

Abstract
Objective:
Insulin-like growth factor I (IGF-1) exerts pleiotropic effects including promotion of cellular growth, differentiation, survival, and anabolism. We have shown that systemic IGF-1 administration reduced atherosclerosis in Apoe−/− mice, and this effect was associated with a reduction in lesional macrophages and a decreased number of foam cells in the plaque. Almost all cell types secrete IGF-1, but the effect of macrophage-derived IGF-1 on the pathogenesis of atherosclerosis is poorly understood. We hypothesized that macrophage-derived IGF-1 will reduce atherosclerosis.
Approach and Results:
We created macrophage-specific IGF-1 overexpressing mice on an Apoe−/− background (MF-IGF1 mice). Macrophage-specific IGF-1 overexpression reduced plaque macrophages, foam cells, and atherosclerotic burden and promoted features of stable atherosclerotic plaque. MF-IGF1 mice had a reduction in monocyte infiltration into plaque, decreased expression of CXC Chemokine Ligand 12 (CXCL12) and upregulation of ATP-Binding Cassette Transporter 1 (ABCA1), a cholesterol efflux regulator, in atherosclerotic plaque and in peritoneal macrophages. IGF-1 prevented oxidized lipid-induced CXCL12 upregulation and foam cell formation in cultured THP-1 macrophages and increased lipid efflux. We also found an increase in cholesterol efflux in MF-IGF1-derived peritoneal macrophages.
Conclusions:
Macrophage IGF-1 overexpression reduced atherosclerotic burden and increased features of plaque stability, likely via a reduction in CXCL12 mediated monocyte recruitment and an increase in ABCA1-dependent macrophage lipid efflux.
Keywords: Growth Factors/Cytokines, Inflammation, Lipids and Cholesterol, Genetically Altered and Transgenic Models, Atherosclerosis
INTRODUCTION
Cardiovascular disease (CVD) is the global leading cause of death and atherosclerosis is the primary cause of CVD1. Atherosclerosis is a chronic inflammatory disease2 in which monocytes and macrophages are the predominant cell types mediating the inflammatory response and disease progression3, 4. Oxidation of subendothelial low density lipoproteins leads to an inflammatory cascade resulting in recruitment of monocytes and proliferation of macrophages that secrete chemokines5 that cause more monocytes to be recruited to the lesion6, resulting in feed forward inflammation that promotes plaque progression7. Atherosclerotic plaque burden and the degree of macrophage infiltration are important contributors to plaque instability and the risk of plaque rupture, which underlies most acute coronary events8–10.
Although Insulin-like growth factor 1 (IGF-1) circulating levels peak at puberty, IGF-1 is continually and almost ubiquitously expressed throughout life11. It plays a critical role in normal growth and development12, 13. In addition to the endocrine effects exerted by liver-derived circulating IGF-1, there are many IGF-1 mediated effects that result from autocrine and paracrine mechanisms. Most cells express both IGF-1 and its receptor, Insulin-like growth factor 1 receptor (IGF1R)14. There is growing evidence that circulating IGF-1 levels are inversely related to the risk of CVD14–16. We have previously demonstrated that IGF-1 exerts anti-atherosclerotic effects in Apolipoprotein E deficient (Apoe−/−) mice, a murine model of atherosclerosis17–21. Systemic IGF-1 administration reduced atherogenesis and decreased plaque macrophages and foam cell formation20. The effects of increased circulating IGF-1 could be mediated by many cell types that express IGF1R, notably monocyte/macrophages, endothelial cells (EC), and smooth muscle cells (SMC). All these cell types secrete IGF-1 and the role of locally produced IGF-1 in the pathogenesis of atherosclerosis is poorly understood. Deletion of monocyte/macrophage-specific IGF1R increased atherogenesis, promoted a proinflammatory macrophage phenotype, increased monocyte recruitment to the plaque, and IGF1R-deficient macrophages had reduced lipid efflux, suggesting that monocyte/macrophage IGF1R signaling had anti-atherogenic effects17.
To determine the role of monocyte/macrophage-derived IGF-1 in atherogenesis, we generated an IGF-1-gain-of-function model specifically in macrophages and investigated potential mechanisms of IGF-1’s anti-atherogenic effects. Our findings indicate that macrophage-derived IGF-1 downregulates expression of the chemokine CXC chemokine ligand 12 (CXCL12), thereby lowering monocyte recruitment to plaques and lipid accumulation within macrophages via elevated lipid efflux. Our results demonstrate a novel link between IGF-1 and CXCL12 suggesting that CXCL12 could play a major role in the anti-atherosclerotic effects of macrophage-derived IGF-1.
MATERIALS AND METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request. Detailed Materials and Methods are available in the supplement.
Animals.
All animal experiments were approved by the Institutional Animal Care and Use Committees of the University of Missouri-Columbia and Tulane University. The rat IGF-1 gene (NC_005106.4) was cloned in a plasmid containing the human Scavenger Receptor-A (SRA) promoter cassette (plasmid kindly provided by Dr. Glass, UCSD22) and GFP cDNA to generate SRA-IGF-1-IRES-GFP vector. Southern blot23 confirmed IGF-1 gene inserts in the genome of founders (Suppl.Fig.I.A). Transgenic lines #12 and #17 were crossed with Apoe−/− mice (Jackson Laboratory #002052) to generate SRA-IGF-1/Apoe−/− mice. Non-transgenic Apoe−/− littermates were used as controls and male and female mice were used equally.
Laser capture microdissection (LCM).
Aortic valve sections were isolated and dissected as previously described24, 25.
Oil Red O assay.
Foam cell formation was assayed using human THP-1 mononuclear cells (ATCC TIB-202) as previously described26.
Isolation of peritoneal macrophages.
Peritoneal macrophages were isolated as previously described17.
Cholesterol Efflux Assay.
THP-1 cells were treated with IGF-1 (100ng/ml, 24 hours) and then IGF-1 and oxidized LDL (oxLDL, 50μg/ml, 24 hours) in serum free media (SFM). Efflux27 was measured according to manufacturer’s instructions (Sigma-Aldrich MAK192).
Isolation of monocytes.
Circulating and splenic monocytes were identified using antibodies from BioLegend as previously described17. Cell identity of monocytes was confirmed by assessment of cell markers (Table 1).
Table 1:
Cells were sorted via flow cytometry and RNA transcripts were measured to confirm monocyte selection by analyzing various cell markers.
| Marker | Type of Marker | Fold Chang to F4/80−/CD11b− Cells | SEM | P value |
|---|---|---|---|---|
| MSF-1 | Monocyte/Macrophage | 19.00 | 7.18 | 0.109 |
| Mac-1 | Monocyte/Macrophage | 47.22 | 13.68 | 0.044 |
| CD36 | Scavenger Receptor | 0.88 | 0.48 | 0.886 |
| Ly6G | Leukocyte | 8.55 | 4.96 | 0.297 |
| Ly6a | Lymphocyte | 0.10 | 0.04 | 0.394 |
| Cd3E | Lymphocyte | 0.10 | 0.05 | 0.224 |
| CD31 | Endothelial | 0.53 | 0.21 | 0.157 |
Monocyte recruitment into atherosclerotic plaque.
Mice were injected intravenously with fluorescent polychromatic red microspheres (Polyscience 19507–5) at 7 and 3 days before sacrifice28, 29.
Circulating Cell Quantification:
Circulating leukocytes and endothelial progenitor cells were quantified via FACs using modified previous gating strategies30–32.
Atherosclerosis quantification.
After 12 weeks of high fat diet, animals were sacrificed, and atherosclerosis was quantified as previously described21.
Atherosclerotic plaque composition.
Plaque composition was assessed by immunostaining of aortic valve cross-sections for Mac3, calponin, and α-smooth muscle actin. Masson’s Trichrome staining was used to quantify collagen. All IgG controls for immunohistochemistry data can be found in Supplementary Figure IX. Cell apoptosis was quantified with TUNEL-Fluorescein kit (Roche 11684795910) with co-staining for Mac3 as described previously20.
Immunoblotting analysis.
This was performed as described previously17. Antibodies for immunohistochemistry and immunoblotting are listed in the major resources table.
Quantitative real-time PCR.
Total RNA extraction and real-time PCR was performed as previously described17.
Statistical analysis.
All numeric data are expressed as mean±SEM. Statistical analyses were performed with GraphPad PRISM (version 8.0) software. Significant differences were determined by unpaired Student t- test with or without the Welch correction, or one-way ANOVA with either a Dunnett’s or Tukey’s post hoc test accordingly with the normality of residuals distribution or sample size. Fisher’s exact test was used to compare frequency distribution differences between groups. The exact test used is mentioned in every figure legend. Differences were considered significant at P<0.05. We declare that the design, execution, and reporting of the current study adheres to the guidelines for experimental atherosclerosis studies described and recommended by the American Heart Association, and we also considered sex as a biological variable as explained by the ATVB Council33, 34.
RESULTS
Generation of macrophage-specific IGF-1 overexpressing mice.
We obtained 13 founder lines in which IGF-1 is overexpressed under the control of the SRA promoter (Suppl.Fig.I.A). Although we utilized internal ribosomal entry site (IRES) mediated bicistronic gene expression to label rat IGF-1 expressing cells by GFP, we failed to observe any GFP positive cells in macrophages from these animals both in vitro and in vivo (data not shown), likely due to the lower expression of a gene downstream of IRES as described previously35. We selected lines #12 and #17 because they were the two founder lines that had multiple tandem transgene copies and were successful breeders (Suppl.Fig.I.B). Because mature rat and mouse IGF-1 differ by only one amino acid36, we expected that IGF-1 signaling would be the same whether using rat or mouse IGF-1. Indeed, IGF-1 signaling (as measured by comparing pAKT/Akt levels in peritoneal macrophages) did not change when either mouse or rat recombinant IGF-1 (0–20 ng/mL) was added to cells (Suppl.Fig.I.C). We also confirmed that insertion of the IGF-1 transgene did not disrupt the CXCL12 gene by amplifying the entire CXCL12 sequence and then amplifying regions within that amplicon using nested PCR (Suppl.Fig.I.D.).
Since mouse and rat mature IGF-1 protein are almost identical36 and exert similar signaling, we measured total IGF-1 levels in all experiments. In almost all mouse models of IGF-1 overexpression, a rat IGF-1 construct was used18, 37–40, and these models do not note any difference in signaling between endogenous and exogenous IGF-1. We found no difference in tissue and serum IGF-1 levels in the #12 and #17 transgenics versus control Apoe−/− mice (Fig.1.A,B). We also did not find a difference in SMC IGF-1 expression between groups (Control 3.0±1.4 pg/ml, MF-IGF1 1.4±0.8 pg/ml, P=NS). However, IGF-1 levels were significantly increased in the conditioned media of peritoneal macrophages isolated from the #17 line (1.8-fold increase) while there was no significant increase in IGF-1 levels in the #12 line (Fig.1.C). After confirming IGF-1 gene overexpression and increased intracellular IGF-1 protein in peritoneal macrophage cell lysates (Fig.1.D, Suppl.Fig.II.A), the #17 transgenic line was chosen for experiments outlined in the current report (henceforth referred to as MF-IGF1 mice). Starting at 8 weeks of age, animals were fed with a high-fat diet for 12 weeks to accelerate atherosclerosis development.
