

Abstract
Hepatitis B virus (HBV) core protein, the building block of the HBV capsid, plays multiple roles in viral replication, and is an attractive target for development of antiviral agents with a new mechanism of action. In addition to the heteroaryldihydropyrimidines (HAPs), sulfamoylbenzamides (SBAs), dibenzothiazepine derivatives (DBTs), and sulfamoylpyrrolamides (SPAs) that inhibit HBV replication by modulation of viral capsid assembly and are currently under clinical trials for the treatment of chronic hepatitis B (CHB), other chemical structures with activity to modulate HBV capsid assembly have also been explored. Here we describe our continued optimization of a benzamide originating from our high throughput screening. A new bicyclic carboxamide lead featuring an electron deficient non-planar core structure was discovered. Evaluations of its ADMET (absorption, distribution, metabolism, excretion and toxicity) and pharmacokinetic (PK) profiles demonstrate improved metabolic stability and good bioavailability.
Graphical Abstract
Targeting various steps in the virus life cycle has been a widely pursued approach in antiviral drug discovery for novel therapies or drug combination.1 In the field of anti-hepatitis B virus (HBV), five nucleos(t)ides analogss (NUCs) had been developed as HBV DNA polymerase inhibitors for the treatment of chronic hepatitis B (CHB).2–3 The NUC therapies can efficiently suppress HBV replication in hepatocytes and significantly reduce viral load, but fail to eliminate the most stable HBV replication intermediate, i.e. covalently closed circular DNA (cccDNA), from infected individuals. As a result, viral replication rebounds after the cessation of NUC therapy and thus, life-long antiviral therapy is required to control the viral replication.4–5 To develop antiviral agents that can potently and durably suppress HBV replication, antiviral agents targeting the steps of HBV replication other than viral DNA synthesis had been extensively explored.6–7 Among them, antiviral agents that disrupt the assembly of nucleocapsids, preventing the subsequent HBV DNA replication, have attracted a lot of attention.8–9 In the last two decades, dozens of structurally diverse core protein assembly modulators (CpAMs) have been discovered (1–10) and several leads from four chemotypes, including heteroaryldihydropyrimidines (HAPs, 1), sulfamoylbenzamides (SBAs, 2–3), dibenzothiazepines (DBTs, 4), and sulfamoylpyrrolamides (SPA, 5), have been evaluated in phase 1 or phase 2 clinical trials for the treatment of CHB (Fig. 1).10–19
Figure 1.

Representative chemotypes of HBV core protein assembly modulators discovered.
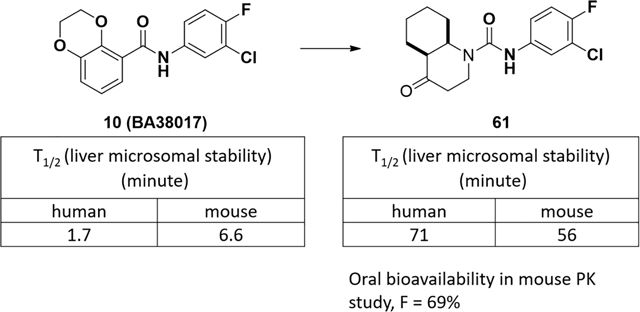
In our early high throughput screening campaign, in addition to the identification of SBA HBV capsid assembly modulators (2),20 which led to the discovery of a phase 1 clinical candidate, AB423,21 another compound (9) with a 2,3-dihydrothieno[3,4-b][1,4]dioxine scaffold was also found to inhibit HBV replication with a moderate EC50. Preliminary structure-activity relationship (SAR) study led to a new lead compound 10 (38017) with sub-micromolar EC50 in a human hepatoma cell line (HepDES19) and an immortalized mouse hepatocyte line (AML12HBV10) supporting robust HBV replication in a tetracycline inducible manner.22 Due to the unique bicyclic structure in which the fused 1,4-dioxane is β to the exocyclic amide moiety, 10 (38017) was further evaluated and continued structural optimization was performed. Here we describe the discovery of a new non-planar bicyclic carboxamide lead featuring an electron deficient core structure with improved ADME properties and good pharmacokinetic profile in mice.
The metabolic stability in liver microsomes was studied first. Compound 10 (38017) was found to be quickly metabolized in both human and mouse microsomes with half-lives that were less than 10 minutes (Figure 2A). Subsequent metabolite identification (MetID) study of 10 (38017) in both human and mouse liver microsomes showed that defluorination and oxidation were the major metabolism pathways, leading to the defluorinated 11 and its oxidized metabolite 12, as well as other oxidized metabolites including 13–14 where oxidation occurring on the bicyclic ring. It is interesting to note that, in contrast, a fluorine atom is usually used to replace a hydrogen atom to improve the metabolic stability.23 The poor stability of 10 (38017) in liver microsomes suggests a liability, and the improvement of stability in liver should help increase the druggability of this novel series of compounds.
Figure 2.

