

Abstract
Genomic disruptions, altered epigenetic mechanisms, and environmental factors contribute to the heterogeneity of congenital heart defects (CHD). In recent years, chromosomal microarray analysis (CMA) has led to the identification of numerous copy number variations (CNV) in patients with CHD. Genes disrupted by and within these CNVs thus represent excellent candidate genes for CHD. Microduplications of 9q (9q+) have been described in patients with CHD, however, the critical gene locus remains undetermined. Here we discuss an infant with tetralogy of Fallot with absent pulmonary valve, fetal hydrops, and a 3.76 Mb de novo contiguous gain of 9q34.2-q34.3 detected by CMA, and confirmed by karyotype and FISH studies. This duplicated interval disrupted RXRA (retinoid X receptor alpha; OMIM #180245) at intron 1. We also review CHD findings among previously reported patients with 9q (9q+) duplication syndrome. This is the first report implicating RXRA in CHD with 9q duplication, providing additional data in understanding the genetic etiology of tetralogy of Fallot, CHD, and disorders linked to 9q microduplication syndrome. This report also highlights the significance of CMA in the clinical diagnosis and genetic counseling of patients and families with complex CHD.
Keywords: tetralogy of Fallot with absent pulmonary valve, retinoid X receptor alpha, RXRA
INTRODUCTION
Congenital heart defects (CHD) are the most common congenital malformations and affect approximately 8 per 1,000 live births [Reller et al., 2008]. The incidence of CHD is even higher among fetuses, as many fetuses with complex lesions do not survive to term birth [Hunter and Simpson, 2014]. Tetralogy of Fallot (TOF; OMIM #187500), the most common cyanotic congenital heart defect, is a conotruncal malformation characterized by four abnormalities: pulmonary infundibular stenosis, ventricular septal defect (VSD), overriding aorta, and right ventricular hypertrophy [Ho et al., 2001]. Tetralogy of Fallot with absent pulmonary valve (TOF/APV) is an uncommon CHD accounting for only 3–6% of patients with TOF-spectrum defects [Moon-Grady et al., 2002]. TOF/APV shares the key features of the classical form of TOF as well as an incompetent or absent pulmonary valve.
Genetic factors, epigenetic changes, and environmental influences contribute to the multifactorial nature of CHD. Numerical and structural chromosome anomalies as well as point mutations in over 50 monogenic loci encoding transcription factors, cell signaling proteins, and epigenetic regulators have been identified among infants with isolated or syndromic CHD [Wilson et al., 1985; Kleefstra et al., 1993; Gijsbers et al., 2008; Bittel et al., 2011; Andersen et al., 2014; Hunter and Simpson, 2014]. Murine models of targeted monogenic deletions have identified over 500 genes which when disrupted contribute to the development of CHD [Andersen et al., 2014] (Mouse Genome Informatics; http://www.informatics.jax.org/). For example, homozygous (Rxra –/–) and heterozygous (Rxra +/–) deletions result in abnormal cardiac structure and function in mice [Kastner et al., 1994; Sucov et al., 1994; Dyson et al., 1995; Gruber et al., 1996], and disruption of RXRA may contribute to the pathogenesis of CHD in humans (retinoid X receptor, alpha; OMIM #180245).
Copy number variants have also been identified among individuals with CHD [Hitz et al., 2012; Tomita-Mitchell et al., 2012; Andersen et al., 2014; Bansal et al., 2014; Soemedi et al., 2014]. In humans, microduplications of the long arm of chromosome 9 (9q+) have been associated with CHD as well as low birth weight, failure-to-thrive, craniofacial dysmorphic features (microcephaly, prominent nasal bridge, microstomia, micrognathia), neurologic findings (hypotonia, intellectual disability, developmental delay, and psychomotor delay), skeletal defects (arachnodactyly, joint contractures, abnormally positioned digits), and urogenital malformations [Turleau et al., 1975; Allderdice et al., 1983]. Chromosome microarray analysis (CMA) has identified various overlapping 9q+ breakpoints thereby expanding the clinical spectrum and severity of phenotypic findings. Smaller duplications of 9q34 (9q34+) tend to correlate with milder phenotypes, while larger duplications correlate with more severe malformations and neurologic impairment [Allderdice et al., 1983; Spinner et al., 1993; Papadopoulou et al., 2010]. However, the critical region and candidate genes that impart risk for CHD among patients with 9q microduplications remain to be established [Gelb, 2004; Greenway et al., 2009; Silversides et al., 2012; Glessner et al., 2014; Warburton et al., 2014].
We discuss an infant with TOF/APV, fetal hydrops, and de novo contiguous gain in 9q34.2-q34.3, disrupting RXRA at intron 1 and duplicating exons 2–10, and review the reported patients with 9q+ duplications. This is the first report of disrupted RXRA and complex 9q+ in a patient with TOF or CHD, suggesting that RXRA at 9q34.2 may be a critical locus in 9q+-associated CHD. This report highlights the importance of CMA in genomic and epigenomic characterization, as well as in genetic diagnosis of complex congenital malformations and clinical features associated with 9q microduplication syndrome.
CLINICAL REPORT
A 2,135 g (10th percentile) infant female was born at 35 weeks gestation to a 26-year-old primigravida mother who was rubella immune. The pregnancy was complicated by the second trimester ultrasound finding of a single umbilical artery. Follow-up fetal ultrasound performed at 35 weeks gestation revealed new-onset cardiomegaly, abdominal ascites, and scalp edema consistent with fetal hydrops. Fetal echocardiogram revealed marked cardiomegaly with a cardiothoracic area ratio of 60% (normal <35%). Also noted were a large malaligned VSD with overriding aorta and an enlarged pulmonary outflow tract over three times greater in size than the aortic outflow tract with to-and-fro flow by color Doppler. Rudimentary pulmonary valve leaflets were noted in the annular position and the main and branch pulmonary arteries were markedly dilated. A ductus arteriosus was not present. These findings were consistent with TOF/APV. The left and right ventricles were dilated with moderately decreased and severely decreased systolic function, respectively. Given the poor prognosis of the underlying cardiac defect in the setting of decreased ventricular function and fetal hydrops, the decision was made to provide comfort care upon delivery. The infant was limp and cyanotic at birth with poor respiratory effort. Her physical examination was notable for diminished breath sounds, a distended abdomen, elongated fingers and toes, and loose skin on her legs. Facial exam was notable for triangular facies with a broad forehead, but no other dysmorphic features including hypertelorism, down-slanting palpebral fissures, coarsening of facies, or abnormal helices. Philtrum, nasal bridge, and mouth were normal in appearance. She took a few spontaneous, shallow breaths after delivery and died within minutes of birth. Cord blood was collected at the time of delivery for chromosome microarray and the family agreed to postmortem magnetic resonance imaging (MRI) and genetic testing.
