

Abstract
Aberrant hepatocyte Notch activity is critical to the development of nonalcoholic steatohepatitis (NASH)–induced liver fibrosis, but mechanisms underlying Notch reactivation in developed liver are unclear. Here, we identified that increased expression of the Notch ligand Jagged1 (JAG1) tracked with Notch activation and nonalcoholic fatty liver disease (NAFLD) activity score (NAS) in human liver biopsy specimens and mouse NASH models. The increase in Jag1 was mediated by hepatocyte Toll-like receptor 4 (TLR4)–nuclear factor κB (NF-κB) signaling in pericentral hepatocytes. Hepatocyte-specific Jag1 overexpression exacerbated fibrosis in mice fed a high-fat diet or a NASH-provoking diet rich in palmitate, cholesterol, and sucrose and reversed the protection afforded by hepatocyte-specific TLR4 deletion, whereas hepatocyte-specific Jag1 knockout mice were protected from NASH-induced liver fibrosis. To test therapeutic potential of this biology, we designed a Jag1-directed antisense oligonucleotide (ASO) and a hepatocyte-specific N-acetylgalactosamine (GalNAc)–modified siRNA, both of which reduced NASH diet–induced liver fibrosis in mice. Overall, these data demonstrate that increased hepatocyte Jagged1 is the proximal hit for Notch-induced liver fibrosis in mice and suggest translational potential of Jagged1 inhibitors in patients with NASH.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD), also referred to as metabolic-associated fatty liver disease (MAFLD) (1, 2), is one of the most common metabolic complications of obesity. NAFLD begins as steatosis, defined simply as excess hepatic lipid content. A subset of these patients develops considerable hepatocellular injury, liver inflammation, and fibrosis, which define the transition from simple steatosis to nonalcoholic steatohepatitis (NASH) (3, 4). Multiple theories have been posited to explain NAFLD/NASH pathogenesis, but one of the most commonly observed abnormalities is alteration in the gut-liver axis (5–7). Increased intestinal permeability and impaired gut-vascular barrier lead to microbiota or bacterial products such as lipopolysaccharide (LPS) translocating to the liver through the portal venous circulation. Subsequent binding to Toll-like receptors (TLRs) leads to nuclear factor κ light chain enhancer of activated B cell (NF-κB) activity. Active NF-κB, in turn, induces liver inflammation, which exacerbates lipotoxicity and promotes liver fibrosis due to hepatic stellate cell (HSC) activation (8). Ongoing hepatocellular injury and progressive fibrosis can eventually lead to life-threatening cirrhosis and hepatocellular carcinoma (4, 9). Although simple steatosis is potentially reversible with lifestyle changes, fibrosis regression takes substantial, and rarely achievable, weight loss. There are no currently approved NASH therapeutics, leading to prevalent unmet clinical need.
Notch is an evolutionarily conserved regulatory signaling pathway typically associated with developmental processes. First identified by mutations that disrupt wing development in Drosophila, Notch signaling has subsequently proven critical for cell fate decisions and normal development of most mammalian organs and tissues (10–13). Juxtacrine Notch activation arises from Jagged (Jagged1 and Jagged2) or Delta-like (Dll1, Dll3, and Dll4) ligand expression on a “signal-sending” cell that binds Notch receptors (Notch1, Notch2, Notch3, and Notch4) on a neighboring signal-receiving cell. Ligand binding triggers a series of cleavage events that culminate in the release and nuclear entry of the Notch intracellular domain (NICD), leading to the transcriptional activation of Notch targets. These targets include hairy/enhancer-of-split family basic helix-loop-helix transcription factor 1 (Hes1) and Hes related with YRPW motif (Hey) families, which are transcription factors that further refine the Notch signal (14, 15).
In liver development, Jagged1 expression in portal mesenchyme drives hepatoblast Notch activation, which prompts biliary lineage specification; the absence of this signal leads to paucity of intrahepatic bile ducts (16–20). An important corollary to this finding is that surrounding hepatoblasts, with repressed Notch signaling, differentiate into hepatocytes (16–18). Although quiescent hepatocyte Notch signaling typically persists into adulthood, we observed aberrant hepatocyte Notch reactivation in patients and NASH mouse models (21–23). This increase in hepatocyte Notch activity drives expression of secreted phosphoprotein 1 (Spp1) that encodes osteopontin (OPN), HSC activation, and liver fibrosis in mouse models of NASH (24).
