

Abstract
Desmosomal cadherins are a recent evolutionary innovation that make up the adhesive core of highly specialized intercellular junctions called desmosomes. Desmosomal cadherins, which are grouped into desmogleins and desmocollins, are related to the classical cadherins, but their cytoplasmic domains are tailored for anchoring intermediate filaments instead of actin to sites of cell–cell adhesion. The resulting junctions are critical for resisting mechanical stress in tissues such as the skin and heart. Desmosomal cadherins also act as signaling hubs that promote differentiation and facilitate morphogenesis, creating more complex and effective tissue barriers in vertebrate tissues. Interference with desmosomal cadherin adhesive and supra-adhesive functions leads to a variety of autoimmune, hereditary, toxin-mediated, and malignant diseases. We review our current understanding of how desmosomal cadherins contribute to human health and disease, highlight gaps in our knowledge about their regulation and function, and introduce promising new directions toward combatting desmosome-related diseases.
Keywords: desmosomes, desmogleins, skin diseases, cardiomyopathy, cell junctions, cancer
1. INTRODUCTION
The cadherin superfamily comprises calcium-dependent transmembrane proteins that are essential for cell–cell adhesion during embryogenesis and tissue homeostasis (1). Classical cadherins, including E-, N-, and P-cadherin (type I) and VE-cadherin (type II), have ancient origins. Their appearance during evolution and their role as building blocks in primitive adherens junctions (AJs) were crucial for establishing multicellularity in metazoans (2, 3). Structurally related desmosomal cadherins appeared later in vertebrates, with further expansion in mammals, coinciding with the development of stronger barriers against environmental stress (4). While the role of classical cadherins in development and cancer progression is well established, desmosomal cadherins and their associated proteins have more recently emerged as important disease targets in epithelial and cardiac tissues.
Desmosomal components structurally and functionally interact with each of the four major types of vertebrate intercellular junctions, but desmosome origins and structural framework are most related to AJs (5). In AJs, transmembrane classical cadherins associate extracellularly through their ectodomains and anchor actin to the plasma membrane indirectly through armadillo (β- and p120-catenin) and cytoskeletal adaptor (α-catenin) proteins (Figure 1a). In contrast to classical cadherins, there are multiple members of the family of desmosomal cadherins that are grouped into two types, desmogleins (Dsg1–Dsg4) and desmocollins (Dsc1–Dsc3), which are expressed in a cell-type and differentiation-dependent manner (Figure 1) (6). Dsgs and Dscs localize to the plasma membrane where they collaborate with associated cytoplasmic proteins to anchor intermediate filaments (IFs) and form a functional desmosome. Of the associated proteins, plakoglobin, an armadillo family member related to the AJ protein β-catenin, directly interacts with the intracellular cadherin segment of Dsgs and the longer a isoform of Dscs, a region homologous to the β-catenin binding domain in classical cadherins (Figure 1b) (4). Plakophilins, desmosomal armadillo proteins related to p120-catenin in AJs, assist in clustering desmosomal proteins within ultrastructurally electron-dense fibrous plaques subjacent to the plasma membranes of two interacting cells (5). These interacting membranes are separated by a 30-nm space bisected by a distinct midline between two cells, absent in AJs (7). While desmosomal and classical cadherins and armadillo proteins share genetic sequence homology, the main cytoskeletal adaptors of these two complexes are distinct. In desmosomes, the plakin family member desmoplakin is structurally unrelated to the actin binding protein α-catenin but plays an analogous role by anchoring the IF cytoskeleton to the desmosome (5).
Figure 1.
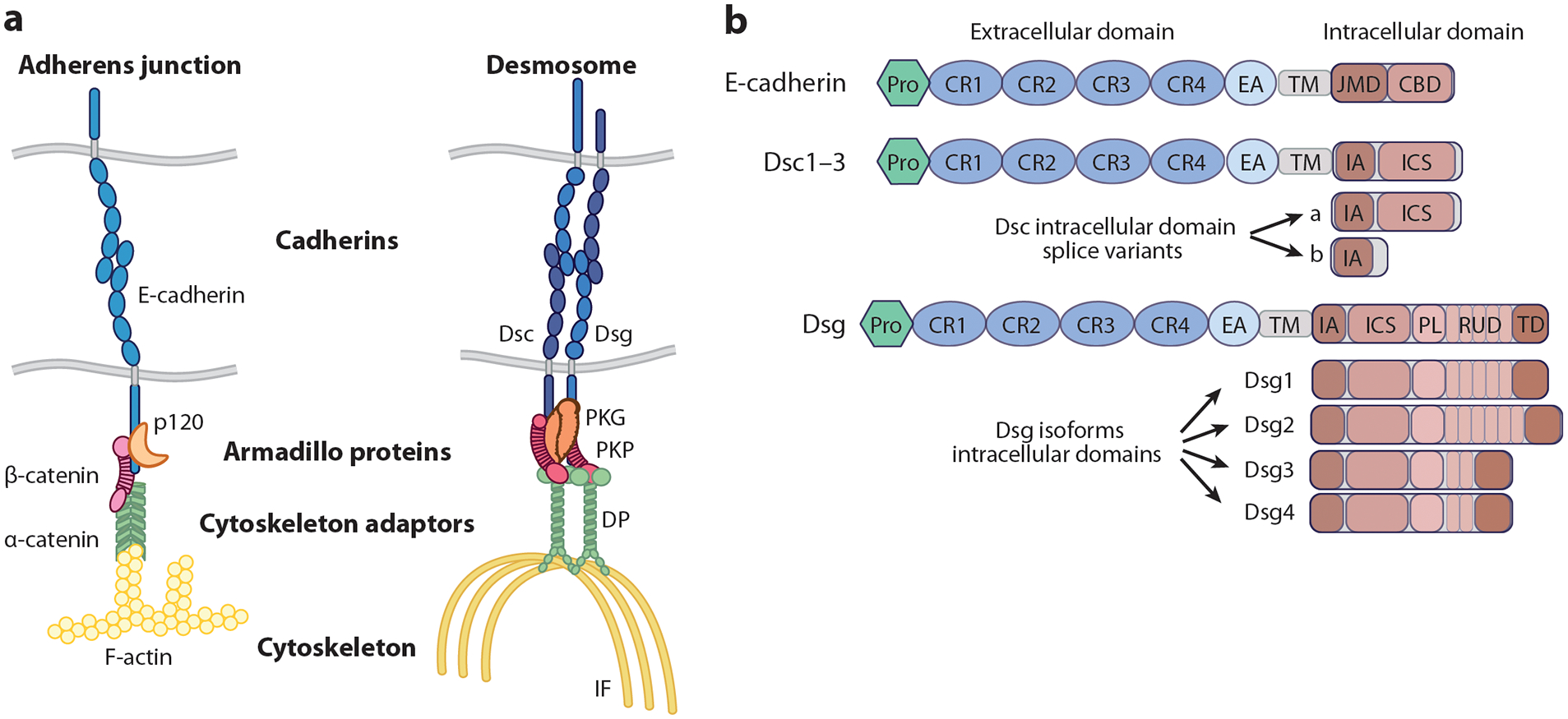
Structure of adherens junctions and desmosomes. (a) A schematic representation of the structural makeup of adherens junctions and desmosomes is shown. Both junctions are composed of transmembrane cadherins (blue), armadillo family proteins (orange and pink), and a cytoskeletal linker protein (green), which connects the junctional complex to a specific cytoskeleton (yellow). (b) The cadherin proteins from both adherens junctions and desmosomes are structurally related but differ in their specific domain makeup. Desmocollin genes have two splice variants, a and b, which differ in the presence of the intracellular cadherin-like sequence, which is truncated in the b form and terminates instead with an additional 11 amino acids in Dsc1 and Dsc2 and eight residues in Dsc3. Dsg isoforms have a larger intracellular domain than classical cadherins or desmocollins with individual isoforms differing by the number of repeat unit subdomains within their intracellular domain. Abbreviations: CBD, catenin binding domain; CR, cadherin repeat; DP, desmoplakin; Dsc, desmocollin; Dsg, desmoglein; EA, extracellular anchor; IA, intracellular anchor; ICS, intracellular cadherin-like segment; IF, intermediate filament; JMD, juxtamembrane domain; PKG, plakoglobin; PKP, plakophilin; PL, proline-rich linker region; Pro, protodomain; RUD, repeat unit subdomain; TD, termination domain; TM, transmembrane domain.
In spite of their similarities to AJs, desmosomal cadherins and desmosomes exhibit several distinguishing features not shared by AJs. As described above, desmosomes are built from members of two cadherin subfamilies, Dsgs and Dscs. Cadherins that contained features of desmosomal cadherins first appeared in jawless fishes, but it was not until later in jawed vertebrates that Dsgs and Dscs emerged as separate entities (4). While in vitro structural evidence suggests that desmosomal cadherins associate through heterophilic interactions of Dsc and Dsg ectodomains, experimental evidence for both homophilic and heterophilic interactions between neighboring cells (trans-interaction) exists (8). For instance, both heterophilic and homophilic binding of Dsgs and Dscs was observed in cell-free atomic force microscopy (AFM). However, cell-based AFM and extracellular cross-linking studies revealed a preference for homophilic interactions. On the other hand, cell-free surface plasmon resonance assays found exclusively heterophilic interactions with KD values between 3.6 and 43.9 μM depending on the specific Dsg:Dsc pairing (9). Interestingly, differentiation-specific pairs tended toward higher affinity interactions.
Another property that distinguishes mature desmosomes from AJs is their spatially and biochemically distinct localization in lipid-rich and detergent-resistant membranes (10–12). It was proposed that the longer transmembrane domain (TMD) of desmosomal cadherins governs the partitioning of desmosomal cadherins into these lipid domains. As described in detail below, a human mutation in this transmembrane region interferes with the incorporation of Dsgs into desmosomes and lipid rafts in vitro and leads to a severe skin and allergic condition in vivo (12).
Desmosomes also exhibit a unique ability to transition between a dynamic calcium-dependent state to a calcium-independent hyperadhesive state. First observed in simple epithelial MDCK cells, desmosomes that were allowed to mature in culture over time eventually became resistant to calcium chelation and maintained their intercellular connections when other intercellular junctions dissociated (13). Desmosomes both in keratinocytes in vitro and within the epidermis in vivo display this behavior, which was associated with the ability of desmosomal cadherins to become highly ordered in the presence of calcium (14). Cadherin-associated cytoplasmic proteins including plakoglobin, plakophilins, and desmoplakin have also been proposed to contribute to this property (14–16). Most recently, fluorescence polarization microscopy was employed to demonstrate that cadherin order is not required for hyperadhesion; instead, this strong adhesive state is associated with reduced protein exchange of cadherins and their interacting proteins, plakoglobin and desmoplakin (17). Strengthening desmoplakin’s association with IF by preventing its phosphorylation through mutation of a C-terminal serine, S2849G, reduces desmosomal protein exchange and increases intercellular adhesion, highlighting the importance of IF-desmosome interactions in hyperadhesion (16, 17). Overall, the ability to transition between dynamic calcium-dependent and stable hyperadhesive states provides cells with the abilities to fine-tune cell–cell interactions during embryonic development and wound healing and to adapt to changing forces and stressors.
The expansion of desmosomal cadherins that occurred during evolution is another feature that sets them apart from classical cadherins. With the emergence of complex tissues in mammals came seven different Dsg and Dsc genes and their spliced isoforms. At least one isoform of Dsgs and Dscs is present in all epithelial tissues and cardiomyocytes in the heart, as well as a subset of specialized tissues such as in the arachnoid layer of the brain meninges and the dendritic reticulum cells in lymphoid follicles (Figure 2a) (18). Whereas simple epithelial tissues generally express only Dsg2 and Dsc2, complex tissues such as the epidermis express multiple Dsgs and Dscs in a patterned manner, such that certain cadherins are expressed in progenitor cells, while others are expressed more abundantly during differentiation (Figure 2b). These observations raise the possibility that desmosomal cadherins have nonredundant functions in cells and tissues (4). In support of this idea, recently discovered supra-adhesive roles of desmosomal cadherins illustrate that these molecules are more than just adhesion molecules (19). Interfering with the adhesive and supra-adhesive functions of desmosomal cadherins through gene mutations, autoimmune antibodies, and bacterial toxin-mediated ectodomain cleavage causes a variety of human epithelial and cardiac diseases (20). In the following sections, we compare the expression patterns, functions, and dynamics of desmosomal cadherins; discuss their associated human diseases; and highlight emerging questions in the field of desmosomal cadherin research.