Laboratory/Imaging Studies
Postmortem MRI was notable for marked cardiomegaly with biventricular hypertrophy (Fig. 1). The main pulmonary artery and branch pulmonary arteries were markedly enlarged. Pleural effusion and large volume abdominal ascites were noted as well as small kidneys. No other defects were identified on the MRI.
FIG. 1.
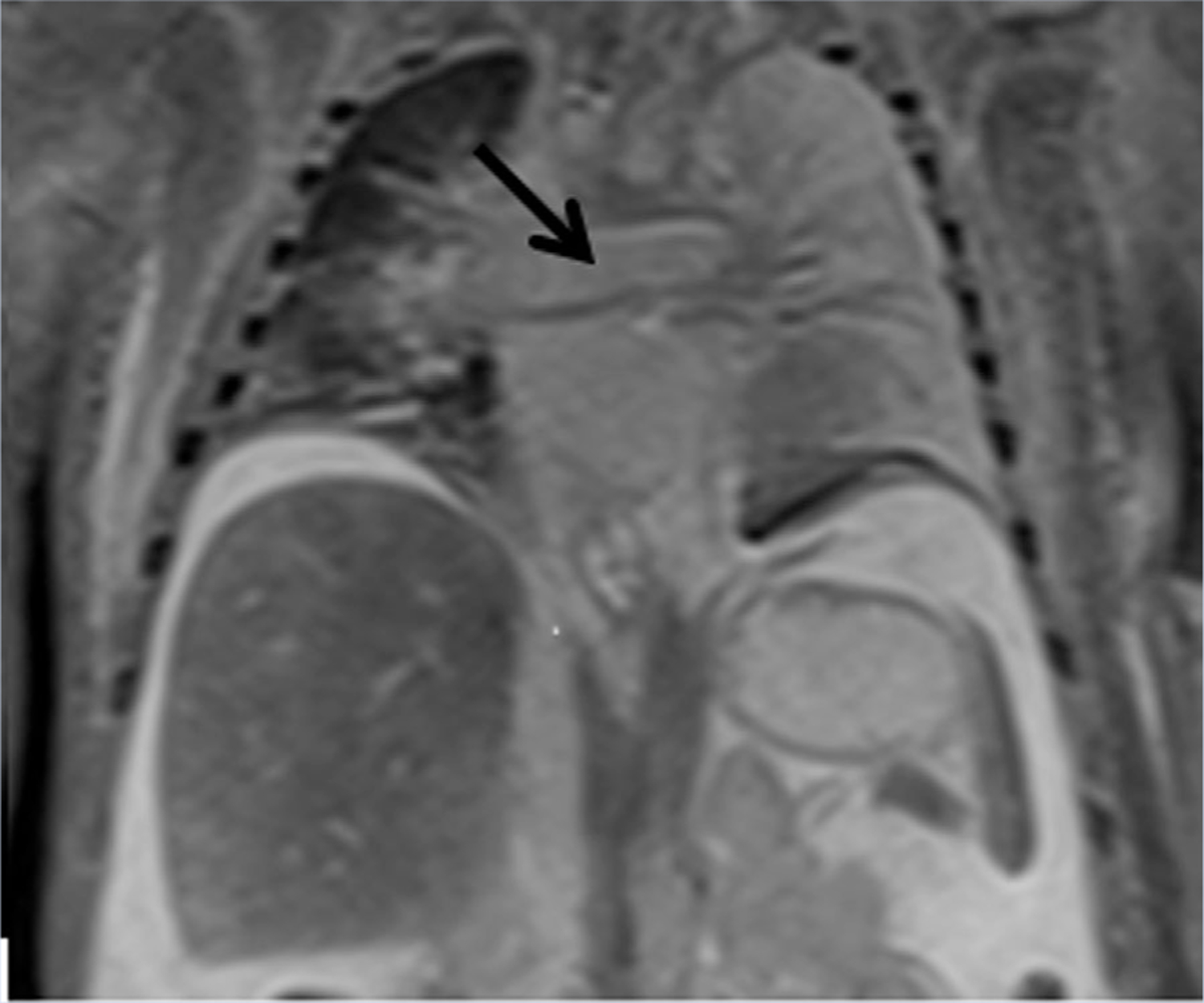
Postmortem MRI demonstrating cardiomegaly, enlarged right pulmonary artery (black arrow), atelectasis especially of left lung, and abdominal ascites.
MATERIALS AND METHODS
Genomic DNA was isolated from cord blood lymphocytes of the infant (proband) for CMA and data output was analyzed using CytoScan HD (Affymterix, Inc., Santa Clara, CA). Lymphocytes from the proband and parental blood samples were propagated in culture and metaphase spreads were obtained for G-banding chromosome analysis and HIRA (22q11.21) FISH studies. Post-CMA FISH studies were also performed on interphase nuclei and metaphase chromosomes using the following probes: RP11–644H13 BAC Clone for 9q34.3 locus (185 Kb; [hg19] chr9:140,535,102–140,719,726) and 9q21-specific control probe.
RESULTS
CMA revealed three contiguous copy number gains (triplication-duplication-triplication) at 9q34.2-q34.3 (3.76 Mb; hg19; 137,259,675–141,020,389) (Fig. 2A). This interval includes 2,730 probes and 66 OMIM genes. Karyotype analysis showed extra chromosome material on 9q34 (Figs. 2B and C, arrow). FISH studies revealed an enhanced signal at 9q+ on metaphase chromosomes (Fig. 2D) and approximately five copies (with one enhanced signal) (Fig. 2E) on interphase nuclei, consistent with the copy number gain (>3 copies) detected by CMA. Parental cytogenetic studies were normal and thus the 9q+ finding in the proband was a de novo event. The proximal breakpoint revealed disruption at intron 1 and duplication of exons 2–10 of RXRA (Figs. 3 and 4B). There was no evidence of deletion of HIRA by FISH studies and CMA.
FIG. 2.