However, these observations left a key unknown as to how hepatocyte Notch signaling is activated—that is, by which Notch ligand, expressed by what type of cell in liver, and mechanism regulating this ligand. Here, we aimed to answer these questions by assessing Notch receptor and ligand expression in human cross-sectional studies and paired biopsy specimens from the Pioglitazone versus Vitamin E versus Placebo for the Treatment of Nondiabetic Patients with Nonalcoholic Steatohepatitis (PIVENS) trial (25). We then tested therapeutic potential of Notch ligand inhibition using gain- and loss-of-function mice as well as ligand-specific inhibitors.
RESULTS
JAG1 expression tracks with liver Notch activity and NASH severity in patients
Notch activity correlates with biochemical and pathologic markers of NASH severity in patients (23). As a first step to understanding the mechanism of increased Notch activity, we conducted an ancillary study to the PIVENS trial (25). In PIVENS, the NASH Clinical Research Network (CRN) randomized adult, nondiabetic patients with NASH to placebo, vitamin E, or pioglitazone treatment arms, with “responders” defined as patients with improved hepatocellular ballooning without an increase in fibrosis, and either a 2+ point decrease in NAFLD activity score (NAS) or to a score of ≤3 points (25, 26). We surveyed Notch signaling components and found that the only Notch receptor or ligand that differed between responders and nonresponders was JAG1 (Fig. 1, A and B), providing a potential explanation for lower Notch activity in PIVENS responders (24).
Fig. 1. JAG1 expression tracks with liver Notch activity and NASH severity in patients.

(A) NOTCH receptor and (B) ligand expression in end-of-treatment biopsies in nonresponders (n = 49) and responders (n = 69), regardless of treatment group, from the PIVENS trial. In 157 consecutive patients undergoing liver biopsy for suspected NASH or severe obesity, (C) correlation between liver expression of JAG1 and Notch target HES1, and (D and E) expression of JAG1 and Notch targets in patients with increased NAS and fibrosis scores. (F) Representative image of Jagged1 (red) and HNF4α (green) staining. In a subset of patients with available paired baseline and 96-week end-of-treatment liver biopsy specimens from PIVENS patients (n = 10 to 11 per group), (G) DLL4 and JAG1 expression, and (H) association between change in JAG1 (ΔJAG1) and ΔNAS. *P < 0.05 as compared to the indicated controls by two-tailed t tests (two groups) or one-way ANOVA followed by post hoc t tests (multiple groups). AU, arbitrary unit. All data are shown as means ± SEM.
To confirm these results in a different population, we performed a cross-sectional study in 157 consecutive patients that had standard-of-care percutaneous liver biopsies for suspected NASH. In this cohort, liver Notch activity positively correlated with NAS and fibrosis stage even when adjusted for demographic or metabolic confounders (24). Consistent with the above results, we found that liver JAG1 positively correlated with liver HES1 (Fig. 1C). Liver JAG1, HES1, and HEYL expression were also higher in patients with increased NAS and fibrosis stage (Fig. 1, D and E). Jagged1 staining showed a similar correlation with NAS (Fig. 1F), with an apparent preference for pericentral liver zones (fig. S1, A and B).
One limitation of the cross-sectional approach, however, is the ability to detect gene expression and NASH pathology at only one moment in time. To overcome this, we analyzed a subset of PIVENS subjects that had paired baseline and end-of-treatment at 96 weeks complementary DNA (cDNA) available for analysis. Using these cDNA pairs, we found that Notch activity is reduced from baseline in responders, but unchanged in nonresponders (24), which we found was associated with a marked reduction in JAG1 in responders (but not nonresponders) without change in other Notch ligands (Fig. 1G). Change in JAG1 from baseline to end-of-treatment visits (ΔJAG1) correlated with change in HES1 and HEYL expression (fig. S1, C and D) and explained more than 37% of the variance in NAS (Fig. 1H). In summary, these data show that liver JAG1 tracks with liver Notch activity and NASH severity in patients.
Hepatocyte Jag1 expression is increased in livers of mice fed a NASH-provoking diet
These patient data suggested that Jagged1 may be limiting for liver Notch reactivation in NASH pathogenesis. As a first step to establish causality, we used a dietary mouse model (composed of high palmitate, sucrose, and cholesterol, coupled with fructose-containing drinking water) that mimics human NASH by inducing liver steatosis, inflammation, and progressive fibrosis in the setting of weight gain and insulin resistance (27). We fed wild-type (WT) mice this NASH-provoking diet for 8 weeks—before the onset of liver fibrosis (27) and when we noted only modest increase in liver Notch activity (fig. S2A)—and found that of Notch ligands and receptors, only Jag1 expression was increased compared to normal chow diet–fed controls (Fig. 2, A and B). Jagged1 protein was similarly increased (Fig. 2C). With prolonged NASH diet feeding (16 weeks of diet), other Notch pathway components also showed increased expression (fig. S2, B to D), but absolute expression of all other Notch ligands combined remained lower than that of Jag1.
