

Abstract
Background:
Trauma increases susceptibility to secondary bacterial infections. The events suppressing antimicrobial immunity are unclear. Neutrophils (PMN) migrate towards bacteria using chemotaxis (CTX), trap them in extracellular NETs and kill them using respiratory burst (RB). We hypothesized plasma and wound fluids from trauma patients alter PMN function.
Methods:
Volunteer PMN were incubated in plasma or wound fluids from trauma patients (Days 0–1, 2–3) and their functions compared with PMN incubated in volunteer plasma. CTX was assessed in transwells. Luminometry assessed total and intracellular RB responses to receptor-dependent and independent stimulants. NET formation (NETosis) was assessed using elastase assays. The role of tissue necrosis in creating functionally suppressive systemic PMN environments was assessed using a novel pig model where PMN were incubated in uninjured pig plasma or plasma from pigs undergoing intraperitoneal instillation of liver slurry.
Results:
Both plasma and wound fluids from trauma patients markedly suppress total PMN RB. Intracellular RB is unchanged, implicating suppression of extracellular RB. Wound fluids are more suppressive than plasma. Biofluids suppressed RB maximally early after injury and their effects decayed with time. CTX and NETosis were suppressed by biofluids similarly. Last, plasma from pigs undergoing abdominal liver slurry instillation suppressed PMN RB, paralleling suppression by human trauma biofluids.
Conclusions:
Trauma plasma and wound fluids suppress RB and other key PMN antimicrobial functions. Circulating suppressive signals can be derived from injured or necrotic tissue at wound sites, suggesting a key mechanism by which tissue injuries can put the host at risk for infection.
Study type:
Basic science (Level of evidence: III)
Keywords: Infection, Neutrophil, Respiratory Burst, Neutrophil Extracellular Traps, Chemotaxis
Background
Trauma is the leading cause of death in individuals under 45 years old and was responsible for 4.4 million deaths worldwide in 2019 (1, 2). Trauma increases susceptibility to secondary infection and nosocomial infections are a common cause of morbidity and mortality in patients who survive their initial trauma (3). Yet the biologic events linking injury to suppression of anti-microbial immunity remain unclear. Injury releases “danger associated molecular patterns” (DAMPs) from cells. These elicit a systemic inflammatory response syndrome (SIRS) (4) characterized by high plasma and wound levels of inflammatory mediators. We have shown that DAMPs in the SIRS environment modify polymorphonuclear neutrophil (PMN) function (5, 6). PMN migrate towards areas of injury or infection by chemotaxis (CTX) down chemical gradients towards such agonists (7). Once there, they can form neutrophil extracellular traps (NETs) that trap bacteria and then use respiratory burst (RB) to kill them (8, 9). We hypothesized that the injury-associated SIRS environment would modulate PMN functions, and we therefore studied the effects of biofluids obtained directly from trauma patients on PMN functions relevant to these antimicrobial responses.
Prior studies have been conflicted as to the effects of injury on RB. Specifically, there is confusion as to the roles of PMN ‘priming’ by injury on intracellular vs. extracellular RB with many model-dependent differences noted. We therefore set out to create a clinical sample-based model to determine the effects of injury on PMN function in a translational fashion. To do this, we used authentic clinical plasma and wound fluids to ‘prime’ PMN. Using such complex biologic fluids yields highly consistent results. Moreover, this approach should be more relevant to in-vivo PMN function than ex-vivo priming using single agonists like lipopolysaccharide (LPS) or tumor necrosis factor alpha (TNFα). Then using these same conditions, we assessed CTX and NET formation. Last, we created a porcine model in which to use the same methods for the study of therapeutic approaches to PMN functional deficits.
Methods
Ethical statements:
All studies were done in compliance with equator network guidelines. Human samples were obtained after informed consent was signed by the patients (or their legally authorized representatives) or by the volunteers and placed in a biorepository. Human studies were approved by our local IRB as well as by the Armed Forces human subjects protection office (HSPO). All animal studies were approved by the local animal use committee (IACUC) as well as by the Department of Defense ACURO.
PMN preparation:
Fresh human PMN for functional assays were isolated from freshly withdrawn peripheral blood of healthy volunteers as described elsewhere (10). Briefly, heparinized (10 U/mL) whole blood was layered onto 1-Step Polymorph (AN221725, Accurate Chemical & Scientific Corp, Carle Place, NY), followed by centrifugation (500 ×g, 30 min). The separated PMN layer was washed in RPMI 1640 and RBC were briefly lysed with ice-cold 0.2% NaCl. PMN were resuspended in physiological solutions appropriate to the functional studies of interest.
Receptor-dependent and independent PMN stimulation:
Receptor-dependent RB was elicited in human PMN using surface G-protein coupled receptor-based stimulation with f-met-leu-phe (fMLF). Porcine experiments were done slightly differently since pig PMN respond poorly to fMLF. Receptor-dependent pig PMN RB was therefore elicited by LTB4, a lipid chemoattractant that acts through similar receptor systems in both pig and human PMN. Receptor-independent RB was uniformly stimulated by application of 100nM phorbol myristate acetate (PMA), which acts directly on Protein Kinase C.
Total and intracellular RB assays:
Reactive oxygen species (ROS) production was measured by luminol-dependent chemiluminescence in a 96-well plate luminometer (LB 960, Berthold) (10). After incubation with biofluids or vehicle, PMN (4 × 106 cells/mL) were mixed 1:1 with 150 nM luminol (Sigma, 123072) and 37 U/μL HRP in DPBS+ (14040117, Gibco), warmed at 37°C for 5 min, transferred to a 96-well plate and chemiluminescent detection was started. At T=0, 20 μL of DPBS (resting cells), PMA (100 nM), fMLF (100 nM) or LTB4 (100 nM) were injected automatically. RB luminescence was displayed in relative light units per second (RLU/sec) and then quantified as the area under curve (AUC) of the RLU trace. Intracellular RB was detected using the same methodology in the presence of superoxide dismutase (SOD – 15 U/μL) and catalase (312 mU/μL) added to the final reaction solution to quench extracellular RB.
