

Abstract
Asymmetric catalysis is a major theme of research in contemporary synthetic organic chemistry. The discovery of general strategies for highly enantioselective photochemical reactions, however, has been a relatively recent development, and the variety of photoreactions that can be conducted in a stereocontrolled manner is consequently somewhat limited. Asymmetric photocatalysis is complicated by the short lifetimes and high reactivities characteristic of photogenerated reactive intermediates; the design of catalyst architectures that can provide effective enantiodifferentiating environments for these intermediates while minimizing the participation of uncontrolled racemic background processes has proven to be a key challenge for progress in this field. This review provides a summary of the chiral catalyst structures that have been studied for solution-phase asymmetric photochemistry, including chiral organic sensitizers, inorganic chromophores, and soluble macromolecules. While some of these photocatalysts are derived from privileged catalyst structures that are effective for both ground-state and photochemical transformations, others are structural designs unique to photocatalysis and offer an insight into the logic required for highly effective stereocontrolled photocatalysis.
Graphical Abstact
1. Introduction
The chirality of a molecule can have a profound influence on its biological and physical properties. Methods that produce enantiomerically enriched products from achiral starting materials are therefore of particular importance for the synthesis of a variety of materials, including drug molecules, agrochemicals, and polymers. The 2001 Nobel Prize for Chemistry, awarded for the development of the field of asymmetric catalysis, underscores the centrality of this problem in contemporary synthetic chemistry.1–2 3 Several decades of sustained interest in enantioselective reaction method development has resulted in the elucidation of a large number of structurally diverse chiral catalyst architectures that effectively control stereochemical outcomes in almost every class of synthetically important transformation.
Photochemical reactions offer a conspicuous exception to this general trend. The emergence of general strategies for enantioselective catalytic photoreactions is a relatively recent development, and the variety of organic photoreactions that can be conducted in a highly enantioselective manner remains comparatively limited. This is despite the long fascination chemists have had with light-promoted reactions, both because of the distinctive reactive intermediates that are most readily available by photochemical activation, and because photochemical reactions often result in topologically complex molecular architectures that can be synthesized in no other way.4–5 6 It stands to reason, therefore, that there exist complications specific to enantioselective catalytic photochemical reactions in comparison to better-established asymmetric ground-state reactions. Correspondingly, the chiral catalyst structures that have proven to be most effective in asymmetric photochemistry have often been structurally unique.
One central challenge common to all areas of asymmetric catalysis is the problematic participation of racemic background processes. Product-forming reaction pathways that occur outside of the stereodifferentiating environment of a chiral catalyst necessarily result in racemic products. Thus, no matter how enantioselective a catalytic process might be, the enantioselectivity of the overall reaction is diminished if background processes are competitive. There are two features specific to photochemical activation that are uniquely challenging in this regard. The first is the possibility of competitive direct photoexcitation. If an achiral substrate molecule directly absorbs light under the conditions of a catalytic photoreaction, the resulting photoexcited species can react to give racemic product prior to interacting with the chiral catalyst. Second, many of the most widely exploited photocatalytic systems operate via collisional activation mechanisms. When a photocatalyst activates an organic substrate by a diffusional electron- or energy-transfer event, the encounter complex is typically short-lived. If dissociation of the deactivated photocatalyst and activated substrate is faster than the rate of subsequent bond formation, any chiral information associated with the photocatalyst cannot influence the stereochemical outcome of the reaction.
Thus, early investigations of catalytic asymmetric photochemistry that introduced chiral elements into well-studied organic sensitizers met with limited success.7, 8 Examples that resulted in measurable stereoinduction generally involved the formation of an exciplex, where a donor–acceptor interaction between the substrate and excited-state photocatalyst results in the formation of a longer-lived excited-state complex. Because the exciplex extends the lifetime of the encounter complex, product-formation can occur within the chiral environment of the photocatalyst. These studies provided a valuable proof-of-principle for the possibility of photocatalytic stereoinduction but suffered from low enantiomeric excess (ee) due to ill-defined substrate–catalyst interactions and relatively short exciplex lifetimes.
Highly enantioselective catalytic photoreactions have become increasingly common over the past two decades. A guiding strategy that underlies many of the most successful methods in asymmetric photocatalysis is the preassociation principle originally articulated for macromolecular photoactive hosts by Inoue9 and expanded for small molecule asymmetric photocatalysis by Krische.10 This principle argues that the participation of background product-forming pathways will be minimized when a chiral photocatalyst is capable of binding the substrate in the ground state. If the substrate in the complex is excited — either by direct excitation or sensitization — more readily than the free substrate, and if the rate of the photoreaction is faster than the rate of disassociation of the complex, then product formation will predominantly occur within the chiral environment of the photocatalyst with minimal competition from racemic background processes. Although Krische’s seminal example utilized hydrogen-bonding interactions as a strategy to enforce preassociation, many other strategies have subsequently been reported. These include the formation of electron donor–acceptor (EDA) complexes, where the formation of a ground-state association is driven by charge transfer between the catalyst and substrate; chromophore activation, where coordination of a chiral catalyst to a substrate results in a new compound with altered light-absorbing properties; association-induced photoinduced electron transfer (PET), where photon absorption is followed by a redox cascade generating reactive radicals that react with a ground-state activated substrate; and organometallic photocatalysis, where the formation of a new coordination complex engenders product-forming photochemical processes within the coordination sphere of the metal center (Scheme 1). The research area of asymmetric photochemistry has been the subject of several previous reviews.11–12 13 14 15 16 17 18 19 20
Scheme 1.
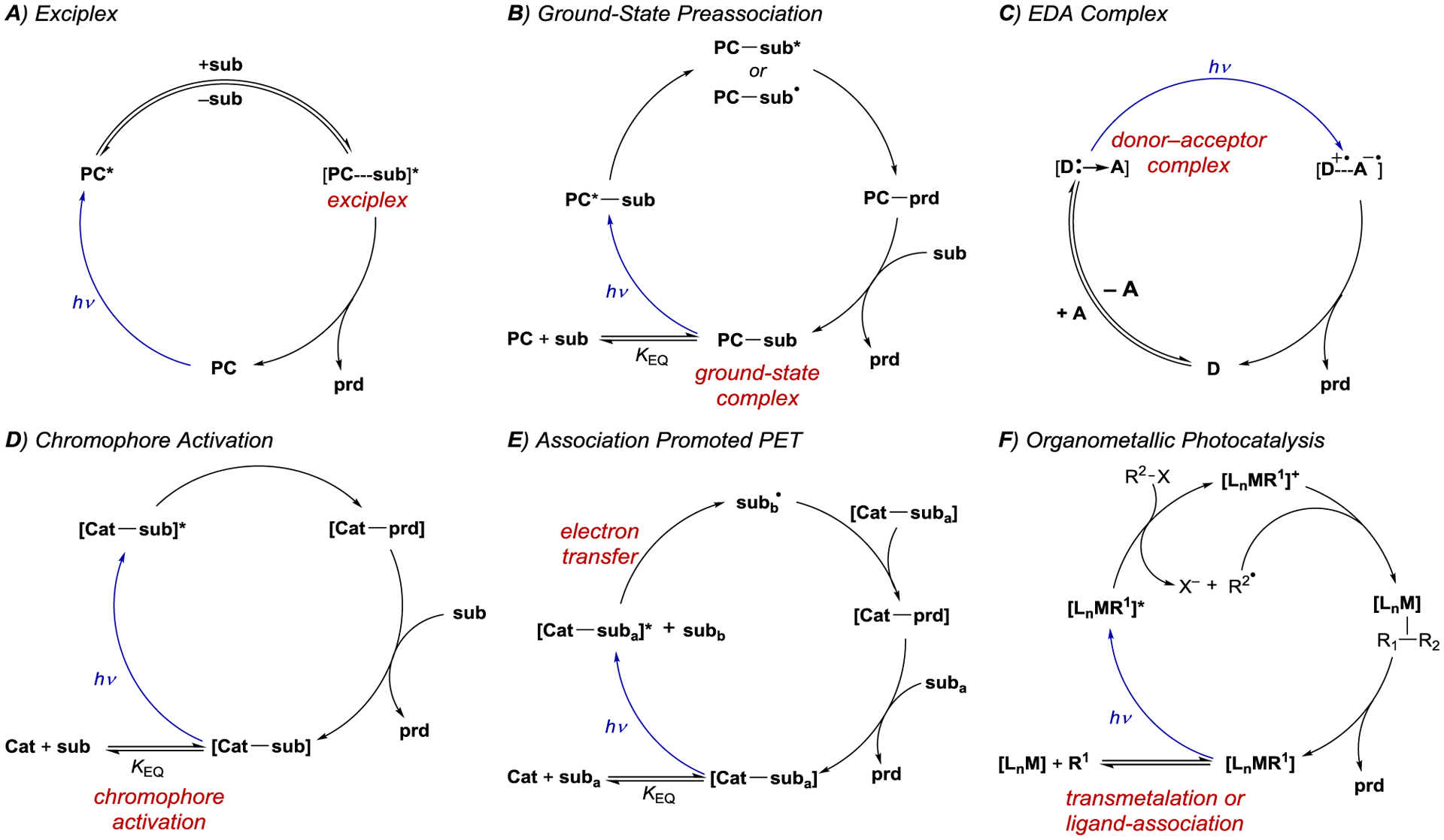
General Mechanisms of Enantiocontrol by Chiral Chromophores in Asymmetric Photocatalysis
While some privileged catalyst structures have proven to be equally applicable to both ground-state and excited-state asymmetric reactions, the challenges specific to photochemical activation have required the invention of novel catalyst structures. This review seeks to provide an examination of the field of catalytic enantioselective photochemistry through the lens of the structures that have proven to be useful as chiral photocatalysts.
The term “photocatalysis” has been used in a somewhat ambiguous fashion throughout the literature of synthetic photochemistry. For the purposes of this review, we adopt a broad definition of photocatalysis and cover any enantioselective catalytic reaction where the stereoinducing chiral catalyst forms part of the light-absorbing complex, regardless of the photophysical properties of the species in isolation. We exclude from this treatment dual-catalytic reactions where the chromophore and the chiral stereoinducing co-catalyst are separate molecular entities; this topic has been reviewed previously.21 The scope of this review is also limited to solution-phase photoreactions; stereoselective photochemistry in the solid state is a broad topic and merits separate treatment.22–23 24
There are also important distinctions that can be made among the mechanisms of photocatalytic reactions involving the nature of the reactive intermediates participating in the key bond-forming events. Primary photoreactions are those that involve bond-forming processes of organic compounds in their electronically excited states. These are distinct from secondary photoreactions, in which the key bond-forming processes are those of photochemically generated reactive intermediates such as radicals or radical ions in their electronic ground states (Scheme 2). While the reactivity patterns available from primary and secondary photoreactions differ significantly, it can sometimes be challenging to unambiguously determine which is operative in a given photoreaction. This review thus covers both classes of photocatalytic reactions, and the organization of the topics centers on the structure of the chiral catalyst rather than on mechanistic considerations.
Scheme 2.
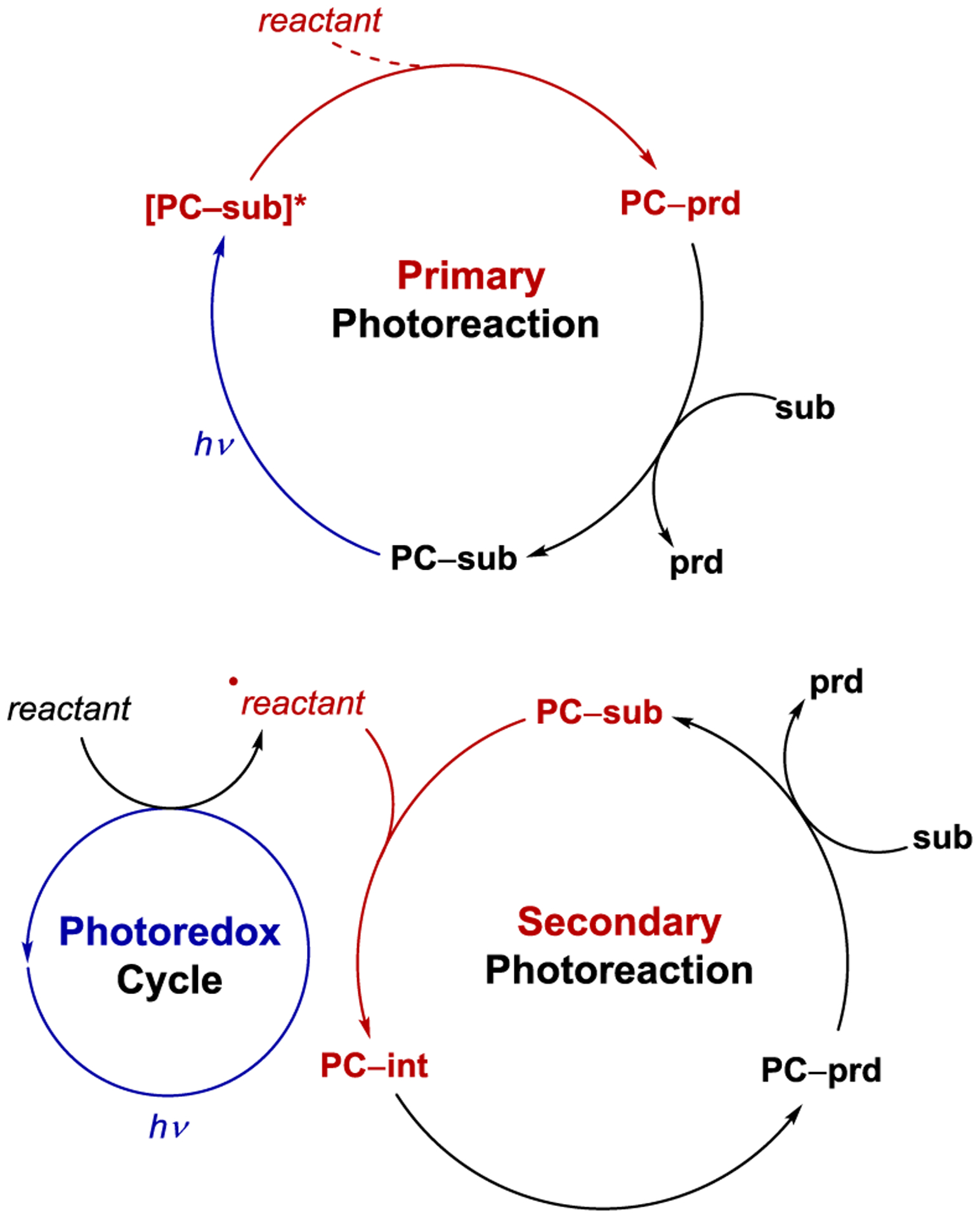
Primary Versus Secondary Photoreactions
2. Organic Chromophores
2.1. Arenes
Much of the early work in asymmetric photochemistry involved the use of chiral arene sensitizers capable of forming exciplexes. An exciplex is an electronically excited molecular complex formed between two species that are not associated in the ground state but are held together by charge-transfer interactions in the excited state (Scheme 3).25 Exciplexes have unique photophysical properties relative to the individual sensitizer and substrate. They have the potential to fluoresce (singlet exciplex) and phosphoresce (triplet exciplex), undergo radiationless decay, or chemically react to form a new species.26 Hence, the best spectroscopic evidence for the formation of an exciplex is the observation of quenching of the sensitizer fluorescence and the appearance of a new emission band that does not correspond to the emission of either component of the exciplex in isolation. Notably, substrate activation via exciplex formation is mechanistically distinct from energy transfer. In an exciplex, the exciton is shared between the sensitizer and the substrate, while during energy transfer, the exciton is transferred from the sensitizer to the substrate, resulting in a ground-state sensitizer.
Scheme 3.
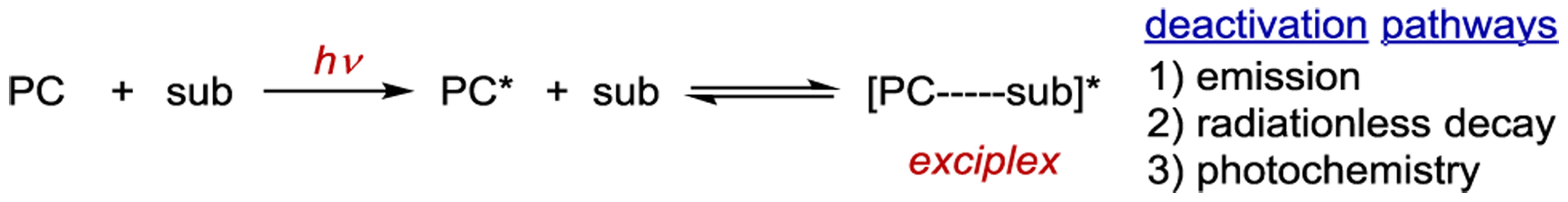
Exciplex Formation and Deactivation Pathways
Hammond reported one of the first examples of an enantioselective photoreaction in 1965 (Scheme 4).27 An optically active naphthylamide sensitizer (2) effects the isomerization of cyclopropane 1 to a mixture of cis- and optically enriched trans-1,2-diphenylcyclopropanes upon irradiation with UV light. Notably, only a 12 mol% loading of 2 gives 1 in 7% ee.28, 29 Naphthylamides bound to silica were also tested as isomerization catalysts, but the enantioselectivity was reduced to 1% ee.30 Hammond initially proposed a triplet sensitization mechanism where different energy-transfer rate constants from the chiral sensitizer to (R)- and (S)-1 lead to enantioenrichment.31–32 33 However, it was later shown that the reaction proceeds through a singlet exciplex.34 Interconversion of (R)- and (S)-1 likely occurs via cleavage of the excited cyclopropane to a 1,3-diradical followed by ring-closure.
Scheme 4.
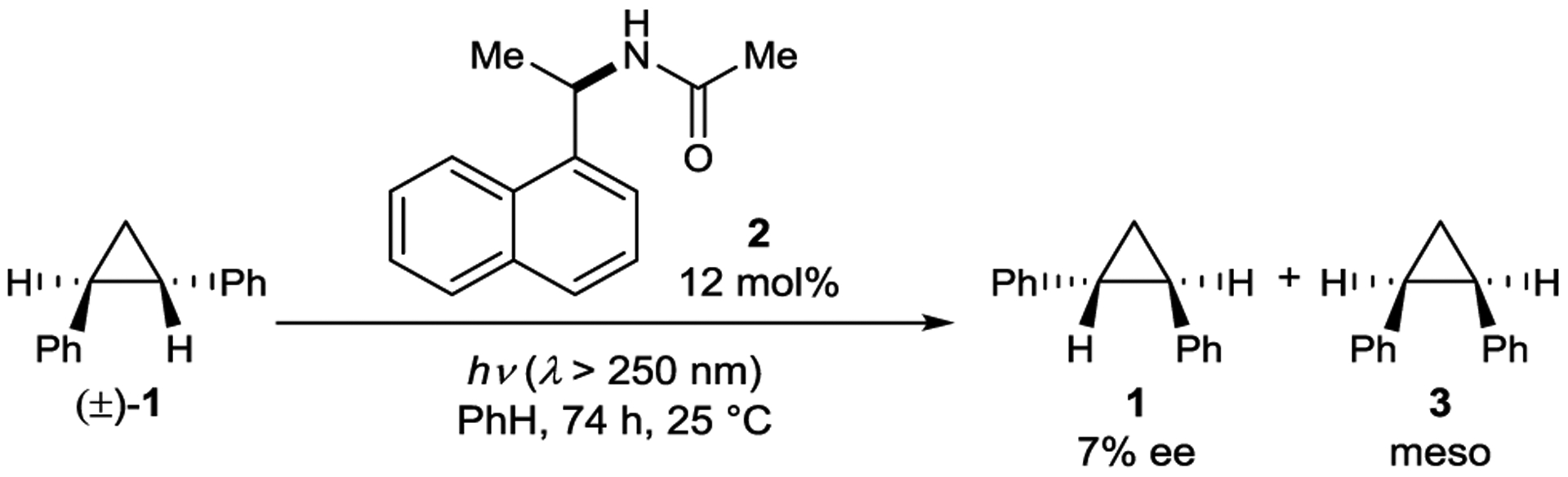
Cyclopropane Isomerization Catalyzed by a Naphthylamide Sensitizer
Naphthylamide sensitizers were also studied by Kagan for their ability to deracemize sulfoxides (Scheme 5). When racemic sulfoxide 4 is irradiated in the presence of 5, the optical purity of the recovered substrate is modestly enriched (12% ee).35, 36 Mechanistic studies on sulfoxide racemization performed by Hammond suggest that this deracemization proceeds through a singlet exciplex.37–38 39 The excited-state configurational lability of the sulfoxide could arise either from direct inversion via a planar electronically excited sulfoxide or from α-cleavage and subsequent radical–radical recombination.40, 41
Scheme 5.
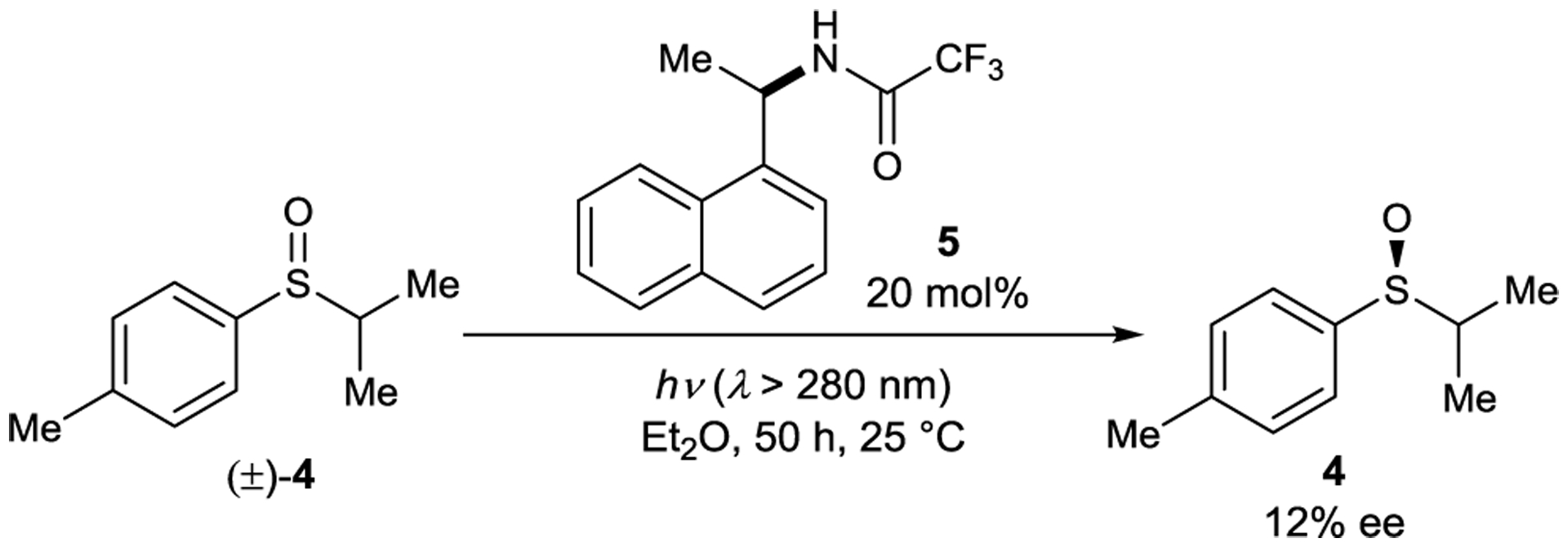
Sulfoxide Deracemization Catalyzed by a Naphthylamide Sensitizer
Several chiral naphthylamide sensitizers were also examined in the 1,5-aryl shift, di-π-methane cascade rearrangement of racemic oxepinones. The ratio of sensitization rate constants (kR/kS = 1.04) suggests an enantioselective transformation, but the ee of the product was not measured.42 Weiss reported the deracemization of 2,3-pentadiene (6) catalyzed by chiral aromatic steroid 7 (Scheme 6).43 Allenes are axially chiral and configurationally stable in the ground state but can undergo stereochemical inversion via a planar, achiral excited state. The authors did not fully elucidate the mechanism but did note that singlet and triplet sensitization from the chiral sensitizer is thermodynamically endergonic.
Scheme 6.
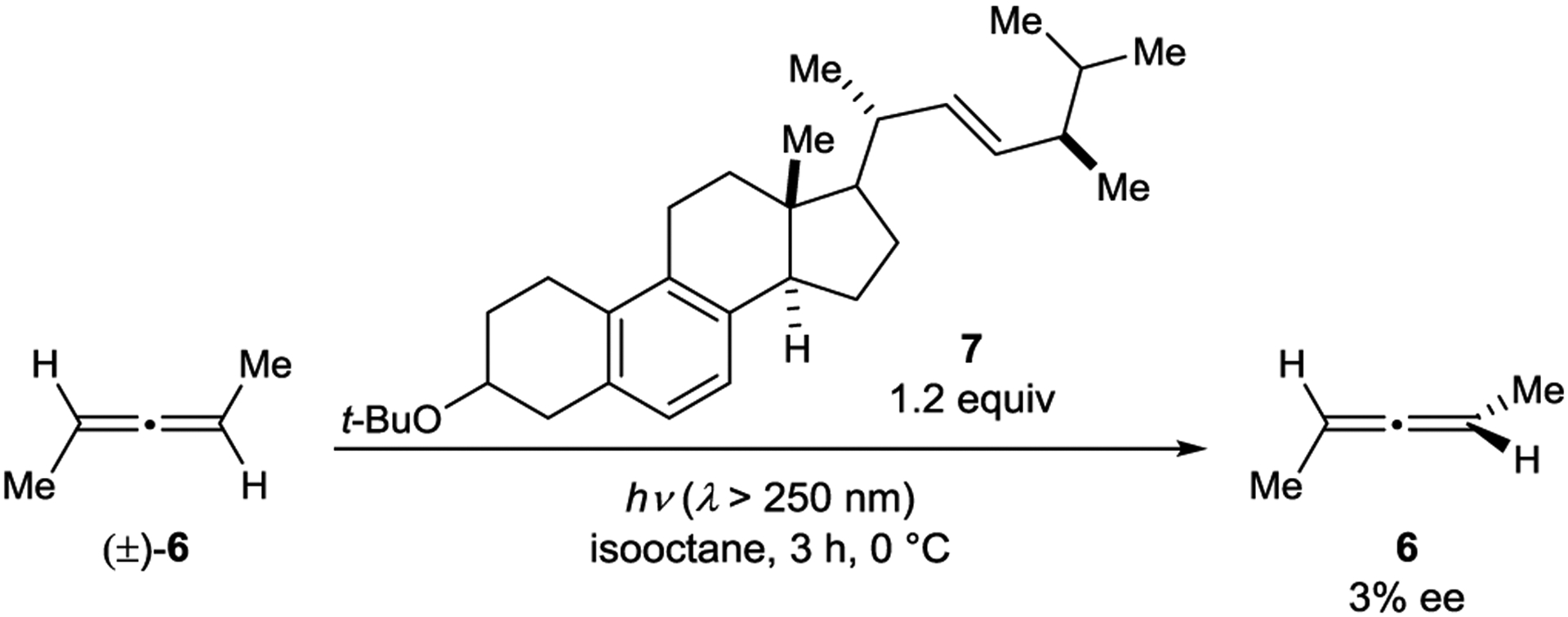
Allene Deracemization Catalyzed by a Steroid Sensitizer
Inoue studied the photochemical isomerization of achiral (Z)-cyclooctene (14) to chiral (E)-cyclooctene (13) using benzenecarboxylates as singlet exciplex sensitizers (Figure 1).44, 45 Initial reports described the production of (E)-cyclooctene in low ee (4% ee), but this process provided the opportunity to study the mechanism of stereoinduction in detail.46, 47 The isomerization proceeds through a singlet exciplex consisting of the excited photocatalyst and cyclooctene (Scheme 7). Rotational relaxation within the initial exciplex produces one of two diastereomeric exciplexes featuring a chiral twisted excited cyclooctene ((S) or (R)-1T). While the exciplex-bound (Z)-cyclooctene may decay to either twisted diastereomeric complex, exciplex-bound (S)- or (R)-cyclooctene decay to the respective (S)- or (R)-1T. After sensitizer dissociation, the excited twisted cyclooctene decays to either (Z)- or (E)-cyclooctene. An enantioselective transformation is achieved as there exists no route for the direct interconversion between the diastereomeric (R)- and (S)-twisted-cyclooctene exciplexes without first reforming (Z)-cyclooctene. Thus, the enantiodetermining step of this process could be either (1) sensitizer quenching by (E)-cyclooctene to form diastereomeric exciplexes (kqS vs. kqR) or (2) stereoselective rotational relaxation from (Z)-cyclooctene to the diastereomeric twisted exciplex intermediates (kS vs. kR). A kinetic resolution of (E)-cyclooctene produced in situ from (Z)-cyclooctene was attempted to distinguish these possibilities. If different sensitizer quenching rates by (E)-cyclooctene dictate enantioselectivity, then an increase in ee over the course of the reaction is expected. On the other hand, if the ee is invariant with time the enantioselectivity is the result of different rates of rotational relaxation from the (Z)-cyclooctene exciplex to the twisted-cyclooctene exciplexes. No change in the ee was observed over the course of the reaction, implying that rotational relaxation of the (Z)-exciplex is the enantiodetermining step.
Figure 1.
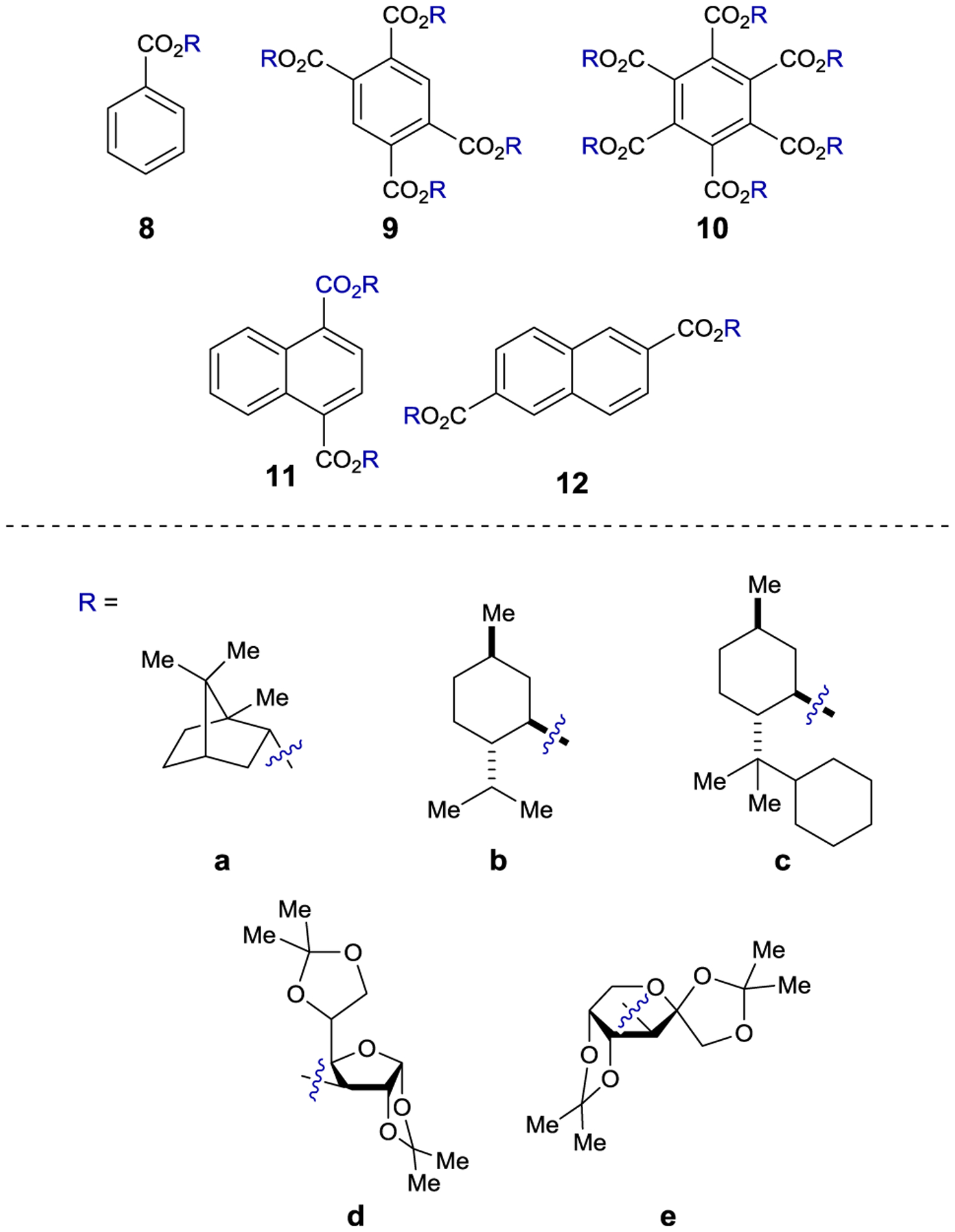
Selected Examples of Chiral Benzenecarboxylate Sensitizers
Scheme 7.
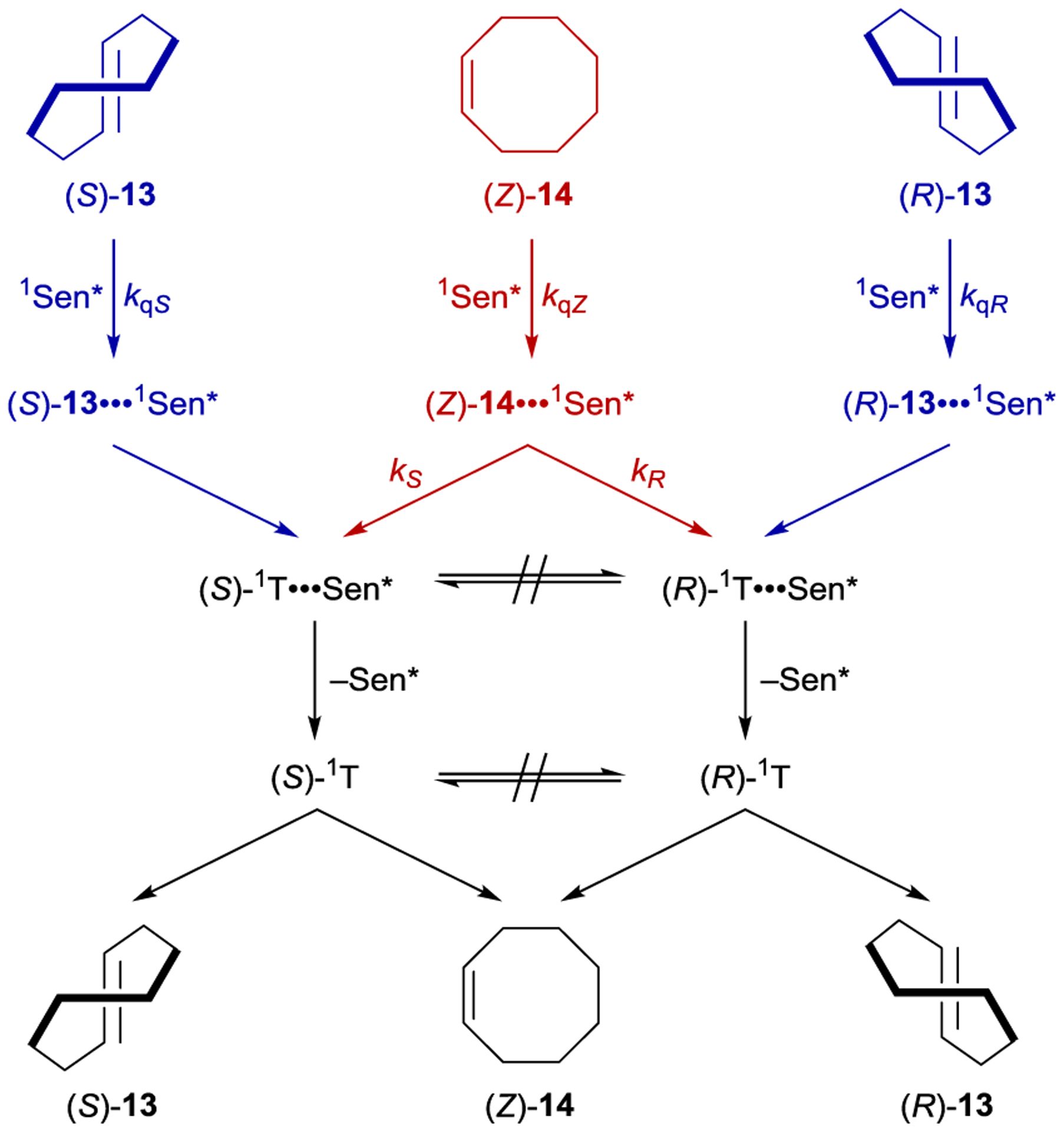
Mechanism of Cyclooctene Isomerization Catalyzed by Benzenecarboxylate Sensitizers
The importance of the singlet exciplex for obtaining high levels of enantioselectivity was investigated using benzyl ether sensitizers, which form singlet exciplexes with (Z)-cyclooctene at high substrate concentrations but undergo collisional triplet energy transfer at low substrate concentrations. The ee obtained with the benzyl ethers decreases with decreasing substrate concentration, suggesting that the singlet process is more selective than the triplet. While the catalyst–substrate interaction is long-lived in the singlet exciplex due to the charge-transfer exciplex interaction, the triplet sensitized process likely only involves a fleeting interaction between the catalyst and alkene. Thus, the triplet alkene isomerizes after dissociation from the catalyst, leading to negligible asymmetric induction.48
Extensive screening of benzenepolycarboxylate, aromatic amide, and phosphoryl ester sensitizers gave improved enantioselectivity and revealed a surprising relationship between ee and reaction temperature (Scheme 8).49–50 51 52 For bornyl sensitizer 9a, the ee of (E)-cyclooctene increases with decreasing temperature, affording 13 in 41% ee at −88 °C. For menthyl sensitizer 9b, the opposite enantiomer of 13 is favored at 25 °C, but the enantioselectivity decreases with decreasing temperatures, and at −19 °C 13 is formed as a racemate. Below this equipodal temperature, the product chirality inverts, and the ee of the antipodal enantiomer increases with decreasing temperature. This runs contrary to the common assumption that enantioselectivity and temperature should be inversely related, which is an oversimplification of the factors controlling enantioselectivity.
Scheme 8.
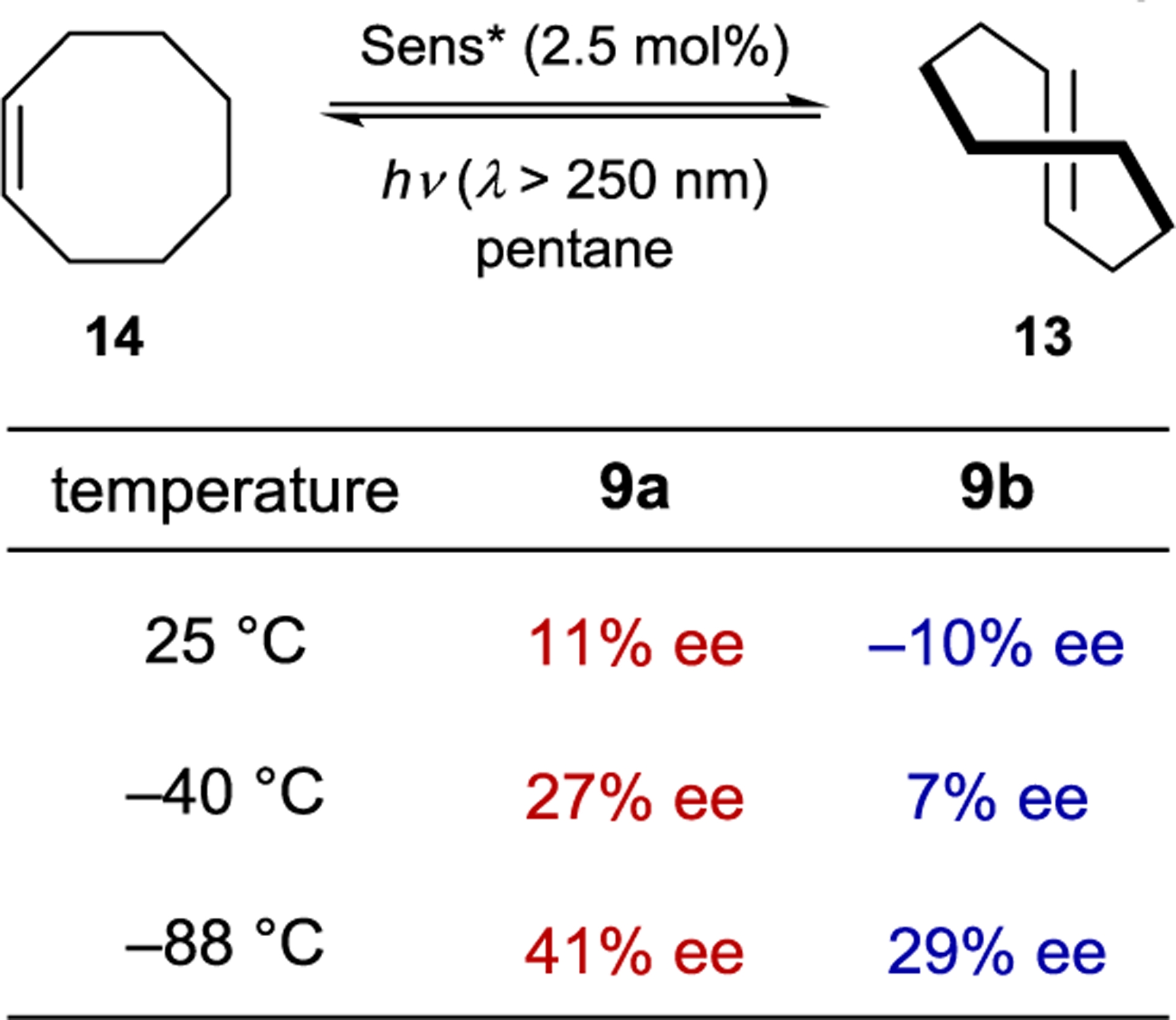
Effect of Temperature on Photosensitized Cyclooctene Isomerization
The differential Eyring equation (eq 1) provides a more complete explanation of the temperature-dependent enantioselectivity.53 The differential activation entropy () and enthalpy () for a given reaction are determined by plotting ln(kS/kR) vs. T−1.54 Here, (kS/kR) is the ratio of rate constants leading to the enantiomeric products and can be calculated from the enantiomeric ratio (e.r.). When the entropy and enthalpy terms have opposite signs, they favor formation of the same enantiomer; if their signs are the same, the terms favor opposite enantiomers. Notably, if the differential activation entropy is large and the entropy and enthalpy terms favor opposite enantiomers, which is the case with 9b, the favored enantiomer will switch with a change in temperature.
| eq. 1 |
For cyclooctene photoisomerization catalyzed by 9b, the differential thermodynamic terms are kcal mol−1 and cal mol−1 K−1. Therefore, at low temperatures, the enthalpy term dominates, favoring (S)-cyclooctene, while at higher temperatures the entropy term dominates, favoring (R)-cyclooctene. Further studies showed that the temperature-switching phenomenon is characteristic of ortho-benzencarboxylates, and an extensive screening of these sensitizers showed 9c to be optimal, giving 64% ee at −89 °C.55, 56
Pressure can also be used as an entropy-related tool to control enantioselectivity.57, 58 The pressure dependence of a reaction at constant temperature is given by eq 2:
| eq. 2 |
where is the difference in activation volume between the diastereomeric transition states. Using eq 2, Inoue determined for a variety of sensitizers.59, 60 In general, ortho-benzenecarboxylates show the greatest pressure dependence on enantioselectivity, and several instances where the product chirality switches as a function of pressure were reported. With 9b, (R)-cyclooctene was obtained in 11% ee at atmospheric pressure, while (S)-cyclooctene was obtained in 18% ee at 400 MPa. For most of the sensitizers examined, the pressure effect becomes discontinuous above 200–400 MPa, suggesting a change in mechanism of the isomerization.61 There is no observed correlation between and , implying that both temperature and pressure can be optimized independently for high enantioselectivity. The optimal conditions were predicted by mathematically modeling as a function of P and T−1. In theory, the maximum of the fitted three-dimensional plot corresponds to the pressure and temperature that should provide the highest ee.59
The enantioselectivity of the reaction is relatively insensitive to solvent for most sensitizers; however, an unusual relationship was observed using saccharide sensitizer 9d (Scheme 9).62 The isomerization was conducted in pentane and diethyl ether at several temperatures between −110 °C and 25 °C. At 25 °C, (R)-cyclooctene is formed in approximately 5% ee in both solvents. In pentane, the ee increases with decreasing temperatures, reaching 40% at −78 °C. On the other hand, the chirality inverts in diethyl ether with an equipodal point at −19 °C, ultimately reaching 73% ee for (S)-cyclooctene at −110 °C. These results can be rationalized by the signs of the differential activation entropy and enthalpy, which for pentane are both positive, while for diethyl ether are both negative. The switching of product chirality as a function of solvent was attributed to the solvation of the ether groups of the saccharide esters. Given the solvent effect on enantioselectivity, the reaction was also conducted in supercritical carbon dioxide because the solvent properties can be dramatically tuned within a relatively narrow range of pressure and temperature near the critical density. The differential activation volume was determined from eq 2; however, the relationship between ln(kS/kR) and pressure is not linear over the measured pressure range, suggesting that different values exist in the near-critical and high-pressure regions.63 Together, these results demonstrate how mechanistic understanding can be used to tune catalyst structure in combination with reaction conditions to optimize the enantioselectivity of a photocatalytic reaction.
Scheme 9.
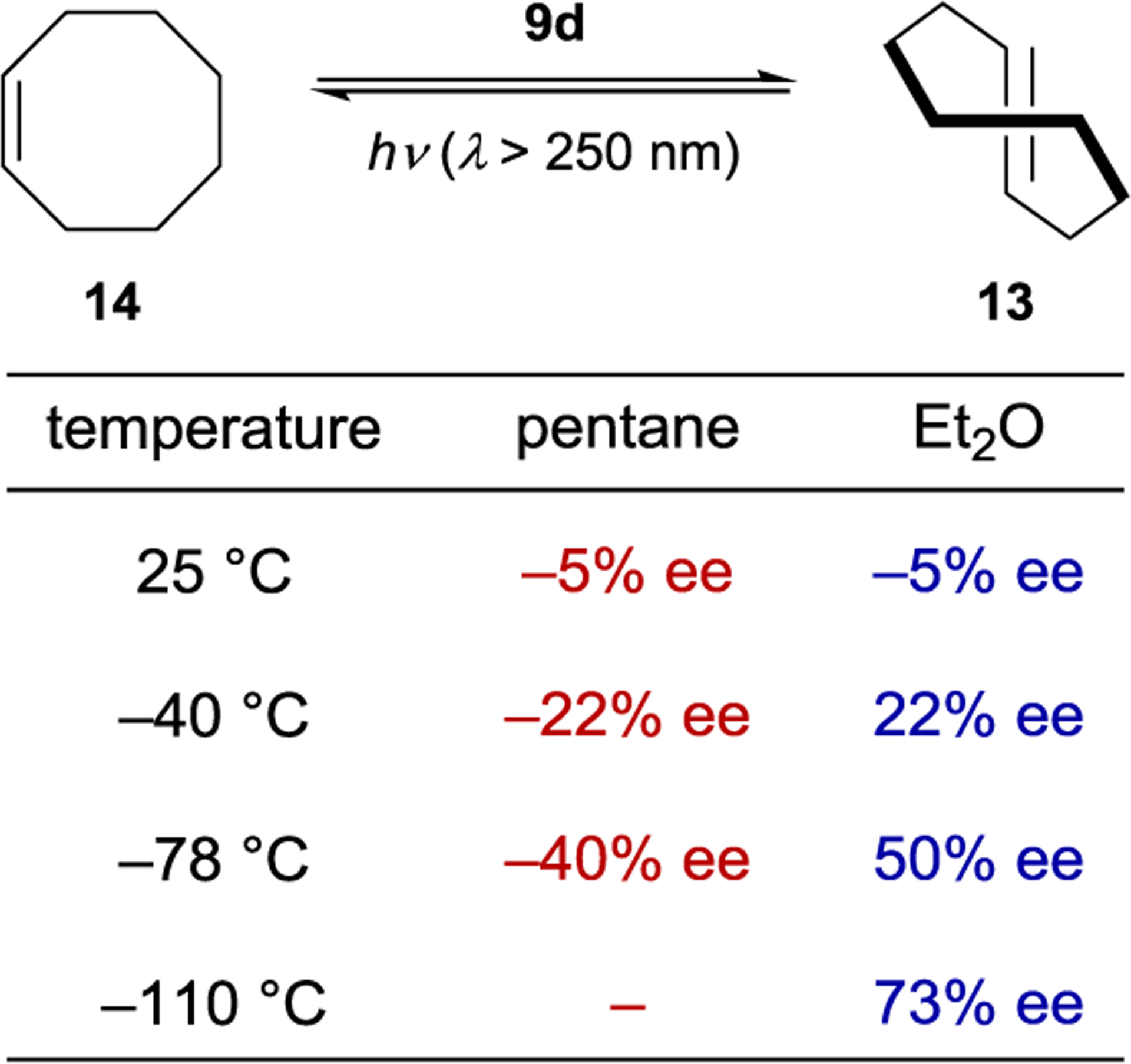
Effect of Solvent on Photosensitized Cyclooctene Isomerization
Using the same class of benzenepolycarboxylate sensitizers, several other cyclic alkenes were subjected to the photoisomerization conditions. In the case of (Z)-1-methylcyclooctene, the ee does not exceed 7%.64, 65 Compared to (Z)-cyclooctene, the E/Z ratio at the photostationary state (PSS) is low, which was attributed to greater steric destabilization in the exciplex. The E/Z ratios could be improved by tethering the sensitizers to the substrate in a diastereodifferentiating isomerization.66–67 68 (Z,Z)-1,3-Cyclooctadiene undergoes photoinduced isomerization to the E,Z-isomer in 18% ee using 10b;69 however, the photoisomerization of 1,5-cyclooctadiene provides only 5% ee.70 The latter reaction is pressure-dependent, favoring (−)-(E,Z)-1,5-cyclooctadiene in 4% ee at atmospheric pressure and (+)-(E,Z)-1,5-cyclooctadiene in 4% ee at 300 MPa.61
Inoue also examined planar–chiral paracyclophanes as exciplex-forming sensitizers in the photoisomerization of cyclooctenes (Scheme 10).71 Using sensitizer 16, the isomerization proceeded to give photostationary E/Z ratios of approximately 0.01, which is smaller than observed with simpler arenecarboxylate sensitizers (0.1–0.4). The authors hypothesized that steric hindrance in the exciplex of (E,Z)-17 compared to (Z,Z)-15 with the bulkier paracyclophane accounts for the lower E/Z ratios. Spectroscopic experiments showed that (Z,Z)-15 efficiently quenched sensitizer 16 with exciplex formation confirmed by the appearance of a new emission feature. The enantioselectivity increased with decreasing temperatures, affording (E,Z)-17 in 87% ee at −140 °C.
Scheme 10.
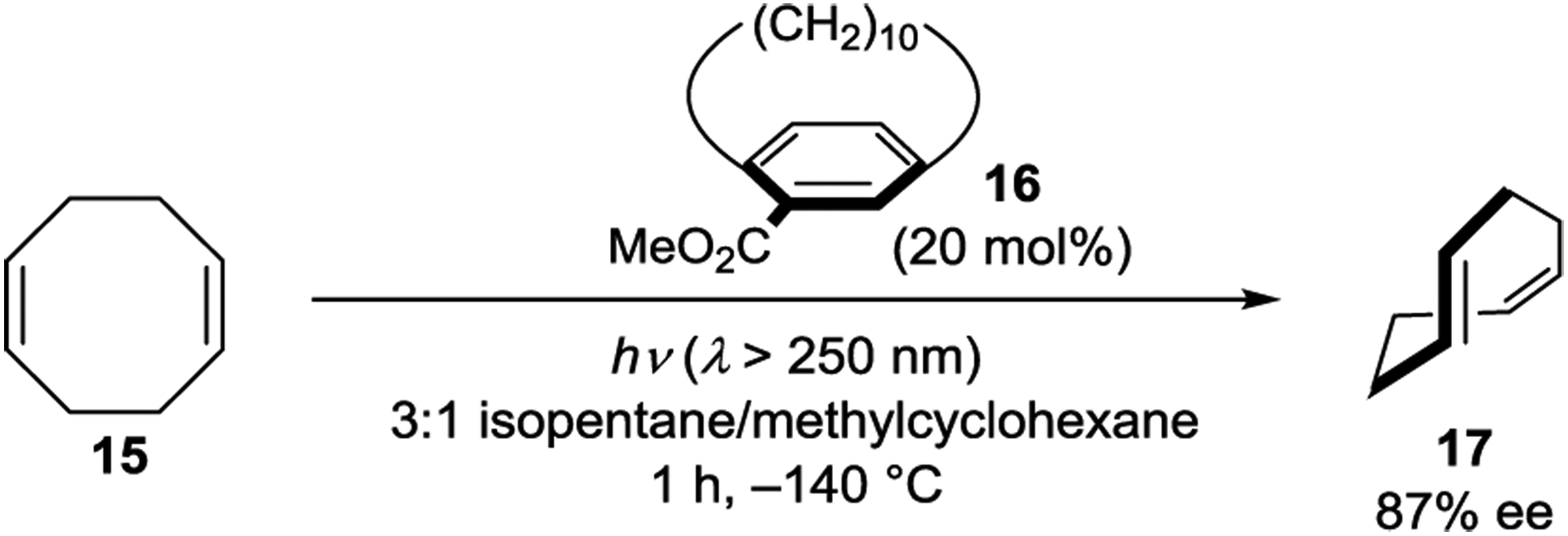
Cyclooctadiene Isomerization Catalyzed by a Cyclophane Sensitizer
Cycloheptene (18) can also be photochemically isomerized using chiral arene sensitizers; however, due to the thermal instability of (E)-cycloheptene, the enantioselectivity was assessed by trapping with 1,3-diphenylisobenzofuran in a stereospecific Diels–Alder cycloaddition (Scheme 11).72 The best enantioselectivity (77% ee) was obtained with 9a in hexane at −80 °C. With most sensitizers, the ee was greater for (E)-cycloheptene than (E)-cyclooctene. This trend was reflected in the calculated and values, which are typically greater by a factor of 2–3 for cycloheptene than for cyclooctene. The authors concluded that the approach of cycloheptene to the photocatalyst is less hindered, enabling a more intimate interaction in the exciplex and consequently greater stereocontrol.
Scheme 11.
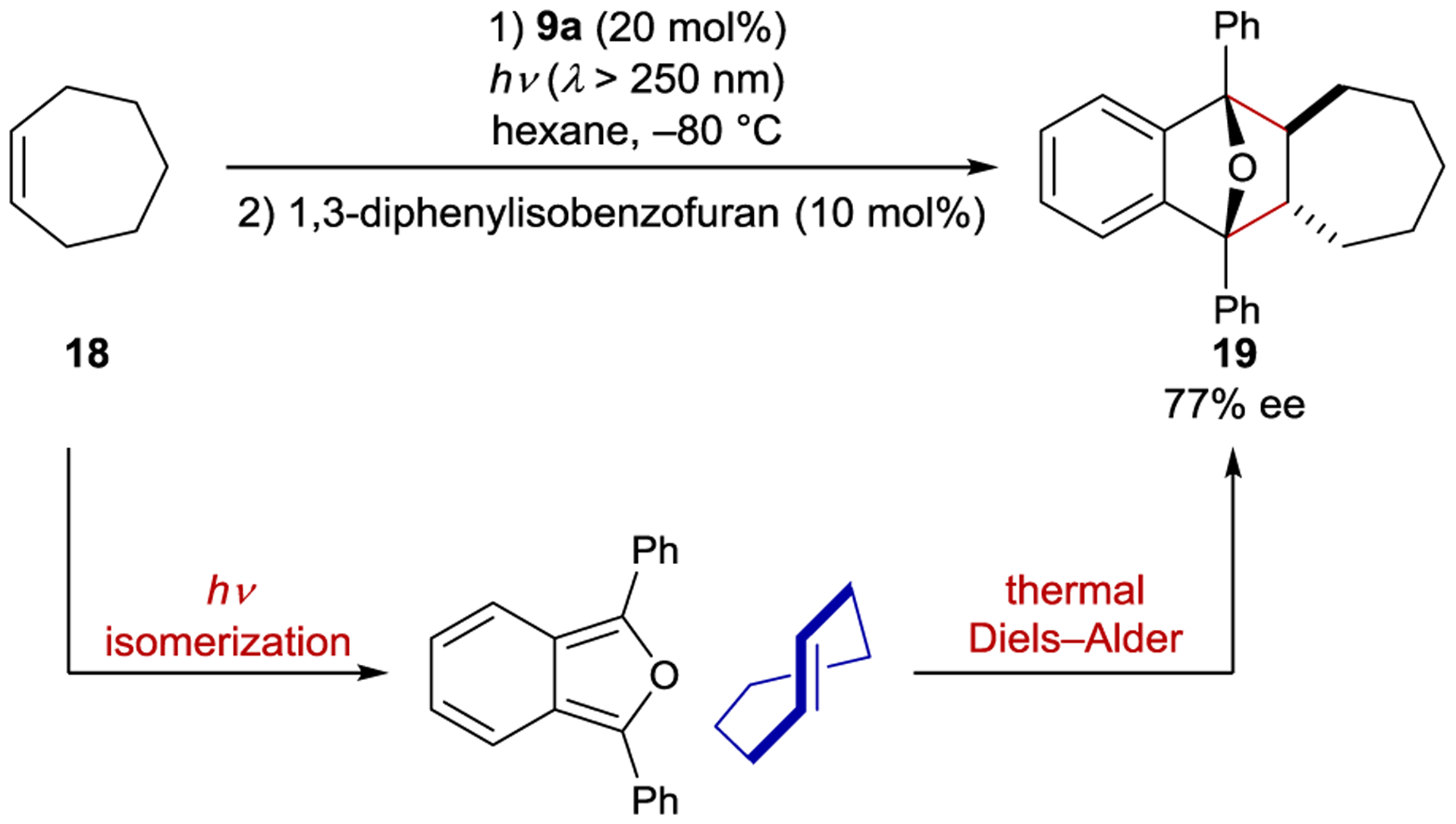
Cycloheptene Isomerization and Cycloaddition Catalyzed by Benzenecarboxylate Sensitizers
Under similar photoisomerization conditions, (Z)-cyclohexene (20) forms a mixture of [2+2]-cyclodimer diastereomers via initial photochemical isomerization to the (E)-isomer followed by thermal cycloaddition (Scheme 12).73 The cycloaddition may occur by two mechanisms: a concerted, stereospecific dimerization, or a stepwise stereoablative radical dimerization. The plot of ln(kS/kR) vs. T−1 is not linear at high temperatures, which was attributed to the contribution of the stepwise mechanism. At low temperatures, however, the relationship is linear. At −78 °C, the trans-anti-trans isomer (21) is obtained in 68% ee using sensitizer 8e. Photochemically produced (E)-cyclohexene also reacts with 1,3-cyclohexadiene in a thermal [4+2] cycloaddition. Complex mixtures of dimeric products are formed, and only the exo-[4+2] product is obtained in appreciable ee (8%).74
Scheme 12.
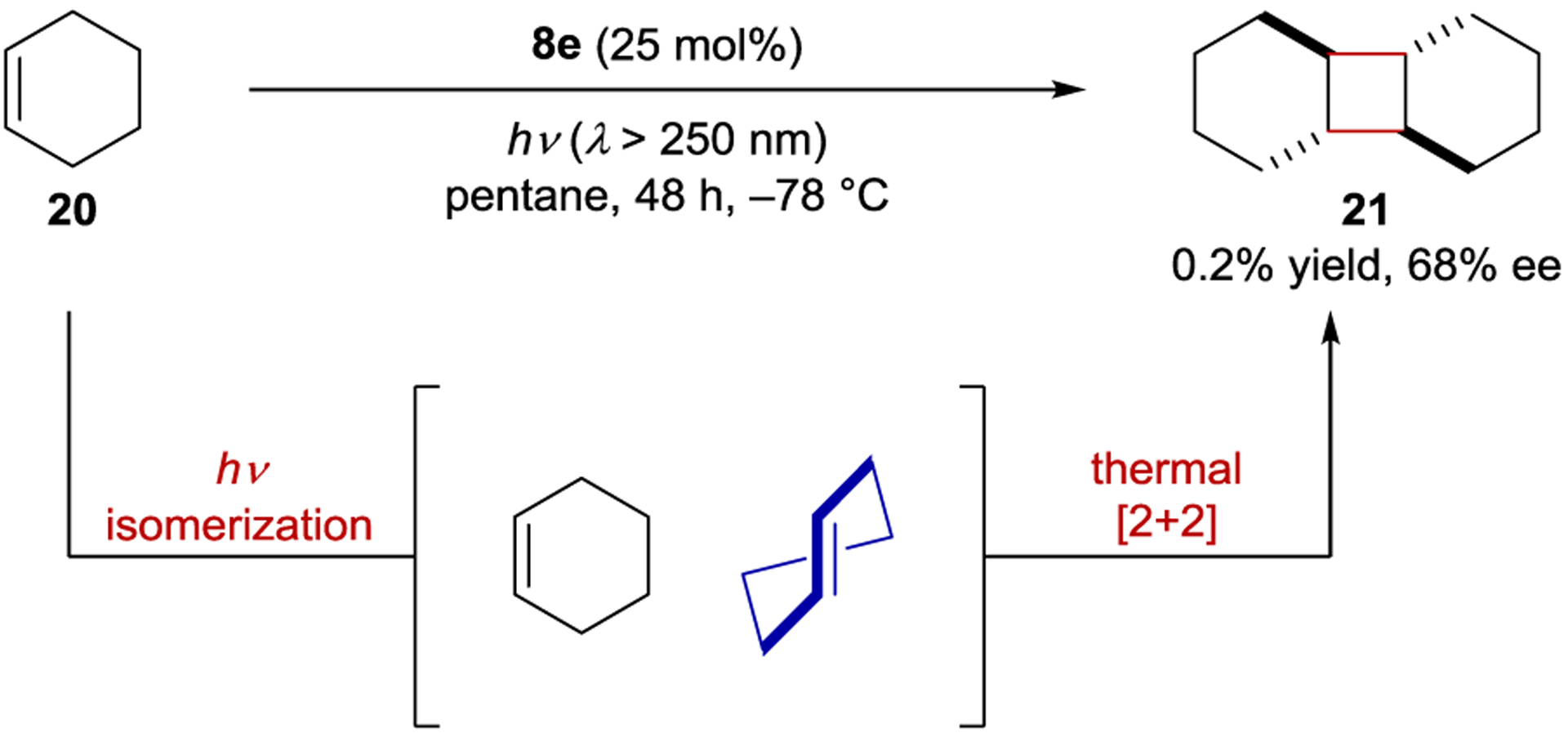
Cyclohexene Isomerization and Cycloaddition Catalyzed by Benzenecarboxylate Sensitizers
With the insights gained from the studies of cyclic alkene isomerization discussed above, Inoue revisited the asymmetric isomerization of 1,2-diarylcyclopropanes originally reported by Hammond.27 Several arenecarboxylate sensitizers were tested, but the highest reported ee for trans-1 was 10%.75–76 77
Bimolecular photoreactions are particularly difficult to control through an exciplex mechanism because the enantiodetermining step must occur in a ternary complex comprising the sensitizer and both substrates. This requirement can be satisfied by the attack of a reactant on a substrate–catalyst exciplex. Because the exciplex possesses a significant degree of charge transfer, it is often essential to conduct the reaction in nonpolar solvents to avoid dissociation into a solvent-separated radical-ion pair from which any subsequent reaction is likely to be racemic. This is problematic for electron-transfer reactions, which are slow under these conditions. Hence, it can be challenging to obtain both good yield and high ee simultaneously. This was the case in a [2+2] cycloaddition of electron-rich styrenes reported by Inoue.78 While the reaction catalyzed by 9b proceeded to high yield in CH3CN, the negligible enantioselectivity observed was likely due to racemic reactivity from the free styrene radical cation. In pentane and ether, which both favor exciplex formation, no product was observed.
A similar effect was observed in the anti-Markovnikov photoaddition of methanol to 1,1-diphenylpropene (22). Here, nonpolar solvents give the ether product in low yield (< 10%) and high ee (up to 27%), while polar solvents afford the product in high yield (up to 60%) and low ee (< 1%).79 As a potential solution to this problem, saccharide esters of napthalenecarboxylic acids (11d, 11e, and 12e) were used as sensitizers.80 The authors proposed that these sensitizers provide microenvironmental polarity control where the saccharide moiety creates a high-polarity region in the direct vicinity of the sensitizer, allowing for electron transfer, within a low-polarity bulk solution, ensuring that the sensitizer and substrate do not dissociate. Using these sensitizers, high enantioselectivity is maintained, while the yield is improved to 20–40%. Less hindered alcohols generally give higher yields but lower enantioselectivity (Scheme 13). The best ee (58%) was achieved with sensitizer 12e using i-PrOH as the nucleophile, but the yield was only 1%.81 Based on computational and fluorescence quenching studies, the authors proposed that the difference in free energy between the diastereomeric sensitizer–substrate exciplexes is the primary determinant for enantioselectivity. The temperature and pressure effects on the reaction were evaluated, and similar effects were observed as with the cyclooctene isomerization.82–83 84 85 86 When the reaction is conducted in supercritical carbon dioxide, there is a sudden increase in the product ee when transitioning from the near-critical to supercritical state, indicating a substantial difference in solvent environment in this pressure region. At the critical state, there is significant solvent clustering, where the local density of CO2 is greater around the exciplex than in the bulk solution. Further, the solvent environments for the diastereomeric exciplexes can be significantly different due to this clustering, increasing the difference in free energy. In a related intramolecular photoaddition of a tethered alcohol, this clustering behavior was manipulated by adding cosolvents to reactions conducted in supercritical CO2. When ether was added to tune the cluster polarity, the ee increased from 30% to 45%.87–88 89
Scheme 13.
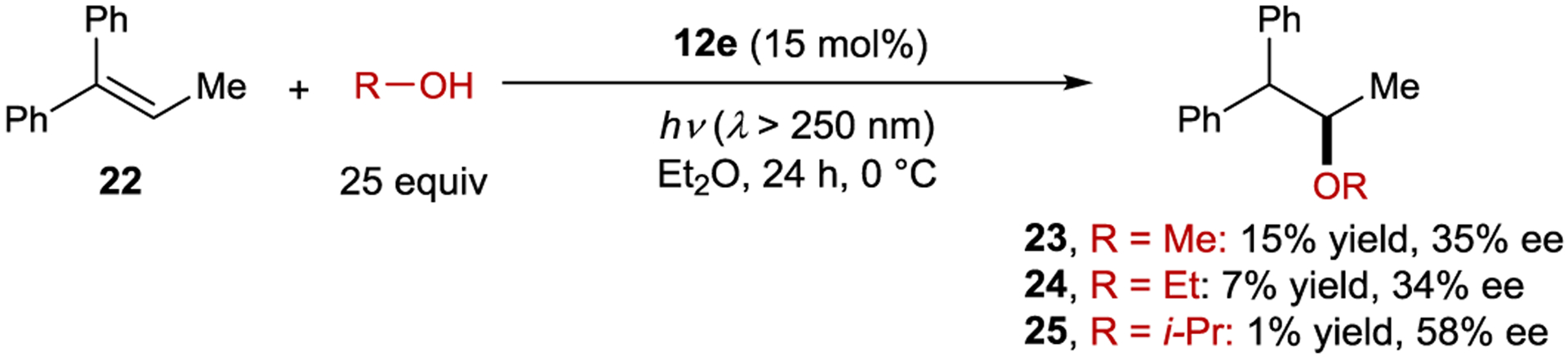
Polar Photoaddition of Alcohols to Alkenes Catalyzed by Naphthalenecarboxylate Sensitizers
Schuster reported an enantioselective [4+2] photocycloaddition catalyzed by axially chiral cyanoarene sensitizer 28 (Scheme 14).90 The discovery built on prior work showing that electron-deficient photocatalysts promote radical cation Diels–Alder reactions between electron-rich dienes and dienophiles in polar solvents.91–92 93 While the racemic [4+2] product is formed when 26 and 27 are sensitized with 28 in MeCN, the cycloadduct 29 is formed in 15% ee in toluene.94 Transient absorption experiments suggest that electron-transfer quenching of the excited sensitizer in polar solvents produces a radical-ion pair, while an exciplex forms in nonpolar solvents. The existence of the exciplex was corroborated by a new fluorescence feature when the singlet excited-state photocatalyst is quenched by styrene. At low temperature, two discrete, diastereomeric catalyst–styrene exciplexes were detected using time-resolved fluorescence experiments. Addition of cyclohexadiene resulted in further quenching of the exciplex fluorescence, leading the authors to propose the formation of a ternary excited-state complex, or triplex, comprised of the sensitizer, diene, and dienophile. The enantioselectivity in this reaction was attributed to a difference in excited-state lifetimes for the two diastereomeric catalyst-styrene exciplexes.
Scheme 14.
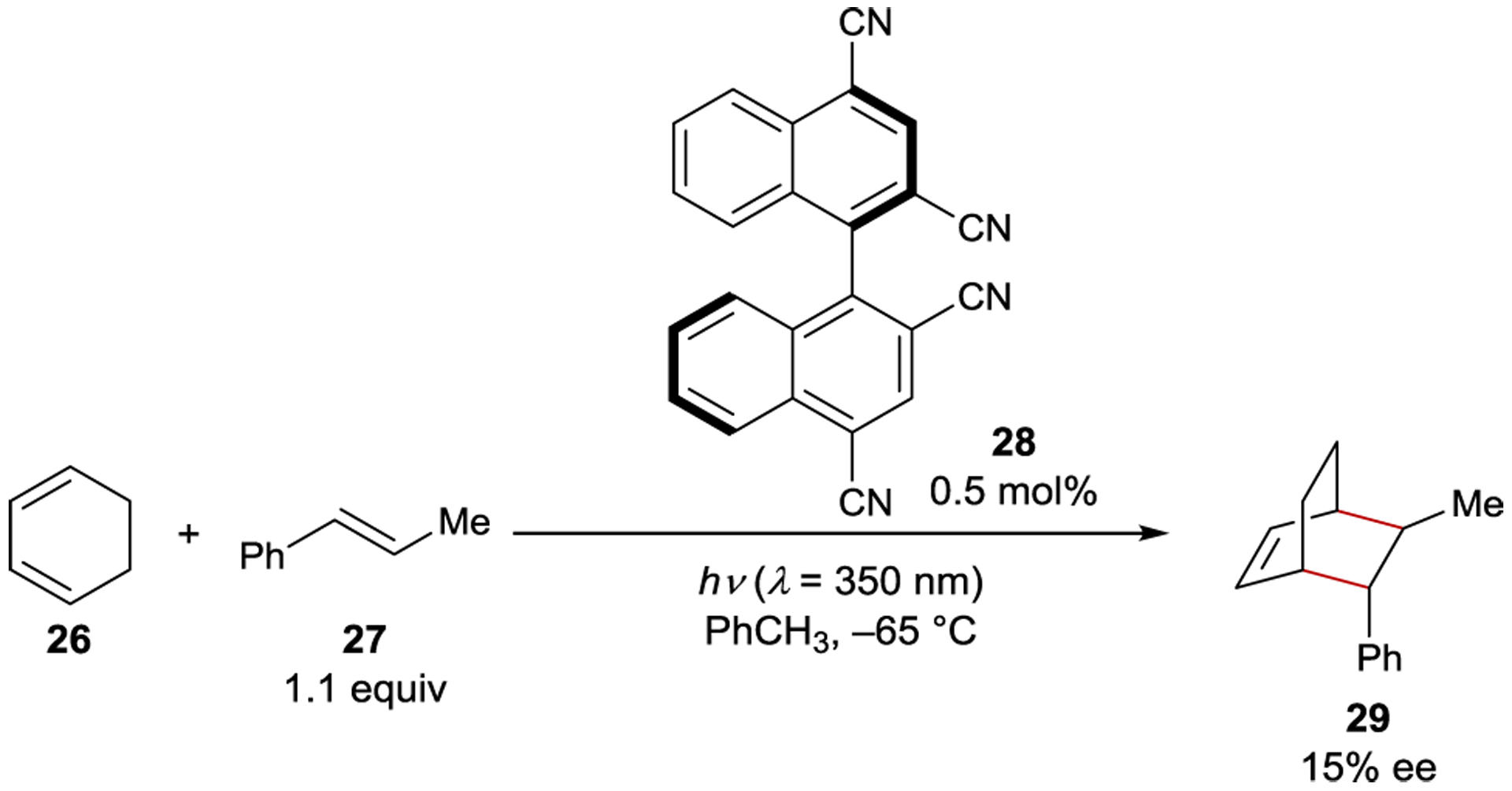
Diels–Alder Cycloaddition Catalyzed by a Cyanoarene Sensitizer
Mattay showed that a similar electron-deficient sensitizer (30) also catalyzes the enantioselective cis-trans isomerization of cyclopropanes (Scheme 15).95, 96 Mechanistic experiments performed with radical cation quencher 1,2,4-trimethoxybenzene showed solvent-dependent reactivity analogous to that observed by Schuster. In toluene, in which an exciplex is formed, enantioenriched trans-1 is obtained in 4% ee. Racemic product is obtained in MeCN, suggesting the existence of an electron-transfer mechanism that produces solvent-separated radical-ion pairs.
Scheme 15.
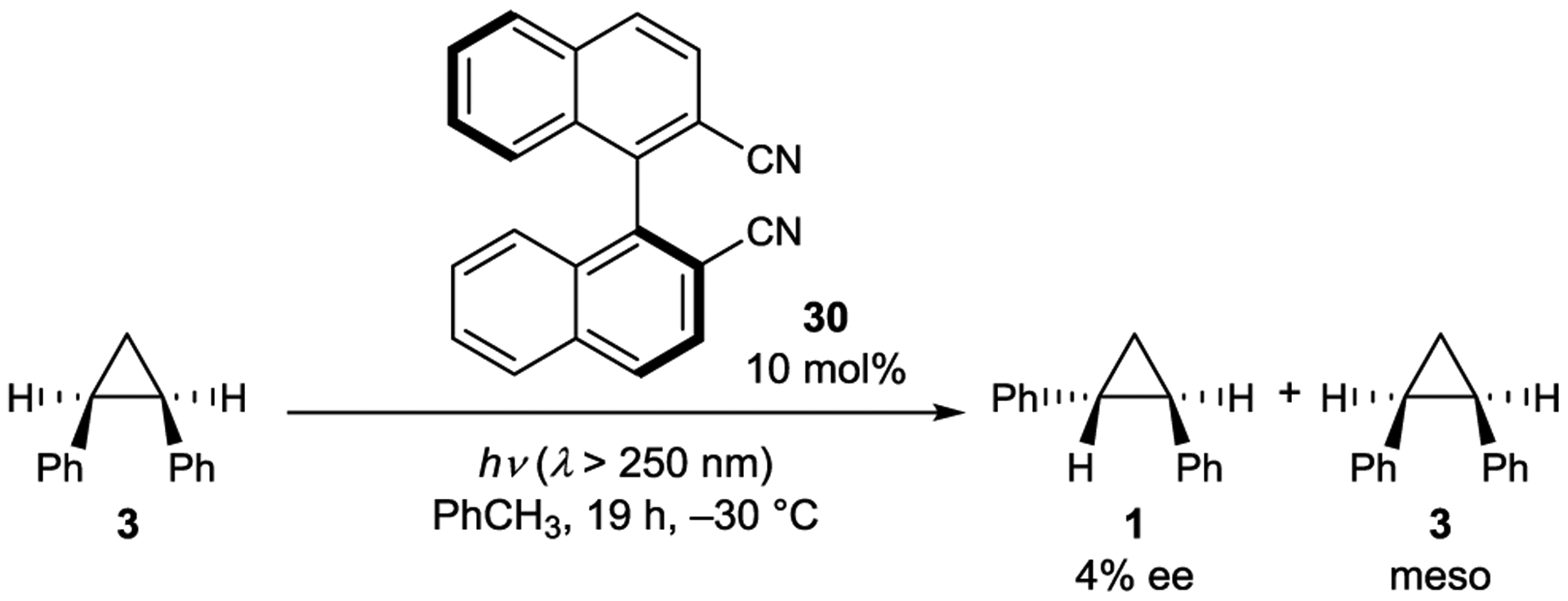
Cyclopropane Isomerization Catalyzed by a Cyanoarene Sensitizer
Hanson studied axially chiral VANOL-derived catalysts as excited-state proton-transfer reagents for the stereoselective protonation of silyl enol ethers (Scheme 16).97 The shift of electron density upon excitation of the chromophore increases the acidity of the hydroxyl protons, enabling a protonation event that would be thermodynamically unfavorable in the ground state.98 Stoichiometric loadings of 32 afford ketone 33 in 64% yield and 35% ee. The presence of a bromine substituent on the arene backbone is necessary for productive reactivity, which was attributed to its ability to facilitate intersystem crossing. When only 1 mol% of 32 is used with one equiv of phenol as a sacrificial proton source, racemic product is formed. The authors hypothesized that the loss of enantioselectivity is due to excited-state proton transfer from 32 to phenol to create PhOH2+, which then protonates the substrate.
Scheme 16.
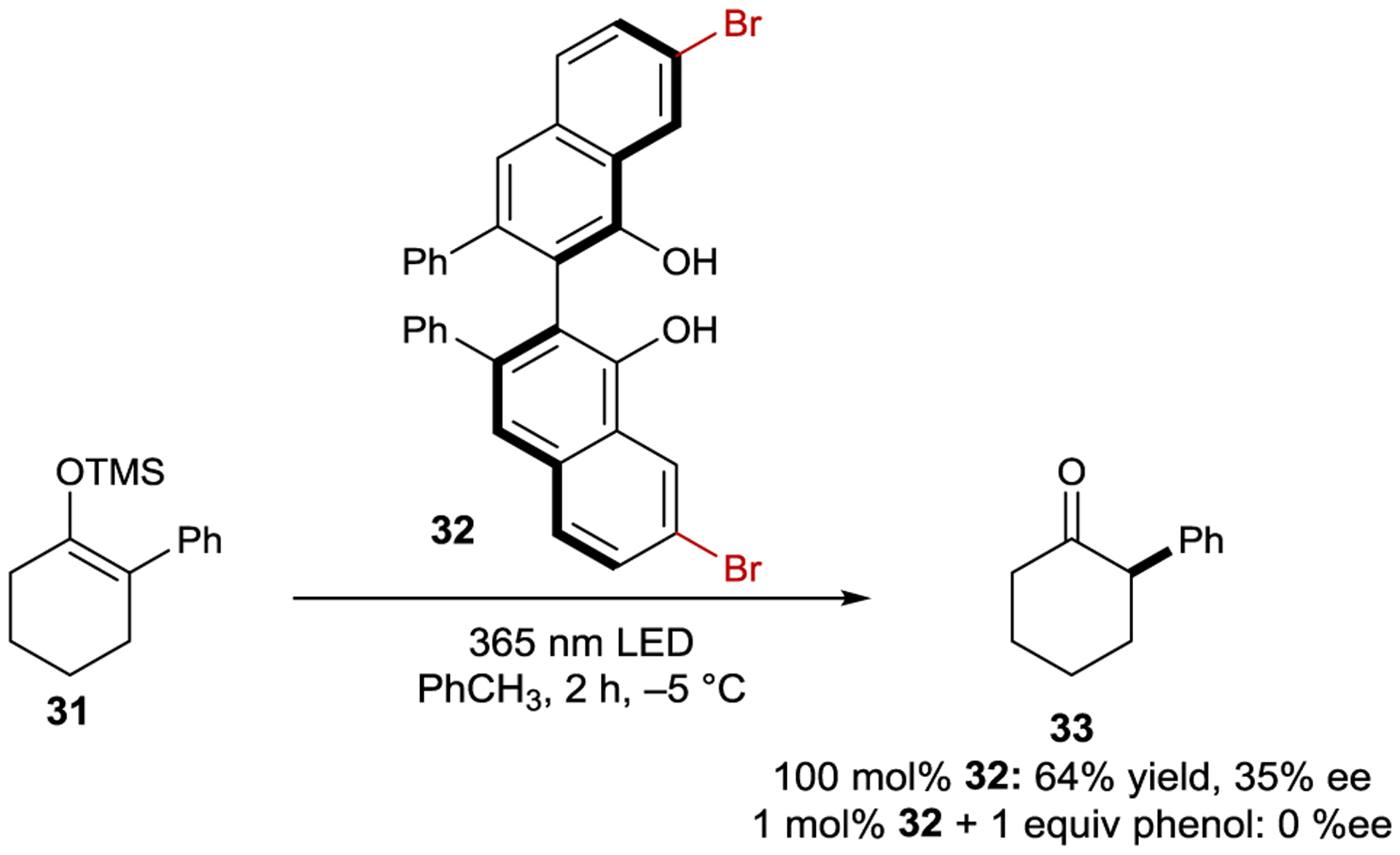
Excited-State Protonation Catalyzed by a VANOL-derived Sensitizer
Sabater studied chiral pyridinium photoredox catalysts for the cyclization of 34 to afford a mixture of saturated lactone 36 and unsaturated lactone 37 (Scheme 17). Optimal results were achieved with sensitizer 35, which gave 7% ee for 36.99 The reaction proceeds through photoinduced electron transfer (PET) oxidation of the alkene to the radical cation, followed by enantiodetermining nucleophilic attack of the pendant alcohol.
Scheme 17.
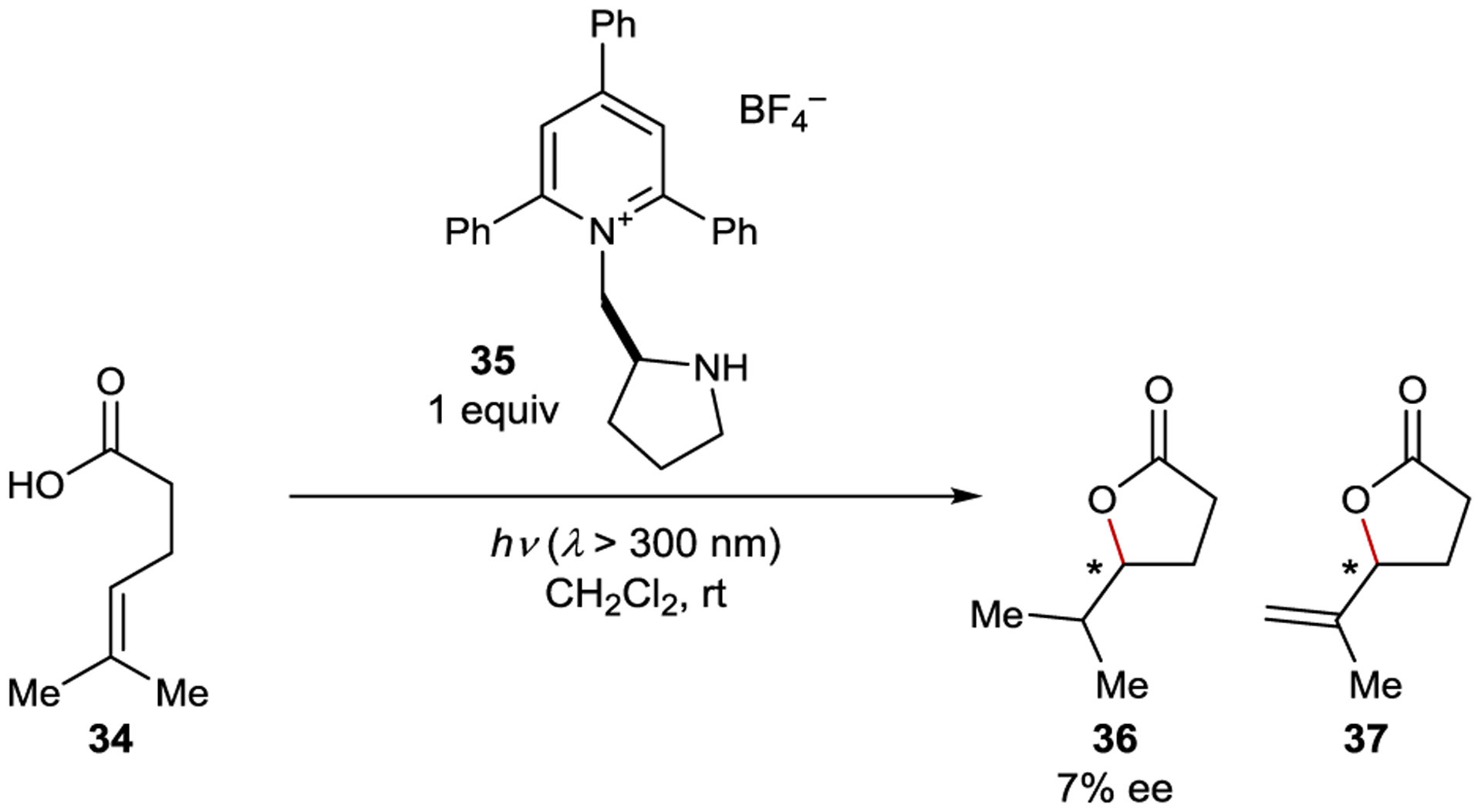
Polar Photoaddition Catalyzed by an Acridinium Sensitizer
Sivaguru, Sibi, and coworkers developed an intramolecular [2+2] photocycloaddition of 4-alkenyl coumarins (38) catalyzed by chiral thiourea 39 (Scheme 18).100–101 102 The photoadducts are produced with 77–96% ee using only 10 mol% chiral catalyst. Hydrogen bonding between the thiourea and carbonyl moieties on the catalyst and substrate organize the substrate within the chiral environment of the catalyst. Both the substrate and sensitizer efficiently absorb light, but the reaction without catalyst is slow, accounting for the lack of a significant racemic background reaction. Stern–Volmer quenching studies revealed that both static and dynamic quenching of the catalyst by the substrate is operative, and the authors proposed that both pathways lead to enantioenriched product. In the static quenching mechanism, the ground-state substrate-catalyst complex absorbs light, and the substrate undergoes cyclization. In the dynamic quenching mechanism, unbound catalyst is excited and forms a triplet exciplex with the substrate, which can undergo the cycloaddition. Sivaguru also reported the enantioselective 6π-cyclization of acrylanilides catalyzed by chiral thioureas, but the highest enantioselectivity obtained was 13% ee.103
Scheme 18.
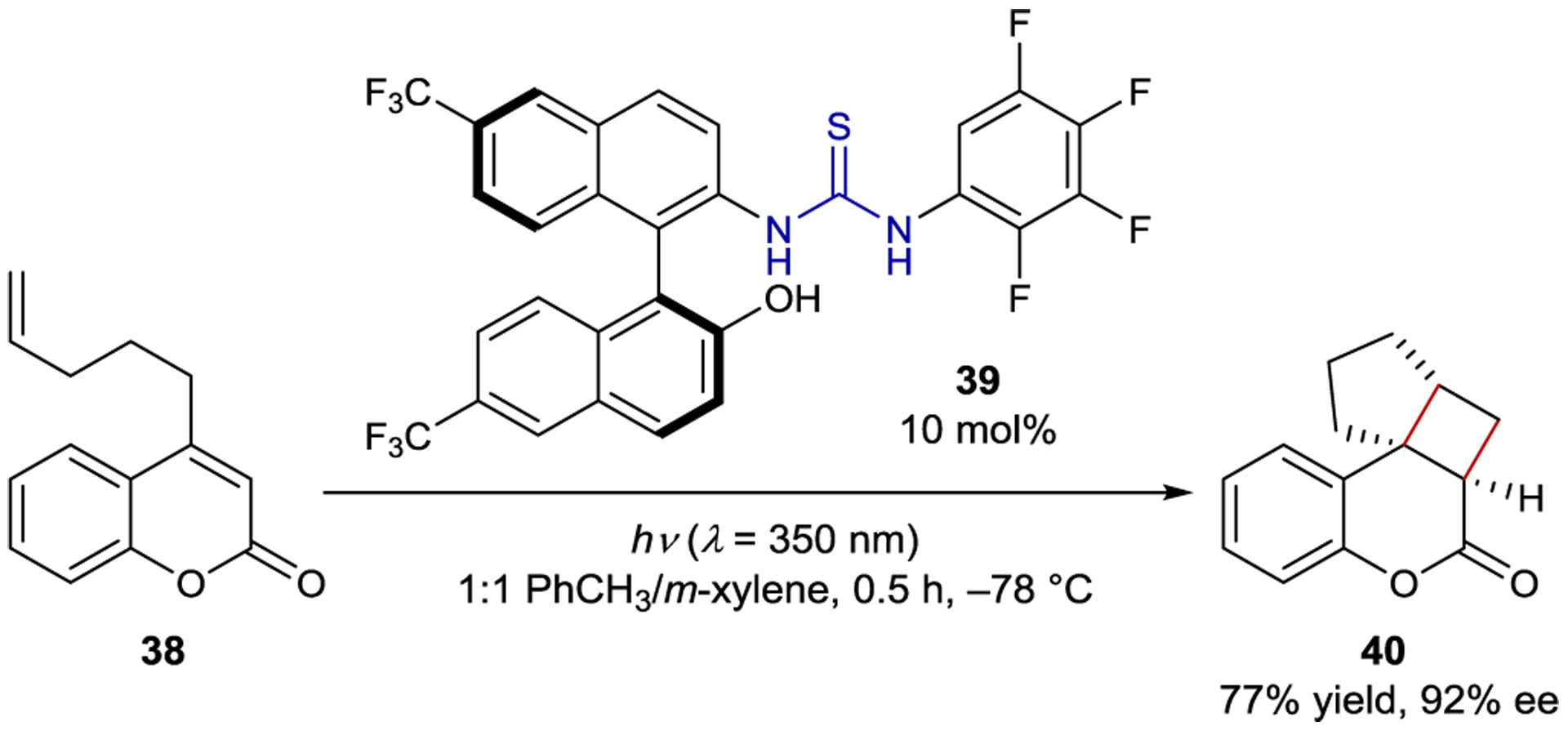
Intramolecular [2+2] Cycloaddition Catalyzed by an Atropisomeric Thiourea Sensitizer
2.2. Ketones
2.2.1. Ketones without Hydrogen-Bonding Domains
Aromatic ketones have fast rates of intersystem crossing and are often employed as triplet sensitizers.25 Despite the high efficiency with which they sensitize a variety of organic transformations, chiral aromatic ketones without hydrogen-bonding domains have not promoted highly enantioselective reactions. This is likely because the sensitizer quickly dissociates from the excited substrate after sensitization, leading to poor stereocontrol.104, 105 For instance, Ouannès and coworkers reported the asymmetric isomerization of 1 using chiral indanone triplet sensitizer 41 (Scheme 19). After 70 hours of UV irradiation, the product distribution reached a photostationary state consisting of a 3:1 ratio of cis:trans isomers and 3% ee for 1.106
Scheme 19.
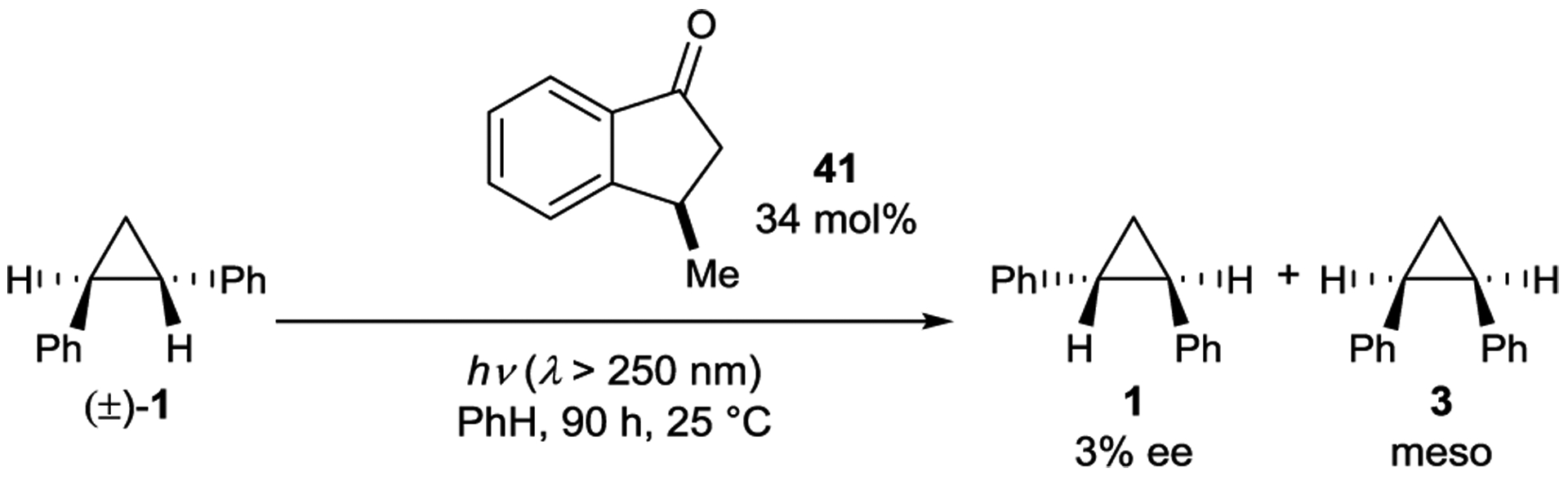
Cyclopropane Isomerization Catalyzed by an Aryl Ketone Sensitizer
Chiral indanone sensitizer 43 catalyzed the kinetic resolution of ketone 42 via an oxa-di-π-methane rearrangement (Scheme 20). At low temperature and low levels of conversion, the rearranged product (44) was formed in 10% ee.107
Scheme 20.