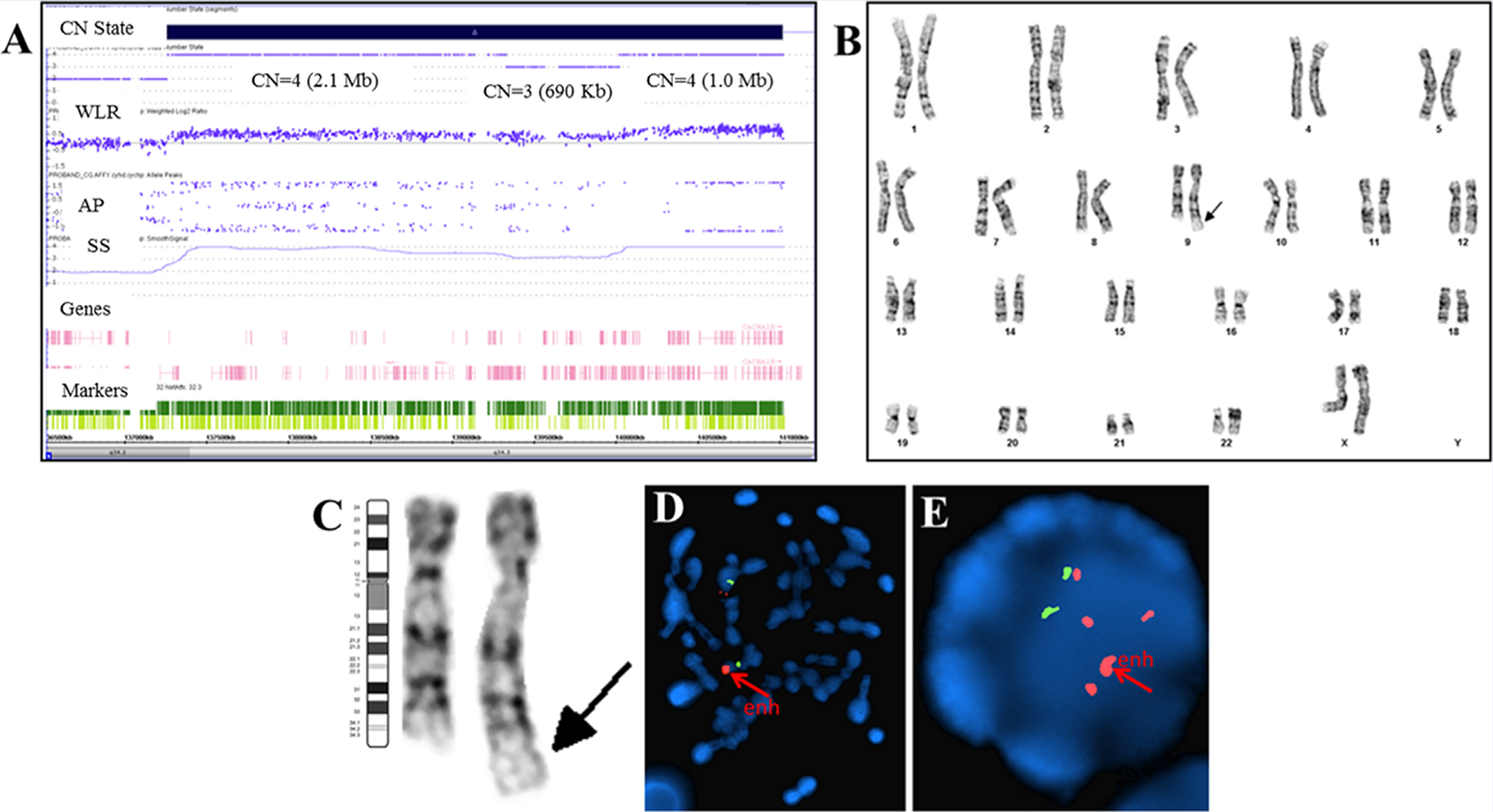
Cytogenomic studies in our patient with 9q34.2-q34.3 gains. A: CMA (Affymetrix ChAS Browser) detected contiguous copy number gains; 9q34.2q34.3(137,259,675–139,328,332) x4 (2.1 Mb; 5’ breakpoint at intron 1 of RXRA and gains of exons 2–10; 3’ breakpoint at intron 3 of INPP5E and gains of exons 1–3), 9q34.3(139,328,793–140,018,897) x3 (690 Kb; no gene disruption), 9q34.3(140,018,931–141,020,389) x4 (1.0 Mb; no gene disruption); allele peaks (AP) (0.5, 0, –0.5); weighted log ratio (WLR) (0.5, 0, –0.5); smooth signal (SS) (1–4); oligo and SNP probes (dark and light green). The nearest normal marker is 4.4 Kb from 5’ of gain at intron 1 of RXRA. B and C: Post-CMA karyotype result supported the copy number gains on 9qter (magnified in C). D and E: FISH studies using RP11–644H13 BAC Clone (185 Kb) for 9q34.3 locus; [hg19] chr9:140,535,102–140,719,726) and 9q21-specific control probe revealed enhanced 9q34.3 signal in metaphase (D) and interphase (E) cells; and at least five 9q34.3 signals in interphase nuclei (E).
FIG. 3.
