

Abstract
For more than seventy years, nitrogen-centered radicals have been recognized as potent synthetic intermediates. This review is a survey designed for use by chemists engaged in target-oriented synthesis. This review summarizes the recent paradigm-shift in access to and application of N-centered radicals enabled by visible-light photocatalysis. This shift broadens and streamlines access to many small molecules because the conditions are mild. Explicit attention is paid to innovative advances in N–X bonds as radical precursors, where X = Cl, N, S, O, and H. For clarity, key mechanistic data is noted, where available. Synthetic applications and limitations are summarized to illuminate the tremendous utility of photocatalytically-generated nitrogen-centered radicals.
Graphical Abstract
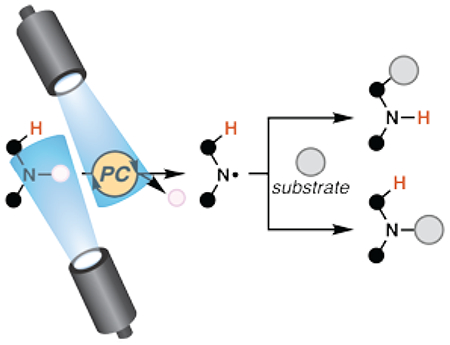
1. INTRODUCTION
Nitrogen-centered radical species are highly reactive, promising synthetic intermediates.1 Given their potential, Zard remarked that paucity in early reports detailing the use of nitrogen-centered radical species as synthetic tool can be attributed to “… a dearth of convenient routes for generating these reactive species and a lack of awareness concerning their reactivity.”1 Indeed, harsh thermal or photonic reaction conditions, or a toxic radical initiator were necessary to generate these intermediates. In the late 2000s, investigations into reactions that rely on nitrogen-centered radical species underwent a tectonic shift when organic chemists adopted visible light-mediated photocatalysis as a practical strategy to construct organic molecules.2
Reactions involving photocatalytically-generated, nitrogen-centered radicals may proceed mechanistically through: a closed, formal catalytic cycle where each molecule of product formed requires direct interaction with a photocatalyst and may include a second interaction for closure of the photocatalytic cycle (Scheme 1, path A), a radical chain where the radical of one molecule of substrate engages another to perpetuate a chain reaction (Scheme 1, path B), or some combination thereof.3,4 To distinguish between these processes, measurements of a reaction’s quantum yield can provide evidence supporting some extent of radical chain propagation.5 Evidence for or against the kinetic feasibility of a given reaction step can be derived from characterization of the excited state photocatalyst and its interactions with reaction components, often by transient absorption spectroscopy (TAS) and Stern-Volmer quenching experiments.6,7Additional data can support or refute proposed mechanistic steps based on electrochemical measurements of oxidation and reduction potentials for reaction components. Published values for a broad array of organic molecules are available; a compilation has been included of known ground and excited state potentials for relevant photocatalysts (Figures 1–4). In many cases, data is unavailable to distinguish between these processes. So several processes described herein as “photoredox catalyzed”47,48 may not technically be catalyzed, but instead simply depend on the combination of visible light irradiation and inclusion of low- to sub-mol-percent amounts of photocatalysis-capable compound.
Scheme 1.
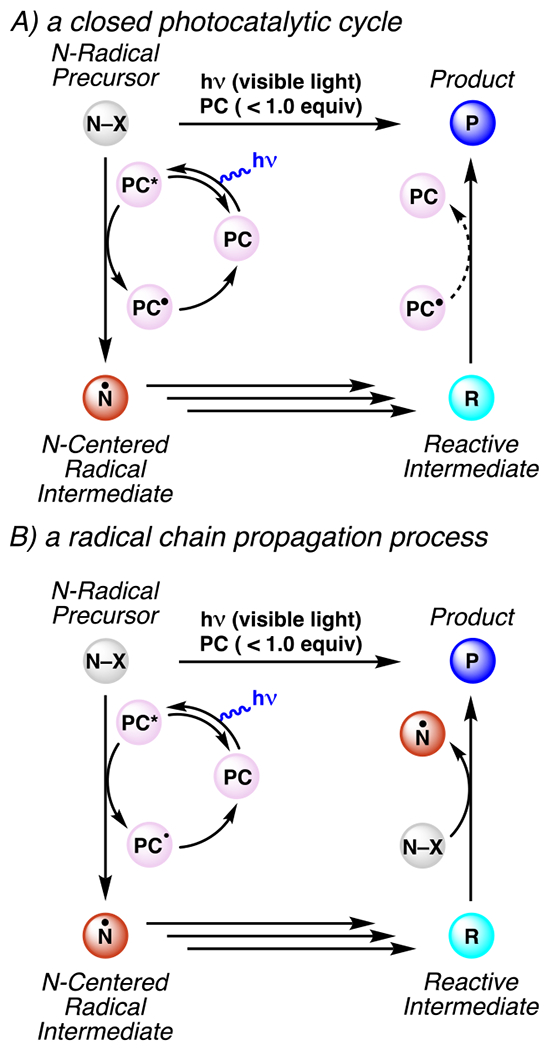
Comparison of Mechanisms That Proceed by Radical Chain Propagation Versus Closed Reaction Cycle
Figure 1.
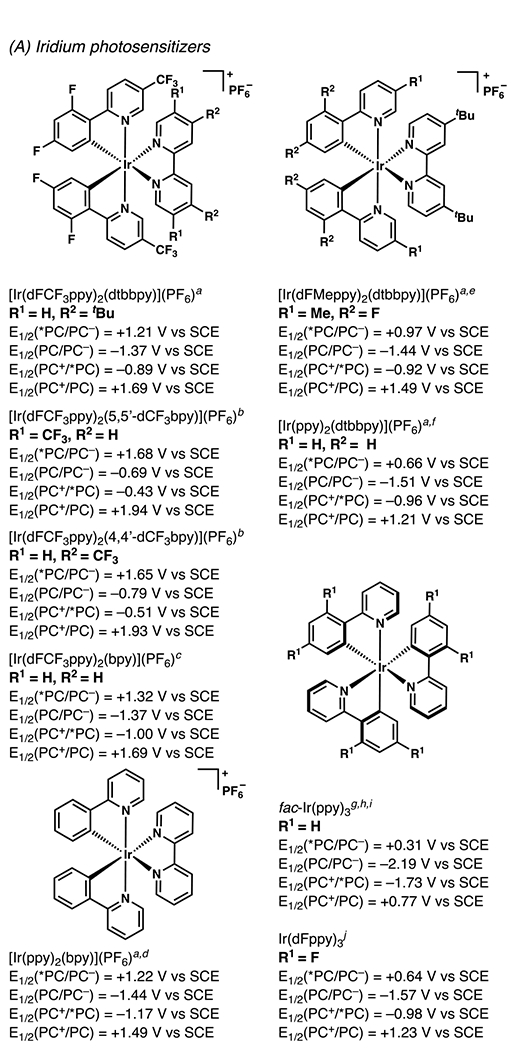
Iridium-Based Photocatalysts and Their Associated Photochemical and Electrochemical Data (All potentials are measured in acetonitrile). aSee ref 8.8 bSee ref 9.9 cSee ref 10.10 dSee ref 1111. eSee ref 12.12. fSee ref 13.13. gSee ref 14.14 hSee ref 15.15 iSee ref 16.16 jSee ref 17.17
Figure 4.
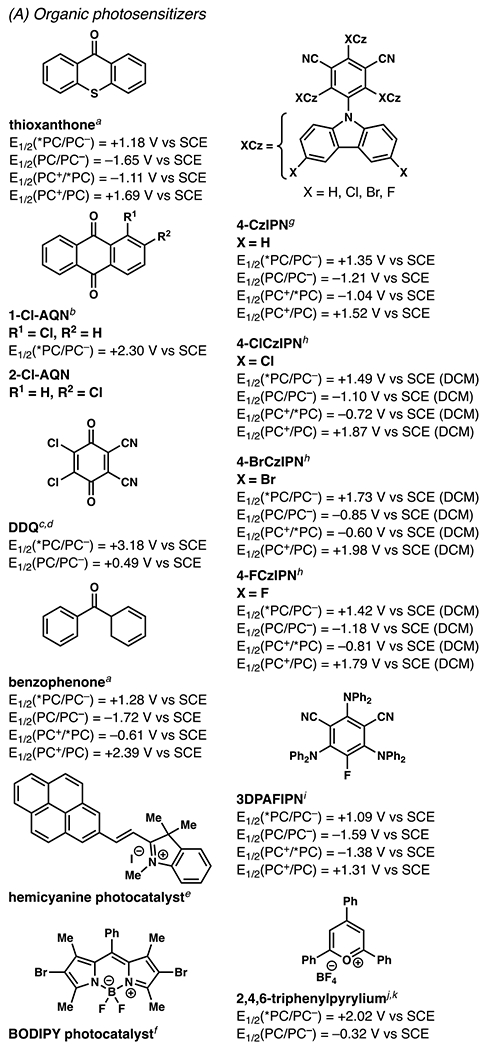
Organic Photocatalysts and Their Associated Photochemical and Electrochemical Data (All potentials are measured in acetonitrile unless otherwise noted.). aSee ref 33.33 bSee ref 34.34 cSee ref 35.35 dSee ref 36.36 eSee ref 37.37 fSee ref 38.38 gSee ref 39.39 hSee ref 40.40 iSee ref 41.41 jSee ref 24.24 kSee ref 42.42
Regardless of their role in a particular reaction, photocatalysis-capable compounds have broadly enabled both novel reactivity and new pathways to known reactivity. Often capable of serving as potent oxidants, reductants, and energy-transfer agents, photocatalysts are generally bench-stable with little to no toxicity and available for commercial purchase. Moreover, photocatalytic reactions are applicable in a vast array of settings, from process-scale and flow-reactor syntheses2,9,43 to picomolar high-throughput screening.44,45 Development of these technologies continues to accelerate their broader adoption.
Ultimately, the glow of photoredox catalysis has illuminated new corridors through which the energy of visible-light can be used to mediate access to versatile nitrogen-centered radical intermediates. Photocatalysis has enabled otherwise rare reaction manifolds, including: the C–H functionalization of (hetero)arenes (Section 2, Section 5, Section 6.3), intermolecular, anti-Markovnikov amino-difunctionalization of alkenes (Sections 2–6.3, Section 6.5), and guided, intermolecular Giese conjugate addition reactions (Section 5.5, Section 6.2, Section 6.4). Furthermore, the array of new bond disconnections leading to nitrogen-centered radicals enabled by photoredox catalysis (Section 4, Section 5, Section 6) has generated a veritable menu of options for viable precursors, each with unique properties capable of enabling reactivity in diverse reaction environments, while tremendous strides have been made in the fundamental understanding of processes such as proton-coupled electron transfer (PCET) (Sections 6.2–6.3). As reports of this type continue to proliferate, they further catalyze interest and innovation in the important field of photocatalysis.
This review constitutes a survey of transformations that may capitalize on visible-light photoredox catalysis to access nitrogen-centered radical intermediates through the direct interaction of an excited-state photosensitizer and a substrate. The reactions discussed throughout this review are selected to include those expected to generate nitrogen-centered radicals under conditions requiring both visible light irradiation and catalytic quantities of a photocatalysis-capable compound. Generally, manuscripts where fulfillment of these criteria is ambiguous or contradicted by the available data are not included. The photochemical and photophysical underpinnings of these processes have been well-reviewed by others,2,46,47,48 so, when available, mechanistic data has been included to allow readers to understand the strength of associated mechanistic proposals.
This review is organized by radical precursor, focusing on the pioneering inventions of viable precursors that rely on cleavage of N–Cl, N–S, N–N, N–O or N–H bonds to generate the critical putative nitrogen-centered radical intermediates. For each addressed technology, attention is paid to the process of nitrogen-centered radical generation, any information that can be used to distinguish between radical-chain propagation processes and truly catalytic processes, and the synthetic advantages and limitations afforded by the associated method. This review focuses principally on reactions that are thought to initially rely on free-radical intermediates, and, for the most part, does not discuss photosensitized or photocatalyzed nickel-mediated transformations,49,50 many of which appear to rely on nitrogen-centered radicals as intermediates.51,52,53,54,55,56,57 Additionally, it does not address photoredox-mediated reactions that employ tertiary amines as reagents.58,59 Notably, related, focused review articles cover a subset of similar topics;60, 61, 62, 63, 64, 65 however, there is no comprehensive review article that unites these topics. Accordingly, this review aims to deliver a comprehensive overview of investigations on this topic.
2. NITROGEN-CENTERED RADICALS CAN BE GENERATED FROM NITROGEN–CHLORINE BONDS IN THE PRESENCE OF PHOTOREDOX CATALYSTS
For more than a hundred years, nitrogen–halogen bonds have been known precursors to δ-functionalized products as in the course of Hofmann-Löffler-Freytag (HLF) reactions.66,67 Investigations into the mechanism of this class of reactions were not reported until the mid-twentieth century with the work of Wawzonek and Thellan, who proposed a radical mechanism based on the importance of radical initiators like hydrogen peroxide or visible light irradiation.68 Their proposal of a radical-chain propagation mechanism was later confirmed through extensive investigations by Corey and Hertler.69 While HLF reactions have found broad synthetic utility, the ability to derive the key nitrogen-centered radical under neutral and room temperature conditions has been the subject of continuing research in the intervening decades. Pre-installation of the nitrogen-halogen bond has allowed for solely visible-light-initiated HLF-type reactions for a variety of nitrogen-containing functional groups, including amides and sulfamate esters.70, 71, 72 Other functional groups, such as N-chlorosulfonamides, are not reactive under the same room temperature, solely visible-light-initiated conditions, instead requiring ultraviolet irradiation to induce photolysis.70, 73, 74 In 2015, S. Yu and Qin disclosed a system for δ-C(sp3)–H chlorination (Scheme 2) that relies on a mild light source and iridium photocatalyst, and may be photoredox-initiated or mediated.75 Indeed, in the presence of visible light irradiation, the photoexcited iridium catalyst [Ir(ppy)2bpy]PF6 can be quenched by N-chlorosulfonamide 1 to produce a chloride anion, oxidized Ir(IV), and N-centered radical intermediate 3. After this point, the N-centered radical intermediate 3 is expected to rapidly proceed through a 1,5-HAT to give the carbon-centered radical intermediate 4, which can feasibly engage in one or more productive reaction pathways depending on the particular substrate. From the carbon-centered radical intermediate 4, the reaction can proceed directly to the chlorinated product 2 by radical-chain propagation (Scheme 2, path a)69 through abstraction of the chlorine present in another molecule of substrate 1. While “light-on, light off” experiments conducted by Qin and Yu showed light to be required for significant reaction progress, it is noteworthy that this does not preclude a radical chain propagation mechanism.3 With more oxidatively labile carbon-centered radical intermediates, a mechanistic scenario has not been excluded: it may be theoretically possible that these alkyl radicals are oxidized to cations, which are then trapped by chloride in radical-polar crossover process. Specifically, both reaction pathways are plausible in the production of δ-chlorinated product 2a, as the resonance stabilization afforded by the benzylic position should result in a relatively low oxidation potential (oxidation of secondary benzylic radical PhCH•CH3 Ep/2ox = +0.37 V vs SCE). 76 By contrast, the demonstrated reactivity under the same conditions to give the primary δ-chlorinated product 2b could indicate an efficient radical chain propagation mechanism, as the oxidation of primary alkyl radical of type 4 where R = H would be thermodynamically challenging.
Scheme 2.
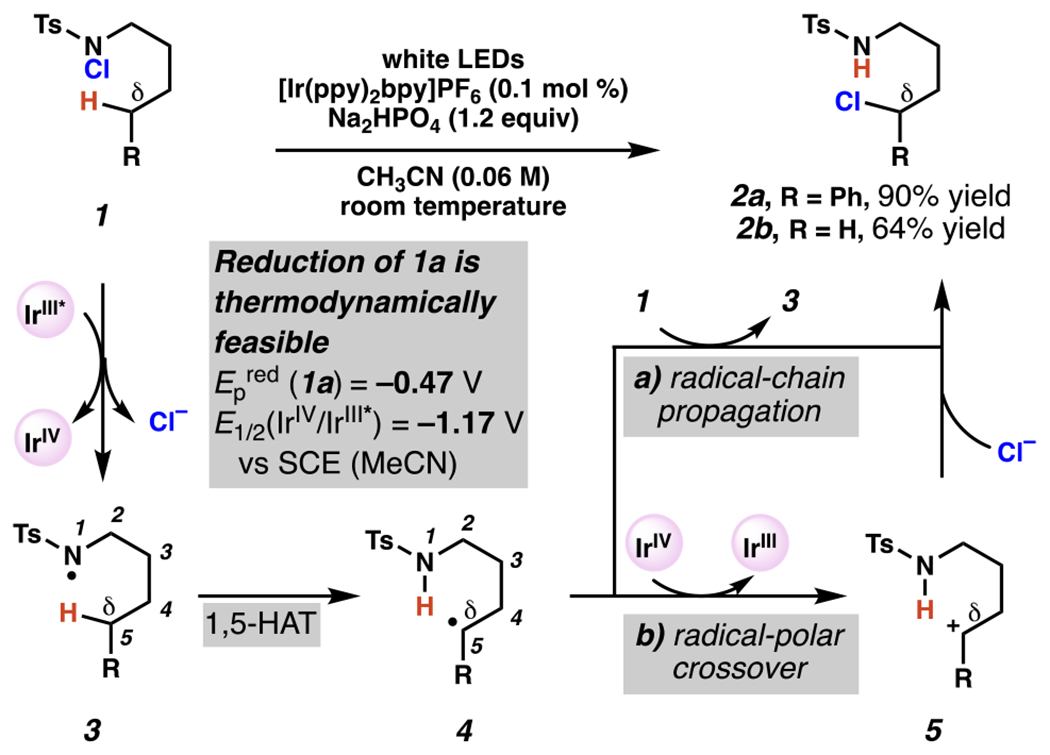
N-Chlorosulfonamides Undergo C(sp3)–H Chlorination in the Presence of Photoredox Catalysts
Subsequent research from other laboratories suggests a radical chain propagation mechanism in photoredox-mediated, sulfonamide-guided chlorination reactions (Scheme 3). 77 In W. Yu and co-workers recent disclosure of a procedure for in situ-generation the nitrogen-chlorine bond, the quantum yield of the sulfonamide-guided chlorination when employing a preformed N–Cl bond was found to be (ϕ) = 3.23, meaning an average of 3.23 molecules of product were formed per absorbed photon. This result provides substantial evidence for a radical-chain propagation mechanism in sulfonamide guided-chlorination reactions.
Scheme 3.
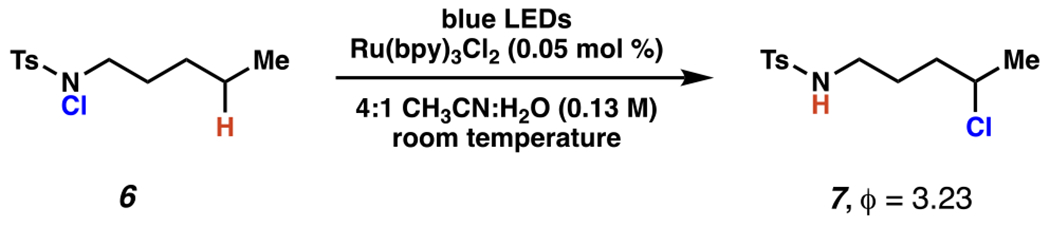
W. Yu and Co-workers’ Quantum Yield Calculation Supports a Radical Chain Propagation Mechanism in Sulfonamide-Guided Chlorination Reactions
N-chlorosulfonamides have also been employed to chlorosulfonamidate unactivated terminal olefins (Scheme 4).78 Under visible-light irradiation and in the presence of an iridium photocatalyst, N-chloro-N-methyl toluenesulfonamide 9 engages in chlorosulfonamidation of terminal olefins 8 and ind-1-ene, with sulfonamidation occurring exclusively at the terminal carbon. Some variation in substitution at the olefin is tolerated, with styrene yielding chlorosulfonamidated product 10a in 78% yield and oct-1-ene yielding product 10b in 64% yield. This reaction, however, relies on secondary N-chlorosulfonamide substrates, and does not engage electron-deficient olefins. As will be observed in numerous examples throughout this review, amidyl radicals, and the electronically similar sulfonamidyl radicals, are electrophilic, which influences reaction selectivity, particularly when engaged in intermolecular 1,2-difunctionalization of alkenes. The electrophilic character of amidyl radicals has been investigated by Newcomb and co-workers, and the reality of this character is borne out through the complete regioselectivity reported in this investigation and others like it.79,80
Scheme 4.
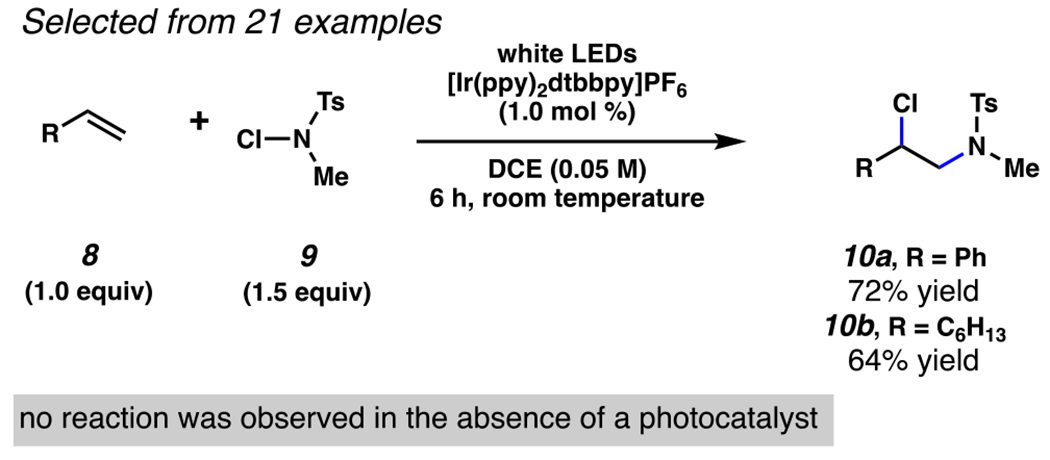
S. Yu and Co-workers Protocol to Access Chlorosulfonamidation of Olefins with N-Chlorosulfonamides by Photoredox Catalysis
While N-chlorosulfoximines have long been known to engage in intermolecular 1,2-difunctionalization of electron-rich olefins via UV-irradiation or radical initators,81 recent methods employing photoredox catalysts have enabled similar reactivity under room-temperature, visible-light irradiation(Scheme 5).82 Using a photo-excited ruthenium complex and organic base in acetonitrile, N-chlorosulfoximine 11 reacts with an excess of styrene 12 to furnish 1,2-difunctionalized products, like 13a. Across substrates 13a–13c, reaction efficiency decreases in parallel with the extent of alkane fluorination, underscoring the apparent necessity of an S-fluoroalkyl substituent for efficient application of this process.
Scheme 5.

Photoredox-Catalysis Enables 1,2-Difunctionalization of Electron-Rich Olefins by N-Chlorosulfoximines Bearing S-Fluoroalkyl Substituents
Photoredox-capable complexes appear important to a visible light-induced N-arylation utilizing N-chloroamines as substrates and benzoxazoles as radical trapping agents (Table 1).83 The mechanism for this reaction is not well explored.
Table 1.
C(sp2)–H Amination Relies on N-Chlorinated Amine
| ||
|---|---|---|
|
| ||
| entry | [Ir(dtbpy)(ppy)2]PF6 (present/absent) | yield (%) |
|
| ||
| 1 | present | 80 |
| 2 | absent | trace |
|
| ||
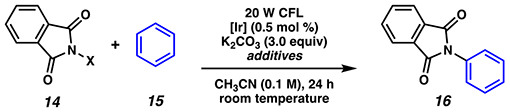
Photoredox catalysis driven by visible light irradiation has also been implemented to convert N-chlorophthalimides 14 to imidyl radicals for use in an arene imidation reaction (Table 2, entries 1–2).84 The conditions can be adapted to permit in situ formation of N-chlorophthalimide (entries 3–4) with a slightly diminished yield. This use of N-chlorophthalimide was disclosed within months of analogous methods for arene imidation that rely on an N-trifluoroacetoxyphthalimide85 and an N-succinimidyl perester86 under photoredox and electrochemical regimes, respectively. Consistent with other investigations, N-acetoxyphthalimide is an inefficient nitrogen source under these reaction conditions (entry 5).85 Similarly, sulfonate-based leaving groups (entries 6–8), as well as other halides (entries 9–10), are inferior substitutes for the chlorine atom.
Table 2.
C(sp2)–H Imidation Proceeds with In Situ-Generated N-Chlorophthalimide-Derived Radicals
| ||||
|---|---|---|---|---|
|
| ||||
| entry | X | [Ir] | additives | yield (%) |
|
| ||||
| 1 | Cl | Ir(ppy)3 | – | 43 |
| 2 | Cl | Ir(dFppy)3 | AcOH (20 mol %) | 65 |
| 3 | H | Ir(dFppy)3 | tBuOH, tBuOCl (1 equiv each) | 50 |
| 4 | H | Ir(dFppy)3 | aq. NaOCl, tBuOH, AcOH (1 equiv each) | 47 |
|
| ||||
| 5 | OAc | Ir(ppy)3 | – | n.r. |
| 6 | OTs | Ir(ppy)3 | – | 22 |
| 7 | OTf | Ir(ppy)3 | – | 13 |
| 8 | OMs | Ir(ppy)3 | – | 20 |
| 9 | Br | Ir(ppy)3 | – | 4 |
| 10 | I | Ir(ppy)3 | – | n.d. |
|
| ||||
These reactions84, 85 are proposed to rely on strongly reducing photocatalyst Ir(dFppy)3 or Ir(ppy)3 to access critical phthalimidyl radical 17 with a chloride or trifluoroacetoxy anion as a byproduct (Scheme 6). With N-chlorophthalimide (14a), the viability of this mechanistic step is supported by data showing that N-chlorophthalimide (14a) quenches the photoexcited [IrIII]* in a concentration dependent manner (Kq = 2.6 × 108 M−1 s−1)84 and is thermodynamically feasible (Ep/2(IrIV/IrIII*) = −1.57 V vs SCE;17 Ep (14a) = −0.45 V vs SCE in CH3CN87). Resultant N-centered radical 17 is proposed to add into the arene’s π-system. Additional support for this mechanism comes from kinetic isotope experiments employing a 1:1 ratio of C6H6:C6D6 which gave a kinetic isotope effect measurement of KH/D = 1.13;84 this value is consistent with those measured in electrophilic aromatic substitution and related radical aromatic substitution reactions, indicating the deprotonation step likely occurs after the rate-determining step.88 The authors propose the photocatalytic cycle to be closed by oxidation of the resonance-stabilized, carbon-centered radical 18 to give cationic Wheland intermediate 19, which is poised for deprotonation to give desired product 16 in modest to good yields. Absent iridium catalyst and acetic acid, only 8% yield of the desired product is isolated, indicating that use of a photoredox catalyst is necessary to access the desired product in synthetically useful yields.
Scheme 6.
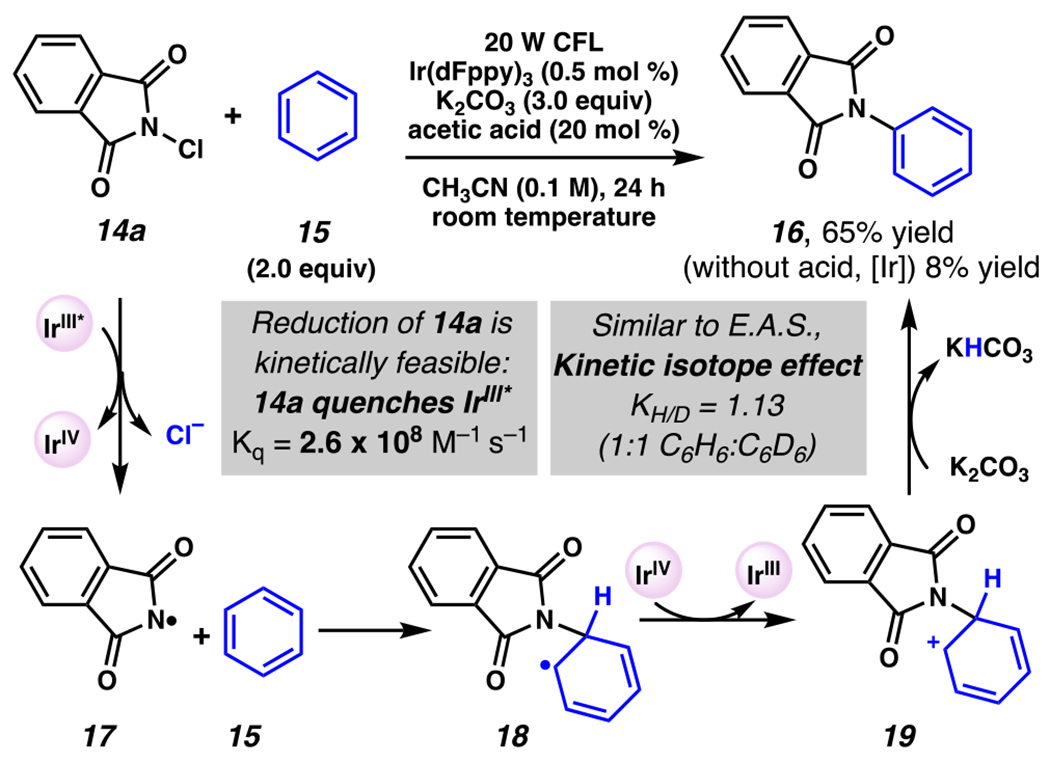
A Plausible Mechanism By Which Photoredox Catalysis Enables Efficient C–H Imidation of Arenes
Whereas substrates such as N-chlorosulfonamides were previously only engaged under ultraviolet irradiation, with the introduction of photocatalysts to initiate and perpetuate reactions, similar transformation can be affected at ambient temperature and with visible light irradiation. Owing to these mild conditions, radical-mediated reactions have become more broadly synthetically viable. Under visible light, photosensitized or catalyzed processes harness N-chlorinated precursors to nitrogen-centered radicals in alkene 1,2-chloroamination reactions, directed C–H functionalization reactions of alkanes, and intermolecular C–H imination of arenes. These constitute a small subset of the technologies unlocked through photocatalytic or photosensitized generation of nitrogen-centered radicals.
3. NITROGEN-CENTERED RADICALS CAN BE GENERATED FROM NITROGEN–NITROGEN BONDS WITHIN N-AMINOPYRIDINIUM SALTS IN THE PRESENCE OF PHOTOREDOX CATALYSTS89,90
Nitrogen–nitrogen bonds are not often formed or cleaved in fine chemical synthesis, yet nitrogen–nitrogen bond cleavage is a viable path to a variety of N-centered radicals. In 2005, Studer and co-workers reported carbamate-masked 3-amino-1,4-cyclohexadienes 20 as stable precursors to nitrogen-centered radicals (Figure 5).91 These were further developed into 1-amino-Hantzsch-type dihydropyridine esters 21.92 Both of these were, however, employed using traditional radical initiators. Photosensitizers were used to engage this category of nitrogen-radical precursor by the 2015 development of aminopyridinium salts 22a.
Figure 5.
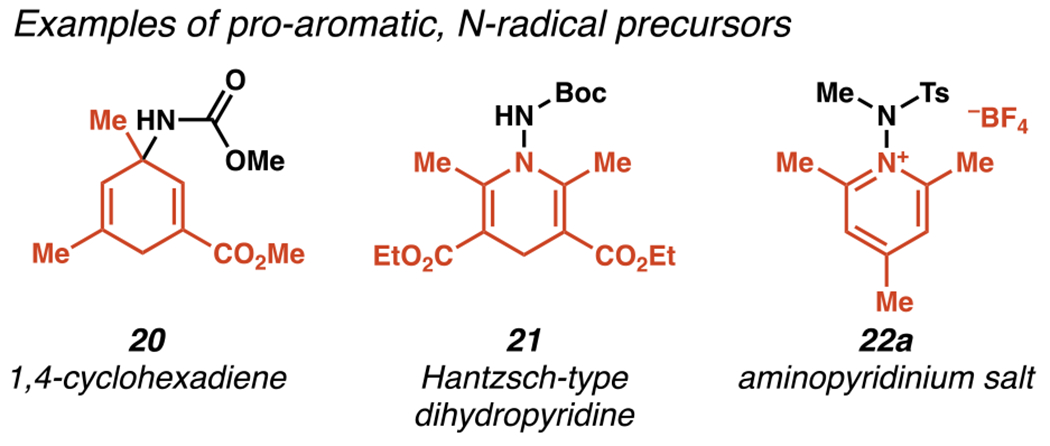
Nitrogen-Centered Radicals Derived from Pro-aromatic Compounds Inspired a Photo-mediated Strategy to Access Nitrogen-Centered Radicals Based on Nitrogen–Nitrogen Bond Cleavage
In one of the first known examples of photoredox-driven generation of a nitrogen-centered radicals from N–N bond cleavage, N-aminopyridinium salts 14b serve as precursors for radical C–H amination of (hetero)arenes (Scheme 7–8).93 These salts have a relatively low reduction of potential (Epred (14b) = −0.75 V vs SCE) and their reduction by photoexcited ruthenium catalyst (E1/2 (RuII*/RuIII) = −0.81 V vs SCE) is thermodynamically favorable.94 Under visible, blue light irradiation, slightly elevated temperatures, and in the presence of 4Å molecular sieves, amino-pyridinium salt 14b can be reduced, releasing 2,4,6-collidine and phthalimide radical 17. Radical species 17 can then react with the excess benzene 15, as had already been observed in related systems with N-acyloxyphthalimides (vide infra)85,95and N-chlorophthalimides (vide supra),84 to access dearomatized radical species 18. Subsequent oxidation to 19 and deprotonation, likely by 2,4,6-collidine, provides N-phenylphthalimide (16) in modest yield. A unique benefit of this approach is the concurrent, stoichiometric production of pyridyl base, which eliminates the need to add exogenous base in this reaction (compare to Scheme 6). 84
Scheme 7.
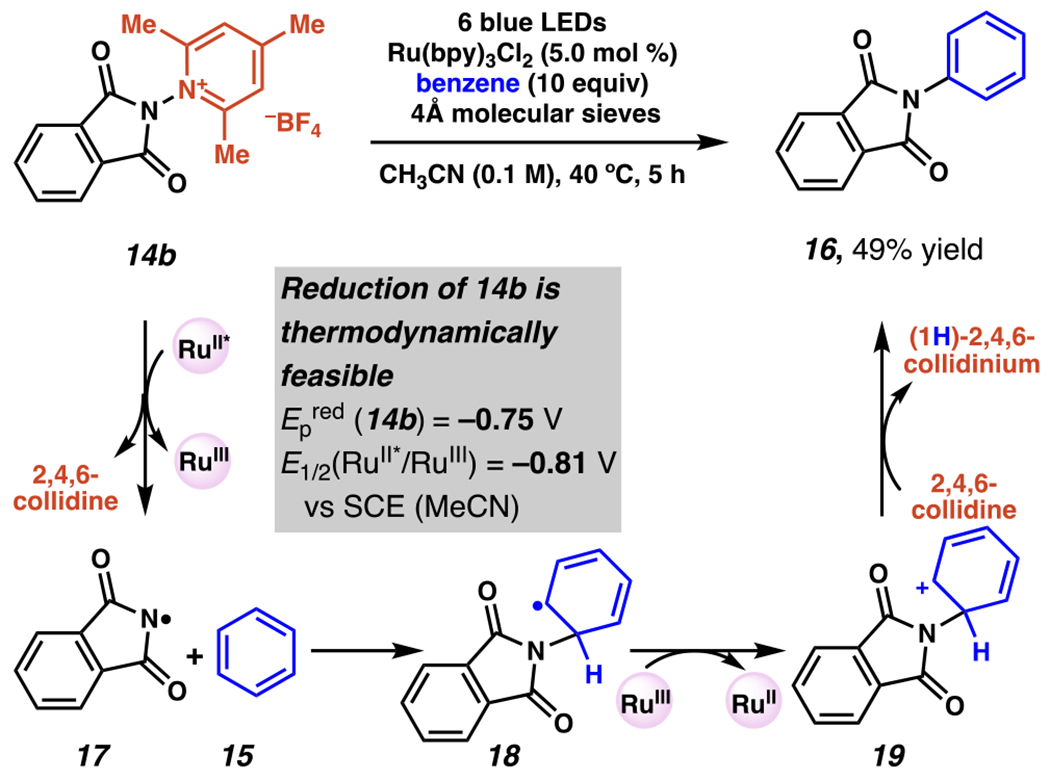
Light Enables Nitrogen-Centered Radical Generation from Pyridinium Salts in the Course of C–H Amination of Arenes
Scheme 8.
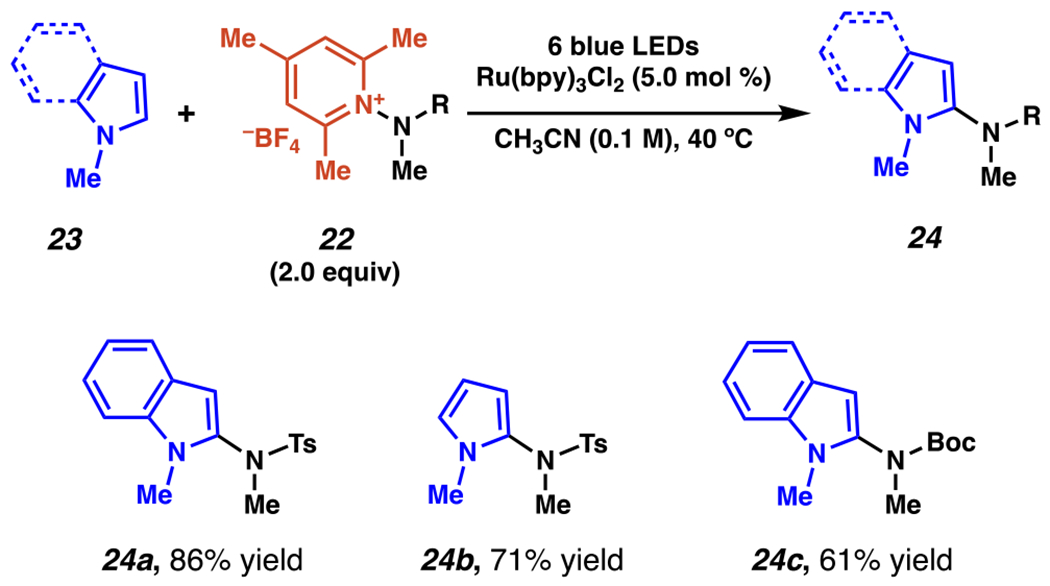
Radical C(2)-Amidation of N-Methyl Indoles and Pyrroles Can Engage N-Aminopyridinium Salts as Nitrogen-Centered Radical Precursors
Under related conditions, Studer and co-workers extended their N-aminopyridinium salt-generated radicals to engage in direct C(2)-amidation of N-methyl indoles and pyrroles (Scheme 8).93 Employing the same ruthenium photocatalyst in acetonitrile, use of two equivalents of N-aminopyridinium salts based on N-(methyl)toluenesulfonamide or N-methyl-Boc-amine to give indole-derived products 24a and 24c, as well as pyrrole-derived 24b. These reactions (Scheme 7–8) are the first reported example of photoredox-enabled generation of nitrogen-centered radicals from the controlled, unsymmetric breaking of N–N bonds. Moreover, the successful use of an N-Boc-amine as a nitrogen-centered radical agent was unprecedented in photoredox chemistry prior to this publication.
Nitrogen-centered radicals, derived from aminopyridinium salts engage in regioselective aminohydroxylation of olefins in the presence of visible light and a photosensitizer, with amination occurring at the less substituted carbon of the parent olefin, yielding products 25. (Scheme 9).96 With a slight excess of aminopyridinium salt 22b and in a 9:1 mixture of acetone and water, photoexcited iridium species is proposed to engage in a single-electron reduction of the N–N bond to produce sulfamyl radical 26 and pyridine. This step is both thermodynamically feasible (Epred (22b) = −0.80 V vs SCE) for photoexcited iridium (E1/2(IrIII*/IrIV) = −1.73 V vs SCE)16 and kinetically feasible (Kq = 6.0 × 103 M−1s−1) based on Stern-Volmer quenching analysis. The nitrogen-centered radical then adds to the less-substituted carbon of the olefin to give carbon-centered radical intermediate 27. Benzylic radical 27 may then be converted by oxidized iridium species to aromatic-stabilized carbocation intermediate 28. In the presence of pyridine and water, a combination of nucleophilic attack by water or hydroxide and deprotonation by pyridine yields desired product 25 in 85% yield.
Scheme 9.
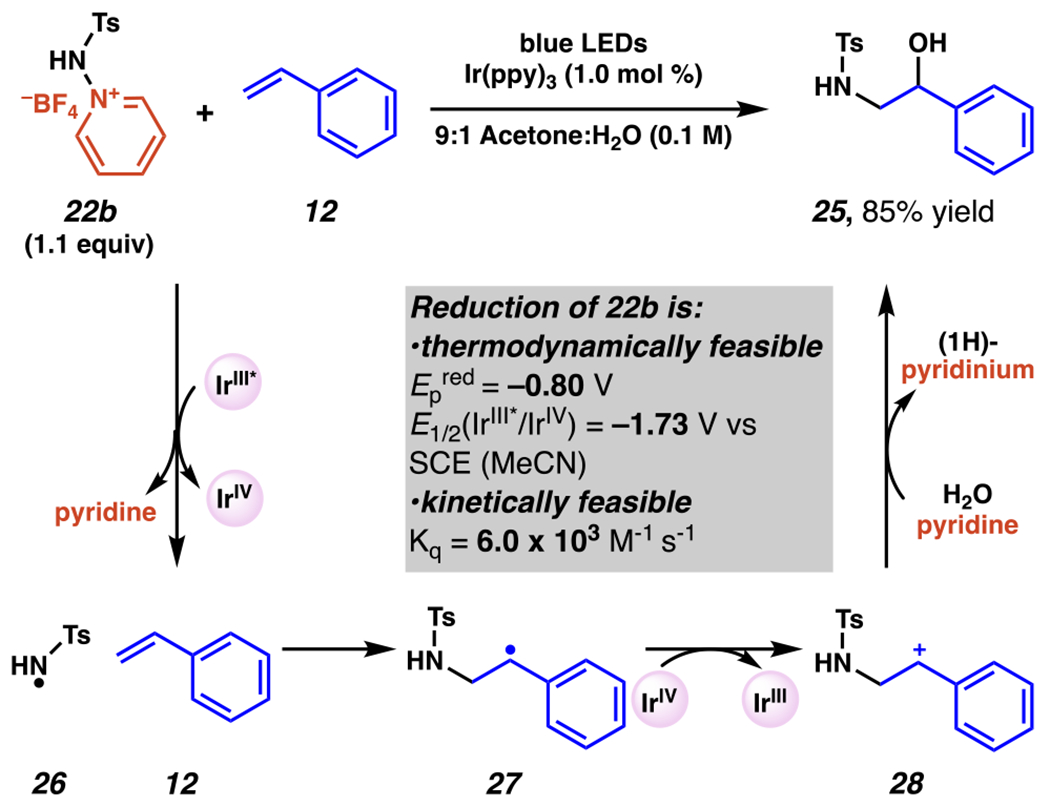
Sulfonamide-Based Aminopyridinium Salts Engage in Photoredox-Mediated Aminohydroxylation of Olefins
Expanding from this reaction (Scheme 9), amidohydroxylation is feasible using amidopyridinium salts 29 as nitrogen sources to furnish amide- and carbamate-functionalized products, 30a and 30b, respectively. (Scheme 10).97 These nitrogen sources are viable owing to inclusion in the reaction of a catalytic amount of scandium (III) triflate, which is proposed to act as a Lewis acid, complexing with amidopyridines 29 and lowering the thermodynamic barrier for reduction of the nitrogen–nitrogen bond. The reaction does not proceed in the absence of light, iridium photocatalyst, or scandium (III) triflate, which is consistent with the proposed roles of these reagents and conditions in the reaction.
Scheme 10.
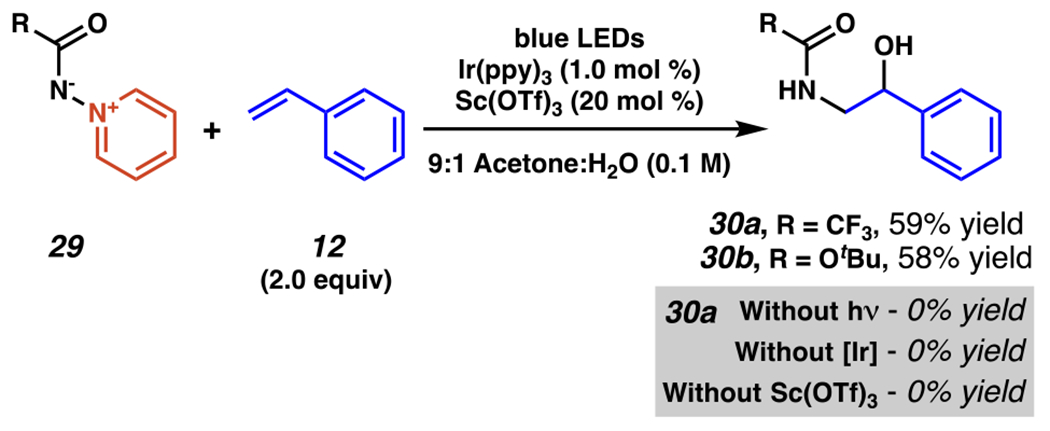
Amidopyridinium Salts Engage in Photo-driven Amidohydroxylation of Olefins
In 2018, Xu, Hu, and co-workers disclosed a set of conditions for aminohalogenation of styrenes employing sulfonamide-based aminopyridinium salt 22b and nucleophilic halogen sources (Scheme 11). Under blue light irradiation at room temperature, the photoexcited iridium catalyst ([Ir(ppy)2dtbbpy]PF6) can generate intermediates analogous to those described above (Scheme 9, intermediates 26, 27, and 28). In the presence of nucleophilic halides, such as Olah’s reagent (pyridine•9(HF)), aminofluorination product 31a can be produced in very good yields; while similarly, pyridine•(HCl) reacts to give aminochlorination product 31b in a somewhat reduced 66% yield. Substitution of the halide sources with pyridine•(HBr) does produce aminobromination product 31c, however only at a heavily reduced 17% yield
Scheme 11.
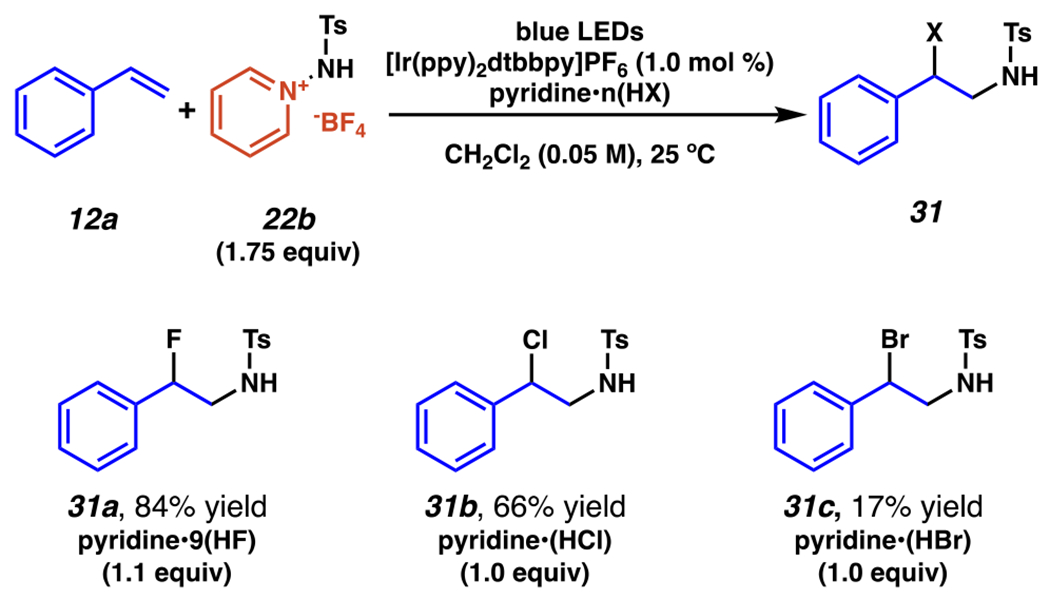
Aminopyridinium Salt-Derived Sulfamyl Radicals Enable Regiospecific Aminohalogenation of Styrenes in the Presence of Pyridinium Halides
Under related conditions, aminopyridinium salt 22b engages in diastereoselective aziridination of β-methylstyrenes 32 (Scheme 12).98 Using a modest excess of dibasic potassium phosphate and aminopyridinium salt 22b in dichloromethane with an iridium photocatalyst under blue light irradiation, mixtures of E- and Z-styrenes are aziridinated in good yields with >20:1 dr for trans-aziridines containing an inductively deactivating group (i.e., 33a and 33b). Somewhat unexpectedly, a product derived from styrenes bearing an inductively donating tolyl group (33c), however, does not react as readily, with substantially reduced yields and a modest 1.5:1 dr biased toward the cis-aziridine product instead.
Scheme 12.
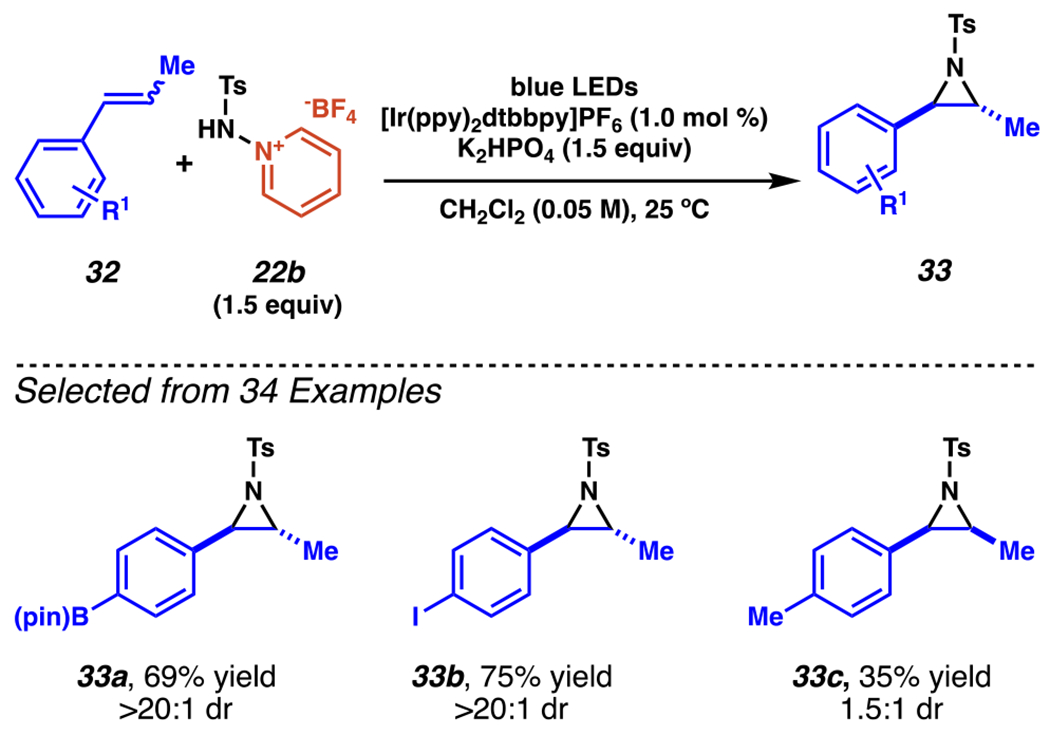
Sulfamyl Radicals Engage in Diastereoselective Aziridination of Substituted Styrenes
Aminopyridinium salts have also been employed to produce α-sulfamyl-containing carbonyl compounds from O-protected enolates (Scheme 13).99 Using primarily O-acyl enolates 34, the reaction proceeds with photoexcited Ir(ppy)3 in acetonitrile under blue light irradiation, providing access to the title compounds. A variety of aromatic ketones can be synthesized in good to excellent yield, including those bearing potentially reactive aryl substituents, such as aryl iodide-containing product 35a at 83% yield. Aliphatic ketones are produced, including ketone 35b containing a newly-formed quaternary sulfamyl center in 52% yield, and cyclic ketone 35c, which, impressively, contains two unreacted alkenes. Finally, several aldehydes are synthesized, including α-sulfamyl undec-10-enal 35d which is produced at 66% yield.
Scheme 13.
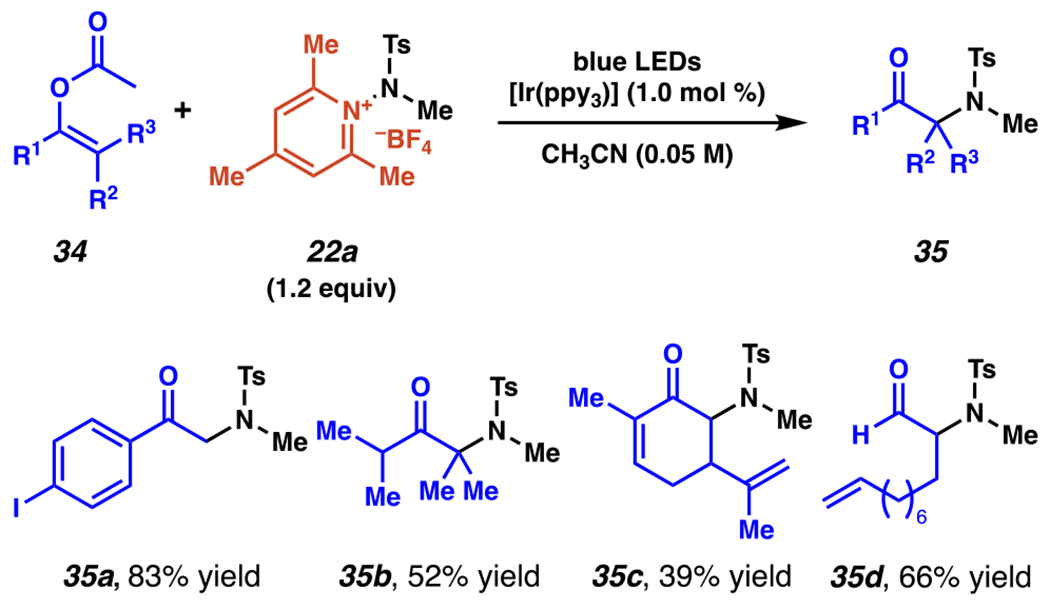
Regioselective Amination of O-Protected Enolates Furnishes α-Sulfamyl-Containing Carbonyl Compounds
Leveraging the pyridine liberated from this class of reactants as a heteroarene source rather than as a base, Hong and co-workers have recently disclosed a series of reactions which produce new pyridyl-C(sp2)–C(sp3) bonds, beginning with an aminopyridylation of vinyl ethers and amides (Scheme 14–15).100 Mechanistically, to initiate the reaction, photo-excited eosin Y reacts with an excess of aminopyridinium salt 37a in a thermodynamically and kinetically feasible single-electron reduction (Ep/2(37a) = −0.62 V vs SCE in CH3CN; E1/2(PC*/PC•+) = −1.11 V vs SCE in 1:1 H2O:CH3CN;101 Stern-Volmer experiments found Kq•(τ) = 11.87 M−1) to release pyridine and produce nitrogen-centered radical intermediate 39. As noted previously (vide supra, Scheme 4),79,80 electrophilic sulfamyl radical intermediate 39 adds to more electron-rich terminal carbon of vinyl ether 36a to give intermediate 40. Carbon-centered, nucleophilic radical intermediate 40 then likely adds to the C(4) position of another equivalent of aminopyridinium salt 37a to give radical, cationic species 41. This readily is deprotonated by the excess of tribasic phosphate which can undergo radical decomposition to produce another equivalent of 39 and final product 38a. This reaction proceeds through a radical chain mechanism, consistent with a quantum yield (ϕ) of 33.5, which suggests that on average each absorbed photon of light results in formation of thirty-three and a half equivalents of product. For this transformation, DFT calculations suggest that position selectivity may arise from non-covalent interactions between the negatively-charged sulfamyl oxygens and positively-charged aminopyridinium nitrogen–nitrogen bond, which are spatially proximal when intermediate 41 forms by reaction at C(4) but not at C(2).102
Scheme 14.
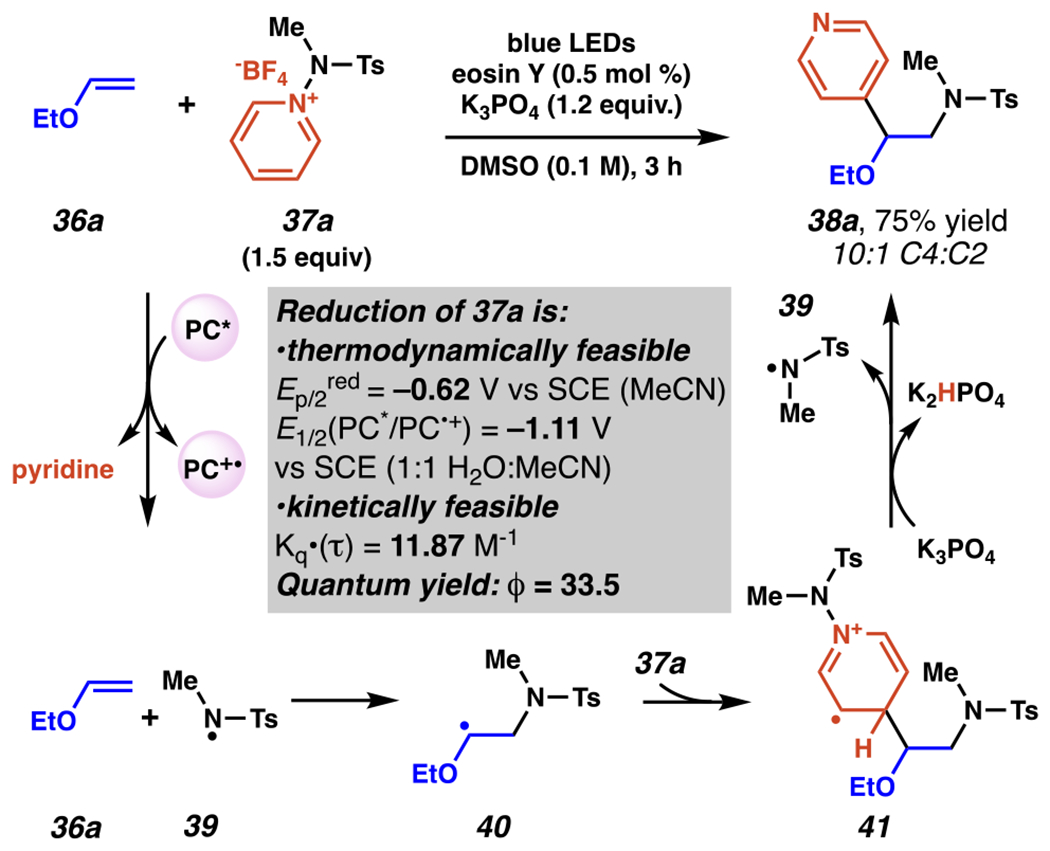
Aminopyridinium Salts Engage in Aminopyridylation of Vinyl Ethers with a Preference for Pyridyl C(4)-Selectivity Likely via Radical Chain Mechanism
Scheme 15.
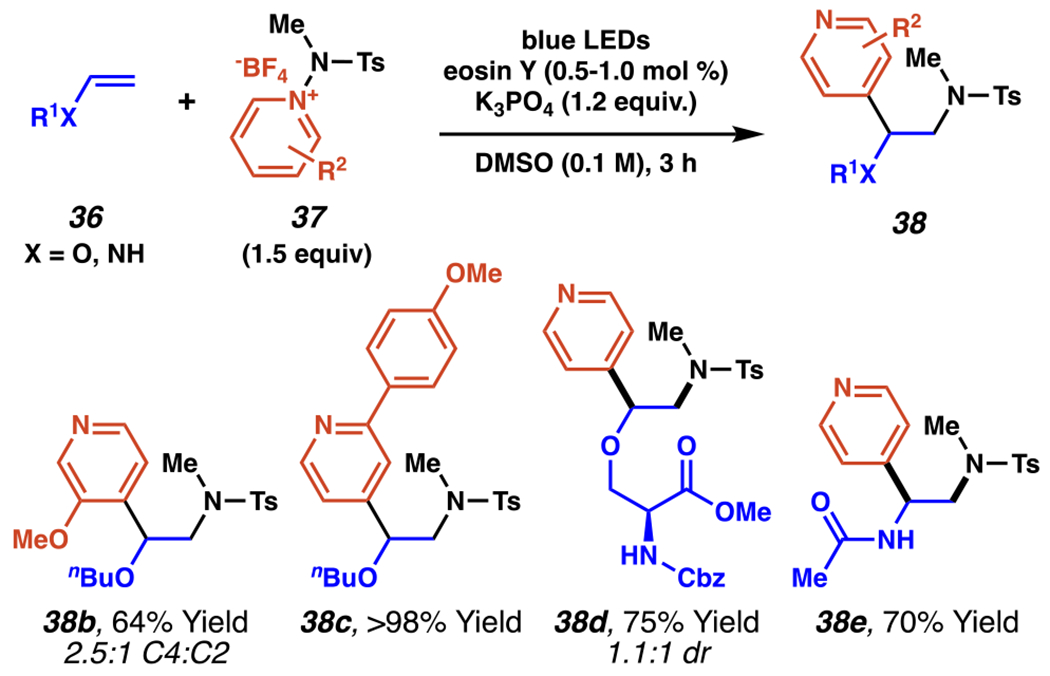
Vinyl Aminopyridylation Tolerates Variations in Vinyl and Pyridyl Substitution
This reaction tolerates variations in both the vinyl component 36 and the pyridyl-component of 37 (Scheme 15). Pyridyl C(4)-selectivity is degraded by substitution to the pyridyl C(3)-position, with 3-methoxypyrid-4-yl product 38b synthesized with a 2.5:1 C(4):C(2) selectivity. Functionalization at C(2) of the pyridine group is frequently beneficial to yields and selectivity with product 38c synthesized in >98% yield. Limited diastereoselectivity is observed in stereocenter-containing vinyl-substrates, such as serine-derived product 38d which is obtained with a 1.1:1 dr. N-vinyl acetamide is also successfully employed under the same reaction conditions to give amide-containing product 38e in 70% yield.
In order to modulate the previously demonstrated C(4)-selectivity (vide supra, Scheme 14–15), Hong and co-workers turned to amidopyridinium reagent 42a and strongly oxidizing mesityl acridinium photocatalyst, which could be generate a nitrogen-centered radical without initial loss of the pyridyl group, in an overall process that induces C(2)-selectivity by radical formal 1,3-dipolar cycloaddition (Scheme 16–17).103 The photocatalytic oxidation of amidopyridinium 42a to nitrogen-centered radical intermediate 44 is kinetically feasible (Kq•(τ) = 370.43 M−1). While some of the alkenes used could be directly oxidized by photocatalyst (E1/2(PC*/PC•−) = +2.09 V vs SCE in CH3CN), such as 32 (Ep/2ox (32) = +1.74 V vs SCE in CH3CN)104, the relatively lower oxidation potential of amidopyridinium 42a (Ep/2ox (42a) = +1.58 V vs SCE in CH3CN) makes the desired pathway highly competitive, if not dominant. Once formed, electrophilic amidyl radical intermediate 44 adds to the more electron-rich carbon of the alkene to give stabilized carbon-centered radical intermediate 45. In a radical formal 1,3-dipolar cycloaddition process, carbon-centered radical 45 transiently produces 5-membered radical cation intermediate 46; this step proceeds with complete diastereoselectivity in all reported instances for an anti-relationship between substituents of unsymmetric alkenes (as well as a 1,2-syn-relationship between the amide and pyridine in the final products). The stereochemical preference is understood to arise from steric interactions between the alkene substituents in the transition state between 45 and 46 (calculated as 2.7 kcal/mol by DFT).105 The final reaction steps, including N–N bond scission, deprotonation of the pyridine C(2), and protonation of the amide, are likely rapid and are proposed to proceed through amidyl radical intermediate 47. Quantum yield experiments (ϕ = 0.141) don’t directly implicate a radical chain propagation mechanism, and kinetic isotope effect for pyridine-d5-derived 42a (KH/D = 1.01) is negligible, indicating the deprotonation of 46 is not rate-limiting. This amidopyridylation reaction engages an array of activated and unactivated alkenes alike, maintaining pyridine C(2)-selectivity throughout (Scheme 17).
Scheme 16.
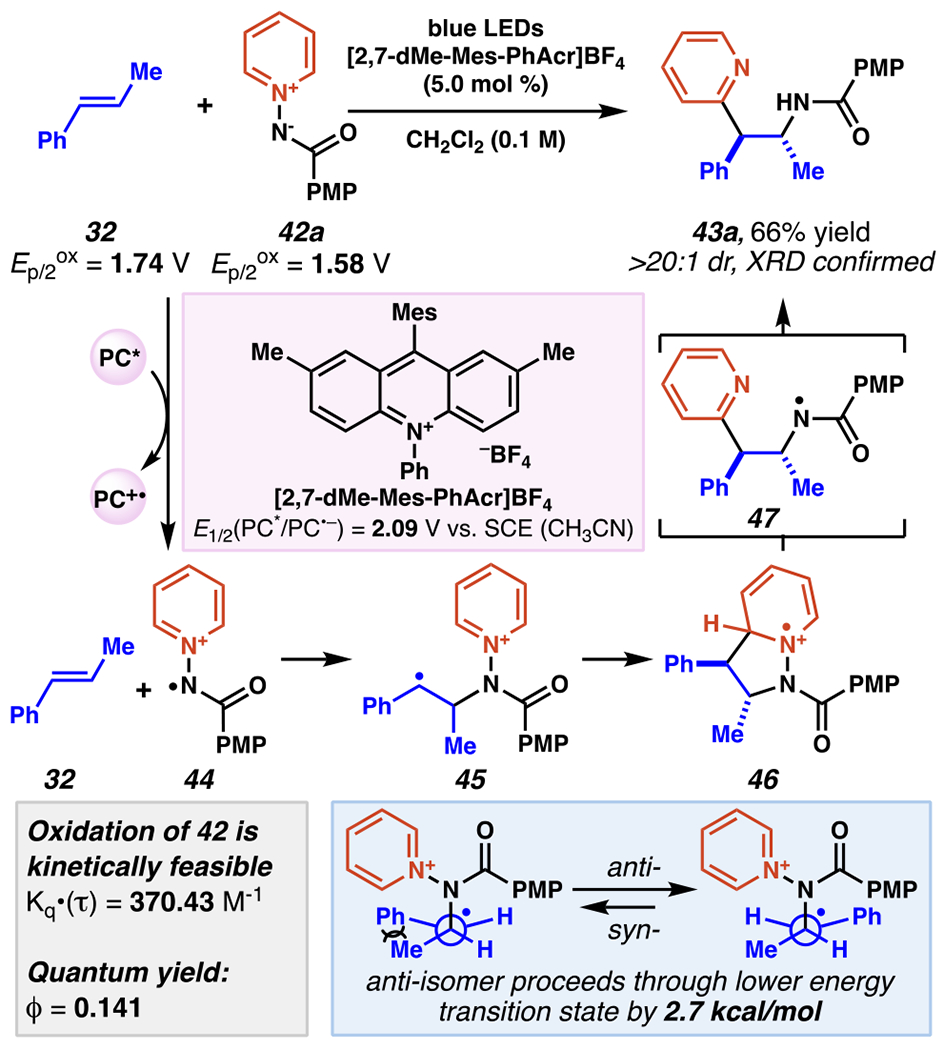
Aminopyridylation Proceeds Selectively at the Pyridyl-C(2) via Radical Formal 1,3-Dipolar Cycloaddition Process
Scheme 17.
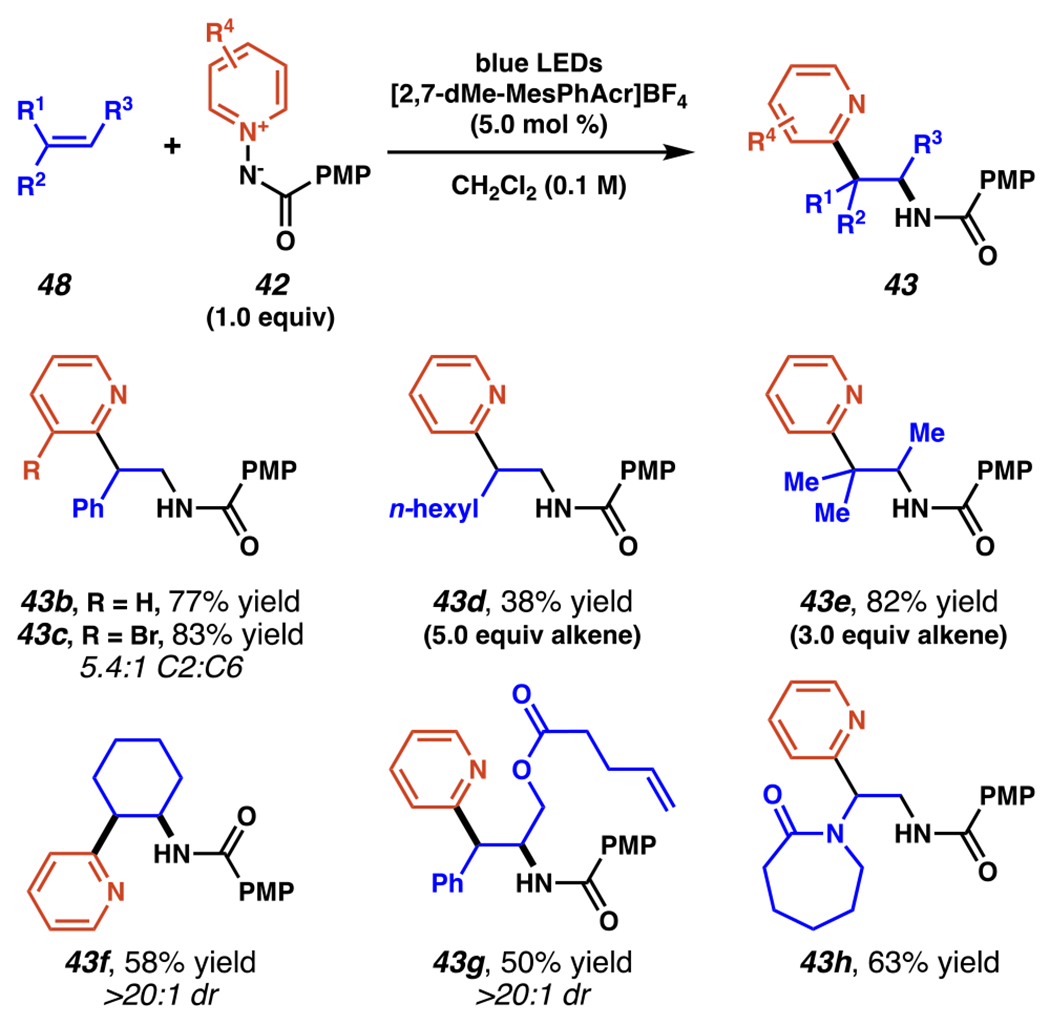
Amidopyridylation Products Are Readily Produced from Both Activated and Unactivated Alkenes
By employing phosphonate-quinonlinone photocatalyst,32 Hong and co-workers convert amino- and amidopyridinium salts via 1,5-HAT process to alkyl pyridines 50, with complete selectivity for addition at pyridine C(4) (Scheme 18).106 The reaction employs blue light irradiation in DMSO at room temperature and is reported to be complete within 30 minutes. Likely beginning with a reduction of starting material 49 (Ep/2red (49) = −0.82 V vs SCE in CH3CN) by excited-state photosensitizer (E1/2(PC*/PC•+) = −1.29 V vs SCE in CH3CN), pyridine is released to form nitrogen-centered radical intermediate 3c. This initial step is further supported by a demonstrated quenching of excited state photosensitizer by starting material 49 (Stern-Volmer experiments found Kq•(τ) = 6.98 M−1). The subsequent 1,5-HAT process furnishes carbon-centered radical intermediate 4c. This intermediate is proposed to engage in nucleophilic attack of another molecule of aminopyridinium salt 49, which occurs selectively at the pyridinium group’s C(4)-position. Pyridyl-radical intermediate 51 is likely then deprotonated and the N–N bond is broken to produce another molecule of nitrogen-centered radical intermediate 3c. The precise mechanism of deprotonation in this step is unclear; however, addition of base slightly decreased NMR yields of the optimization substrate. Quantum yield experiments (ϕ = 41.2) support a proposed radical chain propagation mechanism.
Scheme 18.

Aminopyridinium Salts are Appropriate Substrates for Directed Pyridylation Reactions
With higher concentrations of phosphonate-quinolinone photosensitizer (Scheme 18) and modulating the pyridinium salt’s anion, Hong and co-workers are able to extend their reaction protocol to include amidopyridinium salts, such as 52 (Scheme 19). Reacting at a lower overall concentration and on a longer timescale, pyridylation products 53a and 53b are synthesized with a bias toward pyridine C(4)-selectivity, albeit with meaningful amounts of C(2) minor products. Yields are generally higher for pyridylation at tertiary alkyl radicals, with a stronger bias toward pyridine C(4)-selectivity for these more sterically-hindered radicals as well. Quantum yield experiments (ϕ = 12.7) implicate an analogous radical chain propagation process to that proposed for the sulfonamide-guided pyridylation above (Scheme 18).
Scheme 19.
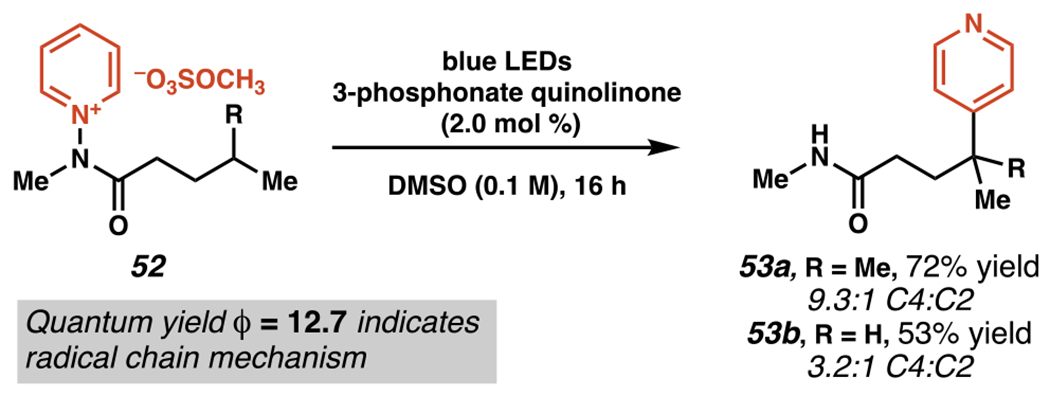
N,N-Dialkylamidopyridinium Salts Engage in 1,5-HAT-Mediated C–H Pyridylation
Introduction of N–N+ salts and N•–N+ zwitterionic species has enabled guided- and unguided-amination reactions. Loss of pyridine from the starting salts upon single-electron reduction by photocatalysts results in the slow addition of a weak base to the reaction, which has simplified systems that might otherwise require an exogenous base (Table 1 and Scheme 6). Moreover, the metered generation of nitrogen-centered radicals, through direct SET or radical chain propagation mechanisms, affords additional control over reaction progress and, in many cases, obviates potential overreaction by preventing N-centered radical generation on the same molecule after its initial reaction. These benefits must be measured against cost of additional steps required to access substrates bearing the requisite N–N bond, and, in instances where these benefits are greater, this method of nitrogen-centered radical generation will surely enable innovative chemistry for years to come.
4. NITROGEN-CENTERED RADICALS MAY BE GENERATED FROM NITROGEN–SULFUR BONDS
Few reports document photocatalyst-mediated reactions that utilize nitrogen–sulfur bonds as precursors to nitrogen-centered radicals; however, a recent example was disclosed in 2018 by Bolm and co-workers (Scheme 20A).107 The dearth of examples may be due in part to N–S bonds with low oxidation-state sulfur groups being relatively labile to direct photolysis, as demonstrated in recent C–H xanthylation reactions.108,109,110 In the presence of Ir(ppy)3, under blue light irradiation, thiocyanate substrates react with an excess of styrene to give good yields of desired, difunctionalized products (Scheme 20A). Though mechanistic information about this reaction is sparce, the observed regioselectivity, lack of additives, and lack of reaction absent photocatalyst implicate a photoredox- or photosensitizer-generated, nitrogen-centered radical mechanism.
Scheme 20.
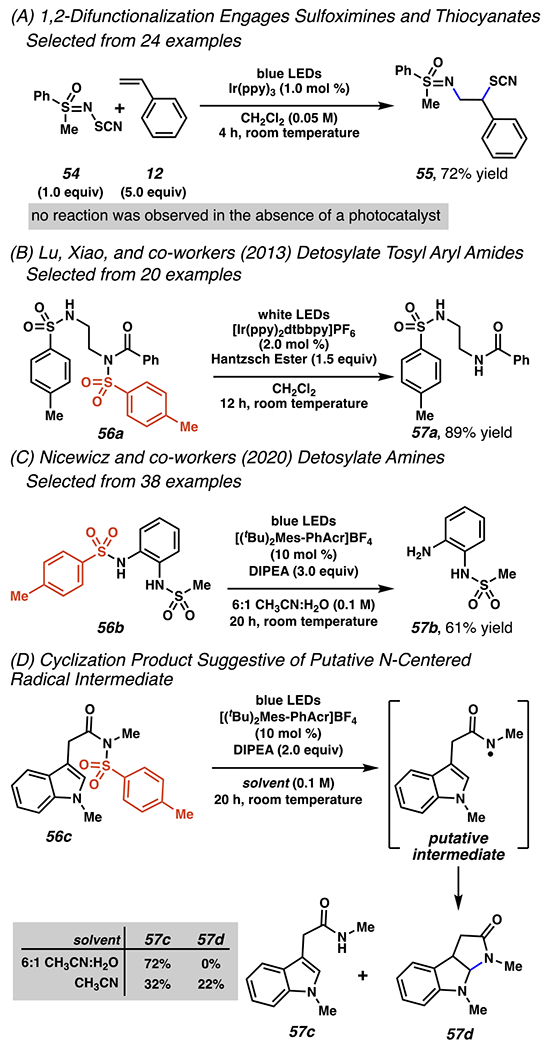
Photo-driven Nitrogen–Sulfur Bond Cleavage
Radical reactions have been described in the presence of a photocatalysis-capable compounds that involve N–S bonds, where the sulfur atom is in higher oxidation state (Scheme 20B–C).111, 112 These detosylation processes differ in terms of substrate applications and conditions. In initial disclosures by Lu, Xiao and co-workers, N,N-disubsituted sulfonamides rely on iridium photocatalyst to reduce the substrate, and Hantzsch ester as a reductant and hydrogen atom source in the course of detosylation. This protocol can enable selective detosylation of a fully-substituted nitrogen centers bearing an aryl amide in the presence of a secondary nitrogen bearing a tosyl group (Scheme 20B).111 More recently, an organic photocatalyst has been applied to affect detosylation of N-aryl, N,N-diaryl and N,N-dialkyl sulfonamides (Scheme 20C). In the presence of an (Mes-PhAcr)-based to mediate substrate reduction and base oxidation, with Hünig’s base as a reductant and hydrogen atom source, detosylation proceeds selectively on a substrate bearing a pendant methylsulfonamide (Scheme 20C).112 Under the standard reaction conditions, detosylation of amidyl 56c furnishes protonated 57c (Scheme 20D). This same substrate provides limited evidence that these reactions may proceed via nitrogen-centered radical intermediates. Specifically, absent water, amidyl 56c furnishes both protonated 57c. and cyclized 57d (Scheme 20D). This byproduct is suggestive of the presence of a nitrogen-centered radical as a putative reaction intermediate.
