

SUMMARY
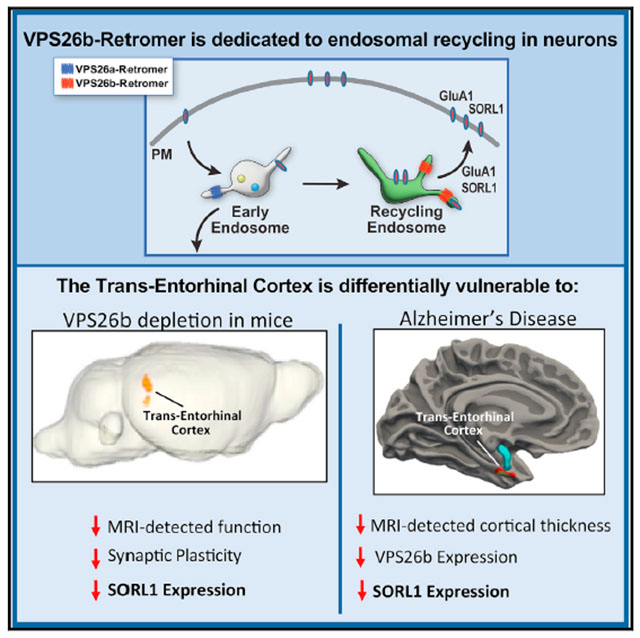
Whether and how the pathogenic disruptions in endosomal trafficking observed in Alzheimer’s disease (AD) are linked to its anatomical vulnerability remain unknown. Here, we began addressing these questions by showing that neurons are enriched with a second retromer core, organized around VPS26b, differentially dedicated to endosomal recycling. Next, by imaging mouse models, we show that the trans-entorhinal cortex, a region most vulnerable to AD, is most susceptible to VPS26b depletion—a finding validated by electrophysiology, immunocytochemistry, and behavior. VPS26b was then found enriched in the trans-entorhinal cortex of human brains, where both VPS26b and the retromer-related receptor SORL1 were found deficient in AD. Finally, by regulating glutamate receptor and SORL1 recycling, we show that VPS26b can mediate regionally selective synaptic dysfunction and SORL1 deficiency. Together with the trans-entorhinal’s unique network properties, hypothesized to impose a heavy demand on endosomal recycling, these results suggest a general mechanism that can explain AD’s regional vulnerability.
Graphical abstract
In brief
Trans-entorhinal cortex neurons are most vulnerable to Alzheimer’s disease. Simoes et al. explain this vulnerability by showing that these neurons are dependent on a distinct VPS26b-retromer core differentially dedicated to endosomal recycling. VPS26b is highly expressed in these neurons, where they regulate synaptic function, GluA1/SORL1 recycling, and disease-associated pathologies
INTRODUCTION
The early endosome, a central trafficking station of the endolysosomal network, has proven to be an organelle fundamental to Alzheimer’s disease (AD) pathophysiology (Nixon, 2005). A combination of genetic (Karch and Goate, 2015) and functional (Small et al., 2017) studies have shown how endosome trafficking can act as a pathogenic biological pathway in the disease. Evidence in support of this conclusion is provided by retromer, a multi-modular protein complex, that is considered a “master conductor” of endosomal trafficking (Burd and Cullen, 2014). While a number of retromer-related genes have been associated with AD (Vardarajan et al., 2012), the strongest genetic link is with the retromer-interacting receptor “sortilin-related receptor 1” (SORL1) (Fjorback et al., 2012; Muhammad et al., 2008; Small and Gandy, 2006; Rogaeva et al., 2007), in which truncating mutations are shown to be highly pathogenic (Holstege et al., 2017; Scheltens et al., 2021). Complementary functional studies have shown that disrupting retromer-dependent trafficking can trigger or mediate AD’s endosomal, amyloid, tau, and synaptic pathologies (Small and Petsko, 2015).
Retromer regulates the two main trafficking pathways out of the endosome (Seaman, 2005; Burd and Cullen, 2014): the retrograde pathway, which traffics cargo from the endosome back to the trans-Golgi network, and the recycling pathway, which recycles cargo to the cell surface. The “cargo recognition core” is retromer’s central module, so named because it is the core assembly to which other retromer modules interact, and, most importantly, because it is the core to which retromer’s endosomal cargo bind (Attar and Cullen, 2010; Seaman, 2012). The “vacuolar protein sorting 35” (VPS35) resides at the center of the heterotrimeric core, to which the other proteins VPS29 and VPS26 bind (Kovtun et al., 2018).
Retromer is critical for synaptic plasticity as it is required for the postsynaptic recycling of glutamate receptors (Choy et al., 2014; Temkin et al., 2017), which is when endosomal recycling regulates the reinsertion of the endocytosed receptor into the postsynaptic membrane (Ehlers, 2000; Park et al., 2004). The loss of glutamate receptor recycling and its levels, caused by retromer depletion, disrupts long-term potentiation (LTP) (Temkin et al., 2017), a synaptic plasticity failure that is thought to act as the initiating event in AD’s neurodegenerative process (Selkoe, 2002). SORL1 is another receptor trafficked by retromer (Small et al., 2005; Rogaeva et al., 2007; Fjorback et al., 2012; Muhammad et al., 2008) that is even more specific to AD pathogenesis. Recent genetic studies have established that SORL1-truncating mutations confer an extremely high AD risk, on par with the extreme high risk conferred by mutations in the amyloid precursor proteins (APP) and the presenilins (PSENs) (Holstege et al., 2017; Scheltens et al., 2021). Complementary functional studies have shown that SORL1 depletion and causal APP/PSEN mutations appear to phenocopy each other by inducing in neurons endosomal traffic jams (Knupp et al., 2020; Kwart et al., 2019), APP misprocessing, and accelerated Aβ and tau secretion (Maia et al., 2013; Andersen et al., 2021).
The brain is enriched with a second VPS26 paralog, which, in contrast to the ubiquitous VPS26a paralog, has been designated VPS26b (Kim et al., 2008; Bugarcic et al., 2011) and evolutionarily emerged in vertebrates (Kerr et al., 2005). A detailed comparison of the two VPS26 paralogs in neurons, their role in endosomal trafficking, and their link to trafficking glutamate receptors and SORL1 has never been reported. Furthermore, we hypothesized that a deeper insight into differential paralog function might clarify regional vulnerability in AD, one of the disease’s most vexing questions. A range of studies have established that the border zone between the entorhinal and perirhinal cortices, termed the trans-entorhinal cortex (TEC) in clinical literature, is selectively vulnerable to AD (Braak and Braak, 1991; Gomez-Isla et al., 1996; Khan et al., 2014; Adams et al., 2019). Interestingly, a distinct network property of the TEC has emerged from recent studies, showing that it has the highest cortical connectivity and functions as a dominant cortical hub upon which synaptic input from the whole cortical mantle converges (Bota et al., 2015). Since high synaptic input is associated with high glutamate receptor stimulation, endocytosis, and trafficking, the TEC has been hypothesized to impose high demands on the endosomal recycling pathway (Small and Swanson, 2019).
Here, we used a range of experimental systems to partly confirm this hypothesis, first by showing that neurons are endowed with a distinct VPS26b-retromer core dedicated to endosomal recycling during synaptic stimulation and then demonstrating that the TEC depends on VPS26b-retromer, where its depletion reduces glutamate receptor levels and induces profound LTP defects. Linking the TEC’s endosomal recycling pathway to AD, we show that VPS26b-retromer is dedicated to SORL1 recycling and maintaining its levels and that SORL1 is deficient in the TEC of Vps26b-depleted mice and in the TEC of AD brains.
RESULTS
VPS26b and VPS26a define distinct retromer cores in cortical neurons
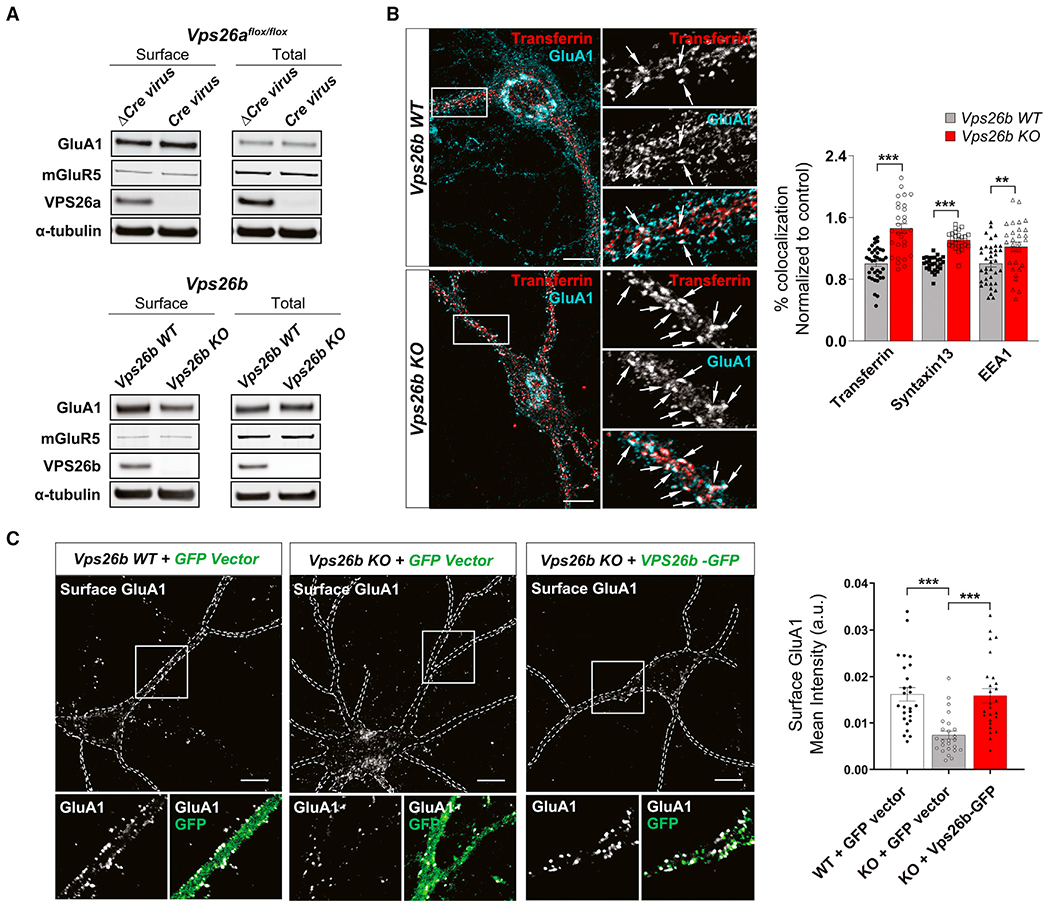
We started by replicating and extending previous studies (Kim et al., 2008; Bugarcic et al., 2011) showing that, compared with VPS26a, VPS26b is not only enriched in the brain, but within the brain, it is differentially expressed in neurons (Figure S1). We next set out to test whether the two VPS26 paralogs are interchangeable in forming retromer cores (Figure 1A, top panel). To begin addressing this question, we performed co-immunoprecipitation experiments in primary hippocampal neurons, showing that while VPS35 pulled down both paralogs, VPS26a pulled down VPS35 but not VPS26b, and VPS26b pulled down VPS35 but not VPS26a (Figure 1A, bottom panel). Providing further evidence for separate cores, confocal microscopy showed that while VPS35 highly colocalized with VPS26b and with VPS26a, both paralogs exhibited low intracellular co-localization (Figure 1B).
Figure 1. VPS26b and VPS26a define distinct retromer cores.
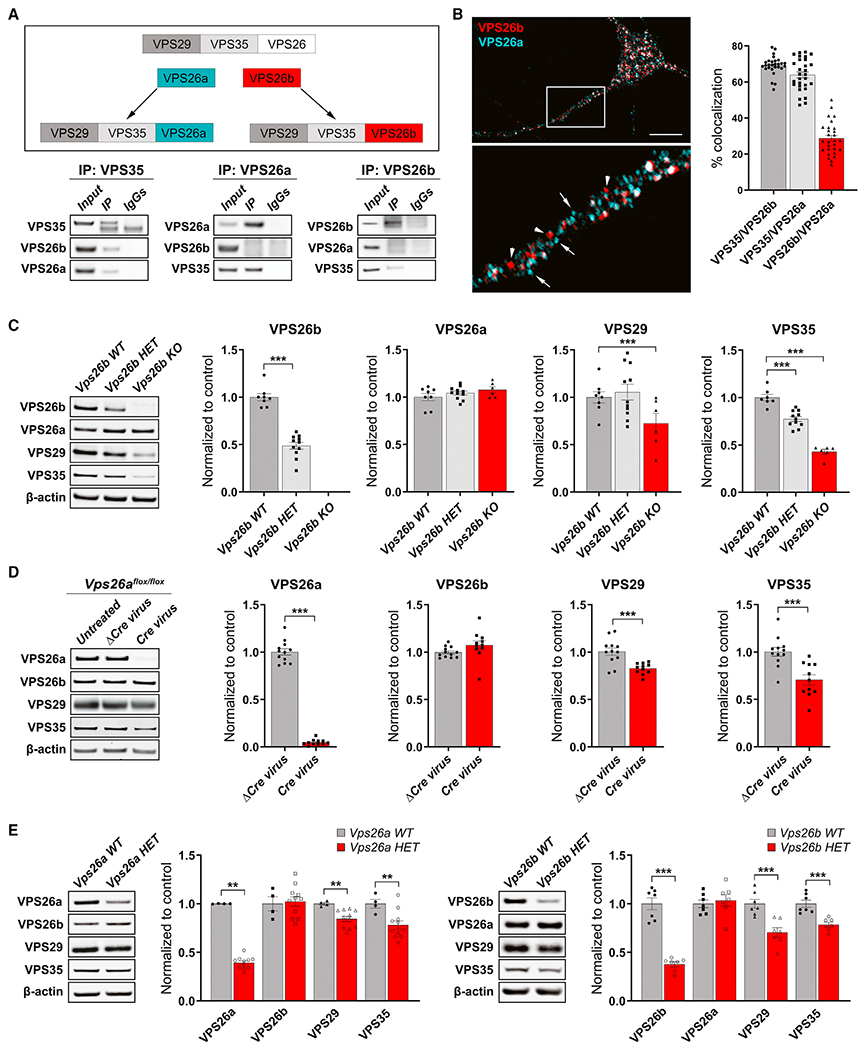
(A) An illustration of the proposed hypothesis for how the VPS26 paralogs form two separate retromer cores (top panel). Co-immunoprecipitation analysis of retromer proteins extracted from primary neuronal cultures using VPS35 (10 μg; left panel), VPS26a (10 μg; middle panel), and VPS26b (10μg; right panel) as baits (bottom panel) support this hypothesis.
(B) A representative confocal image showing partial co-localization of both VPS26 paralogs (left panel), while quantitative colocalization studies, based on Pearson’s correlation, reveal that VPS35 shows a higher percentage of co-localization with each VPS26 paralog (n = 28–29 cells, from three independent experiments) (right panel). Scale bar, 10 μm.
(C) Support for VPS26b forming a distinct retromer core is shown by how a primary depletion of VPS26b, in primary cultures derived from Vps26b heterozygous (HET) mice (n = 6) or Vps26b KO mice (n = 6) compared with Vps26b WT mice (n = 8), has no effect on VPS26a but causes a secondary reduction in VPS29 and VPS35, as summarized in the bar graphs and illustrated by representative immunoblots (in a one-way ANOVA with Tukey’s post hoc test’s two-sided analysis).