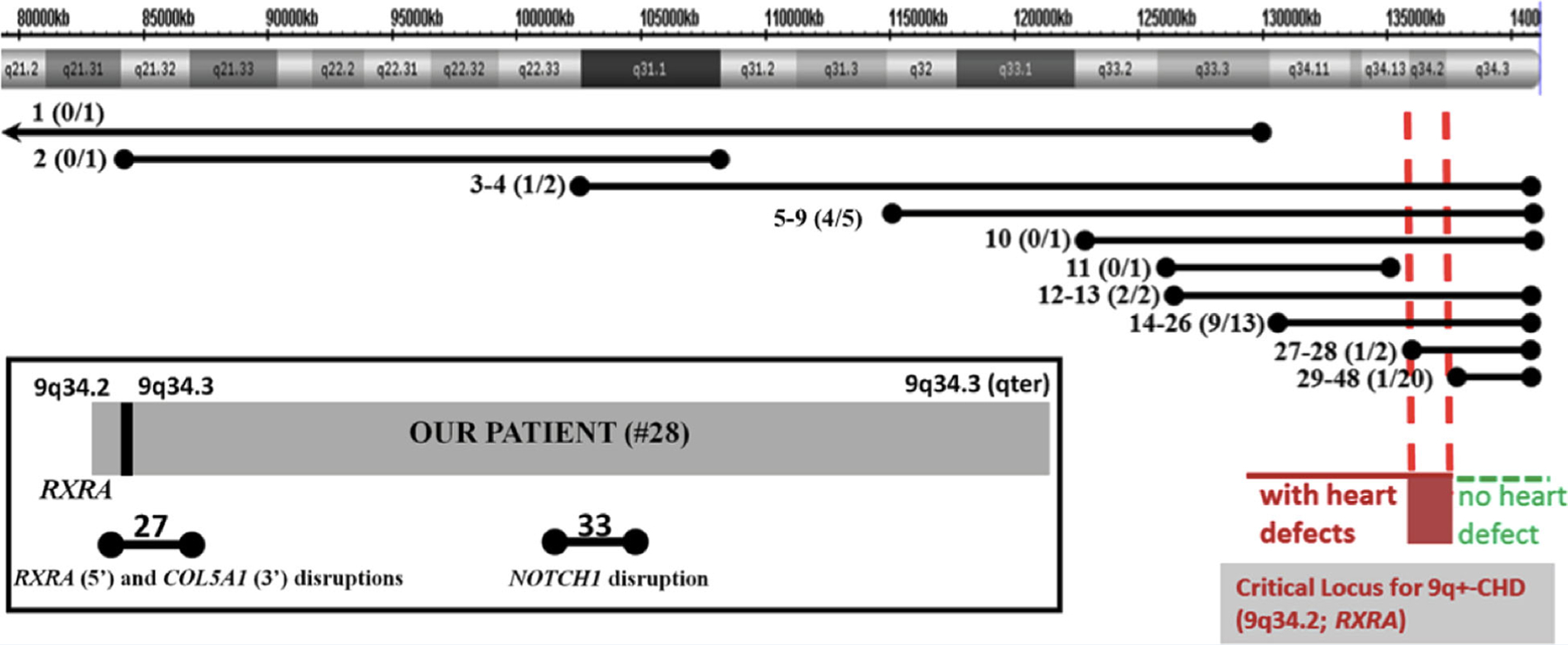
Patients #1–48 with 9q+ detected by karyotype analysis or CMA. Each line represents the extent of 9q+ and corresponds to the patient #, and in parentheses, the ratio of the number of patients with CHD of the total number of patients with the same interval. Breakpoints within 9q21.2-qter end in solid circles, and those outside of 9q21.2-qter end in arrows. While 19/20 patients with only 9q34.3+ do not exhibit CHD (green horizontal dotted interval), patients with 9q34.2 have CHD (red horizontal dotted interval). Patients 1, 2, 11 (no CHD) do not overlap with our patient’s interval. Based on these findings, we suggest that RXRA at 9q34.2 (vertical red dotted lines) may be a critical locus for 9q+-associated CHD. Box: Disruptions in patient #27 (no CHD); and patient #33 at intron 2 of NOTCH1 (with TOF).
FIG. 4.

The genomics and epigenomics landscape of RXRA showing: (A) CpG islands (CGIs) distributed across the gene; (B) the RXRA locus including introns and exons 1–10 and the disruptions (lightning bolt) at introns 1 (our patient; #28) and 7 (patient #27); (C) H3K27Ac marks for predicted active regulatory regions; (D) repeat elements including SINEs (Alu, MIR) (red), LINEs (orange), DNA transposons (blue), long terminal repeats (green), simple repeats (brown). E–I: Each square compares fetal (top) and adult left ventricle (bottom): (E) different intensity peaks (asterisk) in DNase hypersensitivity sites in fetal and adult left ventricle; (F) RNAseq profile; (G) H3K4 methylation; (H) H3K9 methylation; (I) H3K27 methylation.
DISCUSSION
TOF/APV, first described by Royer and Wilson [Royer and Wilson, 1908], is a rare variant of TOF characterized by a dysplastic or absent pulmonary valve resulting in free pulmonary regurgitation which produces an increased volume load on the right ventricle and the pulmonary outflow tract resulting in branch pulmonary artery dilation [Lakier et al., 1974]. Dilation of the pulmonary arteries can be massive and compress the tracheobronchial tree, especially the anterior lower trachea and bronchi [Lakier et al., 1974]. Fetal heart failure may ensue due to the ventricular volume overload. Affected infants often develop respiratory failure at birth and, even with ventilatory support and surgery, perinatal morbidity and mortality remain significant. Additional poor prognostic indicators include an abnormal karyotype or identified genetic syndrome [Volpe et al., 2004], hydrops fetalis [Eronen and Heikkila, 2003], left ventricular dysfunction, and increased pulmonary artery valve-to-aortic valve ratio [Szwast et al., 2014].
The CMA for our patient detected copy number gains on distal 9q that disrupted intron 1 and duplicated exons 2–10 of RXRA at 9q34.2. RXRA is a subclass of RXR (retinoid X receptors) that bind as homodimers, form heterodimers with RARs (retinoic acid receptors) [Mangelsdorf et al., 1992; Zhang et al., 2014], and together, synergistically bind to DNA response elements to promote transcription of genes eliciting specific signal transduction-mediated pathways [Sucov et al., 1994]. Both receptors are important for retinoic acid signaling pathway during cardiac morphogenesis and formation of the outflow tract [Evans, 1988; Green and Chambon, 1988; Mangelsdorf et al., 1992; Chambon, 1996]. Mice with homozygous deletions of Rxra exhibit muscular ventricular septal defects, hypoplasia of the ventricular chamber [Kastner et al., 1994; Sucov et al., 1994; Gruber et al., 1996], and cardiac dysfunction [Dyson et al., 1995]. Heterozygous mice generally have preserved ventricular function, but demonstrate abnormal ventricular chamber structure with abnormal morphology of the trabeculae, myocardium, atrioventricular canal, conotruncal ridges, and papillary muscles [Kastner et al., 1994; Gruber et al., 1996]. These abnormal cardiac findings in mutant Rxra-targeted murine models are considered to be phenocopies for human congenital heart defects [Gruber et al., 1996].