5. NITROGEN-CENTERED RADICALS CAN BE GENERATED FROM NITROGEN–OXYGEN BONDS
Nitrogen-centered radicals are accessible from the nitrogen–oxygen bonds within oximes, oximides, oxyamides, benzenesulfonamides, amidoximes and imidates. The generated nitrogen-centered radicals participate in bond-forming reactions with appropriate radical trapping agents. This section summarizes reactions that propose neutral nitrogen-centered radical species as intermediates. Many complementary reactions are outside of the scope of this review, including promising transformations that rely on photocatalyst-mediated N–O bond cleavage with pyridinium salts to generate pyridyl radical cation intermediates.113, 114
5.1. Iminyl Radicals Can Be Generated from O-Aryl and O-Acyl Oxime Analogues
5.1.1. Radicals Prepared from O-Aryl or O-Acyl Oximes Can Add Across Olefins
Iminyl radicals are important synthetic intermediates,115 and are accessible from O-aryloximes. When incorporated into γ,δ-unsaturated ketoximes, these O-aryloxime precursors can be converted into 1-pyrroline analogues116,117 via iminyl radical intermediates. Within substrate O-aryloximes, a weak N–O bond is amenable to homolytic fission to reveal the corresponding nitrogen-centered iminyl radical.118 The bond homolysis event can be triggered thermally, photochemically, or in the presence of single-electron reductants such as phenolate, Fe- and Cu-salts, SmI2 and SnBu3H.119
At the end of the 20th century, Narasaka and co-workers discovered that iminyl radicals could be generated from O-aryloximes via single-electron reduction.120,121,122 The accessed iminyl radical intermediate is poised to undergo 5-exo-trig cyclization with a pendant olefin to deliver a nucleophilic carbon-centered radical.122 Subsequent trapping of the radical with a hydrogen atom or other electrophile provides 1-pyrroline derivative. Uncovering this photochemical process propelled Narasaka and co-workers to develop a photocatalyst-mediated strategy to convert O-aryloximes to 1-pyrrolines.122 In 2001, Mikami and Narasaka disclosed photoredox catalysis as a tactic to transform γ,δ-unsaturated O-aryl oximes to pyrrolines (Scheme 21A).123,124,125 In this transformation, following photoexcitation of 1,5-dimethoxynaphthalene (1,5-DMN, E1/2 (DMN+/*DMN) = −2.5 V vs. SCE (CH3CN))a the excited state photocatalyst is proposed to reduce an O-aryl oxime to furnish a phenolate and the putative iminyl radical intermediate. The generated iminyl radical intermediate undergoes 5-exo-trig cyclization, and, in more efficient reactions, the resultant carbon-centered radical intermediate is quenched using 1,4-cyclohexadiene as a hydrogen atom source.
Scheme 21.
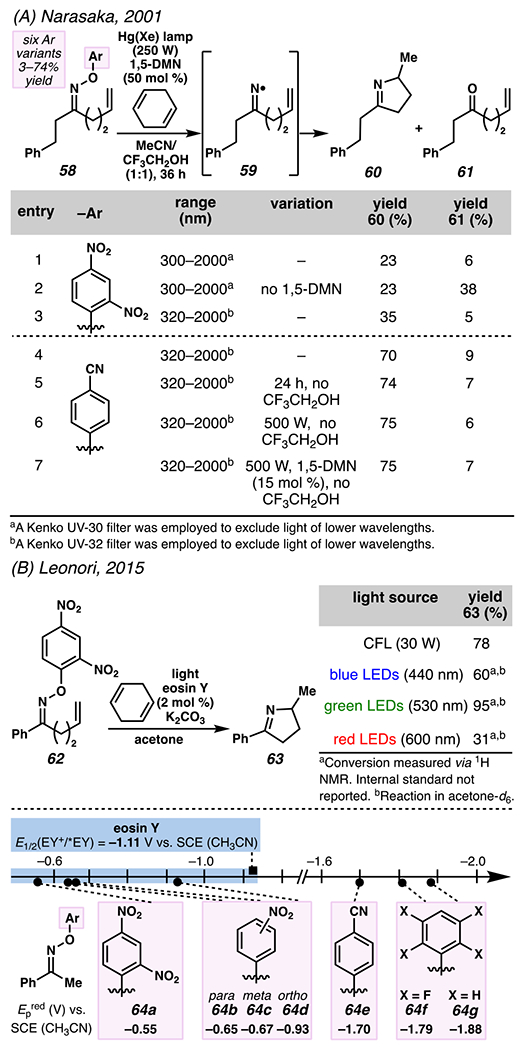
Photoredox Catalysis Transforms O-Aryl Oximes to Iminyl Radicals, Which Participate in 5-exo-trig-Hydroimination Reactions to Afford Pyrrolines
Reaction efficiency is sensitive to the wavelength of the engaged light source, as substrate 58 absorbs irradiation beneath 320 nm. Consequently, absent photosensitizer, O-aryl oxime 58 undergoes homolytic cleavage to generate undesired ketone byproduct 61, along with pyrroline 60 (entry 2). Pyrroline 60 forms more efficiently when light below 320 nm is excluded from the reaction (entry 3), and with O-aryl oxime 58. With more effective O-aryl oximes, lower catalyst loadings can prove equally efficient (entry 7). Ultimately, this practical reaction highlights the promise of photoredox catalysis as a tactic to use O-aryl oximes as stable precursors to iminyl radical intermediates.
Leonori and co-workers126 advanced the technology from Misaka and Narasaka with a synthetic protocol that relies on an O-aryloxime as iminyl radical precursor but requires a lower photocatalyst loading and irradiation using a less intense light source (Scheme 21B). Instead of 1,5-dimethoxynaphthalene, eosin Y (EY) is employed as the photocatalyst, which elicits the desired transformation at 2 mol % catalyst loading. Leonori and co-workers are able to replace the use of a specialized Hg(Xe) lamp, and employ less intense visible light sources. In their research, photoexcitation of eosin Y prompts SET between *EY and O-aryl oxime 62 to generate iminyl radical that participates in a 5-exo-trig hydroimination reaction to furnish 1-pyrroline 63 in 78% yield.
In conjunction with these tactical modifications, Leonori and co-workers evaluated a similar range of aryl groups within the precursor oxime to permit efficient conversion of the O-aryl oxime to the product. Oximes 64a–64d possess the appropriate reduction potential (Epred) to be irreversibly reduced by eosin Y (E1/2[EY+/*EY] = −1.11 V vs. SCE in CH3CN). 2,4-Dinitroaryl oximes, such as 64a, were chosen for more extensive explorations due to their relatively low Epred, which favor irreversible SET between eosin Y and oxime, and because these oximes are relatively easy to purify.
The method advanced by Leonori and co-workers is similar to the method developed by Misaka and Narasaka inasmuch as this reaction is sensitive to the wavelength of the light source. When comparing the efficiency of a reaction under blue, green, or red LEDs, the most efficient process relies on green LEDs (λ = 530 nm), which promote the reaction of starting material 62 to 1-pyrroline 63 in 95% yield. This improvement in efficiency might be predicted, as eosin Y is known to absorb green light (λabs = 539 nm)127 optimally to yield *EY. Consequently, the result from the experiment allows the authors to suggest that the data are consistent with reaction mechanism that is initiated by SET between EY* and O-aryl oxime 62.
Nearly concurrently, a similar cascade sequence was described that, relies on the conversion of O-acyloximes via single-electron reduction through iminyl radical intermediates and 5-exo-trig cyclization onto a pendant olefin to afford a nucleophilic carbon-centered radical. At this point do the processes diverge – the generated radical can be trapped by an electron-rich olefin to furnish pyrroline products with pendant ketones (Scheme 22A).128 This transformation is highly sensitive to the reaction solvent. Broadly, while polar protic and amine solvents (excepting pyridine) greatly hinder or arrest reactivity, polar aprotic solvents are required for product formation. Of these, DMF was chosen as the optimal solvent. Solvents such as DMA and NMP also displayed high conversion of starting material, but favor formation of undesired 67 over desired 66. This reaction manifold is robust to variety of aryl-substituted oximes, such E- and Z-configurated 2-thiophenyl oximes. The reaction is ineffective with styrenyl or phenylacetylynyl oximes, and with tri-substituted and sterically congested silyl enol ethers (not depicted). This reaction is suitable for downstream derivatization of pyrrolines to pyrrolizidine alkaloids (Scheme 22B).128
Scheme 22.
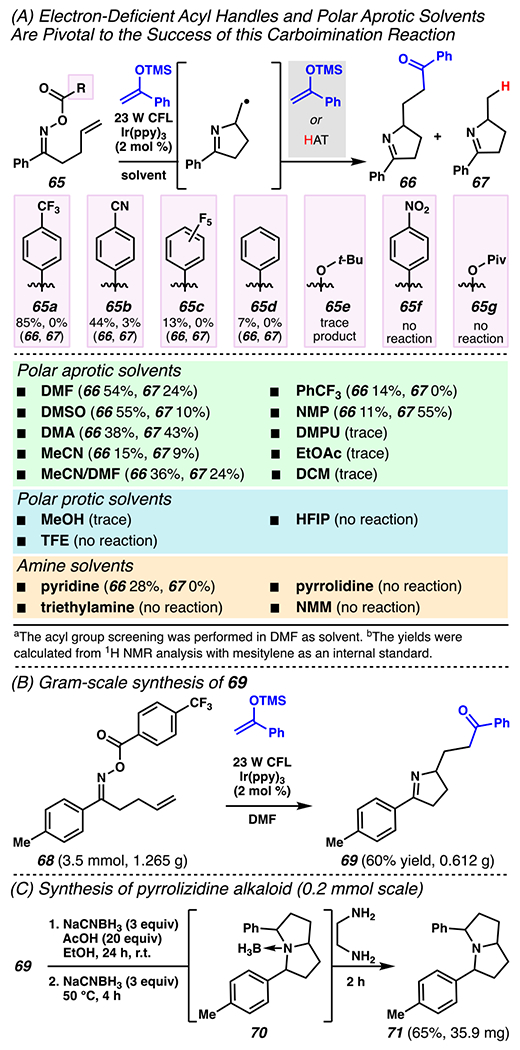
Effective Carboimination Reaction Relies on O-Acyl Oxime in Scaleable Reaction That Provides Rapid Entry to Pyrrolizidine Alkaloid Derivative
Insofar as an iminyl-radical-generation / 5-exo-trig cyclization cascade permits rapid access to pyrroline scaffolds, by analogy, use of an oxime imidate substrate decorated with pendant olefin should provide an oxazoline. This recognition makes the reaction cascade exponentially more powerful than initially envisioned: a masked allyl alcohol can serve as an imidate radical precursor to provide access to oxazolines and masked 1,2-amino alcohols (Scheme 23).129 These reactions rely on an intramolecular cyclization / radical trapping cascade to achieve net aminoalkylation, aminoarylation, and hydroamination reactions. A subsequent acidic work-up can reveal 1,2-amino alcohol products, providing efficient access to high-value targets.
Scheme 23.
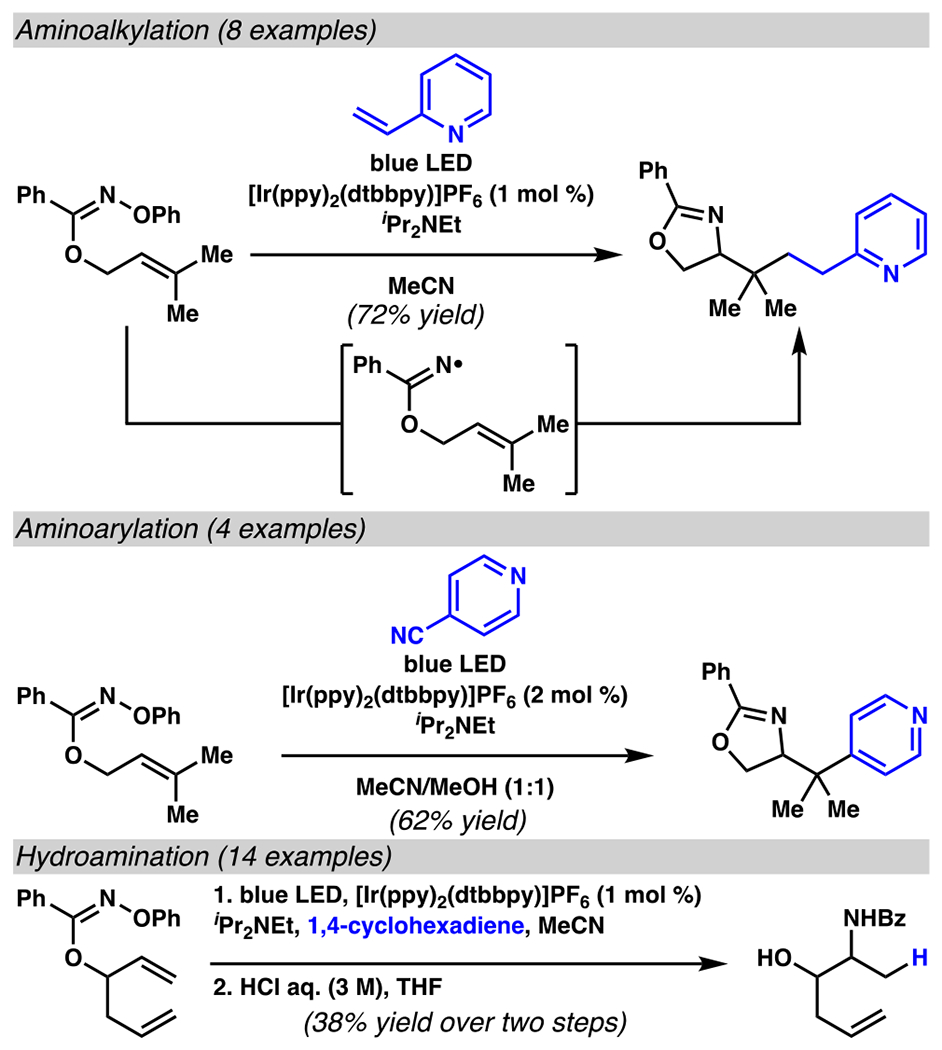
O-Aryl Oximes Can Serve as Imidate Radical Precursors Sequences to Access Masked 1,2-Aminoalcohols
5.1.2. Radicals Prepared from O-Acyl Oximes Can Cyclize onto Arenes or Vinyl Arenes
As a complement to these approaches, iminyl radicals derived from oximes with conjugated π-systems engage in 6-endo-trig cyclization reactions to provide N-heterocycles such as phenanthridines, quinolines, and pyridines (Scheme 24).120,121,130 To access this reactivity, Yu and Zhang activate O-acyl oxime substrates by photoirradiation with visible light in the presence of Ir(ppy)3 photoredox catalyst, ultimately furnishing a series of heterocyclic products.131 These transformations rely on irreversible reduction of starting O-acyl oximes 72 by the photocatalyst (E1/2[IrIV/Ir*III] = −1.73 V versus SCE)16 to reveal iminyl radicals, a process that benefits from the selection of an appropriate acyl precursor (Scheme 24A). Electron-deficient benzoyl groups 72a–72c affect the conversion of O-acyl oximes 72 to phenanthridines 73 in 91% to quantitative yield. By contrast, phenanthridines are not isolated from reactions that rely on more electron-rich, less readily reduced substrates that incorporate benzoyl groups, aryloxy carbonates, or an acetoxy group (i.e., 72d–72h). Presumably, variations in ease of reduction reflect the electronic influence of O-acyl groups on the σ*N–O orbital energy of O-acyl 72.132 While pentafluoro-substituted substrate 72b delivers the desired product in quantitative yield, p-trifluoromethyl 72a is an attractive redox-active handle over 72b due to its slightly lower molecular weight.
Scheme 24.
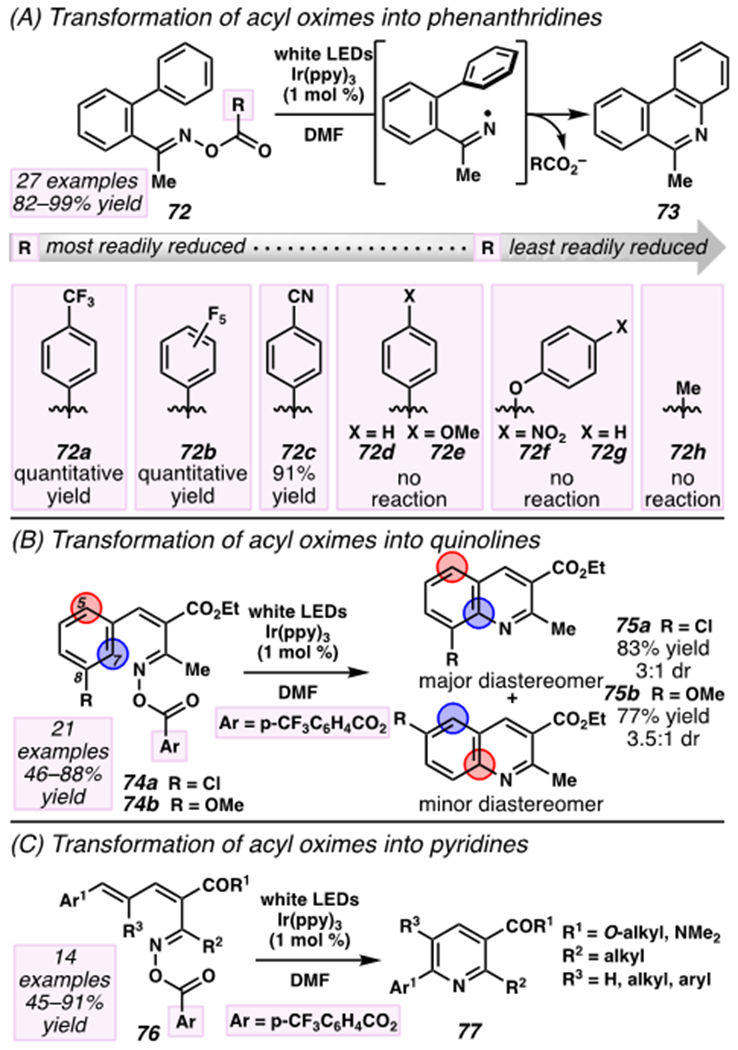
Iminyl Radical with Conjugated π-System Participates in 6-endo-trig Cyclization to Assemble N-Heteroarenes
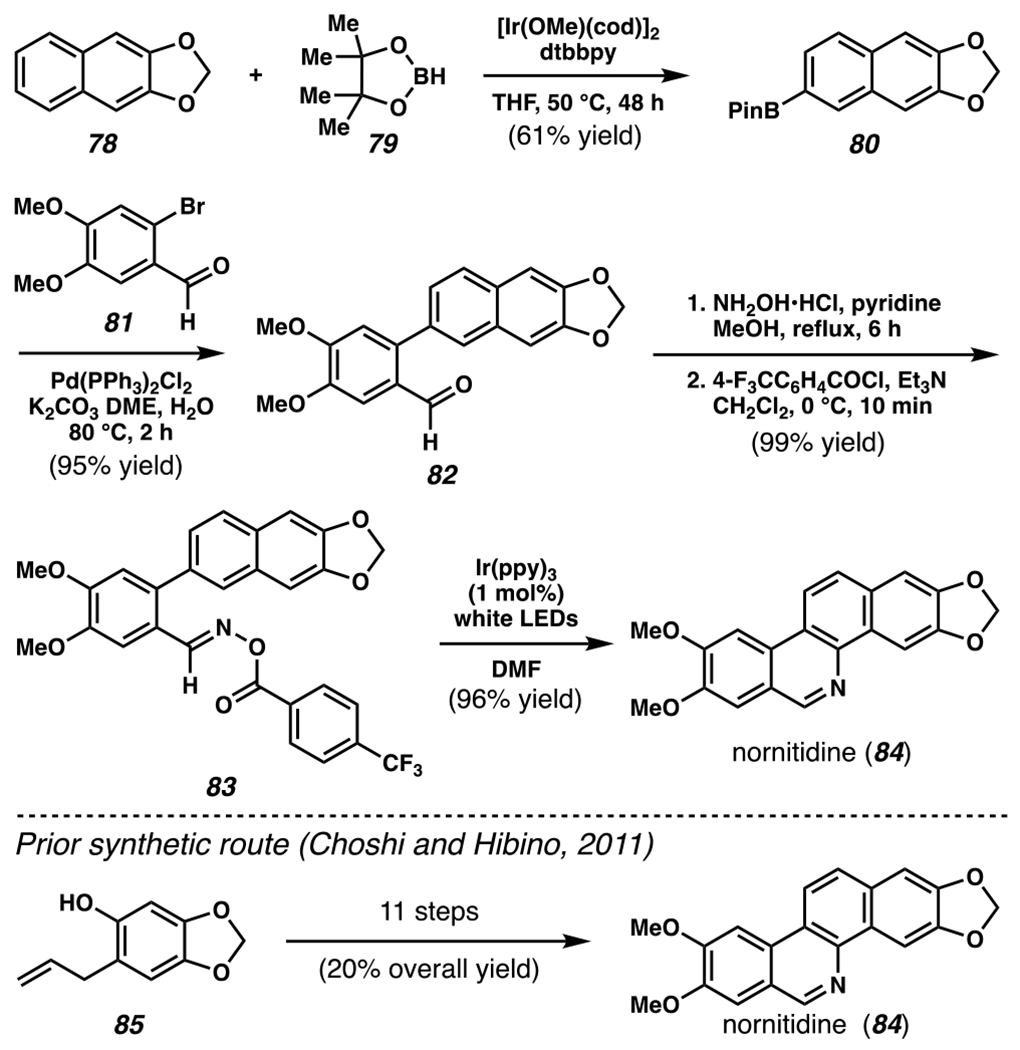
The transformation is robust to a wide variety of O-acyl oximes to provide differentially substituted phenanthridines, quinolines, and pyridines (Scheme 24B–C). Notably, with meta-substituted arene substrates 74a and 74b, accessed iminyl radical intermediates add across the aromatic π-systems at two sites competitively to deliver mixtures of positional isomers as products (i.e., 75a and 75b, Scheme 24B). Importantly, this strategy has been applied to improve access to nornitidine, a benzo[c]phenanthridine derivative with anti-tumor properties (Scheme 25). With these technologies, nornitidine can be accessed in five steps and 55% overall yield, a considerable advance over a previous 11-step route (20% overall yield).133 This achievement underscores the powerful potential of photoredox catalyst and nitrogen-centered radicals for organic synthesis.
Scheme 25.
Yu and Zhao Use This Technology to Accelerate Access to Nornitidine
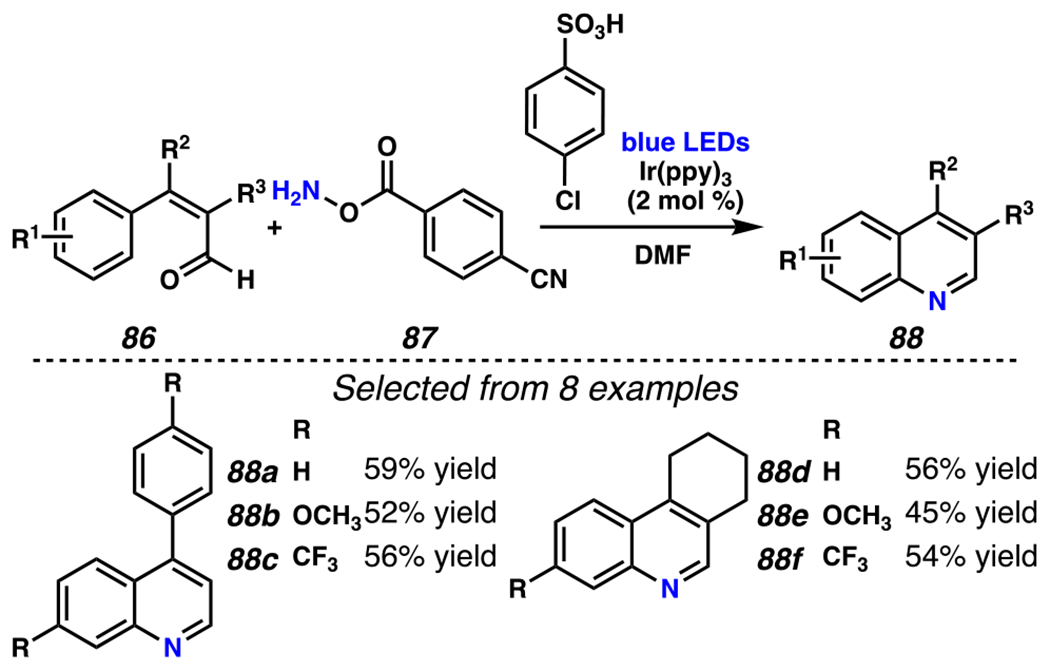
This general approach to preparing quinolines and phenanthridines would be more powerful if it were to rely directly on readily available aldehydes as starting materials, rather than on aldehyde-derived O-acyl oximes. Consequently, Yu and co-workers have disclosed a one-pot protocol to convert aldehydes via O-acyl hydroxylamines into quinolines and tetrahydrophenanthridines (Scheme 26).134 With this method, an aldehyde condenses with an O-acyl hydroxylamine with a 4-cyanobenzoyl group, and the intermediate O-acyl oxime then proceeds through a 6-endo-trig cyclization reaction, obviating the syntheses and isolations of requisite O-acyl oximes. This protocol depends on blue LEDs as the reaction light source, as undesired side products form when a light source is used that emits light across a broader wavelength range. This approach has been employed with biaryl aldehydes and O-4-cyanobenzoyl hydroxylamine to afford phenanthridines, as exemplified by a two-step gram-scale synthesis of trisphaeridine, an alkaloid possessing antitumor and antiretroviral properties (Scheme 27).135,136,137 This procedure represents an improvement over the previous best-in-class synthetic route to trisphaeridine, which requires four synthetic steps from the same aryl bromide 89 and provides trisphaeridine (92) in 29% overall yield.138
Scheme 26.
O-Acyl Hydroxylamines React with Aryl Aldehydes in a One-Pot Quinoline Syntheses
Scheme 27.

O-Acyl Hydroxylamines React with Biaryl Aldehydes to Afford a One-Pot Syntheses of Phenanthridines, Which Is Used in a Two-Step Synthesis of Trisphaeridine
5.1.3. Radicals Prepared from O-Acyl Oximes Can Abstract Hydrogen Atoms
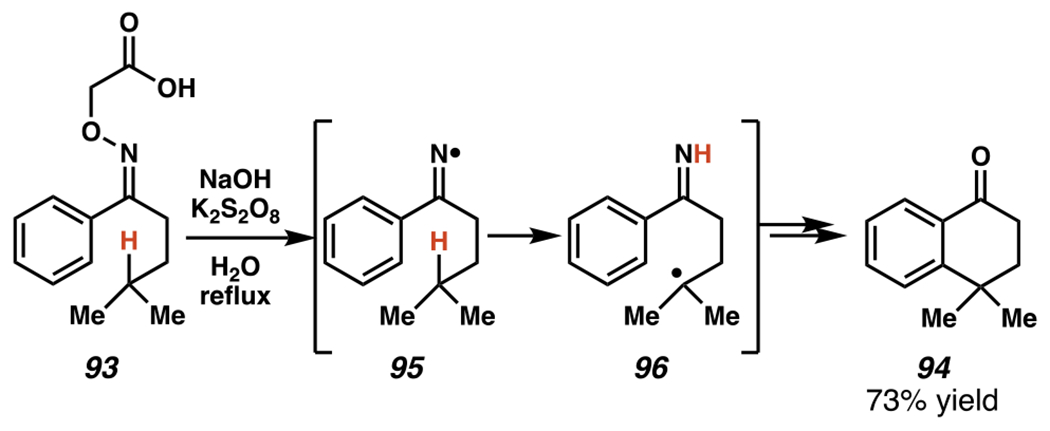
In addition to adding across olefins, or aromatic rings, iminyl radicals can affect C–H abstraction, typically by way of 1,5-hydrogen atom transfer (HAT) based on a six-membered transition state. Indeed, oximes have been used as precursors for iminyl radicals that engage in 1,5-HAT prior to addition across an arene (Scheme 28). These technologies build on seminal resesarch by Forrester and co-workers.139,140 In these reactions, thermolysis of the oxyacetic acid 93 under basic and oxidative conditions triggers a cascade sequence to form iminyl radical 95, which affects 1,5-HAT to generate carbon-centered radical 96. Once formed, this nucleophilic radical adds across an aromatic π-bond to deliver a cyclic imine (not depicted). Upon rearomatization and in situ hydrolysis, this sequence affords 1-tetralone product 94 (Scheme 28). Considering the allure of 1-tetralones as monoamine oxidase inhibitors for Parkinson’s disease therapy141, milder photoredox methods to generate 1-tetralones from oxyacetic acid derivatives have emerged.
Scheme 28.
Forrester and Co-workers Pioneer Cascade Sequence to Convert Oxime through Iminyl Radical Intermediate, Which Engages Sequentially in an HAT Process and Addition Across an Arene Prior to Hydrolysis
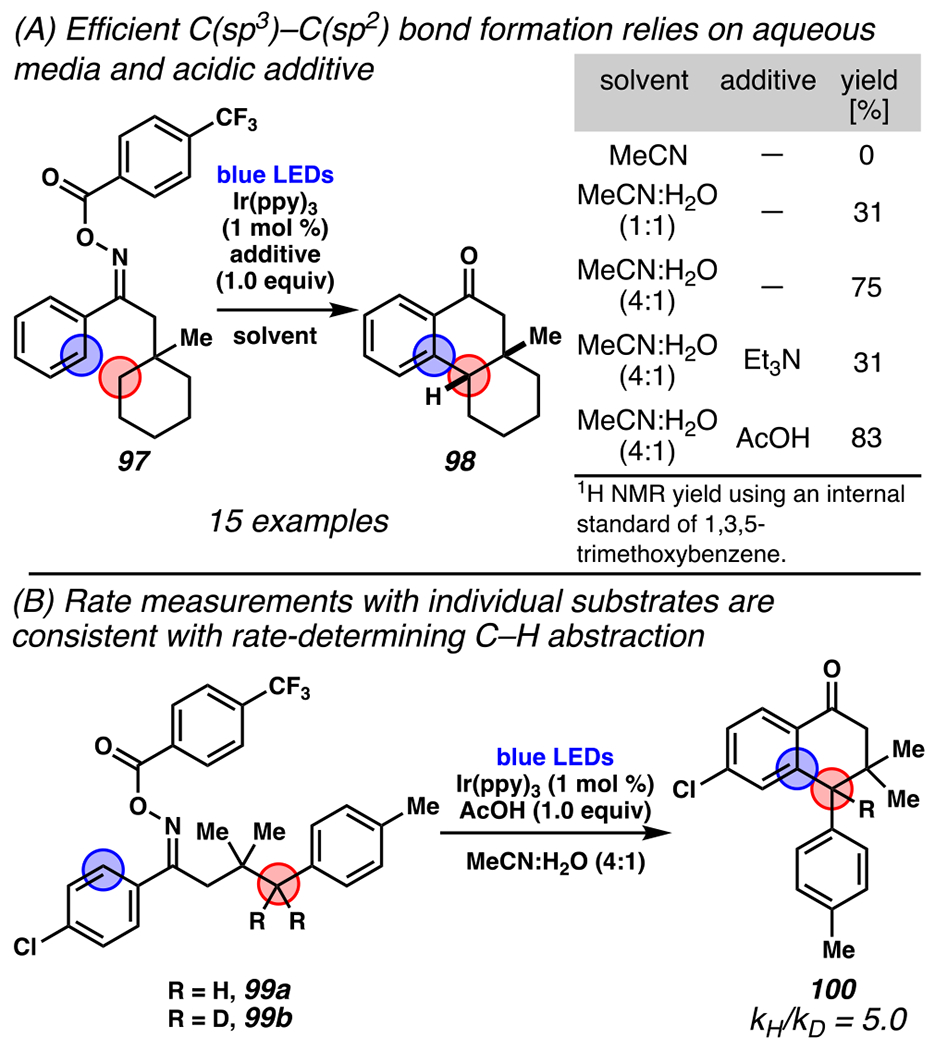
Inspired by this transformation, Nevado and co-workers developed the first photoredox catalyst-mediated process that harnesses this strategy with O-acyl oxime substrates (i.e., 97, Scheme 29). 142 To date, the transformation of O-acyl oximes to 1-tetralones only proceeds in the presence of water as co-solvent, and is most effective in the presence of an acid additive (AcOH), which is thought to facilitate a reversible and rate-determining HAT event (Scheme 29A).140 Competent substrates range from those containing electron-deficient to electron-dense arenes as oxime substituents, benzofurans, or pendant aliphatic rings sizes (not shown). To add complexity to this advance, under basic conditions, in the presence of light, photocatalyst and in a non-aqueous solvent system, these same O-acyl oxime substrates undergo homolytic cleavage and 1,5-HAT, but then subsequently react to provide 1-pyrrolines (Scheme 30). Reaction diastereoselectivity is limited when not reinforced by a cyclic ring.
Scheme 29.
Cyclization Reaction Relies on Aqueous Media and Acidic Additive, and Involves Rate-Determining C–H Abstraction
Scheme 30.
Complementary Reaction Conditions Trigger Chemodivergent C(sp3)–N Bond Formation to Provide 1-Pyrrolines
5.2. Hydroxyacid-Derived Oximes Are Appropriate Precursors to Iminyl Radicals
5.2.1. These Iminyl Radicals Can Serve as Intermediates in Iminofunctionalization Reactions
While O-acyl oximes serve as efficient precursors to iminyl radical intermediates, many similar transformations can be achieved using hydroxyacid-derived oximes. Indeed, nearly fifty years ago, Forrester and co-workers discovered that hydroxyacid-derived oximes are appropriate precursors to iminyl radicals.143,144 Building on this research, thirty years ago, Boivin, Zard and co-workers employ a thusly-derived iminyl radical intermediate in a 5-exo-trig cyclization. The resultant carbon-centered radical reacts with methyl acrylate in a Giese-type reaction to afford alkylated 1-pyrroline.145, 146, 147 These pivotal findings set the stage for analogous technologies that employ milder photoredox-catalyst conditions to streamline access to functionalized 1-pyrroline (Scheme 31–32).
Scheme 31.
Jiang and Studer Demonstrate That Hydroxyacid-Derived γ,δ-Unsaturated Ketoximes Engage in the Giese Reactions
Scheme 32.
A Photocatalyst That Readily Oxidizes the Hydroxyacid Handle Is Critical in the Success of the Iminyl Radical-Initiated Giese-Type Reactions
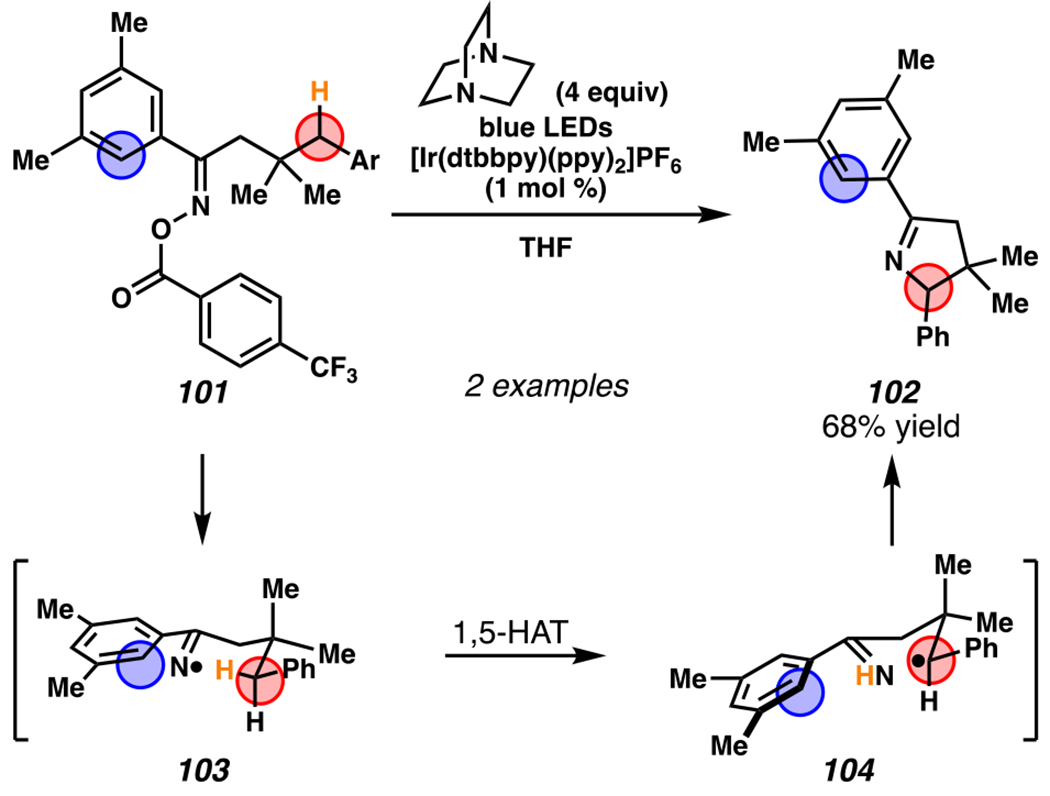
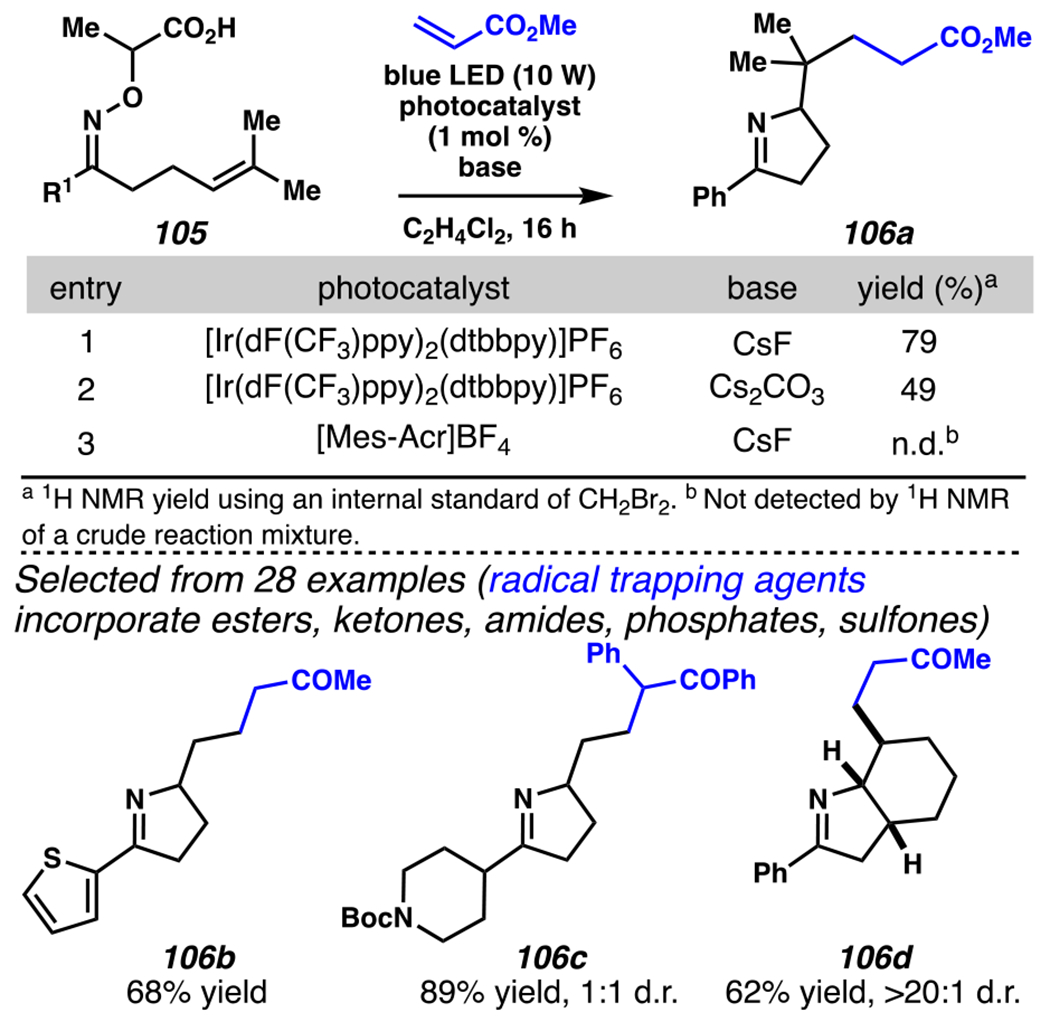
In 2017, Studer, Leonori, and co-workers developed photoredox catalyst-mediated tactics to convert hydroxyacid-derived γ,δ-unsaturated ketoximes, such as 105, to iminyl radicals.148, 149 Following intramolecular 5-exo-trig cyclization, the resultant radicals can be trapped by olefin in intermolecular Giese reactions.145 In Jiang and Studer’s cascading Giese-type reaction, γ,δ-unsaturated ketoxime 105 is transformed to desired product 106a in 68% yield when [Ir(dF(CF3)ppy2)(dtbbpy)]PF6 is employed as photosensitizer and CsF as a base (Scheme 31). Using this strategy, a wide catalogue of Michael acceptors participates in the Giese reaction to access alkylated 1-pyrrolines. For example, thiophene-substituted ketoxime and a pendant piperidine are carried through the reaction to furnish pyrrolines. Furthermore, under these conditions a cascading cyclization reaction proceeds with excellent diastereoselectivity to generate a [5-6]-fused bicyclic system (i.e., 106d).
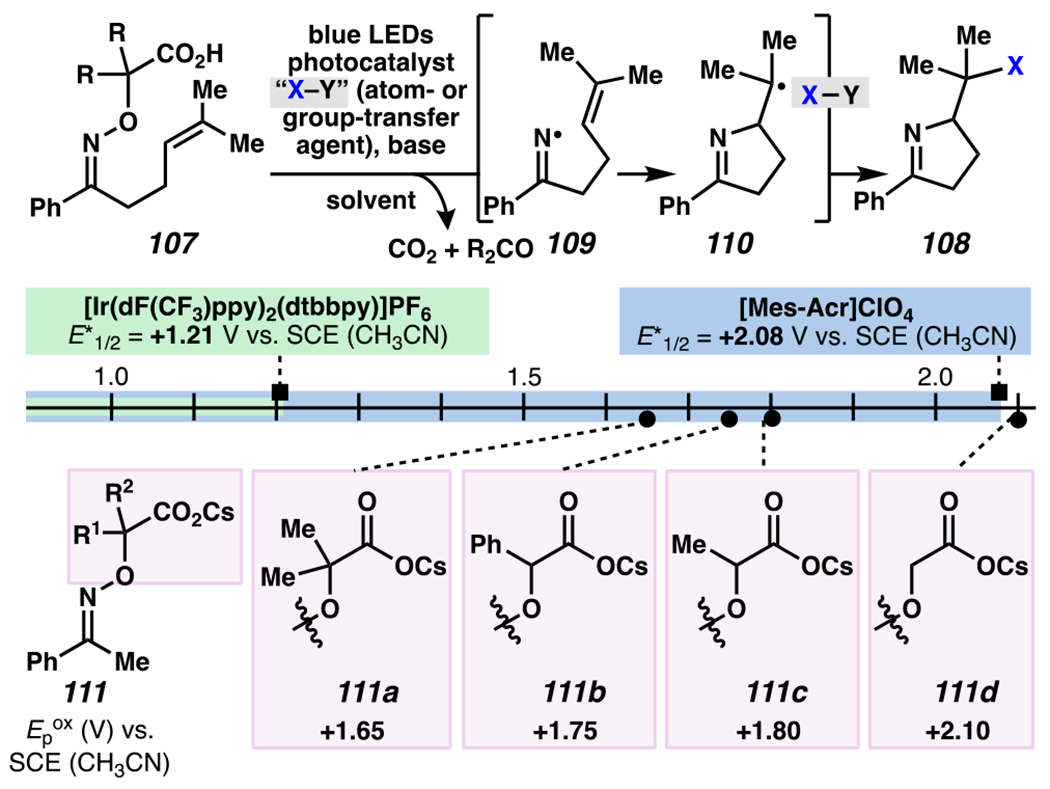
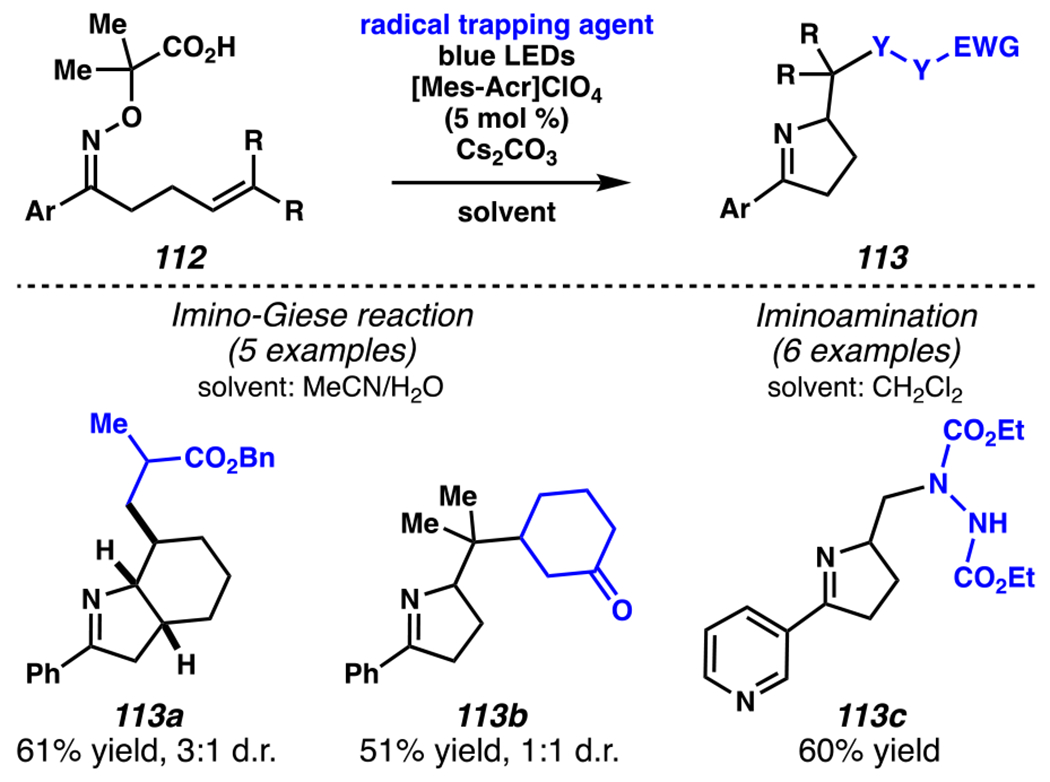
By contrast, Leonori and co-workers rely on [Mes-Acr]ClO4 to affect their iminofunctionalization reactions (Scheme 32). Concurrent with Jiang and Studer’s disclosure, Leonori and co-workers disclosed a very similar approach to iminofunctonalization reactions – one that proceeds through an identical iminyl radical intermediate (Scheme 33). To design an optimal iminyl radical precursor, Leonori and co-workers analyzed the electrochemical half potentials for a series of carboxylates, and determined that α,α-dimethylated carboxylates,150 such as 107, are more readily oxidized than their less substituted analogues (i.e., 111a–111d). Consequently, Leonori and co-workers rely on these more substituted analogues as precursors to their iminofunctionalization reactions. Leonori and co-workers anticipated that these carboxylates would be oxidized in the presence of light by excited state *[Mes-Acr]ClO4 (E*1/2 = +2.08 V vs. SCE in CH3CN),27 a process that is reported to be infeasible with excited state *[Ir(dF(CF3)ppy2)(dtbbpy)]PF6 (E*ox = +0.89 V vs. SCE in CH3CN).8 Upon decarboxylation and loss of acetone, an iminyl radical would form, poised for cyclization onto a pendant olefin. Following cyclization, the resultant carbon-centered radical could trap a broad array of radical electrophores.
Scheme 33.
Concurrently, Leonori and Co-workers Demonstrate That Hydroxyacid-Derived γ,δ-Unsaturated Ketoximes Engage in the Giese Reactions
Like Jiang and Studer, Leonori and co-workers evaluate α,β-unsaturated electrophiles with their novel iminyl radical precursors (Scheme 33). Cascading cyclization reactions proceeded with modest diastereoselectivity to generate a [5-6]-fused bicyclic system (i.e., 113a). Moreover, the reaction tolerates a range of α,β-unsaturated carbonyl electrophiles, including cyclohexenone and diethyl (E)-diazene-1,2-dicarboxylate, to form products such as 113b and 113c, respectively.
Leonori and co-workers’ iminofunctionalization strategy can be generalized to affect iminohalogenation, iminochalcogenation, hydroimination, iminoazidation, and iminoolefination processes by varying the solvent, base, and employed atom- or group-transfer agent (Scheme 34). These reactions rely on a hydroxyacid-handle as an iminyl radical precursor, and [Mes-Acr]ClO4 as a photosensitizer. Halogenated 1-pyrrolines are accessed when NFSI151, NCS152, diethyl bromomalonate153, and NIS154 are employed as halogen-transfer agents. In this context, one limitation is well documented: the iminoiodination reaction results in synthetically useful yields when a terminal olefin is engaged but is not efficient when di- and trisubstituted olefins are targeted as the sites for installation of a relatively weak carbon–iodine bond (not shown). Stronger C(sp3)–S and C(sp3)–Se bonds can be accessed when benzosuccinimide derivatives are used as chalcogen group-transfer agents.155,156 Furthermore, azides can be installed through the use of arylsulfonyl azide as a group-transfer agent.157 If a reductively neutral process is targeted, hydroimination processes proceed in the presence of aryl disulfide, a known catalytic hydrogen-atom donor.158 Alternatively, iodine (V) sources are appropriate precursors to enable group-transfer in the course of carbon–carbon bond-forming reactions.159,160,161 Ultimately, this is a versatile approach to enable targeted iminofunctionalization reactions.
Scheme 34.
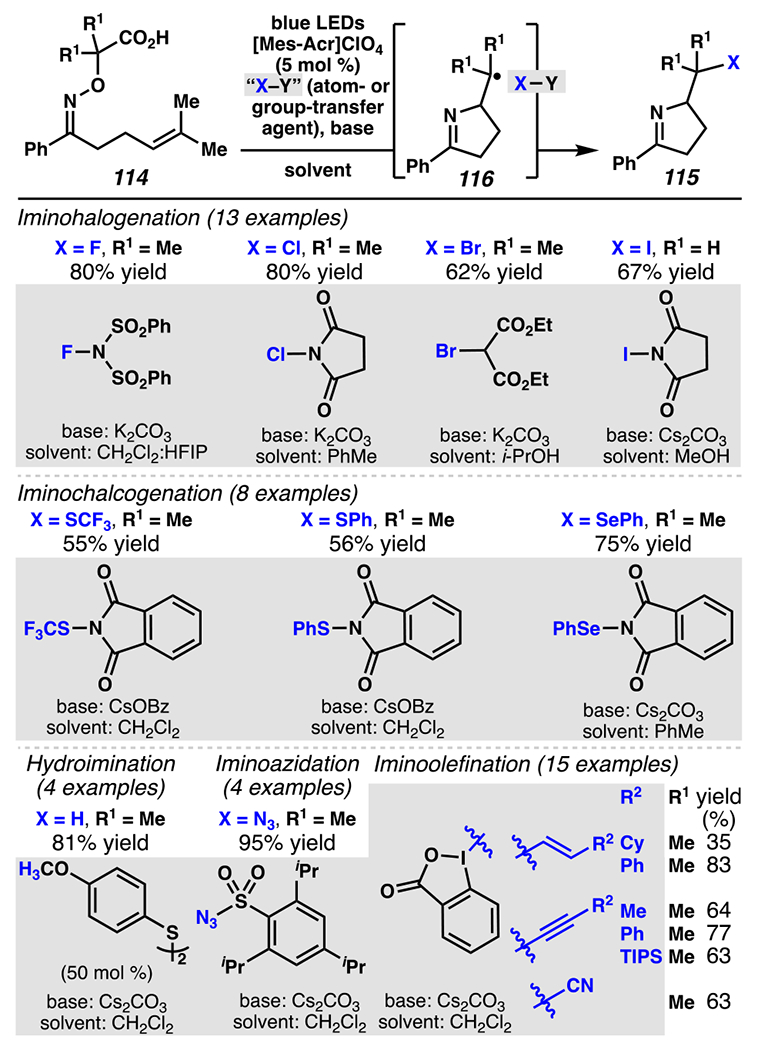
Hydroxyacid-Derived Oximes Are Appropriate Substrates for Iminofunctionalizaton Reactions
Moreover, this approach to iminofunctionalization can enable late-stage reactions with synthetically useful efficiencies. In the late 20th century, Zard and co-workers used a thevinone (117) derivative to showcase an intramolecular hydroiminaton reaction (Scheme 35A). Zard and co-workers converted thevinone to oxime 118, which, following homolysis of the weak N–O bond, cyclizes to furnish imine 119.162 Building upon this precedent, Leonori and co-workers employ thevinone derivative 117 as a precursor for a series of photoredox mediated iminoazidation, iminoamidation and iminoselenylation reactions to access polycyclic imines 121a–121c, respectively (Scheme 35B).149
Scheme 35.
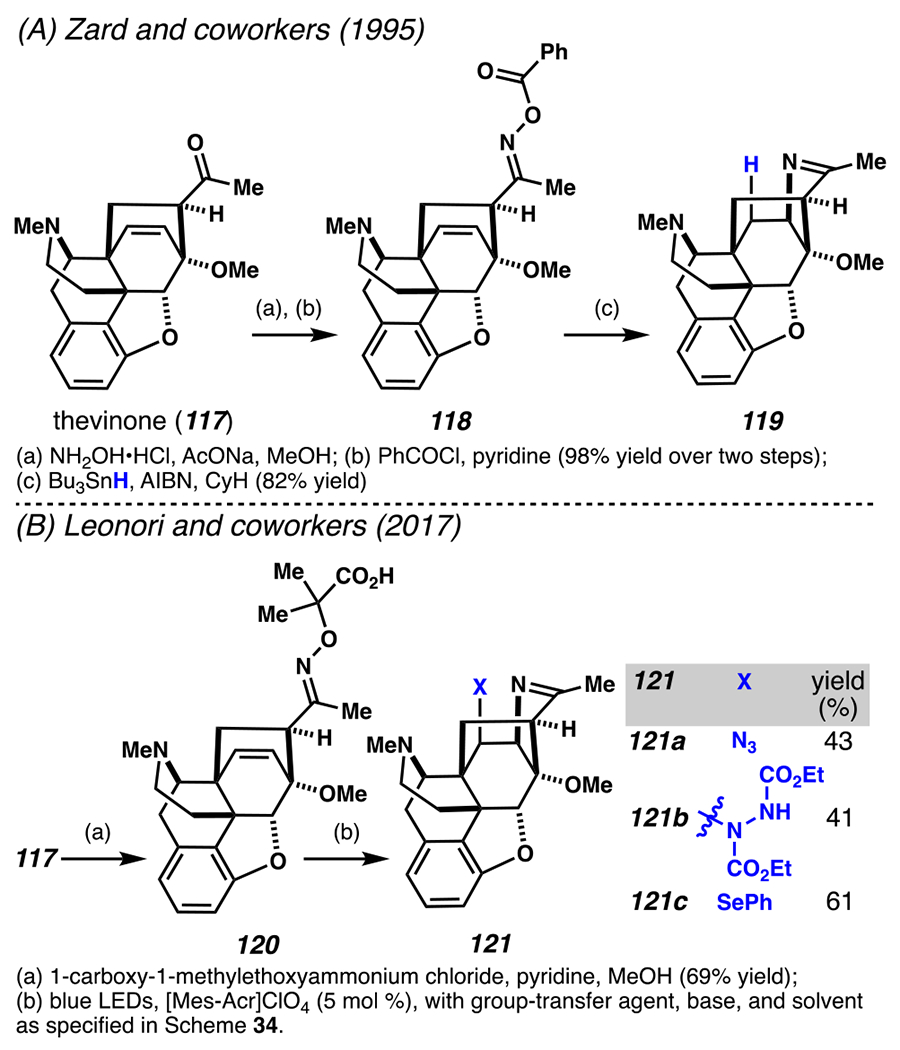
Late-Stage Iminofunctionalization Reactions Highlight the Potential of Radical Intermediates
Similarly designed iminyl-radical precursors can, upon radical-addition to a pendant olefin, be intercepted by a cobalt complex en route to an intramolecular Aza Heck-type cyclization reaction to provide pyrrolines (Scheme 36A).163 In the presence of a [Mes-Acr]ClO4 photoredox catalyst and Co(dmgH)(dmH2)Cl2 complex, oxime ester 122 undergoes cyclization reaction to provide organocobalt intermediate 124, which is poised for β-hydride elimination to furnish product 123. Previously, this pyrroline scaffold was accessed via the Narasaka-Heck cyclization between oxime ester and pendant olefin with a palladium catalyst.164,165 Photocatalytic conditions are relatively mild, so the dual photoredox/Co catalysis manifold offers advantage over the Narasaka-Heck reaction by tolerating oxime esters bearing substituents that are sensitive to Pd or Cu complexes. For example, an oxime ester bearing aryl iodide participates in the reaction to provide pyrroline 123a in 53% yield. This reaction enables rapid construction of polycyclic scaffolds, such as cis-fused tetracyclic product 123b in 49% yield in 1:1 dr. Modifying reaction condition and employing Co(dmgH)2(4-CN-Py)Cl2 complex permits radical Aza-cyclization / intermolecular Heck-type reaction cascade with oxime ester 122 and olefin 125 to provide pyrroline motif 126 (Scheme 36B). Similarly powerful examples come from the synthesis of thiophene-decorated pyrroline 126a and pyrroline bearing 1,1,2-trisubstituted internal olefin 126b in moderate yields and with high selectivity for (E)-isomer.
Scheme 36.
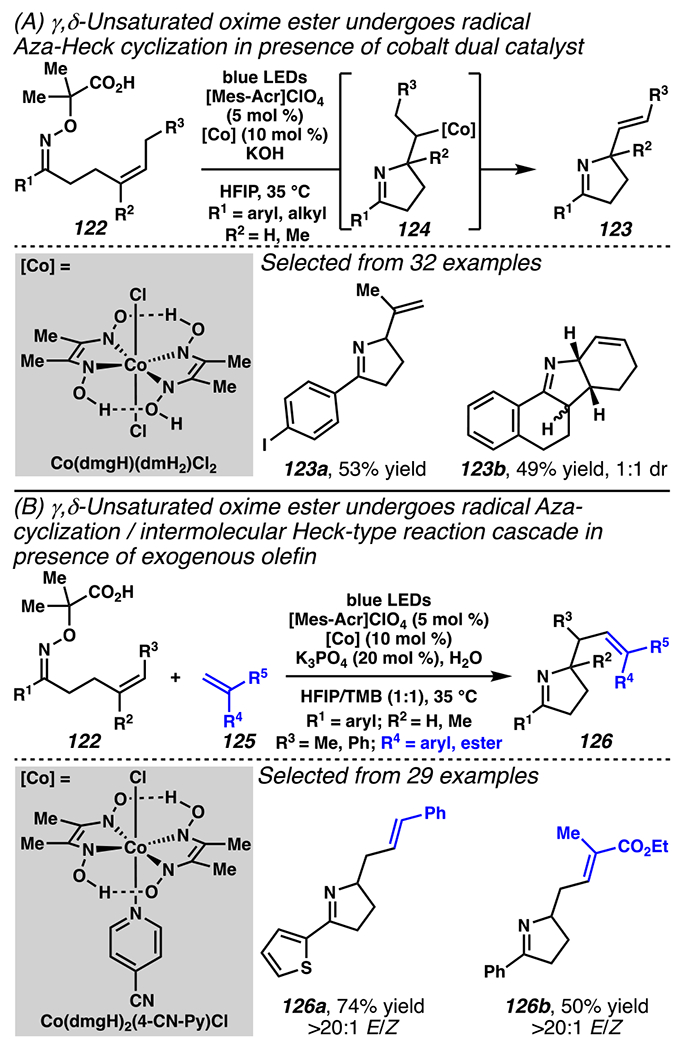
A Photocatalyst and Cobalt Complex Mediate Aza-Heck Cyclization Cascade Sequences
5.2.2. Hydroxyacid-Derived Iminyl Radicals Can React with Chalcogens to Install New Chalcogen–Nitrogen Bonds
A mechanistically analogous iminyl radical formation / intramolecular N-radical addition cascade can be intercepted by a pendant thioether to furnish an isothiazole (Scheme 37).166 In the propose reaction mechanism, generation of radical species 130 triggers intramolecular annulation reaction via N-radical addition to sulfur. Upon thioether fragmentation, this event would provide desired product 129 and methyl radical, which would be oxidized under aerobic condition to provide formaldehyde as a byproduct. During the course of reaction optimization, the net transformation of oxime 127 to isothiazole 129 is viable in presence of photocatalyst with excited oxidation potential lower than the irreversible oxidation potential of sodiated oxime salt 128a (Ep/2ox = +1.58 V versus SCE in acetone). Strongly oxidizing [Mes-Acr]ClO4 (E1/2[PC−/PC*] = +2.08 V versus SCE in CH3CN)27 is empirically identified as the optimal photosensitizer for the reaction. The reaction is less efficient in presence of photosensitizers that are more oxidizing than [Mes-Acr]ClO4. Equally pertinent to the reaction efficiency is the thioether group of α-imino-oxy acid. Within a series of thioethers (i.e., 128b–128e), which all have compatible Ep/2ox values with respect to [Mes-Acr]ClO4, benzyl thioether 128d reacts with the highest efficiency, presumably due to formation of resonance-stabilized benzylic radical en route to product 129. This is a viable strategy to generate varied arenes, including tricyclic 1,3-benzodioxole-containing isothiazole 132a, methylated isothiazolo[5,4-b]pyridine 132b, and furan- (132c) and thiophene-embedded isothiazole (132d) scaffolds (Scheme 38).
Scheme 37.
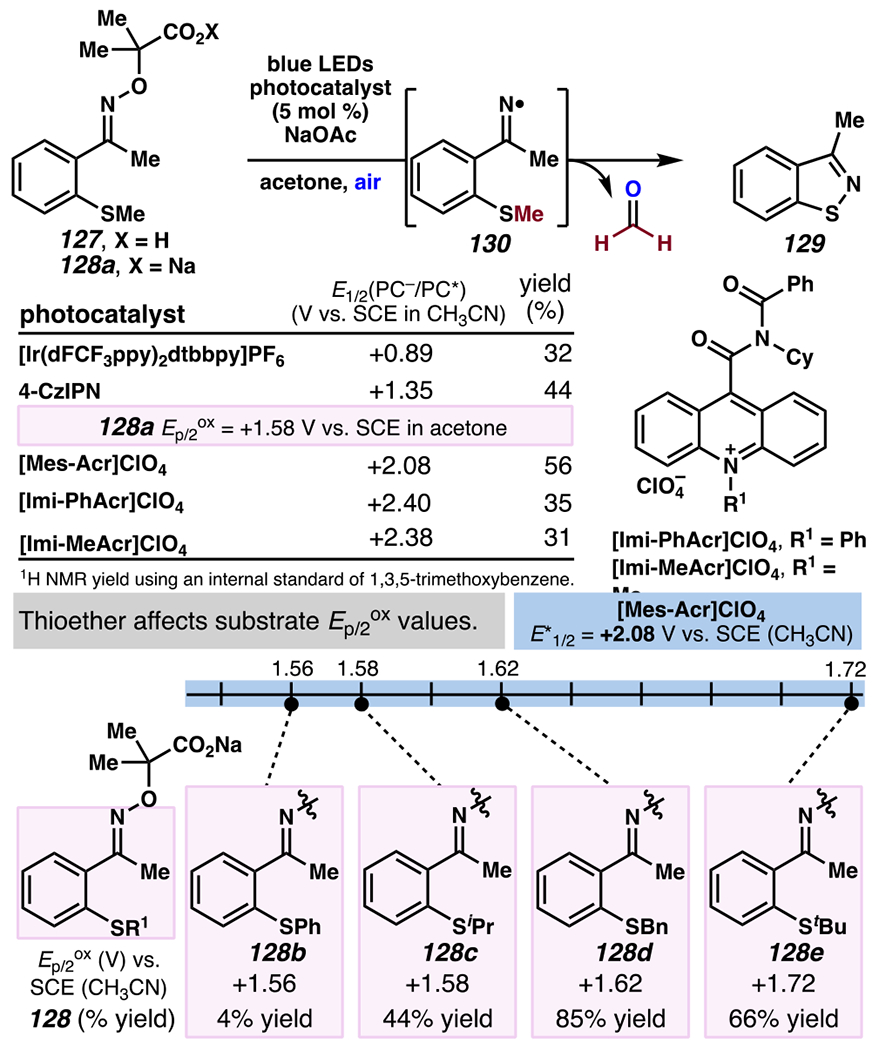
Intramolecular Reactions Between Iminyl Radicals and Thioethers Provide Isothiazoles
Scheme 38.
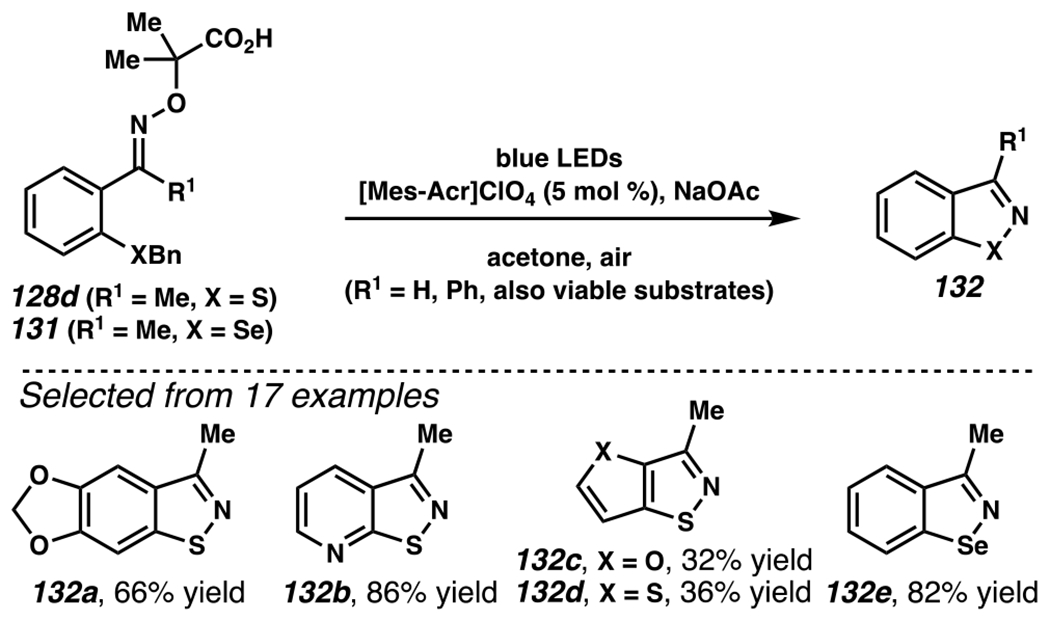
Heterocyclic Isothiazoles Are Constructed from α-Imino Oxy Acids
5.3. Ketoximes and Aldoximes Can Serve as Precursors to Persistent Iminyl Radical Intermediates
Furthermore, with ketoximes such as those derived from acetophenone and benzophenone, conversion to an iminyl radical can procced based on an energy transfer mechanism involving photosensitizer, and thereby prompt an array of radical reactions. This iminyl radical generation strategy was first documented by Glorius and co-workers in 2019 in a reaction in which electron-deficient O-acyl oxime 133 undergoes energy transfer-triggered decarboxylation to provide aryl radical (not depicted) and iminyl radical 135a (Scheme 39A).167 The aryl radical is trapped by iodine-transfer reagent (CH2ICl) to provide aryl iodide 134. Based on the electrochemical potentials of starting material 133 (Ep/2red = −2.00 V versus SCE) and photosensitizer (E1/2[*IrIII/IrIV] = −0.89 V vs. SCE, E1/2[IrIII/IrII] = −1.37 V vs. SCE), a SET pathway is excluded as a mechanistic possibility because it would be a thermodynamically unfavorable process. By contrast, photosensitizer possesses appropriate triplet energy (ET = 60.8 kcal•mol−1) to sensitize oxime 133 (ET = 46.4 kcal•mol−1). Notably, this reaction gives rise to byproducts imine 136, ketone 137, and hydrazine 138a, which are likely to originate from iminyl radical 135a. In fact, these byproducts provide evidence that iminyl radical 135a is a persistent radical species — otherwise, byproducts 136–138a would be unlike to form. Consistent with this hypothesis, under deuterium-transfer conditions, O-acyl oxime 139 produces deuterated arene 140 and 6-phenylphenanthridine 141, presumably by way of intramolecular radical addition of the nitrogen-centerd radical to a neighboring π-system (Scheme 39B).
Scheme 39.
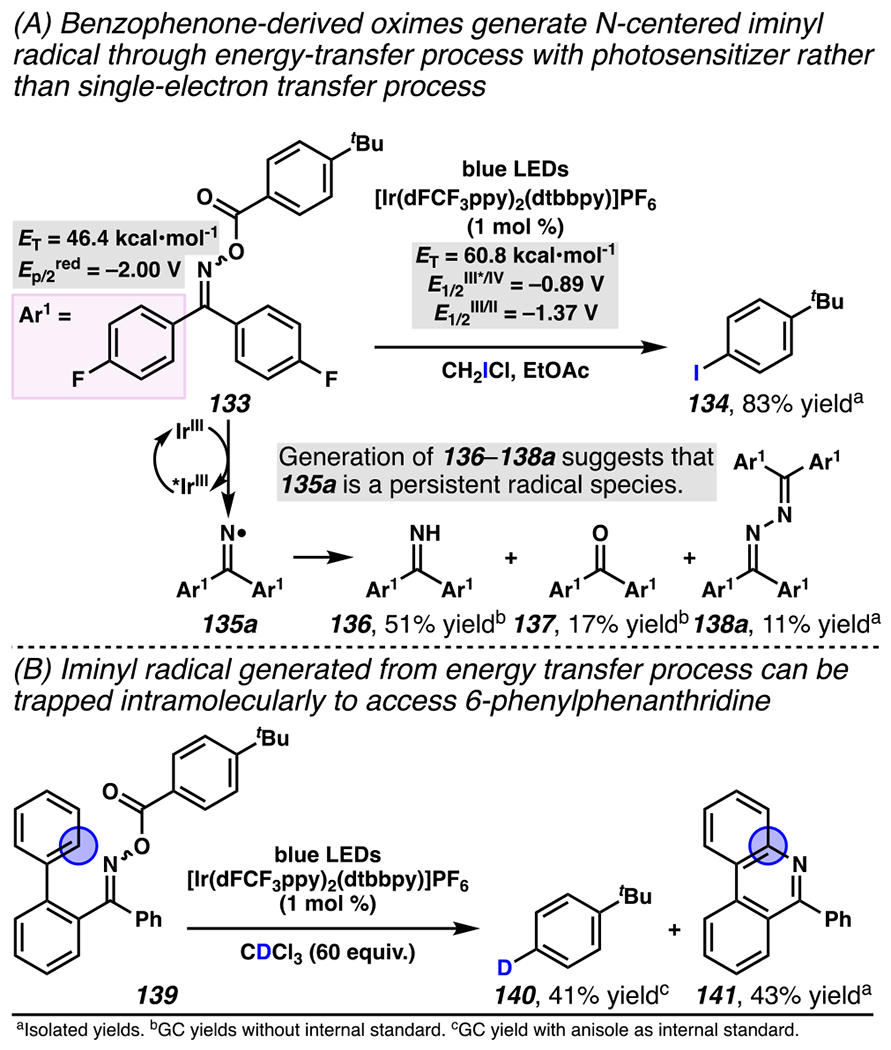
Oxime Analogues of Benzophenone Generate Nitrogen-Centered Iminyl Radicals via an Energy-Transfer Process
A net intramolecular decarboxylative C–N bond forming reaction can be used to access an aliphatic primary amine surrogate, such as alkyl amine 143a, from aliphatic O-acyl oxime 142a (Scheme 40A).18 Mechanistically, this reaction appears to rely on energy transfer between oxime 142a and triplet excited-state iridium photosensitizer to afford triplet excited-state oxime 144 via triplet-triplet energy transfer (TTEP), en route to critical iminyl radical 135b (Scheme 40B). Steady-state and transient electronic transition spectroscopy experiments revealed that the rate of triplet-triplet energy transfer between oxime 142a and photosensitizer (kET = 1.1 × 108 s−1) is faster than the rates of radiative (kr = 3.8 × 105 s−1) and non-radiative decays (knr = 3.8 × 104 s−1) of excited photosensitizer. So, triplet-triplet energy transfer between the two species occurs before the decay of excited photosensitizer back to singlet ground-state. A number of standard methods to access iminyl radical intermediates from O-acyl oxime esters are not viable when using benzophenone-derived O-acyl oxime esters 142a as substrates. For example, Förster-type resonant energy transfer is unlikely, given negligible spectral overlap between the absorption spectrum for oxime 142a and the phosphoresence spectrum for several excited-state iridium (III) complex (Scheme 40C). Additionally, a single electron tranfer mechanism is excluded because it is not thermodynamically favorable, based on the electrochemical potentials of oxime 142a (Epred = −1.83 V versus SCE, Epox = +2.35 V versus SCE, Scheme 40D). Ultimately, once iminyl radical forms, recombination with alkyl radical is proposed to deliver primary amine surrogates.
Scheme 40.
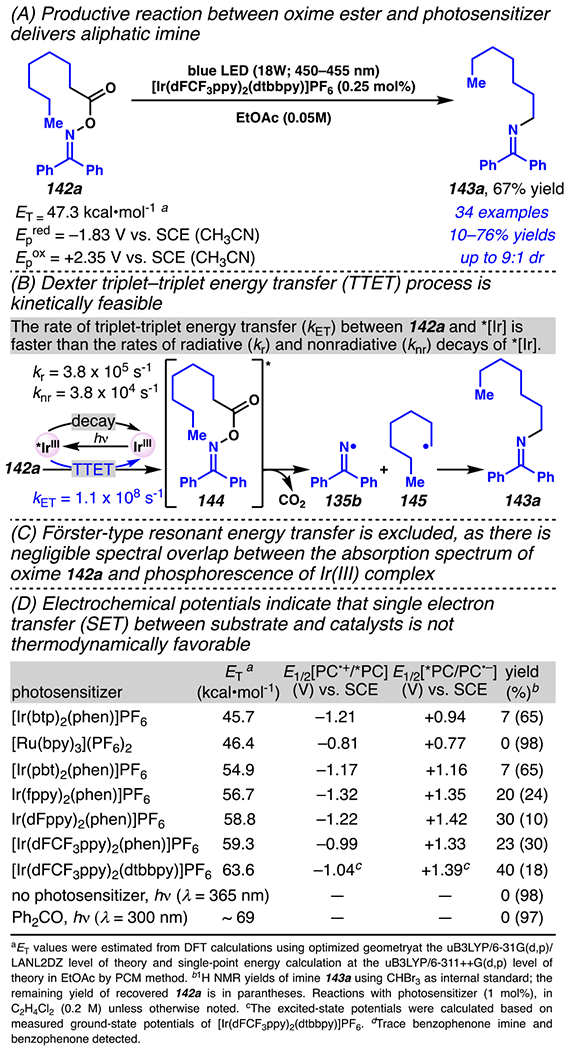
Benzophenone Oxime Esters Engage in Photosensitized Reaction to Deliver Aliphatic Imine
A similar energy transfer mechanism is proposed as the basis for the position-selectivity of a photosensitizer-enabled carboimination reaction between benzophenone-derived O-acyl oxime esters 142b and unactivated olefins 146 to afford α-iminonitriles (Scheme 42).168 Consistent with previous findings,18 a number of standard methods to access iminyl radical intermediates from O-acyl oxime esters are not viable when using benzophenone-derived O-acyl oxime esters 142b as substrates. So, a triplet–triplet energy transfer mechanism is proposed between oxime 142b and photexcited iridium complex to furnish excited oxime 148, which gives way to persistent169 iminyl radical 135b upon decarboxylation. The position-selectivity of this carboimination reaction is predicted to originate from the persistent radical effect169 (Scheme 42B). Relying on long lifetime of iminyl radical 135b, more transient alkyl radical 149 escapes the solvent cage to undergo radical addition to terminal site of olefin 146. In keeping with this hypothesis, radical crossover experiments with oximes 142b, 151a and acrylonitrile 146 furnish non-crossover α-iminonitrile products 147a, 152a and hydrazines 138b, 153, crossover α-iminonitrile products 152b, 147b and hydrazine 154 as detected by GC-MS. This protocol offers position-selectivity that complements that available from previous intermolecular carboamination reactions that were initiated by nitrogen-centered radicals,170 presumably because they did not rely on persistent benzophenone imine radicals.
Scheme 42.