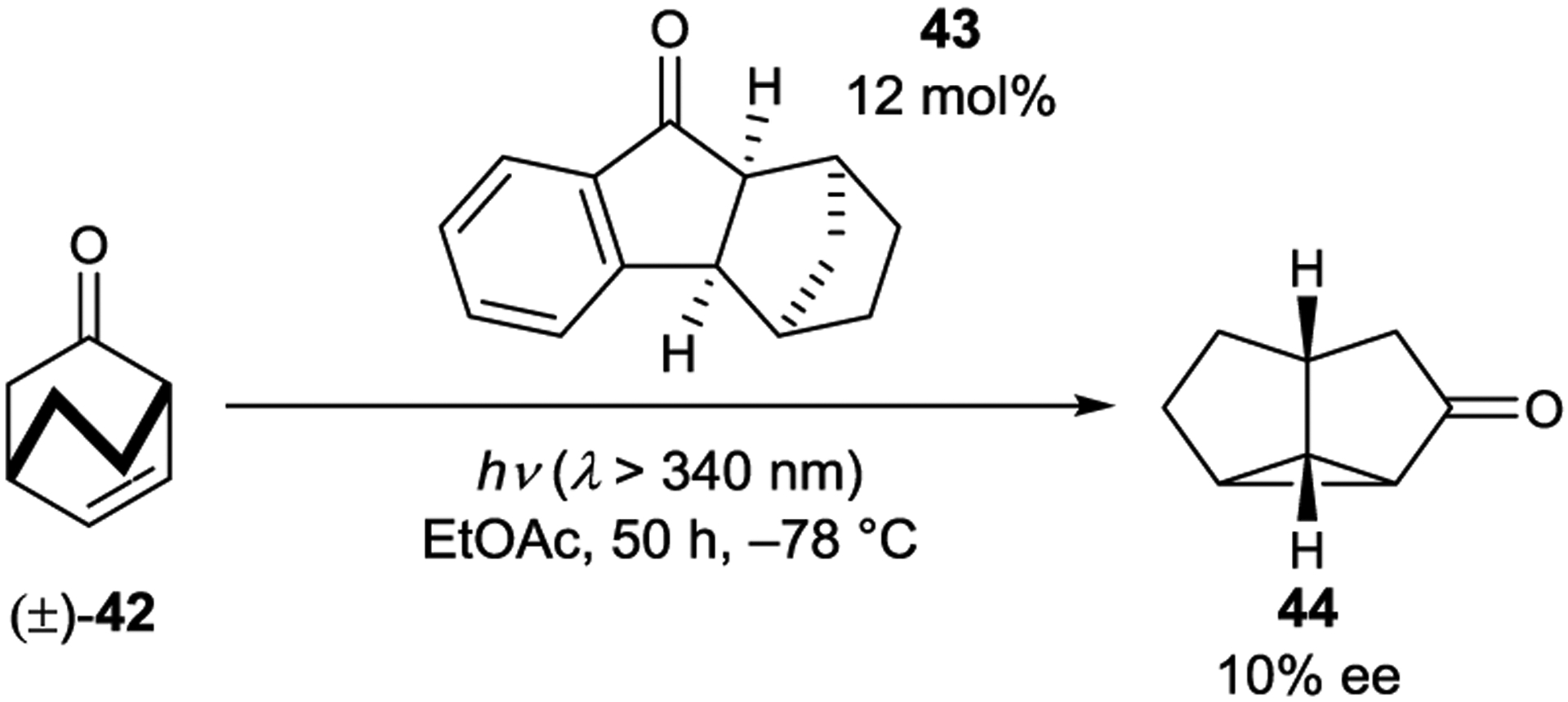
Oxa-di-π-methane Rearrangement Catalyzed by an Aryl Ketone Sensitizer
2.2.2. Chiral Ketones with Hydrogen-Bonding Domains
Prior to 2000, most enantioselective photoreactions involved simple chiral sensitizers where the transfer of chiral information occurs within a transient excited-state interaction with limited organization. Consequently, the enantioselectivities obtained were often low, consistent with the lack of a well-defined substrate–catalyst interaction during the enantiodetermining step. In 2003, Krische proposed that two key criteria would be essential for a highly enantioselective photocatalytic reaction: (1) the substrate must exist in a well-defined chiral environment upon binding to the catalyst, and (2) the catalyst–substrate interaction must confer a kinetic advantage to the photoreaction.10 As a potential solution to the first challenge, Krische proposed that chiral hydrogen-bonding catalysts could bind a substrate in the ground state prior to excitation ensuring a well-defined chiral environment after substrate excitation. Krische hypothesized that the second criterion could also be satisfied by introducing a benzophenone triplet sensitizer within the structure of the catalyst, creating a binding-induced rate enhancement due to the distance dependence of energy transfer. With 2 equiv of catalyst 46, quinolone 45 cyclizes to cyclobutane 47 in 21% ee via a [2+2] intramolecular photocycloaddition (Scheme 21). Notably, the catalyst loading could be lowered to 25 mol% with only a slight loss in enantioselectivity (19% ee). A Job plot and NMR binding studies confirmed complete catalyst-substrate association under the reaction conditions. These results indicate that the catalyst confers a kinetic advantage to the reaction, and that the modest enantioselectivity is the result of poor enantiofacial bias within the hydrogen-bonding complex rather than a contribution from uncatalyzed background reaction.
Scheme 21.
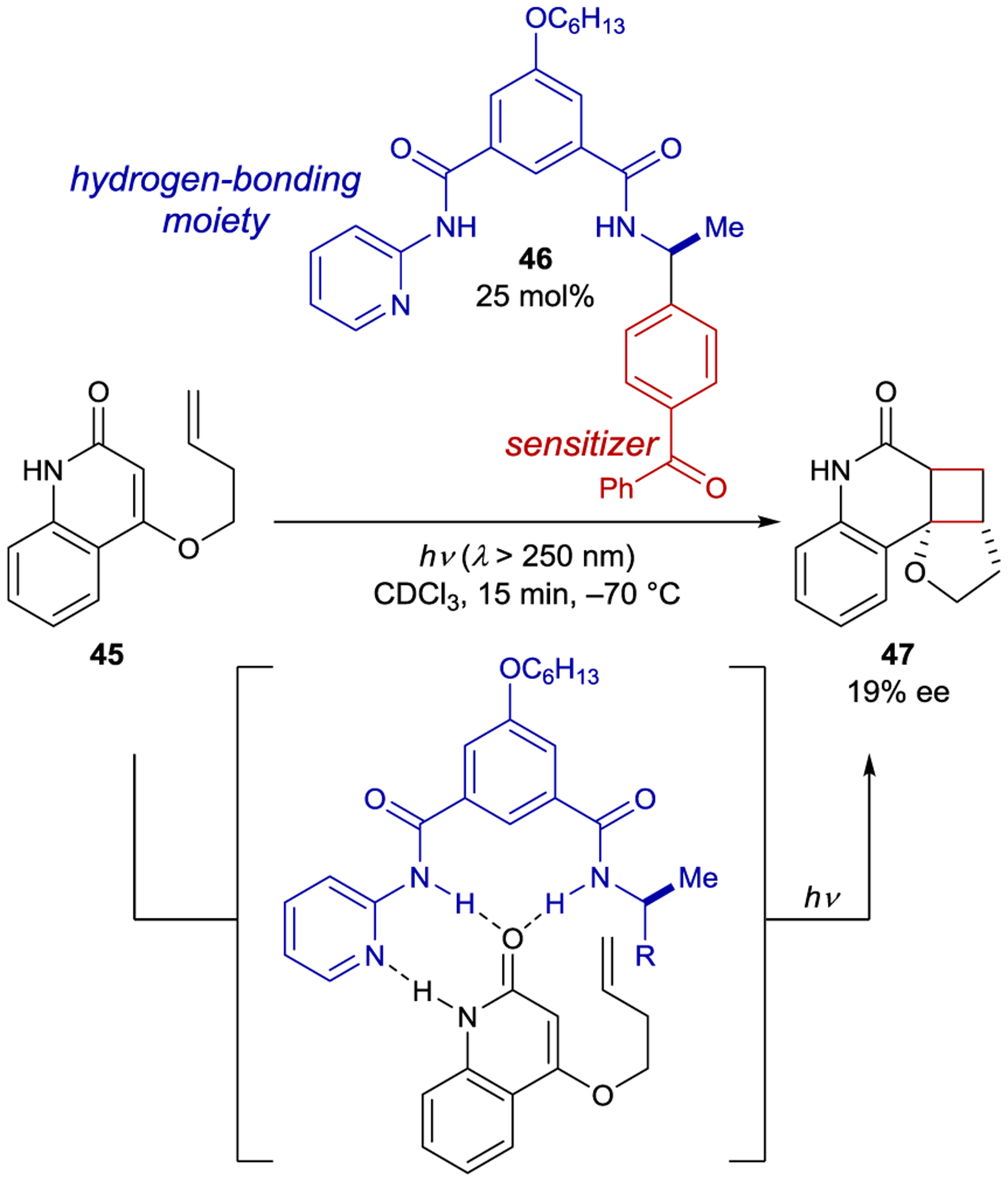
Intramolecular [2+2] Cycloaddition Catalyzed by a Hydrogen-Bonding Ketone Sensitizer
Thus, Krische’s hydrogen-bonding catalyst addressed the second challenge, but did not solve the first. The Bach group, on the other hand, initially solved the first challenge, but not the second. Bach prepared chiral templates derived from Kemp’s triacid that form 1:1 ground-state complexes with amide-containing substrates through hydrogen-bonding interactions.108 In this precomplexation strategy, a superstoichiometric loading of the chiral template ensures that the substrate is always bound within a chiral environment after absorbing light. Bach initially disclosed a diastereoselective Paternò–Büchi reaction using the template strategy.109, 110 Chiral template 49 is optimal for a related intramolecular enantioselective [2+2] photocycloadditon of 2-quinolone 48 (Scheme 22).111, 112 It contains both a rigid cyclohexyl backbone, restricting the conformational flexibility, and a sterically demanding benzoxazole moiety, producing effective facial differentiation in a prochiral substrate. The best enantioselectivity is achieved at low temperatures and in nonpolar solvents, both of which maximize hydrogen-bonding interactions.
Scheme 22.
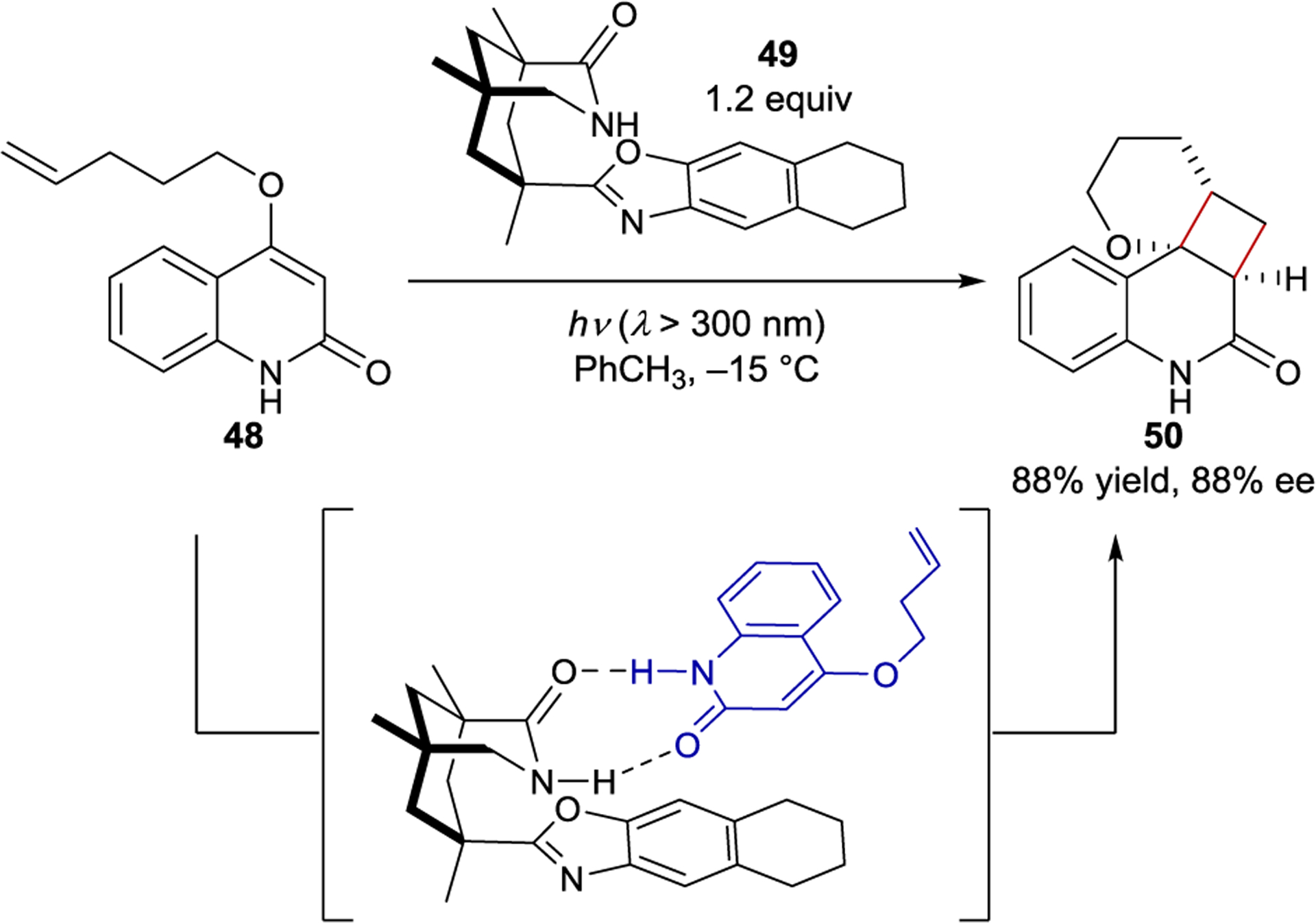
Intramolecular [2+2] Cycloaddition via a Hydrogen-Bonding Amide Template
Chiral hosts related to 49 were examined in several mechanistically diverse photoreactions including intramolecular [2+2]113–114 115, intermolecular [2+2] 116–117 118 119 120, [4+2] 121, 122, and [4+4] 123, 124 cycloadditions, as well as Norrish–Yang cyclizations125,126 and electrocyclizations127. In several cases ee’s greater than 90% were reported; however, in every case superstoichiometric loadings of the template are required because the rate of photoreaction is similar for bound and unbound substrate. In order to ensure high enantioselectivity, nearly all of the substrate must be bound to the template to prevent unbound substrate reacting through a racemic pathway.
In 2005, Bach made a significant contribution to the field of asymmetric photocatalysis by incorporating a sensitizer into the chiral template.128, 129 Adding an aromatic ketone sensitizer to the existing chiral Kemp’s acid motif retained the desired hydrogen-bonding functionality of the original template while introducing the ability to catalyze the photochemical reaction of the bound substrate. Only 30 mol% of 52 was required in the cyclization of pyrrolidine 51 to spirocycle 53 in 70% ee (Scheme 23). The proposed mechanism invokes a ground-state hydrogen-bonding complex between the catalyst and substrate, situating the substrate within the chiral environment of the photocatalyst. Excitation of the ketone catalyst is followed by PET, oxidizing the bound substrate. After intramolecular proton transfer, the resulting α-amino radical cyclizes, setting the stereochemistry and producing 53 after back electron transfer and protonation. Notably, only a catalytic loading of 52 is required in the reaction because cyclization does not occur in the absence of 52.
Scheme 23.
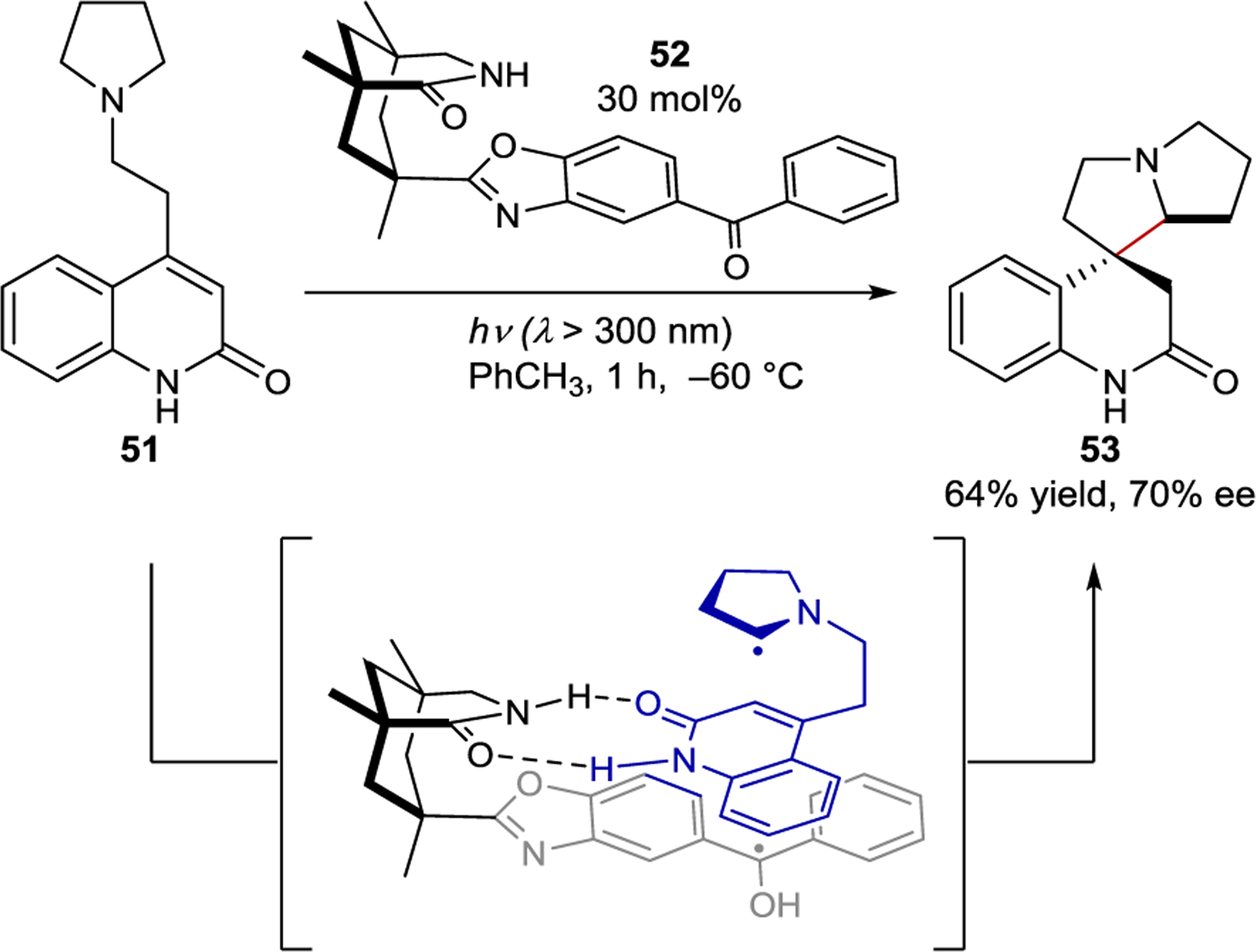
Intramolecular Cyclization Catalyzed by a Hydrogen-Bonding Benzophenone Sensitizer
Extension of ketone catalyst 52 to energy-transfer reactions required further modification of the catalyst structure. In an intramolecular [2+2] cycloaddition of quinolone 45, benzophenone 52 afforded the cyclobutane in 39% ee, while xanthone 54 afforded the cycloadduct in 90% ee (Scheme 24).130 For both 52 and 54, the catalyst is completely bound to the substrate at the start of the reaction, thus the ground-state association does not account for the selectivity change.131 Instead, the increase in selectivity was rationalized based on the efficiency of sensitization and the rate of the subsequent reaction.132 Notably, substrate 45 absorbs at the irradiation wavelength. However, the xanthone photocatalyst 54 has a much larger extinction coefficient than the substrate and absorbs nearly all the photons under the reaction conditions. Thus, the racemic background reaction is attenuated and high enantioselectivity achieved. Conversely, benzophenone 52, which has a much lower extinction coefficient at 366 nm, does not effectively attenuate the competitive racemic background reaction, rationalizing the lowered selectivity.133 Finally, to ensure high levels of stereoinduction, the rate of the enantiodetermining step must outcompete the rate of substrate dissociation. After excitation, dissociation prior to cyclization leads to racemic product. Unlike xanthone, photoexcited benzophenone is not planar, which may facilitate faster substrate dissociation.134 This dissociation hypothesis is bolstered by the lower ee (27%) obtained when substrate 48 is sensitized by the xanthone photocatalyst. Laser flash photolysis experiments revealed that the rate of intramolecular photoreaction for 45 is approximately three times greater than for 48. Hence, while 45 reacts prior to dissociation, Bach proposed that the rates of cyclization and dissociation are similar for 48 and result in the lower enantioselectivity.
Scheme 24.
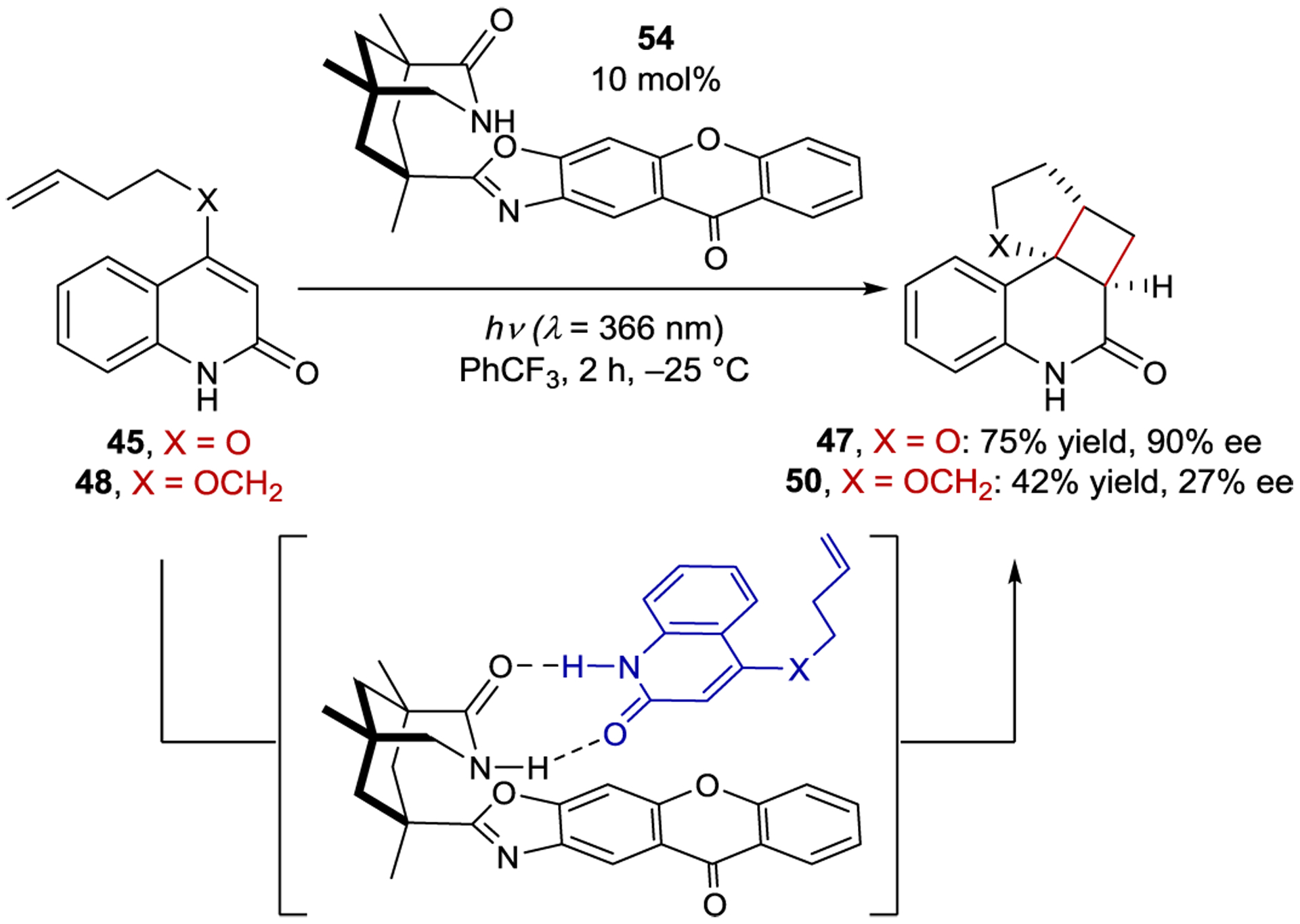
Intramolecular [2+2] Cycloaddition Catalyzed by a Xanthone Sensitizer
Given the competition between dissociation and cyclization, intermolecular photoreactions are considerably more difficult to design than intramolecular variants. Despite this challenge, Bach developed an intermolecular [2+2] photocycloaddition between pyridone 55 and acetylenedicarboxylates that is catalyzed by ent-54 (Scheme 25).135 Because the pyridone does not absorb the irradiated light under the reaction conditions, there is not a significant correlation between catalyst loading and ee, however, there is a correlation between alkyne loading and ee. It was proposed that a higher concentration of coupling partner leads to a faster bimolecular reaction and less possibility of substrate dissociation. The photochemical rearrangement of spirooxindole epoxides was also examined using xanthone 54, yielding product in up to 33% ee.136 Bach similarly attributed the poor selectivity to a slow rate of rearrangement relative to substrate dissociation from the catalyst.
Scheme 25.
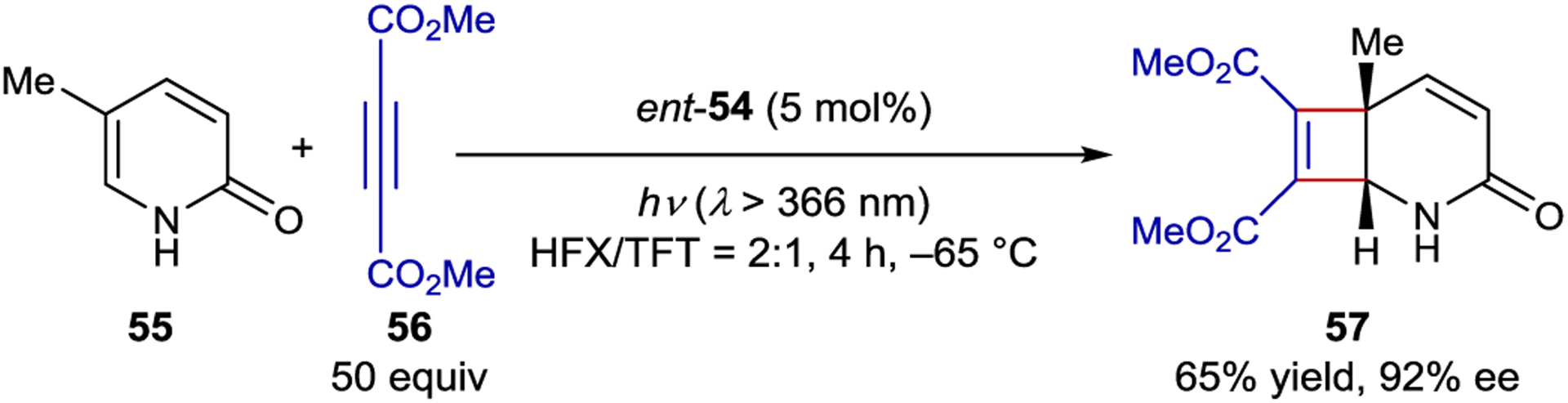
Intermolecular [2+2] Cycloaddition Catalyzed by a Xanthone Sensitizer
Although 54 was successful in promoting highly enantioselective reactions, there are several problems inherent with the xanthone chromophore that limited reaction development. First, photoexcited 54 is very active towards hydrogen atom abstraction. In toluene, the sensitizer decomposes in less than 10 min under 366 nm irradiation, limiting the choice of reaction solvents.137 The sensitizer also does not have a significant absorption in the visible region, necessitating irradiation at UV wavelengths that have a greater possibility of exciting free substrate. Thioxanthone 61 solves both problems: it is more stable under irradiation in toluene and possesses a significant absorption in the visible region. While the triplet energy of thioxanthone 61 (63 kcal/mol) is lower than xanthone 54 (76 kcal/mol), it is high enough to sensitize a variety of substrate molecules. The new sensitizer was evaluated in the canonical quinolone intramolecular [2+2] photoreaction producing cycloadducts in up to 96% ee under visible light irradiation.137 Under similar conditions, 3-alkylquinolones with tethered alkenes and allenes were also amenable to the intramolecular cycloaddition.138
An intermolecular [2+2] photocycloaddition between quinolone 58 and a variety of alkene coupling partners was also catalyzed by 61; however, 50 equiv of the coupling partner were needed to ensure cyclization occurs prior to dissociation from the catalyst (Scheme 26).139 It follows from this analysis that alkenes that react at slower rates should be expected to give lower enantioselectivities. Vinyl acetate (60) reacts more than an order of magnitude slower than ethyl vinyl ketone (59) when sensitized by an achiral thioxanthone, and consequently gave cycloadducts in significantly lower ee (91% ee vs. 58% ee).
Scheme 26.
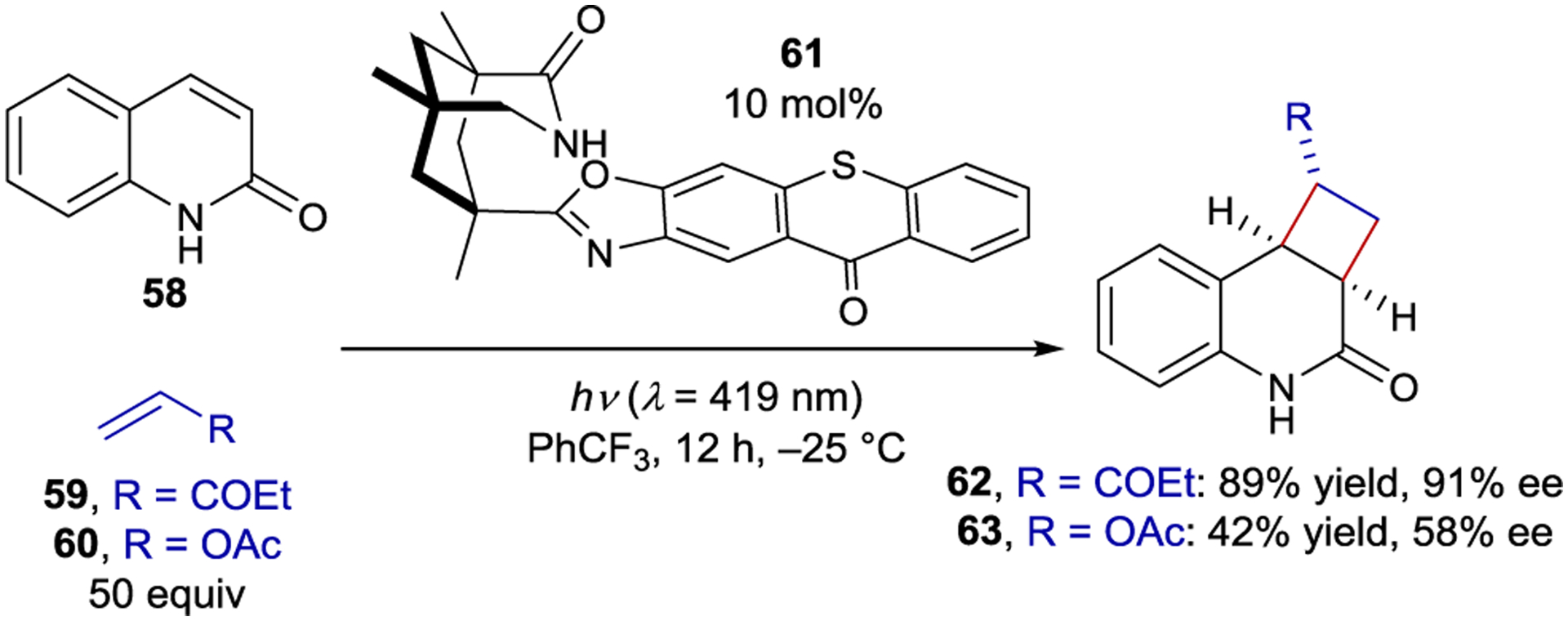
Intermolecular [2+2] Cycloaddition Catalyzed by a Thioxanthone Sensitizer
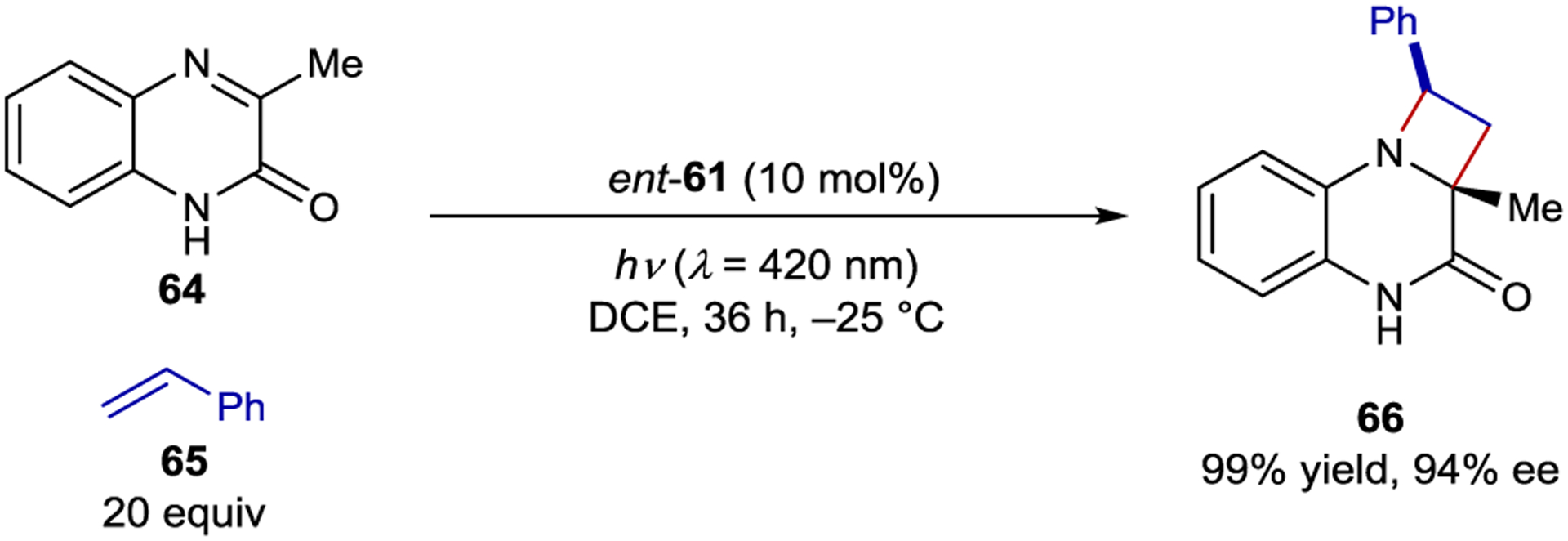
Thioxanthone ent-61 is also an effective photocatalyst for enantioselective aza-Paternò–Büchi reactions that produce chiral azetidines.140 Cyclic imine 64 was tested in the [2+2] reaction shown in Scheme 27, because N-substituted quinoxalinones were previously shown to undergo [2+2] cycloadditions with arylalkenes.141 A variety of styrenes were accommodated as coupling partners in the reaction affording azetidines in high yields and up to 98% ee.
Scheme 27.
Aza-Paternò–Büchi Reaction Catalyzed by a Thioxanthone Sensitizer
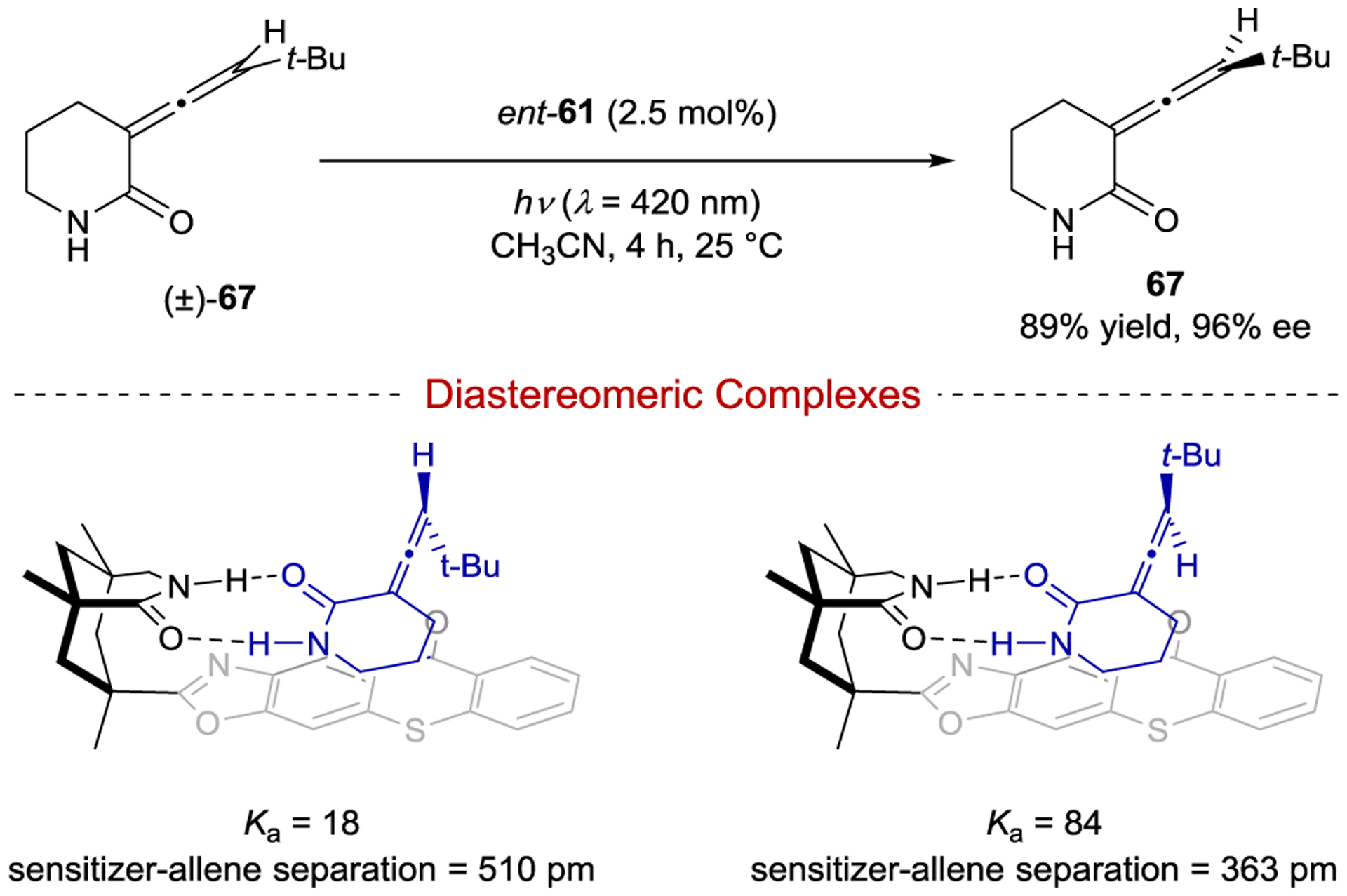
In addition to cycloadditions, thioxanthone 61 has proven to be applicable to photocatalytic deracemization. Under optimized reaction conditions Bach found that allene 67 could be deracemized in up to 97% ee (Scheme 28).142, 143 Mechanistically, triplet energy transfer to chiral allene 67 from thioxanthone 61 produces the achiral triplet-state allene.144 A measured quantum yield of racemization of Φ = 0.52 suggests that the excited achiral triplet allene decays with equal probability to 67 or ent-67. Deracemization is therefore achieved during the sensitization step of the reaction. Because both the allene and sensitizer are chiral, two distinct diastereomeric ground-state sensitizer–allene complexes can form. NMR titrations provided association constants of Ka = 84 and 18 for the respective diastereomeric complexes. The stronger-binding enantiomer is preferentially sensitized and thus selectively depleted over the course of the reaction, enriching the population of the weaker-binding antipode. Notably, the strategy relies on the fact that after sensitization the excited triplet allene dissociates from the catalyst and decays unselectively to the ground state leading to racemization of the stronger binding enantiomer. If, on the other hand, the allene always remained bound to the sensitizer during decay back to the ground state, retention of configuration would likely be expected. Thus, in contrast to cycloaddition reactions where dissociation of excited-state substrate is typically a hurdle in reaction development, dissociation is necessary to achieve high enantioselectivity in deracemization reactions. Computational studies also suggested that the separation between allene and thioxanthone was smaller for the stronger-binding complex, leading to more efficient energy transfer than the other diastereomeric complex. Thus, both the difference in ground-state association constants and the excited-state sensitization efficiencies for the two diastereomeric complexes are responsible for the high selectivity for deracemization.
Scheme 28.
Allene Deracemization Catalyzed by a Thioxanthone Sensitizer
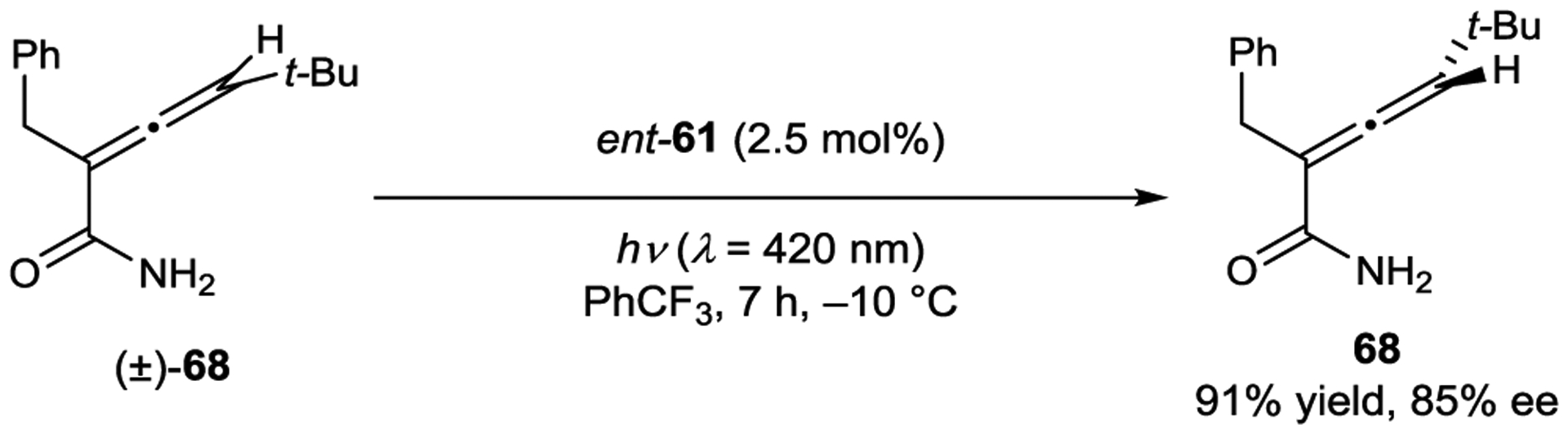
Thioxanthone 61 also catalyzed the deracemization of primary amides in up to 93% ee (Scheme 29).145 A combination of experimental and computational studies again confirmed that both ground-state association constants and excited-state sensitization efficiencies contribute to the enantioselectivity.
Scheme 29.
Primary Allene Amide Deracemization Catalyzed by a Thioxanthone Sensitizer
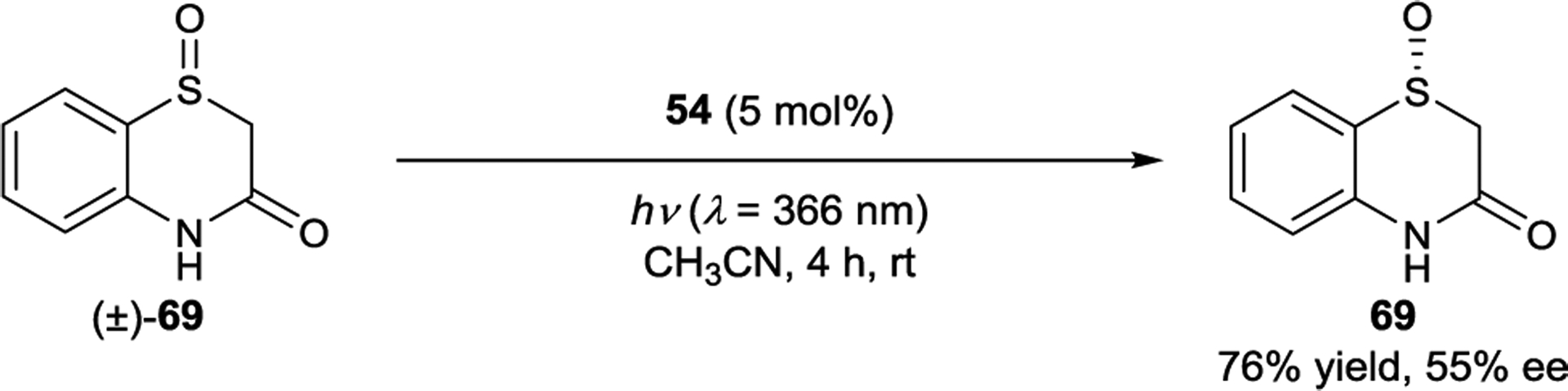
Xanthone 54 catalyzes the deracemization of cyclic sulfoxides in up to 55% ee (Scheme 30).146 An analogous mechanism was proposed in which one enantiomer of 69 binds more strongly to 54 than ent-69, leading to more efficient sensitization for the former. Once excited, the substrate dissociates from the chiral catalyst and can undergo stereochemical inversion prior to relaxation to the ground state. Over time, the enantiomer that binds the catalyst more strongly is preferentially depleted via unselective racemization.
Scheme 30.
Sulfoxide Deracemization Catalyzed by a Xanthone Sensitizer
In theory, any substrates that racemize in the excited state could be amenable to photoderacemization. Bach and coworkers discovered the photoderacemization of 3-cyclopropylquinolones by 61 serendipitously when attempting to develop an enantioselective di-π-methane rearrangement (Scheme 31).147 They observed that the enantioselectivity of the cyclopropane product increased from nearly 0% ee to 55% ee within the first hour of irradiation.
Scheme 31.
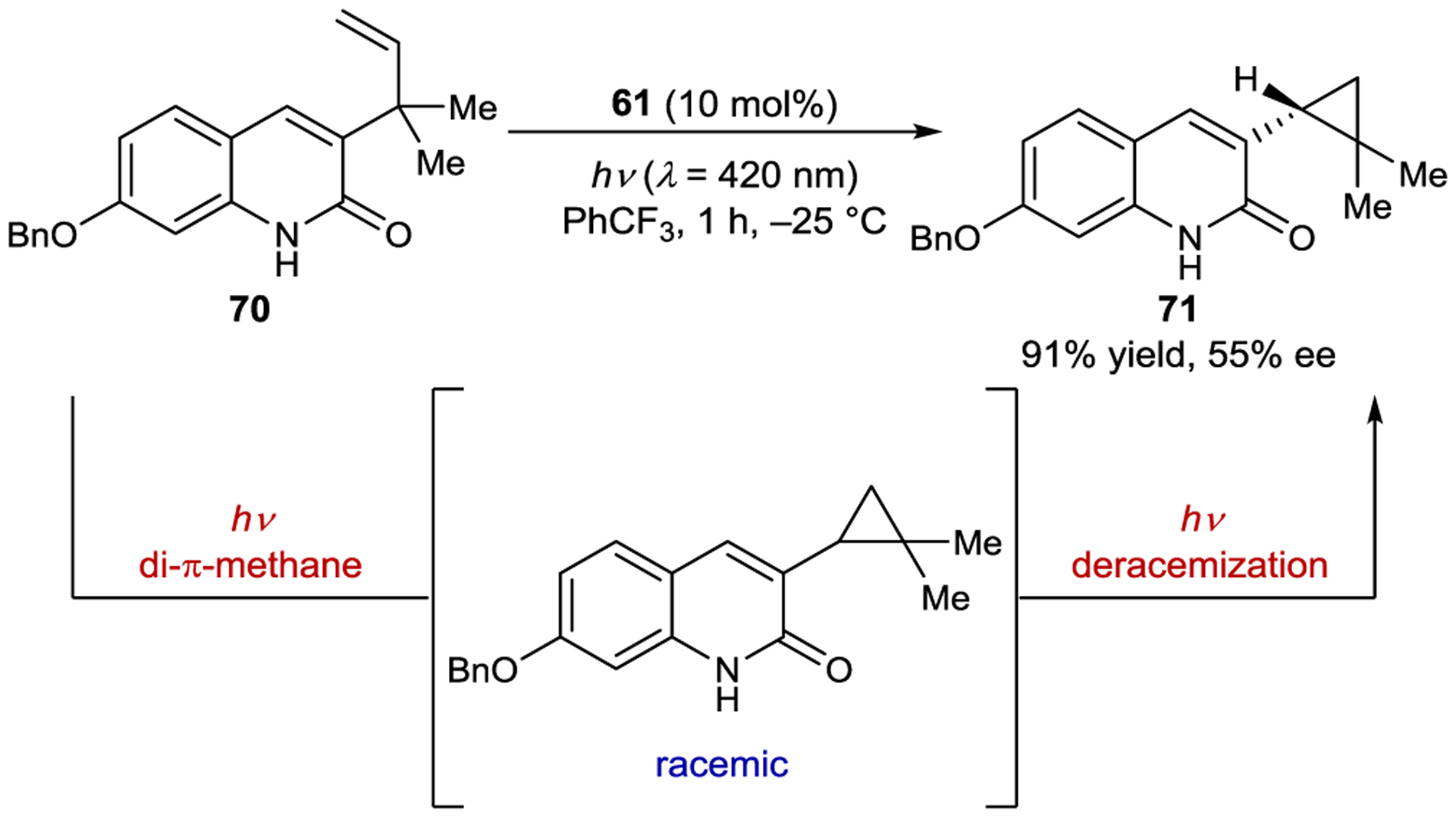
Di-π-Methane Rearrangement–Cyclopropane Deracemization Cascade Catalyzed by a Thioxanthone Sensitizer
Detailed mechanistic studies were undertaken for the related deracemization of spirocyclopropyl oxindoles (Scheme 32).148 Transient absorption studies suggested a triplet energy transfer from the sensitizer to cyclopropyl substrate 72. The spectral position and shape of the transient signal of the triplet substrate also matched that calculated for the expected ring-opened 1,3-diradical. As with allene deracemization, both the ground-state association constants and excited-state sensitization kinetics of the diastereomeric substrate–catalyst complexes were invoked to account for the enantioenrichment of the product. The triplet lifetime of the 1,3-diradical is 22 μs while the lifetime of the substrate–sensitizer complex is below 1 μs, implying that the 1,3-diradical dissociates from the catalyst prior to cyclization. This kinetic regime is desirable since cyclization within the chiral domain of the catalyst would favor retention of configuration.
Scheme 32.
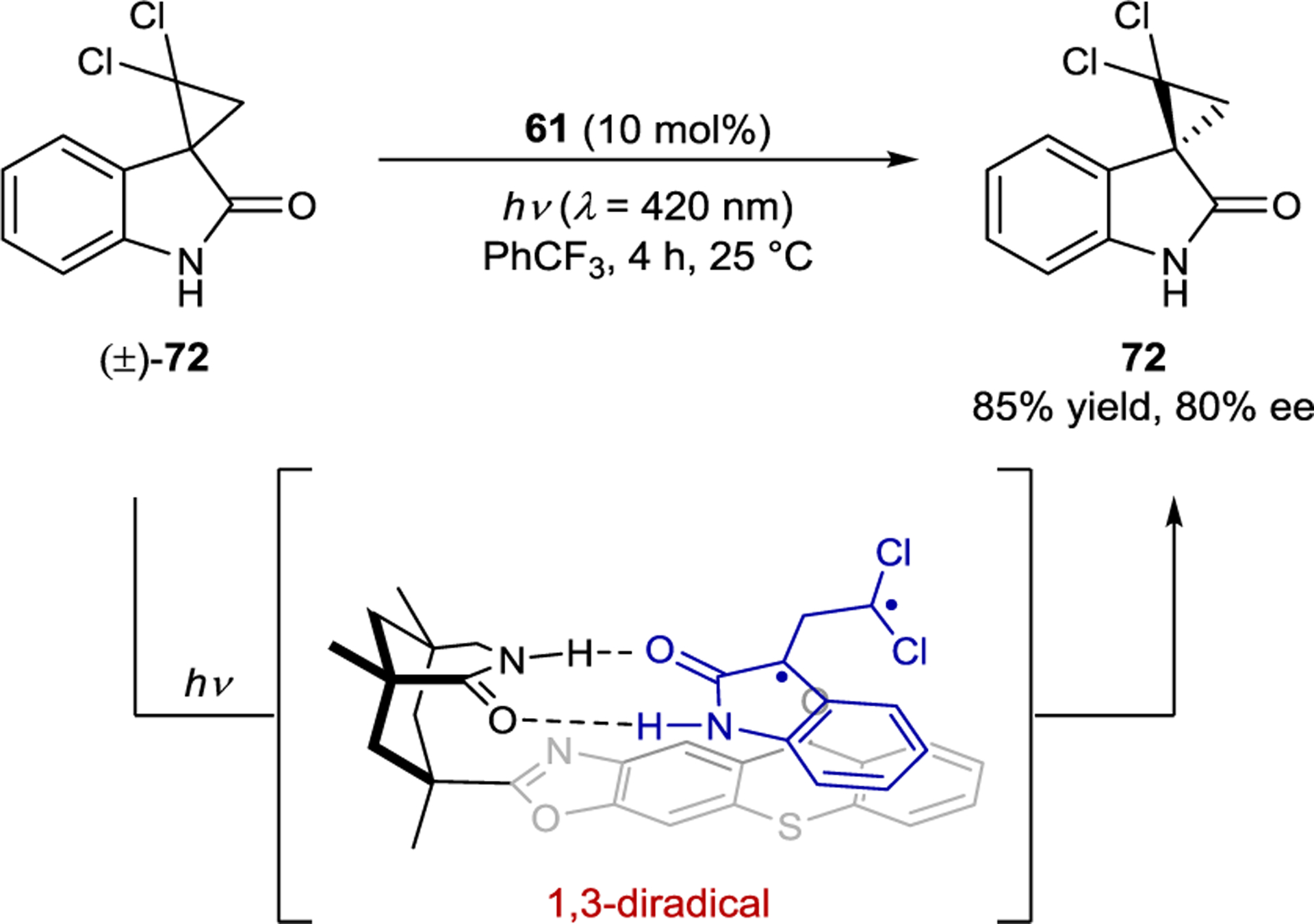
Cyclopropane Deracemization Catalyzed by a Thioxanthone Sensitizer
Despite the wide range of photochemical reactions that can be catalyzed by xanthone 54 and thioxanthone 61, the reactions are limited to lactam-containing substrates.149 In an attempt to expand the range of available binding motifs, Bach and coworkers have recently prepared new catalysts that incorporate a thioxanthone sensitizer into an alternate chiral hydrogen-bonding scaffold (Figure 2). While thiourea-linked thioxanthone 74 does not induce high levels of enantioselectivity in a sensitized 6π-electrocyclization (12% ee),150 BINOL-derived phosphoric acid 73 catalyzes an intermolecular [2+2] cycloaddition between carboxylic acid 75 and cyclopentene (76) in 86% ee (Scheme 33).151 NMR studies demonstrated that the carboxylic acid substrate binds the BINOL catalyst under the reaction conditions. Computational studies support a 1:1 complex but suggest that several binding modes are similar in energy, which may account for the moderate enantioselectivity. Takagi has studied a simpler BINOL-derived phosphoric acid lacking a thioxanthone substituent in asymmetric intramolecular [2+2] photocycloadditions of quinolones. However, in this reaction a stoichiometric loading of the acid template was necessary, presumably because both bound and unbound substrate react at similar rates.152
Figure 2.
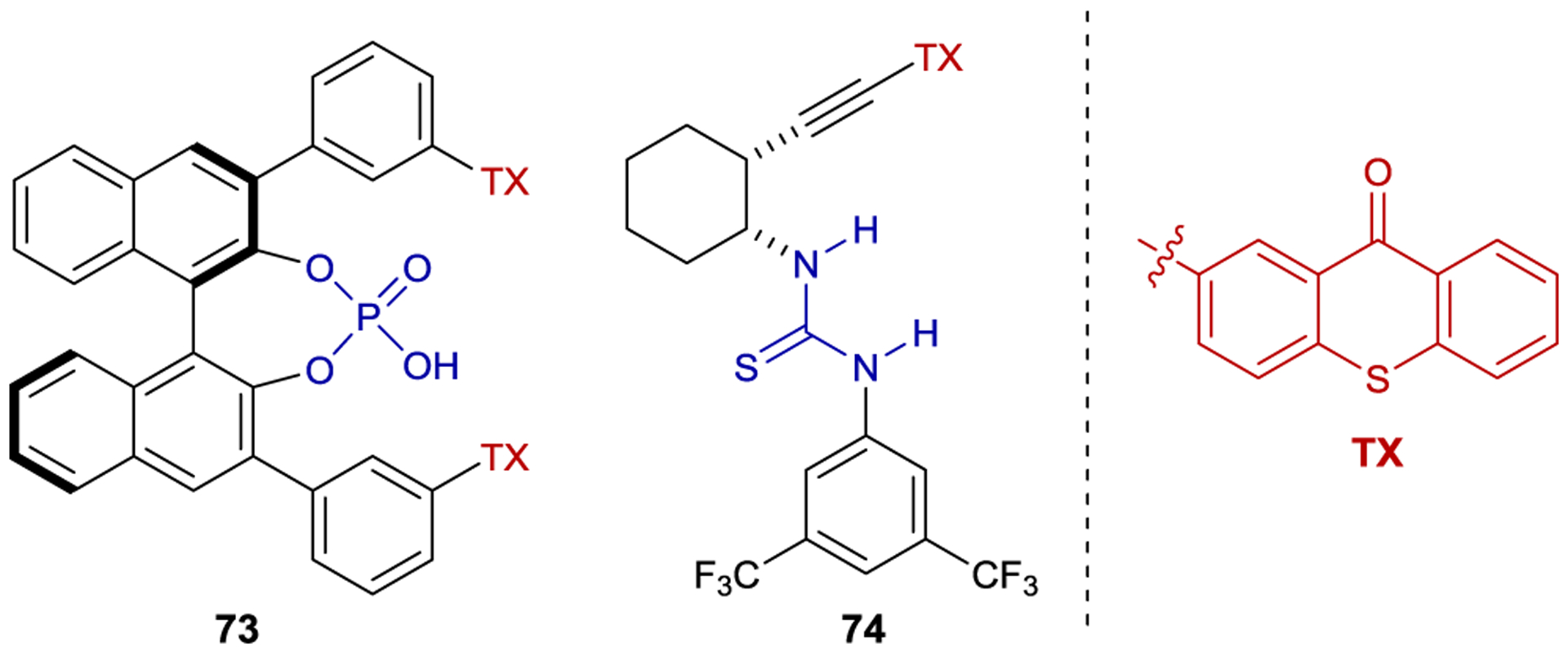
Chiral Thioxanthone-Derived Sensitizers
Scheme 33.
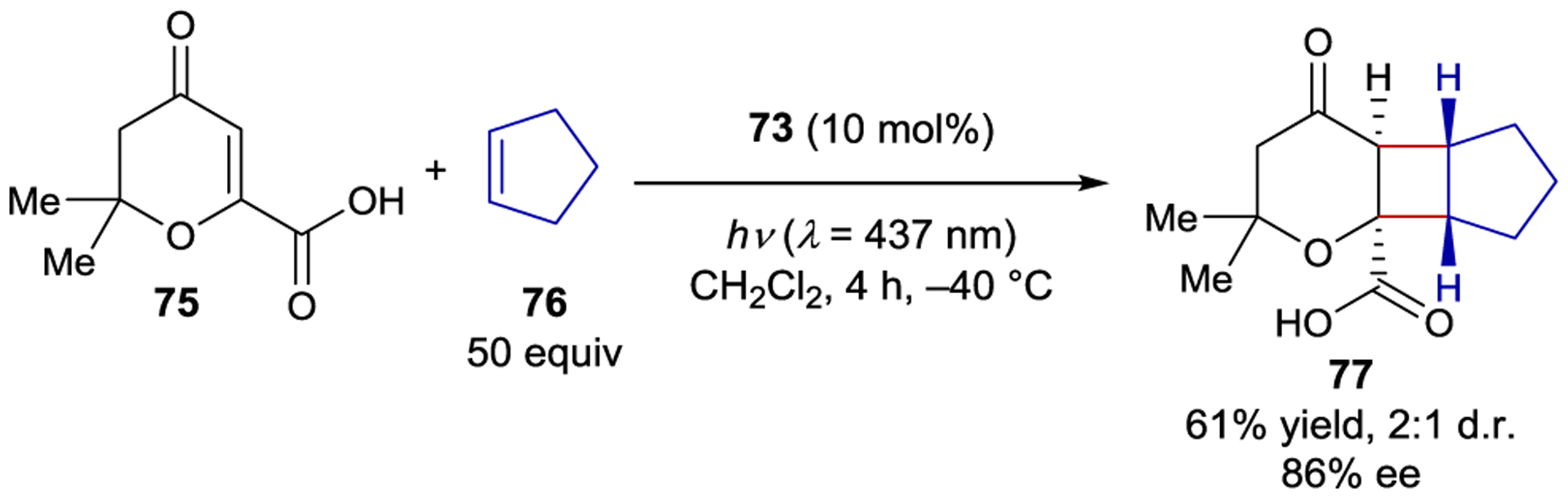
Intermolecular [2+2] Cycloaddition Catalyzed by a BINOL-Derived Thioxanthone Sensitizer
Thioxanthone 73 catalyzes the enantioselective photocycloaddition of N,O-acetal 78 and alkene 79 in 95% ee (Scheme 34). NMR studies revealed that 78 exists as a mixture of the cyclic N,O-acetal and the ring-opened imine. UV-Vis absorption studies showed that a bathochromic shift occurs upon protonation of the imine, while emission studies providing the triplet energies of the iminium ion and thioxanthone catalyst (51 kcal/mol and 56 kcal/mol, respectively) showed that energy transfer to the iminium was exothermic. Conversely, both the N,O-acetal and unprotonated imine are unable to be sensitized by the photoexcited thioxanthone catalyst.
Scheme 34.
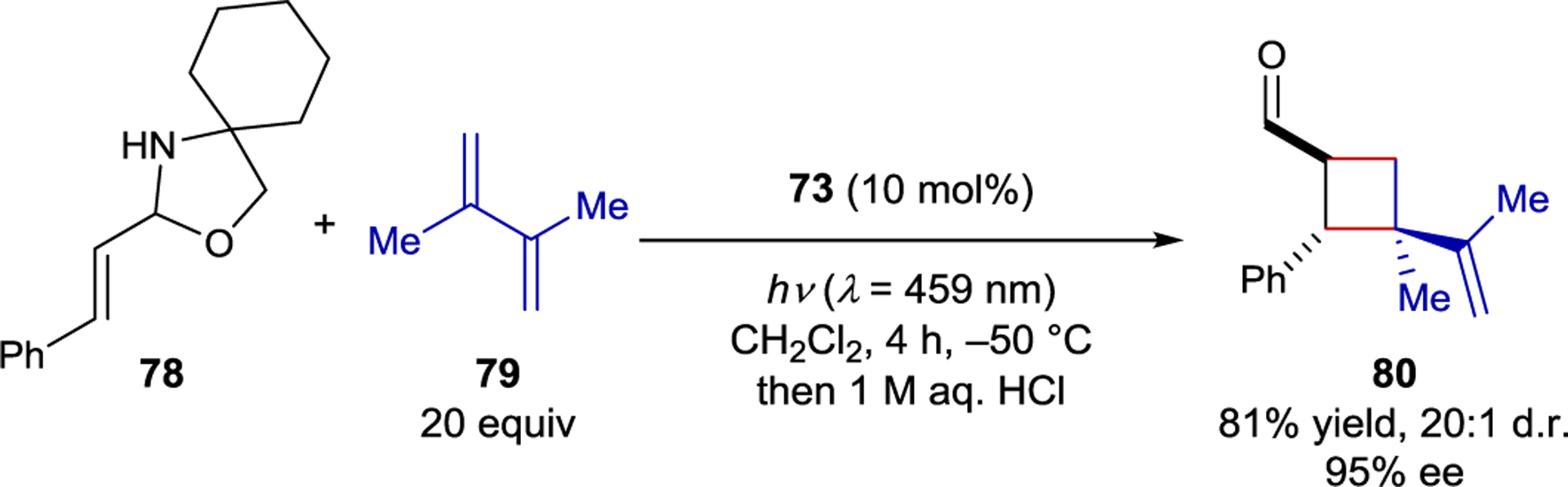
Intermolecular [2+2] Cycloaddition of Iminium Ions Catalyzed by a BINOL-Derived Thioxanthone Sensitizer
Masson developed a non-C2-symmetric BINOL-derived catalyst with a tethered thioxanthone chromophore for the enantioselective synthesis of 1,2-diamines (Scheme 35).153 This reaction occurs in two steps. In the first, the nucleophilic enecarbamate reacts with the electrophilic azodicarboxylate in an enantioselective thermal reaction. The resulting imine is unstable under the reaction conditions; thus ethanethiol is added to generate α-carbamoylsulfide 84. In the second step, pyrazole is added and the reaction is irradiated with blue light. The authors propose that α-carbamoylsulfide 84 is photochemically oxidized to the sulfur radical cation, triggering mesolytic cleavage of thiyl radical and ultimately generating imine 85, which is intercepted by pyrazole in a thermal, diastereodetermining reaction. Notably, both stereodetermining steps in this reaction are thermal, while the only photochemical step regenerates the reactive imine intermediate.
Scheme 35.
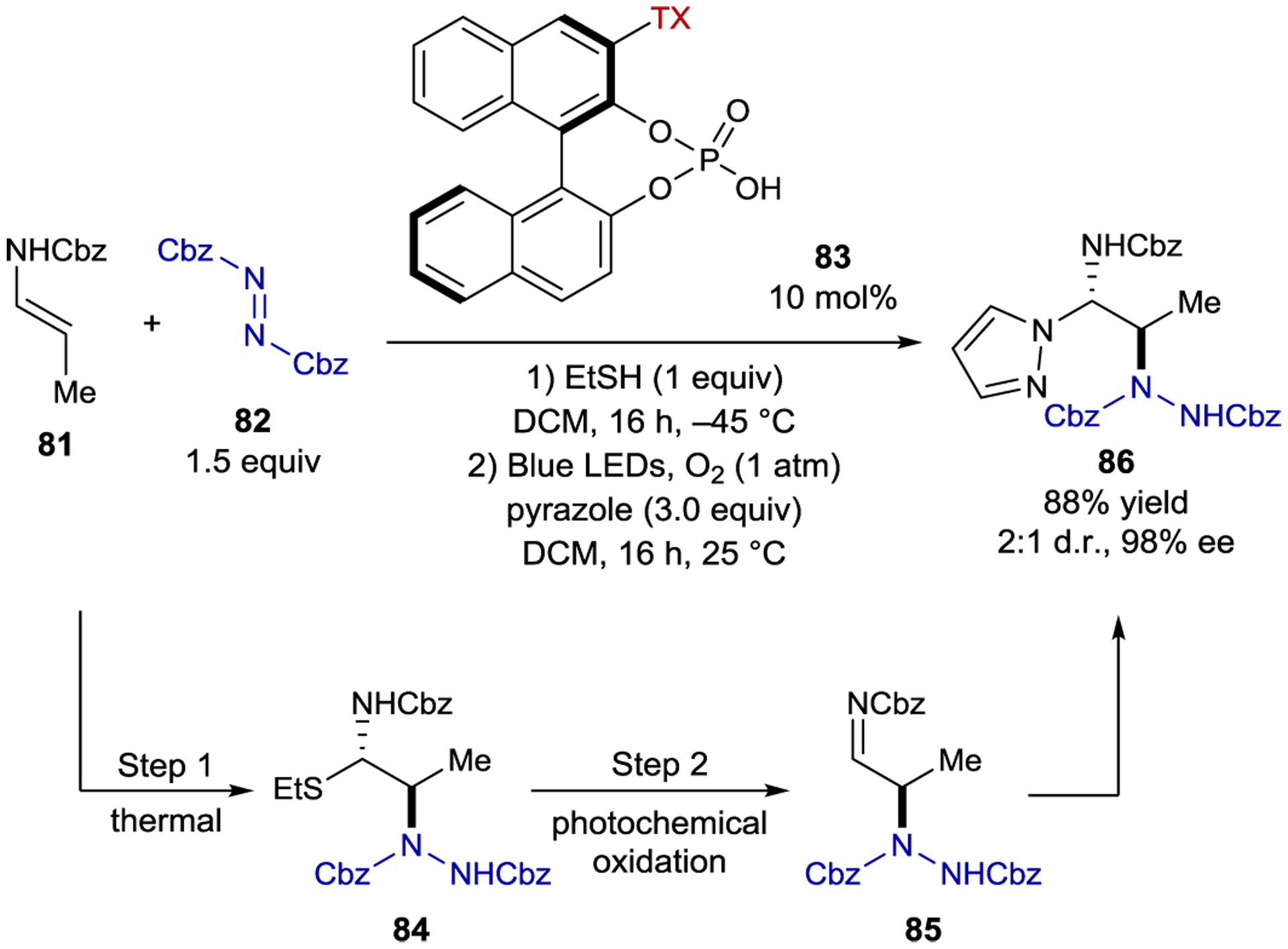
1,2-Diamine Synthesis Catalyzed by a BINOL-Derived Thioxanthone Sensitizer
2.2.3. Enamine and Iminium Chromophores
Melchiorre introduced a new strategy in asymmetric photocatalysis in which the light-absorbing species is an electron donor–acceptor (EDA) complex.154, 155 In this strategy, an electron-rich donor molecule and an electron-poor acceptor molecule associate in the ground state, comprising an EDA complex (Scheme 36).156 The photophysical properties of the complex are distinct from the donor or acceptor in isolation. This association leads to the formation of a new charge-transfer absorption band, which at the simplest level can be understood as an intracomplex electron transfer from the donosr HOMO to the acceptor LUMO. In most cases, orbital mixing between the donor and acceptor changes the relative position of the frontier molecular orbitals, stabilizing the formation of the EDA complex. The appearance of color when mixing two colorless compounds is a hallmark of EDA complexes and was the original observation that led Mulliken to formulate the charge transfer theory.157
Scheme 36.
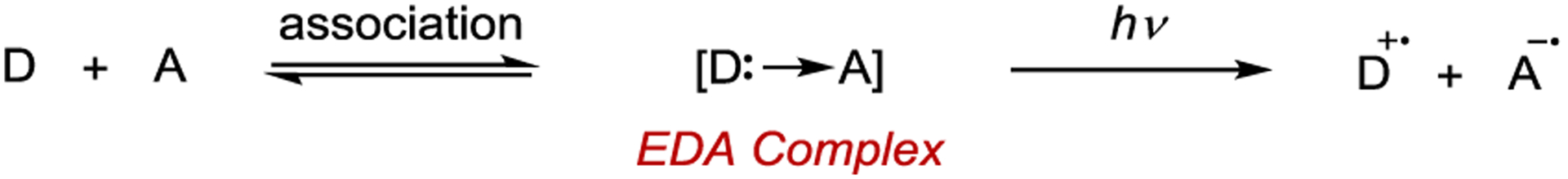
Electron Donor–Acceptor Complex Formation and Excitation
In 2013, Melchiorre exploited this strategy to promote the enantioselective α-alkylation of aldehydes with alkyl halides (Scheme 37).158 This reaction was based on organocatalyzed photoredox reactions developed by MacMillan.159 In both designs, a chiral secondary amine organocatalyst condenses with an aldehyde to form an enamine intermediate. However, unlike the previous organocatalyzed reactions, an exogenous photocatalyst was not needed. Although neither enamine nor alkyl bromide absorb visible light individually, the mixture exhibited a broad absorption feature in the visible region, characteristic of the formation of an EDA complex. Spectrophotometric analyses confirmed the 1:1 ground-state association with an association constant of KEDA = 11.6 determined from Benesi–Hildebrand analysis.160 Upon excitation of the ground-state complex, an electron is promoted from the enamine donor to the alkyl bromide acceptor, forming a radical ion pair (Scheme 38, top). The short-lived radical anion undergoes bromide cleavage to produce an iminium ion and an alkyl radical. The authors first proposed a closed catalytic cycle in which radical-radical recombination forms an iminium intermediate which is then hydrolyzed to afford the α-alkylated product, but further investigations revealed that the quantum yield (Φ) of the reaction with both benzyl and phenacyl bromides is Φ > 20, consistent with a radical chain mechanism.160 Notably, the chiral catalyst is involved in both the photochemical initiation and the ground-state enantiodetermining radical addition into the enamine.
Scheme 37.
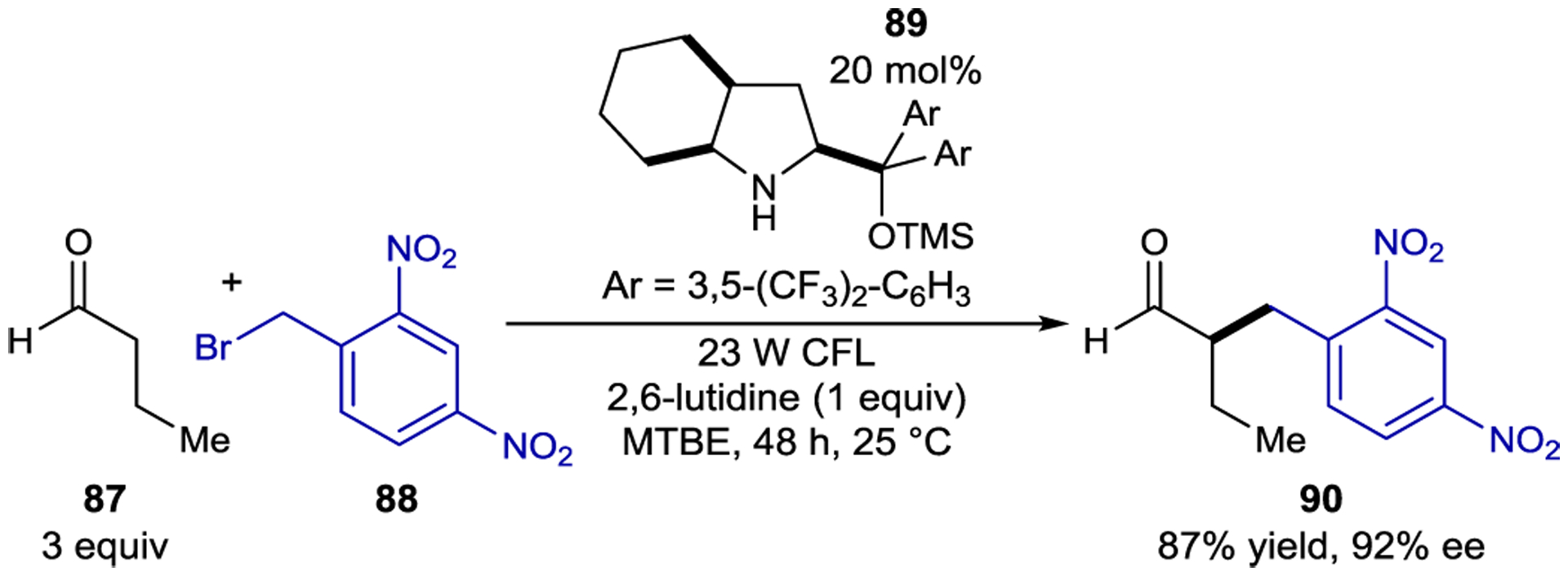
Aldehyde α-Alkylation via EDA Complex Excitation
Scheme 38.
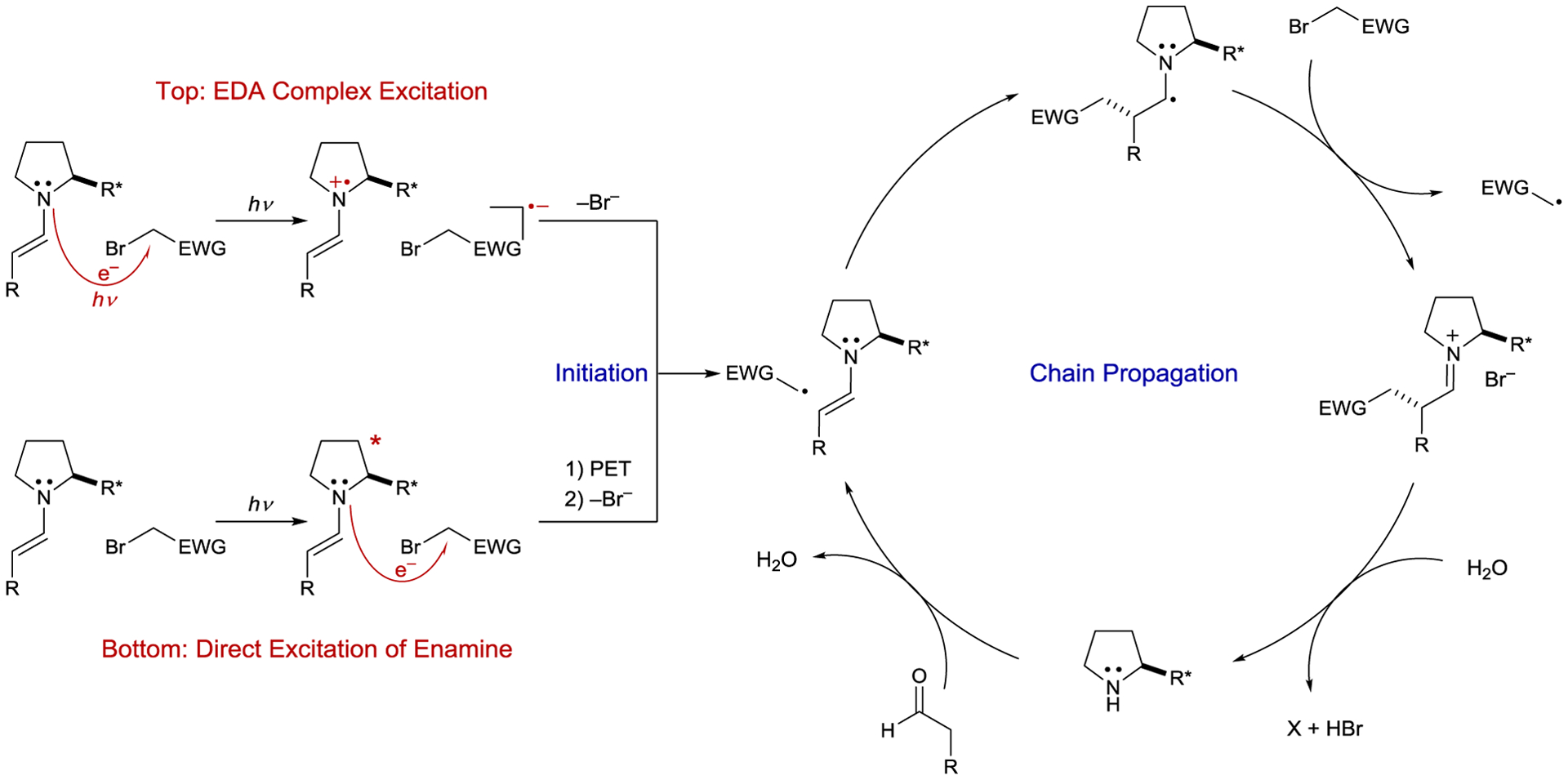
Photoinitiated Enamine Catalysis Mechanisms
A similar photo-organocatalytic strategy proved applicable to ketone substrates when coupled with cinchona-based primary amine catalyst 92 (Scheme 39).161 Both electron poor benzyl bromides (88) and phenacyl bromides were competent electron acceptors, but the reaction was limited to cyclic ketones as precursors to the chiral enamine electron donors.
Scheme 39.
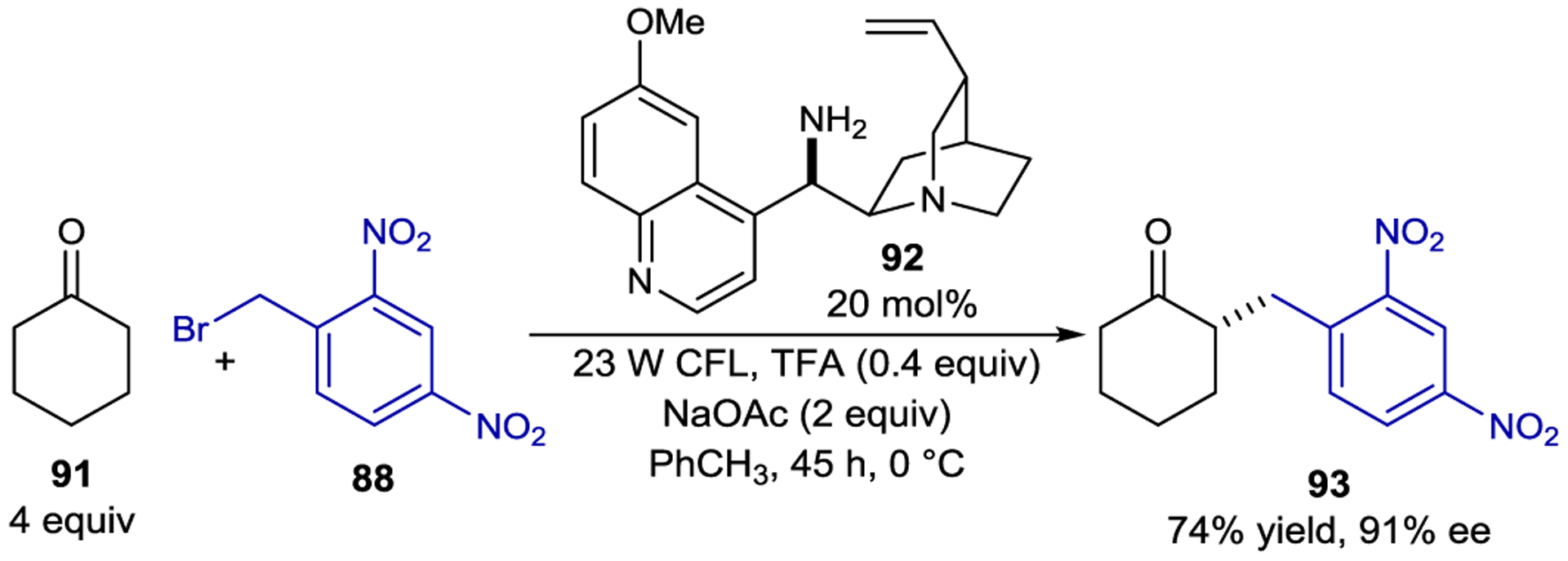
Ketone α-Alkylation via EDA Complex Excitation
Melchiorre and coworkers also showed that bromomalonates (94) are competent electron acceptors in the α-alkylation of aldehydes (87) and enals using amine organocatalysts (Scheme 40).162 Alkylated products were obtained in up to 94% ee, and complete γ-selectivity was observed in radical addition to enals. The authors initially assumed the intermediacy of an EDA complex between the enamine and bromomalonate; however, no charge transfer band was observed in the UV-Vis absorption spectrum of the mixture. Instead, the enamine was the only reaction component that significantly absorbed light under the optimized conditions. A series of Stern–Volmer quenching studies showed that the bromomalonate quenched the excited-state emission of the enamine. From this study, the authors proposed that PET reduction of the bromomalonate by the excited enamine followed by loss of bromide resulted in the formation of an electrophilic malonyl radical that would subsequently react with another equivalent of ground-state enamine (Scheme 38, bottom). The chain propagation step is likely different than in the EDA reaction because the α-aminoradical is incapable of reducing the bromomalonate. Instead, the α-aminoradical abstracts bromine from another equivalent of bromomalonate. Collapse of the bromoamine intermediate expels bromide, forming an iminium intermediate, which is hydrolyzed to the product. Notably, the photochemical step in both the EDA and PET reactions serves only to initiate the radical chain. Other classes of electron acceptors including (phenylsulfonyl)alkyl iodides were also amenable to the PET reaction.163 After desulfonylation, α-methylation and α-benzylation products could be obtained in high yield and enantioselectivity.
Scheme 40.
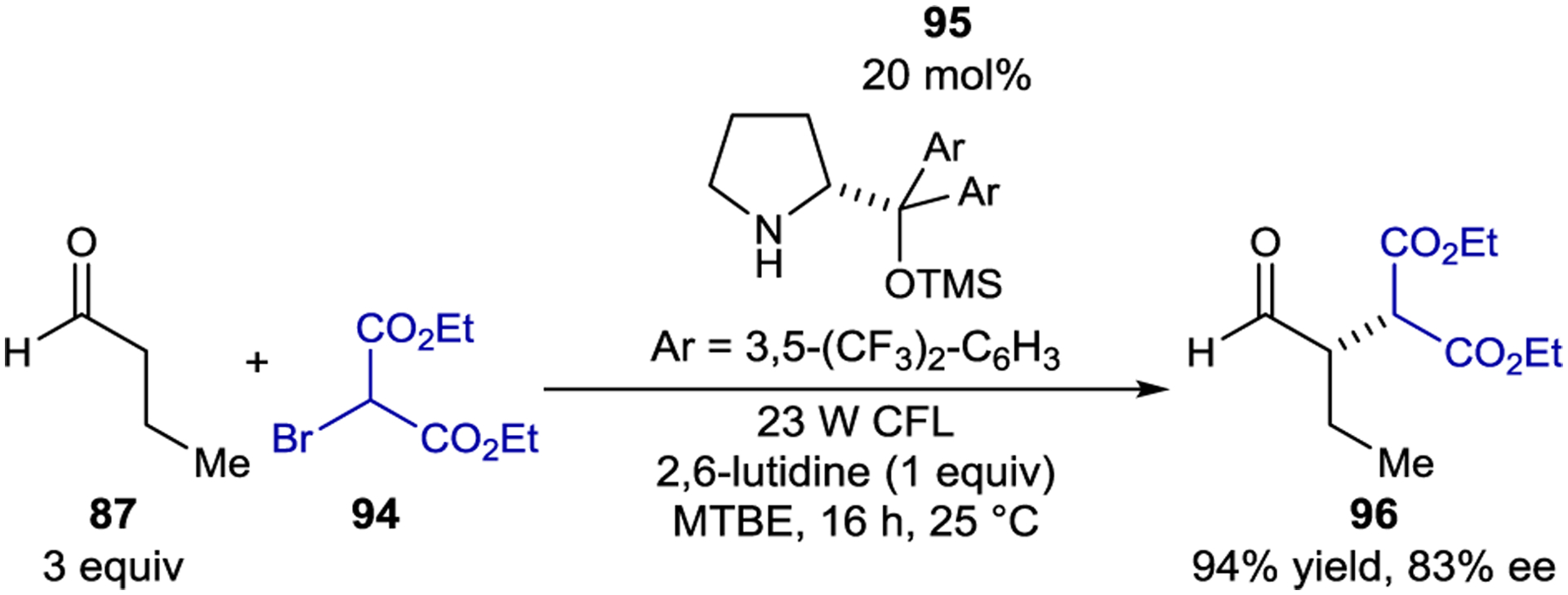
Aldehyde α-Alkylation via Direct Enamine Excitation
A similar reaction was reported by Alemán with a thioxanthone-substituted organocatalyst (98) yielding alkylated aldehydes in high yields and enantioselectivities (Scheme 41). In this case, the bromomalonate is reduced by the thioxanthone catalyst instead of the enamine, initiating the radical chain.164
Scheme 41.
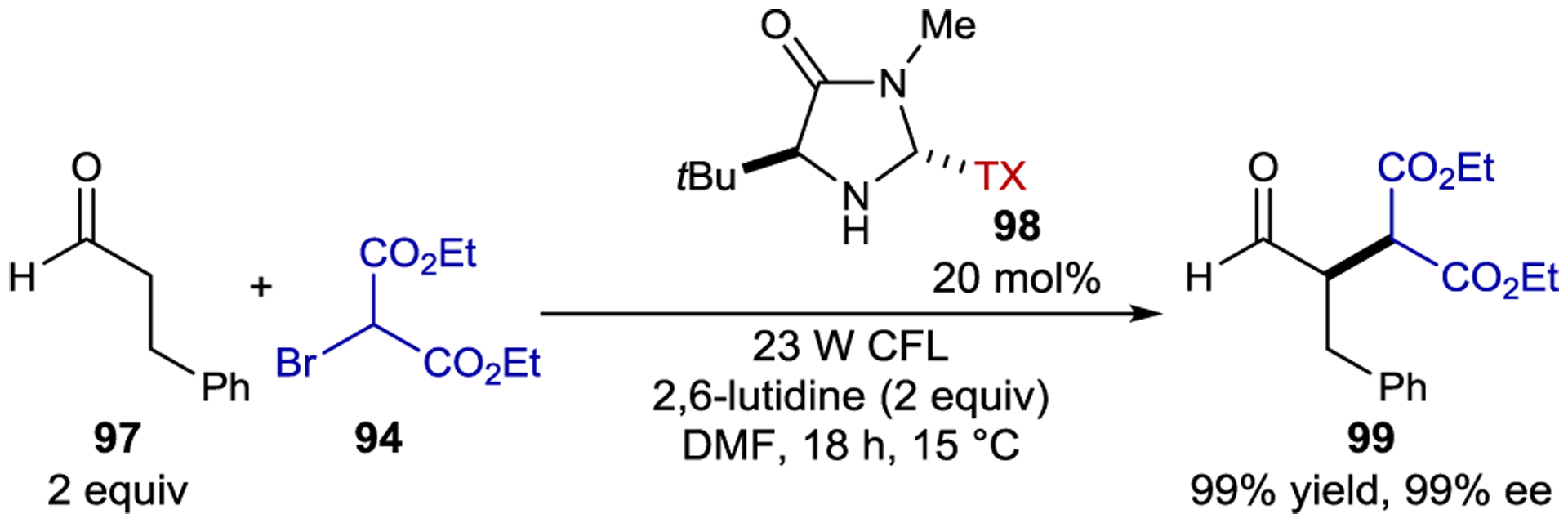
Aldehyde α-Alkylation Promoted by a Bifunctional Photoaminocatalyst
In 2017, Melchiorre and coworkers showed that chiral iminium ions generated in situ by condensation of α,β-unsaturated aldehydes (100) with chiral amine catalysts (102) could promote the asymmetric β-alkylation of enals (Scheme 42).165 Enamine and iminium photocatalysis are similar yet complementary strategies. Electron-rich enamines are nucleophilic in the ground state and become potent single-electron reductants after photoexcitation. In contrast, iminium ions act as electrophiles in the ground state and are strong oxidants after photoexcitation, allowing the use of complementary radical precursors.
Scheme 42.
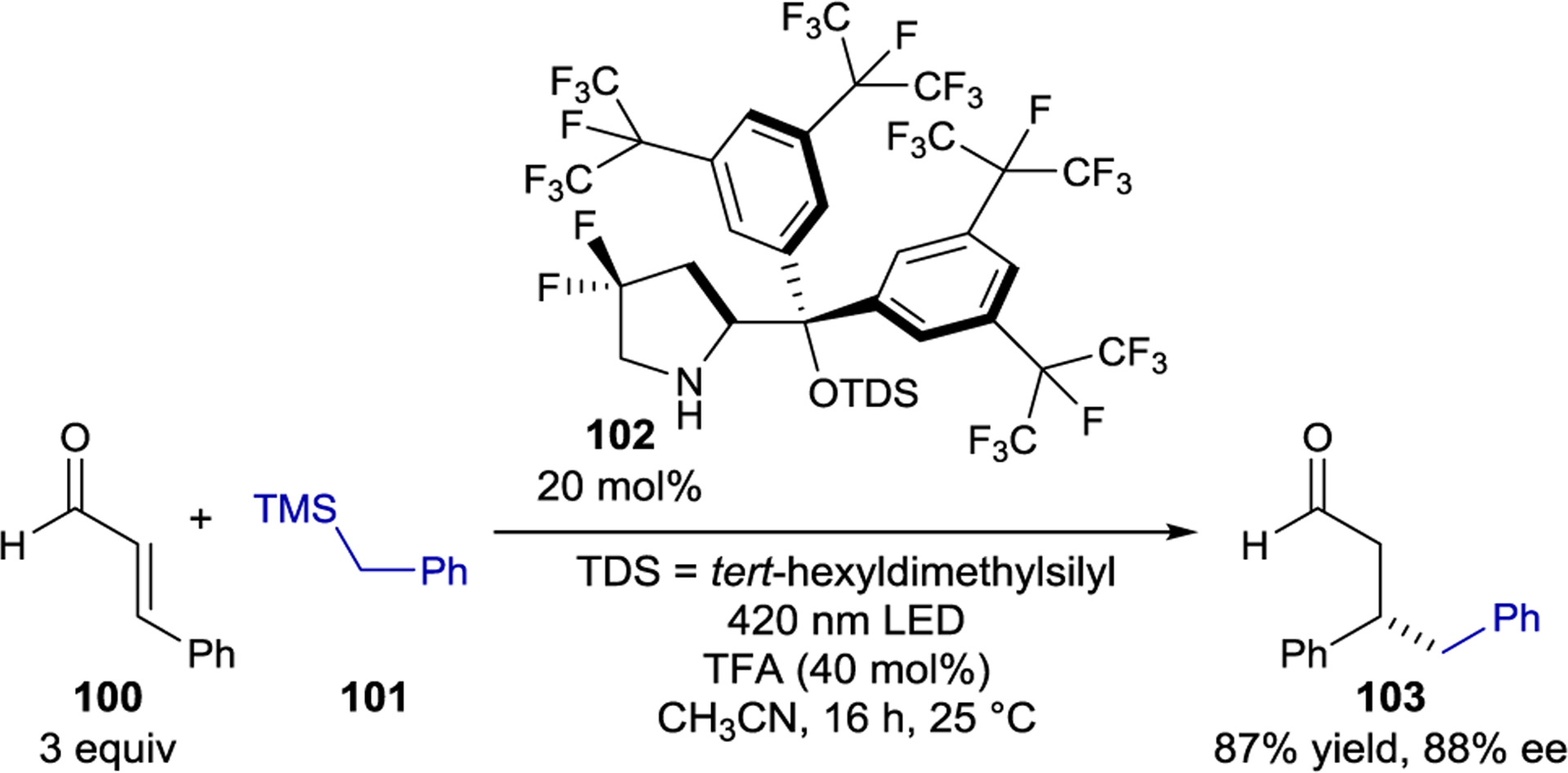
Enal β-Alkylation via Direct Iminium Ion Excitation
Melchiorre proposed a mechanism in which a chiral iminium ion directly absorbs visible light and functions as a single-electron oxidant to an electron-rich alkyl trimethylsilane (101) (Scheme 43). The silyl radical cation undergoes mesolytic fragmentation to afford a stabilized alkyl radical and trimethylsilyl cation. Coupling of the β-enaminyl radical intermediate with the alkyl radical affords an enamine, which after hydrolysis yields the β-alkylated aldehyde. This radical–radical coupling mechanism was proposed instead of a radical chain mechanism because the putative propagation step, single electron transfer (SET) between the silane and α-iminyl radical cation, would be endergonic. Although this method is limited to benzyl radicals or radicals stabilized by an adjacent heteroatom, unstabilized alkyl radicals could be accommodated when using a dihydropyridine radical precursor.166 This approach gave selective 1,4-addition and offers an advance over thermal iminium catalysis with organometallic nucleophiles, which often produces a mixture of 1,2- and 1,4-addition adducts.
Scheme 43.
