiosis by either prolonging metaphase or by preventing the proper formation of meiotic spindles. We conclude that the deposition of D onto mRNAs is a new critical regulator of meiosis.
mRNA dihydrouridylation is conserved in human cells
At this stage, we were interested to learn if DUS-dependent mRNA dihydrouridylation is evolutionarily conserved. To test this possibility, we applied Rho-seq to diploid colon cells (HCT116). A cell line lacking all four DUS genes was generated using CRISPR-Cas9 genome editing (Figure 7A). Each individual deletion was also produced (Figure 7A). In each case, the exon carrying the catalytic motif was precisely excised from the genome on both alleles. The quadruple deleted cell line was confirmed by HPLC to lack any detectable D (Figure 7B). The inactivation of the DUS enzymes was further confirmed at known tRNA positions by primer extension for Dus1 (position D16, Figure 7C), Dus2 (D20, Figure 7D), and Dus4 (D20a, Figure 7E).
Figure 7. D is detected on the tubulin mRNA in human cells.
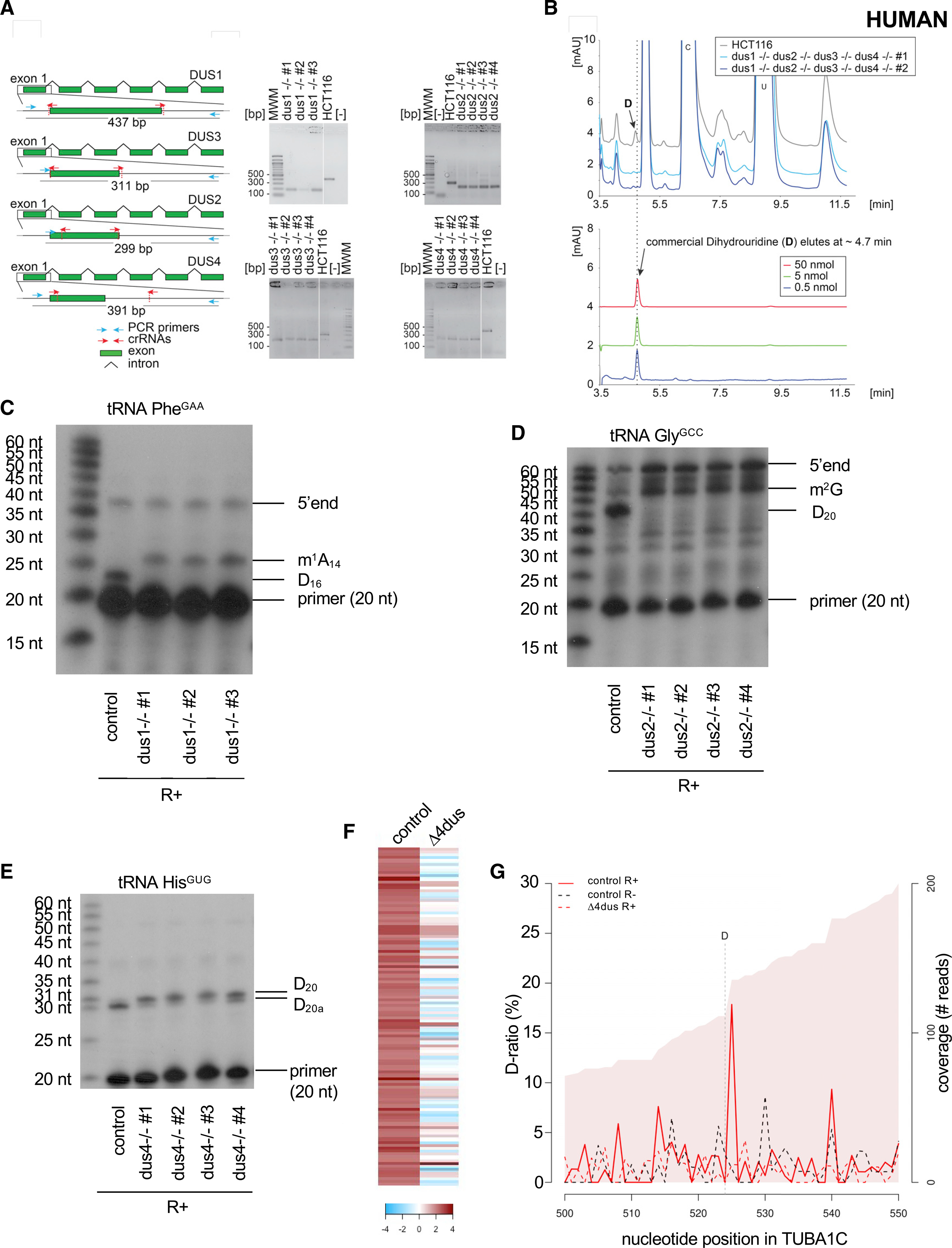
(A) Left: structure of the genomic loci of the four human DUS genes. For all four DUSes, exon 1 containing the catalytic domain was targeted for precise deletion by CRISPR-Cas9 in HCT116 diploid cells. The guide RNAs (crRNAs) used are depicted as red arrows. The primers used for the diagnostic PCR are highlighted in cyan, and the size of the PCR-amplified product is indicated. Right: diagnostic of the cell lines by PCR. Genomic DNA extracted from the indicated purified clones and isogenic reference control (HCT116) was subjected to a PCR amplification with indicated primers (blue arrows). MWM, molecular weight marker; [−], control without DNA. Note the site of deletion was confirmed by DNA sequencing for all cell lines. In all cell lines, both alleles were deleted.
(B) Total RNA extracted from human cells lacking all four DUS genes and from isogenic reference control cells (HCT116) was analyzed by quantitative HPLC for the presence of D. Lower panel: synthetic D was used for calibration (the indicated amounts were used).
(C) Primer extension assays specific to human tRNAPhe(GAA) (containing a Dus1-dependent D16) were performed on total RNA from control cells (HCT116) and three clones of the ΔDUS1. m1A14 and the 5′ end are indicated.
(D) Primer extension assays specific to human tRNAGly(GCC) (containing a Dus2-dependent D20) were performed on total RNA from control cells (HCT116) and four clones of the ΔDUS2. m2G and the 5′ end are indicated.
(E) Primer extension assays specific to human tRNAHis(GUG) (containing a Dus4-dependent D20a and a Dus2-dependent D20) were performed on total RNA from control cells (HCT116) and four clones of the ΔDUS4.
(F) Heatmap highlighting the 112 D-sites detected within human mRNAs sorted by comparison of their respective D-ratios in test (R+) and control (R−) conditions. Dark red highlights more RT terminations in the test condition (D-ratio R+ > D-ratio R−). The columns depict the cell lines used.
(G) Evolution of the D-ratio (%) and the coverage (reads) in the indicated conditions along the TUBA1C (tubulin alpha 1C) mRNA. The R+ reaction generates a high (about 17%) D-ratio coupled to a decrease of coverage, which supports the presence of a D position one nucleotide upstream.
Applying Rho-seq to RNA extracted from human cells identified a set of 112 D-sites within mRNAs (Figure 7F). Remarkably, these include the TUBA1C mRNA (Figure 7G) encoding the human ortholog of fission yeast nda2. Taken together, these observations support the notion that mRNA dihydrouridylation is conserved in eukaryotes, opening the way to studying its role in normal and pathological contexts in human cells.
DISCUSSION
The discovery of RNA modifications as a new layer contributing to gene expression has led to the urgent need for new reliable methods to determine the position of these marks across the transcriptome. Combining the rhodamine-labeling technique with high-throughput sequencing, Rho-seq relies on the specific labeling of modified nucleotides to hinder RT and ultimately allow the determination of the transcriptomic position of the modification. Conceptually, Rho-seq is similar to the method used to detect pseudouridine (ψ) (Schwartz et al., 2014), but it introduces both technical and analytical improvements to address the concerns of false positives that have led to several controversies (Grozhik and Jaffrey, 2018).
Rho-seq is performed in biological duplicates, which allows for sound statistical testing. In addition, the control condition is not limited to a mock-treated sample (R−) but is extended to the use of a mutant lacking D, which arguably allows for distinguishing the Dus-dependent RT drop-off. Such R− and Dus-dependent effects on RT termination are readily modelized in the generalized linear model at the core of the Rho-seq analytic pipeline. Importantly, the pipeline applies p value correction for multiple testing to control for false negatives, a much-needed feature given the number of putative D-sites to be tested. Finally, to maximize statistical power, Rho-seq implements stringent independent pre-filtering on the data to reduce the pool of positions tested (Bourgon et al., 2010). Together, those features allow Rho-seq to call modifications with high confidence, as supported by the fact that 98.5% of all detected D-sites on tRNAs were found on canonical D positions. The Rho-seq mapping method could easily be extended to other modifications, considering the chemical reactivity to sodium borohydride of additional RNA modifications (including ac4C, f6A, m1A, m3C, m7G, S4U, and yW) (Cerutti et al., 1968; Lin et al., 2019).