(A) Stability evaluation of 10 (38017) in human and mouse liver microsomes; (B) Metabolite ID studies of 10 (38017) in both human and mouse liver microsomes.
Analysis of the structure of 10 (38017) revealed that the two oxygen atoms fused to the benzamides could donate their lone pair electrons to the phenyl ring, making it more electron rich and more likely to be oxidized. Therefore, we looked for surrogates with poorer electron donating properties for the bicyclic rings, and amines either without leaving groups or with groups that can interfere with the interaction with the oxidizing enzymes.23–24 The new compounds were evaluated for their anti-HBV activities and cytotoxicities. The antiviral activity of the compounds was tested in an immortalized mouse hepatocyte (AML12) derived stable cell line (AML12HBV10) by a dot blot hybridization assay as described previously.20 The cytotoxicity was determined in AML12HBV10 cells by an MTT assay. This cell-based assay was the platform for our initial high throughput screening of HBV replication inhibitors, and was used to determine EC50 and CC50 of new compounds and direct the structure-activity relationship (SAR) study.20,22, 26–27
Aniline variations.
We prepared these analogs based on the reaction of 2,3-dihydrobenzo[b][1,4]dioxine-5-carboxylic acid with different amines, following the synthetic sequence described in Table 1. Replacement of the fluorine with a hydrophilic morpholine caused the loss of anti-HBV activity in 17. The electron withdrawing trifluoromethyl and cyano group in 18 and 19 led to reduced activities as well. A possible metabolite, 20, was synthesized, but it was not active. A saturated 1,1-difluorocyclobutane was used as a bioisostere of the halogenated phenyl ring, but the resulting product, 21, lost activity. The use of heteroaromatic rings, such as 2-chloro-4-pyridylamine in 22 or 2,6-dichloro-4-pyridylamine in 23, was not particularly effective, as only micromolar EC50s were obtained. linking the meta and para substituents to form five-membered rings in 24, 25, and 26 failed to maintain or improve the activities. Extension of the phenyl ring from the amide through the use of (2,4-difluorophenyl)methanamine did not increase the activity, suggesting the requirement for anilines at this position. In the end, two halogenated anilines, 3-chloro-5-fluoroaniline and 3,4,5-trifluoroaniline in 28 and 29, maintained submicromolar anti-HBV activities.
Table 1.
SAR on anilines based on 10 (38017).

| |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R1 | EC50 (μM) | CC50 (μM) | Cmpd | R1 | EC50 (μM) | CC50 (μM) |
| 10 |

|
0.20 | > 50 | 17 |

|
> 10 | > 25 |
| 18 |

|
1.59 | > 50 | 19 |

|
3.84 | > 50 |
| 20 |

|
9.83 | > 100 | 21 |

|
> 10 | > 100 |
| 22 |

|
4.65 | > 100 | 23 |

|
6.30 | > 33.3 |
| 24 |

|
> 10 | > 50 | 25 |

|
> 10 | > 50 |
| 26 |

|
4.20 | > 100 | 27 |

|
4.62 | > 20 |
| 28 |

|
0.50 | > 50 | 29 |

|
0.28 | > 100 |
Bicyclic core variations.
Next, bicyclic benzoic acids with different electron properties were investigated. Removal of one oxygen atom in the fused dioxane ring in 33 reduced the activity by about 4-fold. Employment of quinoxaline in 34 caused more than a 10-fold activity loss. The 2-(1,4-dioxan-2-yl)acetyl group in 36, a partial structure of the bicyclic ring in 10, had weaker activity compared to 10, indicating the importance of the bicyclic structure. When the fused dioxane ring was shrunk to five-membered rings, 2,2-difluoro-1,3-dioxolane in 37 increased the potency by 2-fold, while three other oxygen containing five-membered rings in 38, 39, and 40 exhibited weaker activities than 37. Sulfonamides were installed at the meta position of the 2,3-dihydrobenzo[b][1,4]dioxine ring28 with the expectation that it would mimic the sulfamoylbenzamides (SBAs, 2–3, Figure 1) and reduce the electron density of the phenyl ring that they were connected due to their electron withdrawing properties. However, all three compounds with this feature did not show additive effects, and instead, negatively reduced the activities (42, 43, and 44, Table 2).
Table 2.
Screening bicyclic moieties based on 10 (38017).