(D) Support for VPS26a forming a distinct retromer core is shown by how a primary depletion of VPS26a, induced by infecting neurons from Vps26aflox/flox mice with a lentivirus-expressing Cre recombinase (Cre, n = 11), compared with neurons infected with a lentivirus expressing a catalytically dead Cre recombinase (ΔCre, n = 12), has no effect on VPS26b (p = 0.1480, in an unpaired t test with Welch’s correction) but causes a secondary reduction in VPS29 and VPS35, as summarized in the bar graphs and illustrated with representative immunoblots. Statistical analyses were performed using either unpaired two-sided Student’s t test, with Welch’s correction when required, or a non-parametric Mann-Whitney t test.
(E) Western blots of hippocampus homogenates from Vps26a and Vps26b HET mice (Vps26a WT, n = 4; Vps26a HET, n = 11; Vps26b WT, n = 7; Vps26b HET, n = 7). Quantitative analysis of the western blots probed for retromer proteins shows that a primary deficiency in VPS26a (p = 0.0015) results in a secondary reduction VPS35 (p = 0.0037) and VPS29 (p = 0.0061) but not VPS26b (p = 0.8101), while a primary deficiency in VPS26b (p < 0.0001) results in a secondary reduction in VPS35 (p = 0.0004) and VPS29 (p = 0.0009) but not VPS26a (p = 0.6373), arguing in favor of separate retromer cores. All statistical analysis were performed using two-sided Student’s t test except for VPS26a in the Vps26a WT versus Vps26a HET analysis. Values denote mean ± SEM, where *p < 0.05, **p < 0.01, and ***p < 0.001.
See also Figures S1 and S2.
These observations suggest that neurons might have two separate retromer cores. To further test this hypothesis, we relied on previous studies showing that a primary knock down of one core protein causes a secondary reduction in other bound members (as reviewed in Small and Petsko, 2015). If VPS26a and VPS26b are part of separate cores, a primary reduction in one should not affect the levels of the other. By examining primary neuronal cultures derived from Vps26b knockout (KO) mice, we find a secondary reduction in VPS35 and VPS29 but no effect on VPS26a (Figure 1C). To overcome the problem of Vps26a KO lethality, we used a Vps26aflox/flox mouse model and infected cultured neurons derived from these mice with a lentivirus expressing either Cre recombinase (Cre) or a catalytically dead version of Cre (ΔCre). A near-complete depletion of VPS26a had no effect on VPS26b (Figure 1D). Further supporting these findings, fluorescent microscopy showed that VPS26b-VPS35 co-localization was unaffected in Vps26a-depleted neurons and that VPS26a-VPS35 co-localization was unchanged in Vps26b KO neurons (Figure S2A).
We next set out to validate these findings in vivo using three genetically engineered retromer-deficient mouse models (Figures 1E and S2B). Since homozygote Vps26a−/− mice are embryonic-lethal, we decided to compare the expression levels of both VPS26a and VPS26b in heterozygote (HET) mice. We harvested the hippocampus from 6-month-old HET mice for Vps26a (Muhammad et al., 2008) (Vps26a HET) and their wild-type (WT) littermates (Vps26a WT) or HET mice for Vps26b (Kim et al., 2010) (Vps26b HET) and their WT littermates (Vps26b WT) and found by western blot analysis that a primary deficiency in VPS26a caused a secondary reduction in VPS35 and VPS29, but had no effect on VPS26b (Figure 1E, left panel). Conversely, a primary deficiency in VPS26b caused a secondary reduction in VPS35 and VPS29, but had no effect on VPS26a (Figure 1E, right panel). We then investigated the protein expression levels of both VPS26 paralogs in Vps35-deficient mice. As Vps35 KO mice are embryonic-lethal, we used a conditional mouse model in which the CamKIIα-Cre/LoxP system was used to KO Vps35 in forebrain neurons (Simoes et al., 2020) (Figure S2B). By harvesting the hippocampus of 6-month-old Vps35 conditional KO (cKO) mice and their WT littermates (Vps35 WT), western blot analysis shows that a primary deficiency in VPS35 caused a secondary reduction in VPS29 and both VPS26 paralogs. Collectively, these studies support the hypothesis that, within the brain, neurons are enriched with a distinct retromer core, VPS26b-VPS35-VPS29.
VPS26b redistributes to recycling endosomes during synaptic plasticity
Based on the studies above, and because previous studies have suggested that the VPS26 paralogs might serve different functions in endosomal trafficking (Bugarcic et al., 2011), we turned to neuronal cell cultures to test the hypothesis that VPS26b and VPS26a might differentially localize to endosomal compartments with which retromer has been associated: the early endosome, the recycling endosome, and the late endosome.
To begin addressing this question, we used confocal microscopy and markers of various organelles to compare the endogenous subcellular localization of the two VPS26 paralogs (Figures 2A and S3A). While overlaps in localization were noted, significant differences were detected. VPS26a was found broadly distributed across all membrane compartments: the early (EEA1) and recycling endosomes (pulse-chase transferrin and Syntaxin13) but also the late endosome (Rab7) and trans-Golgi network (Golgin97). Compared with VPS26a, VPS26b localized more to early and recycling endosomes. An immunoelectron microscopy (IEM) analysis performed in hippocampal cultures further revealed an enrichment of VPS26b in tubulo-vesicular structures most likely derived from the recycling pathway (Figures 2B and S3B).
Figure 2. VPS26b redistributes to recycling endosomes during neuronal stimulation.
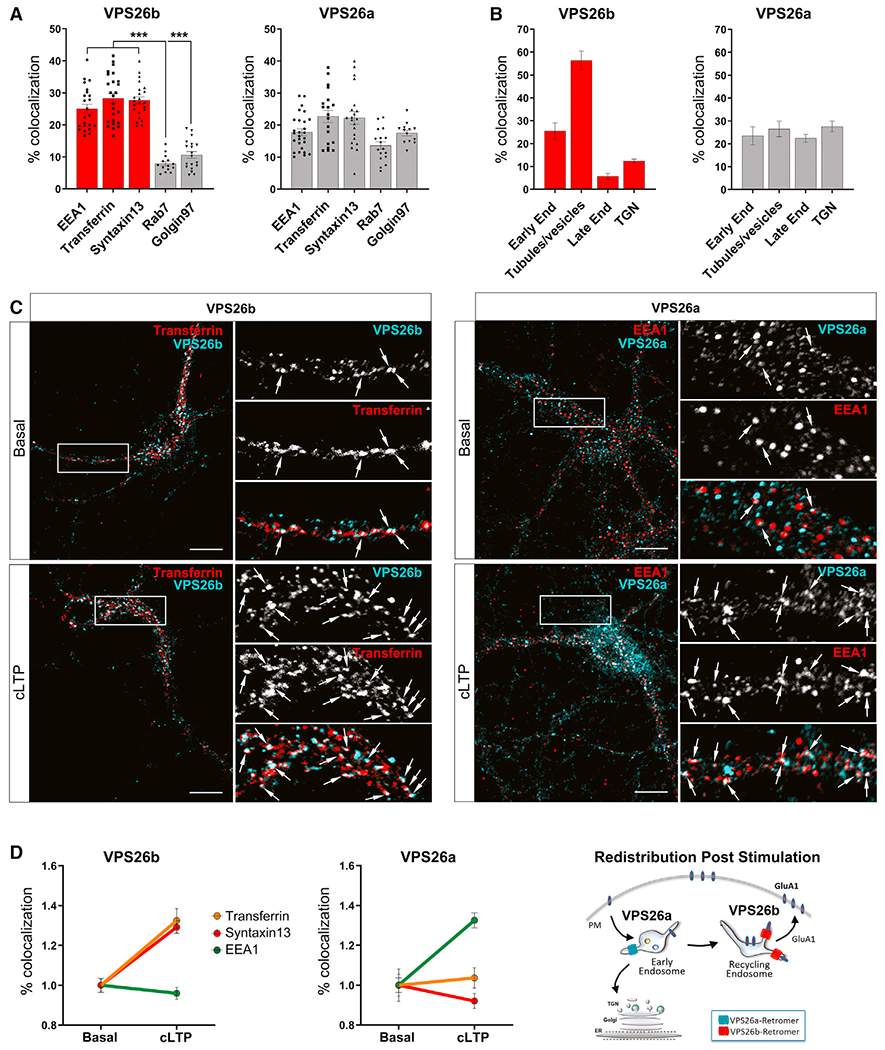
(A and B) Subcellular distribution of both VPS26 paralogs was performed using confocal and ultracryomicrotomy analyses.
(A) Confocal microscopy quantifications based on Pearson’s correlation coefficient were obtained by analysis of 15–27 cells per group/condition. Kruskal-Wallis test with a Dunn’s post hoc test was used for the statistical analysis. Note that while VPS26a is broadly detected in all different compartments, VPS26b is highly enriched in early (EEA1) and recycling (Syntaxin13 and pulse-chase transferrin) endosomes, with less presence in the trans-Golgi network (Golgin97) and late endosomes (Rab7). Data expressed as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
(B) Immunogold labeling quantifications (percetange of gold particles) for VPS26b and VPS26a on ultrathin cryosections shown in Figure S3B. Localization of an equivalent number of gold particles in each sample was assessed relative to the indicated cell compartment. Note that whereas the bulk of VPS26b localizes to tubular-vesicular structures found at the vicinity of endosomes, VPS26a is broadly distributed among the different compartments.
(C) Primary hippocampal neurons were stimulated with glycine for 5 min to induce cLTP, and the subcellular distribution of VPS26b and VPS26a was assessed by confocal microscopy using markers of early (EEA1) and recycling (Syntaxin13 and pulse-chase transferrin) endosomes. Compared to basal conditions, cLTP caused VPS26b to increase its co-localization with markers of the recycling endosomes (in an ANOVA analysis; pulse-chase transferrin: F[1,33] = 20.7, p = 6.9E−5; Syntaxin13: F[1,36] = 38.8, p = 3.5E−7) and caused VPS26a to increase its co-localization with a marker of early endosomes (EEA1: F[1,33] = 24.5, p = 2.1E−5), as illustrated in the representative confocal images of dendritic segments. Arrows indicate sites of co-localization. Scale bar, 10μm (top panel).
(D) Co-localization studies based on Pearson’s correlation coefficient were used to generate the line graphs (n = 31–41 cells per condition, from four independent experiments) (left panels). An illustration of the changes in distribution observed post-stimulation for both VPS26 paralogs is shown (right panel).
See also Figures S3 and S4.
Since retromer is required for synaptic plasticity, in particular LTP (Temkin et al., 2017), which is known to redistribute proteins to endosomal compartments in which they function (Wang et al., 2008), we tested whether there would be redistribution of the two VPS26 paralogs after chemical LTP (cLTP) (Figures 2C and S4). Upon cLTP stimulation, the paralogs showed different patterns of redistribution. While VPS26b increased its localization to recycling endosomes but not to early endosomes, VPS26a increased its localization to early endosomes but not to recycling endosomes (Figure 2D). Collectively, these results show that compared with VPS26a, VPS26b differentially localizes to recycling endosomes, particularly during synaptic plasticity.
The TEC differentially depends on VPS26b
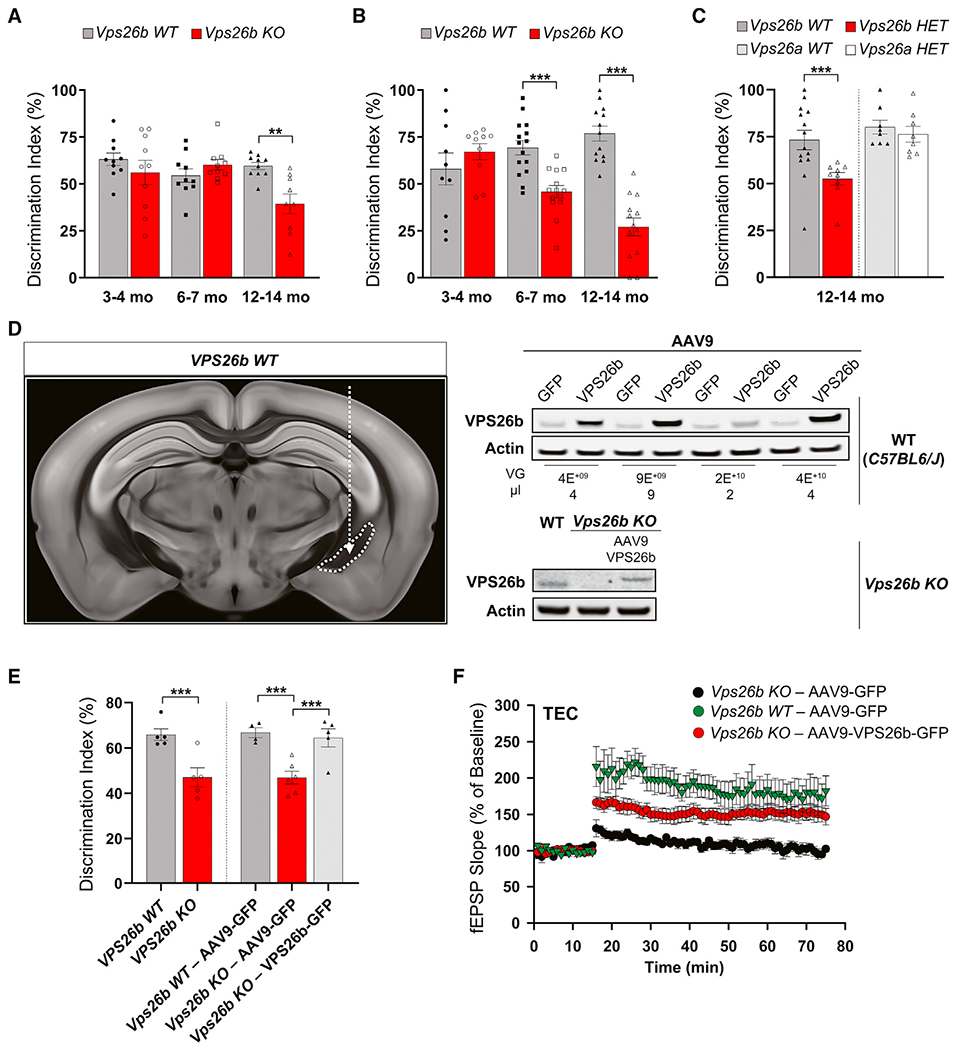
Since Vps26b KO mice live into adulthood, we set out to determine whether there are brain regions differentially sensitive to Vps26b depletion. We began by using an fMRI variant that maps regional levels of cerebral blood volume (CBV), a neuroimaging indicator of basal metabolism (Small et al., 2011) with particularly high spatial resolution and thus useful for interrogating the whole rodent brain in localizing metabolic defects (Khan et al., 2014). We imaged 3- to 4- and 12- to 14-month-old Vps26b KO mice and their Vps26b WT littermates and tested for a genotype X age interaction. Across the whole brain, voxel-based analysis reliably identified a focal age-related defect in the Vps26b KO mice (Figure 3A; Video S1). This metabolic defect was found to localize to the border zone between the perirhinal and the entorhinal cortices, which in the clinical literature is called the TEC (Figure 3A, top right and bottom left panels). A region of interest analysis was used to document that the interaction was driven by age-related dysfunction in the TEC of the Vps26b KO mice (Figure 3A, bottom right panel).
Figure 3. The trans-entorhinal cortex differentially depends on VPS26b.