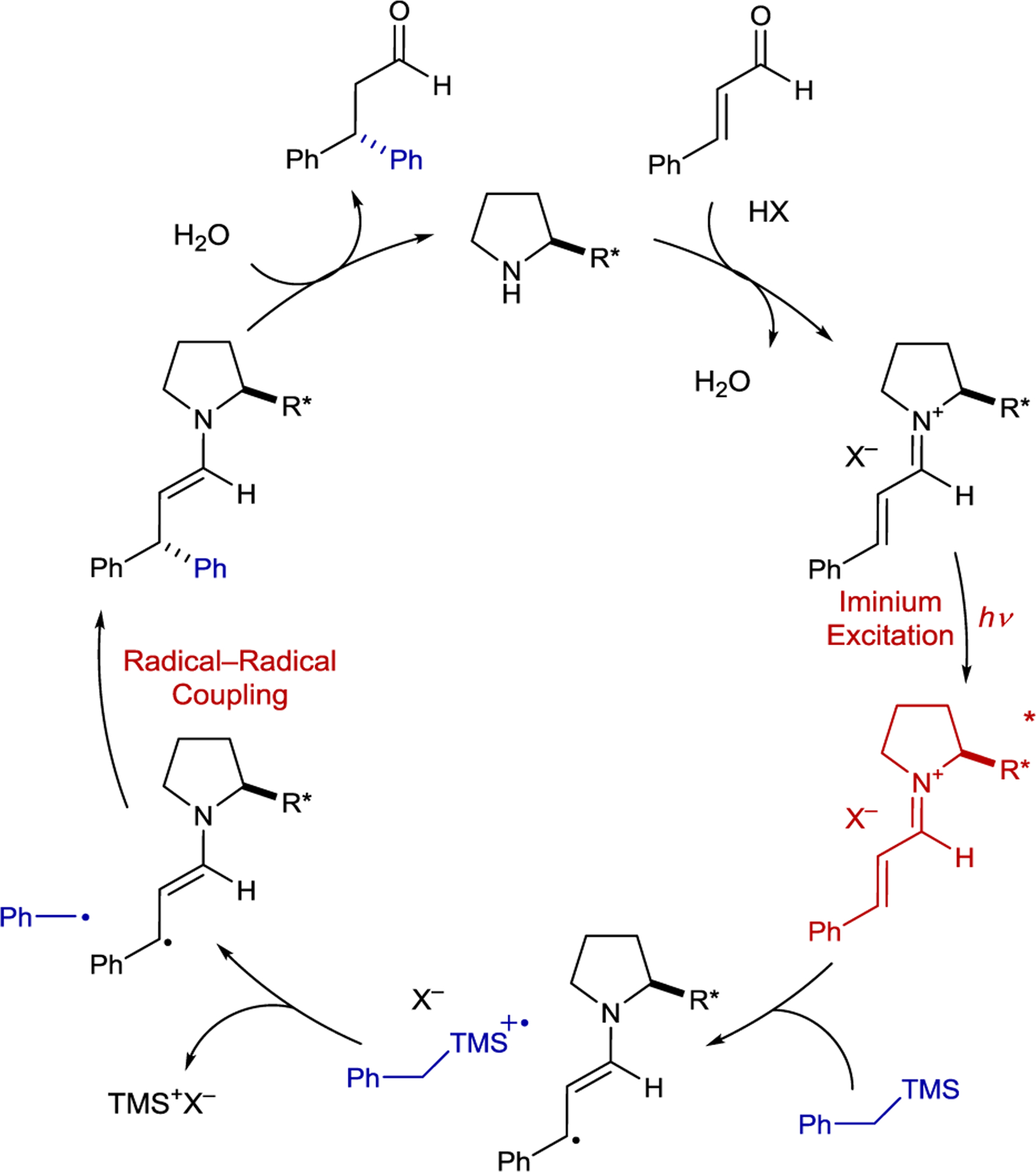
Enal β-Alkylation Mechanism
The initial product of a radical–radical coupling under iminium photocatalysis is an enamine, which can further react with an electrophilic moiety if one is present. Melchiorre and coworkers leveraged this insight to design a photochemical cascade reaction in which the excited-state and ground-state reactivity of organocatalytic intermediates were exploited to form cyclopentanols (106) in excellent diastereo- and enantioselectivity (Scheme 44).167 Cyclopropanols (104) were used as electron donors, reducing the photoexcited iminium, to generate β-keto radical cation intermediates after ring-opening. In analogy to previous work, stereocontrolled radical–radical coupling affords an enamine intermediate. The ground-state enamine nucleophile cyclizes with the electrophilic ketone in a second stereocontrolled step. A redox mediator, 1,1’-biphenyl, was added to the reaction to increase efficiency. Mechanistic experiments revealed that the excellent enantioselectivity of the reaction arises from a kinetic resolution in the thermal cyclization. The major enantiomer formed in the radical–radical coupling cyclizes quickly, leading to stereochemical amplification over the two steps, while the minor enantiomer does not cyclize.
Scheme 44.
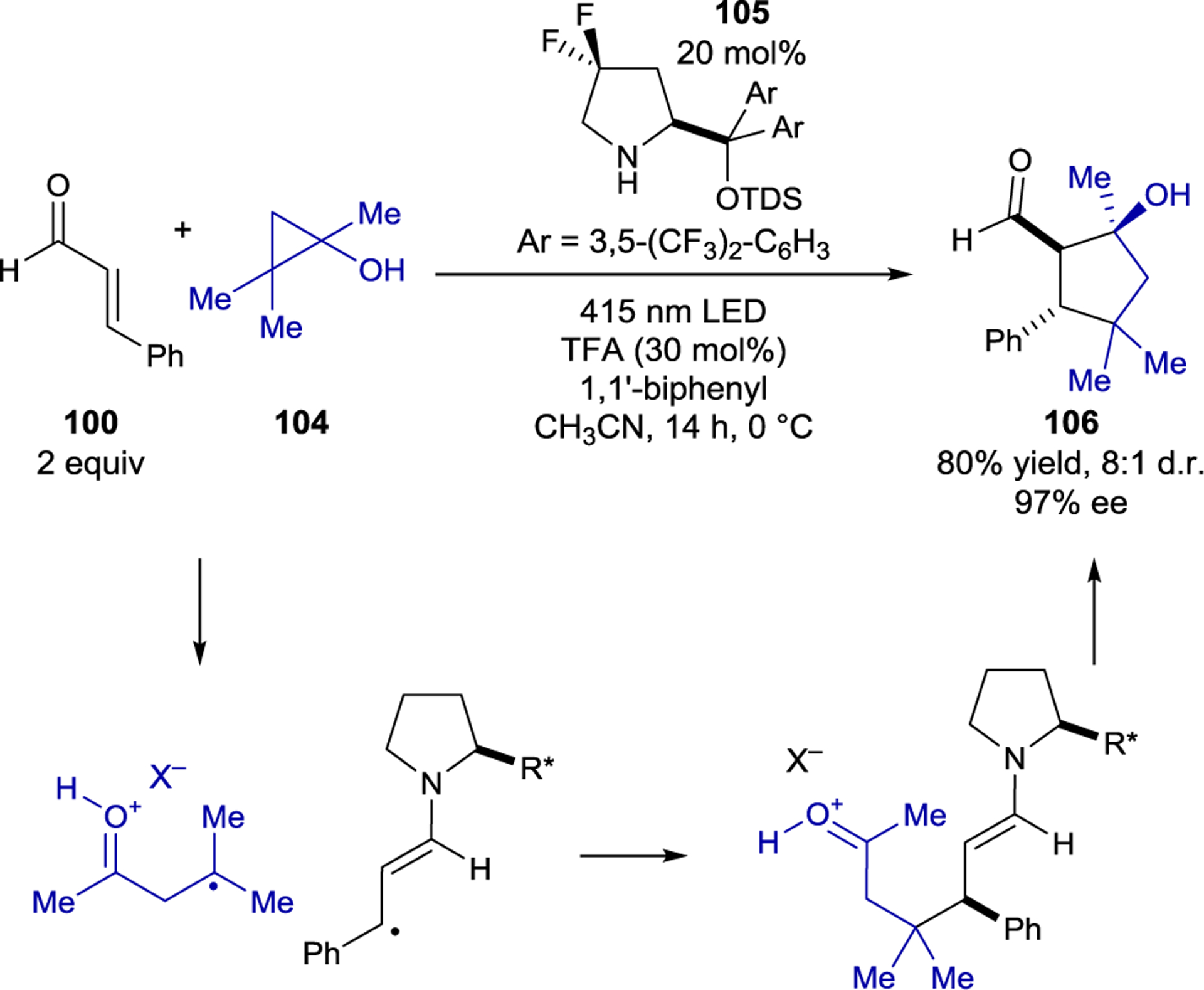
Cyclopentanol Synthesis via Iminium Ion Catalysis
Another example of the cascade strategy enabled the stereoselective construction of complex butyrolactones (108).168 Photoexcitation of an iminium derived from an enal substrate results in oxidation of alkene 107, the appended carboxylic acid moiety of which can cyclize (Scheme 45). Enantioselective radical–radical coupling produces cascade product 108 in high enantioselectivity. Because the first cyclization occurs in the absence of the chiral catalyst, no diastereoselectivity was observed. A similar cascade was subsequently developed with allene-appended carboxylic acids, ultimately yielding bicyclic lactones with moderate enantioselectivity.169
Scheme 45.
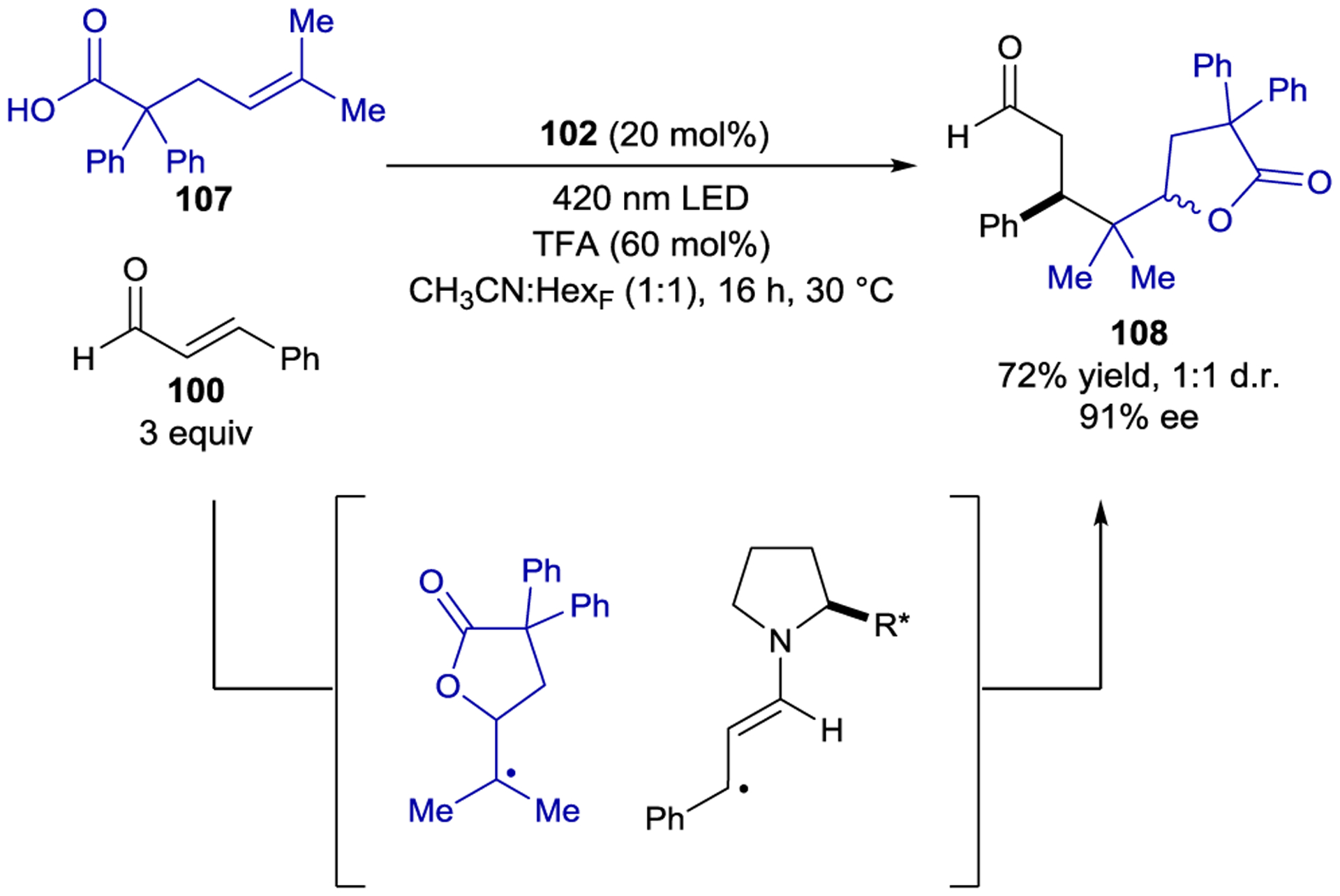
Cascade Reaction via Iminium Ion Catalysis
Toluene derivatives also reductively quench the iminium excited state leading to β-benzylated aldehydes from the corresponding enals (Scheme 46).170 Following oxidation of toluene, the benzylic C–H bond is significantly acidified; the pKa is estimated to be −13 in CH3CN.171 Deprotonation results in a benzyl radical that undergoes radical–radical coupling with the β-enaminyl radical intermediate. Zn(OTf)2 was a necessary additive in this process; the Zn2+ cation was proposed to serve as an acid to promote iminium formation, while the triflate counteranion was proposed to deprotonate the photogenerated toluene radical cation.
Scheme 46.
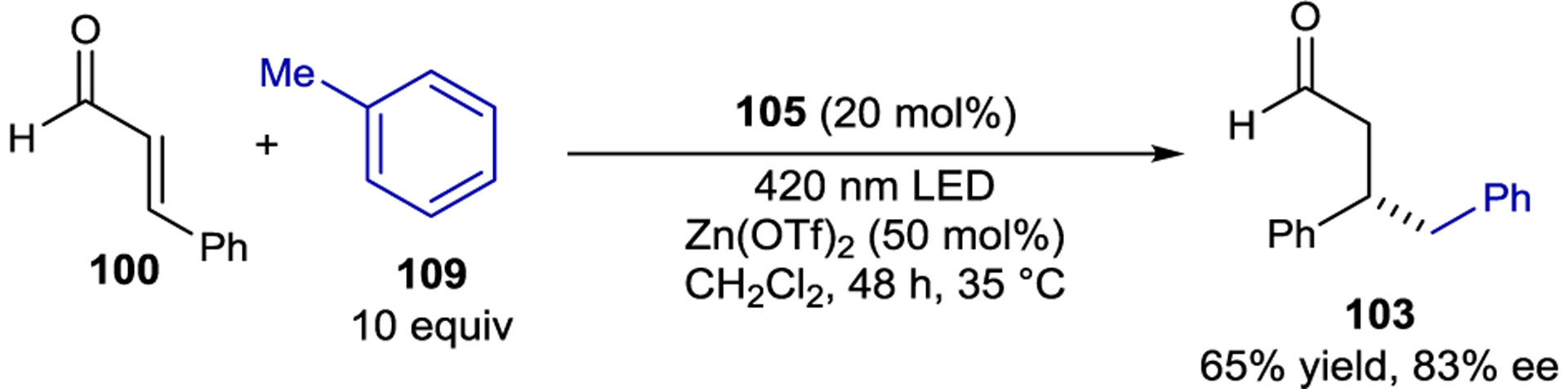
C–H Functionalization of Toluene Derivatives via Iminium Ion Catalysis
Melchiorre also demonstrated that chiral iminium ions can act as electron acceptors in photocatalytically active EDA complexes (Scheme 47).172 Aliphatic eniminium ions typically absorb in the UV regime (< 400 nm). The iminium produced from condensation of carbazole-functionalized amine 112 and cyclohexanones, however, absorbs strongly in the visible region. This absorption band was assigned as an intramolecular charge transfer between the electron-rich carbazole and electron-poor iminium. Excitation of the intramolecular EDA complex furnishes a carbazole radical cation which oxidizes an α-silyl amine (111). The resulting radical is stereoselectively intercepted by a ground state iminium ion. The overall reaction, which proceeds through a chain mechanism, results in the β-alkylation of enones.
Scheme 47.
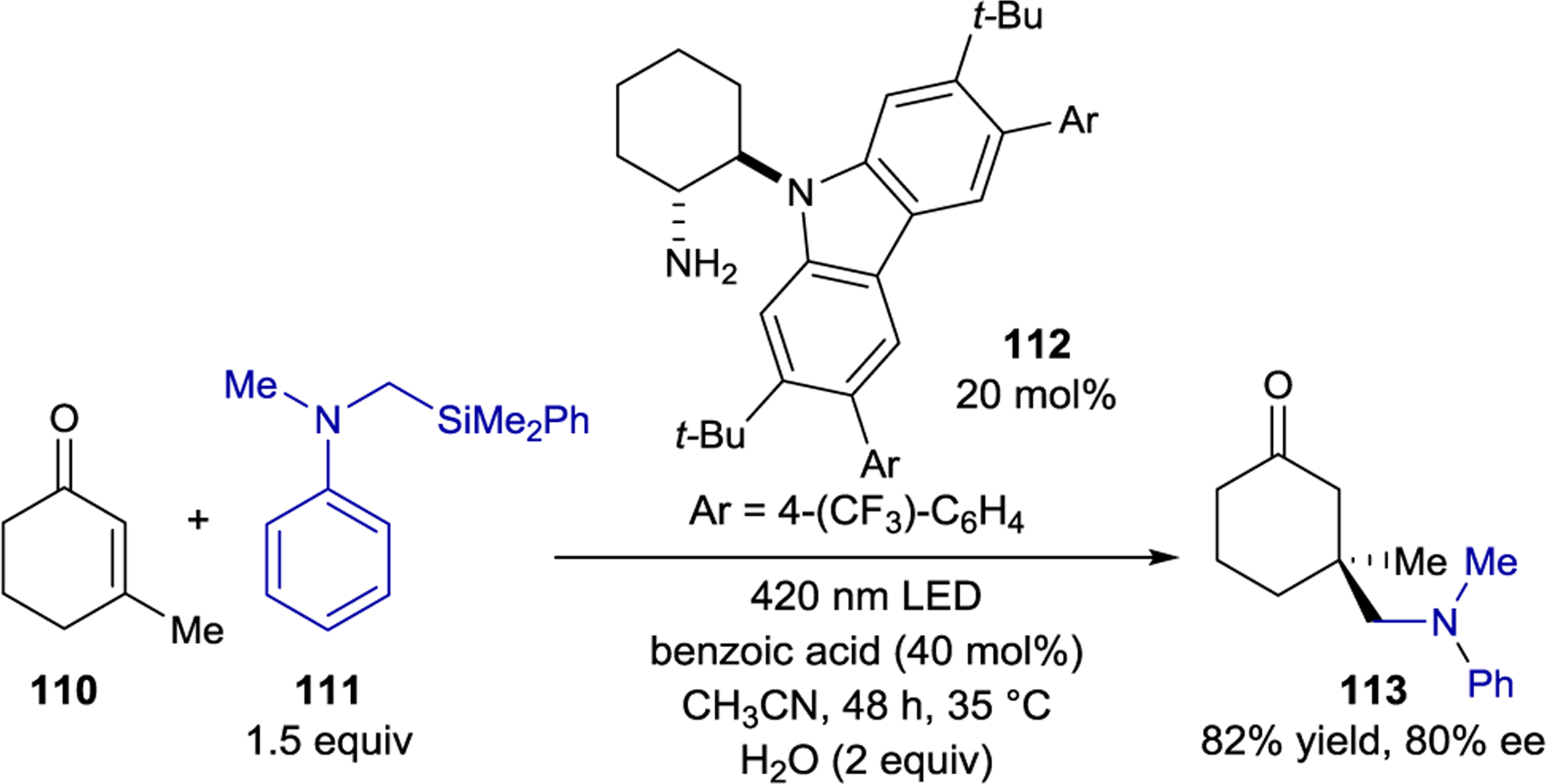
EDA Complex Formation with Iminium Electron Acceptors
Iminium ion catalysis has also been applied to [2+2] photocycloadditions. The absorption profile of enones features an (n,π*) transition that is red-shifted in comparison to the (π,π*) transition characteristic of analogous iminium species.173 Hence, it can be difficult to minimize the participation of racemic background cycloadditions through direct excitation because the iminium ion cannot be selectively excited. However, the lowest triplet excited state is typically lower in energy for iminium ions than for enones, which could allow for their selective photosensitization. Bach developed an enantioselective [2+2] photoreaction using chiral iminium ions and a Ru triplet sensitizer, but catalysis was inefficient, likely due to competing unproductive electron-transfer quenching of the photocatalyst.174 In 2020 Alemán developed a catalytic enantioselective [2+2] cycloaddition that circumvented the racemic reactivity problem by employing amine catalysts with stereogenic naphthyl substituents (Scheme 48).175 These served as electron donors in an intramolecular EDA complex with the electron-accepting imine. The intramolecular charge-transfer band was sufficiently red-shifted compared to the free enone to allow for its selective excitation. With 20 mol% catalyst 115, cycloadduct 116 was produced in 99% yield and 80% ee. Computational analysis confirmed that the lowest energy transition was the expected charge transfer, while the reactive (π,π*) imine excited state lies slightly higher in energy. The authors proposed that thermal population of the (π,π*) state from the charge-transfer excited state produces an equilibrium population able to undergo the [2+2] cycloaddition. This method was also applied to an intramolecular [2+2] cycloaddition, enabling the synthesis of tricyclic products in high enantioselectivity.176
Scheme 48.
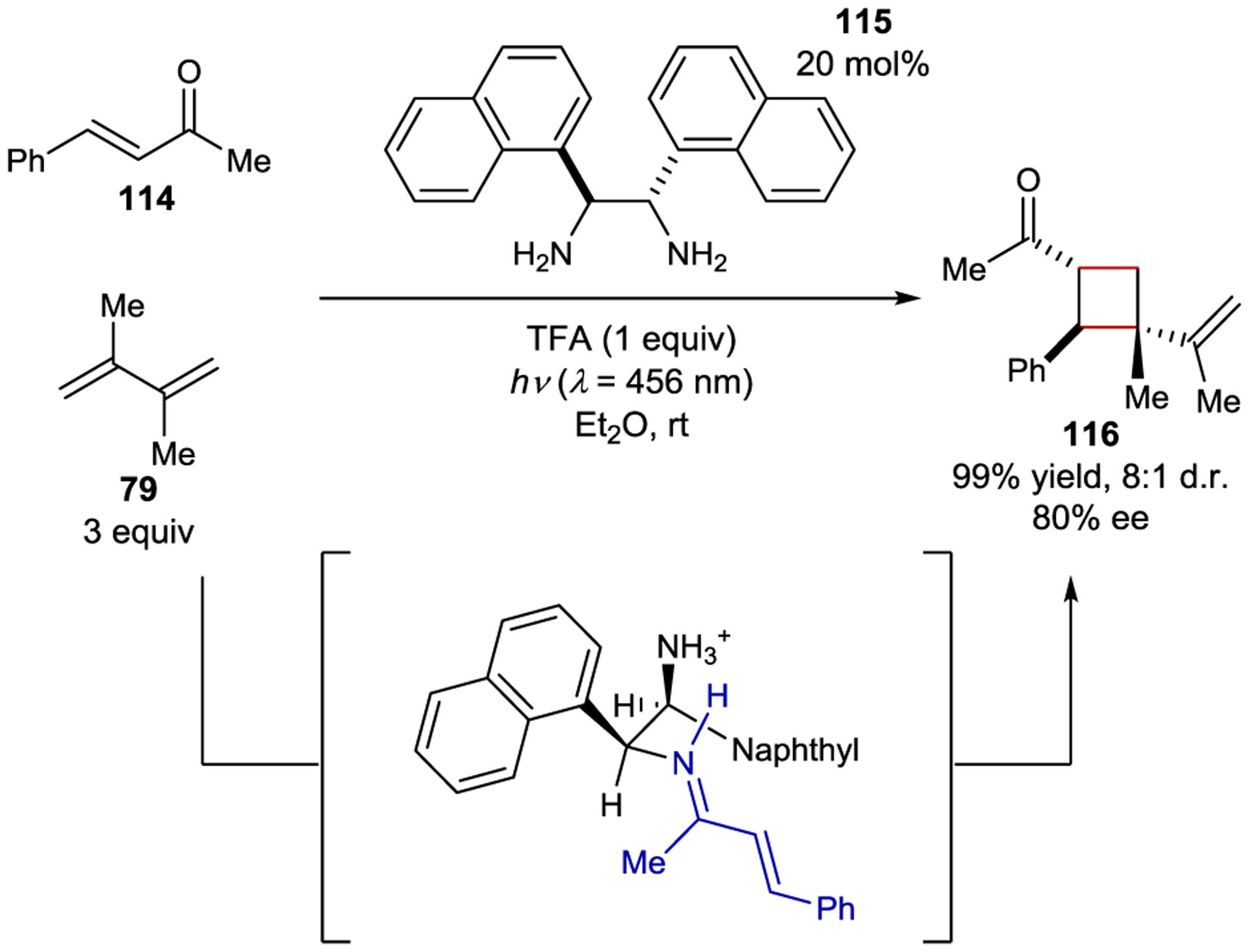
Intermolecular [2+2] Cycloaddition via a Chiral Iminium Chromophore
2.2.4. Chiral Counterions
Recently, several groups have attempted to apply chiral anion catalysis to asymmetric photochemistry.177, 178 Chiral anions are intriguing for applications in photoredox chemistry because they can associate with any cationic intermediate through electrostatic interactions, circumventing the need for specific substrate binding moieties such as carbonyls. Further, many of the most common organic and inorganic photoredox catalysts are cationic, offering a conceptually straightforward means to incorporate a chiral counteranion structure into an asymmetric photoredox reaction (Figure 3).
Figure 3.
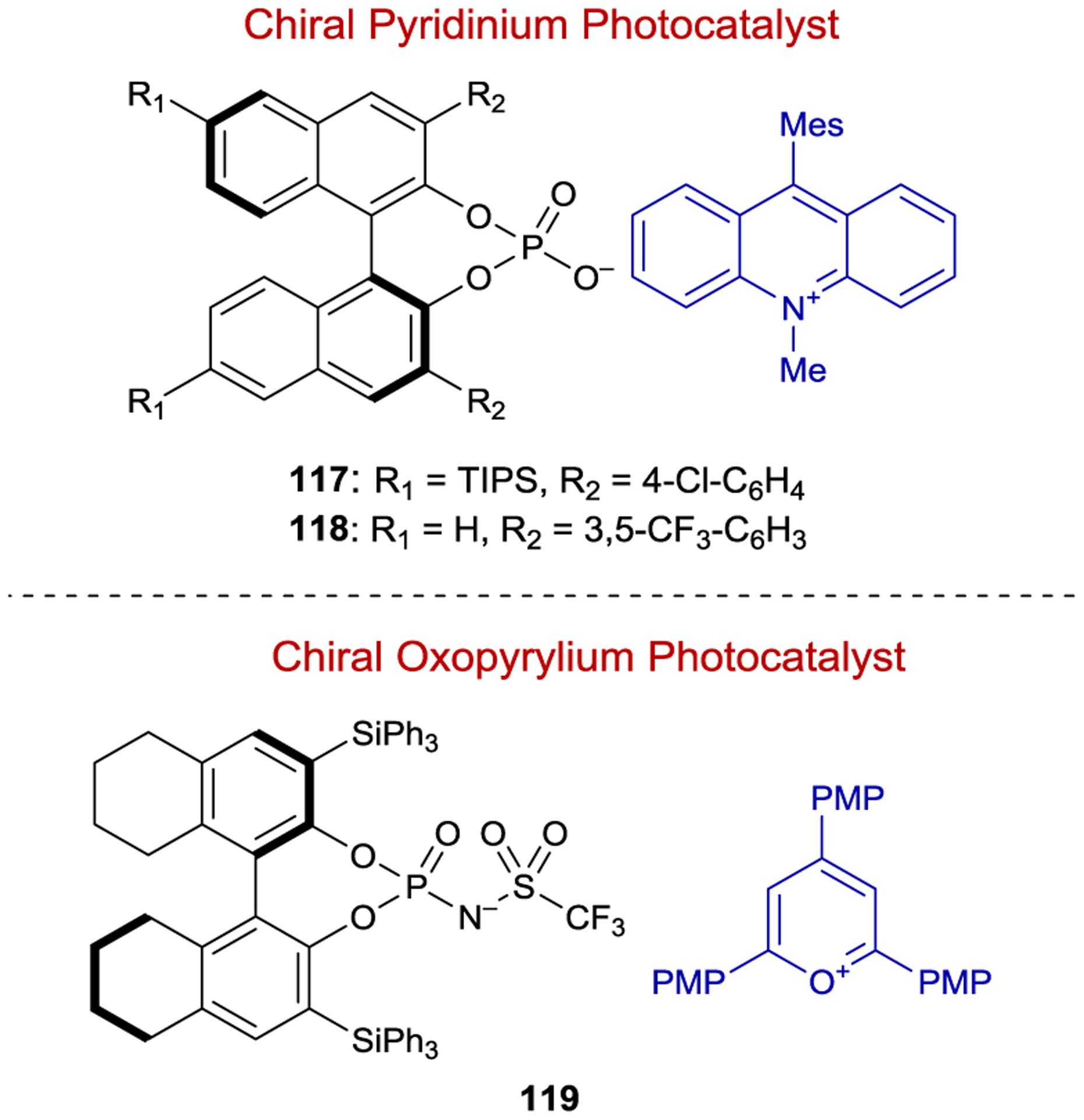
Photocatalysts with Chiral Counteranions
Luo demonstrated the feasibility of this approach by developing an asymmetric version of the anti-Markovnikov hydroetherification originally reported by Nicewicz.179, 180 Under the optimized conditions an acridinium photocatalyst with a BINOL-derived phosphate counteranion (117) afforded cyclic ether 121 in 60% ee (Scheme 49). The reaction proceeds through initial oxidation of alkene 120 to the radical cation, which pairs with the chiral anion, followed by enantiodetermining nucleophilic attack of the pendant alcohol. Hydrogen atom transfer from 2-phenylmalononitrile to the resulting carbon-centered radical yields the cyclic ether product.
Scheme 49.
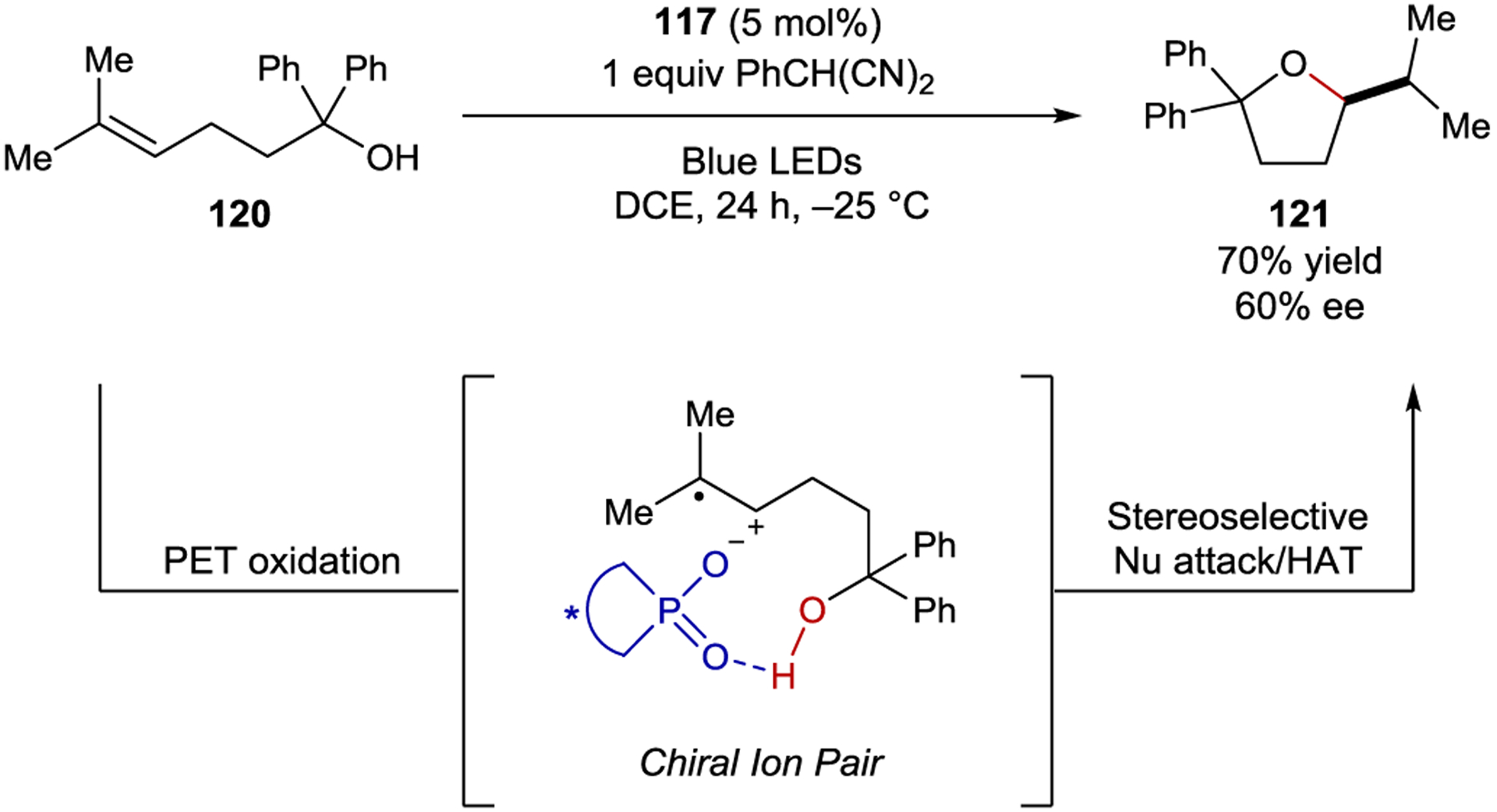
Hydroetherification via Chiral Ion-Pairing Catalysis
Other stereoselective radical cation photoreactions have utilized chiral phosphonate counteranions. Tang examined chiral acridinium catalyst 118 in a [3+2] cycloaddition between 2H-azirine 122 and azodicarboxylate 123; however, the 1,2,4-triazoline product (124) was only formed in 20% ee (Scheme 50).181
Scheme 50.
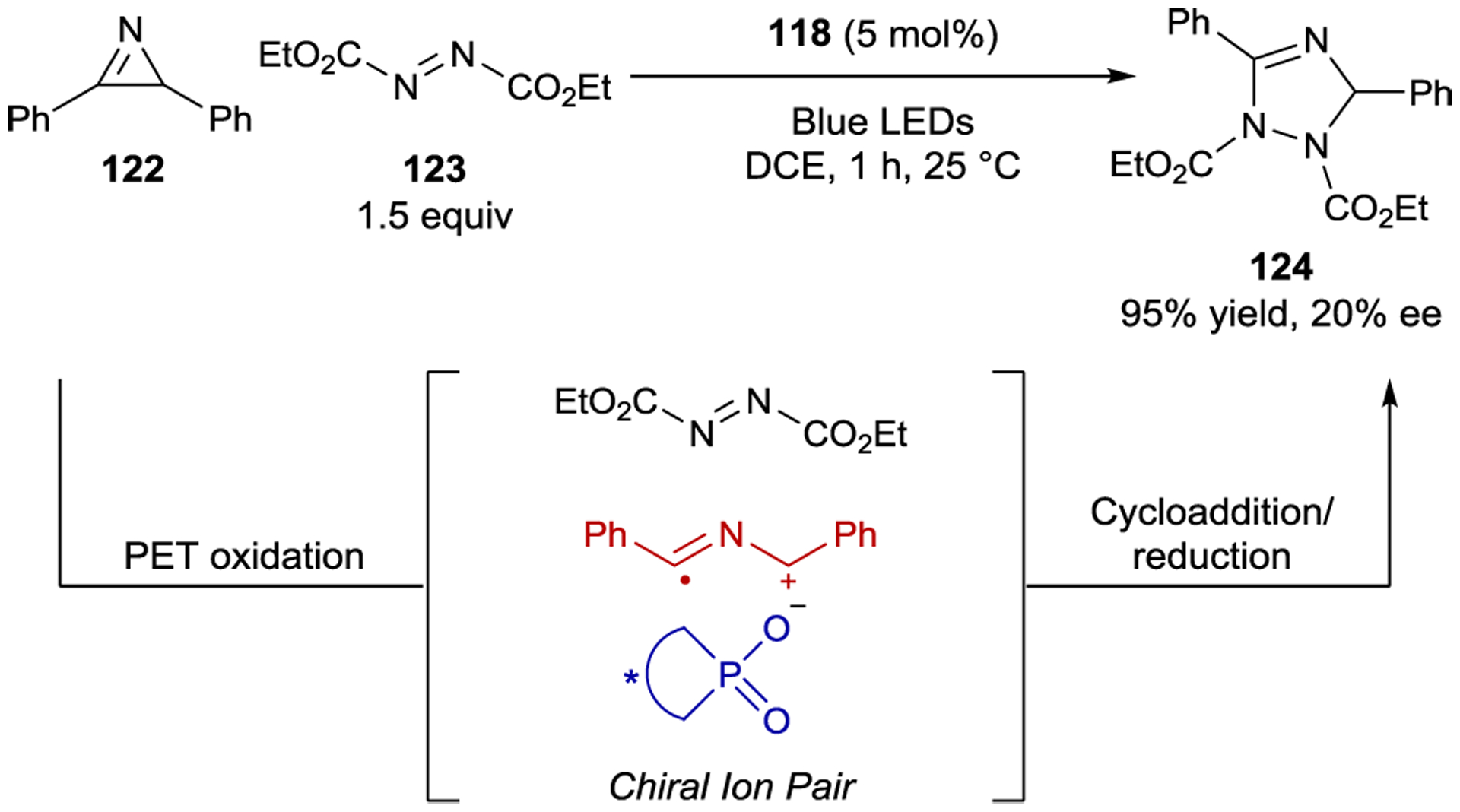
[3+2] Cycloaddition via Chiral Ion-Pairing Catalysis
Nicewicz examined a range of BINOL-derived chiral anions with an oxopyrylium photocatalyst in a radical-cation Diels–Alder reaction, with a maximum selectivity of 50% ee with catalyst 119 (Scheme 51).182 When screening solvents, the authors observed an inverse relationship between enantioselectivity and solvent dielectric constant, consistent with the hypothesis that an intimately interacting ion pair is critical for asymmetric induction. The generally modest enantioselectivities reported to date using this strategy reflect its early stage of development.
Scheme 51.
Diels–Alder Cycloaddition Catalyzed by an Oxopyrylium Sensitizer
Wang developed an asymmetric dicarbofunctionalization of enamides that proceeded in both the presence and absence of a ruthenium photosensitizer (Scheme 52).183 In the absence of a photosensitizer, indole 127, enamide 128, and redox-active ester 129 were coupled in the presence of a chiral phosphate base to form 131 in 47% yield and 89% ee. A Job plot analysis indicated a ground-state preorganization of the phosphate base, enamide 128 and ester 129 into an EDA complex. Photoexcitation of this EDA complex led to reduction of the redox-active ester followed by rapid decarboxylation. The formed benzyl radical and enamide-derived radical combine to form an iminium intermediate. In the presence of the phosphate catalyst, indole 127 then attacks the electrophilic iminium cation via a Friedel–Crafts reaction to provide enantioenriched indole 131.
Scheme 52.
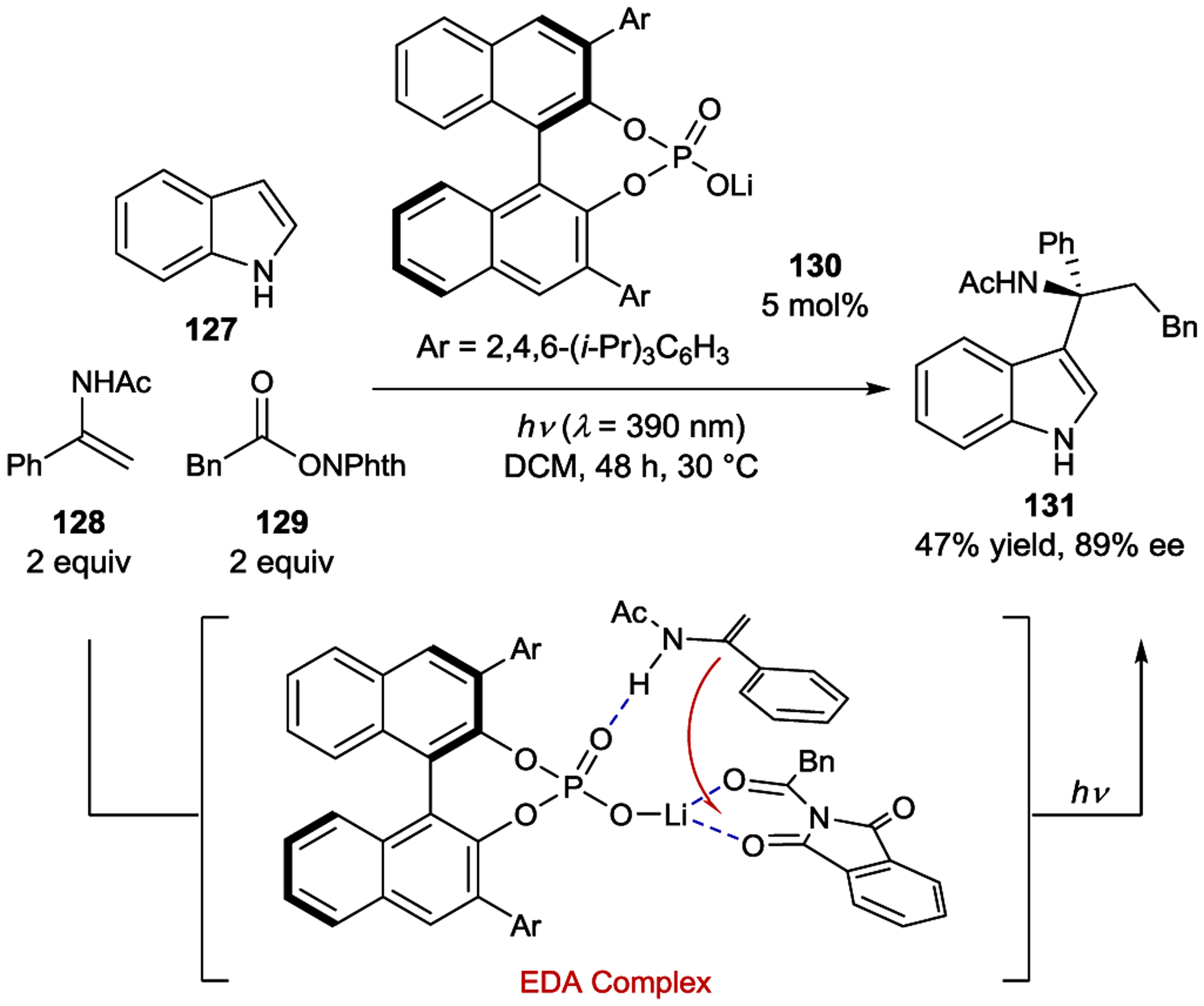
Dicarbofunctionalization of Enamides via Ion-Pairing Catalysis
Terada reported the radical addition of toluene-derived radicals to benzopyrylium intermediates to generate chromene derivatives (Scheme 53).184 Protonation of chromenol 132 by phosphoric acid 133 produces a photoactive benzopyrylium intermediate that can photooxidize toluene. Upon deprotonation, the resulting toluyl radical undergoes addition to another equivalent of benzopyrylium. Reduction of the radical adduct by the reduced benzopyrylium affords the product and regenerates the photooxidant. Because the radical addition occurs in the presence of the conjugate base of the acid, the authors tested chiral phosphoric acid 133 in an enantioselective reaction, obtaining 134 in 60% ee.
Scheme 53.
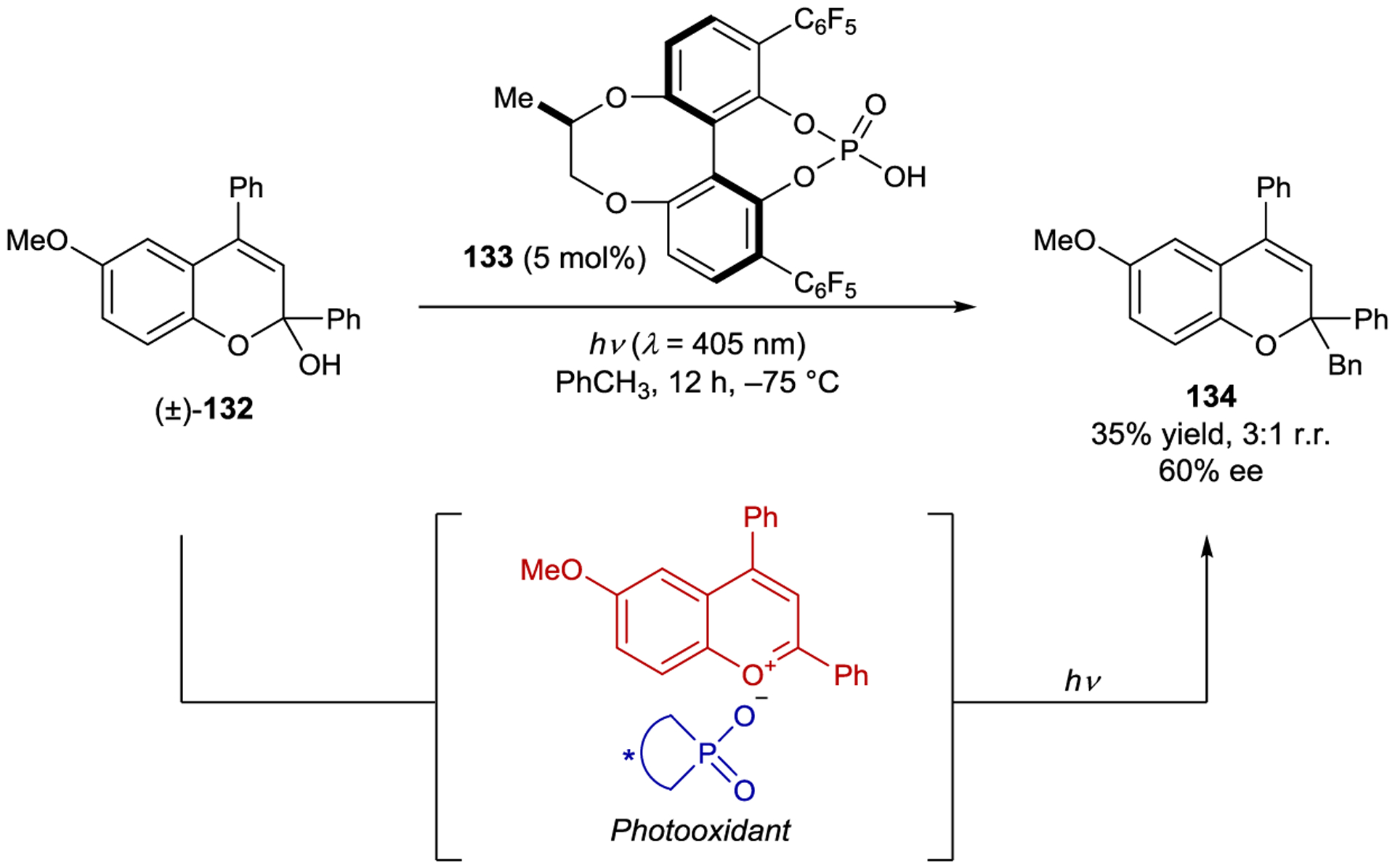
Toluene Functionalization via Excitation of Benzopyrylium Intermediates
Finally, Melchiorre developed an asymmetric perfluoroalkylation of β-ketoesters exploiting an EDA complex activation strategy (Scheme 54).185 In the presence of base and cinchona-derived phase transfer catalyst 137, β-ketoester 136 is converted to the corresponding enolate with a chiral countercation. The electron-rich enolate forms a ground-state association with an electron-poor perfluoroalkyl iodide (135) constituting an EDA complex. After excitation of the EDA complex and cleavage of the alkyl iodide, the pefluoroalkyl radical is proposed to be trapped by another equivalent of the enolate in the enantiodetermining step. The resulting ketyl radical abstracts an iodide from a perfluoroalkyl iodide, regenerating the alkyl radical and propagating a radical chain. Finally, the iodide adduct collapses to form the product. Subsequent computational studies suggested that multiple hydrogen-bonding interaction between the enolate and catalyst account for the high enantioselectivity.186 This work is notably distinct from previous photo-organocatalyzed reports. In prior work the chiral catalyst was covalently bonded to the substrate in the enamine intermediate, while in this study the catalyst is ion paired to the enolate electron donor.
Scheme 54.

Perfluoroalkylation of Enolates via Ion-Pairing Catalysis
In 2018, Meng showed that a cinchona-derived catalyst (141) tethered to a photocatalyst promotes β-ketoester oxidation via sensitization of oxygen in up to 86% ee (Scheme 55).187 In 2019 the same group discovered that the reaction also proceeds with a similar catalyst lacking the photosensitizing unit (142).188 As in the Melchiorre precedent above, an EDA complex was proposed between the deprotonated substrate enolate and the catalyst. The authors proposed that the excited EDA complex sensitizes oxygen, which then reacts with the enolate, eventually affording the hydroxylated product. The reaction was also performed in a flow photomicroreactor where the product was obtained in similar yield and enantioselectivity in under 1 h.
Scheme 55.
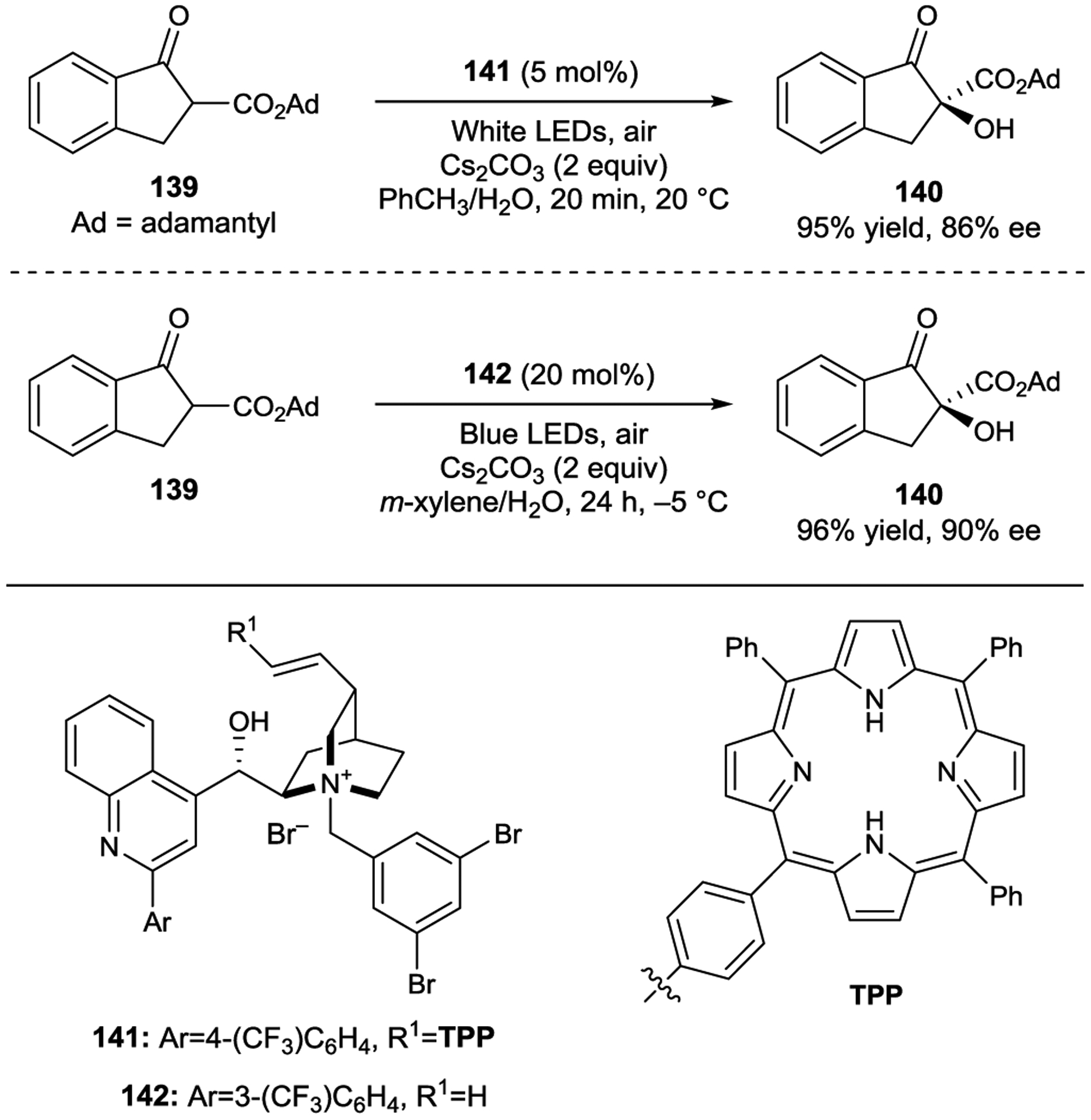
Aerobic Oxidation of β-Ketoesters via Ion-Pairing Catalysis
3. Chiral Inorganic Chromophores
3.1. Boron
Lewis acids have been studied extensively for their ability to influence the outcomes of a wide range of ground-state transformations. Their effect on photochemical reactions has received considerably less attention. In 1910, Praetorius and Korn described a uranyl chloride accelerated [2+2] photodimerization of α,β-unsaturated ketones.189 Alcock later showed that SnCl4 exhibits a similar effect, suggesting that many Lewis acids might have a general effect in photochemical transformations.190 In more recent work, Lewis and Barancyk performed a detailed study of the Lewis acid promoted [2+2] photocycloaddition of coumarin with 2,3-dimethyl-2-butene.191, 192 While little product was formed in the absence of Lewis acid, addition of 50 mol% of various Lewis acid catalysts led to a large enhancement of product formation. The most active catalyst tested was AlBr3, which yielded complete conversion to the cycloadduct. Mechanistically, Lewis proposed that the binding of a Lewis acid alters the electronic structure of the enone, which in the case of coumarin manifests as a bathochromic shift in the complex’s absorption profile (Scheme 56).
Scheme 56.
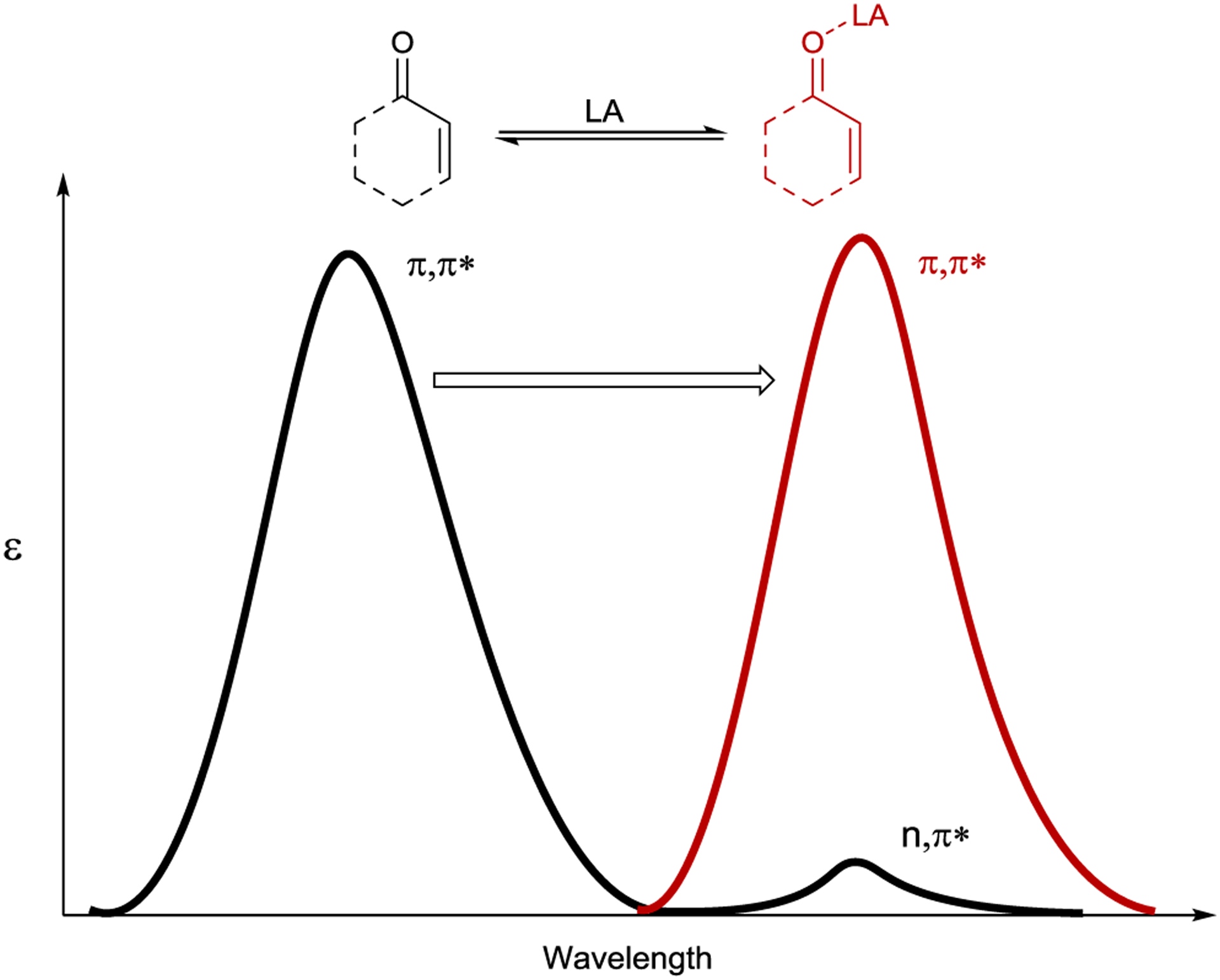
Bathochromic Shift of α,β-Unsaturated Carbonyls upon Lewis Acid Coordination
Bach has described this phenomenon as “chromophore activation”193, 194 and proposed that it could be exploited in the design of Lewis acid catalyzed asymmetric photoreactions. Irradiation at wavelengths that maximize the selective photoexcitation of a chiral catalyst–substrate complex over the free α,β-unsaturated carbonyl in solution ensures that reaction occurs within the chiral environment of the Lewis acid, while minimizing direct racemic photoreaction. The initial report demonstrating the feasibility of this concept examined the intramolecular photocycloaddition of coumarin 38 (Scheme 57).195 The reaction proceeds sluggishly upon irradiation at 366 nm due to the low extinction coefficient of the coumarin. In line with the proposed chromophore activation mechanism, addition of simple boron or aluminum Lewis acids red-shift the (π,π*) absorption, greatly accelerating product formation upon irradiation, highlighting that Lewis acids can indeed increase photoreaction rates. Moreover, the use of chiral oxazaborolidine Lewis acid 143 provides the cycloadduct in 82% yield and a remarkable 62% ee, which could be increased to 82% ee at −75 °C.196 At lower concentrations (20 mol%) of oxazaborolidine catalyst 143, however, the ee was diminished due to direct excitation of the weak (n,π*) absorption of free coumarin.
Scheme 57.
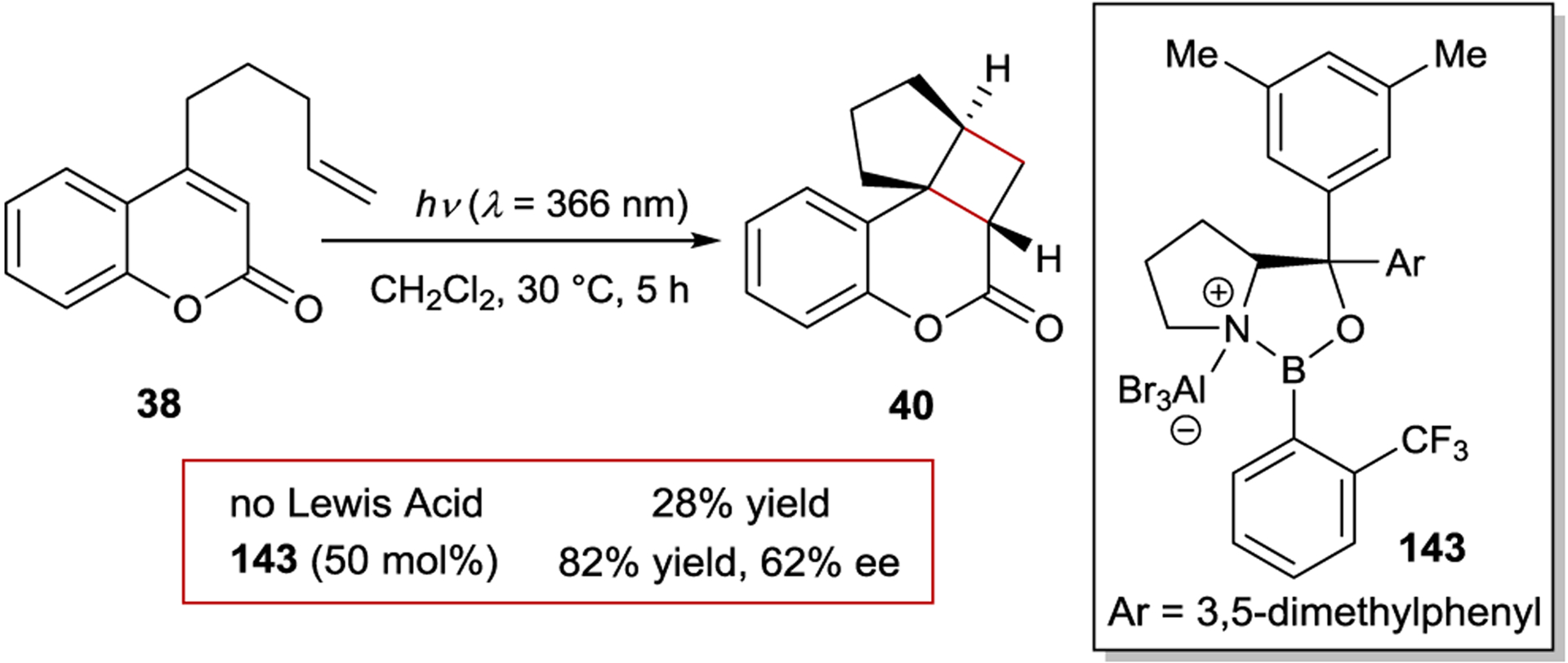
Enantioselective [2+2] Photocycloadditions of Coumarins with a Chiral Oxazaborolidine Catalyst
Mechanistic studies provided further insights into the origins of the catalytic effect.197 When the coumarin substrate is irradiated in the absence of a Lewis acid, the cycloaddition is stereospecific, where starting from either the tethered (E) or (Z)-alkene leads to distinct diastereomers. This is consistent with reaction through a singlet excited state. Upon addition of a Lewis acid, the quantum yield (Φ) of the photocycloaddition increases by an order of magnitude. Importantly, under these conditions the cycloaddition becomes stereoconvergent, indicating that the reaction proceeds through a triplet excited state. The Lewis acid promoted increase in efficiency was attributed to the presence of a heavy atom that facilitates intersystem crossing to the coumarin triplet excited state, which proceeds to the cycloadduct more efficiently than the shorter-lived singlet coumarin.
Exploring the generality of this strategy, Bach and coworkers investigated the enantioselective [2+2] photocycloaddition of the synthetically useful class of 5,6-dihydro-4-pyridones (Scheme 58).198 In contrast to their previous work, enone 144 undergoes a more pronounced bathochromic shift when coordinated to a Lewis acid. The authors reasoned that this red shift in combination with a longer wavelength light source would enable selective excitation of the bound species, thus eliminating racemic background reactivity. This proved correct and yielded highly selective cycloadditions (up to 90% ee). In addition, one of the products (146) was utilized as a key intermediate in the total synthesis of (+)-lupinine and the formal synthesis of (+)-thermopsine.
Scheme 58.
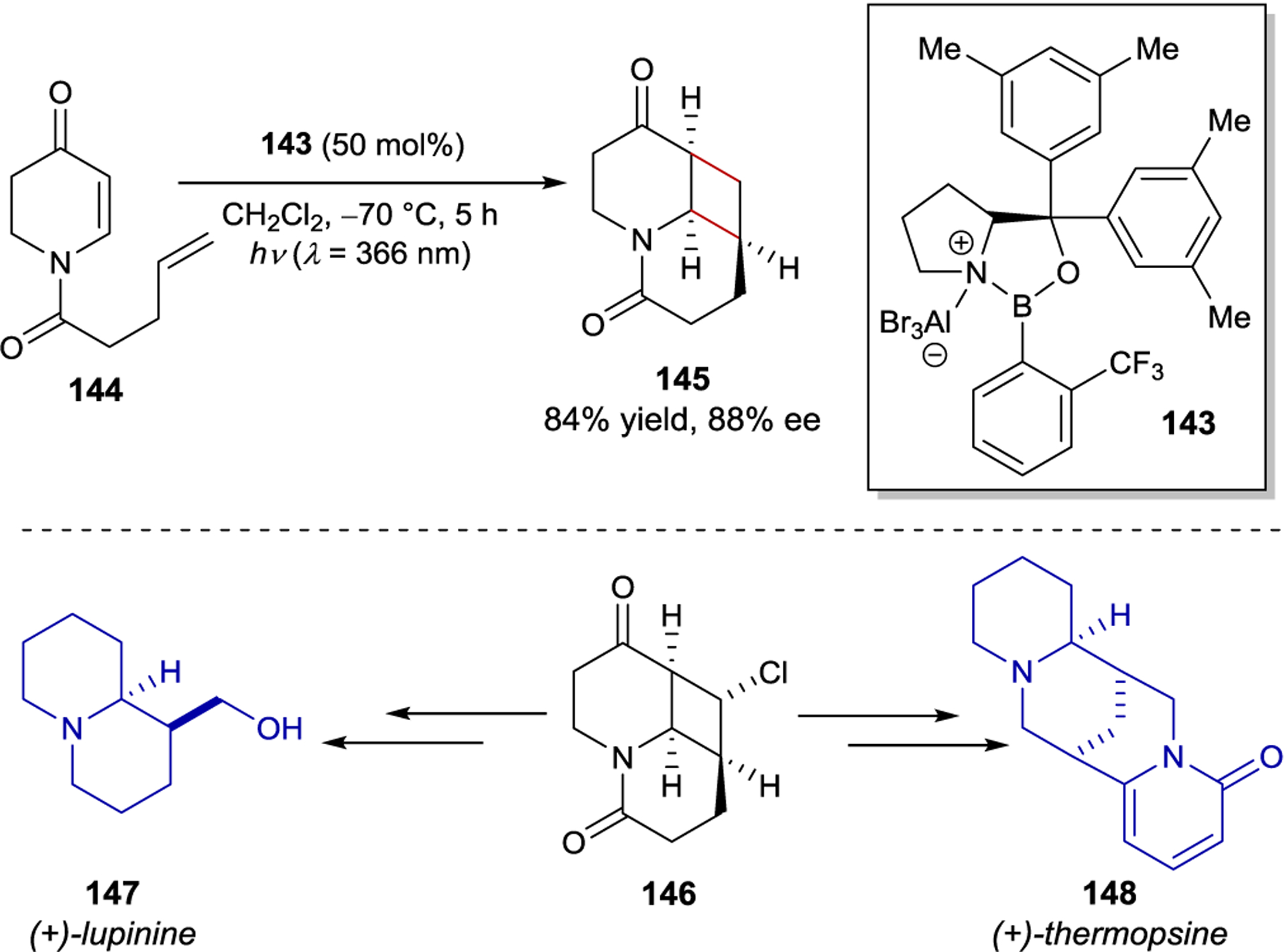
Enantioselective [2+2] Photocycloadditions of 5,6-Dihydro-4-pyridones
Despite the superficial similarities of coumarin 38 and dihydropyridone 144, there are several intriguing differences in the mechanisms of their Lewis acid catalyzed [2+2] photocycloadditions. First, the dihydropyridone substrates undergo stereoconvergent cyclization with and without Lewis acid, consistent with the formation of a triplet excited state in both cases. In contrast to the coumarin substrates, the quantum yield of the dihydropyridone cycloaddition decreases from 0.23 to 4 × 10−3 in the presence of a Lewis acid. It is therefore surprising that high enantioselectivity is achieved given that after excitation, free pyridone cyclizes significantly more efficiently than catalyst-bound pyridone. However, this highlights the importance of the change in the absorption spectrum of the Lewis acid complex. Because almost all photons are absorbed by the Lewis acid-bound dihydropyridone upon irradiation at 366 nm, the relatively inefficient stereoselective cyclization still outcompetes the racemic direct excitation pathway. Bach’s mechanistic proposals for both the coumarin and dihydropyridone cycloadditions were consistent with computational studies conducted by Dolg199 and Chen.200
The ability to afford highly enantioenriched cyclobutane products from a chemo- and regioselective [2+2] photocycloaddition of simple cyclic enones and alkenes is a highly desirable goal. To this end, Bach and coworkers studied the enantioselective [2+2] photocycloaddition of 3-alkenyloxy-2-cycloalkenones (149) (Scheme 59).201 For this [2+2] photocycloaddition, modified AlBr3-activated oxazaborolidine 150 proved to be ideal, delivering the simple aliphatic cyclobutane products in nearly quantitative yield and up to 94% ee. The utility of these products was showcased through acid-catalyzed allylation and ring expansion reactions that afforded 154 and 155 without erosion of enantioselectivity.
Scheme 59.
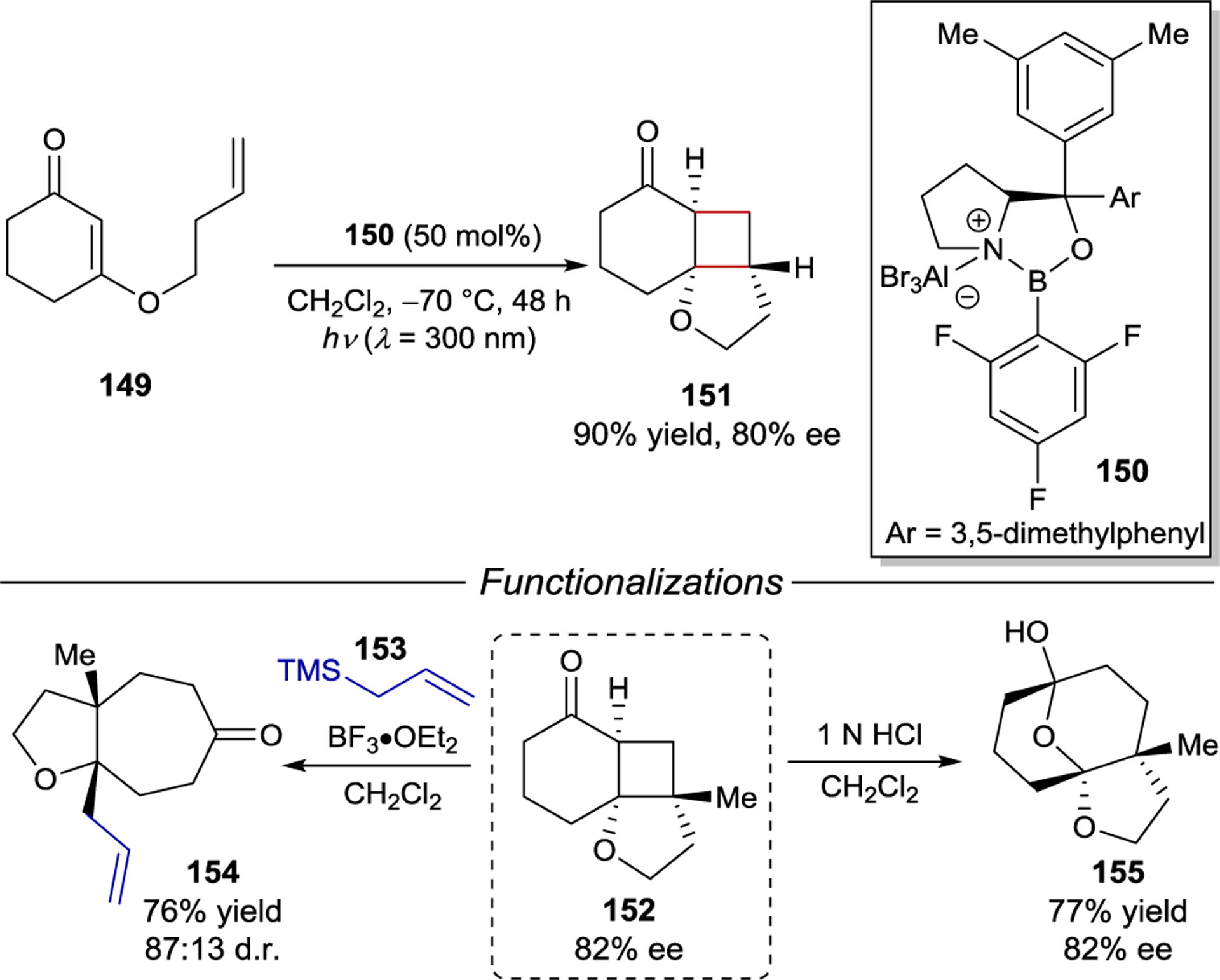
Application of Chromophore Activation to the [2+2] cycloaddition of 3-Alkenyloxy-2-cycloalkenones
The intermolecular photocycloaddition of cyclohexanones with alkenes has played an important role in the history of total synthesis.202 Bach and coworkers expanded the chromophore activation strategy to intermolecular cycloadditions of simple cyclohexenones with a range of terminal alkenes (Scheme 60).203 The synthetic utility of the enantioenriched cyclobutane products was showcased in a concise 6-step synthesis of the enantiopure monoterpene (−)-grandisol. Interestingly, the uncatalyzed reaction affords a mixture of regioisomers, while the use of chiral Lewis acid 156 results in the exclusive formation of the head-to-tail regioisomer. This observation was rationalized as the result of increased polarization of the excited-state enone–Lewis acid complex, which improves the selectivity for initial bond formation.
Scheme 60.
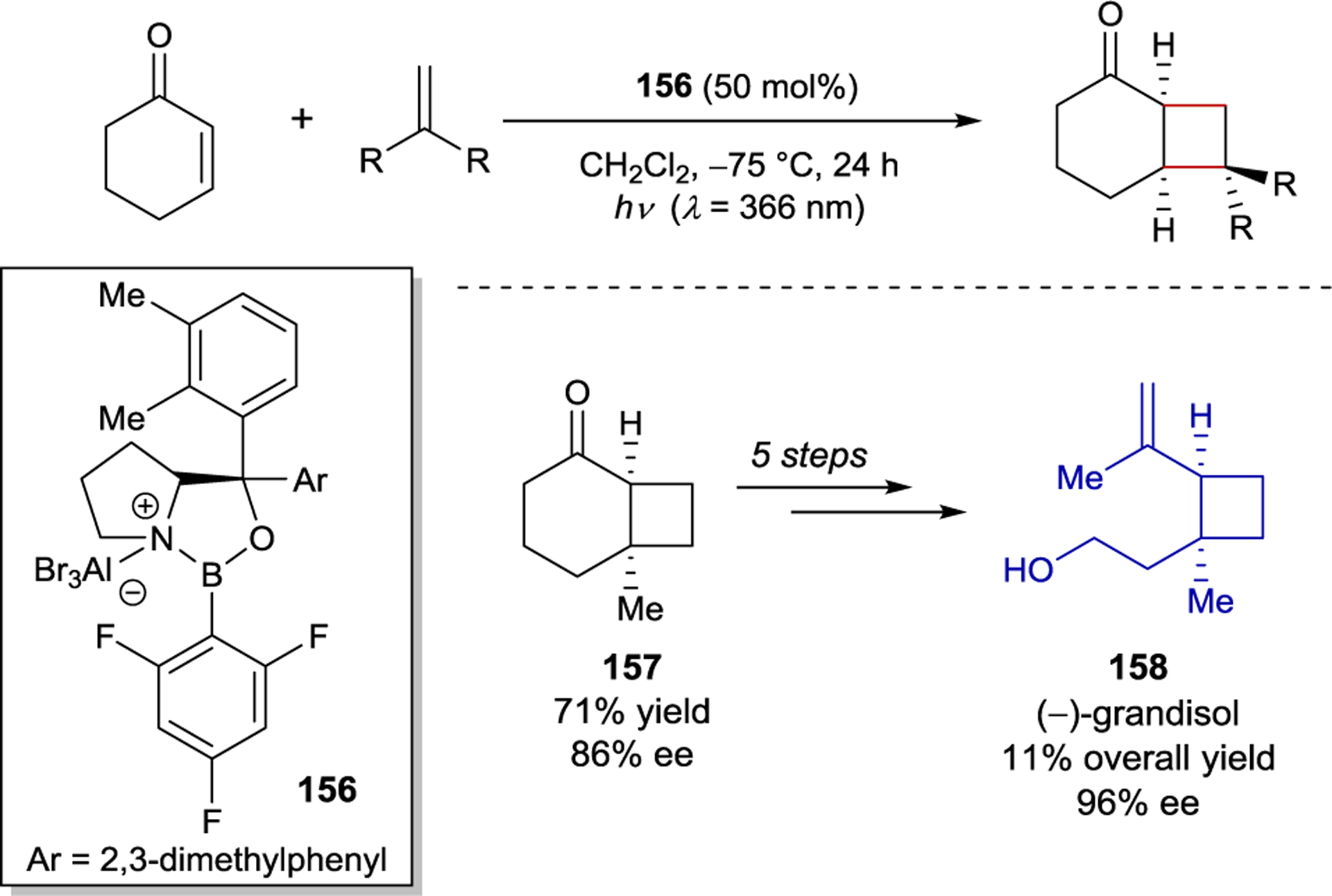
Intermolecular Enantioselective [2+2] Cycloaddition Between Cyclohexenones and Terminal Alkenes
In each of the previous examples, coordination of a Lewis acid to an α,β-unsaturated carbonyl compound results in a bathochromic shift of the (π,π*) absorption band that typically overlaps with the (n,π*) absorption band of the free substrate. Consequently, relatively high catalyst loadings (50 mol%) are required to outcompete racemic uncatalyzed photoreaction of the free substrate. If however the (π,π*) absorption could be further shifted beyond the (n,π*) absorption, one might expect lower catalyst loadings to be effective. Bach noted that the addition of EtAlCl2 to phenanthrene-9-carboxaldehyde 159 results in such a shift. When oxazaborolidine 161 was used to catalyze its photocycloaddition with tetramethylethylene (160), the corresponding ortho-photocycloadduct 162 was produced in high enantioselectivity (94% ee) using only 10 mol% of the catalyst (Scheme 61).204
Scheme 61.

Enantioselective ortho-Photocycloaddition with Low Catalyst Loadings of Chiral Oxazaborolidine Lewis Acids
Many of the enone cycloadditions described above require a relatively slow, symmetry-forbidden S1(π,π*)→T1(π,π*) intersystem crossing (ISC) event.205 The corresponding uncatalyzed photoreactions, on the other hand, involve a faster symmetry-allowed S1(n,π*)→T1(π,π*) ISC. In this scenario, the more efficient ISC of the free excited-state substrate further challenges the ability to outcompete the racemic background reaction. Thus, Bach and coworkers sought to optimize a photochemical enone reaction that occurs from the singlet excited state, removing the complication of slow ISC and in turn allowing for lower Lewis acid catalyst loading. Inspired by work of Griffiths206 and more recent precedent by Ramamurthy,207 they investigated the oxa-di-π-methane rearrangement of 2,4-cyclohexadienone 163 to bicyclic ketone 164 (Scheme 62).208 Indeed, this reaction was shown to proceed with high enantioselectivity (92–97% ee) at low chiral catalyst loadings (10 mol%). Computational experiments suggest that the population of the triplet hypersurface is minimized due to a conical intersection on the singlet surface.
Scheme 62.
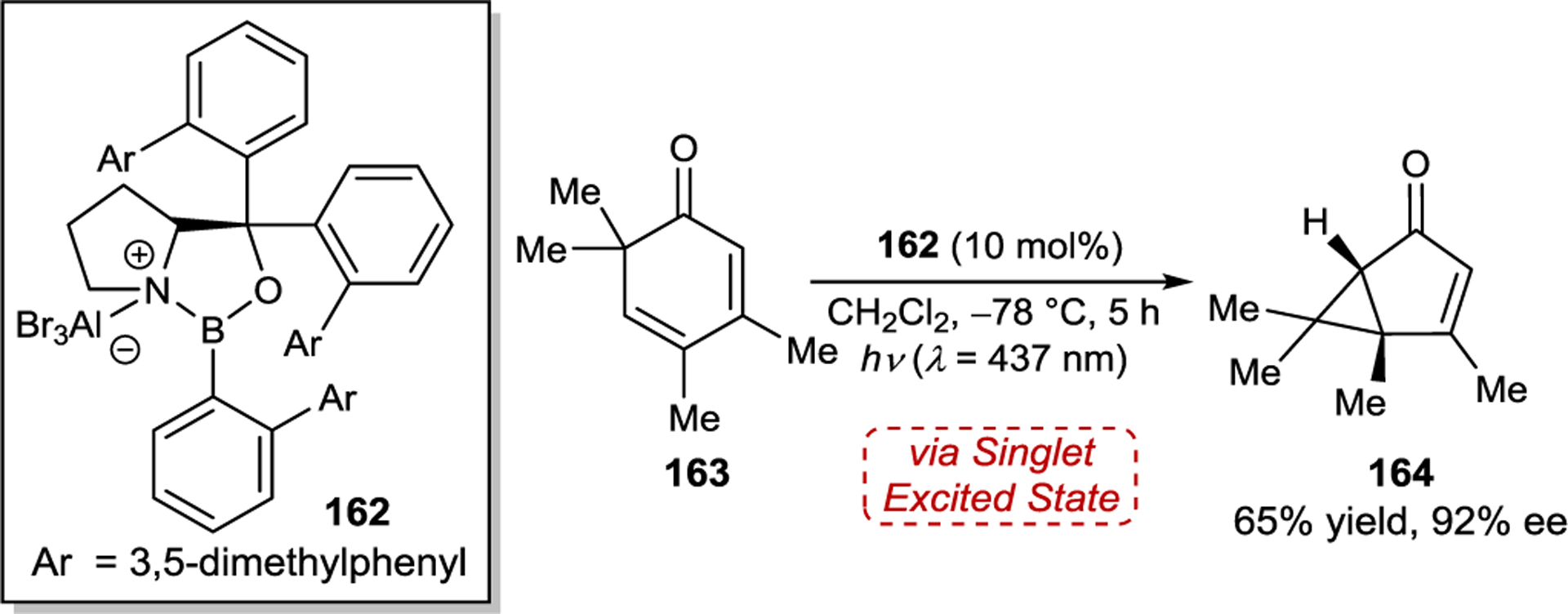
Enantioselective Oxa-di-π-methane Rearrangement via a Singlet Excited State
Coeffard recently described a chiral photocatalyst for stereoselective α-oxygenation reactions that includes boron in the photosensitizing unit.209 The catalyst (165) consists of a BODIPY photosensitizer covalently tethered to a chiral cinchona alkaloid. The quinuclidine moiety of the catalyst is proposed to serve two roles. First, it activates 1,3-dicarbonyl substrate 166 through deprotonation and provides a chiral environment for the resulting enolate through hydrogen-bonding. Additionally, the lone pair of the unprotonated quinuclidine is a competent scavenger for 1O2 when the substrate is not bound. As a result, the local concentration of photogenerated 1O2 is enriched when the substrate is bound and depleted when it is not, further minimizing the possibility of racemic background oxygenation. In practice, this system catalyzed the formation of α-hydroxylation product 167 from the corresponding β-dicarbonyl substrate in 40% ee. Coeffard uses the term “on/off” photooxygenation to reflect the dual roles of the quinuclidine nitrogen (Scheme 63).
Scheme 63.
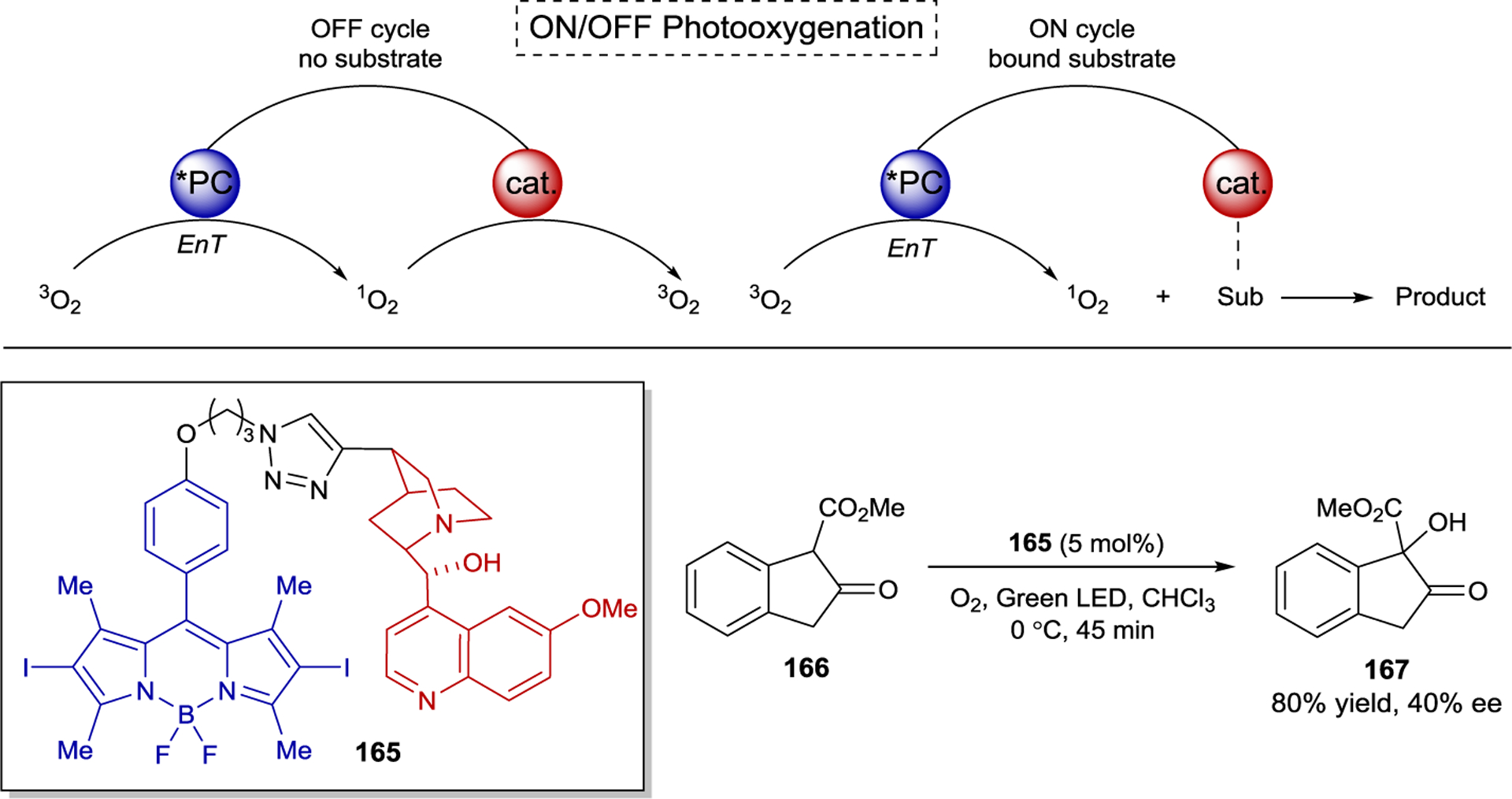
ON/OFF Photooxygenation Catalysis
3.2. Ruthenium
Many structurally diverse photocatalysts have proven to be useful in a range of important transformations. Recent developments in the field of synthetic photochemistry, however, have centered on the use of transition-metal chromophores, including the canonical Ru(bpy)32+ (bpy = 2,2’-bipyridine) photocatalyst. These complexes are attractive because they generally feature large molar extinction coefficients, high intersystem crossing quantum yields, and long-lived excited states. Moreover, the excited-state energies and redox properties of photoactive coordination complexes can often be readily tuned by modulating the structures of the supporting ligands without adversely affecting their photophysical properties. The homoleptic Ru(bpy)32+ chromophore possesses D3 symmetry and can be resolved into two enantiomeric forms, denoted as Λ or Δ (Scheme 64).210 Stereoinduction from the metal-centered stereochemistry of chiral coordination complexes has been the topic of significant recent interest.211,212 Interestingly, despite their widespread use as synthetic photocatalysts, the study of chiral enantiopure transition metal chromophores in asymmetric photocatalysis was limited until quite recently.
Scheme 64.
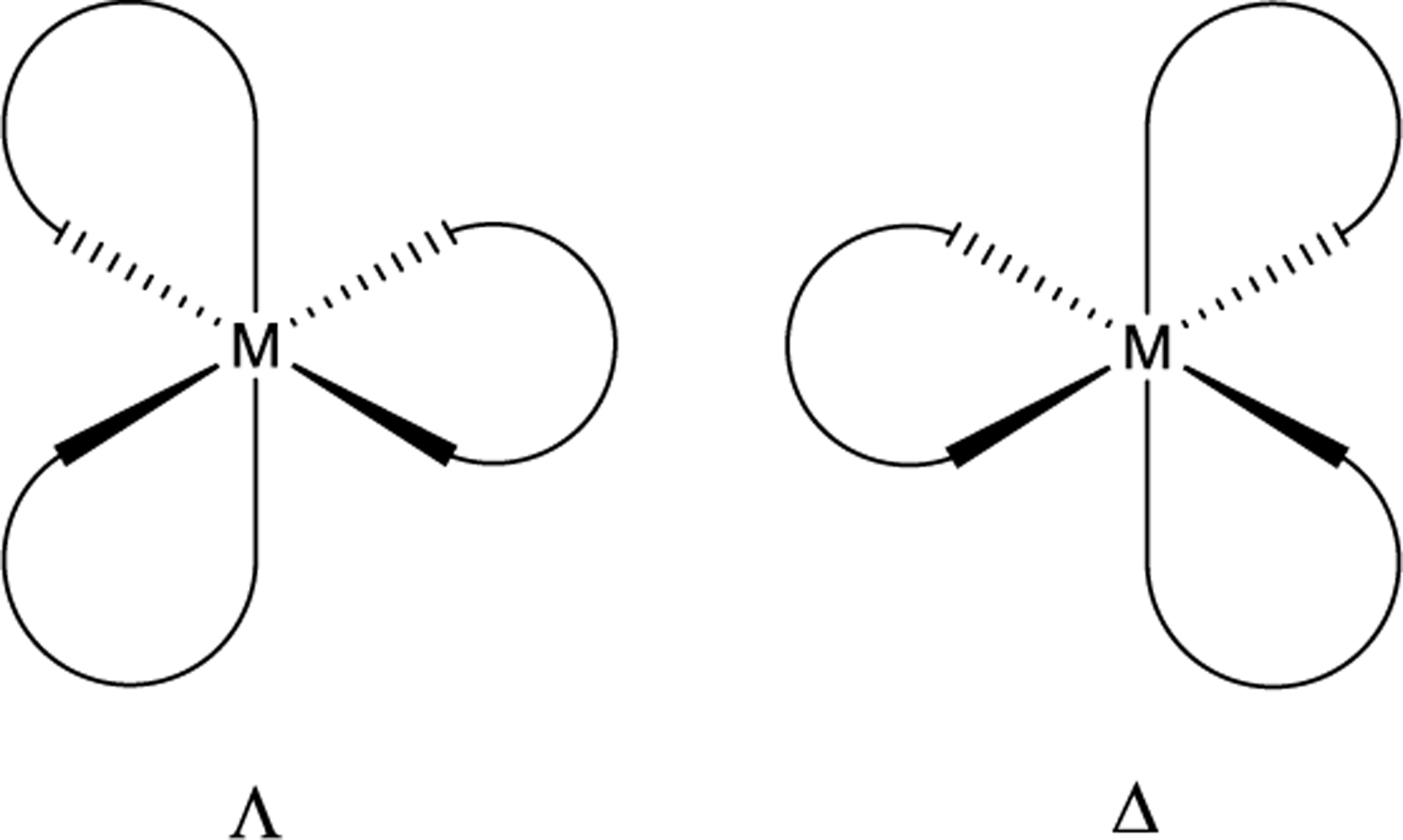
Helical Chirality of Octahedral Transition-Metal Complexes
Early studies by Geselowitz and Taube demonstrated that ground-state, outer-sphere electron transfer from racemic Co(edta)2− (edta = ethylenediaminetetraacetate) to enantiomerically enriched Os(bpy)33+ results in the formation of the oxidized Co(III) complex in 2.9% ee.213 Similar effects were observed using other inorganic complexes of cobalt, iron, and ruthenium.214 These studies provided proof-of-principle examples that the helical chirality of octahedral metal centers could be stereoinducing in electron-transfer processes. The first example of an enantioselective photoinduced electron transfer was reported in 1979 by Porter and Sparks, who found that enantioenriched Δ-Ru(bpy)32+ preferentially reduces Λ-Co(acac)3 (acac = acetylacetone) over the Δ enantiomer.215 The relative rate constants (kr) for the reduction of Λ- and Δ-Co(acac)3 were determined to be 1.08:1 by monitoring the circular dichroism (CD) of the unreacted Co(III) starting material. Kobayashi and coworkers subsequently determined a similar ratio of 1.19:1 from luminescence quenching measurements (Scheme 65).216 The analogous enantioselective photoreduction of Co(oxox)33− (oxox = oxalate) was studied by Kato, Kimura, and coworkers, with a maximum reported selectivity ratio of 1.32:1.217
Scheme 65.
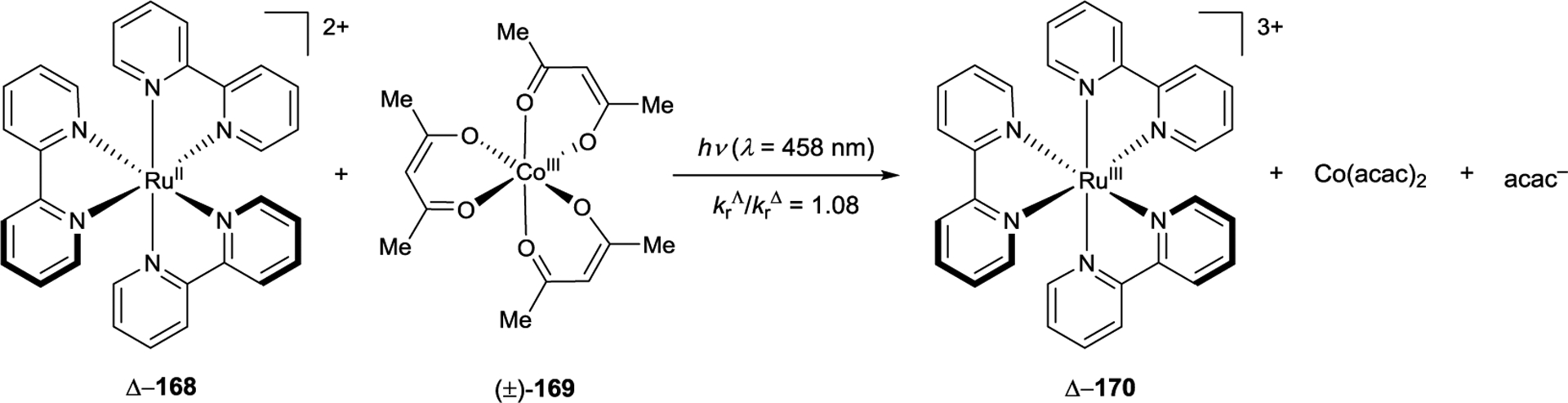
Enantioselective PET to Co(acac)3 from Δ-Ru(bpy)32+
Although the stereochemical configuration of Δ- or Λ-Ru(bpy)32+ is stable to electron transfer218 and to heating,219 photoracemization is known to occur from its excited state (Φrac = 2.88 × 10−4).220 Thus, the reported selectivity for the enantiodiscriminating electron transfer might be artificially diminished due to photoracemization of the catalyst over the course of the reaction. To minimize the influence of competitive photoracemization, Ohkubo and coworkers developed catalysts bearing stable chiral substituents on the bipyridine ligands. These complexes, Ru((−)-menbpy)32+ (171) and Ru(PhEtbpy)32+ (172 and 173), exhibit similar absorption and emission profiles, are slightly more oxidizing compared to Ru(bpy)32+, and notably have racemization quantum yields substantially lower than Ru(bpy)32+ (4.0 × 10−6 and 7.6 × 10−6, respectively).221 Ru(PhEtbpy)32+ proved relatively unselective (1.05:1) for the reduction of Co(acac)3.222 On the other hand, Ru((−)-menbpy)32+ preferentially quenches (kq) Δ-Co(acac)3 in a 1.33:1 ratio (Scheme 66).223
Scheme 66.