| |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R2 | EC50 (μM) | CC50 (μM) | Cmpd | R2 | EC50 (μM) | CC50 (μM) |
| 33 |

|
0.75 | > 12.5 | 34 |

|
2.39 | > 12.5 |
| 35 |

|
> 10 | > 50 | 36 |

|
5.02 | > 100 |
| 37 |

|
0.11 | > 100 | 38 |

|
0.73 | > 100 |
| 39 |

|
2.18 | 4.6 | 40 |

|
1.48 | > 50 |
| 41 |

|
4.48 | > 33.3 | 42 |

|
1.64 | > 33.3 |
| 43 |

|
5.57 | > 50 | 44 |

|
13.36 | - |
Four compounds were selected for a metabolic stability test. Although two of them, 18 and 38, showed some improvement in the human liver microsomal assay compared to 10, the stability in the mouse liver model was still not good enough (Table 3).
Table 3.
Microsomal Stability of Selected Compounds
| Cmpd | 18 | 42 | 37 | 38 | |
|---|---|---|---|---|---|
| Half-life T1/2 (min) | mouse | 6.9 | 16.4 | 11.0 | 6.0 |
| human | 44.4 | 37.4 | 16.4 | 61.2 | |
With the limited success from these two avenues of investigation, we turned our attention to more changes in the fused bicyclic benzamide structure. We envisioned that the acyl group of the benzamide could be translocated to the fused dioxane ring in 10 (38017), with the oxygen atom on the same side being replaced with a nitrogen atom to form new analogs through a urea connection. This change allowed us to test and evaluate new molecules that bring new electron distribution and spatial potentials different from 10 (38017), and which can be synthesized from starting materials that are easily accessible. The resulting products (e.g., 45 and 46, Figure 3) preserved the bicyclic core feature, and could serve as platforms to introduce other functional groups for additional binding (Figure 3).
Figure 3.

(A) Translocation of Acyl Connection to 2,3-Dihydrobenzodioxine: Urea Exploration. (B) Docked 10 and 45 with the capsid residues in the dimer-dimer interface.
Docking studies were performed with the crystal structure 5D7Y with a grid centered on T128 of chain C and a 20Å box for the grid. Compounds 45 and 10 were docked with Glide extra-precision mode as implemented in Maestro29 without any constraints. Both compounds formed hydrogen bonds with W102 (a characteristic feature of CpAM binding from the crystal structure analysis), and the di-halogenated benzene rings were situated in the hydrophobic pocket (consistent with other CpAMs binding) with a docking score of −6.8 kcal/mol for both of them. The results showed tolerance of the target to the translocation of the acyl group.
These compounds were synthesized from a reaction of a bicyclic amine with isocyanate 48 or with a carbamate 49 formed from a reaction of phenyl chloroformate with aniline 31.27 This strategy was first tested on 2,3-dihydroquinolin-4(1H)-one because of the presence of an electron withdrawing carbonyl group. However, the resulting urea 46 only displayed moderate anti-HBV activity (EC50 = 1.32 μM, Table 3), although it was more potent than its close analog 51 with one additional carbon expanding the ring size to a seven-membered ring. Substitutions around the structure of 46 resulted in 52 and 53 but these changes did not improve the activities. The opening of the fused rings provided compounds 54 and 55, but they were less potent in comparison to 46. Bioisosteric replacement of the nitrogen in the 2,3-dihydroquinolin-4(1H)-one with a carbon afforded 56, which exhibited similar activity to 46. This is interesting because 56 is not a planar molecule due to the sp3 hybridization state of the carbon that the acyl group is connected to. Encouraged by this result and attracted to the potential benefit of making non-flat and saturated molecules,30–31 we prepared a saturated version of 46, leading to two racemic isomers, 58 and 59, assigned as cis and trans based on NMR analysis (Figure 4). The cis- isomer 58 was more potent than the trans- isomer 59 (EC50, 0.71 vs. 1.81 μM) and showed better activity than the unsaturated 46. Chiral separation (Chiralpak IF column) of 58 provided two enantiomers, where the second fraction 61 was more potent than the first fraction 60 (EC50, 0.46 vs. 1.86 μM) and comparable to 10 (38017).33 Bicyclic bridged urea derivatives 62 and 63 were also synthesized and evaluated, but no activities (> 10 μM) were observed, suggesting a limited tolerance to spatial molecules. Intolerance to additional polar groups around the bicyclic rings were observed as 64–66 lost activities, although 45 with a fused morpholine had a submicromolar activity (EC50 = 0.52 μM).
Figure 4.