Figure 1: Macrophage-specific IGF-1 overexpressing mice.
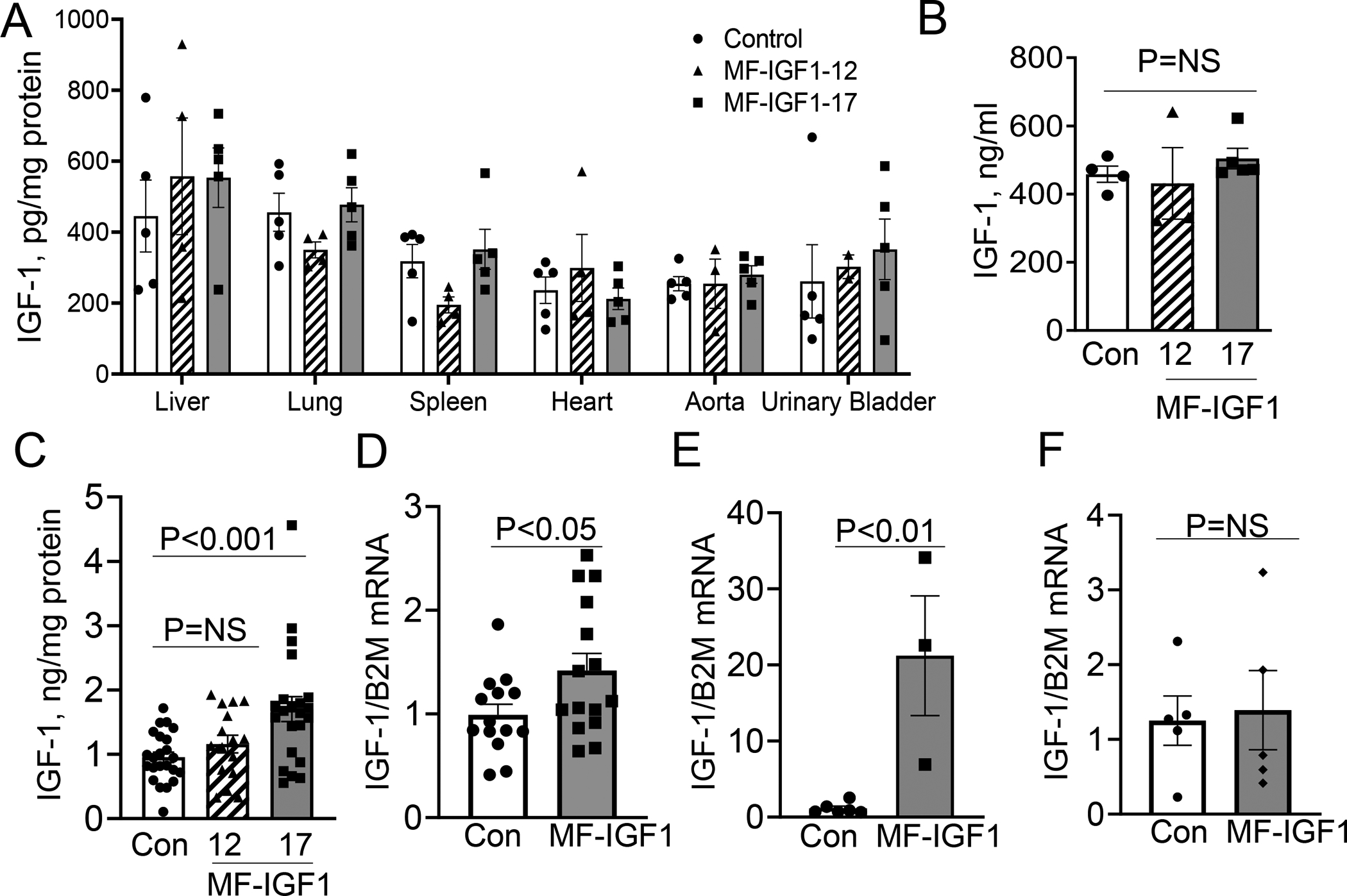
A. IGF-1 protein isolated from mice with macrophage-specific IGF-1 overexpression (#12 and #17 mice) and control (Apoe−/− mice) (N=4– 5 mice per group) showing no significant difference in any tissue. B. Serum IGF-1 levels measured by ELISA (N=4–5 mice per group). C. IGF-1 levels in conditioned media of peritoneal macrophages (N=22–25 mice per group). IGF-1 levels were normalized to total protein levels. D. IGF-1 mRNA levels in peritoneal macrophages in #17 (MF-IGF1) and control mice after 24 hours in SFM (N=12–14 mice per group). E. IGF-1 mRNA levels in plaque samples isolated by LCM in #17 (MF-IGF1) and control mice (N=3–6 mice per group). F. IGF-1 mRNA levels in circulating CD11b+/F4/80+ monocytes (N=5–6 mice per group). A, B, C used one-way ANOVA with a Tukey’s post-hoc test. All other statistical tests are Student’s two-tailed t-test.
To test whether atherosclerotic plaques in MF-IGF1 mice had increased IGF-1 gene expression, we isolated plaques by LCM (see methods) and quantified IGF-1 mRNA levels using primers designed to detect both mouse and rat Igf1 genes18. MF-IGF1 mice had an 18.4-fold-increase in total IGF-1 gene expression in LCM plaque isolates (Fig.1.E). We did not observe increased IGF-1 mRNA levels in circulating CD11b+/F4/80+ monocytes in MF-IGF1 mice, potentially due to low SRA promoter activity in this cell population (Fig.1.F). Taken together, these results strongly suggest that MF-IGF1 mice have a selective increase in macrophage IGF-1 levels.
IGF-1 binds with high affinity to the IGF1R and with lower affinity to the insulin receptor (InsR), although both can mediate intracellular signaling41. We found no difference in IGF1R or InsR expression in peritoneal macrophages isolated from MF-IGF1 mice compared to control mice (Suppl.Fig.II.B). Oxidized low density lipoprotein (oxLDL, 50μg/mL) treatment reduced IGF1R expression in peritoneal macrophages, but the effect was the same in control and MF-IGF1 mice (Suppl.Fig.II.C) which is consistent with the effect of oxLDL on IGF1R expression in smooth muscle cells (SMC)42 and human-derived macrophages (THP-1 cells)17. There was also no difference in IGF1R mRNA levels in circulating monocytes between MF-IGF1 mice and controls (Suppl.Fig.V.A). Under normal laboratory diet (PicoLab Rodent Diet 5053) feeding, both sexes of MF-IGF1 mice showed a small trend of increase in BW compared to control (14.5% increase, P=NS) at 8 weeks age. When fed with high-fat diet, MF-IGF1 mice showed higher body weight (average 24.1% increase) at weeks 10 to 18, whereas the difference was not significant at 20 weeks (Suppl.Fig.II.D). No difference was found in systolic blood pressure (Suppl.Fig.II.E), total cholesterol (Control 17.7±1.8 mmol/L, MF-IGF1 20.2±1.2 mmol/L, P=NS), free cholesterol levels (Control 12.7±2.1 mmol/L, MF-IGF1 15.4±1.1 mmol/L, P=NS), or triglyceride levels (Control 2.6±0.6 mmol/L, MF-IGF1 2.4±0.9 mmol/L, P=NS) between control and MF-IGF1 mice after twelve weeks of high fat diet. Neither cholesterol nor triglyceride levels differed between groups after four weeks of high fat diet (free cholesterol: Control 9.2±0.9 mmol/L, MF-IGF1 11.4±1.3 mmol/L, total cholesterol: Control 16.1±1.8 mmol/L, MF-IGF1, 19.0±1.8 mmol/L, triglycerides: control, 1.7±0.2 mmol/L, MF-IGF1 2.5±0.4 mmol/L, P=NS for all).
Macrophage-specific IGF-1 overexpression reduced atherosclerotic burden and changed atherosclerotic plaque composition toward a stable plaque phenotype.
Macrophage-specific IGF-1 overexpression significantly reduced Oil Red O-positive plaque area by 29.7% (Fig.2.A,C) as well as cross-sectional lesion area by 28.3% (Fig.2.B,D). Total aortic area was not different between MF-IGF1 and control mice (Control 262.4±30.3 mm2, MF-IGF1 268.1±32.6 mm2). Atherosclerotic plaques in MF-IGF1 mice had a significant 29.5% reduction in acellular/necrotic core area compared to control Apoe−/− mice (Fig.2.E). MF-IGF1 mice had a significant reduction in plaque macrophage levels (26.4% decrease in Mac3 immunopositivity, Fig.3.A,E), and the percentage of cells that co-stained for DAPI and Mac3 in the plaque was reduced by 30% in MF-IGF1 mice (Fig.3.F). No change was detected in plaque SMC levels as was quantified by immunohistochemistry for two SMC contractile proteins43, 44: calponin and α-smooth muscle actin (αSMA, Suppl.Fig.IV.A–D). The ability of IGF-1 to reduce atherosclerotic burden, necrotic core area, and macrophage content was not different between sexes (Suppl.Fig.III.A–C). MF-IGF1 mice had a near two-fold increase in plaque collagen (Trichrome staining, Fig.3.B,G) but this increase was statistically significant only in female mice (Suppl.Fig.III.D). Oil Red O is widely used to stain neutral lipids, including those present in lipid-laden (foam) cells in vitro and in vivo45–47. Atherosclerotic plaques in MF-IGF1 mice had a marked reduction in Oil Red O-positive area compared to control (60% reduction, Fig.3.C,H) indicating that macrophage-specific IGF-1 overexpression decreased lesional lipids. Furthermore, MF-IGF1 mice had reduced apoptosis specifically in plaque macrophages, as TUNEL+/Mac3+ cell numbers decreased by 55.4% (Fig.3.D,I), whereas there was no difference in TUNEL+/Mac3− cell number (Suppl.Fig.IV.E). Thus, macrophage-specific IGF-1 overexpression reduced atherosclerotic burden, decreased plaque macrophages, reduced foam cells, and did not change plaque SMC levels. The decrease in plaque macrophage accumulation and necrotic core area, together with the increase in plaque collagen levels10 suggested that macrophage IGF-1 overexpression promoted features of stable atherosclerotic plaque.
Figure 2: Macrophage-specific IGF-1 overexpression reduced atherosclerosis.