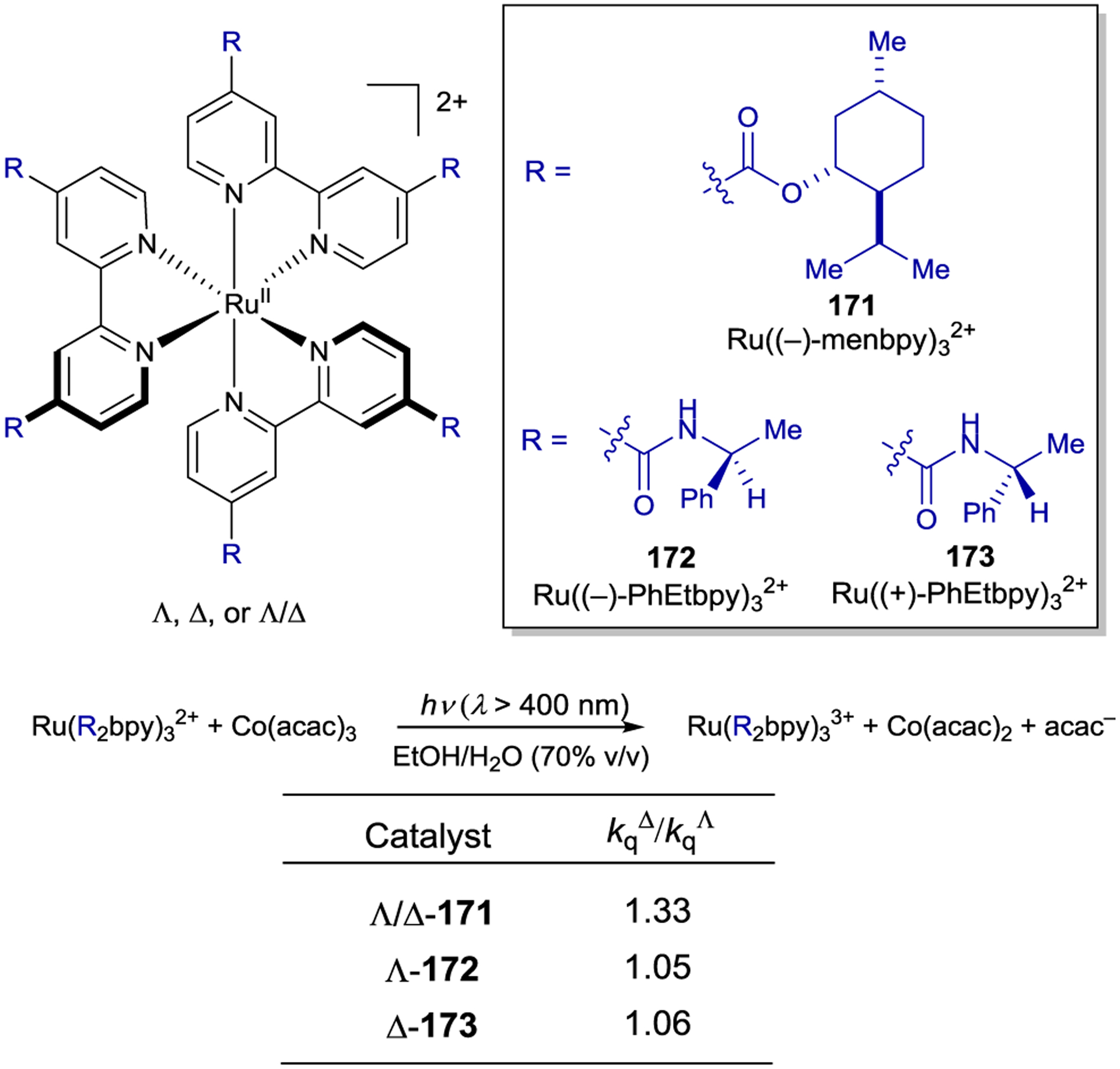
Stereoselective PET to Co(acac)3 with Chiral Ru(bpy)32+ Photocatalysts
An investigation of the influence of the metal stereocenter revealed a significant impact (Scheme 67). Ohkubo prepared the diastereomerically pure Λ-Ru((−)-menbpy)32+ isomer and showed that its selectivity for photoreduction of Co(acac)3 was higher than that of the unresolved 1:1 mixture of Λ- and Δ-Ru((−)-menbpy)32+ (Scheme 67).223, 224 Thus, while some degree of selectivity is controlled by the chirality of the bipyridyl ligands and Ru metal center, the overall selectivity of the reaction is almost entirely driven by the macroscopic helicity of the complex.225 Interestingly, the ee of the overall photoreduction (kr) was found to be substantially larger than that of solely the electron-transfer quenching process (kq ). The authors proposed that while the Ru(III) species may be reduced by acac− or solvent to reform the ground-state Ru(II) photocatalyst, back electron transfer (BET) from Co(II) to Ru(III) is competitive and enantioselective. This strategy was used to develop a kinetic resolution of Co(acac)3, achieving 39% ee at 40% conversion.226 Notably, in a later study Ohkubo discovered that the addition of exogenous acetylacetone (Hacac) had a beneficial effect on the selectivity of BET to reform Λ/Δ-Co(acac)3; a remarkable krΔ/krΛ of 91.9 was obtained in the presence of 10 equiv of Hacac (Scheme 67).227
Scheme 67.
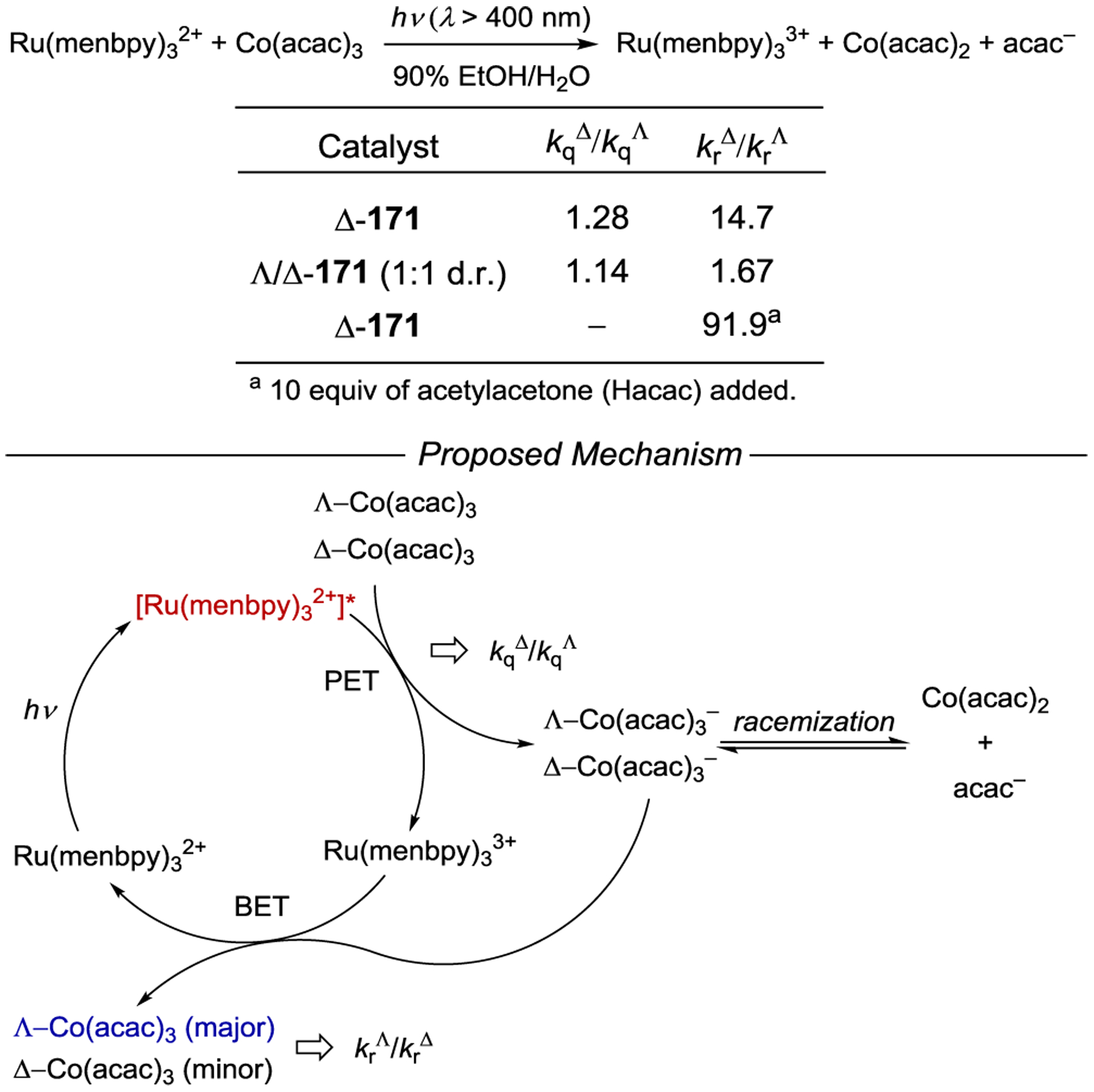
Effect of Helical Chirality on Enantioselective PET and Photoderacemization of Co(acac)3
Methyl viologen, a pyridinium dication, is an efficient quencher of photoexcited Ru(bpy)32+. Several groups have examined the effect of chiral viologens on the rate of electron transfer from Λ- and Δ-Ru(bpy)32+. Using partially enantioenriched Λ- and Δ-Ru(bpy)32+ Rau and Ratz studied the photoreduction of viologen 174, finding a significant difference in the rate of quenching (kqΛ/kqΔ = 1.66).228, 229 In related studies, Tsukahara and coworkers investigated selective quenching using viologens featuring chiral sec-arylethylamine units (Scheme 68).230 Photoexcited Δ-Ru(bpy)32+ is preferentially quenched by viologen (S,S)-175 over its enantiomer (kq(S,S)/kq(R,R) = 1.27). Interestingly, the naphthyl substituted viologens (176, 177) quench the photocatalyst by both static and dynamic pathways, the former arising from attractive π-π interactions with ground-state Δ-Ru(bpy)32+. Despite this, viologens 176 and 177 quenched with lower stereoselectivity (kq(S,S)/kq(R,R) = 1.16 and 1.11 respectively).
Scheme 68.
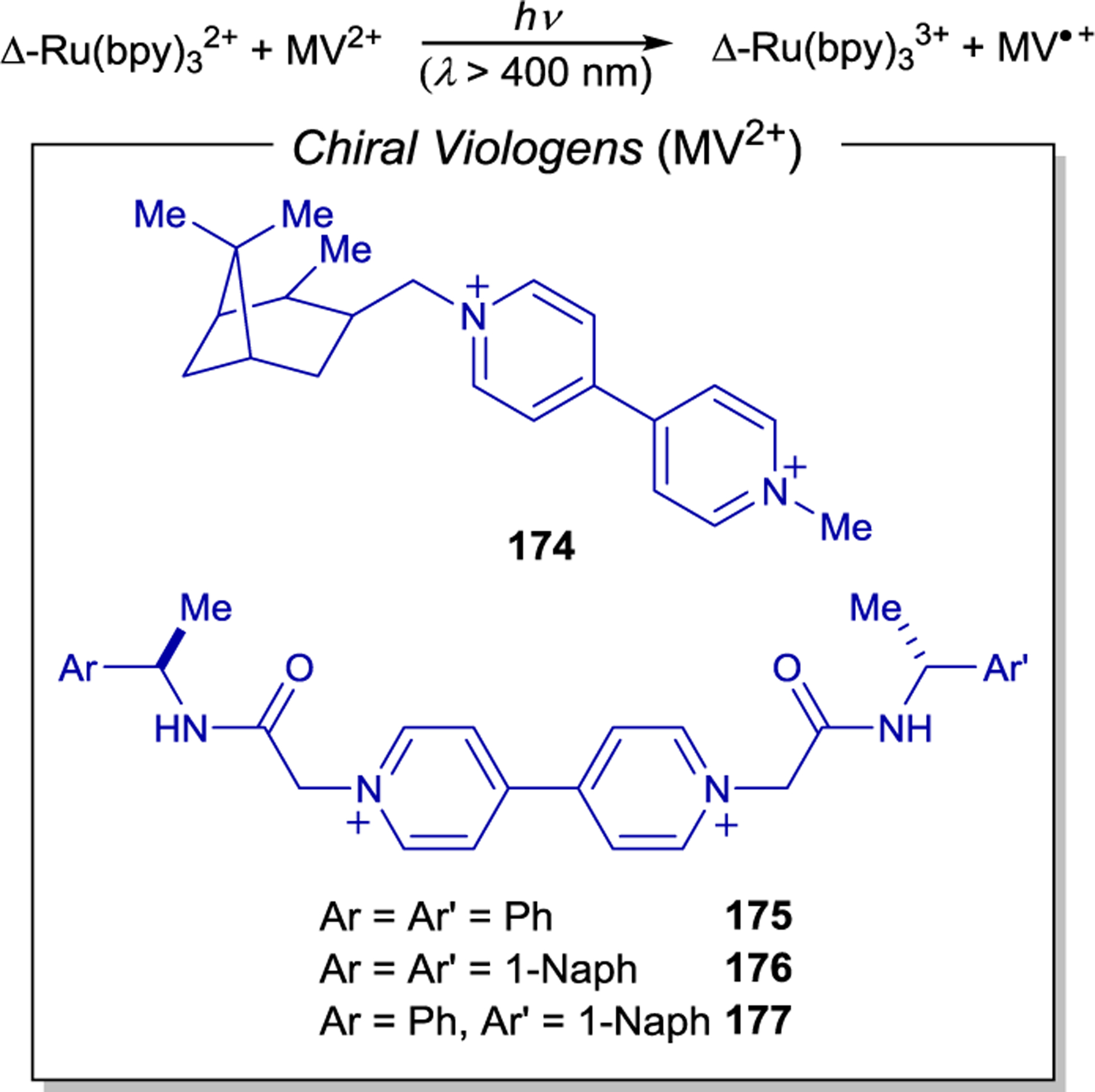
Stereoselective PET to Chiral Methyl Viologens
Ohkubo also demonstrated that the Δ-171 enables the atroposelective synthesis of binaphthols (Scheme 69).231, 232 This reaction proceeds by initial oxidation of the photoexcited Δ-Ru((−)-menbpy)32+ by Co(acac)3 to generate a Ru(III) complex, which subsequently oxidizes naphthol and initiates oxidative coupling to afford binaphthol in 16% ee.
Scheme 69.
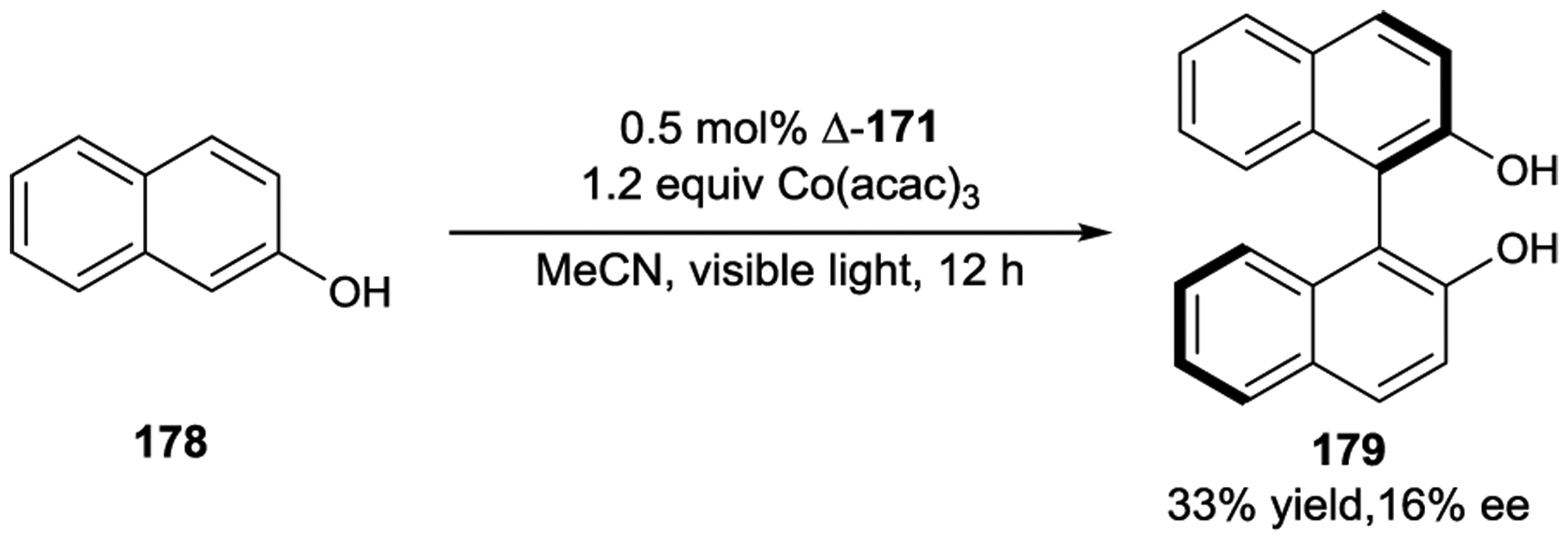
Atroposelective Synthesis of Binapthols
3.3. Lanthanides
Stereoselective energy transfer processes have also been investigated using chiral metal coordination complexes. Brittain observed that energy transfer between amino acid-bound Tb(III) and Eu(III) complexes exhibited different efficiencies depending on the enantiopurity of the amino acid ligand.233, 234 Later, Metcalf, Richardson, and coworkers investigated energy transfer from a racemic Tb(dpa)33− photocatalyst to enantiopure Δ-Ru(phen)32+.235 In these reactions, the Tb(III) complex absorbs UV light, and the resulting enantiomeric excited-state lanthanides are quenched by the enantiopure Ru(II) acceptor at different rates (Scheme 70). The stereoselectivity of the quenching process was monitored by observing the luminescence of the [Tb(dpa)33−]* excited state, which exhibited a significant circular polarization (CP) consistent with preferential quenching of the Λ isomer by Δ-Ru(phen)32+. Subsequent investigations examined chiral discrimination in energy transfer using complexes with varied metal centers236–237, 238 and their supporting ligands,239,240 in which the highest selectivity (kqΔ/kqΛ = 7.21) was recorded for the quenching of photoexcited Δ- and Λ-Eu(dpa)33− by diastereomeric Δ-Co((S,S)-trans-1,2-cyclohexadiamine)33+.241
Scheme 70.
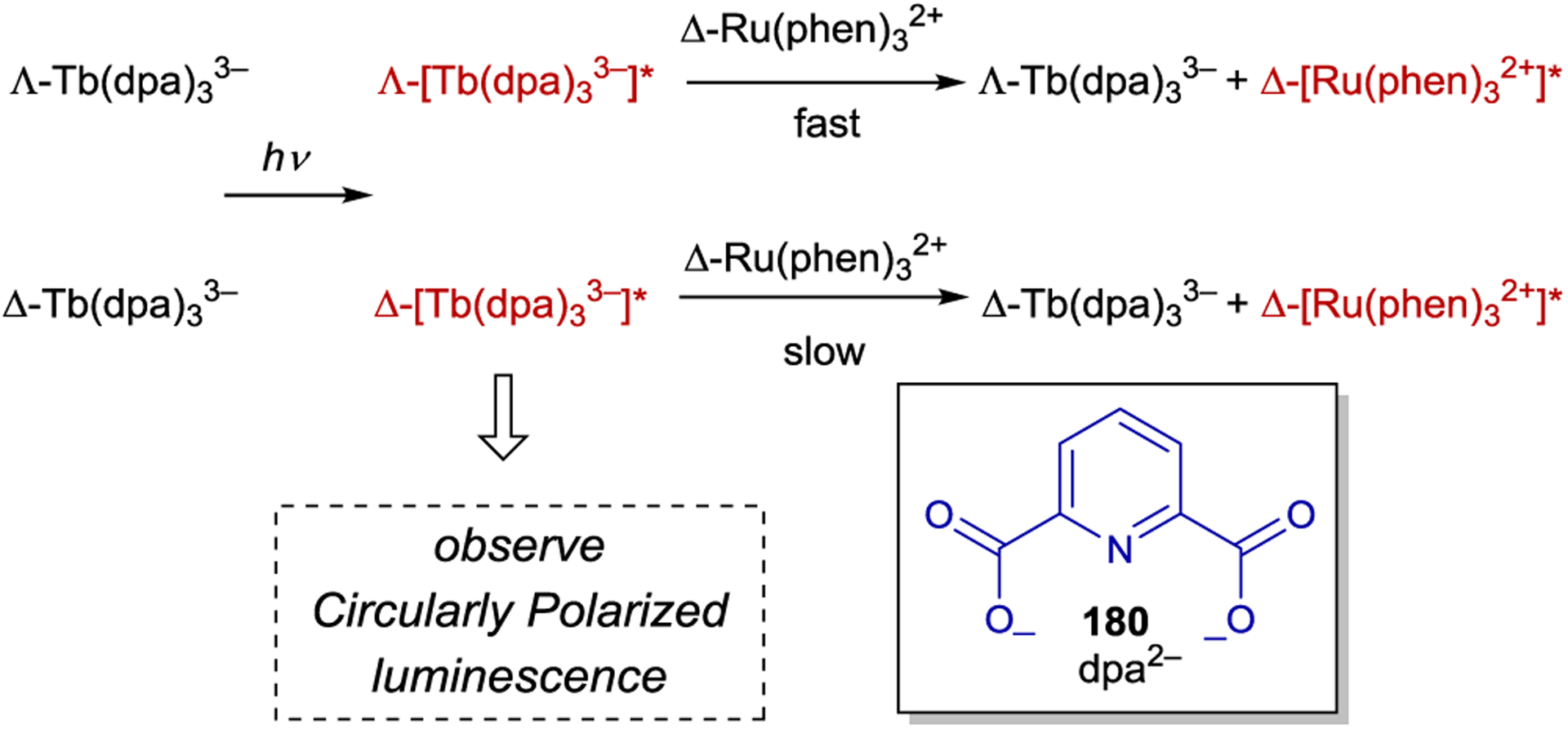
Stereoselective Energy Transfer
More recently, Liu and Feng disclosed a chromophore activation strategy using a chiral Tb(III) complex to promote [2+2]-photocycloadditions of 2-cinnamoylpyridine 181 in excellent enantioselectivity (Scheme 71).242 Mechanistic experiments revealed that addition of a mixture of Tb(OTf)3 and chiral N,N’-dioxide ligand 182 to 2-cinnamoylpyridine 181 results in a bathochromic shift in the UV-Vis absorption spectrum, indicating the formation of a new photoactive species. The authors hypothesized that the rate of intersystem crossing of excited-state 181 is accelerated by paramagnetic and heavy-atom effects from Tb(III).
Scheme 71.
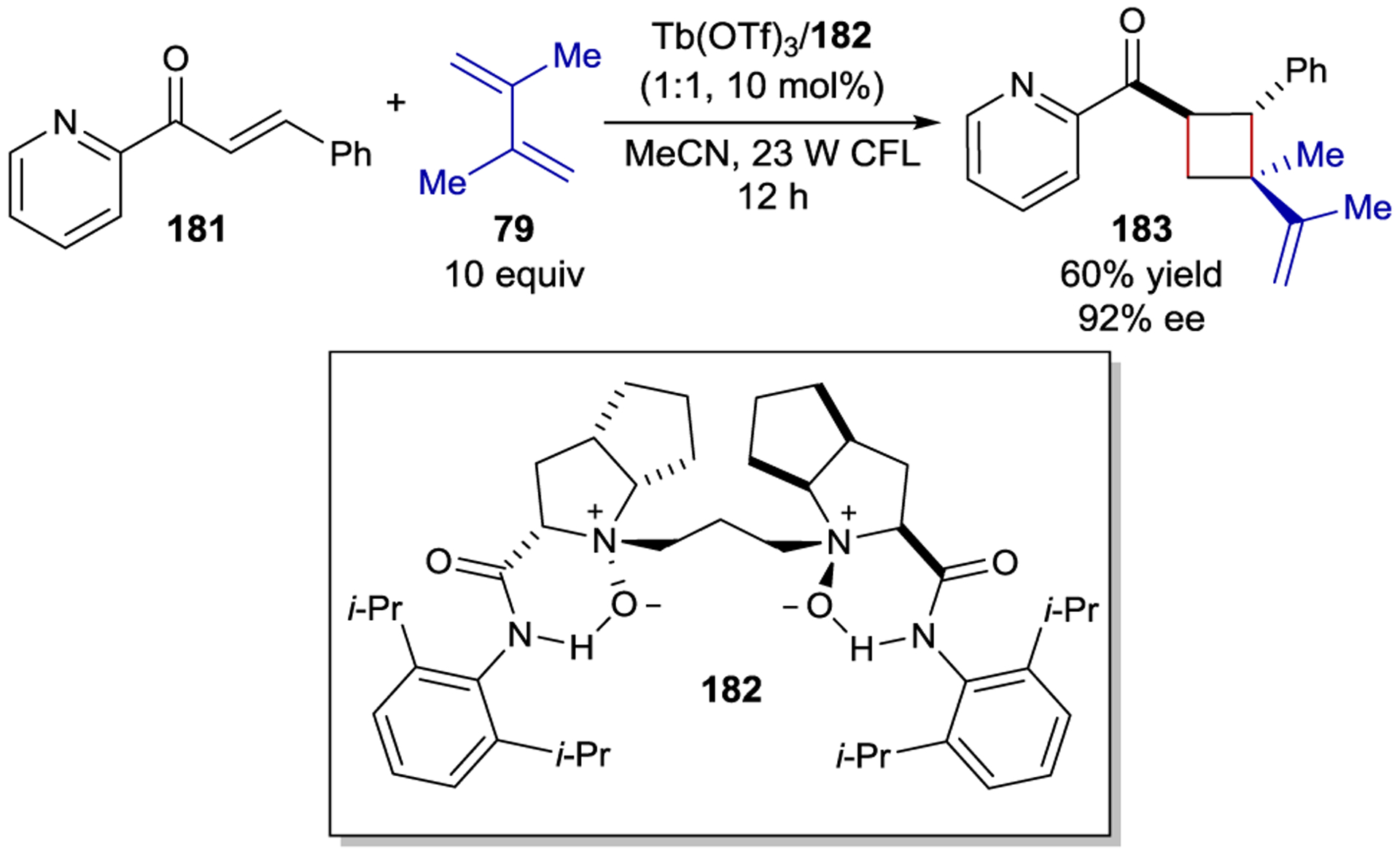
Enantioselective [2+2] Photocycloadditions Using Chiral N,N’-dioxide-Tb(III) Complexes
3.4. Iridium
Meggers pioneered the use of helically chiral iridium(III) polypyridyl complexes as enantioselective catalysts for a variety of synthetically important transformations. These catalysts can be easily prepared in stereochemically pure form using a chiral auxiliary approach243 and have shown to be resistant to racemization.244 Initial studies involved iridium complexes featuring ligands modified with organic functional groups that could act as organocatalysts, using the metal-centered stereocenter as a stereocontrolling element. These investigations resulted in the development of highly enantioselective transfer hydrogenations,245 Friedel–Crafts alkylations,246 sulfa-Michael and aza-Henry reactions,247 and α-amination reactions.248
Cyclometallated Ir(III) complexes are among the most widely utilized photocatalysts in contemporary synthetic photochemistry. Meggers hypothesized that chiral-at-metal Ir(III) complexes could serve a dual purpose as both enantioselective catalysts and photocatalysts.249 Chiral bis(acetonitrile) Ir(III) complex Λ-183b was designed to be a Lewis acid catalyst that becomes photoactive upon coordination of a bidentate Lewis basic substrate. Meggers’ initial investigations demonstrated that Λ-183b successfully mediates the highly enantioselective photoalkylation of 2-acylimidazolyl ketones with benzyl (184) and phenacyl bromide (185) radical precursors, yielding α-alkylated substrates in superb enantioselectivities up to 99% ee (Scheme 72). In subsequent work, Λ-183b and modified analogue Λ-183c proved to be effective chiral photocatalysts for enantioselective α-trichloromethylations250 of both N-phenyl 2-acylimidazoles (186) and 2-acylpyridines (187) and α-perfluoroalkylations (188, 189)251 of 2-acylimidazoles. Moreover, Meggers later showed that similar stereocontrolled radical addition reactions could be carried out in a one-pot, two-step procedure where the chirality is induced during a dimethylpyrazole-accelerated asymmetric transfer hydrogenation reaction.252, 253
Scheme 72.
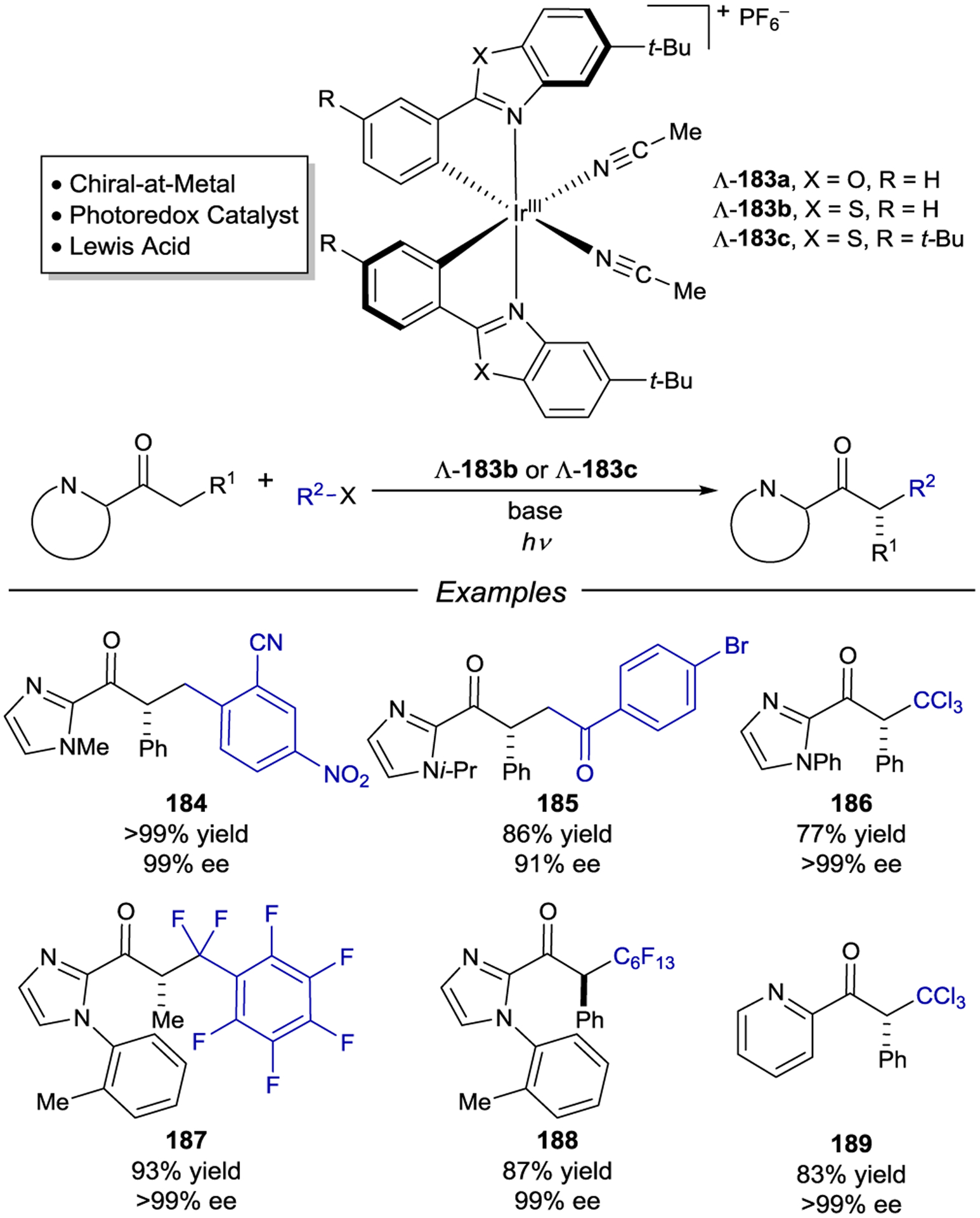
Chiral-at-Metal Ir(III) Photocatalysts Used in Asymmetric Secondary Photoreactions
Several experiments were conducted to understand the mechanism of these reactions (Scheme 73). First, in many cases the benzothiazole complexes (Λ-183b) were found to provide superior asymmetric induction compared to the benzoxazole congeners (Λ-183a). This effect was attributed to the longer C–S bond distance, which results in an increase in the steric profile of the two tert-butyl groups proximal to the site of substrate coordination.244,245 Second, X-ray crystallography verified that the substitutionally labile acetonitrile ligands are displaced by a bidentate substrate without loss of the stereochemical integrity of the metal center; deprotonation by an exogenous base affords the corresponding chiral enolate complex (190). Stern–Volmer luminescence quenching studies and electrochemical measurements were consistent with the role of this complex as the active photocatalyst that absorbs visible light to become a photoreductant capable of one-electron reduction of electron-deficient alkyl bromides. An Ir(III) Lewis acid catalyzed radical addition is followed by ketyl radical oxidation to reform the photoredox catalyst and provide the enantioenriched alkylation products. While a closed catalytic cycle was proposed, the possibility of a radical-chain mechanism was not ruled out.
Scheme 73.
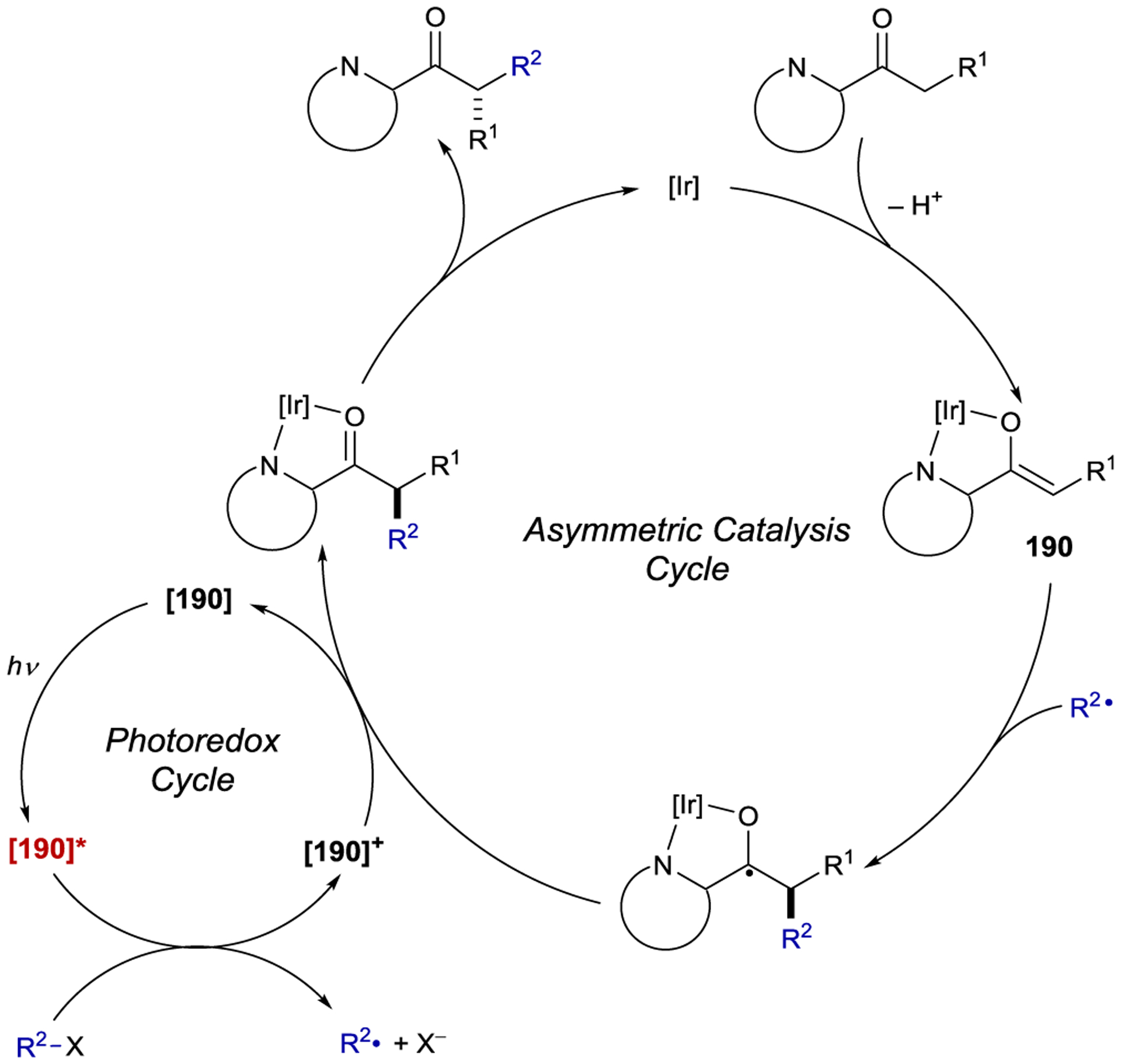
Proposed Catalytic Cycle of Asymmetric Radical Additions with Chiral Bis(acetonitrile) Ir(III) Complexes
In subsequent work, Meggers reported that these substrate-bound chiral-at-metal complexes can also function as photooxidants, facilitating the desilylative oxidation of α-silyl amines (Scheme 74).254 The resulting electron-rich α-aminoradicals undergo oxidation in the presence of oxygen to afford electrophilic iminium cations, which can react with the iridium enolate to afford Mannich adducts (193) in high enantioselectivity (97% ee). In this example, the Λ-183a catalyst proved to be superior to Λ-183b due to its increased oxidizing power. Nucleophilic α-aminoradicals can also be generated from deprotonation of photogenerated amine radical cations; Meggers demonstrated that these could engage in productive enantioselective radical–radical recombination with Ir-bound trifluoromethyl ketyl radicals to afford valuable 1,2-aminoalcohols (Scheme 75).255
Scheme 74.
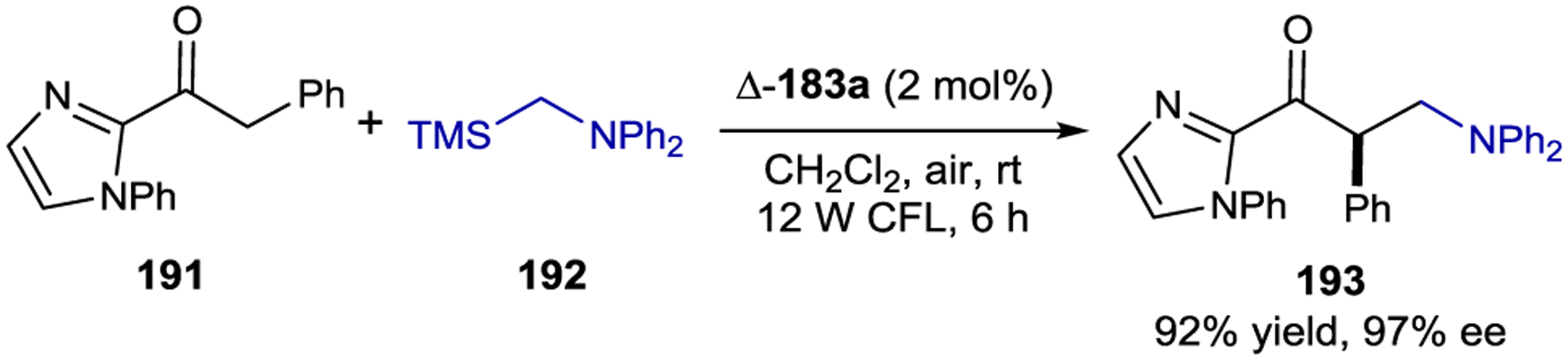
Enantioselective Net-oxidative Mannich Reaction of 2-Acylimidazoles
Scheme 75.
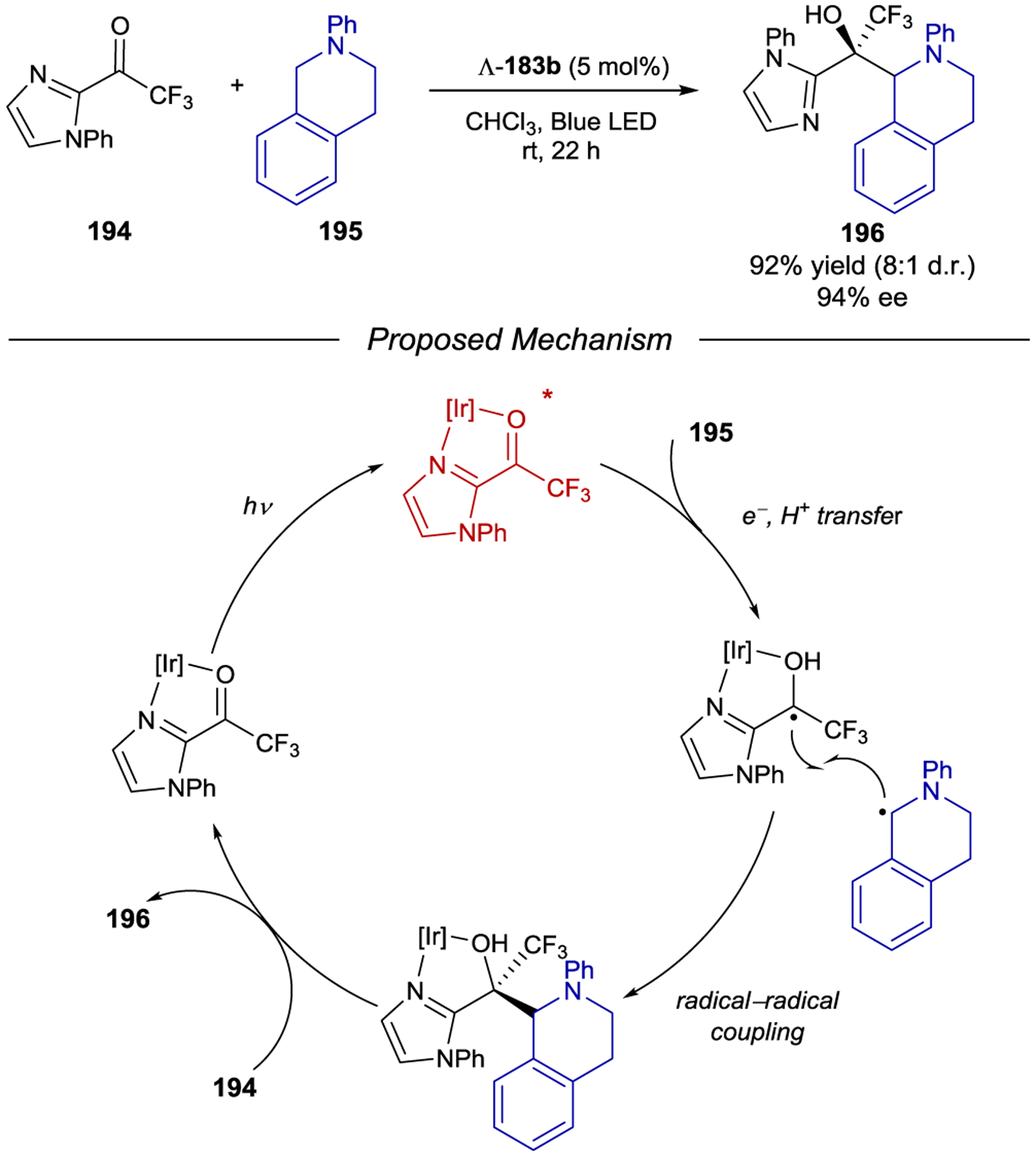
Enantioselective Radical–Radical Couplings of Trifluoromethyl Ketyl Radicals and α-Aminoradicals
As previously discussed, Bach has studied the use of a Kemp’s triacid derived chiral lactam functionalized with a xanthone or thioxanthone sensitizer to develop a variety of enantioselective photoreactions. Bach recently prepared an iridium photocatalyst bearing a modified bipyridine ligand functionalized with a similar chiral lactam moiety and studied its ability to influence the stereochemistry of electron- and energy-transfer photoreactions (Scheme 76).256 In contrast to the work of Meggers, however, the stereochemistry of the Ir(III) center was not controlled, and the photocatalytic assembly was used as a mixture of diastereomers. A model reductive cyclization of tertiary α-bromocarbonyl 197 unfortunately suffered from competitive hydrodebromination, and the expected cyclization product was formed in modest yields. The authors attributed the low enantioselectivity of this reaction (<10% ee) to a non-catalytic radical chain process. An energy-transfer reaction in which a chain mechanism is not feasible was more successful; the photosensitized rearrangement of spiroepoxide 201 afforded ketone 202 in 29% ee.
Scheme 76.
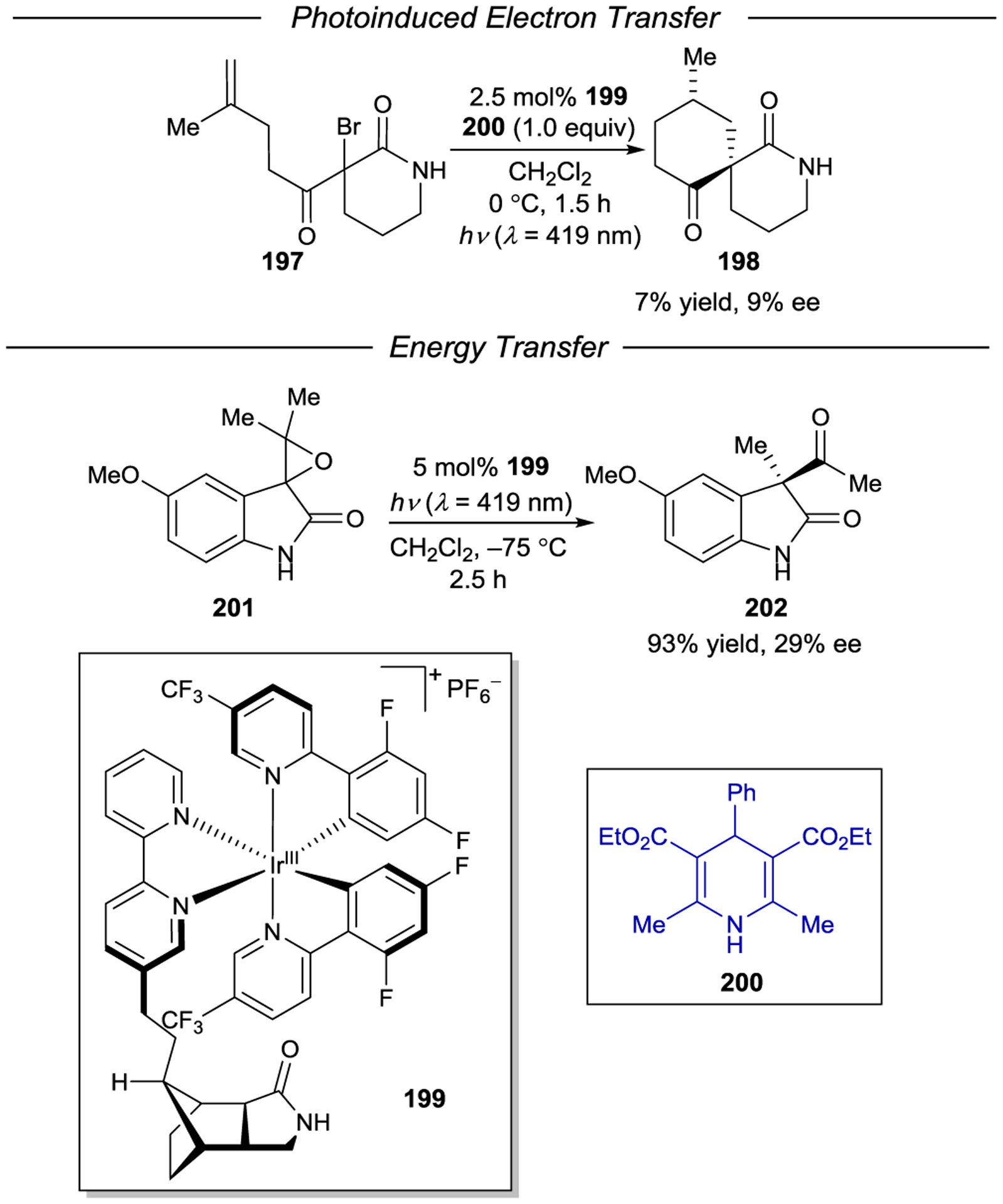
Stereoselective PET and Energy Transfer Reactions using a Chiral Hydrogen-Bonding Ligand
Inspired by MacMillan’s seminal asymmetric organocatalytic photoredox alkylation,159 Ceroni, Lombardo, and Cozzi designed an alternate supramolecular photocatalyst that tethers an imidazolidinone organocatalyst to an iridium sensitizer (Scheme 77).257 As in the previous example, the stereochemistry about the metal center was not controlled; instead, efficient enantiofacial bias relies solely upon the chirality of the amine catalyst . Under optimized conditions photocatalyst 203 afforded product 99 in 84% yield and 70% ee. Interestingly, the authors noted lower yield and enantioselectivity (57% yield and 59% ee) when the corresponding organocatalyst and photocatalyst were separated into their individual components. This decreased performance demonstrates how controlling the spatial proximity of the independent catalysts can improve the efficiency of the overall process.
Scheme 77.
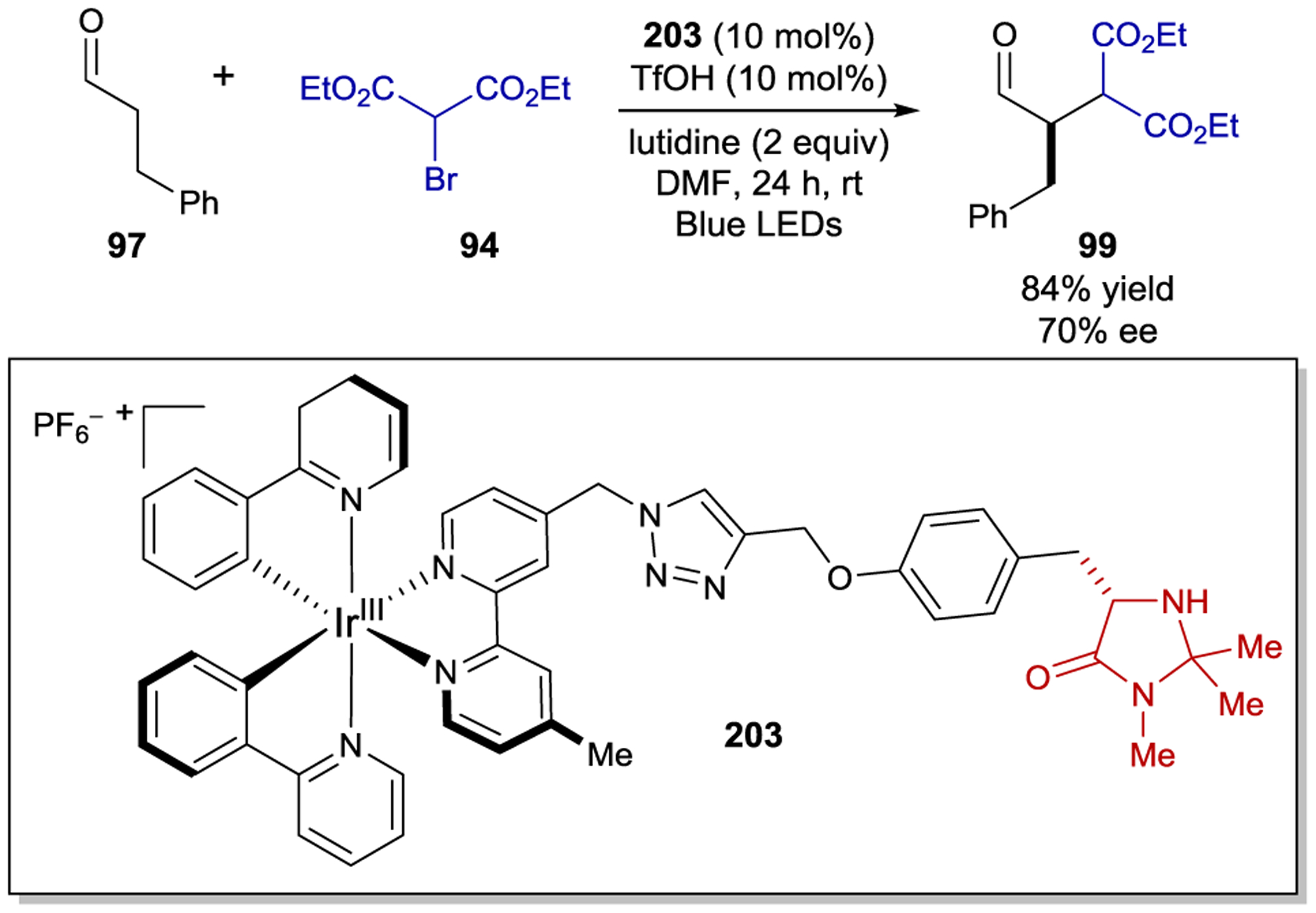
Enantioselective Alkylation of Aldehydes using Supramolecular Bifunctional Iridium Photoaminocatalyst
Inspired by Meggers’ precedential work, Yoon and Baik developed chiral iridium photosensitizer Λ-206 functionalized with a hydrogen-bonding domain that can control the enantioselectivity of an excited-state [2+2] photocycloaddition (Scheme 78).258 The design of this photocatalyst incorporates a pyridylpyrazole ligand capable of recruiting quinolone substrate 204 through a secondary-sphere hydrogen-bonding interaction. Under optimal conditions, cyclobutane product 205 can be isolated in near quantitative yield and 91% ee. Interestingly, computational analysis revealed a stereochemical model involving an unusual hydrogen bond between the amide group of the quinolone and the N–H of the pyrazole moiety, in addition to a π–π interaction between the cyclometalated ligand on the photocatalyst and substrate. Importantly, these non-covalent interactions facilitate both the orbital overlap necessary for triplet energy transfer to the quinolone and rationalize the absolute stereoinduction observed in the intramolecular reaction with the pendant alkene.
Scheme 78.

Enantioselective Intramolecular [2+2] Photocycloaddition with a Chiral Hydrogen-Bonding Iridium Photosensitizer
Following this report, Yoon, Baik, and Meyer extended this approach to the intermolecular [2+2] photocycloaddition of 3-isopropoxy quinolone 207 with maleimide 208. The optimal conditions for this reaction involved modified chiral photocatalyst Δ-210, which results in the formation of cyclobutane 209 in high yield and 97% ee (Scheme 79).259 Interestingly, a combination of NMR, steady-state luminescence quenching, and transient absorption studies revealed an unexpected mechanism in which the chiral photocatalyst selectively binds quinolone 207 but preferentially sensitizes maleimide 208 through a collisional energy-transfer process. The triplet excited state maleimide then reacts with the bound quinolone enantioselectively to form the product in a rebound-like mechanism. DFT calculations suggested that the lowest-energy binding interaction between the photocatalyst and quinolone was substantially different than in the intramolecular case, and that the favored binding geometry lacks the orbital overlap necessary for efficient intracomplex Dexter energy exchange. Consistent with this proposal, an N–Me quinolone (211) incapable of engaging in N–H-to-pyrazole hydrogen bonding affords reasonable enantioselectivity in this reaction.
Scheme 79.
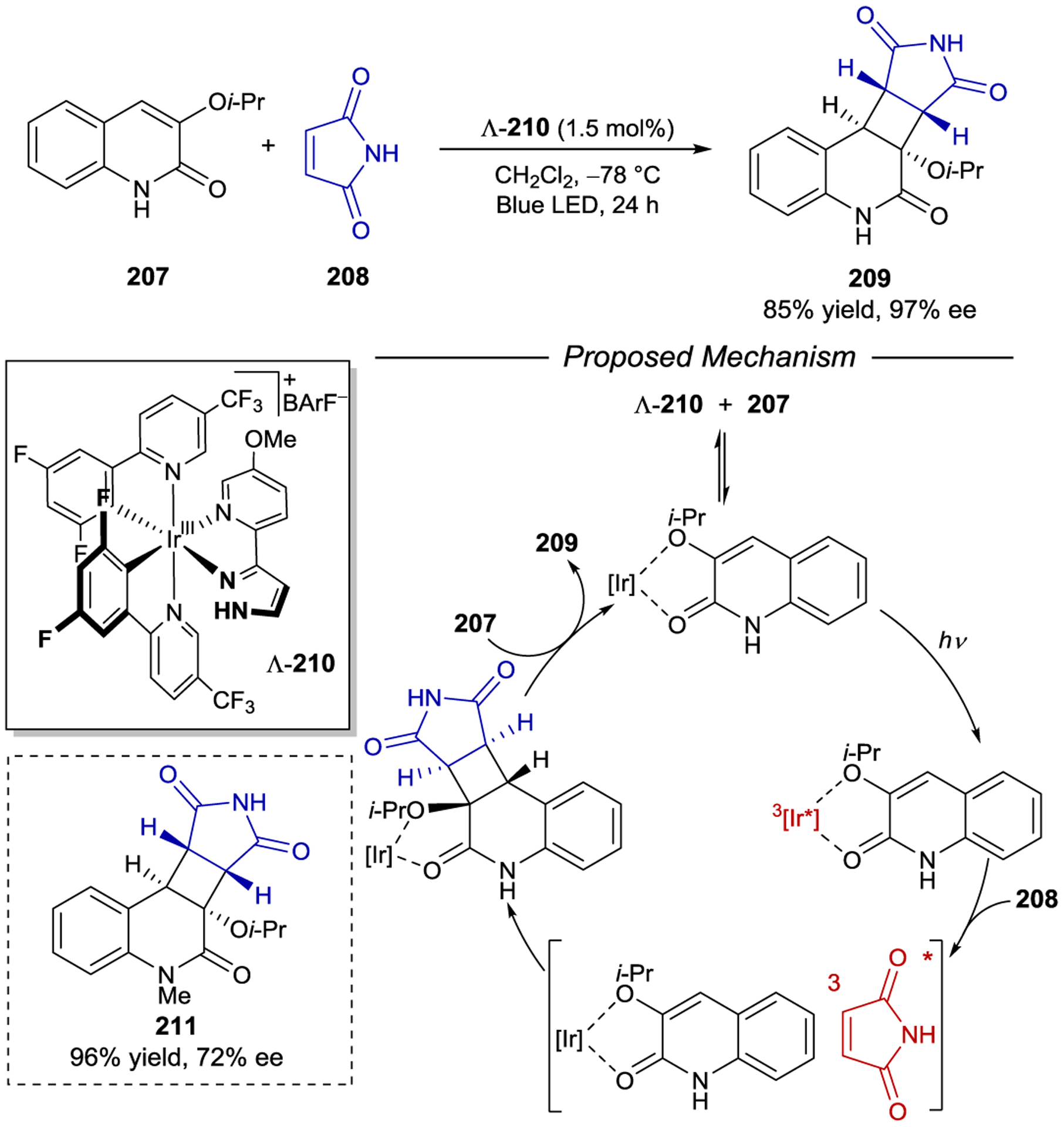
Enantioselective Intermolecular [2+2] Photocycloaddition via an Energy Transfer Rebound Mechanism
In 2020, Ooi developed an asymmetric [3+2] photocycloaddition using a cationic iridium photocatalyst with a chiral hydrogen-bonding borate counteranion (Scheme 80).260 This strategy utilizes the ability of N-cyclopropylurea 212 to associate with chiral borate 215. In addition to preorganizing the substrate within a stereodifferentiating environment, this complexation increases the rate of reaction, as a parallel reaction with [rac-Ir(dFCF3ppy)2(dtbbpy)]PF6 was significantly slower (<10% yield). Notably, the chirality of the cationic Ir center bore no influence on reaction selectivity, as the use of either the Δ- or Λ-isomer of the iridium photocatalyst produced essentially the same results. Photoinduced electron-transfer oxidation of the borate-bound urea substrate produces radical-ion pair 212⊂215 that reacts smoothly and selectively with α-substituted styrenes to provide the chiral cyclopentane products. This methodology was later shown to work equally well with α-substituted acrylates as the alkene partner.261
Scheme 80.
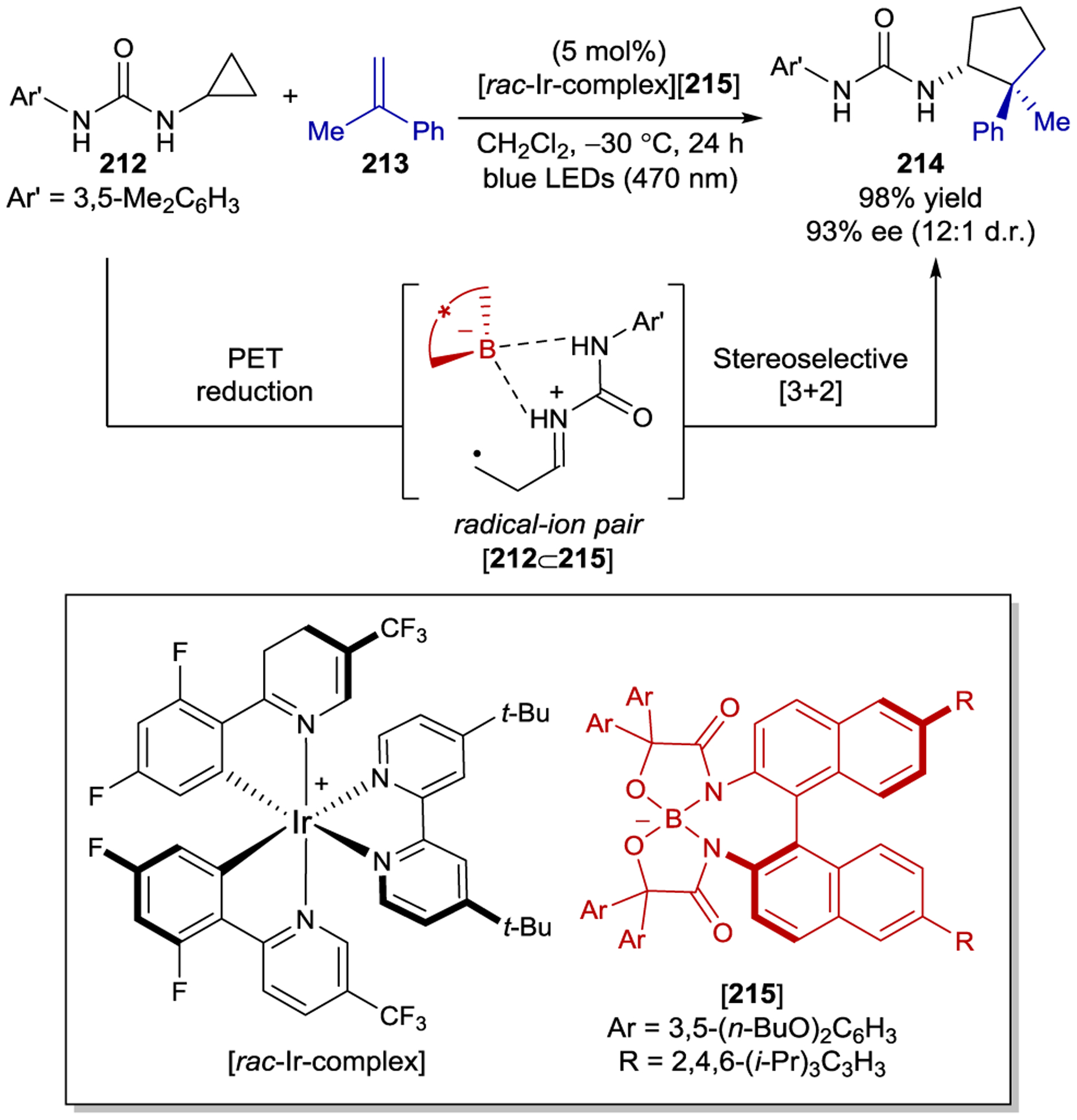
Asymmetric Photocatalysis Using Iridium-Chiral Borate Ion Pairs
More recently, Melchiorre demonstrated that iridium catalyst 222, a complex widely recognized as a privileged organometallic intermediate in enantioselective allylic substitutions,262 is also a competent photoredox catalyst. Photoexcitation of the complex enables the oxidation of carbazoles functionalized with cleavable redox auxiliary (RA) groups (Scheme 81).263 The resulting carbazole-stabilized radicals is captured by an (η3-allyl)iridium(II) complex, which then undergoes stereodetermining reductive elimination to afford the alkyl–alkyl cross-coupled product in high yield and enantioselectivity.
Scheme 81.
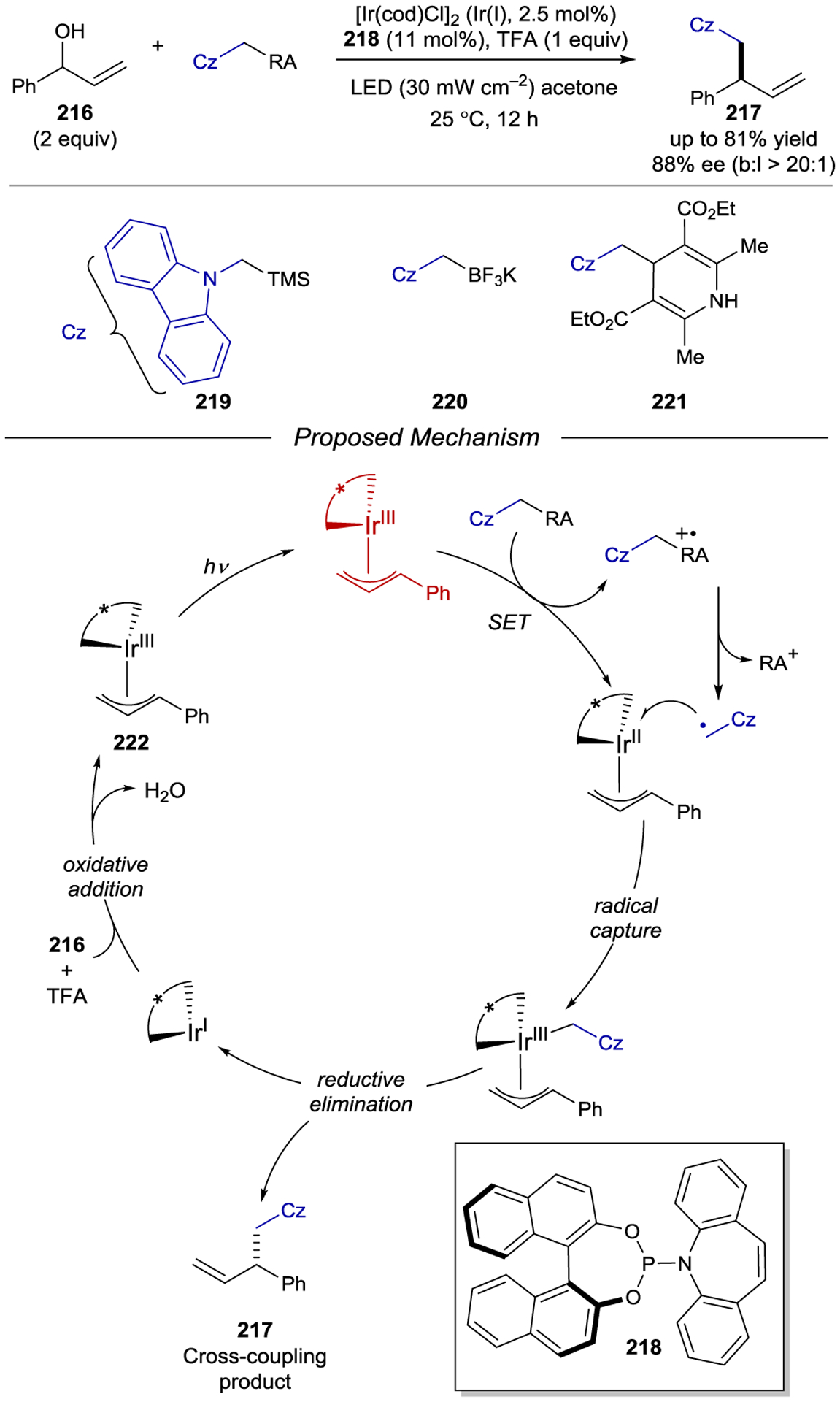
Asymmetric C–C Cross-Couplings Enabled by (η3-allyl)Iridium(III) Photocatalyst
3.5. Rhodium
Meggers has extended the protocol for the synthesis of enantiopure helically chiral metal complexes to several late transition metals. The Rh(III) complexes of this class have proven to be exceptionally effective structures for asymmetric photochemistry (Figure 4). Many of these methods have utilized the Rh(III) complex as a non-photoactive chiral Lewis acid in tandem with exogenous photocatalysts;264 the coverage of the literature in this section, however, is limited to examples in which the chiral Rh(III) fragment forms part of the light-absorbing chromophore.
Figure 4.
Chiral Bis(acetonitrile) Rh(III) Complexes
The first demonstration of chiral-at-rhodium Lewis acids in asymmetric photochemistry involved a study of the α-amination of C-acylimidazole substrates (Scheme 82). Meggers proposed that photoexcitation of the photocatalyst–substrate complex would promote reduction of ODN-carbamates (ODN = 2,4-dinitrophenylsulfonyloxy) producing electrophilic nitrogen-centered radicals. Reaction of the radicals with the chiral Rh–enolate complex in a stereoselective Lewis acid catalyzed chain process provides the α-aminoketone in nearly quantitative yield and 97% ee.265 Interestingly, while Λ-223a is an excellent catalyst for this transformation, the chiral-at-iridium photocatalysts described previously proved ineffective. The improved reaction rate using Λ-223a was attributed to its faster rate of ligand exchange, 266 which can compensate for the short lifetimes of the highly reactive nitrogen-centered radicals. In line with results of studies using the analogous Ir(III) complexes, benzothiazole Λ-223c catalysts provided somewhat superior stereocontrol in comparison to the analogous benzoxazole complexes.267
Scheme 82.
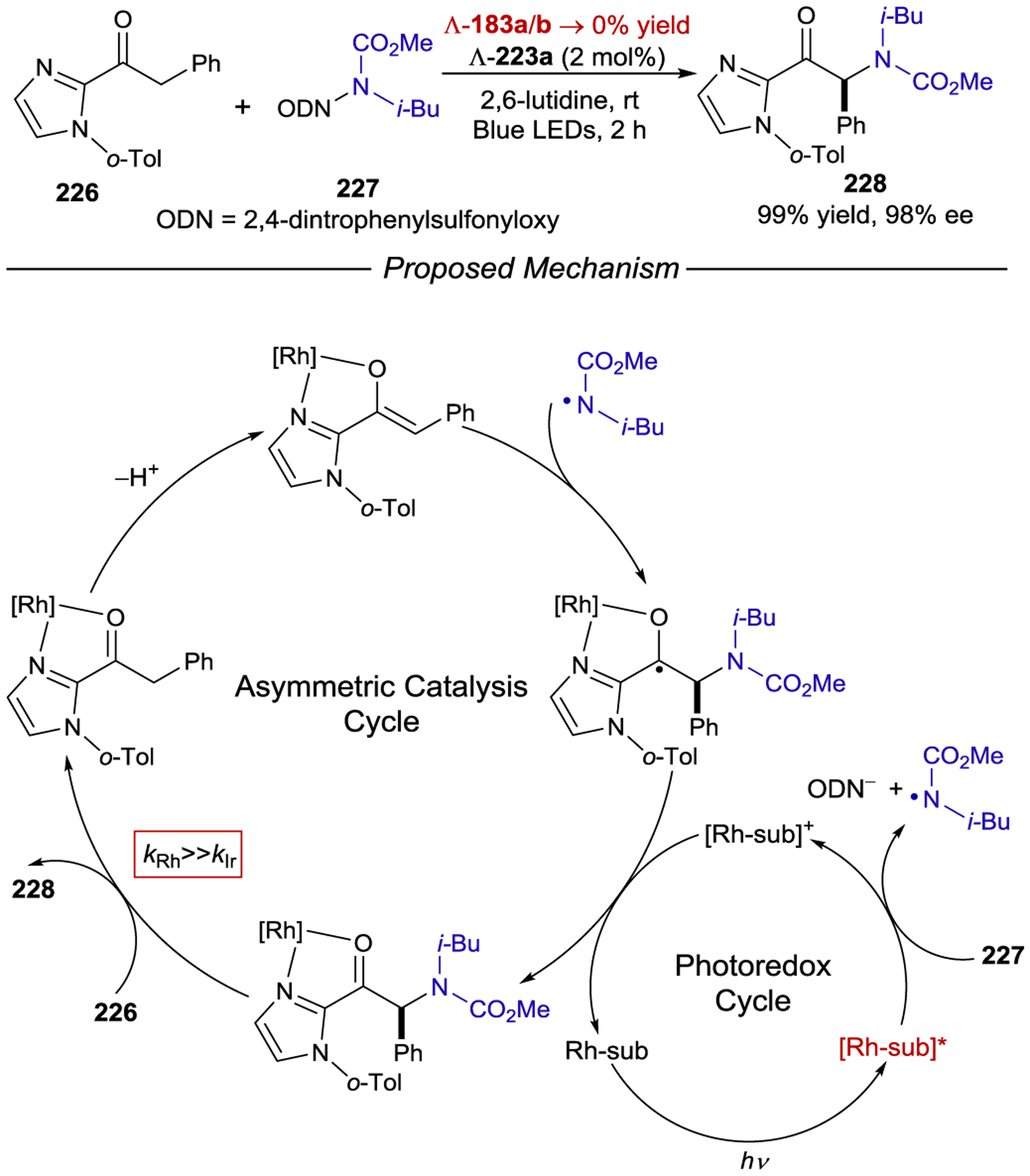
Enantioselective α-Amination using Chiral-at-Metal Rhodium Photocatalysts
Chiral-at-rhodium photocatalysts have proven to be extremely versatile (Scheme 83). Notably, many of these reactions cannot be catalyzed by the first-generation iridium complexes. Meggers reported a photocatalytic oxidative coupling of enolates to photogenerated α-amino radicals that uses air as the terminal oxidant.268 Kang independently developed the asymmetric radical conjugate addition of tetrahydroisoquinolone-derived α-amino radicals into rhodium complexed α,β-unsaturated 2-acylimidazoles.269 Comparable reactivity was thereafter reported by Meggers using Hantzsch ester derivatives as alkyl radical sources.270 Meggers later showed that a similar enantioselective α-amino radical conjugate addition could be initiated through photoinduced reductive decarboxylation of O-phthalimidyl protected amino acids.271 More recently, Su and Kang established an asymmetric Giese addition of p-aminobenzyl radicals from the corresponding carboxylic acids using chiral rhodium catalyst Λ-223b.272 Finally, the synthesis and application of a chiral-at-metal novel bis-cyclometalated indazole catalyst Λ-224 was useful in producing α-cyano radicals and controlling their facially-selective addition into rhodium enolates.273 The mechanisms and origins of enantioselectivity using this class of Rh(III) photocatalysts (Scheme 84) has been studied by Meggers together with Wiest274 revealing a strong rate enhancement induced by the rhodium catalyst to overcome competitive racemic reactivity. An X-ray crystal structure of a Rh(III)–enolate intermediate exposed a steric burden imposed by the tert-butyl groups on the Si face of the prochiral substrate.275 Additional DFT calculations performed in collaboration with Houk276 suggest that distortions of the substrate–catalyst complex is a major contributor in controlling the enantioselectivity.
Scheme 83.
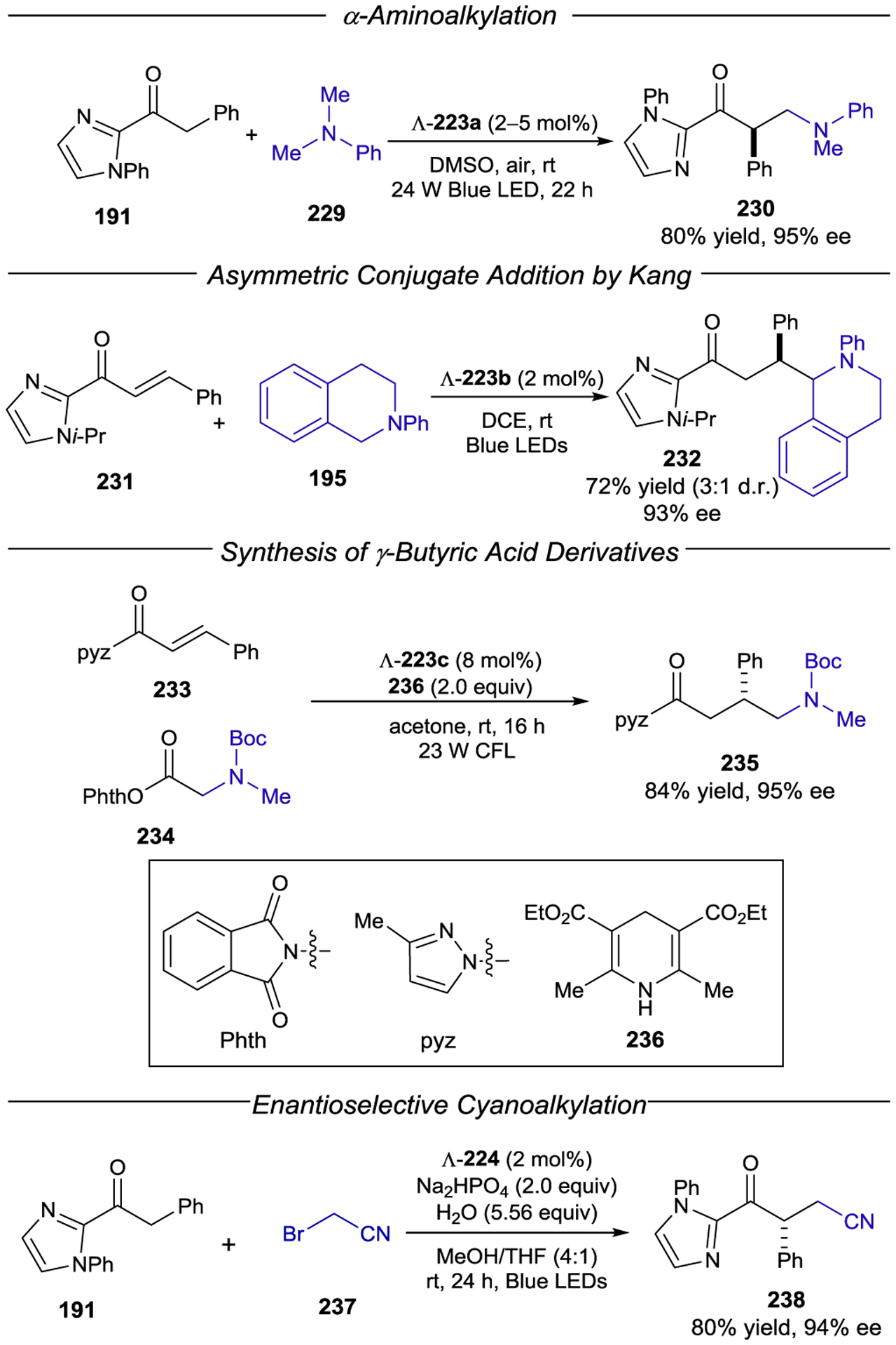
Asymmetric Radical Additions Catalyzed by Chiral Rhodium Catalysts
Scheme 84.
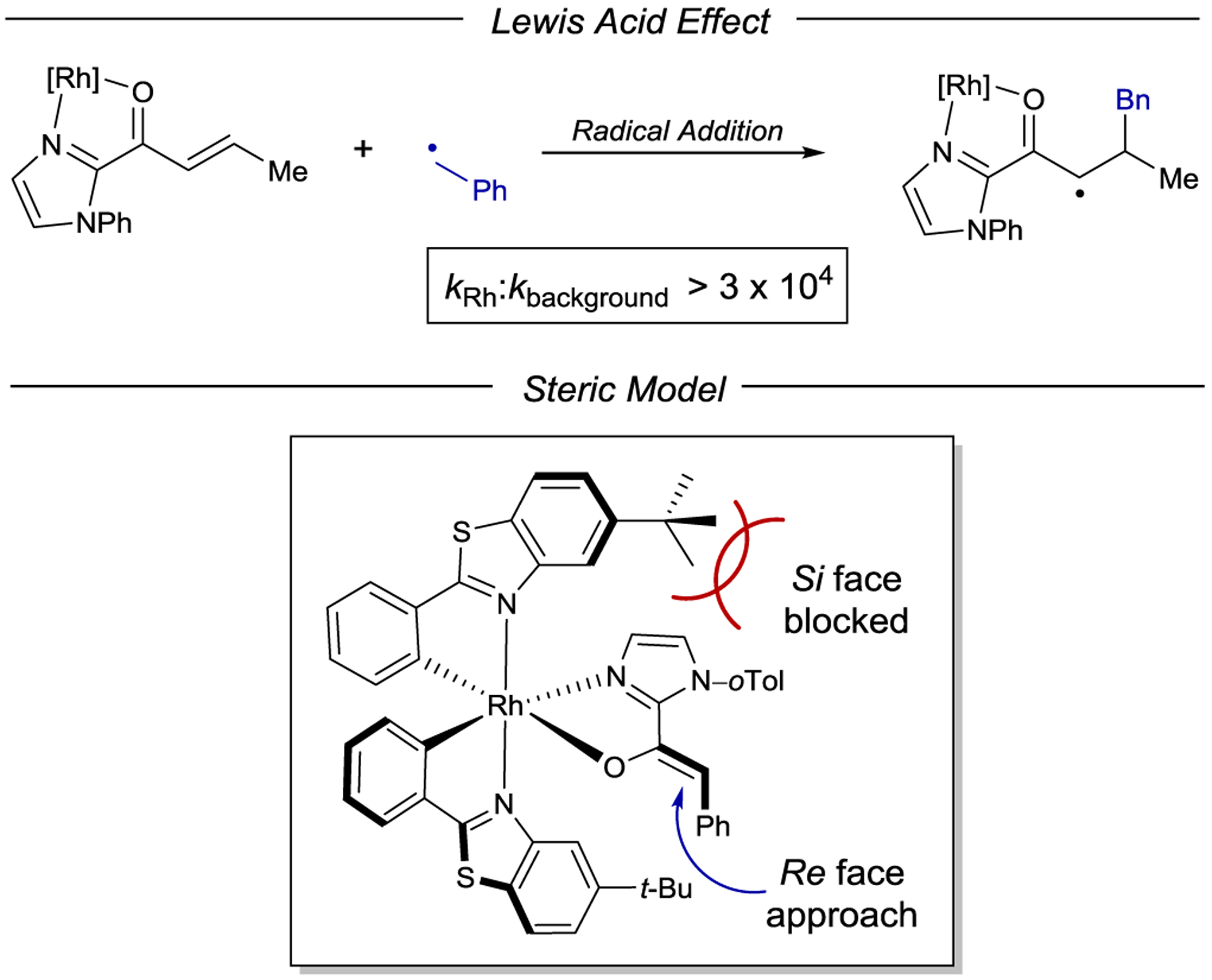
Lewis Acid Effects and Mode of Enantioinduction
Enantioselective radical–radical coupling reactions can also be catalyzed by chiral Rh(III) photocatalysts. In 2017, Weist and Meggers reported the asymmetric β-C–H functionalization of 2-acylimidazoles (239) and 2-acylpyrazoles with α-ketoester 240 (Scheme 85).277 The radical reactants in this process are generated by photoinduced electron transfer from the excited-state Rh(III)-enolate complex to the α-ketoester to afford ketyl radical and α-ketoradical intermediates. Radical coupling occurs with high diastereoselectivity and >90% ee. A complementary study resulted in a method for the stereoconvergent asymmetric coupling of racemic α-chloro-imidazol-2-yl-ketones with N-arylglycines (Scheme 86).278 In this work the radical partners were generated by a photoinduced decarboxylation of the N-arylglycinate 243 and a concomitant photoreductive dehalogenation of the Rh(III)-ketone complex; radical–radical coupling of the resulting α-aminoradical and α-ketoradical affords chiral β-aminoketone 244 in high ee.
Scheme 85.
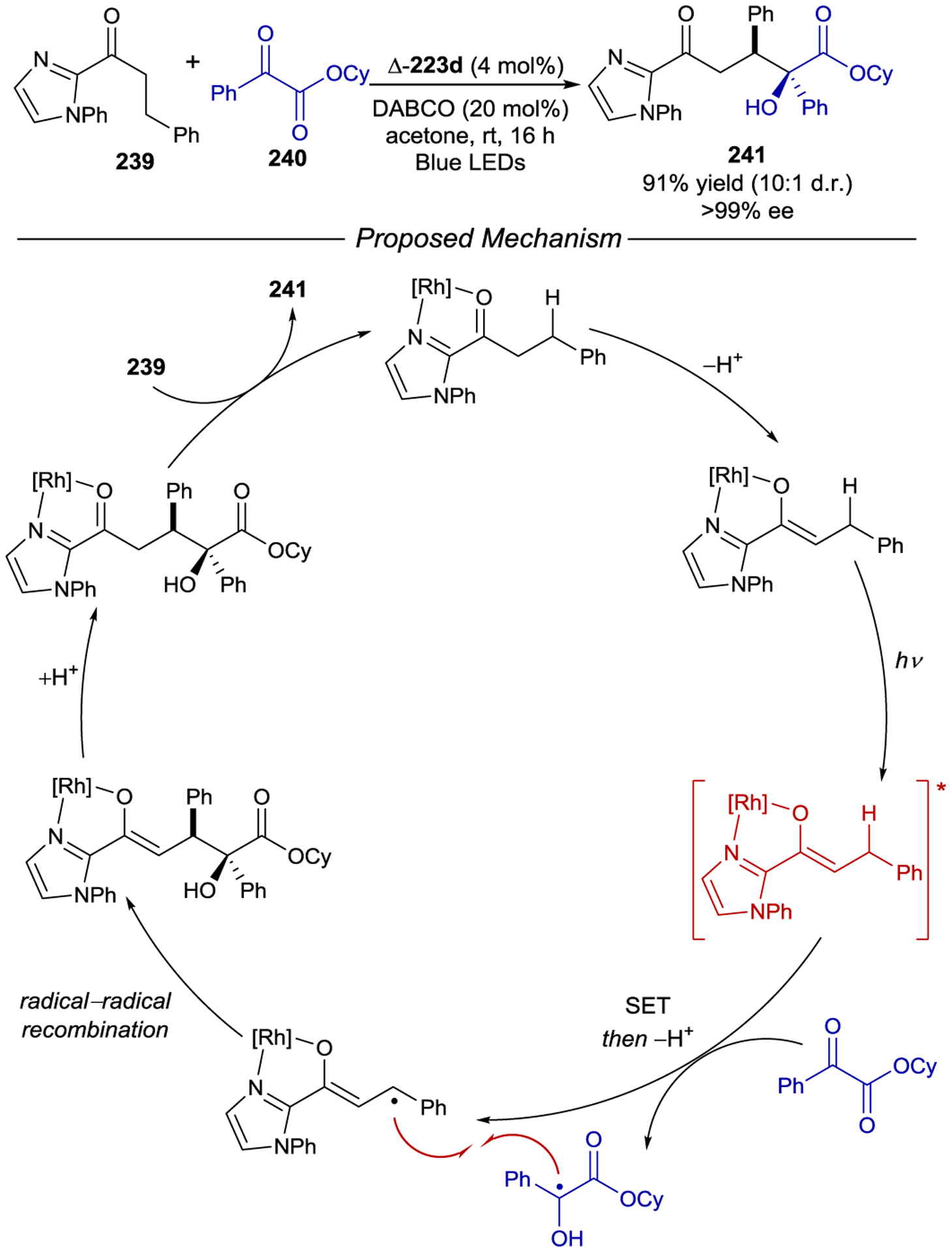
Enantioselective Radical–Radical Coupling Reaction of 2-Acyl-Imidazoles/Pyrazoles and α-Ketoesters
Scheme 86.
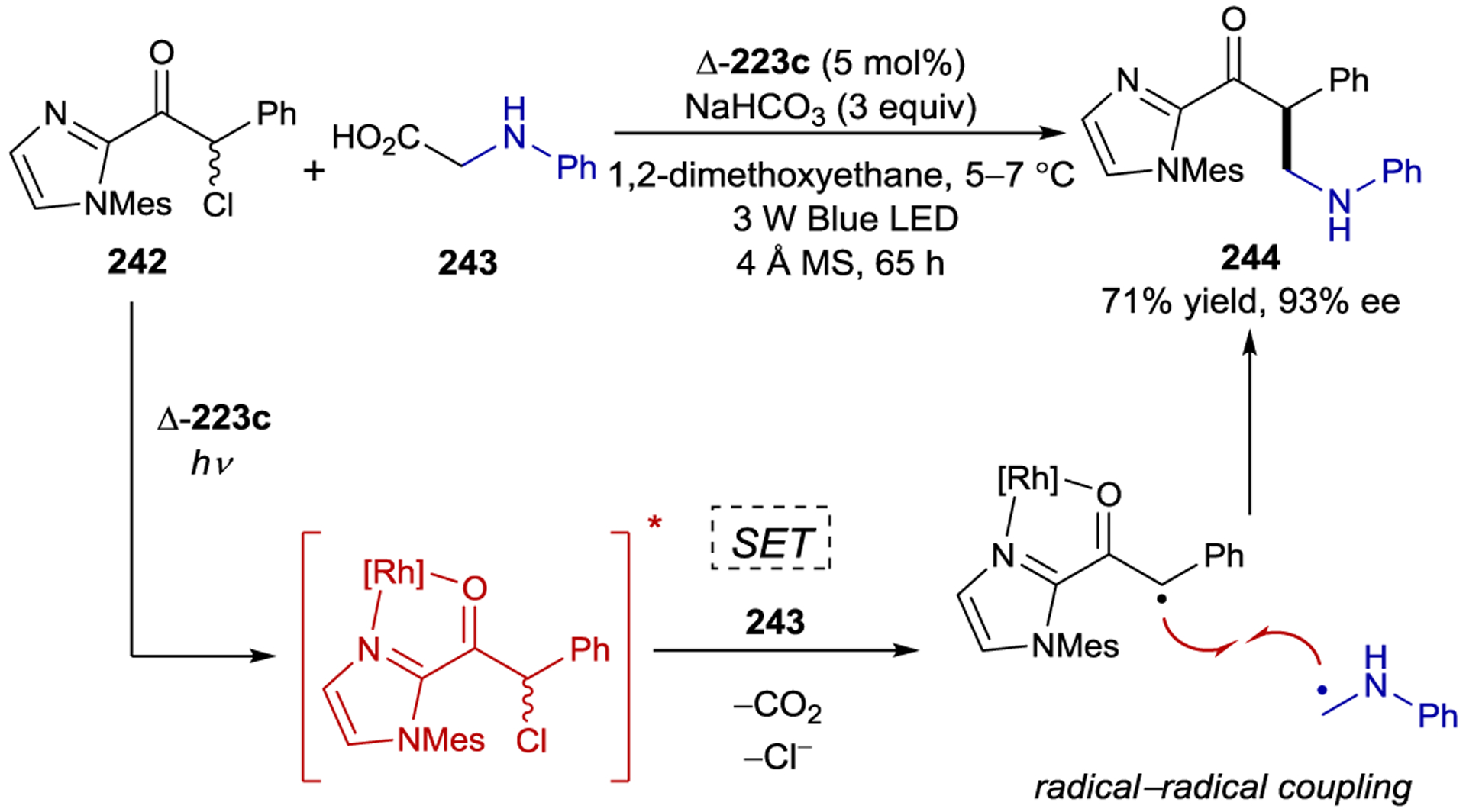
Enantioconvergent Coupling of Racemic α-Chloroketones and N-Arylglycines
In addition to enantioconvergent cross-couplings, Meggers and Chen demonstrated that Rh(III) catalyst Λ-224 in combination with a tertiary amine could accomplish a photochemical deracemization of 2-pyridylketone 245 in 96% ee (Scheme 87).279 A photochemically driven single-electron reduction of Rh-bound 245 by aniline 246 followed by HAT to the oxidized amine yields the achiral rhodium–enolate complex. Diastereoselective proton transfer provides the rhodium coordinated ketone as a single stereoisomer. This mechanistic separation was critical in circumventing the restriction of microscopic reversibility as the elementary steps for enolate formation are distinct from stereoselective protonation. (R)-245 bound to Λ-224 is destabilized compared to (S)-245, and thus iterative SET/HAT/protonation sequences selectively enhance (R)-245. Notably, the protonation step alone does not fully account for the enhanced stereoselectivity. Instead the selective association and photochemical activation of (S)-245 facilitates the enrichment of (R)-245.
Scheme 87.
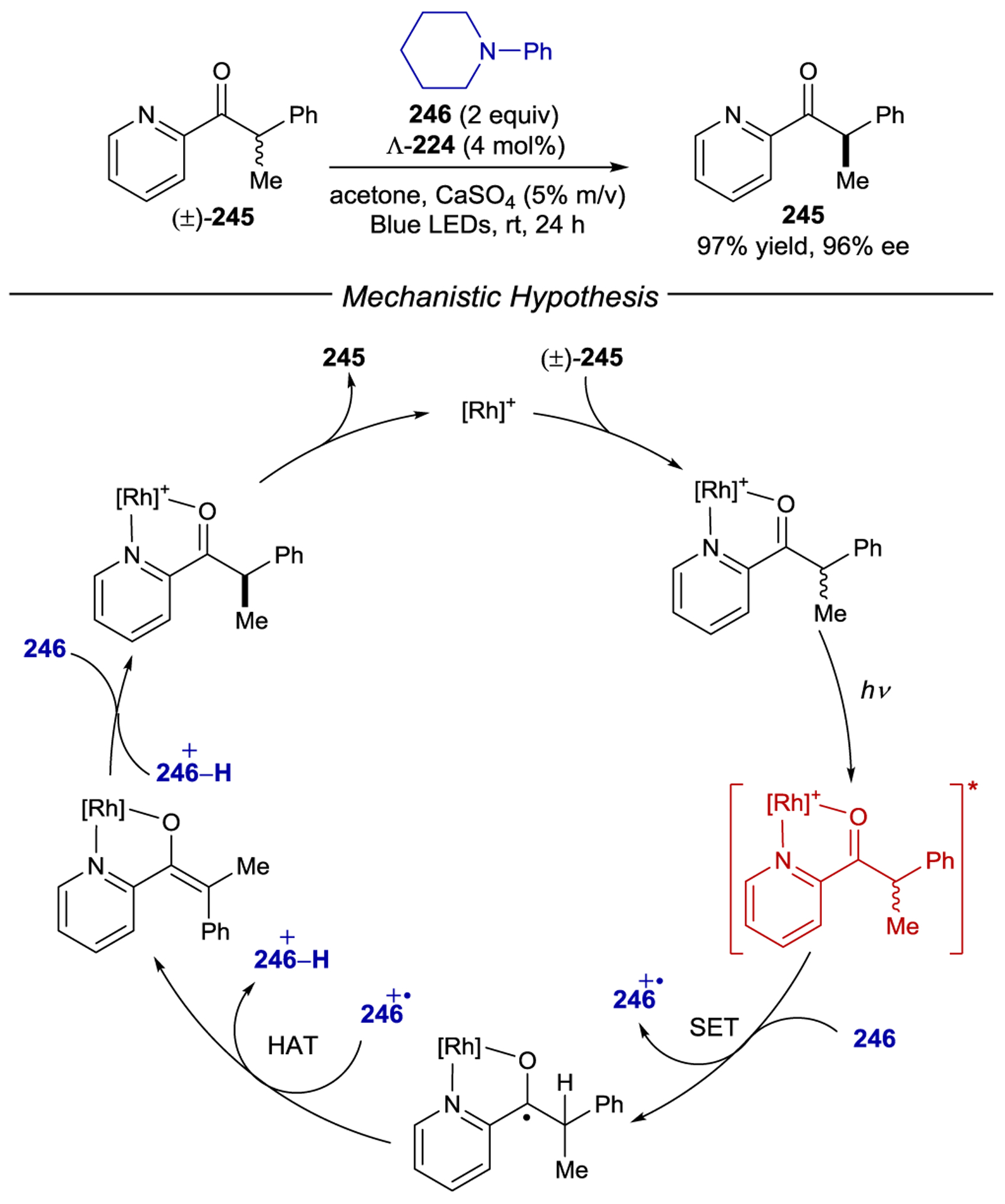
Photocatalytic α-Deracemization of 2-Pyridylketones
Meggers developed an asymmetric [3+2] photocycloaddition that also involves a chiral Rh(III) photocatalyst.280 Cyclopropyl imidazolyl ketones react smoothly with a wide variety of alkenes and alkynes upon irradiation in the presence of the Rh(III) complex Λ-223c to afford highly enantioenriched cyclopentanes (247) and cyclopentenes (248, 249) (Scheme 88). The mechanism of this reaction involves chromophore activation: complexation of the imidazolyl ketone to the Rh(III) Lewis acid results in a bathochromic shift that enables excitation by visible light. The Lewis acid activation also results in an anodic shift in the substrate reduction potential, facilitating formation of the ketyl radical by photoreduction.