Figure 2.

Desmosome expression within tissues and cells. (a) A representation of desmosome expression in select tissues in the human body is shown. The three major desmosome-expressing cell types are labeled as stratified epithelia (purple), cardiomyocytes (blue), and simple epithelia (green). Additional desmosome-expressing cell types are labeled as other (pink) and include arachnoid reticular cells (brain meninges), follicular dendritic cells (lymphatic tissue), and myoepithelia (mammary tissue). (b) Desmosomal cadherin expression patterns vary by cell type. Dsc2 and Dsg2 comprise desmosomes in simple epithelia and cardiomyocytes. In simple epithelia, these desmosomes are localized to the basolateral membranes of the cell, anchoring the intermediate filament network. In cardiomyocytes, desmosomes are found in the hybrid junctions of the area composita containing both desmosomes and adherens junctions, as well as in separate individual desmosome junctions anchoring the intermediate filament network. In stratified epithelia, desmosomal cadherins are differentially expressed in different layers of the tissue, and the patterning differs between mucosal and cutaneous epithelial tissues. Here, the areas in the triangles and rectangles represent relative expression levels across the apical to basal axis of the tissue. The outline color indicates cadherins in desmosomes (dark red) or adherens junctions (pink). Abbreviations: Dsc, desmocollin; Dsg, desmoglein.
2. MULTIPLE DESMOSOMAL CADHERINS FUNCTION IN TISSUE DEVELOPMENT, REGENERATION, AND HOMEOSTASIS
The adhesive and mechanical functions of desmosomal cadherins are reflected by their prominence in tissues that experience high mechanical stress such as the stratified epidermis of the skin and the cardiac muscle of the heart (Figure 2). Desmosomes are also found in other tissues that experience constant environmental stress such as the simple epithelia of the gastrointestinal tract and lung. Desmosomal cadherins and their association with IF through desmosomal plaque proteins provide mechanical resilience to tissues and regulate cellular signaling to counter multiple types of intrinsic and extrinsic stress (21). While Dsg2 and Dsc2 are ubiquitously expressed in all desmosome-containing tissues, multiple desmosomal cadherins are expressed in differentiation-dependent patterns in stratified epithelia (Figure 2b) (22). How these patterns are established is still poorly understood, but as described in more detail below the patterns are functionally important.
2.1. Role of Desmosomal Cadherins and Associated Proteins in Embryogenesis
Loss-of-function and gain-of-function experiments support the idea that desmosomal cadherin expression and patterning are required not only for proper morphogenesis and differentiation of complex tissues but also for multiple stages of embryogenesis, even prior to the appearance of desmosomes. For instance, global knockout of Dsg2 or Dsc3 resulted in embryonic lethality prior to desmosome formation and blastocyst implantation, highlighting a desmosome-independent function for these cadherins during development (23, 24). Desmosomes also play a role in cell fate determination by promoting the apical localization of keratin-18 IF in mammalian blastocysts, which is necessary for asymmetric inheritance of keratin-18 IF and trophectoderm lineage specification in the outer cells of the embryo (25). In zebrafish, depletion of either Dsgs or Dscs impaired cell sheet spreading during the process of epiboly, as well as axis elongation and somite formation (14). In frogs, desmoplakin was required for expansion of radially intercalating cells that move from an inner to outer epidermal layer during embryogenesis, resulting in morphogenetic defects and a reduction in the number of differentiated multiciliated and secretory cells (26). Ablation of cadherin-associated desmosomal plaque components desmoplakin, plakoglobin, and plakophilin-2 resulted in embryonic lethality at embryonic day 6.5 due to impaired egg cylinder formation or at midgestation due to skin and/or heart defects, illustrating that functional desmosomes are also important for embryogenesis postimplantation (27). Collectively, these observations highlight the essential roles desmosomal cadherins and their associated proteins play from the earliest stages of embryogenesis throughout morphogenesis of distinct tissues.
2.2. Desmosomal Cadherin Functions in Stratified Epithelia
Forced misexpression of Dsg2, Dsg3, and Dsc3 in stratified layers of the epidermis of mice using keratin-1 or involucrin promotors (28–30) results in abnormal differentiation and hyperproliferation. Animals in which Dsc3 is misexpressed suprabasally exhibited enhanced β-catenin activity, whereas animals in which Dsg2 was misexpressed suprabasally displayed elevated activation of epidermal growth factor receptor (EGFR), nuclear factor κB (NFκB), and survival signaling, associated with increased susceptibility to carcinogenesis (29, 30). Misexpression of Dsg3 in suprabasal layers of epidermis driven by the early differentiation keratin-1 promoter exhibited features of ichthyosis, such as hyperproliferation, epidermal thickening, and abnormal differentiation (28). However, misexpression of Dsg3 driven by the later differentiation involucrin promoter resulted in features that resembled mucosal epithelium, which exhibits a higher Dsg3:Dsg1 ratio (Figure 2b) (31). Collectively, these studies suggest that forced expression of basally expressed desmosomal cadherins in the superficial layers disrupts epidermal differentiation and that the ratio of desmosomal cadherin isoforms may affect tissue identity.
While hyperproliferation and abnormal differentiation were observed upon aberrant suprabasal expression of basal cadherins, this phenotype was also observed in a Dsc1 knockout animal model, consistent with a role for superficially expressed Dsc1 in differentiation (32). Furthermore, these knockout animals exhibited localized skin blisters and gradual hair loss. However, no observable phenotype was seen in mice lacking the Dsc1 cytoplasmic domain, raising the possibility that this domain is not necessary for its adhesive function nor for promoting differentiation in the suprabasal layers (33). Likewise, forced expression of full-length Dsc1 in the basal layer of animals with a keratin-14 promoter did not promote premature differentiation in the basal layer (34).
Dsg loss-of-function models also revealed supra-adhesive roles for desmosomal cadherins in stratified tissue. For instance, while loss of Dsg1 resulted in neonatal lethality due to epidermal blistering and a disrupted epidermal barrier (35), recent transcriptomic analysis revealed evidence of abnormal differentiation, enhanced mitogen-activated protein kinases (MAPK) signaling, and a Th17-skewed inflammatory signature (36). This inflammatory signature was present in embryos prior to experiencing external stimuli, raising the possibility that loss of Dsg1 predisposes skin to inflammation (36). This study also supports earlier in vitro work showing that Dsg1 dampens EGFR/extracellular signal-regulated kinase 1/2 (ERK1/2) signaling to promote differentiation and that Dsg1 knockdown stimulates cytokine secretion in primary keratinocytes (37–40). Further in vitro work revealed two mechanisms by which Dsg1 regulates EGFR signaling. One mechanism depends on Dsg1’s association with ErbB2 interacting protein (ERBIN) to inhibit Ras/Raf coupling and downstream ERK1/2 activation (38). The second depends on Dsg1’s association with the COP9 signalosome subunit Cops3 to promote EGFR turnover via deneddylation and subsequent ubiquitination (39). These studies illustrate the ability of Dsg1 to act as a signaling hub by associating with molecules that can modulate cell signaling.
Animals with global knockout of Dsg3 or conditional knockout of Dsc3 in squamous epithelia are viable, exhibiting similar phenotypes of hair loss with mucocutaneous or cutaneous blistering, respectively (41–43). Dsg3-depleted mice exhibited increased p38 MAPK activation associated with enhanced cell migration and faster wound closure of skin lesions (43). Dsg3-deficient mice also had increased p53/p21WAF1/CIP1 and cleaved caspase-3 expression in hair follicles, a result suggesting that Dsg3 promotes an antistress response (44). Depletion of Dsc3 in the epidermis did not affect p38 MAPK activity or the ERK, c-Jun N-terminal kinase, and NFκB pathways (45). Therefore, it is unclear if Dscs, which have a significantly smaller cytoplasmic tail compared with Dsgs, have the ability to directly regulate signaling.
Disruption of one desmosomal cadherin can be compensated, at least in part, by upregulation of another desmosomal cadherin. For example, keratin-14-mediated expression of Dsg1 in Dsg3 knockout mice reversed the hair-loss phenotype (46). Forced suprabasal expression of Dsg2 protected against blister formation resulting from autoantibody-mediated and exfoliative toxin-mediated disruption of Dsg1-dependent cell adhesion (47). Although these data suggest that desmosomal cadherins can have redundant functions in adhesion, Dsg1 and Dsg3 knockout animals exhibited increased Dsg3/Dsc1 and Dsg1/2, respectively, which was not sufficient to prevent blistering and abnormal differentiation (36, 48). This raises the possibility that there are nonredundant functions for desmosomal cadherins that might be uncovered through the discovery and analysis of specific binding partners, such as ERBIN’s interaction with Dsg1 to regulate EGFR activity (38). These examples of desmosomal cadherin compensation through altered expression also suggest that feedback mechanisms exist to regulate cadherin patterning, a possibility that warrants further investigation.
2.3. Desmosomal Cadherin Functions in Simple Epithelia
Simple intestinal epithelia normally express only Dsg2 and Dsc2, both of which are important for intestinal barrier function (49, 50). Constitutive silencing of Dsg2, but not Dsc2, resulted in an impaired barrier associated with increased permeability (49). This is consistent with earlier in vitro work showing that antibody-mediated disruption of Dsg2 in intestinal cells disrupted tight junctions despite upregulation of Dsc2 (51). On the other hand, inducible depletion of Dsc2 resulted in intestinal barrier disruption (50). The fact that constitutive loss of Dsc2 did not affect the intestinal barrier might be explained by the compensatory recruitment of galectin-3 to cell–cell junctions, promoting stabilization of Dsg2 to enhance cadherin function (49, 52). While mechanisms by which Dsg2 and Dsc2 influence the barrier are not fully understood, evidence suggests that Dsg2 normally supports adhesion through Src-mediated EGFR phosphorylation and downstream p38 MAPK activity (53, 54) but that loss of Dsg2 or Dsc2 diverts EGFR signaling toward proliferation (54, 55). Thus, Dsg2 and Dsc2 provide intestinal cells with the ability to fine-tune the balance of adhesion and proliferation through EGFR.
2.4. Desmosomal Cadherin Functions in Cardiomyocytes
Cardiac muscle is another tissue in which desmosomes play an essential role in countering the forces of mechanical stress. In the heart, Dsg2 and Dsc2 are the only desmosomal cadherin isoforms expressed. They can be found both in desmosomes and in junctions called area composita, which are unique to postnatal vertebrates and in which desmosome and AJ components are intermixed (Figure 2b). Both junction types are localized along with AJs and gap junctions within specialized regions of the plasma membrane called intercalated discs, which are critical for structural and electrical coupling of cardiomyocytes. The importance of desmosomes in the heart is reflected by the existence of a rare cardiac disease historically called arrhythmogenic right ventricular cardiomyopathy, now referred to simply as arrhythmogenic cardiomyopathy (AC), which can present in patients with and without skin manifestations. A large proportion of AC patients have mutations in desmosome molecules, which can lead to sudden death due to heart arrhythmia (56). Experimental animal models recapitulate some of the pathological anomalies observed in AC. For example, Dsg2-deficent mice and mice expressing a Dsg2 mutant lacking the EC1-EC2 extracellular domains exhibit fibrotic lesions containing necrotic cardiomyocytes and right ventricle dilation by 4–6 weeks of age (57). Dsg2 is not necessary for cardiac development, but it protects against AC by providing mechanical integrity to the maturing heart (57). Although it is unclear if there are direct signaling roles for Dsg2 or Dsc2 in the heart, they interact with the sodium channel NaV1.5 and gap junction protein connexin-43 (Cx43), respectively, which are important for electrical conductance (56). Dsg2/Dsc2 might also indirectly promote cellular signaling by associating with plakophilin-2 and desmoplakin, which regulate protein kinase C-alpha (PKCα) and the Ras/ERK pathway, respectively (58, 59).