Our data do not currently support that D is found on mRNAs in E. coli. However, a peculiarity of the bacterial D landscape is the presence of a site on the 23S rRNA. We show that this D position is installed independently of the DusA-B-C enzymes. To our knowledge, this is the first experimental evidence suggesting that D could be synthesized by a non-canonical D synthase. The pyrimidine catabolism in bacteria and in higher eukaryotes includes the reduction of uracil to dihydrouracil by flavin-dependent enzymes related to the Dus family: dihydroorotate dehydrogenase (DHODH) and dihydropyrimidine dehydrogenase (DHPDH) enzymes. In E. coli, the DHPDH PreA and PreT were shown to catalyze this reaction (Hidese et al., 2011) and could be responsible for D formation on rRNA.
About 40% of D deposited by the Dus enzymes extends beyond tRNAs in fission yeast, raising further questions about their specificity. Schwartz et al. have shown that ψ synthases have a dramatically larger substrate specificity than previously assumed and that mRNA pseudouridylation depends on both site-specific and snoRNA-guided enzymes, which could also apply to the Dus enzymes. We noticed that a predicted D within a mRNA is more likely than randomly selected sites to be located in the loop of a predicted hairpin (binomial test, p value 9.3e–07) reminiscent of the D-loop (Figure S11A). In addition, these predicted secondary structures are more stable around D positions found in mRNAs compared to random sites (Wilcoxon-Mann-Whitney test, p value 0.0009) (Figure S11B) (Lorenz et al., 2011). These findings raise the hypothesis that Dus enzymes may be targeted to mRNAs through substrate mimicry.
We report evidence of the modification of mRNAs by all the yeast Dus enzymes. Mass spectrometry independently revealed the presence of D in mRNA fractions at a level similar to inosine. Determining if this low amount of dihydrouridination is of biological importance is a central question. Promiscuous enzyme activities are not rare and may be the source of ongoing selection for new functions. In that context, Dus3 is of particular interest, as it modifies tRNA outside of the D-loop and targets U47 in the variable loop of tRNATyr.
Here, we show that the deletion of dus3 in fission yeast results in phenotypes similar to the mutation of the D-modified codon on the tubulin-encoding mRNA nda2. In addition, the absence of Dus3 affects the level of α-tubulin, and Dus3 binds the nda2 mRNA in vivo. These data support that Dus3 is at least partially implicated in the modification of the nda2 mRNA.
Another question arising from our work is the role of D within mRNAs. Our in vitro and in vivo data are compatible with D affecting translation kinetics, which in turn may affect co-translational folding. In this respect, it is striking that mRNA encoding cytoskeleton-related proteins that form highly structured complexes are found in the Rho-seq dataset (Buhr et al., 2016; Zhang et al., 2009; Zhou et al., 2013).
Finally, the present work reveals the biological relevance of the D modification of one codon within the nda2 mRNA that encodes α-tubulin by showing that it is critical for meiotic progression, though it does not seem to affect mitotic progression. A similar meiotic-specific defect was also reported for the F200Y mutation in β-tubulin encoded by the nda3 gene. Intriguingly, Rho-seq also identified nda3 as a dihydrouridylated mRNA with the modified codon (U586CU encoding S196) being in the immediate proximity of F200. We show that abolishing the modification of this codon while maintaining a very related amino acid (serine versus threonine) leads to increased protein expression in a Dus3-dependent manner, which is reminiscent of the case of nda2. It has been established that the level of the tubulin monomers is critical for proper cytoskeleton dynamics (Adachi et al., 1986). The concentration of the αβ-tubulin dimer is about 5 μM throughout the cell cycle with an excess pool being detrimental (Loiodice et al., 2019). We propose that marking the relevant mRNAs with a “D mark” plays an essential role in maintaining this homeostasis. The fact that Rho-seq in human cells also identified the nda2 ortholog TUBA1C as a target of mRNA dihydrouridylation suggests a conservation of the process and opens the way for further studies.
In conclusion, the unbiased Rho-seq epitranscriptomic approach revealed the universal D modification as a new internal mRNA modification critical for gamete formation (Figure S12).
Limitations of the study
Beside the compelling evidence supporting that D is installed in mRNAs and is biologically relevant, some limitations remain. We have been unable to detect the modification on specific mRNAs with a method orthogonal to Rho-seq. In addition, we cannot exclude that mutating the target uridine in the noD mutants alters the mRNA beyond impeding dihydrouridylation. Finally, while D negatively affects translation in all individual cases tested, it remains to be established that this effect occurs at a global level.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Damien Hermand (Damien.Hermand@unamur.be)
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
The Rho-seq, Ribo-seq and RNA-seq raw and processed data are available on GEO under the accession number GSE145686 and are publicly available as of the date of publication. Original blot images reported in this paper have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-alpha Tubulin | Sigma | Cat# T5168;RRID:AB_477579 |
| Mouse monoclonal beta-alpha Tubulin | Millipore | Cat# MAB3408; RRID:AB_94650 |
| Rabbit polyclonal anti Histone H3 | Abcam | Cat# ab1791;RRID:AB_302613 |
|
| ||
| Yeast, E. coli and human strains/lines | ||
|
| ||
| Fission yeast h- | Lab stock | yDH94 |
| Fission yeast h- dus1::natR | This work | yDH1491 |
| Fission yeast h- dus2::kanR | This work | yDH1456 |
| Fission yeast h- dus3::hphR | This work | yDH1659 |
| Fission yeast h- dus4::bleR | This work | yDH1631 |
| Fission yeast h- dus1-4 Δ | This work | yDH1754 |
| Fission yeast h- nda2noD | This work | yDH2013 |
| Fission yeast h- nda3noD | This work | yDH2021 |
| Fission yeast h- nda2noD nda3noD | This work | yDH2018 |
| Human cell line HCT116 | ATCC | CCL247 |
| Human cell line HCT116 DUS1-4 Δ | This work | HsΔ4 −/− (#1) |
| E. coli | Lab stock | JC8679 |
| E. coli dusA::cmR dusB-fis::kanR dusC::specR | Lab stock | PS5107 |
|
| ||
| Chemicals | ||
|
| ||
| Dynabeads Oligo(dT)25 | Life Technologies | 61002 |
| sodium borohydride | Sigma | 452882 |
| rhodamine 110 chloride | Sigma | 83695 |
| [Ɣ-32P]-ATP | Perkin Elmer | NEG002A250UC |
| 35S labeled methionine | Perkin Elmer | NEG709A500UC |
|
| ||
| Critical commercial assays | ||
|
| ||
| wheat germ extracts | Promega | L4380 |
| Ribodepletion kit | Thermo | A15020 – K155005 |
|
| ||
| Deposited data | ||
|
| ||
| Rho-seq, Ribo-seq and RNA-seq raw and processed data | This work | GEO: GSE145686; https://doi.org/10.17632/m6v9rcprz6.1 |
| Raw data (Mendeley data) | This work | |
|
| ||
| Oligonucleotides | ||
|
| ||
| tRNAiMet primer extension ACCTACGGGTTATGAGCCCGTCGG | This work | 2540 |
| tRNAAspGUC (SPCTRNAASP.06) primer extension GGGCTGCAAGCGTGAC | This work | 2743 |
| SPMITTRNAARG.02 primer extension CTAACAATTTCGAAGATTGTTAC | This work | 2746 |
| gBLOCK for synthetic RNA TAATACGACTCACTATAGGGGAGGCGAGAACACACCACAACGAAAACGAGCAAAACCCGGTACGCAACACAAAAGC GAACAACGCGAAAAAGGACACCGAAGCGGAAGCAAAGACAACCAACAGAAAACAACCGCAAACAAACGGGACCAGACAACGCACCAGCAAAA | This work | N/A |
| gBLOCK for synthetic RNA with 3 D TAATACGACTCACTATAGGGGAGGCGAGAACACACCACAACGAAAACGAGCAAAACCCTTTACGCACCACAAAAGC GAACAACGCGAAAAAGGACACCGAAGCGGAAGCAAAGACAACCAACAGAAAACAACCGCAAACAAACGGGACCAGA CAACGCACCAGCAAAA | This work | N/A |
| synthetic RNA primer extension GTCTTTGCTTCCGCTTCGGTGTCC | This work | 2567 |
| synthetic RNA with 3 D primer extension CGCGTTGTTCGCTTTTGTGG | This work | 3079 |
| RNA adaptor (Rho-seq) | 5′-P-AGATCGGAAGAGCGTCGTG-ddC | 2661 |
| DNA adaptor (Rho-seq) | 5′-P-AGATCGGAAGAGCACACGTCTG-ddC | 2674 |
|
| ||
| Software and algorithms | ||
|
| ||
| AnGeLi | Bitton et al., 2015 | N/A |
| WebLogo | Crooks et al., 2004 | N/A |
| RNAfold | Lorenz et al., 2011 | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Schizosaccharomyces pombe strains were grown in YES (Formedium PCM0310) or EMM (MP 4110–012) at 32°C to OD595nm 0.5– 0.8. For spot assays, strains were cultured in liquid YES or EMM media at 32°C and harvested at late-exponential phase, spotted (5-fold dilutions) on YES agar plates and incubated up to 5 days at 20, 25, 32 or 37°C.