Scheme 88.
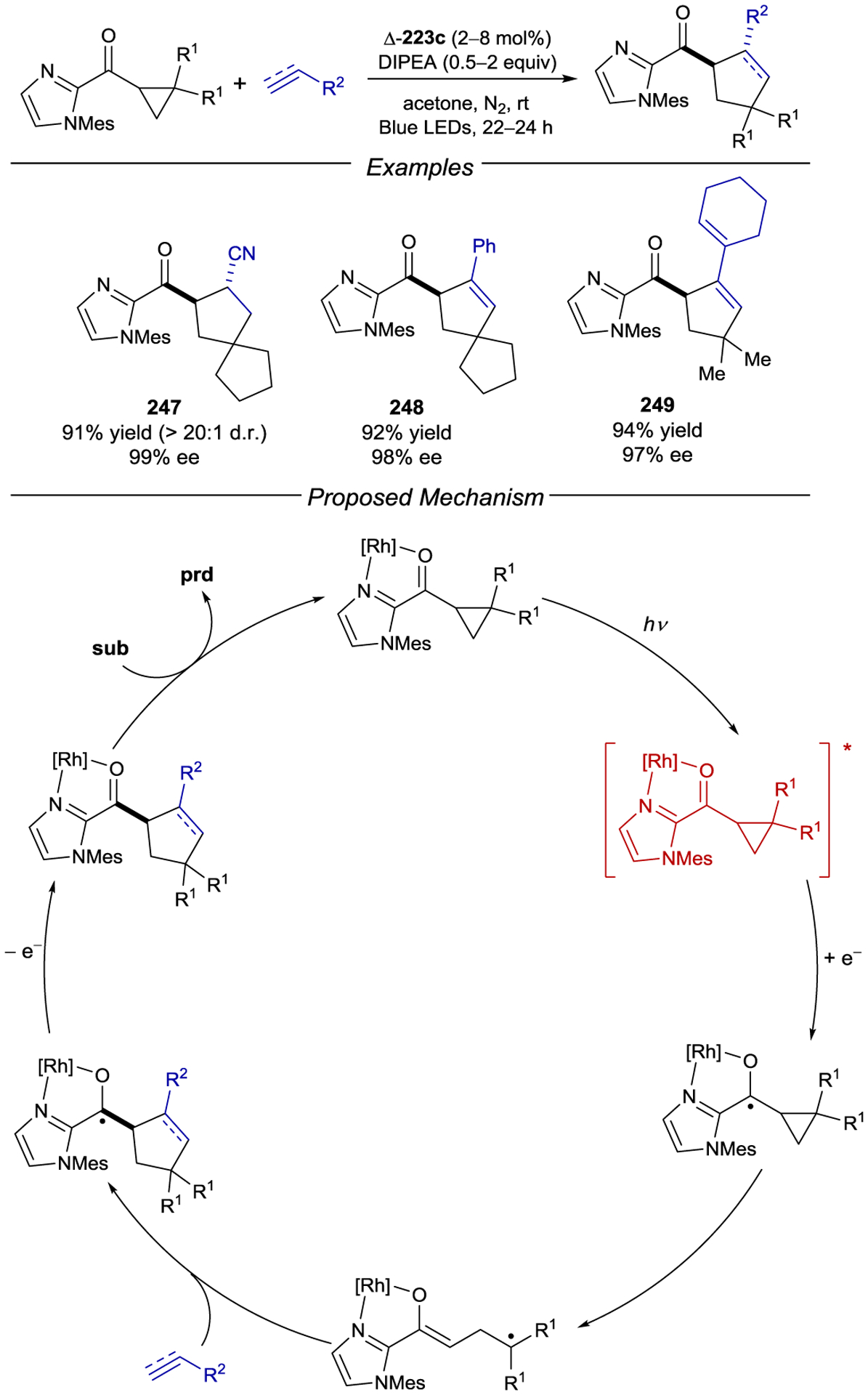
Enantioselective [3+2] Cycloaddition of Cyclopropyl Ketones with Alkenes and Alkynes
Meggers and Wiest extended this concept to the enantioselective [2+2] photocycloaddition of chelating N-phenyl-cinnamoylimidazole 250 with alkenes (Scheme 89).281 As in the previous example, the enhanced visible-light absorption of the substrate–catalyst complexation allows for the selective photoexcitation of the chiral catalyst-bound compound and suppresses racemic background reactivity. Notably, both isomers of the reacting alkene converged to provide the same cyclobutane stereoisomer ratio, suggesting that this reaction proceeds through a triplet excited state. This hypothesis was computationally and spectroscopically validated, which revealed a localization of the triplet spin density across the substrate alkene carbons in the photoexcited Rh-substrate complex.282 In later work, a novel non-C2-symmetric tris-heteroleptic Rh(III) complex (Λ-225) was also shown to be an efficient asymmetric catalyst for the [2+2] photocycloaddition of related substrates.283
Scheme 89.
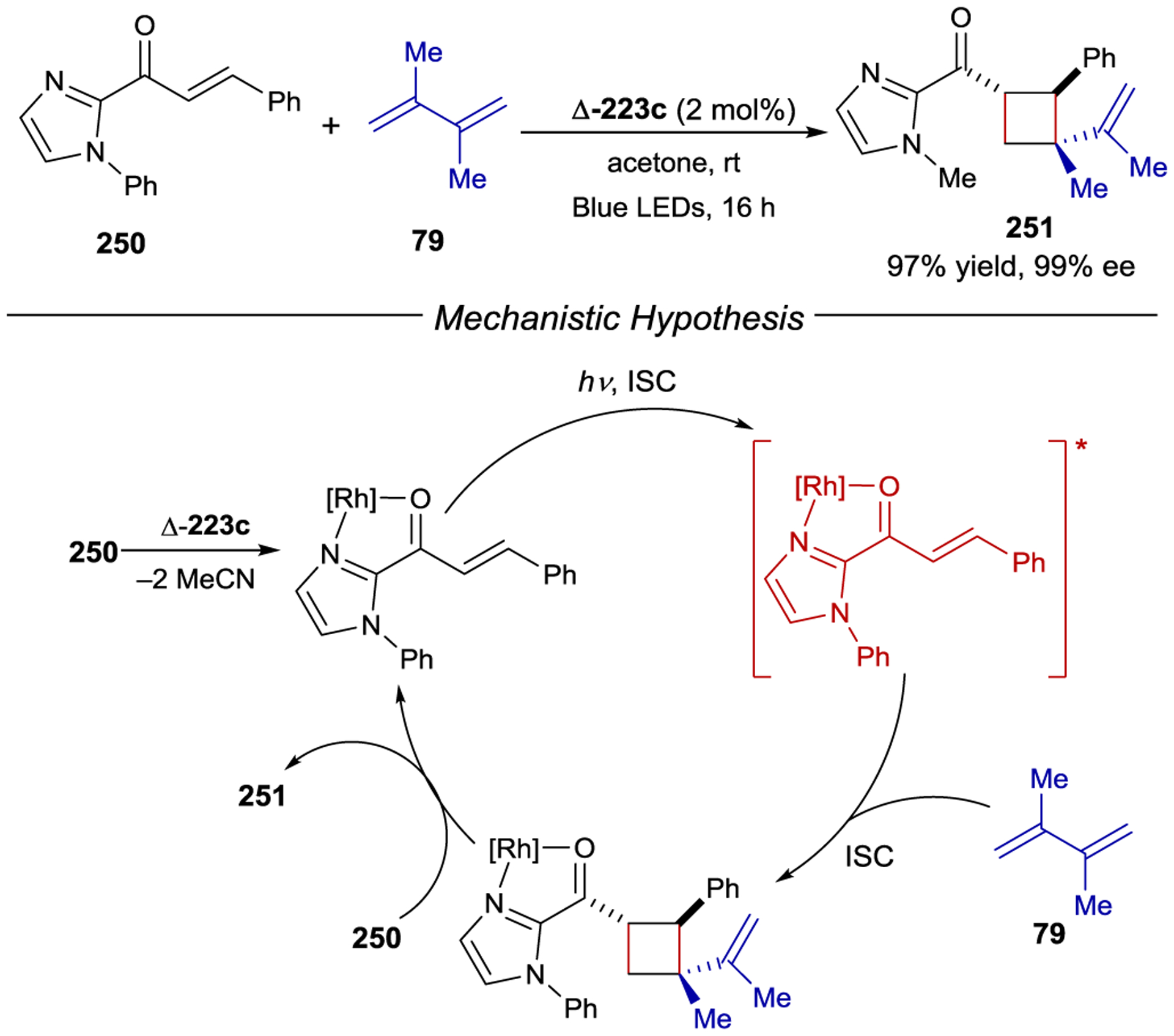
Enantioselective Excited-State [2+2] Cycloaddition with Chiral Rhodium Complexes via Chromophore Activation
This strategy was also applied to the enantioselective dearomative [2+2] photocycloaddition of benzofurans and benzothiofurans (252) with styrenes.284 An N-acylpyrazole moiety was appended at the 2-position of the heteroaromatic substrate to facilitate coordination to the rhodium catalyst. The pyrazole moiety can also be cleaved without eroding the ee of the cycloadduct. Interestingly, this system exclusively produces the head-to-tail regioisomer rather than the tail-to-tail isomer that is commonly formed in the cycloaddition of two styrenic olefins. In collaboration with the Baik group, Meggers reported DFT calculations rationalizing the regiochemistry by considering the relative stability of the two possible regioisomeric 1,4-biradical intermediates. The intermediate of the major product benefits from captodative stabilization by the lone pair of the adjacent oxygen atom and the neighboring carbonyl group, leading to a free energy difference of over 5.0 kcal/mol (Scheme 90).
Scheme 90.
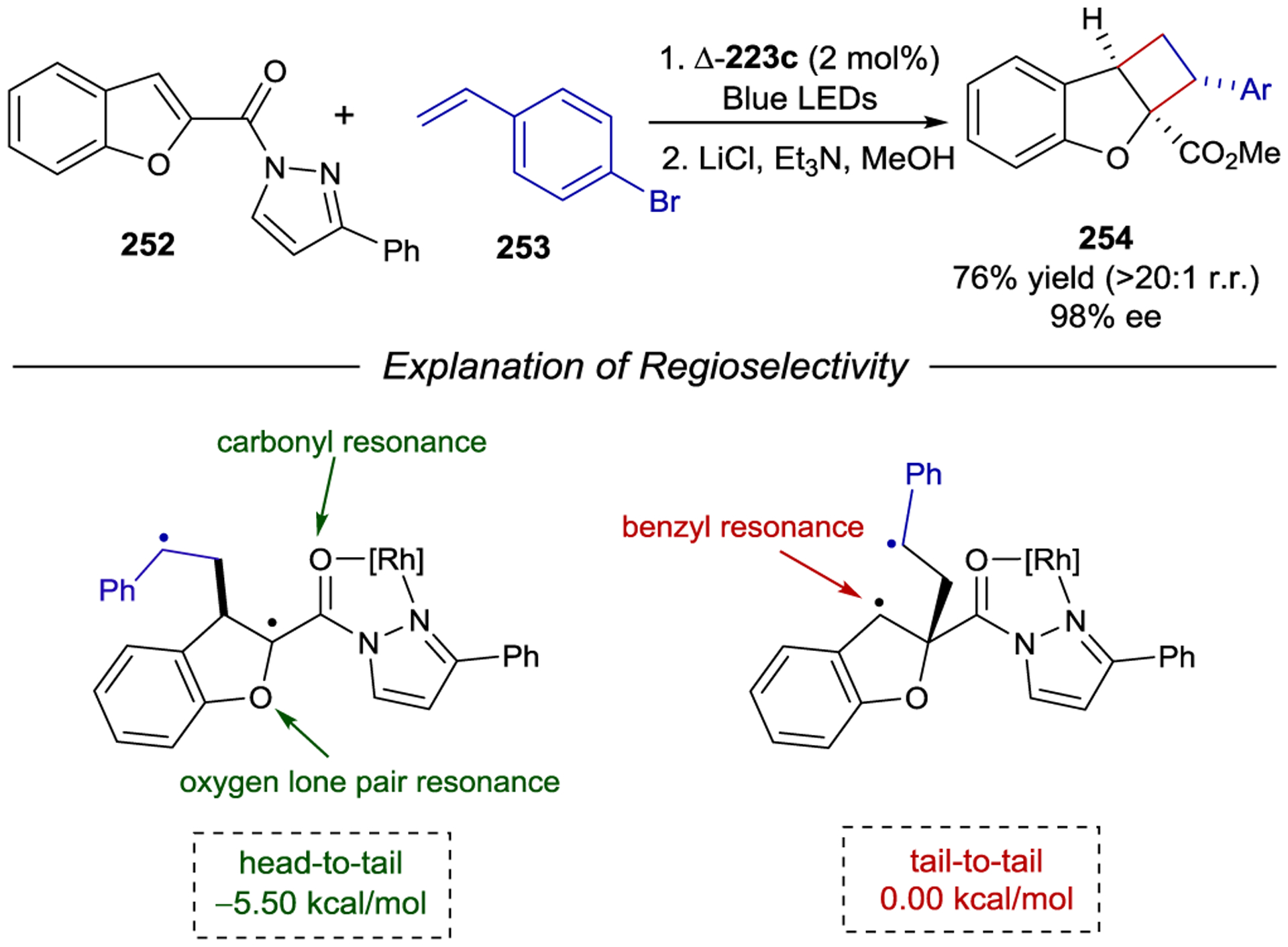
Regio- and Enantioselective Formation of Head-to-Tail Cyclobutanes
Bach recently demonstrated that Λ-223c can influence the stereochemistry of the [2+2] photocycloadditions of 2-benzimidazoyl styryl sulfone 255 (Scheme 91).285 The sulfone substrate binds to the chiral Lewis acid by chelation between the sulfone oxygen and the benzimidazole. During optimization, the authors noted an interesting dependence on the wavelength of the light source used. Longer wavelengths result in low sulfone conversion but higher ee. Irradiation at 420 nm resulted in the best compromise, affording product 256 in 41% yield and 77% ee.
Scheme 91:
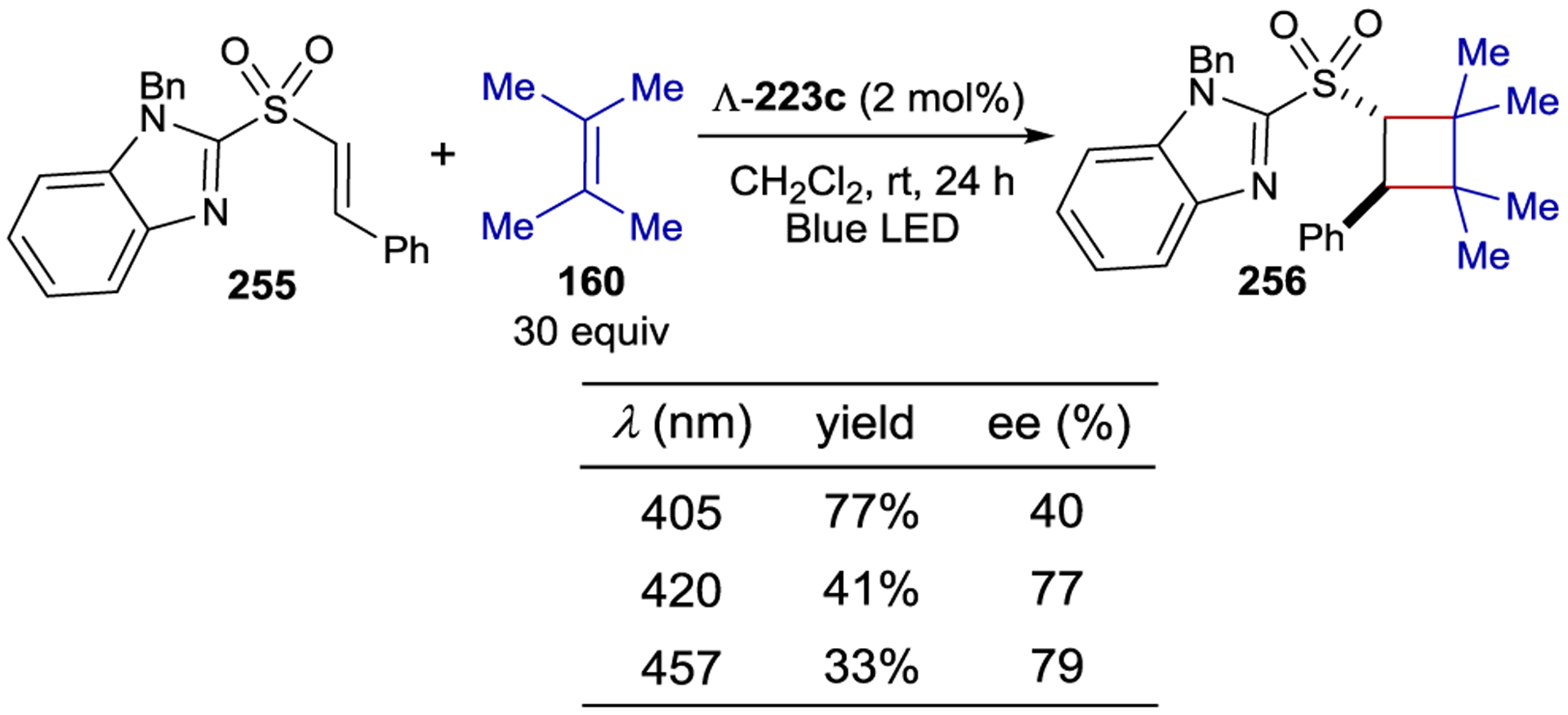
Enantioselective [2+2] Photocycloaddition of α,β-Unsaturated Sulfones
This chromophore activation catalyst is also able to control the stereoselectivity of other classes of excited-state photoreactions. In 2017 the Meggers group reported a catalytic enantioselective synthesis of chiral 1-pyrroline 259 through a [3+2] cycloaddition between N-cinnamoylpyrazole 257 with vinylazide 258 (Scheme 92).286 Mechanistic investigations indicate that the excited-state chiral catalyst–substrate complex reacts directly with ground-state vinylazide, rather than through bimolecular energy transfer, which could result in racemic reactivity. The resulting biradical intermediate undergoes N2 extrusion followed by ring closure to form the enantioenriched product. Interestingly, the chiral Ir(III) analogues are unselective photocatalysts for this cycloaddition. DFT computations indicated that the excited state of the iridium–substrate complex is primarily localized on the metal center rather than the bound alkene. Meggers proposed that the Ir(III) catalyst functions primarily through bimolecular collisional Dexter energy transfer, which sensitizes unbound pyrazole and produces racemic cycloadduct.
Scheme 92:
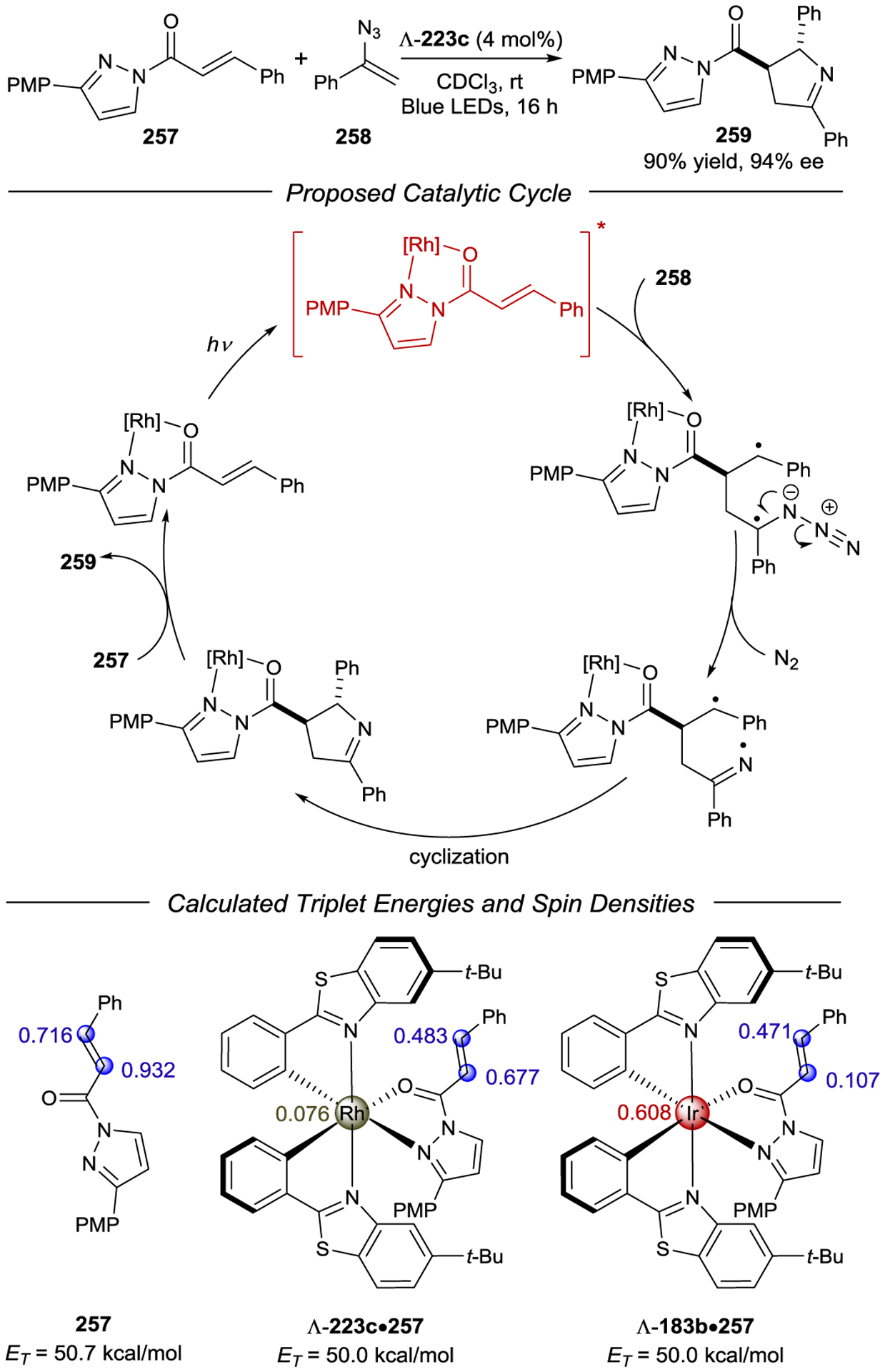
Enantioselective [3+2] Photocycloaddition Between N-Cinnamoylpyrazoles and Vinylazides
In subsequent work, Meggers and Houk reported a unique example of a catalyst–substrate adduct engaging in a visible light-initiated hydrogen atom transfer (HAT) reaction to form a bound radical intermediate (Scheme 93).287 Consistent with previous results, a 2-acylpyrazole auxiliary was necessary for bidentate chelation to the helically chiral rhodium complex. Complexation of the substrate to the metal center constitutes the photoactive species, which upon excitation populates the alkene-centered triplet. This triplet diradical, which is near the aldehydic hydrogen atom, undergoes hydrogen atom transfer and concomitant rearrangement to a ketene intermediate; this intramolecular HAT step is supported by DFT calculations. Finally, the rhodium-bound ground-state ketene engages in a hetero-Diels–Alder cycloaddition with an appended aldehyde to yield fused cyclopropane products in up to 99% ee.
Scheme 93.
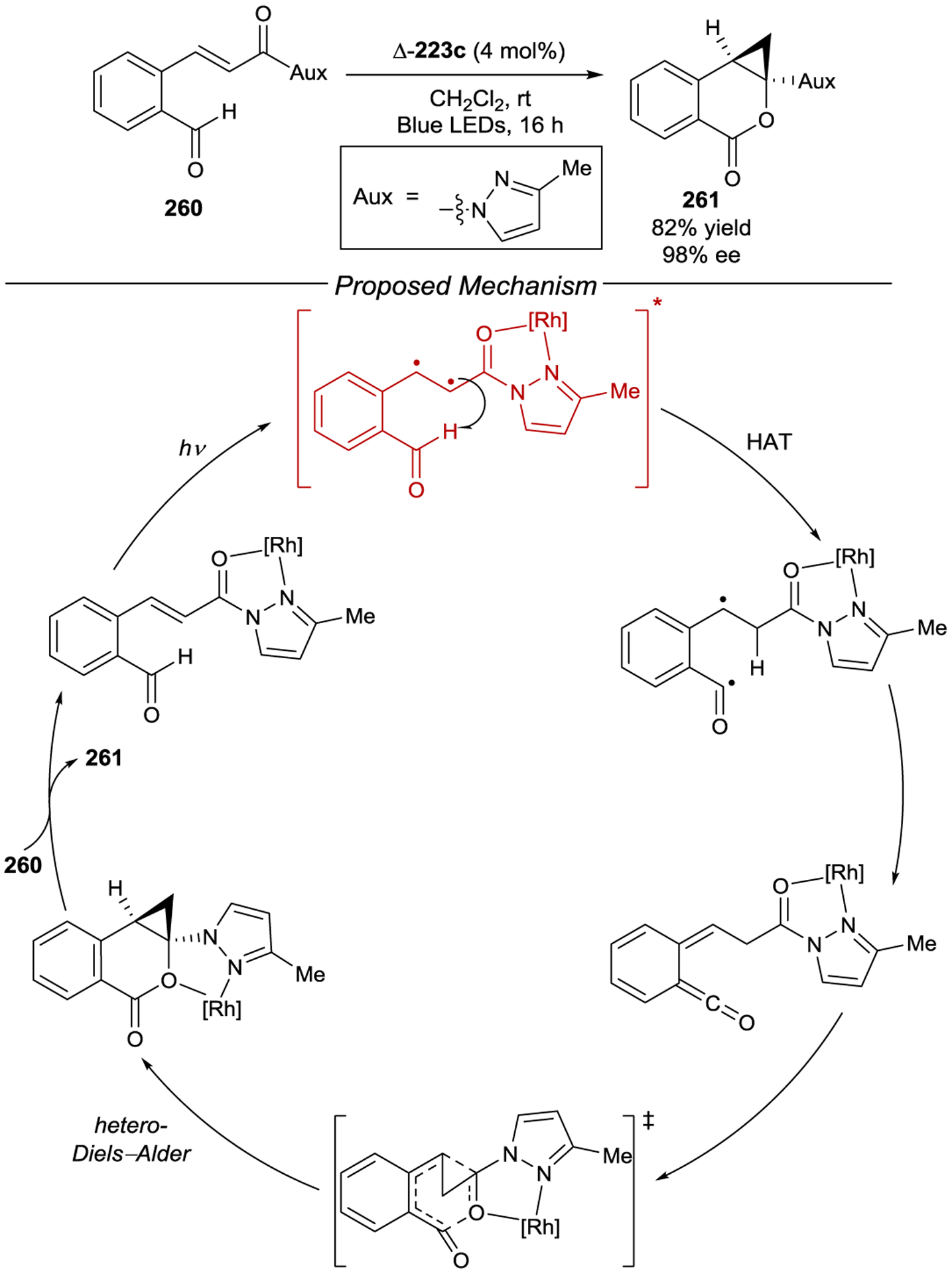
Enantioselective Hetero-Diels–Alder Reaction via Chromophore Activation
3.6. Nickel
The past decade of research in synthetic photochemistry has been dominated by the use of Ru(II) and Ir(III) complexes because of their tunability and attractive photophysical properties. The discovery of photocatalysts with similar efficacy based on less expensive, earth-abundant first-row transition-metal elements is an important challenge. Structurally analogous first-row metal polypyridyl complexes, however, have very different photophysical properties and in general are not effective photocatalysts.288 Hence, there are a few examples of chiral photocatalysts based on first-row transition metals.
In 2016, Xiao described the design of novel chiral BOX ligand 262 in which the central bridging carbon is functionalized with a thioxanthone substituent; the Ni(acac)2 complex of this ligand was used to catalyze the enantioselective oxidation of β-ketoester 139 using oxygen as the terminal oxidant (Scheme 94).289 The thioxanthone moiety acts as a sensitizer for the formation of singlet oxygen, while the Ni(II) center promotes enolization of the β-ketoester substrate and controls the facial selectivity of the C–O bond formation. The absorption and emission spectra of the ligand did not significantly change upon coordination of Ni(II), indicating that the photosensitizer operates independently of the Lewis acidic metal center. Luminescence quenching studies showed no quenching by the complexed substrate but did show a decrease in emission when oxygen was present. The excellent selectivity achieved was attributed to the dynamic steric profile of the covalently attached thioxanthone sensitizer. Interestingly, when the reaction is conducted with separated thioxanthone and Ni(II) BOX catalysts, the yield and ee are diminished, which was attributed to the reduced steric profile of the BOX ligand.
Scheme 94.
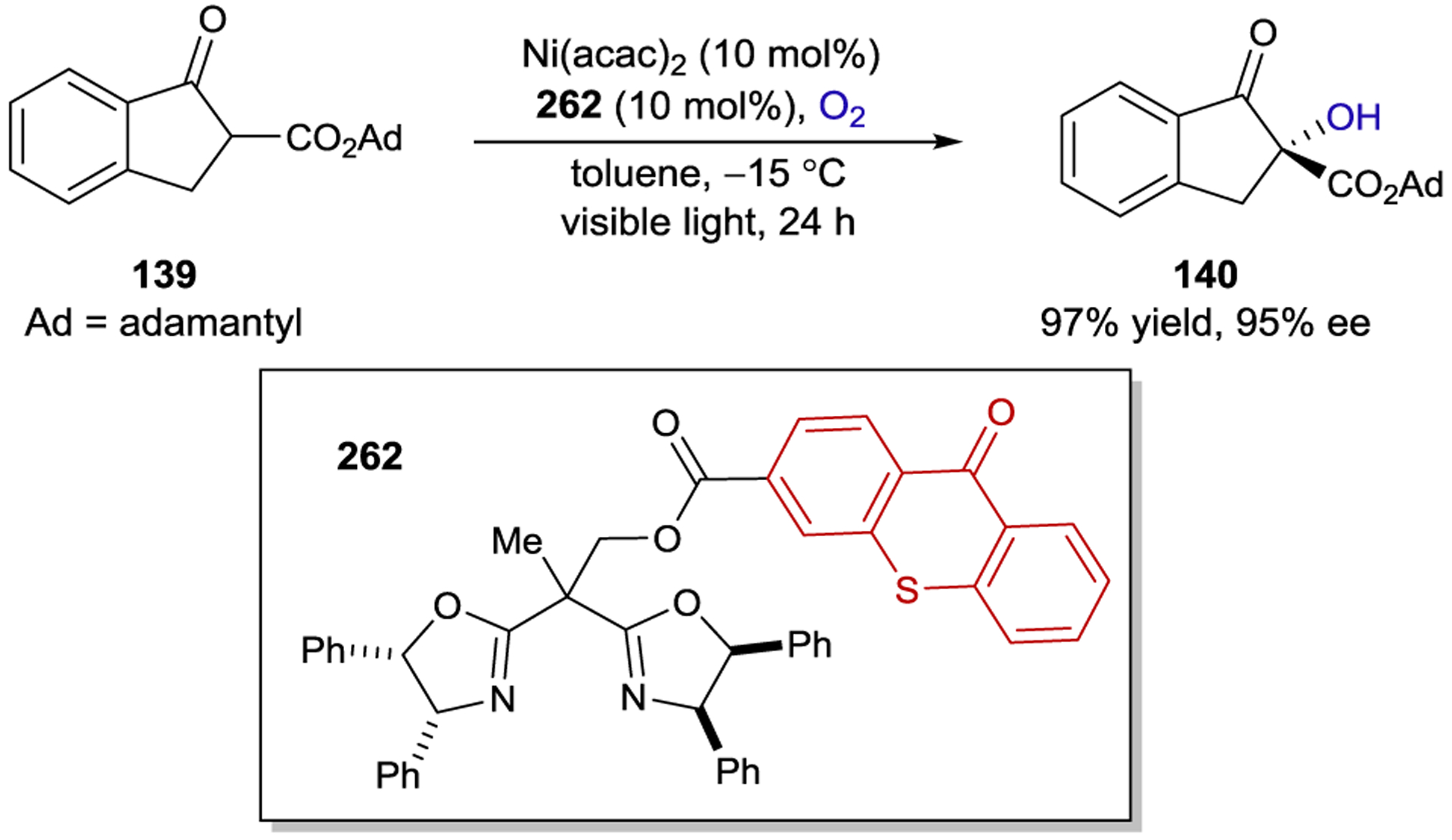
Stereoselective Oxidation of β-Ketoesters by a Thioxanthone-Tethered Nickel Lewis Acid
Shen described the use of a bifunctional Ni(II)-DBFOX complex that serves as both a competent photoredox catalyst and chiral Lewis acid catalyst for the asymmetric addition of α-amino radicals to α,β-unsaturated compounds (Scheme 95).290 The identity of the catalytically relevant photocatalyst was not unambiguously determined but was proposed to either be the Ni(II)-DBFOX complex or its complex with substrate 263, both of which absorbed significantly in the visible range. The luminescence of these complexes was readily quenched by addition of α-silylamine 264, consistent with desilylative photooxidation to generate the key α-amino radical intermediate. While a quantum yield of 0.59 was measured, a radical chain cannot be ruled out based on this reaction’s similarity to other literature.291, 292
Scheme 95.
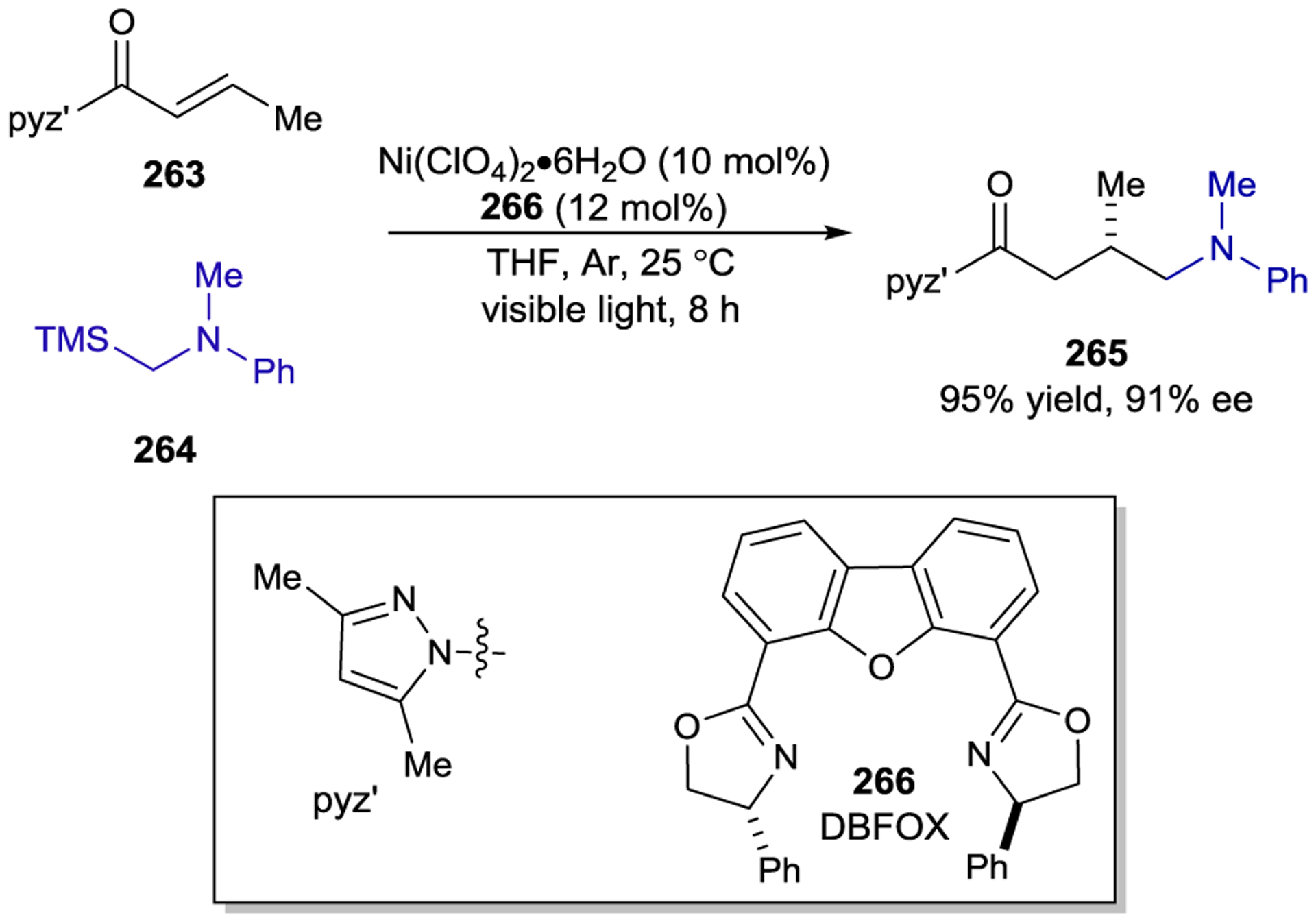
Asymmetric Radical Addition Enabled by Bifunctional Nickel Catalyst
3.7. Copper
Chiral Cu(I) complexes have also been used as asymmetric photocatalysts; however, tetrahedral copper complexes readily undergo fast ligand exchange, which prevents the exploitation of any innate metal-centered chirality.293 Thus, enantiocontrol requires the use of chiral ligands. Ohkubo achieved an enantiodiscriminating photoreduction of the racemic mixture of Λ- and Δ-Co(edta)– using the photocatalyst Cu(dmp)(diop)+ (267), yielding an enantiomer ratio of 1.17:1 (Scheme 96).294 Later studies focused on the use of chiral bpy ligands (268) that position the chiral information closer to the metal center. Under kinetic resolution conditions, 42% ee was obtained at 10% conversion.295 Both the counterion of the Co(II) complex and the edta4− additive had an effect on the outcome of the reaction (Scheme 97).296
Scheme 96.
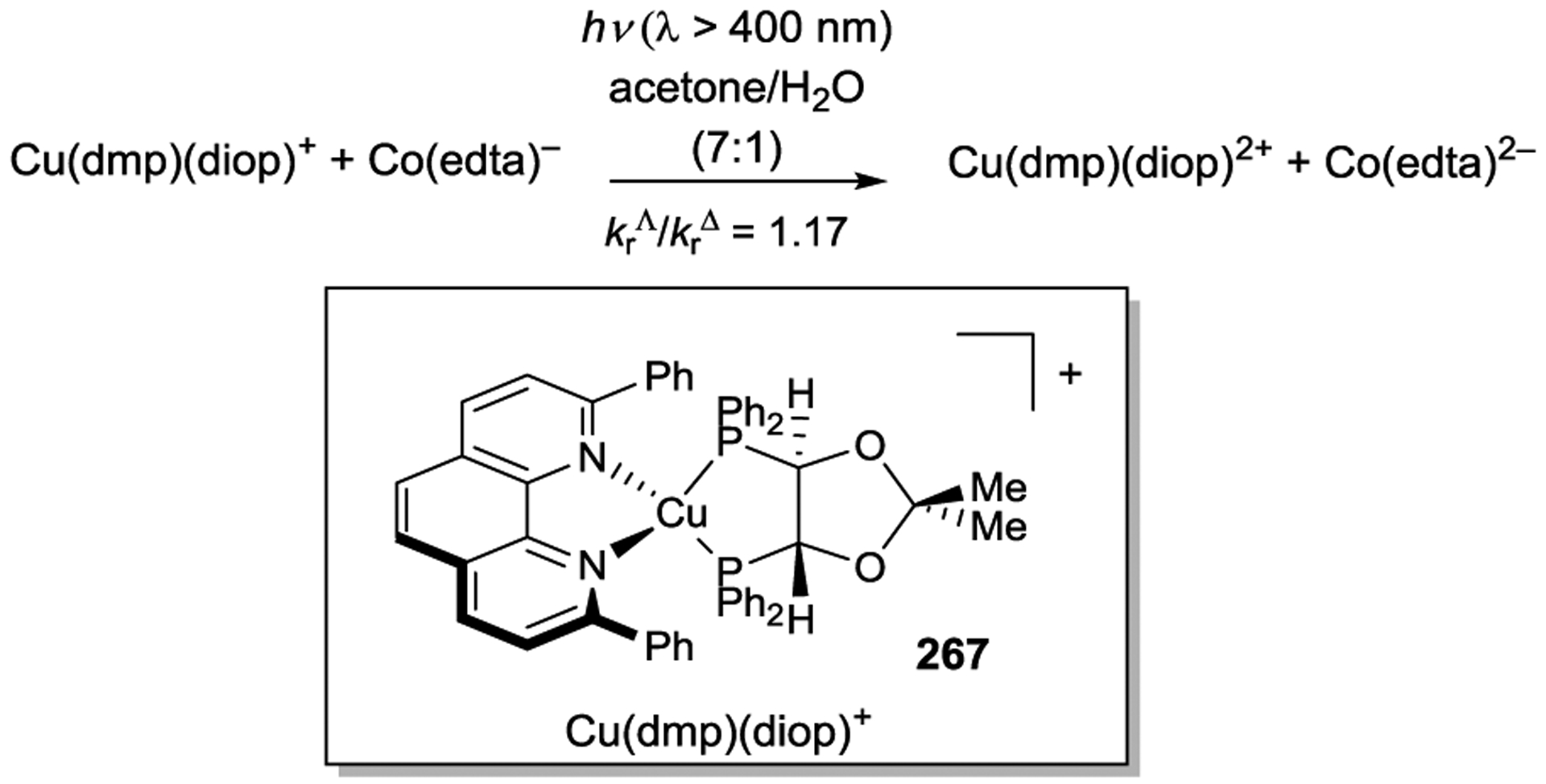
Enantioselective PET Using Chiral Cu(I) Complexes
Scheme 97.
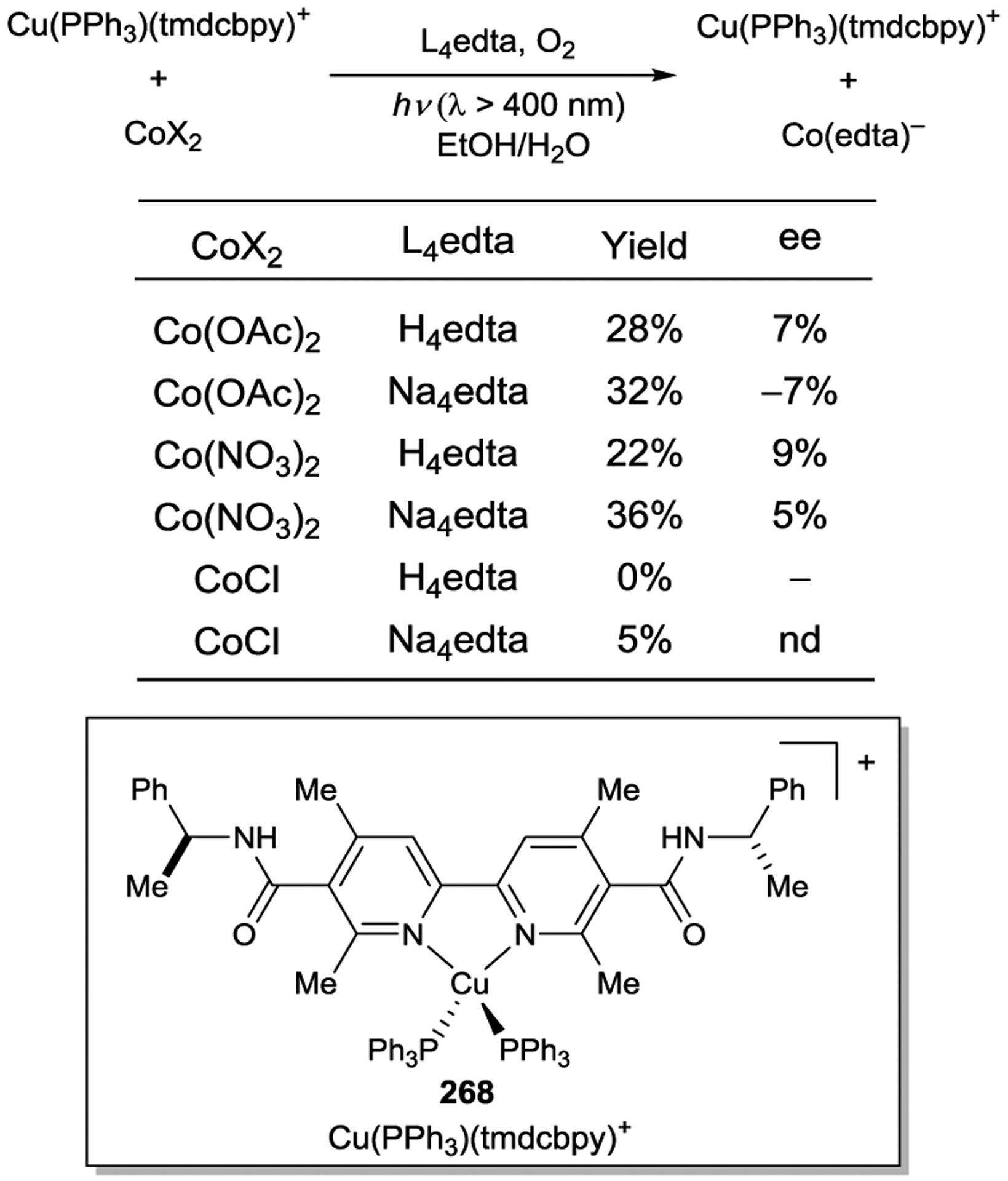
Asymmetric PET to Cobalt Complexes Using Chiral Cu(I) Complexes
Bach observed that a Cu(II) catalyst bearing a chiral BOX ligand could influence the stereochemistry of the 6π-photoelectrocyclization of 2-aryloxycyclohexenone substrate 269 (Scheme 98).297 Consistent with previous examples of chromophore activation, the copper Lewis acid induces a bathochromic shift in the absorption of the substrate, resulting in an intense (π,π*) absorption that overlaps with the weaker (n,π*) absorption of the non-coordinated substrate. The optimized conditions employed 50 mol% Cu(ClO4)2∙6H2O and 60 mol% of C2-symmetric BOX ligand 271, providing the photocyclized product in up to 40% ee.
Scheme 98.
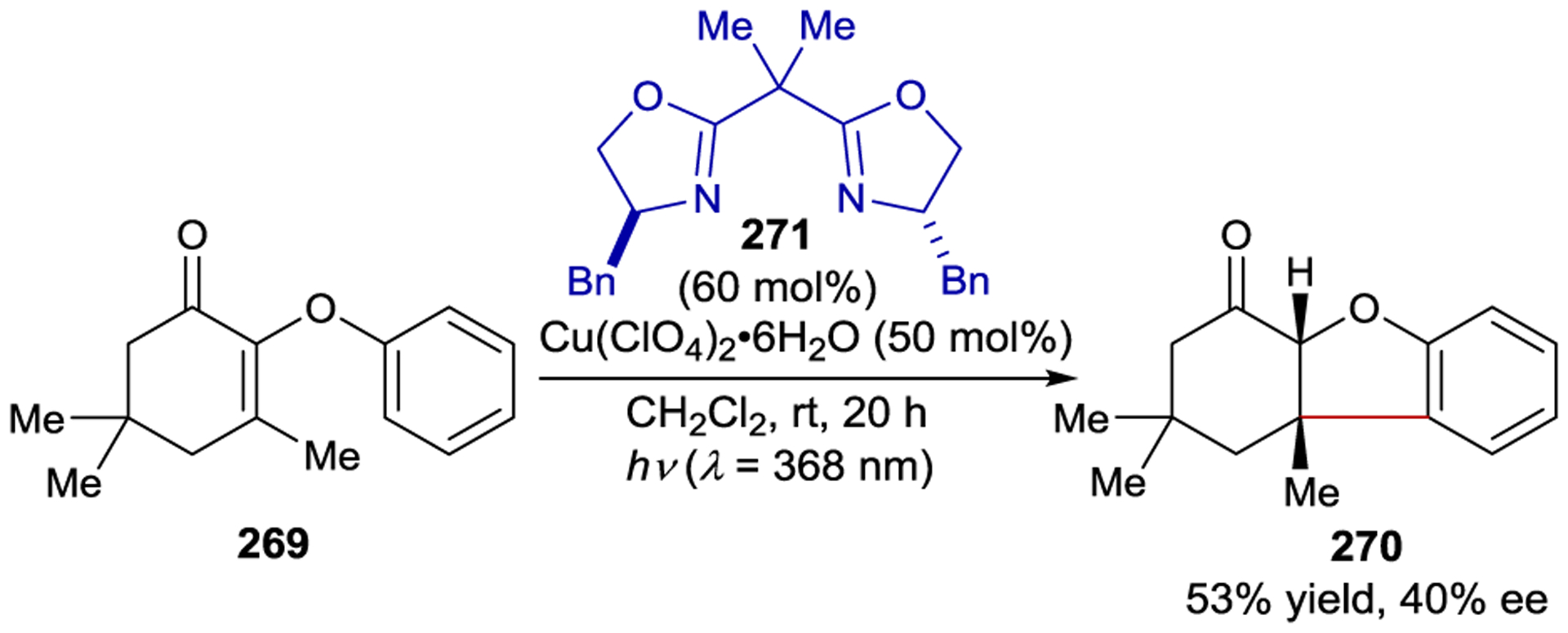
Application of Chromophore Activation to an Enantioselective 6π-Photoelectrocyclization
In 2012 Peters and Fu reported a Cu(I)-carbazolide complex capable of undergoing an ultraviolet light mediated Ullmann C–N coupling with aryl halides via a radical pathway.298 These findings were followed by several reports describing the extension of this chemistry to alkyl halide electrophiles.299–300 ,301 ,302, 303 In 2016, a chiral phosphine–Cu(I) complex was shown to catalyze the stereoconvergent coupling of carbazole 273 to racemic tertiary α-chloroamide 272.304 A kinetic resolution was ruled out because no enantioenrichment of the starting alkyl halide was observed after completion of the reaction (Scheme 99). Later efforts extended this methodology toward the asymmetric amidation of unactivated racemic alkyl chlorides.305
Scheme 99.
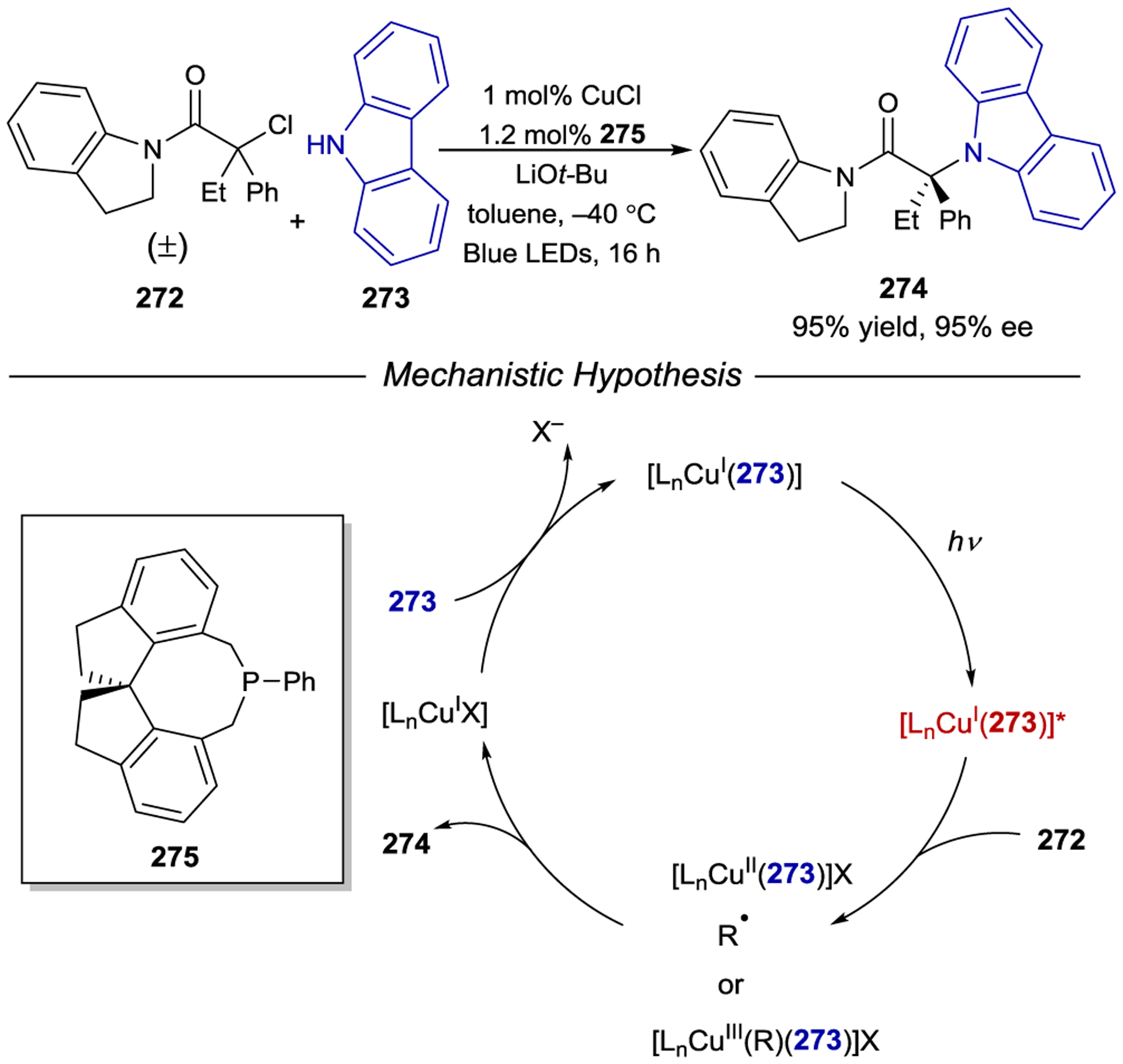
Enantioconvergent Ullman C–N Coupling with a Chiral Phosphine–Cu(I) Catalyst
Xu and Wang reported a photocatalytic protocol for the asymmetric cyanofluoroalkylation of styrenic olefins utilizing a chiral BOX ligand (Scheme 100).306 In contrast to the system developed by Fu and Peters, in this reaction, chiral phosphine ligands slowed product formation and did not result in any enantiomeric enrichment. A combination of UV-Vis and NMR experiments indicated that the active photocatalyst is formed in situ by the coordination of chiral BOX ligand 280 and ligand exchange to afford a (280)CuCN complex. The excited state of this species is a competent reductant (−2.24 V vs SCE) capable of generating the resultant alkyl-radical from the perfluoroalkyl iodide (−1.32 V vs SCE).
Scheme 100.
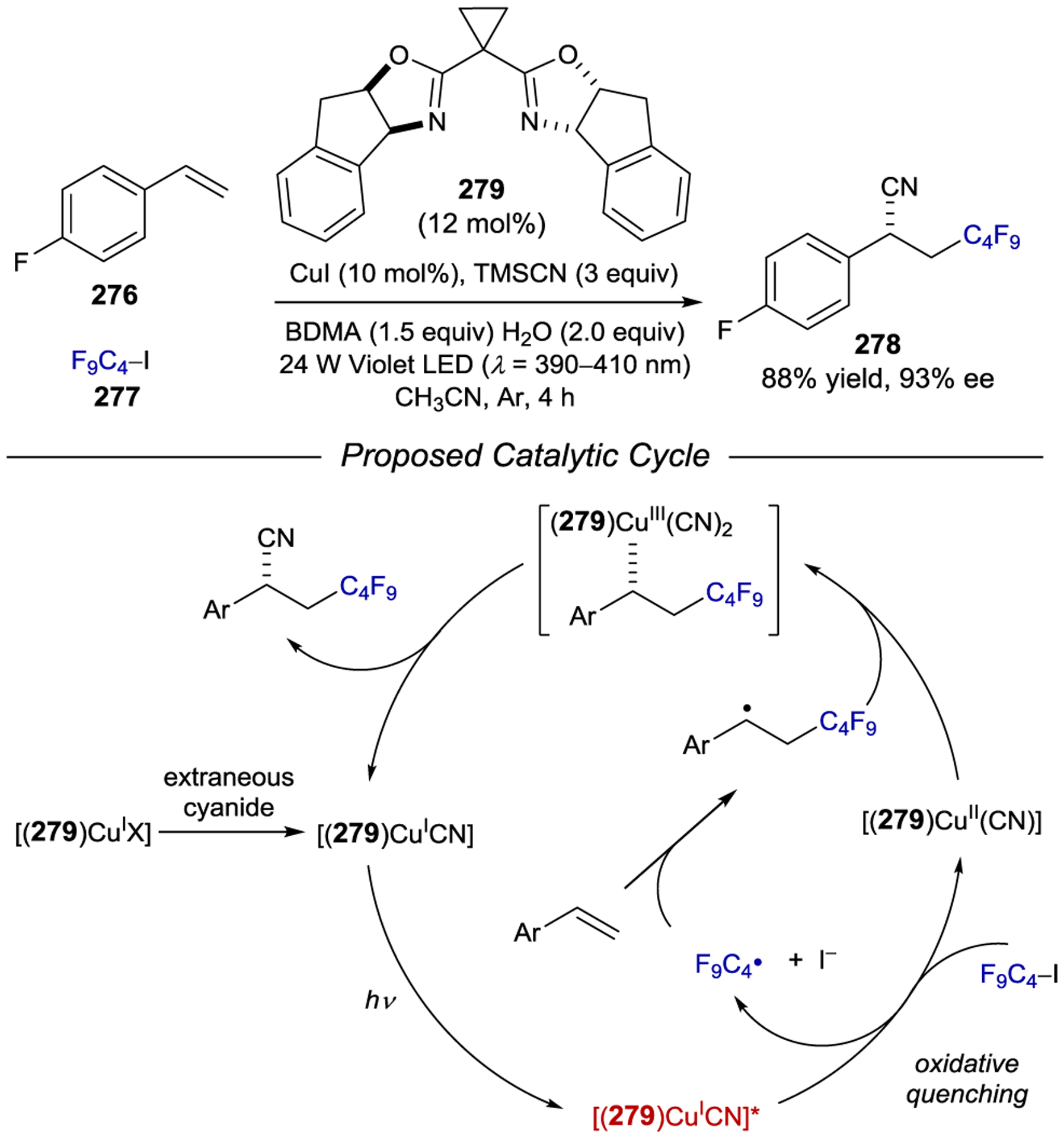
Enantioselective Three-Component Coupling Using a Chiral Copper Photocatalyst
More recently, G. Zhang and D. Zhang described a photoinduced, copper-catalyzed, highly enantioselective dual carbofunctionalization of the same class of alkenes (Scheme 101).307 UV-Vis experiments suggested that the photoactive catalyst is generated from the aggregation of the chiral t-Bu-BOPA ligand 280 and an alkyne nucleophile to the Cu(I)–acetylide species. Moreover, steady-state luminescence quenching indicated that this complex was effectively quenched by the alkyl halide coupling partner.
Scheme 101.
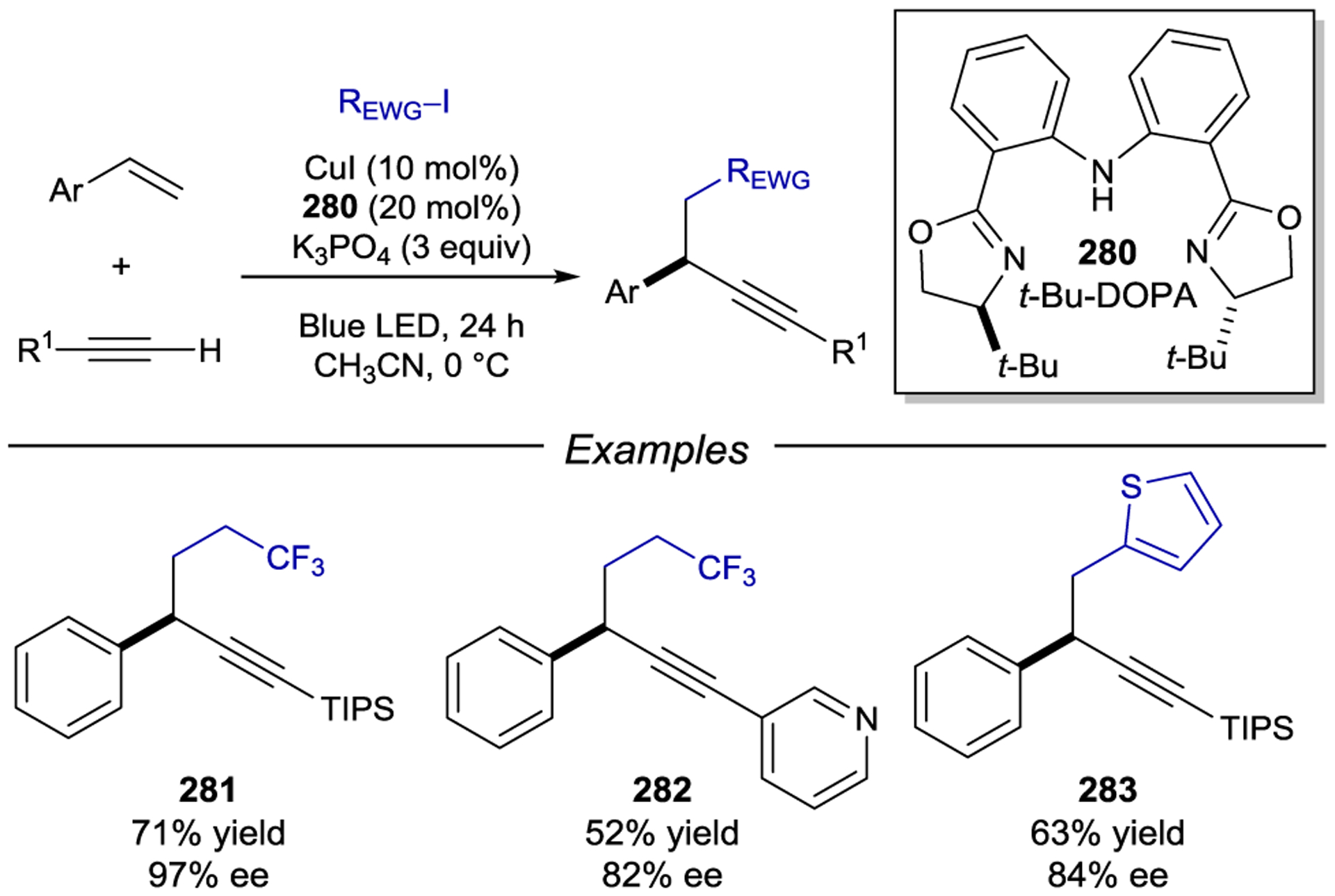
Enantioselective Dual Carbofunctionalization of Styrenes
Liu reported a related approach for the asymmetric alkynylation of N-hydroxypthalimide (NHP)-type esters using a Cu(I)–acetylide complex ligated by cinchona alkaloid-derived N,N,P-ligand 287 as the photoreductant (Scheme 102).308 Critical to their success was the use of naphthyl-NHP ester 284 to suppress the formation of the Glaser homocoupling product of terminal alkyne 285 and radical dimerization of 284. The authors proposed this effect results from a decreased rate of quenching of the excited-state copper complex by 284 compared to commonly employed NHP esters. Finally, stoichiometric experiments demonstrated that in the absence of chiral ligand 287 only 10% of product 286 is formed, indicating that the ligand significantly improves the photocatalytic efficiency of the transformation.
Scheme 102.
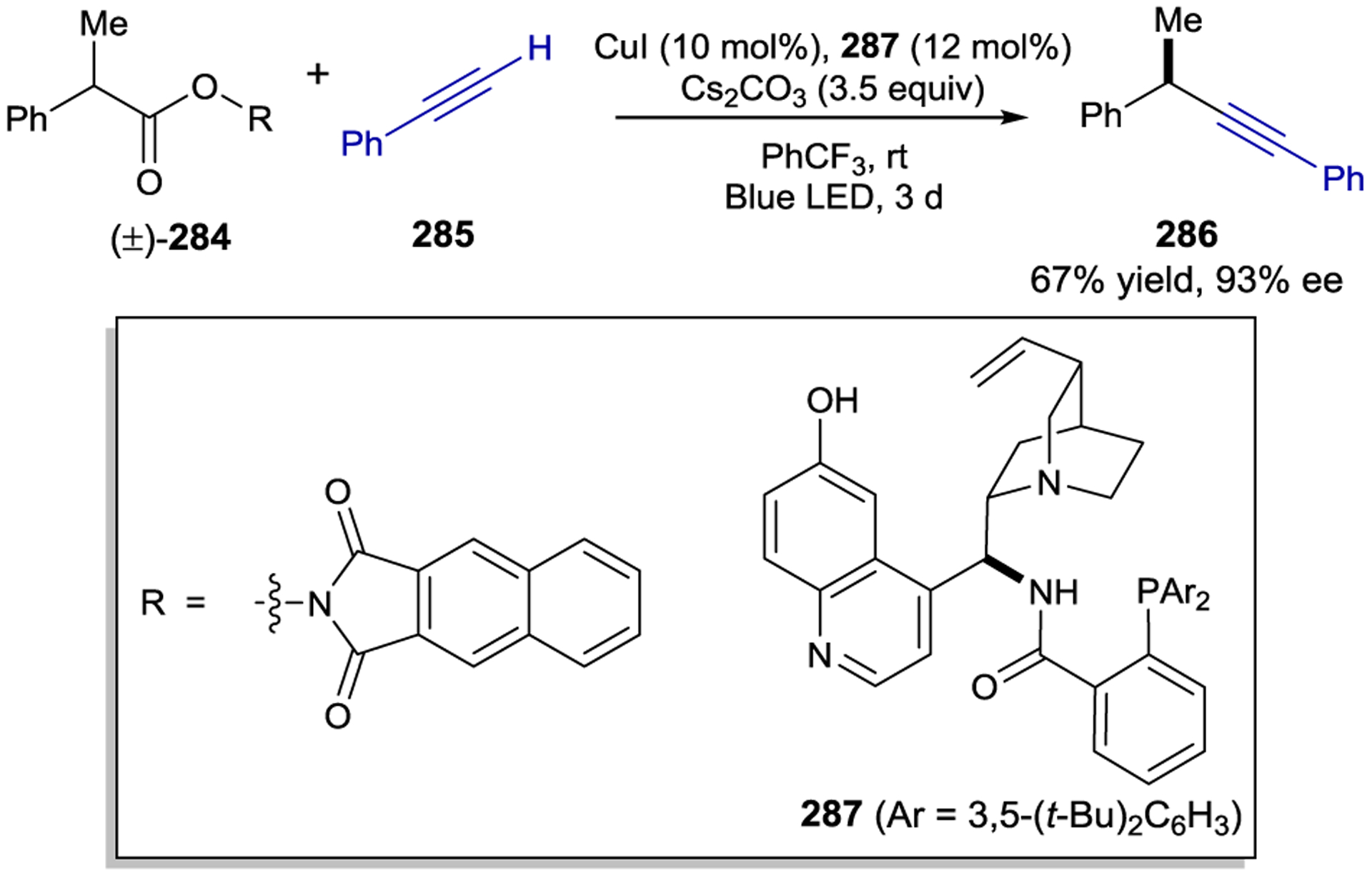
Asymmetric Decarboxylative Alkynylation with Terminal Alkynes
Subsequently, Xu and Wang reported the enantioselective C(sp3)-alkylation of quinolinyl-appended glycinate ester 288 utilizing NHP ester 289 as an alkyl radical precursor (Scheme 103).309 Similar to the previously discussed examples, coordination of substrate 288 to a chiral phosphine–Cu(I) complex results in the in situ generation of the photoactive catalyst. Oxidative quenching by ester 289 to form the corresponding alkyl radical is thermodynamically feasible. Under optimized conditions the cross-coupled product is produced in 88% yield and 94% ee.
Scheme 103.
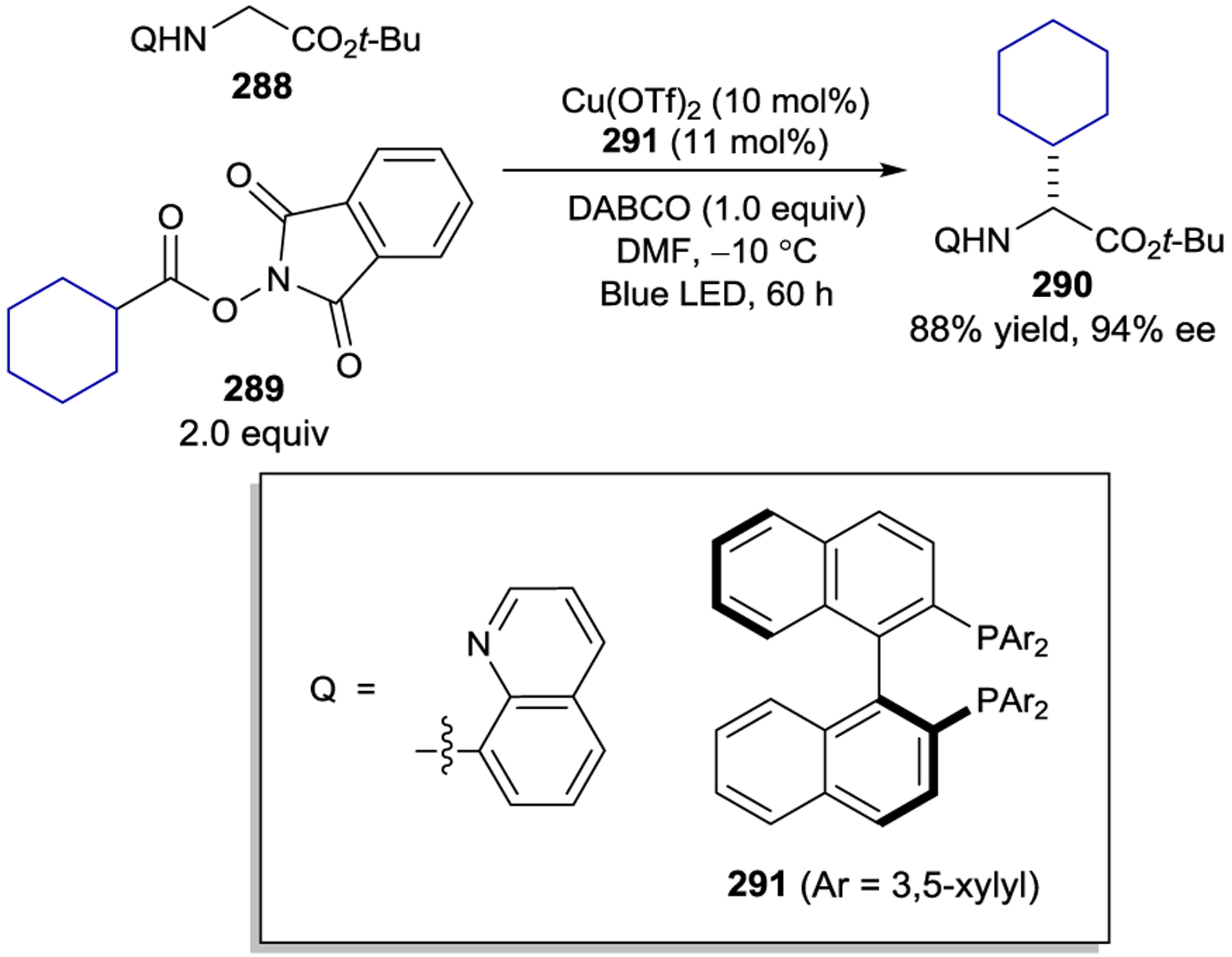
Visible Light Induced Enantioselective C(sp3)-alkylation of Glycine Ester Derivatives
In 2018, Gong reported that a Cu(II) complex ligated by BOX ligand 295 enables the photoinduced alkylation of imine 292 to afford enantioenriched tetrasubstituted carbon centers in up to 98% ee (Scheme 104).310 Mechanistically, the authors suggest a fast transmetallation to form a Cu(II)–alkyl complex that is proposed to undergo a ligand-to-metal charge transfer (LMCT) photoexcitation and subsequent homolysis to liberate the alkyl radical. This nucleophilic radical undergoes addition to the coordinated imine substrate, which produces a nitrogen-centered radical that is stabilized by Cu(II). This species can be reduced by the Cu(I)–295 complex.
Scheme 104.
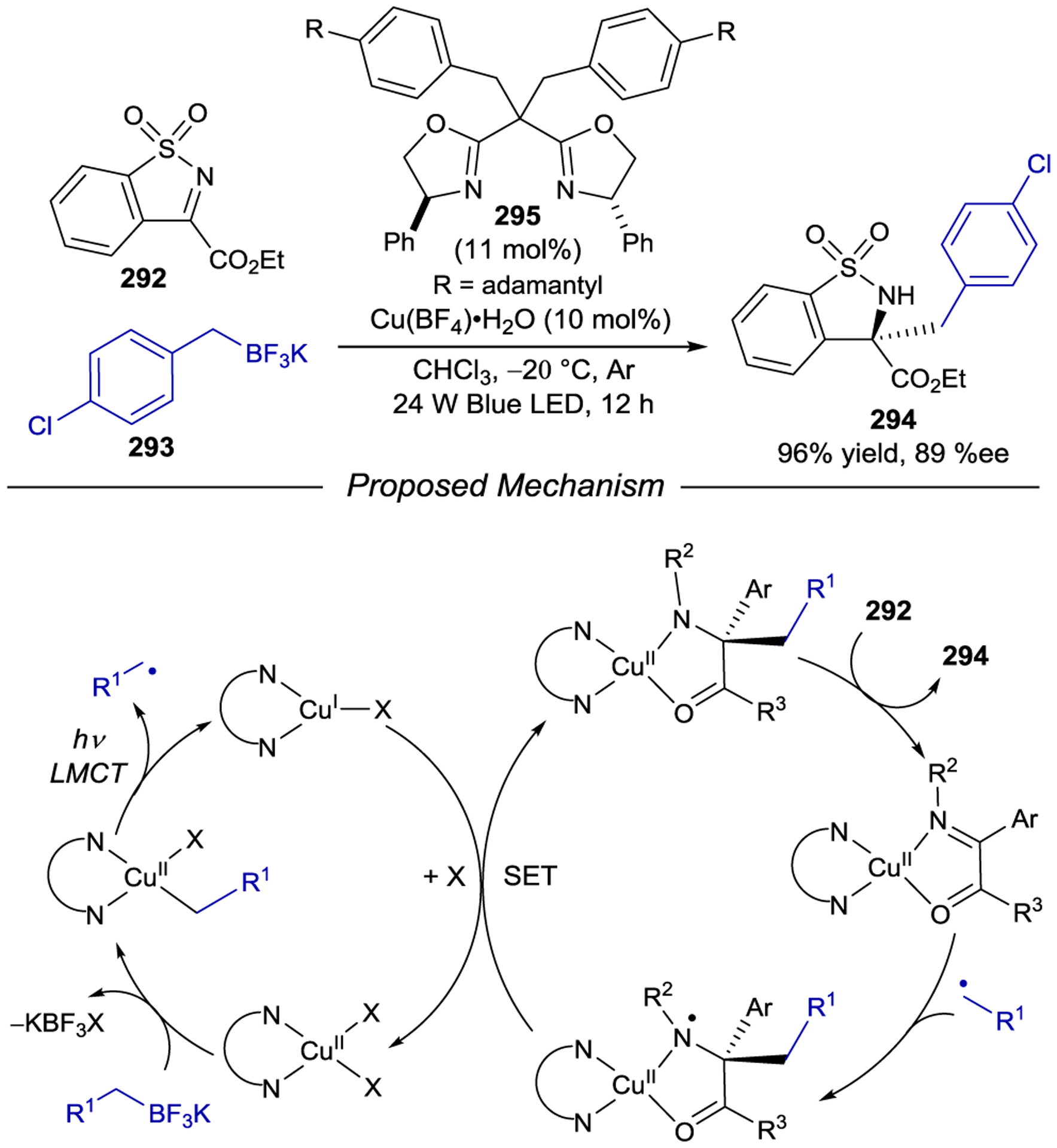
Stereoselective Radical Addition Using a Dual-Purpose Copper Photocatalyst
Following this study, Gong reported that chiral Cu(II)–BOX complexes enable the related addition of α-amino radicals to acyclic imine derivative 296 to afford highly desired enantioenriched vicinal diamine compounds (298) (Scheme 105).311 While the Cu(II)–BOX complex acts as both a photoredox catalyst and Lewis acid producing the alkyl radical and coordinating the prochiral imine, the mechanism of this reaction differs from the previous example because the α-TMS aniline radical precursor is proposed to be directly oxidized by a PET process, rather than by transmetallation and LMCT homolysis.
Scheme 105.
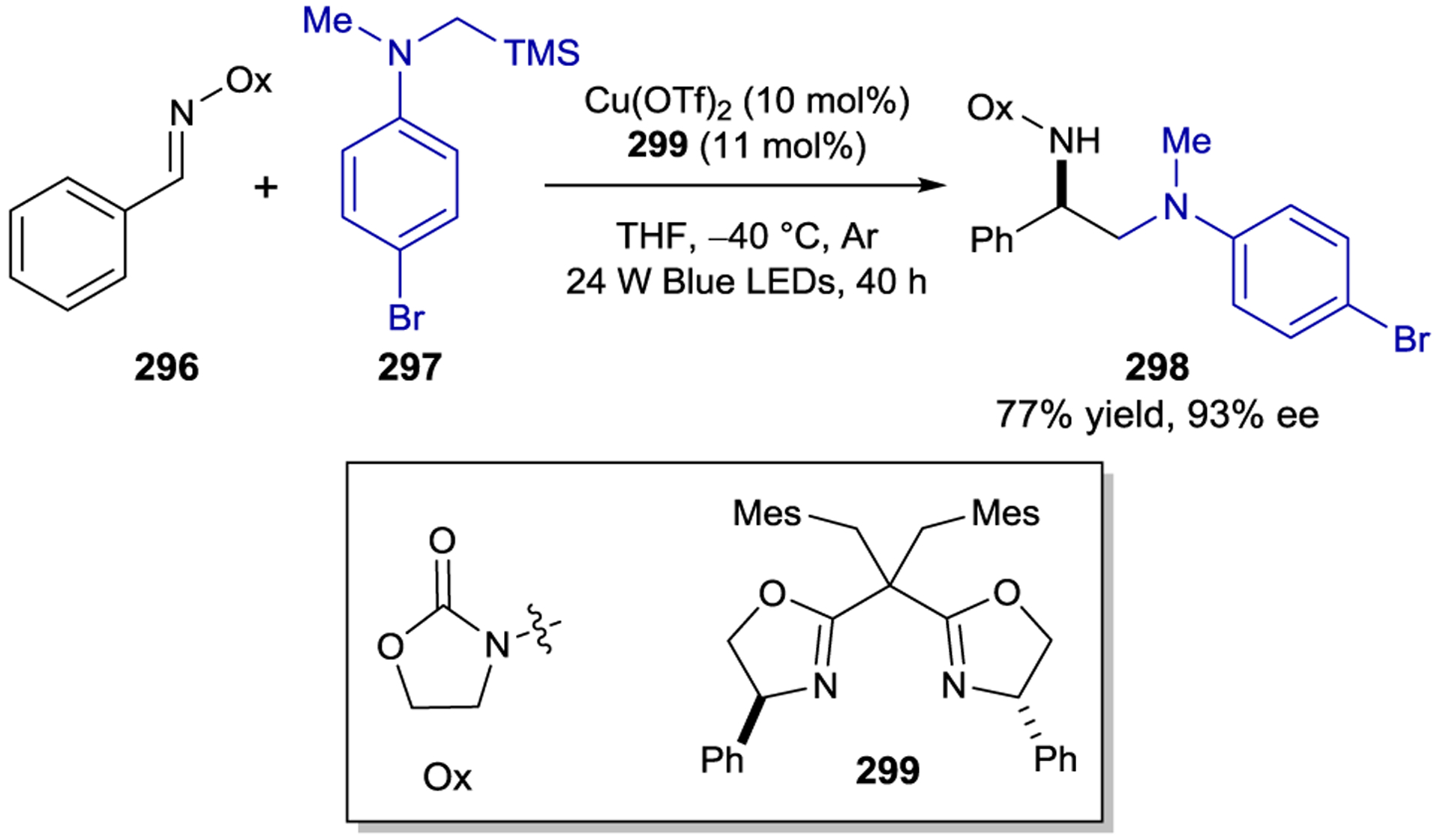
Stereoselective Radical Addition into Imines by a Bifunctional Copper Catalyst.
In later work, Gong extended this methodology to the asymmetric cross-dehydrogenative coupling of 2-acylimidazole 300 with xanthene derivatives, utilizing O2 as a terminal oxidant, to produce 302 in good yield and excellent enantioselectivity (Scheme 106).312 Electrochemical and spectroscopic experiments revealed the Cu(II)–BOX complex as the photoactive species, initiating the reaction via oxidation of xanthene 301. Furthermore, this complex serves to preorganize 300 resulting in efficient stereofacial bias.
Scheme 106.
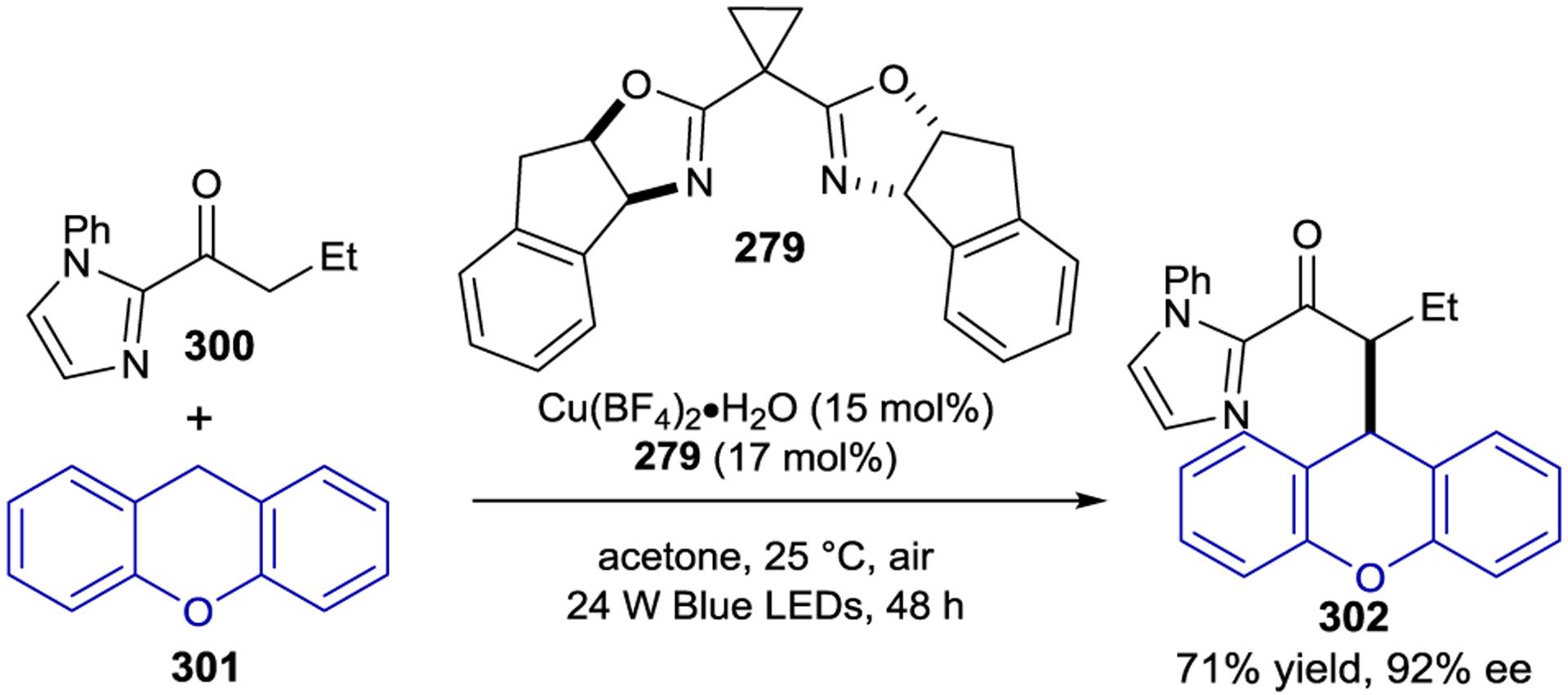
Asymmetric Aerobic Cross-Dehydrogenative Coupling
More recently, Chen, Xiao, and Guan reported an enantioselective C–O cross-coupling of 1,3-dienes and readily available oxime esters catalyzed by Cu(I)–BOX under irradiation with purple light (Scheme 107).313 This photocatalyst serves a dual role in generating an alkyl radical via reduction of oxime ester 303 and as the source of asymmetric induction in the C–O bond formation. Interestingly, this reaction heavily favors 1,2-addition rather than 1,4-substitution. DFT computations revealed a thermodynamic and kinetic preference for the formation of a π-allylcopper complex over the competing π-benzylcopper complex, leading to the experimentally observed 1,2-adduct.
Scheme 107.
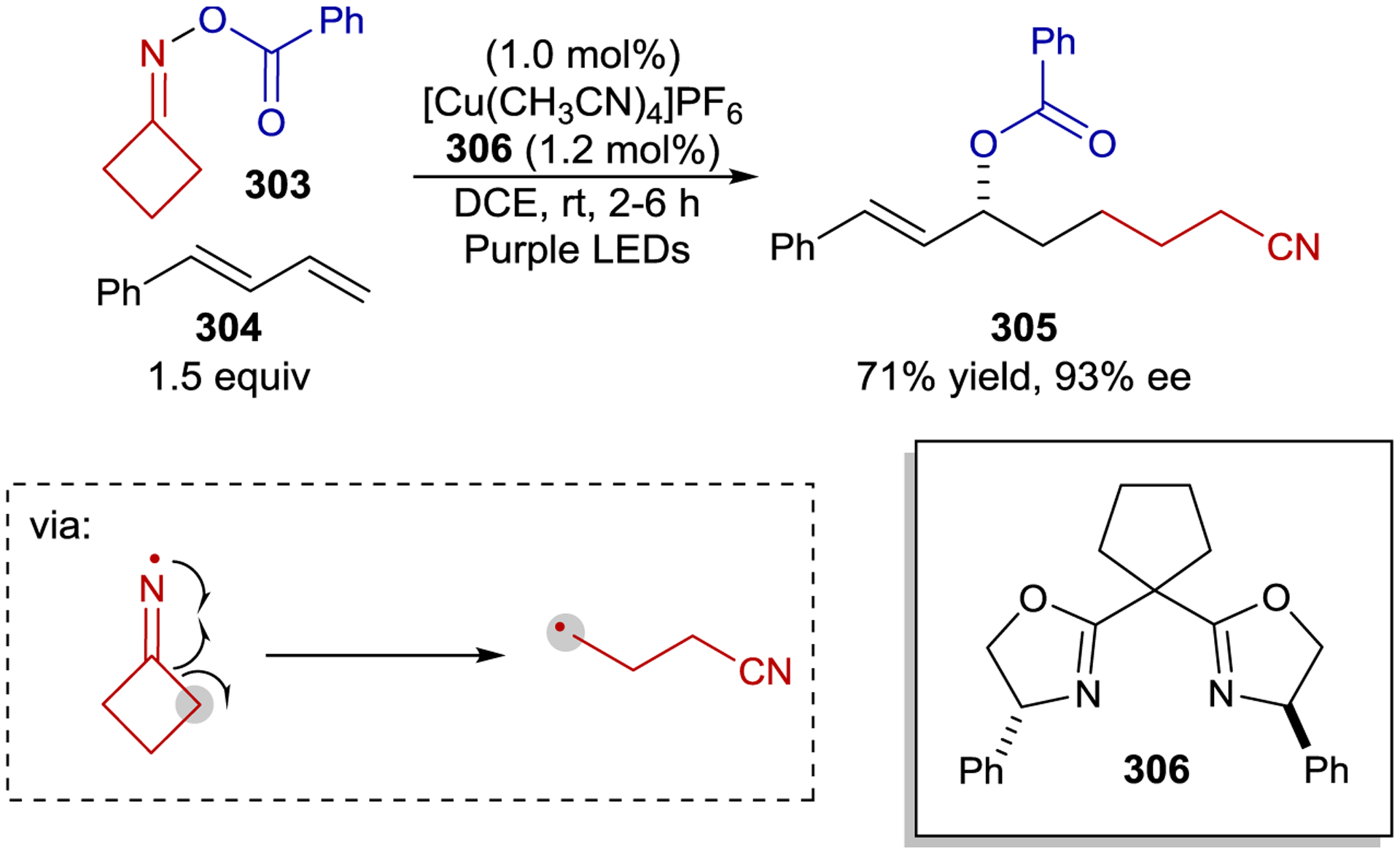
Photoinduced Copper-Catalyzed Asymmetric C–O Cross-Coupling
4. Chiral Macromolecular Photocatalysts
When chiral small organic molecules, organometallic compounds, and transition metal complexes are employed as asymmetric photocatalysts, the reliance on hydrogen bonds, electrostatic interactions, or EDA interactions to direct catalyst-substrate preassociation often necessitates the use of relatively nonpolar organic solvents. Polar protic solvents that weaken these non-covalent interactions often result in lower enantioselectivities.94,95, 314–315 316 An alternate strategy that has been studied for asymmetric photocatalysis involves the use of macromolecular systems that encapsulate (or include) small organic molecules and subsequently alter the outcomes of their reactions. Several excellent reviews on this topic have been published.15,22, 317–318 319
The inclusion of an organic substrate (often referred to as a guest) within the cavity of a macromolecule (host) is primarily driven by hydrophobic interactions in aqueous and other polar protic solvents. A chiral host can influence the stereochemistry of a photochemical reaction that occurs within it; Scheme 108 outlines a generic asymmetric macromolecular photocatalytic mechanism.12, 320–321 322 323 Inclusion of an organic molecule within the macromolecular cavity provides a ground-state association complex. Photoexcitation can either occur through excitation of the macromolecular host followed by electron or energy transfer to the substrate or via direct excitation of the host-bound substrate. Non-covalent interactions between the host environment and the activated substrate can subsequently influence the stereochemistry of the resulting photoreaction.
Scheme 108.
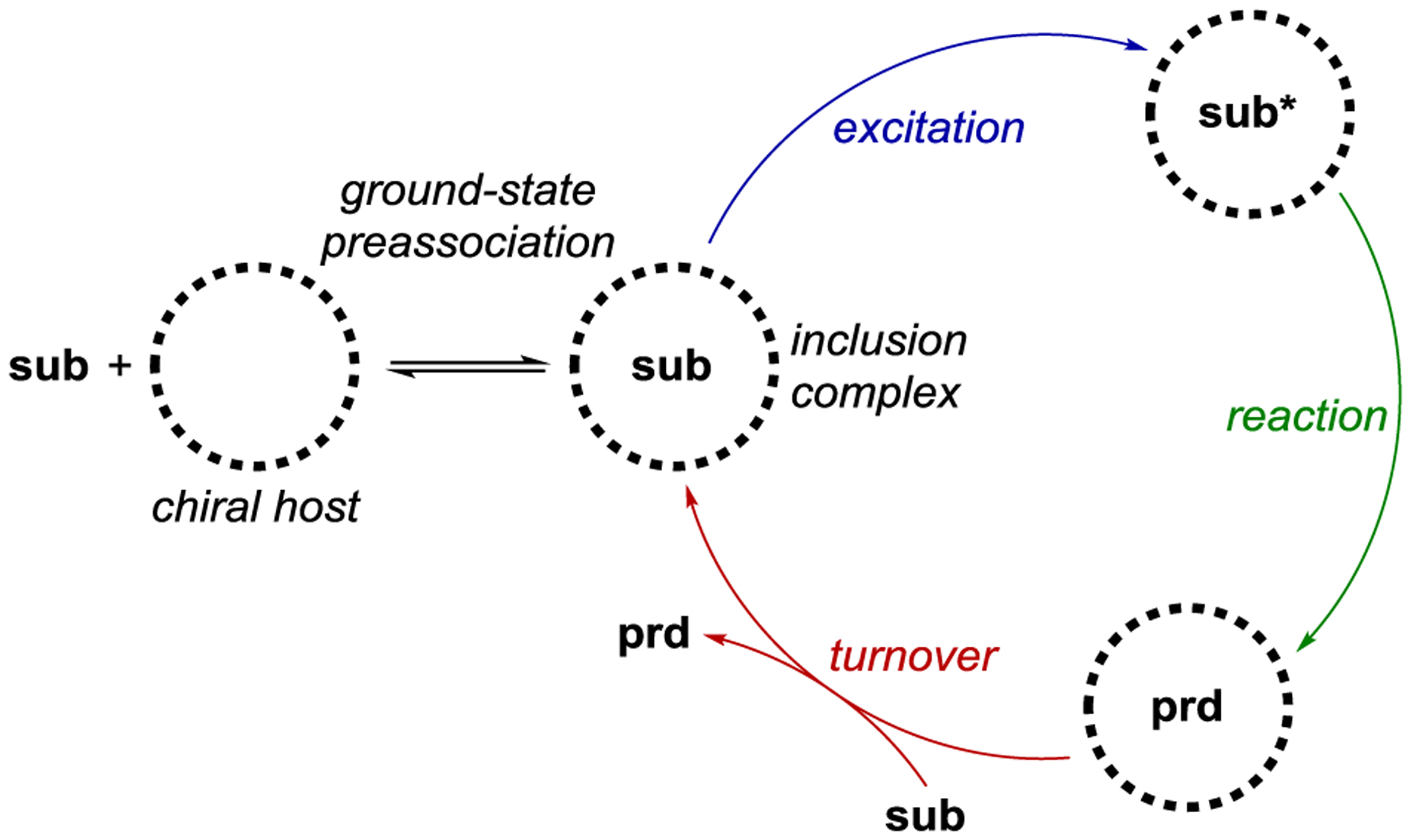
General Mechanism for Asymmetric Photocatalysis by Macromolecular Hosts
4.1. Cyclodextrins
Cyclodextrins (CDs) are cyclic oligosaccharides consisting of ᴅ-glucose subunits linked through α-1,4 glycosidic bonds. The three most common CDs (αCD, βCD, and γCD) referred to as native CDs, are readily isolated from the degradation of starch and differ solely in the number of glucose monomers (Scheme 109).324 These large macrocyclic rings feature hydrophilic primary and secondary faces and a hydrophobic cavity defined by the organic hexose rings (Scheme 109). A network of intramolecular hydrogen bonds among the secondary face hydroxyls rigidifies its conical three-dimensional structure. Organic guest compounds within CDs are typically positioned with their nonpolar constituents within the hydrophobic core, while polar or charged functionalities reside outside, stabilized by the hydrophilic faces.325 Thus, CDs can selectively encapsulate specific components of a complex reaction mixture and geometrically organize them relative to the intrinsic chirality of the glucose moieties.
Scheme 109.