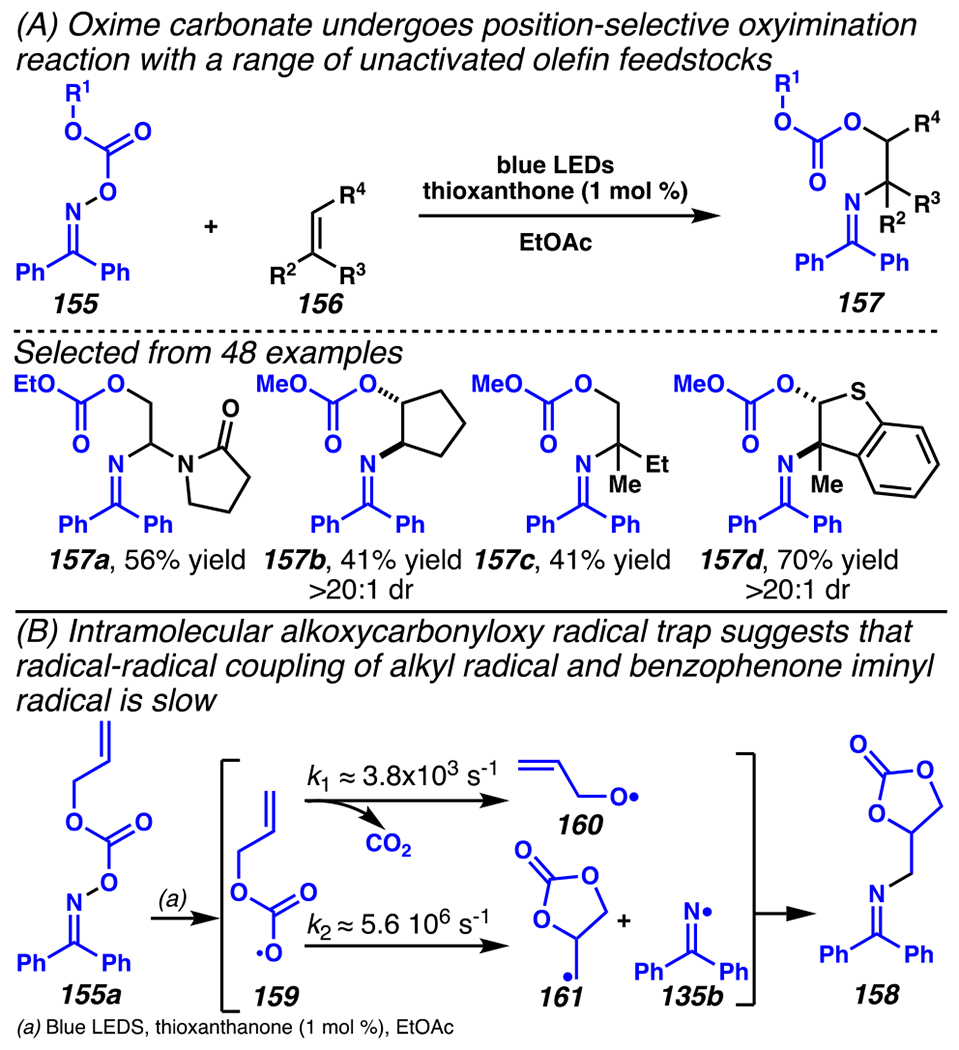
In Oxyimination Reaction, Chemoselectivity May Arise Due to the Low Rate of Radical Recombination Between Alkoxycarbonyloxy and Iminyl Radicals
Intermolecular, radical-radical cross-coupling reactions that rely on a persistent benzophenone iminyl radical as a precursor to a transient alkyl radical can be extended to synthesize 1,2-amino alcohol surrogates (Scheme 42).171 Specifically, oxime carbonates 155 react with olefins 156 in net oxyimination proceses, mediated by a thioxanthone organic photosensitizer. This protocol permits highly atom-economical assembly of 1,2-amino alcohol analogues as every atom of oxime carbonate and mono-, di-, and trisubstituted olefin feedstocks are utilized. Control experiments suggest that chemoselectivity may arise due to the low rate of radical recombination between alkoxycarbonyloxy and iminyl radicals (Scheme 42B), thereby affording 1,2-aminoalcohol surrogates efficiently.
Intermolecular, radical-radical cross-coupling reactions have been disclosed between a persistent benzophenone iminyl radical and an aryl-derived radical, also based on a proposed energy transfer mechanism, and can be used to assemble primary amines bearing tetrasubstituted α-carbon centers (Scheme 43A).172 For example, amine product 164a is isolated in 86% yield upon reaction of acetophenone-derived oxime 162 and 4-cyanopyridine 163 in presence of an iridium photosensitizer, diisopropylamine as a reductant and benzoic acid as a soluble proton source (Scheme 43A). This reaction protocol provides access to primary amines adjacent to a sterically bulky tBu-substituent (164b), and medicinally-relevant cyclopropyl173 (164c) and F3C174 (164d) groups. The transformation is largely restricted to cyanopyridines, which can be more readily reduced to critical radical intermediates (i.e., 172). For example, non-heteroaromatic cyanoarene such as 1-napthonitrile does not engage in two-component coupling reaction with iminyl radical to provide amine 164g.
Scheme 43.
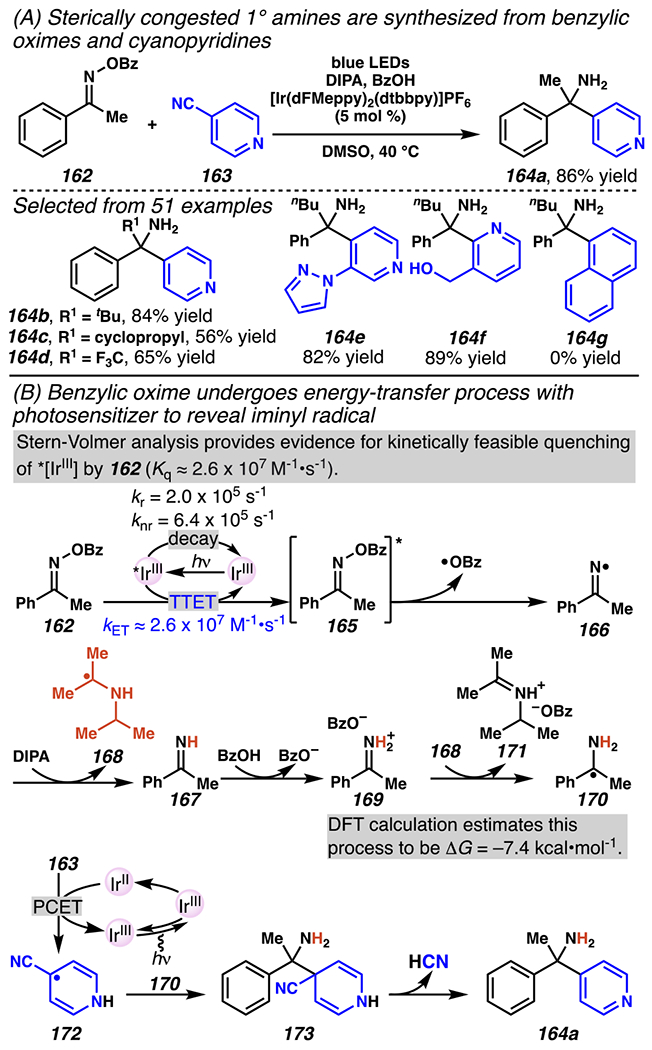
Acetophenone-Derived O-Acyl Oxime and Cyanopyridines Undergo Photosensitized Two-Component Coupling Reaction to Afford Sterically Congested Primary Amines
The reaction is proposed to proceed based on initial energy transfer between photoexcited *[IrIII] and oxime 162 to provide triplet excited-state oxime 165. Stern-Volmer analysis reveals that bimolecular quenching of *[IrIII] by oxime 162 is kinetically favorable (KET = Kq ≈ 2.6 × 107 M−1•s−1) and proceeds faster than decay of *[IrIII] back to ground-state [IrIII] (kr = 2.0 × 105 s−1, knr = 6.4 × 105 s−1).175 Indeed, oxime 162 undergoes facile N–O bond cleavage to generate persistent iminyl radical 166 that is poised to abstract hydrogen-atom from DIPA to form closed-shell imine 167 and carbon-centered radical 168. It is proposed that imine is protonated by benzoic acid to generate iminium benzoate 169, which engages in exergonic single-electron reduction with radical 168 to provide 1-phenylethaneaminyl radical 170 and diisopropylimium benzoate 171 (ΔG = −7.4 kcal•mol−1). Alternatively, 1-phenylethaneaminyl radical 170 can be generated from single-electron transfer between iminium benzoate 169 (Ered ≈ −0.8 V versus SCE in DMSO) and [IrII] (E1/2red = −1.42 V versus SCE in MeCN). Concurrently, 4-cyanopyridine 163 undergoes PCET with [IrII] and proton source to generate persistent radical 172 that undergoes radical-coupling with 1-phenylethaneaminyl radical 170 to afford intermediate 173. Ultimately, rearomatization of intermediate 173 expels hydrogen cyanide and delivers product 164a.
Remarkably, nitrogen-centered radicals can be either nucleophilic or electrophilic, based in part on the pendant functionality, and this polarity influences reaction kinetics. This is the chemical basis for the reaction of sulfonamide 174 with hydrazone 175 to provide hydrazonamide 176 (Scheme 44).176 This C(sp2)–N bond-forming reaction is initiated by photoredox-mediated generation of sulfonamidyl radical species 177, which undergoes radical-addition across C=N bond to provide nitrogen-centered radical intermediate 178. In fact, the unpaired electron and adjacent lone-pair stabilizes the intermediate through three-electron π-bonding interaction.177 Single-electron oxidation of intermediate 178 by [IrIV] complex regenerates ground-state photocatalyst and resonance-stabilized diazenium cation 179 / 180. Ultimately, deprotonation of diazenium cation furnishes desired product 176. Through this reaction, hydrazonamide bearing quinoline 176a and phenyl-enriched hydrazonamide 176b form in 88% yield, and 76% yield, respectively. By contrast, N-Boc and N-Bs hydrazones do not participate in reactions with sulfonamidyl radical intermediates – events that would give rise to products 176c and 176d, respectively.
Scheme 44.
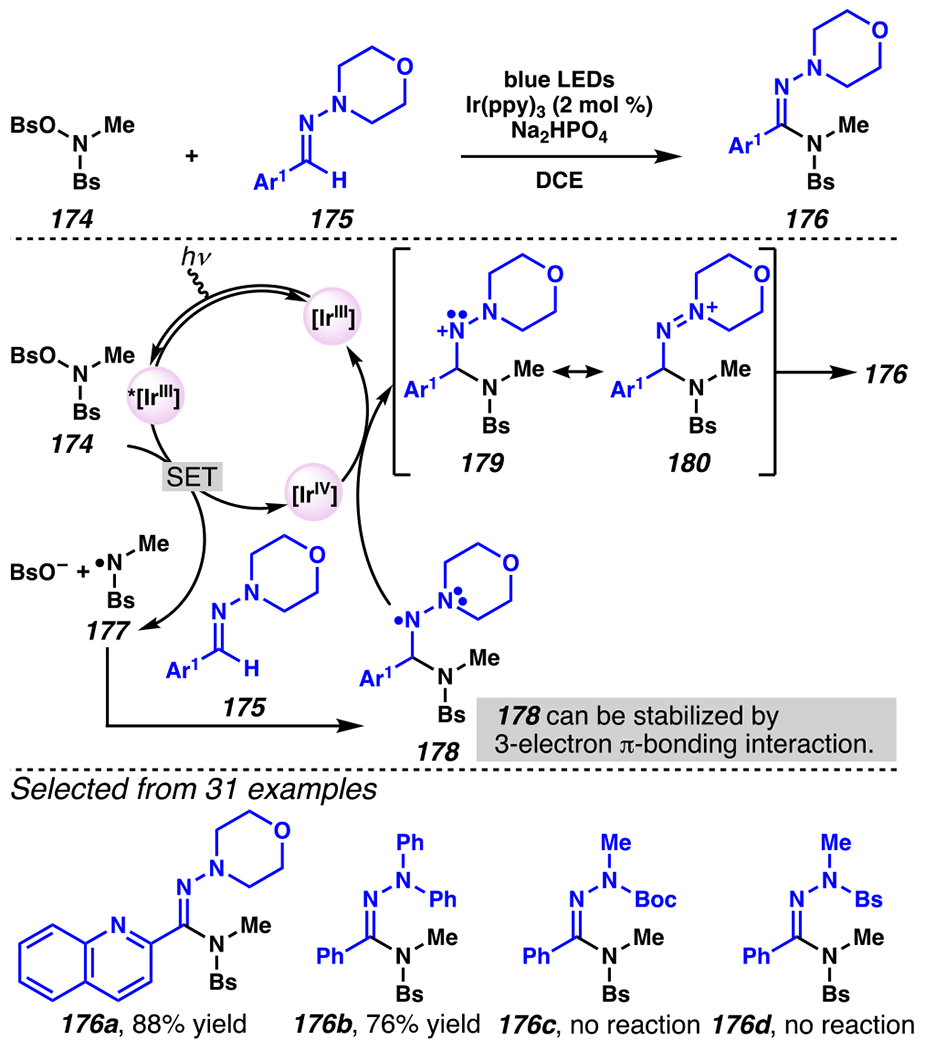
O-Aryloxy Amide and Hydrazones React in a C–H Amidation Process
5.4. Iminyl Radicals That Are Generated from Strained Cyclic Oximes Can Engage in Ring-Opening Cascade Reactions
5.4.1. These Ring-Opening Cascades Are Well-Established Strategies to Affect Distal Carbofunctionalization Reactions Involving Olefins
When the accessed iminyl radicals are substituents on strained cyclic oximes, β-scission can occur to furnish an alkyl radical that is available for subsequent reactions. Zard and co-workers discovered that under visible light, photoreactive cyclobutanone-derived oxime substrate 181 partakes in a β-scission / radical conjugate addition / chalcogenation cascade with a radical trapping agent such as methyl acrylate (Scheme 45).178 The operative mechanism in this transformation is posited to involve activation of cyclic oxime 181 via net photolysis of N–O bond to reveal iminyl radical species 183. Subsequently, β-scission of iminyl radical 183 generates γ-cyanoalkyl radical 184. This radical intermediate is poised for the Giese reaction with methyl acrylate to provide electrophilic radical intermediate 185 that is ultimately sequestered by 2-pyridinethiyl group to give rise to chalconized indane 182. Torsionally strained four-membered cyclic oximes are generally employed as substrates where the ring-opening steps are considerably exothermic (∆G ≈ −26.3 kcal•mol−1 for cyclobutane).179 Five-membered cyclic oximes, which have less exothermic ring-opening steps (∆G ≈ −6.2 kcal•mol−1)179 than their four-membered congeners, are reactive substrates under certain reaction conditions, and they typically require further driving force, such as that available when a resonance- or inductively stabilized alkyl radical species forms through iminyl radical β-scission.
Scheme 45.
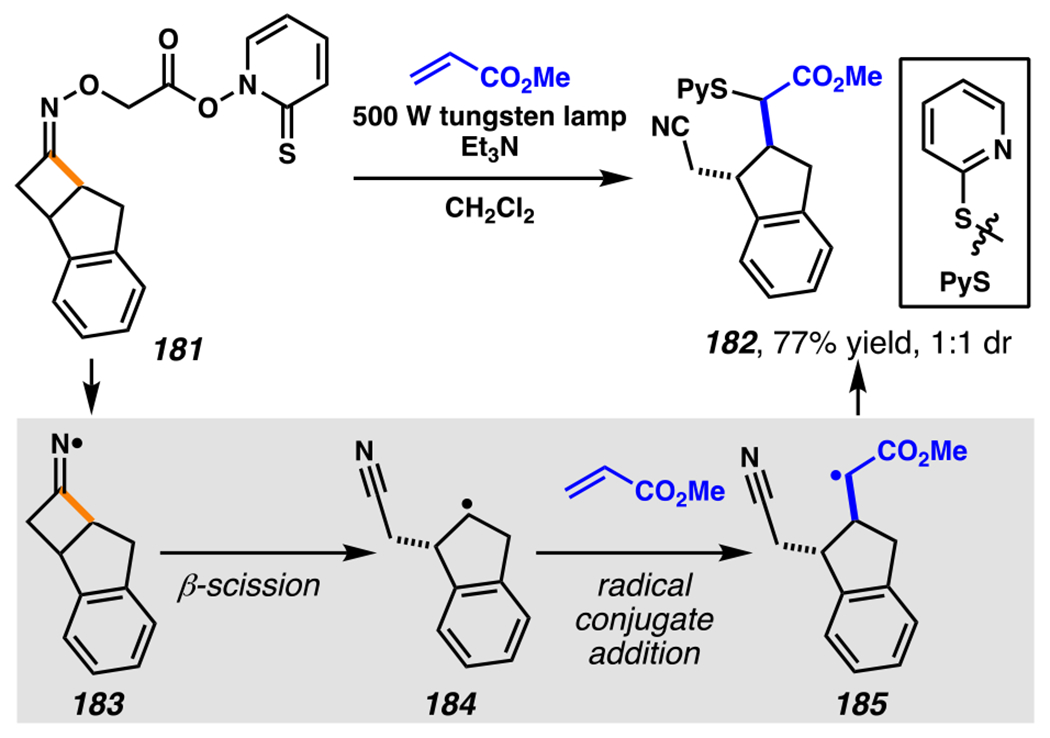
Cyclobutanone-Derived Oxime Triggers β-Scission / Radical Conjugate Addition / Chalcogenation Cascade
5.4.2. By Using Hydroxyacid-Derived Oxime Substrates, Ring-Opening Cascades Can Result in Direct Atom- or Group-Transfer Processes
Leonori, Sheik and co-workers recognized hydroxyacid-derived oximes as iminyl radical precursors amenable to photoredox-mediated iminyl radical formation/ β-scission / fluorine-, chlorine- and azide-transfer cascades.180 Formally, oximes 186 react with electrophilic atom- or group-transfer agents “X–Y”, where X is partially positive atom or group, to furnish distal functionalized aliphatic nitriles 187 (Scheme 46A). The initial reaction mechanism is analogous to the tactics disclosed by Leonori and Sheik to affect intramolecular cyclization of an iminyl radical onto an olefin (Scheme 21B).149 In one plausible mechanism, acid 186 is a precursor to α,α-dimethylhydroxycarboxylate (not depicted), which can reductively quench an excited state photocatalyst to initiates the sequence that forms iminyl radical 188 and triggers β-scission to unveil 3° cyanoalkyl radical intermediate 189. The nucleophilic carbon-centered radical engages in atom- or group-transfer reaction with X–Y to yield desired products 187. Electrophilic reaction byproduct Y• readily accepts an electron from photocatalyst to generate closed-shell species Y and regenerate the ground-state photocatalyst. In fact, the single-electron reduction of electrophilic Y• by photocatalyst is speculated to be exergonic, which could prevent premature oxidation of 3° carbon-centered radical 189. Using this approach, fluorination, chlorination and azidation of aliphatic nitrile are realized (Scheme 46A). Under the appropriate reaction conditions, Selectfluor®181 serves as a competent fluorine-transfer agent to yield fluorinated products, while NCS152 can serve as appropriate chlorine-transfer agent to afford chlorinated products. Furthermore, arylsulfonyl azide traps the putative cyanoalkyl radicals to deliver azide products.157
Scheme 46.
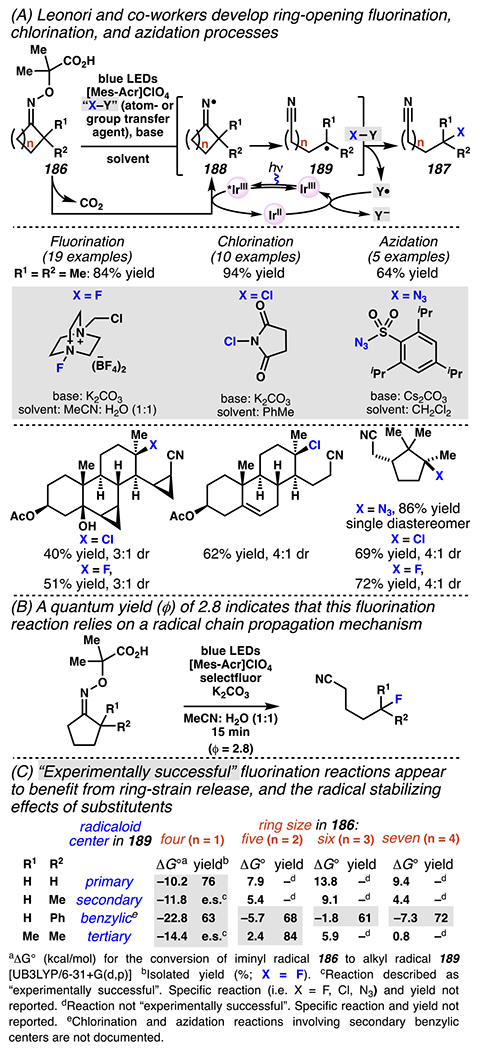
Cascade C–C Cleavage and Functionalization Reactions Afford Aliphatic Nitirles with Fluorine, Chlorine and Azide Functional Handles
Even though this distal fluorination reaction of aliphatic nitriles hinges on productive photocatalyst turnovers by the substrates, a quantum yield experiment indicates that this fluorine-atom transfer process proceeds predominantly via radical chain propagation mechanism (ϕ = 2.8), albeit in tandem with the photoredox reaction manifold (Scheme 46B). To gauge the propensity for substrates to participate in the fluorination reaction, ∆G0 values of the conversion of iminyl radical 188 to alkyl radical 189 are approximated by DFT calculations (Scheme 46C). The quantum mechanical investigation suggests that “experimentally successful” fluorination reaction is broadly driven by thermodynamically favorable ring-strain release of the iminyl radical and the radical stabilizing effects of substituents. Cyclobutanone-derived oximes are speculated to engage in facile fluorination reaction due to the exergonic ring-strain release, while larger cyclic oximes (ring size = 5 ≤ 7) partake in the reaction if they possess substituents that stabilize the concomitant alkyl radicals.
Furthermore, hydroxy acid-derived oximes engage in potentially mechanistically analogous group-transfer reactions with other electrophilic group-transfer agents. Similar to photoredox-mediated alkynyl transfer reaction with hypervalent iodine (V) reagent and unsaturated oxime by Leonori and co-workers,149 Waser and co-workers reported that alkynyl transfer reaction with α,α-dimethylhydroxyacid-derived oxime 186a and ethynyl benziodoxolone-based alkynyl transfer agent160 191a provides alkynylated product 192a (Scheme 47).40 The reaction is not optimal with conventional photocatalysts such as [Ir(dFCF3ppy)2dtbbpy]PF6, 4-CzIPN, [Mes-Acr]ClO4 and 9,10-dicyanoanthracene, presumably because they possess excited state potentials that result in inefficient oxidation of oxime 190, or undesirably efficient oxidation of product. Nevertheless, this reaction proceeds in the presence of more oxidizing 4-CzIPN analogues. Among the 4-CzIPN derivatives, 4-ClCzIPN most effectively converts 190 to 192a, presumably because its oxidation potential is appropriate to oxidize substrate 190 without promoting product oxidation. With this catalyst, diastereoenriched product 192b is accessed in 65% yield, and electron-deficient alkyne 192c is isolated in 71% yield (Scheme 48). In fact, other hypervalent iodine (V) reagents can be employed to permit styrene-transfer reactions (cf. 192d) with good stereoselectivity, as well as nitrile-transfer (cf. 192f) and silyl alkyne-transfer (cf. 192e) reactions, albeit with diminished product yields. Togni’s reagent is found to be the exception, as it does not promote trifluoromethyl-transfer reaction to furnish α-trifluoromethylated amine 192g.
Scheme 47.
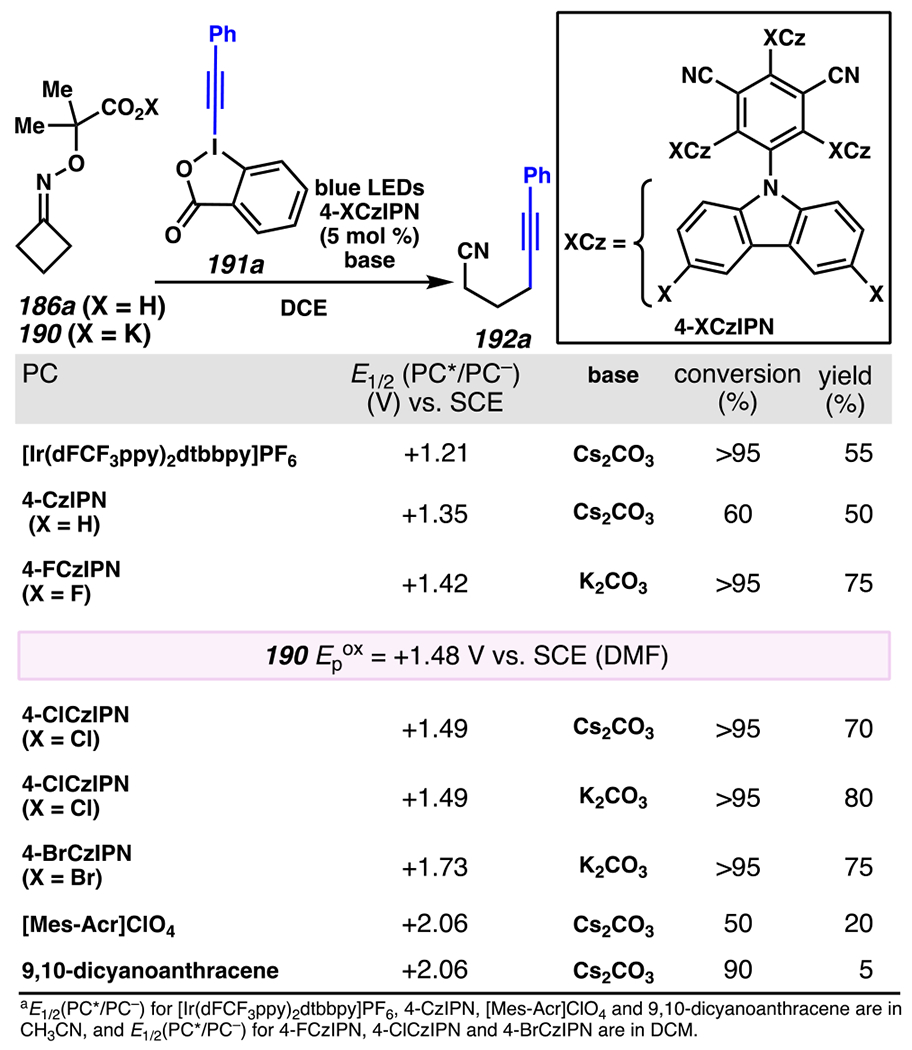
Ethynyl Benziodoxolone-Based Alkyne-Transfer Reagents Facilitates C(sp3)–C(sp) Cross-Coupling Reactions to Furnish Aliphatic Nitriles with Internal Alkyne
Scheme 48.
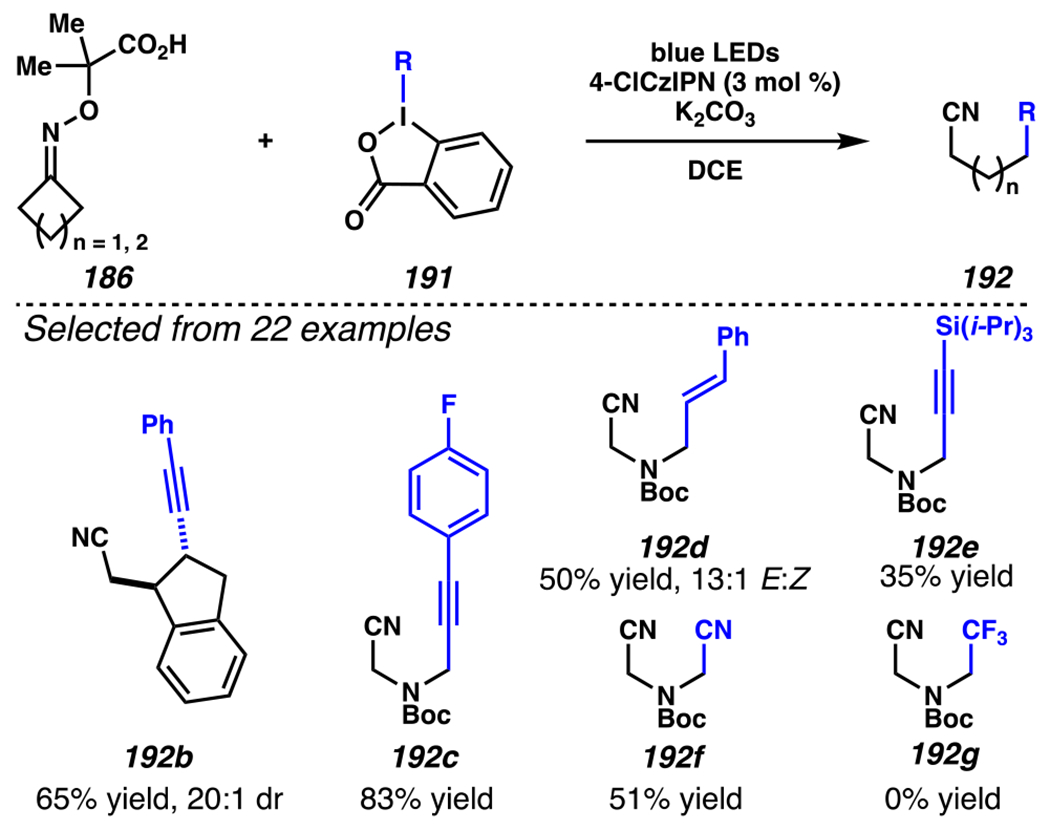
An Iminyl Radical Formation/ β-Scission Cascade Can Be Used to Induce Vinylation, Alkynylation, or Alkylation
5.4.3. Ring-Opening Cascades Can Trigger Alkene Carbofunctionalization Reactions
β-Scission/ring-opening cascade sequences can be intercepted by iminyl radicals derived from cyclic oximes with readily oxidizable electron-dense methoxybenzyl groups, much as γ,δ-unsaturated oximes with PMP handles template iminyl radical generation / intramolecular annulation upon reductive quenching of photocatalyst.34 Indeed, cyclic oximes with PMB ethers engage in the Giese reactions with an array of electron-deficient olefins to furnish alkylated products 193a under UV irradiation with organic photosensitizer 2-Cl-AQN (Scheme 49).182 To activate cyclic oxime 193a, reactions involve a net hydrogen-atom abstraction to furnish benzylic/α-ethereal radical 195 that fragments via β-scission to unveil iminyl radical 196 and p-tolualdehyde. The generation of iminyl radical is speculated to involve benzylic hydrogen-abstraction of 193a by *2-Cl-AQN,183 a postulate that is consistent with KIE experiments (kH/kD = 2.13–2.18). In this reaction, efficiency is correlated with the thermodynamic stability of the generated benzylic radical across a series of readily reducible oximes 193b–193d (Ep/2 = +1.37 to +1.86 V versus SCE in CH3CN, E1/2ox = +2.3V versus SCE in CH3CN). 34,182 Once generated, β-scission of iminyl radical 196 provides alkyl radical intermediate 197 that is trapped by olefin to provide ester-stabilized α-radical species 198. Finally, ester-stabilized radical 198 is converted to desired product 194 through two possible mechanisms. First, radical 198 could engage in direct solvent hydrogen-atom abstraction with 2-butanone to provide 194. Second, radical 198 (Ep/2 ≈ −0.63 V versus SCE in CH3CN)184 could be reduced by 2-Cl-AQN (E1/2 [AQ/AQ•–] = −0.96 V versus SCE in CH3CN)35 to provide enolate (not depicted) that undergoes solvent deprotonation to afford 194. Indeed, deuterium labeling experiments suggest that the latter mechanism should prevail over the hydrogen-atom abstraction mechanism.
Scheme 49.
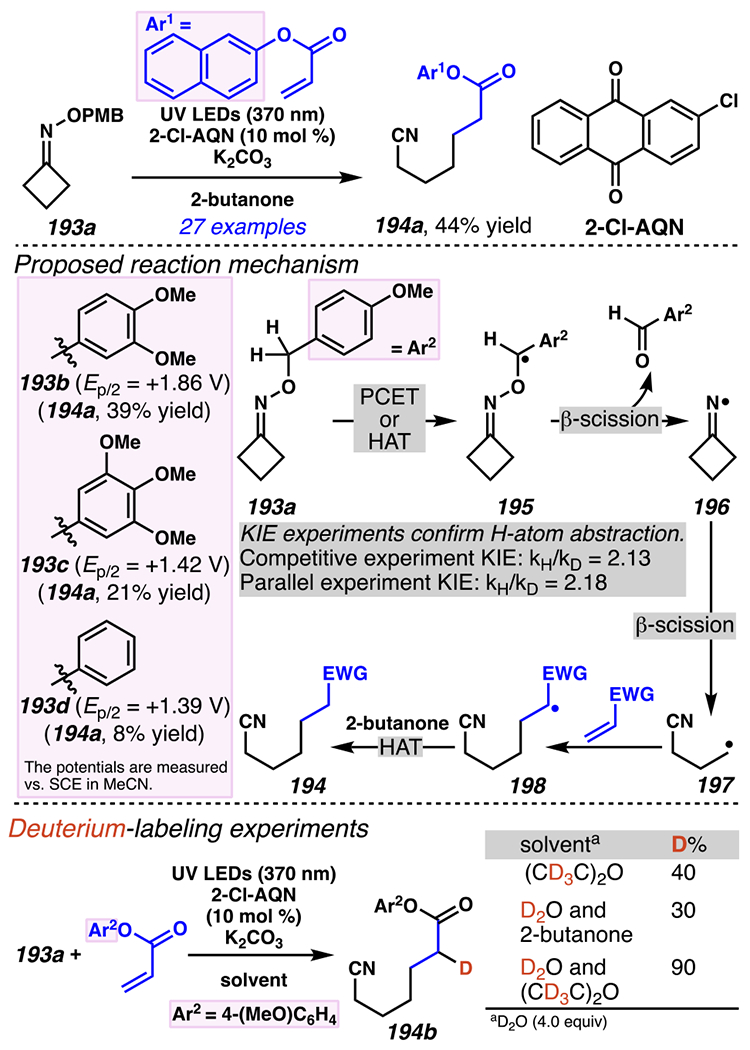
PMB-Substituents Make Oximes Labile to Oxidation, Which Can Facilitate a Photoinitiated Cascade Sequence
Zhou and co-workers developed a related approach to assemble gem-difluoroalkenes from hydroxy-acid-derived oxime 199 and α-trifluoromethyl alkene (Scheme 50).185 Using this approach, a variety of (hetero)aryl-derived α-trifluoromethyl alkenes can engage in the reaction, as exemplified by synthesis of gem-difluoroalkene with 2-methoxypyridine substituent in 80% yield. While larger cyclic oximes tend to be unresponsive to radical cascade reaction paradigm, this reaction condition renders six-membered cyclic oxime reactive to provide the corresponding product 201e in 75% yield with 5:1 dr. Remarkably congested compounds can be assembled efficiently, including gem-difluoroalkene product 201f, which contains two contiguous quaternary centers, and is accessed in 77% yield with 15:1 dr.
Scheme 50.
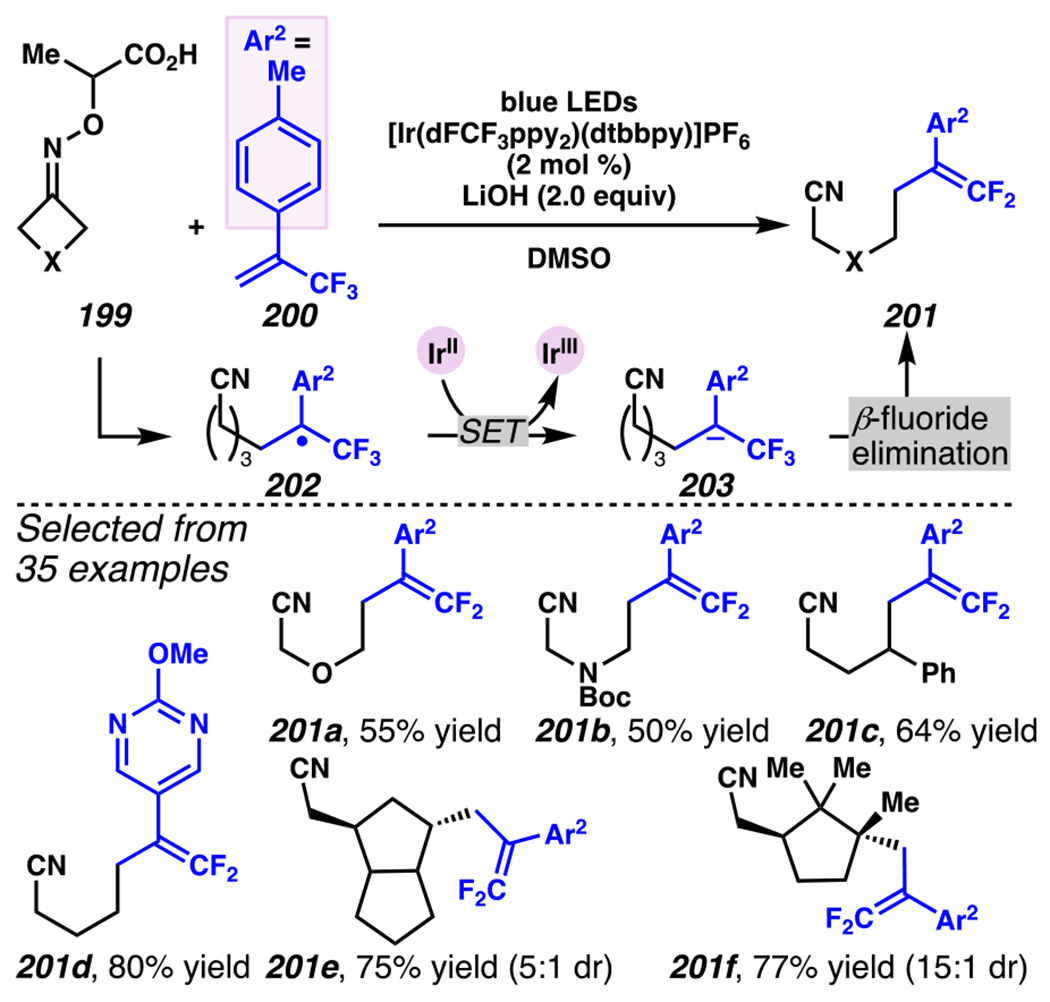
Hydroxy Acid-Derived Oximes Engage in Two-Component Coupling Reactions with α-Trifluoromethyl Alkenes to Furnish gem-Difluoroalkenes
To the best of knowledge, prior to 2019, there were no photo-mediated strategies to allow the productive and direct use of hydroxyoximes as iminyl radical precursors. Hydroxyoximes are used directly with arylsulfinyl chlorides in the Hudson reaction to generate N-sulfonylimines that results in N–O bond fragmentation, and this approach has been leveraged to convert hydroximes to iminyl radicals.186,187 Nevertheless, photo-irradiative reactions of hydroxyoximes have remained unexplored due to unproductive and competitive O–H bond cleavage that arise from weak O–H BDEs of hydroxyoximes.188 In 2019, Xiang, Chen, Yang and co-workers addressed this gap in the literature (Scheme 51). 189 Building on the recognition by the Xie and Zhu research groups that triphenylphosphine-derived phosphoranyl radicals190 could be used to directly activate strong C–O bonds,191, 192, 193 these researchers determined that the N–O bond of hydroxyoximes could be activated to homolytic cleavage by reaction with these phosphoranyl radicals (Scheme 51). Specifically, these researchers accomplish two-component coupling with hydroxime 204 as an iminyl radical precursor, and α,α-diaryl- or α-trifluoromethyl-substituted alkenes as radical trapping agents en route to installation of bis-aryl or gem-difluoroalkene groups under the aegis of Ph3P additive. Triphenylphosphine enjoys a pivotal role in the reaction as an activating group by forming phosphonyl radical,194 Ph3P•+ (E1/2[Ph3P•+/Ph3P] = +0.98 V versus SCE),195 which arises from thermodynamically favorable reductive quenching of the photocatalyst (E1/2red [*IrIII/IrII] = +1.21 V versus SCE).8 Indeed, Stern-Volmer analysis confirms that the photocatalyst can be reductively quenched by Ph3P. The generated phosphonyl radical reacts with hydroxime 204 to furnish phosphoranyl radical 205.196 Thermodynamically driven β-scission197 is followed by radical addition to an alkene form carbon-centered radical 206. The radical is poised for reduction by photocatalyst (E1/2 [IrIII/IrII] = −1.37 V versus SCE)8 to generate carbanion 207. For example, reduction of diphenyl-stabilized methinyl radical species (E1/2[R•/R-] ≈ −1.34 V versus SCE)204 is achieved by strong reductant [Ir]II to provide the desired carbanion intermediate. At this stage, protonation or β-fluoride elimination from anion 207 give rise to diaryl 208 or difluorinated 209, respectively. Thereby, hydroxyoximes can serve as direct precursors to iminyl radicals in productive synthetic transformations.
Scheme 51.
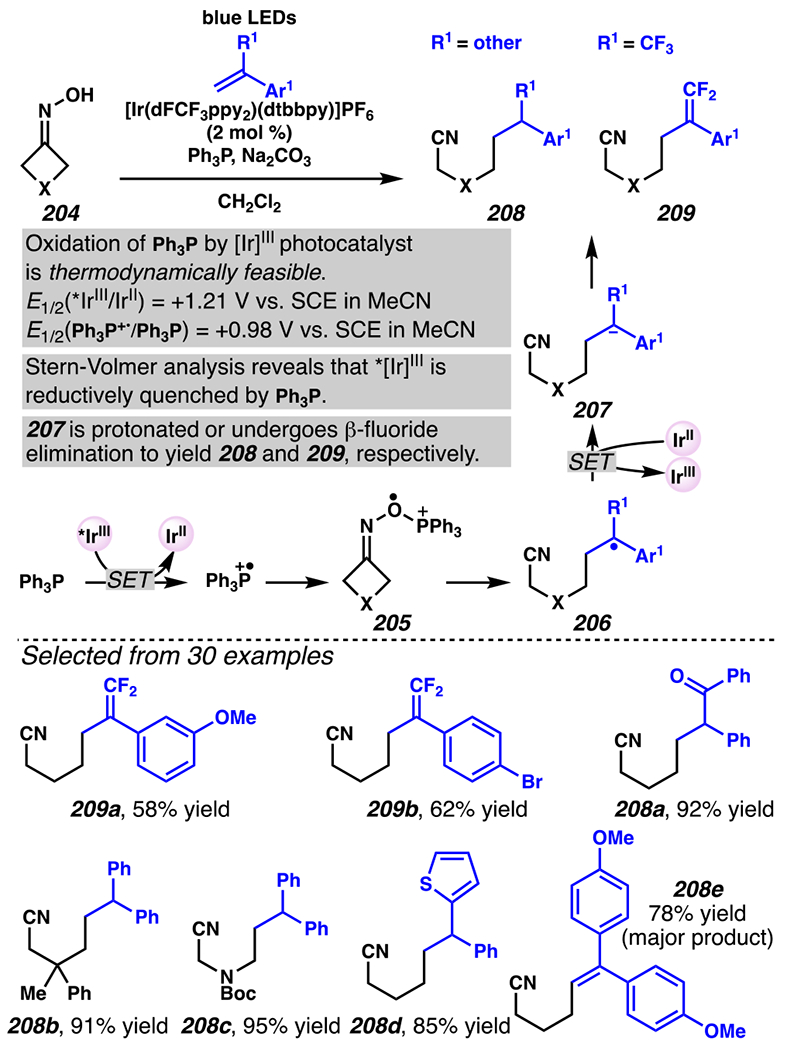
Triphenylphosphine Can Enable Hydroximes to Serve as Direct Precursors to Iminyl Radicals
Using this approach, extremely electronically varied olefins constitute appropriate radical trapping agents (Scheme 51). α-Trifluoromethylated alkenes participate in the coupling reaction to access gem-difluorinated alkenes with resonance donating and electron-withdrawing aryl substituents (i.e., 209a and 209b, respectively).198, 199 Unsurprisingly, a Giese reaction with chalcone proceeds under this reaction conditions, furnishing Giese reaction product 208a in 92% yield. With α,α-diarylolefins, the reaction tolerates pendant quaternary carbon center 208b, and carbamate 208c, and can install alkenes with pendant heterocycles to furnish products such as 2-thiophenyl 208d. Interestingly, bis-p-methoxyphenyl groups prompt the formation of olefinated product 208e as the major product rather than the anticipated net-alkylated product (not depicted). Logically, this reaction pathway could result if generate radical intermediate 206 is more amenable not to the depicted reduction to anion 207, but, instead, to oxidation to a cationic intermediate (not shown), with subsequent base-mediated elimination to install the olefin. Recognizing this minor limitation, this process offers a step-economical approach to directly engage cyclic hydroxyoximes in β-scission / Giese-type reactions with electron-deficient olefins without pre-installing redox-active handle.
5.4.4. Ring-Opening Cascades Can Be Intercepted by Transition-Metal Mediated Bond-Forming Processes
Oximes can be used as alkyl radical precursors in nickel-mediated cross-coupling reactions (Scheme 52),200 merging the above-described approach with known nickel- and photosensitizer-driven processes.146 A process that relies on oxime precursors may entail photocatalyst activation of oxime 186 to reveal cyanoalkyl radical 189, which is speculated to undergo C–C cross-coupling reaction with nickel complex and carbon group transfer agent. Highly related reaction conditions furnish arylated-, alkylated-, and vinylated aliphatic nitriles. (Hetero)arylated aliphatic nitriles are easily installed with an array of (hetero)aryl bromides, as exemplified by cross-coupling reactions with caffeine-derived aryl bromide. Alkyl bromide such as l-β-bromoalanine can be trapped to afford an unnatural amino acid product. Terminal alkynes as trapping agents promotes a net vinylation reaction that delivers gem-disubstituted terminal olefin, as demonstrated with N-(3-butynyl)phthalimide as reaction partner to afford an 1,4-diamine product. These photoredox-mediated nickel-catalyzed cross-coupling reactions provide an approach to access (hetero)arylated, vinylated and alkylated alkanes.
Scheme 52.
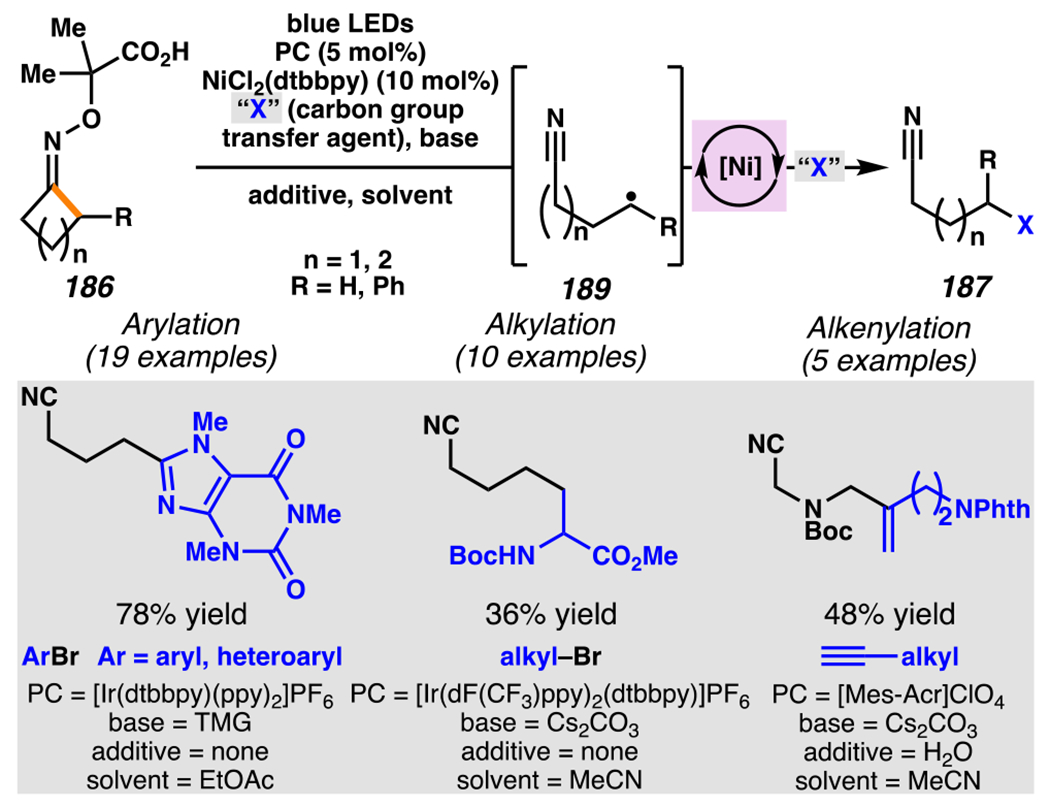
Dual Photoredox and Nickel Catalysts-Mediated Strategy Confers Synthetic Capabilities to Access Nitriles with New C–C Bonds
An alternative strategy to intercept this photoredox-mediated ring opening cascade sequence takes precedent from a photoredox-mediated and Co-catalyzed vinylation reaction between oxime esters and alkenes163 to affect olefination (Scheme 53).201 Indeed, this transformation permits assembly of a diverse catalogue of alkenes from a range of cyclix oxime esters and styrene derivatives. For example, azetidine-derived oxime ester and styrene react to give allylic amine 211a in 63% yield with high stereoselectivity (>20:1 E/Z). This reaction is more broadly relevant, as 2-vinylbenzothiophene is a viable substrate to provide (E)-isomer-enriched desired product 211c in 72% yield. Interestingly, when benzyl methacrylate is subjected to the reaction conditions, a mixture of internal and terminal alkene products 211d and 211e are generated. This observation suggests that β-hydride elimination step of organocobalt complex is not regioselective in the presence of multiple β-hydrogen atoms. Unfortunately, this transformation is not suitable for use with 1,2-disubstituted styrenes or aliphatic olefins. Nevertheless, dual photoredox- and Co-catalyzed Heck-type reactions between cyclic oxime esters and styrenes constitutes complimentary reaction to dual photoredox- and Ni-catalyzed cross-coupling processes that engage cyclic oximes and terminal alkynes to access internal alkenes.
Scheme 53.
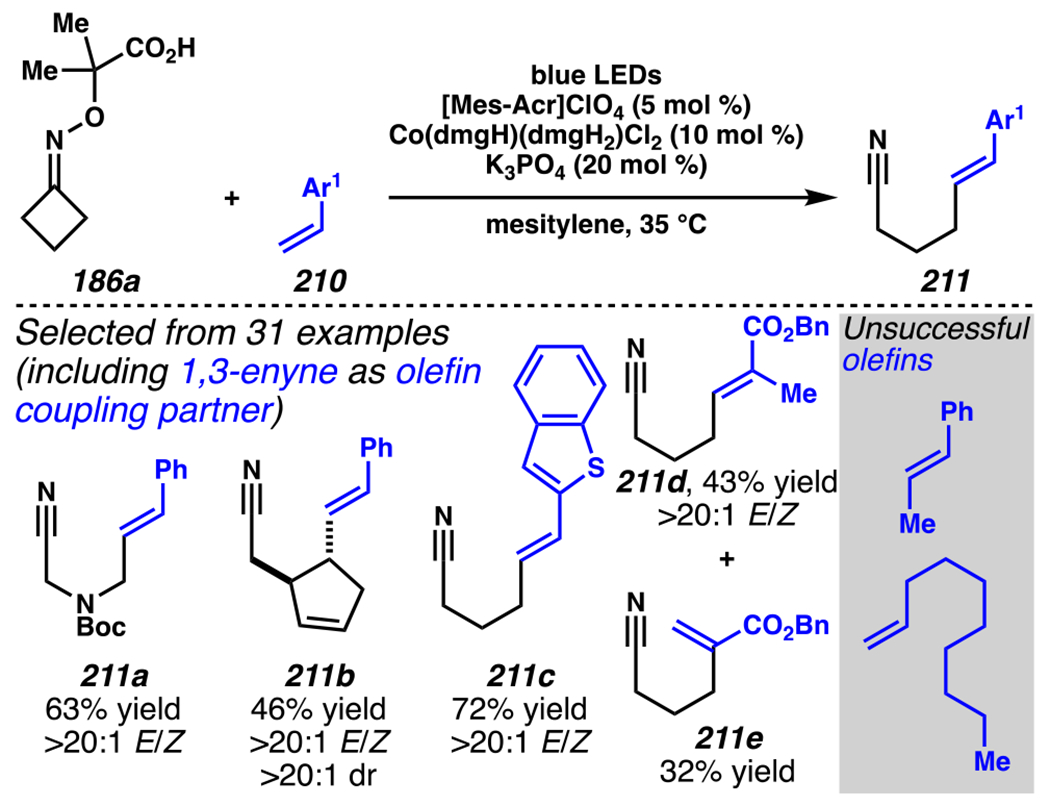
Photoredox-Mediated and Cobalt-Catalyzed Intermolecular Heck-type Coupling Reaction Is Amenable with Cyclic Oxime and Styrenes
5.4.5. Ring-Opening Cascades Can Rely on O-Acyl Cyclic Oxime Substrates to Affect Alkene Carboetherification Reactions
In a seminal report by Uemura, Nishimura and co-workers, cyclobutanone-derived O-benzoyloxime engage in thermally-mediated and iridium-catalyzed N–O bond cleavage to form a nitrogen-centered iminyl radical intermediate.202 This high-energy radical species is predisposed to undergo β-scission to furnish carbon-centered alkylnitrile radical, which is sequestered by a hydrogen-atom source or group-transfer agent to provide a series of aliphatic nitriles. This reaction cascade is mechanistically analogous to the process that engages α-hydroxy acid-derived cyclic oximes as substrates. In principle, O-acyl cyclic oximes should generate iminyl radicals in presence of photoredox-active molecules. More than a decade after the pioneering disclosure, Zhou and co-workers have hypothesized O-acyl cyclic oxime as photoredox-active substrate that undergoes iminyl radical formation / β-scission cascade under photoredox manifold.203
A Giese-type reaction constitutes a complementary strategy to introduce new alkyl groups to alkyl chains, using olefin electrophiles and cyclic oximes as alkylnitrile precursors. Within this broader reaction manifold, Zhou and co-workers have developed a photoredox catalyst-mediated approach to utilize cyclobutanone-derived O-acyl oxime 212a as a precursor to an iminyl radical, which enters a cascade sequence involving β-scission and addition across a styrenyl olefin with high electron density to provide linear alkane 213a (Schemes 54).203 This strategy relies on a strongly reducing photocatalyst for optimal reaction efficiency: only Ir(ppy)3 and eosin Y deliver the desired product 213a in synthetically relevant yields, and each of these photocatalysts possesses excited reduction potentials that are compatible with the reduction potential of oxime 212a (Epred = −0.99 V vs. SCE in CH2Cl2, Scheme 54).
Scheme 54.
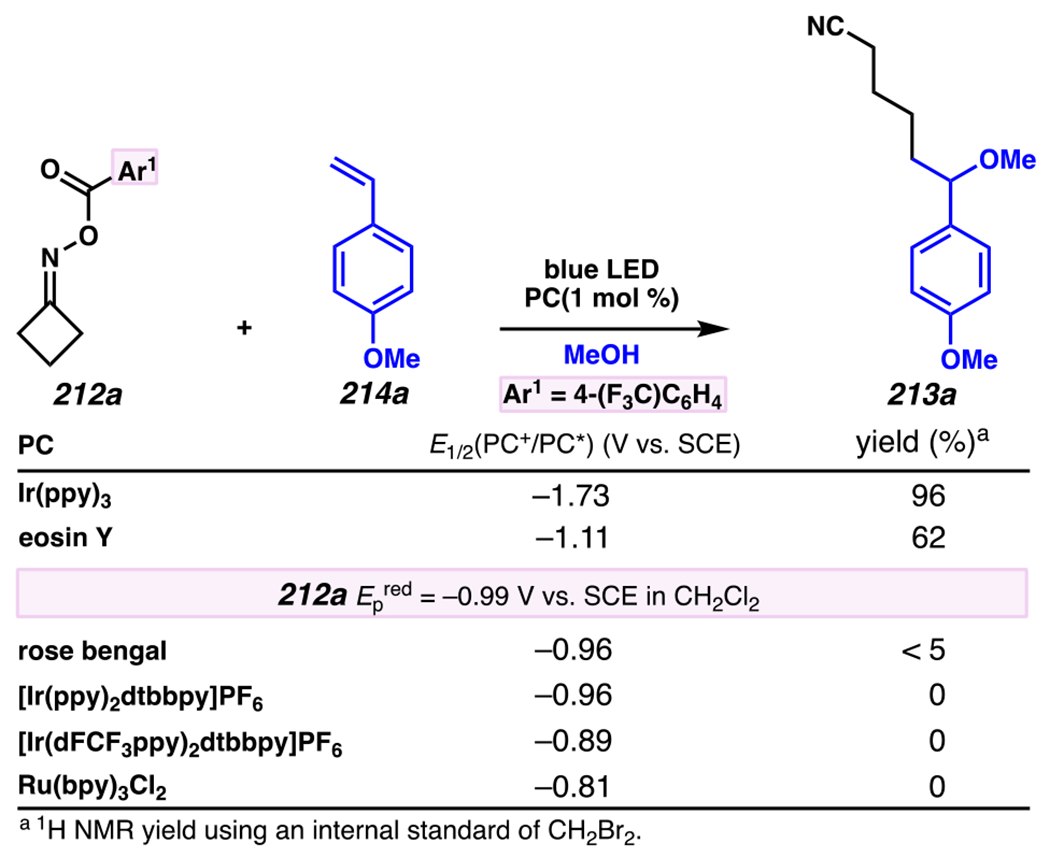
Strongly Reducing Photocatalyst Confers Optimal Reaction Efficiency in Three-Component Coupling Reactions
This approach enables three-component coupling reactions that engage cyclobutanone-derived O-acyloximes 212a as iminyl radical precursors and styrenyl derivatives as radical trapping agents. Following radical trapping, generated radical intermediate 215 is presumed to undergo in situ oxidation furnish a cationic intermediate, which is trapped by the third reaction component: a nucleophilic solvent (Scheme 55). Indeed, the oxidation of benzyl radical 215 (Ep/2 = +0.73 V versus SCE)204 to benzyl cation 216 is thermodynamical feasible with Ir(ppy) (E1/2[IrIV/IrIII] = +0.77 V versus SCE).16 In methanol, this process can provide synthetically useful access to β-alkoxyester derives, a class of compounds that is typically accessed via aldol addition reactions, based on use of the 1,2-disubstituted olefin, ethyl cinnamate as a radical trapping agent. Alternatively, this process can be used to install a tetrasubstituted distal center based on the use of a 1,1-disubstituted alkene, such as isopropenylbenene, as a radical trapping agent. Furthermore, 1,3-amino alcohol analogues may be accessed using a 3-azetidinone-derived oxime as a substrate. To target formation of more complex ethers, other nucleophilic alcoholic solvents can be employed, such as propargylic alcohol and ethylene glycol. Alternatively, to form simpler benzyl alcohols, water is a suitable nucleophilic solvent.
Scheme 55.
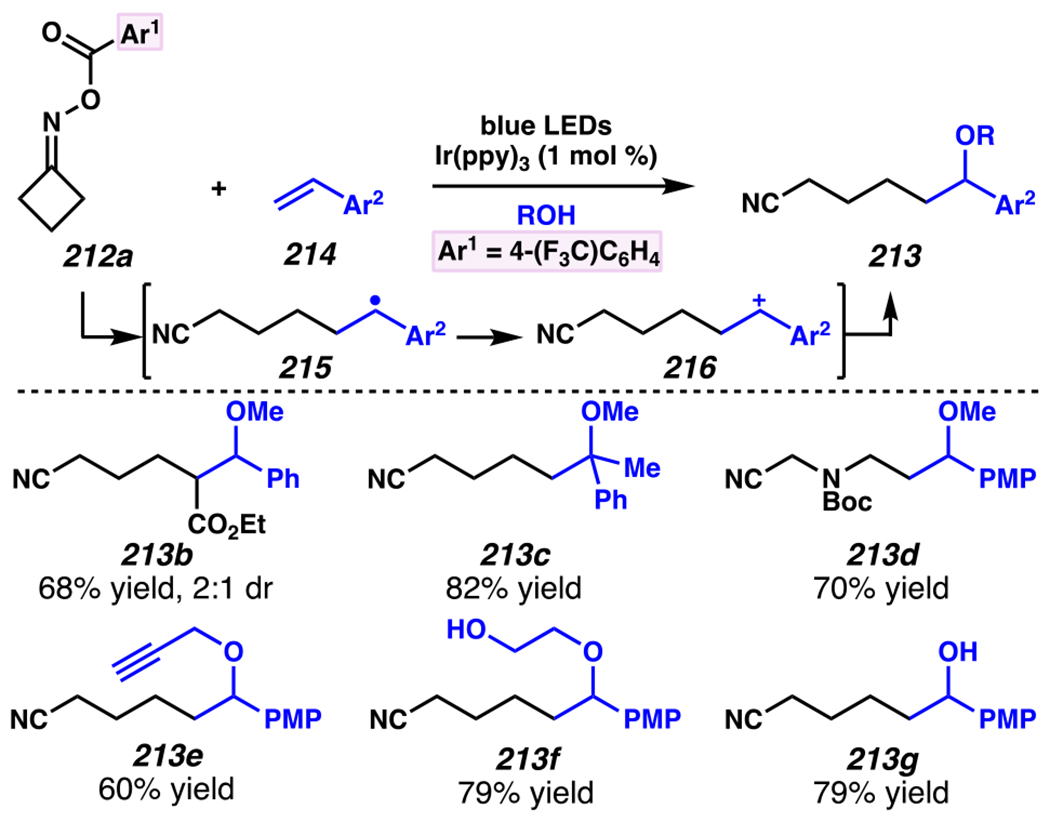
Three-Component Coupling Reactions Furnish Highly Decorated Aliphatic Nitriles
5.4.6. Ring-Opening Cascades Can Rely on O-Acyl Cyclic Oxime Substrates in Net Vinylation Processes
In principle, many alternative approaches could be used to productively engage intermediates generated from cyclobutanone-derived O-acyl oxime 212 through two-component cascades involving iminyl radical formation / β-scission, and addition across a π-system. Specifically, Yu has demonstrated that if the engaged olefin is a styrylboronic acid, the generated benzylic radical 219 is proposed to be oxidized to benzylic carbocation 220, and this intermediate cation can be quenched through deborylation to furnish an (E)-1,2-disubstituted olefin (Scheme 56).205 This strategy permits assembly of biologically relevant scaffolds such as 4,4-disubstituted piperidine,206 which is prepared in 83% yield and in modest stereoselectivity. Additionally, 1,2-disubstituted allylether and 1,3-diarylpropene are accessed in synthetically relevant yield with high stereoselectivity. As such, this type of reactivity can be harnessed to forge new C(sp2)–C(sp3) bonds with appropriate olefin coupling partners.
Scheme 56.
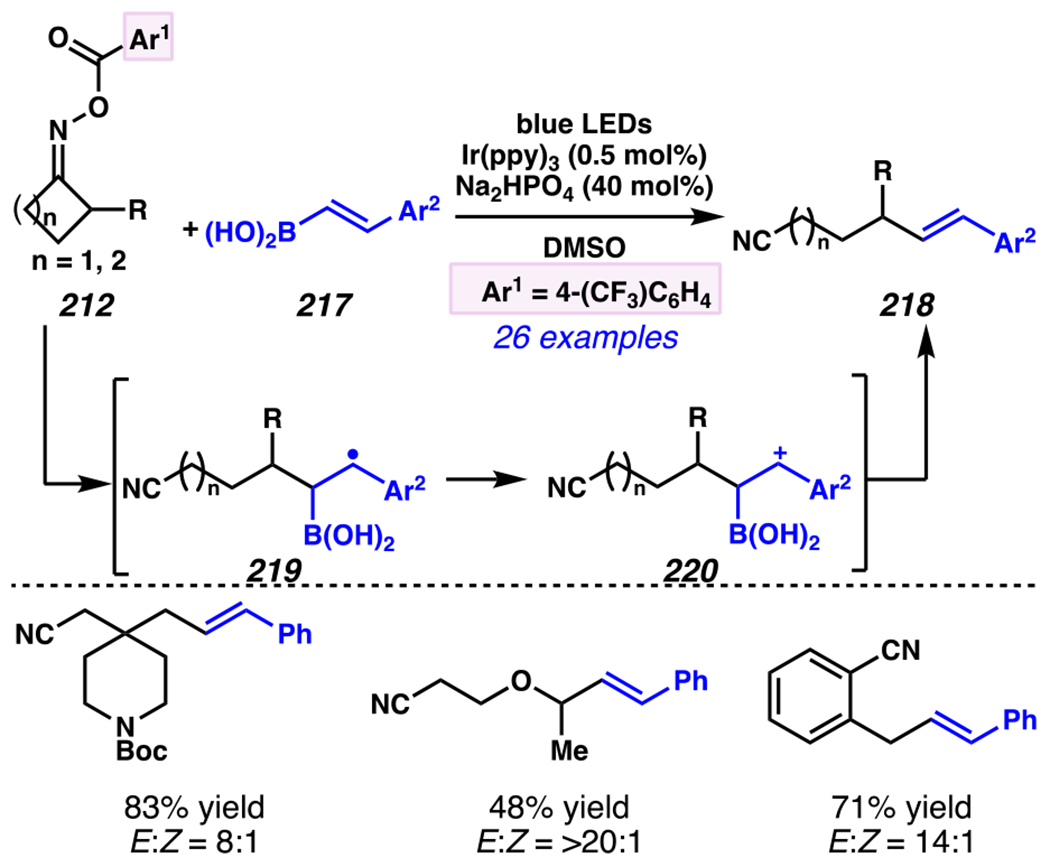
C–C Cleavage and Radical Addition Cascade Proceeds when O-Acyloximes React with Styrylboronic Acids to Affect a Net Vinylation Reaction
A vinylation reaction is also feasible based on initial cyclobutanone-derived O-acyl oxime substrates 221, while relying on the same initial two-component cascade involving iminyl radical formation / β-scission, and addition across an olefin. Indeed, Chen, Xiao and co-workers intercept these cascades to affect several complementary reactions (Schemes 57–58). In a net vinylation sequence, following radical trapping by an olefin, oxidation of a putative benzylic radical provides a benzylic cation, which undergoes base-mediated elimination reaction of the homobenzlic proton to furnish the olefin (Scheme 57A).207 Intriguingly, this reaction manifold is robust to silyl enol ether as radical acceptors (Scheme 57B). For example, O-acyloxime 212a reacts with tri-substituted silyl enol ether to provide the α-substituted ketone 227, a class of radical acceptor that was not previously viable within the context of another iminyl radical cascade.128
Scheme 57.
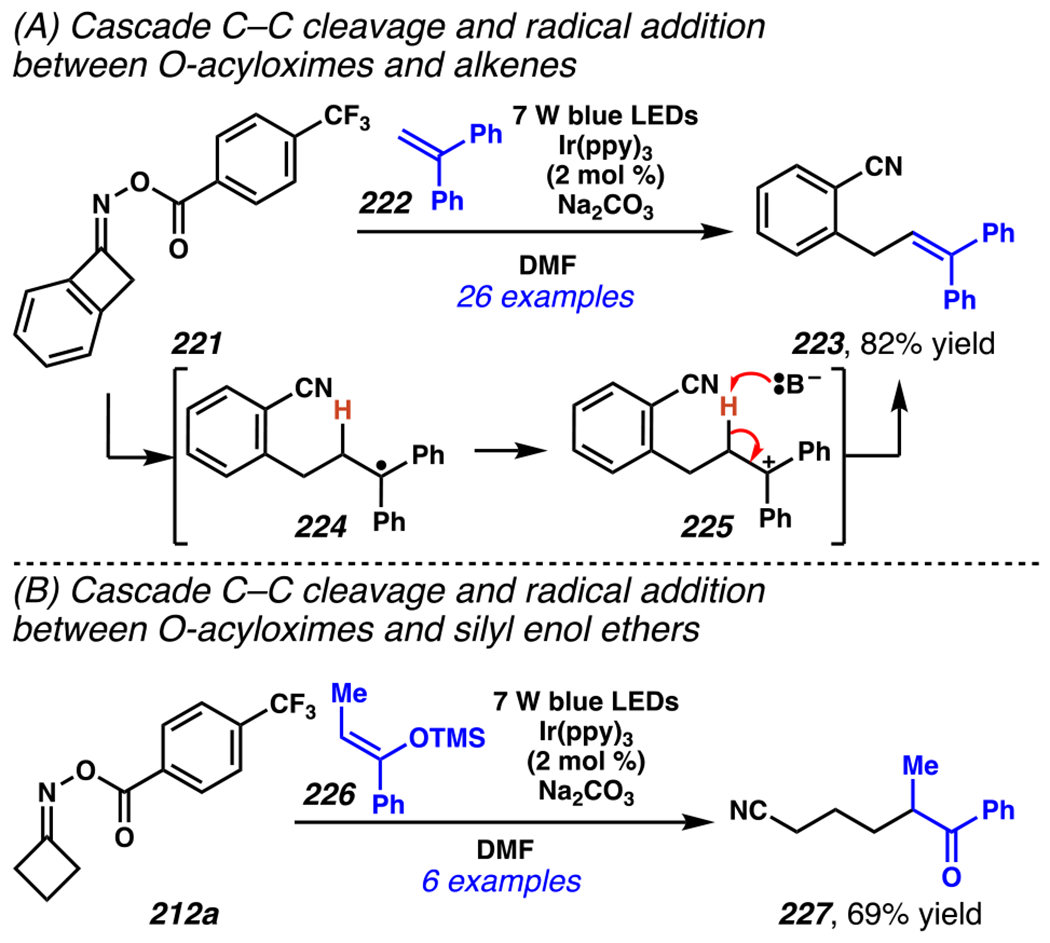
Cascade C–C Cleavage and Radical Addition Between O-Acyloximes and Olefin Radical Acceptors Provide Nitriles with π-Systems
Scheme 58.

When Terminal Alkenyl Cyclobutanols Are Radical Trapping Agents, They Ring Expand to Furnish Cyclopentanone Scaffolds
5.4.7. Ring-Opening Cascades Can Rely on O-Acyl Cyclic Oxime Substrates to Trigger Ring-Expansion Processes
Furthermore, a mechanistically analogous initial two-component cascade can be proposed wherein cyclobutanone-derived O-acyl oxime substrates 212 react to trigger ring expansion of vinyl cyclobutanol radical trapping agents 228 and furnish α-arylated cyclopentanone products 229 (Scheme 58).208 A plausible mechanism to explain this reactivity involves a familiar cascade sequence of iminyl radical formation / β-scission / addition across an olefin / radical oxidation that can furnish a cation that is vicinal to a fully-substituted alcohol. This intermediate would trigger a 1,2-alkyl shift in a process that is analogous to type-1 semipinacol rearrangement and furnish α-arylated cyclopentanone products 229. Consistent with the proposed reaction mechanism, these reactions are more efficient when they engage electron-dense vinyl arenes, which are better poised to resonance stabilize an intermediate cation (i.e., 231). So, anisole-substituted product 229a forms more efficiently (in 92% yield) than 4-fluorophenyl-substituted analogue, which is isolated in 54% yield. An appropriately positioned ester could stabilize radical intermediate 230 (Y = CH2, R1 = CO2Me), and does not appreciably affect the reaction outcome, furnishing ester-substituted 229d in 90% yield in 1:1 dr. By contrast, oxetane-derived oxime reacts to form ether 229c in 54% yield. In this case, it is possible that ring-opening to relatively stable α-oxygenated radical 230 (Y = O, R1 = H) could slow olefin trapping en route to ether 229c, which could be the origin of depressed reaction efficiencies. Oxetanes themselves do not poison the reaction: vinyl oxetanes can engage in the reaction as radical trapping agents, as evidenced by production of dihydrofuranone 229e in 83% yield. Additionally, while this process is sensitive to substrate substitution, this reaction can engage less strained cyclopentanol-derived terminal olefin 232 as viable coupling partner with oxime 212a to deliver cyclohexanone 233 in 69% yield.
5.4.8. Ring-Opening Cascades Can Rely on O-Acyl Cyclic Oxime Substrates to Affect Alkene Carbofunctionalization Reactions
A more practical strategy to introduce carbonyl group to alkyl nitriles from ketone feedstocks is through a radical-polar crossover cascade that couples O-acyloxime 212a and more readily accessible olefin trapping agents (i.e., aryl olefins) in the presence of DMSO to affect a radical addition / Kornblum oxidation cascade. 209,210 This reaction is presumed to proceed through a cascade involving iminyl radical formation / β-scission, and addition across a π-system, yet, the thusly generated intermediate cation (i.e., 236) is expected to be intercepted intermolecularly by DMSO, with generated cation 237 serving as a viable intermediate in a formal Kornblum oxidation (Scheme 59). While the traditional Kornblum oxidations requires exogenous base to convert alkoxysulfonium intermediates to ketones, this reaction does not require exogenous base to obtain the desired ketone product. In fact, the use of base diminishes the reaction performance.b The operative oxidation process is speculated to arise from a photoredox-mediated process.211 Specifically, this research relies on the recognition by Akita and co-workers that an alkoxysulfonium intermediate, such as 237, can be reduced by a photocatalyst in the key step of a radical-mediated formal Kornblum oxidation reaction.211, 212 This mechanistic hypothesis is consistent with the detection by HRMS of dibenzylsulfide in a crude reaction mixture (Scheme 59B). Furthermore, oxygen-isotope labeling experiment with Me2S18O (89% 18O incorporated) delivers unlabeled and labeled products 238a and 238b in 23:73 ratio.210 This mechanistic proposal is also consistent with the reaction outcome when α-arylated oxime 212c is used as a substrate. Specifically, oxime 212c is expected to react through thermodynamically stable benzylic radical 240, which does not trap olefin, and therefore does not form three component coupling product 241, but instead forms ketone 239 directly in 60% yield (Scheme 59C).209
Scheme 59.

Base-Free C–C Cleavage / Radical Addition /Kormblum Oxidation Cascade Reactions Are Triggered by Photoredox Catalysts in the Presence of Vinylarenes and DMSO
While the Kornblum oxidation cascade constitutes a viable strategy to incorporate ketone functionality, sulfonylation can proceed when an SO2 surrogate is included in the reaction. In fact, 1,4-diazabicyclo[2.2.2]octane disulfate (DABSO) has been employed as SO2 surrogates to affect visible-light mediated sulfonylation cascade with O-aryl γ,δ-unsaturated oximes and silyl enolates.213,214 Less expensive, commercially abundant potassium metabisulfite can also be used as an SO2 surrogate to affect sulfonylation215 under photoredox-mediated conditions.216 O-acyl oximes 212, K2S2O5, olefins and alcohols participate in multi-component coupling reactions to provide aliphatic sulfones 242 (Scheme 60). This reaction is speculated proceed through trapping of cyanoalkyl radical 189 by SO2 to provide sulfonyl radical 243, which is trapped by olefin to deliver radical species 244. Subsequent oxidation of benzylic radical 244 to carbocation 245 and nucleophilic attack by alcohols ultimately produces product 242. Presumably, oxidation of the benzylic radical occurs at a rate faster than k ≈ 8.8 × 107 s−1, as a cyclopropyl radical is expected to undergo ring-opening at this rate,217 and product 242a, with an intact cyclopropane forms in 45% yield. This result is difficult to interpret as it is unclear whether cyclopropyl ring-opening products have been detected in this reaction. Reaction conditions are sufficiently mild to enable conversion of 1-methyleneindane to elimination-prone tertiary ether product 242b, without generating potential indene byproduct.216 The reaction benefits from strain-release during cyclobutane opening, as less-strained cyclopentanone-derived O-acyl oxime is an ineffective substrate that does not provide the product 242c. In terms of nucleophiles, while tert-butanol is not an effective trapping agent in this reaction, presumably owing to steric encumbrance, the reaction is flexible enough to allow the use of water and CD3OD as nucleophiles, paving the way to deliver tertiary alcohol 242d, or to assemble deuterium- incorporating sulfones.
Scheme 60.
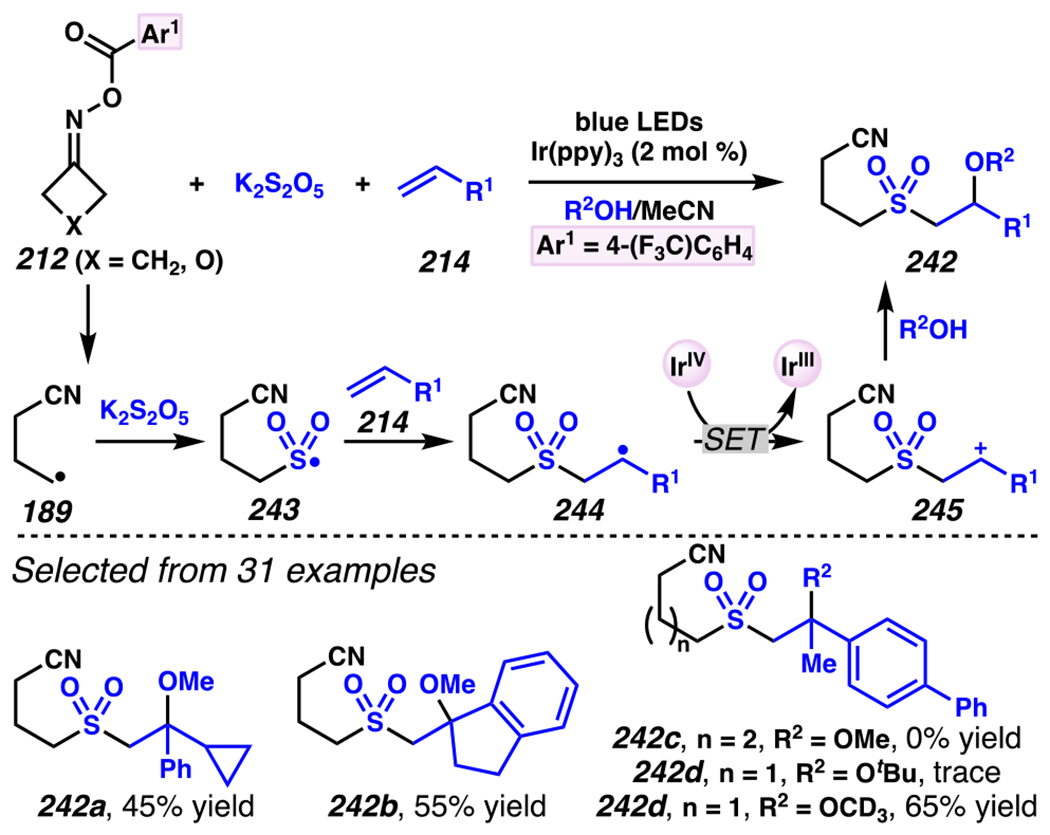
Potassium Metabisulfite Is an Effective SO2 Source to Assemble Benzylic Sulfones
The cascade sequence involving iminyl radical generation / β-scission and SO2 trapping can be invoked to generate alkylsulfonyl radical 243, which can be intercepted by aryl-substituted methylenecyclopropanes 246 to provide 3,4-dihydronaphthalenes 247 (Scheme 61).218 Plausibly, the initial three-component iminyl radical generation / β-scission and SO2 trapping cascade furnishes carbon-centered radical intermediate 248 that undergoes β-scission to form homoallylic radical species 249. This homoallylic radical is trapped intramolecularly by a pendant arene and re-aromatization (not depicted) ultimately yields product 247. Higher yields are observed when methylenecyclopropanes incorporate electron-dense arenes, such as anisole analogues, rather than electron-deficient arenes, such as benzonitrile derivatives 247b and 247f. Sterically, ortho-substituted aryl methylenecyclopropanes (247a and 247b) are the least responsive olefin substrates in the reaction. Aryl methylenecyclopropanes with meta-substitution are more reactive but suffer because radical-addition to arene is not position-selective. For example, the reaction product with meta-substituted benzylic ether is isolated as a mixture of positional isomer, favoring A1,3-strain minimized product 247c over A1,3-strained product 247d. Not surprisingly, para-substituted aryl methylenecyclopropanes (247e and 247f) are the most reactive among the arene series. This three-component coupling reaction is not effective with phenyl-substituted methylelecyclobutane 250 as a radical trapping agent, presumably because it is expected to undergo net endergonic cyclobutyl-ring-opening / seven-membered ring formation sequence (∆G ≈ +4.9 kcal•mol−1).179 This reaction is not effective when the iminyl radical is generated from heterocyclic oximes, such as azetidine and oxetane derivatives (212d and 212e). Lastly, but not surprisingly, less torsionally strained five-membered cyclic oxime 212f does not furnish product under the reaction conditions.