Escherichia coli strains were grown at 37°C in LB (Invitrogen 12780–029) to OD595nm 0.6–0.7. For spot assays, strains were cultured in liquid LB medium at 37°C and harvested at late-exponential phase, spotted (10-fold dilutions) on LB agar plates and incubated up to 2 days at 20, 30, 37 or 42°C.
Human cells: HCT116 p53-positive colon carcinoma cells (ATCC, #CCL-247). Cells were cultured in McCoy’s 5A Medium (Lonza BE12–168F) in a New Brunswick Galaxy 170R incubator at 37°C and under 5% CO2.
METHOD DETAILS
Yeast methods
For mating experiments, S. pombe strains were maintained on YES plates at 25°C. For spore (gamete) quantification, strains were transferred to liquid YES and put on (malt extract) ME-plates at exponential growth and incubated for 56 h at 25°C before imaging. For spindle analysis, strains, that were refreshed one day before on YES plates, were put on ME plates and incubated at 25°C for 16–20 h.
For transgenesis in S. pombe, Expand High Fidelity-generated PCR products (Roche 19012322) were generated by amplification of a nucleotide sequence (consisting of a resistance cassette against an antibiotic or a tag sequence cloned in a pFA6A vector) with primers designed by the Pombe PCR Primer Program (developed by the Bahler lab, http://www.bahlerlab.info/resources/, 3′ end 20nt homology with the vector and 5′ end 60nt homology with the genome). Purified PCR products (QIAGEN 28106) were transformed by following the lithium acetate procedure (Bamps et al., 2004). Gene deletions resulted in [ATG-STOP] replacement and gene tagging resulted in a C-terminal tagging by removal of the endogenous STOP codon and insertion of the TAP (tandem affinity purification tag) sequence. Genome editing at the nda2 locus with CRISPR was performed using the ura4-based cloning-free method as described (Zhang et al., 2018) with the following guide TACCGATTTTGAAACCAGTA(nGG) and repair template: GGCTGCCGTTACTAGCATTAAATCTCGTCGCACCATCCAATTTGCCGATTGGTGtCCTACTGGTTTCAAAATCGGTATTTGCTATGAGCC with the underlined sequence highlighting the mutated codon (GTT -> GCC) and the lowercase “t” indicated the mutated PAM (Cassart et al., 2020).
Genome editing at the nda3 locus with CRISPR was similarly conducted with the following guide TACTCTTTCTATGCAT CAAT(nGG) and repair template: ACCTTACAATGCTACTCTTTCTATGCATCAATTaGTAGAGAACACCGACGAAACATTTTGCATTGATAATGAaGCTCTTTCTTCAATTTT with the underlined sequence highlighting the mutated codon (UCU -> ACC) and the lowercase “a” indicated the mutated PAM.
S. pombe GST-Dus1 was prepared as follows; Phusion High Fidelity-PCR amplification (NEB M0530) of dus1 (without its start codon) was performed on genomic DNA, purified (QIAGEN 28106), enzymatically restricted (BamHI-XhoI), ligated (Promega M180A) into the restricted pGEX-4T1 vector (downstream and in phase with the GST sequence), transformed in E. coli DH10B competent cells and sequenced. S. pombe GST-Dus2 was prepared as follows; Phusion High Fidelity-PCR amplification (NEB M0530) of dus2 cDNA sequence (without its start codon) was performed on a gBLOCK gene fragment (IDT), purified (QIAGEN 28106), enzymatically restricted (BamHI-XhoI), ligated (Promega M180A) into the restricted pGEX-4T1 vector (downstream and in phase with the GST sequence), transformed in E. coli DH10B competent cells and sequenced. Vectors were eventually transformed in E. coli BL21 cells for protein expression in LB + 1mM IPTG at 37°C for 120min and recombinant proteins were purified by combining the Poly-Prep Chromatography Columns (Bio-Rad 731–1550) with Glutathione Sepharose 4B (GE 17–0756-01).
Spore viability was determined as reported (Kucsera et al., 2000). Iodine staining was performed as described (Materne et al., 2015).
RNA extraction
50 mL of S. pombe (OD595nm ~0.5) or E. coli (OD595nm ~0.6) were harvested by centrifugation and washed in DEPC water. The cell pellets were resuspended in 750 μL of TES buffer (10 mM Tris pH 7.5, 10 mM EDTA pH 8, 0.5% SDS) and the RNA was isolated by hot phenol extraction; equal volume of phenol:chloroform 5:1 was added (Sigma P1944), heated and shaken at 65°C for 60 min, the upper phase was collected after centrifugation, mixed with an equal volume of phenol:chloroform:IAA 125:24:1 (Sigma 77619); the upper phase was then collected after centrifugation, mixed with an equal volume of chloroform:IAA 24:1 (Sigma 25666); the upper phase was collected again and the RNA was alcohol-precipitated with 2.5 volumes of 100% ethanol and 0.1 volume of 3M sodium acetate pH 5.2 for minimum 30min at −80°C or overnight at −20°C. After centrifugation, the pellet was washed with 70% ethanol, air- dried and resuspended in DEPC (Sigma D5758) water or pure water (Sigma 900682).
mRNA was purified by two tandem poly-T purification following the instructions of the manufacturer (Dynabeads™ Oligo(dT)25 from Life Technologies 61002).
Microchip analysis of RNA
Total, chemically-treated and ribodepleted RNA samples were visualized by RNA 6000 Pico Assay by using the Bioanalyzer microchip technology according to the manufacturer’s instructions (Agilent 5067–1513). Total RNAs were expected to have a RIN (RNA integrity number) > 8.5 and > 6 for Rho labeled RNAs.
Rhodamine labeling and dot blot assay
Rhodamine labeling was performed as previously described with modifications (Betteridge et al., 2007). Briefly, 30 μg of total RNA were resuspended in a 400 μL volume with 0.04 M Tris-HCl pH 7.4 and 4 mg of sodium borohydride (Sigma 452882, freshly resuspended in 1mM potassium hydroxide) for 60 min at 25°C in dark with constant shaking (750 rpm). Mock samples (R−) were treated similarly in KOH in the absence of sodium borohydride. The reaction was stopped by addition of glacial acetic acid to a final concentration of ~0.3 M. The reduced RNA was then ethanol-precipitated as described above and the dried pellet was resuspended in 5 μL of DEPC water and 85 μL of 0.1 M sodium formate pH 4 (Sigma 71539) by heating at 65°C for 7 min. Fluorescent labeling was performed for 90 min at 37°C in dark with 2.2mM of rhodamine 110 chloride (Sigma 83695). 10X dye concentrates were prepared in advance in 100% methanol and conserved at −20°C in dark for maximum 26 months. The pH of the reaction was then increased to 7.5 with 0.8 M Tris-HCl pH 8.5 and the dye excess was eliminated by phenol extraction (Sigma P1944) for 60min at 65°C. After centrifugation, the labeled RNA-containing upper phase was collected and RNA was ethanol-precipitated as described above. For dot blot assay, RNA was resuspended in < 10 μL of DEPC water by heating at 65°C and 0.2 to 1 μg/μL of RNA was spotted on a Hybond-N+ membrane (GE RPN1210b). The signal intensity was immediately monitored with a Cy3 channel (GE ImageQuant LAS 4000) and the quantification was performed with ImageJ (Schneider et al., 2012). The intensity of a DEPC water drop was considered as the background, subtracted to all other values and the subsequent signal of a 22mM rhodamine 110 in methanol drop was scaled to 1. Methylene blue staining was used as a loading control; the membrane was incubated for 5min at RT with 0.5M sodium acetate and 0.04% methylene blue, then washed twice in 1X PBS-T.
In vitro reduction of uridine to dihydrouridine
S. cerevisiae GST-Dus1 was a gift of E.M. Phizicky (URMC, NY, US). In vitro dihydrouridination was based on a previously published protocol (Xing et al., 2002). 30 μg of total RNA were incubated for 40 min at 30°C with 1 μg GST-Dus, 100mM Tris- HCl pH 8, 100mM ammonium acetate, 5mM magnesium chloride, 2mM DTT, 0.1mM EDTA, 1mM NADH and 1mM NADPH. The mock reaction (depicted as ‘+ buffer’) was done with 10mM glutathione instead of GST-Dus (GE 27–4570-01). The dihydrouridinated RNA was then purified with an equal volume of phenol:chloroform:IAA 125:24:1 (Sigma 77619), ethanol-precipitated as described above and resuspended in DEPC water for subsequent rhodamine labeling.
Synthetic RNA preparation
A gBLOCK gene fragment (IDT) was resuspended in 1X TE buffer. 20 ng were amplified with Phusion High-Fidelity DNA Polymerase (NEB M0530S), the subsequent PCR product was selected on an agarose gel and purified with QIAquick PCR Purification Kit (QIAGEN 28106). For RNA production, 200ng of DNA were in vitro transcribed by following the RiboMAXTM Large Scale RNA Production Systems’ instructions (Promega P1300). For D spike-in, rUTP was replaced by rDTP (Tebu-bio 040N-1035–1). The synthetic RNA was purified with ProbeQuantTM G-50 Micro Columns by following the manufacturer’s instructions (GE 28–9034-08). A total of 30ng of spike-in was added to 30 μg of total RNA for primer extension and 3ng for Rho-seq. The sequence of the gBLOCK is TAATACGACTCACTATAGGGGAGGCGAGAACACACCACAACGAAAACGAGCAAAACCCGGTACGCAACACAAAAGCGAACAACGCGAAAAAGGACACCGAAGCGGAAGCAAAGACAACCAACAGAAAACAACCGCAAACAAACGGGACCAGACAACGCACCAGCAAAA including the T7 promoter (underlined) and the unique T (bold).