2.5. Desmosomal Cadherin Functions in Mechanotransduction
An emerging function for desmosomal cadherins is their ability to sense and respond to mechanical signals from neighboring cells and the environment. Desmosomes regulate tissue mechanics, such as tension and cell stiffness, through desmoplakin-IF associations (60). The development of a Dsg2 Förster resonance energy transfer (FRET)-based tension biosensor enabled investigators to measure mechanical loading in epithelia and contracting cardiac cell lines, revealing that Dsg2 can act as a mechanosensor (61). In contrast to the Dsg2 tension sensor, two desmoplakin FRET-based tension sensors did not exhibit mechanical loading in epithelial cells unless external force was applied (62). These potential differences may be explained by tissue context dependence or cytoskeletal crosstalk mechanisms (63). Using E-cadherin and Dsg2 FRET-based tension sensors, researchers monitored changes in tension on AJs and desmosomes in intestinal epithelial cells upon Dsc2 loss and found increased tension on Dsg2 but not E-cadherin in Dsc2-deficient cells (50). The authors went on to show that under these conditions, desmoplakin phosphorylation at S2849 decreased. This dephosphorylated form of desmoplakin has been shown to enhance interactions with IF (64), which in this context was associated with increased tension on desmosomes. This study suggests that Dsc2 has a role in force transduction by modulating desmoplakin-IF affinity.
Dsg1 also regulates tension distribution in keratinocytes by promoting cortactin/Arp2/3 to facilitate cortical actin reorganization and tension redistribution away from AJs (65). This redistribution of tension is hypothesized to promote basal cell delamination during the formation and maintenance of the multilayered epidermis. In addition to changes in cortical actin organization, coupling between desmosomes and IF was also required to support delamination in multilayered tissues as well as apoptotic extrusion in simple epithelia (66, 67). In further support for the desmosome’s role in mechanosensing and transduction, Dsg3 expression modulates keratinocyte sensitivity to strain and substrate stiffness by regulating the expression and subcellular localization of YAP, a mechanosensory protein (68). More work is needed to understand the role of desmosomal cadherins in regulating tension and force transduction and their impact on tissue morphogenesis.
3. DESMOSOMAL CADHERIN DYNAMICS
While the classic textbook view of desmosomes depicts them as static, stable entities, recent studies show that desmosomes are also dynamic complexes that undergo turnover and remodeling (69). Membrane delivery or trafficking and turnover of cadherins regulate the cell’s adhesive potential to allow for transitions between adhesive and dynamic cellular states (70). Dynamic incorporation of new desmosomal cadherins into existing desmosomes must also occur during epidermal homeostasis to maintain the patterned organization of desmosomal cadherins (71). Studies on classical and desmosomal cadherins illustrate that complex mechanisms exist to regulate the localization and separation of these molecules into distinct compartments. Certain diseases, discussed below, disrupt cell–cell cohesion and cell signaling by interfering with desmosome localization and dynamics, highlighting the importance of understanding how desmosome components are synthesized and properly positioned on the plasma membrane and how their turnover, recycling, and degradation are controlled (72).
Desmosome component biosynthesis and dynamics were first investigated using biochemical methods, which revealed lower solubility and longer half-lives compared with other intercellular junction components (19). The well-established insolubility of Dsgs may be due in part to their assembly into highly ordered, lipid-rich microdomains on the plasma membrane, often called detergent-resistant membranes or lipid rafts (10, 73). Pharmacological disruption of lipid rafts with methyl-β-cyclodextrin impairs the formation and adhesive function of desmosomes but not AJs, underscoring the importance of desmosome localization in these microdomains (11). A key feature that governs the localization of desmosomes but not AJs into lipid rafts is proposed to be the longer TMD present in Dsgs but not classical cadherins, allowing these molecules to partition into cholesterol-rich regions (Figure 1b) (11). In support of this idea, replacement of the Dsg1 TMD with the E-cadherin TMD disrupts Dsg1 detergent-resistant membrane localization (12). Plakoglobin may also have a role in junction segregation. Plakoglobin can bind α-catenin as well as both E-cadherin and the desmosomal cadherins (11). However, as the plakoglobin binding sites for desmosomal cadherins and α-catenin overlap (11), plakoglobin association with desmosomal cadherins promotes their exclusion from AJs by masking the α-catenin binding site. The segregation of desmosomes from AJs may be functionally important for regulating cytoskeletal organization and tension distribution. As an example, expression of Dsg1 in primary keratinocytes promotes an enrichment in cortical actin and reduces tension on AJs, but this Dsg1-mediated tension redistribution is lost when Dsg1 is mispositioned in the detergent-soluble membrane (65). More studies are needed to comprehensively determine additional driving forces and the functional importance of desmosome and AJ segregation.
Although mature AJs and desmosomes are localized in spatially distinct domains on the plasma membrane, evidence suggest that AJ and desmosomal components cooperate to promote junction assembly. AJ assembly precedes desmosomal assembly during embryogenesis and de novo cell–cell contact in vitro (5). Disruption of AJ establishment by depletion of classical cadherins impairs desmosome assembly, illustrating that AJs are necessary for efficient desmosome formation (74). Recent studies identified an interaction between E-cadherin and Dsg2, consistent with the idea that association between these classical and desmosomal cadherins may facilitate initiation of desmosome assembly (75). Of cadherin-associated proteins, plakoglobin has historically been considered a primary mediator of interjunctional crosstalk between AJ and desmosomes due to its ability to interact with both desmosomal and classical cadherins (5). However, plakophilin-2 also participates in this crosstalk by cooperating with E-cadherin to promote RhoA-mediated actin organization and subsequent desmoplakin-IF recruitment to nascent desmosomes (76). Nonjunctional, detergent-soluble Dsg3 was also reported to regulate AJ formation by activating Src and facilitating its association with E-cadherin in keratinocytes (77).
In spite of their stability, imaging analyses reveal desmosomes as dynamic structures both in cultured cells and in tissues. Early immunogold electron microscopy studies demonstrated that desmosomal cadherin composition in desmosomes is constantly changing during the process of epidermal differentiation. Basal Dsc3 is progressively lost and newly synthesized Dsc1 becomes incorporated into existing desmosomes as cells progress to the surface of the epidermis (71). How this replacement occurs in vivo is not well understood.
3.1. Trafficking of Desmosomal Cadherins During Desmosome Assembly
Recent advances in optical live-cell imaging and fluorescence recovery after photobleaching analysis have enabled temporal analysis of desmosomal cadherin dynamics, using in vitro models. Live-cell tracking of fluorescently tagged desmosomal molecules showed that clustering of Dsg2 on the plasma membrane occurs first followed by desmoplakin recruitment and then Dsc2 coclustering with Dsg2 (78). Differences in Dsg2 and Dsc2 behavior and kinetics during desmosome assembly could arise from reported differences in long-range trafficking and use of distinct kinesin motors (79). Furthermore, Dsg2, but not Dsc2, interacts with E-cadherin during desmosome assembly and thus may contribute to the initiation of desmosome assembly prior to coclustering with Dsc2 (75). This stepwise process of desmosome assembly and its integration with classical cadherin dynamics provides an opportunity for coordinating junction assembly and cadherin stoichiometry to regulate adhesion during epithelial remodeling.
Most live-cell imaging analysis of desmosome assembly has been carried out in the context of single cell layers in submerged culture. Mechanisms associated with desmosome assembly in 3D cultures and tissues are not well understood. For instance, it is unclear if desmosomal cadherins expressed superficially in complex stratified tissues utilize similar trafficking mechanisms to those exhibited by Dsg2 and Dsc2 in cultured cells. This is a particularly interesting question as microtubule orientation and positioning of plus and minus ends change during epidermal differentiation. Microtubule minus ends are released from the centrosome and become repositioned near the plasma membrane in suprabasal cells (80). While basal Dsg2 and Dsc2 utilize microtubule plus-end motor kinesin (79), this microtubule reorganization opens the possibility that suprabasal desmosomal cadherins use the minus-end motor, dynein. In support of this idea, Dsg1, but not basal Dsg2 and Dsg3, interacts with the dynein light chain Tctex-1 (65). Depletion of Tctex-1 resulted in loss of Dsg1 from the insoluble membrane without blocking its delivery to the plasma membrane, a result that suggests complex multistep trafficking of Dsg1 or compensation by another dynein light chain (65). Besides microtubule reorganization, epidermal differentiation also induces the enrichment of cortical actin, cell flattening, and Golgi fragmentation (81–83). Therefore, long-range microtubule trafficking may not be needed for initial delivery of desmosomal cadherins to the plasma membrane in differentiated keratinocytes. More studies are needed to understand how desmosomal cadherin trafficking evolves with changes in differentiation.
3.2. Posttranslational Modification of Desmosomal Cadherins
Certain posttranslational modifications can regulate desmosomal cadherin trafficking, dynamics, and turnover, including N-glycosylation, palmitoylation, phosphorylation, and proteolytic cleavage. The addition of oligosaccharides on asparagine residues in the extracellular domain of cadherins in the Golgi apparatus is necessary for both AJ and desmosome formation and function (84–86). Inhibition of this N-glycosylation with tunicamycin resulted in loss of cell–cell adhesion in keratinocytes, possibly due to a decrease in Dsg1 and Dsg3 protein stability (86). While a direct mechanism for decreased stability is unclear, N-glycosylation is known to play a role in protein folding and membrane trafficking. Either of these could be impaired when desmosomal cadherins lack proper carbohydrate modifications, resulting in targeting to degradative pathways.
With the exception of desmoplakin, desmosome components can also be modified by the addition of the palmitate, a 16-carbon fatty acid, to cysteine residues by palmitoyl-acyl transferases (PATs) (87). Fatty acid modification of desmosomal components is another distinction from AJ components, which have not been reported to be palmitoylated. Dsg2 and plakophilin-3 were recently discovered to be substrates of the PAT DHHC5 (88). Although the loss of palmitoylation of Dsg2 results in increased targeting to the lysosome, palmitoylation is not necessary for targeting Dsg2 and Dsg3 to lipid rafts; this result suggests that palmitoylation is important for desmosomal cadherin stabilization on the plasma membrane but not positioning (12, 89). These studies support the existence of palmitoylation-independent mechanisms that target desmosomal cadherins to lipid rafts.
Emerging evidence suggests that the phosphorylation status of desmosomal components is an important way to tune adhesion (90, 91). A good example is the switch between hyperadhesive and dynamic desmosomes. Induction of tyrosine phosphorylation with pervanadate has been reported to induce hyperadhesion (92). Although pervanadate treatment increased tyrosine phosphorylation of Dsg2 and plakoglobin in the soluble and insoluble membrane, it is unclear if the phosphorylation of these desmosomal components contributes to hyperadhesion (92). Additionally, the adhesive state of desmosomes is regulated by the phosphorylation status of desmoplakin mediated by glycogen synthase kinase 3 (GSK3) phosphorylation and PRMT-1-dependent arginine methylation (93). Hypophosphorylation of desmoplakin and PKC inhibition can also promote Ca2+-independent hyperadhesion by reducing desmosomal component exchange on the plasma membrane (16, 17, 94). Finally, inhibition of the tyrosine kinase EGFR decreased Dsg2 and plakoglobin phosphorylation and promoted desmosome assembly and adhesion in a skin cancer cell line (95). Therefore, phosphorylation of desmosomal components can modulate adhesion to allow plasticity during tissue remodeling or when switching between tissue homeostasis and regeneration.
In addition to modification of desmosome molecules through the addition of carbohydrate, fatty acid, and phosphate moieties, classical and desmosomal cadherins undergo proteolytic cleavage of their extracellular domain in a process known as cadherin shedding. Cleavage is facilitated by the a disintegrin and metalloprotease (ADAM) family of sheddases, matrix metalloproteases (MMPs), and kallikrein-related peptidases (KLKs) and results in disruption of cell cohesion by removal of ectodomain sequences required for intercellular interactions (96–99). Disrupting cadherin trans-interactions can also promote cadherin internalization and subsequent lysosomal turnover (98). Understanding the complex mechanisms of desmosomal cadherin regulation may provide insight into pathomechanisms that disrupt cadherin localization and function.