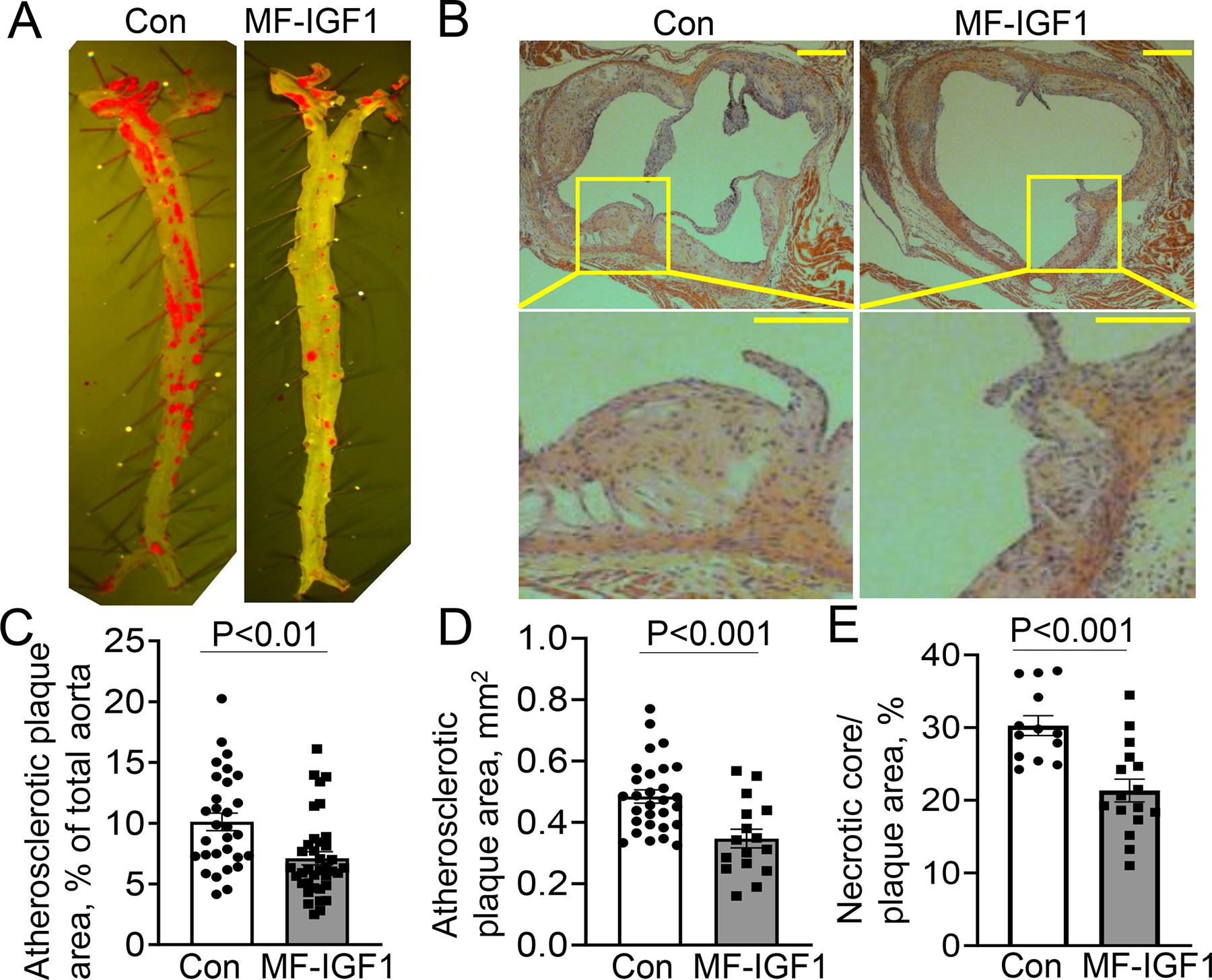
Macrophage-specific IGF-1 overexpressing (MF-IGF1 mice) and control mice (Con) were fed with a high-fat diet. A, C. Enface analysis of atherosclerotic burden (N=30–37 mice per group). B, D. H&E stained cross-sectional aortic valve sections to assess lesional area (N=16–29 mice per group). B Insert: magnified view of lesions showing that plaque in MF-IGF1 mice is less cellular. Scale bar, 100 μm. E. Necrotic core area in atherosclerotic plaque (N=14–15 mice per group). All statistical tests are Student’s two-tailed t-test.
Figure 3: Macrophage-specific IGF-1 overexpression changed atherosclerotic plaque toward a more stable plaque phenotype.
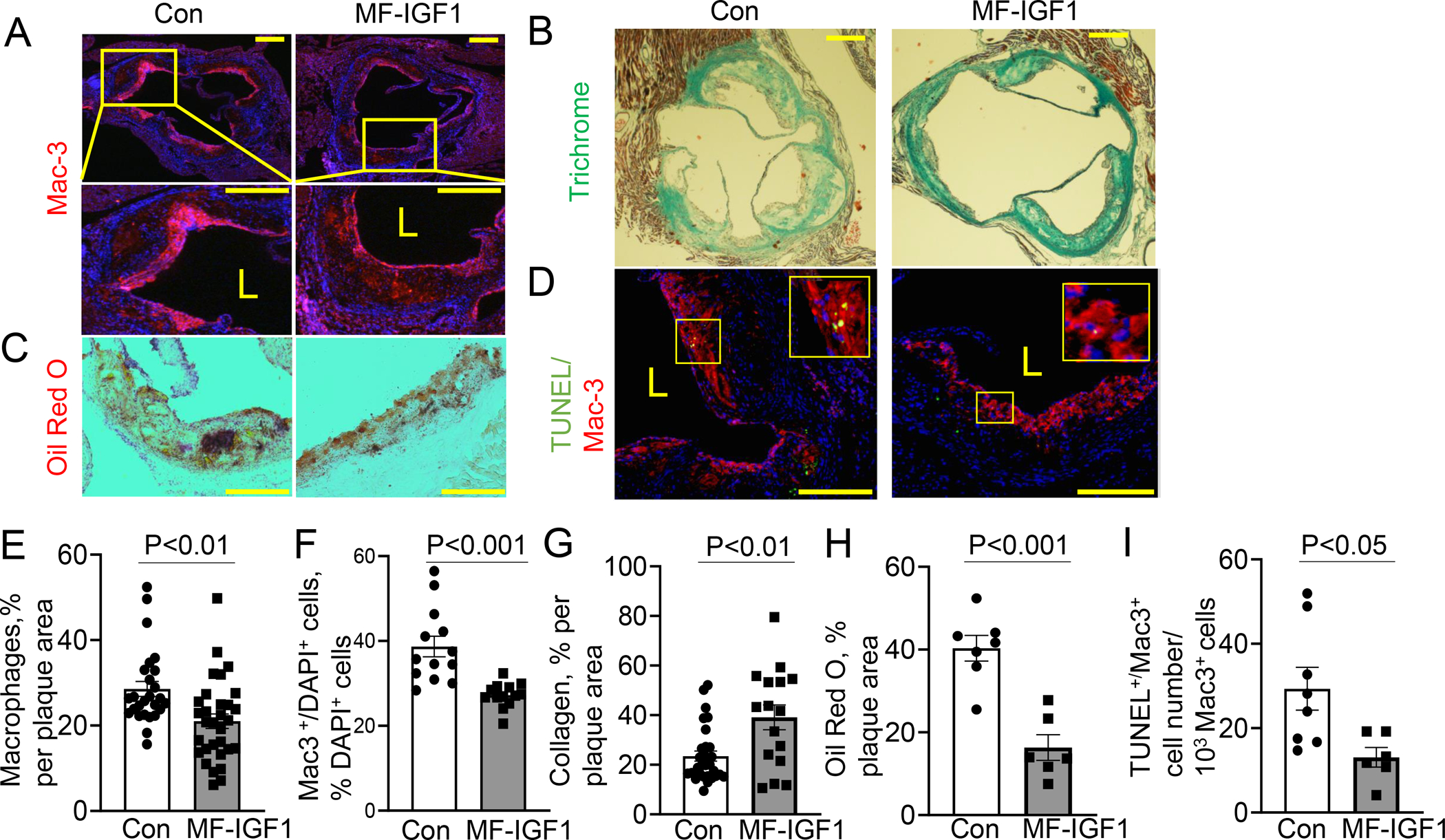
A, E Serial aortic valve cross-sections were stained with Mac3 antibody, (N=16–29 mice per group). B, G. Aortic valve section were stained with trichrome (N=16–29 mice per group). C, H. Snap-frozen aortic valve sections were stained with Oil Red O (N=6–7 mice per group). D, I. TUNEL assay co-stained with Mac3 antibody (N=5–6 mice per group). Data is normalized to 1000 Mac3+ cells. Insert showing TUNEL+/Mac3+-positive plaque cells. F. Mac3/DAPI double positive cell number was normalized to total number of DAPI positive cells in the plaque. (N=13 mice per group). Scale bar, 100 μm. All statistical tests are Student’s two-tailed t-test, except (G), which has a Welch’s correction to account for a difference in SDs. L=Lumen
Macrophage-specific IGF-1 overexpression reduced monocyte recruitment into atherosclerotic plaque and decreased CXCL12 chemokine levels.
As atherosclerosis develops, circulating monocytes infiltrate the plaque to become lesional macrophages, contributing to both formation of lipid-rich foam cells and to the physical bulk of developing plaques48. To quantify monocyte recruitment and infiltration into the atherosclerotic plaque we labeled circulating monocytes with red latex beads and tracked the persistence of this label in plaque49, 50. Red latex microspheres undergo specific uptake only by mononuclear cells28 and this methodology has been validated for experiments with atherosclerotic mice by our group17 and others28, 51. To assess the efficiency of monocyte labeling with red latex beads, we quantified the number of red bead-positive circulating and splenic monocytes in MF-IGF1 and control mice. We found that approximately 20% of circulating monocytes were red bead-positive and there was no difference in labeling efficiency of circulating and splenic monocytes in MF-IGF1 mice versus controls (Suppl.Fig.VI.A–C). We detected a similar number of splenic and circulating monocytes as a subset of all leukocytes (Suppl.Fig.VI.D,E) suggesting no change in monocytosis52. MF-IGF1 mice had a significant reduction in the number of red bead-positive cells in plaques compared to control (74.2% decrease, Fig.4.A,B) indicating that macrophage-specific IGF-1 overexpression reduced monocyte infiltration into atherosclerotic plaque. We also analyzed other leukocyte and endothelial progenitor cell (EPC) populations in the circulation using flow cytometry. There was no change in natural killer, B cell, or T cell (lymphoid-derived cells) populations, nor in neutrophil, eosinophil, or monocyte (myeloid-derived cells) populations, nor in EPC populations (Suppl.Fig.V.C–F).
Figure 4: Macrophage-specific IGF-1 overexpression reduced monocyte recruitment into atherosclerotic plaque and decreased CXCL12 chemokine expression.