Primer extension
Primer extension was performed essentially as described (Sakurai et al., 2010). First, 10 pmol of 5′-unphosphorylated primer were 32P-labeled with 10U of PNK (Promega M4101) and 3 μL of [Ɣ-32P]-ATP (Perkin NEG002A250UC). The reaction was performed at 37°C for 10min and stopped at 90°C for 2min. The resulting labeled primer was diluted to 0.4 pmol/μL. 5 to 25 μg of denatured R+/R− RNA were retro- transcribed for 60min at 50°C with 150 μM dNTPs (Applied biosystems 4368814), 1 μL of 32P-labeled primer and 100U of SSIII in 1X first-strand buffer (Invitrogen 18080–044). The reaction was stopped by addition of 2X sample buffer (98% formamide, 10mM EDTA, 0.1% blue bromophenol, 0.1% xylene cyanol) and incubation for 10 min at 90°C. The samples were immediately resolved on a 15 to 20% PAGE with 7M urea at 225V in 1X TBE buffer (Sigma T4415) and visualized with Super RX-N film (Fujifilm 47410 19289). The ladder was composed of DNA oligonucleotides (0.5 μM each) that were 32P-labeled as described above and mixed with 2X sample buffer. The ladder consists of 32P-labeled DNA oligonucleotides ranging from 15 to 60 nt.
Fluorescent northern blotting
10 μg of R+/R− or untreated total RNA were denatured in 1mM Tris-HCl pH 7.4, 6mM EDTA, 6% glycerol and were electrophoresed on a 1.2% agarose gel (0.67% formaldehyde, 20mM MOPS, 5mM sodium acetate, 1mM EDTA) in 1X FA buffer (20mM MOPS, 5mM sodium acetate, 1mM EDTA, pH 7). The gel was washed once in 50mM sodium hydroxide and twice in neutralization buffer (1.5M sodium chloride, 0.5M Tris-HCl pH 7.4). The nucleic acids were capillary transferred overnight in 1X SSC (Sigma S6639) on a Hybond-XL membrane (GE RPN203S). After crosslinking for 120 min at 80°C, the rhodamine signal was monitored as described above. Methylene blue staining was used as a loading control; the membrane was incubated for 5min at RT with 0.5M sodium acetate and 0.04% methylene blue, then washed twice in 1X PBS-T.
Wheat germ-based in vitro translation assay
Translation assays were performed using wheat germ extracts (Promega L4380) and either 35S labeled methionine (Perkin Elmer NEG709A500UC) or cysteine (Perkin Elmer NEG022T001MC). In order to produce a synthetic 137 amino-acids long protein of 15.571 Dalton, gBLOCK gene fragments (IDT) were ordered with the following 5′-sequence containing the T7 promoter TAATACGACTCACTATAgGGACCAGGGAGAGCCACCATGG where the lowercase g corresponds to the first transcribed nucleotide and the underlined track is the KOZAK sequence with the initiation codon in bold. For RNA production, 500 ng of DNA were in vitro transcribed using the RiboMAX™ Large Scale RNA production kit (Promega P1300). For D-containing mRNAs, rUTP was substituted by rDTP (Tebu-bio 040N-1035–1). The synthetic mRNA was quantified by QUBIT and 1.5 μg was directly used for in vitro translation 2 h at 25°C. The translated products were separated by PAGE (Bio-Rad Mini protean), treated with Amplify (Sigma GENAMP-100) and quantified using a Cyclone phosphoImager.
tRNA northern blotting
Small RNA fraction was enriched from 200–400 μg of total RNA by incubation 30min on ice with 190 mM sodium chloride and 7.3% PEG 8000. The supernatant was carefully collected after centrifugation for 20 min at 15,500g at 4°C and ethanol- precipitated as described above. The pellet was washed with 95% ethanol, air-dried and resuspended in DEPC water. 5 μg of small RNA were denatured in 1X sample buffer (50% formamide, 2.5 mM EDTA, 0.01% blue bromophenol), electrophoresed on a 10% PAGE with 7 M urea at 225V in 1X TBE buffer (Sigma T4415) and transferred on a Hybond-XL membrane (GE RPN203S) by semi-dry transfer for 45 min at 3mA/cm2 (Thermo Owl HEP Series Semidry Electroblotting System). Membrane was rinsed in 2X SSC before UV crosslinking for 30 s at 254nm (Vilbert Lourmat). For probe preparation, 20 pmol of 5′-unphosphorylated primer were 32P-labeled with 10 U of PNK (Promega M4101) and 2 μL of [Ɣ-32P]-ATP (Perkin NEG002A250UC) in a 20 μL- reaction for 60 min at 37°C. The probe was purified using ProbeQuantTM G-50 Micro Columns following the manufacturer’s instructions (GE 28–9034-08), denatured and hybridized to the crosslinked membrane overnight at 42°C in PerfectHybTM Plus Hybridization Buffer (Sigma H7033). The membrane was then washed in 2X SSC 0.1% SDS, twice in 0.1X SSC 0.1% SDS at 42°C and visualized with Super RX-N film (Fujifilm 47410 19289).
Rho-seq library preparation
Libraries for sequencing were prepared by combining previously published methods (Engreitz et al., 2013; Shishkin et al., 2015). 2–5 μg of R+/R− total RNA were ribodepleted (Thermo A15020 and K155005 [modified protocol provided by the manufacturer] for S. pombe and Illumina MRZGN126 for E. coli), quantified with Qubit RNA HS assay (Thermo Q32852) and analyzed by microchip as described above. Zinc-mediated fragmentation was performed at 70°C for 15 min with 90–125 ng of ribodepleted RNA and the reaction was stopped by following the manufacturer’s instructions (Thermo AM8740). Simultaneous DNA digestion and RNA extremities dephosphorylation were performed at 37°C for 30min with 2U of TURBO DNase (Thermo AM2238) and 3U of FastAP Thermosensitive Alkaline Phosphatase (Thermo EF0654) in 10mM Tris-HCl pH 7.4, 1 mM magnesium chloride, 0.12 mM calcium chloride, 10mM potassium chloride, 0.002% Triton X-100 and 2 mM DTT. 40 U of RNase inhibitor were added to prevent RNA degradation (NEB M0314). 20 pmol of RNA adaptor were then added to the sample, followed by denaturation for 2 min at 70°C. 3′ end ligation was performed for 90 min at 25°C with 39 U of T4 RNA ligase (NEB M0437) and 12 U of RNase inhibitor (NEB M0314) in 1X NEB Ligase 1 Buffer, 9% DMSO, 1 mM ATP and 20% PEG 8000. After ligation, 10 pmol of DNA primer were added, the sample was denatured for 2min at 70°C and the reverse transcription was performed as described above with SSIII (Invitrogen 18080–044). The excess of DNA primer was subsequently digested with 2 μL of ExoSAP-IT (Affymetrix 78250) by incubating 4min at 37°C and the enzyme was then inactivated by incubating 1 min at 80°C. The RNA was degraded for 10 min at 70°C with 100mM sodium hydroxide and the solution was neutralized by addition of acetic acid. 40 pmol of DNA adaptor were then added to the sample, followed by denaturation for 2 min at 75°C. The 3′ end ligation was performed overnight at 25°C with 48 U of T4 RNA ligase (NEB M0437) in 1X NEB Ligase 1 Buffer, 4% DMSO, 1mM ATP and 23.75% PEG 8000. Nucleic acid clean-ups were performed between each step by using MyOne SILANE magnetic beads (Thermo 37002D) resuspended in the chaotropic Buffer RLT (QIAGEN 74104). PCR amplification was performed in a 50 μL-reaction by mixing cDNA, 1 μM of barcoded PCR primer, 1 μM of universal PCR primer and 25 μL of NEBNext High- Fidelity 2X PCR Master Mix (NEB M0541) with the following program; 30 s at 98°C, 5 cycles of (10 s at 98°C, 30 s at 67°C, 30 s at 72°C), 13 cycles of (10 s at 98°C, 30 s at 72°C) and 60 s at 72°C as a final extension. Finally, two successive rounds of DNA polymer size selection were performed by mixing PCR products with 1.2 and 1.1 volume of AMPure XP beads (Beckman A63880). The DNA library was quantified (Thermo Q32851), examined on a DNA microchip (Agilent 5067–4626, ~250–300 nt polymers) and sequenced on Illumina HiSeq 2500 [1 flow cell, 2 lanes, 300M clusters] or on Novaseq S1 [100 cycles, 1 flow cell, 2 lanes, 1600 M clusters] (paired-end reads, 2 × 50 nt).