4. AUTOIMMUNE DISEASES TARGETING DESMOSOMAL CADHERINS
Pemphigus is a skin disorder caused by inhibitory autoantibodies targeting one or more desmosomal cadherins resulting in blistering of mucosal and/or cutaneous tissue due to acantholysis or loss of cell cohesion (100). Sera from patients with pemphigus vulgaris (PV), the most common form of pemphigus, predominantly contain autoantibodies targeting Dsg3 or Dsg1/3. These antibodies disrupt keratinocyte cohesion in mucosal or mucocutaneous tissue, respectively. Autoantibodies that target Dsg1 result in pemphigus foliaceus (PF), which has a cutaneous phenotype. Rarer subsets of pemphigus patients have pathogenic antibodies targeting Dsc3 or autoantibodies against proteins in the plakin family.
Differences in the location of pemphigus blisters is thought to be due to the differentiation-specific expression of the Dsg targets along with compensation resulting from higher protein expression levels of Dsg1 and Dsg3 in cutaneous and mucosal tissues, respectively (Figure 2b) (100). Therefore, patients with autoantibodies against Dsg3 (mucosal-dominant PV) only exhibit mucosal blistering while patients with autoantibodies against Dsg1 (PF) exhibit cutaneous blistering. However, a subset of patients with autoantibodies against both Dsg1 and Dsg3 (mucocutaneous PV) present with blistering in both mucosal and cutaneous tissue. The presentation of pemphigus-mediated blistering illustrates how the distinct tissue expression of desmosomal cadherins can affect disease pathology.
Work with patient-derived PV immunoglobulin G, PV monoclonal antibodies, and pathogenic anti-Dsg3 antibodies illustrates that multiple mechanisms contribute to pemphigus-mediated acantholysis (100, 101). These include steric hindrance of Dsg trans-interactions, endocytosis-mediated Dsg depletion from desmosomes, changes in cell signaling, and cytoskeletal reorganization. The discovery of multiple cellular signaling pathways altered by antibodies targeting Dsg1 and Dsg3 provides further evidence that desmosomal cadherins act as signaling hubs during homeostasis. Interestingly, anti-Dsg1 antibodies were found to activate ERK signaling and calcium influx, while anti-Dsg3 antibodies were found to activate p38 MAPK and Src; these findings support the idea that nonredundant signaling pathways are regulated by desmosomal cadherins (102). Pharmacological inhibition of p38 MAPK, c-Myc, GSK3β, phospholipase C, PKC, EGFR, signal transducer and activator of transcription 3 (STAT3), and Src all prevented pemphigus-induced blistering in mouse models, a result that illustrates the complexity of signaling changes linked to cell–cell cohesion (103–106). The current first-line therapy for pemphigus treatment is rituximab-mediated B cell depletion to decrease circulating anti-Dsg antibodies (107). Although rituximab is efficacious in patients, long-term treatment is often required and relapse is likely to occur (107, 108). Promising preclinical studies targeting the signaling pathways discussed above may provide guidance in developing additional therapeutic options for patients who are refractory to immunosuppressive therapies (107).
5. HEREDITARY DISEASES ASSOCIATED WITH MUTATIONS IN DESMOSOMAL CADHERINS
Figure 3 provides an overview of mutations on desmosomal cadherins that are associated with disease (109). As expected on the basis of their tissue expression, Dsg1 mutations are associated with skin disorders, Dsg2 and Dsc2 mutations are associated with cardiac disorders, and Dsg4 mutations are associated with hair diseases. Notably, only a small subset of these disease-associated mutations have been functionally validated, identifying an important area for future research. Not included in the figure is a recently identified Dsg3 nonsense mutation, which resulted in mucosal-specific blistering (110).
Figure 3.

A graphical representation of identified disease-associated alterations in desmosomal cadherins. Shown are all cadherins with multiple mutations identified and registered by March 2021 in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk). Each symbol represents a specific type of alteration, i.e., deletion, insertion, missense mutation, and nonsense mutation (see key). Symbols are color coordinated by the disease with which they are associated, as indicated in the key. Alterations that have been functionally validated to cause the disease are indicated with a black border around the symbol, and the alteration and amino acid position are labeled on the graph. The x-axis represents the amino acid position on the cadherin proteins, with a binning of 20 amino acids per group and major and minor ticks every 500 and 50 amino acids, respectively. The y-axis represents the number of alterations identified within each group. Below the x-axis is a schematic of the cadherin protein, with the domains aligned to correspond to their appropriate amino acid position. Some diseases are grouped together: Hair-related diseases include hypotrichosis, monilethrix, and keratosis pilaris atrophicans; cardiac diseases (noncardiomyopathy) include atrial fibrillation, atrioventricular block, cardiac arrest, and conotruncal heart defects; ARVC/AC diseases include arrhythmogenic right ventricular cardiomyopathy, arrhythmogenic right ventricular dysplasia, and arrhythmogenic cardiomyopathy; and cardiomyopathies (non-ARVC) include cardiomyopathy (both dilated and hypertrophic), monogenic dilated cardiomyopathy, and left ventricular noncompaction cardiomyopathy. Abbreviations: AA, amino acid; AC, arrhythmogenic cardiomyopathy; ARVC, arrhythmogenic right ventricular cardiomyopathy; CR, cadherin repeat; Dsc, desmocollin; Dsg, desmoglein; EA, extracellular anchor; IA, intracellular anchor; ICS, intracellular cadherin-like segment; PL, proline-rich linker region; Pro, protodomain; RUD, repeat unit subdomain; SAM, severe dermatitis, multiple allergies, and metabolic wasting; SPPK, striate palmoplantar keratoderma; TD, termination domain. TM, transmembrane domain.
5.1. Skin Diseases Caused by Dsg1 Mutations
A recently characterized desmosome-related inherited disease is severe dermatitis, multiple allergies, and metabolic wasting (SAM) syndrome, which was first reported in 2013 (111). SAM syndrome is a rare condition that results from mutations in Dsg1 or, in a subset of atypical SAM syndrome patients, mutations in desmoplakin (111–114). Most reported SAM-associated mutations have been described as biallelic loss-of-function Dsg1 mutations resulting in a disruption of Dsg1 plasma membrane localization or decreased mRNA stability (111, 115). Functional characterization of a SAM syndrome patient with a homozygous mutation that causes exon 2 skipping in Dsg1 resulted in cytoplasmic accumulation of Dsg1, possibly due to inefficient trafficking (111). One heterozygous SAM-associated mutation in the Dsg1 TMD prevented its targeting to lipid rafts, thus disrupting its desmosome association (12). Overall, these findings illustrate the importance of Dsg1 plasma membrane localization for epidermal homeostasis. In atypical SAM syndrome patients with desmoplakin mutations, Dsg1 expression is reduced and its localization is also disrupted, appearing as cytoplasmic aggregates (114). This observation raises the possibility that disrupted Dsg1 may be a proximal mediator of disease pathogenesis. This idea is supported by the observation that Dsg1 but not desmoplakin inhibits NFκB-dependent inflammatory responses in vitro (116).
An association between Dsg1 and inflammation has also been reported in two patients with a SAM-like disorder called SAMEC (SAM syndrome, ectodermal dysplasia, arrhythmogenic cardiomyopathy) harboring desmoplakin mutations (116). In SAMEC, the downregulation of Dsg1 is thought to promote an NFκB-mediated inflammation in the skin through the loss of ERBIN accumulation on the plasma membrane (116). Understanding the mechanisms linking desmosomal cadherins and immune regulation is clinically relevant. Transcriptomic analysis of Dsg1-deficent mice and SAM syndrome patients revealed a Th17-skewed immune response (36). Consistent with this finding, targeting the Th17/interleukin 23 (IL-23) nexus with drugs commonly used to treat psoriasis benefited patients with Dsg1 and desmoplakin mutations who have SAM syndrome characteristics (36, 117–119). These studies suggest that suppression of the Th17/IL-23 immune response can be a successful strategy for treating Dsg1-related diseases.
SAM syndrome–associated Dsg1 mutations have also been associated with a decrease in the expression and altered distribution of Cx43, a major gap junction component, in patient tissues. This observation raises the possibility that compromised intercellular gap junction communication contributes to the disease (120). Consistent with this idea, ectopic expression of Dsg1 promoted Cx43-dependent gap junction function as indicated by increased dye transfer between keratinocytes, and genetic ablation of Dsg1 increased gap junction turnover in vitro. In vitro, decreased expression of Cx43 was associated with PKC-dependent Cx43 S368 phosphorylation, which is known to signal Cx43 turnover. Consistent with this finding, lysosomal degradation inhibition restored Cx43 levels in Dsg1-deficient cells. Dsg1 is not the only desmosome molecule reported to support gap junction function and localization. Desmoplakin promotes plasma membrane association of Cx43 in epidermal and cardiac cells through two independent mechanisms: (a) stabilization of microtubules at the plasma membrane through its association with the plus-end molecule EB1 and (b) inhibition of ERK-mediated lysosomal turnover (59, 121). Collectively, these results suggest that desmosome-mediated regulation of gap junction maintenance and function could be important in SAM syndrome pathogenesis.
While a majority of SAM syndrome patients harbor homozygous mutations of Dsg1, heterozygous mutations in Dsg1 (or in some cases desmoplakin) that result in haploinsufficiency cause striate palmoplantar keratodermas characterized by epidermal thickening in the soles of the feet and palms of the hands (69). Contributing to the pathomechanism is reduced Dsg1-ERBIN association, which releases the scaffolding protein Shoc2 to promote Ras-Raf dimerization and consequent ERK1/2 activation. This failure to dampen ERK is associated with abnormal epidermal differentiation (38).
5.2. Heart Diseases Associated with Dsg2 and Dsc2 Mutations
Reflecting their importance in the skin and heart, mutations in desmosomal components can result in cardiocutaneous syndromes with apparent phenotypes in both tissues. As mentioned previously, multiple mutations in Dsg2, Dsc2, and plaque proteins have been associated with the cardiac disease AC. AC is characterized by arrhythmia, chamber dilation, and cardiomyocyte necrosis with fibrofatty replacement and is associated with increased risk of sudden cardiac death (57, 122). About 50% of patients clinically confirmed to have AC have been found to harbor at least one mutation in desmosomal proteins (122). Although plakophilin-2 mutations are most common in AC patients, mutations in desmoplakin, plakoglobin, and both desmosomal cadherins expressed in cardiomyocytes, Dsg2 and Dsc2, have been reported (Figure 3). AC can also be found in conjunction with skin phenotypes in syndromic disorders such as Carvajal or Naxos disease; however, these occur when the pathogenic mutations are found in the desmosomal plaque proteins (122). Severity and phenotypic characteristics in AC vary widely and patient genotypes are not generally predictive of phenotype (123), making a precision medicine approach challenging. Two mutations (R45Q and R48H) have been found in the propeptide domain of Dsg2, suggesting a loss of furin-mediated cleavage of Dsg2 to produce its active form (122). Another mutation in the Dsg2 extracellular domain (D105E) resulted in altered binding kinetics and enhanced cell adhesion in vitro, suggesting that altering desmosomal cadherin trans-interactions may contribute to AC pathogenesis. The pathomechanism for some AC-associated Dsc2 missense mutations is thought to involve trafficking defects. Interestingly, arrhythmia was alleviated in a conditional plakoglobin knockout model using a peptide that promotes Dsg2 stabilization (124). Along with another study showing that a Dsg3 stabilizing peptide ameliorates pemphigus blistering (125), this observation provides a premise for a novel therapeutic approach for desmosome-related diseases via desmosomal cadherin stabilization.