Structure and Nomenclature of Native CDs
4.1.1. Native Cyclodextrins
In 1984, Tamaki studied the photodimerization of substituted anthracenes within native α-, β-, and γCD (307, 308, and 309 respectively).326, 327 Previous studies on naphthalene complexation by 308328 and the observation that a thermal Diels–Alder reaction occurs at an accelerated rate within 308329 inspired the investigation of the [4+4] photodimerization of anthracene within these hosts (Scheme 110). Water-soluble 1- and 2-substituted anthracenes readily form ground-state inclusion complexes with β- and γCD. No interaction was observed between the smaller αCD and the anthracene derivatives. The stoichiometry of the inclusion complexes is influenced by the size of the host (Scheme 111); βCD preferentially forms a ground-state 1:1 CD:anthracene complex at low anthracene concentrations. Notably, the 1:1 complex is proposed to be photochemically unproductive. Photodimerization mediated by βCD instead requires the formation of a higher-order 2:2 CD:anthracene complex at higher anthracene and βCD concentration. In contrast, γCD favors the formation of a 1:2 CD:anthracene complex; here, the association of a second anthracene molecule is highly favored, featuring an association constant a few orders of magnitude larger than the first association.
Scheme 110.
[4+4] Photodimerization of Substituted Anthracenes
Scheme 111.
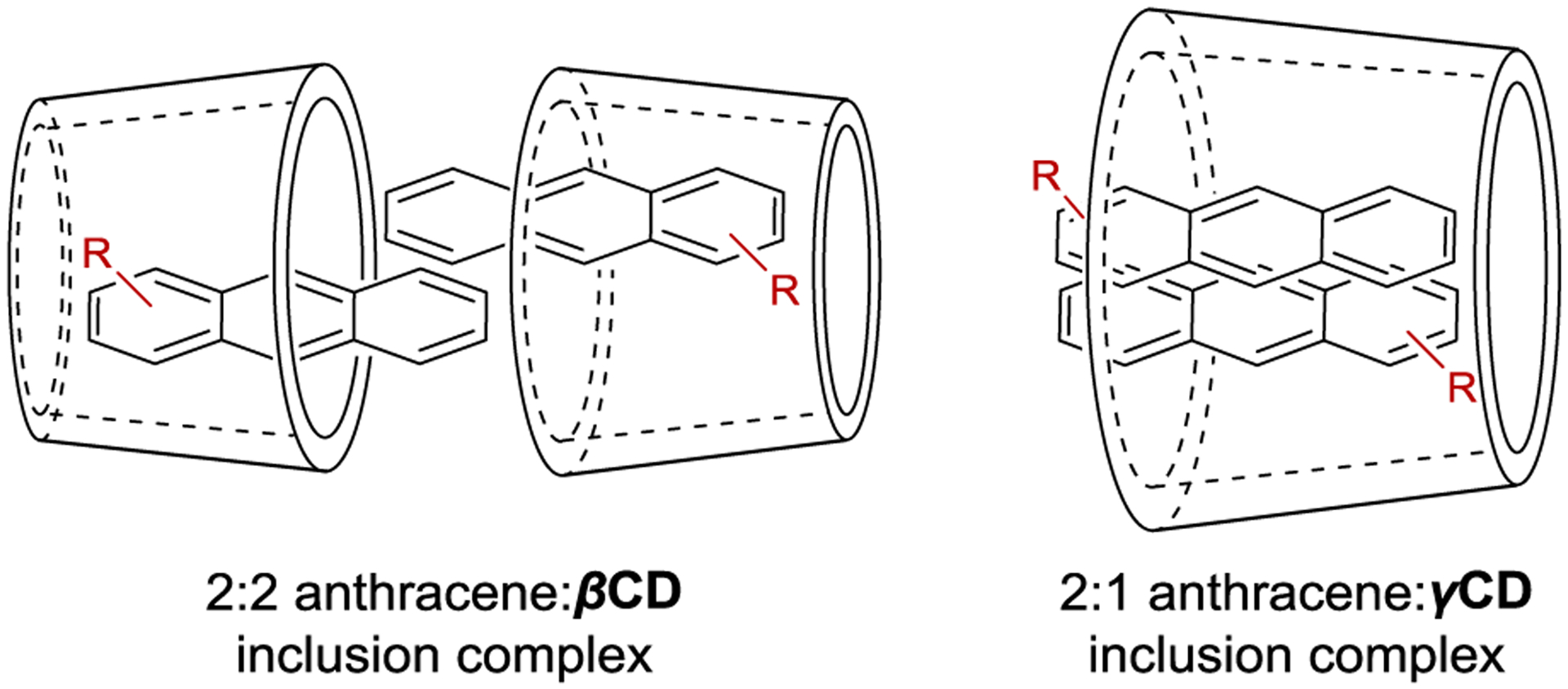
Inclusion Complexes of Substituted Anthracenes and βCD or γCD
Under direct irradiation, the substituted anthracenes can dimerize to produce four possible regioisomers: the syn- and anti-head-to-tail (HT) and head-to-head (HH) dimers (Scheme 110). Of these, the syn-HT and anti-HH are chiral. When the photoreaction is conducted in the presence of superstoichiometric γCD, the dimerization rate of 2-anthracenesulfonate (310) or 2-anthracenecarboxylate (311) is significantly increased while the ratio of the four isomers is unchanged (Scheme 112).326 This rate increase was attributed to an order of magnitude increase in the dimerization quantum yield for anthracene included within γCD, as inclusion both removes the need for diffusion of the excited anthracene and optimally organizes the included anthracene pair for rapid dimerization. In contrast, βCD provided no rate increase but gave a large change in the product ratio, with exclusive formation of the anti-HT isomer.327 The difference in reactivity and regioselectivity between γCD and βCD has been explained by the stoichiometry of the inclusion complex (Scheme 111). Induced circular dichroism (ICD) studies suggested an axial alignment of anthracene within the γCD core with (R)-helicity.330 Asymmetric induction by γCD was investigated, and both the syn-HT and anti-HH cycloadducts were estimated to have been formed in approximately 10% ee from their ICD spectra.
Scheme 112.
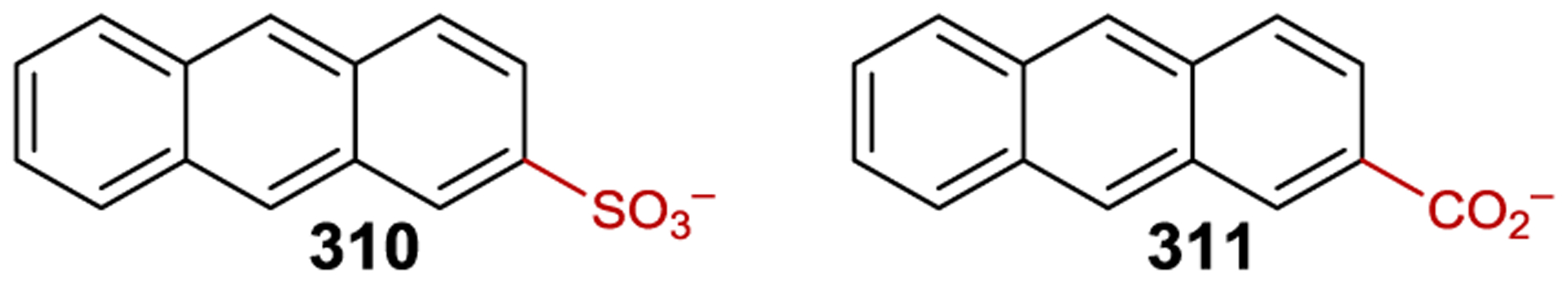
Anthracene Derivatives
Nakamura and Inoue provided the first direct quantification of enantioselectivity in [4+4] anthracene photodimerizations in a study of the reaction of 2-anthracenecarboxylate 311.331 The direct photoreaction of 311 forms the syn- and anti-HT dimers (312 and 313) as the major products, presumably due to minimized repulsion between the anionic carboxylates. The syn- and anti-HH dimers (314 and 315) dimers were formed to a lesser extent. Upon addition of 25 mol% of γCD, syn-256 is formed in 44% relative yield and 28% ee (Scheme 113). Increasing the loading of γCD to 50 mol% slightly increases the selectivity to 31% ee, although further increases did not improve the ee. The anti-257 was nearly racemic under all conditions. The ability to achieve reasonable enantioselectivity with only 25 mol% loading of γCD is attributable to the larger quantum yield for anthracene photodimerization within the chiral cavity. However, the authors estimated that only 65% of the photodimers were produced within the chiral cyclodextrin core, and that the remainder formed through racemic dimerization in bulk solution. Hence, many of the subsequent studies in this area used superstoichiometric loadings of γCD to minimize the concentration of unassociated 311.
Scheme 113.
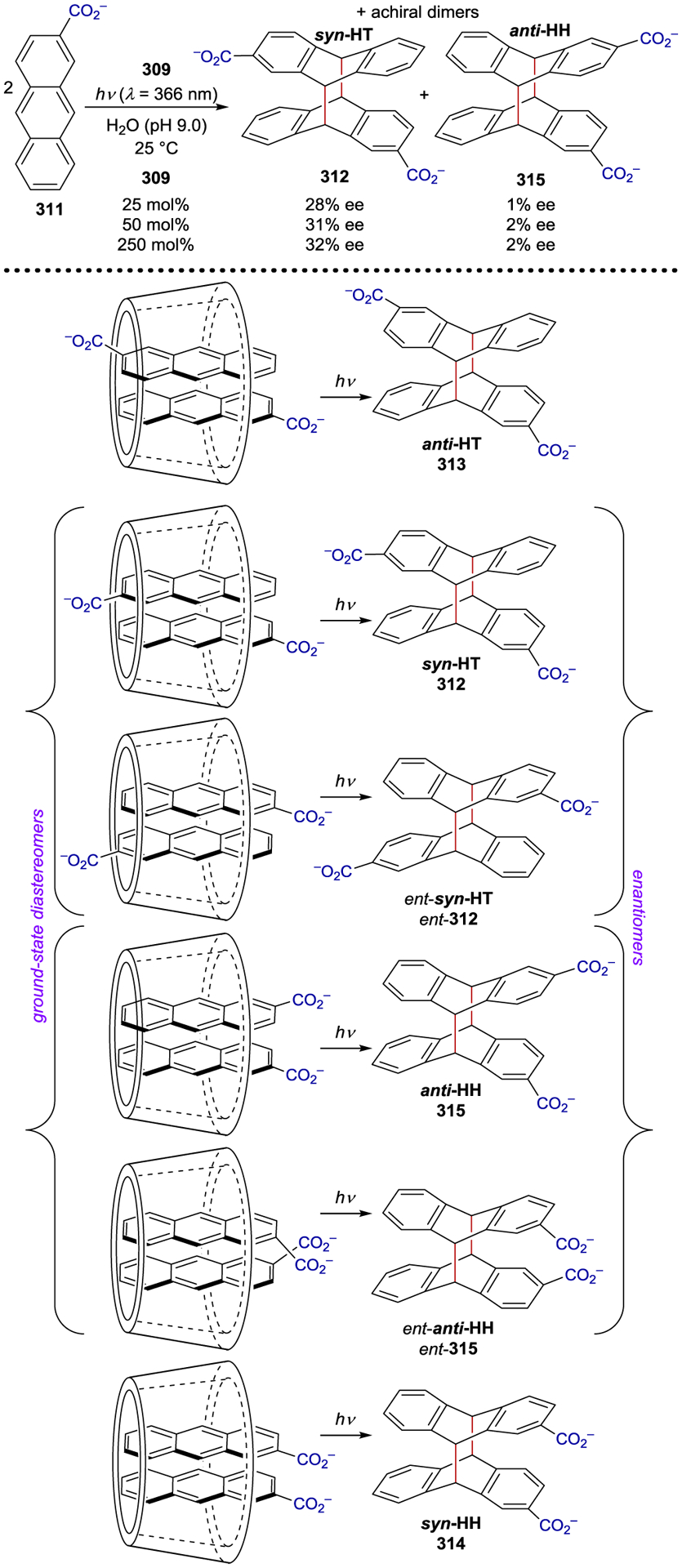
γCD Mediated Photodimerization of Anthracene Carboxylate
The enantioselectivity of the γCD mediated anthracene photodimerization was rationalized by considering the various possible geometries of the stacked anthracenes within the 1:2 ground-state inclusion complex.332 Reorientation is restricted within the inclusion complexes, and dissociation/re-association of anthracene is slower than the excited-state lifetime of photoexcited anthracene. Thus, the orientation is fixed upon photoexcitation, and the included anthracenes dimerize in a geometry predetermined by the ground-state orientation (Scheme 113). The different enantiomers of the syn-HT and anti-HH chiral dimers are produced from diastereomeric ground-state complexes, and the observation of significant ee in these reactions suggests that the difference in free energies of the diastereomeric complexes is appreciable. This mechanism of enantioinduction is common in CD-mediated photochemical dimerization reactions, and studies have been performed to study the influence of external factors such as temperature, pressure, solvent mixtures, and salt additives that could modulate the differential stabilization.333, 334
In an idealized γCD catalyzed photodimerization, the enantiomeric ratio can be influenced by the relative concentrations of the diastereomeric ground-state complexes, the excitation ratio dictated by their respective extinction coefficients, and the relative rates of the diastereomeric photodimerizations. The distinct diastereomeric ground-state complexes have different physical properties, and this can include a difference in their absorption spectra. Hence, variation of the excitation wavelength changes the population of photoexcited complexes, which correlates to the ee of the dimeric products.332,335 In studies of anthracene dimerization in the presence of 3 equiv of γCD, Inoue observed significant changes in both the HT/HH isomeric ratio and the enantioselectivity as a function of the wavelength of irradiation (Scheme 114). A maximum of 41% ee for the syn-312 dimer was obtained at 360 nm, and a significantly lower value of 24% ee was obtained at 440 nm. The anti-HH dimer (315) underwent an inversion in selectivity from −9% ee at 290 nm to 12% ee at 440 nm.
Scheme 114.
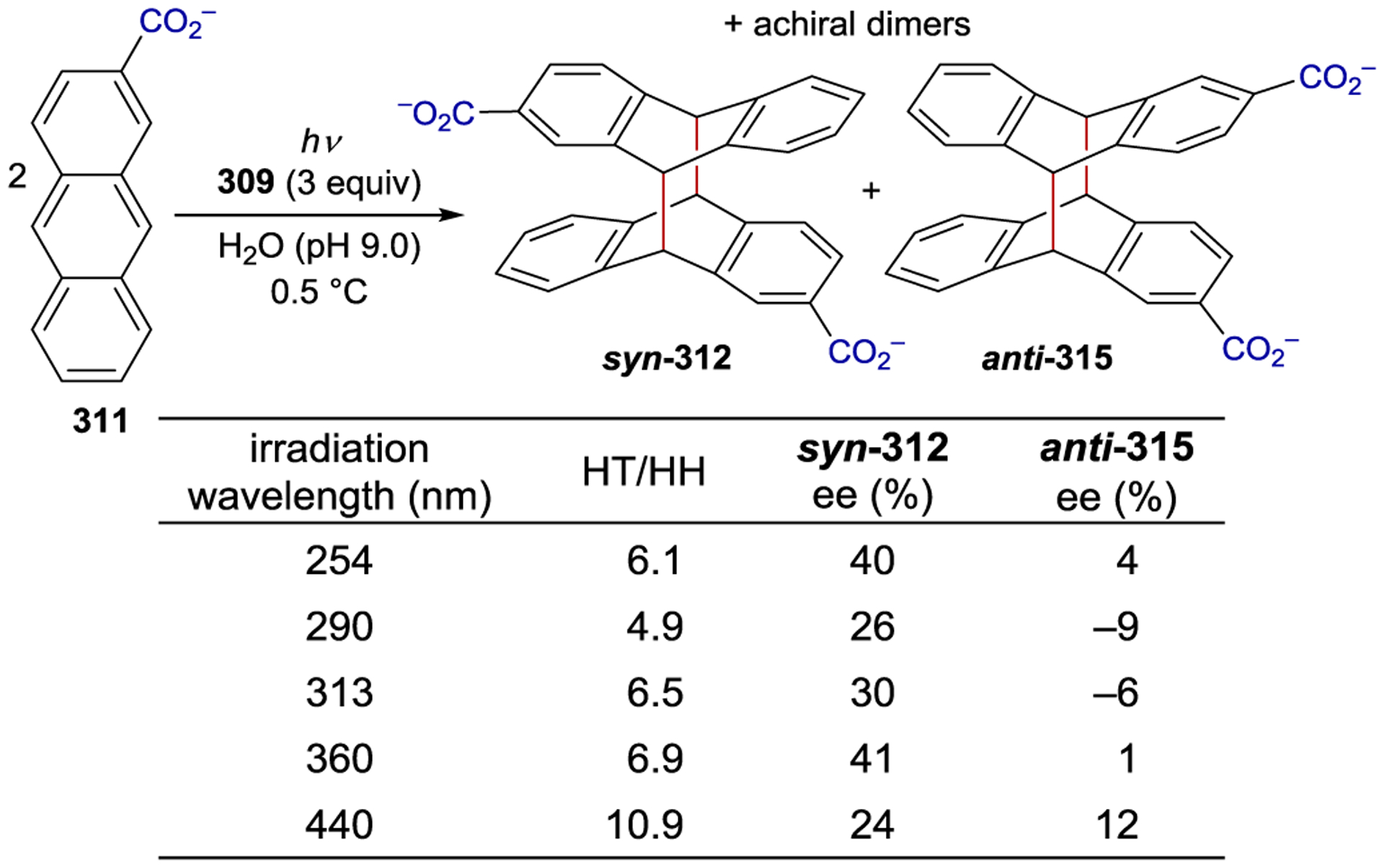
Wavelength Dependance of γCD-Mediated Anthracene Dimerization
Recently, Wang, Inoue, Yang, and Liu studied the [4+4] photodimerization of the doubly anionic 2,6-anthracenedicarboxylate (316).336 In the photodimerization of symmetric 316, two diastereomeric products are possible. The syn-dimer (317) is achiral, but the anti-dimer (318) may be formed as either the (M)- or (P)-enantiomers. In the absence of CDs, the diastereomers are formed in a 1:1 anti/syn ratio. Optimization of the photodimerization in the presence of native γCD showed that 313 nm irradiation provided the (P)-anti-dimer in 18% ee and 1.2 anti/syn ratio (Scheme 115). The anti-dimer was favored at lower energy wavelengths (5.1 anti/syn), though in only 10% ee.
Scheme 115.
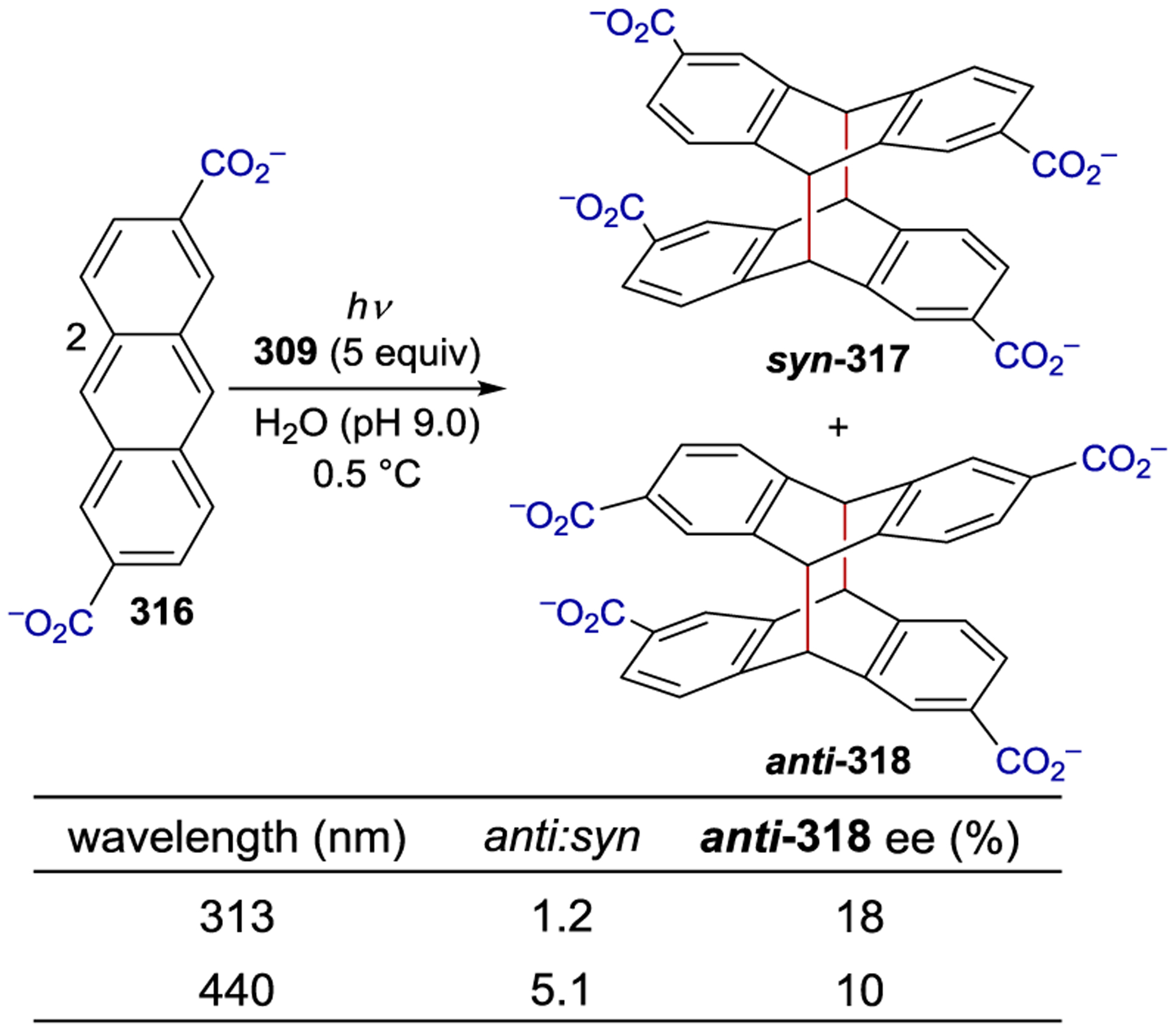
Native γCD-Mediated 2,6-Anthracenecarboxylate Dimerization
When βCD (308) was used as a photocatalyst in the photodimerization of 311, Yang and Inoue observed the formation of unusual chiral “slipped” dimers 319 and 320 (Scheme 116).334 These dimers formed in relatively low yield but with moderate enantioselectivity (45% ee and 32% ee for 319 and 320, respectively). The authors attributed these unusual products to a difference in the selectivity for the formation of the inclusion complexes. Because the central cavity of βCD is somewhat smaller, the most stable complex is a 1:1 complex rather than the 2:1 inclusion complex observed using γCD. Dimerization is not possible directly from the 1:1 complex, and the slipped product is proposed to be formed from a higher-energy 2:2 complex (Scheme 116) from which the transition state for unsymmetrical dimerization is more accessible. The authors further proposed that the formation of this inclusion complex depletes the concentration of free anthracene and slows the rate of formation of the normal symmetrical dimers.
Scheme 116.
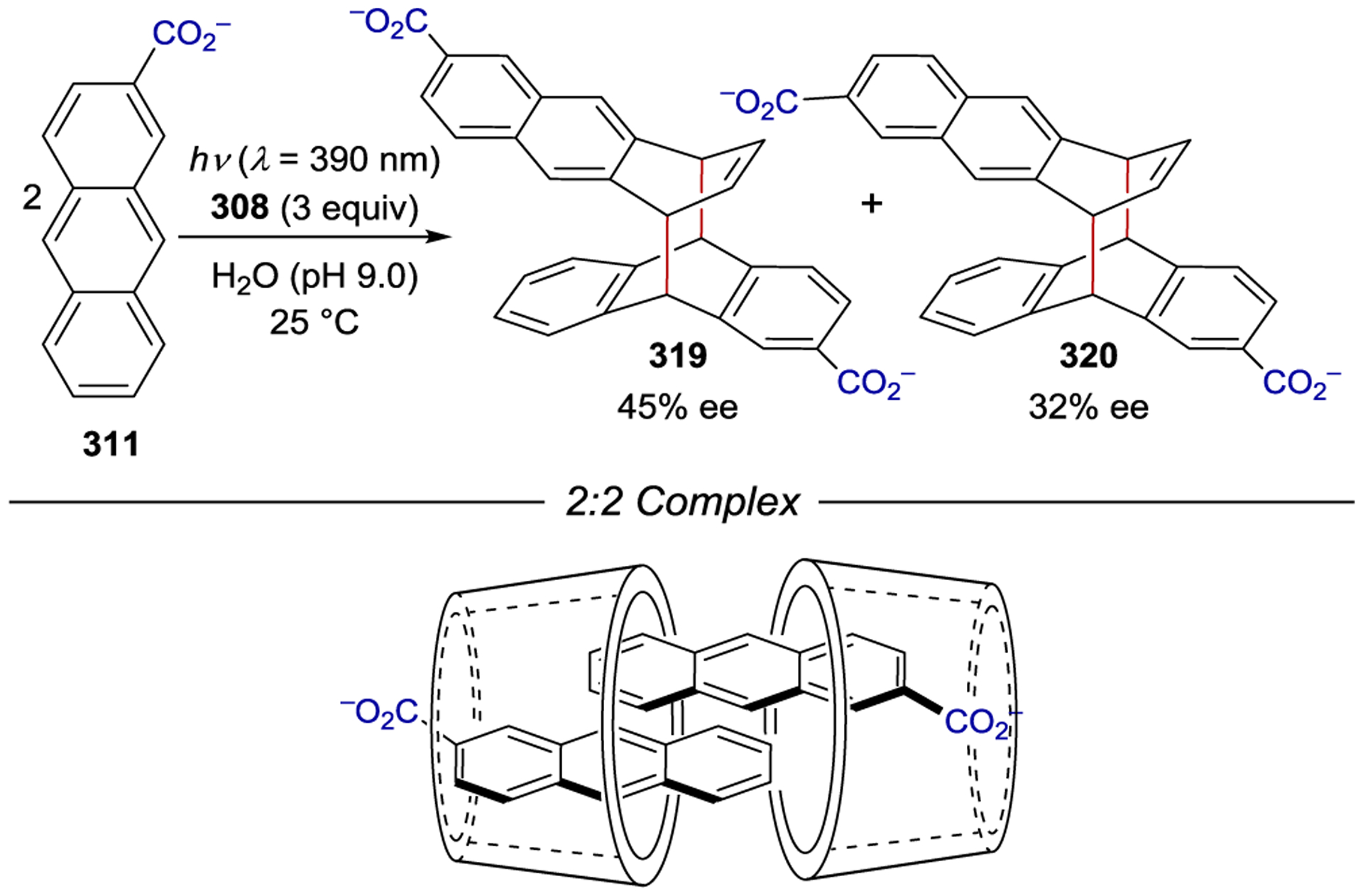
βCD-Mediated Slipped Dimer Formation
Naphthalene derivatives also undergo a [4+4] photodimerization.337 The observation of excimer fluorescence from the 1:2 inclusion complex of γCD and a naphthalene derivative supported the possibility of γCD controlled photodimerization.338 Upon irradiation in the presence of 2 equiv of γCD, photodimerization of methyl-3-methoxyl-2-naphthoate (321) results in the initial formation of [4+4] dimer 322 in 39% ee (Scheme 117).339 Further irradiation promotes a subsequent intramolecular [2+2] cycloaddition to give 323. Notably, a modest enhancement in ee occurred in the second photochemical step, with 323 formed in 48% ee.340
Scheme 117.
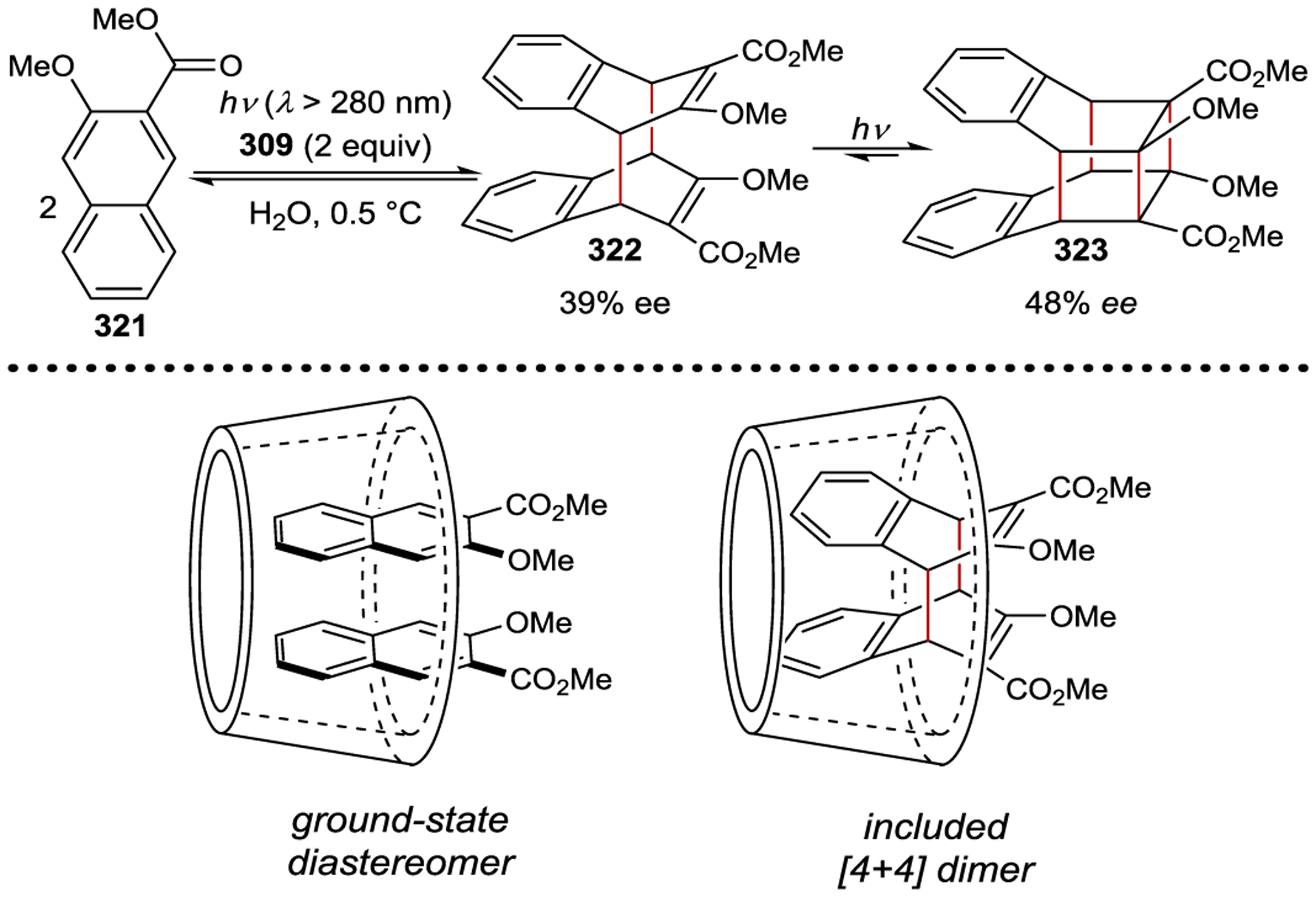
Sequential Asymmetric Dimerization of Naphthoate
The dimerization of anthracene has dominated the study of asymmetric photochemistry within CDs. However, other examples of enantioselective photoreactions have also been studied (Scheme 118). The 4π-disrotatory photoelectrocyclization of substituted tropolones yields chiral bicyclo[3.2.0]hepta-3,6-dien-2-one products.341 Photolysis in the presence of α, β, and γCD produced only modest enantioselectivity.342 In the presence of βCD, irradiation of nitrone 327 produced oxaziridine 328 in good yield, but minimal selectivity (< 1% ee).343 Interestingly, the addition of ᴅ-Ala enhanced both the yield and ee (76% yield and 2% ee). Finally, Ramamurthy reported that irradiation of βCD-encapsulated cis-diphenylcyclopropane (3) produces the enantioenriched 1 in low ee.344
Scheme 118.
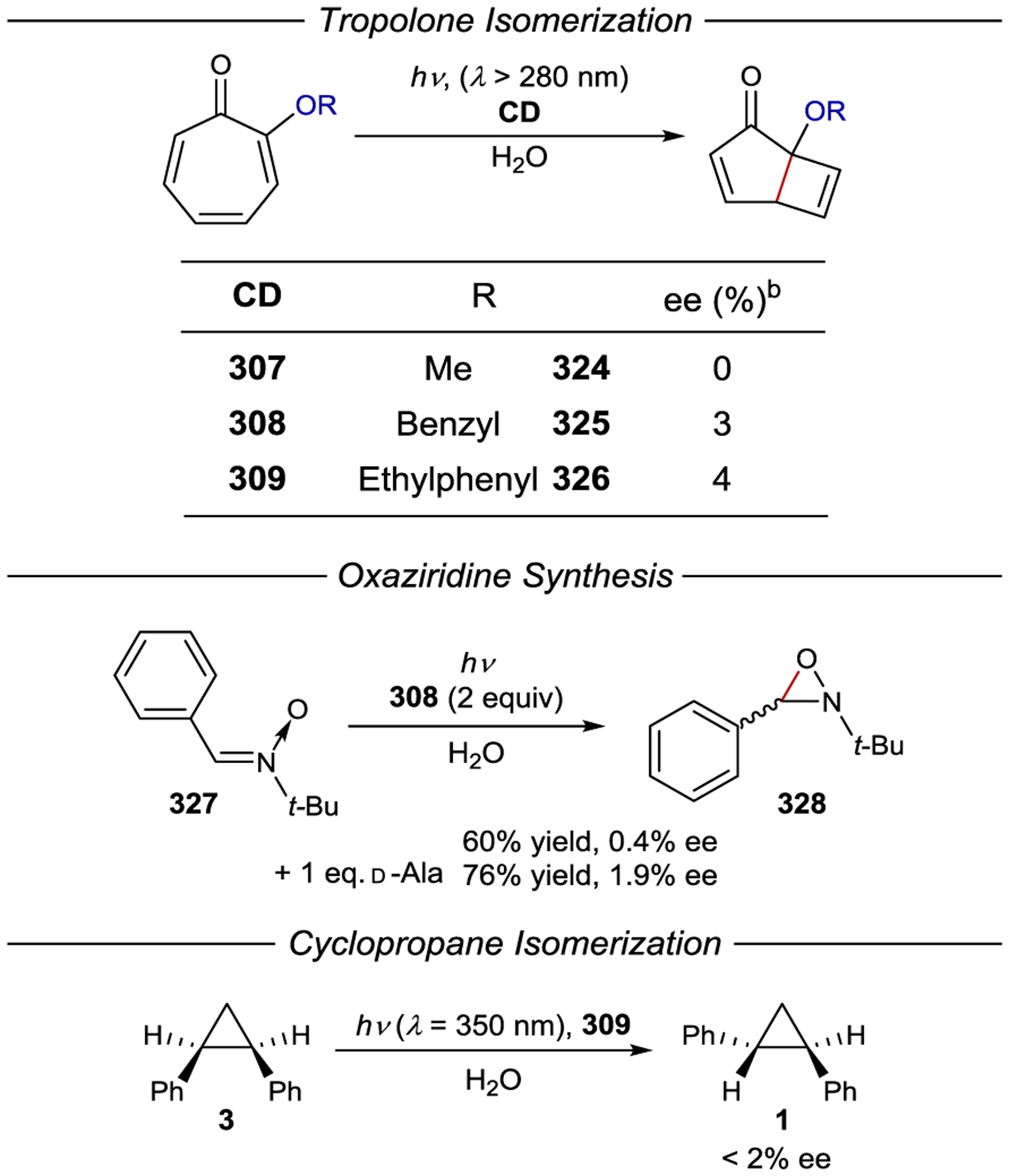
Other Native CD Catalyzed Photoreactions
4.1.2. Functionalized Cyclodextrins
Cyclodextrins can be structurally modified easily, and the introduction of new functional groups provides a means to optimize the rate and selectivity of the photoreactions that occur within their cavities. Functionalization of the primary face, secondary face, or the sugar core of γCD or βCD can induce changes in the orientation of encapsulated substrates, alter the relative stabilization of the diastereomeric ground-state complexes, and introduce photosensitizing moieties to provide access to photoactive CDs.
The first example of a structurally modified photoactive cyclodextrin was developed by Kuroda, who synthesized the porphyrin-modified dimeric βCD 332 in 1991 (Scheme 119).345 The porphyrin moiety sensitizes the formation of singlet oxygen, which promotes the hydroperoxidation of the alkene moieties of linoleic acid (329). Kuroda found that the reaction is essentially non-stereoselective using low catalyst concentrations of 332 (0.5 mol%); however, using 1.1 equiv of 332 resulted in an appreciable 20% ee for 330 and 12% ee for 331.
Scheme 119.
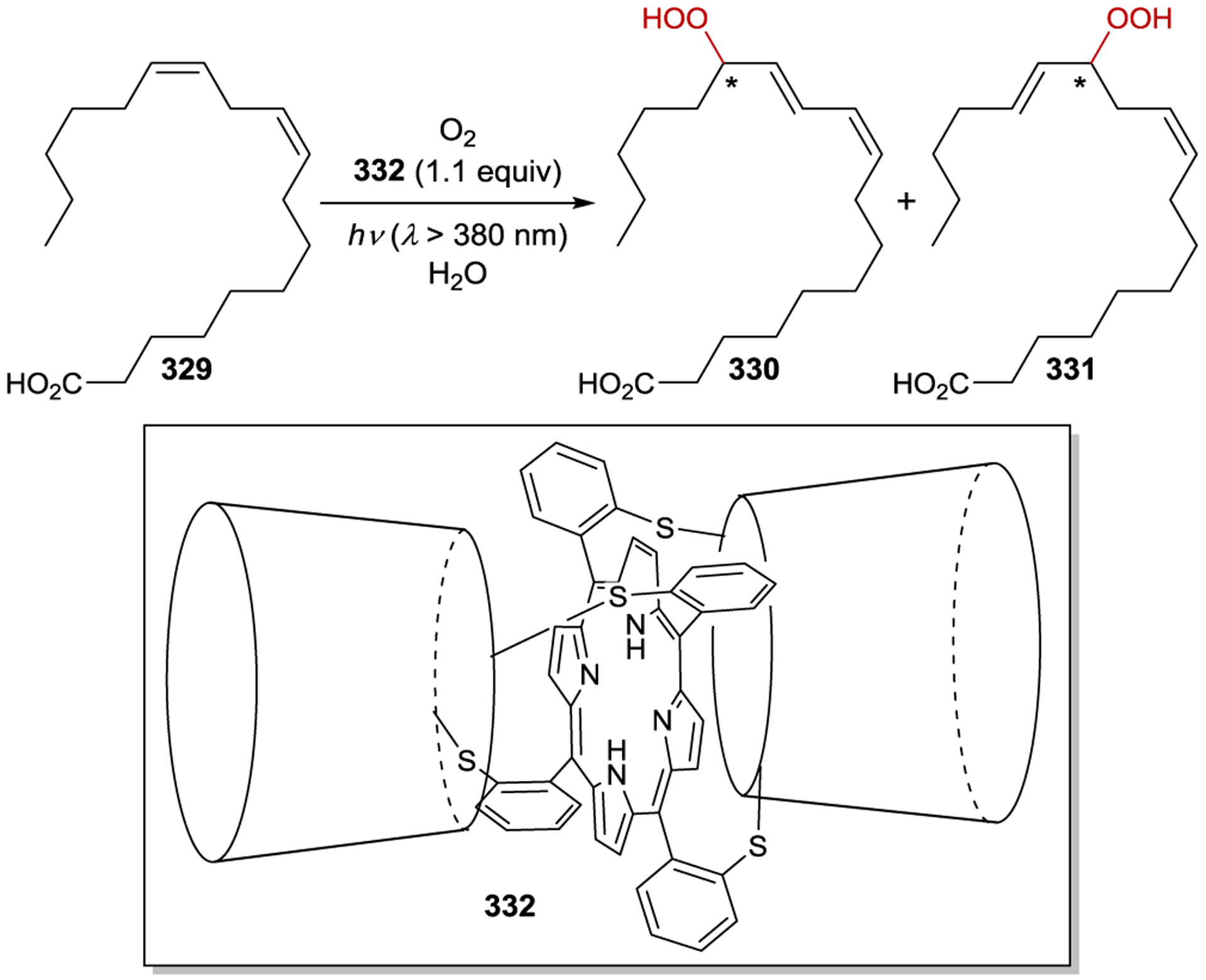
Hydroperoxidation of Linoleic Acid Mediated by a βCD-Derived Porphyrin Photosensitizer
The use of modified cyclodextrins to influence the photoisomerization of (Z)-cyclooctene (14) has been extensively studied. Supramolecular control offers a promising strategy for this classical photoreaction because 14 contains no functional group handles that might bind readily to a small-molecule catalyst. Figure 5 shows a selection of the structures of sensitizing βCDs that have been developed. An important characteristic of these arene-functionalized βCDs is that the hydrophobic sensitizer penetrates the primary face of the βCD cavity in the absence of encapsulated substrate. This feature minimizes the possibility of collisional sensitization of unbound substrates. Upon encapsulation of a substrate within the cyclodextrin cavity, however, the sensitizer is partially displaced, and photoinduced electron or energy transfer can occur directly to the preassociated substrate. The partial inclusion of the sensitizer may also influence the geometry of the associated substrate, and substitution about the sensitizer can have a significant influence on both the sign and magnitude of the ee.
Figure 5.
Selected Aryl-Sensitizer Derived βCDs
In an initial investigation, Inoue prepared a modified βCD functionalized with a benzoyl group (333).9 Inclusion of (Z)-14 within the CD cavity positions it close to the benzoate sensitizer, facilitating energy transfer from its excited state. When (Z)-14 is irradiated in water in the presence of 10 mol% of 333, the system reaches a photostationary state where (R)-(E)-cyclooctene (13) is formed in 11% ee.
The structure of the benzoate sensitizer has been extensively modified in attempts to optimize the enantioselectivity of the isomerization of (Z)-14.346–347 348 349 350 Scheme 120 summarizes the optimal reaction conditions for a selection of these sensitizers. Substitution by either an o-ester (334) or m-methoxy group (336) enhances the resulting ee of 13 (19% and −46% ee, respectively). Notably, these modified CDs favor formation of opposite enantiomers. A m-methylthiol-derived benzoate (341) provided (R)-13 in 47% ee, the highest reported for this series of βCD derivatives. The photoisomerization of 1,3-(Z,Z)-cyclooctadiene in the presence of modified CDs has also been studied. However, achieving high selectivity has proven more difficult than for cyclooctene. Irradiation in the presence of 10 mol% of the 6-O-napthyl-derived βCD 350 resulted in a photostationary state with 4% ee.351 Yang attempted to conduct this reaction using a much more complex γCD-derived rotaxane, but the ee was only marginally improved.352
Scheme 120.
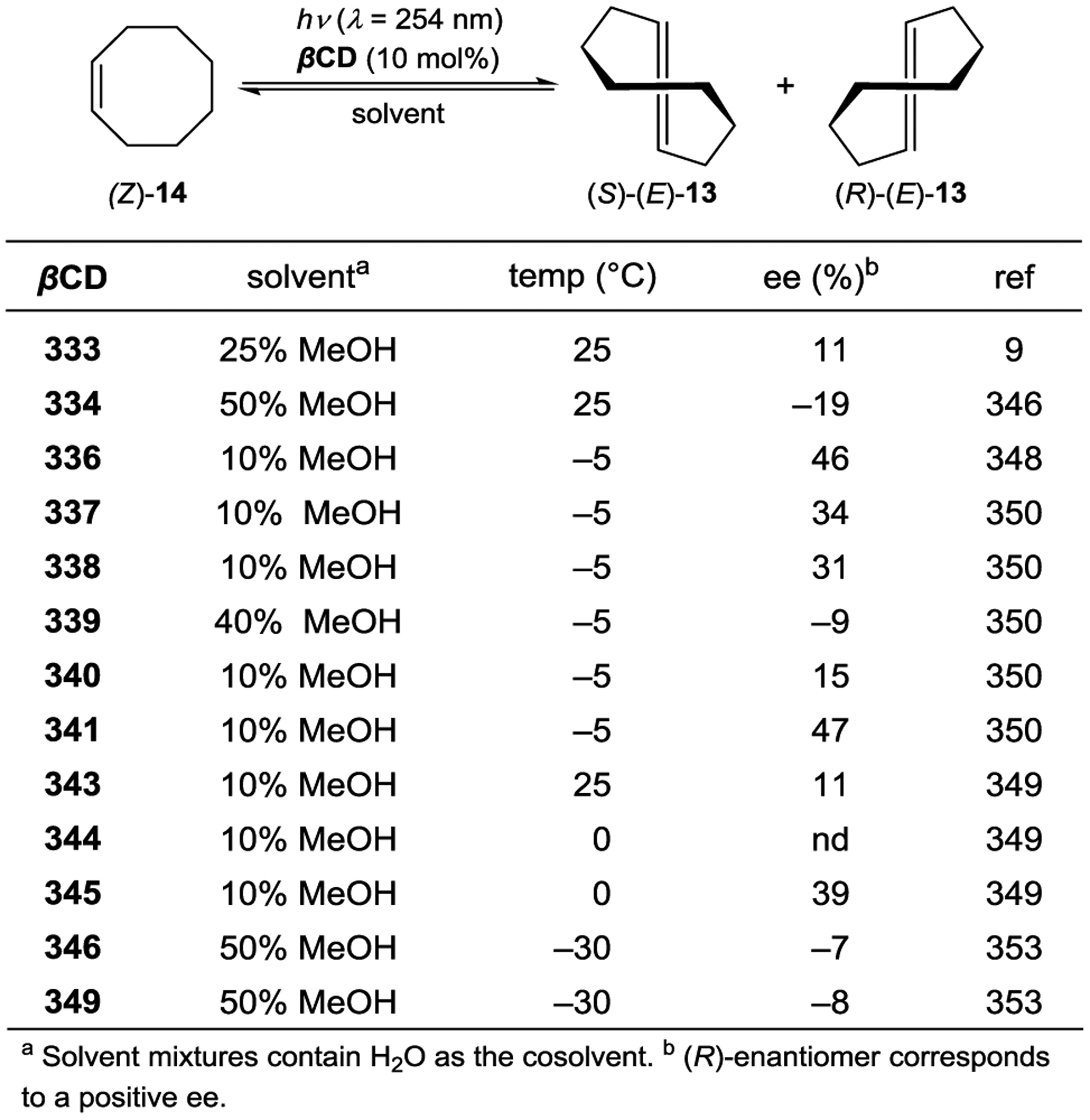
Selection of Asymmetric βCD Mediated Cyclooctene Isomerizations
Inoue noted that the enantioselectivity of photoreactions involving βCD-derived sensitizers have surprisingly little sensitivity to temperature. Because the hydrogen-bonding network at the secondary face of βCD results in a rigid structure with few degrees of freedom, Inoue proposed that the differential activation entropy (ΔΔS‡) for the formation of the enantiomers of 13 was close to zero. To increase the flexibility of the βCD core, Inoue prepared cyclodextrins 346–349 featuring permethylated secondary faces.353,354 While this modification did not enhance the enantioselectivity beyond the previous results, these catalysts resulted in a more pronounced change in enantioselectivity as a function of temperature. The calculated differential activation entropies (ΔΔS‡ = 11 cal mol−1 K−1) were significantly larger than for unmethylated βCDs.
Structurally modified βCD hosts can also act as enantioselective photosensitizers for other organic transformations. Inoue reported the intermolecular anti-Markovnikov hydrofunctionalization of 1,1-diphenylpropene 22 (Scheme 121) using a naphthalene-modified CD (351) 355, 356 Using 1 equiv of 351 in 25% MeOH/H2O at −10 °C, methylether 23 and secondary alcohol 352 were formed in a combined 60% yield and 11% ee and 15% ee, respectively.
Scheme 121.
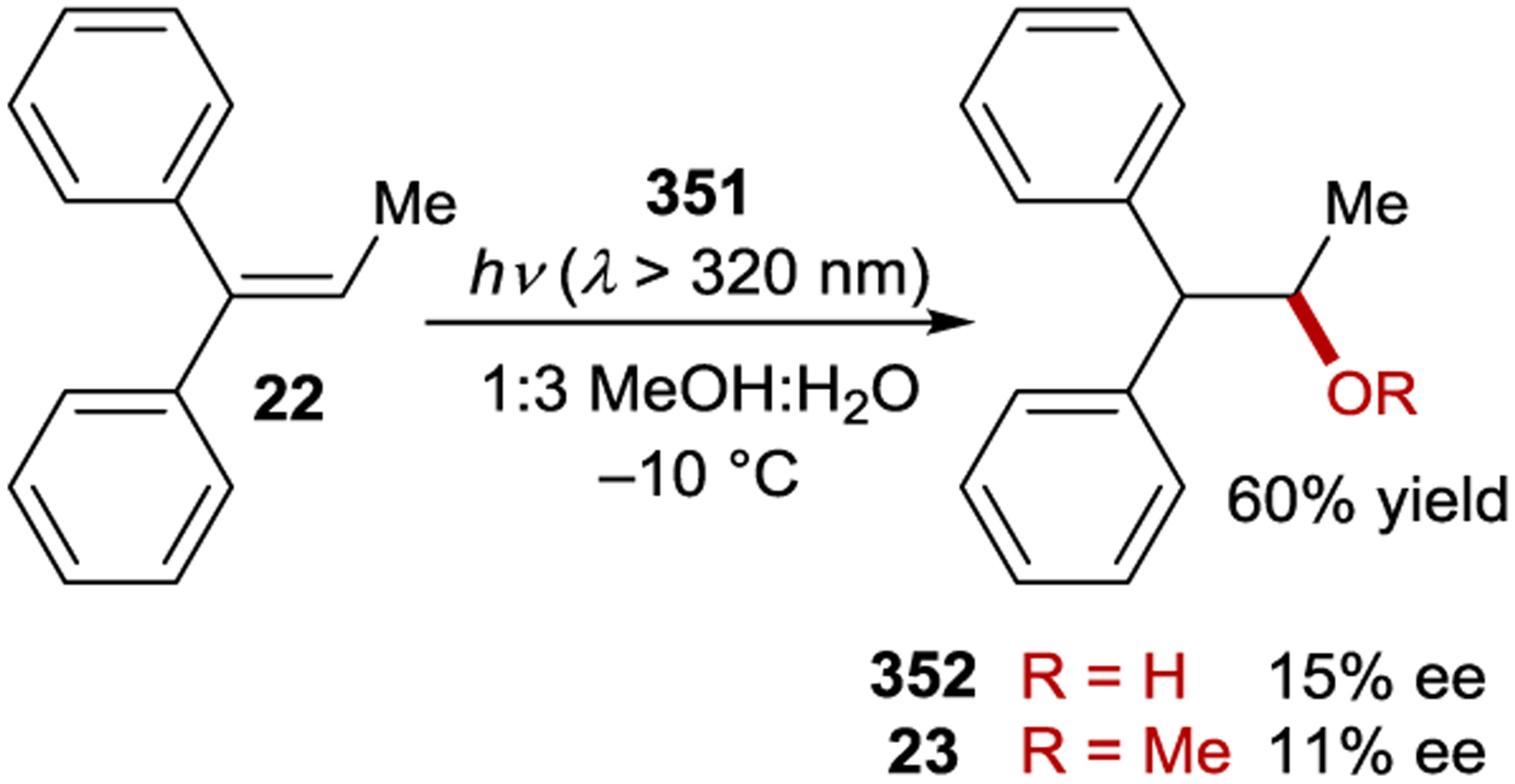
Anti-Markovnikov Hydrofunctionalization of 1,1-Diphenylpropene
Ramamurthy studied the effect of cyclodextrin hosts on the Norrish–Yang photocyclization of functionalized acetophenones to afford chiral cyclobutanols (Scheme 122).357 Interestingly, the ground-state association complex involved two βCDs encapsulating a single acetophenone guest molecule. Irradiation of adamantyl acetophenone 353 in the presence of chiral benzylamine-derived βCD 356 (1 equiv) provided trans-cyclobutanol 354 in 20% ee and cis-355 in 9% ee. As no enantioselectivity is observed using native βCD, the authors suggested that the additional chiral center of the benzylamine plays a role in enantioinduction; however, the nature of this effect was not elucidated.
Scheme 122.
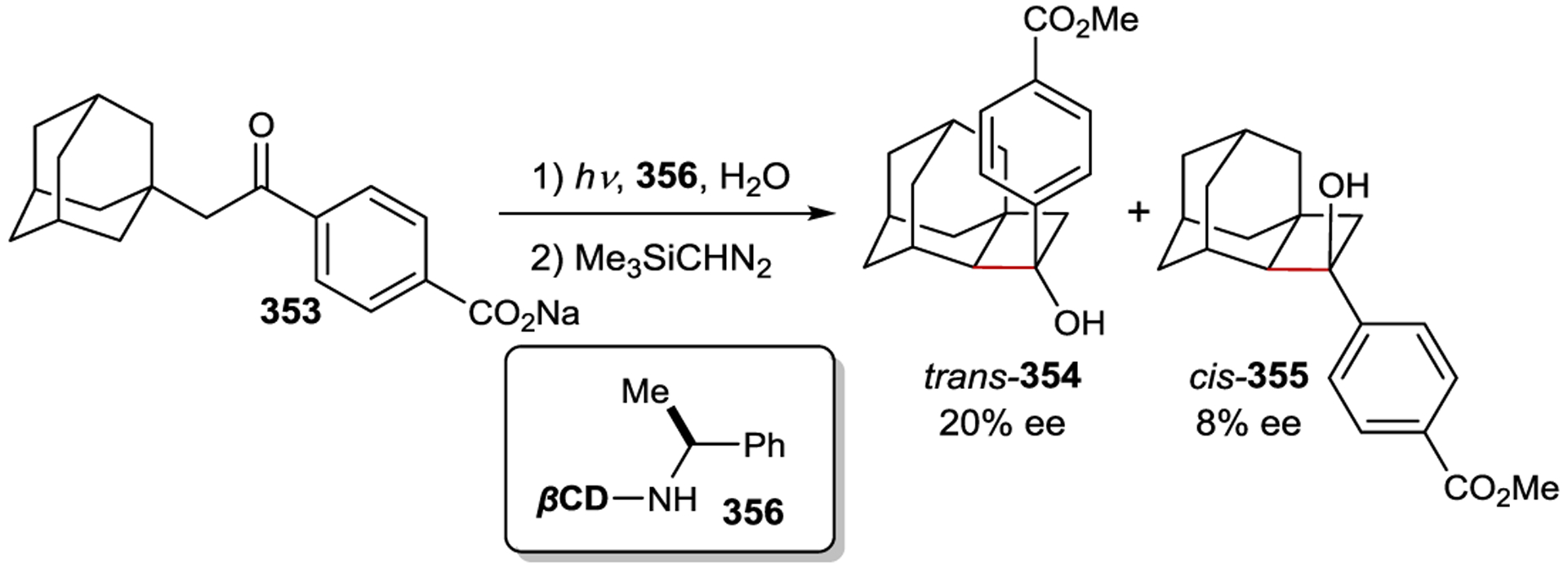
Norish–Yang Photocyclization of Adamantyl Acetophenone
As mentioned previously, the [4+4] photodimerization of 2-anthracenecarboxylate 311 in the presence of βCD produces unusual “slipped” dimers through the formation of a small concentration of a photoactive 2:2 βCD:311 complex. While native βCD produced the slipped dimers in modest ee, the regioselectivity was relatively poor. Yang hypothesized that structurally modified CD hosts might influence the regioselectivity and enantioselectivity of the photodimerization (Scheme 123).334, 358 Functionalization of the βCD primary face with charged functional groups (357–362) significantly enhances slipped dimer formation. Trimethyl ammonium derived 361 provides the largest increase in enantioselectivity; at 80 mol% catalyst loading, anti-slipped dimer 319 is formed in 56% ee and the syn-dimer 320 is formed in 8% ee. The addition of 6 M CsCl enhances the stability of the 2:2 ground-state association complex, increasing both the yield (99%) and selectivity for anti-319 (65% ee). Notably, the enantioselectivity of syn-320 is inverted, providing the opposite enantiomer in 7% ee. Further modification of the 3-hydroxy group to an azide (364) provided syn-320 in 70% ee at −20 °C, though the enantioselectivity of anti-319 decreased to 15% ee using this host.
Scheme 123.
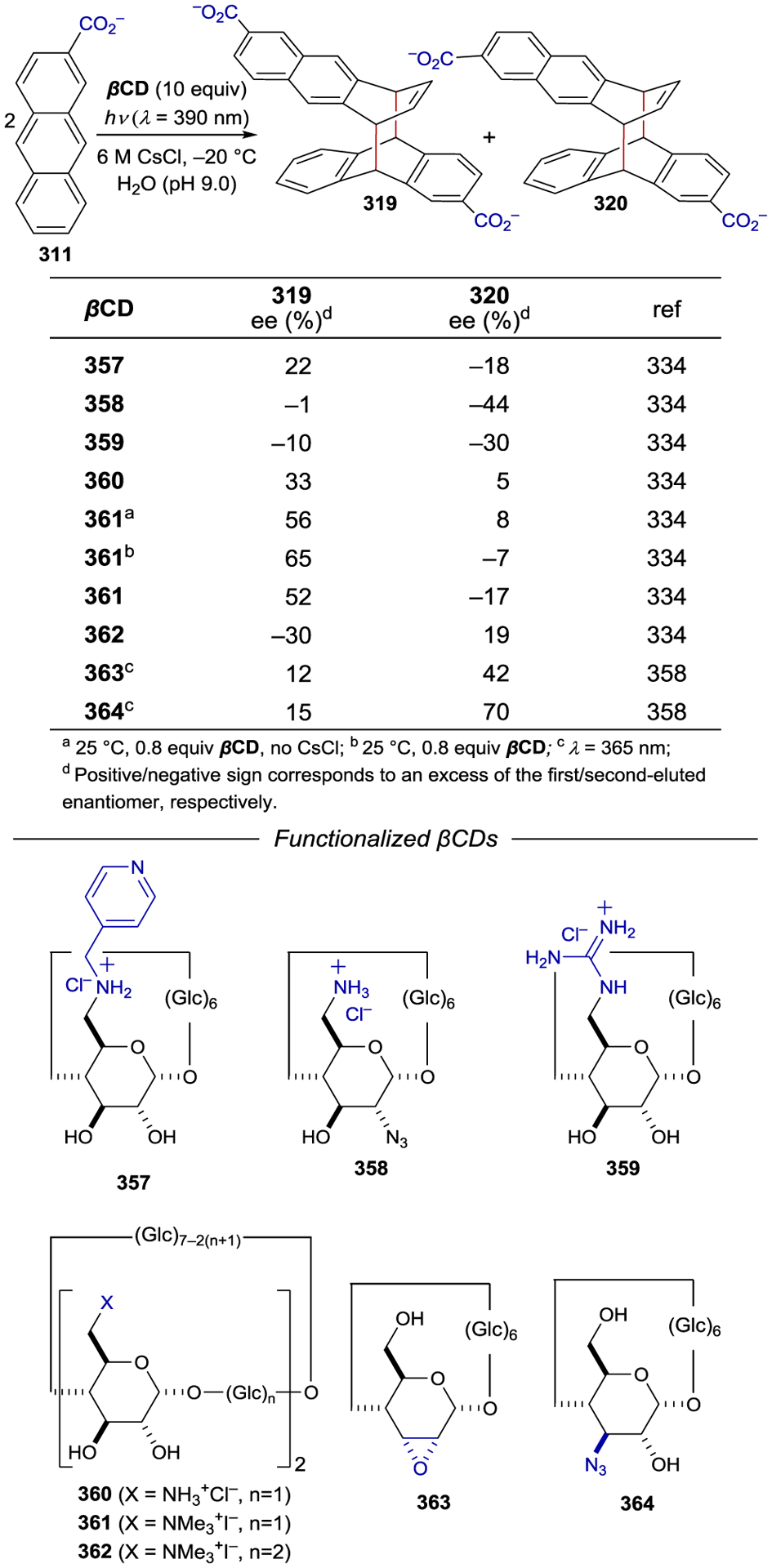
Non-Sensitizing Primary and Secondary Face βCD Structural Modifications
The central core of γCD is larger than in βCD and favors formation of a 1:2 inclusion complex with carboxylate anthracene 311 (Scheme 111). A wide range of primary and secondary face-modified γCDs have been synthesized and studied in the asymmetric [4+4] photodimerization of anthracene. Many of these studies focus on the use of cationic functional groups, including ammonium, guanidinium, and pyridinium groups (Figure 6 and Scheme 124).332, 359–360 361 362 363 364 365 Electrostatic interactions between the cationic moieties and the anionic carboxylate of the substrate increase the formation of HH anthracene dimers, which in certain cases are formed as the major product.360, 361 The maximum enantioselectivity reported for formation of anti-HH dimer 315 using one of these modified γCDs was −86% ee, provided by 370.364, 366 Conversely, the enantioselectivity of syn-HT 312 was degraded versus native γCD for most of the cationic functionalized γCDs, in some cases favoring the opposite enantiomer (Scheme 124).360 Notably, the selectivity of the dimerization is sensitive to the relative location of cationic substituents in difunctionalized γCDs.367 The substitution of two pyridinium moieties onto γCD provides a good example. Substitution of the pyridiniums proximate to each other (366, n = 0, and 367, n = 1), produced lower enantioselectivities than when they were positioned further apart, as in γCD 368 (n = 3).
Figure 6.
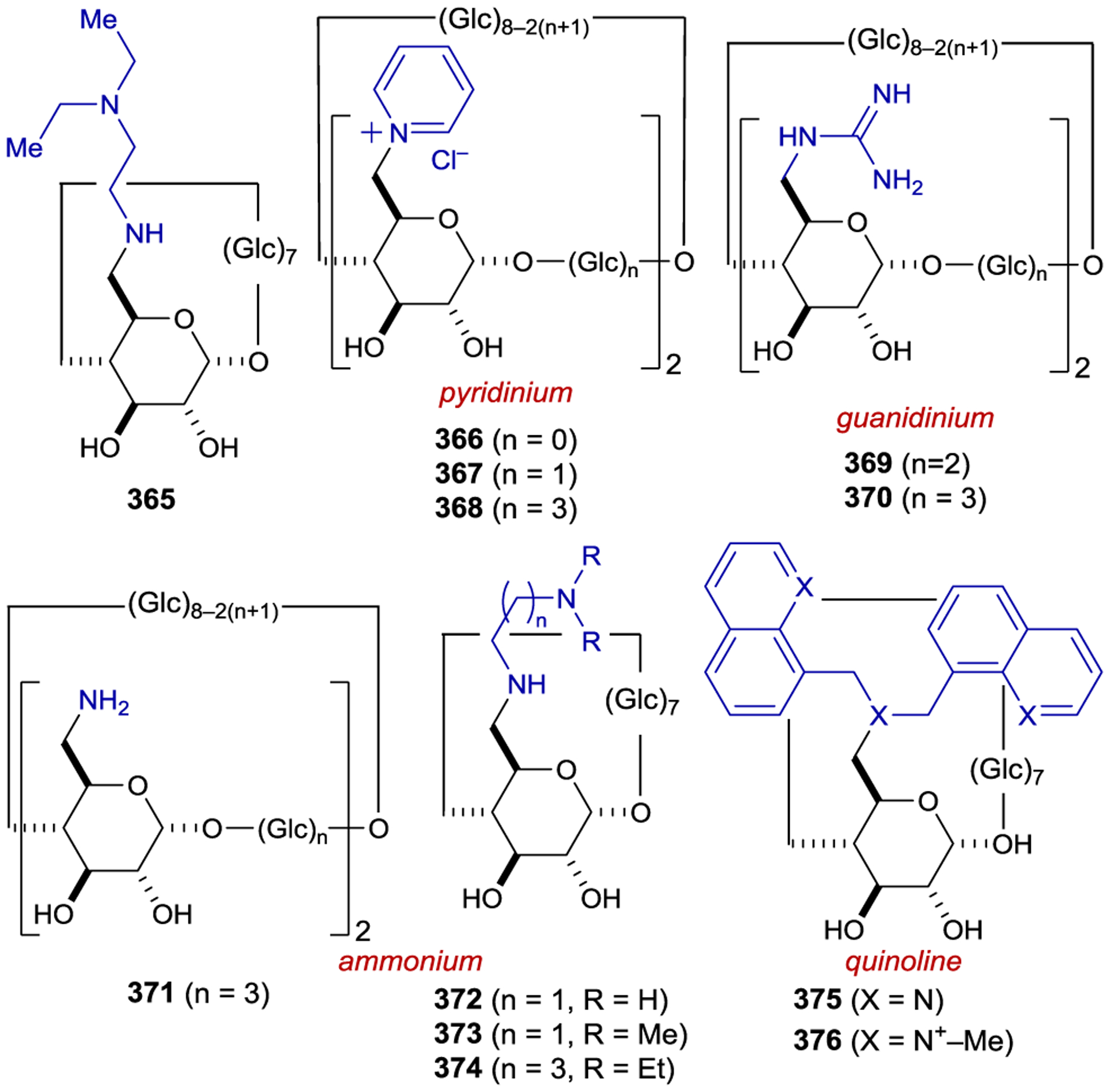
Representative Examples of Primary Face Modifications of γCD. Note that under reaction conditions the amine groups are protonated making the γCDs cationic.
Scheme 124.
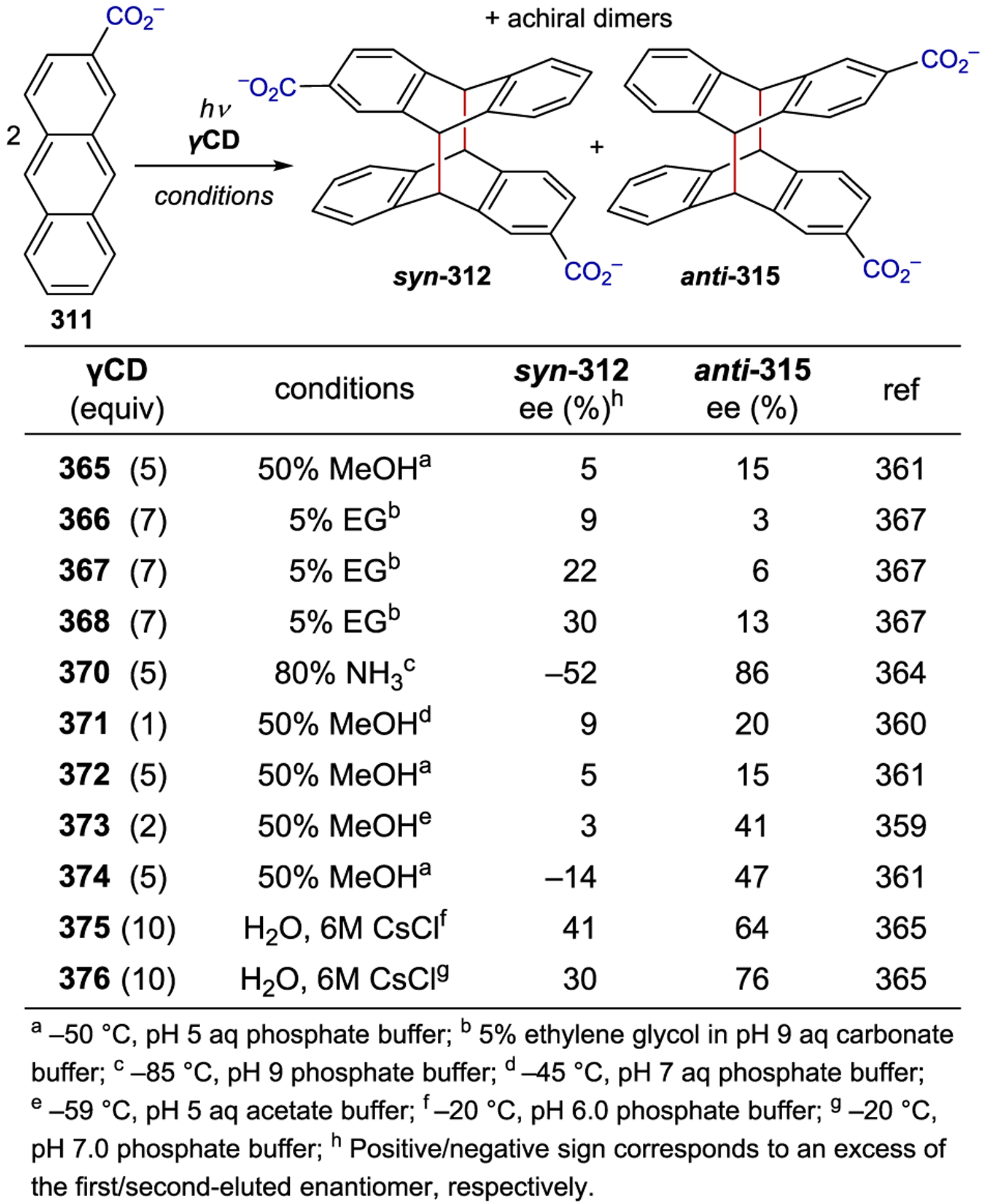
Selected Examples of Anthracene [4+4] Photodimerization Mediated by Cation Functionalized γCDs.
The diamine side chain of γCD 365 readily complexes Cu(II), which positions a Lewis acid functionality at the primary face of the γCD (Scheme 125).361 The addition of Cu(II) slows the rate of anthracene photodimerization but bolsters the regioselectivity for the anti-HH dimer. The enantioselectivity of this reaction is 70% ee using 10 mol% of 365 and 50 mol% Cu(II); lowering the loading of 365 to 1 mol% gave 43% ee. Inoue proposed that coordination of the substrate carboxylates to the ligated Cu(II) Lewis acid increases their organization within the chiral CD core.
Scheme 125.
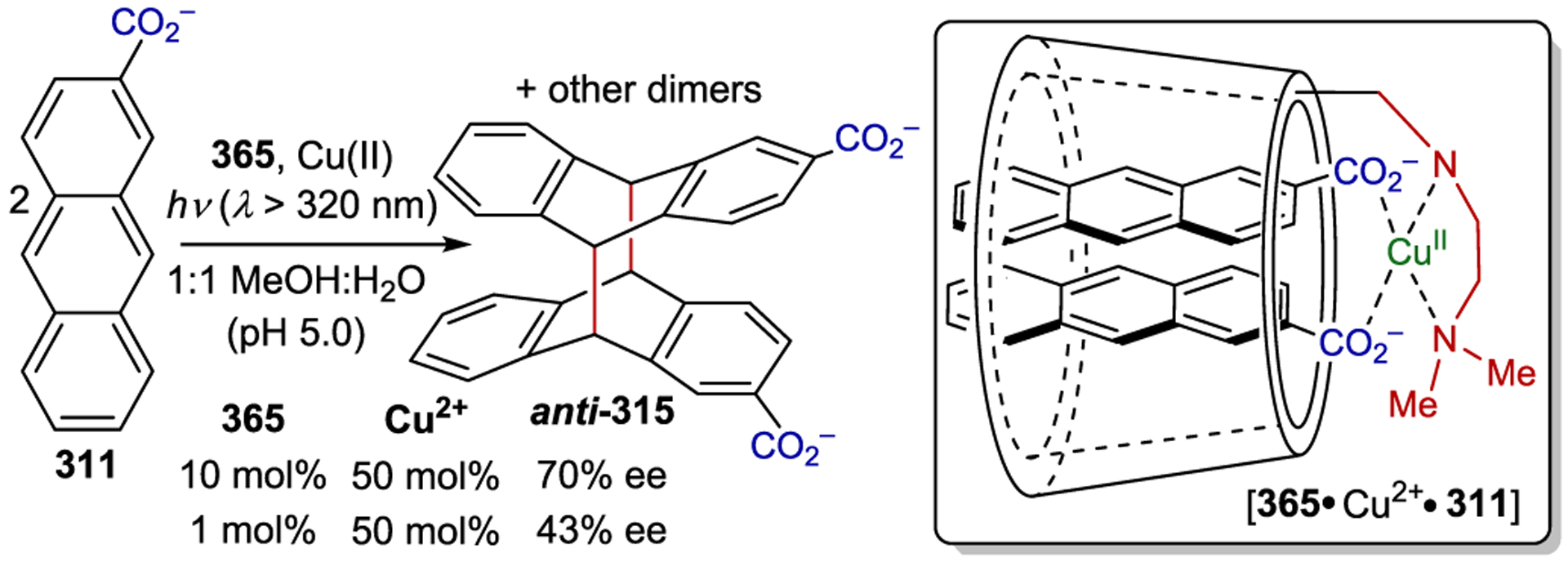
Anthracene Photodimerization Catalyzed by a γCD–Cu2+ Complex
A selection of these primary face modified γCDs were screened as chiral templates for the photodimerization of 2,6-anthracenedicarboxylate (316) (Scheme 126). Temperature, solvent, and irradiation wavelength were optimized to enhance both yield and selectivity. Irradiation of 316 in the presence of the diamine-derived γCD 371 (50% aqueous MeOH, −50 °C, 360 nm) provided the highest enantioselectivity, providing the (P)-anti-dimer 317 in 58% yield, 1.4:1 anti:syn, and 72% ee. Notably, the use of diguanidine-derived γCD 369 along with a change in reaction conditions (28% aqueous NH3, −60 °C, 450 nm) enhanced the selectivity for the anti-dimer to 15:1 anti:syn with minimal decrease in enantioselectivity (69% ee).
Scheme 126.
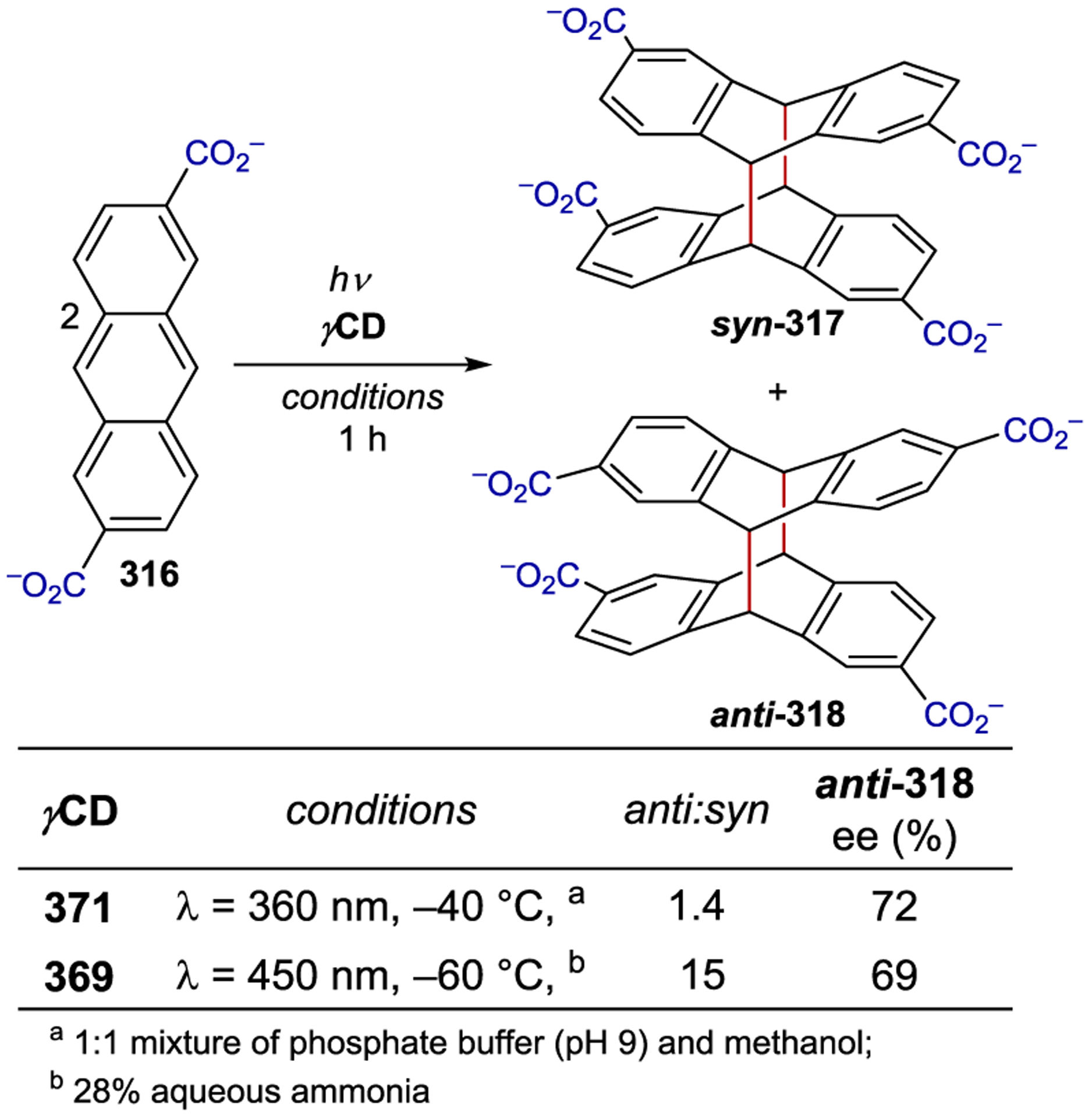
2,6-Anthracenedicarboxylate Photodimerization with Primary Face Modified γCDs.
Cyclodextrin hosts have also been modified with a rigid cap that spans the primary face, alters the encapsulation of substrate, and rigidifies the CD cavity (Figure 7 and Scheme 127)., 368, 369 Irradiation of anthracene 311 with 2.5 equiv of γCD 377 provided syn-312 in 57% ee under aqueous conditions.368 A maximum of 36% ee for the anti-HH isomer could be achieved by changing the solvent to 50% MeOH. The introduction of an ether spacer within the rigid cap (378) led to an inversion of the syn-312 ee, with a concomitant decrease in the ee of anti-315. The measurement of negligible differential activation entropies suggested that the capped CD is restricted in motion and does not alter its conformation upon complexation of anthracene.
Figure 7.
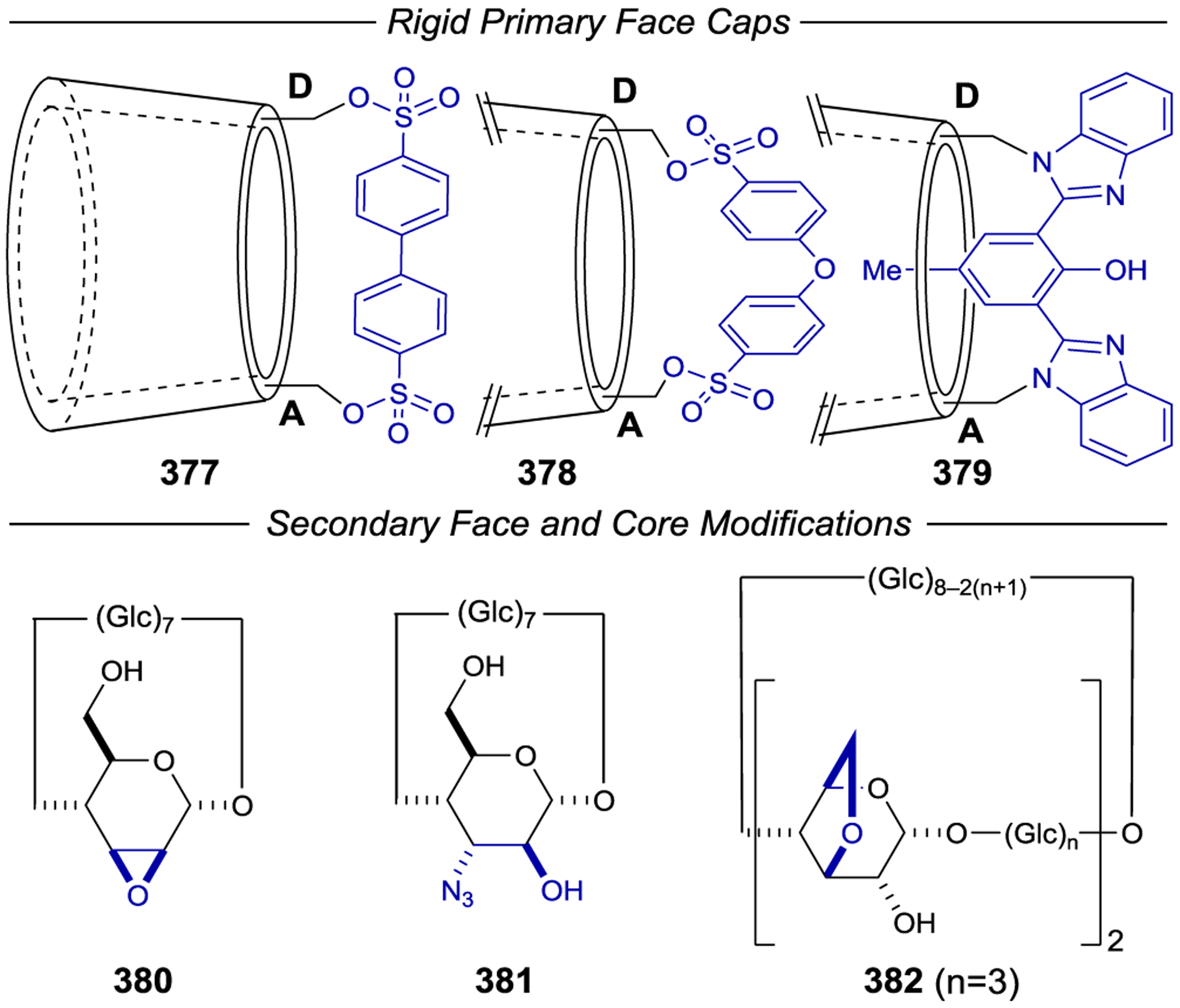
Representative examples of rigid γCD primary face and secondary face or core γCD modifications
Scheme 127.
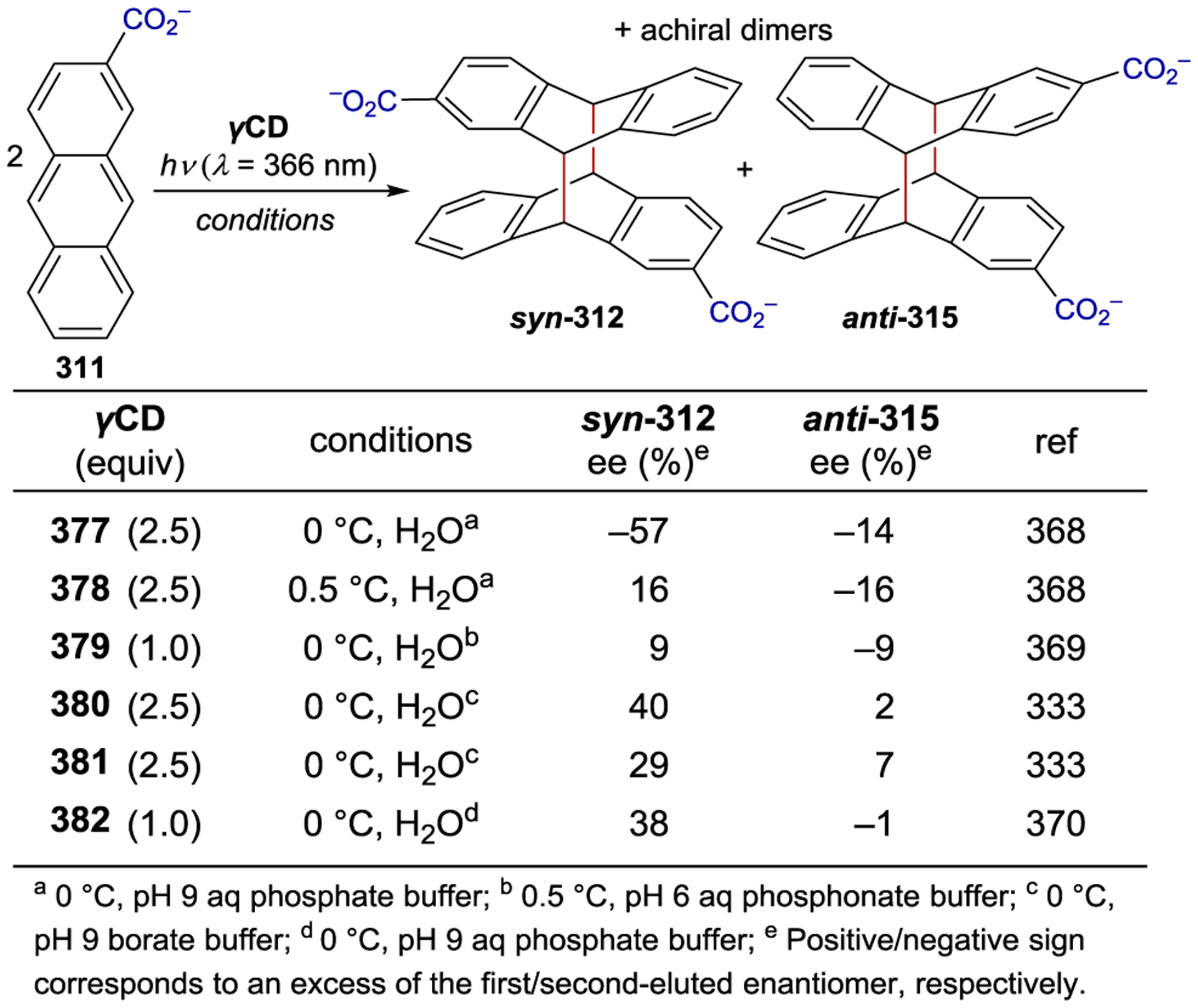
Selected Examples Anthracene [4+4] Photodimerization Mediated by Rigid-Capped and Secondary-Face Modified γCDs
Inoue has also synthesized γCDs in which the structures of the glucose monomers are modified (Figure 7, 380–382).333, 363, 370 In general, these structures do not have the same well-ordered hydrogen bonding network at the secondary face; the resulting increase in structural flexibility can lead to a greater sensitivity to temperature and pressure. However, the optimal enantioselectivities obtained using this class of γCDs did not exceed those available with simpler modifications.
Naphthalene can act as an energy-transfer photosensitizer through Förster resonance energy transfer (FRET). Inoue designed 383 featuring a naphthalene bridge across the γCD primary face, which provides a means to sensitize included anthracene 311 (Figure 8).371 Naphthalene absorbs more strongly than 311 at 300 nm; irradiation at this wavelength minimizes direct excitation of 311 and slows the racemic background reaction. With 30 mol% 383, the syn-312 and anti-315 dimers were formed in reasonable enantioselectivity (24% ee and 30% ee, respectively).
Figure 8.
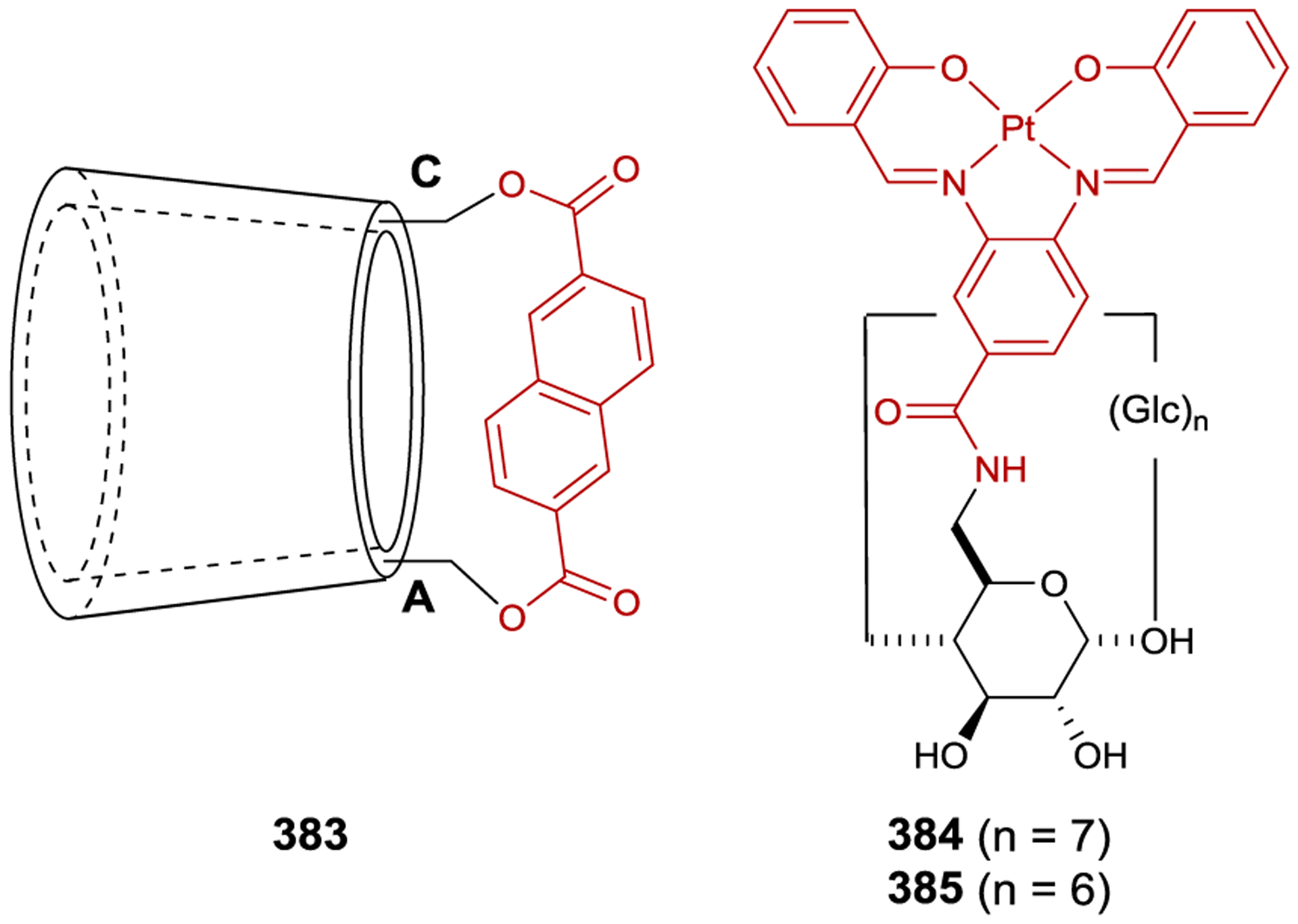
Photosensitizer-derived γCDs
Similarly, Wu and Yang appended a Pt(II) Schiff-base chromophore to the primary face of γCD (Figure 8).372 Excitation of the Pt(II) chromophore leads to Dexter triplet energy transfer (TET) to CD-included 311. Triplet-triplet annihilation within the CD core produces the excited singlet anthracene required for photodimerization. Under irradiation at 532 nm, photodimerization occurs readily in the presence of only 0.5 mol% 384 favoring the HT dimers. The anti-315 dimer was isolated in 34% ee while the syn-312 dimer was racemic. Replacing the γCD core with βCD (385) results in significantly lower ee.373
The highest enantioselectivities yet obtained for anthracene dimerization were recently reported by Yuan, Yang, and Inoue. Dimeric capsules can be assembled by tethering two βCDs together; these can encapsulate two molecules of 311 in a preferred HT orientation.374 A series of these tethered βCDs were synthesized; however, the derivative with a bridging sulfur across the 3-O positions on both βCDs was studied in depth (388, Scheme 128). After extensive optimization, irradiation of 311 at 365 nm in the presence of 10 equiv of 388 cleanly formed the syn-HT dimer 312 in 96% yield, 98:2 regioisomeric ratio, and >99% ee. Impressively, lowering the loading of 388 to 25 mol% maintained a high selectivity of 98% ee, though the yield and regioselectivity were somewhat lower.
Scheme 128. Anthracene Photodimerization Catalyzed by Tethered βCDs.
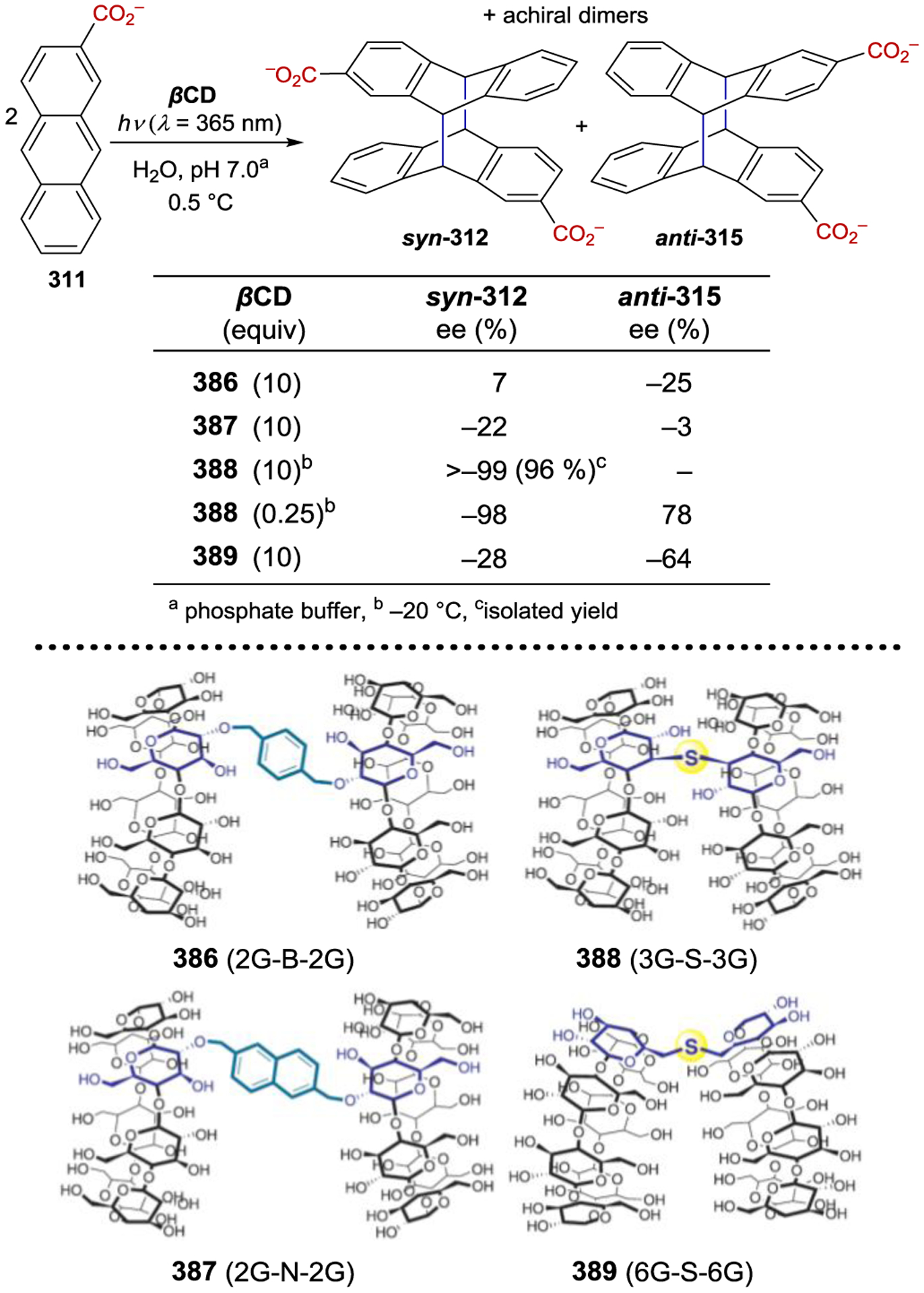
Reproduced from Ref. 374. Copyright 2019 American Chemical Society.
In a follow-up study, Yang targeted the enantioselective formation of the slipped anthracene dimers. A chiral pillar[5]ane spacer was introduced between the tethered βCDs to provide the extra space required to accommodate the slipped dimer transition state (Scheme 129).375 High enantioselectivity can be achieved for anti-319 (87% ee) in the presence of 2 equiv of the chiral host 390, although the symmetric [4+4] dimers are the main products of this reaction. Upon cooling the reaction to −20 °C and adding 6 M CsCl, the relative yield of the slipped dimers increased to 60% with enantioselectivities of 63% ee and 34% ee for anti-319 and syn-320 respectively.
Scheme 129. Anthracene Photodimerization Catalyzed by Tethered βCDs.