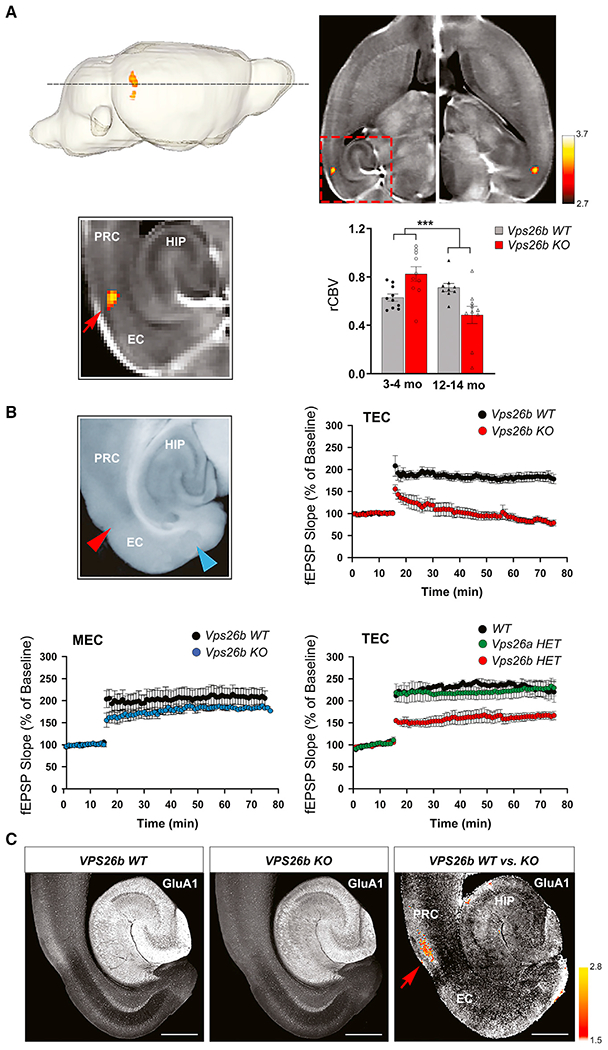
(A) Functional MRI. Cerebral blood volume (CBV) fMRI maps were generated from the whole brain of 3- to 4-month-old Vps26b KO and their control littermates (n = 10 per group) and 12- to 14-month-old Vps26b KO mice and their control littermates (n = 9–10 per group) (top left panel). A voxel-based analysis across the whole brain revealed a significant genotype X age focal defect (voxel-wise p < 0.005; cluster-wise p < 0.05; cluster size >25 voxels) (top right panel), which upon magnification is found to localize to the TEC (bottom left panel) (color bar represents t values of the interaction; PRC, perirhinal cortex; EC, entorhinal cortex, HIP, hippocampus; red arrow indicates the TEC). A region of interest (ROI) analysis of the relative CBV (rCBV) at the TEC between Vps26b WT and Vps26b KO revealed a significant age-dependent worsening of rCBV in the TEC region (in an ANOVA analysis of the genotype × age interaction: F[3,38] = 16.08, p = 0.0003; n = 9–10 animals/genotype) (top right panel).
(B) Electrophysiology. An example of an acute ex vivo brain slice whose precise anatomical coordinates were matched to the neuroimaging defects, illustrating electrode placement (top left panel) (TEC, red arrow; MEC, blue arrow). Mean fEPSP slopes, expressed as the percentage of baseline measured before and after high-frequency stimulation in the TEC (top right panel) and in the MEC (bottom left panel), showed that 12- to 14-month-old Vps26b KO mice, compared with control littermates (n = 6 per group), have LTP defects in the TEC (F(1,14) = 69.2; p < 0.001) but normal LTP in the MEC (F(1,14) = 0.93; p = 0.365) in a repeated-measures ANOVA post hoc Tukey test. Abnormal LTP was also found in the TEC of 18-month-old Vps26b HET mice but not in 18-month-old Vps26a HET mice (Vps26b HET versus WT: F(2,11) = 3.27; p = 0.005; Vps26a HET versus WT: p = 0.42) (post hoc Tukey) (bottom right panel).
(C) By comparing co-registered brain slices of GluA1 immunostainings from 14-month-old Vps26b WT mice (n = 5) (left panel) and Vps26b KO mice (n = 5) (middle panel), a pixel-based analysis (“Vps26b WT versus KO”) showed that Vps26b KO mice have focal TEC reductions in GluA1 immunostaining levels (indicated by the red arrow) (right panel) in a two-sample t test; pixel-wise p < 0.001; color bar represents t values. Scale bar, 500 μm.
See also Figure S5.
To further validate this strikingly focal effect, we performed electrophysiological studies in acute slices of the medial temporal lobe, whose slice locations and orientations were generated and guided by the neuroimaging study (Figure 3B, top left panel). We first characterized the electrophysiological properties of the TEC synapse in this slice orientation (Figures S5A and S5B). Treatment of slices with NBQX, which blocks AMPA-receptor (AMPA-R)-mediated currents, was shown to abolish 81.6% ± 6.4% of the evoked excitatory post-synaptic potential (EPSP), while the remaining current was resistant to GABA-A, NMDA, and adrenergic modulators. Moreover, there was no delay from the stimulus artifact, and the current changed direction when inverting the stimulus, unmasking the fiber volley. Lastly, the delay between the EPSP and the stimulus artifact was 1.28 ± 0.1 ms. Thus, as expected, the TEC synapse in this slice is found to be monosynaptically glutamatergic.
By comparing the TEC synapse of Vps26b KO mice with control littermates, we find a dramatic defect in the LTP in the TEC of Vps26b KO mice (Figures 3B, top right panel, and S5C) with normal basal synaptic transmission (Figure S5D). Confirming the anatomical selectivity of the effect, while visual inspection revealed a mild reduction, no statistically reliable deficits were observed in the medial entorhinal cortex (MEC) of Vps26b KO mice (Figure 3B, bottom left panel). Confirming the paralog selectivity of the effect, a significant LTP defect was observed in the TEC of Vps26b HET mice, but no defects were observed in Vps26a HET mice (Figure 3B, bottom right panel).
The dramatic LTP defects observed in Vps26b deficiency is consistent with a reduction in the AMPA-sensitive glutamate receptor GluA1 (Zamanillo et al., 1999; Park et al., 2004), a glutamate receptor known to be recycled by retromer (Temkin et al., 2017). Accordingly, we immunostained brain slices of the medial temporal lobe for GluA1, whose slice location and orientation were guided by the neuroimaging study. Across all regions analyzed, the most reliable GluA1 reduction was observed in the superficial layers of TEC of Vps26b KO mice (Figures 3C and S5E), precisely in the same location where fMRI identified metabolic dysfunction and electrophysiology identified LTP defects. Collectively, these observations show that VPS26b depletion specifically targets the TEC region, the subregion of the brain most vulnerable to AD (Khan et al., 2014).
Previous studies have established that recycling endosomes are the intracellular reservoirs of GluA1 (Ehlers, 2000; Park et al., 2004) and that GluA1 recycling to the cell surface is retromer-dependent (Temkin et al., 2017; Tian et al., 2015). These studies, however, used VPS35 depletion to establish this retromer-dependency, and, as we show above, VPS35 causes a secondary reduction in both VPS26 paralogs. Interestingly, our findings, reported above, suggest that it is the VPS26b-retromer that is more likely to play a role in GluA1 recycling.
To begin testing this hypothesis, we performed cell-surface biotinylation experiments in cells deficient for each VPS26 paralog. Consistent with the hypothesis, we found that only VPS26b depletion, but not VPS26a deficiency, results in a reduction of surface GluA1 levels (Figures 4A and S6). When the cell-surface level of GluA1 was normalized to the total amount of GluA1 in the cell lysate, we found that the surface GluA1 decreased by 35% in VPS26b-depleted neurons, while the GluA1 surface levels did not change in cells deficient for VPS26a (Figure S6). To further validate our hypothesis, we found by confocal microscopy that VPS26b depletion caused a backlog of GluA1 trafficking, with the greatest accumulation in recycling endosomes and to a lesser degree in early endosomes (Figure 4B). These results show that in absence of VPS26b, GluA1 receptors are trapped in endosomes on their way to the cell surface. Finally, to strengthen this mechanistic link, we performed rescue experiments using a lentivirus to replete VPS26b in Vps26b KO neuronal cultures and found that this repletion fully restored GluA1 localization at the cell surface (Figure 4C). Collectively, these results show that, compared with VPS26a, VPS26b is dedicated more to receptor recycling (Kennedy and Ehlers, 2011).
Figure 4. VPS26b mediates glutamate receptor trafficking from the recycling endosome.
(A) Biotinylated cell-surface proteins were immunoprecipitated from total lysates of cortical neurons depleted for VPS26b (n = 11 biological replicates) or VPS26a (n = 5 biological replicates). Western blot analysis revealed that only VPS26b depletion results in a decrease in GluA1 cell-surface levels.
(B) Representative confocal images of Vps26b KO neurons and Vps26b WT littermates subjected to pulse-chase transferrin uptake and stained for GluA1 and additional endosomal markers. Arrows indicate sites of co-localization. Scale bar, 10 μm (left panel). Quantitative colocalization analysis of GluA1 with early and recycling markers based on Pearson’s correlation coefficients revealed that VPS26b depletion results in an accumulation of GluA1 in recycling endosomes (transferrin: p < 0.0001; Syntaxin13: p < 0.0001) and to a lesser extent in early endosomes (EEA1: p = 0.032) in a non-parametric Kruskal-Wallis with Dunn’s post hoc test (n = 27–37 cells per group/condition, from three separate cultures) (right panel).
(C) Cell-surface GluA1 levels were assessed by confocal microscopy in non-permeabilized cultured neurons incubated with an N-terminal GluA1 antibody in three conditions: Vps26b WT neurons infected with lentivirus expressing GFP alone (“Vps26b WT + GFP vector”), Vps26b KO neurons infected with lentivirus expressing GFP alone (“Vps26b KO + GFP vector”); and Vps26b KO neurons infected with lentivirus expressing VPS26b-GFP (“Vps26b KO + VPS26b-GFP”) (left panel). Scale bar, 10 μm (left panel). Mean fluorescence intensity values revealed that VPS26b repletion fully restored GluA1 surface localization in Vps26b KO neurons, as summarized in the bar graph (p < 0.0001), in a non-parametric Kruskal-Wallis with Dunn’s post hoc test (n = 25–26 neurons/condition, from four independent experiments) (right panel). Data expressed as mean ± SEM., *p < 0.05, **p < 0.01, and ***p < 0.001.
See also Figure S6.
Cognitive profiling supports VPS26b’s regional association
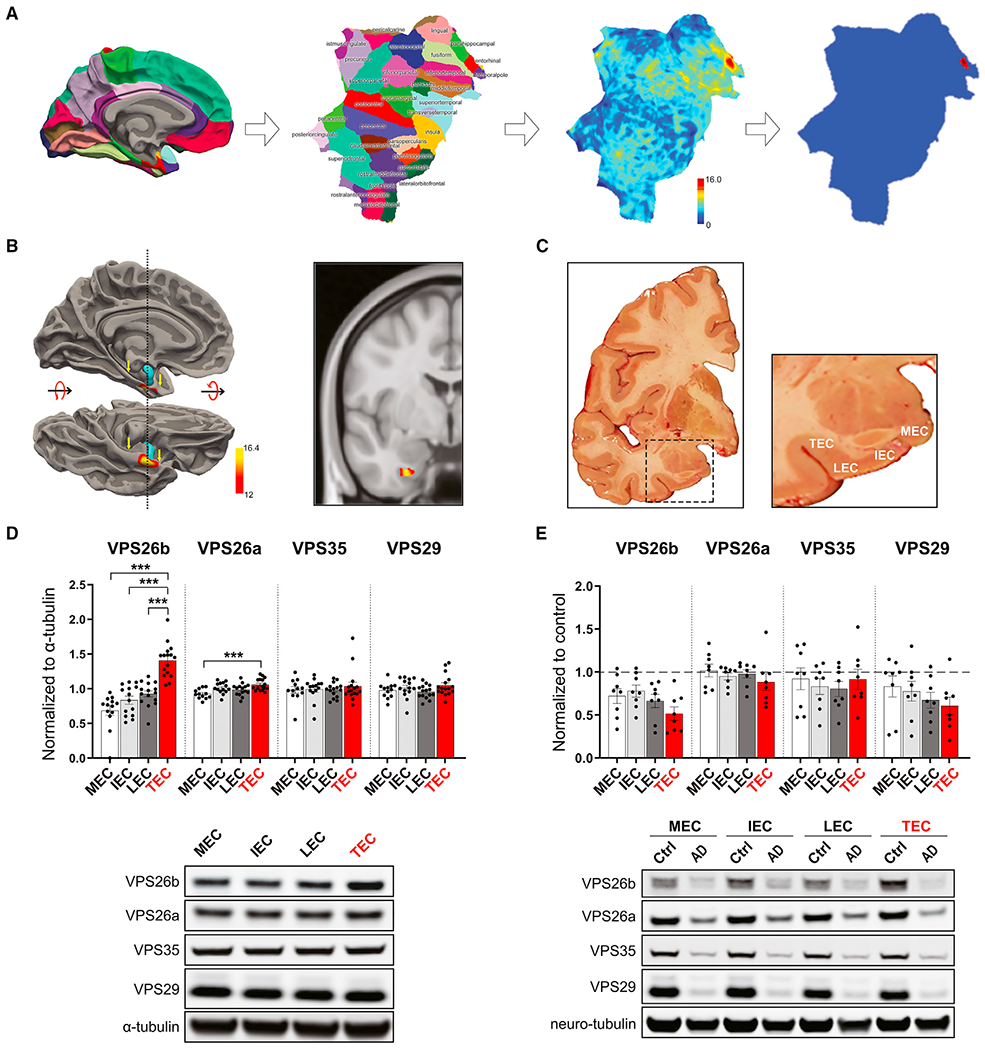
There are no established behavioral tasks whose performance selectively depends on the TEC. Instead, since the TEC is at the border zone of the perirhinal cortex and the lateral entorhinal cortex, we used available tasks whose performance localize to these two parahippocampal regions: the novel object recognition (NOR) task for the perirhinal cortex (Barker and Warburton, 2011) and the object-context recognition (OCR) task for the lateral entorhinal cortex (Wilson et al., 2013). In addition, as the hippocampus is interconnected with the parahippocampus, we also used the modified 2-day radial-arm water maze (2d-RAWM) to evaluate hippocampal function (Alamed et al., 2006). Animals tested on the 2d-RAWM were also tested in a visible platform to rule out impairments of vision, motivation, and motor coordination, as well as cognitive deficits that are not restricted to spatial learning.
We used these tasks to cognitively profile Vps26b KO mice and their WT littermates at 3–4, 6–7, and 12–14 month of age (Figures 5 and S7). While at the oldest ages some defects were observed in both the NOR and the OCR tasks, results showed that OCR performance was most sensitive to VPS26b depletion and was affected first and foremost (Figures 5A and 5B). More specifically, a significant genotype X age interaction was observed, driven by an age-dependent worsening beginning at 6–7 months of age. No defects on the 2d-RAWM task were observed at this age, and only a trend toward a defect was observed at 12–14 months (Figures S7A and S7B).
Figure 5. Cognitive profiling supports VPS26b’s regional association.
(A and B) Vps26b mice were tested in the novel object recognition (NOR) and object-context recognition (OCR) tasks at 3 time points, as described in STAR Methods. Data are expressed as mean ± SEM. A two-way ANOVA with Bonferroni’s post hoc test was used for the analysis.
(A) In the NOR task targeting the perirhinal cortex (n = 10 animals for each of the 6 independent groups), a defect in memory performance was observed only in 12- to 14-month-old mice.
(B) In the entorhinal-sensitive OCR task (n = 10–14 for each of the 6 independent groups), a two-way ANOVA with Bonferroni’s post hoc corrections revealed a significant genotype X age interaction that was driven by age-related worsening in the Vps26b KO mice: F(2,67) = 13.92, p < 0.001.