Modulating PMN functions with biofluids:
Plasma samples collected in heparin were banked from trauma patients with ISS≥15 (11) or from pre-operative volunteer controls admitted for elective surgeries. Wound fluids were obtained from trauma patients with closed drains in injury sites. Fluids were removed as close to the source as possible, aliquotted, spun and frozen as quickly as possible, and banked similarly. No antibiotics were added. In preliminary studies we examined RB in human PMN incubated in high plasma environments. Here we found that RB elicited by surface G-protein coupled receptor-based stimulation using fMLF was profoundly quenched by the presence of either autologous or healthy volunteer plasmas in comparison to incubation in media. We subsequently found however, that by incubating PMN in the presence of relatively low plasma concentrations and then washing the cells twice in PBS (Figure 1) we could completely avoid any ‘quenching’ effect of plasma seen relative to incubation in media. This allowed visualization of the residual PMN-priming effects of exposure to trauma biofluids (Figures 2–5) on subsequent exposure to the terminal agonists. We subsequently used this methodology (incubation in 10% biofluids followed by washing twice in buffer) for all the studies reported here. Plasma samples from patients as described above were drawn at random from the bio-repository for use based upon the time after injury. Prior studies have used 20–30 min as a ‘priming period’ for PMN using TNFα (12–14). We noted here that a 5 minute incubation in biofluids was equivalent to a 25 minute incubation. We chose to use 25 minutes for priming to allow comparison with prior priming experiments.
Figure 1. The effects of varied plasma concentration on RB.
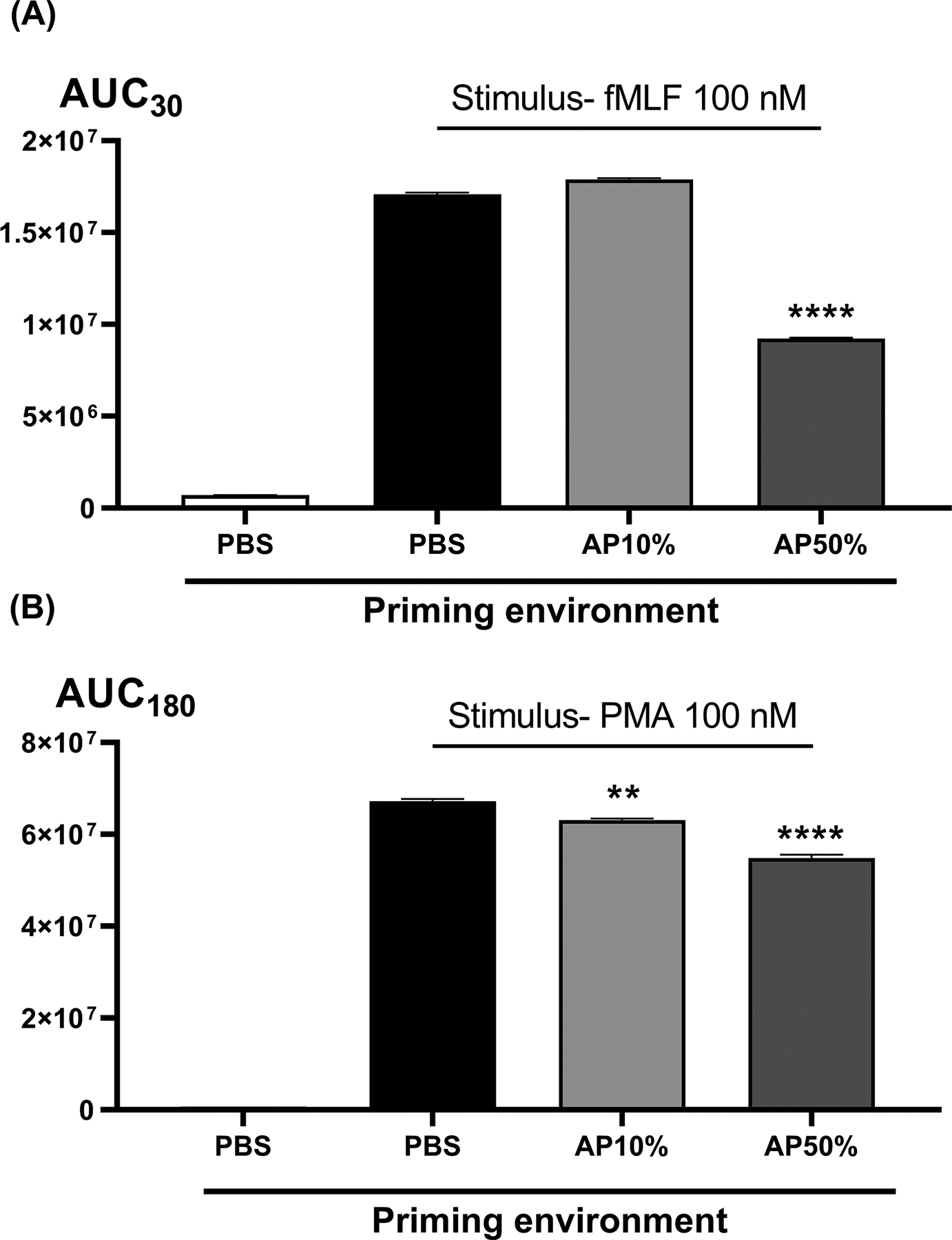
No RB is seen without terminal stimulation by either receptor-mediated fMLF (panel A) or receptor-independent PMA (panel B). Pre-incubation in high concentrations of autologous plasma (AP50%) quench respiratory burst as compared to pre-incubation in PBS media. By using lower concentrations of the autologous plasma to preincubate the cells though, we return RB measurements to the values seen after pre-incubation in PBS (Panel A, PBS vs AP10%). Then by varying the type of biofluid used at the preincubation stage we can see the residual biologic effects of that biofluid environment. For subsequent comparisons with trauma patient plasma we routinely used non-autologous (but healthy) control plasmas since the trauma plasma was always non-autologous. In side-by side comparisons (not shown) no differences in RB were seen after pre-incubation in 10% healthy volunteer (non-autologous) plasma vs 10% autologous plasma. AUCs were compared using one-way ANOVA / Tukey’s test (**p<0.01; ****p<0.001). AP, autologous plasma.
Figure 2. Effect of trauma plasma on fMLF-induced total (Panel A) and intracellular PMN RB.