The human RXRA locus contains multiple regulatory regions (Fig. 4A–D) [Kent et al., 2002; Zhou et al., 2011]. RXRA intron 1 (2.2 Kb) contains short interspersed elements or SINEs (MIRs, Alu sequences) that correlate with predicted regions of active regulatory elements (H3K27Ac marks), simple repeats, long interspersed elements (LINEs), and a DNA transposon (MER5B) [Kent et al., 2002; Zhou et al., 2011]. The 5’ promoter and other regions within intron 1 of RXRA (Fig. 4E, asterisk) are spatiotemporally regulated in fetal and adult hearts as demonstrated by varying peak intensities or patterns of methylation (H3K4, H3K9, H3K27) (Figs. 4E–I). Disruption of this tightly-regulated region, as seen in our patient, may impair RXRA expression and disrupt gene-specific signal transduction pathways. Epigenetic studies in patients with TOF demonstrate elevated promoter and LINE-1 methylation resulting in down-regulation of RXRA expression [Sheng et al., 2012, 2014; Zhang et al., 2014]. The duplicated interval containing exons 2–10 seen in our patient also contains multiple repeat elements including SINEs, LINEs, and DNA transposons (MLT1D, MER103C, Charlie18a) (Fig. 4D). Duplication of this interval may impair transcript splicing, expression, ligand-RXRA binding, signal transduction, or other mechanisms during fetal heart development. Studies overexpressing Rxra in transgenic mice demonstrate abnormal organization of cardiomyocyte contractile elements and dilated cardiomyopathy [Subbarayan et al., 2000]. We speculate that disruption of RXRA or additional copies of this gene may have contributed to our patient’s cardiac phenotype; however, additional studies are needed to determine how genetic perturbations of RXRA contribute to CHD.
Disruption or Duplication of RXRA at 9q34.2 and CHD Among 9q+ Patients
In our review of 47 patients with 9q+ microduplication reported in the literature and in the DECIPHER database (Table I, Fig. 3, Supplementary Table SI), duplications of 9q34.2-q34.3 loci were described for 51% (24/47), 9q34.3 locus only for 43% (20/47), and 9q11-q34.1 loci for 6% (3/47). Approximately half (13/25) of the individuals with 9q34.2-q34.3 duplications including our patient had simple or complex CHD. Of the 20 patients with duplication of only the most distal 9q34.3 locus, 95% (19/20) did not have CHD (Fig. 3). Based on these findings, we suggest that RXRA at 9q34.2 may be an important locus for CHD among individuals with 9q34+ (Fig. 3). Patient #33 (Fig. 3, box; Table I) has TOF and 9q34.3+ (410 Kb) that does not include duplication of RXRA or neighboring EHMT1, but has a breakpoint at intron 2 of NOTCH1 with duplication of exons 3–34. Most described CHD involving NOTCH1 in humans and mice are attributed to haploinsufficiency, rather than duplication of NOTCH1 [Timmerman et al., 2004; Garg et al., 2005; McKellar et al., 2007; McBride et al., 2008; Luna-Zurita et al., 2010; Bosse et al., 2013; Wang et al., 2013]. Additionally, microdeletions or mutations in EHMT1 (9q34.3) result in Kleefstra syndrome as characterized by CHD, developmental delay, intellectual disability, hypotonia, dysmorphic facial features, urogenital defects, and epilepsy [Kleefstra et al., 2006; Willemsen et al., 2012]. Upon careful review of the CMA results from the nineteen other patients with 9q34.3+ (patients 29–48 except #33) one patient’s duplication included only NOTCH1 (#34), three included only EHMT1 (#40, 41, 44), and three included both genes (#31, 32, 48). None of these seven patients manifested CHD, suggesting that duplication of 9q34.3, NOTCH1 or EHMT1 may not contribute to CHD among 9q+ patients. Furthermore, phenotypic variability among patients with cardiac structural defects may be attributed to incomplete penetrance, epigenetic events, or other mechanisms of disruption of a single gene or combination of RXRA, NOTCH1, and EHMT1. Additional expression studies are needed to determine the role of these genes and the region in the development of CHD.
TABLE I.
Summary of Patients with 9q+; Duplications of RXRA, NOTCH1 and EHMT1; Congenital Heart Defects (CHD) and Other Clinical Findings
| 9q+ Bands | RXRA | NOTCH1 | EHMT1 | Test method | Cardiac anomalies | Failure to thrive | Joint contractures | Arachnodactyly | Hypotonia | Craniofacial dysmorphic features | Eye malformations | GI defects | Urogenital malformations | Neurocognitive deficits | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #/N | 14 | 5 | 9 | 23 | 19 | 34 | 18 | 4 | 13 | 43 | |||||
| 1 | q11-q33 | N | N | N | K | ID | |||||||||
| 2 | q21.32-q31.1 | N | N | N | C | + | + | + | DD, ID, SD | ||||||
| 3 | q31-q34.3 | Y | Y | Y | K | + | + | + | − | − | ID | ||||
| 4 | q31-q34.3 | Y | Y | Y | K | Congenital heart disease (CHD) | + | + | + | + | + | + | DD | ||
| 5 | q32-q34.3 | Y | Y | Y | K | Aortic arch duplication, ventricular septal defect | + | + | + | + | ID | ||||
| 6 | q32-q34.3 | Y | Y | Y | K | + | + | + | + | + | MD, DD | ||||
| 7 | q32-q34.3 | Y | Y | Y | K | Complex CHD | + | + | + | DD | |||||
| 8 | q32-q34.3 | Y | Y | Y | K | Left superior vena cava | + | + | + | + | + | DD | |||
| 9 | q32-q34.3 | Y | Y | Y | K | Patent ductus arteriosus | + | + | + | + | + | SD, MD | |||
| 10 | q33.2-q34.3 | Y | Y | Y | C | + | + | ||||||||
| 11 | q33.3-q34.1 | N | N | N | C | + | + | + | + | + | MD, SD, mild ID | ||||
| 12 | q33.3-q34.3 | Y | Y | Y | C | Left superior vena cava, right sided aortic arch, aberrant left subclavian artery | + | + | + | + | + | + | + | + | DD, SD, mild ID |
| 13 | q33.3-q34.3 | Y | Y | Y | C | Ventricular septal defect | − | + | + | ID | |||||
| 14 | q34.1-qter | Y | Y | Y | K | CHD | + | + | DD | ||||||
| 15 | q34.1-qter | Y | Y | Y | K | Ventricular septal defect | + | + | + | + | + | ID | |||
| 16 | q34.1-qter | Y | Y | Y | K | Atrial septal defect | + | + | + | MD, DD | |||||