(C) OCR defects were also found in 12- to 14-month-old Vps26b HET mice versus their WT littermates (n = 9–14 per independent group; p = 0.0016, in unpaired non-parametric Mann-Whitney t test) but not in 12- to 14-month-old Vps26a HET mice versus their WT littermates (n = 8 per group; p = 0.5077, in unpaired two-sided Student’s t test).
(D) Schematic representation of the mouse brain showing the injection site in the lateral recess of the lateral ventricle (image 77, http://atlas.brain-map.org/atlas?atlas=602630314#atlas=602630314&structure=549&resolution=10.26&x=5700&y=4000&zoom=-3&plate=576989860) (left panel). VPS26b overexpression in the EC of WT mice (C57BL6/J): mice were injected at 3 months of age with AAV9-VPS26b at different doses and volume and harvested at 4 months of age (top right panel). VPS26b rescue in EC of Vps26b KO mice: mice were injected at ~3.7 months of age, and brains were harvested at ~7.7 months (bottom right panel).
(E) Vps26b mice were tested in the OCR task. Data are expressed as mean ± SEM. As expected, a defect in memory performance was observed in the Vps26b KO mice compared with the Vps26b WT controls, both injected with AAV9-GFP (n = 4–6 per group, p = 0.0028, in unpaired two-sided Student’s t test). An OCR behavioral rescue was observed in Vps26b KO mice injected with AAV9-VPS26b compared with Vps26b KO mice injected with AAV9-GFP alone (n = 4–6 animals/group, p = 0.0044, in a one-way ANOVA with Tukey’s post hoc test [two-sided]). *p < 0.05, **p < 0.01, and ***p < 0.001.
(F) Mean fEPSP slopes expressed as a percentage of baseline measured before and after high-frequency stimulation in the TEC region of Vps26b WT– AAV9-GFP (n = 5), Vps26b KO – AAV9-GFP (n = 6), and Vps26b KO – AAV9-VPS26b (n = 6) mice. In a two-way repeated measured ANOVA test with a Dunnett’s post hoc, a rescue of LTP defects is observed in Vps26b KO mice injected with AAV9-VPS26b-GFP compared with Vps26b KO mice injected with AAV9-GFP (Vps26b KO – AAV9-VPS26b-GFP: 151.6% ± 0.40% versus Vps26b KO – AAV9-GFP: 103.6% ± 0.95% versus Vps26b WT – AAV9-GFP: 178.3% ± 1.08%, F[2,14] = 6.427, p = 0.0105).
See also Figure S7.
Since Vps26a KOs are embryonic-lethal, we confirmed the paralog specificity of this effect by finding that Vps26b HET mice, but not Vps26a HET mice, have OCR defects (Figure 5C). Collectively, this cognitive profile supports the conclusion that, compared with VPS26a, the general vicinity of the TEC function differentially depends on VPS26b.
To provide further validation that the behavioral phenotype in Vps26b KO mice is indeed caused by VPS26b depletion, we tested whether the behavioral defect can be rescued by AAV9-VPS26b repletion. In preparation for the repletion study, we first completed a dose titration experiment in WT (C57BL6/J) mice to identify the optimal dose for VPS26b expression (Figure 5D, top right panel). We injected AAV9-VPS26b-2A-GFP (herein after AAV9-VPS26b) into the ventral recess of the lateral ventricle (Figure 5D, left panel) to show that we can achieve a high expression in the entorhinal cortex (EC) and at the same time minimize the needle-induced damage associated with direct intraparenchymal injections. The optimal dose was then injected in the Vps26b KO animals (Figure 5D, bottom right panel), which were aged for 3 months post-surgery and behaviorally tested. AAV9-GFP was injected in control animals. Replicating the previous findings, compared with the Vps26b WT animals injected with AAV9-GFP, the Vps26b KO mice injected with AAV9-GFP had an OCR defect, but when they were injected with AAV9-VPS26b, the behavioral defect was completely rescued (Figure 5E). Lastly, to confirm the mechanistic underpinnings of this behavioral rescue, we tested the electrophysiological effect of VPS26b repletion and found that the LTP defect in the TEC was normalized in Vps26b KO mice when injecting AAV9-VPS26b compared with mice injected with AAV9-GFP (Figure 5F). Together, these results suggest that the TEC’s function is differentially dependent on VPS26b.
VPS26b in the AD-targeted trans-EC
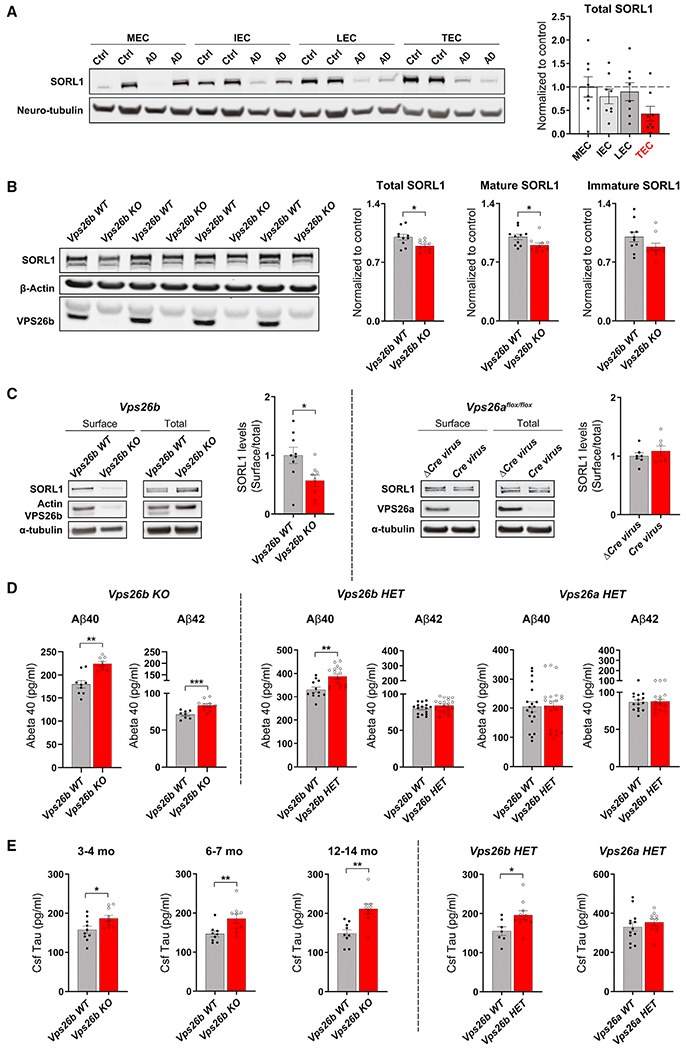
While the TEC has been found to be differentially vulnerable to AD, this subregion, and the EC as a whole, extends for over 4 cm, neighboring the hippocampus in its posterior aspect and the amygdala more anteriorly. In anticipation of our molecular investigation, we needed to pinpoint the epicenter of the AD-targeted TEC. To do so, we performed a large-scale MRI analysis on data acquired from 188 AD cases and 169 healthy controls and generated cortical thickness maps (Figure 6A, two left panels).
Figure 6. VPS26b is the AD-targeted trans-entorhinal cortex.
(A) 3D cortical surface. Cortical map. A 3D rendering of the cortical surface, parcellated into color-coded cortical regions, and shown in a medial view (first panel). Cortical flat map. The 3D surface of a unilateral cortical surface is shown flattened and rendered as a “flat-map” so that all cortical regions can be viewed in a single snapshot. Individual cortical regions are color-coded as labeled (second panel). Raw t value map comparing AD versus controls. A color-coded map of t values was generated comparing the cortical thickness of AD versus controls and co-varying for sex and age (color bar represents t values of the between-group comparisons; the dashed line indicates the threshold t = 12 to generate the thresholded t value map; see STAR Methods) (third panel). Thresholded t value map comparing AD versus controls. By thresholding the t value map, the most reliable cortical thinning in AD versus controls is localized to the vicinity of the TEC (fourth panel).
(B) The Alzheimer’s-targeted TEC was isolated by performing a cortical thickness analysis of MRIs generated from 188 patients with AD and 169 healthy controls. Its longitudinal extent is indicated by the yellow arrows, where the entorhinal cortex abuts the amygdala (the highlighted blue region) (left panel). Pixels with the most reliable volumetric loss compared with controls are indicated in yellow/red (the color bar represents t values of the between-group comparisons), and the TEC defect is also shown on a coronal MRI slice (right panel).
(C) A representative human postmortem brain slice matching the precise anatomical coordinates of the neuroimaging finding (left panel) with the subregions of the entorhinal cortex harvested for protein measurements shown in higher magnification (right panel): the TEC, the lateral EC (LEC), the intermediate EC (IEC), and the medial EC (MEC).
(D) In healthy controls (n = 16), among the four retromer core proteins, the TEC was found differentially enriched in VPS26b (p < 0.0001) in a non-parametric Kruskal-Wallis with Dunn’s post hoc test, as summarized in the bar graph showing mean levels normalized to α-tubulin (top panel) and illustrated with representative immunoblots (bottom panel).
(E) An ANOVA analysis revealed that, compared with age-matched healthy controls (n = 9), AD brains (n = 8) showed the most reliable reduction in the TEC’s VPS26b (F[1,16] = 12.96, p = 0.002), as summarized in the brain graphs showing normalized means (top graph) and illustrated with representative immunoblots (bottom panel) (stippled lines represent the mean of the healthy controls). Data expressed as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
Comparing maps of AD versus controls, besides a global cortical thinning as previously reported (Singh et al., 2006), we observed that the vicinity of the TEC is the cortical area most reliably affected in AD (Figure 6A, two right panels). Moreover, by overlaying the statistical maps onto anatomical images, the epicenter was found to precisely localize to the anterior TEC, where it abuts the amygdala (Figure 6B).
Guided by this anatomical precision, we were able to identify post-mortem samples of healthy subjects, which extended into the anterior medial temporal lobe to include the AD-targeted TEC. From these samples, we dissected the TEC and other subregions of the EC—the lateral EC (LEC), the intermediate EC (IEC), and the medial EC (MEC) (Figure 6C). Western blot analysis revealed that among the 4 retromer core proteins, only VPS26b expression was enriched in the TEC (Figure 6D). In AD brains, retromer proteins showed the greatest reductions in the TEC, with the VPS26b showing the most reliable effect (Figure 6E).
VPS26b mediates Alzheimer’s-associated pathologies
While the previous results differentially link VPS26b to AD’s specific anatomical pathology, we next set out to test whether VPS26b is differentially linked to AD’s known molecular pathologies. First and foremost, we focused on the retromer-related receptor SORL1 because, when deficient, it can act as a pathogenic driver of AD (Scheltens et al., 2021). Examining our human tissue, we found a striking reduction of SORL1 in the TEC of AD brains, at an average level that corresponds to SORL1 haploinsufficiency (Figure 7A). While previous studies have shown that SORL1 is highly expressed in neurons (Thonberg et al., 2017) and that retromer is required for recycling SORL1 and thus maintaining its levels (Nielsen et al., 2007), whether SORL1 is dependent on VPS26b or VPS26a retromers is unknown. To indirectly address this question, we performed a regression analysis including both VPS26b and VPS26a as dependent variables and found that SORL1 levels are differentially linked to VPS26b (t = 4.25, p = 0.001). Next, we mechanistically confirmed that VPS26b is required to maintain SORL1 levels by showing that SORL1 is deficient in the EC of Vps26b KO mice (Figure 7B). Finally, in neuron cultures, we performed cell-surface biotinylation experiments to more formally show that VPS26b, but not VPS26a, is linked to retromer-dependent SORL1 recycling (Figure 7C). Consistently, when cell-surface levels of SORL1 were normalized to the total amount of SORL1 in the cell lysate, we found that surface SORL1 decreased by 44% in Vps26b-depleted neurons (Figure 7C, left panel), while the SORL1 surface levels did not change in cells deficient for VPS26a (Figure 7C, right panel).
Figure 7. VPS26b mediates Alzheimer’s-related pathologies.
(A) Western blotting analysis of different subregions of the entorhinal cortex from healthy controls and patients with AD immunoblotted for SORL1 protein is shown. TEC, trans-EC; LEC; lateral EC; IEC, intermediate EC; MEC, medial EC (left panel). An ANOVA analysis revealed that, compared with age-matched healthy controls (n = 9), AD brains (n = 8) showed a significant and reliable reduction of SORL1 in the TEC region compared with other EC subregions (F[1,16] = 12.32, p = 0.003, as summarized in the brain graphs showing normalized means). Stippled lines represent the mean of the healthy controls (right panel).
(B) Quantitative analysis of SORL1 immunoblots from 7- to 9-month-old mice showing a reduction of SORL1 in the entorhinal cortex of Vps26b KO compared with Vps26b WT mice (total SORL1: p = 0.0160, in an unpaired two-sided Student’s t test; mature SORL1: p = 0.0185, in a on parametric Mann Whitney test; immature SORL1: p = 0.1167, in an unpaired two-sided Student’s t test).
(C) Biotinylated cell-surface proteins were immunoprecipitated from total lysates of cortical neurons depleted for VPS26b (n = 8–9 biological replicates) or VPS26a (n = 7 biological replicates). Western blot analysis revealed that only VPS26b (p = 0.0274, in a two-tailed non-parametric Mann Whitney test), but not VPS26a (p = 0.9755, in a two-tailed non-parametric Mann Whitney test), depletion results in a decrease in SORL1 cell-surface levels.
(D) Bar graph showing the levels of Aβ40 and Aβ42 levels measured by ELISA from entorhinal cortex homogenates harvested from Vps26b KO mice and control littermates at 6–7 months (Aβ40 measurements: Vps26b WT [n = 9] versus Vps26b KO [n = 9], p = 0.0002 in a two-tailed non-parametric Mann Whitney test; for Aβ42 measurements: Vps26b WT [n = 8] versus Vps26b KO [n = 10], p = 0.0013). Data expressed as mean ± SEM from 2–4 independent experiments (left panel). Bar graph showing the levels of entorhinal Aβ40 and Aβ42 measured by ELISA harvested from Vps26b mice (Aβ40 measurements: Vps26b WT [n = 12] versus Vps26b HET [n = 16], p = 0.0011; Aβ42 measurements: Vps26b WT [n = 16] versus Vps26b HET [n = 20], p = 0.1203). Vps26a mice (Aβ40 measurements: Vps26a WT [n = 20] versus Vps26a HET [n = 19], p = 0.8886; Aβ42 measurements: Vps26a WT [n = 15] versus Vps26a HET [n = 15], p = 0.8059). Two-tailed unpaired Student’s t test was used for the statistical analysis (right panel).
(E) Bar graph showing tau CSF levels measured by Simoa technology from Vps26b KO mice and control littermates at 3–4 months: Vps26b WT (n = 10) versus Vps26b KO (n = 10), p = 0.0226, in a two-tailed unpaired Student’s t test; 6–7 months: Vps26b WT (n = 9) versus Vps26b KO (n = 9), in a two-tailed unpaired Student’s t test, p = 0.0056, in a non-parametric Mann Whitney test; and 12–14 months: Vps26b WT (n = 9) versus Vps26b KO (n = 9), **p = 0.0012, in a two-tailed unpaired Student’s t test (left panel). (F) Bar graph showing tau CSF levels measured by Simoa technology from Vps26 HET mice at 9–12 months. Vps26b mice (Vps26b WT[n = 7] versus Vps26b HET [n = 10], p = 0.0270) and Vps26a mice (Vps26a WT [n = 13] versus Vps26a HET [n = 10], p = 0.4169). Two-tailed unpaired Student’s t test was used for all statistical analysis (right panel). Data expressed as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
Retromer has also been linked to AD’s amyloid pathology, with previous studies linking retromer to accelerated APP processing mainly by showing that VPS35 deficiency increases the secretion of both Aβ40 and Aβ42 peptides (Bhalla et al., 2012). Since VPS35 depletion leads to a deficiency of both paralogs (as shown above), we set out to address whether this is differentially mediated by VPS26b. We measured both peptides in the EC of Vps26b KO mice and Vps26a and Vps26b HET mice and found that only VPS26b depletion results in a reliable increase in both Aβ peptides (Figure 7D).