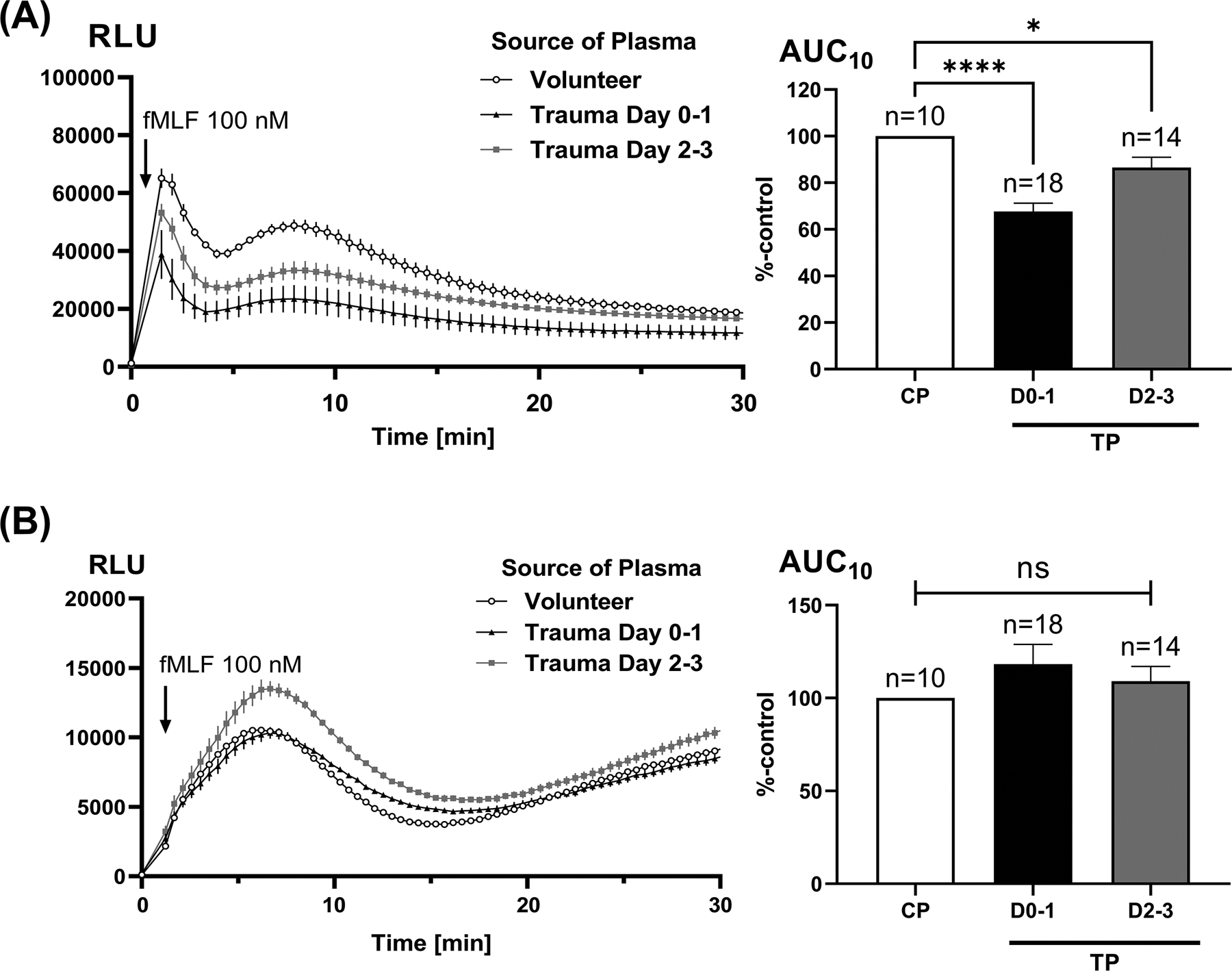
Intracellular RB (Panel B) was assessed by stimulating cells in the presence of the extra-cellular oxidant scavengers SOD and catalase (see methods). Healthy PMN were incubated with 10% trauma plasma (TP) from samples taken on either Day 0 to 1 (D0–1) or Day 2 to 3 (D2–3) for 25 min. fMLF-induced RB was measured for 30 min using a luminometer (left sided traces). Note that total RB values and RB elicited by PMA are typically much larger than intracellular RB and receptor dependent RB. The scales of the luminometry graphs are typically modified to demonstrate differences where needed. The standard error bars are drawn to scale. The areas under the curve (AUC) for luminol activation over 10 min were calculated (right hand graphs) and compared using one-way ANOVA / Tukey’s test (*p<0.05; ****p<0.001). CP, control plasma; TP, trauma plasma; RLU, relative light units.
Figure 5. Effect of trauma biofluids on PMN chemotaxis and NETosis.
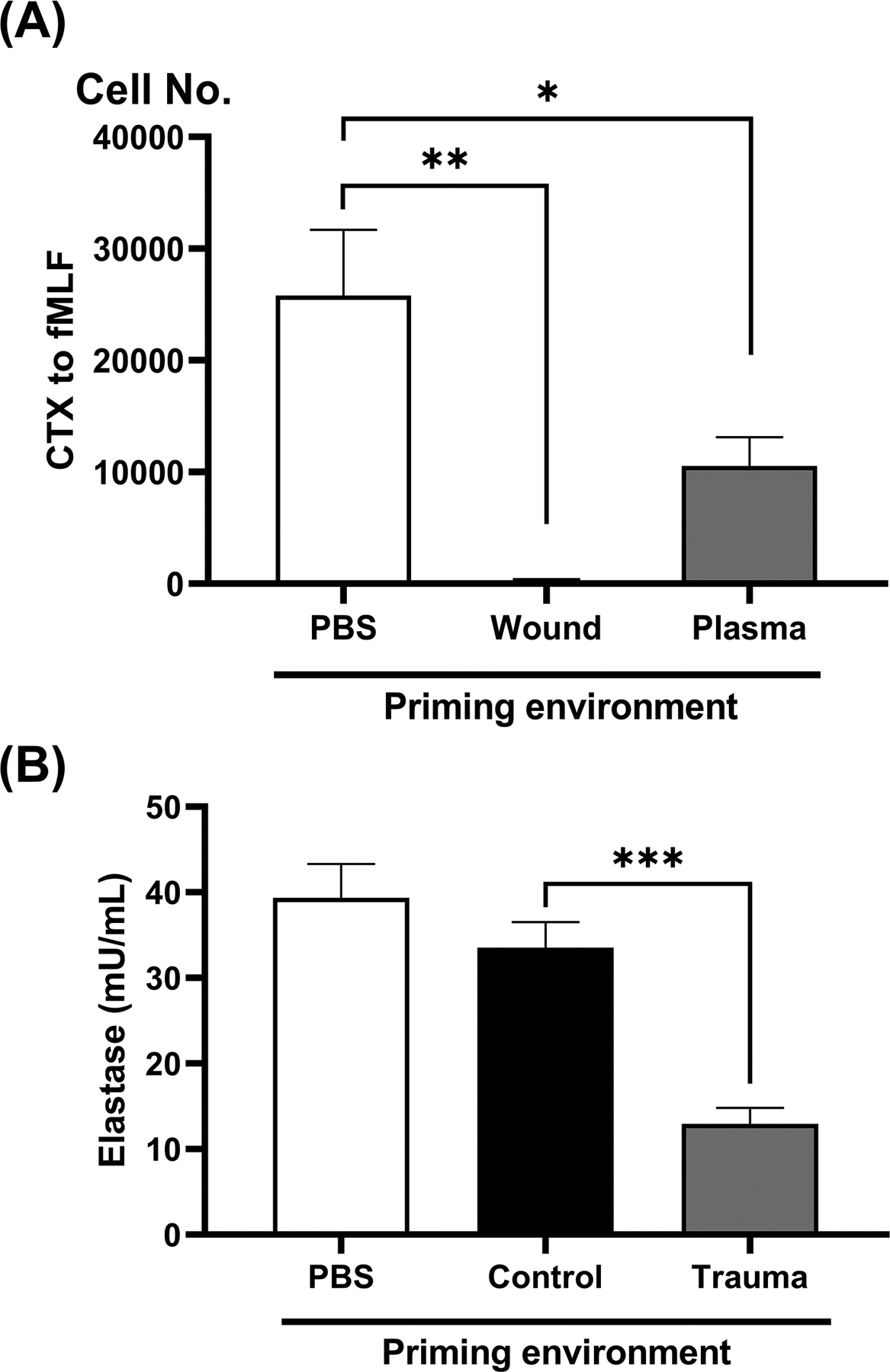
Healthy PMN were incubated with 10% biofluids for 25 min. In Panel A, PMN were pre-incubated in 10% volunteer control plasma, wound fluid or trauma plasma collected from the same patient at the same time as the wound fluid. CTX towards fMLF (100 nM) was then studied in transwells. Migrated PMN were retrieved after 60 min and CyQuant dye was used to quantify cell numbers. Chemokinesis (cells spontaneously migrating without fMLF being present) was subtracted. Both plasma and wound fluids markedly suppress chemotaxis. In Panel B, PMN were pre-incubated in buffer (‘None’) 10% volunteer (Control) or trauma plasma (Trauma). Extracellular trap formation (NETosis) was then induced by PMA over 4 hours and then measured using the elastase technique. Trauma plasma markedly suppressed NETosis as compared to volunteer plasma. Values were compared using one-way ANOVA / Tukey’s test (*p<0.05; **p<0.01; ***p<0.005).
Chemotaxis:
PMN CTX was studied in 3.0 μm-pore-transwells as described previously (15). Briefly, 1 × 105 PMN in 75 μL of RPMI with 2% heat-inactivated FBS were applied to the upper chamber and 150 μL of the same media containing fMLF (100 nM) were applied to the lower chamber. After 60 minutes plates were spun and the PMN lysed. CyQuant dye was then used to assess cell number using a standard curve of pre-counted PMN.
Neutrophil extracellular trap (NET) formation (NETosis) assay:
NETosis was assayed using the elastase technique (16) per manufacturer’s instructions (Kit No. 601010, Cayman Chemical, Ann Arbor, MI, USA). Briefly, PMN were incubated in 24 well plates with biofluids in NET assay buffer (1% bovine serum albumin and 1 mM calcium chloride in RPMI 1640) and subsequently treated with 20 nM PMA for 4 h at 37°C. Buffer was carefully removed and cells were washed twice with NET assay buffer. S7 Nuclease was then added to the wells for 15 min to disrupt NETs. Supernatants were transferred to a microtube, EDTA was added to inactivate nuclease, and debris was removed by centrifugation. Elastase levels (mU/mL) was detected by adding substrate (N-methoxysuccinyl-Ala-Ala-Pro-Val p-nitroanilide) to the samples. Standard curves were generated using neutrophil elastase reagent in the kit.