Numbered structure and key NOESY correlations for 58.
The more active racemate, 58, was characterized by 1D and 2D NMR analysis in order to confirm the relative configuration of the octahydro-4(1H)-quinolinone rings system. Interpretation of the 1H, 13C, HSQC, and HMBC spectra for 58 allowed for the assignment of the connectivity of the structure. The NOESY spectrum of 58 revealed correlations between the axial protons at H-2a (δH 3.58 ppm) and H-9 (δH 1.38 ppm), as well as a correlation between the axial protons H-6a (δH 1.30 ppm) and H-10 (δH 4.40 ppm). Taken together, these correlations were consistent with a cis-fused ring system in the structure of 58, as depicted in Figure 4.
With the saturated bicyclic octahydroquinolin-4(1H)-one on hand, we combined this moiety with other anilines. These compounds were synthesized in the method used in the synthesis of compounds 50 (Table 4). The results are shown in Table 5. Only cis- ureas were selected and tested, since they were more potent than the trans- isomers. Among them, ureas 70, 71, and 74, derived from 3-methyl-4-fluoroaniline, 3-(difluoromethyl)-4-fluoroaniline, and 3-chloroaniline, demonstrated submicromolar EC50s that were comparable to that of 10 (38017).
Table 4.
New bicyclic scaffold Screening.

| |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R2 | EC50 (μM) | CC50 (μM) | Cmpd | R2 | EC50 (μM) | CC50 (μM) |
| 46 |

|
1.32 | > 12.5 | 51 |

|
> 10 | > 100 |
| 52 |

|
4.83 | > 100 | 53 |

|
2.89 | > 50 |
| 54 |

|
5.01 | > 100 | 55 |

|
5.11 | > 20 |
| 56 |

|
1.67 | > 100 | 57 |

|
9.4 | 25 |
| 58 |

|
0.71 | > 100 | 59 |

|
1.81 | > 100 |
| 60 |

|
1.86 | > 100 | 61 |
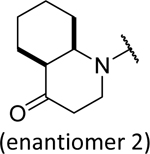
|
0.46 | > 100 |
| 62 |

|
> 10 | > 100 | 63 |

|
> 10 | > 100 |
| 45 |

|
0.52 | > 100 | 64 |

|
> 10 | 26.3 |
| 65 |

|
> 10 | > 100 | 66 |

|
> 10 | > 100 |
Table 5.
Optimization of octahydroquinolin-4(1H)-one-based analogs.

| |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R1 | EC50 (μM) | CC50 (μM) | Cmpd | R1 | EC50 (μM) | CC50 (μM) |
| 70 |