Fig. 2. Pericentral hepatocyte Jag1 expression is increased in livers of mice fed a NASH-provoking diet.
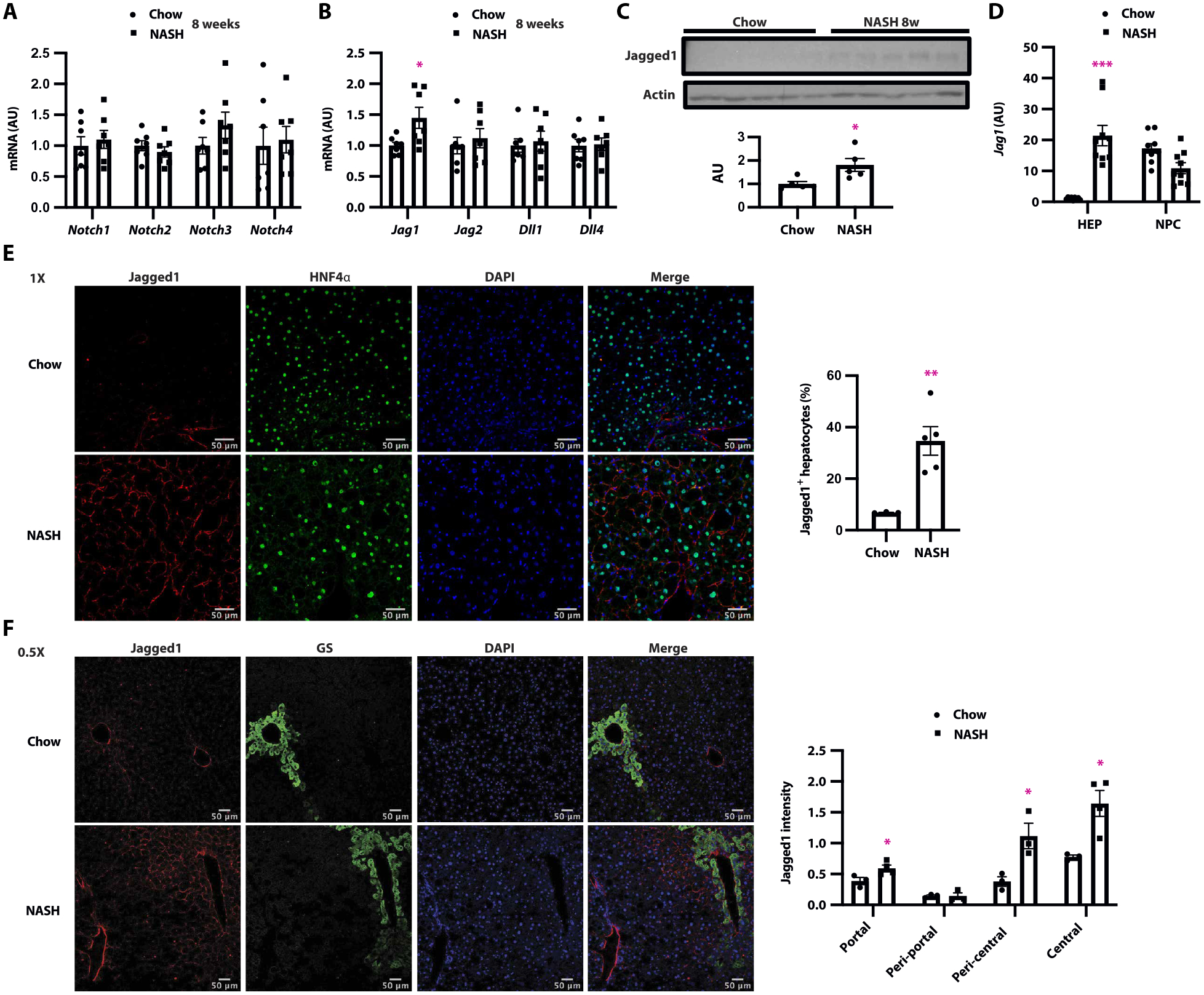
(A) Expression of Notch receptors, (B) ligands, and (C) liver Jagged1 protein in livers from C57BL/6J WT mice fed normal chow diet or NASH diet for 8 weeks (n = 5 to 7 per group). (D) Jag1 expression in hepatocytes and NPC in livers of mice fed either chow or NASH diet for 16 weeks (n = 9 per group). (E) Representative image of Jagged1 (red) and HNF4α (green) staining, and quantitation of HNF4α/Jagged1 double-positive cells, (F) representative image of Jagged1 (red) and GS (green) staining, and quantitation of Jagged1+ cells among GS+ and GS− cells in WT mice fed normal chow or NASH diet for 16 weeks (n = 3 to 5 per group). *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to chow-fed mice by two-tailed t tests (two groups) or one-way ANOVA followed by post hoc t tests (multiple groups). All data are shown as means ± SEM.