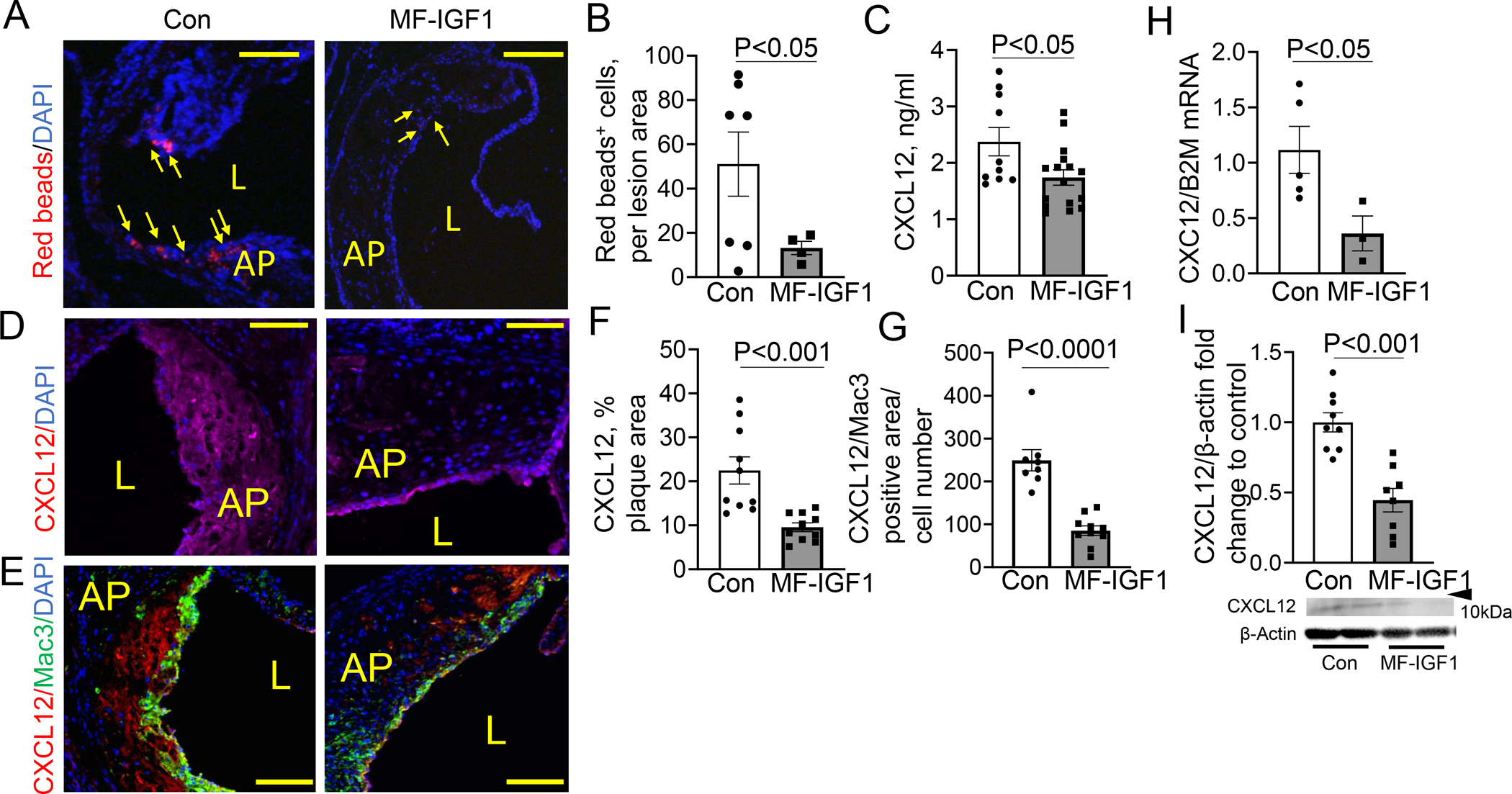
A, B. Monocyte recruitment was measured by quantification of plaque levels of red beads after normalization to plaque size. Arrows, plaque red spots (N=4–7 mice per group). C. Circulating levels of CXCL12 measured by ELISA (N=8–11 mice per group). D, F. CXCL12 positive area was normalized to plaque area (N=10 mice per group). E,G. CXCL12/Mac3 positive area was normalized to cell number (N=8–10 mice per group). H. CXCL12 mRNA levels in LCM plaque isolates (N=3–5 mice per group). I. CXCL12 levels in peritoneal macrophages were quantified by immunoblotting. Predicted weight 7–14kDa. (N=8–9 mice per group). Scale bar, 100 μm. All statistical tests are Student’s two-tailed t-test, except B, which had a Welch’s correction due to differences in SDs. AP=Atherosclerotic Plaque, L=lumen
Leukocytes are trafficked via involvement of chemokines/chemokine receptors, where a concentration gradient of a chemokine will lead a leukocyte to the chemokine’s source6. To investigate potential chemokine candidates mediating reduced monocyte infiltration into atherosclerotic plaque in MF-IGF1 mice and to determine potential changes in chemokines induced by macrophage-specific IGF-1 overexpression, we measured serum levels of 12 pro-atherogenic chemokines in MF-IGF1 and control mice via protein array (Table 2). Only CXCL12 levels were downregulated by macrophage-specific IGF-1 overexpression (P<0.0001). CXCL12, a heparin binding chemokine53 is highly expressed in macrophages, SMC, and ECs in atherosclerotic plaques but not in normal vessels54 and CXCL12 has been implicated in regulation of monocyte-macrophage differentiation55, macrophage-derived foam cell formation56, and in recruitment of macrophages to sites of injury57, and of monocytes to atherosclerotic plaque58.
Table 2.
Serum cytokines levels: Six animals per group were used to measure circulating cytokine levels. Units are arbitrary absorbance units.
| Control | MF-IGF1 | |||||
|---|---|---|---|---|---|---|
| Chemokine | Mean | SEM | Mean | SEM | Fold change | P value |
| CCL5 | 0.19 | 0.00 | 0.22 | 0.01 | 1.13 | 0.052 |
| CCL2 | 1.27 | 0.17 | 1.07 | 0.11 | 0.84 | 0.355 |
| CCL3 | 0.18 | 0.01 | 0.22 | 0.04 | 1.25 | 0.305 |
| CCL4 | 0.19 | 0.02 | 0.20 | 0.02 | 1.07 | 0.613 |
| CXCL12 | 3.27 | 0.15 | 1.56 | 0.18 | 0.48 | 0.00002 |
| IP-10 | 0.32 | 0.04 | 0.31 | 0.03 | 0.99 | 0.938 |
| MIG | 0.25 | 0.01 | 0.30 | 0.02 | 1.22 | 0.016 |
| Eotaxin | 1.76 | 0.04 | 2.08 | 0.16 | 1.18 | 0.083 |
| TARC | 0.34 | 0.03 | 0.37 | 0.02 | 1.07 | 0.485 |
| MDC | 0.33 | 0.03 | 0.44 | 0.03 | 1.34 | 0.017 |
| KC | 0.41 | 0.06 | 0.32 | 0.01 | 0.78 | 0.153 |
| 6CKine | 0.27 | 0.01 | 0.25 | 0.02 | 0.90 | 0.187 |
Downregulation of CXCL12 protein in serum of MF-IGF1 mice was confirmed by ELISA (26.7% reduction in MF-IGF1 mice compared to control, Fig.4.C). We detected strong CXCL12 immunopositivity in aortic valve plaque lesions in control mice (Fig.4.D,F) and CXCL12 signal overlapped with Mac3 positivity (Fig.4.E,G), suggesting that macrophages were the predominant source of CXCL12 in murine plaque. There was a marked reduction in CXCL12 staining in plaque lesions in MF-IGF1 mice (Fig.4.D–G) and a 5-fold decrease in CXCL12 mRNA levels in LCM-isolated plaque samples from MF-IGF1 mice compared to control (Fig.4.H). IGF-1 and CXCL12 mRNA levels in circulating monocytes in MF-IGF1 mice were not different from control (Fig.1.F, Suppl.Fig.V.B), whereas CXCL12 protein levels in peritoneal macrophages from MF-IGF1 mice were decreased compared to control (Fig.4.I). CXCL12 levels in conditioned media from peritoneal macrophages were not detectable (data not shown), likely because the majority of non-circulating CXCL12 is bound to the cellular membrane53, 59. Thus, macrophage-specific IGF-1 overexpression reduced monocyte infiltration into atherosclerotic plaque and downregulated foam cells, and these effects were associated with decreased circulating, plaque, and macrophage CXCL12 expression.
Macrophage-specific IGF-1 overexpression increased ABCA1 in atherosclerotic plaque and in peritoneal macrophages, with an accompanying increase in cholesterol efflux.
Mechanisms mediating the proatherogenic effects of CXCL12 are complex and include promotion of monocyte differentiation, recruitment of macrophages to injured endothelium, and enhancement of foam cell formation60, 61. Furthermore, CXCL12 is known to downregulate expression of the ATP-binding cassette transporter A1 (ABCA1) in macrophages, thereby inhibiting cholesterol efflux and contributing to its proatherogenic effects62. Because we found reduced macrophage CXCL12 expression and reduced foam cells in the plaques of MF-IGF1 mice, we hypothesized that ABCA1 is involved in macrophage-derived IGF-1’s effect on foam cells. ABCA1 gene expression was increased in LCM plaque isolates in MF-IGF-1 mice compared to control (Fig.5.A). ABCA1 protein expression was increased in peritoneal macrophages in MF-IGF1 mice compared to controls (Fig.5.B). Furthermore, blocking IGF-1 signaling with picropodophyllin (PPP) reduced ABCA1 protein levels to control levels in peritoneal macrophages from MF-IGF1 mice (Fig.5.C). Cholesterol efflux in peritoneal macrophages from MF-IGF1 mice was increased by 2.4-fold compared to control when using Apolipoprotein A1 (ApoA1) as the cholesterol acceptor, and unchanged when using High Density Lipoprotein (HDL) as the cholesterol acceptor (Fig.5.D). Taken together, these results suggest that macrophage overexpression of IGF-1, in addition to reducing monocyte recruitment and infiltration into plaque, also upregulates ABCA1 and increases cholesterol efflux, contributing to macrophage-derived IGF-1’s anti-atherogenic effect.
Figure 5: Macrophage-specific IGF-1 overexpression upregulated ABCA1 expression.
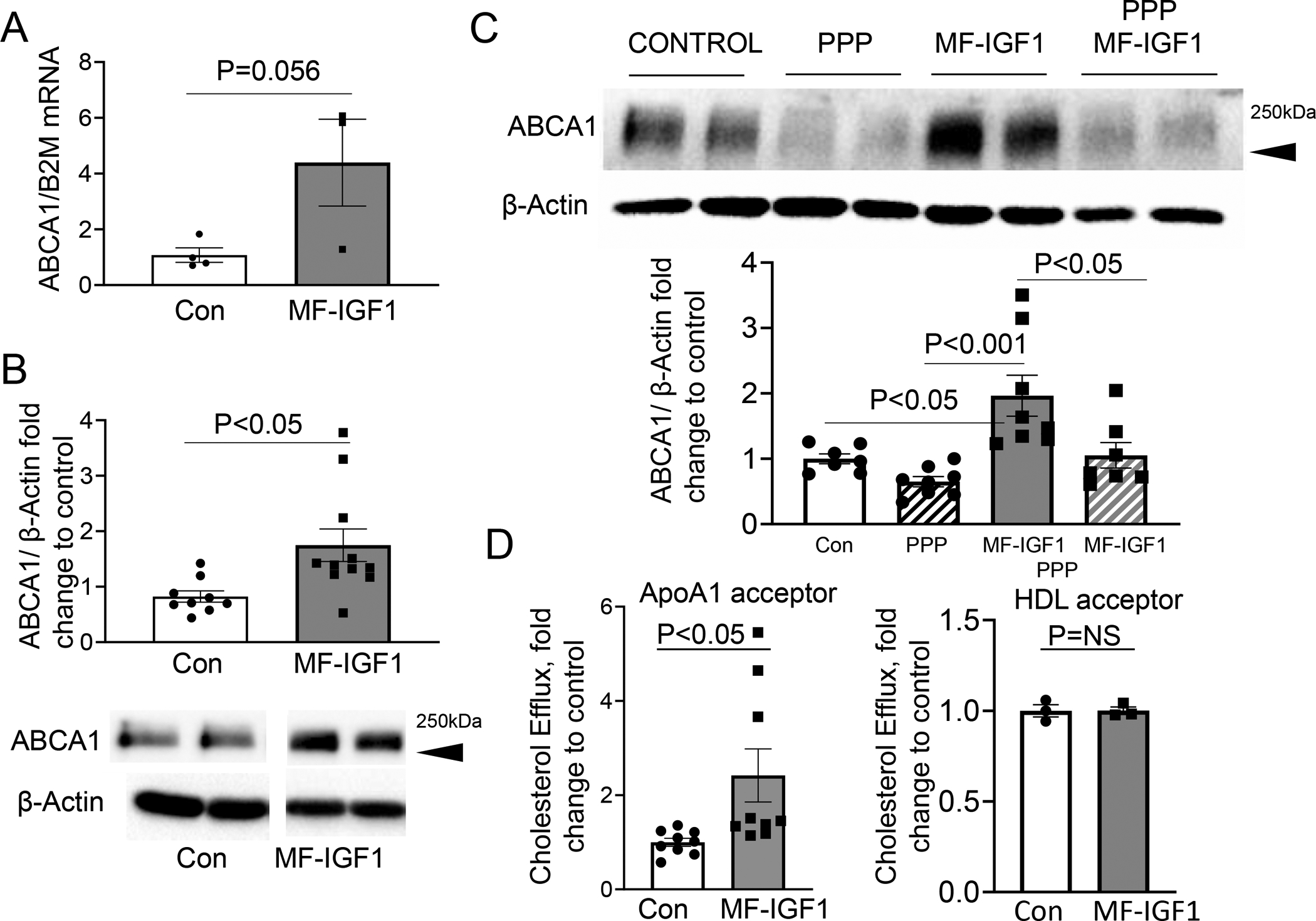
A. ABCA1 mRNA levels in LCM plaque isolates (N=3–5 mice per group). B. ABCA1 protein levels in peritoneal macrophages were quantified by immunoblotting. Cells were incubated in SFM for 48 hours. Predicted weight 254 kDa. (N=10–11 mice per group). C. Peritoneal macrophages were pretreated with PPP and ABCA1 protein levels were measured (N=3–4 wells per group in 2 independent experiments). D. Cholesterol efflux assay with ApoAI as an acceptor (N=3 wells per animal, 3 mice in each group) or with HDL as an acceptor. (N=1 well per animal, 3 mice per group). All statistical tests are Student’s two-tailed t-test.