Pig injury model:
To model the systemic injury environment and enable pre-clinical modeling of PMN function in trauma, we injected liver slurry (10% of the subject animal’s predicted liver mass) harvested from syngeneic Yorkshire pigs (25–30 kg) into the peritoneal cavity of a healthy animal under general anesthesia. We then incubated PMN from uninjured pigs (i.e. those not receiving i.p. liver slurry) in either 10% plasma from control pigs or in 10% plasma from the pigs that had undergone i.p. instillation of liver slurry 1 day prior. RB was then assessed in response to PMA (100nM) as well as LTB4 using identical methods to those used for human PMN except for using LTB4 as the agonist. We used LTB4 because pig PMN respond poorly to fMLF due to differences in their FP receptors (17, 18). Moreover, LTB4 is a potent PMN chemoattractant in humans as well as pigs (19).
Statistical analysis:
Quantitative data were expressed as mean ± standard error of mean (SEM) for 3 or more independent experiments as noted. Statistical analysis was performed using Prism 8 (GraphPad, San Diego, CA). Data were analyzed by analysis of variance (ANOVA) followed by Tukey’s post hoc test except in one instance noted where t-tests were appropriate. P values <0.05 were considered statistically significant.
Results
Trauma plasma suppresses PMN receptor-dependent total RB but not intracellular RB
Incubation of healthy donor PMN in plasma samples from trauma patients (n=14–18) causes significant suppression of total RB response to the bacterial-style formyl peptide fMLF as compared to plasma from control patients (n=10, Figure 2A). The most suppression was noted when using plasmas from earlier (Day 0–1) time points after injury and RB suppression decreased over time. Notably, unlike total RB, intracellular RB was not significantly changed by incubation with trauma plasma (Figure 2B). Taken together, these findings show that suppression of total PMN RB reflects suppression of extracellular RB.
Trauma plasma suppresses receptor-independent PMN RB
We next studied whether the suppressive effects noted were also seen with receptor-independent stimulation using PMA as the RB agonist. Responses to PMA are slower but very complete since it activates Protein Kinase C independent of cell surface receptors (20). Again, trauma plasma specimens suppressed PMN RB, showing receptor-independent RB is also suppressed by trauma biofluids (Figure 3A). Also again, RB suppression was maximal at Day 0–1 post-injury (p<0.01) and tended to fall toward control levels by Day 2–3. Intracellular RB responses to PMA were not significantly affected by trauma plasma (Figure 3B).
Figure 3. Effect of trauma plasma on receptor-independent total versus intracellular PMN RB.