Liver is composed of both hepatocytes and nonparenchymal cells (NPCs). To identify the cellular source of increased liver Jag1, we isolated hepatocytes and NPCs from chow- and NASH diet–fed WT mice. Basal liver Jag1 expression was predominantly seen in NPCs, with low expression in hepatocytes (Fig. 2D). NPC Jag1 expression did not change with NASH diet feeding, but hepatocyte Jag1 increased roughly 20-fold (Fig. 2D). We saw a commensurate increase in Jagged1+ hepatocytes, from ~5% in chow-fed to ~35% of hepatocytes in NASH diet–fed WT mice (Fig. 2E). Consistent with human NASH, most Jagged1+ hepatocytes were found adjacent to the central vein (CV) (Fig. 2F), marked by glutamine synthetase (GS) staining (28, 29). These data suggest that hepatocyte Jag1 expression progressively increases in NASH diet–fed mice, which may explain the greater Notch activity seen in this pathology.
Hepatocyte-specific Jag1 knockout mice are protected from NASH-induced liver fibrosis
To test whether hepatocyte Jagged1 is necessary for NASH diet–induced liver fibrosis, we transduced 8- to 10-week-old Jag1flox/flox mice with AAV8-Tbg-Cre (or AAV8-Tbg-GFP) by tail vein injection to generate hepatocyte-specific Jag1 knockout (L-Jag1KO) and Cre− control mice (Fig. 3A). NASH diet–fed L-Jag1KO showed similar body (fig. S3A), liver, and white adipose tissue (WAT) weight as control littermates (fig. S3B). As expected, L-Jag1KO mice showed reduced liver Jag1 mRNA and protein compared to Cre− controls (Fig. 3, B and C), specifically in hepatocytes (Fig. 3, D to F). Reduced hepatocyte Jagged1 attenuated NASH-induced Notch activity in hepatocytes but not NPCs (Fig. 3, E and F, and fig. S3, C and D). L-Jag1KO mice showed unchanged serum alanine aminotransferase (ALT) (fig. S3E), liver and serum triglyceride (TG) (fig. S3, F and G), markers of liver inflammation (fig. S3H), CD45 staining (fig. S3I), and cholangiocyte staining (fig. S3J). However, phenocopying hepatocyte-specific Notch loss-of-function mice (24), L-Jag1KO mice showed decreased markers of HSC activity, including expression of collagen type 1 α1 (Col1a1) and TIMP metallopeptidase inhibitor 1 (Timp1) (Fig. 3G), and lower liver Sirius red staining than Cre− control mice (Fig. 3H). We have previously shown that Notch inhibition blocks hepatocyte secretion of OPN, which, in turn, reduces HSC activity (24). Consistent with these data, L-Jag1KO mice showed reduced hepatocyte Spp1 expression (fig. S3K). These data suggest that inter-hepatocyte Jagged1-Notch signaling regulates Notch activity and NASH diet–induced liver fibrogenesis.
Fig. 3. Hepatocyte-specific Jag1 KO mice are protected from NASH-induced liver fibrosis.