| 17 | q34.1-qter | Y | Y | Y | K | Atrial septal defect | − | + | + | + | + | + | + | MD, SD, ID | |
| 18 | q34.1-qter | Y | Y | Y | C | Ebstein’s anomaly | − | − | + | + | + | DD, SD, mild ID | |||
| 19 | q34.1-qter | Y | Y | Y | C | − | − | + | + | − | DD, mild ID | ||||
| 20 | q34.1-qter | Y | Y | Y | K | + | + | + | + | + | MD | ||||
| 21 | q34.1-qter | Y | Y | Y | K | + | + | + | + | + | SD, MD | ||||
| 22 | q34.1-qter | Y | Y | Y | K | + | + | + | − | MD, SD, ID | |||||
| 23 | q34.1-qter | Y | Y | Y | K | + | + | + | − | + | MD | ||||
| 24 | q34.1-qter | Y | Y | Y | K | + | + | + | + | MD, SD, ID | |||||
| 25 | q34.1-qter | Y | Y | Y | K | + | + | + | |||||||
| 26 | q34.1-qter | Y | Y | Y | K | + | + | + | + | MD | |||||
| 27 | q34.2-q34.3 | Y | Y | Y | K | + | |||||||||
| 28 | q34.2-q34.3 | Y | Y | Y | C | Tetralogy of Fallot-Absent Pulmonary Valve | + | + | + | ||||||
| 29 | q34.3 | Y | Y | Y | K | mild ID | |||||||||
| 30 | q34.3 | Y | Y | Y | K | + | mild ID | ||||||||
| 31 | q34.3 | N | Y | Y | C | − | + | − | + | + | ID, MD, SD | ||||
| 32 | q34.3 | N | Y | Y | C | − | + | + | + | + | ID, MD | ||||
| 33 | q34.3 | N | Y | N | C | Tetralogy of Fallot | + | ||||||||
| 34 | q34.3 | N | Y | N | C | ID | |||||||||
| 35 | q34.3 | N | N | N | C | ID | |||||||||
| 36 | q34.3 | N | N | N | C | seizures | |||||||||
| 37 | q34.3 | N | N | N | C | + | + | ID, SD | |||||||
| 38 | q34.3 | N | N | N | C | + | ASD | ||||||||
| 39 | q34.3 | N | N | N | C | DD | |||||||||
| 40 | q34.3 | N | N | Y | C | + | + | DD | |||||||
| 41 | q34.3 | N | N | Y | C | + | ID, seizures | ||||||||
| 42 | q34.3 | N | N | N | C | ASD | |||||||||
| 43 | q34.3 | N | N | N | C | ID | |||||||||
| 44 | q34.3 | N | N | Y | C | ID | |||||||||
| 45 | q34.3 | N | N | N | C | + | ID, SD | ||||||||
| 46 | q34.3 | N | N | N | C | seizures | |||||||||
| 47 | q34.3 | N | N | N | C | + | ID, MD, SD, ASD, seizures | ||||||||
| 48 | q34.3 | N | Y | Y | C | + | + | + | ID |
#/N, Patient # (#; vertical, n=48); Total number of patients with congenital heart disease (CHD) and other clinical findings described above (N, horizontal).
Clinical findings: (+) present; (–) absent/normal; () not done/no data available.
Genes within 9q+ interval: RXRA (chr9:137,208,944–137,298,240), NOTCH1 (chr9:139,388,896–139,440,238), EHMT1 (chr9:140,513,444–140,730,578) (Y)—present by Chromosomal Microarray Analysis (CMA) or Karyotype (N)—not present by CMA or Karyotype.
Test Method: (C)—present by CMA, (K)—present by karyotype analysis.
Exact base pair coordinates for each CMA case are in Supplementary Table I.
Neurocognitive deficits: (ID) Intellectual disability, (DD) developmental delay, (MD) motor delay, (SD) speech delay, (ASD) autism spectrum disorder.
References for published reports and documented patients are in Supplementary Table I.
RXRA and Other Abnormal Findings in 9q+
Patient #27 is the only other individual with duplication of 9q34.2–9q34.3 loci that involves 5’ disruption of RXRA, but at intron 7 (exons 8–10 duplication) (Fig. 3, box; Fig. 4B), and 3’ disruption at intron 12 of COL5A1 (OMIM #120215) (exons 1–11 duplication). This patient has joint hypermobility and scoliosis, features compatible with COL5A1-associated Ehlers-Danlos syndrome, and no documentation of CHD (DECIPHER #284048). The 5’ breakpoint in RXRA (intron 7) in this patient did not disrupt any CGIs or promoter regions, however, the duplicated interval overlaps with CpG:22 and a few repeat elements including SINEs (Alu, MIR) and LINEs. This interval also exhibits differential regulatory expression and methylation patterns between fetal and adult hearts (Fig. 4B and E–I). Perhaps the disruption or duplication still produces a functional protein or does not affect RXRA binding and signal-mediated pathways in normal heart development; however, without further genomic investigations, this correlation remains speculative. The remaining 32 patients without CHD reveal duplications distributed along the entire 9q (9q11-q34.3) and a phenotypic spectrum consistent with 9q34 microduplication syndrome including hypotonia, dysmorphic facial features, eye defects, arachnodactyly, joint contractures, and urogenital defects (Table I, Supplementary Table I). In 31% (10/32) of these patients, the duplication includes the RXRA locus. In addition to cardiac structural defects, RXRA-dependent pathways when disrupted are known to result in CNS anomalies, craniofacial defects, limb dysmorphogenesis and eye defects, suggesting that a common precursor gives rise to different tissue types. and structures via RXRA-mediated pathways. While these clinical features are evident in several of the patients reviewed here [Maden, 1982; Dencker et al., 1987; Wedden et al., 1988; Mascrez et al., 2009], our patient did not have significant craniofacial or extremity defects suggesting others genes, epigenetic changes, or environmental factors modify penetrance.
In summary, this report suggests RXRA at 9q34.2 may be a locus for CHD among individuals with 9q microduplications. Although the exact mechanisms remain to be investigated in humans, disruption and/or duplication of this gene may impair the genomic and epigenomic spatiotemporal regulation of RXRA-dependent expression, ligand binding, and signaling pathways associated with normal cardiac development and function. This report highlights the importance of CMA and genomics to identify specific breakpoints and affected gene loci among patients with complex congenital malformations including CHD and to inform genetic counseling of families.
Supplementary Material
ACKNOWLEDGMENTS
We would like to acknowledge Mike Evenson, Jennifer Burke, and the Washington University Cytogenomics Lab for assisting in CMA studies. We also acknowledge Ting Wang from the Washington University Department of Genetics for assisting in epigenomic analysis and interpretation.