6. DESMOSOMAL CADHERINS IN CANCER
It has been well established that alterations in cadherins, integrins, and their associated cellular signaling accompany malignant transformation in most epithelia. Depending on context, these changes can have either a positive or a negative impact on tumor progression (126). One phenomenon reported to occur in both simple and complex epithelia is called cadherin switching. This process results in a switch in the expression of the predominant cadherin type, the most characterized being an E-cadherin to N-cadherin switch. Cadherin switching occurs during epithelialmesenchymal transition of malignant cells and is associated with increased invasion (126). Changes in desmosomal cadherins resembling a cadherin switch also occur in certain cancers. In squamous cell carcinoma (SCC), Dsg1 and Dsc3 are downregulated while Dsg2 and Dsg3 are upregulated (45, 127, 128). Experimental evidence suggests that this desmosomal cadherin switch is functionally important. For instance, Dsg1 expression in SCC cells downregulated EGFR/ERK-mediated matrix degradation and formation of invadopodia, actin-mediated protrusions that facilitate invasion of cancer cells, while Dsg2 was required for formation of these protrusions (129). Additionally, in vivo work discovered inverse roles for Dsg3 and Dsc3 in skin tumorigenesis. Dsg3 depletion inhibited tumor growth in H-Ras V15/p53−/−-transformed allografts, whereas Dsc3 depletion increased incidence of K-Ras transformed skin tumors (45, 48). The mechanism of Dsc3 tumor suppression is not clear; however, Dsg3-mediated cell migration and invasion in SCC were attributed to c-Jun/AP-1 activation and PKC-mediated ezrin phosphorylation (130).
In colon cancer cells, a loss of Dsc2 expression was accompanied by an increase in Dsc1 and Dsc3 expression, two Dscs not normally expressed in simple epithelia (131). Reportedly, the suppression of Dsc2 in colon cancer can be attributed to the loss of the homeobox transcription factors Cdx1 and Cdx2, common regulators of intestinal differentiation. Dsc2 loss was reported to result in increased proliferation through the activation of Akt/β-catenin signaling (55, 132). Distinct roles for desmosomal cadherin isoforms in tumorgenicity are also observed in lung cancer, where Dsg2 expression promoted proliferation through the upregulation of p27Kip1 and CDK2, while Dsc3 expression inactivated EGFR/ERK signaling and inhibited lung cancer growth (133, 134).
The signaling roles of desmosomal cadherins likely contribute to changes in the tumor microenvironment. As previously mentioned, forced expression of Dsg2 in suprabasal mouse epidermis increases MAPK, PI3K, NFκB, and STAT3 signaling, predisposing the mice to skin tumor development (29). Overexpression of Dsg2 in SCC resulted in enhanced extracellular vesicle secretion enriched with the Dsg2 C-terminal fragment and mitogenic proteins. These extracellular vesicles were engulfed by fibroblasts to promote their proliferation, illustrating how overexpression of Dsg2 can modulate the tumor microenvironment (135). Interfering with normal intercellular communication between different cell types in epidermis through alterations in desmosomal cadherin expression could also contribute to cancer. For instance, depletion of Dsg1 in keratinocytes resulted in increased paracrine signaling and promoted melanocyte pagetoid behavior in 3D organotypic cocultures. The appearance of this early feature of melanoma suggests a possible role for keratinocyte Dsg1 in regulating melanocyte transformation (40).
Although Dsg2 exhibits protumorigenic activity and is upregulated in head and neck SCC and pulmonary SCC, Dsg2 is downregulated in colon, gastric, and pancreatic cancers (136–138). The explanation for this tissue-specific difference in Dsg2 tumorigenicity is unclear, but it should be noted that the antitumorigenic role of Dsg2 is observed in nonsquamous epithelia that do not normally express other Dsg isoforms. Interestingly, Dsg2 exhibits a Dsg1-like role of downregulating ERK activity to inhibit invasion in these nonsquamous epithelial cell types (129, 138). Differences in Dsg2 function in squamous versus cuboidal/columnar cells support the idea that desmosomal cadherins interface with signaling machinery in a cell-type-dependent manner. Elucidating how different cadherins engage pro- and antitumorigenic signaling pathways in different cellular contexts will be important if we are to understand the clinical relevance of protein expression changes, particularly as the cancer field moves closer toward personalized medicine approaches to therapy.
7. DESMOSOMAL CADHERINS’ RESPONSE TO STRESS
Desmosomal cadherins are subjected to multiple types of environmental extrinsic and disease-induced intrinsic stressors (Figure 4). Indeed, their appearance late in evolution coincident with the creation of more robust barriers in vertebrates prompted the proposal that desmosomal cadherins may serve as stress sensors (4). Here, we review the importance of both extrinsic and intrinsic stresses in regulating desmosomal cadherins.
Figure 4.

Intrinsic and extrinsic factors and desmosomal cadherin signaling events contributing to homeostasis and disease pathogenesis. The illustration shows select intrinsic (green) and extrinsic (blue) factors leading to Dsg signaling events (brown) that contribute to homeostasis and disease pathogenesis highlighted in this review. Specific upstream and downstream events for Dsg2 (a), Dsg1 (b), and Dsg3 (c) are within the boundaries indicated by the dashed lines. Downstream events that occur with normal Dsg expression are represented by a smooth box, while events that occur due from Dsg loss of function are represented by a rough, dashed box. The type of function of the intrinsic (green) and extrinsic (blue) factors is marked as activating (pointed arrow), inhibitory (blunt-end arrow), or cleavage (circle-end arrow). The downstream events are colored on the basis of the tissue type where the event was observed: skin (purple), heart (pink), and intestines (orange). The citations associated with the downstream events are as follows: (a, clockwise from top left) proliferation/barrier recovery (147), Dsm assembly (75), proliferation/survival (29), barrier function (53), loss of cell adhesion (54), IFN-β antiviral response (144), Wnt signaling (144), apoptosis (73, 148); (b, clockwise from top left) loss of cell adhesion (99, 143, 145), apoptosis of damaged cell (140), tanning response (40), inflammatory response (36, 151), delamination (65), differentiation (37–39), blistering (100–102), invasion (129); (c, clockwise) apoptosis (44, 142), blistering (100–102), membrane protrusion/cell motility/invasion (130), wound closure (43), proliferation (28, 48), AJ and Dsm assembly (77, 146). Abbreviations: AJ, adherens junction; Dsc, desmocollin; Dsg, desmoglein; Dsm, desmosome; ECD, extracellular domain; EGFR, epidermal growth factor receptor; ER, endoplasmic reticulum; ICD, intracellular domain; IFN, interferon; IL, interleukin; PG, plakoglobin; SERCA, sarco/ER Ca2+ ATPase; TNF, tumor necrosis factor; UV, ultraviolet.
7.1. Extrinsic Factors That Affect Desmosomal Cadherins
Ingested factors and compounds are the most common type of environmental stressor for the gastrointestinal tract. The naturally derived poison and chemotherapeutic agent camptothecin induced apoptosis in intestinal cells to promote caspase-3 cleavage of Dsg2 where the resulting intracellular fragment of Dsg2 sensitizes cells to undergo apoptosis (73). Environmental factors are one type of trigger for inflammatory bowel diseases (IBDs), such as ulcerative colitis and Crohn’s disease. In multiple cohort studies, Dsg2 is downregulated in IBD patients (139). Enteropathogenic Escherichia coli infection (EPEC) can also reduce Dsg2 and Dsc2 levels in vitro, hypothesized to occur by EspH toxin-dependent sequestration of RhoA, a small GTPase that is important for desmosome function (139).
Whereas toxins and certain ingested compounds are external stressors for the intestines, ultraviolet (UV) radiation is a major stressor for the epidermis. UV irradiation induced downregulation of Dsg1 and caspase-3 cleavage of its cytoplasmic tail, but did not affect Dsg3 or E-cadherin (140, 141). UV-mediated Dsg1 loss protected against apoptosis in the HaCaT keratinocyte cell line (140). Interestingly, Dsg1 depletion in primary epidermal keratinocytes stimulated melanocyte dendrite growth and pigment secretion, suggesting that acute downregulation of Dsg1 activates a protective tanning response (40). However, Dsg1 expression also enhanced the recovery of 3D organotypic epidermal cultures following UV exposure. Collectively these findings suggest that Dsg1 loss and expression might have protective effects during both the initial and recovery phases following UV exposure, respectively (140, 141). While loss of Dsg1 can be protective against apoptosis in damaged keratinocytes, Dsg3 has a distinct role in inhibiting apoptosis by suppressing p53 activation (44). The role of Dsg3 in apoptosis may have clinical relevance, as it has been proposed that PV autoantibodies induce apoptosis by targeting Dsg3 (142). Therefore, intrinsic factors can also promote disease pathogenesis through targeting desmosomal cadherins.
Bacterial and viral pathogens are another type of extrinsic factor that can alter desmosomal cadherin functions. For example, staphylococcal scalded skin syndrome results from Staphylococcus aureus producing exfoliative toxin A, which cleaves Dsg1’s extracellular domain to disrupt cell adhesion (143). Coxsackievirus B3 (CVB3) causes viral myocarditis and disrupts desmosome integrity by cleaving Dsg2 and Dsc2 via CVB3 protease 3C and virus-activated cellular caspases. CVB3-mediated disruption of desmosomal cadherins in vitro resulted in the downregulation of plakoglobin and subsequent activation of Wnt/β-catenin signaling and downregulation of the interferon-β-mediated antiviral immune response (144). Thus, pathogens have developed mechanisms to target desmosomal cadherins to disrupt cell adhesion, alter cellular signaling, and enhance pathogenicity.
7.2. Intrinsic Factors That Regulate and Are Regulated by Desmosomal Cadherins
Certain skin diseases indirectly affect desmosomal cadherins via upregulation of proteases and endoplasmic reticulum (ER) stress. Netherton syndrome is a disorder caused by homozygous loss-of-function mutations affecting the serine protease inhibitor LEKTI. Loss of LEKTI results in increased activity of kallikrein proteases KLK5/7/14 and consequent cleavage of Dsg1/Dsc1 and corneodesmosin, an adhesive protein in modified so-called corneodesmosomes of the cornified layer (145). Altogether, these cleavage events result in a defective skin barrier.
Inactivating mutations in SERCA2, a sarco/ER Ca2+-ATPase pump, result in Darier’s disease, a disease characterized by a disruption in desmosomal cadherins and E-cadherin localization resulting in the appearance of rough, peeling skin papules (146). Proposed pathomechanisms for loss of cell–cell adhesion in Darier’s disease patients are impaired trafficking of desmoplakin and protein misfolding and ER retention of Dsg3, Dsc3, and E-cadherin. Consistent with a possible role for protein misfolding in the disorder, a pharmacological chaperone, miglustat, was reported to rescue AJ and desmosome formation.
Inflammation is another intrinsic factor that can alter desmosomal cadherin expression and/or function. For example, addition of the proinflammatory cytokines tumor necrosis factor alpha (TNF-α) and interferon gamma (IFN-γ) to intestinal epithelial cells promoted MMP9 and ADAM10-mediated extracellular cleavage of Dsg2 and activated death receptors such as TNFR to promote caspase-8 intracellular cleavage of Dsg2 (147, 148). Extracellular cleavage of Dsg2 was shown to induce HER2/3–mediated cell proliferation of neighboring cells, promoting epithelial barrier recovery during intestinal regeneration, while the subsequent intracellular cleavage of Dsg2 promoted proapoptosis signaling downstream of TNF-α and IFN-γ activation of death receptors (147, 148). This proinflammatory regulation of Dsg2 is clinically relevant since TNF-α is a central cytokine contributing to Crohn’s disease pathogenesis. TNF-α has been proposed to cause p38 MAPK–mediated downregulation of Dsg2 to increase intestinal permeability in vitro (149). A protective role for Dsg2 in the setting of intestinal injury is also supported by in vivo IBD and EPEC mouse model studies in which intestinal specific depletion of Dsg2 had increased susceptibility to dextran sodium sulfate colitis and Citrobacter rodentium–mediated infective colitis (49).
As discussed above, Dsg1 also engages in crosstalk with inflammatory response pathways. While inherited loss-of-function mutations in Dsg1 are associated with a Th17 response, Dsg1 expression can also be dramatically reduced in the skin of patients with more common complex inflammatory disorders including psoriasis, a skin disorder associated with an increased Th17 immune signature (36, 150). Interestingly, Cx43 loss occurred in the Dsg1-deficient regions of psoriatic skin, similar to its observed loss in SAM syndrome (36). These observations raise the possibility that loss of Dsg1 contributes to psoriasis pathogenicity. Additionally, specific downregulation of Dsg1 by the inflammatory cytokine IL-13 is proposed to contribute to eosinophilic esophagitis pathogenicity (151). Loss of Dsg1 may create a feed-forward loop by promoting an increase in the Th2 inflammatory gene signature associated with this disorder, which in turn could further downregulate Dsg1 (151).