|
0.38 | > 20 | 71 |

|
0.26 | > 20 |
| 72 |
|
0.97 | > 20 | 73 |

|
0.85 | > 20 |
| 74 |

|
0.22 | > 20 | 75 |

|
2.41 | > 100 |
| 76 |

|
> 10 | > 50 | 77 |

|
> 10 | > 100 |
Compounds that had EC50 values of less than 2 μM and unique structural features were selected for metabolic stability tests (Table 6). Although urea 46 had an electron withdrawing carbonyl in the bicyclic fused ring and made some improvement in human liver microsomes (T1/2 = 47.2 min), it did not get significant stability in mouse liver microsomes (T1/2 = 10 min). Compound 56, a bioisostere of 46, did not have improved stability in mouse liver microsomes. In comparison, saturated cis- bicyclic ureas 58 and 71 demonstrated notable improvement in both human and mouse liver microsomes ((T1/2 > 56 min (human) and T1/2 > 32 min (mouse)). Interestingly, the cis- isomer 58 is more stable than the trans- isomer 59, and the urea 61, the second enantiomer fraction of 58, exhibited much higher stability in mouse liver microsomes than the first enantiomer fraction 60 (T1/2, 56 vs. 7 min). The effort for determination of the absolute stereochemistry of urea 61 was not successful due to the failure to grow qualified crystals for X-ray crystallography. Compound 45 did not show significant stability in the mouse liver microsomes. This is not surprising likely due to its rich electron density and flat structure.
Table 6.
Evaluation of new compounds in liver microsomes assays.
| Cmpd | 46 | 56 | 58 | 59 | 71 | 60 | 61 | 45 | |
|---|---|---|---|---|---|---|---|---|---|
| T1/2 (min) | mouse | 10.0 | 5.8 | 32.0 | 7.8 | 35 | 7 | 56 | <4.0 |
| human | 47.2 | 56.8 | 5.0 | 69 | 48 | 71 | 14.7 | ||
Because of its submicromolar potency and improved metabolic stability, urea 61 was further assessed in an ADME panel and in mice for its pharmacokinetics profile (Table 7). Besides the improved metabolic stability compared to 10 (38017), urea 61 displayed favorable solubility and plasma protein bindings, good permeability, and only one IC50 to an isoform of CYP enzymes that was slightly less than 10 μM. These parameters were well translated into a mouse PK study, where good absorption was observed and 69% of bioavailability was obtained for an oral route.
Table 7.
A, ADME test results for Urea 61 with non-planar and electron-deficient octahydroquinolin-4(1H)-one core; B, PK profile of urea 61 via IV and PO routes.
| A | Cmpd | Solubility (μM) | Metabolic stability (T1/2, min) | CYP (#/9 of isoforms, > 50% of inhibition at 10 μM) | Plasma Protein Binding | Caco-2 Permeability* | |||||
| PH = 4 | PH = 7 | Human | Mouse | Mouse (%) | Human (%) | Papp(a-b) 10−6 cm/s | Papp(b-a) 10−6 cm/s | Efflux Ratio | |||
| 10 (38017) | 1.7 | 6.6 | |||||||||
| 61 | 239 | 253 | 71 | 56 | 1 (2C19, 56%) | 94 | 90 | 14.7 | 7.8 | 0.5 | |
| B | IV (3 mg/kg) | PO (10 mg/kg) | |||||||||
| Cl_obs (mL/min/kg) | T1/2 (hr) | AUClast (h*ng/mL) | Vss_obs (L/kg) | T1/2 (hr) | Cmax (ng/mL) | AUClast (h*ng/mL) | F (%) | ||||
| 25.5 | 0.6 | 1952 | 0.8 | 0.7 | 2727 | 4516 | 69 | ||||
the Caco-2 data were from a test for racemic 58.
Urea derivatives work as typical type II CpAMs
To determine whether the modified urea derivatives inhibited HBV replication via the same or a distinct mechanism of its parental benzamines and other type II CpAMs, we mapped the HBV replication step inhibited by the compound in HepDES19 cells with a typical type I CpAM (Bay 41–4109), type II CpAM (ENAN-34017), and an HBV DNA polymerase inhibitor (Entecavir, ETV) as controls. As shown in Fig. 5, compared to mock-treated control (DMSO), treatment with all the compounds did not apparently reduce the amounts of HBV RNA (Fig. 5A). However, as anticipated, ETV treatment resulted in the accumulation of encapsidated pgRNA, but both type I and type II CpAMs reduced the amounts of encapsidated pgRNA (Fig. 5B). Also as expected, ETV treatment did not alter the levels of intracellular core protein (Fig. 5C) and capsids (Fig. 5D, upper panel), but reduced the amount of capsid associated HBV DNA (Fig. 5D, lower panel and Fig. 5E). These results suggest that ETV treatment arrested HBV replication at the reverse transcription of encapsidated pgRNA, which is consistent with its mode of action. Consistent with the mode of action of type I CpAMs, particle gel assay revealed that Bay41–4109 treatment completely abolished capsid formation (Fig. 5D) and induced the degradation of core protein (Fig 5C). As a consequence, pgRNA encapsidation (Fig. 5B) and DNA synthesis (Fig. 5D and E) were prevented. However, similar to ENAN-34017, treatment of HepDES19 cells with compound 61 induced the accumulation of capsids with faster electrophoresis mobility and reduced the amounts of encapsidated pgRNA (Fig. 5B) and the capsid-associated HBV DNA (Fig. 5D and E). Therefore, similar to its parental benzamide derivatives, compound 61 is a typical type II CpAM.
Figure 5. Determination of HBV replication step inhibited by compound 61.