Finally, we investigated a link to AD-associated tau pathology. Recent studies have established that accelerated tau secretion and elevation of CSF tau is not only a sensitive and early feature of AD’s tau pathology but is what distinguishes AD’s tauopathies from other non-AD tauopathies (Zetterberg, 2017). Since tau secretion has emerged as the early and specific event in AD’s tau pathology, and since murine tau can be reliably measured in mouse CSF and is already used in AD mouse models (Maia et al., 2013), we focused on CSF murine tau. In fact, we have previously shown that Vps35 cKO mice phenocopy this extracellular manifestation of tau pathology (Simoes et al., 2020), but here again, whether this is differentially mediated by the two different retromer cores is unknown. To address this question, we collected CSF from both Vps26b HET and Vps26a HET mice at 9–12 months of age and, as previously described, used a single-molecule array to measure CSF tau. Consistent with other AD-associated pathologies described above, VPS26b deficiency, but not VPS26a deficiency, resulted in an abnormal accumulation of CSF tau (Figure 7E). Collectively, these studies establish a link between VPS26b and AD-related molecular pathologies.
DISCUSSION
By using a complimentary series of experimental techniques, we achieved the goals that motivated this study. First, we show that, in contrast to other cells in our body and phylogenetically down to yeast, neurons are endowed with a second retromer core organized around VPS26b. We then show that, compared with the ubiquitous VPS26a-retromer, the VPS26b-retromer is dedicated to the endosomal recycling pathway, particularly during synaptic plasticity. Since glutamate receptor recycling is critical for synaptic plasticity, our results provide a plausible explanation for why neurons might require an additional retromer core dedicated to endosomal recycling.
The second goal of this study was to determine whether deeper insight into retromer biology could clarify AD’s anatomical vulnerability. CBV-fMRI mapping, an fMRI variant whose high spatial resolution is particularly useful for mapping metabolic dysfunction across the mouse brain (Khan et al., 2014; Schobel et al., 2013), was notable for finding that across the brain, the TEC is differentially dependent on VPS26b. A selective regional vulnerability map of the kind observed in Vps26b KO mice is rarely found. CBV-fMRI, like all fMRI approaches, relies on hemodynamic coupling to act only as an indirect indicator of regional energy metabolism and activation, and in the case of CBV, the hemodynamic coupling reflects changes in the regional arteriole diameter. Thus, we further validated this regional-selective finding by using tools that are more direct indicators of brain dysfunction, notably electrophysiology and glutamate receptor immunostaining. Behavior is, of course, the ultimate readout of the brain, and we further show a cognitive correlate of these endophenotypic markers of dysfunction. In the absence of a task that specifically localizes to the TEC, we used tasks to evaluate its neighboring regions, the perirhinal (Barker and Warburton, 2011) and the lateral entorhinal cortices (Wilson et al., 2013). While both are ultimately affected, the cognitive profiling showed that the memory task sensitive to the LEC cortex was affected first and foremost by VPS26b depletion. Besides validating the observed dysfunction, these results suggest that the TEC functionally overlaps with that of the LEC.
The observed retromer expression profile in human brains whereby the TEC highly expresses VPS26b generally supports the interpretation that, in humans, the TEC is differentially involved in high rates of endosomal recycling, as hypothesized by the TEC’s unique network properties (Small and Swanson, 2019). The differential deficiency in SORL1 in the TEC of AD brains turned out to be more important for linking retromer to AD’s pathobiology. While a range of previous studies have established that SORL1 is a retromer-related receptor (Small et al., 2005; Rogaeva et al., 2007; Fjorback et al., 2012; Muhammad et al., 2008), our human data suggested that SORL1 in neurons is differentially associated with the VPS26b core, an interpretation that we then mechanistically validate in mouse models and neuronal cultures, where we show that VPS26b regulates SORL1 recycling and its levels.
Collectively taking together the trans-entorhinal’s unique network properties, the results suggest a plausible explanation for AD’s regional vulnerability. Specifically, the evidence suggests that any pathogenic process impairing the neuron’s endosomal recycling pathway will potentially trigger AD’s pathogenic process, first and foremost, in the TEC. Since the common, late-onset form of AD is a complex disorder, the etiological causes that disrupt endosomal recycling in the brain are by definition pleiotropic. They likely include a complex interaction of the class of “endosomal trafficking” genes (Karch and Goate, 2015) with comorbidities such as diabetes and obesity (Morabito et al., 2014), which can converge by imposing high demands on endosomal recycling. This explanation extends even to early-onset AD caused by mutations in APPs, the PSENs, and trisomy 21. It is now known that by affecting APP processing, these genetic abnormalities not only lead to Aβ accumulation but also to endosomal traffic jams upstream of Aβ production (Kwart et al., 2019; Jiang et al., 2010; Pensalfini et al., 2020). Thus, our results suggest a general mechanism for why the TEC is the disease’s most vulnerable anatomical hub in all forms of the disease and from where it can then spread (Khan et al., 2014).
Limitations of the study
The main limitation of this study is that while the suggested mechanism of regional vulnerability is supported by a convergence of experimental findings in model systems and observational findings in the AD brain, it can only be formally confirmed by showing that retromer-enhancing drugs administered to patients with AD can ameliorate entorhinal-sensitive cognitive defects and relevant biomarkers. Notably, studies have already provided the proof-of-concept that retromer can be pharmacologically targeted (Young et al., 2018; Mecozzi et al., 2014), and recent studies have established a CSF biomarker of retromer-dependent neuronal dysfunction (Simoes et al., 2020). Thus, future clinical trials will be able to formally test the mechanistic model and address this limitation. Importantly, our results inform these drug discovery efforts by suggesting that drugs that target retromer’s VPS26b core will enhance efficacy and also, since this distinct core is brain enriched, diminish the potential for side effects.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Scott Small (sas68@cumc.columbia.edu).
Materials availability
The Vps26b mice can be obtained with an MTA from Korea Research Institute of Bioscience and Biotechnology, while the Vps26a and Vps35 lines are available upon request sent to the Lead Contact.
Data and code availability
Source data of graphs plotted in Figures 1, 2, 3, 4, 5, 6, 7, and S1–S8 are available as source data files. The original Western blotting raw data have been deposited on Mendeley at https://doi.org/10.17632/jggdkrpxts.1, and will be publicly available as of the date of publication. Data used in preparation of this article were partially obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu).
Custom Linux bash and MATLAB scripts, developed by our laboratory to conduct mice and human MRI analysis, are available from the Lead Contact upon reasonable request.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECTS DETAILS
Mouse models
All animal procedures and experiments were performed in accordance with national guidelines (National Institutes of Health) and approved by the Institutional Animal Care and Use Committee of Columbia University, and SUNY Downstate Medical Center. Mice were maintained in groups of 5 or less with 12 h on/off light cycles. Behavioral task experiments were performed using 2 to 3 independent groups (separate cohorts) at different time point (3–4, 6–7, 12–14 and 18-month of age) during the light phase. Animals from both sex were randomly allocated to experimental groups. All assessments were performed by someone blind to the genotype of the animals. Mice were sacrificed by cervical dislocation, and tissue samples were dissected immediately and frozen in dry ice before storage at −80°C. Hippocampi and entorhinal cortex samples were dissected from all genotypes described below and analyzed by ELISA and western blotting for retromer proteins.
We have established a colony of Vps26b KO mice originally described by Kim et al.(Kim et al., 2008). This KO line was generated by replacing exons 5 and 6 of the Vps26b gene by a neomycin-resistance cassette. These animals are viable, live into adulthood, and do not display any gross physical abnormalities. Vps26a HET mice were previously described and characterized in Muhammad et al.(Muhammad et al., 2008). Vps26aflox/flox mice, containing exons 3–7 of the Vps26 gene flanked by a LoxP site in both alleles were used to generate primary neuronal cultures. This line was described in (Mecozzi et al., 2014). Lastly, Vps35flox/flox mice previously described and characterized in Simoes et al. (Simoes et al., 2020) were crossed with mice expressing Cre recombinase under the Camk2α promoter to knock-out Vps35 in forebrain neurons. In all experiments, littermate Vps35flox/flox; Camk2α-Cre (Vps35 cKO) were compared to Vps35flox/flox (Control).
Cell cultures
Neuronal cell culture
Hippocampal and cortical cultures (mixed sex) were obtained from P0 mice, trypsinized for 10 min at 37°C, dissociated by passing through a Pasteur pipette and finally plated on poly-Ornithine coated 35 or 100 mm dishes (biochemistry) or poly-L-lysine-coated coverslips (Immunofluorescence). Neurons were incubated with Neurobasal media (Invitrogen) with 10% fetal bovine serum for 2–4 h before being transferred into serum-free Neurobasal-A media supplemented with B27 (Invitrogen), Glutamax, and cultured for 15 or 21 days in vitro (DIV). Media were always changed 24h post-plating to remove any cell debris.
Cell transfection and lentivirus production
Knockout Vps26a−/− or control Vps26aflox/flox primary hippocampal neurons (mixed sex) were generated by infecting cells at DIV 5, for a period of 10 days, with a lentivirus carrying either the catalytically active Cre or catalytically dead Cre recombinase (ΔCre, control), respectively. Both Cre recombinases encoded two nuclear localization sequences (NLS) and were fused to eGFP containing a third NLS for enhanced nuclear targeting. Lentiviruses were produced in HEK-293T cells. Briefly, virus-containing medium was collected 72 h after transfection, centrifuged at 2,800g for 5 min, filtered through a 0.45 mm filter (Millipore), supplemented with 10 μg/mL polybrene (Sigma-Aldrich) and applied at a 1:5 ratio to hippocampal/cortical medium. The KD efficiency was further monitored by Western blot analysis.
Human subjects
The human MRI data was obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). ADNI is a multicenter study aimed to develop AD biomarkers. The details of the ADNI MRI acquisition, preprocessing protocols (Jack et al., 2008) can be found in the project website (http://adni.loni.usc.edu/methods/mri-tool/mri-analysis/). We used a total of 188 patients with AD and 169 normal subjects baseline scans in the current study. The demographic information of the subjects can be found in Table S1.
METHOD DETAILS
Mouse studies
AAV9-VPS26b rescue
AAV9-VPS26b rescue experiments were conducted as described previously (Qureshi et al., 2019, 2020) with some modifications. Briefly the AAV9-CAG-VPS26b-2A-eGFP-WPRE vector was designed and finalized in our laboratory at Columbia University and then manufactured by Vector Biolabs. AAV9-CAG-eGFP-WPRE from Vector Biolabs was used as control. Viruses were injected in the brain as described previously (Qureshi et al., 2019). The injection coordinates were calculated to target the ventral recess of lateral ventricle using mouse brain atlas. Dose titration was performed in WT animals. The optimal dose (4E+09 VG in 4 ul volume) was injected bilaterally in the experimental group. Animals were monitored post-surgery for side effects of the procedure. All procedures were approved by Columbia University’s IACUC.
Murine cerebrospinal fluid (CSF) collection
Due to the well-known circadian rhythm fluctuations in CSF proteins (Bateman et al., 2007), all CSF collections were performed within a specific 4-h time window (generally in the afternoon) that did not vary by cohort or experiment method as described in (Simoes et al., 2020). Briefly, antemortem CSF was collected from anesthetized animals in accordance with Columbia University IACUC guidelines. Anesthetized mice were placed in a prone position and the skin covering the back of the neck was shaved. A cotton swab containing 70% ethanol was used to remove any hair from the exposed skin. Then, a 27-gauge sterile needle (SV*27EL, Terumo Medical Products) attached to a 1-mL syringe (329650, BD Biosciences) was inserted into the cisterna magna allowing flow of CSF into the butterfly needle. After 10–15 s, the needle was removed and the CSF aspirated into microcentrifuge tubes (1605-0000, USA Scientific), followed by a brief centrifugation at 600×g for 6 min at 4°C. Supernatant was transferred to a new tube, immediately placed on dry ice, and further stored at −80°C. Roughly 5–10uL of fluid was collected per mouse. CSF visibly contaminated with blood (pelleted residual erythrocytes) was discarded. All remaining samples underwent more stringent assessment for blood contamination via hemoglobin ELISA (ab157715, Abcam) using 0.5uL CSF with a 1:200 dilution.
CSF tau measurements using single-molecule-array (Simoa) technology
Antemortem CSF tau was quantified by Simoa technology using the mouse ‘total-tau assay (102209, Quanterix), as described in (Simoes et al., 2020). Briefly, individual CSF samples (4uL) were diluted 1:60 in sample buffer and then split into technical duplicates. Standards and internal controls were run according to the manufacturer’s instructions and read on the HD-X analyzer (Quanterix).
In vivo Aβ measurements
Mouse Aβ samples were derived from entorhinal tissue samples of Vps26b and Vps26a mice. Aβ40 and Aβ42 ELISAs were performed according to the manufacturer’s instructions (Life Technologies) and as previously described in Muhammad et al. (Muhammad et al., 2008). Briefly, ELISAs were carried out in duplicates and represented as absolute values.
Electrophysiology
Hippocampal horizontal slices were obtained as previously described (Cepeda-Prado et al., 2012). In short, mice were anesthetized with ketamine/xylazine, brains removed and placed in modified artificial cerebral spinal fluid (aCSF) bubbled with O2 and CO2 (pH 7.4) and sectioned through the ventral hippocampus into 400 μm-thick slices. Recordings were performed at an approximate position A/P −3.8 mm, 3.0 mm, and D/V −5.6 mm, where ~100% of the LEC/TEC superficial layers connectivity are quite prominent found, as described in- (Angulo et al., 2020). The horizontal brain slices contained all the regions of the hippocampal formation: DG, CA3, CA1, Sub, pre/para Sub, LEC, MEC, and perirhinal cortices (slice had an orientation that was similar to the middle hippocampus MRI slice used). Two synapses were tested, TECII to TECIII and MECII to MECI/II. The slice field excitatory postsynaptic potentials (fEPSP) were recorded in the TEC layer III and MEC layer I/II with glass electrodes filled with NaCl 150 mM (2–3 MΩ resistance), which were elicited by stimulating TEC layer II and MEC layer II using a tungsten bipolar electrode. Input-output relationship curves were obtained and a stimulus evoking ~40–50% of the maximum response was selected for the rest of the experiment. For LTP experiments a stimulus as described above was used, a baseline of test responses was obtained (15 min with an inter-stimulus interval of 30 s). LTP was induced on the TEC synapse by high frequency stimulation (HFS, 3 trains of 100 pulses at 100Hz, 10 s interval) as previously described (Criscuolo et al., 2015) and on the MEC synapse by a high frequency stimulation of 2 trains of 100 pulses at 100Hz, 60 s interval (Modified from [Ma et al., 2008]). Responses were recorded for 60 min after HFS. The field-EPSP (fEPSP) slope was measured and expressed as percentage of baseline. The results are expressed as mean ± S.E.M. To ensure that the synapse was AMPA mediated, at the end of each recording, slices were perfused with 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxa-line-2,3-dione (NBQX, 10 μM). After 20 min, a remaining current persisted. To determine if this current was the fiber volley we recorded in the presence of (2R)-amino-5-phosphonovaleric acid (APV, 30 μM), Atropine (10 μM) and Picrotoxin (PTX, 200 μM), to block ionotropic glutamate receptors, GABAA receptors and muscarinic receptors.