Scheme 61.
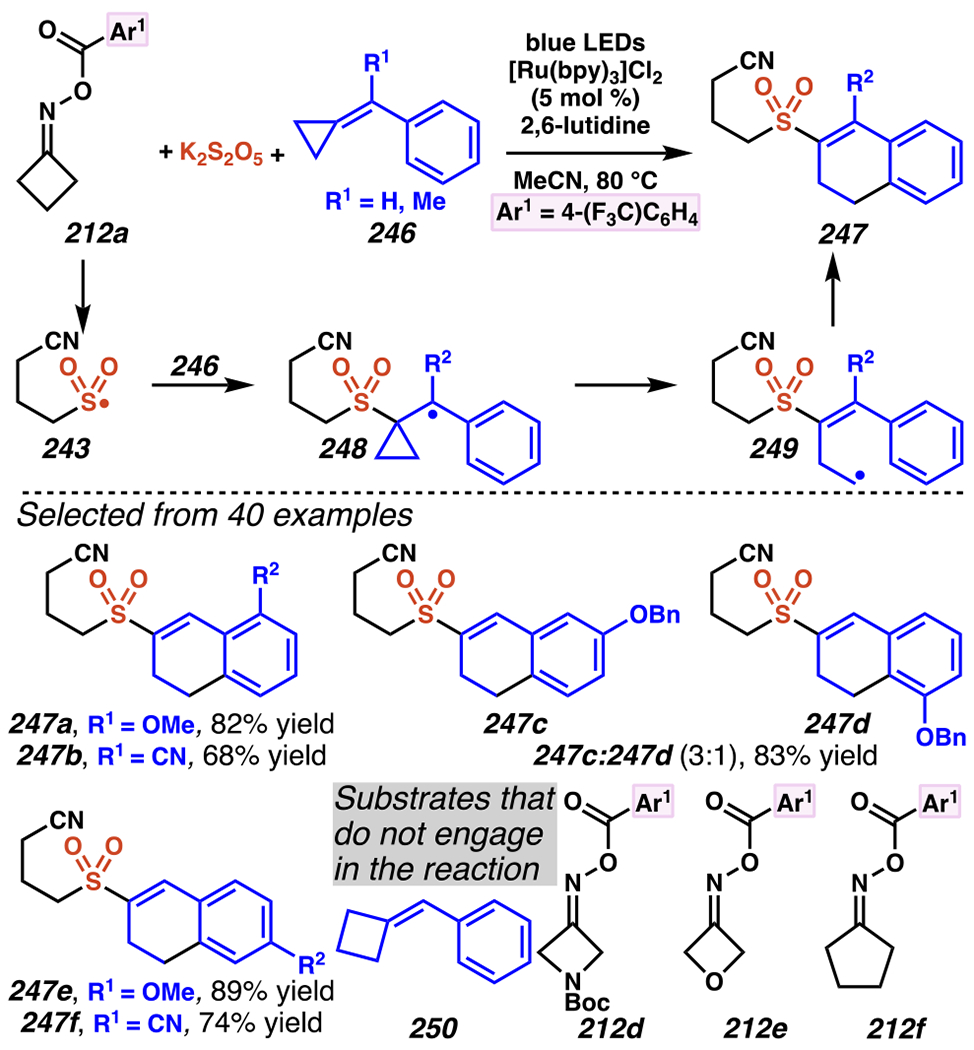
Reactions of Alkylsulfonyl Radicals with 2-Aryl-Substituted Methylenecyclopropanes Leads to Cyanoalkylsulfonylated 3,4-Dihydronaphthalene Scaffolds
By extension, the iminyl radical generation / β-scission / addition to SO2 cascade sequence can be used to generate alkylsulfonyl radical 243, which can be intercepted by cyclic vinyl sulfone 252, which can serve as both an “SO2”-source and radical trapping agent in the preparation of β-ketosulfones 253 (Scheme 62). 219 Mechanistically, this transformation leverages organic triflate’s propensity to fragment towards F3C• and SO2220 and relies on chemoselective alkyl radical addition to SO2 to generate alkylsulfonyl radical species.221 To initiate the reaction, literature precedent suggests that energy transfer between photosensitizer and vinyl sulfone 252 generates enol radical 254 and CF3SO2•.222 A net radical α-trifluoromethylation of 254 liberates SO2, which is poised to react with radical species 189 to form alkylsulfonyl radical 243. Intermolecular trapping of alkylsulfonyl radical 243 by another molecule of cyclic vinyl triflate 252 delivers O-stabilized enol radical 257. This enol radical is poised for β-scission to deliver β-ketosulfone 253 and F3CSO2•, which can fragment into F3C• and SO2 to propagate another cycle of product formation. This transformation tolerates oximes with synthetically useful functional groups, such as ester and nitrile; however, yields erode with phenyl-substituted oxime substrates, unless the benzylic carbon is fully substituted. Branched β-ketosulfones such as 253e and 253f can be synthesized, albeit with reduced isolated yields. Unlike the aforementioned sulfonylation reactions, this reaction renders five-membered cyclic oxime to provide the product 253f in 42% yield.
Scheme 62.
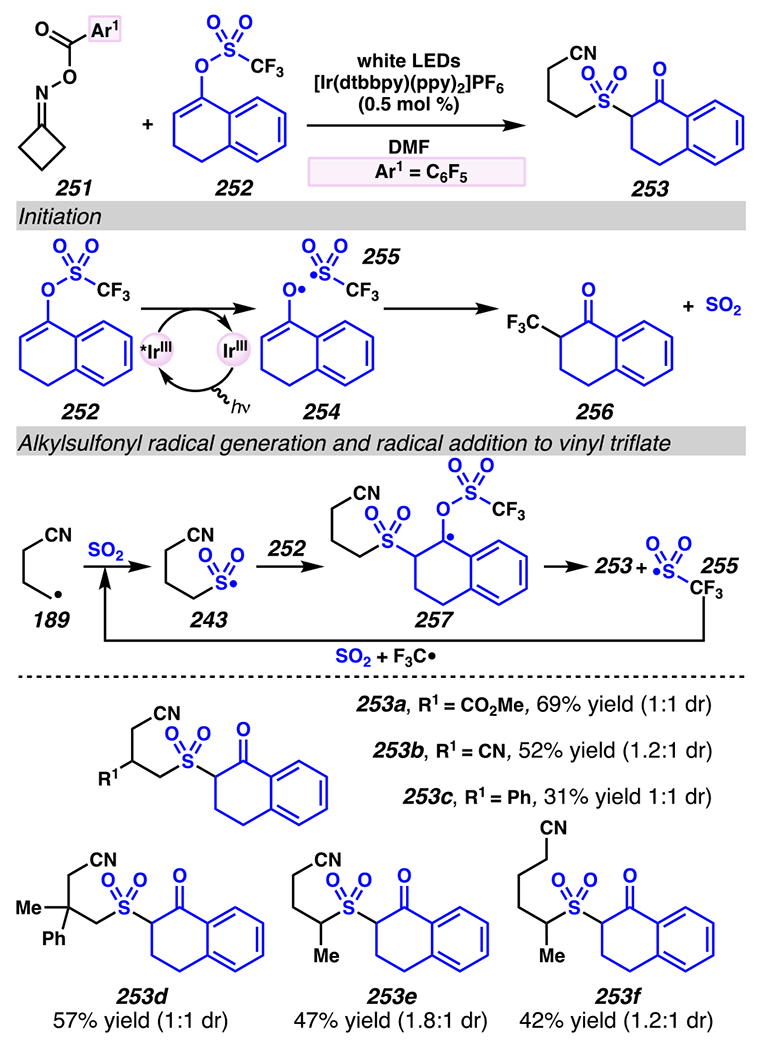
Oximes and Vinyl Triflate Engage in β-Scission / Radical Addition / Radical Fragmentation to Construct β-Ketonesulfones
5.4.9. Ring-Opening Cascades Can Rely on O-Acyl Cyclic Oxime Substrates in (Hetero)arylation Reactions
The related Minisci-type reactions223, 224 permit synthetic chemists to formally alkylate heteroarenes and electron-deficient arenes with alkyl radical species via C(sp2)–H alkylation process. Thus, Minisci-type reactions between cyanoalkyl radicals generated from cyclic oximes and heteroarenes should be possible. In 2017, the Guo laboratory demonstrated that such cyanoalkyl radicals effect Minisci-type reactions with heteroarenes under thermally- and nickel-mediated process.225 The aforementioned research begat a series of milder photoredox-mediated Minisci-type reactions with cyclic oximes as alkyl radical precursors and heteroarenes (Scheme 63).226, 227, 228 Research laboratories led by Kong, Xu, and Wang have disclosed that O-acyloxime 212a reacts with quinoxalinone 258a to deliver alkylated quinoxalinone product 259, with DMF or CH2Cl2 as a reaction solvent (Scheme 63A). 226, 227 Shortly thereafter, Xiang, Yang and co-workers document that perfluoropyridin-4-ol-derived oxime 260 is an appropriate precursor to generate the same product via a Minisci-type reaction with the same quinoxalinone (i.e., 259, Scheme 63B).228 Collectively, this transformation depends on strongly reducing Ir(ppy)3 photocatalyst (E1/2 [*IrIII/IrIV] = −1.73 versus SCE)16 to cleave the redox-active handles to reveal the iminyl radical. The O-perfluoropyridinyl-derived oxime 260 (Ep/2 = −1.49 V versus SCE)227 can be cleaved by oxidative quenching of *Ir(ppy)3, and by thermal N–O bond cleavage in dark. Indeed, the reaction without light furnished the desired product 259 in 60% yield. Sans Ir(ppy)3, the reaction yields trace quantities of product 259. At this point, the role of Ir(ppy)3 is unclear, but, in a less mechanistically ambiguous scenario, the perfluoropyridin-4-ol handle offers additional opportunity to trigger Minisci-type reactions with heteroarenes under thermally mediated conditions.228 Following iminyl radical formation and β-scission, cyanoalkyl radical species 189 is poised to undergo radical addition to quinoxalinone to generate N-centered radical intermediate 261, which is oxidized by photocatalyst to generate highly reactive ammoniumyl species that is poised for base-mediated re-aromatization to give the alkylated quinoxalinone 263 (Scheme 63C).
Scheme 63.
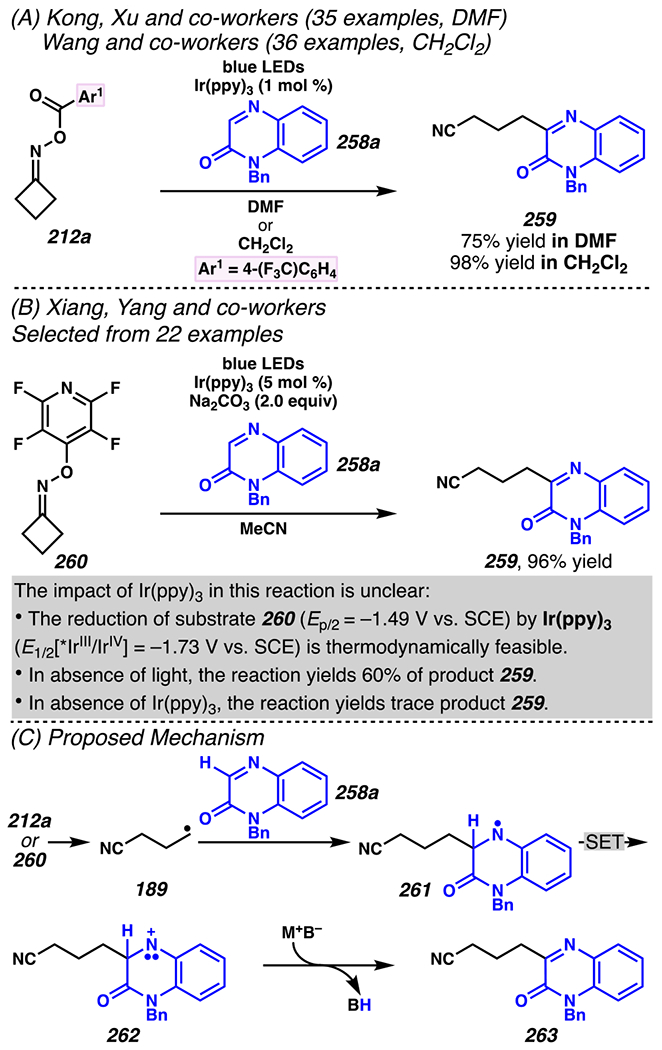
Photoredox-Mediated Minisci-Type Reactions Rely on Cyclic Oximes and Quinoxalinones
Each of these disclosures about potentially photo-mediated Minisci reactions documents reactivity that is distinct. Wang and co-workers show that less-strained α-phenyl substituted six-membered cyclic oxime reacts with quinoxalinone 258a to provide a less compact product 264 in 42% yield (Scheme 64A).227 Concurrently, Kong and Xu laboratories show that chromenone 265 can engage in Minisci-type reaction with oxime 212a to yield the alkylated product 266 in 46% yield,226 which can be thought of as a base-free alternative that compliments the technology pioneered by Chen, Xiao and co-workers (Scheme 57).207
Scheme 64.
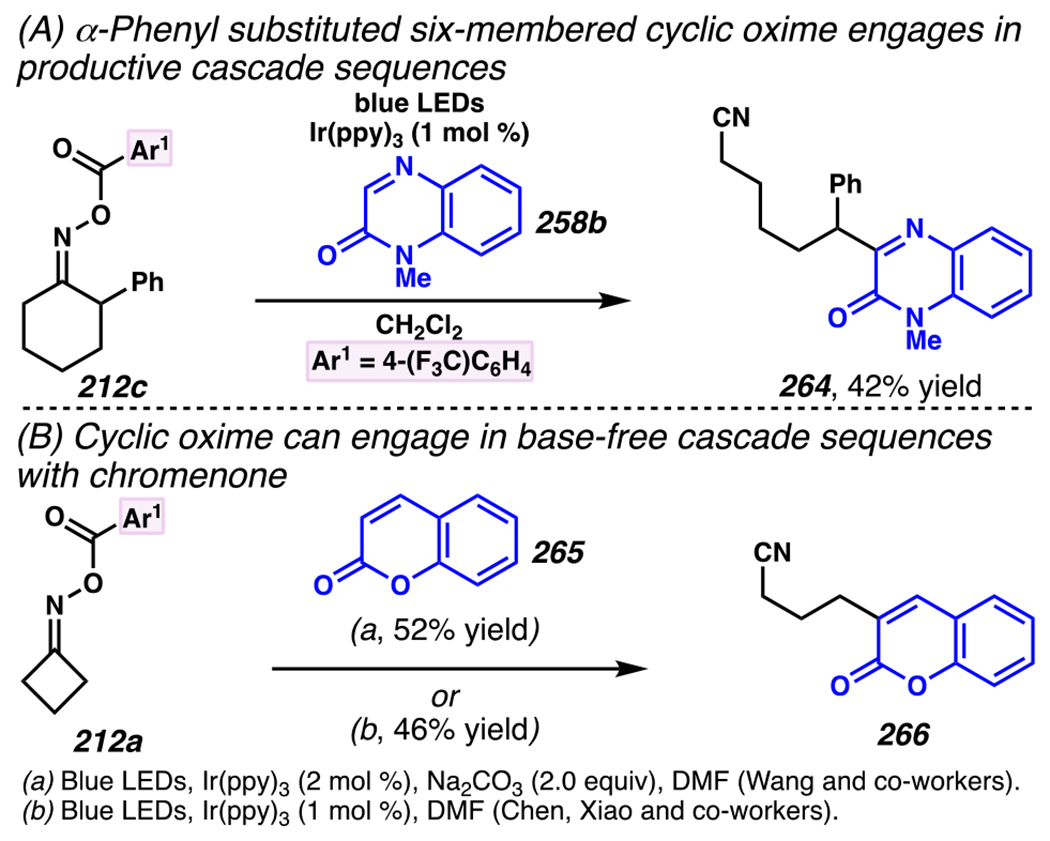
α-Phenyl Substituted Six-Membered Cyclic Oxime and Chromenone are Viable Substrates
Quinoxalinones are not unique as viable heteroarene radical trapping agents, the Minisci-type reaction can engage other heteroarenes to provide isoquinoline 268 in 37% yield (Scheme 65A).227 The sluggish reaction performance is improved by the addition of triflic acid, which presumably protonates isoquinoline to generate a more reactive radical acceptor poised, after radical addition, to undergo single electron oxidation and subsequent elimination, ultimately generating isoquinoline 268 in 94% yield.229 While many heteroarenes are viable, unsubstituted quinoline 269 and 2-phenylpyridine 271, which have an additional electrophilic carbon-center, produce regioisomeric mixtures of products 270a, 270b and 272a, 272b, respectively (Scheme 65B). These photo-mediated reactions compliment a silver-catalyzed process.230
Scheme 65.
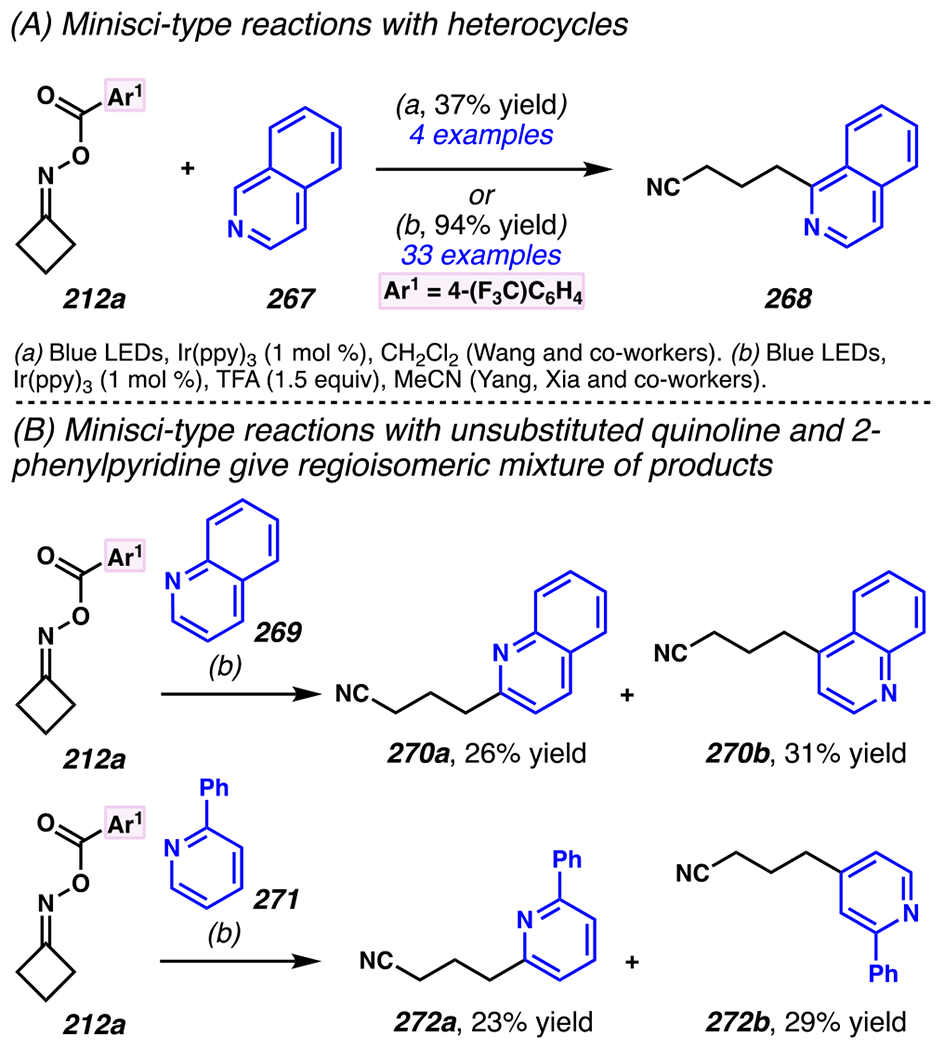
Cyclic Oximes Can Participate in Minisci-Type Reactions with Heterocycles to Furnish Alkylated Isoquinolines, Quinolines and Other Heteroarenes
5.4.10. Ring-Opening Cascades Can Rely on O-Acyl Cyclic Oxime Substrates in Annulation Processes
Logically, these approaches can be extended to allow for net annulation cascades. The laboratories of Zhao, Chen and Xiao, recognized the viability of annulation cascades involving N-phenylacrylamide (273) as a radical trapping agent to assemble 3,3-dialkyloxindole derivative 274 (Scheme 66).203,207 Furthermore, both publications document isonitriles as radical trapping agents in the course of annulation reactions (Scheme 66).203,207 As a complement to these sp-hybridized π-systems, Chen, Xiao and co-workers demonstrated that phenylacetylene derivative 277 traps intermediates arising from 3-phenyl-O-acyl oxime 212g in a net iminyl radical formation/ β-scission / radical formal [4+2]-cycloaddition reaction to access nitrile-decorated 1,2-dihydronaphthalene derivative 278. (Scheme 66).207 These photoredox-mediated radical annulation cascades prompted development of synthetic strategies to assemble polycyclic scaffolds.
Scheme 66.
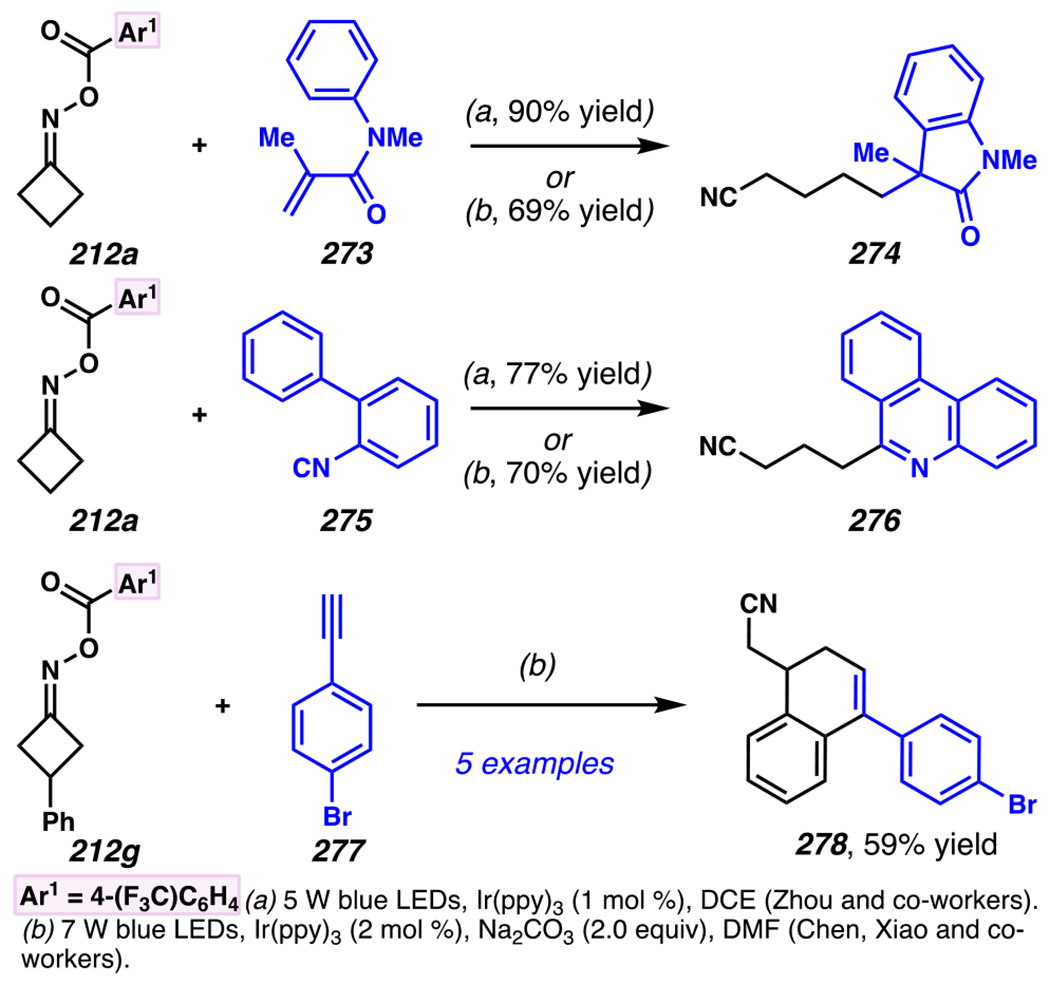
Vinyl Arenes Are Not the Only Radical Acceptors That Engage in Coupling Reactions with O-Acyloximes
The net iminyl radical formation/ β-scission / radical formal [4+2]-cycloaddition reaction of 3-phenyl-O-acyl oxime 212g and phenylacetylene derivative 277 relies on a pendant arene within cyclobutanone-derived O-acyl oxime 212g for annulation (Scheme 66, third reaction).207 Chen and Xiao laboratories have developed complementary synthetic protocol that employs 3-naphthyl-substituted O-acyl oximes 212 and electron-deficient olefins as coupling partners to build 1,2,3,4-tetrahydrophenanthrene derivatives 279 (Scheme 67A).231 The efficiency of this protocol depends on specific nature of the electron-deficient olefin radical trapping agent. These reaction conditions do not efficiently engage β-substituted methyl acrylate, dimethyl fumarate, α,β-unsaturated amide, or styrene in the desired reactions. Fortunately, sterically encumbered 3-methylene-dihydrofuran-2-(3H)-one proves to be a viable radical trapping agent and provides spirocyclic product 279b in 27% yield. Furthermore, reactions with α,α-disubstituted acrylates furnish 1,2,3,4-tetrahydrophenanthrenes with tetrasubstituted carbon-centers. Notably, this strategy affects position-selective radical addition to arenes and avoids the production of site-isomeric product. Site-selectivity may prove to be predictable: DFT calculations are consistent with the hypothesis that kinetically originated selectivity reflects the relative stabilities of the two penultimate carbon-centered radical species 281a and 281b leading to the two positional isomers (Scheme 67B). Indeed, the desired carbon-centered radical 281a is estimated to be 7.0 kcal•mol−1 more stable than its site-isomer 281b.
Schemes 67.

3-Naphthyl Substituted O-Acyl Oximes and Electron-Deficient Olefins Engage in Position-Selective Radical Annulation Cascade to Synthesize 1,2,3,4-Tetrahydrophenanthrene Scaffolds
In the absence of an α- or β-aryl substituent on the cyclobutanone-derived O-acyl oxime, annulation reactions have been reported with aryl isonitriles as radical trapping agents, once again relying on net iminyl radical formation/ β-scission cascades to furnish cyclopenta[b]quinoxalines (Scheme 68). 232 Previously, tin-based radical initiator had been used to synthesize cyclopenta[b]quinoxlines from aryl isonitrile and iodonitrile.233,234 The photoredox manifold obviates the use of toxic stannane reagents. In this transformation, the addition of O-acyl oxime 212-derived cyanoalkyl radical 189 to aryl isonitriles 282a forms carbon-centered radical 284, which is poised to engage in the first annulation sequence (Scheme 68A). In the event, ring closure furnishes iminyl radical 285, which cyclizes onto a well-positioned arene. Rearomatization reveals cyclopenta[b]quinoxaline products 283a. In these reactions, arene substitution pattern impacts regio- and chemoselectivity. For example, meta-substituted aryl isonitriles such as 282b and 282c produce regioisomeric mixture of products 283b, 283c and 283d, 283e, respectively (Scheme 68B). Reasonably, under the reaction conditions, o-thiomethyl substituted 282d delivers alkylated benzothiazole product 286 instead of anticipated product, suggesting that the radical-addition to SMe is favored over the radical-addition to nitrile (Scheme 68C).
Scheme 68.
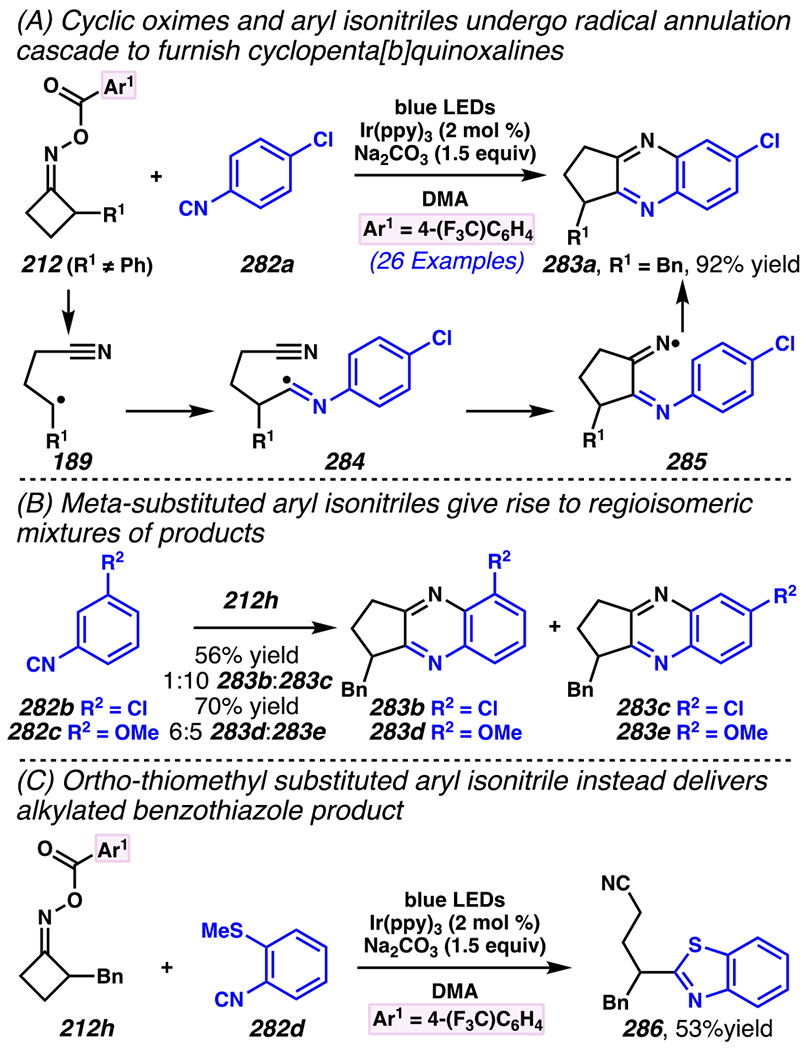
Cyclopenta[b]quinoxaline Derivatives Are Synthesized from Radical Annulation Cascade Reaction Between Cyclic Oximes and Aryl Isonitriles, with Well-Documented Limitations
5.4.11. Ring-Opening Cascades Can Rely on O-Acyl Cyclic Oxime Substrates to Affect Atom- and Group-Transfer and -Trapping Processes
Furthermore, group-transfer agents (Y–X–X–Y) such as disulfides, diphenyldiselenide and diboronates have been identified as radical trapping agents that partake in the cascadic two-component coupling reactions with O-acyl cyclic oximes (Scheme 69). 235 These group-transfer agents are electronically compatible O-acyl cyclic oximes because electron-rich radical byproduct Y–X• can reduce oxidized photocatalyst. Thiolation reactions with azetidine-derived oximes are viable with dimethyldisulfide and a variety of electronically perturbed diaryldisulfides to provide amine-substituted sulfides (288a–288c) in good yields. Analogous selenylation reaction with the same oxime substrate and diphenyldiselenide provides amine 288d in 49% yield. O-acyl cyclic oximes respond to borylation reaction with diboronic acid and subsequent reaction work-up with pinacol affords easily isolatable boronic ester product. For example, benzyl ether substituted oxime is transformed to 288e, which is a scaffold could be carried further to access 1,2-diols, highlighting the broad variety that can be introduced into a molecular scaffold using this approach.
Scheme 69.
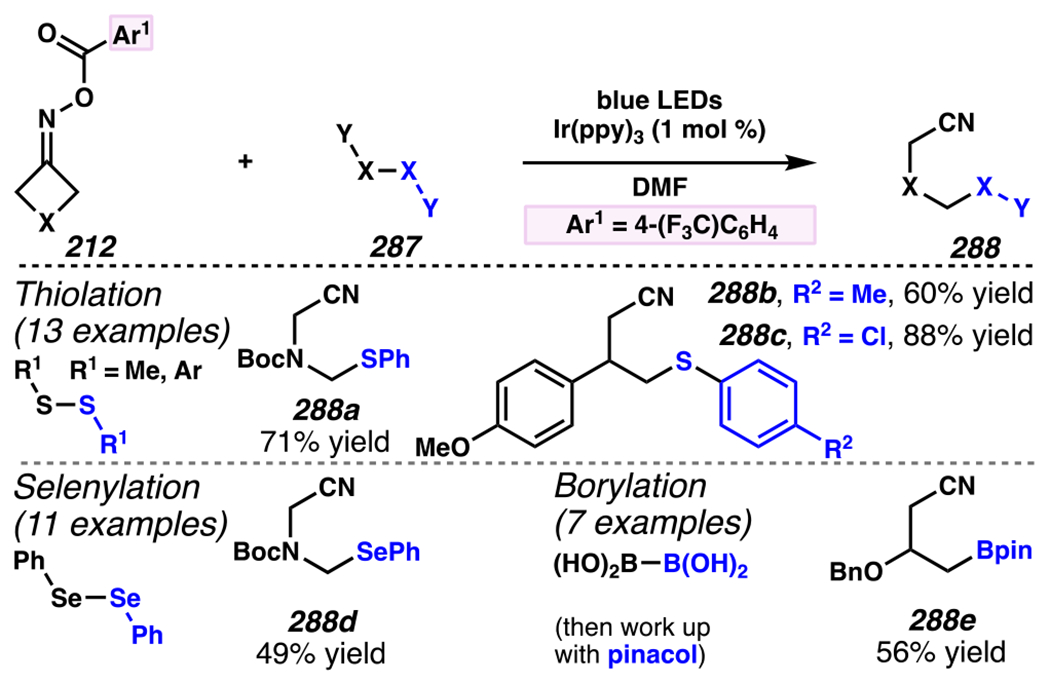
Oximes Participate in Group-Transfer Reactions with Disulfides, Diphenylselenide and Diboronic Acids
Another two-component coupling cascade that begins with O-acyl cyclic oxime substrates involves reductive carboxylation with carbon dioxide.236 Carbon dioxide is an inexpensive feedstock that is poised for nucleophilic attack by carbanion generated from iminyl radical formation / β-scission / single-electron reduction cascade. This type of reactivity is difficult to access from with O-acyl cyclic oximes because substrate reduction by the photocatalyst outcompetes reduction of corresponding alkyl radical to the critical carbanion intermediate. Zhang, Yu and co-workers used this challenge as a testing ground to convert O-acyl cyclic oxime to carbanion using exogenous reductant (Scheme 70).237 Mechanistically, it is proposed that upon excitation of [IrIII] to [IrIII*], Hünig’s base (Epox = +0.68 V versus SCE in benzene)238 triggers reductive quenching of [IrIII*] to [IrII] (E1/2[IrII/*IrIII] = +0.68 V versus SCE in MeCN).8 Resultant [IrII] complex is poised to reduce oxime 289 to generate radical anion 291. In fact, Stern-Volmer analysis reveals that Hünig’s base reductively quenches [IrIII*] more quickly (Kq = 3.3 × 108 M−1•s−1) relative to oxime 289. β-scission of the radical anion species reveals iminyl radical 292, which undergoes a second β-scission process to furnish benzylic radical species 293. It is speculated that [IrII] generated from reductive quenching of [IrIII*] by Hünig’s base reduces 293 to carbanion 294. Indeed, the generation of carbanion is supported by deuterium incorporation with D2O in the reaction of substrate 289 to afford product 296 with 95% deuterium incorporation. Presuming an intermediate radical anion forms, nucleophilic attack on CO2 would occur to deliver carboxylate 295, which could be protonated upon acidic work-up to provide carboxylic acid 290 and complete the ring-opening cascade sequence. Under optimal reaction conditions, oxime 289 is transformed to carboxylic acid 290 in 93% yield. This reaction does not proceed in the absence of light and furnishes trace product absent Ir(ppy)3. In absence of CO2, product 290 is detected in 35% yield, which suggests that sodium carbonate plays dual role of minor source of CO2 source and reaction base. Removing sodium carbonate from the reaction yields product 290 in 70%, which establishes CO2 as the major CO2 source. Intriguingly, absence of Hünig’s base nevertheless affords product 290 in 34% yield. So, many of the reaction components serve multiple roles in the reaction.
Scheme 70.
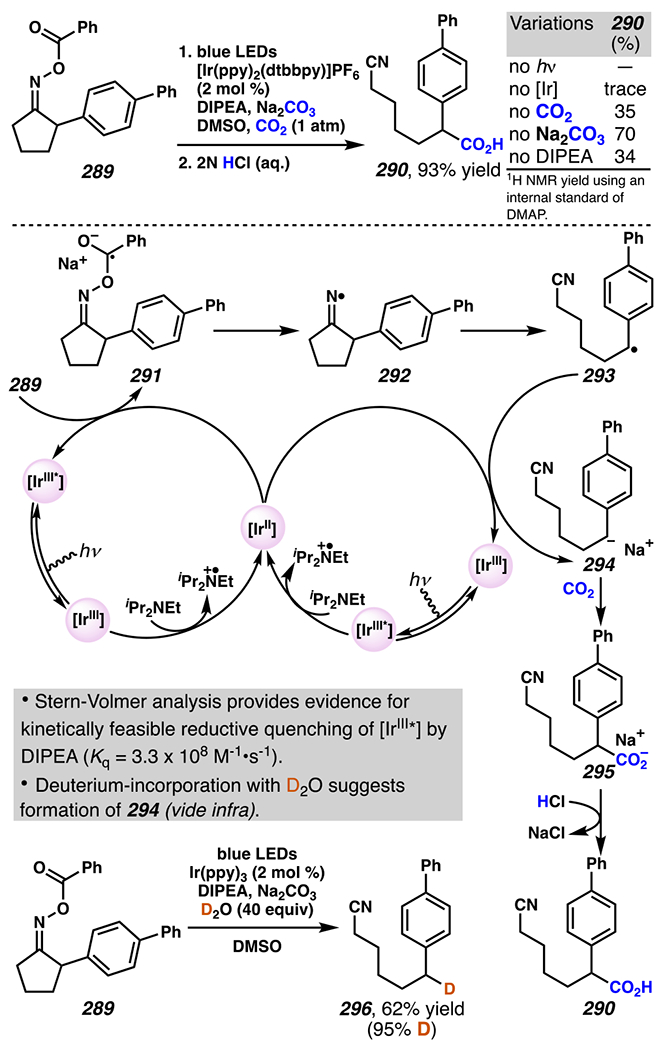
Oxime Can Engage in Carboxylation Reaction with CO2 to Access Carboxylic Acids
Through iminyl radical formation / β-scission cascades, other nucleophiles can be incorporated under the assistance of copper catalyst. This process is exemplified by non-photo-mediated copper catalyzed radical cross-coupling reactions of cyanoalkyl radicals generated from O-acyl oximes and terminal alkynes.239 With the rise in utility of copper under photo-mediated manifolds,240 analogous photo-mediated strategies employing copper catalysts have emerged. It is worth underscoring that despite ongoing discussions on copper’s dual role as a photocatalyst and a transition-metal catalyst,240 dark reactions are viable reaction pathways to elicit desired transformations. The complexity in reaction mechanisms involving copper arises in part because of the redox properties of copper.241 Notwithstanding the mechanistic uncertainty, irradiation with visible light nevertheless imparts improvement on reaction performance within the context of multicomponent coupling reactions involving cyclic oximes, radical trapping agents and organocuprate intermediates.242, 243 For these reactions, reaction efficiency improves in the presence of visible-light. This suggests that either a) the copper catalyst participates in photoredox process in tandem with light-independent pathways or, b) visible-light (blue light ≈ 450 nm or 63.5 kcal•mol−1) promotes oxidation of the copper catalyst by N–O bond scission (N–O BDE ≈ 57 kcal•mol−1).244 In light of these phenomenon, the upcoming discussions will include reactions that are speculated to involve photoredox events and may note reaction mechanisms that occur in the absence of light.
As an alternative strategy to intercept an iminyl radical formation/ β-scission cascade and to install an alkyne, copper catalyst can affect alkynylation in the presence of a terminal alkyne (Scheme 71). Without light, this reaction is possible under thermally mediated conditions.239 Alternatively, Shi and co-workers report a two-component coupling reaction between O-acyloxime 212a and terminal alkyne 297 with Cu(OTf)2 under visible-light irradiation to deliver alkynyl nitrile 298.245 This strategy can be employed to convert sterically encumbered cyclic oxime to the alkynyl nitrile 298a in 77% yield, as well as alkynyl nitrile with quaternary carbon-center 298b in 59% yield. This reaction also permits terminal alkynes such as indole-derived alkyne to provide the corresponding product 298c in 55% yield and can render diacetone D-glucose-derived terminal alkyne susceptible to the desired transformation to access complex alkynyl nitrile in 43% yield. Control experiments reveal that this reaction does not proceed in the absence of light, which suggests that light is critical to the operative reaction mechanism. The authors suggest that the benefits of light to the reaction suggest that the copper catalyst is an active photoredox catalyst240 or that photo-irradiation promotes oxidation of the copper catalyst by N–O bond scission.244
Scheme 71.
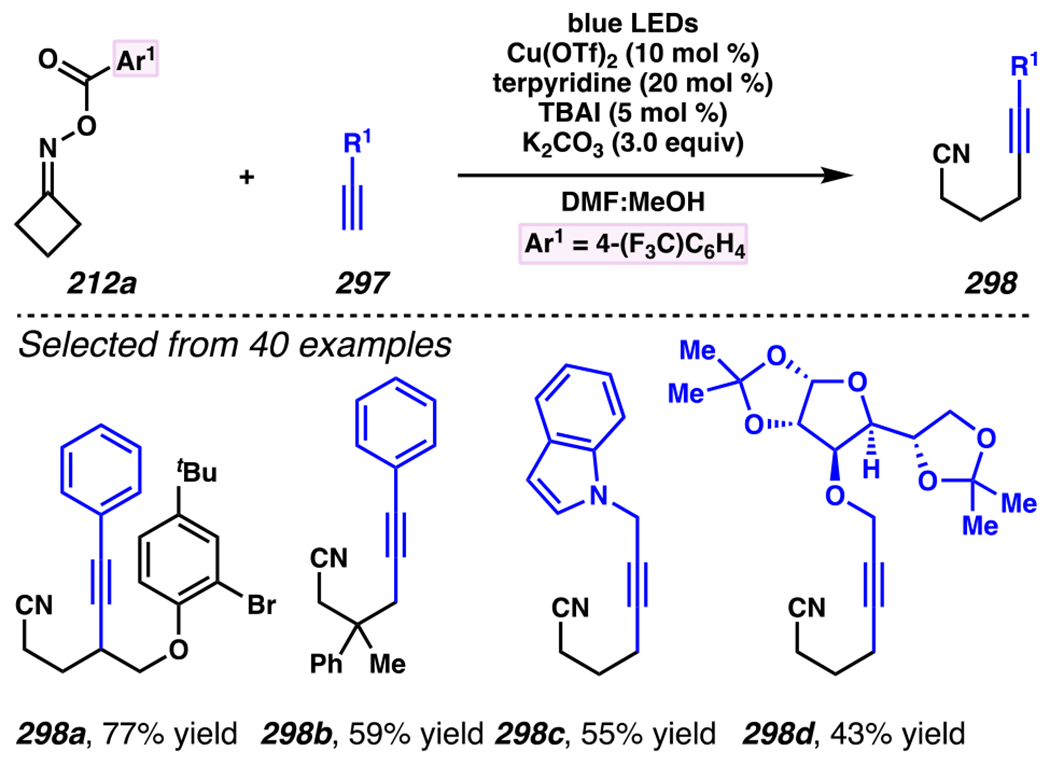
In Principle, Copper Can Serve as a Dual Photoredox- and Transition-Metal Catalyst and Both of These Mechanisms Could Be Operative in the C(sp)–C(sp3) Cross-Coupling Reaction Between Cyclic Oximes and Terminal Alkynes
An alkyl radical, generated from an O-acyl oxime can react with an enantioenriched cyanocuprate complex in a process that ultimately forges a new C(sp)–C(sp3) bonds enantioselectively (Scheme 72).246,247 In the presence of amino indanol-derived chiral spirobisoxaoline ligand248 299, enantioselective cyanation proceeds efficiently between five-membered cyclic oxime 212i and TMSCN, in the presence of blue light and Ir(ppy)3 photocatalyst to deliver (R)-nitrile 300a in 78% yield with 96:4 er. This reaction proceeds inefficiently absent light, or absent photocatalyst. Absent copper pre-catalyst, this reaction affords the racemic product in 14% yield, which confirms that bond-formation is mediated by chiral copper complex. In a concurrently developed reaction, ent-ligand 299 (ent-299) and an organic photocatalyst Ph-PTZ react under purple light to furnish product 300b in (S)-enantiomer quantatively in 95.5:4.5 er (Scheme 72B). Interestingly, this reaction does not proceed without light and the photocatalyst, although trace product is detected when the copper pre-catalyst is omitted.
Scheme 72.
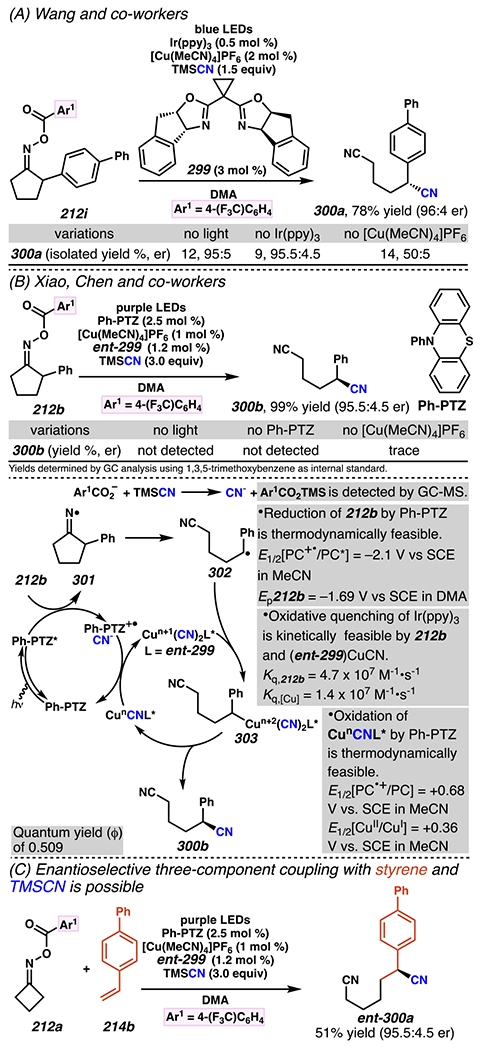
Photo- and Copper-Mediated Enantioselective Cyanation Reactions Rely on Chiral Bisoxazoline Ligands
Mechanistically, both transformations are initiated by generation of nitrogen-centered iminyl radical 301 from photoredox-mediated N–O bond scission involving oxime 212b (Scheme 72B). Indeed, the reduction of oxime 212b (Ep = −1.69 V versus SCE in DMA) by both Ir(ppy)3 (E1/2 [IrIV/*IrIII] = −1.77 V versus SCE in CH3CN) and Ph-PTZ (E1/2 [Ph-PTZ•+/Ph-PTZ*] = −2.1 V versus SCE in CH3CN) are thermodynamically favorable.247 Stern-Volmer experiments indicate that the oxidative quenching of Ir(ppy)3 by oxime 212b is kinetically feasible (Kq,212b = 4.7 × 107 M−1•s−1 in PhMe). c, 249 The experiments further reveals that (ent-299)Cun(CN) complex can oxidatively quench the photocatalyst (Kq,[Cu] = 1.4 × 107 M−1•s−1 in PhMe)c to potentially generate the corresponding reduced [Cu]n-1 species. After the photoredox-mediated activation of oxime, it is proposed that the expelled carboxylate liberates cyanide anion from TMSCN through nucleophilic substitution reaction, a hypothesis that is consistent with the detection of trimethylsilyl carboxylate in reaction mixture by GC-MS analysis.247 The liberated cyanide anion is hypothesized to serve as a counteranion for oxidized photocatalyst. Thermodynamically favorable single-electron oxidation of (ent-299)Cun(CN) (E1/2 [CuI/CuII] = +0.36 V versus SCE in CH3CN)226 by photocatalyst (E1/2 [IrIV/IrIII] = +0.83 V versus SCE in CH3CN, E1/2 [Ph-PTZ•+/Ph-PTZ] = +0.68 V versus SCE in CH3CN)16, 226 provides (ent-299)Cun+1(CN)2.247 This copper complex is poised to trap alkyl radical species 203 generated from the β-scission of 301 to access organocupric intermediate 303. The ultimate reductive elimination step establishes new C(sp3)–C(sp) bond to provide enantioenriched dinitrile 300b and regenerate (ent-299)CunCN complex. It should be underscored that under the optimal reaction developed by Xiao, Chen and co-workers, this reaction possesses low quantum yield (ϕ = 0.509). So, the possibility of this transformation proceeding through radical chain propagation mechanism cannot be ruled out. Overall, these enantioselective two-component coupling reactions between cyclic oximes and TMSCN constute an example from a collection of strategies to affect enantioselective bond-forming reactions through the union of photoredox catalysis with chiral transition-metal catalysis.
5.4.12. Absent an Exogenous Radical-Trapping Agent, Ring-Opening Cascades That Rely on O-Acyl Cyclic Oxime Substrates Engage in Migration/Elimination Sequences, or Trap In Situ-generated Carboxylate Anions
O-Acyl cyclic oxime substrates can engage in a photoredox-mediated reaction cascade in the absence of radical or nucleophilic trapping agents. For example, 3,3-diaryl-substituted O-acyl cyclic oxime 212 is converted to α,β-unsaturated nitrile under the aegis of Ir(ppy)3 photocatalyst and DABCO as exogenous base (Scheme 73).250 Photoredox-mediated N–O bond scission / β-scission cascade of oxime 212 ultimately reveals 1° alkyl radical intermediate 305, which undergoes 1,2-aryl migration to form a secondary benzylic radical. Oxidation of benzylic radical 306 to carbocation 307 and elimination of acidic benzylic proton by DABCO furnishes (E)-alkene 304 as the major product. For oximes with different aryl substituents, the migratory aptitude of aryl group appears to be dictated by stereoelectronic considerations. For example, a more electron-rich thiophene moiety migrates more readily than a phenyl substituent to deliver thiophenyl 304a. Unfortunately, non-selective aryl migration occurs when two differently substituted arenes possess similar electronic and steric profiles. In the case of phenyl and o-xylenyl bearing oxime, both arenes rearrange competitively to provide a mixture of products. Ultimately, this reaction affords α,β-unsaturated nitriles and β-functionalized nitriles under mild reaction conditions.
Scheme 73.
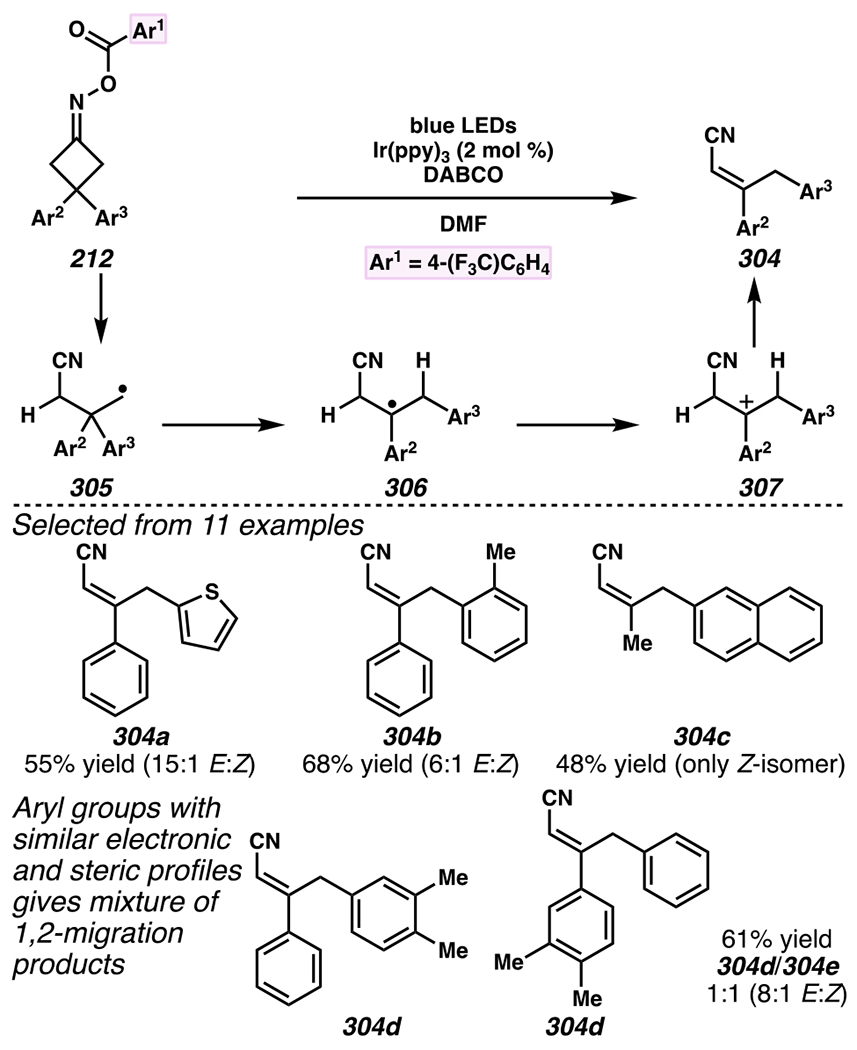
Diaryl Substituted Oximes Engage in β-Scission / 1,2-Aryl Migration / Elimination Cascade
Absent diaryl-substituents, O-acyl oxime 212 engages in β-scission / aryl acetate coupling cascade through intermediacy of benzyl radical 309 and benzyl cation 310 to furnish benzoate 308,251 which opens possibilities for downstream functionalization reactions with nitrile- or benzoate-groups (Scheme 74). Remarkably, compact to large cyclic oximes (n = 1–5) engage in this transformation productively. Strained 4- and 5-membered cyclic oximes proceed through the reaction cascade to provide corresponding desired products 308a and 308b in 54% and 81% yields, respectively. Despite the diminished isolated yield, 6-membered cyclic oxime reacts to provide product 308c in 48% yield. Slightly more strained 7- and 8-membered cyclic oximes are amenable to the reaction, and provide products 308d and 308e in 63% and 69% yield, respectively. This reaction cascade can be applied to acyclic oxime, in which ketoxime 311 was converted to ester 312 in 51% yield.
Scheme 74.
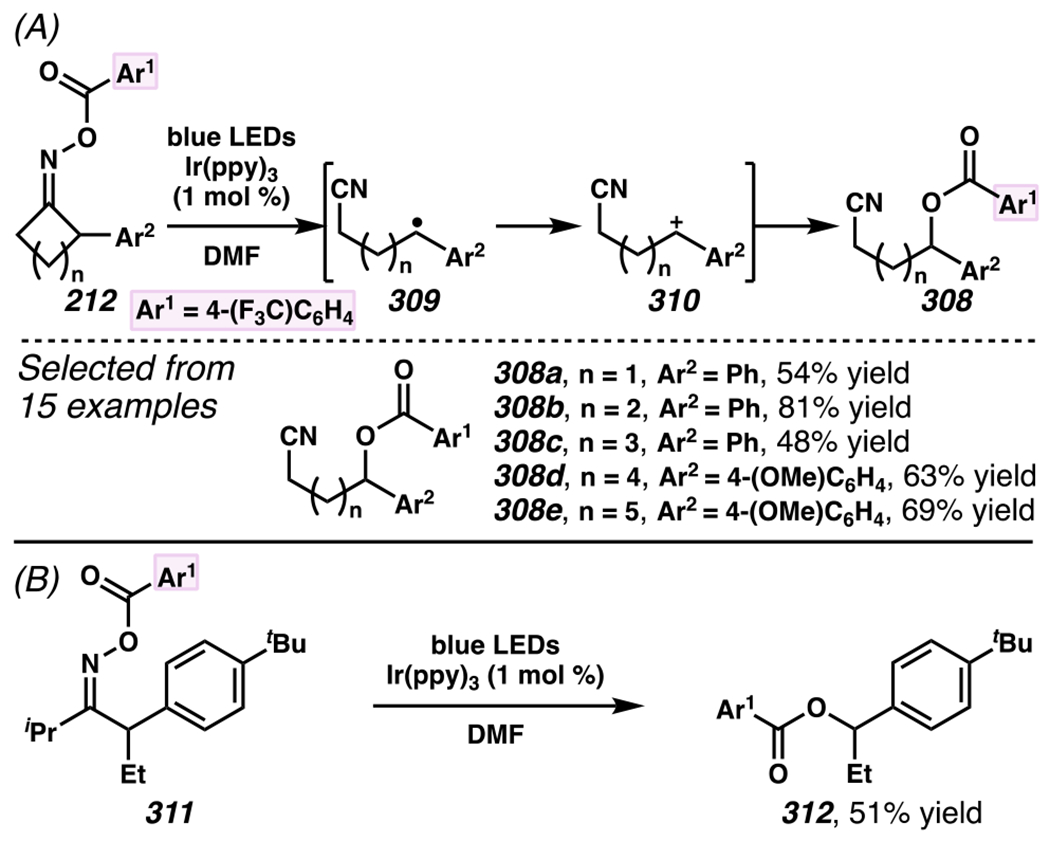
Absent Radical Trapping Agents, Aryl Acetate Quenches Carbocationic Intermediates
Broadly, reactions with O-acyl cyclic oximes as N-centered radical precursors rely on oxidative quenching of photocatalyst to affect β-scission / radical addition cascades. These sequences terminate based on oxidation to form a carbocation intermediate, which is quenched by a nucleophile to foment new C–C or C–heteroatom bonds, or with exogenous base to affect elimination to establish new C=C bonds. Alternatively, by switching the redox-active handles to those that rely on reductive quenching, new reaction modes become available. Specifically, the β-scission / radical addition cascade can be followed directly or indirectly by reduction to install a carbanionic intermediate, and involving electrophilic radical or anion trapping agents, or Brønsted acids. In principle, this approach should enable atom- and group-transfer reactions with electrophilic atom- and group-transfer agents and Giese-type reactions with electron-deficient olefins.
5.5. Iminyl Radicals Guide C–H Functionalization Reactions
Notably, iminyl radical intermediates can induce C–H functionalization reactions, which are expected to proceed through 1,5-hydrogen atom transfer (HAT) processes, prior to trapping a halogen atom to generate a 3° alkyl fluoride or chloride. This is remarkable as intramolecular 1,5-HAT process are not likely to be induced by an iminyl radical, and gas-phase DFT calculations performed at the UB3LYP/6–31+G(d,p) level of theory suggest that this abstraction event is an endergonic process (Scheme 75A).180 Theoretical calculations estimate that the hydrogen-atom abstraction by iminyl radical 313 to generate carbon-centered radical 314 is endergonic by ∆G = 4.2 kcal•mol−1. Fortunately, Forrester and co-workers139 envisioned that this process could be energetically favorable under acidic conditions. In acid, the iminium radical cation generated would be more electrophilic than neutral iminyl radical and would favor hydrogen-atom abstraction.252 Indeed, gas-phase DFT calculations at the same level of theory reveal that the HAT event that converts 315 to 316 is exergonic by ∆G = −24.9 kcal•mol−1. These calculation results suggest that acidic reaction condition could deliver a highly effective reaction system. Unfortunately, traditional protic conditions are incompatible with the mildly basic photoredox-mediated decarboxylative conditions that furnish access to iminyl radical intermediates.253
Scheme 75.
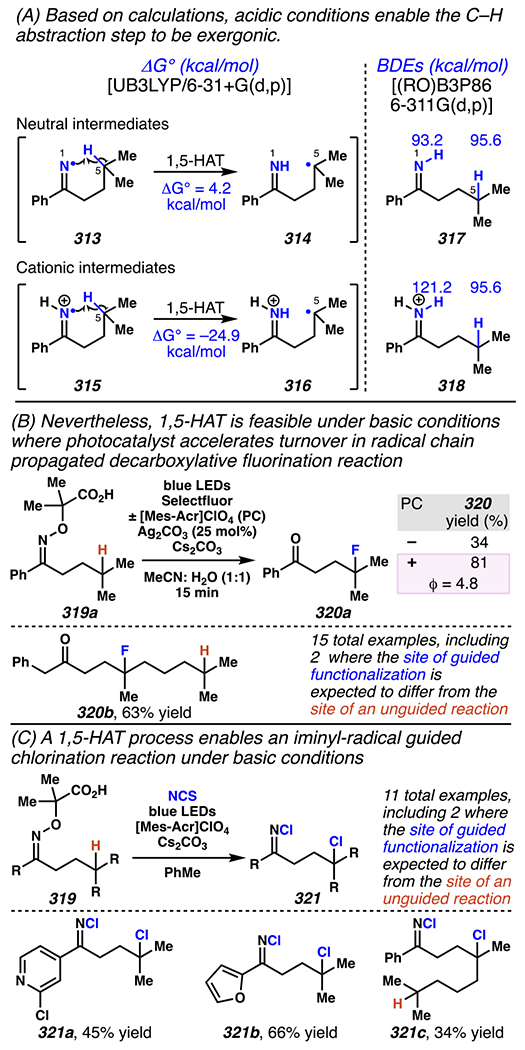
Oximes Are Substrates for C(sp3)–H Fluorination and Chlorination Reactions
To overcome this tactical limitation, Leonori and co-workers identified photo-aided conditions to promote decarboxylative fluorination and chlorination reactions (Scheme 75B–C).180 These photo-mediated fluorination conditions benefit from the addition of a silver salt, which is presumed to facilitate C–F bond formation.254 The fluorination process proceeds less rapidly in the absence of photocatalyst. Under the optimal conditions, the reaction relies on a radical chain propagation mechanism, as indicated through quantum yield (ϕ = 4.8) experiments, which suggest that at least 4.8 equivalents of product form per photon of light absorbed. Nevertheless, at least one product suggests that these reaction conditions overcome the site-selectivity that would be achieved through an unguided reaction. The reaction tolerates pendant esters, phenyl substituents, bromides, azides, nitriles, ethers, and thioethers, and has not been described with heteroaromatic substituents. Surprisingly, the developed chlorination conditions tolerate pendant heteroaromatic substituents, including a chloropyridyl unit and a furan, which is prone to undesired electrophilic aromatic chlorination reactions, and would be expected to react directly with the employed chlorinating agent, N-chlorosuccinimide (NCS), under highly related conditions (Scheme 75C). These guided chlorination reaction conditions, like the γ-fluorination process conditions, have been applied to two substrates that could be used to highlight guided site-selectivity. Subjecting a 4,8-dimethyl-1-phenylnonan-1-one-derived oxime to the reaction condition affords chlorinated product 321c in 34% yield.
Concurrently, Jiang and Studer148 developed an oxime-guided Giese reaction at tertiary and benzylic C(sp3)–H centers (Scheme 76).255 Following hydrolytic aqueous work-up, a C(4)-functionalized ketone is revealed. This process transforms wide scope of oximes to their corresponding functionalized ketones, and suitable radical trapping agents include α,β-unsaturated esters and ketones. The reaction tolerates aryl and heteroaryl-substituted oximes, as demonstrated with thiophenyl-substituted oxime. Furthermore, if the reaction is worked-up with benzoyl chloride and triethylamine, the product incorporates an activated imine handle that is poised for further functionalization.256
Scheme 76.
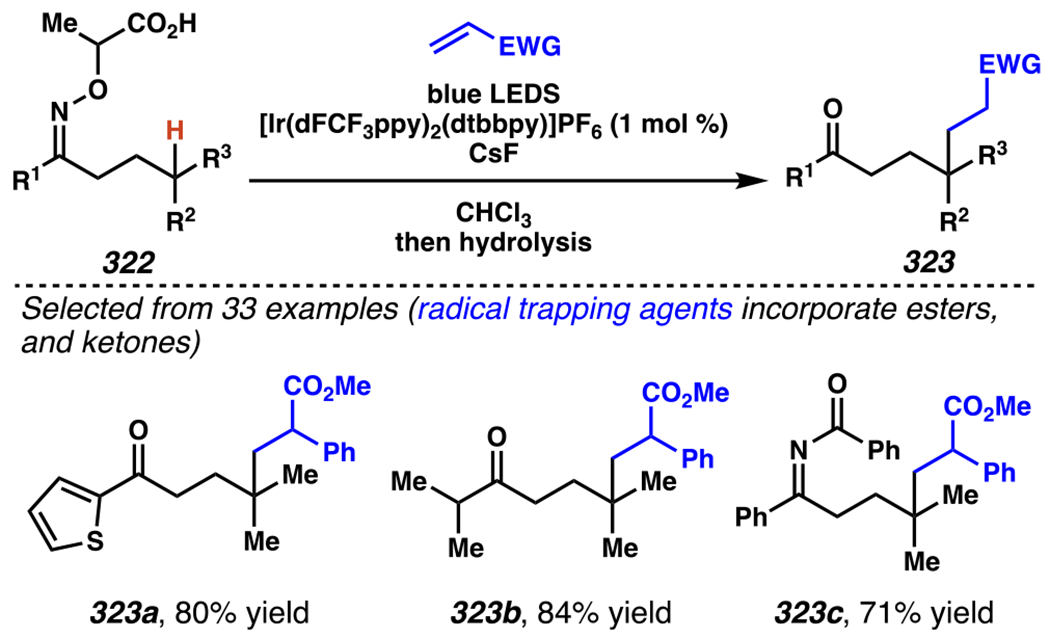
Oximes Are Substrates for Guided Giese Reactions
Aliphatic O-acyl oximes 324 can induce guided net C–H vinylation reactions with styrenyl boronic acids 217a as radical trapping agents under photoredox-mediated conditions to afford aliphatic ketones 325 (Scheme 77).205 The most efficient vinylation reactions rely on tertiary radicals as intermediates. So, when a substrate can undergo competitive reactions at secondary or tertiary centers, vinylation proceeds at the tertiary center. Nevertheless, vinylation of a secondary carbon is possible when the engaged center is stabilized by an adjacent heteroatom (cf. 325b), and even at primary benzylic centers (cf. 325c).
Scheme 77.
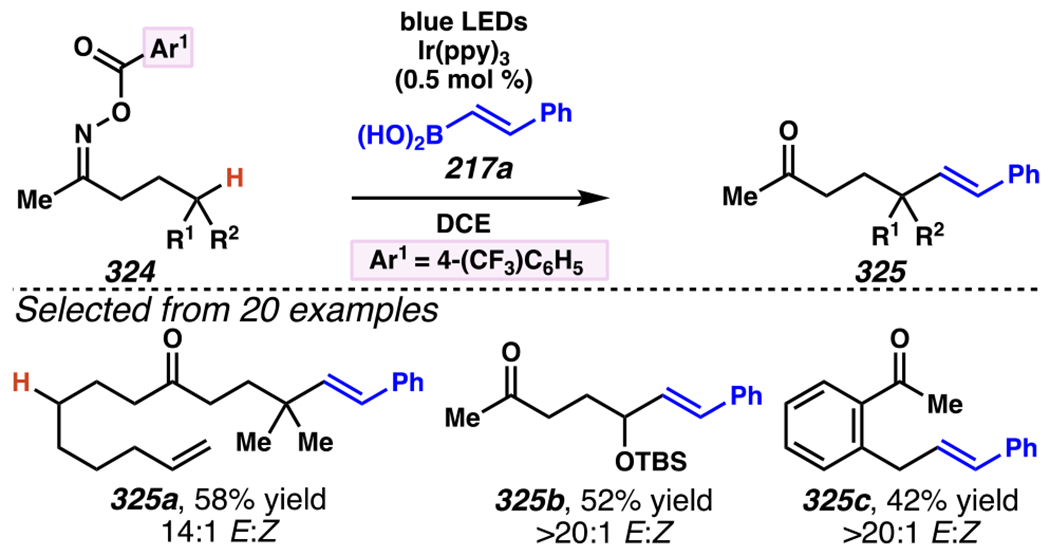
Oximes Can Template Guided Vinylation Reactions with Styrenyl Boronic Acids
A directed vinylation reaction proceeds between O-acyl oxime 326 and olefin 327 to afford vinylated aliphatic ketone 328 by way of proposed intermediate cation 330, which is more readily accessible in the presence of an acidic additive (Scheme 78A).257 Not surprisingly, this transformation exhibits more facile vinylation reactions at tertiary centers relative to secondary centers, where reactions proceed more readily than at primary centers, presumably based on the relative stability of the requisite intermediate radicals. Interestingly, with α-methyl styrene as a radical trapping agent, terminal alkene product 328d predominates relative to internal alkene product (not depicted) in 92% yield and in >99:1 t:i. With coumarin as a radical trapping agent, C–C bond formation proceeds at the α-carbon rather than the β-carbon. Indeed, electron-deficient olefins prove challenging as radical trapping agents, including benzyl acrylate, benzyl crotonate, acrylonitrile, and electron-deficient styrenes. Presumably these electron-withdrawing groups should less readily form requisite carbocation intermediates. Notably, the critical carbocationic intermediate can be trapped by water or a nucleophilic alcohol to afford alcohol or ether products (Scheme 78B).258 In these reactions, when 1,3-diene is used as a radical trapping agent, reactions proceed preferentially at C(2) to provide alcohol 329a, based on the formation of stable benzylic carbocation en route to product. Ultimately, this approach can furnish either vinylation or oxygenation products.
Scheme 78.
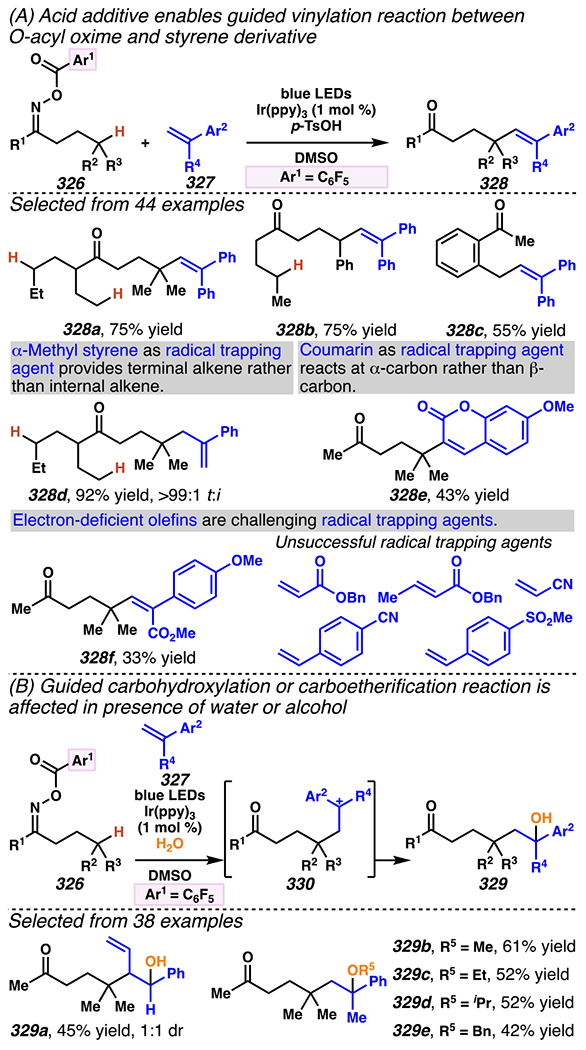
Oximes Guide Vinylation Reactions and Hydroxyalkylation Processes
Iminyl radicals are powerful reaction intermediates that can be harnessed to affect position-selective C(sp3)–H functionalization reactions through the intramolecular 1,5-HAT / alkyl radical trap cascade. Logical future directions for reaction development involve enantio- and position-selective C(sp3)–H functionalization reactions. These formidable processes are challenging because enantiodeterming steps involving alkyl radicals must be faster than unproductive reaction pathways that lead to enantioerosion.259
Fortunately, Nagib and co-workers have reported a guided enantioselective intramolecular C(sp3)–H amination reaction that transforms closely related imidates to enantioenriched oxazolines (Scheme 79). These transformations are proposed to involve initial coordination of the iminyl nitrogen to a chiral Cu-complex. This coordination facilitates N–O bond reduction by a photoexcited iridium complex. The resultant imidate radical is poised for 1,5-HAT to direct formation of an alkyl radical. In the critical bond-forming step, the chiral ligand is able to induce enantioselective C–N bond formation.260 This imidate radical-guided enantioselective C(sp3)–H functionalization could set the stage for the invention of iminyl radical-guided enantioselective C(sp3)–H functionalization reactions that rely on exogenous radical trapping agents.
Scheme 79.
Guided C(sp3)–H Amination Reactions Can Be Enantioselective
5.6. Nitrogen-Centered Radicals Can Be Generated from Oxyimides, Oxyamides, and Benzenesulfonamides
5.6.1. Radicals Prepared from Oxyimides, Oxyamides, and Benzenesulfonamides Can Engage in N-(Hetero)arylation Reactions
Oxyimides are useful synthetic precursors to imidyl radicals by N–O bond reduction, which can be mediated by visible light photocatalysts. In a pioneering research by Sanford and co-workers, N-acyloxyphthalimide 334 bearing an electron-withdrawing trifluoromethyl group engages in N-(hetero)arylation reaction with (hetero)arenes in presence of Ir(ppy)3 photoredox catalyst (Scheme 80).85 The electron-withdrawing trifluoroacyl group is critical to this reaction. In an earlier investigation led by Okada and Oda, N-acyloxyphthalimide with a less electron-deficient acyl group preferentially fragments to imidyl anion and alkyl radical upon photoredox-mediated decarboxylation reaction (Scheme 80A).95 By contrast, with a more electron-deficient trifluoroacyl substituent, excited state [IrIII] catalyst is oxidized to [IrIV] complex, with concommitant reduction of imidate 334 to afford trifluoroacetate 336 and critical imidyl radical 17. The accessed electrophilic imidyl radical 17 is poised to add to arene 15 to afford radical 18, which can be oxidized by the [IrIV] complex to provide Wheland intermediate 19 and regenerate ground-state Ir-photocatalyst. Ultimately, re-aromatization of intermediate 19 by trifluoroacetate 336 yields product 16 and trifluoroacetic acid. Generally, addition of imidyl radical to arene follows position-selectivity and reactivity patterns that have been associated with electrophilic aromatic substutition reactions. For example, p-anisole reacts to provide predominantly ortho-substituted product 335a, along with less significant amounts of meta-substituted and para-substituted products. Similarly, α,α,α-trifluoromethylbenzene reacts to furnish meta-substituted product 335b as the major product. More electron-rich arenes are viable substrates for the reaction, as are 2,6-dichloropyridine and 2,6-dibromopyridine. As the process tolerates halogenated arenes, reaction products can be poised for subsequent manipulations in cross-coupling reactions.
Scheme 80.
Electron-Deficient N-Acyloxyphthalimides Are Imidyl Radical Precursor for Use in N-(Hetero)arylation Reactions
While initially these N-arylation technologies relied on imidyl radical intermediates, this approach has become more versatile. A similar reaction mechanism can be invoked to rationalize photoredox-mediated reactions between hydroxylamine analogue 337 and heteroarenes, such as indole derivative 338 to yield C(2)-functionalized product 339 (Scheme 81A).261 Indeed, this reaction also follows general reactivity patterns previously observed in electrophilic aromatic substitution reactions. Indoles with electron-donating groups are more reactive (339a, 94% yield) than those with electron-withdrawing groups. The power of this process is showcased using a melatonin derivative that can be transformed into product 339f in 97% yield. With more recent advances in nitrogen-source designs, varied electron-deficient O-aryloxy amides 340 can be employed as aminating reagents (Scheme 81B).262
Scheme 81.
Benzenesulfonyl Radical and Amidyl Radical Intermediates React with Arenes
5.6.2. Radicals Prepared from Oxyimides or Oxyamides Can Add Across Double Bonds263
This approach has been extended to enable intramolecular carboamination reactions to access pyrroloindolines (Scheme 82).264,265 In fact, this strategy has been exploited for rapid synthesis of (±)-flustramide B (Scheme 82A) and (±)-flustraminol B (Scheme 82B). To access (±)-flustramide B,264 N-prenylated O-aryloxy amide 346 can be prepared in two steps from brominated indole-3-acetic acid 342 and prenyl bromide 343. Indeed, O-aryloxy amide 346 engages in photoredox-mediated alkylation with α-sulfonyl acrylate 347 to provide alkylated pyrroloindoline 348 in 76% yield. Subsequent vinyl sulfone reduction and cross-metathesis deliver (±)-flustramide B. This strategy has been applied to prepare (±)-flustraminol B,265 albeit with a critical difference. The key transformation converts O-aryloxy amide 346 to tertiary alcohol 352 based on trapping of an alkyl radical intermediate by O2 in air in a net hydroxyamination reaction. In this sequence, amide reduction leads to (±)-flustraminol B. As an alternative, it is possible to intercept the key radical cascade sequence to affect net intramolecular hydroamination, as a powerful approach to access other pyrroloindoline cores (Scheme 82C).265
Scheme 82.