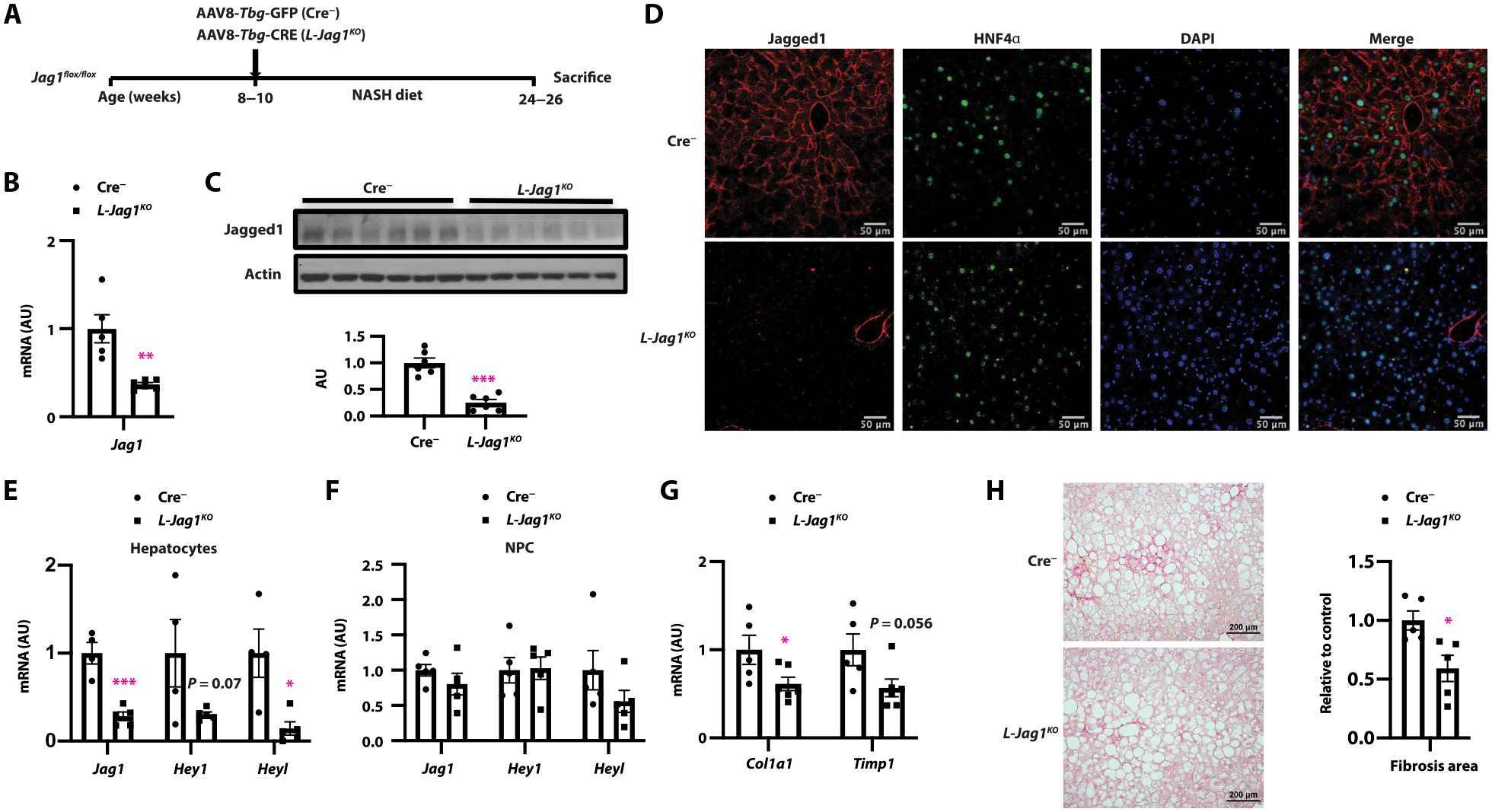
(A) Adult Jag1flox/flox mice were transduced with either AAV8-Tbg-GFP (Cre−) or AAV8-Tbg-CRE (L-Jag1KO) and then fed NASH diet for 16 weeks before sacrifice. (B) Liver Jag1 mRNA and (C) protein expression, (D) representative image of Jagged1 (red) and HNF4α (green) staining, (E and F) Jag1 and Notch target expression in hepatocytes and NPC, and (G) gene expression markers of HSC activation. (H) Sirius red staining. n = 5 to 7 per group. *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to Cre− mice by two-tailed t tests (two groups). All data are shown as means ± SEM.