Rho-seq data analysis
Reads were quality- and adaptor-trimmed using trimmomatic 0.36 with options PE ILLUMINACLIP:TruSeq2-PE.fa:2:30:10 HEAD-CROP:1 SLIDINGWINDOW:4:15 MINLEN:20
The filtered reads were mapped on the spike-in sequence with bowtie2 (options–no-discordant–no-mixed–phred33 -p 4–un-conc) then on the genome using also bowtie2 for the E. coli libraries or tophat2 for the S. pombe libraries (options–mate-std-dev 150 -N 2–read-edit-dist 2–min-intron-length 5–max-intron-length 3000–no-discordant–no-coverage-search -p 4–library-type fr-firststrand).
Properly paired mapped reads were split by strand. Then bedtools (options genomecov -ibam -d −5) was used to calculate the number of RT termination events for each strand-specific position as the number of 5′ end of R2 reads that map at this position. In parallel, the paired reads were computationally extended/merged into fragments that covered the genome from the left end of the leftmost read to the right end of the rightmost reads but not across splice junctions. Those spliced fragments where then used to compute a fragment coverage reflecting the number of time a reverse transcriptase passed through a specific position. Then the ratio between the number of RT termination events and fragment coverage at each position (the D-ratio) was calculated.
After independent pre-filtering against the positions unlikely to have significant differences in D-ratio across conditions, the number of RT termination events out of the number of RT readthrough were modelized in generalized linear model of the binomial family (function glm in R) according to the effect of the treatment (R+ versus R−), the effect of the strain (WT versus Δ4dus), and their interaction. The positions were the interaction between the WT and the Δ4dus lead to a statistically significant (FDR < 0.1) increase of the proportion of RT termination events were retained as putative dihydrouridylation sites. The scripts used in this study are available on https://github.com/cyaguesa/Rho-seq.
Gene list and motif analyses
The S. pombe lists of genes were analyzed by the fission yeast-specific platform called AnGeLi (Bitton et al., 2015). The sequence motif analyses were performed with the web-based application WebLogo (Crooks et al., 2004).
Codon occurrence in the S. pombe transcriptome
The probability to get a D-site on the coding S. pombe transcriptome was calculated based on the codon usage database as follows; there are 27 codons containing a single U, representing 1,099,338 occurrences, 9 codons containing two U residues, representing 612,597 occurrences and 1 codon containing three U residues, representing 92,872 occurrences. There are therefore 2,603,148 possibilities to get a D-site on a coding region [(1 × 1,099,338) + (2 × 612,597) + (3 × 92,872) = 2,603,148]. Expressed in percentages, single U, double U and triple U-containing codons represent 42.2, 47.1 and 10.7% of the possibilities to get a D-site, respectively.
qRT-PCR
0.5 μg of RNeasy-purified total RNA (QIAGEN 74104) was retro-transcribed with the High-Capacity cDNA Reverse Transcription Kit (Thermo 4368813) by following the manufacturer’s instructions. The real-time PCR amplification was performed with SYBR Green Supermix (Bio-Rad 172–5124) in a Bio-Rad CFX96TM Real-Time machine. The PCR program was 3 min at 95°C and 40 cycles of (15 s at 95°C and 30 s at 60°C). Relative RNA quantification relied on the ΔΔCT method.
Western blotting
Proteins were extracted from 10mL of yeasts cultured in YES at 32°C. Cells were pelleted, washed in water and disrupted with 0.3M hydroxide sodium for 10min at RT. The lysate was then centrifuged and the supernatant was discarded. The pellet was resuspended in 70 μL of alkaline extraction buffer (60 mM Tris-HCl pH 6.8, 4% β-mercaptoethanol, 4% SDS, 0.01% bromophenol blue, 5% glycerol) and boiled for 5 min at 95°C. 10 μL of the samples were loaded on a 4%–15% Mini-PROTEAN TGX Precast Protein Gel (Bio-Rad 456–1083), transferred on a nitrocellulose membrane (Bio-Rad 1704158) and blocked for 60min at RT or overnight at 4°C in 50:50 1X PBS:Odyssey Blocking Buffer PBS (Westburg LI 927–40100). The membrane was incubated with the primary antibody for 60 min at RT or overnight at 4°C with Odyssey Buffer containing 0.05% Tween20 (Bio-Rad 161–0781) and a 1:500–1,000 dilution of anti-α-tubulin (Sigma T5168) or anti-Histone H3 (abcam ab1791) primary antibodies. After three washes in PBS-T, the membrane was incubated with a 1:10,000 dilution of anti-mouse (Westburg LI 925–32210) or anti-rabbit (Westburg LI 925–32211) secondary antibodies for 60min at RT in Odyssey Buffer containing 0.05% Tween20. After three washes in PBS-T and three washes in PBS, the membrane was dried for 60 min at 37°C and visualized on a LiCOR scanner on channel 800. The quantification was performed with ImageJ (Schneider et al., 2012).
Dot blotting
Proteins were extracted from 10 mL of yeasts cultured in YES at 32°C. Cells were pelleted, washed in water and disrupted with 0.3 M hydroxide sodium for 10min at RT. The lysate was then centrifuged at 13000 rpm for 30 s and the supernatant was discarded. The pellet was resuspended in 70 μL of alkaline extraction buffer (60 mM Tris-HCl pH 6.8, 4% β-mercaptoethanol, 4% SDS, 0.01% bromophenol blue, 5% glycerol) and boiled for 5 min at 95°C. 5 μL of the samples were mixed with the Amersham Cy5-QuickStain Total Protein Labeling Kit and spotted on a nitrocellulose membrane using the Bio-Rad dot blot system. The Tubulin signal was normalized on the Cy5 total protein labeling signal.
Human cell genome editing
To delete the DUS genes from the genome, CRISPR guide RNAs (crRNA) were designed near the exon–intron junctions and resuspended in IDTE buffer (10 μL in a 2 nmol-containing tube to get 200 μM). crRNAs, tracRNA (IDT, #1072532) and S.p. Cas9 (IDT, #1081058) were purchased from Integrated DNA Technologies (IDT). Cells were transfected with in vitro reconstituted crRNA:tracRNA:Cas9 complex:
For single-gene targeting, 5 μL crRNA forward:tracrRNA and 5 μL crRNA reverse:tracrRNA were used. For quadruple-gene targeting: 4 × 1.25 μL crRNA forward:tracrRNA and 4 × 1.25 μL crRNA reverse:tarcrRNA were used together with electroporation enhancer (IDT, #1075915, final concentration 4 μM) in a nucleofector device (both versions IIb and 4D were used). Cells were incubated for 24 h to allow them to recover and then detached, and cloned. Cells from wells containing a single colony were detached, split into replicate 96-well plates and further incubated. Genomic DNA (gDNA) was extracted from one plate once cells reached confluence. Each target sequence with a deletion was detected by diagnostic PCR. The PCR products were sequenced to establish the exact junctions of each deletion.
Size-exclusion chromatography (SEC) and LC-MS/MS
Large RNA (28S rRNA, 18S rRNA and mRNA) and tRNA portions from total RNA samples were isolated by size-exclusion chromatography (SEC) performed on tandem Agilent SEC-5 column (1000Å, 7.8 × 300 mm, 5 μm) and Agilent SEC-3 column (300 Å, 7.8 × 300 mm, 3 μm). The column was equilibrated with 100 mM ammonium acetate buffer (pH = 7.0) at 0.6 mL/min. The fractions containing the desired RNA (large RNA portions: 10–13 min; tRNA: 16.3–17.7 min) were collected by auto-fraction collector and concentrated to around 50 μL using SpeedVac. 3K Da MWCO spin filters were further used for sample desalting. Large RNA portions were quality checked by an Agilent Bioanalyzer Pico chip.
~8 μg RNA from large RNA, ~800 ng RNA from small RNA portions or ~1.3 μg of poly-T purified mRNA were hydrolyzed in 50 μL digestion cocktail containing 50 U benzonase, 20 U calf intestinal alkaline phosphatase, 0.6 U phosphodiesterase I, 100 μM deferoxamine, 100 μM butylated hydroxytoluene, 5 ng coformycin, 100 nM internal standard [15N]5-deoxyadenosine, 2.5 mM MgCl2 and in 5 mM Tris buffer pH 8.0. The digestion mixture was incubated at 37°C for 6 h. Digestion enzymes were subsequently removed by passing mixture through a 10k Da MWCO spin filter.
Using standards, HPLC retention times for 31 RNA modifications were confirmed on a Phenomenex Synergi Fusion-RP C18 column (100 × 2.0 mm, 2.5 μm) coupled to an Agilent 1290 HPLC system and an Agilent 6490 Triple Quad mass spectrometer. The LC system was conducted at 35°C and a flow rate of 0.35 mL/min, with a gradient starting with 100% solution A (5 mM ammonium acetate, pH 5.3), followed by 0%–10% solution B (acetonitrile) 2–12 min; 10%–40% solution B, 12–16 min; 40%–80% solution B, 16–17 min; 80%–90% solution B, 17–17.1 min; 90% solution B, 17.1–20 min. The HPLC column was coupled to an Agilent 6490 Triple Quad mass spectrometer with an electrospray ionization source in positive mode with the following parameters: gas temperature, 200 degree; gas flow, 11 L/min; nebulizer, 20 psi; sheath gas temperature, 300 degree; sheath gas flow, 12 L/min; capillary voltage, 1800 V; Vcharging, 2000 V. MRM mode was used for detection of product ions derived from the precursor ions for all the RNA modifications with instrument parameters including the collision energy (CE) optimized for maximal sensitivity for the modifications. The level of indicated RNA modifications was determined by pmol of input.