Western blotting
Mouse tissue samples were homogenized using a Glas-Col Homogeneizer in an ice-cold lysis buffer consisting of 20 mM Tris HCl (pH 7.4), 1% Triton X-100,15 mM NaCl and protease and phosphatase inhibitors (Roche) for the extraction of proteins. After centrifugation at 17,000g for 10 min at 4°C, supernatants were collected and the protein concentration was measured using BCA assay (PIERCE) prior to Western blot analysis using the Odyssey blocking buffers (LI-COR Biosciences). For all other immunoblots, hippocampal neurons were lysed for 10 min at 4°C in RIPA buffer with protease and phosphatase inhibitor cocktails, centrifuged for 15 min at 13,000g, and supernatant were processed for SDS–PAGE and immunoblotting. The images were acquired with the Odyssey Infrared Imaging System (LI-COR Biosciences) and analyzed by the software program Image Studio Lite Ver 5.0 as specified in the Odyssey software manual.
Pulse–chase transferrin
Primary hippocampal cultures were starved in warmed Neurobasal-A medium containing 1% HEPES (Invitrogen) for 30 min and incubated with 15 μg/mL of human transferrin -Alexa 555 for 30 min at 37°C. Medium was then replaced and transferrin was chased for 10 min (to load recycling endosomes) before cell fixation.
Glycine stimulation
Glycine stimulation experiments for Immunofluorescence analysis of VPS26 co-stainings with different intracellular markers were performed in DIV 15 primary hippocampal neurons, essentially as described by Park et al. (2004). Very briefly, neurons were incubated at 37°C for 5 min in extracellular solution (ECS) containing 124 mM NaCl, 2 mM CaCl2, 3 mM KCl, 10 mM HEPES (Invitrogen) and 10 mM Glucose (Sigma) (pH 7.4). Activity was induced with E4 supplemented with 20 μM μM (−)-Bicuculline methiodide and 200 μM Glycine (Sigma) for 5 min, and finally in E4 with 20 μM bicuculine for 5 min before fixation and imaging. For transferrin stainings, a transferrin uptake was performed right before glycine treatment. Transferrin was only added to the first step of the glycine treatment (5 min incubation with E4). Neuronal activity was induced in absence of transferrin.
Co-immunoprecipitation (Co-IP)
Co-immunoprecipitation experiments were performed using 500–600 μg of proteins extracted from primary neuronal cultures using a lysis buffer containing 0.5% Triton X-100, 50 mM Tris, 100 mM EDTA, 150 mM NaCl, pH 7.3. Protein extracts were incubated overnight with 10 μg of anti-VPS35 (Bethyl laboratories, A304-727A), anti-VPS26a (Abcam, ab23892) and anti-VPS26b (Novus biologicals, NBP1-92575) antibodies pre-incubated with protein-G beads (Invitrogen). Precipitates were subsequently washed five times with IP buffer and boiled for 30 min at 50°C in 100ul of reducing SDS sample buffer 2X, followed by 5 min at 90°C. Immunoprecipitates were fractionated by SDS-PAGE and immunoblotted using same antibodies as probes. Input samples represent 10 μg of total protein corresponding to 2% of total protein used for each co-immunoprecipitation.
Cell surface biotinylation
Surface biotinylation pulldowns were performed according to the manufacturer’s instructions (Pierce). Briefly, cortical neurons were washed twice with PBS and “live” labeled with 1mg/mL of EZ-Link Sulfo-NHS-LC-Biotin at 4°C for 30 min. After being rapidly washed 3 times with PBS, cells were quenched with 100mM of glycine for 10 min. Cell surface biotinylated proteins were pulled down overnight, at 4°C, from cell lysates by streptavidin precipitation and detected by western blotting analysis. Lysates not subjected to streptavidin beads were analyzed as a measure of total cellular protein levels.
Confocal microscopy
Primary hippocampal neurons cultured on coverslips were fixed in a final concentration of 2% paraformaldehyde (PFA) and 0.5% sucrose (mixed with culture medium) and permeabilized with 0.05% saponin in PBS supplemented with 5% donkey serum. For all intracellular stainings, neurons were incubated overnight with primary antibody and further incubated for 45 min with secondary antibodies (Molecular probes). Images were captured with a Zeiss LSM 700 META confocal microscope equipped with a 63× Plan-Apochromat objective and HeNE1, HeNe2 and argon lasers. Colocalization analysis (% colocalization) was obtained based on Pearson’s correlation, that is, (Pearson’s coefficient)2 × 100, using the JACoP Plugin on ImageJ 1.53e (Bolte and Cordelieres, 2006). z stack images of soma and proximal dendrites (with no thresholds applied), were used for data analysis shown in all Figures, except for Figure 4B. For this latter, regions of interest (ROI’s) were used to investigate the intracellular localization of GluA1 in Vps26b KO and WT cultures. Three to four proximal dendritic segments of 40μm were analyzed and averaged for each cell. Cell surface GluA1 staining intensity was determined using the ‘Multi Measure’ build-in option in ImageJ and the intensity of the staining was normalized to the cell area. For staining, hippocampal neurons were initially “live” incubated with an anti-N-terminal GluA1 antibody (MAB2263; 1:300) diluted in Neurobasal-A media containing 1% HEPES for 30 min at 37°C. After a brief washing in prewarmed Neurobasal A, cells were fixed with 4% PFA. Fixed nonpermeabilized neurons were further incubated with a fluorescent secondary antibody for 1 h at RT. When required, following a short fixation with 2% PFA, cells were permeabilized using PBS-0.1% Triton X-100 for 10 min, and further incubated with MAP2 antibody for the detection of dendrites.
Confocal microscopy GluA1 pixel-based analysis
Confocal microscopy of GluA1 stainings shown in Figure 4C were co-registered into a group-wise template space using an inverse-inconsistent diffeomorphic co-registration algorithm, as described previously (Sabuncu et al., 2009; Avants et al., 2011; Khan et al., 2014). The intensity of the GluA1 staining was normalized to the mean intensity of each brain section. Normalized GluA1 intensity was analyzed using pixel-based analysis with two sample t test implemented in SPM8 (Wellcome Department of Imaging Neuroscience).
Immunohistochemistry analysis
Mice were anesthetized, rinsed by cardiac perfusion with 0.9% saline buffer and further fixed with 4% PFA. Whole brains were excised and post fixed in 4% PFA overnight, followed by cryoprotection treatment in 30% sucrose (wt/vol) in PBS for 16 h. Tissue blocks were then embedded in Optimal Cutting Temperature (OCT) compound and stored at −80°C. Briefly, 30 μm free-floating horizontal sections were washed three times with PBS and incubated at 4°C overnight on a rotator in 1 mL of primary antibody diluted in PBS containing 0.3% Triton X-100 (vol/vol) and 5% normal serum (vol/vol). After three washes with PBS-T (0.1% Triton X-100), the sections were incubated for 30 min with secondary antibodies (Molecular probes). Following three washes with PBS-T, and one wash with PBS, sections were mounted on slides. Fluorescent images were collected using a Zeiss LSM 700 META confocal microscope equipped with a 63× Plan-Apochromat objective and HeNE1, HeNe2 and argon lasers.
Ultracryomicrotomy and immunogold labeling
For ultrathin cryosectioning and immunogold labeling, hippocampal neurons (15 DIV) were fixed with a mixture of 2% (wt/vol) PFA and 0.125% (wt/vol) glutaraldehyde in 0.1 M phosphate buffer (PB), pH 7.4. Cell pellets were washed with PB, embedded in 10% (wt/vol) gelatin and infused in 2.3 M sucrose. Mounted gelatin blocks were frozen in liquid nitrogen and ultrathin sections were prepared with an EM UC6 ultracryomicrotome (Leica). Ultrathin cryosections were collected with 2% (vol/vol) methylcellulose, 2.3 M sucrose and single or double immunogold labeled with antibodies and protein A coupled to −10 or 15-nm gold (PAG10 and PAG15), as previously reported (Truschel et al., 2009). Sections were observed under a Philips CM-12 electron microscope (FEI; Eindhoven, The Netherlands) and photographed with a Gatan (4k ×2.7k) digital camera (Gatan, Inc., Pleasanton, CA). The relative distribution of VPS26b and VPS26b in hippocampal cultures was evaluated by EM analysis of randomly selected cell profiles from two distinct grids. Approximately 100 gold particles for each condition were counted and assigned to the compartment over which they were located. The definition of the distinct compartments was based on their morphology and their previous characterization by immunogold labeling with different organelle markers. Results are presented as a percentage of the total number of gold particles in each compartment and is represented as mean ± S.E.M. A quantitation of non-specific background labeling was performed for VPS26a and VPS26b. Background labeling accounted for less than 2.1% of the total number of gold particles for VPS26b, and to less than 1% for VPS26a labeling. VPS26a labeling was performed as in (Rojas et al., 2008).
Behavioral analysis
Object context recognition (OCR) task
The OCR task was carried out as previously described (Wilson et al., 2013) with minor modifications. Briefly, mice were handled daily for a week prior to the study and then given two habituation sessions. In both sessions they were individually exposed to two distinct environments (context X and context Y) for 5 min. Testing began 1–2 days after the last habituation session. In the OCR task, mice receive two test sessions separated by 24 h. Each session is divided into four sample phases and a test phase, each lasting 3 min. In the sample phases two identical objects (either A1:A2 or B1:B2) are placed in far corners of a context (context X or Y). A 2-min interval separate the four sample phases: phases 1 and 4 comprised objects A1 and A2 in context X, phases 2 and 3 comprised objects B1 and B2 in context Y. There is a 5-min interval between the end of sample phase 4 and the test phase. In the test phase, the animal is tested in either context X or Y with objects A3 and B3. Thus, during the test phase one object is in the same environment as in the sample phase, and the other object is in the different environment to the sample phase. In a second session (24 h later) the order of the sample phases are reversed and the test environment changes for each individual mouse. Thus, in sample phases 1 and 4, a mouse that had previously been exposed to A1:A2 in context X is now exposed to B1:B2 in context Y, and in phases 2 and 3 it is exposed to A1:A2 in context X. This tests the mouse’s memory for object-context associations as one of the objects will have been experienced in this context before, while the other will not. Test object, test context, order of context presentation in the sample phases and side of presentation was counterbalanced across mice. All training and testing sessions were recorded using the automated ANY-maze 6.3 VIDEO tracking software. Discrimination between the objects were calculated using a discrimination ratio, calculated by dividing the mean time exploring the novel object by the mean of the total time exploring the novel and familiar objects during the test session. This value was multiplied by 100 to obtain a percentage preference for the novel object (Tnovel/[Tnovel + Tfamiliar] × 100).
Novel object recognition (NOR)
The NOR task was performed as previously described (Barker and Warburton, 2011). Very briefly, the day prior to training, mice were habituated to experimental apparatus consisting of a white rectangular open field (60 cm × 50 cm x 26 cm) for 10 min in the absence of any objects. On the second day, mice were placed in the experimental apparatus in the presence of two identical objects and allowed to explore them for 10 min. After a retention interval of 24 h, mice were placed again in the apparatus, where one of the objects was replaced by a novel object. Exploration of the objects was defined as the mice facing and sniffing the objects within 2-cm distance and/or touching them. Sitting and turning around on the object were not considered exploratory behavior. All training and testing sessions were recorded using the automated ANY-maze 6.3 VIDEO tracking software. The ability of the mouse to recognize the novel object was determined by dividing the mean time exploring the novel object by the mean of the total time exploring the novel and familiar objects during the test session. This value was multiplied by 100 to obtain a percentage preference for the novel object (Tnovel/[Tnovel + Tfamiliar] × 100). The positions of the objects in the test phase and the objects used as novel or familiar were counterbalanced between the animals.
Modified radial arm water maze (RAWM)
The 2-days RAWM task was carried out as previously described (Alamed et al., 2006). Briefly, the behavioral apparatus consisted of a pool 1.2 m in diameter filled with water made opaque with white paint. Dividers were placed into the pool to generate a contiguous space with six equidistantly spaced arms radiating from the center. A 10-cm escape platform was at the end of a designated goal arm, which remained fixed for each mouse throughout the experiment. Spatial cues were present on the walls of the testing room. On each trial, the mouse started the task from a different randomly chosen arm. Each trial lasts 1 min. Entry into an arm with no platform or failure to select an arm after 10 s was scored as an error. Animals that enter an incorrect arm were guided back to the start arm. At the end of each trial, animals were allowed to rest on the platform for 20 s. Each mouse was tested for 15 trials each day for two consecutive days. On the first day, mice were trained for 15 trials, with the first 12 trials alternating between visible (flagged) and hidden (submerged) platform. The last 3 trials of the first day and all of the 15 trials of the second day were performed with a submerged platform. Results were analyzed by dividing the 30 trials into 10 trial blocks and calculating the average error for each trial block.
Visible platform test
Mice that were subject to the 2-days RAWM were also tested on a visible platform to rule out impairments of vision, motivation, motor coordination, as well as cognitive deficits that are not restricted to spatial learning. Briefly, behavioral tests were carried out in the same pool but without arms and with a visible (flagged) platform. The platform location was randomly modified to eliminate any contribution of external spatial cues. Mice were given two sessions of three trials each day over 2 days. Each animal was allowed to swim for 1 min from a random location. The time taken (latency) and speed to reach the platform were recorded and analyzed using a ceiling-mounted camera, an HVS- 2020 VIDEO tracking system and the EthoVision XT 7.0 software. Failure to reach the platform was scored as 60 s. Results were analyzed by dividing the 12 trials into 4 trial blocks and calculating an average value for each trial block.
MRI analysis
Image acquisition
We used cerebral blood volume (CBV)-fMRI to image two independent groups of mice, 3–4 and 12–14 months Vps26b KO mice and their WT littermates, with the imaging protocol as previously described (Moreno et al., 2006; Khan et al., 2014). A Bruker BioSpec 94/20 (field strength, 9.4 T; bore size, 20 cm) horizontal small animal MRI scanner with software ParaVision 6.0.1 (Bruker BioSpin, Billerica, MA, USA) and a 23-mm 1H circularly polarized transmit/receive capable mouse head volume coil were used for the imaging. Mice were anesthetized using the medical air and isoflurane (3% volume for induction, 1.1–1.5% for maintenance at 1 L/min air flow, via a nose cone). A flowing water heating pad was used to maintain the body temperature at around 37°C. Sterile eye lubricant was applied after each scan. T2-weighted images were acquired before and 36 min after intraperitoneal injections of the contrast agent Gadodiamide (Omniscan; GE Healthcare, Princeton, NJ, USA) at the dosage of 10 mmol/kg. T2-weighted images were acquired with a fast-spin echo acquisition (repetition time, 2,500 ms; effective echo time, 45 ms; rapid acquisition and relaxation enhancement factor, 16; in plane resolution, 60 μm; slice thickness, 250 μm).
Generating CBV maps
As previously described (Moreno et al., 2006; Khan et al., 2014), CBV was mapped according to changes in the transverse relaxation time (ΔR2) induced by gadolinium injection. CBV was derived by normalizing ΔR2 to the mean ΔR2 signal present in the posterior cerebral artery (SCA), as delineated by a blinded rater.