Overall, multiple extrinsic and intrinsic factors impinge upon the desmosome by targeting desmosomal cadherins, resulting in their modulation or loss of function. In some cases, desmosomal cadherin loss is associated with a protective response, as in the case of UV-mediated loss of keratinocyte Dsg1, which may trigger a paracrine tanning response in melanocytes. In this regard, it is important to note that Dsg1 first appeared in terrestrial organisms, which are particularly susceptible to UV-mediated skin damage (40). Thus, new desmosomal cadherins may have been an evolutionary innovation to help vertebrates meet the challenges of a terrestrial existence.
8. CONCLUDING REMARKS
This review highlights the importance of a class of cadherins that became increasingly complex during evolution and comprise more than 10 protein isoforms with differing adhesion-dependent and supra-adhesive roles in terrestrial vertebrates. These adhesion molecules not only have cell autonomous roles in the epithelia and heart but also interact with the immune system and govern paracrine signaling to other cell types in response to environmental stresses. Thus, desmosomal cadherins have both structural and signaling functions that must be properly integrated to function efficiently in the body’s response to protect against stressors.
The functions of the desmosomal cadherins are tissue and differentiation dependent, underscoring the importance of their expression patterns. In spite of the importance of these patterns, it is unknown how desmosomal cadherin expression is regulated in different tissues. It is also not well understood how desmosomal cadherins affect each other’s function. This holds true of Dsgs and Dscs: Both are present in the same desmosomes, but how they work together as a functional whole is still a mystery. It also holds true of the different isoforms of the same subclass, which are coexpressed in complex tissues and are often present in the same junction. Additional information is needed to understand how desmosomal cadherins coordinate to promote and maintain their distinct and tissue-specific functions.
Desmosomal cadherins are targets of intrinsic and extrinsic factors that contribute to epithelial and heart disease by altering cadherin expression, turnover, and/or function to promote pathogenicity. Understanding how desmosomal cadherins, their downstream signaling, and their connection to the surrounding tissue environment are impacted in these disorders will be essential for the development of therapeutic strategies for repairing and strengthening both physical and immune barriers of epithelia.
ACKNOWLEDGMENTS
We apologize to those authors whose original work we were not able to cite due to limitations on the number of references. We thank members of the Green lab, in particular Jennifer Koetsier and Drs. Lisa Godsel, Joshua Broussard, Hope Burks, and Quinn Roth-Carter, for their valuable feedback and discussion. Work in our laboratory is supported by National Institutes of Health grants R37AR043380, R01AR041836, and R01CA228196 and a grant from the Leo Foundation. Sophia A. Svoboda is supported by a gift from the Lee family. Marihan Hegazy was supported by T32 CA009560 and F31AR076188. Abbey L. Perl is supported by T32AR060710.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Niessen CM, Leckband D, Yap AS. 2011. Tissue organization by cadherin adhesion molecules: dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol. Rev 91:691–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris TJ, Tepass U. 2010. Adherens junctions: from molecules to morphogenesis. Nat. Rev. Mol. Cell Biol 11:502–14 [DOI] [PubMed] [Google Scholar]
- 3.Hulpiau P, Gul IS, van Roy F. 2013. New insights into the evolution of metazoan cadherins and catenins. Prog. Mol. Biol. Transl. Sci 116:71–94 [DOI] [PubMed] [Google Scholar]
- 4.Green KJ, Roth-Carter Q, Niessen CM, Nichols SA. 2020. Tracing the evolutionary origin of desmosomes. Curr. Biol 30:R535–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green KJ, Jaiganesh A, Broussard JA. 2019. Desmosomes: essential contributors to an integrated intercellular junction network. F1000Res 8:F1000 Faculty Rev-2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delva E, Tucker DK, Kowalczyk AP. 2009. The desmosome. Cold Spring Harb. Perspect. Biol 1:a002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Owen GR, Stokes DL. 2010. Exploring the nature of desmosomal cadherin associations in 3D. Dermatol. Res. Pract 2010:930401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vielmuth F, Spindler V, Waschke J. 2018. Atomic force microscopy provides new mechanistic insights into the pathogenesis of pemphigus. Front. Immunol 9:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harrison OJ, Brasch J, Lasso G, Katsamba PS, Ahlsen G, et al. 2016. Structural basis of adhesive binding by desmocollins and desmogleins. PNAS 113:7160–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stahley SN, Saito M, Faundez V, Koval M, Mattheyses AL, Kowalczyk AP. 2014. Desmosome assembly and disassembly are membrane raft-dependent. PLOS ONE 9:e87809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimmer SE, Kowalczyk AP. 2020. The desmosome as a model for lipid raft driven membrane domain organization. Biochim. Biophys. Acta Biomembr 1862:183329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis JD, Caldara AL, Zimmer SE, Stahley SN, Seybold A, et al. 2019. The desmosome is a mesoscale lipid raft-like membrane domain. Mol. Biol. Cell 30:1390–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallis S, Lloyd S, Wise I, Ireland G, Fleming TP, Garrod D. 2000. The α isoform of protein kinase C is involved in signaling the response of desmosomes to wounding in cultured epithelial cells. Mol. Biol. Cell 11:1077–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garrod D, Tabernero L. 2014. Hyper-adhesion: a unique property of desmosomes. Cell Commun. Adhes 21:249–56 [DOI] [PubMed] [Google Scholar]
- 15.Fuchs M, Sigmund AM, Waschke J, Vielmuth F. 2020. Desmosomal hyperadhesion is accompanied with enhanced binding strength of desmoglein 3 molecules. Biophys. J 119:1489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hobbs RP, Green KJ. 2012. Desmoplakin regulates desmosome hyperadhesion. J. Investig. Dermatol 132:482–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartle EI, Rao TC, Beggs RR, Dean WF, Urner TM, et al. 2020. Protein exchange is reduced in calcium-independent epithelial junctions. J. Cell Biol 219:e201906153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al-Jassar C, Bikker H, Overduin M, Chidgey M. 2013. Mechanistic basis of desmosome-targeted diseases. J. Mol. Biol 425:4006–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green KJ, Simpson CL. 2007. Desmosomes: new perspectives on a classic. J. Investig. Dermatol 127:2499–515 [DOI] [PubMed] [Google Scholar]
- 20.Najor NA. 2018. Desmosomes in human disease. Annu. Rev. Pathol. Mech. Dis 13:51–70 [DOI] [PubMed] [Google Scholar]
- 21.Hatzfeld M, Keil R, Magin TM. 2017. Desmosomes and intermediate filaments: their consequences for tissue mechanics. Cold Spring Harb. Perspect. Biol 9:a029157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berika M, Garrod D. 2014. Desmosomal adhesion in vivo. Cell Commun. Adhes 21:65–75 [DOI] [PubMed] [Google Scholar]
- 23.Eshkind L, Tian Q, Schmidt A, Franke WW, Windoffer R, Leube RE. 2002. Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur. J. Cell Biol 81:592–98 [DOI] [PubMed] [Google Scholar]
- 24.Den Z, Cheng X, Merched-Sauvage M, Koch PJ. 2006. Desmocollin 3 is required for pre-implantation development of the mouse embryo. J. Cell Sci 119:482–89 [DOI] [PubMed] [Google Scholar]
- 25.Lim HYG, Alvarez YD, Gasnier M, Wang Y, Tetlak P, et al. 2020. Keratins are asymmetrically inherited fate determinants in the mammalian embryo. Nature 585:404–9 [DOI] [PubMed] [Google Scholar]
- 26.Bharathan NK, Dickinson AJG. 2019. Desmoplakin is required for epidermal integrity and morphogenesis in the Xenopus laevis embryo. Dev. Biol 450:115–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garrod D, Chidgey M. 2008. Desmosome structure, composition and function. Biochim. Biophys. Acta 1778:572–87 [DOI] [PubMed] [Google Scholar]
- 28.Merritt AJ, Berika MY, Zhai W, Kirk SE, Ji B, et al. 2002. Suprabasal desmoglein 3 expression in the epidermis of transgenic mice results in hyperproliferation and abnormal differentiation. Mol. Cell. Biol 22:5846–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brennan D, Hu Y, Joubeh S, Choi YW, Whitaker-Menezes D, et al. 2007. Suprabasal Dsg2 expression in transgenic mouse skin confers a hyperproliferative and apoptosis-resistant phenotype to keratinocytes. J. Cell Sci 120:758–71 [DOI] [PubMed] [Google Scholar]
- 30.Hardman MJ, Liu K, Avilion AA, Merritt A, Brennan K, et al. 2005. Desmosomal cadherin misexpression alters β-catenin stability and epidermal differentiation. Mol. Cell. Biol 25:969–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elias PM, Matsuyoshi N, Wu H, Lin C, Wang ZH, et al. 2001. Desmoglein isoform distribution affects stratum corneum structure and function. J. Cell Biol 153:243–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chidgey M, Brakebusch C, Gustafsson E, Cruchley A, Hail C, et al. 2001. Mice lacking desmocollin 1 show epidermal fragility accompanied by barrier defects and abnormal differentiation. J. Cell Biol 155:821–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng X, Mihindukulasuriya K, Den Z, Kowalczyk AP, Calkins CC, et al. 2004. Assessment of splice variant-specific functions of desmocollin 1 in the skin. Mol. Cell. Biol 24:154–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henkler F, Strom M, Mathers K, Cordingley H, Sullivan K, King I. 2001. Trangenic misexpression of the differentiation-specific desmocollin isoform 1 in basal keratinocytes. J. Investig. Dermatol 116:144–49 [DOI] [PubMed] [Google Scholar]
- 35.Kugelmann D, Radeva MY, Spindler V, Waschke J. 2019. Desmoglein 1 deficiency causes lethal skin blistering. J. Investig. Dermatol 139:1596–99.e2 [DOI] [PubMed] [Google Scholar]
- 36.Godsel LM, Roth-Carter QR, Koetsier JL, Tsoi LC, Broussard JA, et al. 2020. Th17-skewed inflammation due to genetic deficiency of a cadherin stress sensor. bioRxiv 2020.12.01.406587 10.1101/2020.12.01.406587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Getsios S, Simpson CL, Kojima S, Harmon R, Sheu LJ, et al. 2009. Desmoglein 1–dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J. Cell Biol 185:1243–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harmon RM, Simpson CL, Johnson JL, Koetsier JL, Dubash AD, et al. 2013. Desmoglein-1/Erbin interaction suppresses ERK activation to support epidermal differentiation. J. Clin. Investig 123:1556–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Najor NA, Fitz GN, Koetsier JL, Godsel LM, Albrecht LV, et al. 2017. Epidermal growth factor receptor neddylation is regulated by a desmosomal-COP9 (constitutive photomorphogenesis 9) signalosome complex. eLife 6:e22599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arnette CR, Roth-Carter QR, Koetsier JL, Broussard JA, Burks HE, et al. 2020. Keratinocyte cadherin desmoglein 1 controls melanocyte behavior through paracrine signaling. Pigment Cell Melanoma Res 33:305–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, et al. 1997. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J. Cell Biol 137:1091–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J, Den Z, Koch PJ. 2008. Loss of desmocollin 3 in mice leads to epidermal blistering. J. Cell Sci 121:2844–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rotzer V, Hartlieb E, Winkler J, Walter E, Schlipp A, et al. 2016. Desmoglein 3-dependent signaling regulates keratinocyte migration and wound healing. J. Investig. Dermatol 136:301–10 [DOI] [PubMed] [Google Scholar]