Ribosome profiling
Ribosome profiling was performed in triplicate on the Δ4 strain and its isogenic WT according to (Duncan and Mata, 2017; Nedialkova and Leidel, 2015; Pinto et al., 2020) with the following modifications: 1) cells were grown to mid-log phase, 2) to avoid potential interference, cycloheximide was omitted at the collection stage, 3) cells were collected on a filter disc in < 1 min by filtration, one third of the cells was immediately set aside in liquid nitrogen (for total RNA preparation used in RNaseq control to establish mRNA abundance), the remaining two-thirds was pulverized under cryogenic conditions in a Planetary Mill PM100 (Retsch), 4) the conditions of monosome preparation were optimized for S. pombe.
Library preparation.
Ribosome-protected footprints were puriðed from monosome fractions by size selection and gel extraction. Briefly, ribosome protected fragments were generated by digesting 100 A260 units of cleared lysate with 600 U RNase I (ThermoFisher) at 22°C. The reaction was stopped after 1 h by addition of 15 μl Superase In (ThermoFisher) and loaded on a 10%–50% (w/v) sucrose gradient in 50 mM Tris-HCl pH 7.5, 50 mM NH4Cl, 12 mM MgCl2, 0.5 mM DTT, and centrifuged for 4 h at 38 krpm and 4°C in a SW41Ti rotor. Monosomes were fractionated and total RNA isolated. Subsequently, RNA was separated on 15% polyacrylamide gels (8 M urea, 1 × TBE buffer) and 28–32 nt ribosome footprints were extracted from the gel. Purified ribosome protected fragments were cloned with primers that contained four randomized nucleotides to reduce ligation biases (Duncan and Mata, 2017; Lecanda et al., 2016).
Bioinformatic analyses.
Libraries were mapped to the S. pombe transcriptome. The sequencing data was analyzed with Xtail (Xiao et al., 2016), subcodon analysis was performed as described in (Nedialkova and Leidel, 2015; Zinshteyn and Gilbert, 2013).
TMT-based proteomic analyses (Creative Proteomics)
Tandem mass tag (TMT) relies on chemical label allowing sample multiplexing in mass spectrometry-based quantification and identification. Isobaric mass tags are molecules with the same mass yielding reporter ions of differing mass after fragmentation. The relative ratio of the measured reporter ions represents the relative abundance of the tagged molecule.
TMT6plex Isobaric Label Reagent Set, Pierce Quantitative Colorimetric Peptide Assay, was purchased from Thermo Fisher Science. An Ultimate 3000 nano UHPLC system (Thermo Scientific, Waltham, MA) coupled online to a Q Exactive HF mass spectrometer (Thermo Scientific) equipped with a Nanospray Flex Ion Source (Thermo Scientific). Frozen cell pellets were resuspended in lysis buffer (Tissuelyser) and centrifugated. The protein concentration of the supernatant was determined by BCA kit. An aliquot of 100 μg protein was reduced by 10 mM TCEP at 56°C for 1 h and alkylated by 20 mM IAA at RT in dark for 1 h.Free trypsin was added into the protein solution at a ratio of 1:50, and the solution was incubated at 37°C overnight. The extracted peptides were dried and redissolved 100 mM TEAB. Immediately before use, the TMT Label Reagents was added and incubated for 1 h at room temperature. Next, 8 μL of 5% hydroxylamine were added to the sample and incubate for 15 min to quench the reaction.The samples were combined at equal amounts in new microcentrifuge tube.
The following labels were used, corresponding to the 18 tested conditions (WT, dus3Δ, Δ4 in vegetative or meiotic growth, all in triplicates: TMT6–126 TMT6–127 TMT6–128 TMT6–129 TMT6–130 TMT6–131 TMT6–126 TMT6–127 TMT6–128 TMT6–126 TMT6–127 TMT6–128 TMT6–129 TMT6–130 TMT6–131 TMT6–129 TMT6–130 TMT6–131.
Nanoflow UPLC: Ultimate 3000 nano UHPLC system (ThermoFisher Scientific, USA); Nanocolumn: trapping column (PepMap C18, 100A, 100 μm × 2 cm, 5 μm) and an analytical column (PepMap C18, 100A, 75 μm × 50 cm, 2 μm). Loaded sample volume: 1 μg. Mobile phase: A: 0.1% formic acid in water; B: 0.1% formic acid in 80% acetonitrile. Total flow rate: 250 nL/minLC linear gradient: from 5 to 7% buffer B in 2 min, from 7% to 20% buffer B in 80 min, from 20% to 40% buffer B in 34 min, then from 40% to 90% buffer B in 4 min.
For TMT-labeled samples, the full scan was performed between 350–1,650 m/z at the resolution 120,000 at 200 Th, the automatic gain control target for the full scan was set to 3e6. The MS/MS scan was operated in Top 15 mode using the following settings: resolution 30,000 at 200 Th; automatic gain control target 1e5; normalized collision energy at 32%; isolation window of 1.2 Th; charge sate exclusion: unassigned, 1, > 6; dynamic exclusion 40 s.
Differential Protein Analysis
The raw MS files were analyzed and searched against fission yeast protein database based on the species of the samples using Max-quant (1.6.2.6). The parameters were set as follows: the protein modifications were carbamidomethylation (C) (fixed), oxidation (M) (variable), TMT-6plex; the enzyme specificity was set to trypsin; the maximum missed cleavages were set to 2; the precursor ion mass tolerance was set to 10 ppm, and MS/MS tolerance was 0.6 Da. In total, 2193 proteins were identified in meiotic growth (WT, dus3Δ, Δ4), and 1974 proteins were identified in vegetative growth (WT, dus3Δ, Δ4). Proteins of relative quantitation were divided into two categories. Fold-change (FC) > 1.2 was considered up- regulation while FC < 0.83 (1/1.2) was considered as down-regulation. False discovery rate < 10%.
RNA-Immunoprecipitation (RIP-qPCR)
Note that no crosslinking was performed. 100 mL of cells were grown at 30°C in YE (Formedium, #PCM0110) supplemented with 150 mg/L leucine, uracil, histidine, adenine and lysine until OD600nm = 1.0–1.2. Following centrifugation, cell pellets were washed in 1X PBS and resuspended in 2 mL lysis buffer (6 mM Na2HPO4, 4 mM NaH2PO4, 150 mM NaC2H3O2, 5 mM MgC2H3O2, 0.25% NP-40, 2 mM EDTA, 1 mM EGTA, 5% glycerol, 1X complete EDTA-free protease inhibitor cocktail (ThermoFisher Scientific, #A32955) and 40 U RNaseOUT Ribonuclease inhibitor (Invitrogen, #10777–019) to make ‘pop-corn’ in liquid nitrogen. Lysis was performed using a Ball Mill (Retsch, MM400) for 15 min at a 10 Hz frequency. Extracts were cleared by centrifugation before precipitation with 1 mg of pre-washed rabbit IgG-conjugated M-270 Epoxy Dynabeads (Invitrogen, #14311D) for 1 h at 4 °C. Beads were then washed twice with IPP150 (10 mM Tris pH8, 150 mM NaCl, 0.1% NP-40). Total and immunoprecipitated RNAs were extracted with phenol:chloroform 5:1 pH4.7 (Sigma, #P1944), precipitated with ethanol and treated with DNase (Ambion, #AM1906). RT reactions were carried out with 100 units Maxima Reverse Transcriptase (ThermoFisher Scientific, #EP0743) at 50°C for 30 min in the presence of 200 ng random hexamers. Following enzyme denaturation at 85°C for 5 min, reactions were diluted to 1:10 ratio. Samples were analyzed by qPCR with SYBR Green Master mix and a LightCycler LC480 apparatus (Roche). Quantification was performed using the ΔCt method.
RNA-sequencing
1 μg of total RNA was ribodepleted as described in the Rho-seq section and analyzed by microchip electrophoresis as described above. 125ng of ribodepleted samples were prepared for strand-specific NGS with the TruSeq Stranded Total RNA Sample Preparation Protocol (Illumina). The library was sequenced on Illumina HiSeq 2500 (paired-end reads, 2 × 50nt). Reads were mapped on the genome using TopHat2. The quantification was made with featureCounts function on R and the differential expression analysis was performed with DESeq2. The genes following these criteria were considered as differentially expressed; false discovery rate < 10% and absolute value of the fold change > 1.5.
HPLC analysis of D content
The HPLC analysis was performed exactly like previously described (Sharma et al., 2015; van Tran et al., 2019), except that total RNA from the indicated bacterial or yeast strains was used instead of purified ribosomal RNAs. Briefly, total RNA was digested to nucleosides with 2 U P1 nuclease (Sigma, N8630) and 10 μl alkaline phosphatase (Sigma, P4252). As calibration control, we used a commercial source of 5,6-dihydrouridine (Apollo Scientific, Ref OR17694). In our HPLC system, the D nucleosides was found to elute at ~4.7 min.