HepDES19 cells were cultured in the presence of tetracycline (tet) to inhibit pgRNA transcription and serve as negative control or in the absence of tet to induce pgRNA transcription and subsequent HBV DNA replication in cytoplasmic capsids. The cells were mock-treated (DMSO) or treated with ETV (1 μM), Bay 41–4109 (2 μM), ENAN-34017 (5 μM) or Compound 61 (5 μM), starting at 24 h post the removal of tet for 6 days. Intracellular total HBV RNA (A) and encapsidated pgRNA (B) were detected by Northern blot hybridization. Ribosomal RNA (rRNA) served as loading control. HBV core protein was detected by Western blot assay with an antibody against C-terminal 14 amino acid residues of core protein (Cp). β-actin served as loading control (C). HBV capsids and core associated DNA were detected by native agarose gel electrophoresis-based particle gel assay (D). Core associated HBV DNA replication intermediates were analyzed by Southern blot assay (E). rcDNA, relaxed circular DNA. dslDNA, double-stranded linear DNA. ssDNA, single-strand DNA.
In conclusion, based on the structure of an early lead compound 10 (38017) and metabolic stability analysis, we have prepared compounds iteratively with structural variations on both the bicyclic side and aniline side, guided by the anti-HBV activities and metabolic stabilities in liver microsomes. A saturated non-planar and electron-deficient octahydroquinolin-4(1H)-one core was identified as a pharmacophore that led to the discovery of 61, a urea modulator, that not only retained anti-HBV activities but also held good ADME profiles and demonstrated 69% of oral bioavailability in a mouse PK study. Moreover, through the mode of action study, compound 61 was determined to be a type II CpAM, which misdirects the Cp dimers to form empty capsids and thus precludes the synthesis of viral DNA.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health, USA (AI113267) and appreciation of The Commonwealth of Pennsylvania through the Hepatitis B Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.De Clercq E Strategies in the design of antiviral drugs. Nat Rev Drug Discov. 2002;1(1):13–25. [DOI] [PubMed] [Google Scholar]
- 2.Fung J, Lai CL, Seto WK, Yuen MF. Nucleoside/nucleotide analogues in the treatment of chronic hepatitis B. J Antimicrob Chemother. 2011;66(12):2715–25. [DOI] [PubMed] [Google Scholar]
- 3.Yuen MF, Lai CL. Treatment of chronic hepatitis B: Evolution over two decades. J Gastroenterol Hepatol. 2011;26 Suppl 1:138–43. [DOI] [PubMed] [Google Scholar]
- 4.Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo JT, Locarnini S, Zoulim F, Chang KM, Lok AS. Present and future therapies of hepatitis B: From discovery to cure. Hepatology. 2015;62:1893–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang J, Guo F, Zhao X, Guo JT. Therapeutic strategies for a functional cure of chronic hepatitis B virus infection. Acta Pharm Sin B. 2014;4:248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Block TM, Gish R, Guo H, Mehta A, Cuconati A, Thomas London W, Guo JT. Chronic hepatitis B: what should be the goal for new therapies? Antiviral Res. 2013;98:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu J, Cheng J, Tang L, Hu Z, Luo Y, Li Y, Zhou T, Chang J, Guo JT. Virological Basis for the Cure of Chronic Hepatitis B. ACS Infect Dis. 2019;5(5):659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viswanathan U, Mani N, Hu Z, Ban H, Du Y, Hu J, Chang J, Guo JT. Targeting the multifunctional HBV core protein as a potential cure for chronic hepatitis B. Antiviral Res. 2020;182:104917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. Core protein: A pleiotropic keystone in the HBV lifecycle. Antiviral Res. 2015;121:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nijampatnam B, Liotta DC. Recent advances in the development of HBV capsid assembly modulators. Curr Opin Chem Biol. 2019;50:73–79. [DOI] [PubMed] [Google Scholar]