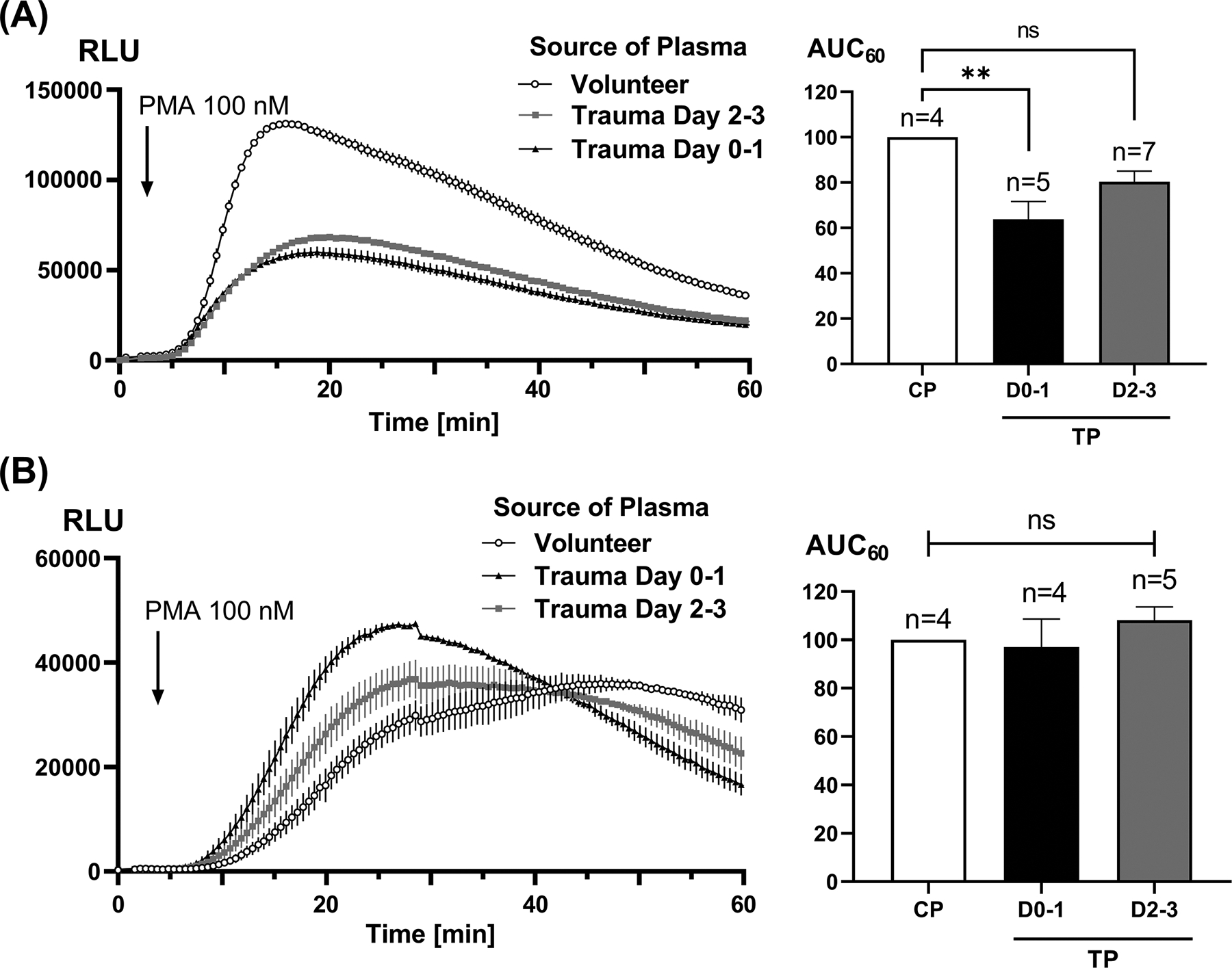
Healthy PMN were incubated with 10% biofluids for 25 min and then PMA-induced RB was measured for 60 min using a luminometer (left hand traces). AUCs were calculated for 60 min (right hand graphs) due to the slower onset of PMA activation. We noted that total RB (panel A) was equally and markedly suppressed by trauma plasmas from Day 0–1 and Day 2–3. Intracellular RB (panel B) is not significantly suppressed. Note that intracellular RB (B) peaks later than total RB (as seen in panel A or in Fig. 2) and is generally much greater than receptor-initiated RB. AUC results were compared using one-way ANOVA / Tukey’s test (**p<0.01). CP, control plasma; TP, trauma plasma; RLU, relative light units.
Trauma wound fluids cause greater PMN RB suppression than trauma plasma
In several cases (n=4) we were able to compare the activity of trauma wound fluids to plasma from the same patients obtained at the same time (Figure 4). fMLF was used as the agonist after standard 10% biofluid incubation. The suppressive effects of wound fluid on PMN RB were uniformly greater than those of contemporaneous plasma specimens. This may suggest that there is a gradient of PMN suppression indicating higher concentrations of suppressive agonists at wound sites that can migrate down their concentration gradients into the circulation.
Figure 4. Effect of trauma wound fluids versus trauma plasma on receptor-induced total PMN RB.