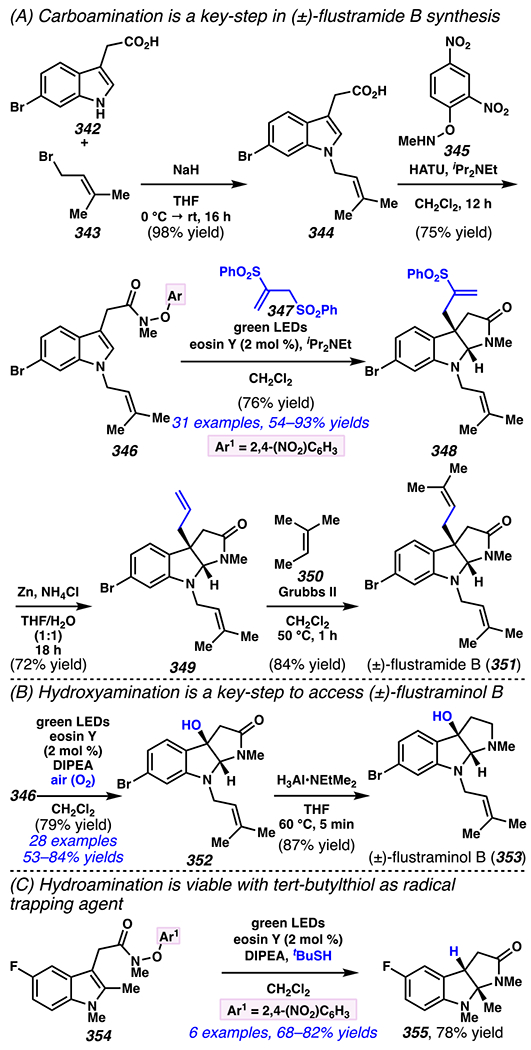
Radical Annulation Sequences Converts Indoles to Higher-Value Products En Route to Natural Products
Amidyl radicals generated upon N–O bond cleavage can add across electron-dense olefins in the course of diamidation and aminooxygenation reactions (Scheme 83). 266 One mechanistic proposal to explain the net diamination of styrene begins with conversion of O-aryloxy amide 356a to amidyl radical, possibly based on oxidative quenching of excited Ir-photocatalyst. The resultant electrophilic amidyl radical 357a is poised to add to styrene 359 to furnish benzylic radical 360. Benzylic radicals (Ep ≈ +0.73 V versus SCE in CH3CN)267 undergo thermodynamically favorable oxidation by [IrIV] complexes (E1/2 [IrIV/IrIII] = +0.77 V versus SCE in MeCN)16 to provide carbocation 361 and ground-state Ir(ppy)3. At this stage, carbocation 361 could engage in a Ritter-type reaction with acetonitrile to generate nitrilium ion 362 and it is attacked by carboxylate 358 to yield intermediate 363. As an alternative, DMSO is another solvent that can serve as carbocation trapping agent, and can be used to affect Kornblum oxidation reaction to afford α-amino ketone 365 from O-aryloxy amide 356b and styrene 359 (Scheme 83B).266 The roles of solvents in these reactions are supported by experiments using isotopically labeled solvents. In both reactions, a variety of styrenes are viable olefin radical trapping agents. In contrast to the diamidation reaction, this Kornblum oxidation reaction can engage heteroarenes such as thiophene, so this value-adding process is more versatile than the mechaistically related diamination reaction.
Scheme 83.
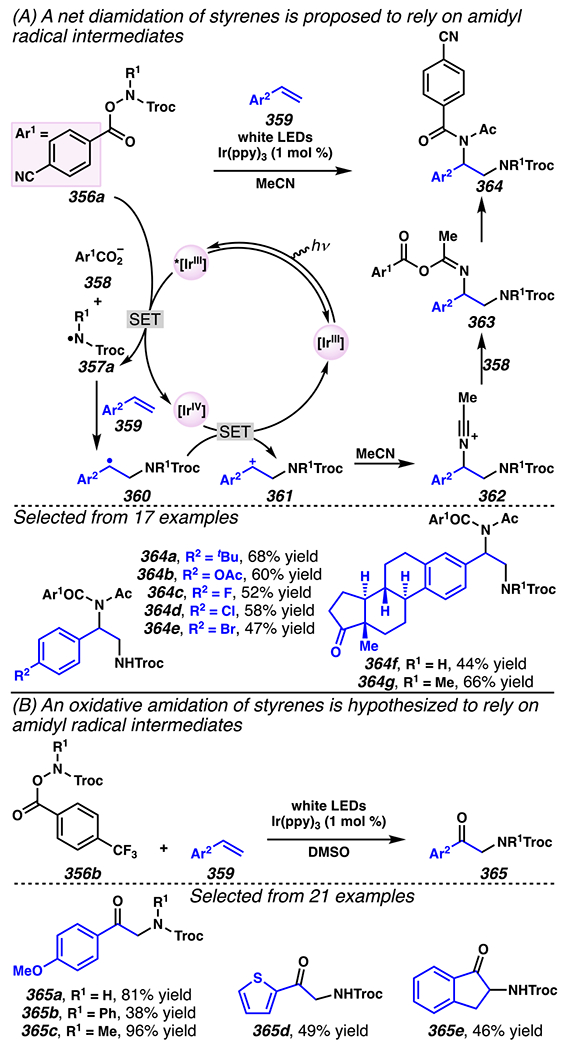
O-Aryloxy Amide and Alkenes React with Acetonitrile and DMSO to Synthesize 1,2-Diamides and α-Amino Ketones
Several complementary mechanisms underpin strategies to induce radical-mediated addition across an olefin. For example, an imidyl radical can be generated from N-phthalimide derivative by way of an energy transfer event between appropriate photosensitizer. Such an energy transfer process is expected to be the basis for a reaction between O-vinylhydroxylamine 366 and alkene 368 in presence of Ir-photosensitizer and H2O to provide ketone 367 (Scheme 84).170 This intermolecular carboamination reaction is promoted by fragmentation of substrate 366 to oxygen-centered radical species 369 and imidyl radical 370 upon energy transfer. At this point, imidyl radical 370 engages in radical addition to alkene 368 at the sterically uncongested carbon-atom to deliver carbon-centered radical intermediate 371. Radical 371 engages in a second radical addition with starting material 366. Finally, β-scission of intermediate 372 affords product 367 and regenerating phthalimidyl radical 370 . As corroboration for this proposed radical chain propagation mechanism, the quantum yield of this reaction is greater than 1 (ϕ = 12.9). Through this methodology an array of ketones is constructed. For example, 1,2-diamine decorated ketone 367a is synthesized in 47%. In contrast to reactions mediated by transition-metal catalysts, substrates containing Lewis basic functional groups are compatible in this photosensitized multi-component carboamination reaction. This benefit is evident when an alkene with pendant free-hydroxyl group is subjected to the reaction to provide amino alcohol derivative 367b in 55% yield.
Scheme 84.
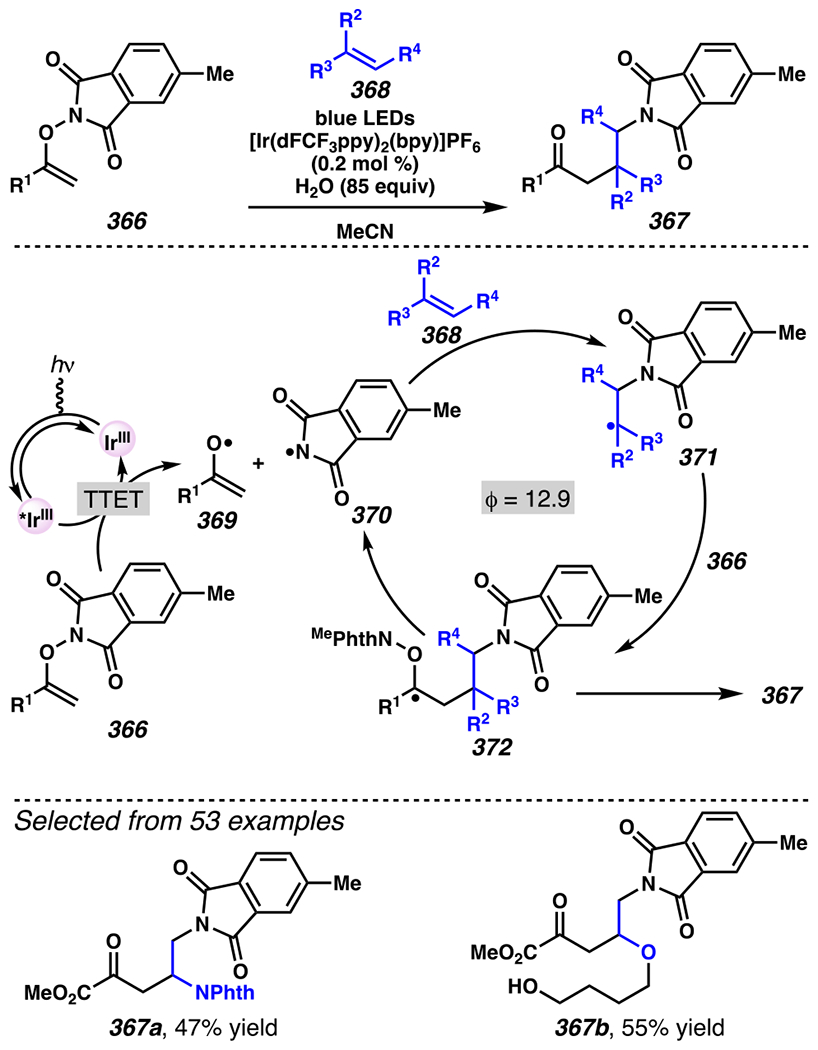
O-Vinylhydroxylamine Derivative Undergoes Group-Transfer Radical Addition with Alkenes in Presence of Photosensitizer
In an effort to identify easily accessible precursors to amidyl radicals, Studer and co-workers identified α-amido oxyacids 373a as amidyl radical precursors (Scheme 85).268 Using these amidyl radical precursors, a cascade sequence involving addition to an olefin can be intercepted by an atom-transfer reagent. To affect fluorination, α-amido oxyacid 373a is proposed to give way to amidyl radical, which can add across olefin 368. The resultant benzylic radical reacts with Selectfluor as a fluorine-atom donor to afford β-fluoroamide 374. This reaction tolerates free hydroxyl groups, as well as bromides (374b), nitro groups (374c), and protected amines (374d). Interestingly, under similar reaction conditions, tetrasubstituted olefin 380a engages in an ene-type fluorination reaction with Selectfluor to provide allylic fluoride 381, which then participates in the amidofluorination reaction with amidyl radical and a second equivalent of Selectfluor to provide difluorinated amide 382 in 85% yield.
Scheme 85.
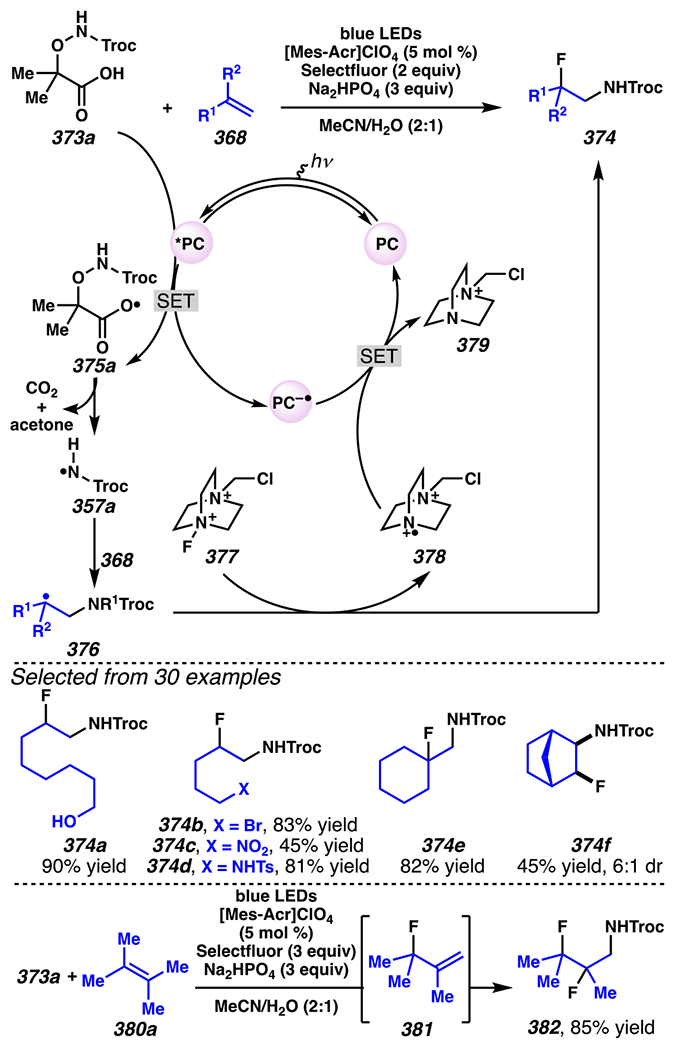
α-Amido-oxy Acids React with Olefins and Selectfluor to Provide β-Fluoroamides
An analogous reaction cascade can be extended to trigger anti-Markovnikov hydro- and deuteroamidation reactions that engage olefins 380 and α-amido oxyacids 373a under conditions that may enable dual photoredox and thiol catalysts in the presence of water or heavy water (Scheme 86).269 In the proposed reaction mechanism, following activation of α-amido oxyacid 373b towards amidyl radical 357b via reductive quenching of a photocatalyst, amidyl radical 357b would react with olefin 380 to form a new C–N bond, and provide carbon-centered radical 364. Concurrently, carbonate or carbonic acid undergoes proton- or deuterium-exchange with H2O or D2O to provide carbonic acid that transfers a proton or deuteron to thiolate 387 to provide thiol or deuterated thiol 385. Hydrogen- or deuterium-atom abstraction delivers hydro- or deuteroamidated product 383. Remarkably, this reaction is so mild that it tolerates pendant trimethylsilyl ethers, as well as free primary alcohols, and can be used to access a range of selectively deuterated products.
Scheme 86.
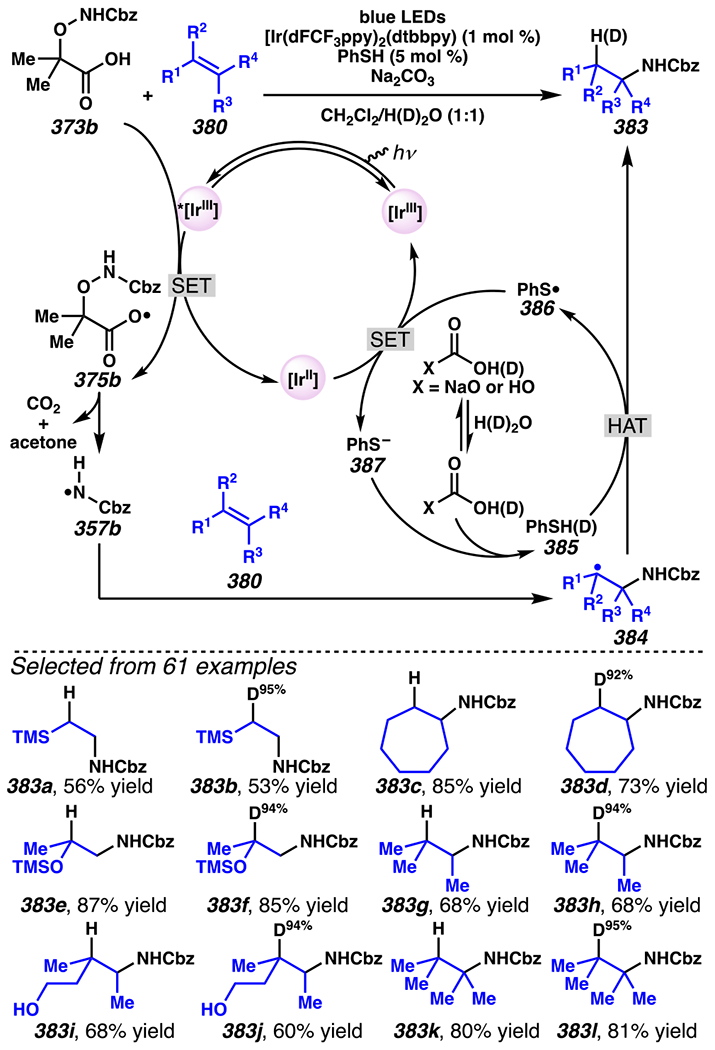
α-Amino-Oxy Acids React with Olefins in Photo-Mediated, Thiol Catalyzed Hydro- and Deuteroamidation Reactions
It is possible to engage amidyl radical precursors in enantioselective reactions with 2-acyl imidazoles 389, which form chiral-at-rhodium complex (Δ-RhO) to provide enantioenriched α-aminated product 390 (Scheme 87).20 This enantioselective transformation relies on Δ-RhO complex to behave as both reaction photosensitizer and enantioselective catalyst, analogous to Δ-Ir complexes employed by Meggers and co-workers to affect enantioselective alkylation between 2-acyl imidazole and alkyl bromides.270 In this enantioselective C–N bond-forming reaction, Δ-RhO complex serves as both photosensitizer to generate amidyl radical 391 from precursor 388 and as an enantio-inducing Rh-catalyst to promote the formation of chiral Rh-enolate 392. Subsequently, amidyl radical 391 undergoes radical-addition to chiral Rh-enolate 392 at si-face to provide Rh-ketyl 393. Ligand-exchange involving starting material 389 ultimately releases product 390 and re-forms Rh-activated 2-acyl imidazole complex 395. Interestingly, this reaction proceeds via radical chain propagation (ϕ = 13.6). It is likely that Rh-enolate 392 behaves as initiatior and reinitiator, or “smart initiator”.271 To re-initiate the reaction via radical chain propagation mechanism, electron-rich Rh-ketyl complex 393 is poised for single-electron reduction with carbamate 388 to provide amidyl radical 391 and Rh-complex 393. Ultimately, this is a powerful approach to enantioselective photosensitized reactions.
Scheme 87.
O-Aryloxy Amide and 2-Acyl Imidazole React in the Presence of Δ-RhO Catalyst to Furnish Enantioenriched Products
5.6.3. Radicals Prepared from Oxyimides or Oxyamides Can Affect C–H Functionalization Reactions
Amidyl radicals can engage in intermolecular C–H functionalization reactions. With the identification of α-amido oxy acids 396 as amidyl radical precursors, an opportunity has emerged to take advantage of the propensity of [1.1.1]propellanes 397 to undergo strain-release reactions with nitrogen-centered radicals, and thereby reveal bicyclo[1.1.1]pentylamines, historically challenging to access motifs that are less readily oxidizable bioisosteres of anilines (Scheme 88).272 This reaction involves mechanistically familiar kinetically feasible reductive quenching of excited state photocatalyst by α-amido oxy acetate 399 (Kq = 3.6 × 106 M−1•s−1) to furnish amidyl radical 401. Once formed, amidyl radical 401 is trapped by [1.1.1]propellane 397 via strain-releasing transition-state geometry 402 to furnish carbon-centered radical 403. DFT calculations estimates this initial two-component coupling step to possess energy barrier of ΔG‡ = 15.3 kcal•mol−1 and is exergonic by −12.4 kcal•mol−1. The formed carbon-centered radical can engage in chlorine-atom transfer to provide chlorinated bicyclo[1.1.1]pentylamine 398a. Alternatively, carbon-centered radical 403 can unproductively react with another molecule of [1.1.1]propellane 397 to provide radical species 408 and undergo oligomerization. Kinetically, the chlorine-atom transfer reaction is predicted to have a lower energy barrier than oligomerization by 0.8 kcal•mol−1, thereby favoring formation of the desired product. Given the relevance of bicyclo[1.1.1]pentane derivatives in medicinal chemistry, chlorinated bicyclo[1.1.1]pentylamine with heteroaryl groups can be incorporated. Indeed, chlorinated bicyclo[1.1.1]pentylamine decorated with 2-chloropyridyl group 398b is synthesized in 64% yield, as well as tolerating furanyl group to access product 398c in 59% yield. As exemplified by product 398d, this reaction can incorporate electron-deficient imidyl group in assembly of chlorinated bicyclo[1.1.1]pentylamine derivative. Unfortunately, primary chlorinated bicyclo[1.1.1]pentylamine 398e cannot be assembled through this reaction protocol. Instead, the reaction with NH-amidyl radical provides complex mixtures of compounds.
Scheme 88.
[1.1.1]Propellane Is Converted to Chlorinated Bicyclo[1.1.1]pentylamine Based on Reactions with Amidyl Radical Precursors
Notably, other atom- and group-transfer reactions can be triggered with appropriate reagents (Scheme 89).272 Brominated bicyclo[1.1.1]pentylamine 409a is accessed with bromotrichloromethane as bromine-atom transfer agent, albeit in 20% yield due to oligomerization. Chalogenation reactions employs phthalimide derivatives as sulfide- and selenyl-transfer agents, providing sulfides 409b and 409c in 66–77% yields and selenylated bicyclo[1.1.1]pentylamine 409d in 72% yield. 2-phenylmalononitrile serves as hydrogen-atom transfer agent to deliver bicyclo[1.1.1]pentylamine 409e in 49% yield. Ultimately, this approach provides access to a broad variety of bicyclo[1.1.1]pentylamines, which are promising aniline isosteres.
Scheme 89.
[1.1.1]Propellane Is Converted to Functionalized Bicyclo[1.1.1]pentylamines Based on Reactions with Amidyl Radical Precursors and Atom- and Group-Transfer Agents
Amidyl radicals can guide C-H functionalization reactions through intramolecular 1,5-HAT events. Accordingly, photoredox-mediated reaction conditions have been developed to transform oxime esters 401 to functionalized amides 411 with atom- or group-transfer agents through the intermediacy of amidyl radical poised for 1,5-HAT reaction (Scheme 90).273 To induce productive 1,5-HAT / guided atom- or group-transfer reaction cascade a variety of nitrogen-centered radical species (413–412d) have been examined through theoretical investigations of local electrophilicity of nitrogen-centered radical (ω+k), N–H and C–H BDEs, as well as ΔG‡ and ΔG of 1,5-HAT processes. Not surprisingly, iminyl radical 413 ranks lowest in electrophilicity (ω+k = 0.77 eV) and 1,5-HAT event is endergonic by 4.2 kcal•mol−1 with energy barrier of 13.7 kcal•mol−1. Carbamoyl radicals 412a, 412b and amidyl radical 412c are more electrophilic than iminyl radical 413 and are estimated to proceed through exergonic 1,5-HAT events with lower energy barriers. Amidyl radical 412d is ranked as the most electrophilic radical intermediate (ω+k = 1.38 eV) and is expected to undergo facile 1,5-HAT with estimated ΔG‡ = 5.7 kcal•mol−1 and ΔG = −16.6 kcal•mol−1. Note that for these nitrogen-centered radicals, the difference in BDEs (ΔBDE = BDEN–H – BDEC–H) correlates to ΔG values of 1,5-HAT events. Relying on previously employed atom- and group-transfer agents and a permutation of photocatalyst, reaction base and solvent, fluorine-,181 chlorine-,152 thiol-,155 cyano-161 and alkynyl-transfer160 reactions are accomplished to provide functionalized amides. For all functionalization reactions under each optimized reaction condition, low quantum yields were observed (ϕ = 0.02 ≤ 0.08). While these low quantum yields suggest closed radical cycle mechanism, they do not rule out the possibility of radical chain propagation mechanism.
Scheme 90.
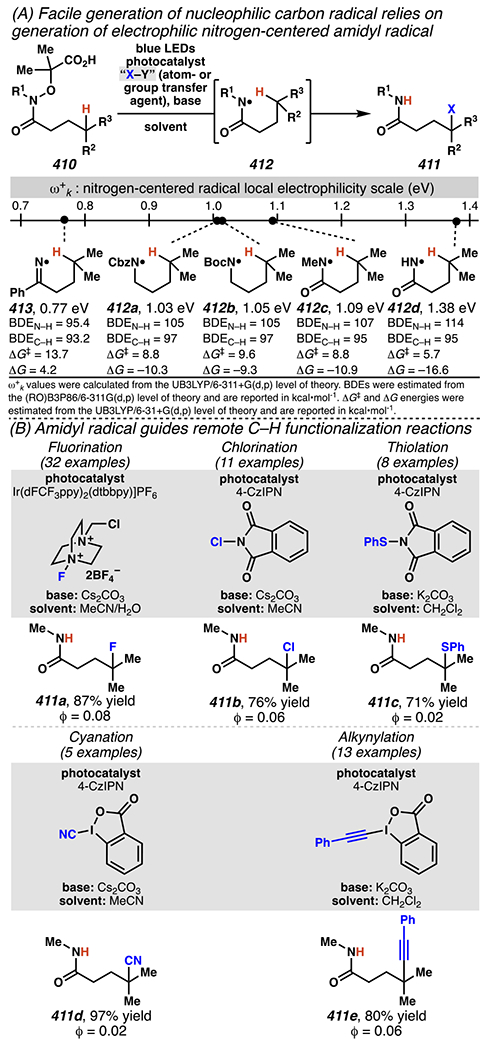
Amidyl Radical Guides Remote Functionalization Reaction to Engage Atom- and Group-Transfer Agents
5.7. Nitrogen-Centered Radicals Can Be Generated from Amidoxime Esters
In 2003, Zard and co-workers found that amidinyl radicals could be employed as intermediates to construct imidazolines and imidazoles, albeit with use of a stannane-diazo initiator at reflux.274 Since this disclosure, various reports have described annulation reactions involving amidinyl radical generation, which include iron-275 and nickel-catalyzed276 radical annulations, iodine(III)-mediated C(sp2)–H imidation of N-arylamidines277,278 and electrolytic N–H bond scission of dihydroquinazoline-derived imines under reflux conditions.279 Drawing inspirations from pivotal discoveries by Zard co-workers, a collaborative effort from the Wang, Liu and Wang research groups developed a mild photoredox-mediated protocol that converts N-aryloxime esters to 2-substituted benzimidazoles (Scheme 91). 280 The reaction suffers predictable substrate scope limitations. For example, tert-Butyl substituted N-aryloxime ester do not engage in the reaction to furnish the product 415c, presumably due to kinetically unfavorable steric repulsion in the transition-state geometry of intramolecular annulation event. Further, when the aniline portion of the substrate contains meta-substituted OMe (414a) and Cl groups (414b), regioisomeric mixtures of corresponding products 415d, 415f and 415e, 415g are obtained, respectively. Nevertheless, this reaction protocol offers mild and benign reaction condition to productively transform N-aryloxime esters to benzimidazoles.
Scheme 91.
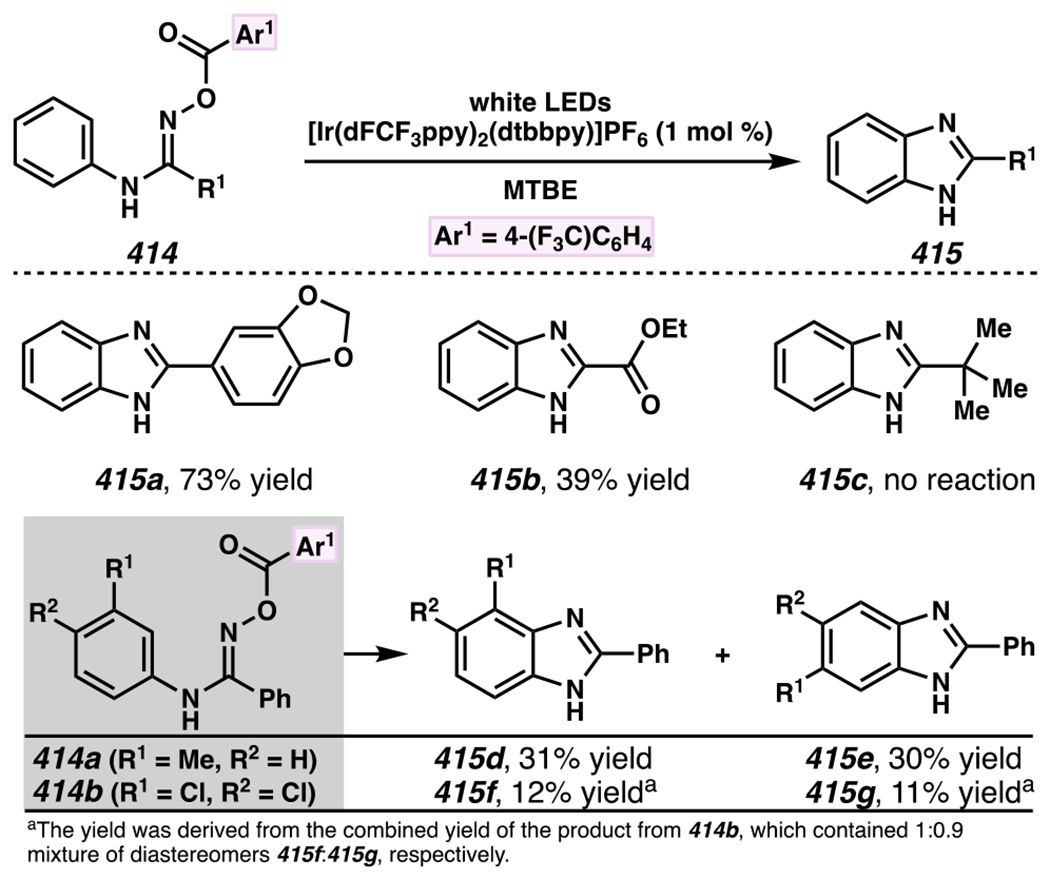
N-Aryl Amidoxime Esters Are Appropriate Precursors to Photo-Mediated Radical Annulation Reactions That Provide 2-Substituted Benzimidazoles
6. NITROGEN-CENTERED RADICALS CAN BE GENERATED FROM NITROGEN–HYDROGEN BONDS
6.1. Nitrogen-Centered Radicals Can Be Generated by Direct Oxidation of Amines
6.1.1. Nitrogen-Centered Aminium Radical Cations That Are Generated from Amines Can Add to Olefins
Nearly 60 years ago, Robert Neale and Richard Hinman disclosed one of the first productive reactions involving the addition of an aminium radical cation across an olefin (Scheme 92).70, 281, 282, This invention relied on a step to stoichiometrically N-halogenate an amine, followed by a second step to process the N-halogenated amine through protonation with a strong Brønsted acid and photolysis with ultraviolet light. Under these conditions, light homolyzes the relatively weak N-X bond (X = halogen or nitrosyl283 groups are among the most common). The generated aminium radical cation is poised to form a C–N bond upon radical addition across an olefin. This process exhibits reliable anti-Markovnikov selectivity, as a consequence of both the positive charge and highly electrophilic nature of the aminium radical cation.281 During the intervening decades, a wide array of methods have been developed for accessing aminium radical cations; none of these reactions, however, enabled direct access to anti-Markovnikov olefin hydroamination without prefunctionalization of amines, forcing reaction conditions, or both. 63, 281, 283, 284
Scheme 92.
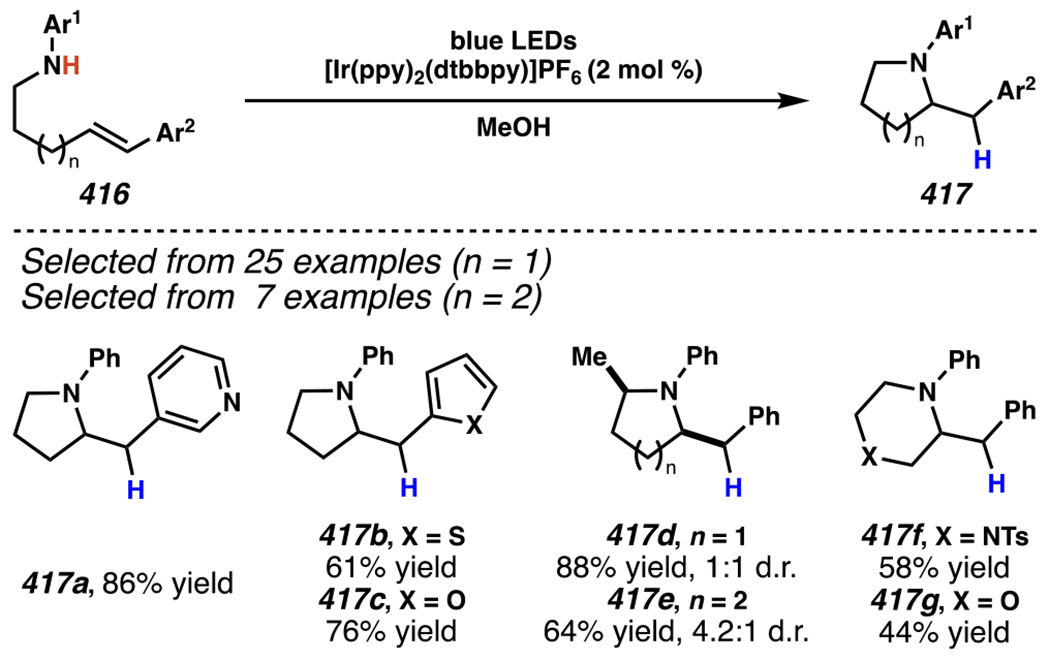
Knowles and Co-workers’ Intramolecular, Anti-Markovnikov Hydroamination Reactions Engage Olefins to Form 5- and 6-Membered, Saturated Heterocycles
Fortunately, Knowles and co-workers have streamlined the reaction and liberated it from relatively harsh reaction conditions. They have developed the first general, aminium radical-based, photocatalyst-driven process for anti-Markovnikov, olefin hydroamination (Schemes 92–93).285,286 In the presence of light and an iridium photocatalyst, N-aryl-N-alkyl amines 416 engage pendant aryl olefin acceptors in 5- and 6-exo-trig cyclization reactions to furnish cyclic N-aryl pyrrolidine, piperidine, morpholine, and piperazine products 417. This advance permits the transformation to generate products that would not be expected to form under traditional conditions, including 3-pyridinyl 417a, which forms in 86% yield, as well as furan- and thiophene-containing products (not depicted). In these types of transformations, 6-exo-trig cyclization reactions to form piperidine products exhibit greater diastereoselectivity than 5-exo-trig cyclization to form pyrrolidine products (cf. 417d vs. 417e). Importantly, this transformation provides access to N-tosyl piperazine 417f and morpholine 417g, albeit in slightly diminished 58% and 44% yields, respectively. This broad substrate tolerance is a testament to the relatively mild and direct conditions that enable this net redox-neutral reaction to proceed.
Scheme 93.
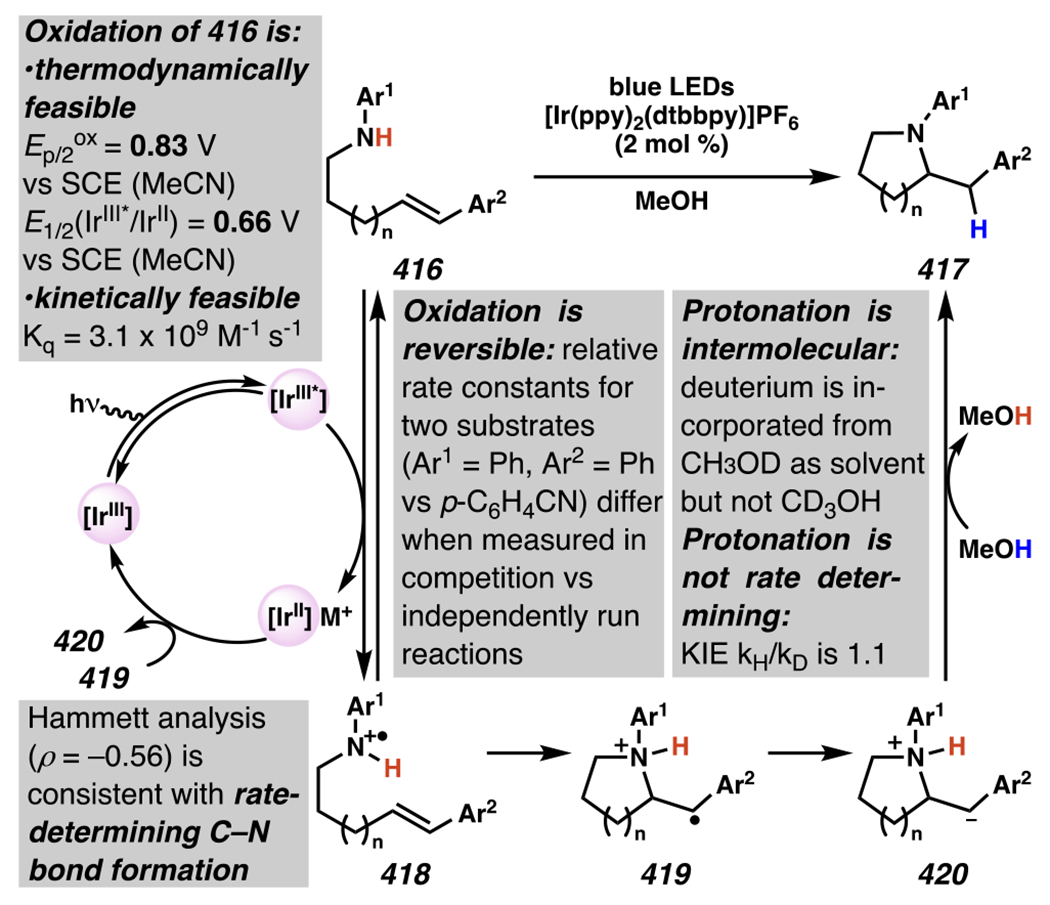
Photoredox Catalysis Can Be Used to Access N-Aryl Aminium Radical Cations That Engage in Intramolecular, Anti-Markovnikov Hydroamination Reactions
Knowles and co-workers have proposed a photoredox-catalyzed mechanism for this transformation that is supported by physicochemical measurements. In the proposed reaction mechanism, initial one-electron oxidation of neutral aniline 416 could generate an aminium radical cation 418, with concurrent reduction of an excited state iridium photocatalyst. Consistent with this proposal, amine 416 has a peak potential (Ep) of +0.83 V versus saturated calomel electrode (SCE), indicating that strongly oxidizing iridium(III) photocatalyst (E1/2 [IrIII*/IrII] = +0.66 V vs. SCE)8 may be slightly thermodynamically disfavored. Nevertheless, electron transfer between substrate 416 and excited state iridium(III) photocatalyst is kinetically facile, as demonstrated by luminescence quenching assays (Kq = 3.1 × 109 M−1 s−1).12 This oxidation is seemingly reversible, as the relative rate constants for two substrates differ when they are measured in reactions run as experiments with substrate competition or as independent transformations. Cyclization of the thusly generated aminium radical cation onto the olefin would produce benzylic radical species 419. This C–N bond-forming step is proposed to be the rate-limiting step, an observation that is supported by Hammett analysis, with a linearly correlated σp (R2 = 0.96) and a negative ρ (ρ = −0.56). Whereas, if the rate-determining step proceeds through addition of a neutral aminyl radical, a positive ρ-value would be expected.287 Moreover, in the subsequent steps, the reaction exhibits a modest, kinetic isotope effect (kH/kD = 1.1, in CH3OH vs. CH3OD), which is consistent with a mechanism in which a new benzylic C–H bond forms to furnish product 417 after the rate-limiting step of the reaction.
In principle, two alternative reaction sequences could convert radical 419 to product: the operative electron-transfer / proton-transfer sequence, or a direct C–H abstraction pathway, a process that is not consistent with deuterium labeling experiments. If the product were to form based on C–H abstraction by radical 419 from the solvent, reactions in CD3OH would be expected to result in benzylic deuteration; however, these experiments do not result in product deuteration. By contrast, an electron-transfer / proton-transfer sequence is consistent with experimental evidence. Initial reduction should be thermodynamically favorable and allow an aryl-stabilized radical (E0[R•/R−] ≈ −1.45 V versus SCE),288 such as 419, to engage a strong reductant (E1/2[IrII/IrIII]= −1.51 V versus SCE)8 to furnish carbon anion 420. Following electron-transfer, a proton-transfer step is consistent with reactions carried out in CH3OD, which result in benzylic deuteration.
Overall, this transformation constitutes a mechanistic complement to the hydroamination system developed by Nicewicz and co-workers, which relies on an organic photocatalyst to oxidize an olefin as the starting point for a net hydroamination reaction.289
Building on the development of this technology, Knowles and co-workers advanced the first general method for the intermolecular, anti-Markovnikov hydroamination of unactivated alkenes with secondary alkyl amines (Scheme 94).290 Investigating dialkyl amine substrates was inspired by research from Lusztyk and co-workers, who observed that piperdine-derived radical cations added to unactivated, internal alkenes with a rate more than 100-fold that of the aniline derivatives previously used by Knowles and co-workers.291, 285 Employing the more strongly oxidizing [Ir(dF(Me)ppy)2(dtbbpy)]PF6 (2 mol%) (E1/2 (IrIII*/IrII) = 0.99 V vs SCE (MeCN)), secondary dialkyl amines (421a), such as piperidine (Epox = 0.94 V vs SCE (MeCN)),104 undergo a single electron transfer to furnish aminium radical cations (424) which engage in facile addition to olefins (422a) resulting in ammonium and radical-carbon bearing intermediate (425). Piperidine was found to quench the photoexcited iridium complex in a concentration-dependent manner (Kq = 1.7 M−1 s−1),12, 290 while among a representative set of alkenes, only N-Boc-2,3-dihydropyrrole was observed to quench the iridium complex (kτ = 160 M−1), thus differentiating this method from those developed by Nicewicz and co-workers.289 In order to close the catalytic cycle and access the neutral, hydroamination product (423a), Knowles and co-workers found it necessary to include 2,4,6-triisopropylphenyl (TRIP) thiol (50 mol%). The thiol likely facilitates both hydrogen abstraction by the carbon-centered radical and the deprotonation of the ammonium ion. Furthermore, the resultant thiol radical can function as an effective electron sink for closing the catalytic cycle and regenerating the Ir(III) photocatalyst. This critical, double role for TRIP thiol is supported by control reactions finding no reaction occurs in its absence, as well as a complete lack of deuterium incorporation when using deuterated solvents indicating that the solvent is like uninvolved in the transfer of protons.
Scheme 94.
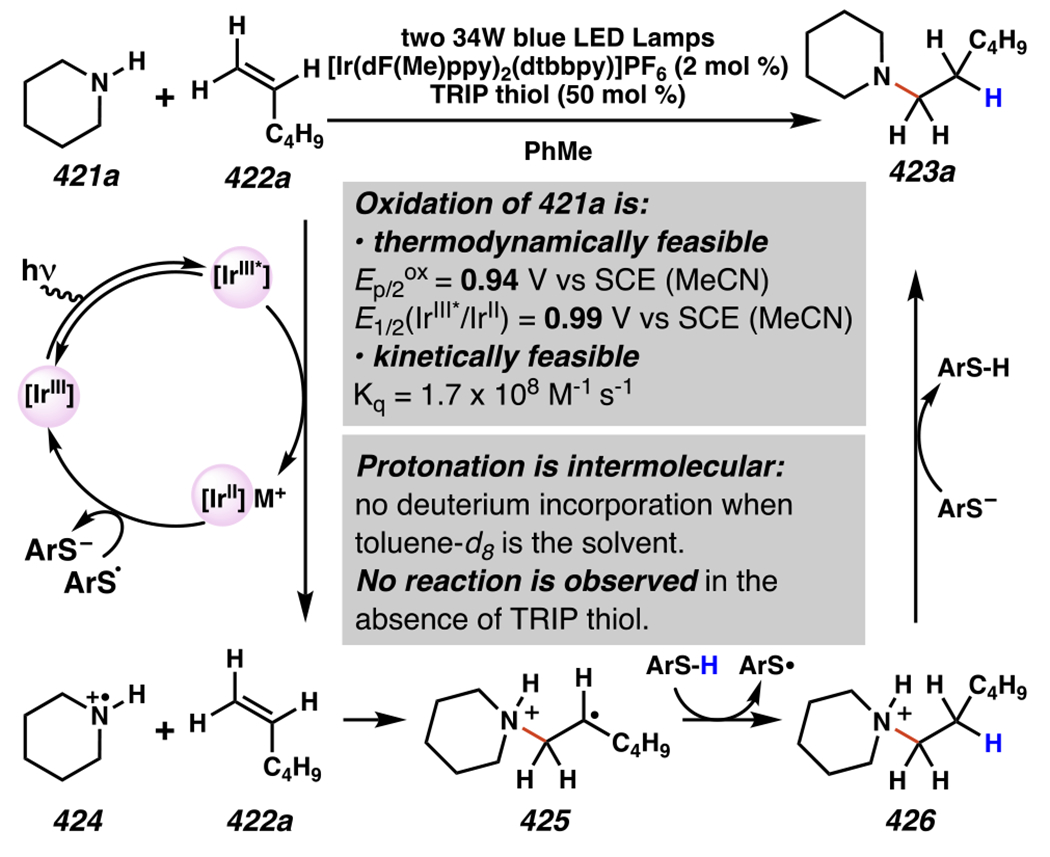
Photoredox Catalysis-Generated Aminium Radicals Engage Unactivated Alkenes in Intermolecular, Anti-Markovnikov Hydroamination in the Presence of an Aryl Thiol HAT-Catalyst
A broad variety of substituted alkenes can be aminated by secondary amines with 47–98% yield (43 examples) and complete anti-Markovnikov selectivity. Monosubstituted alkene products (423b), cyclic and acyclic disubstituted alkene products (423c and 423d), tri-, and tetra-substituted alkene products (423e, 423f, and 423g) were all accessed in 60–96% yields (Scheme 95). The least effective of these was mono-substituted alkene product (423b) which was isolated with 60% yield only after reaction at 45 oC. Conversely, TMS-enol ethers were found to be particularly effective at engaging in this transformation (423d) with a 95% yield. A linalool-derived substrate bearing both a trialkyl-alkene and a terminal allylic alcohol was hydroaminated selectively at the trialkyl alkene in 96% yield (423e). The tetra-substituted alkene 2,3-dimethyl-but-2-ene was aminated (423g) at an impressive 91% yield. The reaction is still effective in the presence of other, potentially reactive heteroatoms on the amine or alkene, including alcohols (423e and 423i), Boc-amines (423f), primary amines (423h), or spirocyclic amides (423l). The benzylic hydroxyl group of a precursor to the antidepressant fluoxetine did result a diminished yield (423j, 55% yield), but was unaffected by the aryl ether present in the API (423k, 80% yield).
Scheme 95.
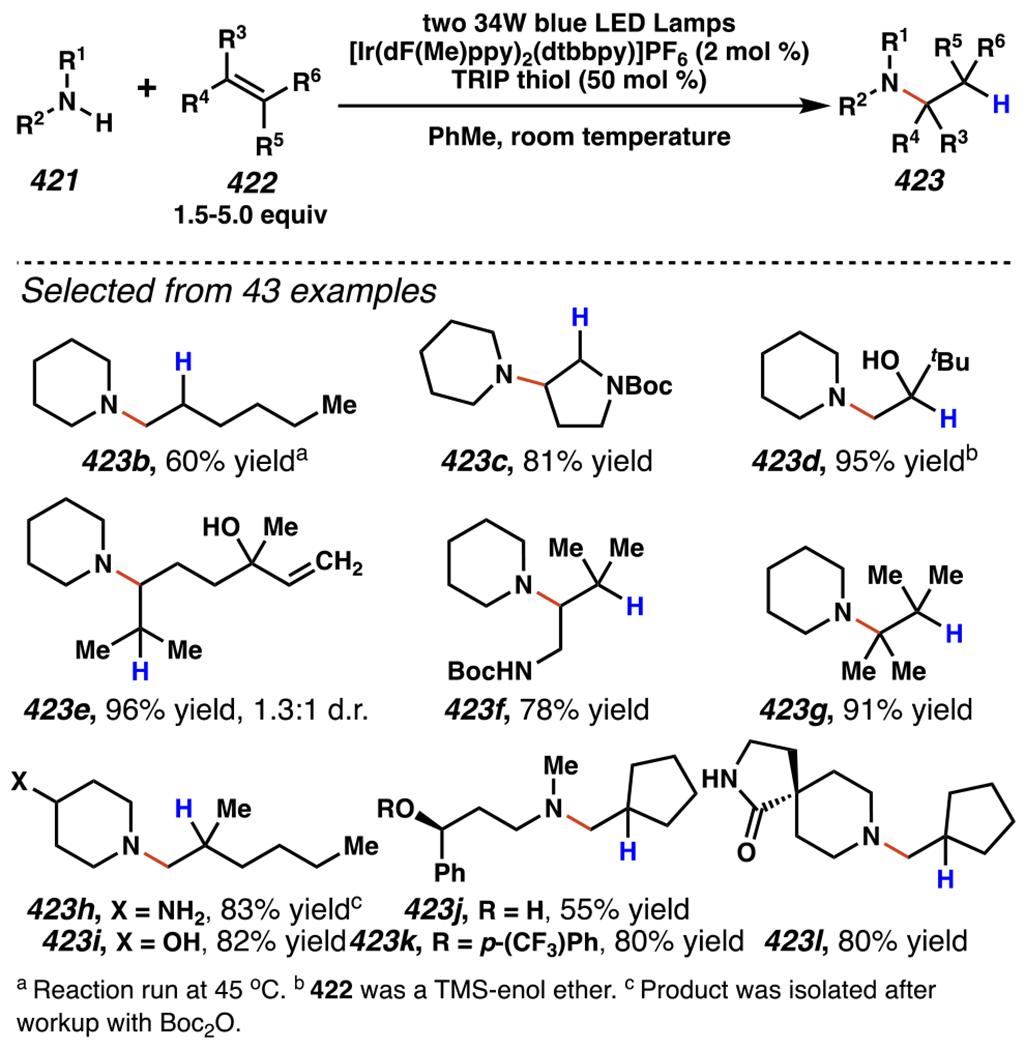
Secondary Amine-Derived Aminium Radicals Engage Unactivated Alkenes in Intermolecular, Anti-Markovnikov Hydroamination
As a further extension of this technology, Knowles and co-workers recently disclosed a new protocol which enables primary amines to engage in intermolecular, anti-Markovnikov hydroamination of unactivated alkenes with 47–98% yields to give secondary amines (Scheme 96).292 Remarkably, overalkylation to give quaternary amines is not observed, and tertiary amines are barely detectable (< 30:1). In terms of position-selectivity, apart from reactions using dihydropyran as a radical trapping agent, Markovnikov products are not observed. Once again, TMS-enol ethers are effective radical trapping agents, giving 80% yield of 1,2-amino alcohol 428a upon acidic workup. Furthermore, a product derived from cumylamine (428b) is synthesized in 69% yield, which is significant because, with an acidic workup, the cumyl group can be removed to give a primary amine at the site of hydroamination. A terminal, primary olefin reacts to furnish primary amine 428c, albeit at reduced yield. The reaction does tolerate other potentially reactive functional groups, such as amides 428d, and engages both cis- and trans-oct-4-ene with a marginally higher yield of desired product 428e when beginning from the cis- isomer.
Scheme 96.
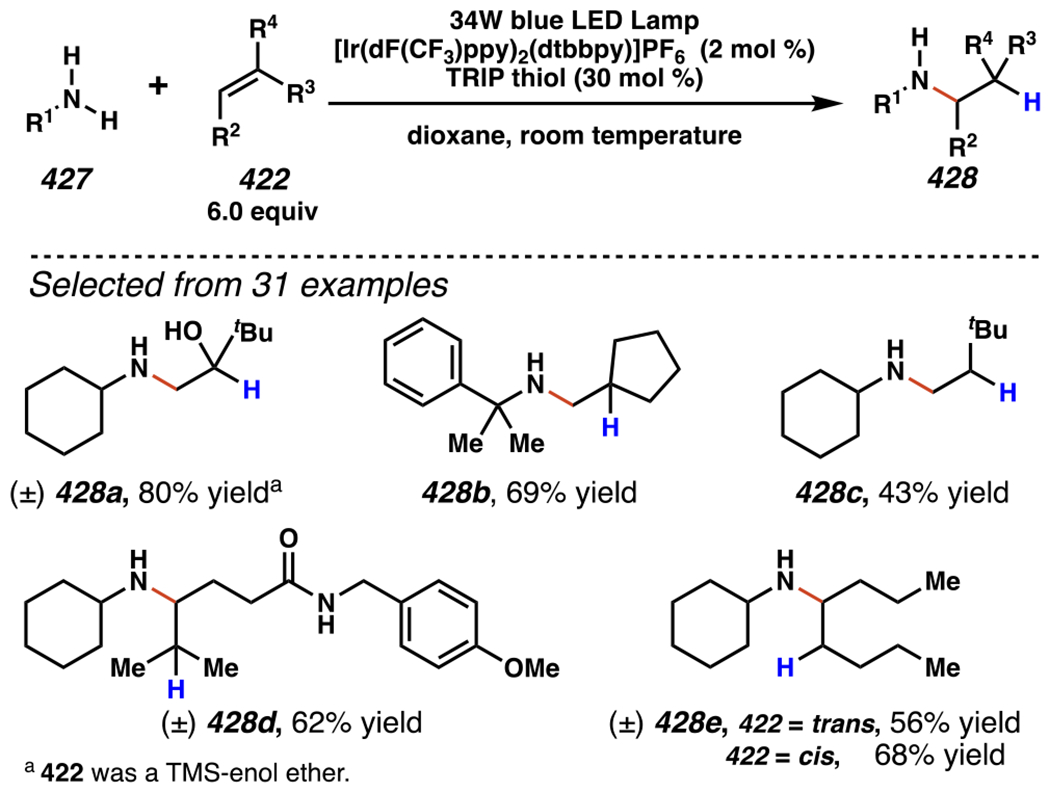
Primary Amine-Derived Aminium Radicals Engage Unactivated Alkenes in Intermolecular, Anti-Markovnikov Hydroamination Reactions
Aminium radical cations generated by photoredox catalysis have enabled anti-Markovnikov hydroamination of alkenes both inter- and intramolecularly. Distinct procedures, as developed by Knowles and co-workers, are applicable to primary and secondary amines for intermolecular hydroamination and without meaningful amounts of over-reaction, produce secondary and tertiary amine products, respectively. Mechanistic insights gained from these developments suggest that the reversal of site-selectivity from that typically observed in transition metal-catalyzed processes arises from the electrophilic nature of the radical cation.
6.1.2. Nitrogen-Centered Aminium Radical Cations That Are Generated from Amines Are Proposed Intermediates in Arylation Reactions
Building off a related photoredox platform that engages direct oxidation of arenes for C–H functionalization,293 Nicewicz and co-workers have disclosed a photo-driven strategy for (hetero)arene C–H amination that is particularly effective with more electron-dense arenes (Scheme 97).294 Employing a powerfully oxidizing mesityl acridinium photocatalyst under blue light irradiation, a broad array of amines can react productively. The position-selectivity of these reactions depends on the arene substituent: isoleucine methyl ester reacts with anisole to form ortho-substituted product preferentially and reacts with an aryl silyl ether to forms para-substituted product preferentially. Interestingly, α-chiral amine products 431a–431d fully retain enantioenrichment during the reaction, suggesting that the reaction does not transiently generate an α-aminyl radical. Notably, this process tolerates sterically encumbered amines, such as adamantylamine, and electron-dense heteroarenes, consistent with the broad substrate scope anticipated for reactions under mild conditions.
Scheme 97.
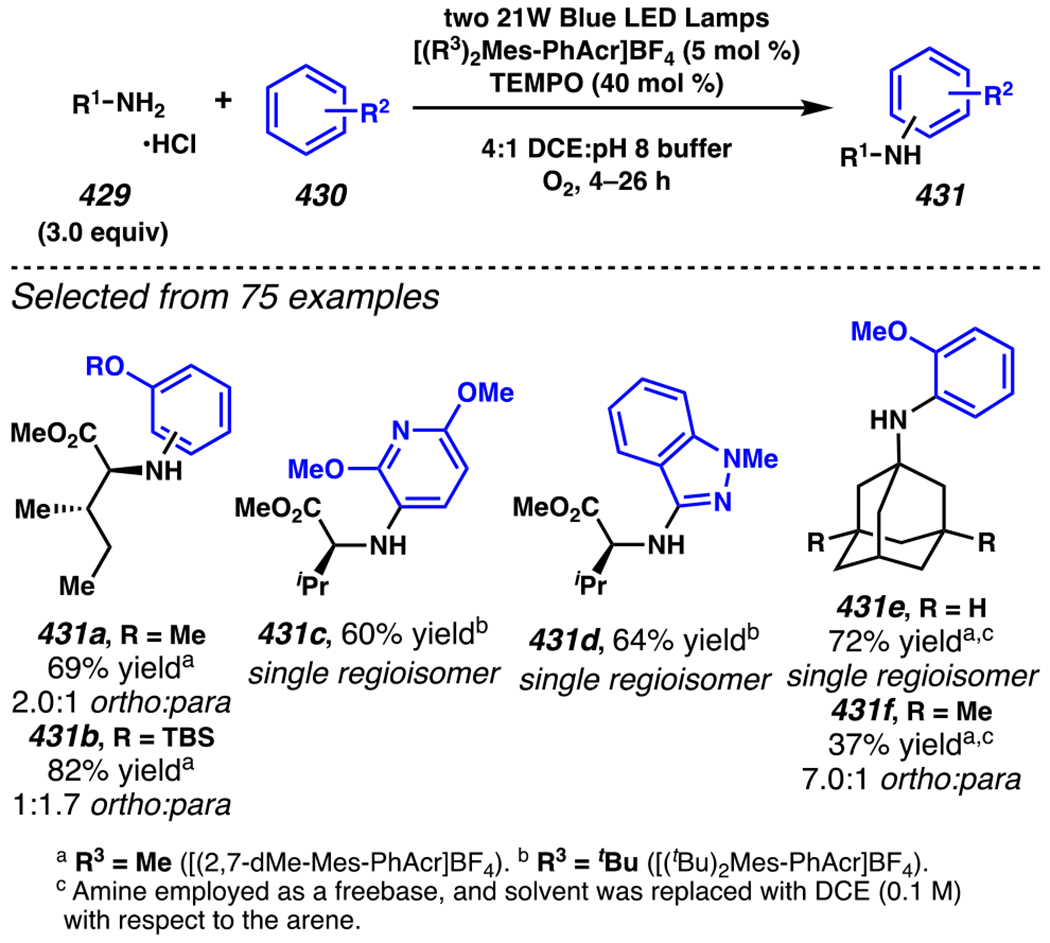
Varied Amines Efficiently Engage in Photoredox-Mediated C–H Amination of (Hetero)arenes
In order to interrogate the mechanism of this reaction, benzene was employed as a co-solvent and coupling partner for the valine methyl ester 429a (Scheme 98A). This simplifies mechanistic investigations, as benzene oxidation by the excited state photocatalyst is not expected to be thermodynamically favored ([(tBu)2Mes-PhAcr]BF4 : E1/2 (PC+*/PC•) = 2.15 V vs SCE (CH3CN);294 PhH: Ep = 2.75 V vs SCE (CH3CN)).294 As product forms under these reaction conditions, this particular set of coupling partners must react through initial amine oxidation, likely to give the aminium radical cation 433, which can add into benzene’s π-system, producing a resonance-stabilized, carbon-centered radical. To close the photocatalytic cycle, Nicewicz and co-workers proposed that adventitious molecular oxygen (O2) is reduced. Superoxide could deprotonate the aminium cation 434, and the resultant peroxy species could again react with carbon-centered radical of 435 to give the desired product and hydrogen peroxide as a byproduct. Consistent with this hypothesis, NMR analysis of the reaction mixture immediately following reaction completion295 reveals a characteristic hydrogen peroxide singlet in two different deuterated solvents.
Scheme 98.
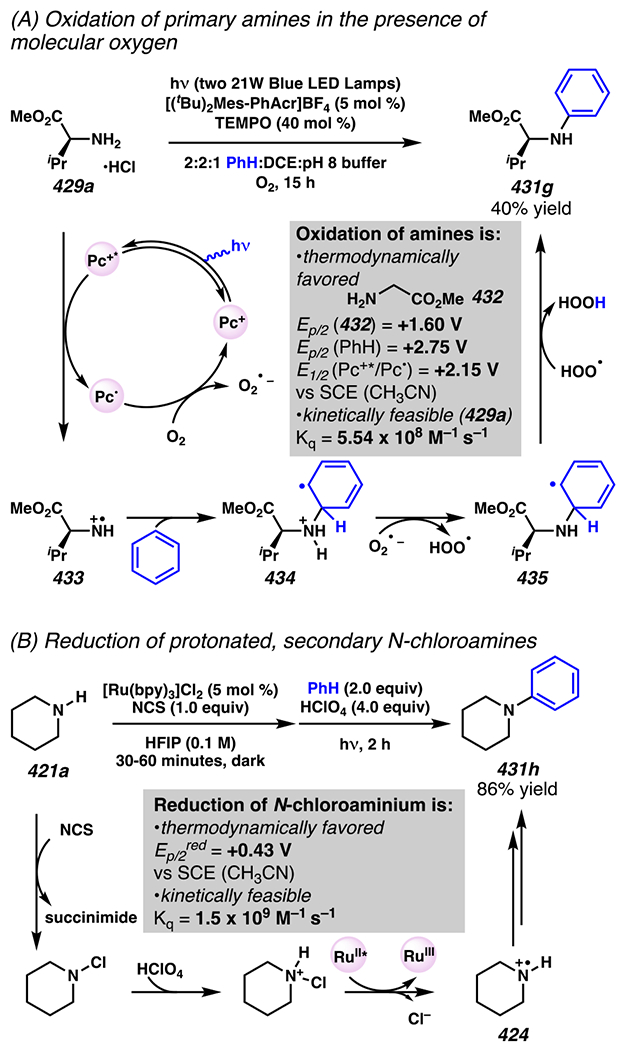
Aryl C–H Amination Can Be Engaged through Photoredox Generation of Aminium Radical Cations
A formally similar, yet mechanistically distinct, overall reaction has been developed by Leonori and co-workers (Scheme 98B).296 They propose that their process involves access to a secondary aminium radical cation that engages in C(sp2)–H amination. The critical aminium radical cation (i.e. 424) is prepared via an in situ generated N-chloroamminium intermediate, generated by in-situ chlorination with NCS followed by activation with an excess of perchloric acid. Once formed, aminium radical cation 424 is readily reduced. Reduction of this cationic intermediate is expected to be thermodynamically favorable (Ep/2 = +0.43 V vs SCE (CH3CN)) and exhibits kinetically significant quenching of the excited state photocatalyst (Kq = 1.5 × 109 M−1 s−1), while quenching of the photocatalyst was not observed with other components of the reaction individually or as mixtures.296 Ultimately, through steps similar to those proposed with primary amines (Scheme 98A), N-aryl amine products such as N-phenylpiperidine 431h, are accessed by C–H amination of relatively electron-dense to electron-neutral areness and heteroarenes. Collectively, the approaches of Nicewicz and Leonori extend the scope of viable nitrogen-centered radical precursors in arylation reactions beyond amides to include varied secondary alkyl amines.
Other stoichiometric oxidants can be used in net aryl amination reactions. Dioxygen has been proposed to perform the same role as a stoichiometric oxidant in the eosin Y-mediated C–N cross-coupling reaction involving primary and secondary alkyl amines and 2-(1H)quinoxalinones (Scheme 99).297 Consistent with this proposal, the reaction is dependent on the presence of molecular oxygen, with no product being produced when run under an atmosphere of nitrogen.
Scheme 99.
Eosin Y Mediates a Dehydrogenative Cross-Coupling Reaction Involving Alkyl Amines and Quinoxalinones
A complementary dehydrogenative cross-coupling reaction relies on catalytic triphenylpyrylium salt with an excess of persulfate in CH2Cl2 under blue light irradiation to affect ortho-selective amination of electron-rich phenols by electron-rich diarylamines (Scheme 100).298 The yield of the desired products decreasing directly with a decrease in electron-donating ability of the arene. Only very electron-dense arenes are viable substrates for this reaction, as phenol and 4-methylphenol do not produce any of the desired product under the reaction conditions.
Scheme 100.

Dehydrogenative Reaction Enables ortho-Selective Coupling of Electron-Dense Diaryl Amines with Electron-Dense Phenols
Nitriles can serve as leaving groups in photo-driven C(sp2)–N bond-forming reactions, which rely on 2,4,6-tris(diphenylamino)-5-fluoroisophthalonitrile (3DPAFIPN) as a photosensitizer or photocatalyst (Scheme 101).299,300,301, 302 Based on transient absorption spectroscopy and Stern-Volmer fluorescence quenching studies, N-methylaniline 441a quenches the excited state photocatalyst (Kq = 9.8 × 107 M−1 s−1), and oxidation of aniline 441a is thermodynamically feasible by photoexcited catalyst (Ep/2 ox (441a) = +0.92 V vs SCE (CH3CN);299 E1/2(PC*/PC•+) = +1.09 V vs SCE (CH3CN).299 While 4-cyanopyridine does not exhibit fluorescence quenching of photoexcited catalyst, transient absorption spectroscopy reveals a concentration dependent quenching of reduced photocatalyst in the presence 4-cyanopyridine (Kq = 7.0 × 107 M−1 s−1). Generated aminyl radical cation 443 and pyridinyl radical anion 444 are then proposed to undergo radical–radical cross-coupling to give zwitterionic species 445 which should rapidly lose cyanide to give protonated product 446. Deprotonation furnishes aminated product 442 in 98% yield. While oxidation of DABCO (Kq = 5.3 × 108 M−1 s−1) is kinetically competitive with oxidation of 441a, this process is not critical to reaction progress as the reaction proceeds with sparingly-soluble, carbonate bases at only slightly reduced yields as well as in the complete absence of base at 21% yield. Notably, other amine sources are viable, including N-alkylhydroxylamines which react in DMSO absent base and, using tribasic potassium phosphate, unsymmetric N-alkyl and N-aryl hydrazines are engaged to prepare N,N-diaryl amine 448c and N-alkyl-N-aryl amine 448d in 70% yield and 45% yield respectively (Scheme 102). The process transforms secondary alkyl amines, and the procedure overall is amenable to reaction at a relatively small number of positions on heteroarenes, particularly nitriles with 1,2- or 1,4-relationship to one or more aromatic nitrogen atoms.
Scheme 101.
Proposed Photoredox-Catalyzed, Radical-Radical Heteroaryl Amination Relies on Loss of Hydrogen Cyanide
Scheme 102.
Amine and Amine-Analogues Engage Heteroarenes to Form New C–N Bonds
Further development of nascent photoredox-generated, aminium radical cation chemistry has enabled the C–H functionalization of (hetero)arenes and the amination of heteroaryl nitriles with amines. Consistent with observations from hydroamination reactions, the C–H functionalization of (hetero)arenes proceeds with preference for amination at relatively electron-dense positions. These organophotocatalyzed reactions provide access to important N-(hetero)aryl products without use of transition-metal catalysis and, in several instances, without addition of stoichiometric reductants, instead engaging adventitious oxygen to close the photocatalytic cycle.
6.1.3. Nitrogen-Centered Aminium Radical Cations That Are Generated from Amines Are Proposed Intermediates in Amide Bond-Forming Reactions
The construction of amide bonds is an important operation in synthetic chemistry, due in large part to their ubiquity in biological and medicinal applications. While a plethora of approaches exist for amide bond formation, the relatively mild, room-temperature, light-driven conditions are appealing for this application, particularly since this approach does not typically result in stoichiometric amounts of byproducts like many state-of-the-art processes.
Beginning in 2014, a wide variety of photocatalysts have been applied in reactions forming amides from secondary alkyl amines, such as 421c, and aryl aldehydes, such as 449a, using ambient oxygen as a critical component and likely oxidant (Scheme 103).21, 26, 37 38 Generally, these reactions employ a visible-light excited photocatalyst and an excess of amine to generate aryl amide products, such as 450a, in good to excellent yields. These reactions, however, rely on the presence of molecular oxygen and most of the inventors of these technologies have shown that the presence of stoichiometric hydrogen peroxide facilitates the reaction with similar efficiency. This significantly muddles the mechanistic investigation for these reactions. While each make important contributions, further discussion thereof is outside of the scope of this paper, the overall substrate scopes are limited to secondary alkyl amines and aryl aldehydes.
Scheme 103.
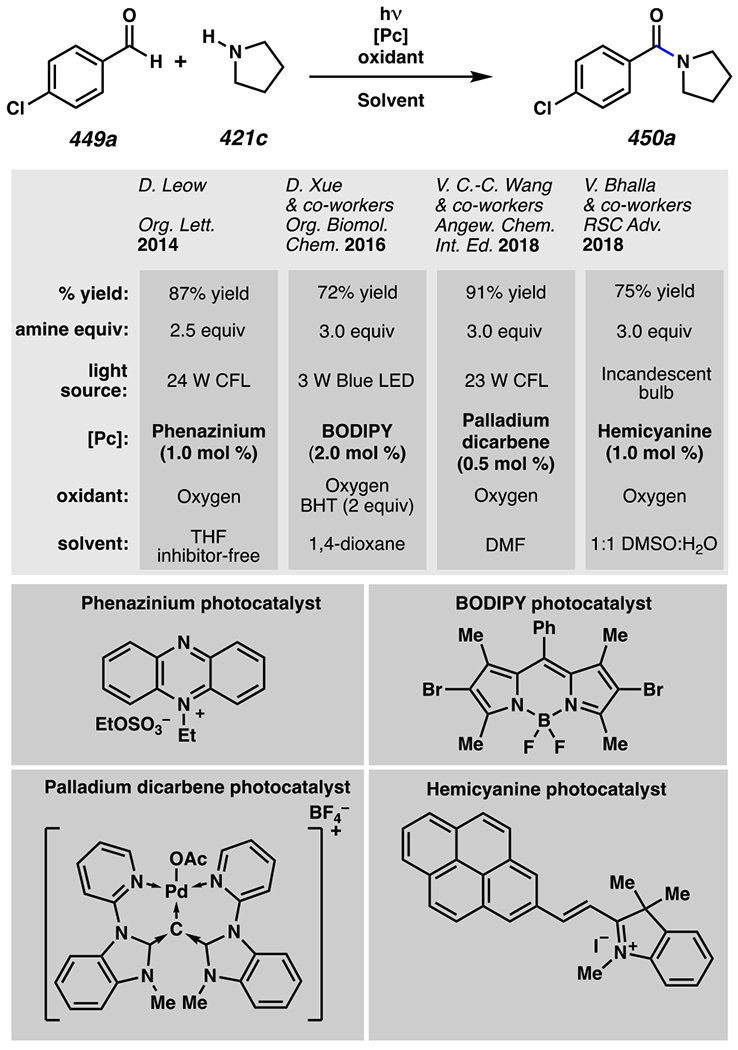
Advances in Amide Bond Formation Reactions Rely on Ambient Molecular Oxygen as an Oxidant
The most recent disclosure of a photo-driven, amide bond-forming reaction, however, has a remarkably broader scope (Scheme 104).303 Utilizing a common iridium photocatalyst and two equivalents of amine 421, aldehydes 449 are coupled to form new C–N amide bonds using CH3CN as the solvent and, in the absence of oxygen, employ bromotrichloromethane as a terminal oxidant. This manuscript provides the first examples of a photoredox-driven amide bond-forming reaction with a primary alkyl amine, furnishing amino ethyl ester product 450f, and the synthesis of a urea product (i.e., 450g).
Scheme 104.
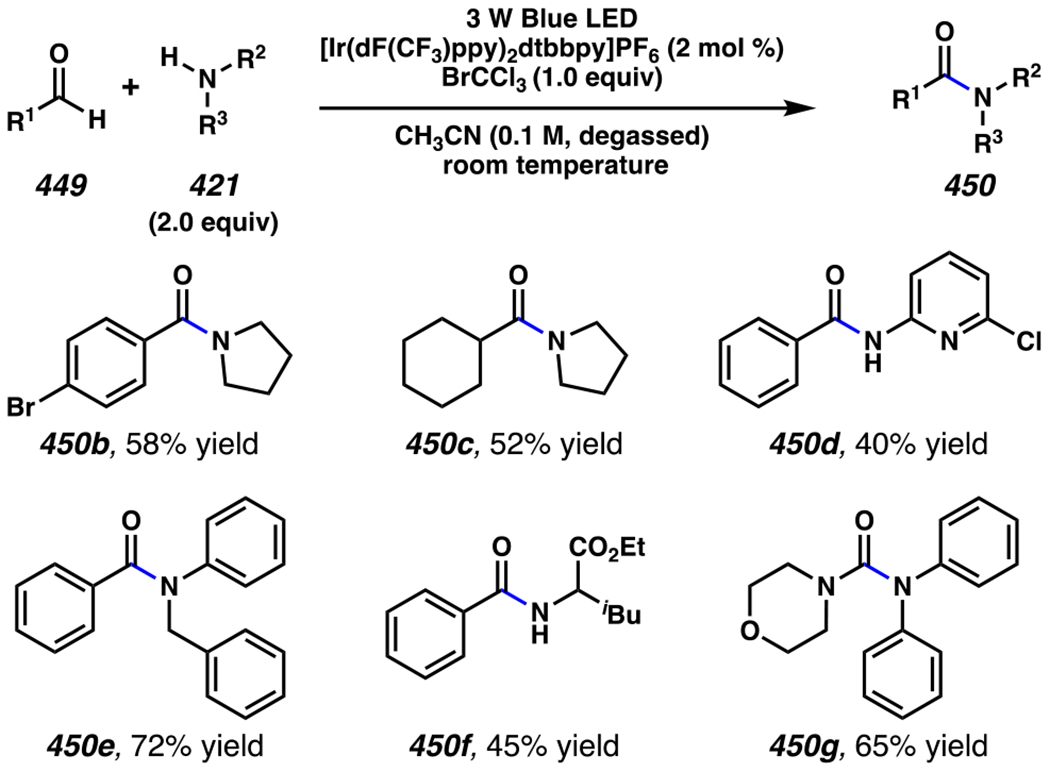
Photo-Driven Reaction Engages Alkyl and Aryl Aldehydes with Primary and Secondary Amines and Anilines to Form Amide Bonds
In contrast to some related technologies, the proposed mechanism reaction is premised on the exclusion of oxygen from the reaction, making the mechanistic analysis and proposal of a photoredox-generated, nitrogen-centered radical more straightforward (Scheme 105).303 Beginning with the photoexcited iridium complex, oxidation of pyrrolidine 421c is thermodynamically feasible (Ep/2ox (421c) = + 0.89 V vs SCE in CH3CN;104 E1/2 (IrIII*/IrII) = + 1.21 V vs SCE in CH3CN),8 while the possibility of iridium-mediated reduction of aldehyde 449b is not thermodynamically feasible (Ep/2ox (449b) = − 1.93 V vs SCE in CH3CN;104 E1/2 (IrIII/IrII) = − 1.37 V vs SCE in CH3CN).8 Oxidized aminium radical cation 451 can then add across the π-bond of aldehyde 449b to give O-radical aminium cation 452. Subsequent deprotonation and C–H abstraction involves bromotrichloromethane en route to desired amide 450h.
Scheme 105.
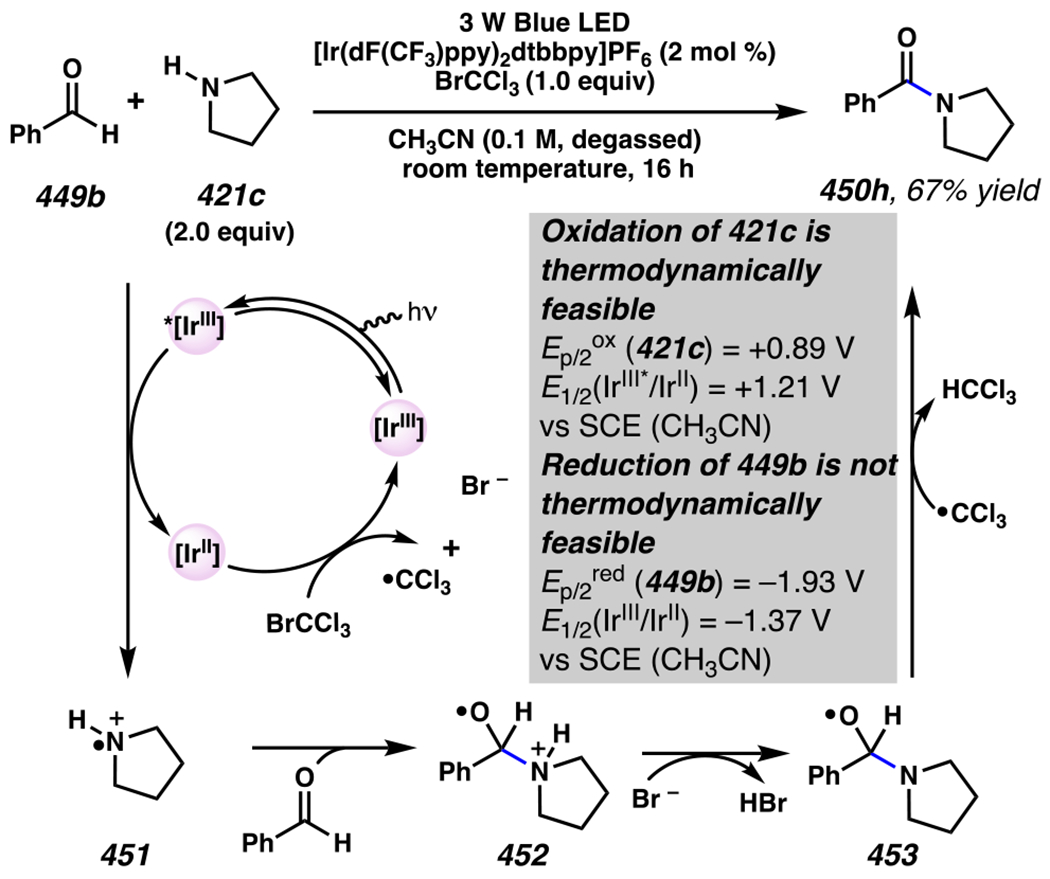
Photo-Driven Reaction Enables Amide Bond Formation from Amines and Aldehydes
While nitrogen-centered radicals derived from other functional groups, such as amides and sulfonamides are resonance stabilized, less stable amine-based, nitrogen-centered radicals can be powerful intermediates. Aminium radical cations engage in synthetically efficient amide bond-forming reactions, C–H functionalization processes involving (hetero)arenes, and hydroamination reactions involving of alkenes. These reactions provide indirect insight into and a richer understanding of the nature of fleeting nitrogen-centered radical intermediates.
6.2. Nitrogen-Centered Amidyl Radicals Can Be Generated from N–H Bonds
6.2.1. Amidyl Radicals Form from Amides via Proton-Coupled Electron Transfer (PCET)
Amidyl radicals have emerged as versatile synthetic intermediates for C–N bond construction, and as directing groups for functionalization reactions, specifically because of the invention of mild technologies to prepare amidyl radicals. Traditionally, amidyl radical intermediates have been inaccessible through direct homolytic activation of amide N–H bonds. Amidyl N–H bond homolysis is challenging because the targeted amidyl N–H bond is strong (BDFE ~100 kcal/mol).304, 305, 306 Consequently, until recently, amidyl radicals were accessed from amides indirectly via N-functionalized precursors,79, 91, 92, 262, 307, 308, 309, 310, 311, 312, 313 or directly with the stoichiometric use of oxidants.314, 315, 316, 317, 318, 319, 320, 321 These methods limit the substrates amenable to reactions involving amidyl radicals, as many functional groups cannot tolerate the harsh conditions necessary to access such intermediates. Fortunately, Knowles and co-workers322 have surmounted these challenges, to access amidyl radical intermediates that engage in productive olefin and C–H functionalization reactions (Schemes 106–109).
Scheme 106.