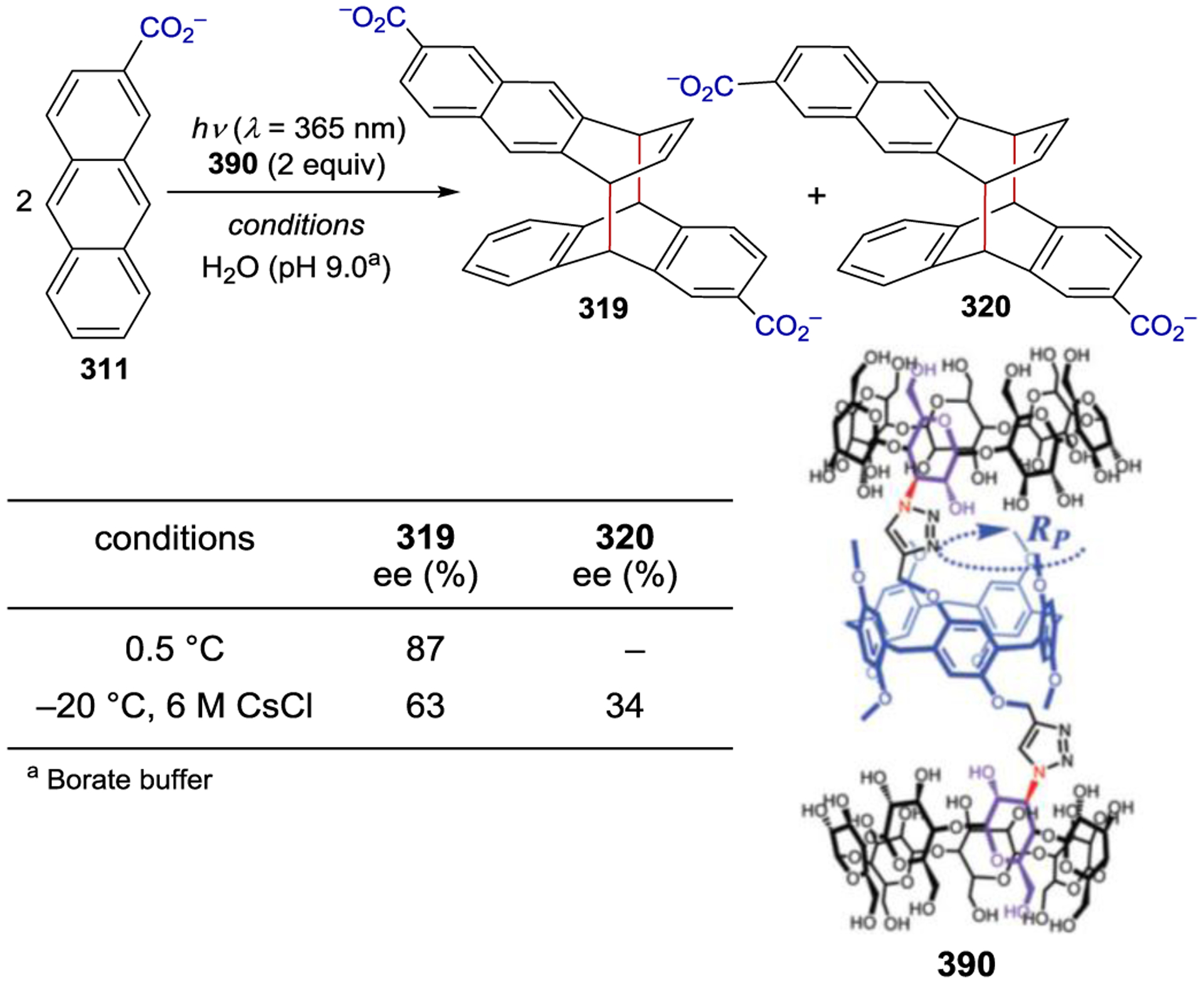
Adapted with permission from Ref. 375. Copyright 2020 The Royal Society of Chemistry.
4.2. Other Polysaccharides
Curdlan, a linear oligosaccharide derived from (1→3)-linked β-d-glucose units, forms a chiral triple helix structure under acidic conditions. The structure of the helix is dynamic, which facilitates the inclusion of small organic molecules through reversible rearrangement of the macroscopic structure. Inoue prepared a modified curdlan (393) randomly functionalized with naphthalene sensitizer on ~8% of the glucose monomers and demonstrated that it readily includes (Z,Z)-cyclooctadiene 391 within its triple helix structure.376 Photosensitization of 391 in the presence of 20 mol% 393 provided (E,Z)-cyclooctadiene 392 in 7% ee (Scheme 130).
Scheme 130.
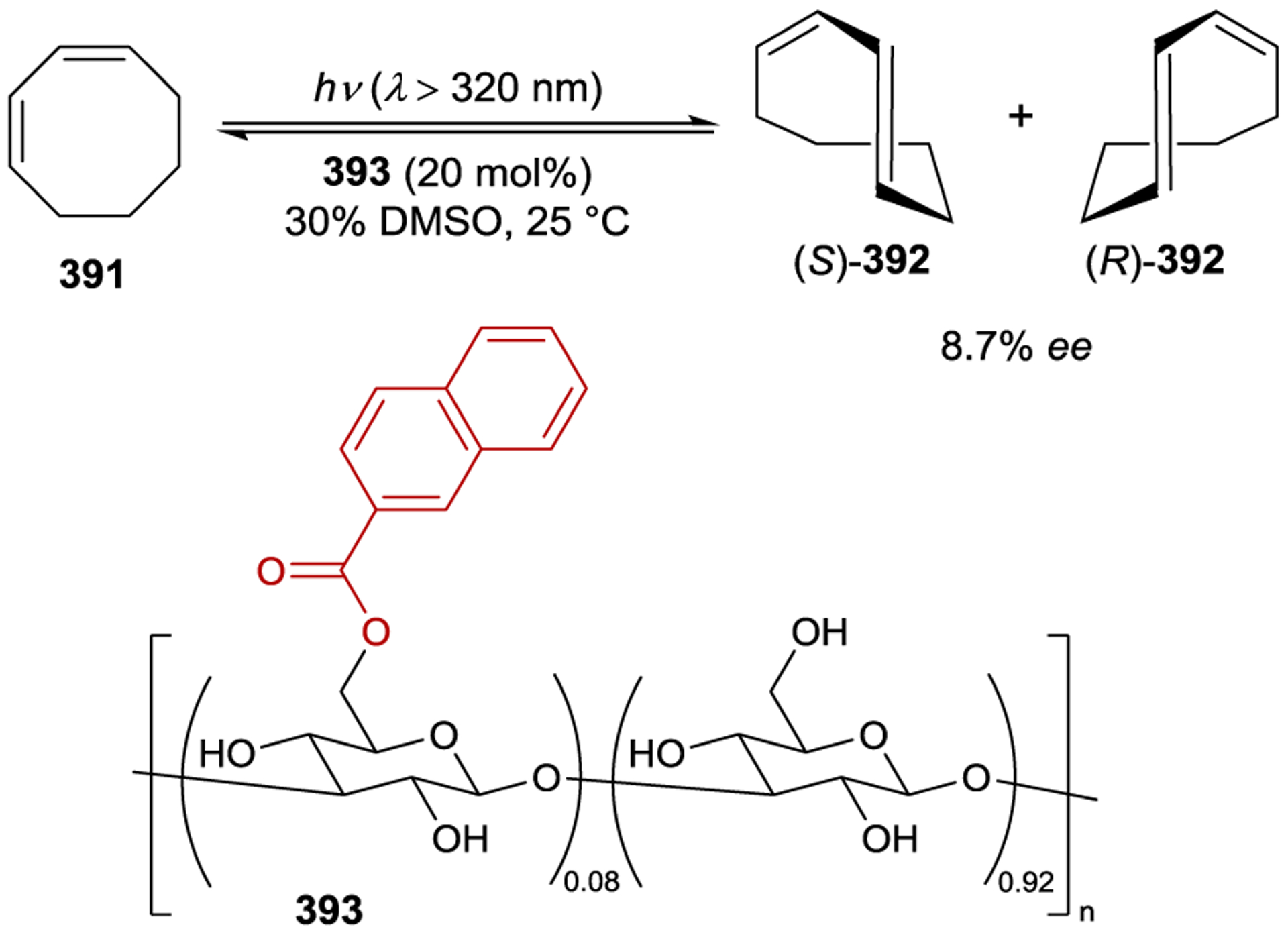
Photoisomerization of Cyclooctadiene Catalyzed by a Naphthalene Curdlan
Cyclic nigerosyl‐(1→6)‐nigerose (CNN) is an inherently chiral cyclic tetrasaccharide with a shallow cavity that can bind small organic molecules through hydrophobic interactions. Yang and Inoue synthesized terephthaloyl and isophthaloyl sensitizer-modified CNNs (394 and 395) and studied their activity in the photoisomerization of (Z)-cyclooctene 14 (Scheme 131).377 Unlike CDs, no clear ground-state interaction between CNN and (Z)-cyclooctene was observed, and photosensitization under aqueous conditions produced negligible enantioselectivity. Counterintuitively, the optimal solvent for this process proved to be diethyl ether, affording 9% ee at −40 °C with sensitizer 394. Interestingly, the isomeric sensitizer 395 favored the opposite cyclooctene enantiomer in 7% ee. Molecular mechanics calculations suggest that the small concave cavity of CNN cannot encapsulate cyclooctene, and that the phthalate sensitizer is positioned relatively far away from the stereogenic centers. The reversal in the absolute sense of stereoinduction was rationalized as an electronic effect, likely involving formation of an exciplex between the photoexcited host and 14.
Scheme 131.
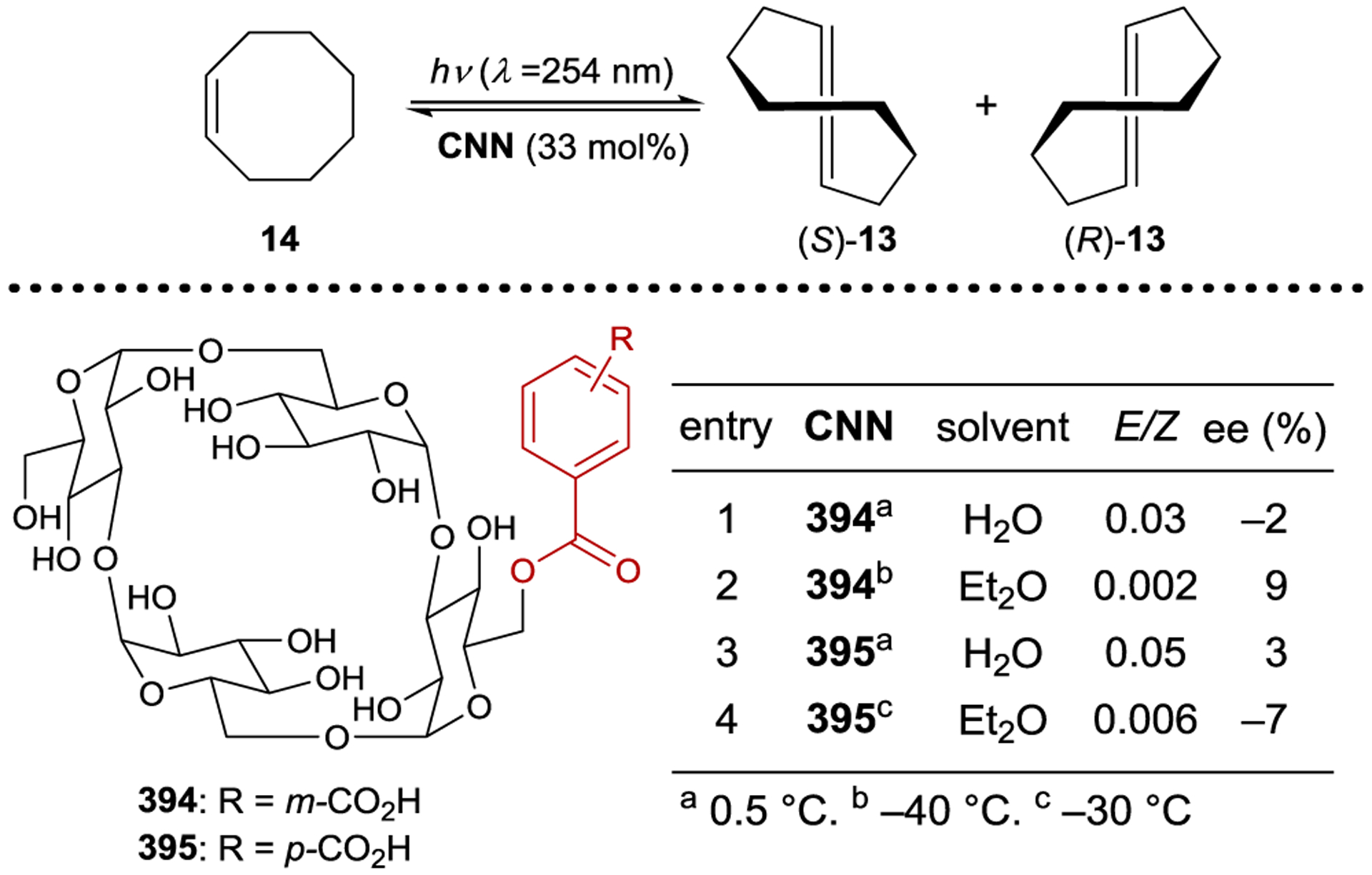
CNN Photosensitized Asymmetric Isomerization of Cyclooctene
4.3. Supramolecular Cages
Recently, chiral macromolecular metal-organic cages (MOCs) have been identified for their potential as asymmetric photocatalysts. Fujita developed an enantiomerically pure chiral diamine capped Pd6L4 cage (396), where L=triazine.378 The chiral information is provided by an external chiral diamine ligand. Fujita initially applied this MOC as an asymmetric host in the crossed [2+2] photodimerization of fluoranthene 397 with maleimide 398 (Scheme 132). Direct irradiation of fluoranthene within the preformed ground-state ternary complex resulted in formation of cyclobutane product 399 in 50% ee. The steric profile of the chiral diamine ligand was shown to directly correlate with the selectivity of the reaction; the ee decreases with less bulky diamines in the order Et(50% ee) > Me(30% ee) > H(10% ee). This observation was supported by molecular mechanics calculations, which indicated that the larger steric profile increased the tilt angle of the triazine ligand, deforming the MOC structure. This MOC catalyst was also applied to the asymmetric [2+2] photocycloaddition of aceanthrylene and 1H-cyclopenta[l ]phenanthrene with maleimide 398.379 Irradiation of these ternary complexes formed within the molecular cage produced the resulting cyclobutane products in 44% ee and 33% ee, respectively.
Scheme 132. [2+2] Photodimerization Catalyzed by a Ru-Pd MOC.
Adapted from Ref. 378. Copyright 2008 American Chemical Society.
Zhang and Yang380 utilized a water-soluble chiral-at-Fe tetrahedral MOC originally synthesized by Nitschke381 to study the photodimerization of anthracene 311. A single molecule of 311 is included within the small cavity formed by the Fe complex, while a second molecule was proposed to weakly associate through π-stacking interactions with the cage. Upon irradiation, the syn-HT and anti-HH dimers are formed in 14% ee and 4% ee, respectively.
Liu and Su identified an enantiopure Ru-Pd MOC that could be used for the photocatalysis of both electron and energy transfer reactions. This cage consists of six Pd termini that are linked through eight chiral Ru-polypyridyl complexes. The polypyridyl ligands define 12 box-like portals around the MOC that can include small organic substrates through hydrophobic and π-stacking interactions. Irradiation of bromonaphthol 400 in the presence of 10 mol% Δ-Ru-MOC 401 (λ = 453 nm) promoted its dimerization to afford 402 in 32% yield and 32% ee (Scheme 133).382 Lowering the photocatalyst loading to 5 mol% enhances the enantioselectivity to 58% ee. The macroscopic MOC structure is required to achieve reasonable enantioselectivity, as sensitization with monomeric Δ-Ru(phen)32+ gives only 10% ee. Notably, the oxidative dimerization of naphthol in solution typically affords the 1,1’-bis(2-naphthol) isomer; however, the 1,4-dimer is the exclusive product of the MOC-mediated process. A mechanism proposed for the reaction is shown in Scheme 133. Reductive quenching of the Ru-polypyridyl excited state by oxygen produces a strongly oxidizing Ru(III) center within the MOC. Oxidation of a bound 2-naphthol affords naphthyl radical 403, which can further oxidize to ortho-naphthoquinone 404. Addition of another equivalent of radical 403 to 404 within the chiral pocket of the MOC results in enantioselective formation of 402.
Scheme 133. MOC Photoredox-Catalyzed Dimerization of Bromonaphthol.
Adapted with permission from Ref. 383. Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
Su expanded the use of Δ-Ru-MOC 401 to the photosensitized dimerization of bromo-substituted acenaphthylene 405, which produces four isomers, two of which are chiral (Scheme 134).383 Irradiation of 405 in the presence of 10 mol% Δ-Ru-MOC 401 exclusively forms the anti-HH dimer 406 in 92% yield and 86% ee. In this reaction, the Ru(II) centers catalyze the photocycloaddition through an energy-transfer mechanism, rather than by electron-transfer.
Scheme 134.
Enantioselective Dimerization of Bromoacenaphthylene.
4.4. Nucleic Acids
DNA and RNA adopt chiral macromolecular helical conformations, the major and minor groves of which are lipophilic and associate organic molecules through hydrophobic interactions. Inoue identified these systems as potential asymmetric photocatalysts for the isomerization of (Z)-cylooctene 14 (Scheme 135).384 This study began with an examination of the ability of the monomeric nucleoside thymidine (407) to sensitize the photoisomerization of (Z)-14 through exciplex formation. The enantioselectivity observed using 50 mol% 407, however, was low (5% ee). Calf thymus DNA (409) gave the opposite enantiomer with a somewhat higher absolute value (−9% ee). The base pair stack of DNA, however, makes the formation of an exciplex unlikely. Instead, Inoue proposed that (Z)-cyclooctene binds within the minor groove of the DNA. Inoue subsequently examined the activity of synthetic oligonucleotides to sensitize the photoisomerization of 14. Interestingly, single-stranded d(T)15 (411) and double-stranded d(T)15 •d(A)15 (412) gave reasonable enantioinduction (14% and 19%, respectively), while d(A)15 alone was unselective (<1% ee).385
Scheme 135.
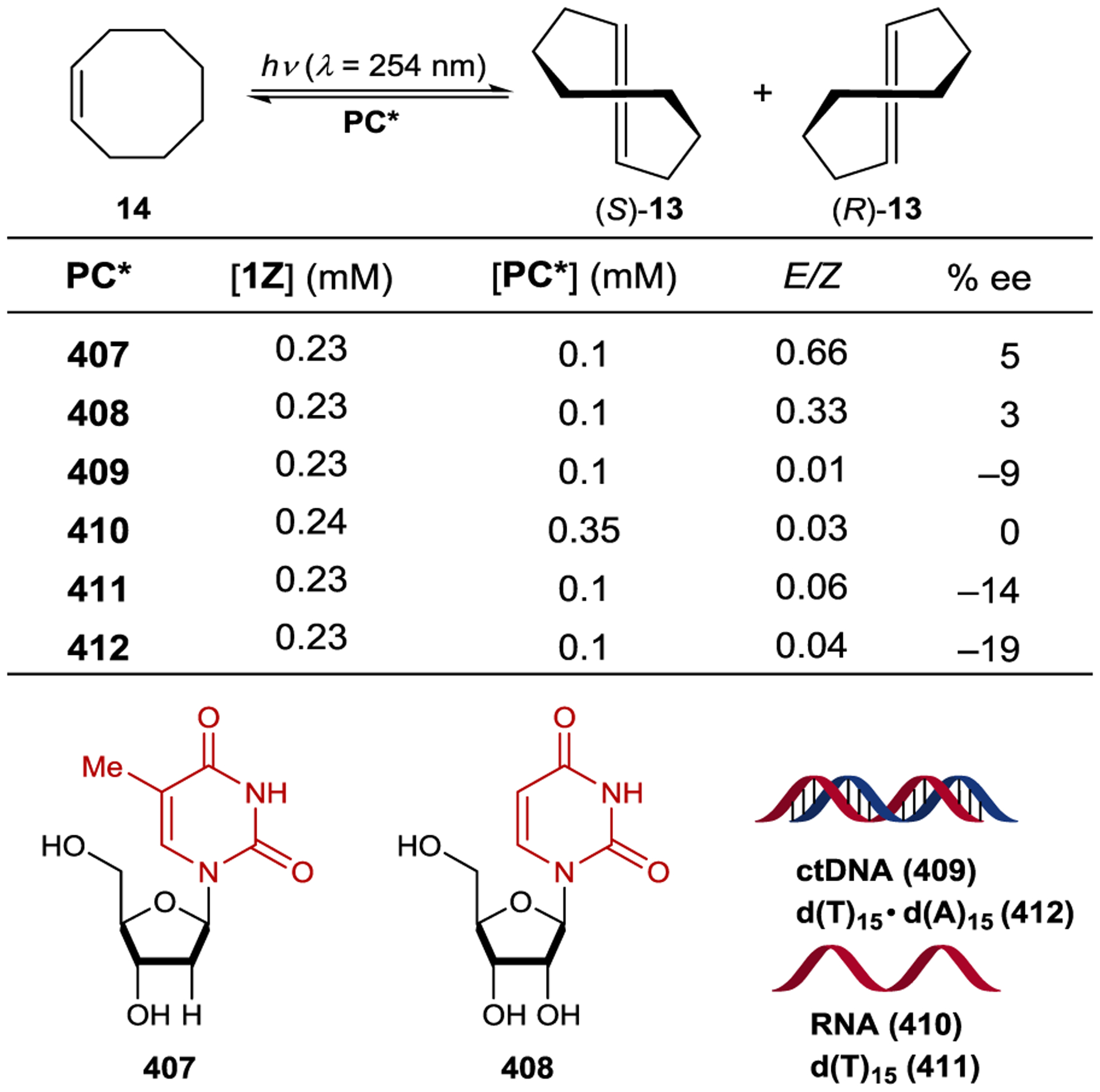
Asymmetric Cyclooctene Isomerization Catalyzed by Nucleosides or Single and Double Stranded DNA
Wagenknecht sought to design a DNAzyme capable of acting as a photocatalytic triplet sensitizer.386–387 388 A combination of the water solubility of DNA and the ability to encapsulate an organic substrate would meet the necessary requirements to provide asymmetric photocatalysis under aqueous conditions. Initial studies examined the [2+2] photocycloaddition of 45 using an artificial benzophenone-derived nucleoside implanted within a strand of DNA.387 However, these studies were hindered by parasitic charge transfer from photoexcited benzophenone to the stacked nucleobases.388 To promote energy transfer over intrastrand charge transfer, the authors positioned the artificial benzophenone nucleoside within a DNA three-way junction directly next to the favored substrate binding site (414 and 415; Scheme 136). Irradiation of quinoline 45 with 20 mol% 414 provided the cyclobutane products 47 and 413 in reasonable yields but with poor regioselectivity. The enantioselectivity of both regioisomers was similar, with the major formed in 28% ee. A methoxybenzophenone analogue (415) provided slightly lower ee.
Scheme 136.
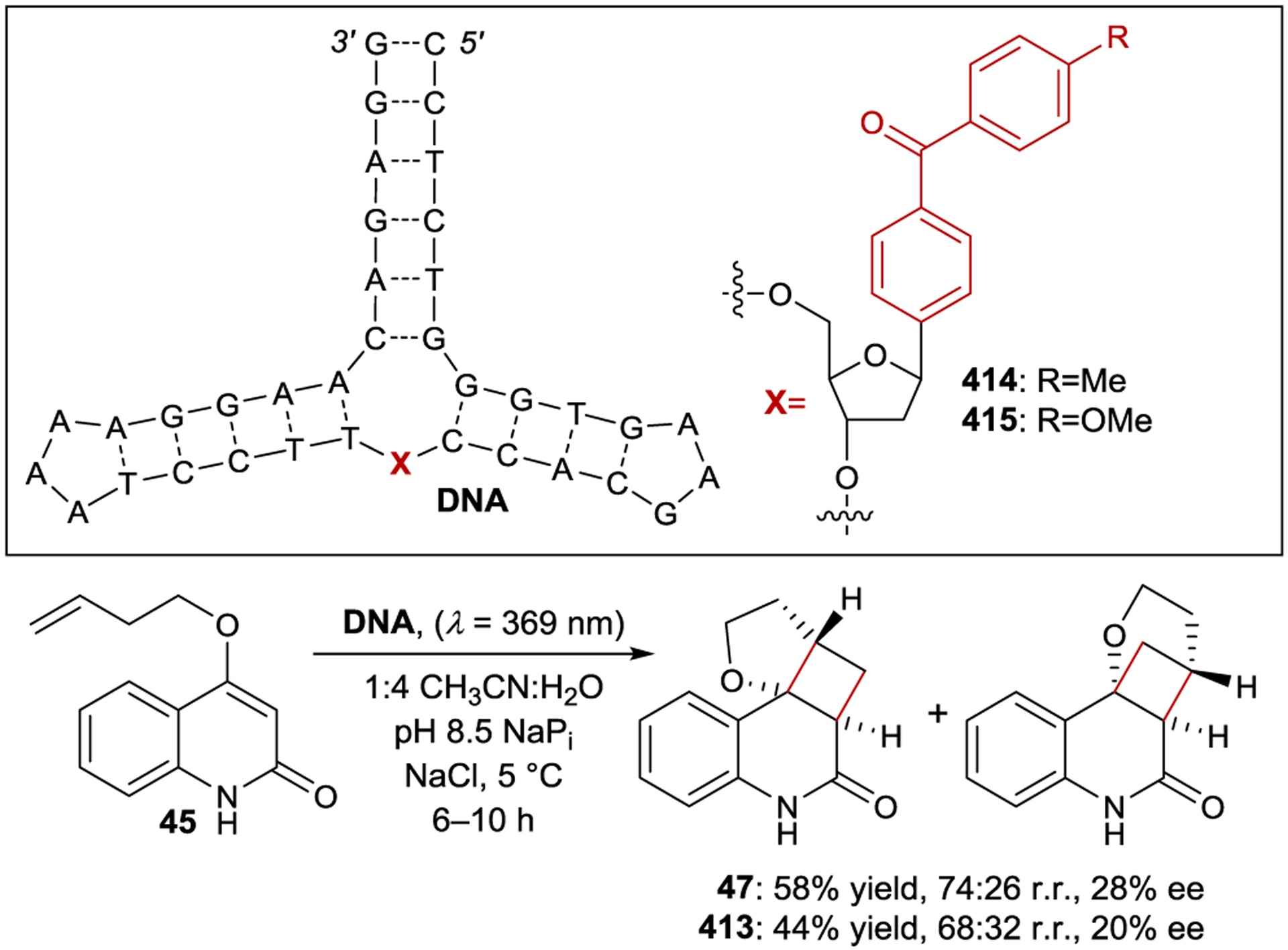
Photosensitized [2+2] Cycloaddition Catalyzed by an Artificially Modified DNA Three-Way Junction
4.5. Photoactive Proteins
There are relatively few photochemical enzymatic reactions known in nature, and of these, only one is an enantioselective organic transformation.389 The enzymatic reduction of protochlorophyllide to chlorophyllide is the penultimate step in the synthesis of chlorophyll; the key photochemical process in this transformation is a photoinduced comformational change that promotes a sequential electron and proton transfer to protochlorophyllide.390, 391 The use of enzymes in asymmetric photochemical synthesis is a relatively recent development.392, 393
4.5.1. Non-sensitizing proteins
Serum albumins (SAs) are the most abundant proteins in the mammalian circulatory system.394, 395 Inoue investigated these proteins as non-photoactive chiral templates for the [4+4] dimerization of anthracene carboxylate 311. As carrier proteins, serum albumins have a variety of binding domains of differing size providing numerous locations to associate organic substrates. The innate chirality of the protein binding domains thus provides a means to control the stereochemistry of photoreactions that occur within them.
Initial studies focused on photodimerizations in the presence of bovine serum albumin (BSA).396, 397 Four binding domains (I–IV) were identified that could include anthracene. Binding domain I associates a single anthracene molecule with the largest equilibrium constant, but the structure of the active site prevents the associated anthracene from reacting with molecules in the bulk solution. Binding domains II–IV are larger and associate three, two, and three molecules of anthracene, respectively, with decreasing affinity. Irradiation of 311 in the presence of 5 mol% BSA under aqueous conditions yields a near equal ratio of the HT and HH anthracene dimers in good conversion. The chiral syn-HT dimer 312 and anti -HH dimer 315 were formed with enantioselectivities of 11% ee and 14% ee, respectively (Scheme 137). Under these conditions, 311 populates each of the four accessible binding domains. Increasing the BSA loading decreases the population of 311 within domains III and IV and leads to an increase in the enantioselectivity of both 312 and 315 (22% ee and 39% ee respectively). Inoue rationalized this observation by proposing that binding domain II provides higher selectivity.
Scheme 137.
Anthracene Photodimerization in the Presence of BSA
Inoue then screened a library consisting of bovine (BSA), human (HSA), sheep (SSA), rabbit (RSA), porcine (PSA), and canine (CSA) serum albumins.398 Binding domain I remained unreactive throughout the series, while subsequent binding domains were more varied. Irradiation with 75 mol% SA (33 mol% for HSA and CSA) provided low conversions due to the inactive domain I (Table 1). Notably, HSA provided anti-HH dimer 315 in 90% ee. CSA proved optimal for the formation of syn-HT dimer 312 (97% ee). PSA provided the opposite enantiomer of 312 with a slightly lower 89% ee.
Table 1.
Anthracene Photodimerization Mediated by Different Serum Albumins
| SA (equiv) | Conversion (%) | syn-312 ee (%)c | anti-315 ee (%) |
|---|---|---|---|
| BSA (0.75) | 2 | −10 | 43 |
| HSAb (0.33) | 13 | 82 | 90 |
| SSA (0.75) | 14 | −11 | 51 |
| RSA (0.75) | 6 | 47 | 28 |
| PSA (0.75) | 4 | −89 | 25 |
| CSA (0.33) | 42 | 97 | 18 |
conditions: 0 °C, pH=7.0, [AC] = 0.6 mM;
5 °C;
positive/nagative sign corresponds to an excess of the first/second-eluted enantiomer, respectively.
Inoue and Bohne provided a more detailed interrogation of HSA in the photodimerization of anthracene 311.399 HSA differs from BSA by 26 amino acids and has two photoinactive binding domains (I and II) that each associate a single anthracene molecule. Photodimerization occurs within binding domains III, IV, and V. Domain III binds up to three anthracene molecules in a well-ordered site, whereas binding domains IV and V are larger and more accessible, associating two or more anthracenes, each with smaller association constants. Irradiation of 311 in the presence of 33 mol% HSA provided anti-HH dimer 315 in 88% ee, with syn-HT dimer 312 in 80% ee (Scheme 137).400 The enantioselectivity of both dimers increased with HSA concentration. This correlates with an increased population of 311 within binding domain III, suggesting domain III provides the highest selectivity for the chiral photodimers. Inoue developed a sequential ‘batch feeding’ process to overcome the need for higher concentration of HSA. With this process, 3.4 turnovers of HSA could be achieved without substantial loss of enantioselectivity; 312 and 315 could be isolated in 73% ee and 82% ee respectively under these conditions.401–402 403
The unproductive binding domains I and II in HSA preclude effective catalysis. Inoue and Otagiri performed selective mutations of arginine, tyrosine, and lysine residues within these domains to destabilize the association of anthracene.404 Circular dichroism titrations confirmed that both a double mutant and a triple mutant showed lowered affinity for anthracene inclusion within domains I and II (Scheme 137). This resulted in a large rate increase in the photodimerization of anthracene 311, with only a small effect on the enantioselectivity.
Yokoyama showed that the asymmetric photoinduced ring closure of a series of bis(thiophen-3-yl)hexafluorocyclopentene based diarylethenes (DTE) (416) could be performed in the presence of HSA (Scheme 138).405,406 Hydrophobic interactions between the small organic substrates and HSA facilitated incorporation within multiple binding domains of HSA. Further modification of the DTEs with polar functional groups promoted hydrogen bonding interactions with accessible peptide side chains within the binding domains. Irradiation at 313 nm under aqueous conditions provided the chiral ring-closed products (417–421) in reasonable conversion ratios (60–90%) and enantioselectivities (up to >98 %ee).
Scheme 138.
HSA Directed Asymmetric Photoisomerization of DTE Derivatives
In a recent report, Yokoyama showed that bis(thiophen-2-yl)hexafluorocyclopentenes also undergo enantioselective photocyclization in the presence of HSA.407 Irradiation of the anionic carboxylate-derived DTE by 366 nm light produces the (S,S)-enantiomer of the ring-closed DTE (422) in 98% ee with complete conversion. Interestingly, the neutral methyl ester DTE favored formation of the opposite (R,R)-enantiomer (423) in 83% ee at 80% conversion. This inversion of chirality was also observed in the thiophen-3-yl analogues previously studied (Scheme 138).
Finally, anthracene dimerization has also been studied in the presence of the molecular chaperone protein prefoldin (PFD).408 Multiple weakly binding domains for anthracene 311 were identified from circular dichroism titrations. Irradiation of 311 in aqueous solution in the presence of 20 mol% PFD resulted in the formation of anti-HH dimer 315 in 10% ee and syn-HT dimer 312 in 16% ee.
4.5.2. Photoredox Enzymes
In 2016, Hyster reported the first example of non-native photoredox catalysis using an enzyme.409 Hyster found that an electron donor–acceptor (EDA) interaction formed between the nicotinamide cofactor (NADPH) and added halogenated lactones (424), which localize within the ketoreductase active site. This EDA complex was characterized by the appearance of a lower-energy visible absorption feature. Selective irradiation of the EDA complex results in photoinduced single-electron reduction of 424. Mesolytic cleavage of the resulting radical anion results in the formation of an α-acyl radical, which undergoes stereoselective hydrogen atom abstraction from the nicotinamide radical cation to afford dehalogenated lactone 425 with high enantioselectivity (Scheme 139). The oxidized NADP is turned over with an external reductant. NADPH fails to promote the reaction in the absence of enzyme, highlighting the importance of preassociation within the active site. Ketoreductase LKADH proved optimal, providing (R)-425 in 81% yield with 96% ee. RasADH was identified from a screen of ketoreductases with large active sites and provided the opposite enantiomer (S)-425 in 51% yield and 85% ee. Notably, enzyme loadings as low as 0.25 mol% still provide good yields and enantioselectivities.
Scheme 139.
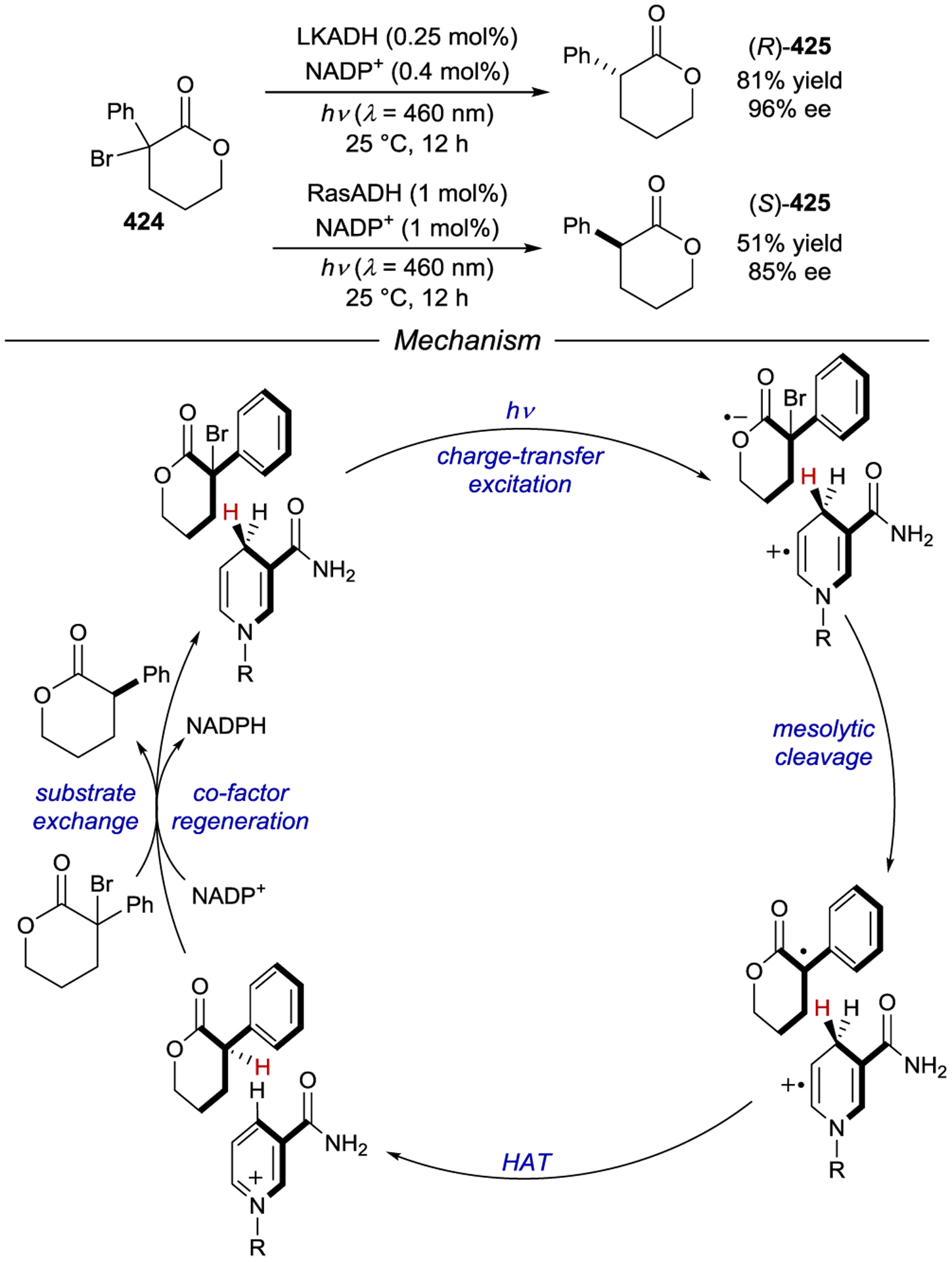
Asymmetric Photocatalytic Dehalogenation by Unnatural Photoactive Enzymes
The formation of EDA complexes within enzyme active sites was expanded to include flavin-dependent ‘ene’-reductases (ERED). Hyster reported that α-chloroamides readily form EDA complexes with flavin mononucleotide (FMN) cofactors in the ERED active site. Here, photoinduced electron transfer and dehalogenation are followed by radical cyclization and hydrogen atom transfer to produce enantioenriched β-stereogenic lactams (Scheme 140).410 Selective mutation of a surface threonine residue produced the optimal enzyme GluER-T36A, which provides lactam 427 in 91% yield and 92% ee. Both the cyclization and hydrogen atom transfer are highly stereoselective, yielding product 429 in excellent diastereo- and enantioselectivity (> 98% ee, 98:2 d.r.). Notably, the high yield and enantioselectivity (92% yield and 88% ee) could be maintained using lyophilized cell-free lysate for a gram-scale reaction. The generally high substrate specificity characteristic of enzymatic catalysis often limits the generality of a given photoactive enzyme. In this study, Hyster performed a screen of alternate EREDs that could broaden the scope of highly selective photocyclizations. NostocER and MorB-Y72F proved to be optimal for the synthesis of enantioenriched lactams 430 and 431 (Scheme 140). The reaction is not limited to α-chloroamides; Hyster showed that the formation of an EDA complex between FMN and alkyliodides facilitates PET reduction of these substrates.411 An ERED with an active site tyrosine mutation (GluER-Y177F), which was required to avoid reduction of the starting alkene, proved optimal for this reaction. Irradiation of the EDA complex gives cyclized product 433 in reasonable yield and 84% ee. An Old Yellow Enzyme (OYE) containing the same active-site tyrosine mutation, OYE2-Y197F, could provide the opposite enantiomer of 433 in slightly lower yield and 72% ee.
Scheme 140.
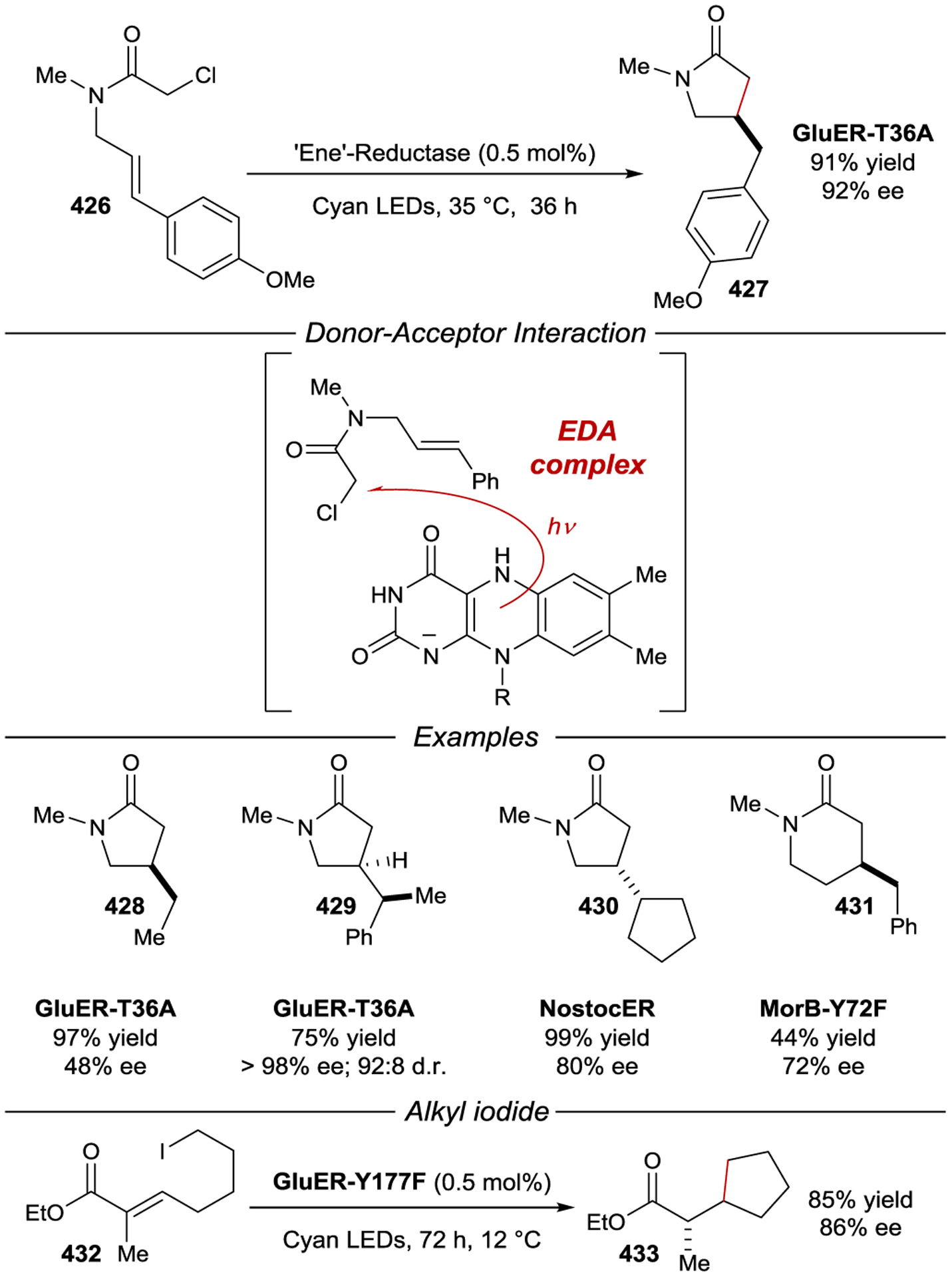
Dehalogentation–Cyclization Photocatalyzed by ‘Ene’-Reductases
Intermolecular enzymatic photoreactions present a significant challenge because they require two or more substrates to be organized near the sensitizing moiety within the active site of the stereocontrolling enzyme. The groups of Hyster412 and Zhou413 contemporaneously described similar photoenzymatic methods for the stereoselective γ-functionalization of α-chlorocarbonyls through an intermolecular reductive dehalogenation/radical addition sequence. Hyster proposed that stereoselectivity arises from the formation of a quaternary charge-transfer EDA complex comprising both substrates, the photoactive cofactor, and the encapsulating enzyme (Scheme 141). UV-vis experiments showed that all four components were required to produce an appreciable low-energy EDA transition.412 The EREDs GluER-T36A and NostocER proved optimal under Hyster’s conditions, providing opposite enantiomers of γ-functionalized 435 in 98% ee and 80% ee, respectively. Zhou identified Old Yellow Enzyme (OYE1) as optimal, providing the γ-functionalized ketone 437 in 88% yield and 96% ee.413 The formation of the quaternary EDA complex positioned the photochemically generated α-radical near the alkene acceptor, which facilitates rapid intermolecular radical addition. This was required to outcompete a parasitic hydrogen atom transfer that leads to the dehalogenated byproduct.
Scheme 141.
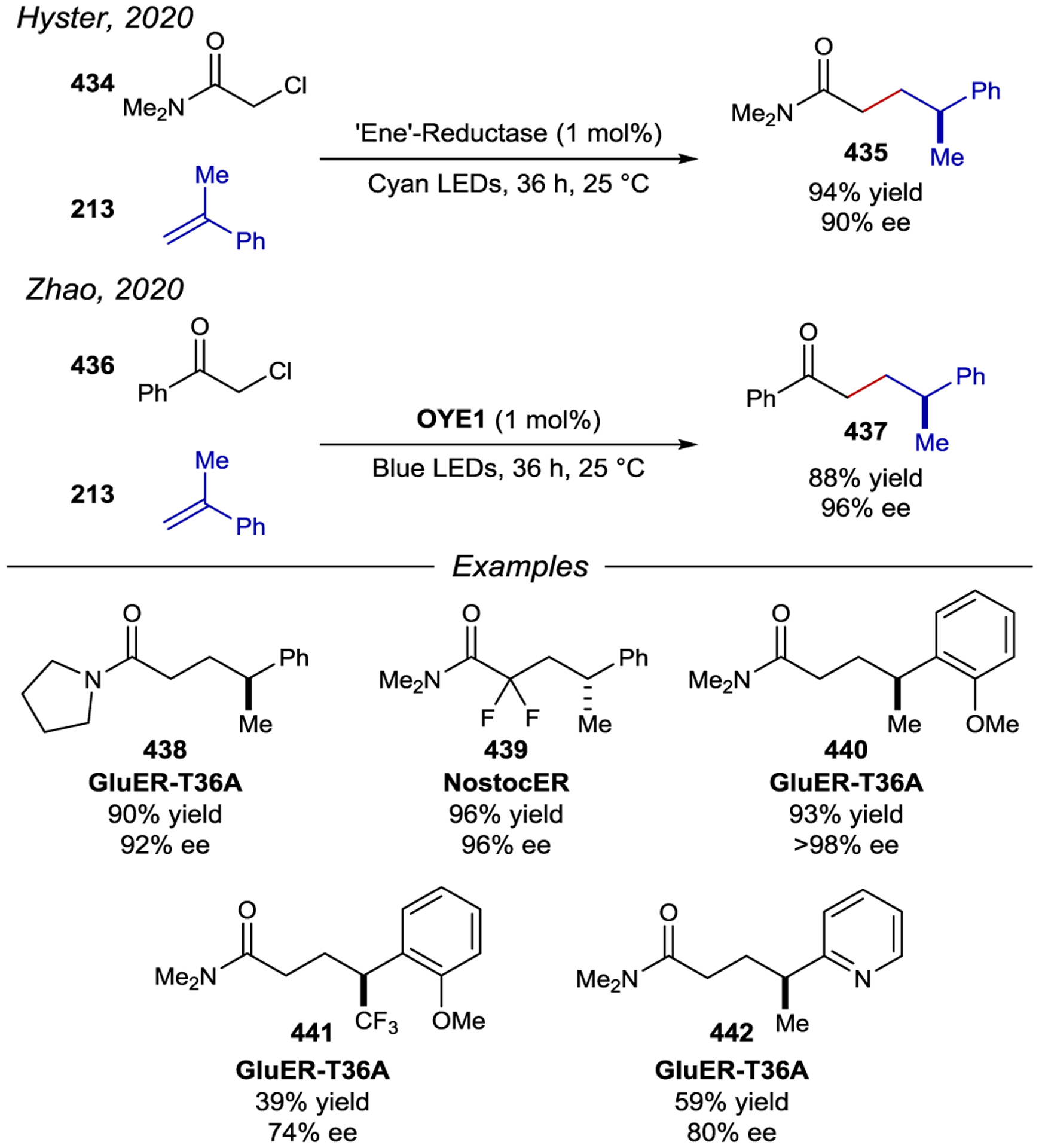
Intermolecular Dehalogenation–Radical Addition Promoted by a Quaternary Charge-Transfer Complex
Hyster also showed that photoactive EREDs can catalyze useful enantioselective photoreactions without forming EDA complexes. This concept was first demonstrated by developing a method for the net redox-neutral cyclization of α-halo-β-amidoesters to afford 3,3-disubstituted oxindoles.414 Irradiation of 443 in the presence of 0.5 mol% of the flavin-dependent ERED enzyme OPR1 resulted in the formation of 444 in 94% yield and 90% ee (Scheme 142). Notably the strongly reducing flavin semiquinone (FMNSQ) was produced through photoinduced oxidation of the tricine buffer and was proposed to be the active one-electron reductant for this transformation.
Scheme 142.
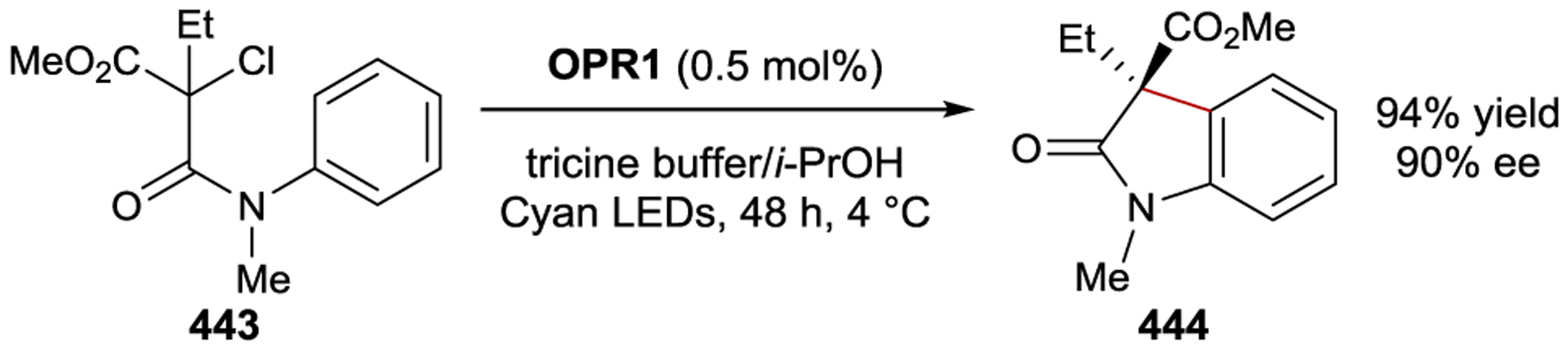
Photoenzymatic Redox-Neutral Cyclization of α-Halo-β-Amidoesters
Acrylamides associate within the active site of EREDs, but no signals attributable to EDA complex formation with the FMN cofactor could be observed. Nevertheless, irradiation of acrylamide 445 in the presence of 0.5 mol% of OYE mutant OYE1-F296G results in the formation of reduced amide 446 in 80% ee (Scheme 143).415 Photoexcitation of the FMN cofactor produces a strongly reducing excited state; subsequent photoinduced electron transfer to the bound acrylamide results in the formation of a radical anion. Transient absorption spectroscopy showed that the photoinduced electron transfer occurs within 10 ps and is followed by rapid, regioselective protonation affording the α-acyl radical. Enantiodetermining hydrogen atom transfer is rate-limiting. Notably, this enzyme also enables the photocatalytic defluorination of 447 in 74% ee.
Scheme 143.
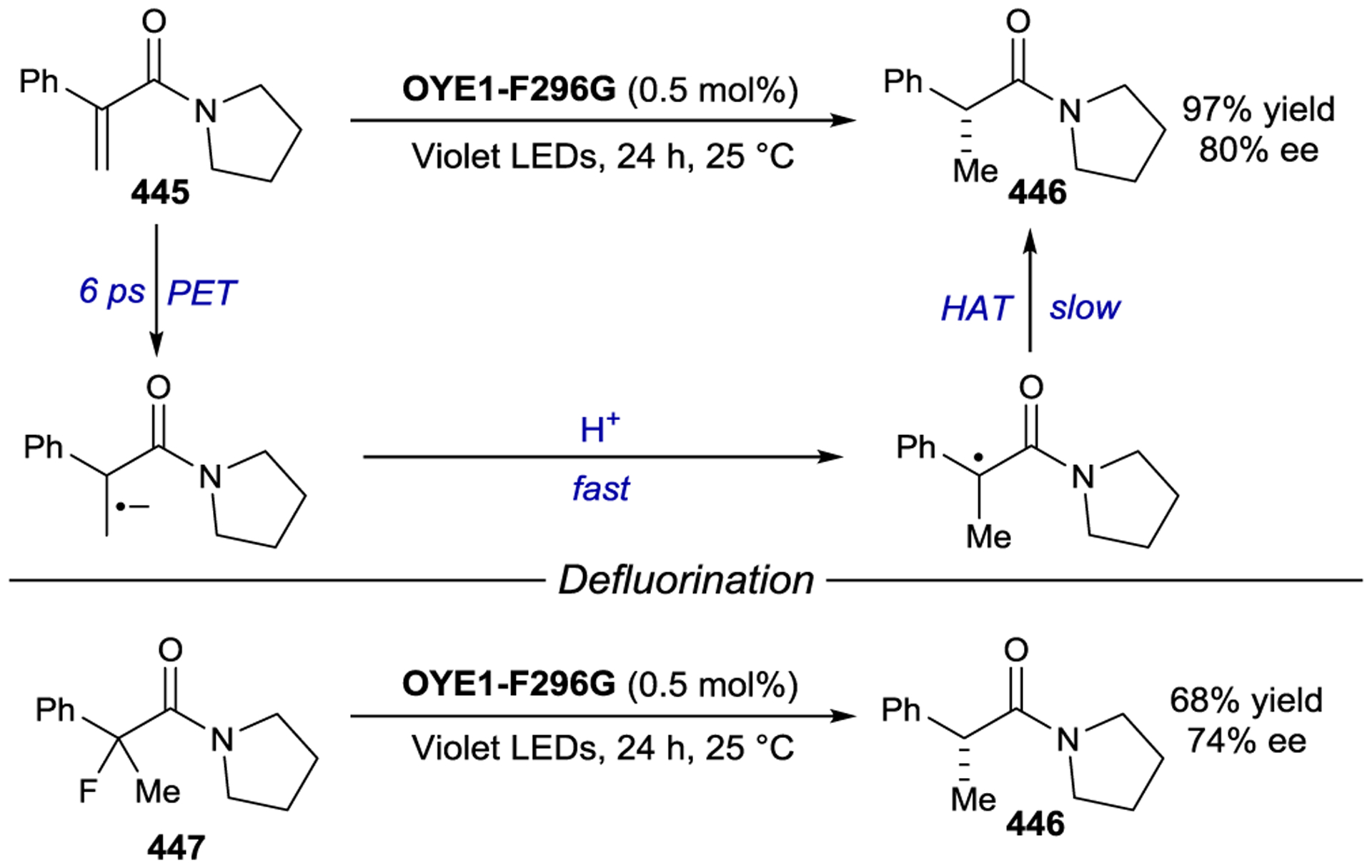
Hydrogenation and Defluorination by Photoexcitation of FMN
Non-native photoactivity can also be introduced into enzymatic reactions by using exogenous photocatalysts that associate with the enzyme through non-covalent or covalent interactions. Park and Hollmann described the spontaneous association of Rose Bengal (448) and its derivatives with the ERED TsOYE (Scheme 144).416 Excitation of the enzyme-bound 448 leads to PET reduction of the FMN cofactor, providing the necessary redox equivalents to promote the highly enantioselective reduction of 2-methylcyclohexenone 449. UV-vis spectroscopy along with electrochemical protein film voltammetry provided clear evidence for a ground-state interaction between 448 and TsOYE. Overall, the photochemical reduction of 449 to (R)-450 proceeds in high yield and >99% ee. The enzyme can also be immobilized on alginate beads, which enables the recycling of the OYE/RB photoenzymatic system.417 Conversions of up to 50% could be maintained over five reaction cycles without loss of enantioselectivity.
Scheme 144.
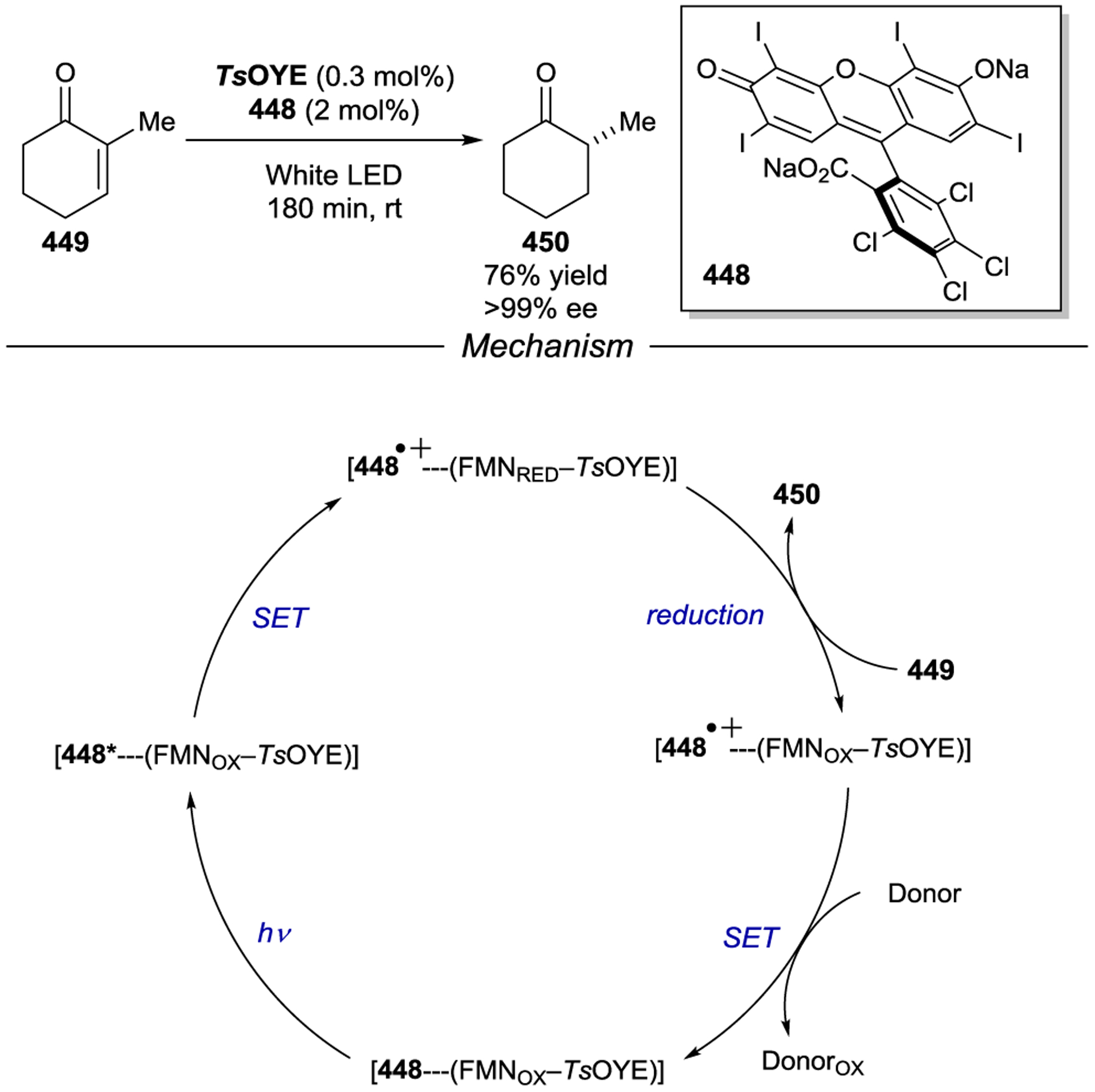
Enantioselective Reduction of Cyclohexenone by a Rose Bengal Associated Enzyme
There has been significant interest in covalently modifying enzymes with artificial photocatalytic cofactors;418 however, it has proven difficult to develop highly enantioselective organic reactions using this strategy.419, 420 Cheruzel described hybrid P450 BM3 enzymes with covalently tethered ruthenium polypyridyl photocatalysts.421 Irradiation of these enzymes was shown to enable the stereoselective hydroxylation of 10-undecenoic acid (451) to (R)-9-hydroxy-10-undecenoic acid (452) in 40% yield and 85% ee (Scheme 145).422 In a follow up study, Cheruzel described a tandem photocatalytic process that began with a non-enzymatic photoredox trifluomethylation of diphenylmethane 453 followed by photoenzymatic hydroxylation affording chiral secondary alcohol 455 in 93% ee.423
Scheme 145.
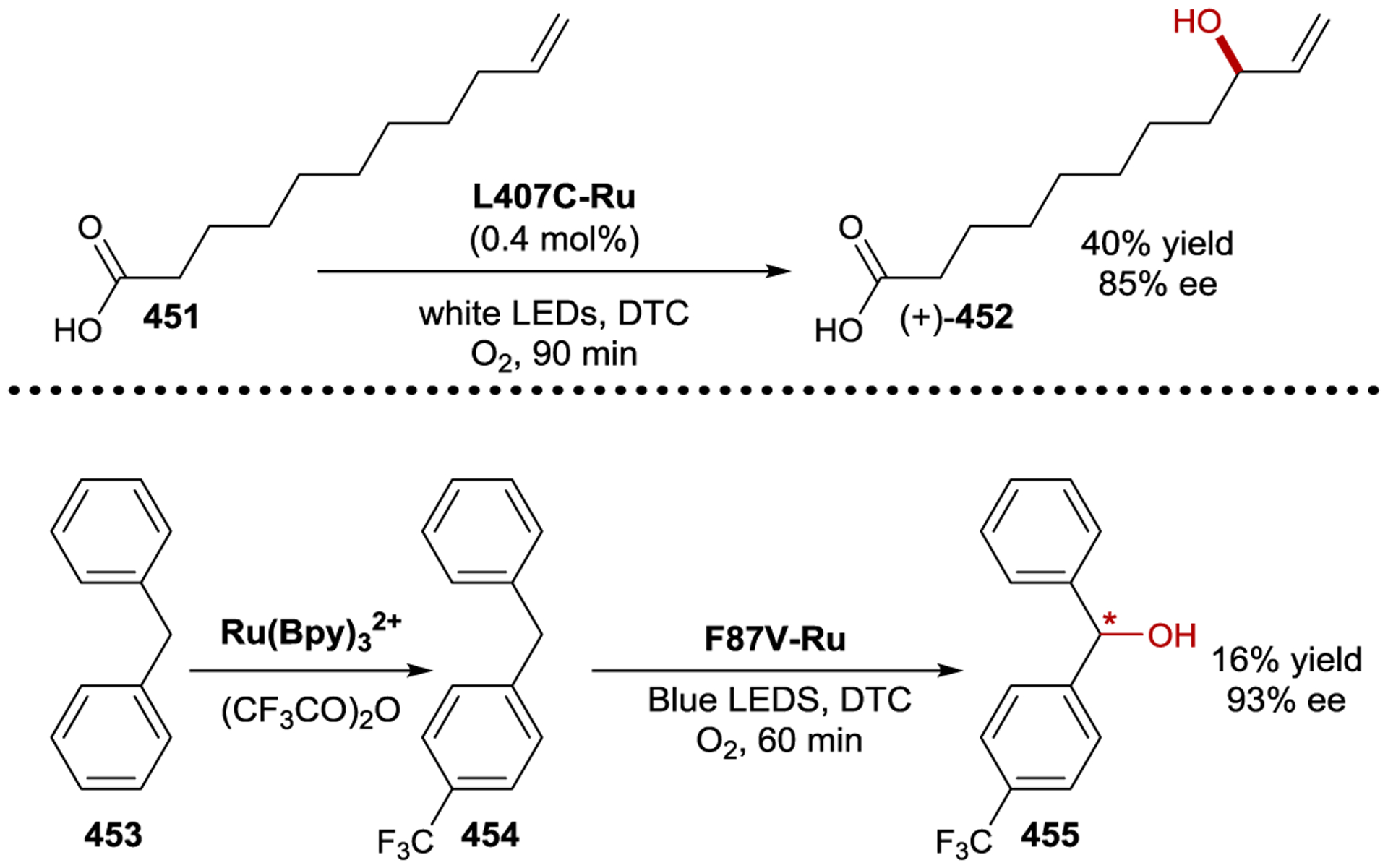
Enantioselective Hydroxylation by a Ru-Polypyridyl Derived P450 Enzyme
5. Conclusion
After several decades of relatively slow development, there has been a recent, rapid increase in the pace of discovery of photocatalytic reactions that deliver enantioselectivities on par with those obtained in modern ground-state asymmetric catalytic reactions. In many cases, chiral photocatalyst structures have been developed by adapting catalyst classes originally designed for use in ground-state transformations. Several of these show promise to become privileged scaffolds within asymmetric photochemistry as well, including the secondary-amine catalysts studied by Melchiorre and oxazaborolidine catalysts investigated by Bach. Various photocatalyst structures unique to excited-state reactions have also been created to address the specific challenges relevant in asymmetric photochemistry. Many of these incorporate a chiral moiety into the structure of a known photosensitizing moiety. Examples include the chiral (thio)xanthone catalysts pioneered by Bach and the chiral transition metal photocatalysts developed by Meggers. Finally, inherently chiral biomolecules have also been adapted as photocatalysts in asymmetric photoreactions, as demonstrated by the research of Inoue, Zhou, and Hyster.
While the structures of these photocatalysts are diverse, and the mechanisms by which they operate varied, a common design principle across multiple catalyst classes is the requirement for ground-state association of the substrate to the photocatalyst prior to excitation. This preassociation is one way to ensure that the catalyst meets the criteria described above. Preassociation sets the substrate within the chiral environment of the catalyst, and in most cases increases the rate of the enantioselective transformation over any competitive racemic background reactivity. However, as a consequence, the structures of these chiral catalysts and the resulting catalyst–substrate interactions have often been rigid and well-defined. This idea is similar to many of the early strategies for asymmetric catalysis in conventional, ground-state reactions; however, there has been a growing recognition that catalyst flexibility can be an important design strategy for thermally activated reactions.424 As the field of asymmetric photocatalysis continues to develop, it stands to reason that the breadth of potential catalyst structures for enantioselective photocatalysis might be similarly broad.
The field of asymmetric photochemistry is poised for significant development over the next few decades. At this early stage in the development of the field, most examples of highly asymmetric photocatalytic reactions have been proof-of-principle academic investigations, and these insights have yet to be translated to commercial applications. As the pharmaceutical industry in particular becomes more interested in the scaleup of photocatalytic processes,425 ,426 issues concerning maximization of quantum yield and light flux427 may become more important than they have been to date. Despite the impressive advances that have emerged in the field of asymmetric photocatalysis in recent years, many challenges remain unsolved, perhaps most important of which is the need for generality. Some photocatalyst structures show promise to develop into privileged structures for enantioselective photochemical synthesis, catalyzing a number of highly enantioselective organic photoreactions with selectivities over 90% ee. These structures, however, are exceptional, and continued investigations are required to both establish their generality and to identify other classes of privileged asymmetric photocatalysts. While asymmetric photoreactions have the potential to greatly expedite the synthesis of complex products in unexplored areas of chemical diversity space, the continued development of more general methods is required before these reactions are used routinely by non-photochemists. We expect that the insights gained in research in this field to date will provide a solid foundation for continued transformative innovation.
Acknowledgements
The authors acknowledge funding from the NSF (CHE‐1954262) and NIH (GM095666, GM127545). M.J.G. and J.B.K. acknowledge personal fellowships from the NSF Graduate Research Fellowship Program (DGE-1747503). W.B.S. acknowledges an NIH Kirschstein-NRSA postdoctoral fellowship (F32GM134611).
Biographies
Matthew J. Genzink completed a B.Sc. in Chemistry from Grove City College in 2018 where he performed research with Charles Kriley. During this time, he performed a research internship for three months at KU Leuven (Belgium) where he studied fluorescent probes with Wim Dehaen. In 2018 Matthew started at UW–Madison, joining the group of Tehshik Yoon where he was an NSF Graduate Research Fellow. Matthew currently studies Brønsted acid-catalyzed asymmetric photochemistry in the Yoon group. In his free time Matthew enjoys making dough and riding bike.
Jesse B. Kidd received a B.Sc. in Chemistry from Concord University in 2016 where he performed research with Prof. Darrel Crick investigating flavone derivatives as antiproliferative agents. During that time, he participated in the CCMR-REU at Cornell University studying the properties of polydimethylsiloxane embedded with photo- and mechanoresponsive spiropyran under the supervision of Prof. Meredith Silberstein. He also conducted research under Prof. Jeremy B. Morgan at the UNC – Wilmington exploring the synthesis of β-substituted tryptamines. In 2016 Jesse began his doctoral studies at UW–Madison where he was an NSF Graduate Research Fellow in Prof. Tehshik Yoon’s lab. His research focused on the application of chiral hydrogen-bonding photocatalysts to enantioselective excited-state reactions and the study of their mechanisms.
Wesley B. Swords completed a B.Sc. in Biochemistry/Chemistry from the University of California, San Diego in 2013. There he performed research in the group of Prof. Joshua Figueroa. He joined the group of Prof. Gerald Meyer Group at UNC – Chapel Hill where he was an NSF Graduate Research Fellow. He spent some time as a visiting researcher at Uppsala University, Sweden, in the group of Prof. Leif Hammarström through the NSF GROW program. His doctoral research covered the use of non-covalent interaction to study halide oxidation and proton-coupled electron transfer in solution and at the semiconductor interface. He received his Ph.D. from UNC in 2018. Wesley is currently an NIH Kirschstein-NRSA funded postdoctoral research fellow in the group of Prof. Tehshik Yoon at UW–Madison where his research has focused on the development of asymmetric photocatalysis and fundamental investigations into photochemical mechanisms.
Tehshik Yoon received his MS and PhD from Caltech under the supervision of Erick Carreira and David MacMillan, respectively. This was followed by an NIH postdoctoral fellowship at Harvard with Eric Jacobsen. He has served on the faculty at the University of Wisconsin–Madison since 2005. Prof. Yoon’s research interests largely focus on the use of photochemistry in organic synthesis. His research and scholarship has been recognized with several awards including the Corporation Cottrell Scholar Award, the Beckman Young Investigator Award, the Amgen Young Investigator Award, an Alfred P. Sloan Research Fellowship, an Eli Lilly Grantee Award, a Friedrich Wilhelm Bessel Award from the Humboldt Foundation, and the ACS Cope Scholar Award.
Footnotes
The authors declare no competing financial interest.