Live microscopy
For live-cell imaging cells were mounted on ME agarose pads, containing 2% agarose (Tran et al., 2004). Imaging was performed at 25°C. Images were acquired on an inverted Spinning Disk Confocal (Roper/Nikon), equipped with Plan Apochromat 100 × /1.4 NA objective lens (Nikon), a PIFOC (perfect image focus) objective stepper, and a charge-coupled device camera (EMCCD 512×512 QuantEM; Photometrics). Stacks of 7 planes spaced 1 μm were acquired for each channel with 100 ms exposure for 561 nm wavelength and 100 ms exposure for 491 nm wavelength, binning one, and an electronic gain of 3 for both wavelengths. For time-lapse movies, generally, an image was taken every minute, or every ten minutes, as it was the case for the nda2noD mutant, for a duration of 240 min.
Image analysis
Using metamorph 7.8, maximum projections of each stack were performed. Spindle dynamics were examined by the length of the mCherry-Atb2 or ENVY-Atb2 signal over time. Prophase, metaphase or anaphase spindle length refers to the maximum spindle length of each phase.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless stated otherwise all quantitative experiments were performed in triplicate and average with SEM as indicated in legends reported. The statistical details of experiments can be found in corresponding figure legends. The data were analyzed by using Student’s t test as described in respective figure legends. p values are indicated as follows: *p < 0.05; **p < 0.01; ***p < 0.001. No additional methods were used to determine whether the data met assumptions of the statistical approach.
Additional details related on the analysis of specific experiments can be found in the corresponding section of the method.
Supplementary Material
Highlights.
Rho-seq detects a range of RNA modifications through differential rhodamine labeling
The dihydrouridine tRNA modification is also present on fission yeast mRNAs
Dihydrouridine on the tubulin mRNA is required for meiotic chromosome segregation
Tubulin mRNA dihydrouridylation is evolutionarily conserved from yeast to human
ACKNOWLEDGMENTS
We are grateful to Eric Phizicky for reagents and Marc Graille for comments on the manuscript. Research in the lab of D.L.J.L. is supported by the Fonds de la Recherche Nationale (F.R.S./FNRS), the Université Libre de Bruxelles (ULB), the European Joint Programme on Rare Diseases (EJP-RD; RiboEurope and DBAGeneCure), the Région Wallone (RiboCancer), and the International Brachet Stiftüng (IBS). High-throughput sequencing was performed by the NGS platform of Institut Curie, supported by grants from the Agence Nationale de la Recherche (investissements d’avenir; ANR-10-EQPX-03 and ANR10-INBS-09–08) and by the Canceropôle Ile-de-France. This work was also supported by NIH grants ES031529 and AG063341 (to P.D.) and PDR T.0012.14, CDR J.0066.16, and PDR T.0112.21 (to D.H.). O. F. and C.Y.-S. were supported by a FRIA fellowship. D.L.J.L and D.H. are FNRS Directors of Research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2021.11.003.
REFERENCES
- Adachi Y, Toda T, Niwa O, and Yanagida M (1986). Differential expressions of essential and nonessential alpha-tubulin genes in Schizosaccharomyces pombe. Mol. Cell. Biol 6, 2168–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamps S, Westerling T, Pihlak A, Tafforeau L, Vandenhaute J, Mäkelä TP, and Hermand D (2004). Mcs2 and a novel CAK subunit Pmh1 associate with Skp1 in fission yeast. Biochem. Biophys. Res. Commun 325, 1424–1432. [DOI] [PubMed] [Google Scholar]
- Betteridge T, Liu H, Gamper H, Kirillov S, Cooperman BS, and Hou YM (2007). Fluorescent labeling of tRNAs for dynamics experiments. RNA 13, 1594–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Xu J, Johnson RC, Schimmel P, and de Crécy-Lagard V (2002). Identification of the tRNA-dihydrouridine synthase family. J. Biol. Chem 277, 25090–25095. [DOI] [PubMed] [Google Scholar]
- Bitton DA, Schubert F, Dey S, Okoniewski M, Smith GC, Khadayate S, Pancaldi V, Wood V, and Bähler J (2015). AnGeLi: A Tool for the Analysis of Gene Lists from Fission Yeast. Front. Genet 6, 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgon R, Gentleman R, and Huber W (2010). Independent filtering increases detection power for high-throughput experiments. Proc. Natl. Acad. Sci. USA 107, 9546–9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhr F, Jha S, Thommen M, Mittelstaet J, Kutz F, Schwalbe H, Rodnina MV, and Komar AA (2016). Synonymous Codons Direct Cotranslational Folding toward Different Protein Conformations. Mol. Cell 61, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candiracci J, Migeot V, Chionh YH, Bauer F, Brochier T, Russell B, Shiozaki K, Dedon P, and Hermand D (2019). Reciprocal regulation of TORC signaling and tRNA modifications by Elongator enforces nutrient-dependent cell fate. Sci. Adv 5, v0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassart C, Drogat J, Migeot V, and Hermand D (2012). Distinct requirement of RNA polymerase II CTD phosphorylations in budding and fission yeast. Transcription 3, 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassart C, Yague-Sanz C, Bauer F, Ponsard P, Stubbe FX, Migeot V, Wery M, Morillon A, Palladino F, Robert V, and Hermand D (2020). RNA polymerase II CTD S2P is dispensable for embryogenesis but mediates exit from developmental diapause in C. elegans. Sci. Adv 6, eabc1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti P, and Miller N (1967). Selective reduction of yeast transfer ribonucleic acid with sodium borohydride. J. Mol. Biol 26, 55–66. [DOI] [PubMed] [Google Scholar]
- Cerutti P, Holt JW, and Miller N (1968). Detection and determination of 5,6-dihydrouridine and 4-thiouridine in transfer ribonucleic acid from different sources. J. Mol. Biol 34, 505–518. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalluge JJ, Hamamoto T, Horikoshi K, Morita RY, Stetter KO, and McCloskey JA (1997). Posttranscriptional modification of tRNA in psychrophilic bacteria. J. Bacteriol 179, 1918–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudin O, Merlini L, Bendezú FO, Groux R, Vincenzetti V, and Martin SG (2017). A systematic screen for morphological abnormalities during fission yeast sexual reproduction identifies a mechanism of actin aster formation for cell fusion. PLoS Genet. 13, e1006721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan C, and Mata J (2017). Ribosome Profiling for the Analysis of Translation During Yeast Meiosis. Methods Mol. Biol 1471, 99–122. [DOI] [PubMed] [Google Scholar]
- Edmonds CG, Crain PF, Gupta R, Hashizume T, Hocart CH, Kowalak JA, Pomerantz SC, Stetter KO, and McCloskey JA (1991). Posttranscriptional modification of tRNA in thermophilic archaea (Archaebacteria). J. Bacteriol 173, 3138–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES, et al. (2013). The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 341, 1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauquenoy S, Migeot V, Finet O, Yague-Sanz C, Khorosjutina O, Ekwall K, and Hermand D (2018). Repression of Cell Differentiation by a cis-Acting lincRNA in Fission Yeast. Curr. Biol 28, 383–391.e3. [DOI] [PubMed] [Google Scholar]
- Grozhik AV, and Jaffrey SR (2018). Distinguishing RNA modifications from noise in epitranscriptome maps. Nat. Chem. Biol. 14, 215–225. [DOI] [PubMed] [Google Scholar]
- Hidese R, Mihara H, Kurihara T, and Esaki N (2011). Escherichia coli dihydropyrimidine dehydrogenase is a novel NAD-dependent heterotetramer essential for the production of 5,6-dihydrouracil. J. Bacteriol 193, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang RC, and Bonner J (1965). Histone-bound RNA, a component of native nucleohistone. Proc. Natl. Acad. Sci. USA 54, 960–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasprzak JM, Czerwoniec A, and Bujnicki JM (2012). Molecular evolution of dihydrouridine synthases. BMC Bioinformatics 13, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Daigo Y, Hayama S, Ishikawa N, Yamabuki T, Ito T, Miyamoto M, Kondo S, and Nakamura Y (2005). A novel human tRNA-dihydrouridine synthase involved in pulmonary carcinogenesis. Cancer Res. 65, 5638–5646. [DOI] [PubMed] [Google Scholar]
- Kaur J, Raj M, and Cooperman BS (2011). Fluorescent labeling of tRNA dihydrouridine residues: Mechanism and distribution. RNA 17, 1393–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse MG, and Wilusz JE (2017). Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev. 31, 1717–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucsera J, Yarita K, and Takeo K (2000). Simple detection method for distinguishing dead and living yeast colonies. J. Microbiol. Methods 41, 19–21. [DOI] [PubMed] [Google Scholar]