Voxel-based analysis
MR images were first skull stripped to yield whole-brain volumes using an automatic rodent brain extraction algorithm based on 3-D pulse-coupled neural networks (PCNN), as described previously (Chou et al., 2011). Mice whole-brain volumes processing was conducted using custom Linux bash and MATLAB R2021a scripts. Volumes were co-registered into a group-wise template space using an inverse-inconsistent diffeomorphic co-registration algorithm, as described previously (Sabuncu et al., 2009; Avants et al., 2011; Khan et al., 2014). CBV maps were analyzed using voxel-based analysis with a general linear model implemented in SPM8 (Wellcome Department of Imaging Neuroscience). Data were modeled in a factorial framework with genotype and age included as the between-subjects factors. The genotype × age interaction effect with the direction showing age-related CBV worsening defects in KO mice was contrasted with a t test. Result was corrected for multiple comparisons using Monte Carlo simulation implemented in AFNI-AlphaSim (http://afni.nimh.nih.gov/afni/).
Human studies
In vivo MRI studies
Image acquisition.
The human MRI data was obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). ADNI is a multicenter study aimed to develop AD biomarkers. The details of the ADNI MRI acquisition, preprocessing protocols (Jack et al., 2008) can be found in the project website (http://adni.loni.usc.edu/methods/mri-tool/mri-analysis/). Briefly, the T1-weighted structural MRI scans were acquired with MP-RAGE (Magnetization-prepared rapid gradient echo) sequence under 1.5T MRI scanner. We used a total of 188 patients with AD and 169 normal subjects baseline scans in the current study. The demographic information of the subjects can be found in Table S1.
Image processing and data analysis.
The human MRI data was obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). The T1-weighted structural images were processed using FreeSurfer, generating individual cortical surfaces and cortical thickness maps (Fischl and Dale, 2000). The individual cortical surfaces were registered into the FreeSurfer fsaverage space for inter-subject analysis. At each vertex, we performed linear regression with cortical thickness as dependent variable, with AD vs. normal diagnosis as testing variable, and with gender and age as covariates. t-statistics of the regression analyses were reported as a raw t-map. We rendered the results in the flat-map representation of the cortical surface using FreeSurfer utilities (Fischl et al., 1999) and custom scripts. The flat-map representation provides unbiased visualization of the whole cortex in one single view, and facilitates observation of the findings, and the identification of clustered parcellation of interest. To localize AD-vulnerable regions, we thresholded the t-map at t < −12 (passing Bonferroni correction at α = 1E−22). The statistics on the surface space were converted to anatomical space using FreeSurfer utilities.
Postmortem studies.
Tissue blocks from healthy controls and AD cases were requested from the Columbia University Alzheimer’s Disease Research Center (ADRC) brain bank (IRB#AAAB0192, Not Human Subjects Research under 45 CFR 46 exemption 4). Neuropathological assessment was performed using methods previously described (Vonsattel et al., 1995). All cases were rated according to Braak and Braak (Braak and Braak, 1991), Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) (Mirra et al., 1991), and National Institute on Aging–Reagan Institute (NIA-RI) criteria. A total of 24 cases were used in this study: healthy controls, n = 16 (age ranging from 36 to 89 years old) and AD, n = 8 (age ranging from 66 to 89 years old). Demographic information can be found in Table S2. Standard brain blocks (SBB8 samples) containing the anterior part of the TEC, where the TEC abuts the amygdala, were used for the Western blot analysis. TEC and neighboring regions harvested from each sample block (Mai et al., 2015) were homogenized using a Bel-Art Micro-Tube Homogenizer in RIPA buffer (10 times the tissue weight in mg) containing protease and phosphatase inhibitors (Roche) for the extraction of proteins. Samples were incubated for 30 min at 4°C in rotation before centrifugation at 13,000 xg for 20 min at 4°C. Protein concentration was measured using BCA assay (PIERCE) prior to Western blot analysis using the Odyssey blocking buffers (LI-COR Biosciences).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are expressed as geometric mean with error bars representing ±SEM. Data normality was analyzed using a D’Agostino-Pearson omnibus normality test. A two-way ANOVA with Bonferroni’s post hoc test was used for the OCR and NOR behavioral tasks (three independent age groups). For the comparison of VPS26 HET mice performance in the OCR task, unpaired two-sided student’s t-test or non-parametric Mann-Whitney t test was used as described in figure legend. To compare the escape latency, swim speed and performance in the 2-days RAWM, a two-way repeated –measures ANOVA with Bonferroni post-tests was used. For the electrophysiology studies, a one-way repeated –measures ANOVA with post hoc Tukey or a two-way repeated –measures ANOVA test with a Dunnett’s post test were performed, as described on each figure legend. Unpaired two-sided student’s t-test was used for some of the biochemical experiments and immunofluorescence analysis using two-tailed distribution with equal variance (p < 0.05). Unpaired t test with Welch’s correction was performed for groups with unequal variances as stated in the figure legend. Differences between more than two groups were evaluated by one-way ANOVA with post hoc Tukey’s multiple comparisons test. All the statistical analysis described above were performed using the GraphPad Prism version 9.00 for Windows (GraphPAd Software). Mice and human MRI statistical analysis was performed using SPM8 (Wellcome Department of Imaging Neuroscience), while the electrophysiology data and human WB analysis were analyzed using SigmaPlot 14.5 and SPSS 24.0, respectively. The “n” indicated on figure legends represent either the number of animals or cells analyzed per condition or group.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-SORL1 | Cell Signaling Technology | Cat#79322S; RRID: AB_2799927 |
| Rabbit polyclonal anti-VPS26b | Novus Biologicals | Cat#NBP1-92575; RRID: AB_11020053 |
| Rabbit polyclonal anti-VPS26a | Abcam | Cat#ab23892; RRID: AB_2215043 |
| Rabbit polyclonal anti-VPS26a | Bonifacino Lab; Rojas et al. (2008) | N/A |
| Rabbit polyclonal anti-Neuro-tubulin | Abcam | Cat#ab18207; RRID: AB_444319 |
| Rabbit polyclonal anti-VPS35 | Bethyl laboratories | Cat#A304-727A; RRID: AB_2620922 |
| Rabbit polyclonal anti-GluA1 C-terminus | MilliporeSigma | Cat#AB1504; RRID: AB_2113602 |
| Rabbit polyclonal anti-mGluR5 | Alomones lab | Cat#AGC-007; RRID: AB_2039991 |
| Rabbit polyclonal anti-Syntaxin13 | Synaptic Systems | Cat#110 133; RRID: AB_2198225 |
| Rabbit polyclonal anti-Rab11 | Thermo Fisher Scientific | Cat#71–5300; RRID: AB_2533987 |
| Rabbit polyclonal anti-Syntaxin13 | Synaptic Systems | Cat#110 132; RRID: AB_67552 |
| Rabbit polyclonal anti-MAP2 | MilliporeSigma | Cat#AB5622, RRID: AB_91939 |
| Goat polyclonal anti-VPS35 | Abcam | Cat#ab10099; RRID: AB_296841 |
| Goat polyclonal anti-VPS29 | MilliporeSigma | Cat#SAB2501105; RRID: AB_10602838 |
| Goat polyclonal anti-EEA1 | Santa Cruz Biotechnology, Inc. | Cat#sc-6415; RRID: AB_2096822 |
| Mouse monoclonal anti-β-actin | Novus Biologicals | Cat#NB600-535; RRID: AB_2222878 |
| Mouse monoclonal anti-α-tubulin | MilliporeSigma | Cat#T6074; RRID: AB_477582 |
| Mouse monoclonal anti-VPS35 | Abcam | Cat#ab57632; RRID: AB_946126 |
| Mouse monoclonal anti-VPS26a | Novus Biologicals | Cat#NBP2-36754 |
| Mouse monoclonal anti-Golgin97 | Thermo Fisher Scientific | Cat#A-21270; RRID: AB_221447 |
| Mouse monoclonal anti-Rab7 | Santa Cruz Biotechnology, Inc. | Cat#sc-376362; RRID: AB_10987863 |
| Mouse monoclonal anti-GluA1 N-terminus | MilliporeSigma | Cat#MAB2263; RRID: AB_11212678 |
| Mouse monoclonal anti-Neuro-tubulin | BioLegend, Inc. | Cat#801202, RRID: AB_10063408 |
| Mouse monoclonal anti-Rab5 | Santa Cruz Biotechnology, Inc. | Cat#sc-46692, RRID: AB_628191 |
| Chicken polyclonal anti-GFAP | Abcam | Cat#ab4674, RRID: AB_304558 |
| Chicken polyclonal anti-MAP2 | Abcam | Cat#ab5392; RRID: AB_2138153 |
| IRDye® 800CW Donkey anti-Rabbit IgG Secondary Antibody | LI-COR Biosciences | Cat#926–32213; RRID: AB_621848 |
| IRDye® 680RD Donkey anti-Mouse IgG Secondary Antibody | LI-COR Biosciences | Cat#926–68072; RRID: AB_10953628 |
| IRDye® 680RD Donkey anti-Goat IgG Secondary Antibody | LI-COR Biosciences | Cat#926–68074; RRID: AB_10956736 |
| IRDye® 800CW Donkey anti-Mouse IgG Secondary Antibody | LI-COR Biosciences | Cat#926–32212; RRID: AB_621847 |
| Donkey anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#21206; RRID: AB_2535792 |
| Donkey anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-31572; RRID: AB_162543 |
| Donkey anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31573; RRID: AB_2536183 |
| Donkey anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21202; RRID: AB_141607 |
| Donkey anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat#31570; RRID: AB_2536180 |
| Donkey anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat#31571; RRID: AB_162542 |
| Donkey anti-Goat IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#11055; RRID: AB_2534102 |
| Donkey anti-Goat IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat#21432; RRID: AB_2535853 |
| Donkey anti-Goat IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat#21447; RRID: AB_141844 |
| Chemicals, peptides, and recombinant proteins | ||
| Transferrin From Human Serum, Alexa Fluor™ 555 Conjugate | Thermo Fisher Scientific | Cat#T35352 |
| (−)-Bicuculline methiodide | Tocris | Cat#2503; CAS Number: 40709-69-1 |
| EZ-Link™ Sulfo-NHS-LC-Biotin | Thermo Fisher Scientific | Cat#21335; CAS Number: 127062-22-0 |
| Picrotoxin | MilliporeSigma | Cat# P1675-1G; CAS Number: 124-87-8 |
| Glycine | MilliporeSigma | Cat# G7126-100G; CAS Number: 56-40-6 |
| 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f] quinoxaline-2,3-dione | MilliporeSigma | Cat#N183-5MG; CAS Number: 118876-58-7 |
| (2R)-amino-5-phosphonovaleric acid | MilliporeSigma | Cat#A5282; CAS Number: 76326-31-3 |
| Atropine | MilliporeSigma | Cat#A0132; CAS Number: 51-55-8 |
| VPS26A Recombinant Protein Antigen | Novus Biologicals | Cat# NBP2-36754PEP |
| Critical commercial assays | ||
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat#23227 |
| Mouse Hemoglobin ELISA Kit | Abcam | Cat#ab157715 |
| Simoa™ Mouse Tau Discovery Kit | Quanterix | Cat#102209 |
| Amyloid beta 42 Mouse ELISA Kit | Thermo Fisher Scientific | Cat#KMB3441 |
| Amyloid beta 40 Mouse ELISA Kit | Thermo Fisher Scientific | Cat#KMB3481 |
| Deposited data | ||
| https://doi.org/10.17632/jggdkrpxts.1 | N/A | |
| Experimental models: Organisms/strains | ||
| Mouse: Vps26b HET/KO mice | Kim et al. (2008) | N/A |
| Mouse: Vps26a HET mice | Muhammad et al. (2008) | N/A |
| Mouse: Vps26aflox/flox mice | Mecozzi et al. (2014) | N/A |
| Mouse: Vps35flox/flox mice | Simoes et al. (2020) | N/A |
| Recombinant DNA | ||
| Second generation lentiviral packaging plasmids VSVg and Δ8.9 | Peter Scheiffele (University of Basel) | N/A |
| EGFP-tagged active, full-length Cre recombinase and catalytically inactive, truncated Cre FUGW lentiviral constructs | Thomas Sudhof (Stanford University, California) | N/A |
| p-Lenti-C-mGFP-P2A-Puro | OriGene Technologies, Inc. | Cat#PS100093 |
| p-Lenti-C-mGFP VPS26b | OriGene Technologies, Inc. | Cat#MR204988L4 |
| Lentiviral Packaging Kits | OriGene Technologies, Inc. | Cat#TR30037 |
| AAV9-CAG-VPS26b-2A-eGFP-WPRE vector | Vector Biolabs | N/A |
| AAV9-CAG-eGFP-WPRE | Vector Biolabs | N/A |
| Software and algorithms | ||
| EthoVision XT 7.0 | Noldus Information Technology | https://www.noldus.com/ethovision-xt |
| SigmaPlot 14.5 | Systat Software Inc. | https://systatsoftware.com/products/ |
| ImageJ 1.53e | NIH | https://imagej.nih.gov/ij/download.html |
| Image Studio Lite 5.0 | LI-COR Biosciences | https://www.licor.com/bio/image-studio-lite/ |
| MATLAB R2021a | The MathWorks, Inc. | https://www.mathworks.com/products/matlab.html |
| GraphPad Prism 9.00 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| SPSS 24.0 | IBM | https://www.ibm.com/products/spss-statistics |
| ANY-maze 6.3 | Stoelting Co. | https://stoeltingco.com/Neuroscience/ANY-maze |
| Other | ||
| Butterfly needle to collect CSF | Terumo Medical Products | Cat#SV*27EL |
| Neurobasal™-A Medium | Thermo Fisher Scientific | Cat#10888022 |
| B-27™ Supplement (50X), serum free | Thermo Fisher Scientific | Cat#17504044 |
Highlights.
Neurons are endowed with a separate VPS26b-retromer dedicated to endosomal recycling
The trans-entorhinal cortex (TEC) differentially depends on VPS26b-retromer
VPS26b in mice regulates entorhinal-based memory, LTP, and SORL1 recycling
VPS26b is enriched in the human TEC, whereas VPS26b and SORL1 are deficient in AD
ACKNOWLEDGMENTS
We thank the DART Microscopy Core Lab of New York University Langone Medical Health for the support of the EM work. We also acknowledge Dr. Ottavio Arancio for support with animal behavioral equipment and thank Dr. Catherine Marquer for helpful discussions. We are also grateful to Drs. Sang-Rae Lee and Y.-H.K. for sharing the Vps26b KO mice. We also want to thank Nicoletta Barolini for her expert help with illustrations and graphics. Additionally, data used in preparation of this article were partially obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, we thank all the investigators within the ADNI who contributed to the design and implementation of ADNI and/or who provided data. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. Additionally, lists of participants and their affiliations appear in the supplemental information. Lastly, this paper is dedicated to the memory of K.-T.C. and our long-term collaborator H.M.
This study was partly supported by NIH R01 grants AG034618, AG035015, and P30AG066462 to S.A.S., NS056049 to G.D.P., and AG051556 to H.M. Human MRI data collection and sharing for this project were funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (NIH grant U01 AG024904) and the Department of Defense ADNI (award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging and the National Institute of Biomedical Imaging and Bioengineering and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche, Ltd., and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO, Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development, LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer, Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110182.
DECLARATION OF INTERESTS
S.A.S. is a co-founder of Retromer Therapeutics, has equity in the company, and is a paid consultant to the company. In addition, S.A.S. has equity in Imij Technologies, an MRI-based company. G.D.P. is a full-time employee of Denali Therapeutics, Inc. O.M.A. has commercial interests in Retromer Therapeutics. Lastly, S.A.S., S.S., and Y.H.Q. are listed as co-inventors on Columbia University-owned patents that relate to retromer biomarkers and retromer drug discovery targets.