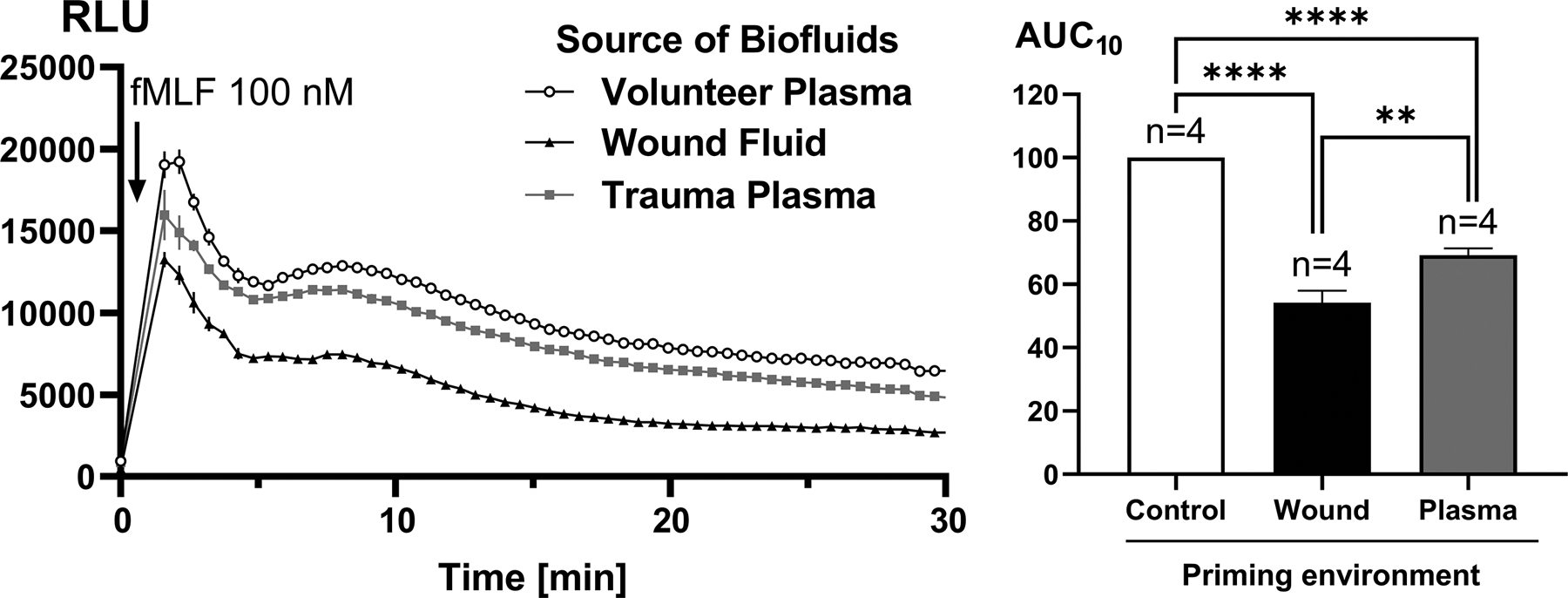
Here, healthy PMN were incubated for 25 minutes with either 10% wound fluid (n=4) or 10% peripheral plasma sampled from the same patient on the same day (n=4). fMLF-induced total RB was then measured for 30 min (left sided graph). RB AUCs for 30 min were then calculated (right hand graph) and compared using one-way ANOVA/Tukey’s test (**p<0.01; ****p<0.001). Both plasma and wound fluids suppressed RB when compared to pre-incubation in control (volunteer) plasmas, but wound fluids were noted to be significantly more suppressive of RB than plasmas collected at the same time point. RLU, relative light units.
Trauma biofluids inhibit PMN chemotaxis towards fMLF
To investigate whether the suppressive effects on PMN function by trauma biofluids are unique to RB or reflect a more global suppression of effector functions, we next assessed PMN chemotaxis after incubation in biofluids. As shown in Figure 5A, trauma plasma significantly suppresses PMN migration towards fMLF. Moreover, incubation in 10% wound fluid completely abolished PMN chemotaxis (p < 0.01).
Trauma plasma suppresses PMA-induced NETosis in PMN
Myeloperoxidase (MPO) ‘decorates’ PMN NETs, and MPO-dependent RB is critical for microbial killing (21). Thus extracellular RB may be closely linked to NET formation and we studied the effects of incubation in trauma plasma on NET formation. As seen in Figure 5B, NETosis was significantly suppressed by trauma plasma as compared to incubation in volunteer plasma.
A porcine injury model mimicking trauma-induced changes in PMN function
Tissue necrosis (modeled by i.p. instillation of liver slurry in pigs) was found to result in similar plasma-induced deficits in receptor-induced (LTB4, Figure 6A) and receptor-independent (PMA, Figure 6B) RB. This finding suggests that causative factors in trauma plasma may either be DAMPs derived directly from necrotic tissues or may be agonists generated by innate immune cells in response to stimulation by DAMPs.
Figure 6. Effect of plasma from pigs challenged by intraperitoneal necrotic tissue on porcine PMN RB.
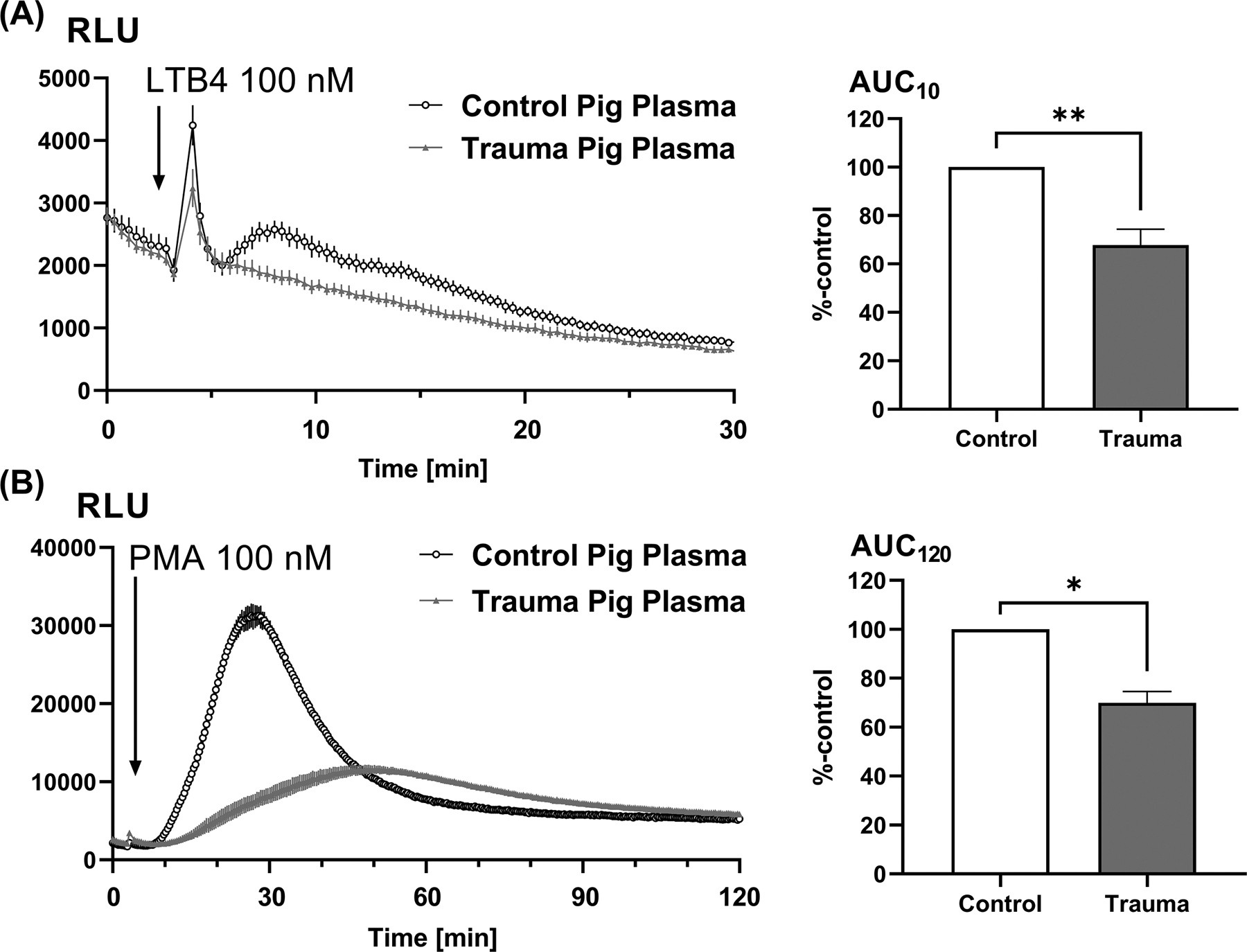
PMN sampled from a healthy pig were incubated for 25 min with either 10% plasma from control (untreated) pigs or from pigs that had undergone i.p. injection of pig-liver slurry 1 day prior. Then receptor-dependent (LTB4-induced, Panel A) and receptor-independent (PMA-induced, Panel B) respiratory burst were measured for the indicated times using the luminometer (left sided traces). Again, receptor-dependent RB is measured for a shorter time than receptor-independent RB. As in human trauma, RB by pig PMN was markedly suppressed by plasma from pigs challenged with DAMPs. AUCs were calculated (right hand graphs) and compared using one-way ANOVA / Tukey’s test (*p<0.05; **p<0.01). RLU, relative light units.