Increased hepatocyte Jagged1 exacerbates NASH diet–induced liver fibrosis
We next hypothesized that the increase in hepatocyte Jagged1 was sufficient to induce liver Notch activity and downstream effects on liver pathology. We crossed mice harboring a tetracycline-inducible Jagged1 allele (Jag1teto) with rtTAflox/flox mice, transduced with AAV8-Tbg-Cre, and then activated expression with ad libitum access to doxycycline-containing drinking water. We examined phenotypes of hepatocyte-specific Jagged1 gain-of-function (L-Jag1TG) mice fed normal chow. L-Jag1TG mice developed normally, with comparable body weight (BW), liver weight, serum ALT, and mild increase in markers of liver inflammation as control mice (fig. S4, A to D). However, as hypothesized, L-Jag1TG mice showed increased Notch activity and commensurately higher liver Spp1 and Col1a1 expression (fig. S4, E to G). Collagen accumulation in liver was unchanged (fig. S4H), suggesting that other factors (such as Notch receptor expression in NASH) may also be necessary for liver fibrosis (23, 24). To test that possibility, we repeated the experiment but in the context of 16 weeks of NASH diet feeding (Fig. 4A). Body, liver, and WAT weight were unchanged (fig. S5, A and B), but increased hepatocyte Jagged1 (Fig. 4, B and C) led to a modest further increase in Notch target expression in NASH diet–fed L-Jag1TG mice (Fig. 4D). Despite unchanged markers of liver injury, lipid content, and inflammation compared to control mice (fig. S5, C to G), liver Spp1 (fig. S5H) and Col1a1 expression (Fig. 4E) were increased in L-Jag1TG mice, leading to increased liver fibrosis (Fig. 4F).
Fig. 4. Forced hepatocyte Jag1 expression exacerbates diet-induced liver fibrosis.

(A) Eight- to 10-week-old Jag1teto/−/rtTAflox/− or rtTAflox/− mice were transduced with AAV8-Tbg-CRE to generate Control (CON) or hepatocyte-specific Jag1 transgenic (L-Jag1TG) mice and then fed NASH diet with ad libitum access to doxycycline-containing drinking water for 16 weeks (n = 5 to 11 per group). (B) Representative image of Jagged1 (red) and HNF4α (green) staining, (C) Jagged1 protein, (D) expression of Notch targets, and (E) markers of HSC activation. (F) Liver Sirius red staining (n = 4 per group). (G) Control and L-Jag1TG mice were fed HFD with ad libitum access to doxycycline-containing drinking water for 16 weeks (n = 7 to 8 per group). (H) Gene expression of markers of HSC activation. (I) Sirius red staining (n = 6 per group). *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to control mice by two-tailed t tests or nonparametric Mann-Whitney U test (two groups). All data are shown as means ± SEM.
As compared to NASH diet feeding, high-fat diet (HFD) feeding induces marked liver steatosis, but not fibrosis. Notch receptor expression is also increased with HFD feeding (23). Consistent with this, transgenic expression of hepatocyte Jagged1 (Fig. 4G) showed similar change of BW (fig. S5I), liver and WAT weight (fig. S5J), and liver TG (fig. S5K). Although serum TG (fig. S5L) was slightly higher in L-Jag1TG mice, markers of liver injury and inflammation remained unchanged (fig. S5, M and N). We also found increased liver Spp1 (fig. S5O), Col1a1 expression (Fig. 4H), and liver collagen content (Fig. 4I) in L-Jag1TG mice compared to HFD-fed controls.
To confirm whether these effects were cell autonomous, we isolated hepatocytes from Jag1teto/ rtTAflox/flox mice and induced a nearly two-fold increase in Jag1 expression by adenoviral transduction of Cre recombinase (fig. S5P). Increased hepatocyte Jagged1 was sufficient to increase Spp1 expression (fig. S5P). Conditioned medium from these hepatocytes showed increased OPN, which was sufficient to activate isolated and plated HSCs (fig. S5, Q and R). In summary, these data indicate that increased hepatocyte Jagged1 expression is sufficient to drive HSC activity, which, in turn, can promote liver fibrosis in obese mice.
TLR4–NF-κB signaling drives hepatocyte Jag1 expression
We next turned our attention to the mechanism of increased hepatocyte Jag1 expression in NASH. We analyzed the Jag1 promoter for transcription factor binding sites (30, 31) and identified an NF-κB binding site [5′-GGGAGTCCC-3′ from −2351 to −2343 base pairs (bp)] that is conserved across species (32–34). NF-κB activity is associated with liver injury caused by gut-derived pathogens and bacterial products such as LPS and LPS-induced cytokines such as tumor necrosis factor–α (TNFα) through TLR4 (8). Consistent with human data (35), NASH diet−fed mice showed higher circulating LPS than chow-fed controls (Fig. 5A). NASH diet–fed mice also showed increased expression of both Tlr4 and NF-κB p65 subunit (Fig. 5, B and C). We found strong TLR4 colocalization with Jagged1 in NASH diet–fed mice (fig. S6A).
Fig. 5. TLR4–NF-κB signaling drives hepatocyte Jag1 expression.