- 44.Rehman A, Cai Y, Hunefeld C, Jedlickova H, Huang Y, et al. 2019. The desmosomal cadherin desmoglein-3 acts as a keratinocyte anti-stress protein via suppression of p53. Cell Death Dis 10:750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, O’Shea C, Fitzpatrick JE, Koster MI, Koch PJ. 2012. Loss of Desmocollin 3 in skin tumor development and progression. Mol. Carcinog 51:535–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanakawa Y, Matsuyoshi N, Stanley JR. 2002. Expression of desmoglein 1 compensates for genetic loss of desmoglein 3 in keratinocyte adhesion. J. Investig. Dermatol 119:27–31 [DOI] [PubMed] [Google Scholar]
- 47.Brennan D, Hu Y, Medhat W, Dowling A, Mahoney MG. 2010. Superficial Dsg2 expression limits epidermal blister formation mediated by pemphigus foliaceus antibodies and exfoliative toxins. Dermatol. Res. Pract 2010:410278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baron S, Hoang A, Vogel H, Attardi LD. 2012. Unimpaired skin carcinogenesis in Desmoglein 3 knockout mice. PLOS ONE 7:e50024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gross A, Pack LAP, Schacht GM, Kant S, Ungewiss H, et al. 2018. Desmoglein 2, but not desmocollin 2, protects intestinal epithelia from injury. Mucosal Immunol 11:1630–39 [DOI] [PubMed] [Google Scholar]
- 50.Raya-Sandino A, Luissint AC, Kusters DHM, Narayanan V, Flemming S, et al. 2021. Regulation of intestinal epithelial intercellular adhesion and barrier function by desmosomal cadherin desmocollin-2. Mol. Biol. Cell 32:753–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schlegel N, Meir M, Heupel WM, Holthofer B, Leube RE, Waschke J. 2010. Desmoglein 2-mediated adhesion is required for intestinal epithelial barrier integrity. Am. J. Physiol. Gastrointest. Liver Physiol 298:G774–83 [DOI] [PubMed] [Google Scholar]
- 52.Jiang K, Rankin CR, Nava P, Sumagin R, Kamekura R, et al. 2014. Galectin-3 regulates desmoglein-2 and intestinal epithelial intercellular adhesion. J. Biol. Chem 289:10510–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ungewiss H, Vielmuth F, Suzuki ST, Maiser A, Harz H, et al. 2017. Desmoglein 2 regulates the intestinal epithelial barrier via p38 mitogen-activated protein kinase. Sci. Rep 7:6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ungewiss H, Rotzer V, Meir M, Fey C, Diefenbacher M, et al. 2018. Dsg2 via Src-mediated transactivation shapes EGFR signaling towards cell adhesion. Cell. Mol. Life Sci 75:4251–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kolegraff K, Nava P, Helms MN, Parkos CA, Nusrat A. 2011. Loss of desmocollin-2 confers a tumorigenic phenotype to colonic epithelial cells through activation of Akt/β-catenin signaling. Mol. Biol. Cell 22:1121–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoorntje ET, Te Rijdt WP, James CA, Pilichou K, Basso C, et al. 2017. Arrhythmogenic cardiomyopathy: pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res 113:1521–31 [DOI] [PubMed] [Google Scholar]
- 57.Kant S, Holthofer B, Magin TM, Krusche CA, Leube RE. 2015. Desmoglein 2–dependent arrhythmogenic cardiomyopathy is caused by a loss of adhesive function. Circ. Cardiovasc. Genet 8:553–63 [DOI] [PubMed] [Google Scholar]
- 58.Bass-Zubek AE, Hobbs RP, Amargo EV, Garcia NJ, Hsieh SN, et al. 2008. Plakophilin 2: a critical scaffold for PKCα that regulates intercellular junction assembly. J. Cell Biol 181:605–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kam CY, Dubash AD, Magistrati E, Polo S, Satchell KJF, et al. 2018. Desmoplakin maintains gap junctions by inhibiting Ras/MAPK and lysosomal degradation of connexin-43. J. Cell Biol 217:3219–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Broussard JA, Yang R, Huang C, Nathamgari SSP, Beese AM, et al. 2017. The desmoplakin-intermediate filament linkage regulates cell mechanics. Mol. Biol. Cell 28:3156–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Baddam SR, Arsenovic PT, Narayanan V, Duggan NR, Mayer CR, et al. 2018. The desmosomal cadherin desmoglein-2 experiences mechanical tension as demonstrated by a FRET-based tension biosensor expressed in living cells. Cells 7:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Price AJ, Cost AL, Ungewiss H, Waschke J, Dunn AR, Grashoff C. 2018. Mechanical loading of desmosomes depends on the magnitude and orientation of external stress. Nat. Commun 9:5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Broussard JA, Jaiganesh A, Zarkoob H, Conway DE, Dunn AR, et al. 2020. Scaling up single-cell mechanics to multicellular tissues – the role of the intermediate filament–desmosome network. J. Cell Sci 133:jcs228031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Godsel LM, Hsieh SN, Amargo EV, Bass AE, Pascoe-McGillicuddy LT, et al. 2005. Desmoplakin assembly dynamics in four dimensions: multiple phases differentially regulated by intermediate filaments and actin. J. Cell Biol 171:1045–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nekrasova O, Harmon RM, Broussard JA, Koetsier JL, Godsel LM, et al. 2018. Desmosomal cadherin association with Tctex-1 and cortactin-Arp2/3 drives perijunctional actin polymerization to promote keratinocyte delamination. Nat. Commun 9:1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thomas M, Ladoux B, Toyama Y. 2020. Desmosomal junctions govern tissue integrity and actomyosin contractility in apoptotic cell extrusion. Curr. Biol 30:682–90.e5 [DOI] [PubMed] [Google Scholar]
- 67.Broussard JA, Koetsier JL, Hegazy M, Green KJ. 2021. Desmosomes polarize mechanical signaling to govern epidermal tissue form and function. bioRxiv 2020.01.21.914176 10.1101/2020.01.21.914176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uttagomol J, Ahmad US, Rehman A, Huang Y, Laly AC, et al. 2019. Evidence for the desmosomal cadherin desmoglein-3 in regulating YAP and phospho-YAP in keratinocyte responses to mechanical forces. Int. J. Mol. Sci 20:6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kottke MD, Delva E, Kowalczyk AP. 2006. The desmosome: cell science lessons from human diseases. J. Cell Sci 119:797–806 [DOI] [PubMed] [Google Scholar]
- 70.Delva E, Kowalczyk AP. 2009. Regulation of cadherin trafficking. Traffic 10:259–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.North AJ, Chidgey MA, Clarke JP, Bardsley WG, Garrod DR. 1996. Distinct desmocollin isoforms occur in the same desmosomes and show reciprocally graded distributions in bovine nasal epidermis. PNAS 93:7701–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stahley SN, Kowalczyk AP. 2015. Desmosomes in acquired disease. Cell Tissue Res 360:439–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nava P, Laukoetter MG, Hopkins AM, Laur O, Gerner-Smidt K, et al. 2007. Desmoglein-2: a novel regulator of apoptosis in the intestinal epithelium. Mol. Biol. Cell 18:4565–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Michels C, Buchta T, Bloch W, Krieg T, Niessen CM. 2009. Classical cadherins regulate desmosome formation. J. Investig. Dermatol 129:2072–75 [DOI] [PubMed] [Google Scholar]
- 75.Shafraz O, Rubsam M, Stahley SN, Caldara AL, Kowalczyk AP, et al. 2018. E-cadherin binds to desmoglein to facilitate desmosome assembly. eLife 7:e37629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Godsel LM, Dubash AD, Bass-Zubek AE, Amargo EV, Klessner JL, et al. 2010. Plakophilin 2 couples actomyosin remodeling to desmosomal plaque assembly via RhoA. Mol. Biol. Cell 21:2844–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tsang SM, Brown L, Lin K, Liu L, Piper K, et al. 2012. Non-junctional human desmoglein 3 acts as an upstream regulator of Src in E-cadherin adhesion, a pathway possibly involved in the pathogenesis of pemphigus vulgaris. J. Pathol 227:81–93 [DOI] [PubMed] [Google Scholar]
- 78.Moch M, Schwarz N, Windoffer R, Leube RE. 2020. The keratin-desmosome scaffold: pivotal role of desmosomes for keratin network morphogenesis. Cell. Mol. Life Sci 77:543–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nekrasova OE, Amargo EV, Smith WO, Chen J, Kreitzer GE, Green KJ. 2011. Desmosomal cadherins utilize distinct kinesins for assembly into desmosomes. J. Cell Biol 195:1185–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lechler T, Fuchs E. 2007. Desmoplakin: an unexpected regulator of microtubule organization in the epidermis. J. Cell Biol 176:147–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lewis L, Barrandon Y, Green H, Albrecht-Buehler G. 1987. The reorganization of microtubules and microfilaments in differentiating keratinocytes. Differentiation 36:228–33 [DOI] [PubMed] [Google Scholar]
- 82.Muroyama A, Lechler T. 2017. A transgenic toolkit for visualizing and perturbing microtubules reveals unexpected functions in the epidermis. eLife 6:e29834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mahanty S, Dakappa SS, Shariff R, Patel S, Swamy MM, et al. 2019. Keratinocyte differentiation promotes ER stress-dependent lysosome biogenesis. Cell Death Dis 10:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liwosz A, Lei T, Kukuruzinska MA. 2006. N-glycosylation affects the molecular organization and stability of E-cadherin junctions. J. Biol. Chem 281:23138–49 [DOI] [PubMed] [Google Scholar]
- 85.Zhao H, Liang Y, Xu Z, Wang L, Zhou F, et al. 2008. N-glycosylation affects the adhesive function of E-Cadherin through modifying the composition of adherens junctions (AJs) in human breast carcinoma cell line MDA-MB-435. J. Cell. Biochem 104:162–75 [DOI] [PubMed] [Google Scholar]
- 86.Jin SP, Chung JH. 2018. Inhibition of N-glycosylation by tunicamycin attenuates cell–cell adhesion via impaired desmosome formation in normal human epidermal keratinocytes. Biosci. Rep 38:BSR20171641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roberts BJ, Johnson KE, McGuinn KP, Saowapa J, Svoboda RA, et al. 2014. Palmitoylation of plakophilin is required for desmosome assembly. J. Cell Sci 127:3782–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Woodley KT, Collins MO. 2019. S-acylated Golga7b stabilises DHHC5 at the plasma membrane to regulate cell adhesion. EMBO Rep 20:e47472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Roberts BJ, Svoboda RA, Overmiller AM, Lewis JD, Kowalczyk AP, et al. 2016. Palmitoylation of desmoglein 2 is a regulator of assembly dynamics and protein turnover. J. Biol. Chem 291:24857–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thomason HA, Scothern A, McHarg S, Garrod DR. 2010. Desmosomes: adhesive strength and signalling in health and disease. Biochem. J 429:419–33 [DOI] [PubMed] [Google Scholar]
- 91.Bertocchi C, Vaman Rao M, Zaidel-Bar R. 2012. Regulation of adherens junction dynamics by phosphorylation switches. J. Signal. Transduct 2012:125295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Garrod DR, Fisher C, Smith A, Nie Z. 2008. Pervanadate stabilizes desmosomes. Cell Adhes. Migr 2:161–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Albrecht LV, Zhang L, Shabanowitz J, Purevjav E, Towbin JA, et al. 2015. GSK3- and PRMT-1–dependent modifications of desmoplakin control desmoplakin–cytoskeleton dynamics. J. Cell Biol 208:597–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kimura TE, Merritt AJ, Garrod DR. 2007. Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J. Investig. Dermatol 127:775–81 [DOI] [PubMed] [Google Scholar]
- 95.Lorch JH, Klessner J, Park JK, Getsios S, Wu YL, et al. 2004. Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J. Biol. Chem 279:37191–200 [DOI] [PubMed] [Google Scholar]