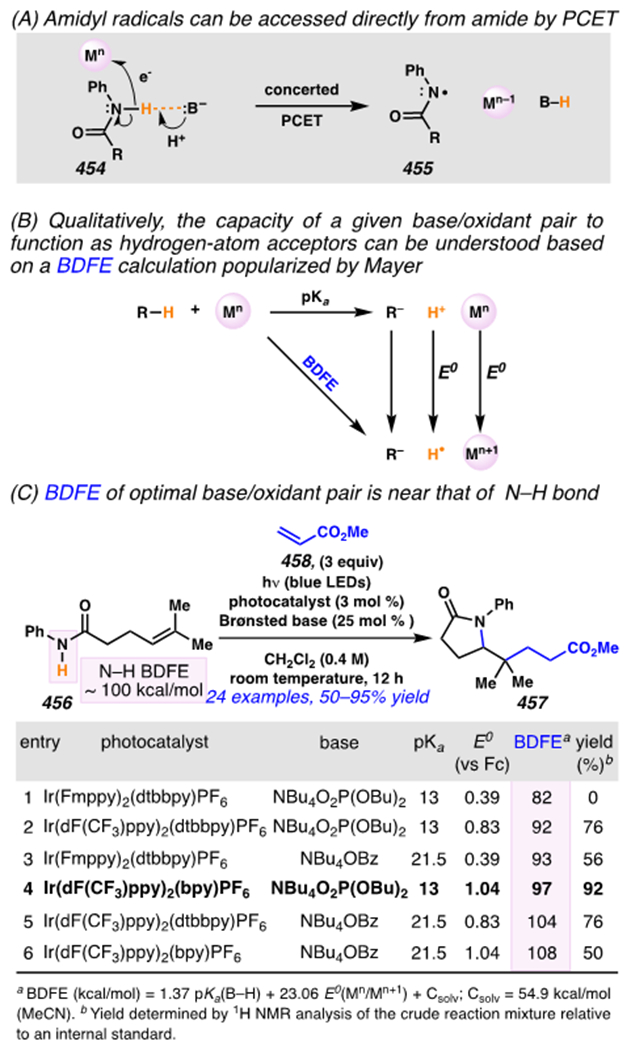
Photo-driven Proton-Coupled Electron Transfer (PCET) Processes Can Provide Access to Amidyl Radicals, Critical Intermediates in Alkene Carboamination Reactions
Scheme 109.
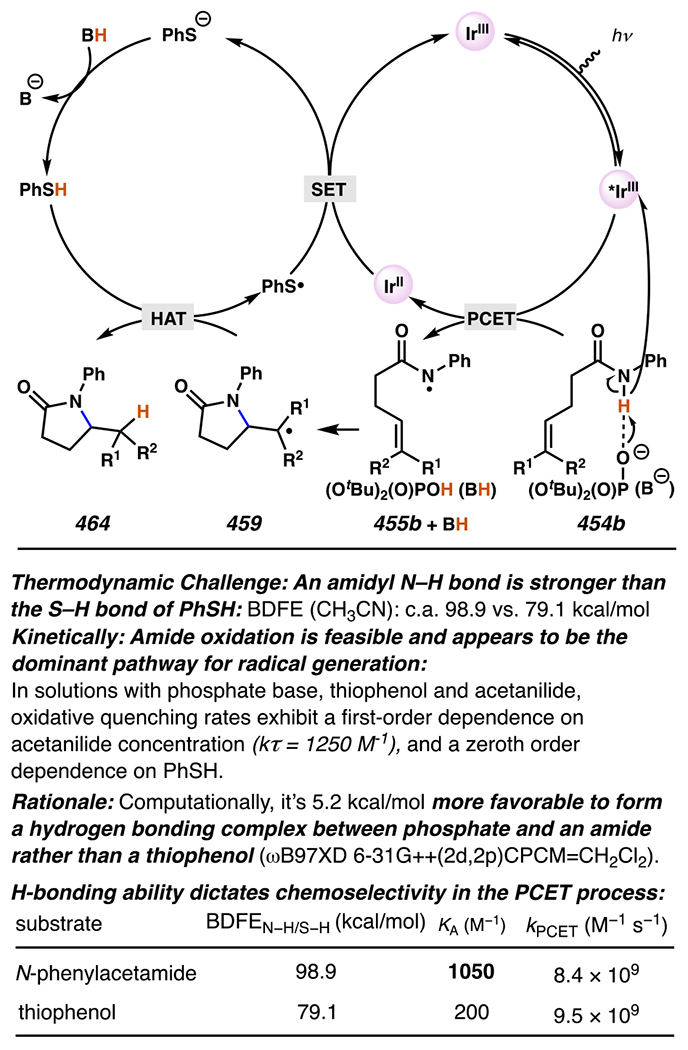
While an Amidyl N–H Bond Is Stronger Than A Thiyl S–H Bond, the Greater Hydrogen-Bonding Ability of an Amide Relative to a Thiol Enables Chemoselective Activation of An Amide to An Amidyl Radical
As a critical insight to enable these processes, Knowles and co-workers demonstrate that the strong N–H bonds of N-arylamides can be formally homolyzed to furnish amidyl radical intermediates directly based on Proton-Coupled Electron Transfer processes (PCET, Scheme 106A). In this PCET process, an electron from the N–H bond and the N–H proton are transferred concurrently to two independent acceptors – a one electron oxidant (Mn) and a Brønsted base (:B−), respectively. In this system, hydrogen-bonding between the base and the amide weakens the N–H bond, and facilitates its oxidation to give an amidyl radical intermediate.
For PCET processes, a thermodynamic formalism can qualitatively predict effective bond strength for engaged bonds (Scheme 106B), which has predictive power. Specifically, a bond dissociation free energy (BDFE) is the energy required to homolytically break a bond in a particularly solvent (Scheme 106B). Formally, a BDFE is the sum of the energy associated with breaking that same bond heterolytically based on deprotonation, as reflected by the acidity of that bond (pKa), with the energies required to oxidize the resulting anion to a neutral radical, to reduce the associated proton to a hydrogen radical.323 In a PCET process, where the proton and electron travel to separate destinations, an additional term is needed. 324,325,326,327
This formalism can be employed to accurately predict the reagents necessary to affect Knowles and co-workers’ intramolecular carboamination reactions with synthetically useful efficiencies (Scheme 106C). Specifically, Knowles envisions that amidyl radical generation will rely on a weakly hydrogen-bonding base, and a photocatalyst, whose excited state is capable of accepting an electron from the amide-base complex. Consequently, the optimal base/oxidant pair (entry 4) must have a calculated BDFE similar to that of the amide N–H bond. Indeed, the optimal iridium photocatalyst (Ir(dF(CF3)ppy)2(bpy)PF6) and phosphate base (NBu4OP(O)(OBu)2) are predicted to engage with a bond with a BDFE of ~97 kcal/mol, near to that of an amide N–H bond (BDFEs ~100 kcal/mol), and furnish the desired carboamination product in 92% yield (entry 4, Scheme 106C).
Mechanistically, Knowles and co-workers envision that PCET-based oxidation of the amide to an amidyl radical involves concerted deprotonation by phosphate base (B−) and electron transfer to photoexcited iridium catalyst (*IrIII) (Scheme 107). Luminescence quenching experiments demonstrate that amide oxidation is kinetically feasible, and requires the presence of base (kτ = 731 M-1). Moreover, the reaction remains viable when the employed base is less acidic than the amide by 20 pKa units, an observation that is consistent with a PCET mechanism, but would not be consistent with a single electron transfer (SET) process. Furthermore, in independent rate experiments, a small kinetic isotope effect (kN–H/N–D = 1.15) suggests that hydrogen-bonding influences the rate of quenching. The formed amidyl radical undergoes 5-exo cyclization to form a nascent nucleophilic carbon centered radical, which further engages in an intermolecular addition with acrylate acceptor 458 to yield an electrophilic, resonance stabilized α-carbonyl radical. This radical is poised for reduction by iridium(II) to regenerate the iridium(III) precatalyst, and form a resonance stabilized anion, which is poised to deprotonate the weak phosphate base and generate the desired carboamination product.
Scheme 107.
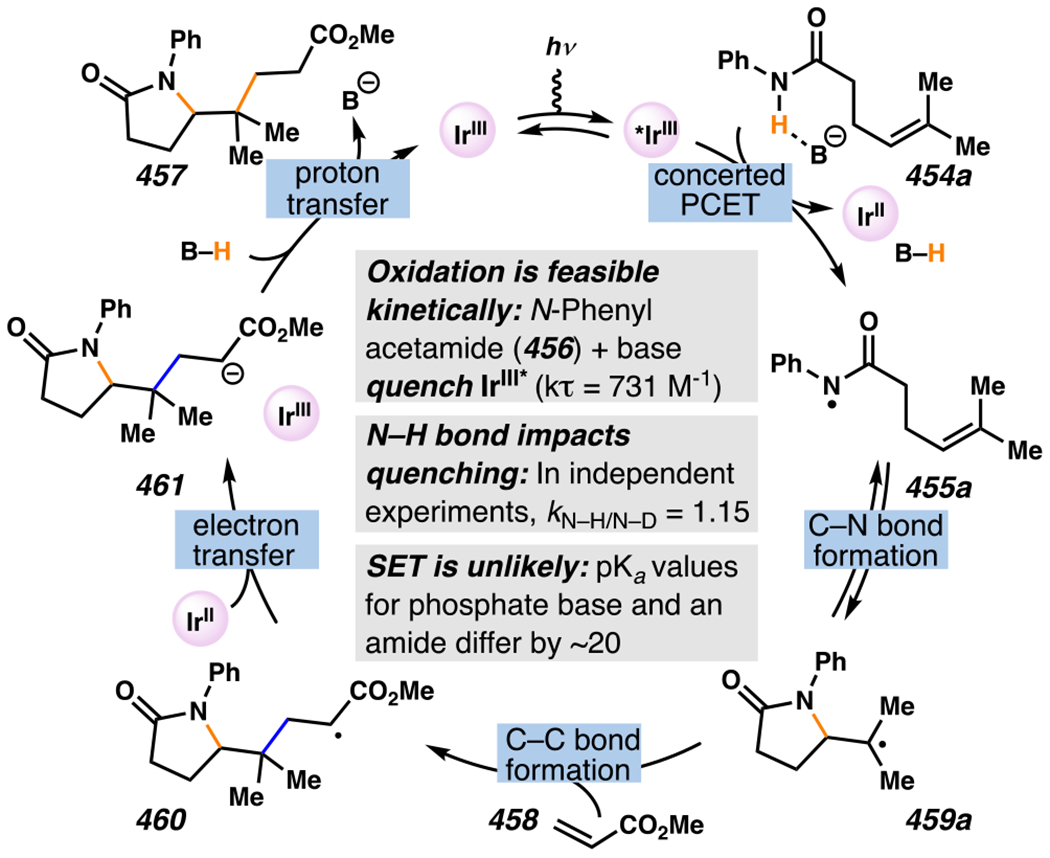
Evidence Is Consistent with the Proposed PCET Mechanism
Building on this new approach to access amidyl radicals, Knowles and co-workers have used this strategy to develop efficient and practical olefin hydroamidation protocols.328, 329, 330, 331 Like other amidyl radical-based functionalization reactions, traditionally, amidyl radical-based olefin hydroamidation reactions have been limited by multistep substrate syntheses, or reliance on hazardous reagents.91, 92, 162, 316, 332, 333, 334 Additionally, hydroamidation processes require selective homolysis of amide N–H bond (BDFE ~99 kcal/mol) in the presence of hydrogen atom donor that incorporates a weaker bond, such as a thiophenol S–H bond (BDFE ~79 kcal/mol). Indeed, while empirically optimizing this reaction, Knowles and co-workers find that background reactivity does not substantially improve through the inclusion of catalytic quantities of many common hydrogen-atom donors, such as some phenols, triphenylsilane, some arylamines, and diphenyl acetonitrile (not depicted). Fortunately, reactions proceed optimally when 10 mol% of thiophenol is included as a hydrogen-atom donor (Scheme 108).
Scheme 108.
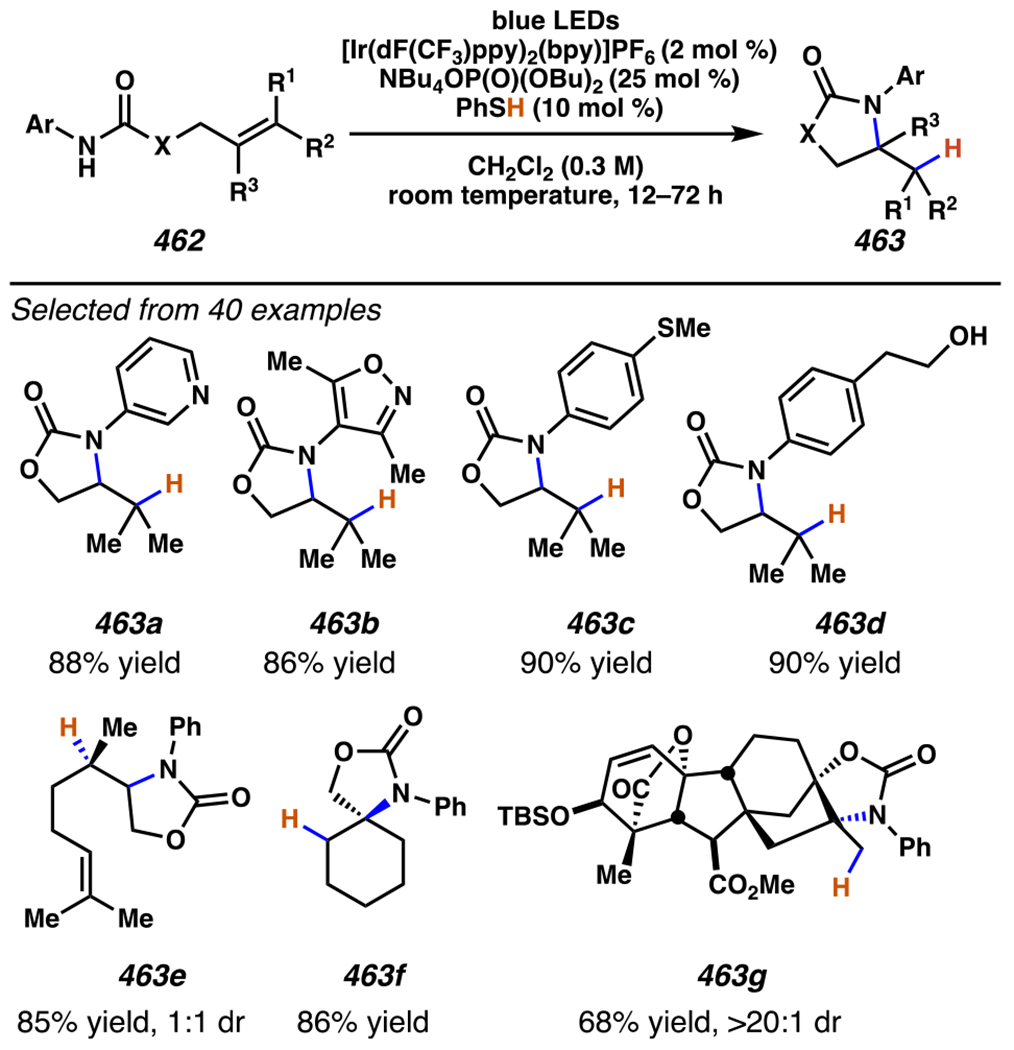
Olefin Hydroamidation is Enabled by Selective Amidyl Radical Generation in the Presence of Thiophenol
This olefin hydroamidation reaction accommodates otherwise oxidatively labile substrates due to the ability of PCET-mediated process to selectively activate amidyl N–H bonds, and can proceed with substrate-induced diastereoselectivity. For example, the reaction tolerates varied N-(hetero)aryl substituents to afford pyridyl 463a, oxazolidinone-containing 463b in excellent yields. Readily oxidizable pendant thioether 463c and primary alcohol 463d form efficiently, overcoming a known limitation of previous hydroamidation technologies, which do not tolerate thioethers or unmasked alcohols. Interestingly, the cyclization reactions involving carbamates derived from the isomeric polyolefins nerol and geraniol give isoxazolidinones 463e in 85–95% yield with 1:1 d.r., and provide evidence that substrate olefin geometry has a limited impact on reaction outcome. As expected, more rigid enantioenriched small molecules undergo hydroamidation with the high levels of diastereoselectivity to furnish products such as gibberellic acid-derived 463g (>20:1 d.r.).
A plausible proposed mechanism for this reaction relies on one processes also critical to the disclosed carboamidation reaction: PCET-based oxidation of the amide to an amidyl radical, with concurrent reduction of the excited state catalyst to Ir(II), and subsequent reaction of the amidyl radical with a pendant olefin to form an exocyclic carbon-centered radical 459 (Scheme 109). Once exocyclic carbon-centered radical 459 forms, hydroamidation product 464 forms via hydrogen-atom abstraction from thiophenol, which also furnishes a thiyl radical. The resultant thiyl radical could be reduced with concurrent oxidation of the Ir(II) photocatalyst to regenerate an Ir(III) photocatalyst and a thiophenyl anion.
Of critical import, this proposal requires the stronger amidyl N–H bond (BDFE ~99 kcal/mol) to undergo direct oxidation by the photocatalyst, even in the presence of 20 kcal/mol weaker thiophenol S–H bond (BDFE ~79 kcal/mol), which is known candidate for PCET activation.335, 336, 337 Although both base-coordinated amide and thiophenol engage in PCET-based activation with similar rates (kPCET = 8.4 × 109 M−1 s−1 for amide vs. 9.5 × 109 M−1 s−1 for thiophenol), chemoselective PCET-based quenching by the amide proceeds owing to the greater hydrogen-bonding ability of an amide relative to a thiophenol (hydrogen-bonding equilibrium constant, KA = 1050 M−1 for amide vs. KA = 200 M−1 for phenol; Scheme 109).329 A preponderance of evidence supports this mechanistic proposal, accumulated through luminescence quenching experiments, and DFT calculations.
To improve this reaction, Nocera and co-workers investigate the reaction mechanism in gory detail, relying on computational, electrochemical, fluorescent quenching, and transient absorption spectroscopy studies (Scheme 110).338 Consistent with Knowles and co-workers’ analysis, after photocatalysts absorb light to produce the excited state photocatalyst, followed by the PCET between excited photocatalyst/base pair and the amide 465 form a key amidyl radical intermediate (kPCET1). The generated amidyl radical can engage in any of at least three possible reactions: (i) Nonproductive back-electron transfer (BET) between the amidyl radical 467, and the reduced photocatalyst/conjugate base (BH) pair to regenerate the starting reagents (kBET), (ii) Nonproductive HAT between the amidyl radical and thiol to produce the starting amide 465 and thiyl radical (kHAT1), or (iii) Productive intramolecular 5-exo cyclization of the amidyl radical 467 with the pendant olefin form a alkyl radical 468 (kc). In the productive reaction, formed alkyl radical 468 abstracts a hydrogen atom from thiol give desired final product 466 and thiyl radical (kHAT2). Eventually, the photoredox cycle is completed by the PCET to regenerate starting reagents (kPCET2). The catalytic cycle could also be closed by the PCET between alkyl radical and reduced photocatalyst/conjugate base pair (kPCET3). In this investigation, seven rate constants (kPCET1, kBET, kHAT1, kc, kHAT2, kPCET2, and kPCET3) associated with the productive and nonproductive pathways are measured using transient absorption spectroscopy.
Scheme 110.
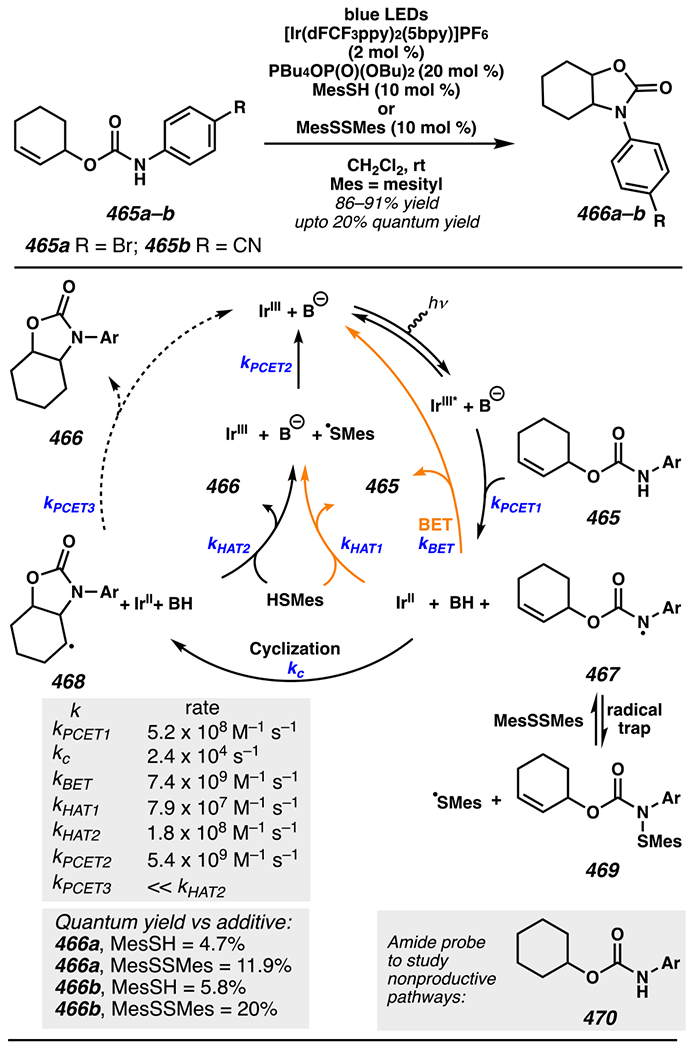
The Off-Cycle Reversible Trapping of Amidyl Radical Intermediates Disrupts Energy-wasting Pathways and Improves the Reaction Quantum Efficiency
Nocera and co-workers identify an additive, 2,4,6-trimethyldiphenyl sulfide (MesSSMes), that can decrease the incidence of nonproductive BET pathways, and increase the photon-efficiency of the productve olefin hydroamidation. To separate the nonproductive BET pathways (kBET and kHAT1) from the productive transformations, the authors rely on amide 470 as a substrate, as, owing to the absence of a pendant olefin, this amide cannot cyclize. These nonproductive pathways consume 91.6% of formed amidyl radical and leave 8.4% for the intramolecular radical cyclization. Furthermore, the slow radical cyclization rate (kc) due to delocalization of the radical over aromatic ring also contribute to the lower quantum efficiency of the reaction. To compete with these nonproductive reaction pathways, disulfide (MesSSMes) is introduced as an additive. This additive reversibly traps the amidyl radical to form an off-cycle equilibrium product thioamide 469, which would be released back to the catalytic cycle to effect the productive radical cyclization. Notably, thioamide 469 can be subjected to olefin hydroamination conditions in the presence of MesSH, and furnishes a mixture of amide 465 and cyclized product 466 in 87% overall yield with ratio of 1:0.2. This experiment supports the presence of reversible equilibria between amidyl radical 467 and thioamide 469 in the olefin hydroamination. Gratifyingly, the use of disulfide (MesSSMes) in place of thiol (MesSH), perturbs the nonproductive BET pathways and indeed provide four-fold increased energy efficiency, for example the quantum yield increased to 11.9% for the para-bromo analog of amide 465a and 20% for para-cyano analog of the amide 465b.
When Knowles’ olefin hydroamidation reaction relies on thiophenol as a hydrogen-atom transfer catalyst,328 N-aryl amides react productively. Unfortunately, the same protocol is not effective with N-alkyl amides, which incorporate stronger N–H bonds (N−H BDFE: N-aryl amides ∼ 100 kcal/mol vs. N-alkyl amides ∼ 110 kcal/mol), 339 and form more reactive amidyl radicals. Such highly reactive amidyl radicals engage in nonproductive pathways that compete kinetically with the desired amidyl radical addition to pendant olefins.60,340,341 Thiol selection can mitigate this limitation: TRIP thiol can perform as an HAT catalyst, with TRIP disulfide as an additive in their photoredox system to effect the olefin hydroamidation of N-alkyl amides (Scheme 111).330
Scheme 111.
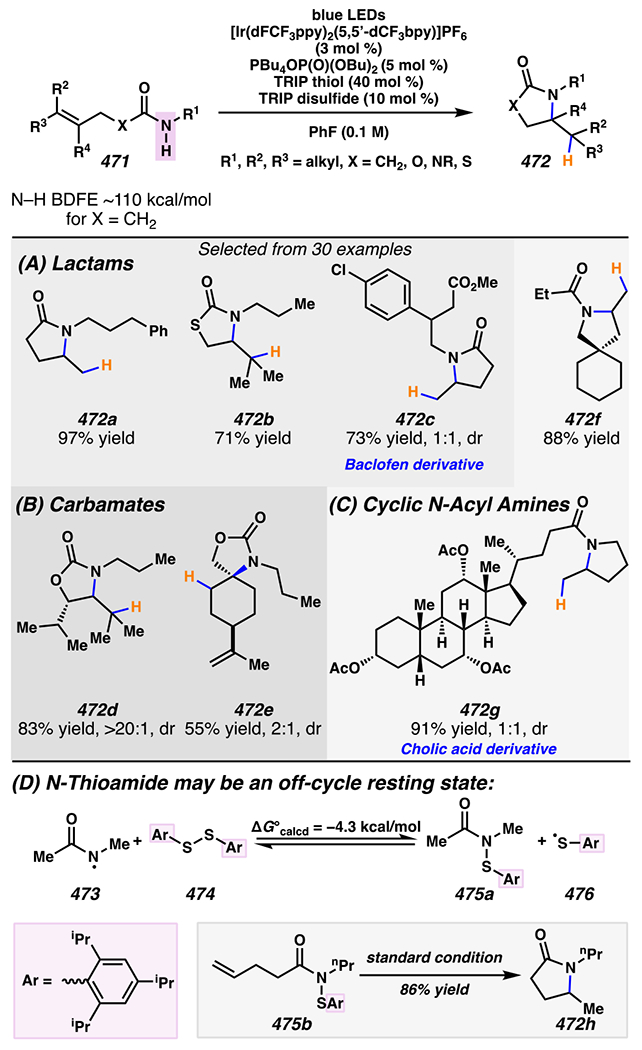
Amidyl Radicals Form from the Strong N–H Bonds of Unactivated N-Alkyl Amides by Excited-State PCET Process and Can Be Employed in Syntheses of Lactams, Cyclic N-Acyl Amines and Carbamates
This protocol converts N-alkyl amides, carbamates, thiocarbamates, and ureas bearing terminal olefins to γ-lactams, carbamates, thiocarbamates, and cyclic N-acyl amine derivatives. It has been applied to functionalize the muscle relaxant, Baclofen to give Baclofen derivative 472c in 73% yield with 1:1 dr. Notably, hydroamidation of carbamates having stereogenic centers adjacent to olefin gives product 472d in good yield and high levels of diastereoselectivity. Predictably, substrates that incorporate multiple olefins exclusively react through a geometrically accessible pathway to furnish five-membered ring-containing product 472e. More elaborate fine chemicals, such as a peracetylated cholic acid derivative, react in excellent yields.
In this reaction, the critical TRIP disulfide may form a stable off-cycle resting state for the amidyl radical, N-thioamide 475a, as reported by Nocera and co-workers.338 This hypothesis is consistent with DFT studies that indicate that the conversion of amidyl radical 473 to N-thioamide 475a is energetically favorable (ΔG°calc. = −4.3 kcal/mol, DFT calculations (UωB97XD/6–311G++(2d,3p)//UB3LYP/6–31+G(d,p))). Additionally, in parallel with Nocera’s findings,338 the olefin hydroamidation of presynthesized N-thioamide 475b affords lactam 472h in 86% yield. Based on these observations, Knowles hypothesizes that the off-cycle intermediate 475b is likely to improve the reaction efficiency by suppressing the nonproductive back electron transfer pathways.
A broader range of radical acceptors have been developed for use reactions that rely on PCET to induce amidyl radical formation. Indeed, Rueping and co-workers employ aryl alkynyl sulfones to trap the carbon centered radical formed from intramolecular 5-exo-trig cyclization of amidyl and carbamoyl radicals (Scheme 112).342 Indeed, aryl amides, carbamates, and ureas 477a–477d participate in the radical addition with internal and terminal alkyne, and alkene acceptors to give the functionalized alkynyl pyrrolidine-2-ones in good to execellent yields.
Scheme 112.
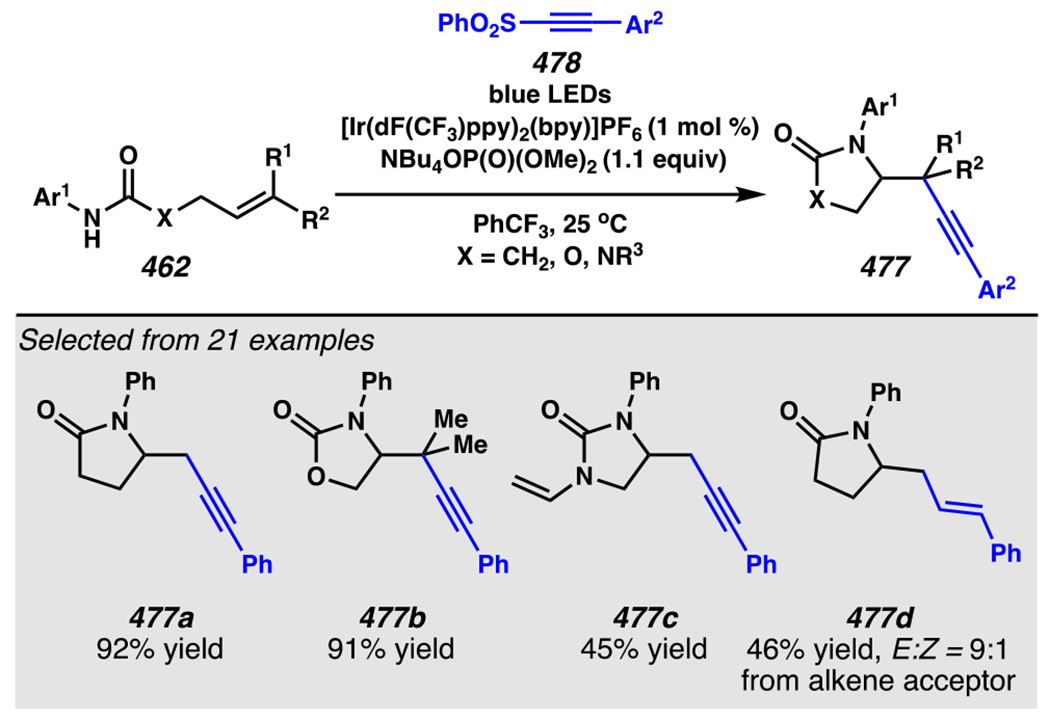
Alkynyl Sulfones Can Be Used as Acceptors in PCET-Mediated Processes
Premised on this approach to access amidyl radicals, arene annulation proceeds with N-phenylbenzamide 478 or N-phenylcinnamamide 480 substrates, using either iridium-based photosensitizers,343 or photoactive organic complexes344 (Scheme 113). In these systems, generated amidyl radicals are, or become, poised to undergo oxidative intramolecular C–H amidation to give phenanthridinones and quinolinones, respectively. With cinnamamide substrates, to achieve this outcome, (E)-olefins must be able to isomerize to (Z)-olefins under the reaction conditions. Indeed, even in the absence of base, (E)-cinnamamide (482) partially isomerizes into (Z)-cinnamamide, which is positioned for annulation.
Scheme 113.
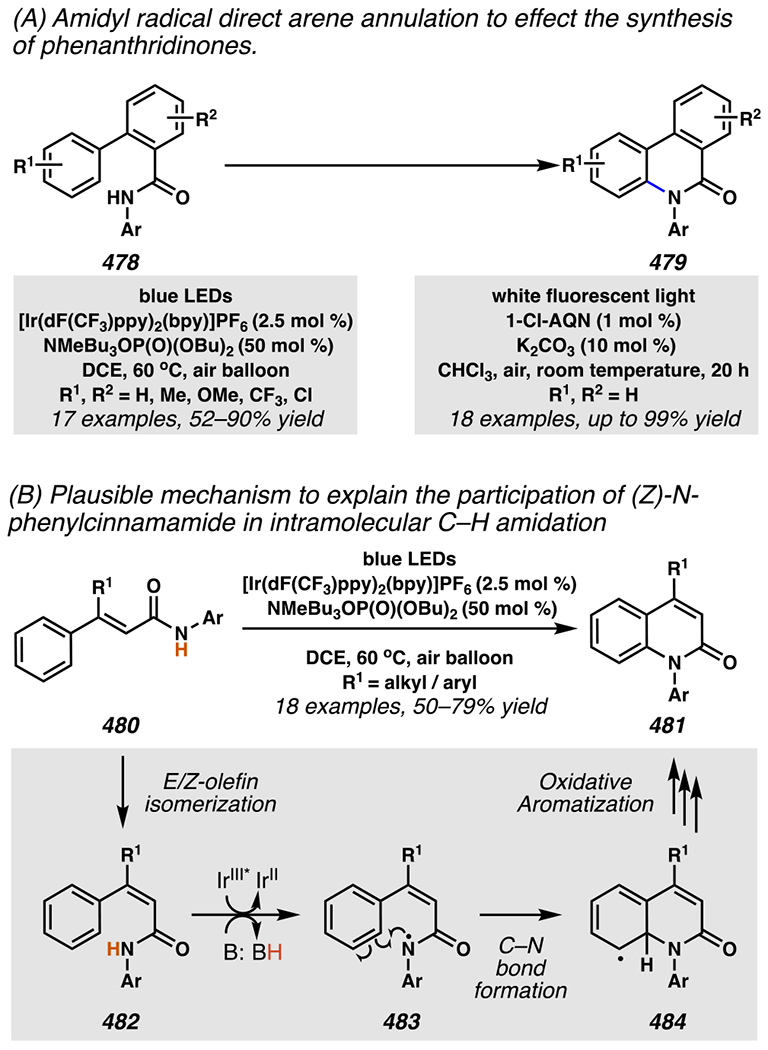
A Cascade Sequence Accesses Phenanthriodinnes and Quinolinones in a PCET-mediated Process
Building upon this PCET-based approach to amidyl radical formation,322, 328 Knowles and co-workers launched a method for a directed site-selective Hofmann-Löffler-Freytag-inspired 67, 307, 345, C–H functionalization reaction (Scheme 114).9 By analogy to the Knowles’ laboratory’s olefin hydroamidation process, this pioneering transformation begins with the PCET-enabled conversion of amide 485 to amidyl radical intermediate 486. Evidence to support the proposed PCET activation mechanism comes from luminescence quenching experiments and prior pKa measurements.346 The generated nitrogen-centered radical is poised for C–H abstraction through a six-membered transition state via a 1,5-hydrogen-atom transfer process (1,5-HAT), which has been well-established in the context of Hofmann-Löffler-Freytag reactions.307 The resultant nucleophilic alkyl radical (i.e., 487) is trapped by intermolecular conjugate addition to olefin acceptor 489. In the proposed reaction mechanism, reduction of this electrophilic radical with IrII, and subsequent proton transfer afford alkylated product 491.
Scheme 114.
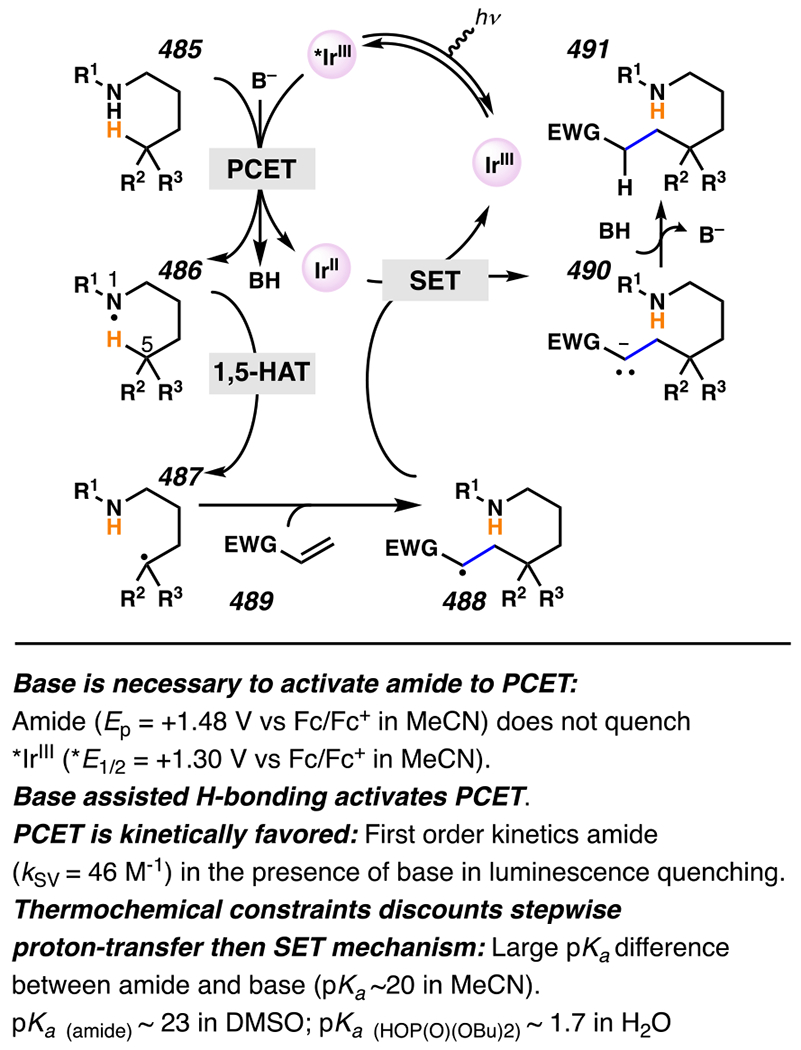
PCET Provides Access to Amidyl Radicals That Guide Alkylation of Remote C–H Bonds
6.2.2. Amidyl Radicals Formed by PCET Engage in Giese-Type Remote Alkylation Reactions
These Giese reactions rely on mild conditions that are compatible with a broad range of functional groups, and can be directed by aryl amides, sulfonamides and carbamates, albeit in slightly lower yields when carbamates guide the reaction (Scheme 115A). These reactions are most effective when directed at tertiary centers, and less efficient when guide to secondary centers. Furthermore, this mechanism is viable as a strategy to affect intermolecular Giese reactions, using a limiting quantity of olefin (Scheme 115B).
Scheme 115.
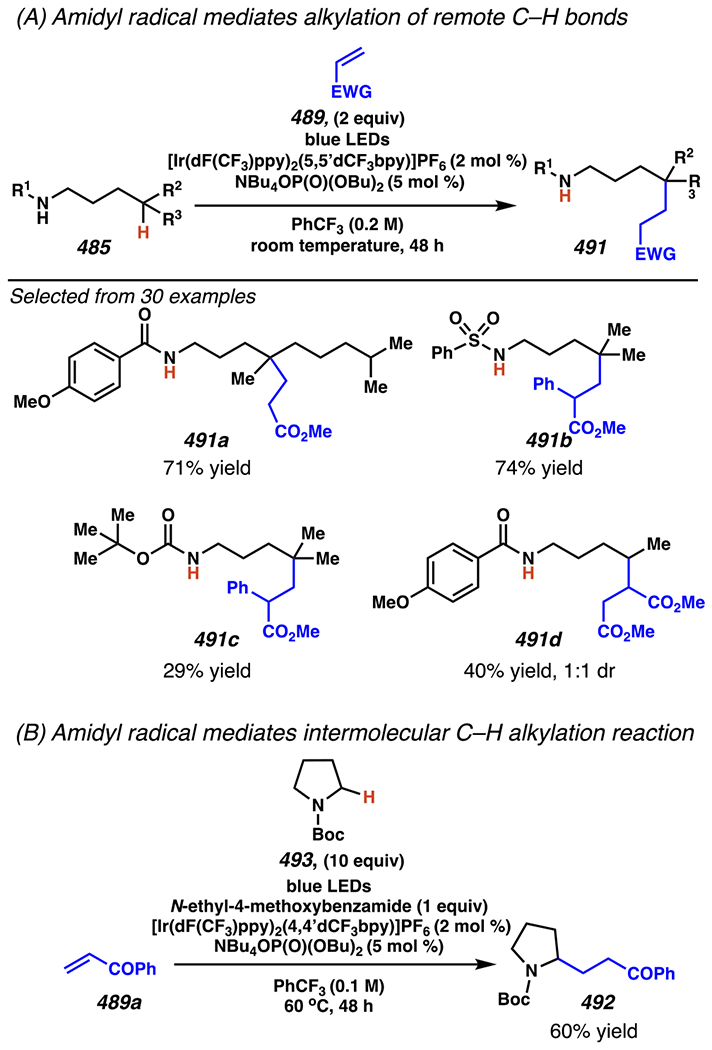
Amidyl Radicals Mediate Alkylation of Remote C–H Bonds
Following these pioneering studies,9, 322, 328 and related investigations by Rovis and co-workers (vida supra),353, 354 Meggers and co-workers have identified synergistic possibilities between their approach to chiral-Rh(III)-based, Lewis acid-mediated asymmetric catalysis347 and Knowles’ and Rovis’ strategies for accessing amidyl radicals to develop an enantioselective, amide-guided C–H bond alkylation reaction.348 Under the optimized conditions, an in situ-generated amidyl radical engages in 1,5-HAT to guide the formation of an alkyl radical. Once formed, the proposed model for asymmetric induction involves radical-radical coupling between the generated alkyl radical and a persistent carbon-centered radical. The requisite persistent radical is proposed to form at the β-position of an α,β-unsaturated carbonyl compound that is coordinated to an enantioenriched rhodium complex, following reduction. In this model, the rhodium complex induces asymmetry at the reactive center (Scheme 116). These reactions rely on α,β-unsaturated 2-acyl imidazoles to generate the rhodium-coordinated complex, and benzamides as amidyl radical precursors, and are sensitive to steric encumbrance during the key bond-forming event. These reactions are ineffective for reactions at secondary centers, but with more activated centers afford products in moderate to good yields with high levels of stereoinduction.
Scheme 116.
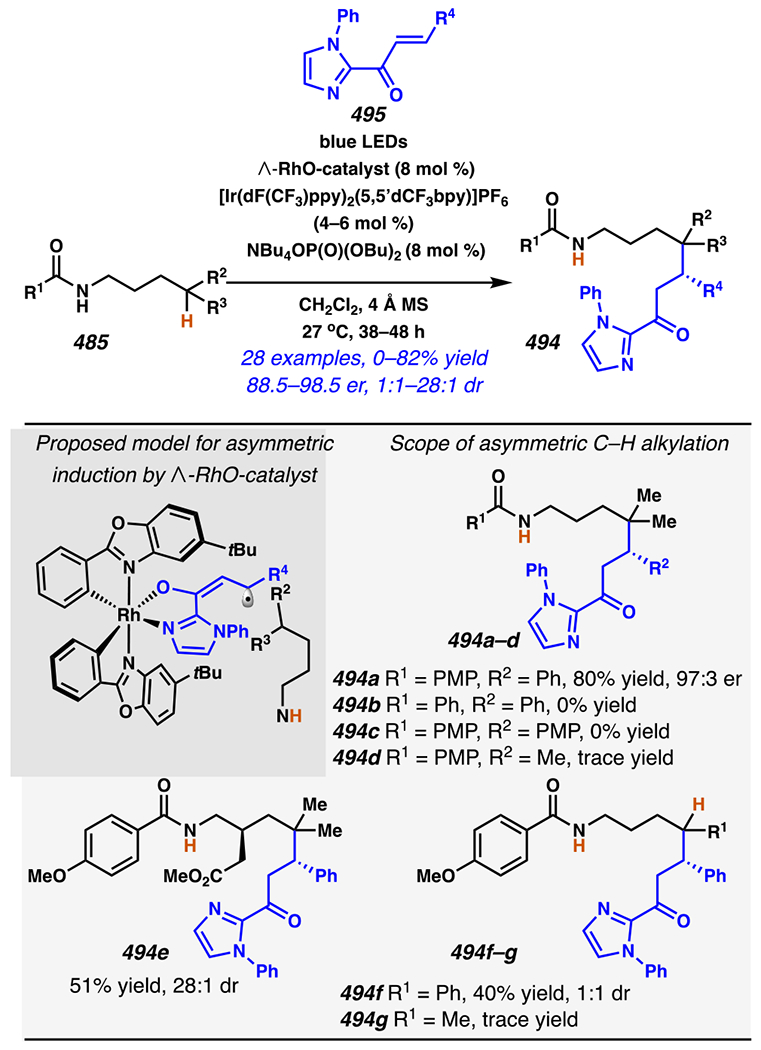
Asymmetric Alkylation of Remote C–H Bonds Proceeds Based on Amidyl Radical Directed 1,5-HAT and Chiral Rh-Controlled Asymmetric Induction
An advantage arises from the limitations of this directed reaction – as they are sensitive to steric encumbrance during the key bond-forming event, and ineffective in directed Giese reactions at secondary centers, carbamoyl radicals that are not positioned to react efficiently in directed reactions can engage directly in asymmetric intermolecular reactions with α,β-unsaturated β-aryl 2-acyl imidazoles to install C–N bonds (Scheme 117).349 By analogy to the C–C bond-forming reactions, the model for asymmetric induction involves radical-radical coupling – in this instance between the generated carbamoyl radical and a persistence carbon-centered radical. Once again, the invoked persistent radical is proposed to form at the β-position of an α,β-unsaturated carbonyl compound that is coordinated to an enantioenriched rhodium complex, following reduction, and the rhodium complex is expected to induce asymmetry at the reactive center (Scheme 117). The optimized conditions benefit from enantioenriched Δ-RhO350, relative to catalysts Δ-RhS351 and Δ-IrO352, which do not promote efficient reactions. Interestingly, absent iridium photoredox catalyst, the reaction proceeds in high enantioselectivity and 39% yield, such that the role of iridium photocatalyst in this reaction may be less clear. Consistent with prior research, N-alkyl carbamate 497b, and amide 497d are not ideal nitrogen-centered radical precursors for this transformation, and β-alkyl enone 497c does not react to furnish much product. Otherwise, this enantioselective reaction tolerates an impressive range of carbamates and α,β-unsaturated 2-acyl imidazoles to give chiral β-amino acyl imidazoles in moderate to good yields as well as in high enantioselectivity.
Scheme 117.
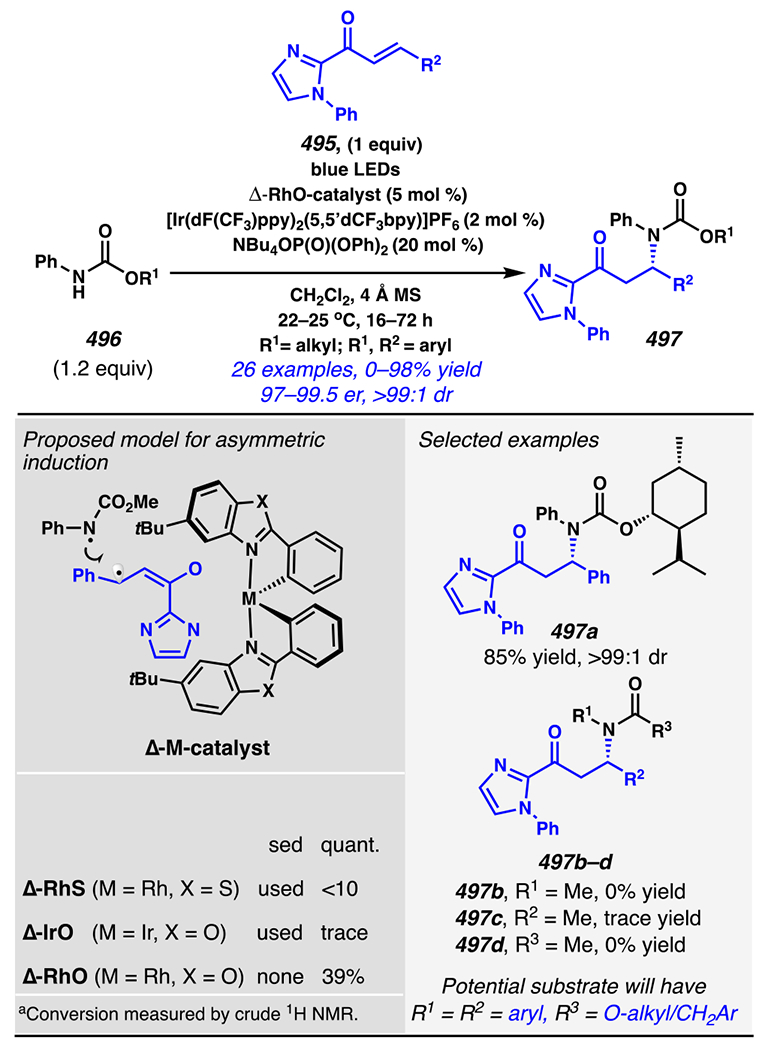
The PCET Enabled Carbamyl Radical Participates in Intermolecular Chiral Rh-Catalyst Controlled Enantioselective β-Amination Reaction
Visible light-mediated PCET technologies are well-developed in other contexts, but have only recently emerged as direct strategies to access to amidyl radical intermediates. This PCET-mediated protocol serves as a useful alternative to the traditional strategies for generating amidyl radicals which rely on the N-prefunctionalization of amides or processes that involve an excess of oxidizing agents or harsh reaction conditions. As initial hydrogen bonding facilitates the PCET process, the chemoselective activation of an amide in the presence of relatively weaker H-bonding thiol can be efficient under PCET reaction conditions. The mild nature of the PCET process enables efficient reactions involving simple to complex small molecules, and even those incorporating otherwise oxidatively labile functional groups.
6.2.3. Amidyl Radicals Form from Amides via Sequential Proton Transfer and Single Electron Transfer (SET) Processes
Concurrent with Knowles and co-workers’ pioneering directed Giese reactions,9 the Rovis laboratory reported a similar strategy to access amidyl and carbamoyl radicals, and enter the same cascade of steps to affect C(sp3)–H bond alkylation.353,354 Critically, the Rovis’ and Knowles’ strategies rely on complementary mechanisms to form amidyl radicals. While Knowles’ protocol relies on a concerted PCET process wherein an amidyl N–H bond relies on hydrogen bonding with a weak phosphate base (∆pKa ≈ 20) to activate the amide to photocatalyzed oxidation, the Rovis laboratory accesses amidyl radicals via sequential deprotonation of acidic amidyl N–H bond with an appropriately strong base (∆pKa ≈ 1.4) with single-electron-transfer-based amidate oxidation (Scheme 118B). With acidic amide and carbamate substrates, deprotonation by K3PO4 is thermodynamically favorable. Subsequent oxidation of the resultant anion with Ir-photocatalyst is kinetically and thermodynamically feasible, as demonstrated by reduction potentials and Stern-Volmer analyses.
Scheme 118.
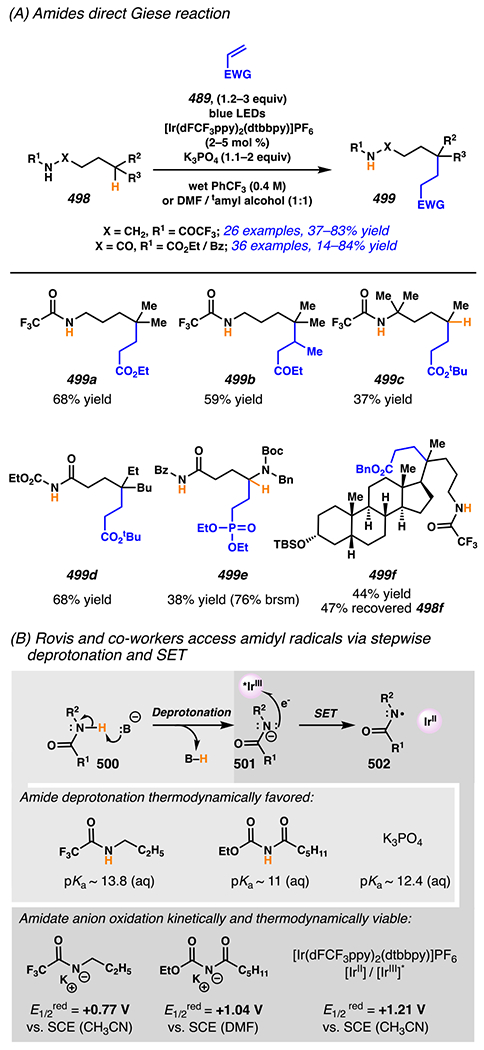
Sequential Proton Transfer and SET Can Provide Access to Amidyl Radicals That Guide Giese Reactions at Unactivated C(sp3)–H Bonds
Rovis’ Giese reactions enables the functionalization of remote tertiary C–H bond of simple to sterically decorated amides/carbamates with a range of electron-deficient alkene acceptors to give products 499a–499f with high efficiency (Scheme 118A). This protocol accommodates the methylene C–H bond of amides/carbamates 499c in lower efficiency. These conditions enable amide alkylation with simple acrylate or acrylamide radical acceptors, which are not tolerated using Knowles and co-workers’ conditions. Consistent with directed 1,5-HAT, steroid-derived acetamide 498f is selectively functionalized at proximal position to N–H bond though multiple distinct tertiary C–H bonds as well as heteroatom activated C–H bonds present in the core.
6.2.4. Catalytic Quantities of Photoactive Organic Compounds Can Engage Weinreib Amides in Radical-Mediated Reactions
With the popularity of Knowles’ and Rovis’ technologies, techniques have emerged that employ organic photoredox catalysts in radical-mediated reactions involving Weinreib amides as substrates which is particularly remarkable as they incorporate relatively weak N–O bonds (Scheme 119,355 BDE of N–H = 90 kcal/mol).304 In this report, an array of methoxyacetamides or benzamides to participate in the intermolecular amidation of primary, secondary, or tertiary benzyl positions in moderate to good efficiency. In a related reaction, DDQ has been proposed as a photocatalyst to mediate the reaction between Weinreib amides and enol ethers (Scheme 120).356
Scheme 119.
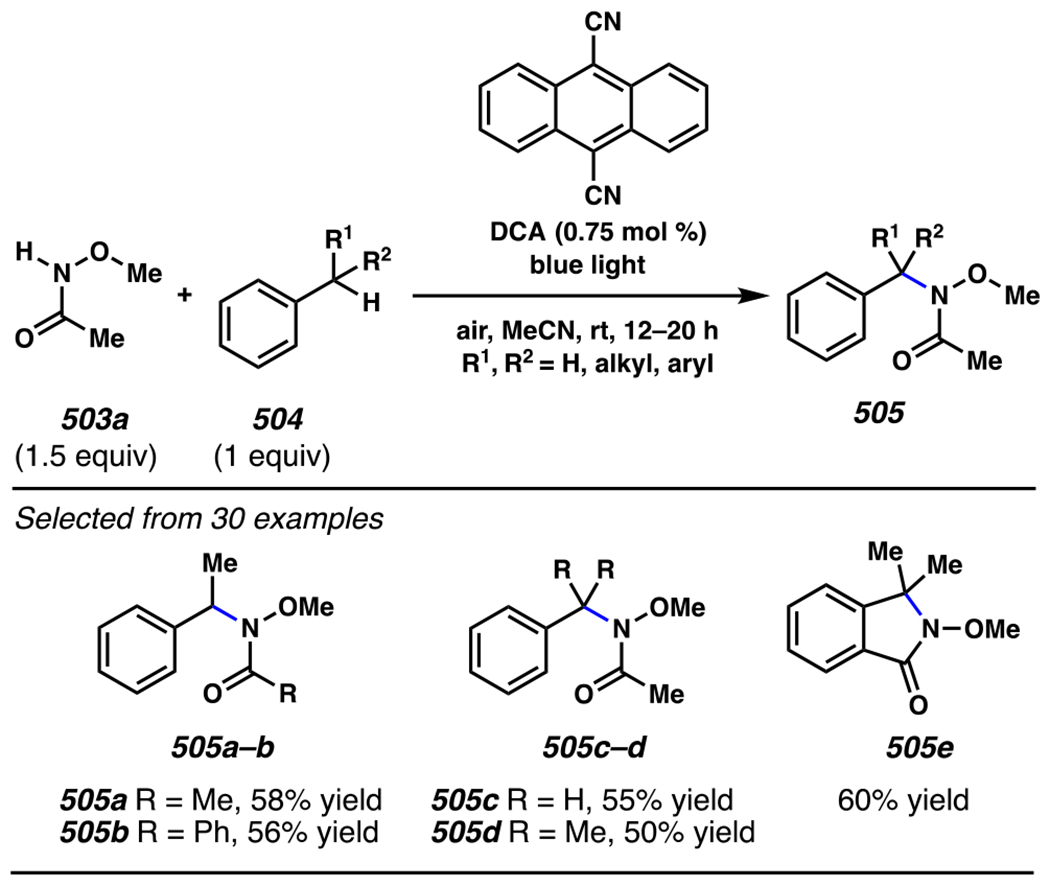
DCA Mediates Radical Reaction Involving Weinreb Amides
Scheme 120.

DDQ-Mediated Radical Reaction Involves Weinreb Amides as Substrates
Overall, photocatalyst-mediated processes surmount the use of strong oxidants, N-functionalized precursors, or harsh reaction conditions during the formation of key amidyl radical intermediates. These mild protocols allow oxidatively sensitive substrates to serve as precursors to amidyl radicals, which can participate in efficient intramolecular alkene carboamination/hydroamination reactions, intra- or intermolecular C–H amidation processes, and guided alkylation reactions involving remote C–H bonds.
6.3. Nitrogen-Centered Sulfamyl Radicals Can Be Prepared from Sulfonamides and Sulfamides
Following their success in both intermolecular hydroamination of alkenes using amines (vide supra, Schemes 94–95)290and intramolecular hydroamination of alkenes by PCET in amides (vide supra, Schemes 106–109),322, 328, 329, 330, 331Knowles and co-workers demonstrated their PCET protocols to engage sulfamyl nitrogen-centered radicals, primarily derived from sulfonamides, in both inter- and intramolecular hydroamination reactions (Scheme 121).331 In the presence of catalytic amounts of strongly oxidizing iridium photocatalyst, phosphate base, and TRIP thiol under visible light irradiation, unsubstituted and mono-substituted sulfonamides 508 engage in intermolecular, anti-Markovnikov addition across unactivated alkenes. The same reaction conditions prove applicable to 5-exo-trig cyclization reactions with sulfonamides bearing a wide array of aryl and alkyl substituents. Of particular note, sulfamide 510 cyclizes onto pendant mono- and tri-substituted alkenes in excellent yields. Sulfamides are a less well-known directing group357 but have been developed substantially over the past several years, 56,358,359 and structurally diverse sulfamides remain highly sought-after in synthetic targets.360
Scheme 121.
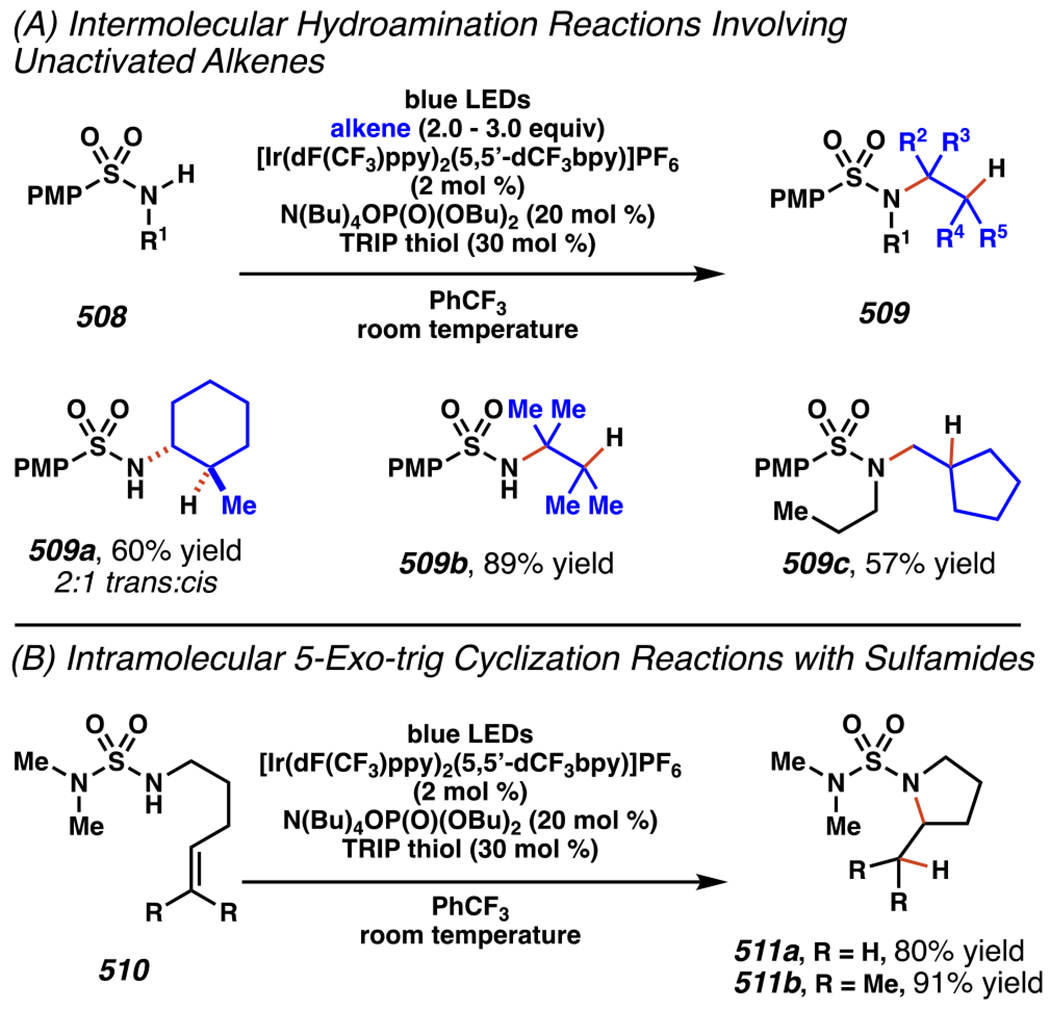
Photoredox Conditions Enable (A) Intermolecular Hydroamination Reactions Involving Unactivated Alkenes and (B) Intramolecular 5-exo-Trig Cyclization Reactions with Sulfamides
Building upon this approach to generate sulfamyl radical intermediates, Knowles and co-workers have developed an enantioselective, intramolecular hydroamination reaction that employs bulky BINOL-derived phosphate base to induce enantioselectivity (Scheme 122).361 At decreased temperatures and in α,α,α-trifluorotoluene, the reaction generates the requisite sulfamyl radical, in substrates of form 512, through PCET with a strongly oxidizing iridium-photocatalyst and phosphate base, also including TRIP thiol as an HAT-cocatalyst (vide supra). Phenol-derived sulfamate ester 513a is accessed in very good yield with 92:8 er, while a cyclobutane-containing sulfonamide 513b is accessed in 96% yield with 95:5 er. As demonstrated by product 513c, the procedure is somewhat sensitive to alkene isomer employed, with yield decreased by 10% when employing a cis-alkene rather than a trans-alkene and displaying a slight erosion of enantioselectivity in the same comparison. This process is relatively mild, and tolerates additional, distal olefins in the substrates, as demonstrated by unsaturated 513d which is synthesized in very good yields with a 96:4 er and 1.5:1 dr.
Scheme 122.
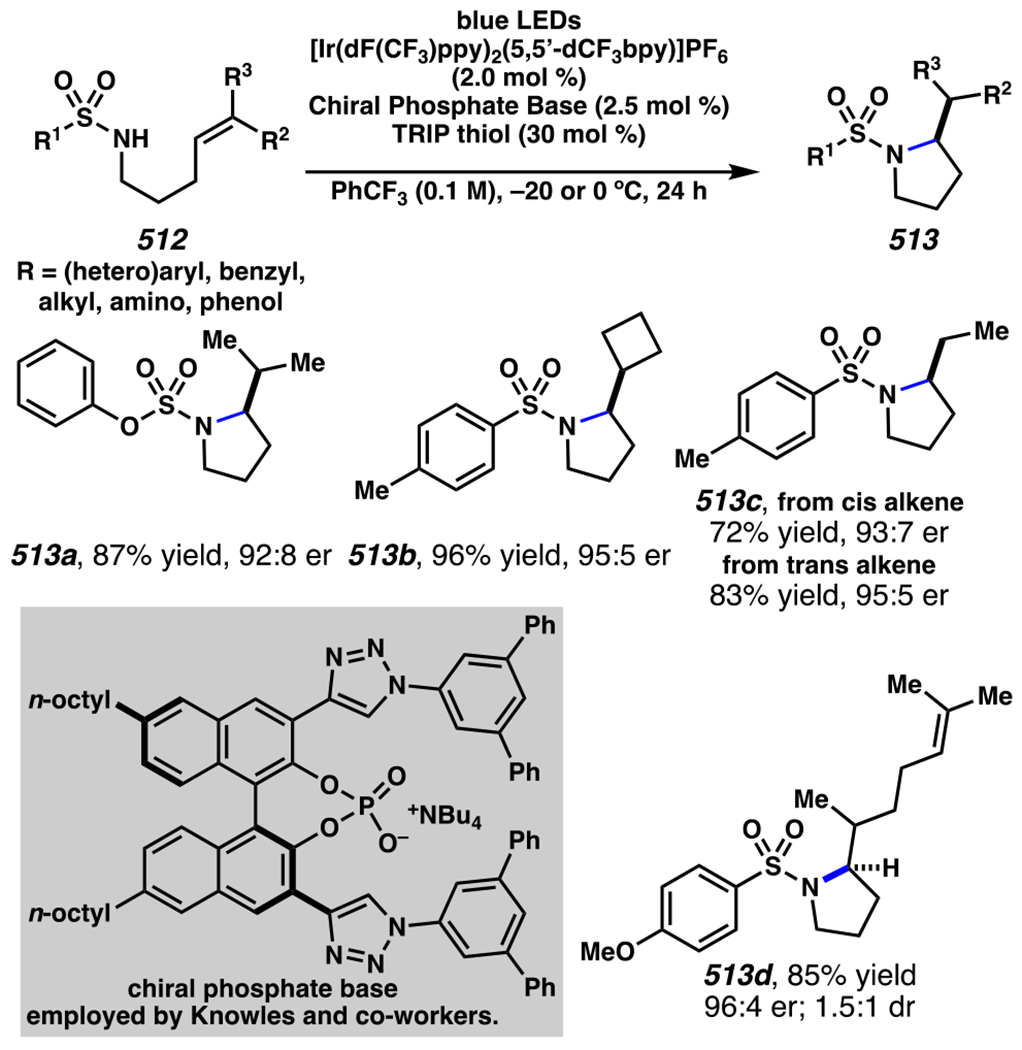
Chiral Phosphate Bases Enable Enantioselective Intramolecular Cyclization Reactions Involving Sulfamyl Radicals
While C–C bond cleavage is not a new concept in synthesis,362,363 trifluoromethanesulfonamides were first reported as competent in carbon to nitrogen aryl migrations by Shi and co-workers in 2015.364 While this initial disclosure employs silver reagents with an excess of strong oxidant at high temperatures, in 2017 Nevado and co-workers published a related method that relies on relatively mild, photoredox conditions (Scheme 123).365 Under blue light irradiation and in the presence of a phosphate base and iridium-based photocatalyst, γ,γ-diphenyl-trifluoromethanesulfonamide 514a reacts to produce nitrogen-centered radical 517, likely by deprotonation and subsequent oxidation by photoexcited [Ir]. The sulfamyl radical can then proceed through spirocyclic transition state 518 which produces the new N-aryl bond and γ-disposed carbon-centered radical through concomitant radical C–N bond formation and C–C bond cleavage. Nevado and co-workers demonstrated the proposed γ-disposed carbon-centered radical 519 could be functionalized by several radical coupling partners, especially phenylsulfone Michael acceptor 515 to give N-aryl γ-alkylated product 516a in good yield. The phenylsulfonyl radical is then putatively reduced to the corresponding anion to close the photocatalytic cycle. Consistent with this mechanistic proposal, an α,α-dimethyl sulfonamide reacts in reduced yield but gave as a byproduct γ-homocoupled disulfonamide 522, the structure of which was confirmed by crystallographic analysis; the most reasonable mechanistic explanations of this byproduct formation would proceed through carbon-centered radicals. Additionally, sulfonamide 514b, which has electronically distinct aryl groups, is representative of Nevado and co-workers observation that more electron-rich aryl groups preferentially migrate (Scheme 124), as would be expected when mediated by electron-deficient sulfamyl radical 517. Notably, by changing the base and replacing the acrylate coupling partner with a thiol as a proton-source or a bromine radical precursor, Nevado and co-workers are able to install a proton or a bromide at the γ-carbon (Scheme 125).
Scheme 123.
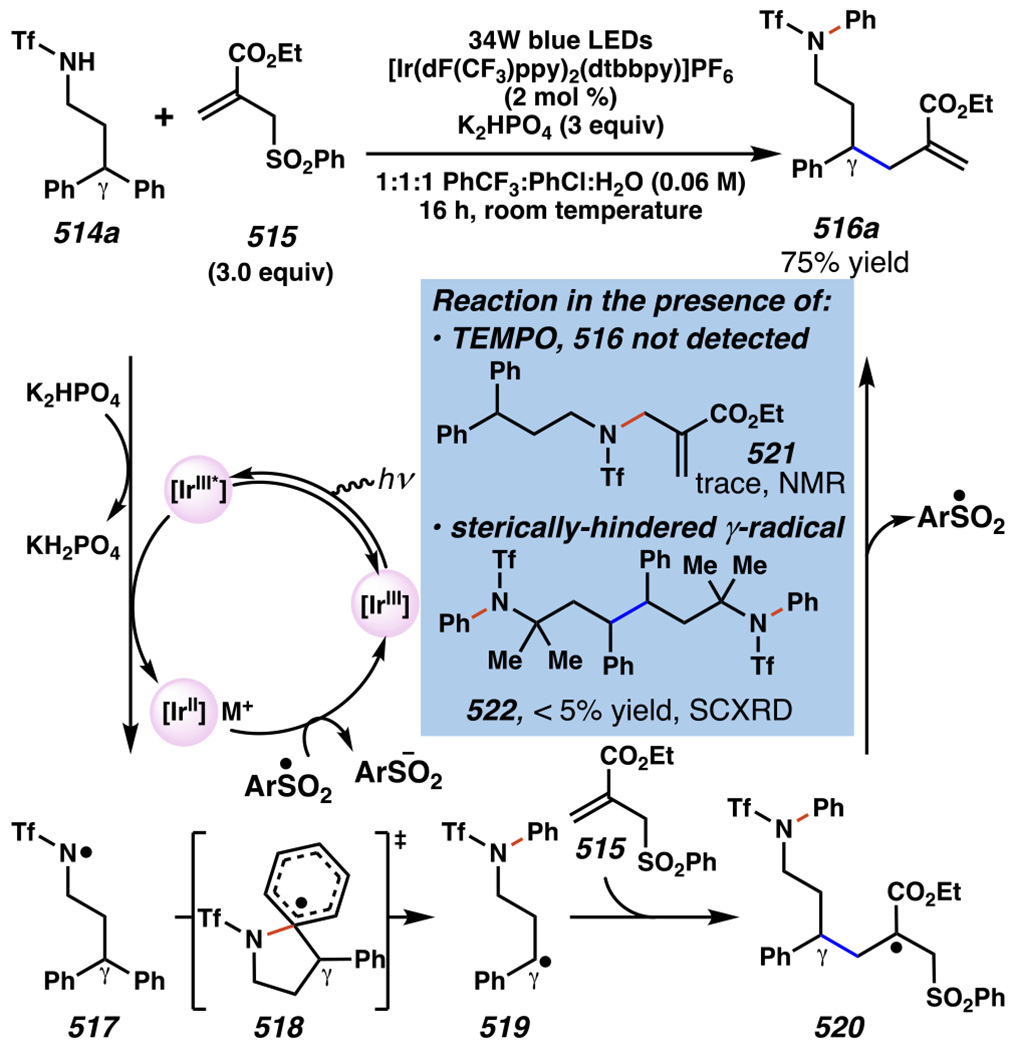
Photoredox Catalysis Enables γ-C(sp3) Functionalization through Aryl C–N Migration, Likely Mediated by N-Centered Sulfamyl Radicals
Scheme 124.
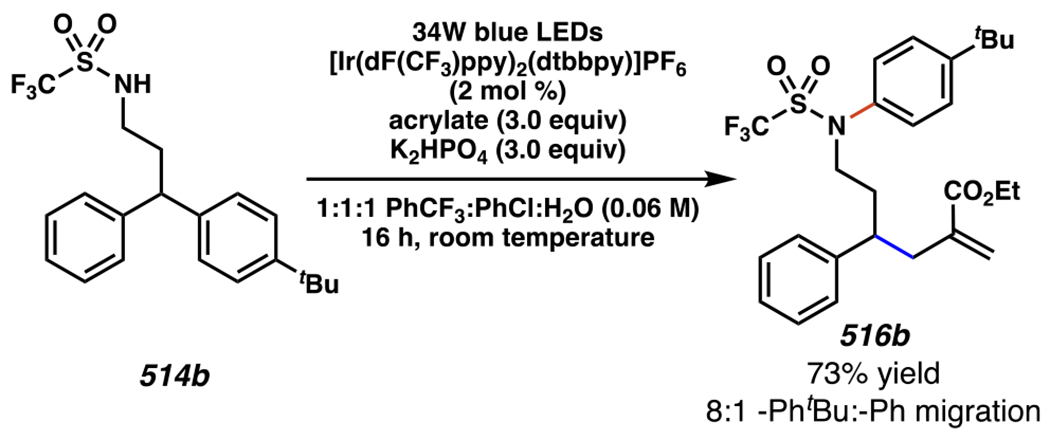
Aryl Migration Is Strongly Biased toward More Electron-Rich Aryl Groups, as Would Be Expected in a Radical Mediated Process
Scheme 125.
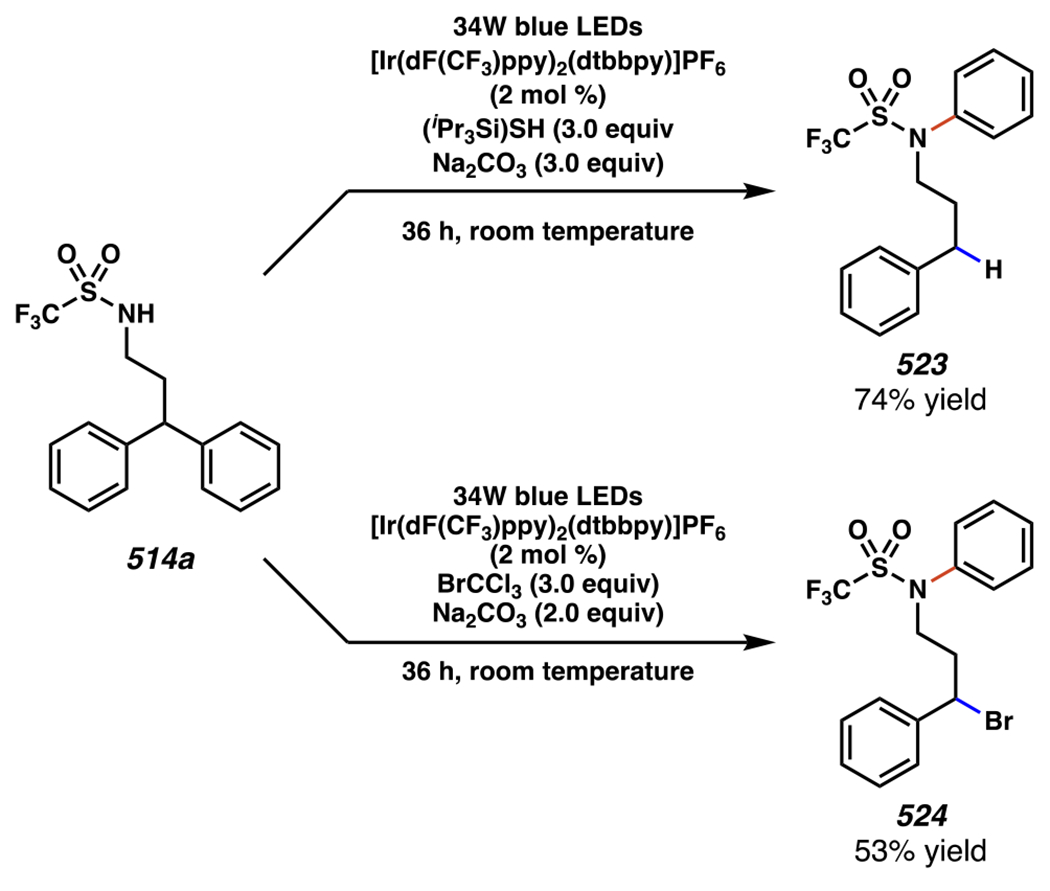
Closely Related Conditions Enable γ-Protodearylation and γ-Bromodearylation Processes
More recently, sulfonamide-guided heteroaryl migration reaction have been developed in conjunction with alcohol oxidation processes, which rely on silver complexes as radical trapping agents (Scheme 126).366 Under strong blue light irradiation at an elevated temperature, a combination of 10-phenylphenothiazine (Ph-PTZ) as photocatalyst and excesses of silver carbonate and monobasic potassium phosphate in 1,4-dioxane enabled the ipso-migration of benzylic heteroarenes, containing two adjacent heteroatoms at tertiary benzylic alcohols to form a new C–N bond and a ketone in an arene migration/oxidation process. The reaction tolerates variation in the sulfonamide moiety and is selective for migration of the electron-dense heteroarenes over other aryl groups.
Scheme 126.
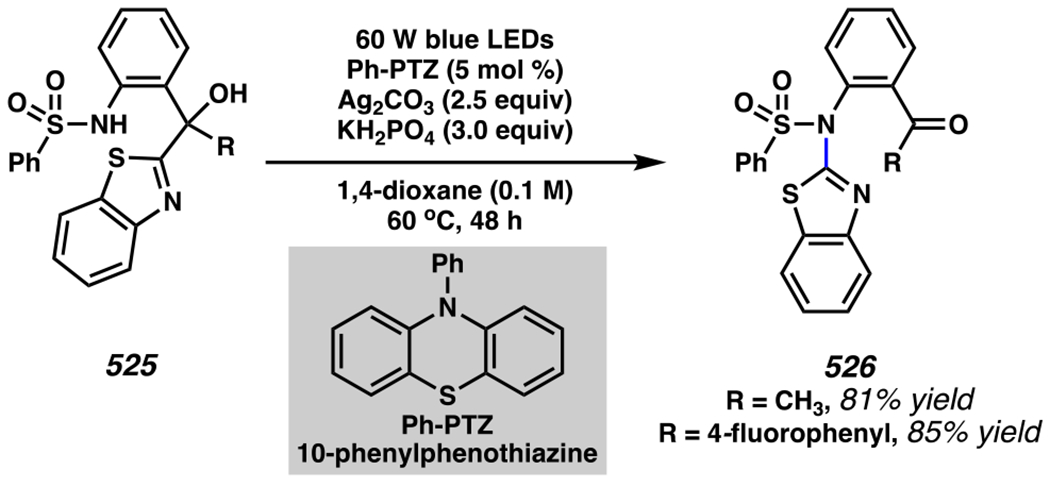
Photoredox Catalysis Enables Remote Heteroaryl Migration from Benzylic Alcohols
Sulfonamides are important functional groups in medicinal and agrochemical chemistry, and their application in mild, light-driven processes further bolsters their usefulness to the larger chemical industry. Though nitrogen-centered radical generation from the N–H bond of sulfonamides is less well-explored, recent research, PCET-enabled sulfonamide activation is amenable to a breadth of potential applications, from reactions involving heteroaryl migration to processes involving enantioselective cyclization. Moreover, the broader adoption of PCET-type activation of N–H bonds may enable a more straightforward application of sulfonamides as directing groups for reactions that have previously been developed using substrates with pre-functionalized nitrogen centers (vide supra: Sections 2-3).