- 11.King RW, Ladner SK, Miller TJ, Zaifert K, Perni RB, Conway SC, Otto MJ. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (−)beta-L-2’,3’-dideoxy-3’-thiacytidine [published correction appears in Antimicrob Agents Chemother 1999;43(3):726]. Antimicrob Agents Chemother. 1998;42(12):3179–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perni RB, Conway SC, Ladner SK, Zaifert K, Otto MJ, King RW. Phenylpropenamide derivatives as inhibitors of hepatitis B virus replication. Bioorg Med Chem Lett. 2000;10:2687–2690. [DOI] [PubMed] [Google Scholar]
- 13.Wang P, Naduthambi D, Mosley RT, Niu C, Furman PA, Otto MJ, Sofia MJ. Phenylpropenamide derivatives: anti-hepatitis B virus activity of the Z isomer, SAR and the search for novel analogs. Bioorg Med Chem Lett. 2011;21(15):4642–4647. [DOI] [PubMed] [Google Scholar]
- 14.Weber O, Schlemmer KH, Hartmann E, Hagelschuer I, Paessens A, Graef E, Deres K, Goldmann S, Niewoehner U, Stoltefuss J, Haebich D, Ruebsamen-Waigmann H, Wohlfeil S. Inhibition of human hepatitis B virus (HBV) by a novel non-nucleosidic compound in a transgenic mouse model. Antiviral Res. 2002;54(2):69–78. [DOI] [PubMed] [Google Scholar]
- 15.Deres K, Schröder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Krämer T, Niewöhner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RN, Reimann A, Jaeger R, Gross R, Beckermann B, Schlemmer KH, Haebich D, Rübsamen-Waigmann H. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science. 2003;299(5608):893–6. [DOI] [PubMed] [Google Scholar]
- 16.Kuduk SD, Lam AM, Espiritu C, Vogel R, Lau V, Klumpp K, Flores OA, Hartman GD. SAR studies in the sulfonyl carboxamide class of HBV capsid assembly modulators, Bioorg Med Chem Lett. 2019;29(16):2405–2409. [DOI] [PubMed] [Google Scholar]
- 17.Schlicksup CJ, Laughlin P, Dunkelbarger S, Wang JC, Zlotnick A. Local Stabilization of Subunit-Subunit Contacts Causes Global Destabilization of Hepatitis B Virus Capsids. ACS Chem Biol. 2020;15(6):1708–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amblard F, Boucle S, Bassit L, Cox B, Sari O, Tao S, Chen Z, Ozturk T, Verma K, Russell O, Rat V, de Rocquigny H, Fiquet O, Boussand M, Di Santo J, Strick-Marchand H, Schinazi RF. Novel Hepatitis B Virus Capsid Assembly Modulator Induces Potent Antiviral Responses In Vitro and in Humanized Mice. Antimicrob Agents Chemother. 2020;64(2):e01701–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu T, Han X, Kou B, Shen H, Yan S, Zhang Z. Pyrazine compounds for the treatment of infectious diseases. WO2016113273.
- 20.Campagna MR, Liu F, Mao R, Mills C, Cai D, Guo F, Zhao X, Ye H, Cuconati A, Guo H, Chang J, Xu X, Block TM, Guo JT. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J Virol. 2013;87(12):6931–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mani N, Cole AG, Phelps JR, Ardzinski A, Cobarrubias KD, Cuconati A, Dorsey BD, Evangelista E, Fan K, Guo F, Guo H, Guo JT, Harasym TO, Kadhim S, Kultgen SG, Lee ACH, Li AHL, Long Q, Majeski SA, Mao R, McClintock KD, Reid SP, Rijnbrand R, Snead NM, Micolochick Steuer HM, Stever K, Tang S, Wang X, Zhao Q, Sofia MJ. Preclinical Profile of AB-423, an Inhibitor of Hepatitis B Virus Pregenomic RNA Encapsidation. Antimicrob Agents Chemother. 2018;62(6):e00082–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu S, Zhao Q, Zhang P, Kulp J, Hu L, Hwang N, Zhang J, Block TM, Xu X, Du Y, Chang J, Guo JT. Discovery and Mechanistic Study of Benzamide Derivatives That Modulate Hepatitis B Virus Capsid Assembly. J Virol. 2017;91(16):e00519–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meanwell NA. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J Med Chem. 2018;61(14):5822–5880. [DOI] [PubMed] [Google Scholar]
- 24.Du Y, Ye H, Guo F, Wang L, Gill T, Khan N, Cuconati A, Guo JT, Block TM, Chang J, Xu X. Design and synthesis of N-alkyldeoxynojirimycin derivatives with improved metabolic stability as inhibitors of BVDV and Tacaribe virus. Bioorg Med Chem Lett. 2013;23(14):4258–62. [DOI] [PubMed] [Google Scholar]