IGF-1 increased THP-1 macrophage cholesterol efflux and reduced foam cell formation.
To determine whether IGF-1 reduces macrophage-derived foam cell formation, we used human THP-1 monocyte-derived macrophages20. Treatment with oxLDL induced a significant increase in CXCL12 mRNA levels compared to native LDL (ntLDL) treatment, and this increase was markedly and dose-dependently inhibited by IGF-1 pretreatment (Fig.6.A; 68.9% decrease with 100ng/ml IGF-1). Pretreatment with IGF-1 also reduced CXCL12 protein expression in oxLDL treated THP-1 cells (Suppl.Fig.VII.A; 56.2% decrease). IGF-1 (100ng/mL) increased ApoA1 mediated cholesterol efflux capacity in THP-1 cells when exposed to oxLDL (28% increase) while there was no change in HDL mediated efflux (Fig.6.B,C, Suppl.FigVII.C). THP-1 cells were pretreated with IGF-1 (0–100 ng/ml) and foam cell formation was induced by exposing cells to oxLDL or ntLDL as control. OxLDL caused an almost 3-fold increase in Oil Red O staining, and this increase was almost completely blunted by IGF-1 (Fig.6.D,E; 76.9% reduction in cells treated with 100ng/ml IGF-1). Importantly, when recombinant CXCL12 pretreatment (80μg/mL) was added to the cells, the IGF-1 mediated reduction in Oil Red O-positive area was completely abolished (Fig.6.F).
Figure 6: IGF-1 reduced formation of THP-1 macrophages-derived foam cell formation.
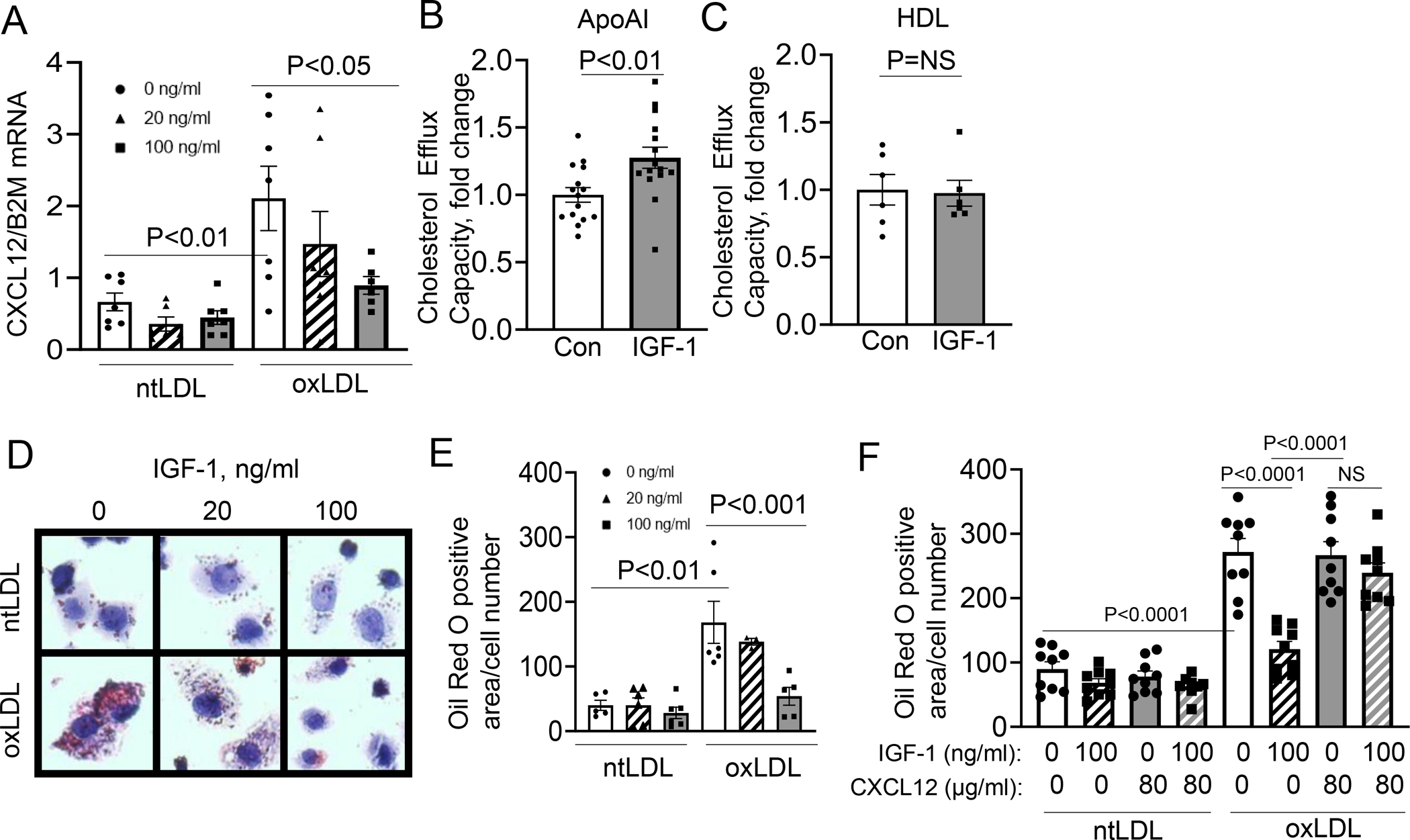
THP-1-derived macrophages were pretreated with IGF-1 and then treated with oxLDL or ntLDL. A. CXCL12 mRNA levels in THP-1 macrophages (N=3 wells per group per experiment, 3 independent experiments). B.10μg/ml ApoA1 was used as a cholesterol acceptor and cells were pretreated with IGF-1 then treated with oxLDL. Cholesterol efflux capacity was normalized to 0ng/ml IGF-1 treatment (Con). (N=4–7 wells per group per experiment, 3 independent experiments). C. 200μg/ml HDL was used as a cholesterol acceptor and cells were pretreated with IGF-1 then treated with oxLDL. Cholesterol efflux capacity was normalized to 0ng/ml IGF-1 treatment (Con). (N=3 wells per group per experiment, 2 independent experiments). D. Representative images of Oil Red O-stained macrophages. E. Quantitative data. (N=3 wells per group per experiment, 3 independent experiments). F. Cells were treated with IGF-1 and/or CXCL12 and then Oil Red O staining was used to quantify neutral lipids. (N=3 wells per group, 3 independent experiments). All statistics are one-way ANOVA except in D and E, which used a Student’s two-tail t-test. B used a Tukey’s post-hoc test, and C used Dunnett’s post -hoc test.
Discussion
Here we report that macrophage-specific IGF-1 overexpression suppressed atherosclerotic burden and promoted features of stable atherosclerotic plaque in Apoe−/− mice. MF-IGF1 transgenic animals had a reduction in total plaque burden in the aorta and in aortic root lesion size after 12 weeks on a high fat diet. Plaques in MF-IGF1 mice had reduced macrophage and foam cell content and necrotic core size, no change in SMC content, and an increase in collagen levels. MF-IGF1 mice had a reduction of monocyte recruitment and infiltration into the plaque and decreased expression of CXCL12 in the plaque, the circulation, and in peritoneal macrophages. Importantly, these mice had increased levels of the cholesterol efflux regulator, ABCA1, in atherosclerotic plaques and in peritoneal macrophages and an increase in ApoA1-dependent macrophage cholesterol efflux. IGF-1 prevented oxLDL-induced increase in CXCL12 expression in cultured macrophages and IGF-1 increased ApoA1-dependent cholesterol efflux. IGF-1 also reduced neutral lipid accumulation in THP-1 macrophages and this effect was blocked by exogenous CXCL12 treatment. Overall, our results indicate that macrophage IGF-1 overexpression downregulates macrophage CXCL12 expression, and that IGF-1 potentially exerts its atheroprotective effect via this mechanism.
To our knowledge, ours is the first report of an animal model of macrophage-specific IGF-1 overexpression. MF-IGF1 mice showed a marked increase in IGF-1 mRNA levels in atherosclerotic plaque, and peritoneal macrophages isolated from these mice secreted 80% more IGF-1 in conditioned medium. As the expression of SRA is selectively confined to macrophages (including splenic, peritoneal and other tissue macrophages63–65) and only minor SRA expression has been reported for freshly isolated monocytes65, 66, our observation of increased IGF-1 expression in both plaque and peritoneal macrophages but not in circulating monocytes in MF-IGF1 mice is consistent with the reported difference in SRA promoter activity in these cell types. SMC do not express SRA basally, but may express SRA upon dedifferentiation to a foam cell phenotype, although not to the same level as macrophages67–69. Of note, however, there was no increase in IGF-1 protein levels in aorta or other SMC-rich tissues like the bladder. Furthermore, we have previously shown that SMC-specific IGF-1 overexpression does not change atherosclerotic burden but does promote features of stable plaque18. MF-IGF1 mice initially gained more weight compared to control mice, although the difference was not significant after 12 weeks of high fat diet. One can speculate that since SRA expression is detected in bone marrow70 and IGF-1 in bone marrow regulates bone density37, 71–75, that perhaps MF-IGF1 mice have increased bone density, resulting in increased weight. Further studies will be required to address this hypothesis.