6.4. Sulfamate Esters Can Direct Photochemically-Mediated Giese Reactions, Likely Guided by Nitrogen-Centered Sulfamyl Radicals
Following the development of sulfamyl radical-guided halogen72, 367 and group transfer reactions,110, 368, 369 Roizen and co-workers,370, 371, 372 Duan and co-workers,373 and Shu and co-workers374 each disclosed methods that, under photochemical conditions, directly engage sulfamate esters375 to guide Giese reactions (Scheme 127). The position-selectivity in these reactions is noteworthy as it complements the selectivity available through other known, radical-mediated C–H functionalization processes. These reactions reliably engage otherwise unactivated secondary and tertiary γ-C(sp3)–H bonds in the presence of more electronically activated C(sp3)–H centers, such as the (ω−1)-C–H bond in substrate 527.
Scheme 127.
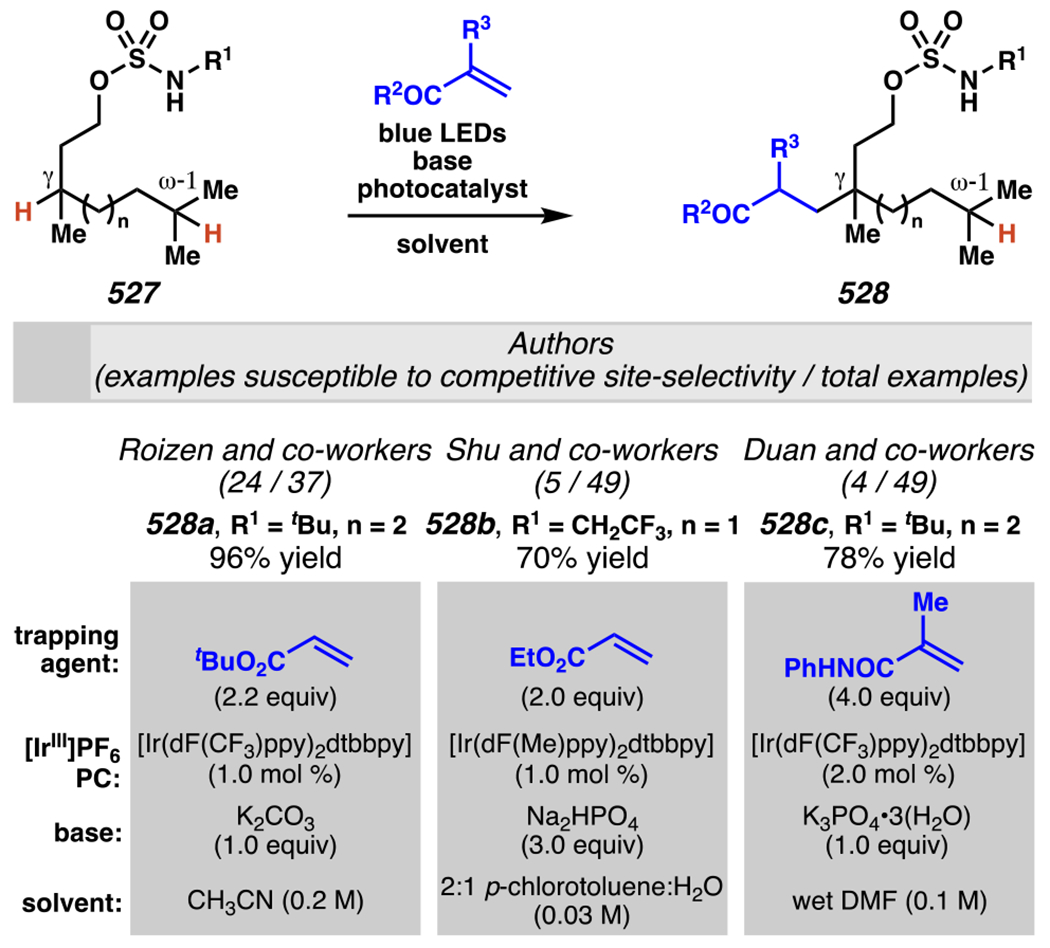
Roizen and Co-workers, Shu and Co-workers, and Duan and Co-workers Develop Sulfamate-Ester Guided Giese Reactions to Selectively Functionalize γ-C(sp3)–H Bonds
While there is insufficient data to elucidate a full mechanism, key aspects of the proposed mechanism are supported by the available information, in particular the sufficiency of the photocatalyst to engage in the proposed electron transfers and the intramolecular, guided proton transfer (Scheme 128). These reactions are proposed to involve initial conversion of sulfamate ester 527a to sulfamyl radical 529. A detailed understanding of the mechanism leading to initial generation of sulfamyl radical 529 cannot be ascertained from current data as both proton-coupled electron transfer (PCET) and serial proton transfer (PT)/single electron transfer (SET) mechanisms are plausible, and there is not yet direct experimental evidence for a nitrogen-centered radical intermediate. Evidence suggests that sequential deprotonation to form an anion and subsequent SET would be both kinetically and thermodynamically viable. Specifically, Roizen and co-workers reported that the electronically similar propyl N-tert-butyl sulfamate anion 532 has a half-peak oxidation potential (Ep/2) of 0.778 V vs. SCE (in CH3CN), so its oxidation would be thermodynamically feasible if driven by photoexcited [Ir] (E1/2III*/II = +1.21 V vs. SCE in CH3CN).370 Once generated, sulfamyl radical 529 would engage in a 1,6 hydrogen-atom-transfer (1,6-HAT) process to produce a carbon-centered radical. This is proposed to be the position-selectivity-determining step and reaction outcomes are consistent with this directed process even when substrates are likely to be subject to alternative (unguided) C–H functionalization based on competitive site-selectivity (cf. Scheme 127). For example, tetrahydrogeraniol-derived substrate 527a reacts at the γ-C–H bond, with no evidence of oxidation of the weaker, more electron-rich (ω−1)-C–H bond, supporting the proposed mechanism as both selective and intramolecular. This 1,6-HAT process is likely rate-determining based on the primary kinetic isotope effect (KIE; kH/kD) of 4.69 as measured in parallel experiments.374 Once this carbon-centered radical is generated, nucleophilic radical addition across an electron-deficient olefin produces an ester-stabilized radical 531, which should readily be reduced by the photocatalyst (E1/2III/II = −1.37 V vs. SCE in CH3CN) and subsequently protonated intermolecularly374 to furnish alkylated sulfamate 528a. Notably, while sulfamates enantioenriched at the site of functionalization are converted to racemic products, sulfamates recovered from these reactions retain their initial enantioenrichment, indicating both that the carbon-centered sulfamyl radical 530 is generated irreversibly and has a radical lifetime sufficient for epimerization to occur (Scheme 129).
Scheme 128.
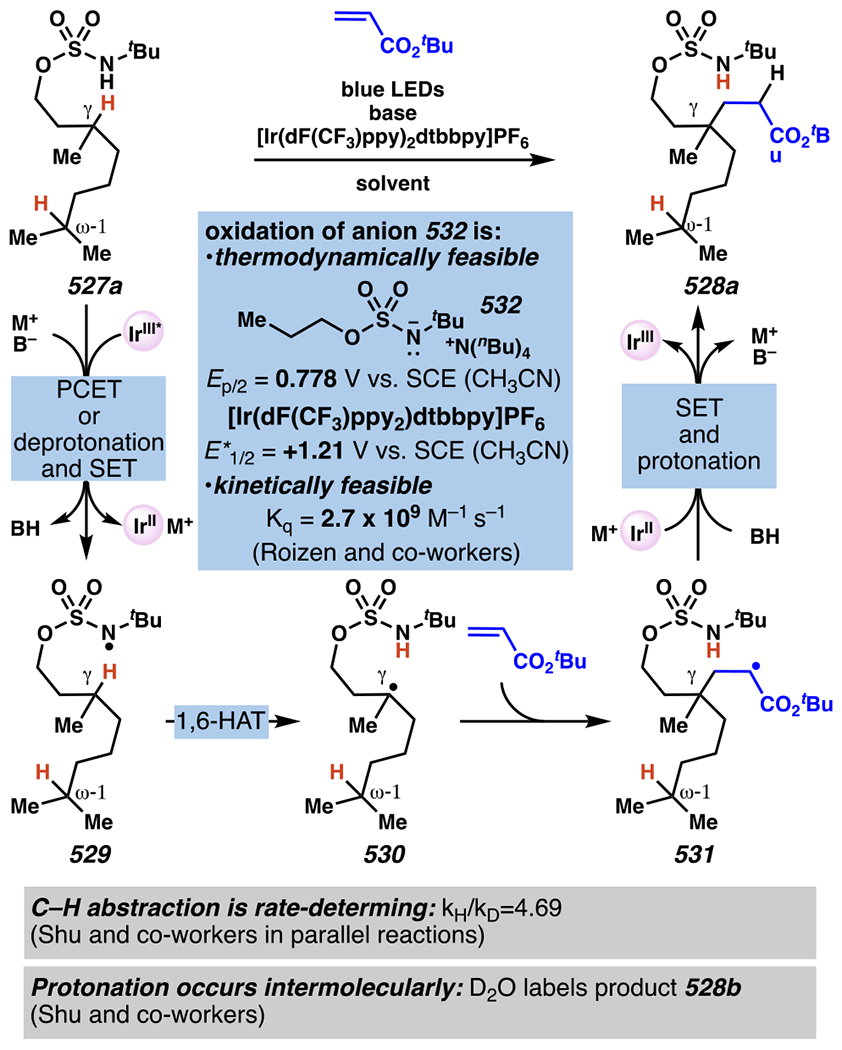
In the Proposed Mechanism, Sulfamyl Radicals Engage in a 1,6-HAT Process to Generate Carbon-Centered Radicals Capable of Trapping Electron-Deficient Alkenes
Scheme 129.
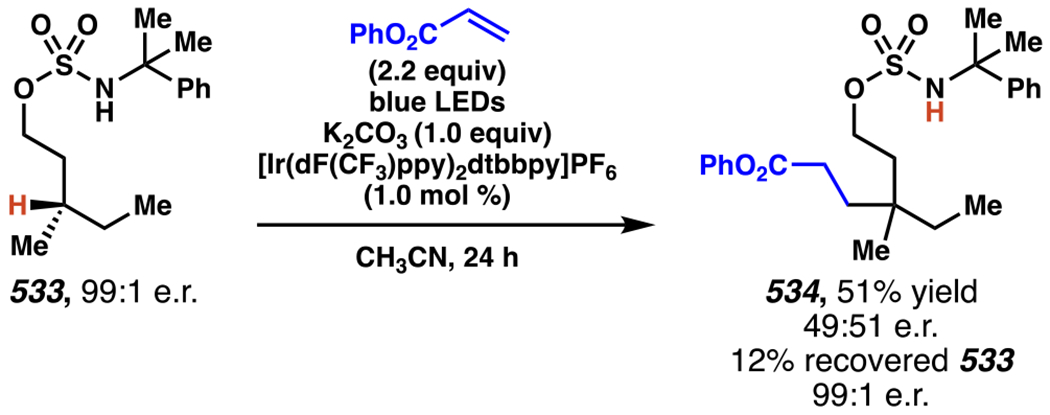
Under Conditions Developed by Roizen and Co-workers, Carbon-Centered Radicals Are Generated Irreversibly
Though a less commonly encountered functional group, the rare γ-carbon selectivity demonstrated by sulfamate ester-derived nitrogen-centered radicals establishes sulfamate esters as an important to the development of C–H functionalization technologies. Furthermore, these reactions are mechanistically interesting, as the exceptionally uncommon 7-membered transition state that is requisite for 1,6-HAT processes. Further development of this γ-carbon selective technology is ongoing.
6.5. Hydrazonyl Radicals Can Be Prepared from Hydrazones
Hydrazonyl radicals are intermediates in organic synthesis that can be easily formed from hydrazones. Intramolecular 5-exo-trig or 6-endo-trig cyclization reactions involving hydrazonyl radicals constitute useful synthetic strategies to prepare pyrazoline, pyrazole, 1,6-dihydropyridazine, and pyradazine analogues. Methods to access hydrazonyl radicals have been developed that rely on the use of copper catalysis,376,377,378 or stoichiometric amounts of oxidants such as azodicarboxylates,379,380 or TEMPO381,382,383 or alkyl peroxides384 (Scheme 130A). By avoiding high temperatures, stoichiometric oxidants, and permissive oxidants, photoredox-mediated processes can convert tosyl hydrazones to hydrazonyl radicals that can participate in 5-exo-trig or 6-endo-trig cyclization reactions to furnish 4,5-dihydropyrazoles,340, 385 or 1,6-dihydropyridazines (Scheme 130B).340 Interestingly, alkene substitution dictates the type of product that forms, with β-unsubstituted or alkyl hydrazones 535a–b furnishing 4,5-dihydropyrazole 536a–b, while β-aryl hydrazones 535c give 1,6-dihydropyridazines 536c (Scheme 130B).340
Scheme 130.
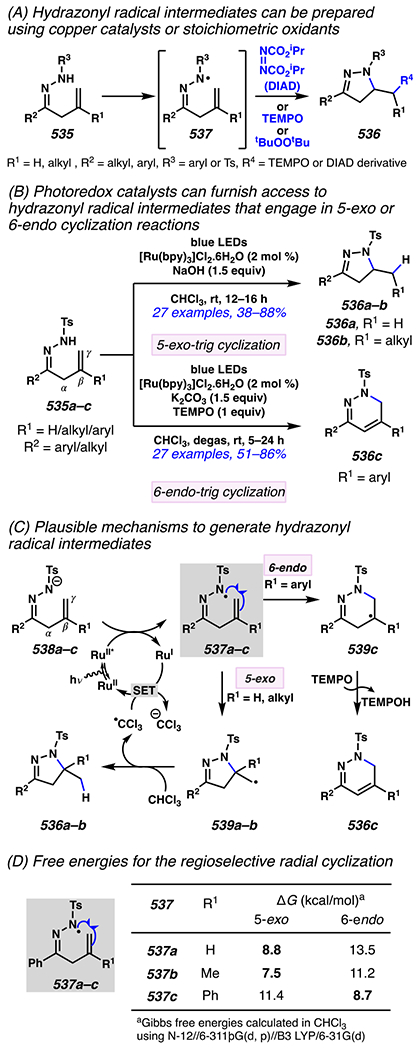
Hydrazonyl Radicals Can Engage in 5-Exo-trig and 6-Endo-trig Cyclization Reactions
A plausible mechanism for this transformation could involve initial deprotonation of the hydrazone by a strong base to afford the hydrazonyl anion 538a–c (Scheme 130C). A single-electron oxidation of hydrazonyl anion (anion 538b, E1/2 = + 0.56 V versus SCE in DMF) by the excited state of the photocatalyst (*RuII, E1/2*II/I = + 0.77 V vs SCE in CH3CN) could afford hydrazonyl radical 537a–c and reduced photocatalyst (RuI). Luminescence quenching experiments reveal that hydrazones 538a–c quench excited photocatalyst only in the presence of base. Subsequently, cyclization can proceed through 5-exo-trig over 6-endo-trig processes. DFT calculations provide insight into the position-selectivity of the cyclization reaction (Scheme 130D), suggesting that, with β-unsubstituted or alkylated intermediates, the activation free energy for 5-exo-trig cyclization of hydrazonyl radicals 537a–b is lower than that for 6-endo-trig cyclization. By contrast, with β-unsubstituted or alkylated intermediates, the activation free energy for 5-exo-trig cyclization of hydrazonyl radicals 537c is higher than that for 6-endo-trig cyclization, presumably because the transition state to access a relatively stable arylic radical, would itself be lower in energy, thereby favoring 6-endo-trig cyclization. Following cyclization, carbon-centered radicals 539a–c could be quenched by a hydrogen atom source or TEMPO to afford products.
To complement this method, it is possible to intercept the generated alkyl radical intermediates to affect net oxyamination reactions (Scheme 131A),386 or net allylation reactions (Scheme 131B).387 For the oxyamination reaction, limited mechanistic investigations are consistent with TEMPO (E1/2 TEMPO•/TEMPO+ = + 0.62 V vs Ag/AgCl) and excited state photocatalyst (*PC, E1/2 = + 2.06 V vs SCE in MeCN) co-operatively catalyzing hydrazonyl radical formation. Once generated, the alkyl radical intermediate intercepts atmospheric oxygen en route to the oxyamination products, as evidenced by an 18O-labeling experiment. This method transforms a series of substituted β,γ-unsaturated hydrazones into its corresponding 4,5-dihydropyrazolyl methanol or 2,3,4,5-tetrahydropyridazinol derivatives in 51–93% yields.
Scheme 131.
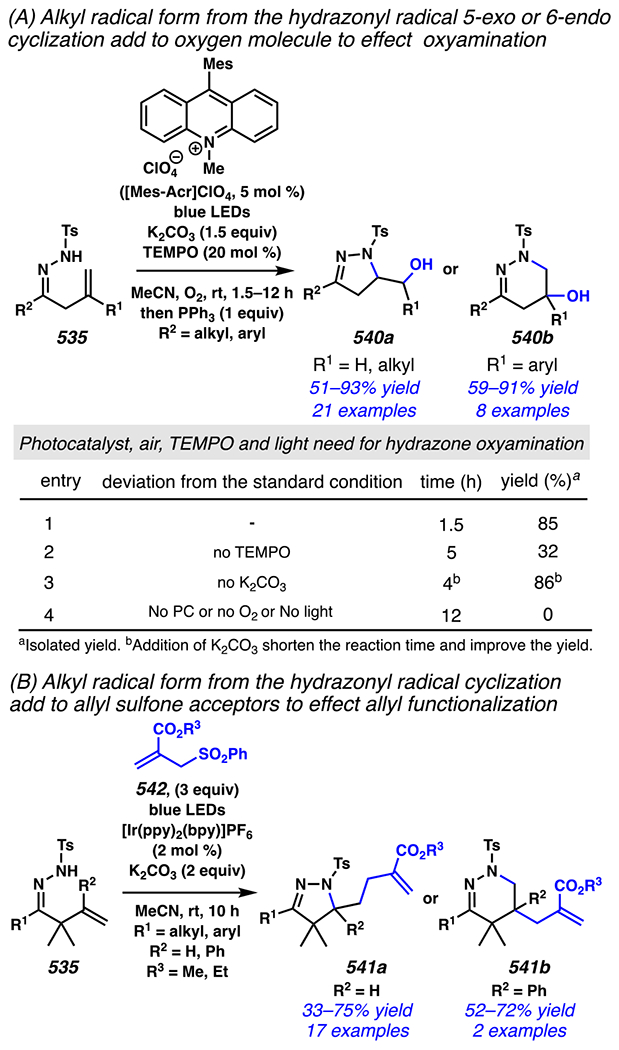
Hydrazonyl Radicals Can Engage In Olefin Functionalization Reactions
With these same substrates, absent an intermolecular radical trapping agent, a hydrazone can be converted to a hydrazonyl radical intermediate, with subsequent cyclization and then undergo a productive intramolecular arylation reaction in the presence of an oxidizing cobalt complex (Scheme 132).388 This research builds on visible-light photoredox/cobalt-mediated reactions disclosed by Lei389,390,391,392 and Wu393,394 research groups. Absent cobalt, hydrazone 535 is converted to unaromatized product 543b. Interestingly, addition of oxidant such as 1,3-dinitrobenzene or tert-butyl perbenzoate provide aromatized product in moderate yield. Gratifyingly, ruthenium/cobalt-mediated reactions furnish aromatized product 543a.
Scheme 132.
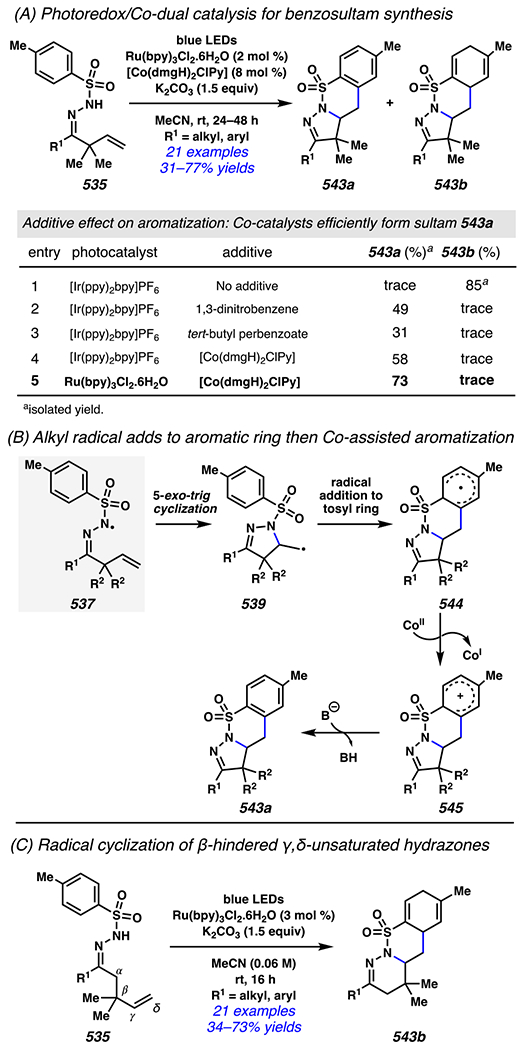
A Sequential Intramolecular 5-Exo-trig Cyclization, Radical Addition, and Aromatization Cascade for the Preparation of Benzosultams
A plausible mechanism for this reaction involves initial converstion of hydrazone 535 to hydrazonyl radical 537 based on deprotonation, and single electron transfer to quench excited state photocatalyst (Scheme 132B). Indeed, electron transfer is a kinetically viable process given relatively rapid luminescence quenching of the excited photocatalyst (RuII*) by hydrazone 535 in the presence of base. Following intramolecular 5-exo-trig radical cyclization by hydrazonyl radical, a generated alkyl radical can add intramolecularly to a pendant aromatic ring to furnish arenium radical 544. In the presence of cobalt, arenium radical oxidation forms arenium cation 545, which is poised for deprotonative aromatization. Under the reaction conditions, alkyl and aryl substituted hydrazones are transformed into the respective benzosultams 543a in moderate to excellent yields. Complementary reactivity can be observed with β,β-disubstituted-γ,δ-unsaturated hydrazones, which engage in a 6-exo-trig cyclization, radical addition to tosyl group and protonation cascade to deliver tetrahydropyridazines 543b in moderate to good yields (Scheme 132C). 395
As a complement to the variety of reactions developed to transform substituted β,γ-alkenyl hydrazones based on intramolecular 5-exo-trig or 6-endo-trig cyclization of hydrazonyl radicals, 6-exo-dig cyclization reaction/Smiles rearrangement396 cascades of hydrazonyl radical intermediates have been invented to transform substrates containing terminal alkynes (Scheme 133).397 A plausible mechanism for this reaction involves base assisted formation of hydrazonyl anion 548 (E1/2 = + 0.54 V vs SCE in EtOH), which is thermochemically capable of being oxidized by excited state photocatalyst (*RuII, E1/2II*/I = + 0.77 V vs SCE in MeCN) to furnish hydrazonyl radical 549. Resultant radical 549 is poised for sequential intramolecular 6-exo-dig cyclization and a radical Smiles rearrangement to form radical species 551. Ultimately, the reduced state photocatalyst (RuI) could reduce alkyl radical 551 to anion 552, which could be protonated by the solvent to yield phthalazine. This method accommodates variety of terminal alkyl or aryl substituted alkynyl hydrazones bearing aryl or heteroaryl migrating groups. Remarkably, authors have also developed an efficient one-pot two-step protocol for the phthalazines starting directly from the corresponding aldehydes and sulfonylhydrazines.
Scheme 133.
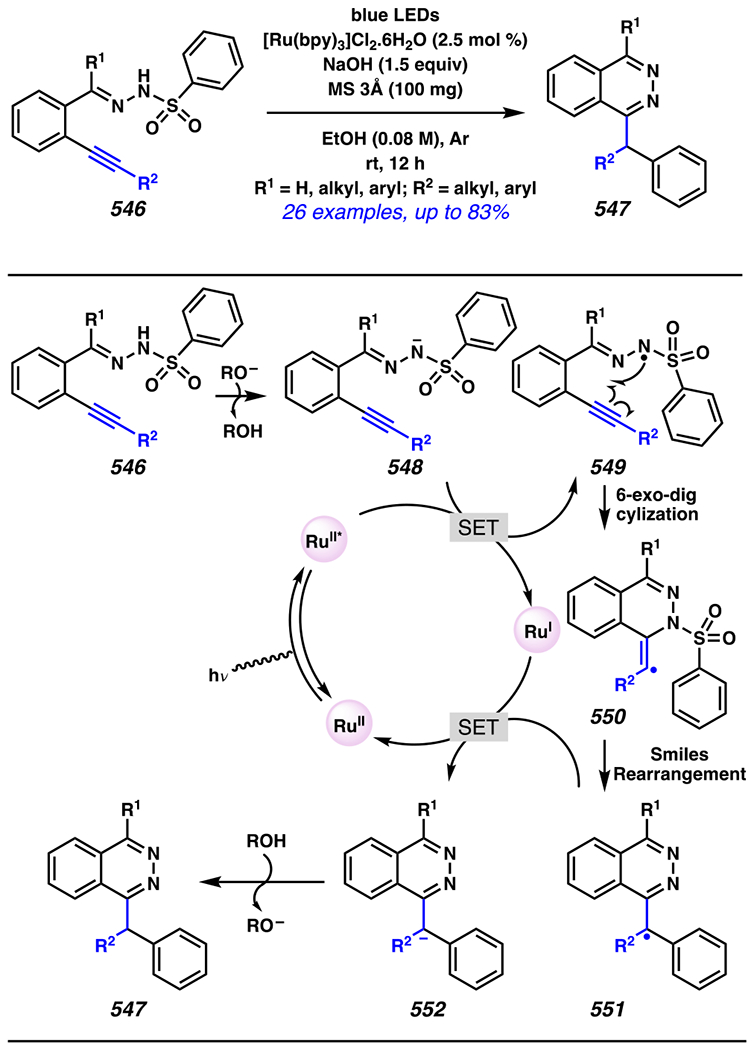
Hydrazonyl Radical Are Proposed to Engage Sequential Intramolecular 6-Exo-dig Cyclization and Radical Smiles Rearrangement
The above described methods to prepare of dihydropyrazoles via hydrazonyl radical cyclization rely on β,γ-unsaturated hydrazones as substrates; however, less elaborate substrates can be viable precursor to dihydropyrazoles based on in situ access to β,γ-unsaturated hydrazones. Recently, Cai and co-workers have used α-bromo hydrazones 553 and β-ketocarbonyls 554 as precursors to dihydropyrazoles (Scheme 134).398 Mechanistically, initial deprotonation of β-ketocarbonyl 554 is followed by nucleophilic displacement of α-bromo hydrazones to provide hydrazonyl anion. In the presence of a visible-light photosensitizer, hydrazonyl anion can be oxidized to hydrazonyl radical 556, which undergoes intramolecular cyclization to give dihydropyrazoles. A hydrazonyl radical intermediate is implicated as, absent photocatalyst, 1,2-dihydropyridazine 557 forms. This method converts a variety of readily accessible alkyl and aryl hydrazones and 1,3-dicarbonyl analogs to furnish dihydropyrazoles.
Scheme 134.
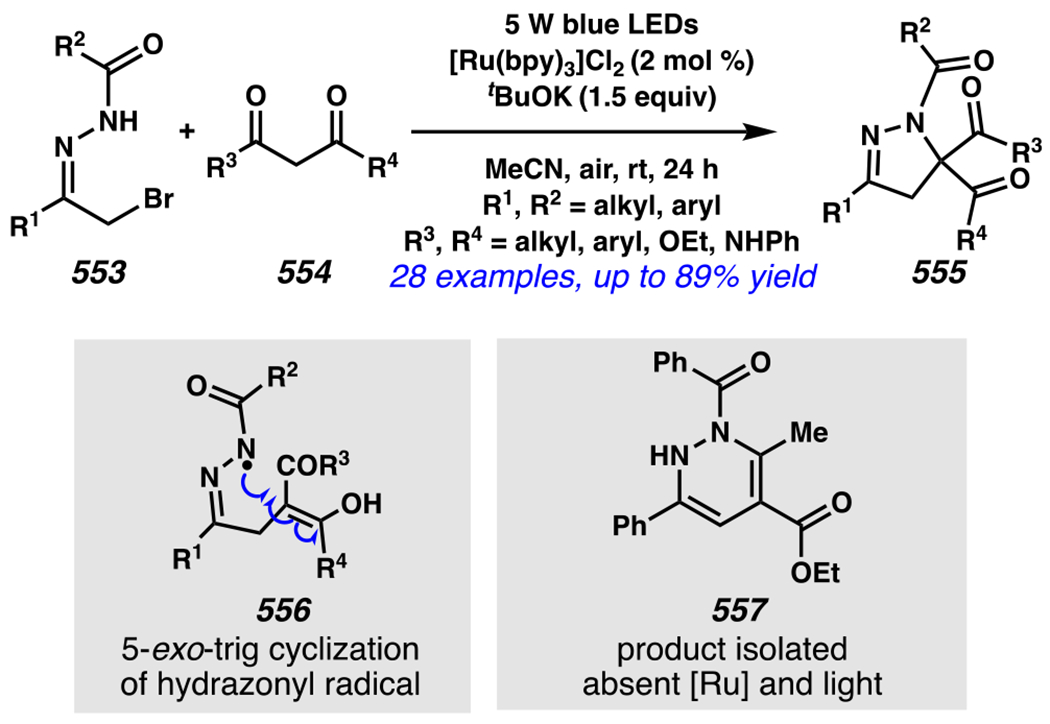
In Situ-Generated Hydrazonyl Radical Provides Access to Dihydropyrazoles
These photocatalyst-mediated process avoid the use of high temperatures, and strong oxidants, and provide a mild protocol for the generation of the hydrazonyl radical intermediates. The intramolecular hydrazonyl radical cyclization process offer straightforward routes functionalized benzosultams, dihydrobenzosultams, 4,5-dihydropyrazoles, and 1,6-dihydropyridazines, with substrate substitution critically influencing product-selectivity.
7. CONCLUSION AND OUTLOOK
With efficient access to nitrogen-centered radicals through photocatalysis, many transformations have become more broadly viable, and more efficient. Among these are C–H functionalizations involving arenes and unactivated aliphatic bonds, as well as olefin functionalization processes and transformations involving bond migration. With typically mild reaction conditions, a variety of otherwise reactive functional groups can be carried through these transformations. Nevertheless, the abundance of publications in this nascent field belies the challenges yet to be overcome that will enable broad adoption of these novel technologies by practitioners of total synthesis, or within polymer chemistry. Among reactions presented herein, stereoselective processes remain underdeveloped, with current strategies relying on exogenous, chiral transition-metal catalysts18, 270, 347, 348, 349, 350, 351, 352 or chiral phosphate bases.361 There are a number of stereoselective reactions that merge photocatalysis and transition-metal catalysts,246, 247, 399, 400 and it is possible that some of these strategies are compatible with currently-racemic reactions, while maintaining the advantages conferred by photocatalytic reactions involving nitrogen-centered radicals. Future developments of broadly generalizable platforms for photocatalytic reactions of nitrogen-centered radicals will hinge upon and be bolstered by continuing efforts to better characterize the underlying physical and photochemical processes in this chemistry. Ultimately, technological developments to date provide a promising foundation for the future of nitrogen-centered radical chemistry.
Figure 2.
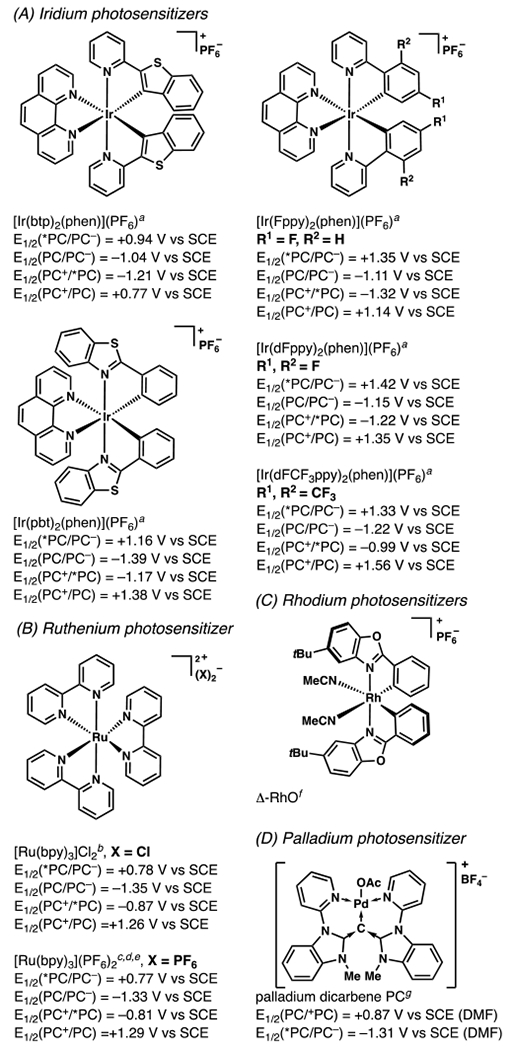
Transition-Metal-Based Photocatalysts and Their Associated Photochemical and Electrochemical Data (All potentials are measured in acetonitrile unless otherwise noted.). aSee ref 18.18 bSee ref 19.19 cSee ref 14.14 dSee ref 15.15 eSee ref 16.16 fSee ref 20.20 gSee ref 21.21
Figure 3.
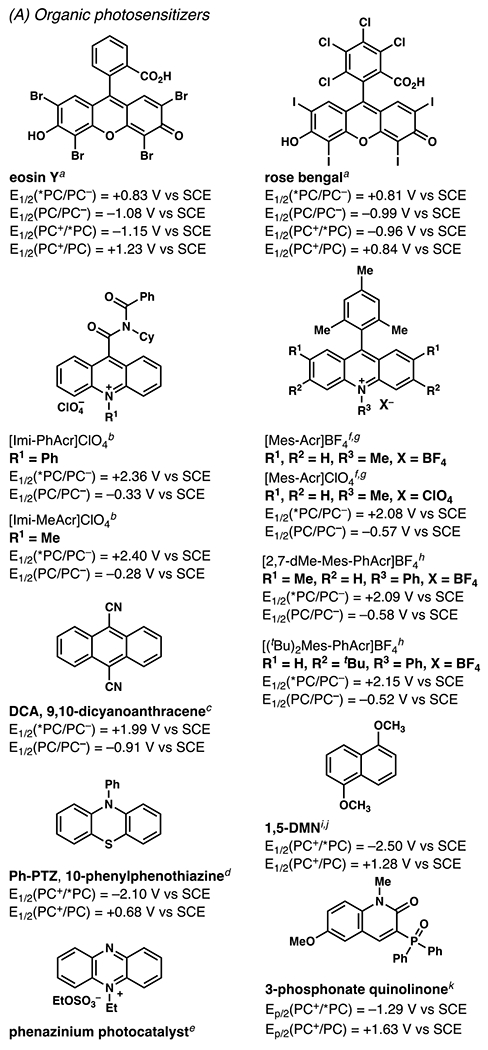
Organic Photocatalysts and Their Associated Photochemical and Electrochemical Data (All potentials are measured in acetonitrile). aSee ref 22.22 bSee ref 23.23. cSee ref 24.24 dSee ref 25.25 eSee ref 26.26 fSee ref 27. 27 gSee ref 28.28 hSee ref 29.29 iSee ref 30.30 jSee ref 31.31 kSee ref 32.32
Scheme 41.
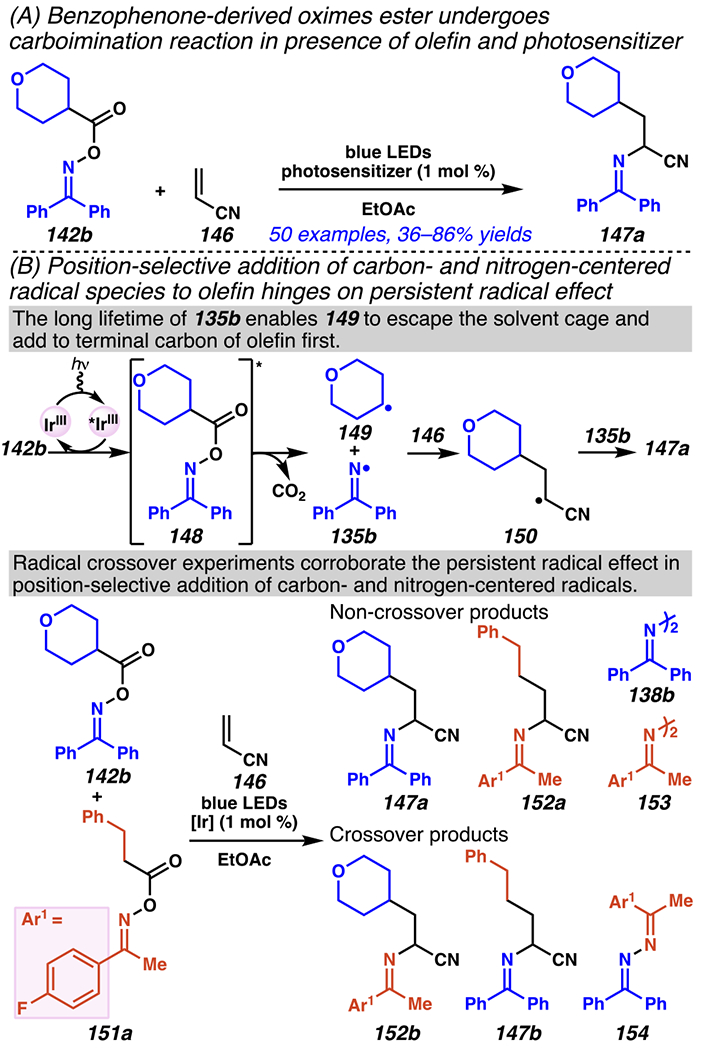
Aliphatic O-Acyl Benzophone-Derived Oxime and Electron-Deficient Olefin Participate in Photosensitized Three-Component Carboimination Reaction
Funding Sources
National Institutes of Health (R35GM128741–01).
ABBREVIATIONS USED
- Å
Ångstrom
- Ac
acetyl
- Acr
acridinium
- AIBN
azobis(isobutyronitrile)
- API
active pharmaceutical ingredient
- aq.
Aqueous
- AQN
anthracene-9,10-dione or anthraquinone
- Ar
aryl
- B:
base
- BET
back-electron transfer
- BDE
bond dissociation energy
- BDFE
bond dissociation free energy
- BH
conjugate base
- BHT
butylhydroxytoluene
- BINOL
1,1′-bi-2-naphthol
- Bn
benzyl
- BNAH
1-benzyl-1,4-dihydronicotinamide
- Boc
tert-butoxycarbonyl
- BODIPY
4,4-Difluoro-4-bora-3a,4a-diaza-s-indacene
- Bpy
2,2’-bipyridine
- Bs
benzenesulfonyl
- Bu
butyl
- Bz
benzoyl
- C
Celsius
- cat
catalytic or catalyst
- calcd
calculated
- Cbz
carboxybenzyl
- CFL
compact fluorescent lamp
- cod
1,5-cyclooctadiene
- 4CzIPN
2,4,5,6-tetrakis(9H-carbazol-9-yl)isophthalonitrile
- DABCO
1,4-diazabicyclo[2.2.2]octane
- DCA
9,10-dicyanoanthracene
- DCE
1,2-dichloroethane
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- dF
difluoro (in reference to photocatalysts)
- dF(CF3)ppy
2-(2,4-difluorophenyl)-5-trifluoromethylpyridine
- dFppy
2-(2,4-difluorophenyl)pyridine
- DFT
density functional theory
- DIPA
diisopropylamine
- DIPEA
N,N-diisopropylethylamine
- DMF
N,N-dimethylforamide
- dmgH
conjugate base of dimethylglyoxime (dmgH2)
- dmgH2
dimethylglyoxime
- DMN
1,5-dimethoxynaphthalene
- DMPU
N,N′-dimethylpropyleneurea
- DMSO
dimethylsulfoxide
- dmTroc
dimethyltrichloroethyl chloroformate
- 3DPAFIPN
2,4,6-tris(diphenylamino)-5-fluoroisophthalonitrile
- dr
diastereomeric ratio
- dtbbpy
4,4′-di-tert-butyl-2,2′-bipyridine
- E 1/2
half-wave potential
- E p
peak potential
- E p/2
half-peak potential
- ee
enantiomeric excess
- E.A.S
- EDG
electron donating group
- EnT
energy transfer
- equiv
equivalent
- Et
ethyl
- ET
electron transfer
- EWG
electron withdrawing group
- EY
eosin Y or 2′,4′,5′,7′-tetrabromofluorescein
- Fc
ferrocene
- FMO
frontier molecular orbital
- g
gram
- GC-MS
gas chromatography–mass spectrometry
- h
hour(s)
- H-Bonding
hydrogen bonding
- HAT
hydrogen atom transfer
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxidehexafluorophosphate
- HFIP
1,1,1,3,3,3-hexafluoroisopropanol
- HLF
Hofmann-Löffler-Freytag
- HRMS
High-resolution mass spectrometry
- ISC
intersystem crossing
- k
kilo
- kcal
kilocalorie
- L
ligand
- LED
light-emitting diode
- M
molar
- m
meta
- m
milli
- Me
methyl
- MeCN
acetonitrile
- Mes
mesityl
- MesSH
2,4,6-trimethylbenzenethiol or mesitylthiol
- mol
mole
- mol %
mole percentage
- MS
molecular sieve
- Ms
methanesulfonyl
- MTBE
methyl tert-butyl ether
- n
normal
- NCR
nitrogen centered radical
- NCS
N-chlorosuccinimide
- NFSI
N-fluorobenzenesulfonimide
- NIS
N-iodosuccinimide
- NMM
N-methylmorpholine
- NMP
N-methyl-2-pyrrolidinone
- NMR
nuclear magnetic resonance
- o
ortho
- ox
oxidation
- p
para
- PC
photocatalyst
- PCET
proton-coupled electron transfer
- Ph
phenyl
- Phth
phthalimido
- Ph-PTZ
10-phenylphenothiazine
- pin
pinacolato
- PMP
para-methoxyphenyl
- ppy
2-phenylpyridinato
- Pr
propyl
- Py
pyridine
- ϕ
quantum yield
- R
undefined group
- red
reduction
- rt
room temperature
- SCE
saturated calomel electrode
- SET
single electron transfer
- τ
excited state lifetime
- t
tert
- TAS
transient absorption spectroscopy
- TBAI
tetra-n-butylammonium iodide
- TEMPO
(2,2,6,6-tetramethyl-piperidin-1-yl)oxyl
- Tf
trifluoromethanesulfonyl
- TFE
2,2,2-trifluoroethanol
- THF
tetrahydrofuran
- TMS
trimethylsilyl
- TMB
1,3,5-trimethoxybenzene
- TRIP
1,3,5-triisopropylbenzene
- Trisyl
2,4,6-triisopropylbenzenesulfonyl
- Ts
para-toluenesulfonyl or tosyl
- TTET
triplet-triplet energy transfer
- UV
ultraviolet
- V
volt
Biographies
Biographies
Kitae Kwon grew up in New York City where he received his bachelor’s degree in Chemical Biology and minor in French from Baruch College. As an undergraduate, he performed theoretical organic chemistry research with Prof. Edyta M. Greer and developed insatiable curiosity for physical organic chemistry. Under the mentorship of Prof. Greer, he published research articles examining reaction mechanisms, a review article on quantum tunnelling, and a book chapter on computational methods relevant to organic chemistry. With an intent to expand his horizon, Kitae chose to pursue Ph.D. research at Duke University under the mentorship of Prof. Jennifer L. Roizen to gain training in synthetic organic chemistry. During his tenure in the Roizen laboratory, he uses principles of physical organic chemistry to invent photoredox-mediated and transition-metal catalyzed C–H functionalization reactions, and to synthesize biologically relevant small molecules.
Nandakumar Meganathan received his bachelor’s degree from Pachaiyappas’ College, Kanchipuram, and a master’s degree from Anna University, India, before choosing to pursue a Ph.D. at the University of Madras, India with Prof. A. K. Mohanakrishnan as a CSIR-Junior Research Fellow. Nandakumar earned his Ph.D. in 2015 for investigations to develop methods to synthesize 1,2-diaroylbenzenes, dibenzothiophene sulfones, fluorenones and vinylens. After graduation, Nandakumar moved to National Tsing Hua University, Taiwan to peruse his postdoctoral research with Prof. Yun Chi where he explored the synthesis of Ru(II) and Pt(II) complexes for optoelectronic applications. Collectively, these research experiences helped him to secure a highly prestigious Marie-Curie Postdoctoral Research Fellowship in 2017 to support research with Prof. Varinder. K. Aggarwal (University of Bristol, UK). At Bristol, he prepared and further functionalized tertiary trifluoromethyl boronic esters using lithiation-borylation chemistry. Pursuing an interest in photoredox-mediated radical chemistry, motivated Nandakumar to pursue further postdoctoral research with Prof. Jennifer L. Roizen at Duke University, where he is currently developing photoredox- and nickel-driven directed arylation strategies.
R. Thomas Simons is an alumnus of Indiana Wesleyan University and the John Wesley Honors College, where he earned a Bachelor of Science degree, with majors in Chemistry and Physics. As an undergraduate, he developed Biochemistry laboratories for undergraduates with Prof. Benjamin Linger and conducted Organic Chemistry research with Prof. Stephen Leonard. In 2018, Thomas began his graduate studies at Duke University, joining the laboratory of Prof. Roizen. His doctoral research has focused on photocatalytic reactions with sulfamides and sulfamates esters, particularly in relation to nickel catalysis and radical-mediated, C–H functionalization processes.
Jennifer Roizen first entered a synthetic chemistry research laboratory as an undergraduate at Williams College in Williamstown, MA. There, Professors J. Hodge Markgraf and Thomas E. Smith ignited her passion for total synthesis, as she pursued syntheses of benzoisocanthenones and hennoxazole A, partially sponsored as a Pfizer SURF. She followed this interest to Prof. Brian M. Stoltz’s laboratory at the California Institute of Technology where she earned a Ph.D. in 2010 based on progress toward a total synthesis of ineleganolide. These efforts rely on a cyclopropanation/Cope rearrangement cascade. With emergent interests in late-stage C–H functionalization reactions, Dr. Roizen moved to Stanford University to learn with a leader in amination technologies, Prof. Justin Du Bois, as an NIH and CMAD Postdoctoralf Fellow. Building on this interest, Dr. Roizen led a laboratory at Duke University, known for photo-driven, sulfamate ester and sulfamide-guided C–H functionalization processes and cross-coupling technologies. Dr. Roizen remains a curiosity-driven chemist.
Footnotes
The authors declare no competing financial interest.
A version of Sections 1–4, 6.1, 6.3–6.4, and 7 have been included in R. Thomas Simons’ Thesis following initial submission of this manuscript.401
The E1/2(DMN+/*DMN) was calculated from the Rehm-Weller equation, where E1/2(DMN+/*DMN) = E1/2 (DMN+/DMN) – E0-0. The E1/2 (DMN+/DMN) and E0-0 values were obtained from Reference 31.
The reaction mass balance was not disclosed to contextualize the investigation of base, so it is unclear whether diminished yields in reactions involving exogenous base result owing to a competitive unproductive elimination reaction to furnish alkene byproducts, which are the dominant reaction products generated in reference 207 under similar reaction conditions.
REFERENCES
- 1.Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centered Radicals. Chem. Soc. Rev, 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]
- 2.Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cismesia MA; Yoon TP Characterizing Chain Processes in Visible Light Photoredox Catalysis. Chem. Sci. 2015, 6, 5426–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majek M; Filace F; von Wangelin AJ On the Mechanism of Photocatalytic Reactions with Eosin Y. Beilstein J. Org. Chem. 2014, 10, 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Julliard M; Chanon M Photoelectron-Transfer Catalysis: Its Connections with Thermal and Electrochemical Analogues. Chem. Rev. 1983, 83, 425–506. [Google Scholar]
- 6.Fraij LK; Hayes DM; Werner TC Static and Dynamic Fluorsence Quenching Experiments for the Physical Chemistry Laboratory. J. Chem. Ed. 1992, 69, 424–428. [Google Scholar]
- 7.Arias-Rotondo DM; McCusker JK The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design. Chem. Soc. Rev. 2016, 45, 5803–5820. [DOI] [PubMed] [Google Scholar]
- 8.Lowry MS; Goldsmith JI; Slinker JD; Rohl R; Pascal RA Jr.; Malliaras GG; Bernhard S Single-Layer Electroluminescent Devices and Photoinduced Hydrogen Production from an Ionic Iridium(III) Complex. Chem. Mater. 2005, 17, 5712–5719. [Google Scholar]
- 9.Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Catalytic Alkylation of Remote C–H Bonds Enabled by Proton-coupled Electron Transfer. Nature 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanss D; Freys JC; Bernardinelli G; Wenger OS Cyclometalated Iridium(III) Complexes as Photosensitizers for Long-Range Electron Transfer: Occurence of a Coulomb Barrier. Eur. J. Inorg. Chem. 2009, 4850–4859. [Google Scholar]
- 11.Excited potential calculated from E00 (In MeCN) from:; Ochola JR; Wolf MO The Effect of Photocatalyst Excited State Lifetime On the Rate of Photoredox Catalysis. Org. Biomol. Chem. 2016, 14, 9088–9092. [DOI] [PubMed] [Google Scholar]
- 12.Ladouceur S; Fortin D; Zysman-Colman E Enhanced Luminescent Iridium(III) Complexes Bearing Aryltriazole Cyclometallated Ligands. Inorg. Chem. 2011, 50, 11514–11526. [DOI] [PubMed] [Google Scholar]
- 13.Slinker JD; Gorodetsky AA; Lowry MS; Wang J; Parker S; Rohl R; Bernhard S; Malliaras GG Efficient Yellow Electroluminescence from a Single Layer of a Iridium Complex. J. Am. Chem. Soc. 2004, 126, 2763–2767. [DOI] [PubMed] [Google Scholar]
- 14.Kalyanasundaram K Photophysics, Photochemistry and Solar Energy Conversion with Tris(bipyridyl)ruthenium(II) and Its Analogues. Coord. Chem. Rev. 1982, 46, 159–244. [Google Scholar]
- 15.Juris A; Balzani V; Belser P; von Zelewsky A Characterization of the Excited State Properties of Some New Photosensitizers of the Ruthenium (Polypyridine) Family. Helv. Chim. Acta 1981, 64, 2175–2182. [Google Scholar]
- 16.Flamigni L; Barbieri A; Sabatini C; Ventura B; Barigelletti F Photochemistry and Photophysics of Coordination Compounds: Iridium. Top. Curr. Chem. 2007, 281, 143–203. [Google Scholar]
- 17.Singh A; Teegardin K; Kelly M; Prasad KS; Krishnan S; Weaver JD Facile Synthesis and Complete Characterization of Homoleptic and Heteroleptic Cyclometalated Iridium(III) Complexes for Photocatalysis. J. Organomet. Chem. 2015, 776, 51–59. [Google Scholar]
- 18.Soni VK; Lee S; Kang J; Moon YK; Hwang HS; You Y; Cho EJ Reactivity Tuning for Radical-Radical Cross-Coupling via Selective Photocatalytic Energy Transfer: Access to Amine Building Blocks. ACS Catal. 2019, 9, 10454–10463. [Google Scholar]
- 19.Tucker JW; Stephenson CRJ Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. J. Org. Chem. 2012, 77, 1617–1622 [DOI] [PubMed] [Google Scholar]
- 20.Shen X; Harms K; Marsch M; Meggers E A Rhodium Catalyst Superior to Iridium Congeners for Enantioselective Radical Amination Activated by Visible Light. Chem. Eur. J. 2016, 22, 9102–9105. [DOI] [PubMed] [Google Scholar]
- 21.Hsu Y-C; Wang VC-C; Au-Yeung K-C; Tsai C-Y; Chang C-C; Lin B-C; Chan Y-T; Hsu C-P; Yap GPA; Jurca T; Ong T-G One-pot Tandem Photoredox and Cross-coupling Catalysis with a Single Palladium Carbodicarbene Complex. Angew. Chem. Int. Ed. 2018, 57, 4622–4626. [DOI] [PubMed] [Google Scholar]
- 22.The potential reported here was determined in MeOH:; Shen T; Zhao Z-G; Yu Q; Xu H-J Photosensitized Reduction of Benzil by Heteroatom-Containing Anthracene Dyes. J. Photochem. Photobiol. A 1989, 47, 203–212. [Google Scholar]
- 23.Gini A; Uygur M; Rigotti T; Alemán J; Mancheño OG Novel Oxidative Ugi Reaction for the Synthesis of Highly Active, Visible-Light, Imide-Acridinium Organophotocatalysts. Chem. Eur. J. 2018, 24, 12509–12514. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y; Haze O; Dinnocenzo JP; Farid S; Farid RS; Gould IR Bonded Exciplexes. A New Concept in Photochemical Reactions. J. Org. Chem. 2007, 72, 6970–6981. [DOI] [PubMed] [Google Scholar]
- 25.Treat NJ; Sprafke H; Kramer JW; Clark PG; Barton BE; Read de Alaniz J; Fors BP; Hawker CJ Metal-Free Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 16096–16101. [DOI] [PubMed] [Google Scholar]
- 26.Leow D Phenazinium Salt-catalyzed Aerobic Oxidative Amidation of Aromatic Aldehydes. Org. Lett. 2014, 16, 5812–5815. [DOI] [PubMed] [Google Scholar]
- 27.Reduction potential for the ground-state of 9-mesityl-10- methylacridinium cation:; Benniston AC; Harriman A; Li P; Rostron JP; van Ramesdonk HJ; Groeneveld MM; Zhang H; Verhoeven JW Charge Shift and Triplet State Formation in the 9-Mesityl-10- Methylacridinium Cation. J. Am. Chem. Soc. 2005, 127, 16054–16064. [DOI] [PubMed] [Google Scholar]
- 28.Reduction potential for the excited-state of 9-mesityl-10- methylacridinium cation:; Ohkubo K; Fukuzumi S 100 Selective Oxygenation of p-Xylene to p-Tolualdehyde via Photoinduced Electron Transfer. Org. Lett. 2000, 2, 3647–3650. [DOI] [PubMed] [Google Scholar]
- 29.Romero NA; Margrey KA; Tay NE; Nicewicz DA Site-selective arene C-H amination via photoredox catalysis. Science, 2015, 349, 1326. [DOI] [PubMed] [Google Scholar]
- 30.Reduction potential for the ground-state of 1,5-DMN:; Zweig A; Maurer AH; Roberts BG Oxidation, Reduction, and Electrochemiluminescence of Donor-substituted Polycyclic Aromatic Hydrocarbons. J. Org. Chem. 1967, 32, 1322–1329. [Google Scholar]
- 31.Reduction potential (in MeCN) for the excited state of 1,5-DMN calc. from E00 from:; Hamada T; Nishida A; Yonemitsu O Selective Removal of Electron-accepting p-Toluene- and Naphthalenesulfonyl Protecting Groups for Amino Function via Photoinduced Donor Acceptor Ion Pairs with Electron-donating Aromatics. J. Am. Chem. Soc. 1986, 108, 140–145. [Google Scholar]
- 32.Kim I; Park B; Kang G; Kim J; Jung H; Lee H; Baik M-H; Hong S Visible-Light-Induced Pyridylation of Remote C(sp3)–H Bonds by Radical Translocation of N-Alkoxypyridinium Salts. Angew. Chem. Int. Ed. 2018, 57, 15517–15522. [DOI] [PubMed] [Google Scholar]
- 33.Timpe H-J; Kronfeld K-P Light-Induced Polymer and Polymerization Reactions XXXIII: Direct Photoinitiation of Methyl Methacrylate Polymerization by Excited States of Ketones. J. Photochem. Photobiol. A 1989, 46, 253–267. [Google Scholar]
- 34.Usami K; Yamaguchi E; Tada N; Itoh A Visible-Light-Mediated Iminyl Radical Generation from Benzyl Oxime Ether: Synthesis of Pyrroline via Hydroimination Cyclization. Org. Lett. 2018, 20, 5714–5717. [DOI] [PubMed] [Google Scholar]
- 35.Reduction potential for the ground-state of DDQ:; Yamago S; Miyazoe H; Iida K; Yoshida J-I Highly Efficient and Chemoselective Reductive Bis-silylation of Quinones by Silyltellurides. Org. Lett. 2000, 2, 3671–3673. [DOI] [PubMed] [Google Scholar]
- 36.Reduction potential (in MeCN) for the excited state of DDQ:; Ohkubo K; Fujimoto A; Fukuzumi S Visible-Light-Induced Oxygenation of Benzene by the Triplet Excited State of 2,3-Dichloro- 5,6-Dicyano-P-Benzoquinone. J. Am. Chem. Soc. 2013, 135, 5368–5371. [DOI] [PubMed] [Google Scholar]
- 37.Deol H; Kumar M; Bhalla V Exploring Organic Photosensitizers Based on Hemicyanine Derivatives: a Sustainable Approach for Prepareation of Amide Linkages. RSC Adv. 2018, 8, 31237–31245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X-F; Yu S-S; Wang C; Xue D; Xiao J BODIPY Catalyzed Amide Synthesis Promoted by BHT and Air Under Visible Light. Org. Biomol. Chem. 2016, 14, 7028–7037. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J; Luo J Donor–Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp2)–C(sp3) Cross-Coupling. ACS Catal. 2016, 6, 873–877. [Google Scholar]
- 40.Le Vaillant F; Garreau M; Nicolai S; Gryn’ova G; Corminbouef C; Waser J Fine-Tuned Organic Photoredox Catalysts for Fragmentation-Alkynylation Cascades of Cyclic Oxime Ethers. Chem. Sci, 2018, 9, 5883–5889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Speckmeier E; Fischer TG; Zeitler K A Toolbox Approach To Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor–Acceptor Cyanoarenes. J. Am. Chem. Soc. 2018, 140, 15353–15365. [DOI] [PubMed] [Google Scholar]
- 42.Reduction potential (in MeCN) for the excited state of 2,4,6-triphenylpyrylium calc. from E00 from:; Searle R; Williams JLR; DeMeyer DE; Doty JC The Sensitization of Stilbene Isomerization. Chem. Commun. 1967, 1165–1165. [Google Scholar]
- 43.Cambié D; Bottecchia C; Straathof NJW; Hessel V; Noël T Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 116, 10276–10341. [DOI] [PubMed] [Google Scholar]
- 44.Halperin SD; Kwon D; Holmes M; Regalado EL; Campeau L-C; DiRocco DA; Britton R Development of a Direct Photocatalytic C–H Fluorination for the Preparative Synthesis of Odanacatib. Org. Lett. 2015, 17, 5200–5203. [DOI] [PubMed] [Google Scholar]
- 45.Cline ED; Bernhard S The Transformation and Storage of Solar Energy: Progress Towards Visible-Light Induced Water Splitting. Chimia 2009, 63, 709–713. [Google Scholar]
- 46.Romero A; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- 47.Strieth-Kalthoff F; James MJ; Teders M; Pitzer L; Glorius F Energy Transfer Catalysis Mediated by Visible Light: Principles, Applications, Directionis. Chem. Soc. Rev. 2018, 47, 7190–7202. [DOI] [PubMed] [Google Scholar]
- 48.Michelin C; Hoffmann N Photosensitization and Photocatalysis—Perspectives in Organic Synthesis. ACS Catal. 2018, 8, 12046–12055. [Google Scholar]
- 49.For pioneering examples of this class of transformation, see:; Zuo Z; Ahneman DT; Chu L; Terret JA; Doyle AG; MacMillan DWC Merging Photoredox with Nickel Catalysis: Coupling of α-Carboxyl sp3-Carbons with Aryl Halides. Science 2014, 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.For an early review on this topic, see:; Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC The Merger of Transition Metal and Photocatalysis. Nature Reviews Chemistry 2017, 1, 52. [Google Scholar]
- 51.For a recent example of a trifluoroacetamide-directed remote C(sp3)–H allylation, see:; Xu B; Tambar UK Remote Allylation of Unactivated C(sp3)–H Bonds Triggered by Photogenerated Amidyl Radicals. ACS Catal. 2019, 9, 4627–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.For a recent example of an intramolecular olefin amidoarylation involving amides, carbamates and ureas as substrates, see:; Zheng S; Gutiérrez-Boent Á; Molander GA Merging Photoredox PCET with Ni-Catalyzed Cross-Coupling: Cascade Amidoarylation of Unactivated Olefins. Chem 2019, 5, 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.For a recent example of an intramolecular diastereoselective olefin amidoacylation of amides and carbamates as substrates, see:; Zheng S; Zhang S-Q; Saeednia B; Zhou J; Anna JM; Hong X; Molander GA Diastereoselective Olefin Amidoacylation via Photoredox PCET/Nickel-dual Catalysis: Reaction Scope and Mechanistic Insights. Chem. Sci. 2020, 11, 4131–4137. [Google Scholar]
- 54.For an amide-directed alkylative cross-coupling reaction, see:; Thullen SM; Treacy SM; Rovis T Regioselective Alkylative Cross-Coupling of Remote Unactivated C(sp3)−H Bonds. J. Am. Chem. Soc. 2019, 141, 14062–14067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blackburn JM; Kanegusuku ALG; Scott GE; Roizen JL Photochemically-Mediated, Nickel-Catalyzed Synthesis of N-(Hetero)aryl Sulfamate Esters. Org. Lett. 2019, 21, 7049–7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simons RT; Scott GE; Kanegusuku AG; Roizen JL Photochemically Mediated Nickel-Catalyzed Synthesis of N-(Hetero)aryl Sulfamides. J. Org. Chem. 2020, 85, 6380–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Till NA; Tian L; Dong Z; Scholes GD; MacMillan DWC Mechanistic Analysis of Metallaphotoredox C–N Coupling: Photocatalysis Initiates and Perpetuates Ni(I)/Ni(III) Coupling Activity. J. Am. Chem. Soc. 2020, 142, 15830–15841. [DOI] [PubMed] [Google Scholar]
- 58.Beatty JW; Stephenson CRJ Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc. Chem. Res. 2015, 48, 1474–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.For review on this topic, see:; Hu J; Wang J; Nguyen TH; Zheng N The Chemistry of Amine Radical Cations Produced by Visible Light Photoredox. Beilstein J. Org. Chem. 2013, 9, 1977–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.For brief tutorial review on photoredox-mediated generations and reactions of nitrogen-centered radicals and radical ions from N–Cl, N–Br, N–O, N–S, N–N, and N–H bonds, see:; Chen J-R; Hu X-Q; Lu L-Q; Xiao W-J Visible Light Photoredox-Controlled Reactions of N-Radicals and Radical Ions. Chem. Soc. Rev, 2016, 45, 2044–2056. [DOI] [PubMed] [Google Scholar]
- 61.For mini review on oxidatively generated nitrogen-centered radicals from photoredox catalysis, see:; Jiang H; Studer A Chemistry With N-Centered Radicals Generated by Single-Electron Transfer-Oxidation Using Photoredox Catalysis. CCS Chem. 2019, 1, 38–49. [Google Scholar]
- 62.For focus review on photochemically generated amidyl, hydrazonyl and imidyl radicals, see:; Kärkäs MD Photochemical Generation of Nitrogen-Centered Amidyl, Hydrazonyl, and Imidyl Radicals: Methodology Developments and Catalytic Applications. ACS Catal. 2017, 7, 4999–5022. [Google Scholar]
- 63.For perspective on phocatalytically generated aminium radical cations for C–N bond forming reactions, see:; Ganley JM; Murray PRD; Knowles RR Photocatalytic Generation of Aminium Radical Cations for C–N Bond Formation. ACS Catal. 2020, 10, 11712–11738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.For an account summarizing reactions involving photocatalytically generated nitrogen-centered radical research led by Chen and co-workers, see:; Yu X-Y; Zhao Q-Q; Chen J; Xiao W-J; Chen J-R When Light Meets Nitrogen-Centered Radicals: From Reagents to Catalysts. Acc. Chem. Res. 2020, 53, 1066–1083. [DOI] [PubMed] [Google Scholar]
- 65.For focus review summarizing photocatalyst-mediated nitrogen-radical cyclization reactions, see:; Wang P; Zhao Q; Xiao W; Chen J Recent Advances in Visible-Light Photoredox-Catalyzed Nitrogen Radical Cyclization. Green Synth. Catal, 2020, 1, 42–51. [Google Scholar]
- 66.Hofmann AW Zur Kenntiss des Piperidins und Pyridins. Ber. Dtsch. Chem. Ges. 1879, 12, 984–990. [Google Scholar]
- 67.Löffler K; Freytag C Über eine neue Bildunsweise von N-alkylierten Pyrrolidinen. Ber. Dtsch. Chem. Ges. 1909, 42, 3427–3431. [Google Scholar]
- 68.Wawzonek S; Thelen PJ Preparation of N-methylgranatanine. J. Am. Chem. Soc. 1950, 72, 5, 2118–2120. [Google Scholar]
- 69.Corey EJ; Hertler WR A Study of the Formation of Haloamines and Cyclic Amines by the Free Radical Chain Decomposition of N-Haloammonium Ions (Hofmann-Loffler Reaction). J. Am. Chem. Soc. 1960, 82, 1657–1668. [Google Scholar]
- 70.Neale RS Nitrogen Radicals as Synthesis Intermediates. N-Halamide Rearrangements and Additions to Unsaturated Hydrocarbons. Synthesis, 1971, 1, 1–15. [Google Scholar]
- 71.Baldwin SW; Doll RJ Synthesis of the 2-aza-7-oxatricyclo[4.3.2.04,8]undecane Nucleus of some Gelsemium Alkaloids. Tetrahedron Letters, 1979, 35, 3275–3278. [Google Scholar]
- 72.Short MA; Blackburn JM; Roizen JL Sulfamate Esters Guide Selective Radical-mediated Chlorination of Aliphatic C–H Bonds. Angew. Chem. Int. Ed. 2018, 57, 296–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Okahara M; Ohashi T; Komori S Synthesis of γ- and δ-Chloroalkanesulfonamides via the Photorearrangement of N-Chlorosulfonamides. J. Org. Chem. 1968, 33, 3066–3070. [DOI] [PubMed] [Google Scholar]
- 74.Neale RS; Marcus NL The Chemistry of Nitrogen Radicals. IX. Reactions of N-Halocyanamides and N-Halosulfonamides. J. Org. Chem. 1969, 34, 1808–1816. [Google Scholar]
- 75.Qin Q; Yu S Visible-Light-Promoted Remote C(sp3)–H Amidation and Chlorination. Org. Lett. 2015, 17, 1894–1897. [DOI] [PubMed] [Google Scholar]
- 76.Wayner DDM; McPhee DJ; Griller D Oxidation and Reduction Potentials of Transient Free Radicals. J. Am. Chem. Soc. 1988, 110, 132–137. [Google Scholar]
- 77.Zhu Y; Wang J-J; Wu D; Yu W Visible-Light-Driven Remote C–H Chlorination of Aliphatic Sulfonamides with Sodium Hypochlorite. Asian J. Org. Chem. 2020, 9, 1650–1654. [Google Scholar]
- 78.Qin Q; Ren D; Yu S Visible-light-promoted Chloramination of Olefins with N-chlorosulfonamide as both Nitrogen and Chlorine Sources. Org. Biomol. Chem. 2015, 13, 10295–10298. [DOI] [PubMed] [Google Scholar]
- 79.Esker JL; Newcomb M Chemistry of Amidyl Radicals Produced from N-Hydroxypyridine-2-thione Imidate Esters. J. Org. Chem. 1993, 58, 4933–4940. [Google Scholar]
- 80.Horner JH; Musa OM; Bouvier A; Newcomb M Absolute Kinetics of Amidyl Radical Reactions. J. Am. Chem. Soc. 1998, 120, 7738–7748. [Google Scholar]
- 81.Akasaka T; Furukawa N; Oae S Sulfoximidoyl Radical. Homolytic Addition of N-halosulfoximides to Olefins. Tetrahedron Letters, 1979, 22, 2035–2038. [Google Scholar]
- 82.Prieto A; Diter P; Toffano M; Hannedouche J; Magnier E Photoredox-initiated 1,2-difunctionalization of Alkenes with N-chloro, S-fluoroalkyl Sulfoximines. Adv. Synth. Catal. 2019, 361, 436–440. [Google Scholar]
- 83.Wang J-D; Liu Y-X; Xue D; Wang C; Xiao J Amination of Benzoxazoles by Visible-Light Photoredox Catalysis. Synlett 2014, 25, 2013–2018. [Google Scholar]
- 84.Kim H; Kim T; Lee DG; Roh SW; Lee C Nitrogen-centered Radical-mediated C–H Imidation of Arenes and Heteroarenes via Visible Light Induced Photocatalysis. Chem. Commun. 2014, 50, 9273–9276. [DOI] [PubMed] [Google Scholar]
- 85.Allen LJ; Cabrera PJ; Lee M; Sanford MS N-Acyloxyphthalimides as Nitrogen Radical Precursors in the Visible Light Photocatalyzed Room Temperature C–H Amination of Arenes and Heteroarenes. J. Am. Chem. Soc. 2014, 136, 5607–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Foo K; Sella E; Thomé I; Eastgate MD; Baran PS A Mild Ferrocene-Catalyzed C–H Imidation of (Hetero)Arenes. J. Am. Chem. Soc. 2014, 136, 5279–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pluta R; Nikolaienko P; Rueping M Direct Catalytic Trifluoromethylthiolation of Boronic Acids and Alkynes Employing Electrophilic Shelf-Stable N-(trifluoro-methylthio)phthalimide. Angew. Chem. Int. Ed. 2014, 53, 1650–1653. [DOI] [PubMed] [Google Scholar]
- 88.Minisci F Novel Applications of Free-Radical Reactions in Preparative Organic Chemistry. Synthesis, 1973, 1, 1–24. [Google Scholar]
- 89.This manuscript does not summarize publications with dates after February 1, 2021. Nevertheless, pertinent manuscripts have been published since this date, see as an example:; Lee W; Jung S; Kim M; Hong S Site-Selective Direct C–H Pyridylation of Unactivated Alkanes by Triplet Excited Anthraquinone. J. Am. Chem. Soc. 2021, 143, 3003–3012. [DOI] [PubMed] [Google Scholar]
- 90.This manuscript does not summarize publications with dates after February 1, 2021. Nevertheless, pertinent manuscripts have been published since this date, see as an example:; Shin S; Lee S; Choi W; Kim N; Hong S Visible-Light-Induced 1,3-Aminopyridylation of [1.1.1]Propellane with N-Aminopyridinium Salts. Angew. Chem. Int. Ed. 2021, 60, 7873–7879. [DOI] [PubMed] [Google Scholar]
- 91.Kemper J; Studer A Stable Reagents for the Generation of N-Centered Radicals: Hydroamination of Norbornene. Angew. Chem. Int. Ed. 2005, 44, 4914–4917. [DOI] [PubMed] [Google Scholar]
- 92.Guin J; Fröhlich R; Studer A Thiol-catalyzed Stereoselective Transfer Hydroamination of Olefins with N-Aminated Dihydropyridines. Angew. Chem. Int. Ed. 2008, 47, 779–782. [DOI] [PubMed] [Google Scholar]
- 93.Greulich TW; Daniliuc CG; Studer A N-Aminopyridinium Salts as Precursors for N-Centered Radicals – Direct Amidation of Arenes and Heteroarenes. Org. Lett. 2015, 17, 254–257. [DOI] [PubMed] [Google Scholar]
- 94.Bock CR; Connor JA; Gutierrez AR; Meyer JT; Whitten DG; Sullivan BP; Nagle JK Estimation of Excited-state Redox Potentials by Electron-transfer Quenching. Application of Electron-transfer Theory to Excited-state Redox Processes. J. Am. Chem. Soc. 1979, 101, 4815–4824. [Google Scholar]
- 95.Okada K; Okamoto K; Morita N; Okubo K; Oda M Photosensitized Decarboxylative Michael Addition through N-(Acyloxy)phthalimides via an Electron-Transfer Mechanism. J. Am. Chem. Soc. 1991, 113, 9401–9402. [Google Scholar]
- 96.Miyazawa K; Koike T; Akita M Regiospecific Intermolecular Aminohydroxylation of Olefins by Photoredox Catalysis. Chem. Eur. J. 2015, 21, 11677–11680. [DOI] [PubMed] [Google Scholar]
- 97.Miyazawa K; Koike T; Akita M Aminohydroxylation of Olefins with Iminopyridinium Ylides by Dual Ir Photocatalysis and Sc(OTf)3 catalysis. Tetrahedron 2016, 72, 7813–7820. [Google Scholar]
- 98.Yu W-L; Chen J-Q; Wei Y-L; Wang Z-Y; Xu P-F Alkene Functionalization for the Stereospecific Synthesis of Substituted Aziridines by Visible-light Photoredox Catalysis. Chem. Commun. 2018, 54, 1948–1951. [DOI] [PubMed] [Google Scholar]
- 99.Goliszewska K; Rybicka-Jasinska K; Szurmak J Gryko, D. Visible-Light-Mediated Amination of π-Nucleophiles with N-aminopyridinium Salts. J. Org. Chem. 2019, 84, 15834–15844. [DOI] [PubMed] [Google Scholar]
- 100.Moon Y; Park B; Kim I; Kang G; Shin S; Kang D; Baik M-H; Hong S Visible Light Induced Alkene Aminopyridylation using N-aminopyridiniuim Salts as Bifunctional Reagents. Nat. Commun 2019, 10, 4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lazarides T; McCormick T; Du P; Luo G; Lindley B; Eisenberg R Making Hydrogen from Water Using a Homogeneous System without Noble Metals. J. Am. Chem. Soc. 2009, 131, 9192–9194. [DOI] [PubMed] [Google Scholar]
- 102.Distortion-interaction energies were calculated in the Jaguar 9.1 suite with the PW6B95-D3/def2-QZVP level of theory and the atomic charges from electrostatic potential were examined using the CHELPG program as implemented in ORCA.
- 103.Moon Y; Lee W; Hong S Visible-Light-Enabled Ortho-selective Aminopyridylation of Alkenes with N-Aminopyridinium Ylides. J. Am. Chem. Soc. 2020, 142, 12420–12429. [DOI] [PubMed] [Google Scholar]
- 104.Roth HG; Romero NA; Nicewicz DA Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar]
- 105.Geometry optimization and energies were calculated in the Jaguar 9.1 suite with the B3LYP-D3 level of theory using the LACVP** basis set.