(A) Serum LPS, (B) liver Tlr4 mRNA, and (C) liver p65 protein expression in WT mice fed chow or NASH diet for 16 weeks (n = 3 to 6 per group). (D) Jag1 promoter luciferase activity in WT primary hepatocytes cotransfected with p65 (n = 4 per group), and (E) ChIP assay for NF-κB occupancy at the Jag1 promoter in livers from mice fed normal chow or NASH diet for 16 weeks (n = 4 per group). (F) Eight- to 10-week-old Tlr4flox/flox mice were transduced with AAV8-Tbg-GFP (Cre−) or AAV8-Tbg-CRE (L-Tlr4) and then fed NASH diet for 16 weeks (n = 12 to 13 per group). (G) Jagged1 protein expression, (H) expression of Notch targets, and (I) markers of HSC activation. (J) Liver Sirius red staining (n = 4 to 9 per group). (K) Control (pLive-con) or Jag1 expression (pLive-Jagged1) vectors were hydrodynamically administered to adult Tlr4flox/flox mice. Two weeks later, all mice were transduced with AAV8-Tbg-CRE and then fed NASH diet for 16 weeks (n = 8 per group). (L) Expression of Jag1 mRNA and (M) markers of HSC activity. (N) Sirius red staining (n = 5 per group). *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to the indicated controls by two-tailed t tests (two groups). All data are presented as means ± SEM.
These correlations prompted us to test whether Jag1 is a direct downstream target of NF-κB. We generated a luciferase construct containing the proximal 2.8 kb of the mouse Jag1 promoter sequence containing WT or a disrupted NF-κB binding site. In mouse primary hepatocytes, exogenous NF-κB transfection or treatment with LPS and TNFα activated Jag1-luciferase, dependent on an intact NF-κB binding site (Fig. 5D and fig. S6, B and C). LPS also increased endogenous hepatocyte Jag1 expression, which was partially blocked by cotreatment with the NF-κB inhibitor caffeic acid phenethyl ester (CAPE) or by Tlr4 knockdown (fig. S6, D to H). We also observed higher liver Jag1 in vivo after LPS (fig. S6, I and J) and increased p65 occupancy at the Jag1 promoter in NASH diet–fed mice (Fig. 5E).
We next tested the hypothesis that hepatocyte Jag1 expression is mediated by TLR4-induced NF-κB activity in vivo (36) by generating hepatocyte-specific Tlr4 KO (L-Tlr4) mice (Fig. 5F). NASH diet–fed L-Tlr4 mice showed lower liver Jag1 and Notch activity than Cre− controls (Fig. 5, G and H), leading to attenuated HSC activity and liver collagen content (Fig. 5, I and J). To test whether reduced Jagged1 was responsible for decreased fibrosis in L-Tlr4 mice, we “rescued” Jag1 expression in these mice by hydrodynamic injection of Jagged1-endcoding plasmid (Fig. 5K). As hypothesized, a forced but modest increase in hepatocyte Jagged1 reversed HSC activation and collagen content in L-Tlr4 mice (Fig. 5, L to N). When combined with earlier observations, these data suggest that inflammatory stimuli activate hepatocyte TLR4–NF-κB signaling, leading to hepatocyte Jag1 transcription, which, in turn, is necessary for hepatocyte Notch activity and NASH-induced liver fibrosis.
Liver-directed pharmacologic Jagged1 inhibitors ameliorate NASH-induced fibrosis
Pharmacologic agents are sorely needed for NASH. To determine therapeutic tractability of hepatocyte Jagged1, we designed Jag1-directed antisense oligonucleotides (ASO) and applied this re-agent to NASH diet–fed mice (Fig. 6A). Jag1 ASO did not change body or liver weight as compared to control ASO, although WAT weight was slightly lower (fig. S7, A and B). As expected, liver Jag1 mRNA and protein expression were reduced in Jag1 ASO-treated mice (Fig. 6, B and C), with a reduction of hepatocyte Jagged1 staining in liver (Fig. 6D). We also observed lower liver Notch activity (Fig. 6B), lower Spp1 and Timp1 expression, and liver collagen content (Fig. 6, E and F, and fig. S7C), without a change in serum ALT (fig. S7D).
Fig. 6. Jag1 inhibitors protect from NASH-induced liver fibrosis.