REFERENCES
- Adams JN, Maass A, Harrison TM, Baker SL, and Jagust WJ (2019). Cortical tau deposition follows patterns of entorhinal functional connectivity in aging. Elife 8, e49132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamed J, Wilcock DM, Diamond DM, Gordon MN, and Morgan D (2006). Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat. Protoc 1, 1671–1679. [DOI] [PubMed] [Google Scholar]
- Andersen OM, Bogh N, Landua AM, Ploen GG, Jensen AM, Monti G, Ulhon BP, Nyengaard JR, Jacobsen K, Jorgensen M, et al. (2021). In vivo evidence that SORL1, encoding the endosomal recycling receptor SORLA, can function as a casual gene in Alzheimer’s Disease. bioRxiv. 10.1101/2021.07.13.452149. [DOI] [Google Scholar]
- Angulo SL, Henzi T, Neymotin SA, Suarez MD, Lytton WW, Schwaller B, and Moreno H (2020). Amyloid pathology-produced unexpected modifications of calcium homeostasis in hippocampal subicular dendrites 16, 251–261. [DOI] [PubMed] [Google Scholar]
- Attar N, and Cullen PJ (2010). The retromer complex. Adv. Enzyme Regul 50,216–236. [DOI] [PubMed] [Google Scholar]
- Avants BB, Tustison NJ, Song G, Cook PA, Klein A, and Gee JC (2011). A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage 54, 2033–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker GR, and Warburton EC (2011). When is the hippocampus involved in recognition memory? J. Neurosci 31, 10721–10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Wen G, Morris JC, and Holtzman DM (2007). Fluctuations of CSF amyloid-beta levels: Implications for a diagnostic and therapeutic biomarker. Neurology 68, 666–669. [DOI] [PubMed] [Google Scholar]
- Bhalla A, Vetanovetz CP, Morel E, Chamoun Z, Di Paolo G, and Small SA (2012). The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol. Dis 47, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, and Cordelieres FP (2006). A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc 224, 213–232. [DOI] [PubMed] [Google Scholar]
- Bota M, Sporns O, and Swanson LW (2015). Architecture of the cerebral cortical association connectome underlying cognition. Proc. Natl. Acad. Sci. U S A 112, E2093–E2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, and Braak E (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (Berl) 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Bugarcic A, Zhe Y, Kerr MC, Griffin J, Collins BM, and Teasdale RD (2011). Vps26A and Vps26B subunits define distinct retromer complexes. Traffic 12, 1759–1773. [DOI] [PubMed] [Google Scholar]
- Burd C, and Cullen PJ (2014). Retromer: A master conductor of endosome sorting. Cold Spring Harb. Perspect. Biol 6, a016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda-Prado E, Popp S, Khan U, Stefanov D, Rodriguez J, Menalled LB, Dow-Edwards D, Small SA, and Moreno H (2012). R6/2 Huntington’s disease mice develop early and progressive abnormal brain metabolism and seizures. J. Neurosci 32, 6456–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou N, Wu J, Bai Bingren J, Qiu A, and Chuang KH (2011). Robust automatic rodent brain extraction using 3-D pulse-coupled neural networks (PCNN). IEEE Trans. Image Process 20, 2554–2564. [DOI] [PubMed] [Google Scholar]
- Choy RW, Park M, Temkin P, Herring BE, Marley A, Nicoll RA, and von Zastrow M (2014). Retromer mediates a discrete route of local membrane delivery to dendrites. Neuron 82, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo C, Fabiani C, Bonadonna C, Origlia N, and Domenici L (2015). BDNF prevents amyloid-dependent impairment of LTP in the entorhinal cortex by attenuating p38 MAPK phosphorylation. Neurobiol. Aging 36, 1303–1309. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2000). Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28, 511–525. [DOI] [PubMed] [Google Scholar]
- Fischl B, and Dale AM (2000). Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc. Natl. Acad. Sci. U S A 97, 11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Sereno MI, and Dale AM (1999). Cortical surface-based analysis. II: Inflation, flattening, and a surface-based coordinate system. Neuroimage 9, 195–207. [DOI] [PubMed] [Google Scholar]
- Fjorback AW, Seaman M, Gustafsen C, Mehmedbasic A, Gokool S, Wu C, Militz D, Schmidt V, Madsen P, Nyengaard JR, et al. (2012). Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J. Neurosci 32, 1467–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Isla T, Price JL, Mckeel DW Jr., Morris JC, Growdon JH, and Hyman BT (1996). Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J. Neurosci 16, 4491–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege H, van der Lee SJ, Hulsman M, Wong TH, van Rooij JG, Weiss M, Louwersheimer E, Wolters FJ, Amin N, Uitterlinden AG, et al. (2017). Characterization of pathogenic SORL1 genetic variants for association with Alzheimer’s disease: A clinical interpretation strategy. Eur. J. Hum. Genet 25, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, Borowski B, Britson PJ, J LW, Ward C, et al. (2008). The Alzheimer’s disease neuroimaging initiative (ADNI): MRI methods. J. Magn. Reson. Imaging 27, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, and Nixon RA (2010). Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. U S A 107, 1630–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, and Goate AM (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, and Ehlers MD (2011). Mechanisms and function of dendritic exocytosis. Neuron 69, 856–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr MC, Bennetts JS, Simpson F, Thomas EC, Flegg C, Gleeson PA, Wicking C, and Teasdale RD (2005). A novel mammalian retromer component, Vps26B. Traffic 6, 991–1001. [DOI] [PubMed] [Google Scholar]
- Khan UA, Liu L, Provenzano FA, Berman DE, Profaci CP, Sloan R, Mayeux R, Duff KE, and Small SA (2014). Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat. Neurosci 17, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Lee JW, Baek DC, Lee SR, Kim MS, Kim SH, Imakawa K, and Chang KT (2008). Identification of novel retromer complexes in the mouse testis. Biochem. Biophys. Res. Commun 375, 16–21. [DOI] [PubMed] [Google Scholar]
- Kim E, Lee Y, Lee HJ, Kim JS, Song BS, Huh JW, Lee SR, Kim SU, Kim SH, Hong Y, et al. (2010). Implication of mouse Vps26b-Vps29-Vps35 retromer complex in sortilin trafficking. Biochem. Biophys. Res. Commun 403, 167–171. [DOI] [PubMed] [Google Scholar]
- Knupp A, Mishra S, Martinez R, Braggin JE, Szabo M, Kinoshita C, Hailey DW, Small SA, Jayadev S, and Young JE (2020). Depletion of the AD risk gene SORL1 selectively Impairs neuronal endosomal traffic independent of amyloidogenic APP processing. Cell Rep. 31, 107719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun O, Leneva N, Bykov YS, Ariotti N, Teasdale RD, Schaffer M, Engel BD, Owen DJ, Briggs JAG, and Collins BM (2018). Structure of the membrane-assembled retromer coat determined by cryo-electron tomography. Nature 561, 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwart D, Gregg A, Scheckel C, Murphy E, Paquet D, Duffield M, Fak J, Olsen O, Darnell R, and Tessier-Lavigne M (2019). A large panel of isogenic APP and PSEN1 mutant human iPSC Neurons reveals shared endosomal abnormalities mediated by APP beta-CTFs, not abeta. Neuron 104, 256–270.e5. [DOI] [PubMed] [Google Scholar]
- Ma L, Alonso A, and Dickson CT (2008). Differential induction of long-term potentiation in the horizontal versus columnar superficial connections to layer II cells of the entorhinal cortex. Neural Plast. 2008, 814815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai J, Majtanik M, and Paxinos G (2015). Atlas of the Human Brain (Academic Press; ). [Google Scholar]
- Maia LF, Kaeser SA, Reichwald J, Hruscha M, Martus P, Staufenbiel M, and Jucker M (2013). Changes in amyloid-beta and Tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci. Transl. Med 5, 194re2. [DOI] [PubMed] [Google Scholar]
- Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, Schneider RT, Petsko GA, Ringe D, and Small SA (2014). Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol 10, 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, Mckeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, and Berg L (1991). The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486. [DOI] [PubMed] [Google Scholar]
- Morabito MV, Berman DE, Schneider RT, Zhang Y, Leibel RL, and Small SA (2014). Hyperleucinemia causes hippocampal retromer deficiency linking diabetes to Alzheimer’s disease. Neurobiol. Dis 65, 188–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno H, Hua F, Brown T, and Small S (2006). Longitudinal mapping of mouse cerebral blood volume with MRI. NMR Biomed. 19, 535–543. [DOI] [PubMed] [Google Scholar]
- Muhammad A, Flores I, Zhang H, Yu R, Staniszewski A, Planel E, Herman M, Ho L, Kreber R, Honig LS, et al. (2008). Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc. Natl. Acad. Sci. U S A 105, 7327–7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen MS, Gustafsen C, Madsen P, Nyengaard JR, Hermey G, Bakke O, Mari M, Schu P, Pohlmann R, Dennes A, and Petersen CM (2007). Sorting by the cytoplasmic domain of the amyloid precursor protein binding receptor SorLA. Mol. Cell. Biol 27, 6842–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA (2005). Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol. Aging 26, 373–382. [DOI] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, and Ehlers MD (2004). Recycling endosomes supply AMPA receptors for LTP. Science 305,1972–1975. [DOI] [PubMed] [Google Scholar]
- Pensalfini A, Kim S, Subbanna S, Bleiwas C, Goulbourne CN, Stavrides PH, Jiang Y, Lee JH, Darji S, Pawlik M, et al. (2020). Endosomal dysfunction induced by directly overactivating Rab5 recapitulates prodromal and neurodegenerative features of Alzheimer’s disease. Cell Rep. 33,108420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi YH, Berman DE, Klein RL, Patel VM, Simoes S, Kannan S, Cox R, Waksal SD, Stevens B, Petsko GA, and Small SA (2019). Retromer repletion with AAV9-VPS35 restores endosomal function in the mouse hippocampus. bioRxiv, 618496. [Google Scholar]
- Qureshi YH, Patel VM, Kannan S, Waksal SD, Petsko GA, and Small SA (2020). Retromer combinatorials for gene-therapy across a spectrum of neurological diseases. bioRxiv. 10.1101/2020.09.03.282327. [DOI] [Google Scholar]
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, et al. (2007). The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet 39, 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas R, van Vlijmen T, Mardones GA, Prabhu Y, Rojas AL, Mohammed S, Heck AJ, Raposo G, van der Sluijs P, and Bonifacino JS (2008). Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J. Cell Biol 183, 513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas R, van Vlijmen T, Mardones GA, Prabhu Y, Rojas AL, Mohammed S, Heck AJR, Raposo G, van der Suijs P, and Bonifacino JS (2008). Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J Cell Biol. 183, 513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabuncu MR, Yeo BTT, van Leemput K, Vercauteren T, and Golland P (2009). Asymmetric Image-Template Registration. Med. Image Comput. Comput. Assist. Interv 12, 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheltens P, de Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE, Cummings J, and van der Flier WM (2021). Alzheimer’s disease. Lancet 397, 1577–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, Inbar BP, Corcoran CM, Lieberman JA, Moore H, and Small SA (2013). Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron 78, 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN (2005). Recycle your receptors with retromer. Trends Cell Biol. 15, 68–75. [DOI] [PubMed] [Google Scholar]
- Seaman MN (2012). The retromer complex - endosomal protein recycling and beyond. J. Cell Sci 125, 4693–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. [DOI] [PubMed] [Google Scholar]
- Simoes S, Neufeld JL, Triana-Baltzer G, Moughadam S, Chen EI, Kothiya M, Qureshi YH, Patel V, Honig LS, Kolb H, and Small SA (2020). Tau and other proteins found in Alzheimer’s disease spinal fluid are linked to retromer-mediated endosomal traffic in mice and humans. Sci. Transl. Med 12,eaba6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Chertkow H, Lerch JP, Evans AC, Dorr AE, and Kabani NJ (2006). Spatial patterns of cortical thinning in mild cognitive impairment and Alzheimer’s disease. Brain 129, 2885–2893. [DOI] [PubMed] [Google Scholar]
- Small SA, and Gandy S (2006). Sorting through the cell biology of Alzheimer’s disease: Intracellular pathways to pathogenesis. Neuron 52, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel JP, and Kim TW (2005). Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann. Neurol 58, 909–919. [DOI] [PubMed] [Google Scholar]
- Small SA, and Petsko GA (2015). Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat. Rev. Neurosci 16, 126–132. [DOI] [PubMed] [Google Scholar]
- Small SA, Schobel SA, Buxton RB, Witter MP, and Barnes CA (2011). A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci 12, 585–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Simoes-Spassov S, Mayeux R, and Petsko GA (2017). Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimer’s disease. Trends Neurosci. 40, 592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, and Swanson LW (2019). A network explanation of Alzheimer’s regional vulnerability. Cold Spring Harb. Symp. Quant Biol 83, 193–200. [DOI] [PubMed] [Google Scholar]
- Temkin P, Morishita W, Goswami D, Arendt K, Chen L, and Malenka R (2017). The retromer supports AMPA receptor trafficking during LTP. Neuron 94, 74–82.e5. [DOI] [PubMed] [Google Scholar]
- Thonberg H, Chiang HH, Lilius L, Forsell C, Lindstrom AK, Johansson C, Bjorkstrom J, Thordardottir S, Sleegers K, van Broeckhoven C, et al. (2017). Identification and description of three families with familial Alzheimer disease that segregate variants in the SORL1 gene. Acta Neuropathol. Commun 5, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Tang F-L, Sun X, Wen L, Mei L, Tang B-S, and Xiong W-C (2015). VPS35-deficiency results in an impaired AMPA receptor trafficking and decreased dendritic spine maturation. Mol. Brain 8, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truschel ST, Simoes S, Setty SR, Harper DC, Tenza D, Thomas PC, Herman KE, Sackett SD, Cowan DC, Theos AC, et al. (2009). ESCRT-I function is required for Tyrp1 transport from early endosomes to the melanosome limiting membrane. Traffic 10, 1318–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardarajan BN, Bruesegem SY, Harbour ME, St George-Hyslop P, Seaman MN, and Farrer LA (2012). Identification of Alzheimer disease-associated variants in genes that regulate retromer function. Neurobiol. Aging 33, 2231.e15–e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, Aizawa H, Ge P, Difiglia M, Mckee AC, Macdonald M, Gusella JF, Landwehrmeyer GB, Bird ED, Richardson EP Jr., et al. (1995). An improved approach to prepare human brains for research. J. Neuropathol. Exp. Neurol 54, 42–56. [DOI] [PubMed] [Google Scholar]
- Wang Z, Edwards JG, Riley N, Provance DW Jr., Karcher R, LI XD, Davison IG, Ikebe M, Mercer JA, Kauer JA, and Ehlers MD (2008). Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell 135, 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DI, Langston RF, Schlesiger MI, Wagner M, Watanabe S, and Ainge JA (2013). Lateral entorhinal cortex is critical for novel object-context recognition. Hippocampus 23, 352–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JE, Fong LK, Frankowski H, Petsko GA, Small SA, and Goldstein LSB (2018). Stabilizing the retromer complex in a human stem cell model of Alzheimer’s disease reduces TAU phosphorylation independently of amyloid precursor protein. Stem Cell Reports 10, 1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Koster HJ, Borchardt T, Worley P, et al. (1999). Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science 284, 1805–1811. [DOI] [PubMed] [Google Scholar]
- Zetterberg H (2017). Review: Tau in biofluids - relation to pathology, imaging and clinical features. Neuropathol. Appl. Neurobiol 43, 194–199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data of graphs plotted in Figures 1, 2, 3, 4, 5, 6, 7, and S1–S8 are available as source data files. The original Western blotting raw data have been deposited on Mendeley at https://doi.org/10.17632/jggdkrpxts.1, and will be publicly available as of the date of publication. Data used in preparation of this article were partially obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu).
Custom Linux bash and MATLAB scripts, developed by our laboratory to conduct mice and human MRI analysis, are available from the Lead Contact upon reasonable request.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.