- 106.Kim N; Lee C; Kim T; Hong S Visible-Light-Induced Remote C(sp3)–H Pyridylation of Sulfonamides and Carboxamides. Org. Lett. 2019, 21, 9719–9723. [DOI] [PubMed] [Google Scholar]
- 107.Zhang D; Wang H; Bolm C Photocatalytic Difunctionalisations of Alkenes with N-SCN Sulfoximines. Chem. Commun. 2018, 54, 5772–5775. [DOI] [PubMed] [Google Scholar]
- 108.Soto A; Yorimitsu H; Oshima K O-Alkyl S-3,3-Dimethyl-2-oxobutyl Dithiocarbonates as Versatile Sulfur-Transfer Agents in Radical C(sp3)–H Functionalization. Chem. Asian J. 2007, 2, 1568–1573. [DOI] [PubMed] [Google Scholar]
- 109.Czaplyski WL; Na CG; Alexanian EJ C–H Xanthylation: A Synthetic Platform for Alkane Functionalization. J. Am. Chem. Soc. 2016, 138, 13854–13857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ayer SK; Roizen JL Sulfamate Esters Guide C(3)-selective Xanthylation of Alkanes. J. Org. Chem. 2019, 84, 3508–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xuan J; Li B-J; Feng Z-J; Sun G-D; Ma H-H; Yuan Z-W, Chen J-R Lu L-Q Xiao W-J Desulfonylation of Tosyl Amides Through Catalytic Photoredox Cleavage of N–S Bond Under Visible-Light Irradiation. Chem. Asian J. 2013, 8, 1090–1094. [DOI] [PubMed] [Google Scholar]
- 112.MacKenzie IA; Wang L; Onuska NPR; Williams OF; Begam K; Moran AM; Dunietz BD; Nicewicz DA Discovery and Characterization of an Acridine Radical Photoreducant. Nature 2020, 580, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.For an example, see:; Rössler SL; Jelier BJ; Tripet PF; Shemet A; Jeschke G; Togni A; Carreira EM Pyridyl Radical Cation for C–H Amination of Arenes. Angew. Chem. Int. Ed. 2019, 58, 526–531. [DOI] [PubMed] [Google Scholar]
- 114.For an example, see:; Ham WS; Hillenbrand J; Jacq J; Genicot C; Ritter T Divergent Late-Stage (Hetero)aryl C–H Amination by the Pyridinium Radical Cation. Angew. Chem. Int. Ed. 2019, 58, 532–536. [DOI] [PubMed] [Google Scholar]
- 115.Yin W; Wang X Recent Advances in Iminyl Radical-Mediated Catalytic Cyclizations and Ring-Opening Reactions. New J. Chem, 2019, 43, 3254–3264. [Google Scholar]
- 116.For review that summarizes early research into O-aryloximes as precusors to 1-pyrrolines, see:; Kitamura M; Narasaka K Catalytic Radical Cyclization of Oximes Induced by One-Electron Transfer. Bull. Chem. Soc. Jpn. 2008, 81, 539–547. [Google Scholar]
- 117.For review that highlights 1-pyrrolines in medicinal chemistry, see:; Dannhardt G; Kiefer W 1-Pyrrolines (3,4-Dihydro-2H-pyrroles) as a Template for New Drugs. Arch. Pharm. Pharm. Med. Chem, 2001, 334, 183–188. [DOI] [PubMed] [Google Scholar]
- 118.Blake JA; Pratt DA; Lin S; Walton JC; Mulder P; Ingold KU Thermolyses of O-Phenyl Oxime Ethers. A New Source of Iminyl Radicals and a New Source of Aryloxyl Radicals. J. Org. Chem. 2004, 69, 3112–3120. [DOI] [PubMed] [Google Scholar]
- 119.Krylov IB; Segida OO; Budnikov AS; Terent’ev AO Oxime-Derived Iminyl Radicals in Selective Processes of Hydrogen Atom Transfer and Addition to Carbon–Carbon π–Bonds. Adv. Synth. Catal. 2021, 363, 2502–2528. [Google Scholar]
- 120.Uchiyama K; Hayashi Y; Narasaka K Synthesis of 8-Hydroxyquinolines by the Cyclization of m-Hydroxyphenthyl Ketone O-2,4-Dinitrophenyloximes. Synlett 1997, 445–446. [Google Scholar]
- 121.Katsuya U; Ayako O; Yujiro H; Koichi Narasaka. Regioselective Synthesis of Quinolin-8-ols and 1,2,3,4-Tetrahydroquinolin-8-ols by the Cyclization of 2-(3-Hydroxyphenyl)ethyl Ketone O-2,4-Dinitrophenyloximes. Bull. Chem. Soc. Jpn. 1998, 71, 2945–2955. [Google Scholar]
- 122.Uchiyama K; Hayashi Y; Narasaka K Synthesis of Dihydropyrroles by the Intramolecular Addition of Alkylideneaminyl Radicals Generated from O-2,4-Dinitrophenyloximes of γ,δ-Unsaturated Ketones. Chem. Lett. 1998, 27, 1261–1262. [Google Scholar]
- 123.Tetsuhiro M; Narasaka K Photochemical Transformation of γ,δ-Unsaturated Ketone O-(p-Cyanophenyl)oximes to 3,4-Dihydro-2H-pyrrole Derivatives. Chem. Lett. 2000, 29, 338–339. [Google Scholar]
- 124.Mikami T; Narasaka K Photochemical Transformation of γ,δ-Unsaturated Ketone O-(p-cyanophenyl)oximes to 3,4-Dihydro-2H-Pyrrole Derivatives. C. R. Acad. Sci. Paris, Chimie / Chemistry 2001, 4, 477–485. [Google Scholar]
- 125.In 2005, Narasaka and co-workers published their research on UV-light-mediated and 1,5-DMN-catalyzed iminyl radical 5-exo-trig hydroamination reactions in Tetrahdron Letter:; Kitamura M; Mori Y; Narasaka K Photochemical Radical Cyclization of γ,δ-Unsaturated Ketone Oximes to 3,4-Dihydro-2H-pyrroles. Tetrahedron Lett. 2005, 46, 2373–2376. [Google Scholar]
- 126.Davies J; Booth SG; Essafi S; Dryfe RAW; Leonori D Visible-Light-Mediated Generation of Nitrogen-Centered Radicals: Metal-Free Hydroimination and Iminohydroxylation Cyclization Reactions. Angew. Chem. Int. Ed. 2015, 54, 14017–14021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Srivastava V; Singh PP Eosin Y Catalyzed Photoredox Synthesis: A Review. RSC Adv, 2017, 7, 31377–31392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cai S-H; Xie J-H; Song S; Ye L; Feng C; Loh T-P Visible-Light-Promoted Carboimination of Unactivated Alkenes for the Synthesis of Densely Functionalized Pyrroline Derivatives. ACS Catal. 2016, 6, 5571–5574. [Google Scholar]
- 129.Nakafuku KM; Fosu SC; Nagib DA Catalytic Alkene Difunctionalization via Imidate Radicals. J. Am. Chem. Soc. 2018, 140, 11202–11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.For review on employing oximes as precursors to access the N-heterocycles, see:; Walton J The Oxime Portmanteau Motif: Released Heteroradicals Undergo Incisive EPR Interrogation and Deliver Diverse Heterocycles. Acc. Chem. Res. 2014, 47, 1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jiang H; An X; Tong K; Zheng T; Zhang Y; Yu S Visible-Light-Promoted Iminyl-Radical Formation from Acyl Oximes: A Unified Approach to Pyridines, Quinolines, and Phenanthridines. Angew. Chem. Int. Ed. 2015, 54, 4055–4059. [DOI] [PubMed] [Google Scholar]
- 132.Alonso R; Campos PJ; Rodrıguez MA; Sampedro D Photocyclization of Iminyl Radicals: Theoretical Study and Photochemical Aspects. J. Org. Chem. 2008, 73, 2234–2239. [DOI] [PubMed] [Google Scholar]
- 133.Ishihara Y; Azuma S; Choshi T; Kohno K; Ono K; Tsutsumi H; Ishizu T; Hibino S Total Synthesis of Benzo[c]phenanthridine Alkaloids Based on a Microwave-Assisted Electrocyclic Reaction of the Aza 6π-Electron System and Structural revision of Broussonpapyrine. Tetrahedron 2011, 67, 1320–1333. [Google Scholar]
- 134.An X -D.; Yu, S. Visible-Light-Promoted and One-Pot Synthesis of Phenanthridines and Quinolines from Aldehydes and O-Acyl Hydroxylamine. Org. Lett. 2015, 17, 2692–2695. [DOI] [PubMed] [Google Scholar]
- 135.Istvan Z; Rethy B; Hohmann J; Molnar J; Imre O; Falkay G Antitumor Activity of Alkaloids Derived from Amaryllidaceae Species. In Vivo 2009, 23, 41–48. [PubMed] [Google Scholar]
- 136.Caballero A; Campos PJ; Rodríguez MA Fluorescence sensing of Cu(II) based on triphaeridine derivatives. Tetrahedron 2013, 69, 4631–4635. [Google Scholar]
- 137.Szlávik L; Gyuris A; Minárovits J; Forgo P; Molnár J; Hohmann J Alkaloids from Leucojum vernum and Antiretroviral Activity of Amaryllidaceae Alkaloids. Planta Med. 2004, 70, 871–873. [DOI] [PubMed] [Google Scholar]
- 138.Chen W-L; Chen C-Y; Chen Y-F; Hsieh J-C Hydride-Induced Anionic Cyclization: An Efficient Method for the Synthesis of 6-H-Phenanthridines via a Transition-Metal-Free Process. Org. Lett. 2015, 17, 1613–1316. [DOI] [PubMed] [Google Scholar]
- 139.Forrester AR; Gill M; Napier RJ; Thomson RH Iminyls. Part 5. Intramolecular Hydrogen Atom Abstraction by Alkyl(aryl)-iminyls. A New Tetralone Synthesis. J. Chem. Soc. Perkin 1 1978, 632–636. [Google Scholar]
- 140.Forrester AR; Napier RJ; Thomson RH Iminyls. Part 7. Intramolecular Hydrogen Atom Abstraction; Synthesis of Heterocyclic Analogues of α-Tetralone. J. Chem. Soc. Perkin 1 1981, 984–987. [Google Scholar]
- 141.For representative reference, see:; Legoabe LJ; Peltzer A; Petzer JP α-Tetralone derivatives as inhibitors of monoamine oxidase. Bioorg. Med. Chem. Lett. 2014, 24, 2758–2763. [DOI] [PubMed] [Google Scholar]
- 142.Shu W; Nevado C Visible-Light-Mediated Remote Aliphatic C–H Functionalizations through a 1,5-Hydrogen Transfer Cascade. Angew. Chem. Int. Ed. 2017, 56, 1881–1884. [DOI] [PubMed] [Google Scholar]
- 143.Forrester AR; Gill M; Sadd JS; Thomson RH New Chemistry of Iminyl Radicals. J. Chem. Soc. Chem. Comm 1975, 291–291. [Google Scholar]
- 144.Forrester AR; Gill M; Meyer CJ; Sadd JS; Thomson RH Iminyls. Part 1. Generations of Iminyls. J. Chem. Soc. Perkin 1 1979, 601–611. [Google Scholar]
- 145.Boivin J; Fouquet E; Zard SZ; A New and Synthetically Useful Source of Iminyl Radicals. Tetrahedron Lett. 1991, 32, 4299–4302. [Google Scholar]
- 146.For a review summarizing the development of photomediated Giese reactions, see:; Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.For an updated review of photomediated Giese reactions, see:; Kanegusuku ALG; Roizen JL Recent Advances in Photoredox-Mediated Radical Conjugate Addition Reactions: An Expanding Toolkit for the Giese Reaction. Angew. Chem. Int. Ed. 10.1002/anie.202016666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jiang H; Studer A Iminyl-Radicals by Oxidation of α-Imino-oxy Acids: Photoredox-Neutral Alkene Carboimination for the Synthesis of Pyrrolines. Angew. Chem. Int. Ed. 2017, 56, 12273–12276. [DOI] [PubMed] [Google Scholar]
- 149.Davies J; Sheikh NS; Leonori D Photoredox Imino Functionalizations of Olefins. Angew. Chem. Int. Ed. 2017, 56, 13361–13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.For pioneering research that employs α,α-dimethylated carboxylate as iminyl radical precursor, see:; Forrester AR; Gill M; Meyer CJ; Sadd JS; Thomson RH Iminyls. Part 1. Generation of Iminyls. J. Chem. Soc. Perkin 1 1979, 606–611 J. Chem. Soc. Perkin 1 1978, 632–636. [Google Scholar]
- 151.Rueda-Becerril M; Sazepin CC; Leung JCT; Okbinoglu T; Kennepohl P; Paquin J-F; Sammis GM Fluorine Transfer to Alkyl Radicals. J. Am. Chem. Soc. 2012, 134, 4026–4029. [DOI] [PubMed] [Google Scholar]
- 152.Gołębiewski WM; Gucma M Applications of N-Chlorosuccinimide in Organic Synthesis. Synthesis, 2007, 3599–3619. [Google Scholar]
- 153.Gleicher GJ; Mahiou B; Aretakis AJ Selectivities in the Addition of Radicals Generated from Derivatives of 2-Bromomalonic Acid to Alkene Pairs. J. Org. Chem. 1989, 54, 308–311. [Google Scholar]
- 154.Kulbitski K; Nisnevich G; Gandelman M Metal-Free Efficient, General and Facile Iododecarboxylation Method with Biodegradable Co-Products. Adv. Synth. Catal. 2011, 353, 1438–1442 [Google Scholar]
- 155.For seminal report on N(SCF3)-benzosuccinimide as electrophilic SCF3 transfer reagent, see:; Haas A; Möller G Preparation and Reactivity of Tris(trifluoromethylselanyl)carbenium [(CF3Se)3C+) and Trifluoromethylsulfanylacetic Acid Derivatives [(CF3S)3–nC2n(O)R]. Chem. Ber. 1996, 129, 1383–1388. [Google Scholar]
- 156.For seminal report on N(SePh)-benzosuccinimide as electrophilic SePh transfer reagent, see:; Nicolau KC; Claremon DA; Barnette WE; Seitz SP N-Phenylselenophthalimide (N-PSP) and N-Phenylselenosuccinimide (N-PSS). Two Versatile Carriers of the Phenylseleno Group. Oxyselenation of Olefins and a Selenium-Based Macrolide Synthesis. J. Am. Chem. Soc. 1979, 101, 3704–3706. [Google Scholar]
- 157.Ollivier C; Renaud P A Novel Approach for the Formation of Carbon–Nitrogen Bonds: Azidation of Alkyl Radicals with Sulfonyl Azides. J. Am. Chem. Soc. 2001, 123, 4717–4727. [DOI] [PubMed] [Google Scholar]
- 158.Griffin JD; Zeller MA; Nicewicz DA Hydrodecarboxylation of Carboxylic and Malonic Acid Derivatives via Organic Photoredox Catalysis: Substrate Scope and Mechanistic Insight. J. Am. Chem. Soc. 2015, 137, 11340–11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.For seminal report on vinylbenziodoxolones as electrophilic vinylating transfer agent, see:; Stridfeldt E; Seemann A; Bouma MJ; Dey C; Ertan A; Olofsson B Synthesis, Characterization and Unusual Reactivity of Vinylbenziodoxolones—Novel Hypervalent Iodine Reagents. Chem. Eur. J. 2016, 22, 16066–16070. [DOI] [PubMed] [Google Scholar]
- 160.For seminal report on alkynylbenziodoxolones as electrophilic alkynylating transfer agent, see:; Frei R; Wodrich MD; Hari DP; Broin P-A; Chauvier C; Waser J Fast and Highly Chemoselective Alkynylation of Thiols with Hypervalent Iodine Reagents Enabled through a Low Energy Barrier Concerted Mechanism. J. Am. Chem. Soc. 2014, 136, 16563–16573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.For seminal report on cyanobenziodoxolones as electrophilic cyano transfer agent, see:; Le Vaillant F; Wodrich M; Waser J Room temperature decarboxylative cyanation of carboxylic acids using photoredox catalysis and cyanobenziodoxolones: a divergent mechanism compared to alkynylation. Chem. Sci, 2017, 8, 1790–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Boivin J; Callier-Dublanchet A-C; Quiclet-Sire B; Schiano A-M; Zard SZ Iminyl, Amidyl, and Carbamyl Radicals from O-Benzoyl Oximes and O-Benzoyl Hydroxamic Acid Derivatives. Tetrahedron 1995, 51, 6517–6528. [Google Scholar]
- 163.Tu J-L; Liu J-L; Tang W; Su M; Liu F Radical Aza-Cyclization of α-Imino-oxy Acids for Synthesis of Alkene-Containing N-Heterocycles via Dual Cobaloxime and Photoredox Catalysis. Org. Lett. 2020, 22, 1222–1226. [DOI] [PubMed] [Google Scholar]
- 164.For seminal report on Narasaka-Heck reaction, see:; Tsutsui H; Narasaka K Synthesis of Pyrrole Derivatives by the Heck-Type Cyclization of γ,δ-Unsaturated Ketone O-Pentafluorobenzoyloximes. Chem. Lett. 1999, 28, 45–46. [Google Scholar]
- 165.For review on the Narasaka-Heck reaction, see:; Race NJ; Hazelden IR; Faulkner A; Bower JF Recent Developments in the Use of Aza-Heck Cyclizations for the Synthesis of Chiral N-Heterocycles. Chem. Sci, 2017, 8, 5248–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cabrera–Afonso MJ; Cembellín S; Halima-Salem A; Berton M; Marzo L; Miloudi A; Maestro MC; Alemán J Metal-Free Visible Light-Promoted Synthesis of Isothiazoles: A Catalytic Approach for N–S Bond Formation from Iminyl Radicals Under Batch and Flow Conditions. Green Chem. 2020, 22, 6792–6797. [Google Scholar]
- 167.Patra T; Satobhisha M; Ma J; Strieth-Kalthoff F; Glorius F Visible-Light-Photosensitized Aryl and Alkyl Decarboxylative Functionalization Reactions. Angew. Chem. Int. Ed. 2019, 58, 10514–10520. [DOI] [PubMed] [Google Scholar]
- 168.Patra T; Bellotti P; Strieth-Kalthoff F; Glorius F Photosensitized Intermolecular Carboimination of Alkenes through the Persistent Radical Effect. Angew. Chem. Int. Ed. 2020, 59, 3172–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Leifert D; Studer A The Persistent Radical Effect in Organic Synthesis. Angew. Chem. Int. Ed. 2020, 59, 74–108. [DOI] [PubMed] [Google Scholar]
- 170.Zhang Y; Liu H; Tang L; Tang H-J; Wang L; Zhu C; Feng C Intermolecular Carboamination of Unactivated Alkenes. J. Am. Chem. Soc. 2018, 140, 10695–10699. [DOI] [PubMed] [Google Scholar]
- 171.Patra T; Das M; Daniliuc CG; Glorius F; Metal-Free Photosensitized Oxyimination of Unactivated Alkenes with Bifunctional Oxime Carbonates. Nat. Catal. 2021, 4, 54–61. [Google Scholar]
- 172.Nicastri MC; Lehnherr D; Lam Y-H; DiRocco DA; Rovis T Synthesis of Sterically Hindered Primary Amines by Concurrent Tandem Photoredox Catalysis. J. Am. Chem. Soc. 2020, 142, 987–998. [DOI] [PubMed] [Google Scholar]
- 173.Talele TT The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem. 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- 174.Meanwell NA Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- 175.The lifetime (τ0), the rate of radiative decay (kr) and the rate of non-radiative decay (knr) of excited-state of [Ir(dMeppy)2(dtbbpy)]PF6 were obtained from:; Ladouceur S; Swanick KN; Gallagher-Duval S; Ding Z; Zysman-Colman E Strongly Blue Luminescent Cationic Iridium(III) Complexes with an Electron-Rich Ancillary Ligand: Evaluation of Their Optoelectronic and Electrochemiluminescence Properties. Eur. J. Inorg. Chem. 2013, 5329–5343. [Google Scholar]
- 176.Zhang M; Duan Y; Li W; Xu P; Cheng J; Yu S; Zhu C A Single Electron Transfer (SET) Approach to C–H Amidation of Hydrazones via Visible-Light Photoredox Catalysis. Org. Lett. 2016, 18, 5356–5359. [DOI] [PubMed] [Google Scholar]
- 177.For discussion on electronic structures of three-electron bonds, see:; Danovich D; Foroutan-Nejad C; Hiberty PC; Shaik S Nature of the Three-Electron Bond. J. Phys. Chem. A 2018, 122, 1873–1885. [DOI] [PubMed] [Google Scholar]
- 178.Boivin J; Fouquet E; Schiano A-M; Zard SZ Iminyl Radicals: Part III. Further Synthetically Useful Sources of Iminyl Radicals. Tetrahedron 1994, 50, 1769–1776. [Google Scholar]
- 179.Anslyn EV; Dougherty DA Modern Physical Organic Chemistry, 1st ed.; University Science Books: Sausalito, 2006. [Google Scholar]
- 180.Dauncey EM; Morcillo SP; Douglas JJ; Sheikh NS; Leonori D Photoinduced Remote Functionalisations by Iminyl Radical Promoted C–C and C–H Bond Cleavage Cascades. Angew. Chem. Int. Ed. 2018, 57, 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yan H; Zhu C SelectFluor Radical Fluorination for Preparing Alkyl Fluorides. In Synthetic Organofluorine Chemistry [Online]; Hu J, Umemoto T, Series Eds.; Fluorination. Springer: Singapore, 2018; 3, pp 1–9. 10.1007/978-981-10-1855-8 (accessed December 10, 2020). [DOI] [Google Scholar]
- 182.Wang P-Z; He B-Q; Cheng Y; Chen J-R; Xiao W-J Radical C–C Bond Cleavage/Addition Cascade of Benzyl Cycloketone Oxime Ethers Enabled by Photogenerated Cyclic Iminyl Radicals. Org. Lett. 2019, 21, 6924–6929. [DOI] [PubMed] [Google Scholar]
- 183.For related mechanism on excited 2-Cl-AQN engaging in hydrogen-atom abstraction, see:; Matsui K; Ishigami T; Yamaguchi T; Yamaguchi E; Tada N; Miura T; Itoh A Aerobic Photooxidative Synthesis of Phenols from Arylboronic Acids Using 2-Propanol as Solvent. Synlett 2014, 25, 2613–2616. [Google Scholar]
- 184.Bortolamei N; Isse AA; Gennaro A Estimation of Standard Reduction Potentials of Alkyl Radicals Involved in Atom Transfer Radical Polymerization. Electrochim. Acta 2010, 55, 8312– 8318. [Google Scholar]
- 185.He Y; Anand D; Sun Z; Zhou L Visible-Light-Promoted Redox Neutral γ,γ-Difluoroallylation of Cycloketone Oxime Ethers with Trifluoromethyl Alkenes via C–C and C–F Bond Cleavage. Org. Lett. 2019, 21, 3769–3773. [DOI] [PubMed] [Google Scholar]
- 186.For seminal report on the Hudson reaction, see:; Brown C; Hudson RF; Record KAF The Reaction Between Oximes and Sulphinyl Chlorides: A Ready, Low-Temperature Radical Rearrangement Process. J. Chem. Soc., Perkin Trans. 2 1978, 822–828. [Google Scholar]
- 187.Lin X-C; Stien D; Weinreb SM A New Method for the Generation and Cyclization of Iminyl Radicals via the Hudson Reaection. Org. Lett. 1999, 1, 637–639. [DOI] [PubMed] [Google Scholar]
- 188.Pratt DA; Blake JA; Mulder P; Walton JC; Korth H-G; Ingold KU O–H Bond Dissociation Enthapies in Oximes: Order Restored. J. Am. Chem. Soc. 2004, 126, 10667–10675. [DOI] [PubMed] [Google Scholar]
- 189.Xia P-J; Ye Z-P; Hu Y-Z; Song D; Xiang H-Y; Chen X-Q Yang H Photocatalytic, Phosphoranyl Radical-Mediated N–O Cleavage of Strained Cycloketone Oximes. Org. Lett. 2019, 21, 2658–2662. [DOI] [PubMed] [Google Scholar]
- 190.For an electron spin resonance spectroscopy investigation of phosphoranyl radicals from phosphines, see:; Kochi JK; Krusic PJ Displacement of Alkyl Groups from Organophosphorus Compounds Studied by Electron Spin Resonance. J. Am. Chem. Soc. 1969, 91, 3944– 3946. [Google Scholar]
- 191.For the pioneering literature in this area, see:; Zhang M-L; Xie J; Zhu C-J A General Deoxygenation Approach For Synthesis Of Ketones From Aromatic Carboxylic Acids And Alkenes. Nat. Commun. 2018, 9, 3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang M; Yuan X-A; Zhu C; Xie J Deoxygenative Deuteration of Carboxylic Acids with D2O. Angew. Chem. Int. Ed. 2019, 58, 312–316. [DOI] [PubMed] [Google Scholar]
- 193.Stache EE; Ertel AB; Rovis T; Doyle AG Generation of Phosphoranyl Radicals via Photoredox Catalysis Enables Voltage-Independent Activation of Strong C–O Bonds. ACS Catal. 2018, 8, 11134–11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Single-crystal X-ray diffraction structure of triarylphosphine radicals have been characterized. See:; Pan X; Chen X; Li T; Li Y; Wang X Isolation and X-ray Crystal Structures of Triarylphosphine Radical Cations. J. Am. Chem. Soc. 2013, 135, 3414– 3417. [DOI] [PubMed] [Google Scholar]
- 195.Padney G; Pooranchand D; Bhalerao UT Photoinduced single electron transfer activation of organophosphines: Nucleophilic trapping of phosphine radical cation. Tetrahedron 1991, 47, 1745–1752. [Google Scholar]
- 196.Phosphoranyl radicals have emerged as popular synthetic intermediates in the photoredox community. For general review on this topic, see:; Ashton-Rossi JA; Clarke AK; Unsworth WP; Taylor RJK Phosphoranyl Radical Fragmentation Reactions Driven by Photoredox Catalysis. ACS Catal. 2020, 10, 7250–7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.For pioneering literature on β-scission of phosphoranyl-radical, see:; Bentrude WG; Hansen ER; Khan WA; Rogers PE α vs. β Scission in Reactions of Alkoxy and Thiyl Radicals with Diethyl Alkylphosphonites. J. Am. Chem. Soc. 1972, 94, 2867– 2868. [Google Scholar]
- 198.For the first reported use of trifluoromethyl alkene as an electrophile in an SN2′ reaction with carbon-nucleophile, see:; Kendrick DA; Kolb M A New, Short, Synthetic Route to α-Substituted 5,5-Difluoro-4-Pentenoic Acid Esters. J. Fluorine Chem. 1989, 45, 265–272. [Google Scholar]
- 199.For the pioneering literature that discloses photoredox-mediated strategy to employs trifluoromethyl alkene to install gem-difluoroalkene moiety, see:; Xiao T; Li L; Zhou L Synthesis of Functionalized gem-Difluoroalkenes via a Photocatalytic Decarboxylative/Defluorinative Reaction. J. Org. Chem. 2016, 81, 7908–7916. [DOI] [PubMed] [Google Scholar]
- 200.Dauncey EM; Dighe SU; Douglas JJ; Leonori D A dual photoredox-nickel strategy for remote functionalization via iminyl radicals: radical ring-opening-arylation, -vinylation and -alkylation cascades. Chem. Sci, 2019, 10, 7728–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tu J-L; Tang W; Xu W; Liu F Iminyl-Radical-Promoted C–C Bond Cleavage/Heck-Like Coupling via Dual Cobaloxime and Photoredox Catalysis. J. Org. Chem. 2021, 86, 2929–2940. [DOI] [PubMed] [Google Scholar]
- 202.Nishimura T; Yoshinaka T; Nishiguchi Y; Maeda Y; Uemura S Iridium-Catalyzed Ring Cleavage Reaction of Cyclobutanone O-Benzoyloximes Providing Nitriles. Org. Lett. 2005, 7, 2425–2427. [DOI] [PubMed] [Google Scholar]
- 203.Li L; Chen H; Mei M; Zhou L Visible-light promoted γ-cyanoalkyl radical generation: three-component cyanopropylation/etherification of unactivated alkenes. Chem. Commun, 2017, 53, 11544–11547. [DOI] [PubMed] [Google Scholar]
- 204.Wayner DDM; McPhee DJ; Griller D Oxidation and Reduction Potentials of Transient Free Radicals. J. Am. Chem. Soc. 1988, 110, 132–137. [Google Scholar]
- 205.Shen X; Zhao J-J; Yu S Photoredox-Catalyzed Intermolecular Remote C–H and C–C Vinylation via Iminyl Radicals. Org. Lett. 2018, 20, 5523–3327. [DOI] [PubMed] [Google Scholar]
- 206.Källström S; Leino R Synthesis of pharmaceutically active compounds containing a disubstituted piperidine framework. Bioorg. Med. Chem. 2008, 16, 601–635. [DOI] [PubMed] [Google Scholar]
- 207.Yu X-Y; Chen J-R; Wang P-Z; Yang M-N; Lian D; Xiao W-J A Visible-Light-Driven Iminyl Radical-Mediated C–C Single Bond Cleavage/Radical Addition Cascade of Oxime Esters. Angew. Chem. Int. Ed. 2018, 57, 738–743. [DOI] [PubMed] [Google Scholar]
- 208.Yao S; Zhang K; Zhou Q-Q; Zhao Y; Shi D-Q; Xiao W-J Photoredox-Promoted Alkyl Radical Addition/Semipinacol Rearrangement Sequences of Alkenylcyclobutanols: Rapid Access to Cyclic Ketones. Chem. Commun, 2018, 54, 8096–8099. [DOI] [PubMed] [Google Scholar]
- 209.He B-Q; Yu X-Y; Wang P-Z; Chen J-R; Xiao W-J A photoredox catalyzed iminyl radical-triggered C–C bond cleavage/addition/Kornblum oxidation cascade of oxime esters and styrenes: synthesis of ketonitriles. Chem. Commun, 2018, 54, 12262–12265. [DOI] [PubMed] [Google Scholar]
- 210.Zhao B; Tan H; Chen C; Jiao N; Shi Z Photoinduced C–C Bond Cleavage and Oxidation of Cycloketoxime Esters. Chin. J. Chem. 2018, 36, 995–999. [Google Scholar]
- 211.Tomita R; Yasu Y; Koike T; Akita M Combining Photoredox-Catalyzed Trifluoromethylation and Oxidation with DMSO: Facile Synthesis of α-Trifluoromethylated Ketones from Aromatic Alkenes. Angew. Chem. Int. Ed. 2014, 53, 7144–7148. [DOI] [PubMed] [Google Scholar]
- 212.Notably, radical-mediated formal Kornblum oxidation reactions relied primarily on peroxide substrates in Kornblum–LaMer reactions. For a pioneering example, see:; Kornblum N; DeLaMare HE The Base Catalyzed Decomposition of a Dialkyl Peroxide. J. Am. Chem. Soc. 1951, 73, 880–881. [Google Scholar]
- 213.Mao R; Yuan Z; Li Y; Wu J N-Radical-Initiated Cyclization through Insertion of Sulfur Dioxide under Photoinduced Catalyst-Free Conditions. Chem. Eur. J. 2017, 23, 8176–8179. [DOI] [PubMed] [Google Scholar]
- 214.Li Y; Mao R; Wu J N-Radical Initiated Aminosulfonylation of Unactivated C(sp3)–H Bond through Insertion of Sulfur Dioxide. Org. Lett. 2017, 19, 4472–4475. [DOI] [PubMed] [Google Scholar]
- 215.Ye S; Wu J A Palladium-Catalyzed Reaction of Aryl Halides, Potassium Metabisulfite, and Hydrazines. Chem. Commun, 2012, 48, 10037–10039. [DOI] [PubMed] [Google Scholar]
- 216.Zhang J; Li X; Xie W; Ye S; Wu J Photoredox-Catalyzed Sulfonylation of O-Acyl Oximes via Iminyl Radicals with the Insertion of Sulfur Dioxide. Org. Lett. 2019, 21, 4950–4954. [DOI] [PubMed] [Google Scholar]
- 217.Bowry VW; Lusztyk J; Ingold KU Calibration of a New Horologery of Fast Radical “Clocks”. Ring-Opening Rates for Ring- and α-Alkyl-Substituted Cyclopropylcarbinyl Radicals and for the Bicyclo[2.1.0]pent-2-yl Radical. J. Am. Chem. Soc. 1991, 113, 5687–5698. [Google Scholar]
- 218.Liu Y; Wang Q-L; Chen Z; Li H; Xiong B-Q; Zhang P-L; Tang K-W Visible-Light Photoredox-Catalyzed Dual C–C Bond Cleavage: Synthesis of 2-Cyanoalkylsulfonylated 3,4-Dihydronaphthalenes Through the Insertion of Sulfur Dioxide. Chem. Commun, 2020, 56, 3011–3014. [DOI] [PubMed] [Google Scholar]
- 219.Zheng M; Li G; Lu H Photoredox- or Metal-Catalyzed in Situ SO2-Capture Reactions: Synthesis of β-Ketosulfones and Allylsulfones. Org. Lett. 2019, 21, 1216–1220. [DOI] [PubMed] [Google Scholar]
- 220.Huang W; Hu L The Chemistry of Perfluoroalkanesulfonyl Iodides. J. Fluorine Chem. 1989, 44, 25–44. [Google Scholar]
- 221.Sulfone byproduct from radical addition to SO2 generated from organic triflate fragmentation has been documented. See:; Uenoyama Y; Fukuyama T; Morimoto K; Nobuta O; Nagai H; Ryu I Trifluoromethyl-Radical-Mediated Carbonylation of Alkanes Leading to Ethynyl Ketones. Helv. Chim. Acta 2006, 89, 2483–2494. [Google Scholar]
- 222.Liu S; Jie J; Yu J; Yang X Visible light induced Trifluoromethyl Migration: Easy Access to α-Trifluoromethylated Ketones from Enol Triflates. Adv. Synth. Catal. 2018, 360, 267–271. [Google Scholar]
- 223.For pioneering literature on the Minisci reaction, see:; Minisci F; Galli R; Cecere M; Malatesta V; Caronna MT Nucleophilic character of alkyl radicals: new syntheses by alkyl radicals generated in redox processes. Tetrahedron 1968, 8, 5609–5612. [Google Scholar]
- 224.For review on Minisci reactions in organic synthesis, see:; Proctor RSJ; Phipps RJ Recent Advances in Minisci-Type Reaections. Angew. Chem. Int. Ed. 2019, 58, 13666–13699. [DOI] [PubMed] [Google Scholar]
- 225.Gu Y-R; Duan X-H; Yang L; Guo L-N Direct C–H Cyanoalkylation of Heteroaromatic N-Oxides and Quinones via C–C Bond Cleavage of Cyclobutanone Oximes. Org. Lett. 2017, 19, 5908–5911. [DOI] [PubMed] [Google Scholar]
- 226.Zhao B; Kong X; Xu B Visible-light-driven cyanoalkylation of quinoxalinones using cyclobutanone oxime esters as the radical precursors. Tetrahedron Lett. 2019, 60, 2063–2066. [Google Scholar]
- 227.Zhang -W; Pan Y-L; Yang C; Li X; Wang B Ring-opening C(sp3)–C coupling of cyclobutanone oxime esters for the preparation of cyanoalkyl containing heterocycles enabled by photocatalyst. Org. Chem. Front, 2019, 6, 2765–2770. [Google Scholar]
- 228.Xia P-J; Hu Y-Z; Ye Z-P; Li X-J; Xiang H-Y; Yang H O-Perfluoropyridin-4-yl Oximes: Iminyl Radical Precursors for Photo- or Thermal-Induced N–O Cleavage in C(sp2)–C(sp3) Bond Formation. J. Org. Chem. 2020, 85, 3538–3547. [DOI] [PubMed] [Google Scholar]
- 229.Jian Y; Chen M; Yang C; Xia W-J Minisci-Type C–H Cyanoalkylation of Heteroarenes Through N–O/C–C Bonds Cleavage. Eur. J. Org. Chem. 2020, 1439–1442. [Google Scholar]
- 230.Li X; Yan X; Wang Z; He X; Dai Y; Yan X; Zhao D; Xu X Complementary Oxidative Generation of Iminyl Radicals from α-Imino-oxy Acids: Silver-Catalyzed C–H Cyanoalkylation of Heterocycles and Quinones. J. Org. Chem. 2020, 85, 2504–2511. [DOI] [PubMed] [Google Scholar]
- 231.Wang P-Z; Yu X-Y; Li C-Y; He B-Q; Chen J-R; Xiao W-J A Photocatalytic Iminyl Radical-Mediated C–C Bond Cleavage/Addition/Cyclization Cascade for the Synthesis of 1,2,3,4-Tetrahydrophenanthrenes. Chem. Commun, 2018, 54, 9925–9928. [DOI] [PubMed] [Google Scholar]
- 232.Yuan Y; Dong W-H; Gao X-S; Xie X-M; Zhang Z-G Visible-light-induced radical cascade cyclization of oxime esters and aryl isonitriles: synthesis of cyclopenta[b]quinoxalines. Chem. Commun, 2019, 55, 11900–11903. [DOI] [PubMed] [Google Scholar]
- 233.Lenoir I; Smith ML Vinyl isonitriles in radical cascade reactions: formation of cyclopenta-fused pyridines and pyrazines. J. Chem. Soc., Perkin Trans. 1 2000, 641–643. [Google Scholar]
- 234.Camaggi CM; Leardini R; Nanni D; Zanardi G Radical Annulations with Nitriles: Novel Cascade Reactions of Cyano-Substituted Alkyl and Sulfanyl Radicals with Isonitriles. Tetrahedron, 1998, 54, 5587–5598. [Google Scholar]
- 235.Anand D; He Y; Li L; Zhou L A Photocatalytic sp3 C–S, C–Se and C–B Bond Formation Through C–C Bond Cleavage of Cycloketone Oxime Esters. Org. Biomol. Chem. 2019, 17, 533–540. [DOI] [PubMed] [Google Scholar]
- 236.Zhang Z; Ye J-H; Ju T; Liao L-L; Huang H; Gui Y-Y; Zhou W-J; Yu D-G Visible-Light-Driven Catalytic Reductive Carboxylation with CO2. ACS Catal. 2020, 10, 10871–10885. [Google Scholar]
- 237.Jiang Y-X; Chen L; Ran C-K; Song L; Zhang W; Liao L-L; Yu D-G Visible-Light Photoredox-Catalyzed Ring-Opening Carboxylation of Cyclic Oxime Esters with CO2. ChemSusChem 2020, 13, 6312–6317. [DOI] [PubMed] [Google Scholar]
- 238.Pischel U; Zhang X; Hellrung B; Haselbach E; Muller P-A; Nau WM Fluorescence Quenching of n,π*-Excited Azoalkanes by Amines: What Is a Sterically Hindered Amine? J. Am. Chem. Soc. 2000, 122, 2027–2034. [Google Scholar]
- 239.Li Z; Torres-Ochoa RO; Wang Q; Zhu J Functionalization of Remote C(sp3)–H Bonds Enabled by Copper-Catalyzed Coupling of O-acyloximes with Terminal Alkynes. Nat. Commun, 2020, 11, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Hossain A; Bhattacharyya A; Reiser O Copper’s Rapid Ascent in Visible-Light Photoredox Catalysis. Science 2019, 364, 450. [DOI] [PubMed] [Google Scholar]
- 241.Pintauer T; Matyjaszewski K Atom Transfer Radical Addition and Polymerization Reactions Catalyzed by ppm Amounts of Copper Complexes. Chem. Soc. Rev, 2008, 37, 1087–1097. [DOI] [PubMed] [Google Scholar]
- 242.Yu X-Y; Zhao Q-Q; Chen J; Chen J-R Xiao W-J Copper-Catalyzed Radical Cross-Coupling of Redox-Active Oxime Esters, Styrenes, and Boronic Acids. Angew. Chem. Int. Ed. 2018, 57, 15505–15509. [DOI] [PubMed] [Google Scholar]
- 243.Chen J; He B-Q; Wang P-Z; Yu X-Y; Zhao Q-Q; Chen J-R; Xiao W-J Photoinduced, Copper-Catalyzed Radical Cross-Coupling of Cycloketone Oxime Esters, Alkenes, and Terminal Alkynes. Org. Lett. 2019, 21, 4359–4364. [DOI] [PubMed] [Google Scholar]
- 244.Huang H; Ji X; Wu W; Jiang H Transition Metal-Catalyzed C–H Functionalization of N-Oxyenamine Internal Oxidants. Chem. Soc. Rev, 2015, 44, 1155–1171. [DOI] [PubMed] [Google Scholar]
- 245.Zhao B; Wu Y; Yuan Y; Shi Z Copper-catalysed Csp3–Csp cross-couplings between cyclobutanone oxime esters and terminal alkynes induced by visible light. Chem. Commun, 2020, 56, 4676–4679. [DOI] [PubMed] [Google Scholar]
- 246.Wang T; Wang Y-N; Wang R; Zhang B-C; Yang C; Li Y-L; Wang X-S Enantioselective Cyanation via Radical-Mediated C–C Single Bond Cleavage For Synthesis of Chiral Dinitriles. Nat. Commun. 2019, 10, 5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Chen J; Wang P-Z; Lu B; Liang D; Yu X-Y; Xiao W-J Chen J-R Enantioselective Radical Ring-Opening Cyanation of Oxime Esters by Dual Photoredox and Copper Catalysis. Org. Lett. 2019, 21, 9763–9768. [DOI] [PubMed] [Google Scholar]
- 248.Davies IA; Gerena L; Castonguay L; Senanayake CH; Larsen RD; Verhoeven TR; Reider PJ The Influence of Ligand Bite Angle On the Enantioselectivity of Copper(II)-Catalysed Diels-Alder Reactions. Chem. Commun, 1996, 1753–1754. [Google Scholar]
- 249.Holzer W; Penzkofer A; Tsuboi T Absorption And Emission Spectroscopic Characterization of Ir(ppy)3. Chem. Phys. 2005, 308, 93–102. [Google Scholar]
- 250.Yu X-Y; Wang P-Z; Yan D-M; Lu B; Chen J-R; Xiao W-J Photocatalytic Neophyl Rearrangement and Reduction of Distal Carbon Radicals by Iminyl Radical-Mediated C–C Bond Cleavage. Adv. Synth. Catal. 2018, 360, 3601–3606. [Google Scholar]
- 251.Zhao B; Chen C; Lu J; Li Z; Yuan Y; Shi Z Photoinduced Fragmentation-Rearrangement Sequence of Cycloketoxime Esters. Org. Chem. Front. 2018, 5, 2719–2722. [Google Scholar]
- 252.It is reported that aminium radical cations are more electrophilic than their neutral aminyl radical counterparts and can rapidly engage in HAT events. For a physical-organic investigation on this matter, see:; Hioe J; Šakić D; Vrček V; Zipse H The stability of nitrogen-centered radicals. Org. Biomol. Chem, 2015, 13, 157–169. [DOI] [PubMed] [Google Scholar]
- 253.Xuan J; Zhang Z-G; Xiao W-J Visible-Light-Induced Decarboxylative Functionalization of Carboxylic Acids and Their Derivatives. Angew. Chem. Int. Ed. 2015, 54, 15632–15641. [DOI] [PubMed] [Google Scholar]
- 254.Liang T; Neumann CN; Ritter T Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [DOI] [PubMed] [Google Scholar]
- 255.Jiang H; Studer A α-Aminoxy-Acid-Auxillary-Enabled Intermolecular Radical γ-C(sp3)–H Functionalization of Ketones. Angew. Chem. Int. Ed. 2018, 57, 1692–1696. [DOI] [PubMed] [Google Scholar]
- 256.Xie J; Jin H; Hashmi ASK The recent achievements of redox-neutral radical C–C cross-coupling enabled by visible-light. Chem. Soc. Rev, 2017, 46, 5193–5203. [DOI] [PubMed] [Google Scholar]
- 257.Chen L; Guo L-N; Ma Z-Y; Gu Y-R; Zhang J; Duan X-H Iminyl Radical-Triggered 1,5-Hydrogen-Atom Transfer/Heck-Type Coupling by Visible-Light Photoredox Catalysis. J. Org. Chem. 2019, 84, 6475–6482. [DOI] [PubMed] [Google Scholar]
- 258.Ma Z-Y; Guo L-N; Gu Y-R; Chen L; Duan X-H Iminyl Radical-Mediated Controlled Hydroxyalkylation of Remote C(sp3)–H Bond via Tandem 1,5-HAT and Difunctionalization of Aryl Alkenes. Adv. Synth. Catal. 2018, 360, 4341–4347. [Google Scholar]
- 259.For mechanistic discussions about radical enantioselective C(sp3)–H reactions, see:; Zhang C; Li Z-L; Gu Q-S; Liu X-Y Catalytic Enantioselective C(sp3)–H Functionalization Involving Radical Intermediates. Nat. Commun 2021, 12, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Nakafuku KM; Zhang Z; Wappes EA; Stateman LM; Chen AD; Nagib DA Enantioselective Radical C–H Amination for the Synthesis of β-Amino Alcohols. Nat. Chem 2020, 12, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Qin Q; Yu S; Visible-Light-Promoted Redox Neutral C–H Amidation of Heteroarenes with Hydroxylamine Derivatives. Org. Lett. 2014, 16, 3504–3507. [DOI] [PubMed] [Google Scholar]
- 262.Davies J; Svejstrup TD; Reina DF; Sheikh NS; Leonori D Visible-Light-Mediated Synthesis of Amidyl Radicals: Transition-Metal-Free Hydroamination and N-Arylation Reactions. J. Am. Chem. Soc. 2016, 138, 8092–8095. [DOI] [PubMed] [Google Scholar]
- 263.Pertinent manuscripts were published contemporaneously with the submission of this review, see as an example:; Zhang Z; Ngo DT; Nagib DA Regioselective Radical Amino-Functionalizations of Allyl Alcohols via Dual Catalytic Cross-Coupling. ACS Catal. 2021, 11, 3473–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Wu K; Du Y; Wang T Visible-Light-Mediated Construction of Pyrroloindolines via an Amidyl Radical Cyclization/Carbon Radical Addition Cascade: Rapid Synthesis of (±)-Flustramide B. Org. Lett. 2017, 19, 5669–5672. [DOI] [PubMed] [Google Scholar]
- 265.Wu K; Du Y; Wei Z; Wang T Synthesis of Functionalized Pyrroloindolines via a Visible-Light-Induced Radical Cascade Reaction: Rapid Synthesis of (±)-Flustraminol B. Chem. Commun, 2018, 54, 7443–7446. [DOI] [PubMed] [Google Scholar]
- 266.Qin Q; Han Y-Y; Jiao Y-Y; He Y; Yu S Photoredox-Catalyzed Diamidation and Oxidative Amidation Alkenes: Solvent-Enabled Synthesis of 1,2-Diamides and α-Amino Ketones. Org. Lett. 2017, 19, 2909–2912. [DOI] [PubMed] [Google Scholar]
- 267.Sim BA; Milne PH; Griller D; Wayner DDM Thermodynamic Significance of ρ+ and ρ– from Substituent Effects on the Redox Potentials of Arylmethyl Radicals. J. Am. Chem. Soc. 1990, 112, 6635–6638. [Google Scholar]
- 268.Jiang H; Studer A Amidyl Radicals by Oxidation of α-Amido-oxy acids: Transition-Metal-Free Amidofluorination of Unactivated Alkenes. Angew. Chem. Int. Ed. 2018, 57, 10707–10711. [DOI] [PubMed] [Google Scholar]
- 269.Jiang H; Studer A Anti-Markovnikov Radical Hydro- and Deuteroamidation of Unactivated Alkenes. Chem. Eur. J. 2019, 25, 7105–7109. [DOI] [PubMed] [Google Scholar]
- 270.Huo H; Shen X; Wang C; Zhang L; Röse P; Chen L-A; Harms K; Marsch M; Hilt G; Meggers E Asymmetric Photoredox Transition-Metal Catalysis Activated by Visible Light. Nature, 2014, 515, 100–103. [DOI] [PubMed] [Google Scholar]
- 271.For review on the concept of “smart initiator”, see:; Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem. Int. Ed. 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]
- 272.Kim JH; Ruffoni A; Al-Faiyz YSS; Sheikh NS; Leonori D Divergent Strain-Release Amino-Functionalization of [1.1.1]Propellane with Electrophilic Nitrogen-Radicals. Angew. Chem. Int. Ed. 2020, 59, 8225–8231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Morcillo SP; Dauncey EM; Kim JH; Douglas JJ; Sheikh NS; Leonori D Photoinduced Remote Functionalization of Amides and Amines Using Electrophilic Nitrogen Radicals. Angew. Chem. Int. Ed. 2018, 57, 12945–12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Gennet D; Zard SZ; Zhang HW Amidinyl radicals: new and useful intermediates for the synthesis of imidazolines and imidazoles. Chem. Commun. 2003, 15, 1870–1871. [DOI] [PubMed] [Google Scholar]
- 275.Yang HB; Selander N Divergent iron-catalyzed coupling of O-acyloximes with silyl enol ethers. Chem. Eur. J. 2017, 23, 1779–1783. [DOI] [PubMed] [Google Scholar]
- 276.Yang HB; Pathipati SR; Selander N Nickel-Catalyzed 1,2-Aminoarylation of Oxime Ester-Tethered Alkenes with Boronic Acids. ACS Catal. 2017, 7, 8441–8445. [Google Scholar]
- 277.Huang J; He Y; Wang Y; Zhu Q Synthesis of Benzimidazoles by PIDA-Promoted Direct C(sp2)–H Imidation of N-Arylamidines. Chem. Eur. J. 2012, 18, 13964–13967. [DOI] [PubMed] [Google Scholar]
- 278.Alla SK; Kumar RK; Sadhu P; Punniyamurthy T Iodobenzene Catalyzed C−H Amination of N-Substituted Amidines Using m-Chloroperbenzoic Acid. Org. Lett. 2013, 15, 1334–1337. [DOI] [PubMed] [Google Scholar]
- 279.Zhao HB; Hou ZW; Liu ZJ; Zhou ZF; Song JS; Xu HT Amidinyl Radical Formation Through Anodic N−H Bond Cleavage And Its Application in Aromatic C−H Bond Functionalization. Angew. Chem. Int. Ed. 2017, 56, 587–590. [DOI] [PubMed] [Google Scholar]
- 280.Li G; He R; Liu Q; Wang Z; Liu Y; Wang Q Formation of Amidinyl Radicals via Visible-Light-Promoted Reduction of N-Phenyl Amidoxime Esters and Application to the Synthesis of 2-Substituted Benzimidazoles. J. Org. Chem. 2019, 84, 8646–8660. [DOI] [PubMed] [Google Scholar]
- 281.Neale RS; Hinman RL The Chemistry of Ion Radicals. The Free Radical Addition of N-Chlorodialklamines to Butadiene. J. Am. Chem. Soc. 1963, 85, 2666–2667. [Google Scholar]
- 282.Neale RS The Chemistry of Ion Radicals. The Free-Radical Addition of N-Chloramines to Olefinic and Acetylenic Hydrocarbons. J. Am. Chem. Soc. 1964, 86, 5340–5342. [Google Scholar]
- 283.N-nitrosylated analogues have been employed. See:; Chow L Nitrosamine Photochemistry. Reactions of Aminium Radicals. Acc. Chem. Res. 1973, 6, 354–360. [Google Scholar]
- 284.Chow YC; Danen WC; Nelsen SF; Rosenblatt D Nonaromatic Aminium Radicals. Chem. Rev. 1978, 78, 243–274. [Google Scholar]
- 285.Musacchio AJ; Nguyen LQ; Beard GH; Knowles RR Catalytic Olefin Hydroamination with Aminium Radical Cations: A Photoredox Method for Direct C–N Bond Formation. J. Am. Chem. Soc. 2014, 136, 12217–12220. [DOI] [PubMed] [Google Scholar]
- 286.For the first known, photocatalytic, aminium radical process for C–N bond formation, see:; Maity S; Zheng N A Visible-Light-Mediated Oxidative C–N Bond Formation/Aromatization Cascade: Photocatalytic Preparation of N-Arylindoles. Angew. Chem. Int. Ed. 2012, 51, 9562–9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Michejda CJ; Campbell DH Addition of Complexed Amino Radicals to Conjugated Alkenes. J. Am. Chem. Soc. 1979, 101, 7687–7693. [Google Scholar]
- 288.Wayner DDM; Griller D Oxidation and Reduction Potentials of Transient Free Radicals. J. Am. Chem. Soc. 1985, 107, 7764–7765. [Google Scholar]
- 289.Nguyen TM; Nicewicz DA Anti-Markovnikov Hydroamination of Alkenes Catalyzed by an Organic Photoredox System. J. Am. Chem. Soc. 2013, 135, 9588–9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Musacchio AJ; Lainhart BC; Zhang X; Naguib SG; Sherwood TC; Knowles RR Catalytic Intermolecular Hydroaminations of Unactivated Olefins with Secondary Alkyl Amines. Science 2017, 355, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Wagner BD; Ruel G; Lusztyk J Absolute Kinetics of Aminium Radical Reactions with Olefins in Acetonitrile Solution. J. Am. Chem. Soc. 1996, 118, 13–19. [Google Scholar]
- 292.Miller DC; Ganley JM; Musacchio AJ; Sherwood TC; Ewing WR; Knowles RR Anti-Markovnikov Hydroamination of Unactivated Alkenens with Primary Alkyl Amines. J. Am. Chem. Soc. 2019, 141, 16590–16594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Romero NA; Margrey KA; Tay NE; Nicewicz DA Site-Selective Arene C–H Amination via Photoredox Catalysis. Science 2015, 349, 1326–1330. [DOI] [PubMed] [Google Scholar]
- 294.Margrey KA; Levens A; Nicewicz DA Direct C–H Amination with Primary Amines Using Organic Photoredox Catalysis. Angew. Chemie. Int. Ed. 2017, 129, 15850–15854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Ohkubo K; Mizushima K; Iwata R; Fukuzumi S Selective Photocatalytic Aerobic Bromination with Hydrogen Bromide via an Electron-transfer State of 9-mesityl-10-methylacridinium Ion. Chem. Sci. 2011, 2, 715–722. [Google Scholar]
- 296.Ruffoni A; Juliá F; Svejstrup TD; McMillan AJ; Douglas JJ; Leonori D Practical and Regioselective Amination of Arenes Using Alkyl Amines. Nature Chemistry 2019, 11, 426–433. [DOI] [PubMed] [Google Scholar]
- 297.Wei W; Wang L; Bao P; Shao Y; Yue H; Yang D; Yang X; Zhao X; Wang H Metal-free C(sp2)–H/N–H Cross-dehydrogenative Coupling of Quinoxalinones with Aliphatic Amines under Visible-light Photoredox Catalysis. Org. Lett. 2018, 20, 7125–7130. [DOI] [PubMed] [Google Scholar]
- 298.Zhao Y; Huang B; Yang C; Li B; Gou B; Xia W Photocatalytic Cross-dehydrogenative Amination Reactions Between Phenols and Diarylamines. ACS Catal. 2017, 7, 2446–2451. [Google Scholar]
- 299.Zhou C; Lei T; Wei X-Z; Ye C; Liu Z; Chen B; Tung C-H; Wu L-Z Metal-Free, Redox-Neutral, Site-Selective Access to Heteroarylamine via Direct Radical–Radical Cross-Coupling Powered by Visible Light Photocatalysis. J. Am. Chem. Soc. 2020, 142, 16805–16813. [DOI] [PubMed] [Google Scholar]
- 300.For an early example of transition metal-catalyzed aryl amination employing nitrile groups as a pseudo-halide leaving-group, see:; Miller JA; Dankwardt JW; Penney JM Nickel-Catalyzed Cross-Coupling and Amination Reactions of Aryl Nitriles. Synthesis 2003, 11, 1643–1648. [Google Scholar]
- 301.Carbon–carbon cross-coupling via loss of nitrile from a (hetero)arene has been previously demonstrated, see:; Zuo Z; MacMillan DWC Decarboxylative Arylation of α-Amino Acids via Photoredox Catalysis: A One-step Conversion of Biomass to Drug Pharmacophore. J. Am. Chem. Soc. 2014, 136, 5257–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.For a recent review of metal-mediated C–CN bond activation, see:; Nakao Y Metal-mediated C–CN Bond Activation in Organic Synthesis. Chem. Rev. 2021, 121, 327–344. [DOI] [PubMed] [Google Scholar]
- 303.Pandey G; Koley S; Talukdar R; Sahani PK Cross-dehydrogenating Coupling of Aldehydes with Amines/R-OTBS Ethers by Visible-light Photoredox Catalysis: Synthesis of Amides, Esters, and Ureas. Org. Lett. 2018, 20, 5861–5865. [DOI] [PubMed] [Google Scholar]
- 304.Bordwell FG; Harrelson JA; Lynch TY Homolytic Bond Dissociation Energies for the Cleavage of Alpha-nitrogen-hydrogen Bonds in Carboxamides, Sulfonamides, and Their Derivatives. The Question of Synergism in Nitrogen-centered Radicals. J. Org. Chem. 1990, 55, 3337–3341. [Google Scholar]
- 305.Bordwell FG; Ji G-Z Effects of Structural Changes on Acidities and Homolytic Bond Dissociation Energies of the H–N bonds in Amidines, Carboxamides, and Thiocarboxamides. J. Am. Chem. Soc. 1991, 113, 8398–8401. [Google Scholar]
- 306.Bordwell FG; Zhang S; Zhang X-M; Liu W-Z Homolytic Bond Dissociation Enthalpies of the Acidic H–A Bonds Caused by Proximate Substituents in Sets of Methyl Ketones, Carboxylic Esters, and Carboxamides Related to Changes in Ground State Energies. J. Am. Chem. Soc. 1995, 117, 7092–7096. [Google Scholar]
- 307.Wolff ME Cyclization of N-Halogenated Amines (The Hofmann-Löffler Reaction). Chem. Rev. 1963, 63, 55–64. [Google Scholar]
- 308.Lin X; Stien D; Weinreb SM A Mild New Procedure for Production, Cyclization and Trapping of Amidyl Radicals: Application to a Formal Total Synthesis of Peduncularine. Tetrahedron Lett. 2000, 41, 2333–2337. [Google Scholar]
- 309.Gagosz F; Moutrille C; Zard SZ A New Tin-Free Source of Amidyl Radicals. Org. Lett. 2002, 4, 2707–2709. [DOI] [PubMed] [Google Scholar]
- 310.Fuentes N; Kong W; Fernandez-Sanchez L; Merino E; Nevado C Cyclization Cascades via N-Amidyl Radicals Toward Highly Functionalized Heterocyclic Scaffolds. J. Am. Chem. Soc. 2015, 137, 964–973. [DOI] [PubMed] [Google Scholar]
- 311.Fallis AG; Brinza IM Free Radical Cyclizations Involving Nitrogen. Tetrahedron 1997, 53, 17543–17594. [Google Scholar]
- 312.Esker JL; Newcomb M Amidyl Radicals from N-(Phenylthio)amides. Tetrahedron Lett. 1993, 34, 6877–6880. [Google Scholar]
- 313.Chen K; Richter JM; Baran PS 1,3-Diol Synthesis via Controlled, Radical-Mediated C−H Functionalization. J. Am. Chem. Soc. 2008, 130, 7247–7249. [DOI] [PubMed] [Google Scholar]
- 314.Zhou L; Tang S; Qi X; Lin C; Liu K; Liu C; Lan Y; Lei A Transition-metal-assisted Radical/radical Cross-coupling: A New Strategy to the Oxidative C(sp3)-H/N-H Cross-coupling. Org. Lett. 2014, 16, 3404–3407. [DOI] [PubMed] [Google Scholar]
- 315.Nicolaou KC; Baran PS; Zhong YL; Sugita K Iodine(V) Reagents in Organic Synthesis. Part 1. Synthesis of Polycyclic Heterocycles via Dess–Martin Periodinane-Mediated Cascade Cyclization: Generality, Scope, and Mechanism of the Reaction. J. Am. Chem. Soc. 2002, 124, 2212–2220. [DOI] [PubMed] [Google Scholar]
- 316.Nicolaou KC; Baran PS; Zhong YL; Barluenga S; Hunt KW; Kranich R; Vega JA Iodine(V) Reagents in Organic Synthesis. Part 3. New Routes to Heterocyclic Compounds via o-Iodoxybenzoic Acid-Mediated Cyclizations: Generality, Scope, and Mechanism. J. Am. Chem. Soc. 2002, 124, 2233–2244. [DOI] [PubMed] [Google Scholar]
- 317.Nicolaou KC; Baran PS; Kranich R; Zhong Y-L; Sugita K; Zou N Mechanistic Studies of Periodinane-Mediated Reactions of Anilides and Related Systems. Angew. Chem. Int. Ed. 2001, 40, 202–206. [DOI] [PubMed] [Google Scholar]
- 318.Martín A; Pérez-Martín I; Suárez E Intramolecular Hydrogen Abstraction Promoted by Amidyl Radicals. Evidence for Electronic Factors in the Nucleophilic Cyclization of Ambident Amides to Oxocarbenium Ions. Org. Lett. 2005, 7, 2027–2030. [DOI] [PubMed] [Google Scholar]
- 319.Janza B; Studer A Stereoselective Cyclization Reactions of IBX-Generated Alkoxyamidyl Radicals. J. Org. Chem. 2005, 70, 6991–6994. [DOI] [PubMed] [Google Scholar]
- 320.Hernández R; Medina MC; Salazar JA; Suárez E; Prangé T Intramolecular Functionalization of Amides Leading to Lactams. Tetrahedron Lett. 1987, 28, 2533–2536. [Google Scholar]
- 321.Freire R; Martín A; Pérez-Martín I; Suárez E Synthesis of Oxa-aza Spirobicycles by Intramolecular Hydrogen Abstraction Promoted by N-Radicals in Carbohydrate Systems. Tetrahedron Lett. 2002, 43, 5113–5116. [Google Scholar]
- 322.Choi GJ; Knowles RR Catalytic Alkene Carboaminations Enabled by Oxidative Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2015, 137, 9226–9229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Bordwell FG; Cheng J-P; Harrelson JA Homolytic bond dissociation energies in solution from equilibrium acidity and electrochemical data. J. Am. Chem. Soc. 1988, 110, 1229–1231. [Google Scholar]
- 324.Mayer JM Proton-coupled Electron Transfer: A Reaction Chemist’s View. Annu. Rev. Phys. Chem. 2004, 55, 363–390. [DOI] [PubMed] [Google Scholar]
- 325.Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev. 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Waidmann CR; Miller AJM; Ng C-WA; Scheuermann ML; Porter TR; Tronic TA; Mayer JM Using Combinations of Oxidants and Bases as PCET Reactants: Thermochemical and Practical Considerations. Energy Environ. Sci 2012, 5, 7771–7780. [Google Scholar]
- 327.Miller DC; Tarantino KT; Knowles RR Proton-Coupled Electron Transfer in Organic Synthesis: Fundamentals, Applications, and Opportunities. Top. Curr. Chem. 2016, 374, 30, DOI: 10.1007/s41061-016-0030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Miller DC; Choi GJ; Orbe HS; Knowles RR Catalytic Olefin Hydroamidation Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2015, 137, 13492–13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Qiu G; Knowles RR Understanding Chemoselectivity in Proton-Coupled Electron Transfer: A Kinetic Study of Amide and Thiol Activation. J. Am. Chem. Soc. 2019, 141, 16574–16578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Nguyen ST; Zhu Q; Knowles RR PCET-Enabled Olefin Hydroamidation Reactions with N-Alkyl Amides, ACS Catal. 2019, 9, 4502–4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Zhu Q; Graff DE; Knowles RR Intermolecular Anti-Markovnikov Hydroamination of Unactivated Alkenes with Sulfonamides Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2018, 140, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Newcomb M; Esker JL Facile Production and Cyclizations of Amidyl Radicals. Tetrahedron Lett. 1991, 32, 1035–1038. [Google Scholar]
- 333.Chou CM; Guin J; Muck-Lichtenfeld C; Grimme A; Studer A Radical-Transfer Hydroamination of Olefins with N-Aminated Dihydropyridines. Chem. - Asian J 2011, 6, 1197–1209. [DOI] [PubMed] [Google Scholar]
- 334.Tang Y; Li C Facile 5-Endo Amidyl Radical Cyclization Promoted by Vinylic Iodine Substitution. Org. Lett. 2004, 6, 3229–3231. [DOI] [PubMed] [Google Scholar]
- 335.Medina-Ramos J; Oyesanya O; Alvarez JC Buffer Effects in the Kinetics of Concerted Proton-Coupled Electron Transfer: The Electrochemical Oxidation of Glutathione Mediated by [IrCl6]2− at Variable Buffer pKa and Concentration. J. Phys. Chem. C 2013, 117, 902–912. [Google Scholar]
- 336.Kuss-Petermann M; Wenger OS Mechanistic Diversity in Proton-Coupled Electron Transfer between Thiophenols and Photoexcited [Ru(2,2′-Bipyrazine)3]2+. J. Phys. Chem. Lett 2013, 4, 2535–2539. [Google Scholar]
- 337.Gagliardi CJ; Murphy CF; Binstead RA; Thorp HH; Meyer TJ Concerted Electron–Proton Transfer (EPT) in the Oxidation of Cysteine. J. Phys. Chem. C 2015, 119, 7028–7038. [Google Scholar]
- 338.Ruccolo S; Qin Y; Schnedermann C; Nocera DG General Strategy for Improving the Quantum Efficiency of Photoredox Hydroamidation Catalysis. J. Am. Chem. Soc. 2018, 140, 14926–14937. [DOI] [PubMed] [Google Scholar]
- 339.Zhao Y; Bordwell FG; Cheng J-P; Wang D Equilibrium Acidities and Homolytic Bond Dissociation Energies (BDEs) of the Acidic H−N Bonds in Hydrazines and Hydrazides. J. Am. Chem. Soc. 1997, 119, 9125–9129. [Google Scholar]
- 340.Hu X-Q; Qi X; Chen J-R; Zhao Q-Q; Wei Q; Lan Y; Xiao W-J Catalytic N-Radical Cascade Reaction of Hydrazones by Oxidative Deprotonation Electron Transfer and TEMPO Mediation. Nat. Commun. 2016, 7, 11188–11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Rand AW; Yin H; Xu L; Giacoboni J; Martin-Montero R; Romano C; Montgomery J; Martin R Dual Catalytic Platform for Enabling sp3 α C–H Arylation and Alkylation of Benzamides. ACS Catal. 2020, 10, 4671–4676. [Google Scholar]
- 342.Jia J; Ho YA; Bülow RF; Rueping M Brønsted Base Assisted Photoredox Catalysis: Proton Coupled Electron Transfer for Remote C–C Bond Formation Via Amidyl Radicals. Chem. Eur. J. 2018, 24, 14054–14058. [DOI] [PubMed] [Google Scholar]
- 343.Moon Y; Jang E; Choi S; Hong S Visible-Light-Photocatalyzed Synthesis of Phenanthridinones and Quinolinones Via Direct Oxidative C–H Amidation. Org. Lett. 2018, 20, 240–243. [DOI] [PubMed] [Google Scholar]
- 344.Usami K; Yamaguchi E; Tada N; Itoh A Transition-Metal-Free Synthesis of Phenanthridinones Through Visible-Light-Driven Oxidative C–H Amidation. Eur. J. Org. Chem. 2020, 1496–1504. [Google Scholar]
- 345.Hofmann AW Chem. Ber. 1883, 16, 558–560. [Google Scholar]
- 346.Kumler WD; Eiler JJ The Acid Strength of Mono and Diesters of Phosphoric Acid. The n-Alkyl Esters from Methyl to Butyl, the Esters of Biological Importance, and the Natural Guanidine Phosphoric Acids. J. Am. Chem. Soc. 1943, 65, 2355–2361. [Google Scholar]
- 347.Zhang L; Meggers E Steering Asymmetric Lewis Acid Catalysis Exclusively with Octahedral Metal-Centered Chirality. Acc. Chem. Res. 2017, 50, 320–330. [DOI] [PubMed] [Google Scholar]
- 348.Yuan W; Zhou Z; Gong L; Meggers E Asymmetric Alkylation of Remote C(sp3)–H Bonds by Combining Proton-coupled Electron Transfer with Chiral Lewis Acid Catalysis. Chem. Commun. 2017, 53, 8964–8967. [DOI] [PubMed] [Google Scholar]
- 349.Zhou Z; Li Y; Han B; Gong L; Meggers E Enantioselective Catalytic β-Amination Through Proton-coupled Electron Transfer Followed by Stereocontrolled Radical–Radical Coupling. Chem. Sci 2017, 8, 5757–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Wang C; Chen L-A; Huo H; Shen X; Harms K; Gong L; Meggers E Asymmetric Lewis Acid Catalysis Directed by Octahedral Rhodium Centrochirality. Chem. Sci. 2015, 6, 1094–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Ma J; Shen X; Harms K; Meggers E Expanding the Family of Bis-cyclometalated Chiral-at-metal Rhodium(III) Catalysts with a Benzothiazole Derivative. Dalton Trans. 2016, 45, 8320–8323. [DOI] [PubMed] [Google Scholar]
- 352.Huo H, Fu C, Harms K and Meggers E, Asymmetric Catalysis with Substitutionally Labile yet Stereochemically Stable Chiral-at-Metal Iridium(III) Complex. J. Am. Chem. Soc. 2014, 136, 2990–2993. [DOI] [PubMed] [Google Scholar]
- 353.Chu JCK; Rovis T Amide-directed photoredox-catalysed C–C bond formation at unactivated sp3 C–H bonds. Nature 2016, 539, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Chen D-F; Chu JCK; Rovis T Directed γ-C(sp3)–H Alkylation of Carboxylic Acid Derivatives through Visible Light Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 14897–14900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Pandey G; Laha R Visible-Light-Catalyzed Direct Benzylic C(sp3)–H Amination Reaction by Cross-Dehydrogenative Coupling. Angew. Chem. Int. Ed. 2015, 54, 14875–14879. [DOI] [PubMed] [Google Scholar]
- 356.Zhang Y; Chen W; Wang L; Li P Visible-light-induced Selective Amination of Enol Ethers with N-Alkoxyamides by Using DDQ as a Photoredox Catalyst. Org. Chem. Front. 2018, 5, 3562–3566. [Google Scholar]
- 357.Short MA; Shehata MF; Sanders MA; Roizen JL Sulfamides Direct Radical-mediated Chlorination of Aliphatic C–H Bonds. Chem. Sci. 2020, 11, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Shehata MF; Short MA; Sanders MA; Roizen JL Efficient Synthesis of Unsymmetrical Sulfamides with Sulfamic Acid Salts by Activation with Triphenylphosphine Ditriflate. Tetrahedron 2019, 75, 3186–3194. [Google Scholar]
- 359.Bafaluy D; Georgieva Z; Muniz K Iodine Catalysis for C(sp3)–H Fluorination with a Nucleophilic Fluorine Source. Angew. Chem. Int. Ed. 2020, 59, 14241–14245. [DOI] [PubMed] [Google Scholar]
- 360.For a recent review of sulfamide-related chemistry, see:; Spillane W; Malaubier J-B Sulfamic Acid and Its N- and O-Substituted Derivatives. Chem. Rev. 2014, 114, 2507–2586. [DOI] [PubMed] [Google Scholar]
- 361.Roos CB; Demaerel J; Graff DE; Knowles RR Enantioselective Hydroamination of Alkenes with Sulfonamides Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2020, 142, 5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Nairoukh Z; Cormier M; Marek I Merging C–H and C–C bond Cleavage in Organic Synthesis. Nature Reviews. Chemistry 2017, 1, 35. [Google Scholar]
- 363.Chen Z-M; Zhang X-M; Tu Y-Q Radical Aryl Migration Reactions and Synthetic Applications. Chem. Soc. Rev. 2015, 44, 5220–5245. [DOI] [PubMed] [Google Scholar]
- 364.Zhou T; Luo F-X; Yang M-Y; Shi Z-J Silver-Catalyzed Long-Distance Aryl Migration from Carbon Center to Nitrogen Center. J. Am. Chem. Soc. 2015, 137, 14586–14589. [DOI] [PubMed] [Google Scholar]
- 365.Shu W; Genoux A; Li Z; Nevado C γ-Functionalizations of Amines through Visible-Light-Mediated, Redox-Neutral C–C Bond Cleavage. Angew. Chem. Int. Ed. 2017, 56, 10521–10524. [DOI] [PubMed] [Google Scholar]
- 366.Liu C; Cai C; Yuan C; Jiang Q; Fang Z; Guo K Visible-light-promoted N-centered Radical Generation for Remote Heteroaryl Migration. Org. Biomol. Chem. 2020, 18, 7663–7670. [DOI] [PubMed] [Google Scholar]
- 367.Sathyamoorthi S; Banerjee S; Du Bois J; Burns N; Zare RN Site-selective bromination of sp3 C–H bonds. Chem. Sci. 2018, 9, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.These investigations have been reviewed:; Short MA, Blackburn J. Miles, Roizen JL Modifying Positional Selectivity in C–H Functionalization Reactions with Nitrogen-Centered Radicals: Generalizable Approaches to 1,6-Hydrogen-Atom Transfer Processes. Synlett 2020, 31, 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.For related studies of sulfamides as directing groups for exogenous atom-transfer, see:; Short MA, Shehata MF, Sanders MA, Roizen JL Sulfamides Direct Radical-Mediated Chlorination of Aliphatic C–H Bonds. Chem. Sci. 2020, 11, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Kanegusuku ALG; Castanheiro T; Ayer SK; Roizen JL Sulfamyl Radicals Direct Photoredox-Mediated Giese Reactions at Unactivated C(3)–H Bonds. Org. Lett. 2019, 21, 6089–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.A preprint of this manuscript was published on May 31, 2019:; Kanegusuku ALG; Castanheiro T; Ayer SK; Roizen JL Sulfamyl Radicals Direct Photoredox-Mediated Giese Reactions at Unactivated C(3)−H Bonds. ChemRxiv 2019, Preprint. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.The bulk of this research was initially disclosed in August 19, 2018:; Kanegusuku ALG; Castanheiro T; Roizen JL Sulfamate esters guide selective alkylation at traditionally non-reactive gamma-C(sp3)–H bonds. ABSTRACTS OF PAPERS OF THE AMERICAN CHEMICAL SOCIETY 256. [Google Scholar]
- 373.Ma Z-Y; Guo L-N; You Y; Yang F; Hu M; Duan X-H Visible Light Driven Alkylation of C(sp3)−H Bonds Enabled by 1,6-Hydrogen Atom Transfer/Radical Relay Addition. Org. Lett. 2019, 21, 5500–5504. [DOI] [PubMed] [Google Scholar]
- 374.Shu W; Zhang H; Huang Y γ-Alkylation of Alcohols Enabled by Visible-Light Induced 1,6-Hydrogen Atom Transfer. Org. Lett. 2019, 21, 6107–6111. [DOI] [PubMed] [Google Scholar]
- 375.Publications describing these Giese reactions rely, at least in part, on sulfamate ester preparation methods disclosed in; Blackburn JM, Short MA, Castanheiro T, Ayer SK, Muellers TD, Roizen JL Synthesis of N-Substituted Sulfamate Esters from Sulfamic Acid Salts by Activation with Triphenylphosphine Ditriflate. Org. Lett. 2017, 19, 6012–6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.These copper-mediated reactions oxidize 1,6-dihydropyridazine intermediates to afford pyrazole products.
- 377.Pünner F; Sohtome Y; Sodeoka M Solvent-dependent Copper-catalyzed Synthesis of Pyrazoles under Aerobic Conditions. Chem. Commun. 2016, 52, 14093–14096. [DOI] [PubMed] [Google Scholar]
- 378.Fan Z; Feng J; Hou Y; Rao Min.; Cheng J Copper-Catalyzed Aerobic Cyclization of β,γ-Unsaturated Hydrazones with Concomitant C=C Bond Cleavage. Org. Lett. 2020, 22, 7981–7985. [DOI] [PubMed] [Google Scholar]
- 379.Zhu MK; Chen YC; Loh TP Radical-Mediated Diamination of Alkenes with Phenylhydrazine and Azodicarboxylates: Highly Diastereoselective Synthesis of trans-Diamines from Cycloalkenes. Chem. Eur. J. 2013, 19, 5250–5254. [DOI] [PubMed] [Google Scholar]
- 380.Duan X-Y; Yang XL; Fang R; Peng XX; Yu W; Han B Hydrazone Radical Promoted Vicinal Difunctionalization of Alkenes and Trifunctionalization of Allyls: Synthesis of Pyrazolines and Tetrahydropyridazines. J. Org. Chem. 2013, 78, 10692–10704. [DOI] [PubMed] [Google Scholar]
- 381.Zhu X; Wang Y; Ren W; Zhang F-L; Chiba S TEMPO-Mediated Aliphatic C–H Oxidation with Oximes and Hydrazones. Org. Lett. 2013, 15, 3214–3217. [DOI] [PubMed] [Google Scholar]
- 382.Zhu X; Chiba S TEMPO-mediated allylic C–H amination with hydrazones. Org. Biomol. Chem. 2014, 12, 4567–4570. [DOI] [PubMed] [Google Scholar]
- 383.Duan X-Y; Zhou N-N; Fang R; Yang X-L; Yu W; Han B Transition from π Radicals to σ Radicals: Substituent-Tuned Cyclization of Hydrazonyl Radicals. Angew. Chem., Int. Ed. 2014, 53, 3158–3162. [DOI] [PubMed] [Google Scholar]
- 384.Duan X-Y; Yang X-L, Jia P-P; Zhang M; Han B Hydrazonyl Radical-Participated Tandem Reaction: A Strategy for the Synthesis of Pyrazoline-Functionalized Oxindoles. Org. Lett. 2015, 17, 6022–6025. [DOI] [PubMed] [Google Scholar]
- 385.Hu X-Q; Chen J-R; Wei Q; Liu F-L; Deng Q-H; Beauchemin AM; Xiao W-J Photocatalytic Generation of N-Centered Hydrazonyl Radicals: A Strategy for Hydroamination of β, γ -Unsaturated Hydrazones. Angew. Chem., Int. Ed. 2014, 53, 12163–12167. [DOI] [PubMed] [Google Scholar]
- 386.Hu X-Q; Chen J; Chen J-R; Yan D-M; Xiao W-J Organophotocatalytic Generation of N- and O-Centred Radicals Enables Aerobic Oxyamination and Dioxygenation of Alkenes. Chem. Eur. J. 2016, 22, 14141–14146. [DOI] [PubMed] [Google Scholar]
- 387.Zhao Q-Q; Chen J; Yan D-M; Chen J-R; Xiao W-J Photocatalytic Hydrazonyl Radical-Mediated Radical Cyclization/ Allylation Cascade: Synthesis of Dihydropyrazoles and Tetrahydropyridazines. Org. Lett. 2017, 19, 3620–3623. [DOI] [PubMed] [Google Scholar]
- 388.Zhao Q-Q; Hu XQ; Yang M-N; Chen J-R; Xiao W-J A Visible-light Photocatalytic N-Radical Cascade of Hydrazones for the Synthesis of Dihydropyrazole-fused Benzosultams. Chem. Commun. 2016, 52, 12749–12752. [DOI] [PubMed] [Google Scholar]
- 389.Zhang G; Liu C; Yi H; Meng Q; Bian C; Chen H; Jian J-X; Wu L-Z; Lei A External Oxidant-Free Oxidative Cross-Coupling: A Photoredox Cobalt-Catalyzed Aromatic C–H Thiolation for Constructing C–S Bonds. J. Am. Chem. Soc. 2015, 137, 9273–9280. [DOI] [PubMed] [Google Scholar]
- 390.Zhang G; Zhang L; Yi H; Luo Y; Qi X; Tung C-H; Wu L-Z; Lei A Visible-light Induced Oxidant-free Oxidative Cross-Coupling for Constructing Allylic Sulfones from Olefins and Sulfinic Acids. Chem. Commun. 2016, 52, 10407–10410. [DOI] [PubMed] [Google Scholar]
- 391.Niu L; Yi H; Wang S; Liu T; Liu J; Lei A Photo-induced Oxidant-free Oxidative C–H/N–H Cross-coupling between Arenes and Azoles. Nat. Commun 2017, 8, 14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Yi H; Niu L; Song C; Li Y; Dou B; Singh AK; Lei A Photocatalytic Dehydrogenative Cross-Coupling of Alkenes with Alcohols or Azoles without External Oxidant. Angew. Chem. Int. Ed. 2017, 56, 1120–1124. [DOI] [PubMed] [Google Scholar]
- 393.Gao X-W; Meng Q-Y; Li J-X; Zhong J-J; Lei T; Li X-B; Tung C-H; Wu L-Z Visible Light Catalysis Assisted Site-Specific Functionalization of Amino Acid Derivatives by C–H Bond Activation without Oxidant: Cross-Coupling Hydrogen Evolution Reaction. ACS Catal. 2015, 5, 2391–2396. [Google Scholar]
- 394.Xiang M; Meng Q-Y; Li J-X; Zheng Y-W; Ye C; Li Z-J; Chen B; Tung CH; Wu L-Z Activation of C–H Bonds through Oxidant-Free Photoredox Catalysis: Cross-Coupling Hydrogen-Evolution Transformation of Isochromans and β-Keto Esters. Chem. Eur. J. 2015, 21, 18080–18084. [DOI] [PubMed] [Google Scholar]
- 395.Azzi E; Ghigo G; Parisatto S; Pellegrino F; Priola E; Renzi P; Deagostino A Visible Light Mediated Photocatalytic N-Radical Cascade Reactivity of γ,δ-Unsaturated N-Arylsulfonylhydrazones: A General Approach to Structurally Diverse Tetrahydropyridazines. J. Org. Chem. 2021, 86, 3300–3323. [DOI] [PubMed] [Google Scholar]
- 396.Allart-Simon I; Gérard S; Sapi J Radical Smiles Rearrangement: An Update. Molecules, 2016, 21, 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Brachet E; Marzo L; Selkti M; König B; Belmont P Visible Light Amination/Smiles Cascade: Access to Phthalazine Derivatives. Chem. Sci. 2016, 7, 5002–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Yu J-M; Lu G-P; Cai C; Photocatalytic Radical Cyclization of α-Halo Hydrazones with β-Ketocarbonyls: Facile Access to Substituted Dihydropyrazoles. Chem. Commun. 2017, 53, 5342–5345. [DOI] [PubMed] [Google Scholar]
- 399.For review on stereoselective reactions that merge photocatalysis and cobalt-catalysis, see:; Kojima M; Matsunaga S The Merger of Photoredox and Cobalt Catalysis. Trends Chem. 2020, 2, 410–426. [Google Scholar]
- 400.For review on stereoselective reactions that merge photocatalysis and palladium- or nickel-catalysis, see:; Zhang H-H; Chen H; Zhu C; Yu S A Review of Enantioselective Dual Transition Metal/Photoredox Catalysis. Sci. China: Chem 2020, 63, 637– 647. [Google Scholar]
- 401.Simons RT Photo-Enabled Synthesis of Carbon–Nitrogen and Remote Carbon–Carbon Bonds. Master’s Thesis, Duke University, Durham, NC, 2021. [Google Scholar]