Macrophages are important for resolution of inflammation and for tissue repair in multiple tissues including the heart and skeletal muscle76–82, in some cases via IGF-1-dependent mechanisms. Indeed, macrophage-derived IGF-1 plays a role in regeneration of skeletal muscle82 and in protection against atrophy81. In atherosclerotic plaques, although macrophages may have anti-inflammatory effects, they are thought to play a critical role in plaque progression via secretion of proinflammatory cytokines, accumulation of lipoproteins to form foam cells, and via apoptosis and release of cellular debris promoting necrotic core formation83. The role of macrophage IGF-1 signaling in these processes was virtually unknown until the recent demonstration by our group that deletion of monocyte/macrophage IGF1R in Apoe−/− mice promoted a proinflammatory macrophage phenotype and increased atherosclerotic burden17, suggesting that IGF-1 could exert beneficial anti-inflammatory effects on macrophages in atherosclerotic plaques. Many of the effects on atherosclerosis that resulted from monocyte/macrophage-specific knockout of the IGF1R are mirrored in our current findings in this novel model of macrophage-specific IGF-1 overexpression. These include effects on plaque burden, plaque stability, recruitment of monocytes, plaque macrophage content, and lipid efflux. This suggests that the ability of macrophage-derived IGF-1 to exert these effects is primarily via autocrine mechanisms, although additional effects on surrounding cells such as SMC or ECs cannot be formally excluded. Furthermore, our current results uncover a novel mediator of these effects, CXCL12. Of note, in our prior model of monocyte/macrophage specific IGF1R knockout, CXCL12 levels were not assessed.
In addition to an overall reduction of plaque burden, we found that MF-IGF1 mice have plaques that are characterized by features of increased stability, namely increased collagen content and smaller necrotic cores. There is evidence that features of unstable plaque (such as necrotic core size, cellular composition, and collagen levels) are important determinants of adverse cardiovascular events, including acute coronary syndromes84. In more stable plaques, collagen can confer a thicker fibrotic cap, lessening the chance of rupture10. Our finding of increased collagen in the plaques of MF-IGF-1 mice could result from multiple mechanisms, including increased collagen synthesis or reduced collagen degradation by matrix metalloproteinases (MMPs), which are known to be highly expressed in plaque macrophages85. We have preliminary data that peritoneal macrophages in MF-IGF1 mice had a significant reduction in MMP8 and MMP14 expression (Suppl.Fig.VIII), suggesting that reduced collagen breakdown could contribute to their increase in collagen content.
Macrophage levels in the plaque are regulated via several mechanisms such as changes in monocyte recruitment, macrophage efferocytosis, proliferation, and/or apoptosis48. Plaque macrophage levels (as measured by Mac3 immunopositivity) were significantly reduced in MF-IGF1 mice. Mac3 been used as a specific macrophage marker in atherosclerosis studies86, but if SMC dedifferentiate into foam cells, they can also express the marker87–89, so it is possible that some cells we have considered as macrophages are actually phenotypically modified SMC. We hypothesized that the change in macrophage number was due to a reduction of monocyte recruitment. It is of note that it has been reported that a reduction in monocyte recruitment to plaque results in reduced macrophage content90, 91. We found a striking 70% reduction in the number of monocytes recruited to the plaques of MF-IGF1 mice. Chemotaxis of leukocytes is a key function of chemokines, and monocyte infiltration is regulated by chemokine secretion from multiple cell types28. Among the twelve circulating chemokines analyzed, only CXCL12 was reduced in MF-IGF1 mice. It is of note that two chemokines (T cell attractants MDC and Mig) were significantly, yet slightly, elevated so we cannot rule out a potential contribution of these chemokines to the phenotype of MF-IGF1 mice. CXCL12, originally discovered in the stromal cells of bone and to be responsible for homing and retention of hemopoietic stem and progenitor cells in the bone marrow57, has been recently implicated in atherosclerosis60, 62, 92–94. Atherosclerotic plaque CXCL12 mRNA and protein expression were significantly reduced in MF-IGF1 mice. Importantly, MF-IGF1 peritoneal macrophages also showed a reduction of CXCL12 expression. These results suggest that decreased CXCL12 expression in the lesions of MF-IGF1 mice caused reduced monocyte recruitment, leading to a lower number of macrophages in the plaque55. It is of note that CXCL12 expression is increased within human plaque compared to its near absence in normal arteries93. Most reports, but not all95, have indicated that CXCL12 is proatherogenic56, 58, 62, 96. This study suggests that CXCL12 is a pro-atherogenic molecule when the source is macrophages.
Mishandled lipid metabolism in plaque macrophages results in lipid accumulation and ultimately macrophage apoptosis, resulting in a less stable plaque with a large necrotic core8. CXCL12 reduces expression of ABCA162, the predominant lipid transporter mediating cholesterol efflux5, resulting in increased macrophage lipid accumulation and foam cell formation. We found increased ABCA1 mRNA levels in MF-IGF1 mouse plaques and increased ABCA1 protein in MF-IGF1 peritoneal macrophages compared to control. Of note, HDL can bind to a number of receptors such as CD36 to mediate cholesterol efflux5, whereas a component of HDL, ApoA1, specifically binds to ABCA1 to mediate efflux97. ApoA1/ABCA1 binding is considered the major cholesterol efflux pathway98. Our finding that IGF-1 increased ApoA1, but not HDL-dependent cholesterol efflux, is consistent with our data indicating that IGF-1 upregulates ABCA1 expression. Thus, our findings suggest that increased macrophage IGF-1 in MF-IGF1 mice increases ABCA1 expression in plaque, and this increase likely contributes to the reduction in lipid accumulation and foam cell formation.
It is important to note that IGF-1 and its receptor are downregulated in human atherosclerotic plaques and in SMC exposed to oxLDL42, 99. People with higher levels of bioavailable IGF-1 have a significant decrease in CVD14–16, 100. Results from the PRIME study showed that baseline IGF-1 is significantly lowered in patients that were developing an acute coronary syndrome100. A number of chemokines are involved in promoting atherogenesis101, including CXCL1260, 62, 92–94. Studies have implicated CXCL12 as a biomarker of heart failure and all around mortality102 and a genome wide association study identified CXCL12 as a potential therapeutic target in coronary disease92. Increased plasma CXCL12 due to genetic diversity has been linked to increased risk of coronary disease96. It is of note that Döring et al.58 found that only EC-derived CXCL12 contributes to atherogenesis. However, that study used a model of CXCL12 deficiency in bone marrow-derived hematopoietic cells which could have masked macrophage specific CXCL12 effects.
Our finding that an increase in macrophage IGF-1 reduces levels of CXCL12 in atherosclerotic plaques, the circulation, and macrophages establishes a potentially important mechanism whereby macrophage-derived IGF-1 can reduce atherosclerotic plaque and promote features of plaque stability. To our knowledge this is the first report of an animal model in which a local increase in plaque IGF-1 levels leads to a reduction in atherosclerosis. Our findings provide strong evidence that an increase in macrophage-derived IGF-1 may have significant anti-atherosclerotic effects and provide potential new therapeutic targets.
Supplementary Material
Highlights.
We have generated a novel mouse model that overexpresses IGF-1 in macrophages (MF-IGF1 mice).
These animals have reduced atherosclerosis and increased features of plaque stability.
These animals have reduced monocyte recruitment into the plaque.
These animals have reduced CXCL12 expression in plaque, serum, and peritoneal macrophages.
MF-IGF1 mice have increased macrophage cholesterol efflux.
IGF-1 treatment increases THP-1 macrophage cholesterol efflux.
IGF-1 treatment reduces Oil Red O-positive area in THP-1 macrophages and CXCL12 blocks IGF-1’s effect.
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Christopher Glass (University of California-San Diego) for the gift of SRA promoter-containing plasmid. We also thank Daniel Jackson for cell sorting services and Alexander Jurkevich for LCM training (University of Missouri at Columbia). This work was supported by funding from the NIH (R01HL070241, 3R01HL070241-16S1 PD and R01HL142796, SS) and the AHA (19TPA34850165 and 15SDG25240022, TY).
Non-standard Abbreviations and Acronyms
- αSMA
α-Smooth Muscle Actin
- ABCA1
ATP-Binding Cassette Transporter 1
- ApoA1
Apolipoprotein A 1
- Apoe−/−
Apolipoprotein E Deficient
- CVD
Cardiovascular Disease
- CXCL12
CXC Chemokine Ligand 12
- EC
Endothelial Cell
- EPC
Endothelial Progenitor Cell
- HDL
High Density Lipoprotein
- IGF-1
Insulin-Like Growth Factor-1
- IGF1R
Insulin-Like Growth Factor-1 Receptor
- InsR
Insulin Receptor
- LCM
Laser Capture Microdissection
- MF-IGF1
Macrophage-Specific Insulin-Like Growth Factor-1 Overexpressor (mouse model)
- ntLDL
Native Low Density Lipoprotein
- oxLDL
Oxidized Low Density Lipoprotein
- PPP
Picropodophyllin
- SFM
Serum Free Media
- SRA
Scavenger Receptor-A
Footnotes
DISCLOSURE
The authors have nothing to disclose.