Discussion
Patients with major injuries are at high risk for bacterial infections and the functional alterations in immunity that underly this susceptibility are not well understood. Alterations in PMN RB and other effector functions related to microbial killing have been suggested as underlying factors (22, 23). But the effects of injury on PMN RB are controversial with different methodologies leading to different findings. Thus, PMN from burn injury patients (24) and children with severe injuries (25) were found to show decreased RB whereas circulating PMN from traumatic brain injury patients were found to have increased oxidative burst (26). Barrett et al. suggested trauma plasma could ‘prime’ PMN by potentiating extracellular RB (27). But non-adherent PMN from patients with severe injuries showed decreased oxidant production where adherent PMN showed the opposite (28). Moreover, prior research has centered more on increases in RB as potential sources of inflammatory organ injury than on decreases in RB as predictors of infection. But clearly, much work still needs to be done to fully understand the underlying mechanisms by which injury alters PMN function.
Here, we used plasma and wound fluids to ‘prime’ PMN function and found both plasma and wound fluids caused clear suppression (i.e. ‘negative priming’) of RB. Of methodologic importance, we found that even normal plasma suppressed RB in luminometry studies but that by using lower plasma concentrations than have been previously reported and washing PMN carefully after exposure, we can clearly delineate the residual effects of priming in healthy volunteer plasma, plasma from trauma patients and wound fluids on PMN. Moreover, using these techniques, we think we can more clearly define the evolution of changes in PMN function elicited by the humoral environment after injury.
Also using these methods, we find that injury suppresses PMN total RB, but by studying total and intracellular RB, we clearly demonstrate that these changes represent diminished extracellular RB. This effect occurs in parallel with decreased NETosis, which is consistent with the concept that RB occurring in NETs is a key mechanism for PMN bacterial killing. Moreover, the PMA results demonstrate that some of this effect must be non-receptor based, reflecting changes in cell signaling that occur downstream to PKC and are able to suppress NET formation. It is also possible that the suppression of NET formation may be a critical reason for the observed suppression of respiratory burst.
We also find that wound fluids have even more suppressive effects on PMN RB than plasma sampled from the same patients concurrently. This suggests that the mediators of inflammation causing suppressed PMN function may arise at wound sites and enter the circulation. Moreover, the data shows that the humoral environment after injury returns towards normal over several days, although individuals varied considerably in the time course of resolution. We also demonstrate that the plasma humoral environment after injury suppresses chemotaxis with wound fluids, again, being more potent suppressants than trauma plasma.
Last, we created a porcine model to enable study of therapeutic interventions potentially aimed at improving PMN anti-microbial function after injury. This entailed instillation of necrotic liver slurry into the peritoneal cavity to mimic necrosis due to a blunt liver injury. We found again, that receptor-dependent and independent RB were markedly suppressed by plasma from pigs challenged with necrotic tissue. This may suggest an important mechanism by which retained necrotic tissues can place the host at risk for infection. Although such necrotic tissues are sterile, they are rich sources of DAMPs that include important innate immune mediators like formyl peptides (FPs) and mitochondrial DNA (mtDNA). The effects noted on receptor-independent RB suggest that cell signaling in the trauma environment acts on intracellular NADPH oxidase function as well as having effects on the expressions and function of cell surface receptors. All these are important for PMN antimicrobial function in-vivo.
The exact molecular species generated by trauma that alter PMN function are unclear. Others have suggested that complement fragments may be key (27), and we have noted that mtDNA suppresses PMN CTX (10). FPs from necrotic tissue also suppress chemotactic receptors that activate PMN (5, 6, 29). We suspect though, that a wide variety of mediators are generated in trauma environments. These will ligate multiple receptors leading to integrated cellular responses. Such ‘cognate’ responses can be functionally adaptive or maladaptive depending upon the individual clinical circumstances. For instance, a group of stimuli that cause PMN to become immobilized at an injury site might be important for local phagocytosis of debris and subsequent wound healing. But the same set of stimuli might suppress the ability of circulating PMN to migrate into areas of bacterial challenge. Further investigations should center on delineation of the particular mediators or groups of mediators that are functional and/or dysfunctional in specific clinical circumstances. These data can then inform future therapies.
Supplementary Material
Fundings:
This study was supported by the U.S. Department of Defense Focused Program Award grant W81XWH-16-1-0464 (The Harvard-Longwood ‘HALO’ collaborative) (C.J.H. [P.I.] and L.E.O.), National Institutes of Health grants NIH R43GM125430 and R01DK119202-01 (L.E.O.), and the National Research Foundation of Korea grants NRF-2020R1A6A3A03040631 (H.I.K.) and NRF-2020R1C1C1009721 (J.P.).
Footnotes
Conflict of interest: The authors all declare no conflicts of interest.
References
- 1.The American Association for the Surgery of Trauma. Trauma Facts [cited 2021 July 28]. Available from: https://www.aast.org/resources/trauma-facts.
- 2.World Health Organization. Global health estimates: Leading causes of death 2020. [cited 2021 July 06]. Available from: https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-leading-causes-of-death.