Footnotes
Conflict of interest: none.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
REFERENCES
- Allderdice PW, Eales B, Onyett H, Sprague W, Henderson K, Lefeuvre PA, Pal G. 1983. Duplication 9q34 syndrome. Am J Hum Genet 35:1005–1019. [PMC free article] [PubMed] [Google Scholar]
- Andersen TA, Troelsen Kde L, Larsen LA. 2014. Of mice and men: Molecular genetics of congenital heart disease. Cell Mol Life Sci 71:1327–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal V, Dorn C, Grunert M, Klaassen S, Hetzer R, Berger F, Sperling SR. 2014. Outlier-based identification of copy number variations using targeted resequencing in a small cohort of patients with Tetralogy of Fallot. PloS ONE 9:e85375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittel DC, Butler MG, Kibiryeva N, Marshall JA, Chen J, Lofland GK, O’Brien JE Jr. 2011. Gene expression in cardiac tissues from infants with idiopathic conotruncal defects. BMC Med Genomics 4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, LaHaye S, Tao G, Lucchesi PA, Lincoln J, Lilly B, Garg V. 2013. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol 60:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambon P. 1996. A decade of molecular biology of retinoic acid receptors. FASEB J 10:940–954. [PubMed] [Google Scholar]
- Dencker L, d’Argy R, Danielsson BR, Ghantous H, Sperber GO. 1987. Saturable accumulation of retinoic acid in neural and neural crest derived cells in early embryonic development. Dev Pharmacol Ther 10:212–223. [DOI] [PubMed] [Google Scholar]
- Dyson E, Sucov HM, Kubalak SW, Schmid-Schonbein GW, DeLano FA, Evans RM, Ross J Jr., Chien KR. 1995. Atrial-like phenotype is associated with embryonic ventricular failure in retinoid X receptor alpha −/− mice. Proc Natl Acad Sci USA 92:7386–7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eronen M, Heikkila P. 2003. Absent aortic and dysplastic pulmonary valves associated with ventricular septal defect in fetal hydrops. Pediatr Cardiol 24:400–402. [DOI] [PubMed] [Google Scholar]
- Evans RM. 1988. The steroid and thyroid hormone receptor superfamily. Science 240:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. 2005. Mutations in NOTCH1 cause aortic valve disease. Nature 437:270–274. [DOI] [PubMed] [Google Scholar]
- Gelb BD. 2004. Genetic basis of congenital heart disease. Curr Opin Cardiol 19:110–115. [DOI] [PubMed] [Google Scholar]
- Gijsbers AC, Bijlsma EK, Weiss MM, Bakker E, Breuning MH, Hoffer MJ, Ruivenkamp CA. 2008. A 400kb duplication, 2.4Mb triplication and 130kb duplication of 9q34.3 in a patient with severe mental retardation. Eur J Med Genet 51:479–487. [DOI] [PubMed] [Google Scholar]
- Glessner JT, Bick AG, Ito K, Homsy JG, Rodriguez-Murillo L, Fromer M, Mazaika E, Vardarajan B, Italia M, Leipzig J, DePalma SR, Golhar R, Sanders SJ, Yamrom B, Ronemus M, Iossifov I, Willsey AJ, State MW, Kaltman JR, White PS, Shen Y, Warburton D, Brueckner M, Seidman C, Goldmuntz E, Gelb BD, Lifton R, Seidman J, Hakonarson H, Chung WK. 2014. Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circ Res 115:884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green S, Chambon P. 1988. Nuclear receptors enhance our understanding of transcription regulation. Trends Genet 4:309–314. [DOI] [PubMed] [Google Scholar]
- Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM, McCarroll SA, Gorham JM, Gabriel S, Altshuler DM, Quintanilla-Dieck Mde L, Artunduaga MA, Eavey RD, Plenge RM, Shadick NA, Weinblatt ME, De Jager PL, Hafler DA, Breitbart RE, Seidman JG, Seidman CE. 2009. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet 41:931–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber PJ, Kubalak SW, Pexieder T, Sucov HM, Evans RM, Chien KR. 1996. RXR alpha deficiency confers genetic susceptibility for aortic sac, conotruncal, atrioventricular cushion, and ventricular muscle defects in mice. J Clin Invest 98:1332–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitz MP, Lemieux-Perreault LP, Marshall C, Feroz-Zada Y, Davies R, Yang SW, Lionel AC, D’Amours G, Lemyre E, Cullum R, Bigras JL, Thibeault M, Chetaille P, Montpetit A, Khairy P, Overduin B, Klaassen S, Hoodless P, Awadalla P, Hussin J, Idaghdour Y, Nemer M, Stewart AF, Boerkoel C, Scherer SW, Richter A, Dube MP, Andelfinger G. 2012. Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet 8:e1002903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S, McCarthy KP, Josen M, Rigby ML. 2001. Anatomic-echocardiographic correlates: An introduction to normal and congenitally malformed hearts. Heart 86:I3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter LE, Simpson JM. 2014. Prenatal screening for structural congenital heart disease. Nat Rev Cardiol 11:323–334. [DOI] [PubMed] [Google Scholar]
- Kastner P, Grondona JM, Mark M, Gansmuller A, LeMeur M, Decimo D, Vonesch JL, Dolle P, Chambon P. 1994. Genetic analysis of RXR alpha developmental function: Convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell 78:987–1003. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, Magee A, Genevieve D, Cormier-Daire V, van Esch H, Fryns JP, Hamel BC, Sistermans EA, de Vries BB, van Bokhoven H. 2006. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet 79:370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra T, Nillesen WM, Yntema HG. 1993. Kleefstra syndrome. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. GeneReviews(R) Seattle, Washington: University of Washington, Seattle. [Google Scholar]
- Lakier JB, Stanger P, Heymann MA, Hoffman JI, Rudolph AM. 1974. Tetralogy of Fallot with absent pulmonary valve. Natural history and hemodynamic considerations. Circulation 50:167–175. [DOI] [PubMed] [Google Scholar]
- Luna-Zurita L, Prados B, Grego-Bessa J, Luxan G, del Monte G, Benguria A, Adams RH, Perez-Pomares JM, de la Pompa JL. 2010. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J Clin Invest 120:3493–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maden M. 1982. Vitamin A and pattern formation in the regenerating limb. Nature 295:672–675. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong ES, Oro AE, Kakizuka A, Evans RM. 1992. Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev 6:329–344. [DOI] [PubMed] [Google Scholar]