SUPPLEMENTAL MATERIALS
REFERENCES
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–e360. [DOI] [PubMed] [Google Scholar]
- 2.Ross R Atherosclerosis is an inflammatory disease. American heart journal. 1999;138:S419–20. [DOI] [PubMed] [Google Scholar]
- 3.Gregersen I, Holm S, Dahl TB, Halvorsen B and Aukrust P. A focus on inflammation as a major risk factor for atherosclerotic cardiovascular diseases. Expert Review of Cardiovascular Therapy. 2016;14:391–403. [DOI] [PubMed] [Google Scholar]
- 4.Flynn MC, Pernes G, Lee MKS, Nagareddy PR and Murphy AJ. Monocytes, Macrophages, and Metabolic Disease in Atherosclerosis. Frontiers in Pharmacology. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yvan-Charvet L, Wang N and Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol. 2010;30:139–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin T, Xu X and Hereld D. Chemotaxis, chemokine receptors and human disease. Cytokine. 2008;44:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Randolph GJ. Mechanisms that regulate macrophage burden in atherosclerosis. Circ Res. 2014;114:1757–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otsuka F, Kramer MC, Woudstra P, Yahagi K, Ladich E, Finn AV, de Winter RJ, Kolodgie FD, Wight TN, Davis HR, Joner M and Virmani R. Natural progression of atherosclerosis from pathologic intimal thickening to late fibroatheroma in human coronary arteries: A pathology study. Atherosclerosis. 2015;241:772–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, Loke P and Fisher EA. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest. 2017;127:2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Virmani R, Burke AP, Kolodgie FD and Farb A. Pathology of the thin-cap fibroatheroma: a type of vulnerable plaque. J Interv Cardiol. 2003;16:267–72. [DOI] [PubMed] [Google Scholar]
- 11.Puche JE and Castilla-Cortazar I. Human conditions of insulin-like growth factor-I (IGF-I) deficiency. J Transl Med. 2012;10:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu JL, Grinberg A, Westphal H, Sauer B, Accili D, Karas M and LeRoith D. Insulin-like growth factor-I affects perinatal lethality and postnatal development in a gene dosage-dependent manner: manipulation using the Cre/loxP system in transgenic mice. Mol Endocrinol. 1998;12:1452–62. [DOI] [PubMed] [Google Scholar]
- 13.Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B and LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 1999;96:7324–7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higashi Y, Gautam S, Delafontaine P and Sukhanov S. IGF-1 and cardiovascular disease. Growth Horm IGF Res. 2019;45:6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juul A, Scheike T, Davidsen M, Gyllenborg J and Jørgensen T. Low Serum Insulin-Like Growth Factor I Is Associated With Increased Risk of Ischemic Heart Disease. Circulation. 2002;106:939–944. [DOI] [PubMed] [Google Scholar]
- 16.Laughlin GA, Barrett-Connor E, Criqui MH and Kritz-Silverstein D. The Prospective Association of Serum Insulin-Like Growth Factor I (IGF-I) and IGF-Binding Protein-1 Levels with All Cause and Cardiovascular Disease Mortality in Older Adults: The Rancho Bernardo Study. The Journal of Clinical Endocrinology & Metabolism. 2004;89:114–120. [DOI] [PubMed] [Google Scholar]
- 17.Higashi Y, Sukhanov S, Shai SY, Danchuk S, Tang R, Snarski P, Li Z, Lobelle-Rich P, Wang M, Wang D, Yu H, Korthuis R and Delafontaine P. Insulin-Like Growth Factor-1 Receptor Deficiency in Macrophages Accelerates Atherosclerosis and Induces an Unstable Plaque Phenotype in Apolipoprotein E-Deficient Mice. Circulation. 2016;133:2263–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shai SY, Sukhanov S, Higashi Y, Vaughn C, Kelly J and Delafontaine P. Smooth muscle cell-specific insulin-like growth factor-1 overexpression in Apoe−/− mice does not alter atherosclerotic plaque burden but increases features of plaque stability. Arterioscler Thromb Vasc Biol. 2010;30:1916–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shai SY, Sukhanov S, Higashi Y, Vaughn C, Rosen CJ and Delafontaine P. Low circulating insulin-like growth factor I increases atherosclerosis in ApoE-deficient mice. Am J Physiol Heart Circ Physiol. 2011;300:H1898–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sukhanov S, Snarski P, Vaughn C, Lobelle-Rich P, Kim C, Higashi Y, Shai SY and Delafontaine P. Insulin-like growth factor I reduces lipid oxidation and foam cell formation via downregulation of 12/15-lipoxygenase. Atherosclerosis. 2015;238:313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sukhanov S, Higashi Y, Shai S-Y, Vaughn C, Mohler J, Li Y, Song Y-H, Titterington J and Delafontaine P. IGF-1 Reduces Inflammatory Responses, Suppresses Oxidative Stress, and Decreases Atherosclerosis Progression in ApoE-Deficient Mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:2684–2690. [DOI] [PubMed] [Google Scholar]
- 22.Moulton KS, Semple K, Wu H and Glass CK. Cell-specific expression of the macrophage scavenger receptor gene is dependent on PU.1 and a composite AP-1/ets motif. Mol Cell Biol. 1994;14:4408–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Southern E Southern blotting. Nature Protocols. 2006;1:518–525. [DOI] [PubMed] [Google Scholar]
- 24.Sukhanov S, Higashi Y, Shai S-Y, Snarski P, Danchuk S, D’Ambra V, Tabony M, Woods TC, Hou X, Li Z, Ozoe A, Chandrasekar B, Takahashi S-I and Delafontaine P. SM22α (Smooth Muscle Protein 22-α) Promoter-Driven IGF1R (Insulin-Like Growth Factor 1 Receptor) Deficiency Promotes Atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2018;38:2306–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vink A, Schoneveld AH, Poppen M, de Kleijn DPV, Borst C and Pasterkamp G. Morphometric and immunohistochemical characterization of the intimal layer throughout the arterial system of elderly humans. J Anat. 2002;200:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Titterington JS, Sukhanov S, Higashi Y, Vaughn C, Bowers C and Delafontaine P. Growth hormone-releasing peptide-2 suppresses vascular oxidative stress in ApoE−/− mice but does not reduce atherosclerosis. Endocrinology. 2009;150:5478–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Low H, Hoang A and Sviridov D. Cholesterol efflux assay. J Vis Exp. 2012:e3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ and Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haka AS, Potteaux S, Fraser H, Randolph GJ and Maxfield FR. Quantitative Analysis of Monocyte Subpopulations in Murine Atherosclerotic Plaques by Multiphoton Microscopy. PLOS ONE. 2012;7:e44823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z, Gu Y, Shin A, Zhang S and Ginhoux F. Analysis of Myeloid Cells in Mouse Tissues with Flow Cytometry. STAR Protocols. 2020;1:100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Urbich C and Dimmeler S. Endothelial Progenitor Cells. Circulation Research. 2004;95:343–353. [DOI] [PubMed] [Google Scholar]
- 32.Becker CM, Beaudry P, Funakoshi T, Benny O, Zaslavsky A, Zurakowski D, Folkman J, D’Amato RJ and Ryeom S. Circulating endothelial progenitor cells are up-regulated in a mouse model of endometriosis. Am J Pathol. 2011;178:1782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinet P, Milewicz Dianna M, Cassis Lisa A, Leeper Nicholas J, Lu Hong S and Smith Jonathan D. Consideration of Sex Differences in Design and Reporting of Experimental Arterial Pathology Studies—Statement From ATVB Council. Arteriosclerosis, Thrombosis, and Vascular Biology. 2018;38:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daugherty A, Tall Alan R, Daemen Mat JAP, Falk E, Fisher Edward A, García-Cardeña G, Lusis Aldons J, Owens AP, Rosenfeld Michael E and Virmani R. Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement From the American Heart Association. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017;37:e131–e157. [DOI] [PubMed] [Google Scholar]
- 35.Mizuguchi H, Xu Z, Ishii-Watabe A, Uchida E and Hayakawa T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol Ther. 2000;1:376–82. [DOI] [PubMed] [Google Scholar]
- 36.Rotwein P Diversification of the insulin-like growth factor 1 gene in mammals. PloS one. 2017;12:e0189642–e0189642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, L Sweeney H and Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nature Genetics. 2001;27:195–200. [DOI] [PubMed] [Google Scholar]
- 38.Palazzolo I, Stack C, Kong L, Musaro A, Adachi H, Katsuno M, Sobue G, Taylor JP, Sumner CJ, Fischbeck KH and Pennuto M. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron. 2009;63:316–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elis S, Courtland HW, Wu Y, Rosen CJ, Sun H, Jepsen KJ, Majeska RJ and Yakar S. Elevated serum levels of IGF-1 are sufficient to establish normal body size and skeletal properties even in the absence of tissue IGF-1. J Bone Miner Res. 2010;25:1257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J, Niu W, Nikiforov Y, Naito S, Chernausek S, Witte D, LeRoith D, Strauch A and Fagin JA. Targeted overexpression of IGF-I evokes distinct patterns of organ remodeling in smooth muscle cell tissue beds of transgenic mice. J Clin Invest. 1997;100:1425–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stachelscheid H, Ibrahim H, Koch L, Schmitz A, Tscharntke M, Wunderlich FT, Scott J, Michels C, Wickenhauser C, Haase I, Brüning JC and Niessen CM. Epidermal insulin/IGF-1 signalling control interfollicular morphogenesis and proliferative potential through Rac activation. Embo j. 2008;27:2091–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scheidegger KJ, James RW and Delafontaine P. Differential effects of low density lipoproteins on insulin-like growth factor-1 (IGF-1) and IGF-1 receptor expression in vascular smooth muscle cells. J Biol Chem. 2000;275:26864–9. [DOI] [PubMed] [Google Scholar]
- 43.Skalli O, Pelte MF, Peclet MC, Gabbiani G, Gugliotta P, Bussolati G, Ravazzola M and Orci L. Alpha-smooth muscle actin, a differentiation marker of smooth muscle cells, is present in microfilamentous bundles of pericytes. J Histochem Cytochem. 1989;37:315–21. [DOI] [PubMed] [Google Scholar]
- 44.Miano JM and Olson EN. Expression of the Smooth Muscle Cell Calponin Gene Marks the Early Cardiac and Smooth Muscle Cell Lineages during Mouse Embryogenesis (∗). Journal of Biological Chemistry. 1996;271:7095–7103. [DOI] [PubMed] [Google Scholar]
- 45.Nunnari JJ, Zand T, Joris I and Majno G. Quantitation of oil red O staining of the aorta in hypercholesterolemic rats. Exp Mol Pathol. 1989;51:1–8. [DOI] [PubMed] [Google Scholar]
- 46.Conn’s biological stains : a handbook of dyes, stains and fluorochromes for use in biology and medicine. 10th ed. ed. Oxford: BIOS; 2002. [Google Scholar]
- 47.Mehlem A, Hagberg CE, Muhl L, Eriksson U and Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nature Protocols. 2013;8:1149–1154. [DOI] [PubMed] [Google Scholar]
- 48.Randolph GJ. Mechanisms that regulate macrophage burden in atherosclerosis. Circulation research. 2014;114:1757–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams JW, Martel C, Potteaux S, Esaulova E, Ingersoll MA, Elvington A, Saunders BT, Huang L-H, Habenicht AJ, Zinselmeyer BH and Randolph GJ. Limited Macrophage Positional Dynamics in Progressing or Regressing Murine Atherosclerotic Plaques—Brief Report. Arteriosclerosis, Thrombosis, and Vascular Biology. 2018;38:1702–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harmsen AG, Muggenburg BA, Snipes MB and Bice DE. The role of macrophages in particle translocation from lungs to lymph nodes. Science. 1985;230:1277–80. [DOI] [PubMed] [Google Scholar]
- 51.Tacke F, Ginhoux F, Jakubzick C, van Rooijen N, Merad M and Randolph GJ. Immature monocytes acquire antigens from other cells in the bone marrow and present them to T cells after maturing in the periphery. J Exp Med. 2006;203:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Combadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A and Mallat Z. Combined Inhibition of CCL2, CX3CR1, and CCR5 Abrogates Ly6Chi and Ly6Clo Monocytosis and Almost Abolishes Atherosclerosis in Hypercholesterolemic Mice. Circulation. 2008;117:1649–1657. [DOI] [PubMed] [Google Scholar]