- 96.Devaux CA, Mezouar S, Mege JL. 2019. The E-cadherin cleavage associated to pathogenic bacteria infections can favor bacterial invasion and transmigration, dysregulation of the immune response and cancer induction in humans. Front. Microbiol 10:2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ponnuchamy B, Khalil RA. 2008. Role of ADAMs in endothelial cell permeability: cadherin shedding and leukocyte rolling. Circ. Res 102:1139–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Klessner JL, Desai BV, Amargo EV, Getsios S, Green KJ. 2009. EGFR and ADAMs cooperate to regulate shedding and endocytic trafficking of the desmosomal cadherin desmoglein 2. Mol. Biol. Cell 20:328–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jiang R, Shi Z, Johnson JJ, Liu Y, Stack MS. 2011. Kallikrein-5 promotes cleavage of desmoglein-1 and loss of cell-cell cohesion in oral squamous cell carcinoma. J. Biol. Chem 286:9127–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, et al. 2017. Pemphigus. Nat. Rev. Dis. Primers 3:17026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, et al. 2012. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLOS ONE 7:e50696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Walter E, Vielmuth F, Wanuske MT, Seifert M, Pollmann R, et al. 2019. Role of Dsg1- and Dsg3-mediated signaling in pemphigus autoantibody-induced loss of keratinocyte cohesion. Front. Immunol 10:1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sharma P, Mao X, Payne AS. 2007. Beyond steric hindrance: the role of adhesion signaling pathways in the pathogenesis of pemphigus. J. Dermatol. Sci 48:1–14 [DOI] [PubMed] [Google Scholar]
- 104.Sayar BS, Ruegg S, Schmidt E, Sibilia M, Siffert M, et al. 2014. EGFR inhibitors erlotinib and lapatinib ameliorate epidermal blistering in pemphigus vulgaris in a non-linear, V-shaped relationship. Exp. Dermatol 23:33–38 [DOI] [PubMed] [Google Scholar]
- 105.Mao X, Cho MJT, Ellebrecht CT, Mukherjee EM, Payne AS. 2017. Stat3 regulates desmoglein 3 transcription in epithelial keratinocytes. JCI Insight 2:e92253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kugelmann D, Rotzer V, Walter E, Egu DT, Fuchs MT, et al. 2019. Role of Src and cortactin in pemphigus skin blistering. Front. Immunol 10:626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Didona D, Maglie R, Eming R, Hertl M. 2019. Pemphigus: current and future therapeutic strategies. Front. Immunol 10:1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang HH, Liu CW, Li YC, Huang YC. 2015. Efficacy of rituximab for pemphigus: a systematic review and meta-analysis of different regimens. Acta Derm. Venereol 95:928–32 [DOI] [PubMed] [Google Scholar]
- 109.Stenson PD, Mort M, Ball EV, Chapman M, Evans K, et al. 2020. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum. Genet 139:1197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim JH, Kim SE, Park HS, Lee SH, Lee SE, Kim SC. 2019. A homozygous nonsense mutation in the DSG3 gene causes acantholytic blisters in the oral and laryngeal mucosa. J. Investig. Dermatol 139:1187–90 [DOI] [PubMed] [Google Scholar]
- 111.Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, et al. 2013. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat. Genet 45:1244–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Taiber S, Samuelov L, Mohamad J, Barak EC, Sarig O, et al. 2018. SAM syndrome is characterized by extensive phenotypic heterogeneity. Exp. Dermatol 27:787–90 [DOI] [PubMed] [Google Scholar]
- 113.Has C, Jakob T, He Y, Kiritsi D, Hausser I, Bruckner-Tuderman L. 2015. Loss of desmoglein 1 associated with palmoplantar keratoderma, dermatitis and multiple allergies. Br. J. Dermatol 172:257–61 [DOI] [PubMed] [Google Scholar]
- 114.McAleer MA, Pohler E, Smith FJ, Wilson NJ, Cole C, et al. 2015. Severe dermatitis, multiple allergies, and metabolic wasting syndrome caused by a novel mutation in the N-terminal plakin domain of desmoplakin. J. Allergy Clin. Immunol 136:1268–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cheng R, Yan M, Ni C, Zhang J, Li M, Yao Z. 2016. Report of Chinese family with severe dermatitis, multiple allergies and metabolic wasting syndrome caused by novel homozygous desmoglein-1 gene mutation. J. Dermatol 43:1201–4 [DOI] [PubMed] [Google Scholar]
- 116.Polivka L, Hadj-Rabia S, Bal E, Leclerc-Mercier S, Madrange M, et al. 2018. Epithelial barrier dysfunction in desmoglein-1 deficiency. J. Allergy Clin. Immunol 142:702–6.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Frommherz LH, Schempp CM, Has C. 2021. Secukinumab for the treatment of SAM syndrome associated with desmoglein-1 deficiency. Br. J. Dermatol 184:770–72 [DOI] [PubMed] [Google Scholar]
- 118.Paller AS, Czarnowicki T, Renert-Yuval Y, Holland K, Huynh T, et al. 2018. The spectrum of manifestations in desmoplakin gene (DSP) spectrin repeat 6 domain mutations: immunophenotyping and response to ustekinumab. J. Am. Acad. Dermatol 78:498–505.e2 [DOI] [PubMed] [Google Scholar]
- 119.Hernandez-Martin A, Kennedy-Batalla R, Canedo E, Bernaldo-de-Quiros E, Carazo-Gallego B, et al. 2019. Imbalance in T-helper 17 cells and targeted therapy in an infant with SAM-like syndrome. N. Engl. J. Med 381:2176–78 [DOI] [PubMed] [Google Scholar]
- 120.Cohen-Barak E, Godsel LM, Koetsier JL, Hegazy M, Kushnir-Grinbaum D, et al. 2020. The role of desmoglein 1 in gap junction turnover revealed through the study of SAM syndrome. J. Investig. Dermatol 140:556–67.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Patel DM, Dubash AD, Kreitzer G, Green KJ. 2014. Disease mutations in desmoplakin inhibit Cx43 membrane targeting mediated by desmoplakin–EB1 interactions. J. Cell Biol 206:779–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vimalanathan AK, Ehler E, Gehmlich K. 2018. Genetics of and pathogenic mechanisms in arrhythmogenic right ventricular cardiomyopathy. Biophys. Rev 10:973–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Addison J, Taylor MRG, Mestroni L. 2019. Genotype-phenotype correlations in ARVC: toward a precision medicine approach. Int. J. Cardiol 286:115–16 [DOI] [PubMed] [Google Scholar]
- 124.Schinner C, Erber BM, Yeruva S, Schlipp A, Rotzer V, et al. 2020. Stabilization of desmoglein-2 binding rescues arrhythmia in arrhythmogenic cardiomyopathy. JCI Insight 5:e130141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Spindler V, Rotzer V, Dehner C, Kempf B, Gliem M, et al. 2013. Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J. Clin. Investig 123:800–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Janiszewska M, Primi MC, Izard T. 2020. Cell adhesion in cancer: beyond the migration of single cells. J. Biol. Chem 295:2495–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wong MP, Cheang M, Yorida E, Coldman A, Gilks CB, et al. 2008. Loss of desmoglein 1 expression associated with worse prognosis in head and neck squamous cell carcinoma patients. Pathology 40:611–16 [DOI] [PubMed] [Google Scholar]
- 128.Dusek RL, Attardi LD. 2011. Desmosomes: new perpetrators in tumour suppression. Nat. Rev. Cancer 11:317–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Valenzuela-Iglesias A, Burks HE, Arnette CR, Yalamanchili A, Nekrasova O, et al. 2019. Desmoglein 1 regulates invadopodia by suppressing EGFR/Erk signaling in an Erbin-dependent manner. Mol. Cancer Res 17:1195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Brown L, Waseem A, Cruz IN, Szary J, Gunic E, et al. 2014. Desmoglein 3 promotes cancer cell migration and invasion by regulating activator protein 1 and protein kinase C-dependent-Ezrin activation. Oncogene 33:2363–74 [DOI] [PubMed] [Google Scholar]
- 131.Khan K, Hardy R, Haq A, Ogunbiyi O, Morton D, Chidgey M. 2006. Desmocollin switching in colorectal cancer. Br. J. Cancer 95:1367–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Funakoshi S, Ezaki T, Kong J, Guo RJ, Lynch JP. 2008. Repression of the desmocollin 2 gene expression in human colon cancer cells is relieved by the homeodomain transcription factors Cdx1 and Cdx2. Mol. Cancer Res 6:1478–90 [DOI] [PubMed] [Google Scholar]
- 133.Cai F, Zhu Q, Miao Y, Shen S, Su X, Shi Y. 2017. Desmoglein-2 is overexpressed in non-small cell lung cancer tissues and its knockdown suppresses NSCLC growth by regulation of p27 and CDK2. J. Cancer Res. Clin. Oncol 143:59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cui T, Chen Y, Yang L, Knosel T, Huber O, et al. 2012. The p53 target gene desmocollin 3 acts as a novel tumor suppressor through inhibiting EGFR/ERK pathway in human lung cancer. Carcinogenesis 33:2326–33 [DOI] [PubMed] [Google Scholar]
- 135.Overmiller AM, Pierluissi JA, Wermuth PJ, Sauma S, Martinez-Outschoorn U, et al. 2017. Desmoglein 2 modulates extracellular vesicle release from squamous cell carcinoma keratinocytes. FASEB J 31:3412–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang T, Gu X, Jia L, Guo J, Tang Q, et al. 2021. DSG2 expression is low in colon cancer and correlates with poor survival. BMC Gastroenterol 21:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Biedermann K, Vogelsang H, Becker I, Plaschke S, Siewert JR, et al. 2005. Desmoglein 2 is expressed abnormally rather than mutated in familial and sporadic gastric cancer. J. Pathol 207:199–206 [DOI] [PubMed] [Google Scholar]
- 138.Hutz K, Zeiler J, Sachs L, Ormanns S, Spindler V. 2017. Loss of desmoglein 2 promotes tumorigenic behavior in pancreatic cancer cells. Mol. Carcinog 56:1884–95 [DOI] [PubMed] [Google Scholar]
- 139.Schlegel N, Boerner K, Waschke J. 2021. Targeting desmosomal adhesion and signalling for intestinal barrier stabilization in inflammatory bowel diseases—lessons from experimental models and patients. Acta Physiol 231:e13492. [DOI] [PubMed] [Google Scholar]
- 140.Dusek RL, Getsios S, Chen F, Park JK, Amargo EV, et al. 2006. The differentiation-dependent desmosomal cadherin desmoglein 1 is a novel caspase-3 target that regulates apoptosis in keratinocytes. J. Biol. Chem 281:3614–24 [DOI] [PubMed] [Google Scholar]
- 141.Johnson JL, Koetsier JL, Sirico A, Agidi AT, Antonini D, et al. 2014. The desmosomal protein desmoglein 1 aids recovery of epidermal differentiation after acute UV light exposure. J. Investig. Dermatol 134:2154–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schmidt E, Waschke J. 2009. Apoptosis in pemphigus. Autoimmun. Rev 8:533–37 [DOI] [PubMed] [Google Scholar]
- 143.Amagai M 2003. Desmoglein as a target in autoimmunity and infection. J. Am. Acad. Dermatol 48:244–52 [DOI] [PubMed] [Google Scholar]
- 144.Zhao G, Zhang HM, Qiu Y, Ye X, Yang D. 2020. Cleavage of desmosomal cadherins promotes γ-catenin degradation and benefits Wnt signaling in coxsackievirus B3-induced destruction of cardiomyocytes. Front. Microbiol 11:767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Petrova E, Hovnanian A. 2020. Advances in understanding of Netherton syndrome and therapeutic implications. Expert Opin. Orphan Drugs 8:455–87 [Google Scholar]
- 146.Park K, Lee SE, Shin KO, Uchida Y. 2019. Insights into the role of endoplasmic reticulum stress in skin function and associated diseases. FEBS J 286:413–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kamekura R, Nava P, Feng M, Quiros M, Nishio H, et al. 2015. Inflammation-induced desmoglein-2 ectodomain shedding compromises the mucosal barrier. Mol. Biol. Cell 26:3165–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yulis M, Quiros M, Hilgarth R, Parkos CA, Nusrat A. 2018. Intracellular desmoglein-2 cleavage sensitizes epithelial cells to apoptosis in response to pro-inflammatory cytokines. Cell Death Dis 9:389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Spindler V, Meir M, Vigh B, Flemming S, Hutz K, et al. 2015. Loss of desmoglein 2 contributes to the pathogenesis of Crohn’s disease. Inflamm. Bowel Dis 21:2349–59 [DOI] [PubMed] [Google Scholar]
- 150.Li B, Huang L, Lv P, Li X, Liu G, et al. 2020. The role of Th17 cells in psoriasis. Immunol. Res 68:296–309 [DOI] [PubMed] [Google Scholar]
- 151.Sherrill JD, Kc K, Wu D, Djukic Z, Caldwell JM, et al. 2014. Desmoglein-1 regulates esophageal epithelial barrier function and immune responses in eosinophilic esophagitis. Mucosal Immunol 7:718–29 [DOI] [PMC free article] [PubMed] [Google Scholar]