- Mascrez B, Ghyselinck NB, Chambon P, Mark M. 2009. A transcriptionally silent RXRalpha supports early embryonic morphogenesis and heart development. Proc Natl Acad Sci USA 106:4272–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KL, Riley MF, Zender GA, Fitzgerald-Butt SM, Towbin JA, Belmont JW, Cole SE. 2008. NOTCH1 mutations in individuals with left ventricular outflow tract malformations reduce ligand-induced signaling. Hum Mol Genet 17:2886–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKellar SH, Tester DJ, Yagubyan M, Majumdar R, Ackerman MJ, Sundt TM 3rd. 2007. Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J Thorac Cardiovasc Surg 134:290–296. [DOI] [PubMed] [Google Scholar]
- Moon-Grady AJ, Tacy TA, Brook MM, Hanley FL, Silverman NH. 2002. Value of clinical and echocardiographic features in predicting outcome in the fetus, infant, and child with tetralogy of Fallot with absent pulmonary valve complex. Am J Cardiol 89:1280–1285. [DOI] [PubMed] [Google Scholar]
- Papadopoulou E, Sismani C, Christodoulou C, Ioannides M, Kalmanti M, Patsalis P. 2010. Phenotype-genotype correlation of a patient with a “balanced” translocation 9;15 and cryptic 9q34 duplication and 15q21q25 deletion. Am J Med Genet Part A 152A:1515–1522. [DOI] [PubMed] [Google Scholar]
- Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT, Correa A. 2008. Prevalence of congenital heart defects in metropolitan Atlanta, 1998– 2005. J Pediatr 153:807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer B, Wilson J. 1908. Incomplete heterotaxy with unusual heart malformations: Case report. Arch Pediatr 25:881. [Google Scholar]
- Sheng W, Qian Y, Zhang P, Wu Y, Wang H, Ma X, Chen L, Ma D, Huang G. 2014. Association of promoter methylation statuses of congenital heart defect candidate genes with Tetralogy of Fallot. J Transl Med 12:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, Wang H, Ma X, Qian Y, Zhang P, Wu Y, Zheng F, Chen L, Huang G, Ma D. 2012. LINE-1 methylation status and its association with tetralogy of fallot in infants. BMC Med Genomics 5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silversides CK, Lionel AC, Costain G, Merico D, Migita O, Liu B, Yuen T, Rickaby J, Thiruvahindrapuram B, Marshall CR, Scherer SW, Bassett AS. 2012. Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet 8:e1002843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soemedi R, Vega H, Belmont JM, Ramachandran S, Fairbrother WG. 2014. Genetic variation and RNA binding proteins: Tools and techniques to detect functional polymorphisms. Adv Exp Med Biol 825:227–266. [DOI] [PubMed] [Google Scholar]
- Spinner NB, Lucas JN, Poggensee M, Jacquette M, Schneider A. 1993. Duplication 9q34->qter identified by chromosome painting. Am J Med Genet 45:609–613. [DOI] [PubMed] [Google Scholar]
- Subbarayan V, Mark M, Messadeq N, Rustin P, Chambon P, Kastner P. 2000. RXRalpha overexpression in cardiomyocytes causes dilated cardiomyopathy but fails to rescue myocardial hypoplasia in RXRalpha-null fetuses. J Clin Invest 105:387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sucov HM, Dyson E, Gumeringer CL, Price J, Chien KR, Evans RM. 1994. RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev 8:1007–1018. [DOI] [PubMed] [Google Scholar]
- Szwast A, Tian Z, McCann M, Soffer D, Combs J, Donaghue D, Rychik J. 2014. Anatomic variability and outcome in prenatally diagnosed absent pulmonary valve syndrome. Ann Thorac Surg 98:152–158. [DOI] [PubMed] [Google Scholar]
- Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, de la Pompa JL. 2004. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 18:99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita-Mitchell A, Mahnke DK, Struble CA, Tuffnell ME, Stamm KD, Hidestrand M, Harris SE, Goetsch MA, Simpson PM, Bick DP, Broeckel U, Pelech AN, Tweddell JS, Mitchell ME. 2012. Human gene copy number spectra analysis in congenital heart malformations. Physiol Genomics 44:518–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turleau C, de Grouchy J, Chavin-Colin F, Roubin M, Brissaud PE, Repesse G, Safar A, Borniche P. 1975. Partial trisomy 9q: A new syndrome. Humangenetik 29:233–241. [DOI] [PubMed] [Google Scholar]
- Volpe P, Paladini D, Marasini M, Buonadonna AL, Russo MG, Caruso G, Marzullo A, Arciprete P, Martinelli P, Gentile M. 2004. Characteristics, associations and outcome of absent pulmonary valve syndrome in the fetus. Ultrasound Obstet Gynecol 24:623–628. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wu B, Chamberlain AA, Lui W, Koirala P, Susztak K, Klein D, Taylor V, Zhou B. 2013. Endocardial to myocardial notch-wnt-bmp axis regulates early heart valve development. PloS ONE 8:e60244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton D, Ronemus M, Kline J, Jobanputra V, Williams I, Anyane-Yeboa K, Chung W, Yu L, Wong N, Awad D, Yu CY, Leotta A, Kendall J, Yamrom B, Lee YH, Wigler M, Levy D. 2014. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum Genet 133:11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedden SE, Ralphs JR, Tickle C. 1988. Pattern formation in the facial primordia. Development 103:31–40. [DOI] [PubMed] [Google Scholar]
- Willemsen MH, Vulto-van Silfhout AT, Nillesen WM, Wissink-Lindhout WM, van Bokhoven H, Philip N, Berry-Kravis EM, Kini U, van Ravenswaaij-Arts CM, Delle Chiaie B, Innes AM, Houge G, Kosonen T, Cremer K, Fannemel M, Stray-Pedersen A, Reardon W, Ignatius J, Lachlan K, Mircher C, Helderman van den Enden PT, Mastebroek M, Cohn-Hokke PE, Yntema HG, Drunat S, Kleefstra T. 2012. Update on Kleefstra syndrome. Mol Syndromol 2:202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GN, Raj A, Baker D. 1985. The phenotypic and cytogenetic spectrum of partial trisomy 9. Am J Med Genet 20:277–282. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ma X, Wang H, Ma D, Huang G. 2014. Elevated methylation of the RXRA promoter region may be responsible for its downregulated expression in the myocardium of patients with TOF. Pediatr Res 75:588–594. [DOI] [PubMed] [Google Scholar]
- Zhou X, Maricque B, Xie M, Li D, Sundaram V, Martin EA, Koebbe BC, Nielsen C, Hirst M, Farnham P, Kuhn RM, Zhu J, Smirnov I, Kent WJ, Haussler D, Madden PA, Costello JF, Wang T. 2011. The Human Epigenome Browser at Washington University. Nat Methods 8:989–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.