- 53.Murphy JW, Cho Y, Sachpatzidis A, Fan C, Hodsdon ME and Lolis E. Structural and functional basis of CXCL12 (stromal cell-derived factor-1 alpha) binding to heparin. J Biol Chem. 2007;282:10018–10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abi-Younes S, Sauty A, Mach F, Sukhova GK, Libby P and Luster AD. The Stromal Cell–Derived Factor-1 Chemokine Is a Potent Platelet Agonist Highly Expressed in Atherosclerotic Plaques. Circulation Research. 2000;86:131–138. [DOI] [PubMed] [Google Scholar]
- 55.Sánchez-Martín L, Estecha A, Samaniego R, Sánchez-Ramón S, Vega MÁ and Sánchez-Mateos P. The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood. 2011;117:88–97. [DOI] [PubMed] [Google Scholar]
- 56.Chatterjee M, von Ungern-Sternberg SN, Seizer P, Schlegel F, Büttcher M, Sindhu NA, Müller S, Mack A and Gawaz M. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis. 2015;6:e1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim YH and Tabata Y. Recruitment of mesenchymal stem cells and macrophages by dual release of stromal cell-derived factor-1 and a macrophage recruitment agent enhances wound closure. J Biomed Mater Res A. 2016;104:942–56. [DOI] [PubMed] [Google Scholar]
- 58.Döring Y, van der Vorst EPC, Duchene J, Jansen Y, Gencer S, Bidzhekov K, Atzler D, Santovito D, Rader DJ, Saleheen D and Weber C. CXCL12 Derived From Endothelial Cells Promotes Atherosclerosis to Drive Coronary Artery Disease. Circulation. 2019;139:1338–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barinov A, Luo L, Gasse P, Meas-Yedid V, Donnadieu E, Arenzana-Seisdedos F and Vieira P. Essential role of immobilized chemokine CXCL12 in the regulation of the humoral immune response. Proceedings of the National Academy of Sciences. 2017;114:2319–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao J-H, Yu X-H and Tang C-K. CXC chemokine ligand 12 (CXCL12) in atherosclerosis: An underlying therapeutic target. Clinica Chimica Acta. 2019;495:538–544. [DOI] [PubMed] [Google Scholar]
- 61.Bakogiannis C, Sachse M, Stamatelopoulos K and Stellos K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine. 2019;122:154157. [DOI] [PubMed] [Google Scholar]
- 62.Gao JH, He LH, Yu XH, Zhao ZW, Wang G, Zou J, Wen FJ, Zhou L, Wan XJ, Zhang DW and Tang CK. CXCL12 promotes atherosclerosis by downregulating ABCA1 expression via the CXCR4/GSK3β/β-catenin(T120)/TCF21 pathway. J Lipid Res. 2019;60:2020–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borges da Silva H, Fonseca R, Pereira RM, Cassado AdA, Álvarez JM and D’Império Lima MR. Splenic Macrophage Subsets and Their Function during Blood-Borne Infections. Frontiers in Immunology. 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kelley JL, Ozment TR, Li C, Schweitzer JB and Williams DL. Scavenger receptor-A (CD204): a two-edged sword in health and disease. Crit Rev Immunol. 2014;34:241–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winther MPJd, Dijk KWv, Havekes LM and Hofker MH. Macrophage Scavenger Receptor Class A. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:290–297. [DOI] [PubMed] [Google Scholar]
- 66.Geng Y, Kodama T and Hansson GK. Differential expression of scavenger receptor isoforms during monocyte-macrophage differentiation and foam cell formation. Arterioscler Thromb. 1994;14:798–806. [DOI] [PubMed] [Google Scholar]
- 67.Yan P, Xia C, Duan C, Li S and Mei Z. Biological characteristics of foam cell formation in smooth muscle cells derived from bone marrow stem cells. Int J Biol Sci. 2011;7:937–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rong JX, Shapiro M, Trogan E and Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100:13531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Allahverdian S, Pannu PS and Francis GA. Contribution of monocyte-derived macrophages and smooth muscle cells to arterial foam cell formation. Cardiovascular Research. 2012;95:165–172. [DOI] [PubMed] [Google Scholar]
- 70.Lin Y-L, de Villiers WJS, Garvy B, Post SR, Nagy TR, Safadi FF, Faugere MC, Wang G, Malluche HH and Williams JP. The Effect of Class A Scavenger Receptor Deficiency in Bone. Journal of Biological Chemistry. 2007;282:4653–4660. [DOI] [PubMed] [Google Scholar]
- 71.Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, Ooi GT, Setser J, Frystyk J, Boisclair YR and LeRoith D. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest. 2002;110:771–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elis S, Wu Y, Courtland HW, Cannata D, Sun H, Beth-On M, Liu C, Jasper H, Domené H, Karabatas L, Guida C, Basta-Pljakic J, Cardoso L, Rosen CJ, Frystyk J and Yakar S. Unbound (bioavailable) IGF1 enhances somatic growth. Dis Model Mech. 2011;4:649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stratikopoulos E, Szabolcs M, Dragatsis I, Klinakis A and Efstratiadis A. The hormonal action of IGF1 in postnatal mouse growth. Proceedings of the National Academy of Sciences. 2008;105:19378–19383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kawai M and Rosen CJ. The insulin-like growth factor system in bone: basic and clinical implications. Endocrinol Metab Clin North Am. 2012;41:323–33, vi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao G, Monier-Faugere MC, Langub MC, Geng Z, Nakayama T, Pike JW, Chernausek SD, Rosen CJ, Donahue LR, Malluche HH, Fagin JA and Clemens TL. Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology. 2000;141:2674–82. [DOI] [PubMed] [Google Scholar]
- 76.Troidl C, Möllmann H, Nef H, Masseli F, Voss S, Szardien S, Willmer M, Rolf A, Rixe J, Troidl K, Kostin S, Hamm C and Elsässer A. Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. J Cell Mol Med. 2009;13:3485–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wynn TA and Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016;44:450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, Sun Y, Silva ND, Panizzi P, Laan AMvd, Swirski FK, Weissleder R and Nahrendorf M. Differential Contribution of Monocytes to Heart Macrophages in Steady-State and After Myocardial Infarction. Circulation Research. 2014;115:284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gallego-Colon E, Sampson RD, Sattler S, Schneider MD, Rosenthal N and Tonkin J. Cardiac-Restricted IGF-1Ea Overexpression Reduces the Early Accumulation of Inflammatory Myeloid Cells and Mediates Expression of Extracellular Matrix Remodelling Genes after Myocardial Infarction. Mediators of Inflammation. 2015;2015:484357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heinen A, Nederlof R, Panjwani P, Spychala A, Tschaidse T, Reffelt H, Boy J, Raupach A, Gödecke S, Petzsch P, Köhrer K, Grandoch M, Petz A, Fischer JW, Alter C, Vasilevska J, Lang P and Gödecke A. IGF1 Treatment Improves Cardiac Remodeling after Infarction by Targeting Myeloid Cells. Molecular Therapy. 2019;27:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dumont N and Frenette J. Macrophages protect against muscle atrophy and promote muscle recovery in vivo and in vitro: a mechanism partly dependent on the insulin-like growth factor-1 signaling molecule. Am J Pathol. 2010;176:2228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tonkin J, Temmerman L, Sampson RD, Gallego-Colon E, Barberi L, Bilbao D, Schneider MD, Musarò A and Rosenthal N. Monocyte/Macrophage-derived IGF-1 Orchestrates Murine Skeletal Muscle Regeneration and Modulates Autocrine Polarization. Mol Ther. 2015;23:1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu H, Jiang J, Chen W, Li W and Chen Z. Vascular Macrophages in Atherosclerosis. J Immunol Res. 2019;2019:4354786–4354786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Shay CM, Spartano NL, Stokes A, Tirschwell DL, VanWagner LB and Tsao CW. Heart Disease and Stroke Statistics—2020 Update: A Report From the American Heart Association. Circulation. 2020;141:e139–e596. [DOI] [PubMed] [Google Scholar]
- 85.Newby AC. Metalloproteinase production from macrophages - a perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp Physiol. 2016;101:1327–1337. [DOI] [PubMed] [Google Scholar]
- 86.Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston A-T, Clement M, Dussiot M, Levillain O, Graff-Dubois S, Nicoletti A and Caligiuri G. Macrophage Plasticity in Experimental Atherosclerosis. PLOS ONE. 2010;5:e8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chaabane C, Coen M and Bochaton-Piallat ML. Smooth muscle cell phenotypic switch: implications for foam cell formation. Curr Opin Lipidol. 2014;25:374–9. [DOI] [PubMed] [Google Scholar]
- 88.Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Jaw JE, Sin DD, Seidman MA, Leeper NJ and Francis GA. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2019;39:876–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cochain C and Zernecke A. Macrophages in vascular inflammation and atherosclerosis. Pflügers Archiv - European Journal of Physiology. 2017;469:485–499. [DOI] [PubMed] [Google Scholar]
- 90.Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG and Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J Clin Invest. 2011;121:2025–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gautier EL, Jakubzick C and Randolph GJ. Regulation of the migration and survival of monocyte subsets by chemokine receptors and its relevance to atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:1412–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Farouk SS, Rader DJ, Reilly MP and Mehta NN. CXCL12: a new player in coronary disease identified through human genetics. Trends Cardiovasc Med. 2010;20:204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Merckelbach S, van der Vorst EPC, Kallmayer M, Rischpler C, Burgkart R, Döring Y, de Borst GJ, Schwaiger M, Eckstein HH, Weber C and Pelisek J. Expression and Cellular Localization of CXCR4 and CXCL12 in Human Carotid Atherosclerotic Plaques. Thromb Haemost. 2018;118:195–206. [DOI] [PubMed] [Google Scholar]
- 94.Gencer S, Evans BR, van der Vorst EPC, Döring Y and Weber C. Inflammatory Chemokines in Atherosclerosis. Cells. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Döring Y, Noels H, van der Vorst EPC, Neideck C, Egea V, Drechsler M, Mandl M, Pawig L, Jansen Y, Schröder K, Bidzhekov K, Megens RTA, Theelen W, Klinkhammer BM, Boor P, Schurgers L, van Gorp R, Ries C, Kusters PJH, van der Wal A, Hackeng TM, Gäbel G, Brandes RP, Soehnlein O, Lutgens E, Vestweber D, Teupser D, Holdt LM, Rader DJ, Saleheen D and Weber C. Vascular CXCR4 Limits Atherosclerosis by Maintaining Arterial Integrity: Evidence From Mouse and Human Studies. Circulation. 2017;136:388–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Döring Y, Vorst EPCvd, Duchene J, Jansen Y, Gencer S, Bidzhekov K, Atzler D, Santovito D, Rader DJ, Saleheen D and Weber C. CXCL12 Derived From Endothelial Cells Promotes Atherosclerosis to Drive Coronary Artery Disease. Circulation. 2019;139:1338–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang N, Silver DL, Costet P and Tall AR. Specific Binding of ApoA-I, Enhanced Cholesterol Efflux, and Altered Plasma Membrane Morphology in Cells Expressing ABC1. Journal of Biological Chemistry. 2000;275:33053–33058. [DOI] [PubMed] [Google Scholar]
- 98.Phillips MC. Molecular mechanisms of cellular cholesterol efflux. The Journal of biological chemistry. 2014;289:24020–24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Delafontaine P, Song Y-H and Li Y. Expression, Regulation, and Function of IGF-1, IGF-1R, and IGF-1 Binding Proteins in Blood Vessels. Arteriosclerosis, Thrombosis, and Vascular Biology. 2004;24:435–444. [DOI] [PubMed] [Google Scholar]
- 100.Ruidavets JB, Luc G, Machez E, Genoux AL, Kee F, Arveiler D, Morange P, Woodside JV, Amouyel P, Evans A, Ducimetière P, Bingham A, Ferrières J and Perret B. Effects of insulin-like growth factor 1 in preventing acute coronary syndromes: The PRIME study. Atherosclerosis. 2011;218:464–469. [DOI] [PubMed] [Google Scholar]
- 101.Gautier EL, Jakubzick C and Randolph GJ. Regulation of the migration and survival of monocyte subsets by chemokine receptors and its relevance to atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1412–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Subramanian S, Liu C, Aviv A, Ho JE, Courchesne P, Muntendam P, Larson MG, Cheng S, Wang TJ, Mehta NN and Levy D. Stromal cell-derived factor 1 as a biomarker of heart failure and mortality risk. Arterioscler Thromb Vasc Biol. 2014;34:2100–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.