- 25.St Jean DJ Jr, Fotsch C. Mitigating heterocycle metabolism in drug discovery. J Med Chem. 2012;55(13):6002–20. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Cheng J, Ma J, Hu Z, Wu S, Hwang N, Kulp J, Du Y, Guo JT, Chang J. Discovery of Novel Hepatitis B Virus Nucleocapsid Assembly Inhibitors. ACS Infect Dis. 2019;5(5):759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang N, Ban H, Chen J, Ma J, Liu H, Lam P, Kulp J, Menne S, Chang J, Guo JT, Du Y. Synthesis of 4-oxotetrahydropyrimidine-1(2H)-carboxamides derivatives as capsid assembly modulators of hepatitis B virus. Med Chem Res. 2021;1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salituro FG, Saunders JO. Preparation of piperazinylcarbonyl benzenesulfonamides for use in the treatment of cancer characterized as having an IDH mutation. WO2011072174
- 29.Schrödinger Release 2021–3: Maestro, Schrödinger, LLC, New York, NY, 2021 [Google Scholar]
- 30.Lopez OD, Nguyen VN, St Laurent DR, Belema M, Serrano-Wu MH, Goodrich JT, Yang F, Qiu Y, Ripka AS, Nower PT, Valera L, Liu M, O’Boyle DR 2nd, Sun JH, Fridell RA, Lemm JA, Gao, Good AC, Meanwell NA, Snyder LB. HCV NS5A replication complex inhibitors. Part 3: discovery of potent analogs with distinct core topologies. Bioorg Med Chem Lett. 2013;23(3):779–84. [DOI] [PubMed] [Google Scholar]
- 31.Cox B, Zdorichenko V, Cox PB, Booker-Milburn KI, Paumier R, Elliott LD, Robertson-Ralph M, Bloomfield G. Escaping from Flatland: Substituted Bridged Pyrrolidine Fragments with Inherent Three-Dimensional Character. ACS Med Chem Lett. 2020;11(6):1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lovering F, Bikker J, Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J Med Chem. 2009. Nov 12;52(21):6752–6. [DOI] [PubMed] [Google Scholar]
- 33.Synthesis of enantiomer fraction 2 of cis-N-(3-chloro-4-fluorophenyl)-4-oxooctahydroquinoline-1(2H)-carboxamide (61): 3-chloro-4-fluoroaniline (123 mg, 0.84 mmol, 1.0 eq) was dissolved in 1:1 EtOAc (5 mL) and saturated NaHCO3 aqueous solution. phenyl chloroformate (132 mg, 1.0 eq) was added dropwise, and the reaction was stirred overnight. The two phases were separated, and the organic phase was washed with brine before being purified by Combi-Flash with a gradient of ethyl acetate from 0:1 to 3:7 to provide the intermediate, phenyl (3-chloro-4-fluorophenyl)carbamate 49 as a white solid (206 mg, 92%); calculated for C13H9ClFNO2, 265.0; observed (M + H) + 266.2. To the intermediate 49 (40 mg, 0.15 mmol, 1.0 eq) in DCM (2 mL), octahydro-4(1H)-quinolinone (23 mg, 0.15 mmol, 1.0 eq) and excess triethylamine was added. Upon completion, the reaction was diluted with EtOAc and washed with 2N HCl twice, saturated NaHCO3, and brine. Purification with Combi-Flash afforded two isomers (32.9 mg and 9 mg respectively) as off-white solids after silica gel separation with a gradient of ethyl acetate : hexanes from 0:1 to 3:7, of which the first peak was the desired cis-N-(3-chloro-4-fluorophenyl)-4-oxooctahydroquinoline-1(2H)-carboxamide (58, 68%). 1H NMR (600 MHz, CDCl3): δ (ppm) 7.53 (dd, J1 = 6.4 Hz, J2 = 2.6 Hz, H-2”a), 7.19 (ddd, J1 = 8.8 Hz, J2 = 4.0 Hz, J3 = 2.8 Hz, H-6”a), 7.06 (t, J1 = 8.7 Hz, H-5”a), 4.4 (m, H-10a), 4.08 (ddd, J1 = 13.2 Hz, J2 = 6.6 Hz, J3 = 5.0 Hz, H-2b), 3.58 (ddd, J1 = 13.1 Hz, 9.8 Hz, 4.5 Hz, H-2a), 2.82 (bs, H-5), 2.58 (ddd, J1 = 15.7 Hz, J2 = 9.6 Hz, J3 = 6.3 Hz, H-3b), 2.48 (ddd, J1 = 15.7 Hz, J2 = 4.7 Hz, J3 = 4.7 Hz, H-3a), 2.38 (dq, J1 = 10.1 Hz, J2 = 2.2 Hz, H-6b), 1.85 (m, H-9b), 1.82 (m, H-8b), 1.53 (m, H-7b), 1.38 (m, H-9a), 1.35 (m, H-8a), 1.30 (m, H-7a), 1.30 (m, H-6a); 13C NMR (151 MHz): δ (ppm) 208.11 (C-4), 154.6 (C-1’), 153.57 (d, JC-F = 245.4 Hz, C-4”), 135.51 (d, JC-F = 3.3 Hz, C-1”), 122.74 (C-2”), 121.13 (d, JC-F = 18.7 Hz, C-3”), 120.15 (d, JC-F = 6.6 Hz, C-6”), 116.62 (d, JC-F = 21.9 Hz, C-5”), 55.68 (C-10), 49.09 (C-5), 39.99 (C-3), 39.86 (C-2), 29.65 (C-9), 25.34 (C-8), 24.67 (C-6), 21.33 (C-7); HRMS, Calculated for C16H18ClFN2NaO2, 347.0933; observed (M+Na)+ 347.0925. 58 (77.3 mg) was further purified on CHIRALPAK IF column eluted with acetonitrile-ethanol (7:3) to provide two chiral factions, 32.1 mg (42%) and 28.7 mg (37%) respectively both with ee > 99%. Fraction 2 is the compound 61.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.