- 3.Sperry JL, Guyette FX, Brown JB, Yazer MH, Triulzi DJ, Early-Young BJ, Adams PW, Daley BJ, Miller RS, Harbrecht BG, et al. Prehospital Plasma during Air Medical Transport in Trauma Patients at Risk for Hemorrhagic Shock. N Engl J Med. 2018;379(4):315–26. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itagaki K, Kaczmarek E, Kwon WY, Chen L, Vlková B, Zhang Q, Riça I, Yaffe MB, Campbell Y, Marusich MF, et al. Formyl Peptide Receptor-1 Blockade Prevents Receptor Regulation by Mitochondrial Danger-Associated Molecular Patterns and Preserves Neutrophil Function After Trauma. Crit Care Med. 2020;48(2):e123–e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaczmarek E, Hauser CJ, Kwon WY, Riça I, Chen L, Sandler N, Otterbein LE, Campbell Y, Cook CH, Yaffe MB, et al. A subset of five human mitochondrial formyl peptides mimics bacterial peptides and functionally deactivates human neutrophils. J Trauma Acute Care Surg. 2018;85(5):936–43. [DOI] [PubMed] [Google Scholar]
- 7.de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. 2016;16(6):378–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17(3):151–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piacenza L, Trujillo M, Radi R. Reactive species and pathogen antioxidant networks during phagocytosis. J Exp Med. 2019;216(3):501–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Konecna B, Park J, Kwon WY, Vlkova B, Zhang Q, Huang W, Kim HI, Yaffe MB, Otterbein LE, Itagaki K, et al. Monocyte exocytosis of mitochondrial danger-associated molecular patterns in sepsis suppresses neutrophil chemotaxis. J Trauma Acute Care Surg. 2021;90(1):46–53. [DOI] [PubMed] [Google Scholar]
- 11.Champion HR. Trauma scoring. Scand J Surg. 2002;91(1):12–22. [DOI] [PubMed] [Google Scholar]
- 12.Fu H, Bylund J, Karlsson A, Pellmé S, Dahlgren C. The mechanism for activation of the neutrophil NADPH-oxidase by the peptides formyl-Met-Leu-Phe and Trp-Lys-Tyr-Met-Val-Met differs from that for interleukin-8. Immunology. 2004;112(2):201–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Onnheim K, Bylund J, Boulay F, Dahlgren C, Forsman H. Tumour necrosis factor (TNF)-alpha primes murine neutrophils when triggered via formyl peptide receptor-related sequence 2, the murine orthologue of human formyl peptide receptor-like 1, through a process involving the type I TNF receptor and subcellular granule mobilization. Immunology. 2008;125(4):591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009;20(2):289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Itagaki K, Sandler N, Gallo D, Galenkamp A, Kaczmarek E, Livingston DH, Zeng Y, Lee YT, Tang IT, et al. Mitochondrial damage-associated molecular patterns from fractures suppress pulmonary immune responses via formyl peptide receptors 1 and 2. J Trauma Acute Care Surg. 2015;78(2):272–9; discussion 9–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yizengaw E, Getahun M, Tajebe F, Cruz Cervera E, Adem E, Mesfin G, Hailu A, Van der Auwera G, Yardley V, Lemma M, et al. Visceral Leishmaniasis Patients Display Altered Composition and Maturity of Neutrophils as well as Impaired Neutrophil Effector Functions. Front Immunol. 2016;7:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fletcher MP, Stahl GL, Longhurst JC. In vivo and in vitro assessment of porcine neutrophil activation responses to chemoattractants: flow cytometric evidence for the selective absence of formyl peptide receptors. J Leukoc Biol. 1990;47(4):355–65. [DOI] [PubMed] [Google Scholar]
- 18.el-Awar FY, Ochs DL, Pyle RH, Misra HP. Lack of formyl-methionyl-leucyl-phenylalanine receptors on porcine neutrophils. Am J Vet Res. 1990;51(10):1561–4. [PubMed] [Google Scholar]
- 19.Störmann P, Auner B, Schimunek L, Serve R, Horst K, Simon TP, Pfeifer R, Köhler K, Hildebrand F, Wutzler S, et al. Leukotriene B4 indicates lung injury and on-going inflammatory changes after severe trauma in a porcine long-term model. Prostaglandins Leukot Essent Fatty Acids. 2017;127:25–31. [DOI] [PubMed] [Google Scholar]
- 20.Wymann MP, von Tscharner V, Deranleau DA, Baggiolini M. The onset of the respiratory burst in human neutrophils. Real-time studies of H2O2 formation reveal a rapid agonist-induced transduction process. J Biol Chem. 1987;262(25):12048–53. [PubMed] [Google Scholar]
- 21.Porto BN, Stein RT. Neutrophil Extracellular Traps in Pulmonary Diseases: Too Much of a Good Thing? Front Immunol. 2016;7:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hampson P, Dinsdale RJ, Wearn CM, Bamford AL, Bishop JRB, Hazeldine J, Moiemen NS, Harrison P, Lord JM. Neutrophil Dysfunction, Immature Granulocytes, and Cell-free DNA are Early Biomarkers of Sepsis in Burn-injured Patients: A Prospective Observational Cohort Study. Ann Surg. 2017;265(6):1241–9. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front Cell Infect Microbiol. 2017;7:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenthal J, Thurman GW, Cusack N, Peterson VM, Malech HL, Ambruso DR. Neutrophils from patients after burn injury express a deficiency of the oxidase components p47-phox and p67-phox. Blood. 1996;88(11):4321–9. [PubMed] [Google Scholar]
- 25.Sikora JP, Sobczak J, Zawadzki D, Przewratil P, Wysocka A, Burzyńska M. Respiratory Burst and TNF-α Receptor Expression of Neutrophils after Sepsis and Severe Injury-Induced Inflammation in Children. Int J Environ Res Public Health. 2021;18(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao Y, Liu P, Guo F, Zhang ZY, Zhang Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS One. 2013;8(7):e68963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrett CD, Hsu AT, Ellson CD, B YM, Kong YW, Greenwood JD, Dhara S, Neal MD, Sperry JL, Park MS, et al. Blood clotting and traumatic injury with shock mediates complement-dependent neutrophil priming for extracellular ROS, ROS-dependent organ injury and coagulopathy. Clin Exp Immunol. 2018;194(1):103–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quaid G, Cave C, Williams MA, Hennigan RF, Bokoch G, Solomkin JS. Mechanisms of human neutrophil oxidant production after severe injury. Surgery. 2001;130(4):669–75; discussion 75–6. [DOI] [PubMed] [Google Scholar]
- 29.Kwon WY, Suh GJ, Jung YS, Park SM, Oh S, Kim SH, Lee AR, Kim JY, Kim H, Kim KA, et al. Circulating mitochondrial N-formyl peptides contribute to secondary nosocomial infection in patients with septic shock. Proc Natl Acad Sci U S A. 2021;118(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.