- Lecanda A, Nilges BS, Sharma P, Nedialkova DD, Schwarz J, Vaquerizas JM, and Leidel SA (2016). Dual randomization of oligonucleotides to reduce the bias in ribosome-profiling libraries. Methods 107, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Liu Q, Jiang YZ, and Gregory RI (2019). Nucleotide resolution profiling of m7G tRNA modification by TRAC-Seq. Nat. Protoc. 14, 3220–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loiodice I, Janson ME, Tavormina P, Schaub S, Bhatt D, Cochran R, Czupryna J, Fu C, and Tran PT (2019). Quantifying Tubulin Concentration and Microtubule Number Throughout the Fission Yeast Cell Cycle. Biomolecules 9, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011). ViennaRNA Package 2.0. Algorithms Mol. Biol 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka MA, Olchowik A, Grosjean H, and Bujnicki JM (2014). Distribution and frequencies of post-transcriptional modifications in tRNAs. RNA Biol. 11, 1619–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Materne P, Anandhakumar J, Migeot V, Soriano I, Yague-Sanz C, Hidalgo E, Mignion C, Quintales L, Antequera F, and Hermand D (2015). Promoter nucleosome dynamics regulated by signalling through the CTD code. eLife 4, e09008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y, Muller S, Behm-Ansmant I, and Branlant C (2007). Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol. 425, 21–53. [DOI] [PubMed] [Google Scholar]
- Nedialkova DD, and Leidel SA (2015). Optimization of Codon Translation Rates via tRNA Modifications Maintains Proteome Integrity. Cell 161, 1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluh JL, Killilea AN, Detrich HW 3rd, and Downing KH (2004). Meiosis-specific failure of cell cycle progression in fission yeast by mutation of a conserved beta-tubulin residue. Mol. Biol. Cell 15, 1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto R, Vågbø CB, Jakobsson ME, Kim Y, Baltissen MP, O’Donohue MF, Guzmán UH, Małecki JM, Wu J, Kirpekar F, et al. (2020). The human methyltransferase ZCCHC4 catalyses N6-methyladenosine modification of 28S ribosomal RNA. Nucleic Acids Res. 48, 830–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova AM, and Williamson JR (2014). Quantitative analysis of rRNA modifications using stable isotope labeling and mass spectrometry. J. Am. Chem. Soc 136, 2058–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, Kline N, Olson S, Weinberg D, Baker KE, Graveley BR, and Coller J (2015). Codon optimality is a major determinant of mRNA stability. Cell 160, 1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai M, Yano T, Kawabata H, Ueda H, and Suzuki T (2010). Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat. Chem. Biol 6, 733–740. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, et al. (2014). Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159, 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Yang J, Düttmann S, Watzinger P, Kötter P, and Entian KD (2014). Identification of novel methyltransferases, Bmt5 and Bmt6, responsible for the m3U methylations of 25S rRNA in Saccharomyces cerevisiae. Nucleic Acids Res. 42, 3246–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Langhendries JL, Watzinger P, Kötter P, Entian KD, and Lafontaine DL (2015). Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res. 43, 2242–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih TY, and Bonner J (1969). Chromosomal RNA of calf thymus chromatin. Biochim. Biophys. Acta 182, 30–35. [DOI] [PubMed] [Google Scholar]
- Shishkin AA, Giannoukos G, Kucukural A, Ciulla D, Busby M, Surka C, Chen J, Bhattacharyya RP, Rudy RF, Patel MM, et al. (2015). Simultaneous generation of many RNA-seq libraries in a single reaction. Nat. Methods 12, 323–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Torres AG, and Ribas de Pouplana L (2021). Inosine in Biology and Disease. Genes (Basel) 12, 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PT, Paoletti A, and Chang F (2004). Imaging green fluorescent protein fusions in living fission yeast cells. Methods 33, 220–225. [DOI] [PubMed] [Google Scholar]
- Vallin J, and Grantham J (2019). The role of the molecular chaperone CCT in protein folding and mediation of cytoskeleton-associated processes: implications for cancer cell biology. Cell Stress Chaperones 24, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P, Bohnsack KE, Bohnsack MT, Jaffrey SR, Graille M, and Lafontaine DLJ (2019). The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 47, 7719–7733. 10.1093/nar/gkz619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Zou Q, Liu Y, and Yang X (2016). Genome-wide assessment of differential translations with ribosome profiling data. Nat. Commun 7, 11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing F, Martzen MR, and Phizicky EM (2002). A conserved family of Saccharomyces cerevisiae synthases effects dihydrouridine modification of tRNA. RNA 8, 370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing F, Hiley SL, Hughes TR, and Phizicky EM (2004). The specificities of four yeast dihydrouridine synthases for cytoplasmic tRNAs. J. Biol. Chem 279, 17850–17860. [DOI] [PubMed] [Google Scholar]
- Zhang G, Hubalewska M, and Ignatova Z (2009). Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol 16, 274–280. [DOI] [PubMed] [Google Scholar]
- Zhang XR, He JB, Wang YZ, and Du LL (2018). A Cloning-Free Method for CRISPR/Cas9-Mediated Genome Editing in Fission Yeast. G3 (Bethesda) 8, 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Guo J, Cha J, Chae M, Chen S, Barral JM, Sachs MS, and Liu Y (2013). Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 495, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinshteyn B, and Gilbert WV (2013). Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS Genet. 9, e1003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Rho-seq, Ribo-seq and RNA-seq raw and processed data are available on GEO under the accession number GSE145686 and are publicly available as of the date of publication. Original blot images reported in this paper have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-alpha Tubulin | Sigma | Cat# T5168;RRID:AB_477579 |
| Mouse monoclonal beta-alpha Tubulin | Millipore | Cat# MAB3408; RRID:AB_94650 |
| Rabbit polyclonal anti Histone H3 | Abcam | Cat# ab1791;RRID:AB_302613 |
|
| ||
| Yeast, E. coli and human strains/lines | ||
|
| ||
| Fission yeast h- | Lab stock | yDH94 |
| Fission yeast h- dus1::natR | This work | yDH1491 |
| Fission yeast h- dus2::kanR | This work | yDH1456 |
| Fission yeast h- dus3::hphR | This work | yDH1659 |
| Fission yeast h- dus4::bleR | This work | yDH1631 |
| Fission yeast h- dus1-4 Δ | This work | yDH1754 |
| Fission yeast h- nda2noD | This work | yDH2013 |
| Fission yeast h- nda3noD | This work | yDH2021 |
| Fission yeast h- nda2noD nda3noD | This work | yDH2018 |
| Human cell line HCT116 | ATCC | CCL247 |
| Human cell line HCT116 DUS1-4 Δ | This work | HsΔ4 −/− (#1) |
| E. coli | Lab stock | JC8679 |
| E. coli dusA::cmR dusB-fis::kanR dusC::specR | Lab stock | PS5107 |
|
| ||
| Chemicals | ||
|
| ||
| Dynabeads Oligo(dT)25 | Life Technologies | 61002 |
| sodium borohydride | Sigma | 452882 |
| rhodamine 110 chloride | Sigma | 83695 |
| [Ɣ-32P]-ATP | Perkin Elmer | NEG002A250UC |
| 35S labeled methionine | Perkin Elmer | NEG709A500UC |
|
| ||
| Critical commercial assays | ||
|
| ||
| wheat germ extracts | Promega | L4380 |
| Ribodepletion kit | Thermo | A15020 – K155005 |
|
| ||
| Deposited data | ||
|
| ||
| Rho-seq, Ribo-seq and RNA-seq raw and processed data | This work | GEO: GSE145686; https://doi.org/10.17632/m6v9rcprz6.1 |
| Raw data (Mendeley data) | This work | |
|
| ||
| Oligonucleotides | ||
|
| ||
| tRNAiMet primer extension ACCTACGGGTTATGAGCCCGTCGG | This work | 2540 |
| tRNAAspGUC (SPCTRNAASP.06) primer extension GGGCTGCAAGCGTGAC | This work | 2743 |
| SPMITTRNAARG.02 primer extension CTAACAATTTCGAAGATTGTTAC | This work | 2746 |
| gBLOCK for synthetic RNA TAATACGACTCACTATAGGGGAGGCGAGAACACACCACAACGAAAACGAGCAAAACCCGGTACGCAACACAAAAGC GAACAACGCGAAAAAGGACACCGAAGCGGAAGCAAAGACAACCAACAGAAAACAACCGCAAACAAACGGGACCAGACAACGCACCAGCAAAA | This work | N/A |
| gBLOCK for synthetic RNA with 3 D TAATACGACTCACTATAGGGGAGGCGAGAACACACCACAACGAAAACGAGCAAAACCCTTTACGCACCACAAAAGC GAACAACGCGAAAAAGGACACCGAAGCGGAAGCAAAGACAACCAACAGAAAACAACCGCAAACAAACGGGACCAGA CAACGCACCAGCAAAA | This work | N/A |
| synthetic RNA primer extension GTCTTTGCTTCCGCTTCGGTGTCC | This work | 2567 |
| synthetic RNA with 3 D primer extension CGCGTTGTTCGCTTTTGTGG | This work | 3079 |
| RNA adaptor (Rho-seq) | 5′-P-AGATCGGAAGAGCGTCGTG-ddC | 2661 |
| DNA adaptor (Rho-seq) | 5′-P-AGATCGGAAGAGCACACGTCTG-ddC | 2674 |
|
| ||
| Software and algorithms | ||
|
| ||
| AnGeLi | Bitton et al., 2015 | N/A |
| WebLogo | Crooks et al., 2004 | N/A |
| RNAfold | Lorenz et al., 2011 | N/A |