References
- 1.Sharpless KB Searching for New Reactivity (Nobel Lecture). Angew. Chem. Int. Ed 2002, 41, 2024–2032. [PubMed] [Google Scholar]
- 2.Noyori R Asymmetric Catalysis: Science and Opportunities (Nobel Lecture 2001). Adv. Synth. Catal 2003, 345, 15–32. [PubMed] [Google Scholar]
- 3.Knowles WS Asymmetric Hydrogenations (Nobel Lecture 2001). Adv. Synth. Catal 2003, 345, 3–13. [Google Scholar]
- 4.Ciamician G The Photochemistry of the Future. Science 1912, 36, 385–394. [DOI] [PubMed] [Google Scholar]
- 5.Roth HD The Beginnings of Organic Photochemistry. Angew. Chem. Int. Ed 1989, 28, 1193–1207. [Google Scholar]
- 6.Inoue Y; Ramamurthy V Molecular and Supramolecular Photochemistry, Volume 11: Chiral Photochemistry; Marcel Dekker, 2004. [Google Scholar]
- 7.Inoue Y Asymmetric Photochemical Reactions in Solution. Chem. Rev 1992, 92, 741–770. [Google Scholar]
- 8.Sherbrook EM; Yoon TP Asymmetric Catalysis of Triplet-State Photoreactions. In Specialist Periodical Reports: Photochemistry; Albini A, Protti S, Eds.; Royal Society of Chemistry: Croydon, UK, 2019; Vol. 46, pp 432–448. [Google Scholar]
- 9.Inoue Y; Dong F; Yamamoto K; Tong LH; Tsuneishi H; Hakushi T; Tai A Inclusion-Enhanced Optical Yield and E/Z Ratio in Enantiodifferentiating Photoisomerization of Cyclooctene Included and Sensitized by β-Cyclodextrin Monobenzoate. J. Am. Chem. Soc 1995, 117, 11033–11034. [Google Scholar]
- 10.Cauble DF; Lynch V; Krische MJ Studies on the Enantioselective Catalysis of Photochemically Promoted Transformations: “Sensitizing Receptors” as Chiral Catalysts. J. Org. Chem 2003, 68, 15–21. [DOI] [PubMed] [Google Scholar]
- 11.Griesbeck AG; Meierhenrich UJ Asymmetric Photochemistry and Photochirogenesis. Angew. Chem. Int. Ed 2002, 41, 3147–3154. [DOI] [PubMed] [Google Scholar]
- 12.Yang C; Inoue Y Supramolecular Photochirogenesis. Chem. Soc. Rev 2014, 43, 4123–4143 [DOI] [PubMed] [Google Scholar]
- 13.Brimioulle R; Lenhart D; Maturi MM; Bach T Enantioselective Catalysis of Photochemical Reactions. Angew. Chem. Int. Ed 2015, 54, 3872–3890. [DOI] [PubMed] [Google Scholar]
- 14.Garrido-Castro AF Asymmetric Induction in Photocatalysis – Discovering a New Side to Light-Driven Chemistry. Tetrahedron Lett. 2018, 59, 1286–1294. [Google Scholar]
- 15.Rao M; Wu W; Yang C Recent Progress on the Enantioselective Excited-State Photoreactions by Pre-Arrangement of Photosubstrate(s). Green Synth. Catal 2021, 2, 131–144. [Google Scholar]
- 16.Rigotti T; Alemán J Visible Light Photocatalysis – From Racemic to Asymmetric Activation Strategies. Chem. Commun 2020, 56, 11169–11190. [DOI] [PubMed] [Google Scholar]
- 17.Nijland A; Harutyunyan SR Light on the Horizon? Catalytic Enantioselective Photoreactions. Catal. Sci. Technol 2013, 3, 1180–1189. [Google Scholar]
- 18.Zhao J-J; Zhang H-H; Yu S Enantioselective Radical Functionalization of Imines and Iminium Intermediates via Visible-Light Photoredox Catalysis. Synthesis 2021, 53, 1706–1718. [Google Scholar]
- 19.Prentice C; Morrisson J; Smith AD; Zysman-Coleman E Recent Developments in Enantioselective Photocatalysis. Beilstein J. Org. Chem 2020, 16, 2363–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong B-C Enantioselective Synthesis Enabled by Visible Light Photocatalysis. Org. Biomol. Chem 2020, 18, 4298–4353. [DOI] [PubMed] [Google Scholar]
- 21.Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramamurthy V; Sivaguru J Supramolecular Photochemistry as a Potential Synthetic Tool: Photocycloaddition. Chem. Rev 2016, 116, 9914–9993. [DOI] [PubMed] [Google Scholar]
- 23.Ramamurthy V; Mondal B Supramolecular Photochemistry Concepts Highlighted with Select Examples. J. Photochem. Photobiol. C 2015, 23, 68–102. [Google Scholar]
- 24.Ramamurthy V; Venkatesan K Photochemical Reactions of Organic Crystals. Chem. Rev 1987, 87, 433–481. [Google Scholar]
- 25.Turro NJ Modern Molecular Photochemistry; The Benjamin/Cummings Publishing Company, Inc., 1978. [Google Scholar]
- 26.Förster T Excimers. Angew. Chem. Int. Ed 1969, 8, 333–343. [Google Scholar]
- 27.Hammond GS; Cole RS Asymmetric Induction During Energy Transfer. J. Am. Chem. Soc 1965, 87, 3256–3257. [Google Scholar]
- 28.Aratani T; Nakanisi Y; Nozaki H The Absolute Configuration of cis-2-Phenylcyclopropanecarboxylic Acid. Tetrahedron Lett. 1969, 10, 1809–1810. [Google Scholar]
- 29.Aratani T; Nakanisi H; Nozaki H The Absolute Configuration of cis-2-Phenylcyclopropanecarboxylic Acid and Related Compounds. Tetrahedron, 1970, 26, 1675–1684. [Google Scholar]
- 30.Horner L; Klaus J Photochemisch Induzierte Reaktionen mi Grenzflächengebundenen Sensibilisatoren. J. Liebigs Ann. Chem 1981, 792–810. [Google Scholar]
- 31.Hammond GS; Saltiel J Mechanisms of Photoreactions in Solution. XVIII. Energy Transfer with Nonvertical Transitions. J. Am. Chem. Soc 1963, 85, 2516–2517. [Google Scholar]
- 32.Hammond GS; Wyatt P; DeBoer CD; Turro NJ Photosensitized Isomerization Involving Saturated Centers. J. Am. Chem. Soc 1964, 86, 2532–2533. [Google Scholar]
- 33.Hammond GS; Saltiel J; Lamola AA; Turro NJ; Bradshaw JS; Cowan DO; Counsell RC Vogt V; Dalton C Mechanisms of Photochemical Reactions in Solution. XXII. Photochemical cis–trans Isomerization. J. Am. Chem. Soc 1964, 86, 3197–3217. [Google Scholar]
- 34.Murov SL; Cole RS; Hammond GS Mechanisms of Photochemical Reactons in Solution. LIV. A New Mechanism for Photosensitization. J. Am. Chem. Soc 1968, 90, 2957–2958. [Google Scholar]
- 35.Balavoine G; Jugé S; Kagan HB Photoactivation Optique du Methyl p-Tolyl Sulfoxide Racemique par Emploi d’un Sensibilisateur Chiral. Tetrahedron Lett. 1973, 14, 4159–4162. [Google Scholar]
- 36.Kagan HB; Fiaud JC New Approaches in Asymmetric Synthesis. Top. Stereochem 1978, 10, 175–285. [Google Scholar]
- 37.Mislow K; Axelrod M; Rayner DR; Gotthardt H; Coyne LM; Hammond GS Light-Induced Pyramidal Inversion of Sulfoxides. J. Am. Chem. Soc 1965, 87, 4958–4959. [Google Scholar]
- 38.Cooke RS; Hammond GS Mechanisms of Photochemical Reaction in Solution. LV. Naphthalene-Sensitized Photoracemization of Sulfoxides. J. Am. Chem. Soc 1968, 90, 2958–2959. [Google Scholar]
- 39.Cooke RS; Hammond GS Mechanisms of Photochemical Reactions in Solution. LXII. Naphthalene-Sensitized Photoracemization of Sulfoxides. J. Am. Chem. Soc 1970, 92, 2739–2745. [Google Scholar]
- 40.Kropp PJ; Fryxell GE; Tubergen MW; Hager MW; Harris GD Jr.; McDermott TP Jr.; Tornero-Velez R Photochemistry of Phenyl Thioethers and Phenyl Selenoethers. Radical vs. Ionic Behavior. J. Am. Chem. Soc 1991, 113, 7300–7310. [Google Scholar]
- 41.Vos BW; Jenks WS Evidence for a Nonradical Pathway in the Photoracemization of Aryl Sulfoxides. J. Am. Chem. Soc 2002, 124, 2544–2547. [DOI] [PubMed] [Google Scholar]
- 42.Hoshi N; Furukawa Y; Hagiwara H; Uda H; Sato K Enantiomer Differentiation in the Photoinduced 1,5-Phenyl Shift of 3-Methyl-3-Phenyl-2(3H)-Oxepinone Utilizing Chiral Sensitizers. Chem. Lett 1980, 9, 47–50. [Google Scholar]
- 43.Drucker CS; Toscano VG; Weiss RG A General Method for the Determination of Steric Effects during Collisional Energy Transfer. Partial Photoresolution of Penta-2,3-Diene. J. Am. Chem. Soc 1973, 95, 6482–6484. [Google Scholar]
- 44.Swenton JS Photoisomerization of cis-Cyclooctene to trans-Cyclooctene. J. Org. Chem 1969, 34, 3217–3218. [Google Scholar]
- 45.Cope AC; Ganellin CR; Johnson HW; Van Auken TV; Winkler HJS Molecular Asymmetry of Olefins. I. Resolution of trans-Cyclooctene. J. Am. Chem. Soc 1963, 85, 3276–3279. [Google Scholar]
- 46.Inoue Y; Kunitomi Y; Takamuku S; Sakurai H Asymmetric cis–trans Photoisomerization of Cyclo-octene Sensitized by Chiral Aromatic Esters. J. Chem. Soc., Chem. Commun 1978, 1024–1025. [Google Scholar]
- 47.Inoue Y; Takamuku S; Kunitomi Y; Sakurai H Singlet Photosensitization of Simple Alkenes. Part 1. cis–trans-Photoisomerization of Cyclo-octene Sensitized by Aromatic Esters. J. Chem. Soc., Perkin Trans 2 1980, 1672–1678. [Google Scholar]
- 48.Tsuneishi H; Hakushi T; Inoue Y Singlet- versus Triplet-Sensitized Enantiodifferentiating Photoisomerization of Cyclooctene: Remarkable Effects of Spin Multiplicity upon Optical Yield. J. Chem. Soc., Perkin Trans 2 1996, 1601–1605. [Google Scholar]
- 49.Inoue Y; Yokoyama T; Yamasaki N; Tai A An Optical Yield that Increases with Temperature in a Photochemically Induced Enantiomeric Isomerization. Nature, 1989, 341, 225–226. [Google Scholar]
- 50.Inoue Y; Yokoyama T; Yamasaki N; Tai A Temperature Switching of Product Chirality upon Photosensitized Enantiodifferentiating Cis–Trans Isomerization of Cyclooctene. J. Am. Chem. Soc 1989, 111, 6480–6482. [Google Scholar]
- 51.Shi M; Inoue Y Enantiodifferentiating Photoisomerization of (Z)-Cyclooctene and (Z,Z)-Cycloocta-1,3-diene Sensitized by Chiral Aromatic Amides. J. Chem. Soc., Perkin Trans 2 1998, 1725–1729. [Google Scholar]
- 52.Shi M; Inoue Y Geometrical Photoisomerization of (Z)-Cyclooctene Sensitized by Aromatic Phosphate, Phosphonate, Phosphinate, Phosphine Oxide and Chiral Phosphoryl Esters. J. Chem. Soc., Perkin Trans 2 1998, 2421–2427. [Google Scholar]
- 53.Mori T; Relevance of the Entropy Factor in Stereoselectivity Control of Asymmetric Photoreactions. Synlett 2020, 31, 1259–1267. [Google Scholar]
- 54.Inoue Y; Wada T; Asaoka S; Sato H; Pete J-P Photochirogenesis: Multidimensional Control of Asymmetric Photochemistry. Chem. Commun 2000, 251–259. [Google Scholar]
- 55.Inoue Y; Yamasaki N; Yokoyama T; Tai A Enantiodifferentiating Z–E Photoisomerization of Cyclooctene Sensitized by Chiral Polyalkyl Benzenepolycarboxylates. J. Org. Chem 1992, 57, 1332–1345. [Google Scholar]
- 56.Inoue Y; Yamasaki N; Yokoyama T; Tai A Highly Enantiodifferentiating Photoisomerization of Cyclooctene by Congested and/or Triplex-Forming Chiral Sensitizers. J. Org. Chem 1993, 58, 1011–1018. [Google Scholar]
- 57.Asano T; Le Noble WJ Activation and Reaction Volumes in Solution. Chem. Rev 1978, 78, 407–489. [DOI] [PubMed] [Google Scholar]
- 58.Chung WS; Turro NJ; Mertes J; Mattay J Radical Ions and Photochemical Charge-Transfer Phenomena. 22. Pressure-Induced Diastereoselectivity in Photoinduced Diels–Alder Reactions. J. Org. Chem 1989, 54, 4881–4887. [Google Scholar]
- 59.Inoue Y; Matsushima E; Wada T Pressure and Temperature Control of Product Chirality in Asymmetric Photochemistry. Enantiodifferentiating Photoisomerization of Cyclooctene Sensitized by Chiral Benzenepolycarboxylates. J. Am. Chem. Soc 1998, 120, 10687–10696. [Google Scholar]
- 60.Kaneda M; Nakamura A; Asaoka S; Ikeda H; Mori T; Wada T; Inoue Y Pressure Control of Enantiodifferentiating Photoisomerization of Cyclooctenes Sensitized by Chiral Benzenepolycarboxylates. The Origin of Discontinuous Pressure Dependence of the Optical Yield. Org. Biomol. Chem 2003, 1, 4435–4440. [DOI] [PubMed] [Google Scholar]
- 61.Kaneda M; Asaoka S; Ikeda H; Mori T; Wada T; Inoue Y Discontinuous Pressure Effect upon Enantiodifferentiating Photosensitized Isomerization of Cyclooctene. Chem. Commun 2002, 1272–1273. [DOI] [PubMed] [Google Scholar]
- 62.Inoue Y; Ikeda H; Kaneda M; Sumimura T; Everitt SRL; Wada T Entropy-Controlled Asymmetric Photochemistry: Switching of Product Chirality by Solvent. J. Am. Chem. Soc 2000, 122, 406–407. [Google Scholar]
- 63.Saito R; Kaneda M; Wada T; Katoh A; Inoue Y First Asymmetric Photosensitization in Supercritical Fluid. Exceptionally High Pressure/Density of Optical Yield in Photosensitized Enantiodifferentiating Isomerization of Cyclooctene. Chem. Lett 2002, 31, 860–861. [Google Scholar]
- 64.Tsuneishi H; Hakushi T; Tai A; Inoue Y Enantiodifferentiating Photoisomerization of 1-Methylcyclooct-1-ene Sensitized by Chiral Alkyl Benzenecarboxylates: Steric Effects upon Stereodifferentiation. J. Chem. Soc., Perkin Trans 2 1995, 2057–2062. [Google Scholar]
- 65.Tsuneishi H; Inoue Y; Hakushi T; Tai A Direct and Sensitized Geometrical Photoisomerization of 1-Methylcyclooctene. J. Chem. Soc., Perkin Trans 2 1993, 457–462. [Google Scholar]
- 66.Sugimura T; Shimizu H; Umemoto S; Tsuneishi H; Hakushi T; Inoue Y; Tai A Efficient Diastereodifferentiating E-Z Photoisomerization of Cyclooctene Tethered to Intramolecular Sensitizer through Optically Active Pentane-2,4-diyl Unit. Chem. Lett 1998, 27, 323–324. [Google Scholar]
- 67.Inoue T; Matsuyama K; Inoue Y Diastereodifferentiating Z-E Photoisomerization of 3-Benzoyloxycyclooctene: Diastereoselectivity Switching Controlled by Substrate Concentration through Competitive Intra- vs. Intermolecular Photosensitization Processes. J. Am. Chem. Soc 1999, 121, 9877–9878. [Google Scholar]
- 68.Matsuyama K; Inoue T; Inoue Y Self-Sensitized Diastereodifferentiating Z-E Photoisomerization of 3-, 4-, and 5-Benzoyloxycyclooctenes: Intra- versus Intermolecular Photosensitization. Synthesis 2001, 1167–1174. [Google Scholar]
- 69.Inoue Y; Tsuneishi H; Hakushi T; Tai A Optically Active (E,Z)-1,3-Cyclooctadiene: First Enantioselective Synthesis through Asymmetric Photosensitization and Chiroptical Property. J. Am. Chem. Soc 1997, 119, 472–478. [Google Scholar]
- 70.Inoue Y; Hakushi T; Goto S; Takamuku S; Sakurai H Singlet Photosensitization of Simple Alkenes. Part 2. Photochemical Transformation of Cyclo-octa-1,5-dienes Sensitized by Aromatic Ester. J. Chem. Soc., Perkin Trans 2 1980, 1678–1682. [Google Scholar]
- 71.Maeda R; Wada T; Mori T; Kono S; Kanomata N; Inoue Y Planar-to-Planar Chirality Transfer in the Excited State. Enantiodifferentiating Photoisomerization of Cyclooctenes Sensitized by Planar-Chiral Paracyclophane. J. Am. Chem. Soc 2011, 133, 10379–10381. [DOI] [PubMed] [Google Scholar]
- 72.Hoffmann R; Inoue Y Trapped Optically Active (E)-Cycloheptene Generated by Enantiodifferentiating Z–E Photoisomerization of Cycloheptene Sensitized by Chiral Aromatic Esters. J. Am. Chem. Soc 1999, 121, 10702–10710. [Google Scholar]
- 73.Asaoka S; Horiguchi H; Wada T; Inoue Y Enantiodifferentiating Photocylization of Cyclohexene Sensitized by Chiral Benzenecarboxylates. J. Chem. Soc., Perkin Trans 2 2000, 737–747. [Google Scholar]
- 74.Asaoka S; Ooi M; Jiang P; Wada T; Inoue Y Enantiodifferentiating Photocyclodimerization of Cyclohexa-1,3-diene Sensitized by Chiral Arenecarboxylates. J. Chem. Soc., Perkin Trans 2 2000, 77–84. [Google Scholar]
- 75.Inoue Y; Shimoyama H; Yamasaki N; Tai A Enantiodifferentiating Photoisomerization of 1,2-Diphenylcyclopropane Sensitized by Chiral Aromatic Esters. Chem. Lett 1991, 20, 593–596. [Google Scholar]
- 76.Inoue Y; Yamasaki N; Shimoyama H; Tai A Enantiodifferentiating Cis–Trans Photoisomerizations of 1,2-Diarylcyclopropanes and 2,3-Diphenyloxirane Sensitized by Chiral Aromatic Esters. J. Org. Chem 1993, 58, 1785–1793. [Google Scholar]
- 77.Roth HD Electron-Transfer Photochemistry of cis- and trans-1,2-Diphenylcyclopropane with Singlet Acceptors: Recombination of Radical Ion Pairs of Singlet and Triplet Multiplicity. J. Phys. Chem. A 2003, 107, 3432–3437. [Google Scholar]
- 78.Inoue Y; Okano T; Yamasaki N; Tai A Enantiodifferentiating Photocyclodimerization of Phenyl Vinyl Ethers and 4-Methoxystyrene Sensitized by Chiral Aromatic Esters. J. Photochem. Photobiol. A 1992, 66, 61–68. [Google Scholar]
- 79.Inoue Y; Okano T; Yamasaki N; Tai A First Photosensitized Enantiodifferentiating Polar Addition: Anti-Markovnikov Methanol Addition to 1,1-Diphenylpropene. J. Chem. Soc., Chem. Commun 1993, 718–720. [Google Scholar]
- 80.Asaoka S; Kitazawa T; Wada T; Inoue Y Enantiodifferentiating Anti-Markovnikov Photoaddition of Alcohols to 1,1-Diphenylalkenes Sensitized by Chiral Naphthalenecarboxylates. J. Am. Chem. Soc 1999, 121, 8486–8498. [Google Scholar]
- 81.Asaoka S; Wada T; Inoue Y Microenvironmental Polarity Control of Electron-Transfer Photochirogenesis. Enantiodifferentiating Polar Addition of 1,1-Diphenyl-1-alkenes Photosensitized by Saccharide Naphthalenecarboxylates. J. Am. Chem. Soc 2003, 125, 3008–3027. [DOI] [PubMed] [Google Scholar]
- 82.Takehara Y; Ohta N; Shiraishi S; Asaoka S; Wada T; Inoue Y External Electric Field Effects on Exciplex Formation of 1,1-Diphenylpropene with Chiral 1,4-Naphthalenedicarboxylate in PMMA Polymer Films. J. Photochem. Photobiol. A 2001, 145, 53–60. [Google Scholar]
- 83.Kaneda M; Nishiyama Y; Asaoka S; Mori T; Wada T; Inoue Y Pressure Control of Enantiodifferentiating Polar Addition of 1,1-Diphenylpropene Sensitized by Chiral Naphthalenecarboxylates. Org. Biomol. Chem 2004, 2, 1295–1303. [DOI] [PubMed] [Google Scholar]
- 84.Nishiyama Y; Kaneda M; Saito R; Mori T; Wada T; Inoue Y Enantiodifferentiating Photoaddition of Alcohols to 1,1-Diphenylpropene in Supercritical Carbon Dioxide: Sudden Jump of Optical Yield at the Critical Density. J. Am. Chem. Soc 2004, 126, 6568–6569. [DOI] [PubMed] [Google Scholar]
- 85.Nishiyama Y; Kaneda M; Asaoka S; Saito R; Mori T; Wada T; Inoue Y Mechanistic Studies on the Enantiodifferentiating Anti-Markovnikov Photoaddition of Alcohols to 1,1-Diphenyl-1-alkenes in Near-Critical and Supercritical Carbon Dioxide. J. Phys. Chem. A 2007, 111, 13432–13440. [DOI] [PubMed] [Google Scholar]
- 86.Yasuhiro N; Takehiko W; Tadashi M; Inoue Y Critical Control by Temperature and Pressure of Enantiodifferentiating Anti-Markovnikov Photoaddition of Methanol to Diphenylpropene in Near Critical and Supercritical Carbon Dioxide. Chem. Lett 2007, 36, 1488–1489. [Google Scholar]
- 87.Nishiyama Y; Wada T; Asaoka S; Mori T; McCarty TA; Kraut ND; Bright FV; Inoue Y Entrainer Effect on Photochirogenesis in Near- and Supercritical Carbon Dioxide: Dramatic Enhancement of Enantioselectivity. J. Am. Chem. Soc 2008, 130, 7526–7527. [DOI] [PubMed] [Google Scholar]
- 88.Nishiyama Y; Wada T; Kakiuchi K; Inoue Y Entrainer Effects on Enantiodifferentiating Photocyclization of 5-Hydroxy-1,1-diphenylpentane in Near-Critical and Supercritical Carbon Dioxide. J. Org. Chem 2012, 77, 5681–5686. [DOI] [PubMed] [Google Scholar]
- 89.Nishiyama Y; Wada T; Kakiuchi K; Inoue Y Microenvironmental Control of Enantiodifferentiating Photocyclization of 5-Hydroxy-1,1-diphenylpentene through Selective Solvation. Chirality 2012, 24, 400–405. [DOI] [PubMed] [Google Scholar]
- 90.Kim J-I; Schuster GB Enantioselective Catalysis of the Triplex Diels–Alder Reaction: Addition of trans-β-Methylstyrene to 1,3-Cyclohexadiene Photosensitized with (−)-1,1’-Bis(2,4-dicyanonaphthalene). J. Am. Chem. Soc 1990, 112, 9635–9637. [Google Scholar]
- 91.Calhoun GC; Schuster GB Radical Cation and Triplex Diels–Alder Reaction of 1,3-Cyclohexadiene. J. Am. Chem. Soc 1984, 106, 6870–6871. [Google Scholar]
- 92.Calhoun GC; Schuster GB The Triplex Dies–Alder Reaction of Indene and Cyclic Dienes. J. Am. Chem. Soc 1986, 108, 8021–8027. [Google Scholar]
- 93.Akbulut N; Hartsough D; Kim J-I; Schuster GB The Triplex Diels–Alder Reaction of 1,3-Dienes with Enol, Alkene, and Acetylenic Dienophiles: Scope and Utility. J. Org. Chem 1989, 54, 2549–2556. [Google Scholar]
- 94.Kim J-I; Schuster GB Enantioselective Catalysis of the Triplex Diels–Alder Reaction: A Study of Scope and Mechanism. J. Am. Chem. Soc 1992, 114, 9309–9317. [Google Scholar]
- 95.Vondenhof M; Mattay J 1,1’-Binaphthalene-2,2’-Dicarbonitrile in Photochemically Sensitized Enantiodifferentiating Isomerizations. Chem. Ber 1990, 123, 2457–2459. [Google Scholar]
- 96.Vondenhof M; Mattay J Sulfonic Acid Esters Derived from 1,1’-Binaphthalene as New Axially Chiral Photosensitizers. Tetrahedron Lett. 1990, 31, 985–988. [Google Scholar]
- 97.Das A; Ayad S; Hanson K Enantioselective Protonation of Silyl Enol Ether Using Excited State Proton Transfer Dyes. Org. Lett 2016, 18, 5416–5419. [DOI] [PubMed] [Google Scholar]
- 98.Das A; Banerjee T; Hanson K Protonation of Silylenol Ether via Excited State Proton Transfer Catalysis. Chem. Commun 2016, 52, 1350–1353. [DOI] [PubMed] [Google Scholar]
- 99.Àlvaro M; Formentín P; García H; Palomares E; Sabater MJ Chiral N-Alkyl-2,4,6-triphenylpyridiniums as Enantioselective Triplet Photosensitizers. Laser Flash Photolysis and Preparative Studies. J. Org. Chem 2002, 67, 5184–5189. [DOI] [PubMed] [Google Scholar]
- 100.Vallavoju N; Selvakumar S; Jockusch S; Sibi MP; Sivaguru J Enantioselective Organo-Photocatalysis Mediated by Atropisomeric Thiourea Derivatives. Angew. Chem. Int. Ed 2014, 53, 5604–5608. [DOI] [PubMed] [Google Scholar]
- 101.Vallavoju N; Selvakumar S; Jockusch S; Prabhakaran MT; Sibi MP; Sivaguru J Evaluating Thiourea Architecture for Intramolecular [2+2] Photocycloaddition of 4-Alkenylcoumarins. Adv. Synth. Catal 2014, 356, 2763–2768. [Google Scholar]
- 102.Vallavoju N; Selvakumar S; Pemberton BC; Jockusch S; Sibi MP; Sivaguru J Organophotocatalysis: Insights into the Mechanistic Aspects of Thiourea-Mediated Intermolecular [2+2] Photocycloadditions. Angew. Chem. Int. Ed 2016, 55, 5446–5451. [DOI] [PubMed] [Google Scholar]
- 103.Raghunathan R; Jockusch S; Sibi MP; Sivaguru J Evaluating Thiourea/Urea Catalyst for Enantioselective 6π-Photocyclization of Acrylanilides. J. Photochem. Photobiol. A 2016, 331, 84–88. [Google Scholar]
- 104.Rau H; Hörmann M Kinetic Resolution of Optically Active Molecules and Asymmetric Chemistry: Asymmetrically Sensitized Photolysis of trans-3,5-Diphenylpyrazoline. J. Photochem 1981, 16, 231–247. [Google Scholar]
- 105.Becker E; Weiland R; Rau H Photochemistry of Bichromophoric Molecules with Camphor Structure I: Energy Transfer and Asymmetry Effects. J. Photochem. Photobiol. A 1988, 41, 311–330. [Google Scholar]
- 106.Ouannès C; Beugelmans R; Roussi G Asymmetric Induction during Transfer of Triplet Energy. J. Am. Chem. Soc 1973, 95, 8472–8474. [Google Scholar]
- 107.Demuth M; Raghavan PR; Carter C; Nakano K; Schaffner K Photochemical High-Yield Preparation of Tricyclo[3.3.0.0]Octan-3-ones. Potential Synthons for Polycyclopentanoid Terpenes and Prostacyclin Analogs. Helv. Chim. Acta 1980, 63, 2434–2439. [Google Scholar]
- 108.Kemp DS; Petrakis KS Synthesis and Conformational Analysis of cis,cis-1,3,5-Trimethylcyclohexane-1,3,5-tricarboxylic Acid. J. Org. Chem 1981, 46, 5140–5143. [Google Scholar]
- 109.Bach T; Bergmann H; Harms K High Facial Diastereoselectivity in the Photocycloaddition of a Chiral Aromatic Aldehyde and an Enamide Induced by Intermolecular Hydrogen Bonding. J. Am. Chem. Soc 1999, 121, 10650–10651. [Google Scholar]
- 110.Bach T; Bergmann H; Brummerhop H; Lewis W; Harms K The [2+2]-Photocycloaddition of Aromatic Aldehydes and Ketones to 3,4-Dihydro-2-pyridones: Regioselectivity, Diastereoselectivity, and Reductive Ring Opening of the Product Oxetanes. Chem. - Eur. J 2001, 7, 4512–4521. [DOI] [PubMed] [Google Scholar]
- 111.Bach T; Bergmann H; Harms K Enantioselective Intramolecular [2+2]-Photocycloaddition Reaction in Solution. Angew. Chem. Int. Ed 2000, 39, 2302–2304. [PubMed] [Google Scholar]
- 112.Bach T; Bergmann H; Grosch B; Harms K; Herdtweck E Synthesis of Enantiomerically Pure 1,5,7-Trimethyl-3-azabicyclo[3.3.1]nonan-2-ones as Chiral Host Compounds for Enantioselective Photochemical Reactions in Solution. Synthesis 2001, 1395–1405. [Google Scholar]
- 113.Bach T; Bergmann H; Grosch B; Harms K Highly Enantioselective Intra- and Intermolecular [2+2] Photocycloaddition Reactions of 2-Quinolones Mediated by a Chiral Lactam Host: Host–Guest Interactions, Product Configuration, and the Origin of the Stereoselectivity in Solution. J. Am. Chem. Soc 2002, 124, 7982–7990. [DOI] [PubMed] [Google Scholar]
- 114.Albrecht D; Vogt F; Bach T Diastereo- and Enantioselective Intramolecular [2+2] Photocycloaddition Reactions of 3-(ω’-Alkenyl)- and 3-(ω’-Alkenyloxy)-Substituted 5,6-Dihydro-1H-pyridin-2-ones. Chem. - Eur. J 2010, 16, 4284–4296. [DOI] [PubMed] [Google Scholar]
- 115.Austin KAB; Herdtweck E; Bach T Intramolecular [2+2] Photocycloaddition of Substituted Isoquinolones: Enantioselectivity and Kinetic Resolution Induced by a Chiral Template. Angew. Chem. Int. Ed 2011, 50, 8416–8419. [DOI] [PubMed] [Google Scholar]
- 116.Bach T; Bergmann H Enantioselective Intermolecular [2+2]-Photocycloaddition Reactions of Alkenes and a 2-Quinolone in Solution. J. Am. Chem. Soc 2000, 122, 11525–11526. [Google Scholar]
- 117.Selig P; Bach T Photochemistry of 4-(2’-Aminoethyl)quinolones: Enantioselective Synthesis of Tetracyclic Tetrahydro-1aH-pyrido[4’,3’:2,3]-cyclobuta[1,2-c] Quinoline-2,11(3H,8H)-diones by Intra- and Intermolecular [2 + 2]-Photocycloaddition Reactions in Solution. J. Org. Chem 2006, 71, 5662–5673. [DOI] [PubMed] [Google Scholar]
- 118.Coote SC; Bach T Enantioselective Intermolecular [2+2] Photocycloadditions of Isoquinolone Mediated by a Chiral Hydrogen-Bonding Template. J. Am. Chem. Soc 2013, 135, 14948–14951. [DOI] [PubMed] [Google Scholar]
- 119.Mayr F; Wiegand C; Bach T Enantioselective, Intermolecular [2+2] Photocycloaddition Reactions of 3-Acetoxyquinolone: Total Synthesis of (−)-Pinolinone. Chem. Commun 2014, 50, 3353–3355. [DOI] [PubMed] [Google Scholar]
- 120.Coote SC; Pöthig A; Bach T Enantioselective Template-Directed [2 + 2] Photocycloadditions of Isoquinolones: Scope, Mechanism and Synthetic Applications. Chem. - Eur. J 2015, 21, 6906–6912. [DOI] [PubMed] [Google Scholar]
- 121.Grosch B; Orlebar CN; Herdtweck E; Massa W; Bach T Highly Enantioselective Diels–Alder Reactions of a Photochemically Generated o-Quinodimethane with Olefins. Angew. Chem. Int. Ed 2003, 42, 3693–3696. [DOI] [PubMed] [Google Scholar]
- 122.Grosch B; Orlebar CN; Herdtweck E; Kaneda M; Wada T; Inoue Y; Bach T Enantioselective [4+2]-Cycloaddition Reaction of a Photochemically Generated o-Quinodimethane: Mechanistic Details, Association Studies, and Pressure Effects. Chem. - Eur. J 2004, 10, 2179–2189. [DOI] [PubMed] [Google Scholar]
- 123.Bach T; Bergmann H; Harms K Enantioselective Photochemical Reactions of 2-Pyridones in Solution. Org. Lett 2001, 3, 601–603. [DOI] [PubMed] [Google Scholar]
- 124.Maturi MM; Fukuhara G; Tanaka K; Kawanami Y; Mori T; Inoue Y Bach T Enantioselective [4+4] Photodimerization of Anthracene-2,6-dicarboxylic Acid Mediated by a C2-Symmetric Chiral Template. Chem. Commun 2016, 52, 1032–1035. [DOI] [PubMed] [Google Scholar]
- 125.Bach T; Aechtner T; Neumüller B Intermolecular Hydrogen Binding of a Chiral Host and a Prochiral Imidazolidinone: Enantioselective Norrish–Yang Cyclisation in Solution. Chem. Commun 2001, 607–608. [Google Scholar]
- 126.Bach T; Aechtner T Neumüller B Enantioselective Norrish–Yang Cyclization Reactions of N-(ω-Oxo-ω-phenylalkyl)-Substituted Imidazolidinones in Solution and in the Solid State. Chem. - Eur. J 2002, 8, 2464–2475. [DOI] [PubMed] [Google Scholar]
- 127.Bach T; Grosch B; Strassner T; Herdtweck E Enantioselective [6π]-Photocyclization Reaction of an Acrylanilide Mediated by a Chiral Host. Interplay between Enantioselective Ring Closure and Enantioselective Protonation. J. Org. Chem 2003, 68, 1107–1116. [DOI] [PubMed] [Google Scholar]
- 128.Bauer A; Westkämper F; Grimme S; Bach T Catalytic Enantioselective Reactions Driven by Photoinduced Electron Transfer. Nature, 2005, 436, 1139–1140. [DOI] [PubMed] [Google Scholar]
- 129.Inoue Y Light on Chirality. Nature, 2005, 436, 1099–1100. [DOI] [PubMed] [Google Scholar]
- 130.Müller C; Bauer A; Bach T Light-Driven Enantioselective Organocatalysis. Angew. Chem. Int. Ed 2009, 48, 6640–6642. [DOI] [PubMed] [Google Scholar]
- 131.Bakowski A; Dressel M; Bauer A; Bach T Enantioselective Radical Cyclisation Reactions of 4-Substituted Quinolones Mediated by a Chiral Template. Org. Biomol. Chem 2011, 9, 3516–3529. [DOI] [PubMed] [Google Scholar]
- 132.Müller C; Bauer A; Maturi MM; Cuquerella MC; Miranda MA Bach T Enantioselective Intramolecular [2+2]-Photocycloaddtion Reactions of 4-Substituted Quinolones Catalyzed by a Chiral Sensitizer with a Hydrogen-Bonding Motif. J. Am. Chem. Soc 2011, 133, 16689–16697. [DOI] [PubMed] [Google Scholar]
- 133.Maturi MM; Wenninger M; Alonso R; Bauer A; Pöthig A; Riedle E; Bach T Intramolecular [2+2] Photocycloaddition of 3- and 4-(But-3-enyl)oxyquinolones: Influence of the Alkene Substitution Pattern, Photophysical Studies, and Enantioselective Catalysis by a Chiral Sensitizer. Chem. - Eur. J 2013, 19, 7461–7472. [DOI] [PubMed] [Google Scholar]
- 134.Hoffmann Roald.; Swenson JR Ground- and Excited-State Geometries of Benzophenone. J. Phys. Chem 1970, 74, 415–420. [Google Scholar]
- 135.Maturi MM; Bach T Enantioselective Catalysis of the Intermolecular [2+2] Photocycloaddition between 2-Pyridones and Acetylenedicarboxylates. Angew. Chem. Int. Ed 2014, 53, 7661–7664. [DOI] [PubMed] [Google Scholar]
- 136.Maturi MM; Pöthig A; Bach T Enantioselective Photochemical Rearrangements of Spirooxindole Epoxides Catalyzed by a Chiral Bifunctional Xanthone. Aust. J. Chem 2015, 68, 1682–1692. [Google Scholar]
- 137.Alonso R; Bach T A Chiral Thioxanthone as an Organocatalyst for Enantioselective [2+2] Photocycloaddition Reactions Induced by Visible Light. Angew. Chem. Int. Ed 2014, 53, 4368–4371. [DOI] [PubMed] [Google Scholar]
- 138.Li X; Jandl C; Bach T Visible-Light-Mediated Enantioselective Photoreactions of 3-Alkylquinolones with 4-O-Tethered Alkenes and Allenes. Org. Lett 2020, 22, 3618–3622. [DOI] [PubMed] [Google Scholar]
- 139.Tröster A; Alonso R; Bauer A; Bach T Enantioselective Intermolecular [2 + 2] Photocycloaddition Reactions of 2(1H)-Quinolones Induced by Visible Light Irradiation. J. Am. Chem. Soc 2016, 138, 7808–7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li X; Großkopf J; Jandl C; Bach T Enantioselective, Visible Light Mediated Aza Paternò–Büchi Reactions of Quinoxalinones. Angew. Chem. Int. Ed 2021, 60, 2684–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nishio T; Omote Y Photocycloaddition of Quinoxalin-2-ones and Benzoxazin-2-ones to Aryl Alkenes. J. Chem. Soc., Perkin Trans 1 1987, 2611–2615. [Google Scholar]
- 142.Hölzl-Hobmeier A; Bauer A; Silva AV; Huber SM; Bannwarth C; Bach T Catalytic Deracemization of Chiral Allenes by Sensitized Excitation with Visible Light. Nature, 2018, 564, 240–243. [DOI] [PubMed] [Google Scholar]
- 143.Plaza M; Jandl C; Bach T Photochemical Deracemization of Allenes and Subsequent Chirality Transfer. Angew. Chem. Int. Ed 2020, 59, 12785–12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yuan K; Wang P; Li H-X; Liu Y-Z; Lv L-L Theoretical Survey of the Photochemical Deracemization Mechanism of Chiral Allene 3-(3,3-Dimethyl-1-Buten-1-Ylidene)-2-Piperidinone. Org. Chem. Front 2020, 7, 3656–3663. [Google Scholar]
- 145.Plaza M; Großkopf J; Breitenlechner S; Bannwarth C; Bach T Photochemical Deracemization of Primary Allene Amides by Triplet Energy Transfer: A Combined Synthetic and Theoretical Study. J. Am. Chem. Soc 2021, 143, 11209–11217. [DOI] [PubMed] [Google Scholar]
- 146.Wimberger L; Kratz T; Bach T Photochemical Deracemization of Chiral Sulfoxides Catalyzed by a Hydrogen-Bonding Xanthone Sensitizer. Synthesis 2019, 51, 4417–4424. [Google Scholar]
- 147.Tröster A; Bauer A; Jandl C; Bach T Enantioselective Visible-Light-Mediated Formation of 3-Cyclopropylquinolones by Triplet-Sensitized Deracemization. Angew. Chem. Int. Ed 2019, 58, 3538–3541. [DOI] [PubMed] [Google Scholar]
- 148.Li X; Kutta RJ; Jandl C; Bauer A; Nuernberger P; Bach T Photochemically Induced Ring Opening of Spirocyclopropyl Oxindoles: Evidence for a Triplet 1,3-Diradical Intermediate and Deracemization by a Chiral Sensitizer. Angew. Chem. Int. Ed 2020, 59, 21640–21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Burg F; Bach T Lactam Hydrogen Bonds as Control Elements in Enantioselective Transition-Metal-Catalyzed and Photochemical Reactions. J. Org. Chem 2019, 84, 8815–8836. [DOI] [PubMed] [Google Scholar]
- 150.Mayr F; Mohr L-M; Rodriguez E; Bach T Synthesis of Chiral Thiourea-Thioxanthone Hybrids. Synthesis 2017, 49, 5238–5250. [Google Scholar]
- 151.Pecho F; Zou Y-Q; Gramüller J; Mori T; Huber SM; Bauer A; Gschwind RM; Bach T A Thioxanthone Sensitizer with a Chiral Phosphoric Acid Binding Site: Properties and Applications in Visible Light-Mediated Cycloadditions. Chem. - Eur. J 2020, 26, 5190–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Takagi R; Tabuchi C Enantioselective Intramolecular [2+2] Photocycloaddition using Phosphoric Acid as a Chiral Template. Org. Biomol. Chem 2020, 18, 9261–9267. [DOI] [PubMed] [Google Scholar]
- 153.Lyu J; Clarez A; Vitale MR; Allain C; Masson G Preparation of Chiral Photosensitive Organocatalysts and Their Application for the Enantioselective Synthesis of 1,2-Diamines. J. Org. Chem 2020, 85, 12843–12855. [DOI] [PubMed] [Google Scholar]
- 154.Crisenza GE; Mazzarella D; Melchiorre P Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes. J. Am. Chem. Soc 2020, 142, 5461–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Silvi M; Melchiorre P Enhancing the Potential of Enantioselective Organocatalysis with Light. Nature 2018, 554, 41–49. [DOI] [PubMed] [Google Scholar]
- 156.Lima CG; Lima T; Duarte M; Jurberg ID; Paixão MW Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal 2016, 6, 1389–1407. [Google Scholar]
- 157.Mulliken RS Molecular Compounds and their Spectra. III. The Interaction of Electron Donors and Acceptors. J. Phys. Chem 1952, 56, 801–822. [Google Scholar]
- 158.Arceo E; Jurberg ID; Álvarez-Fernández A; Melchiorre P Photochemical Activity of a Key Donor-Acceptor Complex can Drive Stereoselective Catalytic α-Alkylation of Aldehydes. Nat. Chem 2013, 5, 750–756. [DOI] [PubMed] [Google Scholar]
- 159.Nicewicz DA; MacMillan DWC Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science, 2008, 322, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bahamonde A; Melchiorre P Mechanism of the Stereoselective α-Alkylation of Aldehydes Driven by the Photochemical Activity of Enamines. J. Am. Chem. Soc 2016, 138, 8019–8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Arceo E; Bahamonde A; Bergonzini G; Melchiorre P Enantioselective Direct α-Alkylation of Cyclic Ketones by Means of Photo-Organocatalysis. Chem. Sci 2014, 5, 2438–2442. [Google Scholar]
- 162.Silvi M; Arceo E; Jurberg ID; Cassani C; Melchiorre P Enantioselective Organocatalytic Alkylation of Aldehydes and Enals Driven by the Direct Photoexcitation of Enamines. J. Am Chem. Soc 2015, 137, 6120–6123. [DOI] [PubMed] [Google Scholar]
- 163.Filippini G; Silvi M; Melchiorre P Enantioselective Formal α-Methylation and α-Benzylation of Aldehydes by Means of Photo-Organocatalysis. Angew. Chem. Int. Ed 2017, 56, 4447–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rigotti T; Casado-Sánchez A; Cabrera S; Pérez-Ruiz R; Liras M; O’Shea VA; Alemán J A Bifunctional Photoaminocatalyst for the Alkylation of Aldehydes: Design, Analysis, and Mechanistic Studies. ACS Catal 2018, 8, 5928–5940. [Google Scholar]
- 165.Silvi M; Verrier C; Rey Y; Buzzetti L; Melchiorre P Visible-Light Excitation of Iminium Ions Enables the Enantioselective Catalytic β-Alkylation of Enals. Nat. Chem 2017, 9, 868–873. [DOI] [PubMed] [Google Scholar]
- 166.Verrier C; Alandini N; Pezzetta C; Moliterno M; Buzzetti L; Hepburn HH; Vega-Peñaloza A; Silvi M; Melchiorre P Direct Stereoselective Installation of Alkyl Fragments at the β-Carbon of Enals via Excited Iminium Ion Catalysis. ACS Catal 2018, 8, 1062–1066. [Google Scholar]
- 167.Woźniak Ł; Magagnano G; Melchiorre P Enantioselective Photochemical Organocascade Catalysis. Angew. Chem. Int. Ed 2018, 57, 1068–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bonilla P; Rey YP; Holden CM; Melchiorre P Photo-Organocatalytic Enantioselective Radical Cascade Reactions of Unactivated Olefins. Angew. Chem. Int. Ed 2018, 57, 12819–12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Perego LA; Bonilla P; Melchiorre P Photo-Organocatalytic Enantioselective Radical Cascade Enabled by Single-Electron Transfer Activation of Allenes. Adv. Synth. Catal 2020, 362, 302–307. [Google Scholar]
- 170.Mazzarella D; Crisenza GEM; Melchiorre P Asymmetric Photocatalytic C–H Functionalization of Toluene and Derivatives. J. Am. Chem. Soc 2018, 140, 8439–8443. [DOI] [PubMed] [Google Scholar]
- 171.Nicholas AM; Arnold DR Thermochemical Parameters for Organic Radicals and Radical Ions. Part 1. The Estimation of the pKa of Radical Cations based on Thermochemical Calculations. Can. J. Chem 1982, 60, 2165–2179. [Google Scholar]
- 172.Cao Z-Y; Ghosh T; Melchiorre P Enantioselective Radical Conjugate Additions Driven by a Photoactive Intramolecular Iminium-Ion-Based EDA Complex. Nat. Commun 2018, 9, 3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hörmann FM; Chung TS; Rodriguez E; Jakob M; Bach T Evidence for Triplet Sensitization in the Visible-Light-Induced [2+2] Photocycloaddition of Eniminium Ions. Angew. Chem. Int. Ed 2018, 57, 827–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hörmann FM; Kerzig C; Chung TS; Bauer A; Wenger OS; Bach T Triplet Energy Transfer from Ruthenium Complexes to Chiral Eniminium Ions: Enantioselective Synthesis of Cyclobutanecarbaldehydes by [2+2] Photocycloaddition. Angew. Chem. Int. Ed 2020, 59, 9659–9668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rigotti T; Mas-Ballesté R; Alemán J Enantioselective Aminocatalytic [2 + 2] Cycloaddition through Visible Light Excitation. ACS Catal 2020, 10, 5335–5346. [Google Scholar]
- 176.Martínez-Gualda AM; Domingo-Legarda P; Rigotti T; Díaz-Tendero S; Fraile A; Alemán J Asymmetric [2+2] Photocycloaddition via Charge Transfer Complex for the Synthesis of Tricyclic Chiral Ethers. Chem. Commun 2021, 57, 3046–3049. [DOI] [PubMed] [Google Scholar]
- 177.Mahlau M; List B Asymmetric Counteranion-Directed Catalysis: Concept, Definition, and Applications. Angew. Chem. Int. Ed 2013, 52, 518–533. [DOI] [PubMed] [Google Scholar]
- 178.Phipps RJ; Hamilton GL; Toste FD The Progression of Chiral Anions from Concepts to Applications in Asymmetric Catalsis. Nat. Chem 2012, 4, 603–614. [DOI] [PubMed] [Google Scholar]
- 179.Yang Z; Li H; Li S; Zhang M-T; Luo S A Chiral Ion-Pair Photoredox Organocatalyst: Enantioselective Anti-Markovnikov Hydroetherification of Alkenols. Org. Chem. Front 2017, 4, 1037–1041. [Google Scholar]
- 180.Hamilton DS; Nicewicz DA Direct Catalytic Anti-Markovnikov Hydroetherification of Alkenols. J. Am. Chem. Soc 2012, 134, 18577–18580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wang H; Ren Y; Wang K; Man Y; Xiang Y; Li N; Tang B Visible Light-Induced Cyclization Reactions for the Synthesis of 1,2,4-Triazolines and 1,2,4-Triazoles. Chem. Commun 2017, 53, 9644–9647. [DOI] [PubMed] [Google Scholar]
- 182.Morse PD; Nguyen TM; Cruz CL; Nicewicz DA Enantioselective Counter-Anions in Photoredox Catalysis: The Asymmetric Cation Radical Diels–Alder Reaction. Tetrahedron, 2018, 74, 3266–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Shen Y; Shen M-L; Wang P-S Light-Mediated Chiral Phosphate Catalysis for Asymmetric Dicarbofunctionalization of Enamides. ACS Catal 2020, 10, 8247–8253. [Google Scholar]
- 184.Kikuchi J; Kodama S; Terada M Radical Addition Reaction Between Chromenols and Toluene Derivatives Initiated by Brønsted Acid Catalyst Under Light Irradiation. Org. Chem. Front 2021, 8, 4153–4159. [Google Scholar]
- 185.Woźniak Ł; Murphy JJ; Melchiorre P Photo-organocatalytic Enantioselective Perfluoroalkylation of β-ketoesters. J. Am. Chem. Soc 2015, 137, 5678–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yang C; Zhang W; Li Y-H; Xue X-S; Li X; Cheng J-P Origin of Stereoselectivity of the Photoinduced Asymmetric Phase-Transfer-Catalyzed Perfluoroalkylation of β-Ketoesters. J. Org. Chem 2017, 82, 9321–9327. [DOI] [PubMed] [Google Scholar]
- 187.Tang X-F; Feng S-H; Wang Y-K; Yang F; Zheng Z-H; Zhao J-N; Wu Y-F; Yin H; Liu G-Z; Meng Q-W Bifunctional Metal-Free Photo-Organocatalysts for Enantioselective Aerobic Oxidation of β-Dicarbonyl Compounds. Tetrahedron, 2018, 74, 3624–3633. [Google Scholar]
- 188.Tang X-F; Zhao J-N; Wu Y-F; Feng S-H; Yang F; Yu Z-Y; Meng Q-W Visible-Light-Driven Enantioselective Aerobic Oxidation of β-Dicarbonyl Compounds Catalyzed by Cinchona-Derived Phase Transfer Catalysts in Batch and Semi-Flow. Adv. Syn. Catal 2019, 361, 5245–5252. [Google Scholar]
- 189.Praetorius P; Korn F Ber. Dtsch. Chem. Ges 1910, 43, 2744–2746. [Google Scholar]
- 190.Alcock NW; Herron N; Kemp TJ; Shoppee CW Orientation of Photo-Dimerization by Metal Ions: The Crystal Structure of Bis(dibenzylideneacetone)uranyl Dichloride. J. Chem. Soc., Chem. Commun 1975, 785–786. [Google Scholar]
- 191.Lewis FD; Howard DK; Oxman JD Lewis Acid Catalysis of Coumarin Photodimerization. J. Am. Chem. Soc 1983, 105, 3344–3345. [Google Scholar]
- 192.Lewis FD; Barancyk SV Lewis Acid Catalysis of Photochemical Reactions. 8. Photodimerization and Cross-Cycloaddition of Coumarin. J. Am. Chem. Soc 1989, 111, 8653–8661. [Google Scholar]
- 193.Brenninger C; Jolliffe JD; Bach T Chromophore Activation of α,β-Unsaturated Carbonyl Compounds and Its Application to Enantioselective Photochemical Reactions. Angew. Chem. Int. Ed 2018, 57, 14338–14349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Schwinger DP; Bach T Chiral 1,3,2-Oxazaborolidine Catalysts for Enantioselective Photochemical Reactions. Acc. Chem. Res 2020, 53, 1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Guo H; Herdtweck E; Bach T Enantioselective Lewis Acid Catalysis in Intramolecular [2+2] Photocycloaddition Reactions of Coumarins. Angew. Chem. Int. Ed 2010, 49, 7782–7785. [DOI] [PubMed] [Google Scholar]
- 196.Brimioulle R; Guo H; Bach T Enantioselective Intramolecular [2+2] Photocycloaddition Reactions of 4-Substituted Coumarins Catalyzed by a Chiral Lewis Acid. Chem. - A Eur. J 2012, 18, 7552–7560. [DOI] [PubMed] [Google Scholar]
- 197.Brimioulle R; Bauer A; Bach T Enantioselective Lewis Acid Catalysis in Intramolecular [2 + 2] Photocycloaddition Reactions: A Mechanistic Comparison between Representative Coumarin and Enone Substrates. J. Am. Chem. Soc 2015, 137, 5170–5176. [DOI] [PubMed] [Google Scholar]
- 198.Brimioulle R; Bach T Enantioselective Lewis Acid Catalysis of Intramolecular Enone [2+2] Photocycloaddition Reactions. Science 2013, 342, 840–843. [DOI] [PubMed] [Google Scholar]
- 199.Wang H; Cao X; Chen X; Fang W; Dolg M Regulatory Mechanism of the Enantioselective Intramolecular Enone [2+2] Photocycloaddition Reaction Mediated by a Chiral Lewis Acid Catalyst Containing Heavy Atoms. Angew. Chem. Int. Ed 2015, 54, 14295–14298. [DOI] [PubMed] [Google Scholar]
- 200.Wang H; Fang WH; Chen X Mechanism of the Enantioselective Intramolecular [2 + 2] Photocycloaddition Reaction of Coumarin Catalyzed by a Chiral Lewis Acid: Comparison with Enone Substrates. J. Org. Chem 2016, 81, 7093–7101. [DOI] [PubMed] [Google Scholar]
- 201.Brimioulle R; Bach T [2+2] Photocycloaddition of 3-Alkenyloxy-2-cycloalkenones: Enantioselective Lewis Acid Catalysis and Ring Expansion. Angew. Chem. Int. Ed 2014, 53, 12921–12924. [DOI] [PubMed] [Google Scholar]
- 202.Bach T; Hehn J Photochemical Reactions as Key Steps in Natural Product Synthesis. Angew. Chem. Int. Ed 2011, 50, 1000–1045. [DOI] [PubMed] [Google Scholar]
- 203.Poplata S; Bach T Enantioselective Intermolecular [2+2] Photocycloaddition Reaction of Cyclic Enones and Its Application in a Synthesis of (−)-Grandisol. J. Am. Chem. Soc 2018, 140, 3228–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Stegbauer S; Jandl C; Bach T Enantioselective Lewis Acid Catalyzed ortho Photocycloaddition of Olefins to Phenanthrene-9-carboxaldehydes. Angew. Chem. Int. Ed 2018, 57, 14593–14596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.El-Sayed MA Triplet state. Its Radiative and Nonradiative Properties. Acc. Chem. Res 1968, 1, 8–16. [Google Scholar]
- 206.Griffiths J; Hart H A New General Photochemical Reaction of 2,4-Cyclohexadienones. J. Am. Chem. Soc 1968, 90, 5296–5298. [Google Scholar]
- 207.Uppili S; Ramamurthy V Enhanced Enantio- and Diastereoselectivities via Confinement: Photorearrangement of 2,4-Cyclohexadienones Included in Zeolites. Org. Lett 2002, 4, 87–90. [DOI] [PubMed] [Google Scholar]
- 208.Leverenz M; Merten C; Dreuw A; Bach T Lewis Acid Catalyzed Enantioselective Photochemical Rearrangements on the Singlet Potential Energy Surface. J. Am. Chem. Soc 2019, 141, 20053–20057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Fischer J; Mele L; Serier-Brault H; Nun P; Coeffard V Controlling Photooxygenation with a Bifunctional Quinine-BODIPY Catalyst: Towards Asymmetric Hydroxylation of β-Dicarbonyl Compounds. Eur. J. Org. Chem 2019, 37, 6352–6358 [Google Scholar]
- 210.Werner A Zur Kenntnis des Asymmetrischen Kobaltatoms. I. Chem. Ber 1911, 44, 1887–1898. [Google Scholar]
- 211.Bauer EB Chiral-at-Metal Complexes and Their Catalytic Applications in Organic Synthesis. Chem. Soc. Rev 2012, 41, 3153–3167. [DOI] [PubMed] [Google Scholar]
- 212.Cao Z-Y; Brittain WDG; Fossey JS; Zhou F Recent Advances in the use of Chiral Metal Complexes with Achiral Ligands for Application in Asymmetric Catalysis. Catal. Sci. Technol 2015, 5, 3441–3451. [Google Scholar]
- 213.Geselowitz DA; Taube H Stereoselectivity in Electron-Transfer Reactions. J. Am. Chem. Soc 1980, 102, 4525–4526. [Google Scholar]
- 214.Lappin AG; Marusak RA Stereoselectivity in Electron Transfer Reactions Involving Metal Ion Complexes. Coord. Chem. Rev 1991, 109, 125–180. [Google Scholar]
- 215.Porter GB; Sparks RH Generation of Optical Activity by Photoinduced Chiroselective Electron Transfer. J. Chem. Soc., Chem. Commun 1979, 1094–1095. [Google Scholar]
- 216.Kaizu Y; Mori T; Kobayashi H Photoinduced Electron-Transfer Reactions between Optical Isomers. J. Phys. Chem 1985, 89, 332–335. [Google Scholar]
- 217.Kato M; Sasagawa T; Ishihara Y; Yamada S; Fujitani S; Kimura M Stereoselective Photosensitized Decomposition of Tris(oxalate)cobaltate(III) and Tris(acetylacetanato)cobalt(III) by Tris(2,2’-bipyridine)ruthenium(II). J. Chem. Soc., Dalton Trans 1994, 583–587. [Google Scholar]
- 218.Dwyer FP; Gyarfas EC The Chemistry of Ruthenium. Part VI. The Existence of the Tris-o-Phenanthroline Ruthenium II and Tris-o-Phenanthroline Ruthenium III Ions in Enantiomorphous Forms. Proc. Royal Soc. N. S. W 1949, 83, 170–173. [Google Scholar]
- 219.Brandt WW; Dwyer FP; Gyarfas EC Chelate Complexes of 1,10-Phenanthroline and Related Compounds. Chem. Rev 1954, 54, 959–1017. [Google Scholar]
- 220.Porter GB; Sparks RH Photoracemization of Ru(bipyridine)32+. J. Photochem 1980, 13, 123–131. [Google Scholar]
- 221.Ohkubo K; Hamada T; Ishida H Novel Enantioselective Photocatalysis by Chiral, Helical Ruthenium(II) Complexes. J. Chem. Soc., Chem. Commun 1993, 1423–1425. [Google Scholar]
- 222.Ohkubo K; Ishida H; Hamada T; Inaoka T Enantioselective Electron Transfer Reaction Catalyzed by a Novel Photosensitizer, [Ru(S(−) or R(+)-PhEt*bpy)3]2+. Chem. Lett 1989, 18, 1545–1548. [Google Scholar]
- 223.Ohkubo K; Hamada T; Inaoka T; Ishida H Photoinduced Enantioselective and Catalytic Reduction of Co(acac)3 with a Chiral Ruthenium Photosensitizer. Inorg. Chem 1989, 28, 2021–2022. [Google Scholar]
- 224.Ohkubo K; Hamada T; Ishida H; Fukushima M; Watanabe M Stereoselective Molecular Recognition of Enantiomeric Cobalt(III) Complexes by Novel Photosensitizers of Helical Ruthenium(II) Complexes. J. Mol. Catal 1994, 89, L5–L10. [Google Scholar]
- 225.Ohkubo K; Hamada T; Watanabe M Novel Photoinduced Asymmetric Synthesis of Λ-Co[(acac)3] from Co(acac)2(H2O)2 and Hacac Catalysed by Racemic Complexes of Δ- and Λ-[Ru(menbpy)3] 2+ {menbpy = 4,4’-Di[(1R,2S,5R)-(−)-menthoxycarbonyl)]-2,2’-bipyridine; Hacac = Pentane-2,4-dione}. J. Chem. Soc., Chem. Commun 1993, 1070–1072. [Google Scholar]
- 226.Ohkubo K; Hamada T; Ishida H; Fukushima M; Watanabe M; Kobayashi H Stereoselective Photocatalysis of Helical Ruthenium(II) Complexes in the Reduction of Racemic Tris(acetylacetonato)cobalt(III). J. Chem. Soc., Dalton Trans 1994, 239–241. [Google Scholar]
- 227.Ohkubo K; Fukushima M; Ohta H; Usui S Extremely High Stereoselectivity of Novel Helical Ruthenium(II) Complexes for Photoinduced Reduction of Racemic-[Co(acac)3] (Hacac = Pentane-2,4-dione). J. Photochem. Photobiol. A 1996, 95, 137–140. [Google Scholar]
- 228.Rau H; Ratz R Asymmetry of Emission Quenching and Product Formation in an Asymmetrically Sensitized Photoreaction. Angew. Chem. Int. Ed 1983, 22, 550–551. [Google Scholar]
- 229.Wörner M; Greiner G; Rau H Asymmetric Electron Transfer from a Chiral Ru Complex Donor to an Atropisomeric Chiral 2,2’-Bipyridine Acceptor. J. Phys. Chem 1995, 99, 14161–14166. [Google Scholar]
- 230.Tsukahara K; Kaneko J; Miyaji T; Hara T; Kato M; Kimura M Stereoselective Luminescence Quenching in the Complex of Excited Triplet State of Δ-Tris(2,2’-bipyridine)ruthenium(II) with Optically Active Viologens in an Aqueous Solution. Chem. Lett 1997, 26, 6455–456. [Google Scholar]
- 231.Hamada T; Ishida H; Usui S; Watanabe Y; Tsumura K; Ohkubo K A Novel Photocatalytic Asymmetric Synthesis of (R)-(+)-1,1’-Bi-2-naphthol Derivatives by Oxidative Coupling of 3-Substituted-2-naphthol with Δ-[Ru(menbpy)32+] [menbpy = 4,4’-di(1R,2S,5R)-(−)-menthoxycarbonyl-2,2’-bipyridine], which Possesses Molecular Helicity. J. Chem. Soc., Chem. Commun 1993, 909–911. [Google Scholar]
- 232.Hamada T; Ishida H; Usui S; Tsumura K; Ohkubo K Enantioselective and Photocatalytic Oxidation of 1,1’-Bi-2-naphthol with a Chiral Ruthenium Complex which Includes Molecular Helicity. J. Mol. Catal 1994, 88, L1–L5. [Google Scholar]
- 233.Brittain HG Intermolecular Energy Transfer between Lanthanide Complexes in Aqueous Solution. 4. Stereoselectivity in the Transfer from Terbium(III) to Europium(III) Complexes of Aspartic Acid. Inorg. Chem 1979, 18, 1740–1745. [Google Scholar]
- 234.Brittain HG Intermolecular Energy Transfer Between Lanthanide Complexes in Aqueous Solution—V: Stereoselectivity in the Transfer from Terbium(III) to Europium(III) Complexes of Malic Acid. J. Inorg. Nucl. Chem 1979, 41, 721–724. [Google Scholar]
- 235.Metcalf DH; Snyder SW; Wu S; Hilmes GL; Riehl JP; Demas JN; Richardson FS Excited-State Chiral Discrimination Observed by Time-Resolved Circularly Polarized Luminescence Measurements. J. Am. Chem. Soc 1989, 111, 3082–3083. [Google Scholar]
- 236.Metcalf DH; Snyder SW; Demas JN; Richardson FS Chiral Discrimination in Electronic Energy-Transfer Processes between Dissymmetric Metal Complexes in Solution. Time-Resolved Chiroptical Luminescence Measurements of Enantioselective Excited-State Quenching Kinetics. J. Am. Chem. Soc 1990, 112, 5681–5695. [Google Scholar]
- 237.Richardson FS; Metcalf DH; Glover DP Intermolecular Chiral Recognition Probed by Enantioselective Quenching Kinetics. A Mechanistic Model for Dissymmetric Metal Complexes in Solution. J. Phys. Chem 1991, 95, 6249–6259. [Google Scholar]
- 238.Metcalf DH; Bolender JP; Driver MS; Richardson FS Chiral Discrimination in Electronic Energy Transfer between Dissymmetric Lanthanide(III) and Cobalt(III) Complexes in Solution. Effects of Ligand Size, Shape, and Configuration in the Acceptor Complexes. J. Phys. Chem 1993, 97, 553–564. [Google Scholar]
- 239.Metcalf DH; Stewart JM McD.; Snyder SW; Grisham CM Chiral Recognition between Dissymmetric Ln(dpa)33− and Co(III)-Nucleotide Complexes in Aqueous Solution. Enantioselective Luminescence Quenching as a Probe of Intermolecular Chiral Discrimination. Inorg. Chem 1992, 31, 2445–2455. [Google Scholar]
- 240.Meskers SCJ; Dekkers HPJM Enantioselective Excited-State Quenching of Racemic Tb (III) and Eu (III) Tris (pyridine-2,6-dicarboxylate) by Vitamin B12 Derivatives. Spectrochim. Acta A 1999, 55, 1857–1874. [Google Scholar]
- 241.Bolender JP; Metcalf DH; Richardson FS Chirality-Dependent Intermolecular Interactions Probed by Time-Resolved Chiroptical Luminescence Measurements of Enantio-differential Excited-State Quenching Kinetics. Chem Phys. Lett 1993, 213, 131–138. [Google Scholar]
- 242.Yu H; Dong S; Yao Q; Chen L; Zhang D; Liu X; Feng X Enantioselective [2+2] Photocycloaddition Reactions of Enones and Olefins with Visible Light Mediated by N,N’-Dioxide–Metal Complexes. Chem. Eur. J 2018, 24, 19361–19367. [DOI] [PubMed] [Google Scholar]
- 243.Ma J; Zhang X; Huang X; Luo S; Meggers E Preparation of Chiral-at-Metal Catalysts and Their use in Asymmetric Photoredox Chemistry. Nat. Protoc 2018, 13, 605–632. [DOI] [PubMed] [Google Scholar]
- 244.Zhang L; Meggers E Steering Asymmetric Lewis Acid Catalysis Exclusively with Octahedral Metal-Centered Chirality. Acc. Chem. Res 2017, 50, 320–330. [DOI] [PubMed] [Google Scholar]
- 245.Chen L-A; Xu W; Huang B; Ma J; Wang L; Xi J; Harms K; Gong L; Meggers E Asymmetric Catalysis with an Inert Chiral-at-Metal Iridium Complex. J. Am. Chem. Soc 2013, 135, 10598–10601. [DOI] [PubMed] [Google Scholar]
- 246.Chen L-A; Tang X; Xi J; Xu W; Gong L; Meggers E Chiral-at-Metal Octahedral Iridium Catalyst for the Asymmetric Construction of an All-Carbon Quaternary Stereocenter. Angew. Chem. Int. Ed 2013, 52, 14021–14025. [DOI] [PubMed] [Google Scholar]
- 247.Ma J; Ding X; Hu Y; Huang Y; Gong L; Meggers E Metal-Templated Chiral Brønsted Base Organocatalysis. Nat. Commun 2014, 5, 4531. [DOI] [PubMed] [Google Scholar]
- 248.Huo H; Fu C; Wang C; Harms K; Meggers E Metal-Templated Enantioselective Enamine/H-Bonding Dual Activation Catalysis. Chem. Commun 2014, 50, 10409–104111. [DOI] [PubMed] [Google Scholar]
- 249.Huo H; Shen X; Wang C; Zhang L; Röse P; Chen LA; Harms K; Marsch M; Hilt G; Meggers E Asymmetric Photoredox Transition-Metal Catalysis Activated by Visible Light. Nature 2014, 515, 100–103. [DOI] [PubMed] [Google Scholar]
- 250.Huo H; Wang C; Harms K; Meggers E Enantioselective, Catalytic Trichloromethylation through Visible-Light-Activated Photoredox Catalysis with a Chiral Iridium Complex. J. Am. Chem. Soc 2015, 137, 9551–9554. [DOI] [PubMed] [Google Scholar]
- 251.Huo H; Huang X; Shen X; Harms K; Meggers E Visible-Light-Activated Enantioselective Perfluoroalkylation with a Chiral Iridium Photoredox Catalyst. Synlett 2016, 27, 749–753. [Google Scholar]
- 252.Zhang X; Qin J; Huang X; Meggers E Sequential Asymmetric Hydrogenation and Photoredox Chemistry with a Single Catalyst. Org. Chem. Front 2018, 5, 166–170. [Google Scholar]
- 253.Zhang X; Qin J; Huang X; Meggers E One-Pot Sequential Photoredox Chemistry and Asymmetric Transfer Hydrogenation with a Single Catalyst. Eur. J. Org. Chem 2018, 2018, 571–577. [Google Scholar]
- 254.Wang C; Zheng Y; Huo H; Röse P; Zhang L; Harms K; Hilt G; Meggers E Merger of Visible Light Induced Oxidation and Enantioselective Alkylation with a Chiral Iridium Catalyst. Chem. - Eur. J 2015, 21, 7355–7359. [DOI] [PubMed] [Google Scholar]
- 255.Wang C; Qin J; Shen X; Riedel R; Harms K; Meggers E Asymmetric Radical-Radical Cross-Coupling through Visible-Light-Activated Iridium Catalysis. Angew. Chem. Int. Ed 2016, 55, 685–688. [DOI] [PubMed] [Google Scholar]
- 256.Böhm A; Bach T Synthesis of Supramolecular Iridium Catalysts and Their Use in Enantioselective Visible-Light-Induced Reactions. Synlett 2016, 27, 1056–1060. [Google Scholar]
- 257.Gualandi A; Calogero F; Martinelli A; Quintavalla A; Marchini M; Ceroni P; Lombardo M; Cozzi PG A Supramolecular Bifunctional Iridium Photocatalyst for the Enantioselective Alkylation of Aldehydes. Dalton Trans 2020, 49, 14497–14505. [DOI] [PubMed] [Google Scholar]
- 258.Skubi KL; Kidd JB; Jung H; Guzei IA; Baik MH; Yoon TP Enantioselective Excited-State Photoreactions Controlled by a Chiral Hydrogen-Bonding Iridium Sensitizer. J. Am. Chem. Soc 2017, 139, 17186–17192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Zheng J; Swords WB; Jung H; Skubi KL; Kidd JB; Meyer GJ; Baik MH; Yoon TP Enantioselective Intermolecular Excited-State Photoreactions Using a Chiral Ir Triplet Sensitizer: Separating Association from Energy Transfer in Asymmetric Photocatalysis. J. Am. Chem. Soc 2019, 141, 13625–13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Uraguchi D; Kimura Y; Ueoka F; Ooi T; Urea as a Redox-Active Directing Group under Asymmetric Photocatalysis of Iridium-Chiral Borate Ion Pairs. J. Am. Chem. Soc 2020, 142, 19462–19467. [DOI] [PubMed] [Google Scholar]
- 261.Kimura Y; Uraguchi D; Ooi T Catalytic Asymmetric Synthesis of 5-Membered Alicyclic α-Quaternary β-amino acids via [3+2]-photocycloaddition of α-substituted acrylates. Org. Biomol. Chem 2021, 19, 1744–1747. [DOI] [PubMed] [Google Scholar]
- 262.Rössler SL, Petrone DA & Carreira EM Iridium-catalyzed Asymmetric Synthesis of Functionally Rich Molecules Enabled by (Phosphoramidite,olefin) Ligands. Acc. Chem. Res 2019, 52, 2657–2672. [DOI] [PubMed] [Google Scholar]
- 263.Crisenza GEM; Faraone A; Gandolfo E; Mazzarella D; Melchiorre P Catalytic Asymmetric C–C Cross-couplings Enabled by Photoexcitation. Nat. Chem 2021, 13, 575–580. [DOI] [PubMed] [Google Scholar]
- 264.Huang X; Meggers E Asymmetric Photocatalysis with Bis-Cyclometalated Rhodium Complexes. Acc. Chem. Res 2019, 52, 833–847. [DOI] [PubMed] [Google Scholar]
- 265.Shen X; Harms K; Marsch M; Meggers E A Rhodium Catalyst Superior to Iridium Congeners for Enantioselective Radical Amination Activated by Visible Light. Chem. - Eur. J 2016, 22, 9102–9105. [DOI] [PubMed] [Google Scholar]
- 266.Wang C; Chen L-A; Huo H; Shen X; Harms K; Gong L; Meggers E Asymmetric Lewis Acid Catalysis Directed by Octahedral Rhodium Centrochirality. Chem. Sci 2015, 6, 1094–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Ma J; Shen X; Harms K; Meggers E Expanding the Family of Bis-cyclometalated Chiral-at-Metal Rhodium(III) Catalysts with a Benzothiazole Derivative. Dalton Trans 2016, 45, 8320–8323. [DOI] [PubMed] [Google Scholar]
- 268.Tan Y; Yuan W; Gong L; Meggers E Aerobic Asymmetric Dehydrogenative Cross-Coupling between Two Csp3—H Groups Catalyzed by a Chiral-at-Metal Rhodium Complex. Angew. Chem. Int. Ed 2015, 54, 13045–13048. [DOI] [PubMed] [Google Scholar]
- 269.Lin SX; Sun GJ; Kang Q A Visible-Light-Activated Rhodium Complex in Enantioselective Conjugate Addition of α-Amino Radicals with Michael Acceptors. Chem. Commun 2017, 53, 7665–7668. [DOI] [PubMed] [Google Scholar]
- 270.de Assis FF; Huang X; Akiyama M; Pilli RA; Meggers E Visible-Light-Activated Catalytic Enantioselective β-Alkylation of α,β-Unsaturated 2-Acyl Imidazoles Using Hantzsch Esters as Radical Reservoirs. J. Org. Chem 2018, 83, 10922–10932. [DOI] [PubMed] [Google Scholar]
- 271.Ma J; Lin J; Zhao L; Harms K; Marsch M; Xie X; Meggers E Synthesis of β-Substituted γ-Aminobutyric Acid Derivatives through Enantioselective Photoredox Catalysis. Angew. Chem. Int. Ed 2018, 57, 11193–11197. [DOI] [PubMed] [Google Scholar]
- 272.Chen L; Hu L; Du Y; Su W; Kang Q Asymmetric Photoinduced Giese Radical Addition Enabled by a Single Chiral-at-Metal Rhodium Complex. Chin. J. Org. Chem 2020, 40, 3944–3952. [Google Scholar]
- 273.Steinlandt PS; Zuo W; Harms K; Meggers E Bis-Cyclometalated Indazole Chiral-at-Rhodium Catalyst for Asymmetric Photoredox Cyanoalkylations. Chem. - Eur. J 2019, 25, 15333–15340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Tutkowski B; Meggers E; Wiest O Understanding Rate Acceleration and Stereoinduction of an Asymmetric Giese Reaction Mediated by a Chiral Rhodium Catalyst. J. Am. Chem. Soc 2017, 139, 8062–8065. [DOI] [PubMed] [Google Scholar]
- 275.Huang X; Webster RD; Harms K; Meggers E Asymmetric Catalysis with Organic Azides and Diazo Compounds Initiated by Photoinduced Electron Transfer. J. Am. Chem. Soc 2016, 138, 12636–12642. [DOI] [PubMed] [Google Scholar]
- 276.Chen S; Huang X; Meggers E; Houk KN Origins of Enantioselectivity in Asymmetric Radical Additions to Octahedral Chiral-at-Rhodium Enolates: A Computational Study. J. Am. Chem. Soc 2017, 139, 17902–17907. [DOI] [PubMed] [Google Scholar]
- 277.Ma J; Rosales AR; Huang X; Harms K; Riedel R; Wiest O; Meggers E Visible-Light-Activated Asymmetric β-C–H Functionalization of Acceptor-Substituted Ketones with 1,2-Dicarbonyl Compounds. J. Am. Chem. Soc 2017, 139, 17245–17248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Zhou Z; Nie X; Harms K; Riedel R; Zhang L; Meggers E Enantioconvergent Photoredox Radical-Radical Coupling Catalyzed by a Chiral-at-Rhodium Complex. Sci. China Chem 2019, 62, 1512–1518. [Google Scholar]
- 279.Zhang C; Gao AZ; Nie X; Ye C-X; Ivlev S; Chen.; Meggers E Catalytic α-Deracemization of Ketones Enabled by Photoredox Deprotonation and Enantioselective Protonation. J. Am. Chem. Soc 10.1021/jacs.1c06637. [DOI] [PubMed] [Google Scholar]
- 280.Huang X; Lin J; Shen T; Harms K; Marchini M; Ceroni P; Meggers E Asymmetric [3+2] Photocycloadditions of Cyclopropanes with Alkenes or Alkynes through Visible-Light Excitation of Catalyst-Bound Substrates. Angew. Chem. Int. Ed 2018, 57, 5454–5458. [DOI] [PubMed] [Google Scholar]
- 281.Huang X; Quinn TR; Harms K; Webster RD; Zhang L; Wiest O; Meggers E Direct Visible-Light-Excited Asymmetric Lewis Acid Catalysis of Intermolecular [2+2] Photocycloadditions. J. Am. Chem. Soc 2017, 139, 9120–9123. [DOI] [PubMed] [Google Scholar]
- 282.Jung H; Hong M; Marchini M; Villa M; Steinlandt PS; Huang X; Hemming M; Meggers E; Ceroni P; Park J; Baik M–H Understanding the Mechanism of Direct Visible-light-activated [2+2] Cycloadditions Mediated by Rh and Ir photocatalysts: Combined Computational and Spectroscopic Studies. Chem. Sci 2021, 12, 9673–9681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Grell Y; Hong Y; Huang X; Mochizuki T; Xie X; Harms K; Meggers E Chiral-at-Rhodium Catalyst Containing Two Different Cyclometalating Ligands. Organometallics 2019, 38, 3948–3954. [Google Scholar]
- 284.Hu N; Jung H; Zheng Y; Lee J; Zhang L; Ullah Z; Xie X; Harms K; Baik MH; Meggers E Catalytic Asymmetric Dearomatization by Visible-Light-Activated [2+2] Photocycloaddition. Angew. Chem. Int. Ed 2018, 57, 6242–6246. [DOI] [PubMed] [Google Scholar]
- 285.Jeremias N; Mohr L–M; Bach T Intermolecular [2+2] Photocycloaddition of α,β-Unsaturated Sulfones: Catalyst-Free Reaction and Catalytic Variants. Org. Lett 2021, 23, 5674–5678. [DOI] [PubMed] [Google Scholar]
- 286.Huang X; Li X; Xie X; Harms K; Riedel R; Meggers E Catalytic Asymmetric Synthesis of a Nitrogen Heterocycle through Stereocontrolled Direct Photoreaction from Electronically Excited State. Nat. Commun 2017, 8, 2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Zhang C; Chen S; Ye C; Harms K; Zhang L; Houk KN; Meggers E Asymmetric Photocatalysis by Intramolecular Hydrogen‐Atom Transfer in Photoexcited Catalyst–Substrate Complex. Angew. Chem. Int. Ed 2019, 58, 14462–14466. [DOI] [PubMed] [Google Scholar]
- 288.Woodhouse M; McCusker JK Mechanistic Origin of Photoredox Catalysis Involving Iron(II) Polypyridyl Chromophores. J. Am. Chem. Soc 2020, 142, 16229–16233. [DOI] [PubMed] [Google Scholar]
- 289.Ding W; Lu LQ; Zhou QQ; Wei Y; Chen JR; Xiao WJ Bifunctional Photocatalysts for Enantioselective Aerobic Oxidation of β-Ketoesters. J. Am. Chem. Soc 2017, 139, 63–66. [DOI] [PubMed] [Google Scholar]
- 290.Shen X; Li Y; Wen Z; Cao S; Hou X; Gong L A Chiral Nickel DBFOX Complex as a Bifunctional Catalyst for Visible-Light-Promoted Asymmetric Photoredox Reactions. Chem. Sci 2018, 9, 4562–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Ruiz Espelt L; Wiensch EM; Yoon TP Brønsted Acid Cocatalysts in Photocatalytic Radical Addition of α-Amino C–H Bonds across Michael Acceptors. J. Org. Chem 2013, 78, 4107–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Ruiz Espelt L; McPherson IS; Wiensch EM; Yoon TP Enantioselective Conjugate Additions of α-Amino Radicals via Cooperative Photoredox and Lewis Acid Catalysis. J. Am. Chem. Soc 2015, 137, 2452–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Endo K; Liu Y; Ube H; Nagata K; Shionoya M Asymmetric Construction of Tetrahedral Chiral Zinc with High Configurational Stability and Catalytic Activity. Nat. Commun 2020, 11, 6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Sakaki S; Satoh T; Ohkubo K Stereo-Selective Photo-Induced Electron Transfer Reaction from [Cu(dmp)((R, R)-diop)]+ ((R, R)-Diop = (R, R)-2,3-o-Isopropylidene-2,3-dihdydroxy-1,4-bis(diphenylphosphino)butane, dmp = 2,9-Dimethyl-1,10-phenanthroline) to [Co(edta)]−. New J. Chem 1986, 10, 145–147. [Google Scholar]
- 295.Sakaki S; Ishikura H; Kuraki K-I; Tanaka K-J; Satoh T; Arai T; Hamada T Synthesis of a New Chiral Copper(I) Complex and its Application to Stereoselective Photoreduction of [Co(edta)]– (H4edta = Ethylenedinitrioltetraacetic Acid). J. Chem. Soc., Dalton Trans 1997, 1815–1820. [Google Scholar]
- 296.Sakaki S; Horita R; Kuraki K; Hamada T Photoasymmetric Synthesis of [Co(edta)]– from Co(II) Salt and L4edta (L = H+ or Na+), using a Chiral Cu(I) Complex. Chem. Lett 1998, 27, 827–828. [Google Scholar]
- 297.Edtmüller V; Pöthig A; Bach T Enantioselective Photocyclisation Reactions of 2-Aryloxycyclohex-2-enones Mediated by a Chiral Copper-Bisoxazoline Complex. Tetrahedron 2017, 73, 5038–5047. [Google Scholar]
- 298.Creutz SE; Lotito KJ; Fu GC; Peters JC Photoinduced Ullmann C–N Coupling: Demonstrating the Viability of a Radical Pathway. Science 2012, 338, 647–651. [DOI] [PubMed] [Google Scholar]
- 299.Uyeda C; Tan Y; Fu GC; Peters JC A New Family of Nucleophiles for Photoinduced, Copper-Catalyzed Cross-Couplings via Single-Electron Transfer: Reactions of Thiols with Aryl Halides Under Mild Conditions (0 °C). J. Am. Chem. Soc 2013, 135, 9548–9552. [DOI] [PubMed] [Google Scholar]
- 300.Tan Y; Muñoz-Molina JM; Fu GC; Peters JC Oxygen Nucleophiles as Reaction Partners in Photoinduced, Copper-Catalyzed Cross-Couplings: O-Arylations of Phenols at Room Temperature. Chem. Sci 2014, 5, 2831–2835. [Google Scholar]
- 301.Ahn JM; Peters JC; Fu GC Design of a Photoredox Catalyst that Enables the Direct Synthesis of Carbamate-Protected Primary Amines via Photoinduced, Copper-Catalyzed N-Alkylation Reactions of Unactivated Secondary Halides. J. Am. Chem. Soc 2017, 139, 18101–18106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Matier CD; Schwaben J; Peters JC; Fu GC Copper-Catalyzed Alkylation of Aliphatic Amines Induced by Visible Light. J. Am. Chem. Soc 2017, 139, 17707–17710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Zhao W; Wurz RP; Peters JC; Fu GC Photoinduced, Copper-Catalyzed Decarboxylative C–N Coupling to Generate Protected Amines: An Alternative to the Curtius Rearrangement. J. Am. Chem. Soc 2017, 139, 12153–12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric Copper-Catalyzed C–N Cross-Couplings Induced by Visible Light. Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Chen C; Peters JC; Fu GC Photoinduced Copper-Catalysed Asymmetric Amidation via Ligand Cooperativity. Nature 2021, 596, 250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Guo Q; Wang M; Peng Q; Huo Y; Liu Q; Wang R; Xu Z Dual-Functional Chiral Cu-Catalyst-Induced Photoredox Asymmetric Cyanofluoroalkylation of Alkenes. ACS Catal. 2019, 9, 4470–4476. [Google Scholar]
- 307.Zhang Y; Sun Y; Chen B; Xu M; Li C; Zhang D; Zhang G Copper-Catalyzed Photoinduced Enantioselective Dual Carbofunctionalization of Alkenes. Org. Lett 2020, 22, 1490–1494. [DOI] [PubMed] [Google Scholar]
- 308.Xia H; Li Z-L; Gu Q-S; Dong X-Y; Fang J-H; Du X-Y; Wang L-L; Liu X-Y Photoinduced Copper-Catalyzed Asymmetric Decarboxylative Alkynlation with Terminal Alkynes. Angew. Chem. Int. Ed 2020, 59, 16926–16932. [DOI] [PubMed] [Google Scholar]
- 309.Qi R; Wang C; Huo Y; Chai H; Wang H; Ma Z; Liu L; Wang R; Xu Z Visible Light Induced Cu–Catalyzed Asymmetric C(sp3)–H Alkylation. 10.1021/jacs.1c05890. [DOI] [PubMed] [Google Scholar]
- 310.Li Y; Zhou K; Wen Z; Cao S; Shen X; Lei M; Gong L Copper(II)-Catalyzed Asymmetric Photoredox Reactions: Enantioselective Alkylation of Imines Driven by Visible Light. J. Am. Chem. Soc 2018, 140, 15850–15858. [DOI] [PubMed] [Google Scholar]
- 311.Han B; Li Y; Yu Y; Gong L Photocatalytic Enantioselective α-Aminoalkylation of Acyclic Imine Derivatives by a Chiral Copper Catalyst. Nat. Commun 2019, 10. 3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Zhou K; Yu Y; Lin Y-M; Li Y; Gong L Copper-catalyzed Aerobic Asymmetric Cross-dehydrogenative Coupling of C(sp3)–H Bonds Driven by Visible Light. Green Chem. 2020, 22, 4597–4603. [Google Scholar]
- 313.Chen J; Liang Y-J; Wang P-Z; Li G-Q; Zhang B; Qian H; Huan X-D; Guan W; Xiao W-J; Chen J-R Photoinduced Copper -Catalyzed Asymmetric C–O Cross-Coupling. 10.1021/jacs.1c06535. [DOI] [PubMed]
- 314.Roos CB; Demaerel J; Graff DE; Knowles RR Enantioselective Hydroamination of Alkenes with Sulfonamides Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2020, 142, 5974–5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Brak K; Jacobsen EN Asymmetric Ion-Pairing Catalysis. Angew. Chem. Int. Ed 2013, 52, 534–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Wynberg H; Greijdanus B Solvent Effects in Homogeneous Asymmetric Catalysis. J. Chem. Soc., Chem. Commun 1978, 427–428. [Google Scholar]
- 317.Ayitou A; Pemberton BC; Kumarasamy E; Vallavoju N; Sivaguru J Fun with Photons: Selective Light Induced Reactions in Solution and in Water Soluble Nano-Containers. Chimia 2011, 65, 202–209. [DOI] [PubMed] [Google Scholar]
- 318.Morimoto M; Bierschenk SM; Xia KT; Bergman RG; Raymond KN; Toste FD Advances in Supramolecular Host-Mediated Reactivity. Nat. Catal 2020, 3, 969–984. [Google Scholar]
- 319.Yan Z; Wu W; Yang C; Inoue Y Catalytic Supramolecular Photochirogenesis. Supramol. Catal 2015, 2, 9–24. [Google Scholar]
- 320.Mori T; Fukuhara G; Wada T Yoshihisa Inoue—A Researcher’s Quest for Photochirogenesis. J. Photochem. Photobiol. A 2016, 331, 2–7. [Google Scholar]
- 321.Yang C Recent Progress in Supramolecular Chiral Photochemistry. Chinese Chem. Lett 2013, 24, 437–441. [Google Scholar]
- 322.Yao J; Xu M; Yan Z; Wu W; Yang C Supramolecular Photochirogenesis with Cyclodextrins. Chinese J. Org. Chem 2014, 34, 26–35. [Google Scholar]
- 323.Inoue Y; Wada T; Asaoka S; Sato H; Pete JP Photochirogenesis: Multidimensional Control of Asymmetric Photochemistry. Chem. Commun 2000, 251–259. [Google Scholar]
- 324.Crini G Review: A History of Cyclodextrins. Chem. Rev 2014, 114, 10940–10975. [DOI] [PubMed] [Google Scholar]
- 325.Rekharsky MV; Inoue Y Complexation Thermodynamics of Cyclodextrins. Chem. Rev 1998, 98, 1875–1918. [DOI] [PubMed] [Google Scholar]
- 326.Tamaki T Reversible Photodimerization of Water-Soluble Anthracenes included in γ-Cyclodextrin. Chem. Lett 1984, 13, 53–56. [Google Scholar]
- 327.Tamaki T; Kokubu T Acceleration of the Photodimerization of Water-Soluble Anthracenes Included by β- and γ-Cyclodextrins. J. Incl. Phenom 1984, 2, 815–822. [Google Scholar]
- 328.Hamai S Association of Inclusion Compounds of β-Cyclodextrin in Aqueous Solution. Bull. Chem. Soc. Jpn 1982, 55, 2721–2729. [Google Scholar]
- 329.Rideout DC; Breslow R Hydrophobic Acceleration of Diels-Alder Reactions. J. Am. Chem. Soc 1980, 102, 7816–7817. [Google Scholar]
- 330.Tamaki T; Tomokuni K; Ichimura K Regio- and Stereoselective Photodimerization of Anthracene Derivatives Included by Cyclodextrins. Tetrahedron 1987, 43, 1485–1494. [Google Scholar]
- 331.Nakamura A; Inoue Y Supramolecular Catalysis of the Enantiodifferentiating [4 + 4] Photocyclodimerization of 2-Anthracenecarboxylate by γ-Cyclodextrin. J. Am. Chem. Soc 2003, 125, 966–972. [DOI] [PubMed] [Google Scholar]
- 332.Yang C; Wang Q; Yamauchi M; Yao J; Zhou D; Nishijima M; Fukuhara G; Mori T; Liu Y; Inoue Y Manipulating γ-Cyclodextrin-Mediated Photocyclodimerization of Anthracenecarboxylate by Wavelength, Temperature, Solvent and Host. Photochem. Photobiol. Sci 2014, 13, 190–198. [DOI] [PubMed] [Google Scholar]
- 333.Yang C; Nakamura A; Fukuhara G; Origane Y; Mori T; Wada T; Inoue Y Pressure and Temperature-Controlled Enantiodifferentiating [4+4] Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Secondary Face- and Skeleton-Modified γ-Cyclodextrins. J. Org. Chem 2006, 71, 3126–3136. [DOI] [PubMed] [Google Scholar]
- 334.Wei X; Wu W; Matsushita R; Yan Z; Zhou D; Chruma JJ; Nishijima M; Fukuhara G; Mori T; Inoue Y; et al. Supramolecular Photochirogenesis Driven by Higher-Order Complexation: Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate to Slipped Cyclodimers via a 2:2 Complex with β-Cyclodextrin. J. Am. Chem. Soc 2018, 140, 3959–3974. [DOI] [PubMed] [Google Scholar]
- 335.Wang Q; Yang C; Ke C; Fukuhara G; Mori T; Liu Y; Inoue Y Wavelength-Controlled Supramolecular Photocyclodimerization of Anthracenecarboxylate Mediated by γ-Cyclodextrins. Chem. Commun 2011, 47, 6849–6851. [DOI] [PubMed] [Google Scholar]
- 336.Wang Q; Liang W; Wei X; Wu W; Inoue Y; Yang C; Liu Y A Supramolecular Strategy for Enhancing Photochirogenic Performance through Host/Guest Modification: Dicationic γ-Cyclodextrin-Mediated Photocyclodimerization of 2,6-Anthracenedicarboxylate. Org. lett 2020, 22, 9757–9761. [DOI] [PubMed] [Google Scholar]
- 337.Teitei T; Wells D; Sasse W Photochemical Syntheses. X. Photodimers of Derivatives of Naphthalenecarboxylic Acids. Aust. J. Chem 1976, 29, 1783–1790. [Google Scholar]
- 338.Ikeda H; Iidaka Y; Ueno A Remarkably Enhanced Excimer Formation of Naphthylacetate in Cation-Charged γ-Cyclodextrin. Org. Lett 2003, 5, 1625–1627. [DOI] [PubMed] [Google Scholar]
- 339.Luo L; Liao G-H; Wu X-L; Lei L; Tung C-H; Wu L-Z γ-Cyclodextrin-Directed Enantioselective Photocyclodimerization of Methyl 3-Methoxyl-2-Naphthoate. J. Org. Chem 2009, 74, 3506–3515. [DOI] [PubMed] [Google Scholar]
- 340.Luo L; Cheng S-F; Chen B; Tung C-H; Wu L-Z Stepwise Photochemical-Chiral Delivery in γ-Cyclodextrin-Directed Enantioselective Photocyclodimerization of Methyl 3-Methoxyl-2-Naphthoate in Aqueous Solution. Langmuir 2010, 26, 782–785. [DOI] [PubMed] [Google Scholar]
- 341.Dauben WG; Koch K; Smith SL; Chapman OL Photoisomerizations in the α-Tropolone Series: The Mechanistic Path of the α-Tropolone Methyl Ether to Methyl 4-Oxo-2-Cyclopentenylacetate Conversion. J. Am. Chem. Soc 1963, 85, 2616–2621. [Google Scholar]
- 342.Koodanjeri S; Joy A; Ramamurthy V Asymmetric Induction With Cyclodextrins: Photocyclization of Tropolone Alkyl Ethers. Tetrahedron 2000, 56, 7003–7009. [Google Scholar]
- 343.Nakamura I; Sugimoto T; Oda J; Inouye Y Asymmetric Photocyclization of Nitrone at Chiral Binding Site of Cyclodextrin in the Presence of Amino Acid. Agric. Biol. Chem 1981, 45, 309–310. [Google Scholar]
- 344.Koodanjeri S; Ramamurthy V Cyclodextrin Mediated Enantio and Diastereoselective Geometric Photoisomerization of Diphenylcyclopropane and Its Derivatives. Tetrahedron Lett 2002, 43, 9229–9232. [Google Scholar]
- 345.Kuroda Y; Sera T; Ogoshi H Regioselectivities and Stereoselectivities of Singlet Oxygen Generated by Cyclodextrin-Sandwiched Porphyrin Sensitization. Lipoxygenase-Like Activity. J. Am. Chem. Soc 1991, 113, 2793–2794. [Google Scholar]
- 346.Inoue Y; Wada T; Sugahara N; Yamamoto K; Kimura K; Tong LH; Gao XM; Hou ZJ; Liu Y Supramolecular Photochirogenesis. 2. Enantiodifferentiating Photoisomerization of Cyclooctene Included and Sensitized by 6-O-Modified Cyclodextrins. J. Org. Chem 2000, 65, 8041–8050. [DOI] [PubMed] [Google Scholar]
- 347.Gao Y; Inoue M; Wada T; Inoue Y Supramolecular Photochirogenesis. 3. Enantiodifferentiating Photoisomerization of Cyclooctene Included and Sensitized by 6-O-Mono(o-Methoxybenzoyl)-β-Cyclodextrin. J. Incl. Phenom 2004, 50, 111–114. [Google Scholar]
- 348.Lu R; Yang C; Cao Y; Wang Z; Wada T; Jiao W; Mori T; Inoue Y Supramolecular Enantiodifferentiating Photoisomerization of Cyclooctene with Modified β-Cyclodextrins: Critical Control by a Host Structure. Chem. Commun 2008, 10, 374–376. [DOI] [PubMed] [Google Scholar]
- 349.Lu R; Yang C; Cao Y; Tong L; Jiao W; Wada T; Wang Z; Mori T; Inoue Y Enantiodifferentiating Photoisomerization of Cyclooctene Included and Sensitized by Aroyl-β-Cyclodextrins: A Critical Enantioselectivity Control by Substituents. J. Org. Chem 2008, 73, 7695–7701. [DOI] [PubMed] [Google Scholar]
- 350.Li G; Wang Z; Lu R; Tang Z Enantiodifferentiating Photoisomerization of Cyclooctene Included and Sensitized by Benzoate Modified β-Cyclodextrin Derivatives: Switching of Product Chirality by Solvent. Tetrahedron Lett. 2011, 52, 3097–3101. [Google Scholar]
- 351.Yang C; Mori T; Wada T; Inoue Y Supramolecular Enantiodifferentiating Photoisomerization of (Z,Z)-1,3-Cyclooctadiene Included and Sensitized by Naphthalene-Modified Cyclodextrins. New J. Chem 2007, 31, 697–702. [Google Scholar]
- 352.Yan Z; Huang Q; Liang W; Yu X; Zhou D; Wu W; Chruma JJ; Yang C Enantiodifferentiation in the Photoisomerization of (Z,Z)-1,3-Cyclooctadiene in the Cavity of γ-Cyclodextrin-Curcubit[6]Uril-Wheeled [4]Rotaxanes with an Encapsulated Photosensitizer. Org. Lett 2017, 19, 898–901. [DOI] [PubMed] [Google Scholar]
- 353.Fukuhara G; Mori T; Wada T; Inoue Y Entropy-Controlled Supramolecular Photochirogenesis: Enantiodifferentiating Z-E Photoisomerization of Cyclooctene Included and Sensitized by Permethylated 6-O-Modified β-Cyclodextrins. J. Org. Chem 2006, 71, 8233–8243. [DOI] [PubMed] [Google Scholar]
- 354.Fukuhara G; Mori T; Wada T; Inoue Y Entropy-Controlled Supramolecular Photochirogenesis: Enantiodifferentiating Z-E Photoisomerization of Cyclooctene Included and Sensitized by Permethylated 6-O-Benzoyl-β-Cyclodextrin. Chem. Commun 2005, 4199–4201. [DOI] [PubMed] [Google Scholar]
- 355.Fukuhara G; Mori T; Wada T; Inoue Y The First Supramolecular Photosensitization of Enantiodifferentiating Bimolecular Reaction: Anti-Markovnikov Photoaddition of Methanol to 1,1-Diphenylpropene Sensitized by Modified β-Cyclodextrin. Chem. Commun 2006, 1712–1714. [DOI] [PubMed] [Google Scholar]
- 356.Fukuhara G; Mori T; Inoue Y Competitive Enantiodifferentiating Anti-Markovnikov Photoaddition of Water and Methanol to 1,1-Diphenylpropene Using a Sensitizing Cyclodextrin Host. J. Org. Chem 2009, 74, 6714–6727. [DOI] [PubMed] [Google Scholar]
- 357.Kaliappan R; Ramamurthy V Chiral Photochemistry within Natural and Functionalized Cyclodextrins: Chiral Induction in Photocyclization Products from Carbonyl Compounds. J. Photochem. Photobiol. A 2009, 207, 144–152. [Google Scholar]
- 358.Wei X; Yu X; Zhang Y; Liang W; Ji J; Yao J; Rao M; Wu W; Yang C Enhanced Irregular Photodimers and Switched Enantioselectivity by Solvent and Temperature in the Photocyclodimerization of 2-Anthracenecarboxylate with Modified β-Cyclodextrins. J. Photochem. Photobiol. A 2019, 371, 374–381. [Google Scholar]
- 359.Nakamura A; Inoue Y Electrostatic Manipulation of Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate within γ-Cyclodextrin Cavity through Chemical Modification. Inverted Product Distribution and Enhanced Enantioselectivity. J. Am. Chem. Soc 2005, 127, 5338–5339. [DOI] [PubMed] [Google Scholar]
- 360.Yang C; Fukuhara G; Nakamura A; Origane Y; Fujita K; Yuan D-Q; Mori T; Wada T; Inoue Y Enantiodifferentiating [4 + 4] Photocyclodimerization of 2-Anthracenecarboxylate Catalyzed by 6A,6X-Diamino-6A,6X-Dideoxy-γ-Cyclodextrins: Manipulation of Product Chirality by Electrostatic Interaction, Temperature and Solvent in Supramolecular Photochirogenesis. J. Photochem. Photobiol. A 2005, 173, 375–383. [Google Scholar]
- 361.Ke C; Yang C; Mori T; Wada T; Liu Y; Inoue Y Catalytic Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylic Acid Mediated by a Non-Sensitizing Chiral Metallosupramolecular Host. Angew. Chemie Int. Ed 2009, 48, 6675–6677. [DOI] [PubMed] [Google Scholar]
- 362.Ke C; Yang C; Liang W; Mori T; Liu Y; Inoue Y Critical Stereocontrol by Inter-Amino Distance of Supramolecular Photocyclodimerization of 2-Anthracenecarboxylate Mediated by 6-(ω-Aminoalkylamino)-γ-Cyclodextrins. New J. Chem 2010, 34, 1323. [Google Scholar]
- 363.Liang W; Zhang HH; Wang JJ; Peng Y; Chen B; Yang C; Tung CH; Wu LZ; Fukuhara G; Mori T; et al. Supramolecular Complexation and Photocyclodimerization of Methyl 3-Methoxy-2-Naphthoate with Modified γ-Cyclodextrins. Pure Appl. Chem 2011, 83, 769–778. [Google Scholar]
- 364.Yao J; Yan Z; Ji J; Wu W; Yang C; Nishijima M; Fukuhara G; Mori T; Inoue Y Ammonia-Driven Chirality Inversion and Enhancement in Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Diguanidino-γ-Cyclodextrin. J. Am. Chem. Soc 2014, 136, 6916–6919. [DOI] [PubMed] [Google Scholar]
- 365.Kanagaraj K; Liang W; Rao M; Yao J; Wu W; Cheng G; Ji J; Wei X; Peng C; Yang C pH-Controlled Chirality Inversion in Enantiodifferentiating Photocyclodimerization of 2-Antharacenecarboxylic Acid Mediated by γ-Cyclodextrin Derivatives. Org. Lett 2020, 22, 5273–5278. [DOI] [PubMed] [Google Scholar]
- 366.Yi J; Liang W; Wei X; Yao J; Yan Z; Su D; Zhong Z; Gao G; Wu W; Yang C Switched Enantioselectivity by Solvent Components and Temperature in Photocyclodimerization of 2-Anthracenecarboxylate with 6A,6X-Diguanidio–γ-Cyclodextrins. Chinese Chem. Lett 2018, 29, 87–90. [Google Scholar]
- 367.Ikeda H; Nihei T; Ueno A Template-Assisted Stereoselective Photocyclodimerization of 2-Anthracenecarboxylic Acid by Bispyridinio-Appended γ-Cyclodextrin. J. Org. Chem 2005, 70, 1237–1242. [DOI] [PubMed] [Google Scholar]
- 368.Yang C; Mori T; Inoue Y Supramolecular Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Capped γ-Cyclodextrins: Critical Control of Enantioselectivity by Cap Rigidity. J. Org. Chem 2008, 73, 5786–5794. [DOI] [PubMed] [Google Scholar]
- 369.Yang C; Ke C; Kahee F; Yuan D-Q; Mori T; Inoue Y pH-Controlled Supramolecular Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate with Capped γ-Cyclodextrins. Aust. J. Chem 2008, 61, 565–568. [Google Scholar]
- 370.Yang C; Nishijima M; Nakamura A; Mori T; Wada T; Inoue Y A Remarkable Stereoselectivity Switching upon Solid-State versus Solution-Phase Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylic Acid Mediated by Native and 3,6-Anhydro-γ-Cyclodextrins. Tetrahedron Lett. 2007, 48, 4357–4360. [Google Scholar]
- 371.Wang Q; Yang C; Fukuhara G; Mori T; Liu Y; Inoue Y Supramolecular FRET Photocyclodimerization of Anthracenecarboxylate with Naphthalene-Capped γ-Cyclodextrin. Beilstein J. Org. Chem 2011, 7, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Rao M; Kanagaraj K; Fan C; Ji J; Xiao C; Wei X; Wu W; Yang C Photocatalytic Supramolecular Enantiodifferentiating Dimerization of 2-Anthracenecarboxylic Acid through Triplet-Triplet Annihilation. Org. Lett 2018, 20, 1680–1683. [DOI] [PubMed] [Google Scholar]
- 373.Rao M; Wu W; Yang C Effects of Temperature and Host Concentration on the Supramolecular Enantiodifferentiating [4 + 4] Photodimerization of 2-Anthracenecarboxylate through Triplet-Triplet Annihilation Catalyzed by Pt-Modified Cyclodextrins. Molecules 2019, 24, 1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Ji J; Wu W; Liang W; Cheng G; Matsushita R; Yan Z; Wei X; Rao M; Yuan DQ; Fukuhara G; et al. An Ultimate Stereocontrol in Supramolecular Photochirogenesis: Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Sulfur-Linked β-Cyclodextrin Dimers. J. Am. Chem. Soc 2019, 141, 9225–9238. [DOI] [PubMed] [Google Scholar]
- 375.Ji J; Wu W; Wei X; Rao M; Zhou D; Cheng G; Gong Q; Luo K; Yang C Synergetic Effects in the Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylic Acid Mediated by β-Cyclodextrin-Pillar[5]Arene-Hybridized Hosts. Chem. Commun 2020, 56, 6197–6200. [DOI] [PubMed] [Google Scholar]
- 376.Fukuhara G; Imai M; Yang C; Mori T; Inoue Y Enantiodifferentiating Photoisomerization of (Z,Z)-1,3-Cyclooctadiene Included and Sensitized by Naphthoyl-Curdlan. Org. Lett 2011, 13, 1856–1859. [DOI] [PubMed] [Google Scholar]
- 377.Yang C; Liang W; Nishijima M; Fukuhara G; Mori T; Hiramatsu H; Dan-oh Y; Tsujimoto K; Inoue Y Supramolecular Photochirogenesis with Novel Cyclic Tetrasaccharide: Enantiodifferentiating Photoisomerization of (Z)-Cyclooctene with Cyclic Nigerosylnigerose-Based Sensitizers. Chirality 2012, 24, 921–927. [DOI] [PubMed] [Google Scholar]
- 378.Nishioka Y; Yamaguchi T; Kawano M; Fujita M Asymmetric [2 + 2] Olefin Cross Photoaddition in a Self-Assembled Host with Remote Chiral Auxiliaries. J. Am. Chem. Soc 2008, 130, 8160–8161. [DOI] [PubMed] [Google Scholar]
- 379.Murase T; Peschard S; Horiuchi S; Nishioka Y; Fujita M Remote Chiral Transfer into [2 + 2] and [2 + 4] Cycloadditions within Self-Assembled Molecular Flasks. Supramol. Chem 2011, 23, 199–208. [Google Scholar]
- 380.Alagesan M; Kanagaraj K; Wan S; Sun H; Su D; Zhong Z; Zhou D; Wu W; Gao G; Zhang H; et al. Enantiodifferentiating [4 + 4] Photocyclodimerization of 2-Anthracenecarboxylate Mediated by a Self-Assembled Iron Tetrahedral Coordination Cage. J. Photochem. Photobiol. A 2016, 331, 95–101. [Google Scholar]
- 381.Mal P; Schultz D; Beyeh K; Rissanen K; Nitschke JR An Unlockable–Relockable Iron Cage by Subcomponent Self-Assembly. Angew. Chem. Int. Ed 2008, 47, 8297–8301. [DOI] [PubMed] [Google Scholar]
- 382.Guo J; Xu YW; Li K; Xiao LM; Chen S; Wu K; Chen XD; Fan YZ; Liu JM; Su CY Regio- and Enantioselective Photodimerization within the Confined Space of a Homochiral Ruthenium/Palladium Heterometallic Coordination Cage. Angew. Chem. Int. Ed 2017, 56, 3852–3856. [DOI] [PubMed] [Google Scholar]
- 383.Guo J; Fan YZ; Lu YL; Zheng SP; Su CY Visible-Light Photocatalysis of Asymmetric [2+2] Cycloaddition in Cage-Confined Nanospace Merging Chirality with Triplet-State Photosensitization. Angew. Chem. Int. Ed 2020, 59, 8661–8669. [DOI] [PubMed] [Google Scholar]
- 384.Wada T; Sugahara N; Kawano M; Inoue Y First Asymmetric Photochemistry with Nucleosides and DNA: Enantiodifferentiating Z-E Photoisomerization of Cyclooctene. Chem. Lett 2000, 29, 1174–1175. [Google Scholar]
- 385.Sugahara N; Kawano M; Wada T; Inoue Y Enantiodifferentiating Z-E Photoisomerization of Cyclooctene Sensitized by DNA and RNA. Nucleic Acids Symp. Ser 2000, 44, 115–116. [DOI] [PubMed] [Google Scholar]
- 386.Gaß N; Gebhard J; Wagenknecht HA Photocatalysis of a [2+2] Cycloaddition in Aqueous Solution Using DNA Three-Way Junctions as Chiral PhotoDNAzymes. ChemPhotoChem 2017, 1, 48–50. [Google Scholar]
- 387.Weinberger M; Wagenknecht HA Synthesis of a Benzophenone C-Nucleoside as Potential Triplet Energy and Charge Donor in Nucleic Acids. Synthesis 2012, 44, 648–652. [Google Scholar]
- 388.Merz T; Wenninger M; Weinberger M; Riedle E; Wagenknecht HA; Schütz M Conformational Control of Benzophenone-Sensitized Charge Transfer in Dinucleotides. Phys. Chem. Chem. Phys 2013, 15, 18607–18619. [DOI] [PubMed] [Google Scholar]
- 389.Björn LO Photoactive Proteins. In Photobiology; Springer New York: New York, NY, 2015; pp 139–150. [Google Scholar]
- 390.Gabruk M; Mysliwa-Kurdziel B Light-Dependent Protochlorophyllide Oxidoreductase: Phylogeny, Regulation, and Catalytic Properties. Biochemistry 2015, 54, 5255–5262. [DOI] [PubMed] [Google Scholar]
- 391.Heyes DJ; Hunter CN Making Light Work of Enzyme Catalysis: Protochlorophyllide Oxidoreductase. Trends Biochem. Sci 2005, 30, 642–649. [DOI] [PubMed] [Google Scholar]
- 392.Hyster TK Radical Biocatalysis: Using Non-Natural Single Electron Transfer Mechanisms to Access New Enzymatic Functions. Synlett 2020, 31, 248–254. [Google Scholar]
- 393.Sandoval BA; Hyster TK Emerging Strategies for Expanding the Toolbox of Enzymes in Biocatalysis. Curr. Opin. Chem. Biol 2020, 55, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Kragh-Hansen U Molecular Aspects of Ligand Binding to Serum Albumin. Pharmacol. Rev 1981, 33, 17–53. [PubMed] [Google Scholar]
- 395.Peters T; All about Albumin: Biochemistry, Genetics, and Medical Applications, Academic Press, San Diego, CA, 1996 [Google Scholar]
- 396.Wada T; Nishijima M; Fujisawa T; Sugahara N; Mori T; Nakamura A; Inoue Y Bovine Serum Albumin-Mediated Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate. J. Am. Chem. Soc 2003, 125, 7492–7493. [DOI] [PubMed] [Google Scholar]
- 397.Nishijima M; Pace TCS; Nakamura A; Mori T; Wada T; Bohne C; Inoue Y Supramolecular Photochirogenesis with Biomolecules. Mechanistic Studies on the Enantiodifferentiation for the Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Bovine Serum Albumin. J. Org. Chem 2007, 72, 2707–2715. [DOI] [PubMed] [Google Scholar]
- 398.Nishijima M; Goto M; Fujikawa M; Yang C; Mori T; Wada T; Inoue Y Mammalian Serum Albumins as a Chiral Mediator Library for Bio-Supramolecular Photochirogenesis: Optimizing Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate. Chem. Commun 2014, 50, 14082–14085. [DOI] [PubMed] [Google Scholar]
- 399.Wada T; Nishijima M; Sakamoto S; Murakami M; Sugawara Y; Araki Y; Inoue Y Novel Strategy of Supramolecular Asymmetric Photochirogenesis with Tailor-Made Biopolymers as Chiral Reaction Fields. J. Photopolym. Sci. Technol 2011, 24, 595–596. [Google Scholar]
- 400.Nishijima M; Wada T; Mori T; Pace TCS; Bohne C; Inoue Y Highly Enantiomeric Supramolecular [4 + 4] Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Human Serum Albumin. J. Am. Chem. Soc 2007, 129, 3478–3479. [DOI] [PubMed] [Google Scholar]
- 401.Nishijima M; Kato H; Yang C; Fukuhara G; Mori T; Araki Y; Wada T; Inoue Y Catalytic Bio-Supramolecular Photochirogenesis: Batch-Operated Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate with Human Serum Albumin. ChemCatChem 2013, 5, 3237–3240. [Google Scholar]
- 402.Nishijima M; Pace TCS; Bohne C; Mori T; Inoue Y; Wada T Highly Enantiodifferentiating Site of Human Serum Albumin for Mediating Photocyclodimerization of 2-Anthracenecarboxylate Elucidated by Site-Specific Inhibition/Quenching with Xenon. J. Photochem. Photobiol. A 2016, 331, 89–94. [Google Scholar]
- 403.Pace TCS; Nishijima M; Wada T; Inoue Y; Bohne C Photophysical Studies on the Supramolecular Photochirogenesis for the Photocyclodimerization of 2-Anthracenecarboxylate within Human Serum Albumin. J. Phys. Chem. B 2009, 113, 10445–10453. [DOI] [PubMed] [Google Scholar]
- 404.Nishijima M; Kato H; Fukuhara G; Yang C; Mori T; Maruyama T; Otagiri M; Inoue Y Photochirogenesis with Mutant Human Serum Albumins: Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate. Chem. Commun 2013, 49, 7433–7435. [DOI] [PubMed] [Google Scholar]
- 405.Fukagawa M; Kawamura I; Ubukata T; Yokoyama Y Enantioselective Photochromism of Diarylethenes in Human Serum Albumin. Chem. - A Eur. J 2013, 19, 9434–9437. [DOI] [PubMed] [Google Scholar]
- 406.Kawamura K; Osawa K; Watanobe Y; Saeki Y; Maruyama N; Yokoyama Y Photocyclization of Photoswitches with High Enantioselectivity in Human Serum Albumin in an Artificial Environment. Chem. Commun 2017, 53, 3181–3184. [DOI] [PubMed] [Google Scholar]
- 407.Saeki Y; Kayanuma M; Nitta A; Shigeta Y; Kawamura I; Nakagawa T; Ubukata T; Yokoyama Y On-Demand Chirality Transfer of Human Serum Albumin to Bis(thiophen-2-yl)hexafluorocyclopentenes through Their Photochromic Ring Closure. J. Org. Chem 2021. DOI: 10.1021/acs.joc.1c00849. [DOI] [PubMed] [Google Scholar]
- 408.Bando K; Zako T; Sakono M; Maeda M; Wada T; Nishijima M; Fukuhara G; Yang C; Mori T; Pace TCS; et al. Bio-Supramolecular Photochirogenesis with Molecular Chaperone: Enantiodifferentiating Photocyclodimerization of 2-Anthracenecarboxylate Mediated by Prefoldin. Photochem. Photobiol. Sci 2010, 9, 655–660. [DOI] [PubMed] [Google Scholar]
- 409.Emmanuel MA; Greenberg NR; Oblinsky DG; Hyster TK Accessing Non-Natural Reactivity by Irradiating Nicotinamide-Dependent Enzymes with Light. Nature 2016, 540, 414–417. [DOI] [PubMed] [Google Scholar]
- 410.Biegasiewicz KF; Cooper SJ; Gao X; Oblinsky DG; Kim JH; Garfinkle SE; Joyce LA; Sandoval BA; Scholes GD; Hyster TK Photoexcitation of Flavoenzymes Enables a Stereoselective Radical Cyclization. Science. 2019, 364, 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Clayman PD; Hyster TK Photoenzymatic Generation of Unstabilized Alkyl Radicals: An Asymmetric Reductive Cyclization. J. Am. Chem. Soc 2020, 142, 15673–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Page CG; Cooper SJ; Dehovitz JS; Oblinsky DG; Biegasiewicz KF; Antropow AH; Armbrust KW; Ellis JM; Hamann LG; Horn EJ; et al. Quaternary Charge-Transfer Complex Enables Photoenzymatic Intermolecular Hydroalkylation of Olefins. J. Am. Chem. Soc 2021, 143, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Huang X; Wang B; Wang Y; Jiang G; Feng J; Zhao H Photoenzymatic Enantioselective Intermolecular Radical Hydroalkylation. Nature 2020, 584, 69–74. [DOI] [PubMed] [Google Scholar]
- 414.Black MJ; Biegasiewicz KF; Meichan AJ; Oblinsky DG; Kudisch B; Scholes GD; Hyster TK Asymmetric Redox-Neutral Radical Cyclization Catalysed by Flavin-Dependent ‘Ene’-Reductases. Nat. Chem 2020, 12, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Sandoval BA; Clayman PD; Oblinsky DG; Oh S; Nakano Y; Bird M; Scholes GD; Hyster TK Photoenzymatic Reductions Enabled by Direct Excitation of Flavin-Dependent “Ene”-Reductases. J. Am. Chem. Soc 2021, 143, 1735–1739. [DOI] [PubMed] [Google Scholar]
- 416.Lee SH; Choi DS; Pesic M; Lee YW; Paul CE; Hollmann F; Park CB Cofactor-Free, Direct Photoactivation of Enoate Reductases for the Asymmetric Reduction of C=C Bonds. Angew. Chem. Int. Ed 2017, 56, 8681–8685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Yoon J; Lee SH; Tieves F; Rauch M; Hollmann F; Park CB Light-Harvesting Dye-Alginate Hydrogel for Solar-Driven, Sustainable Biocatalysis of Asymmetric Hydrogenation. ACS Sustain. Chem. Eng 2019, 7, 5632–5637. [Google Scholar]
- 418.Winkler JR; Gray HB Electron Flow through Metalloproteins. Chem. Rev 2014, 114, 3369–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Gu Y; Ellis-Guardiola K; Srivastava P; Lewis JC Preparation, Characterization, and Oxygenase Activity of a Photocatalytic Artificial Enzyme. ChemBioChem 2015, 16, 1880–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Schwochert TD; Cruz CL; Watters JW; Reynolds EW; Nicewicz DA; Brustad EM Design and Evaluation of Artificial Hybrid Photoredox Biocatalysts. ChemBioChem 2020, 21, 3146–3150. [DOI] [PubMed] [Google Scholar]
- 421.Dwaraknath S; Tran NH; Dao T; Colbert A; Mullen S; Nguyen A; Cortez A; Cheruzel L A Facile and Versatile Methodology for Cysteine Specific Labeling of Proteins with Octahedral Polypyridyl D6 Metal Complexes. J. Inorg. Biochem 2014, 136, 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Kato M; Nguyen D; Gonzalez M; Cortez A; Mullen SE; Cheruzel LE Regio- and Stereoselective Hydroxylation of 10-Undecenoic Acid with a Light-Driven P450 BM3 Biocatalyst Yielding a Valuable Synthon for Natural Product Synthesis. Bioorganic Med. Chem 2014, 22, 5687–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Sosa V; Melkie M; Sulca C; Li J; Tang L; Li J; Faris J; Foley B; Banh T; Kato M; et al. Selective Light-Driven Chemoenzymatic Trifluoromethylation/Hydroxylation of Substituted Arenes. ACS Catal. 2018, 8, 2225–2229. [Google Scholar]
- 424.Crawford J; Sigman M Conformational Dynamics in Asymmetric Catalysis: Is Catalyst Flexibility a Design Element? Synthesis 2019, 51, 1021–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Lévesque F; Di Maso MJ; Narsimhan K; Wismer MK; Naber JR Design of a Kilogram Scale, Plug Flow Photoreactor Enabled by High Power LEDs. Org. Process Res. Dev 2020, 24, 2935–2940. [Google Scholar]
- 426.Harper K; Grieme T; Towne T; Mack D; Diwan M; Ku Y-Y; Griffin J; Miller R; Zhang E-X; Liu Z-W; Zheng S-Y; et al. Commercial-Scale Visible-light Trifluoromethylation of 2-Chlorothiophenol using CF3I gas. ChemRxiv 2021, DOI: 10.33774/chemrxiv-2021-z3xm8. [DOI] [Google Scholar]
- 427.Cambié D; Bottecchia C; Straathof NJW; Hessel V; Noël T Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev 2016, 116, 10276–10341. [DOI] [PubMed] [Google Scholar]