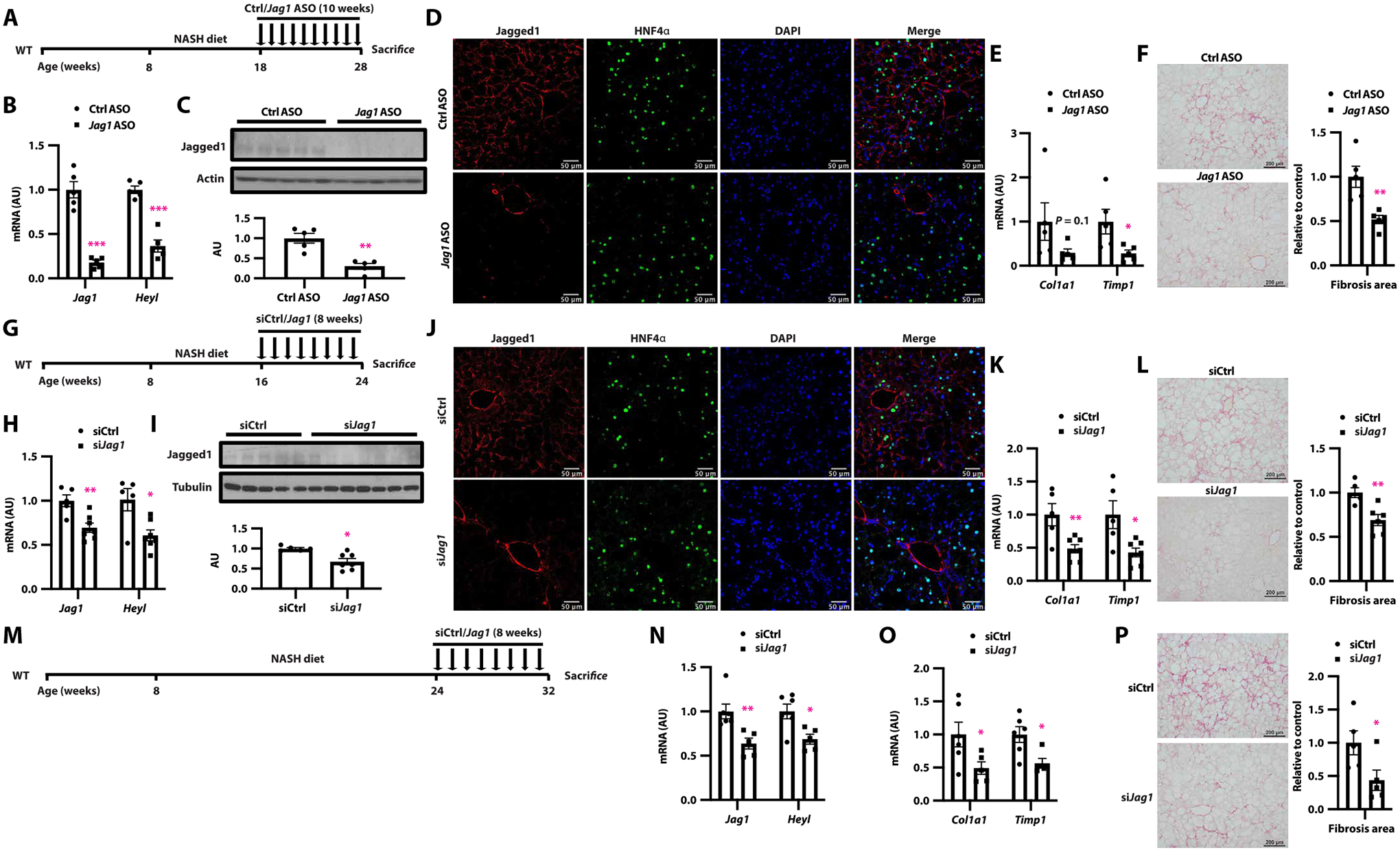
(A) NASH diet–fed WT mice were administered weekly intraperitoneal injections of control (Ctrl ASO) or Jag1-directed (Jag1 ASO) antisense oligonucleotides (ASOs), (B) Jag1 and Notch target gene expression, (C) Jagged1 protein expression, (D) representative image of Jagged1 (red) and HNF4α (green) staining, (E) expression of markers of liver HSC activation, and (F) Sirius red staining (n = 5 per group). (G) NASH diet–fed WT mice were administered weekly subcutaneous injections of control (siCtrl) or Jag1-directed (siJag1) GalNAc-modified siRNA, (H) Jag1 and Notch target gene expression, (I) liver Jagged1 protein, (J) representative image of Jagged1 (red) and HNF4α (green) staining, (K) expression of markers of liver HSC activation, and (L) Sirius red staining (n = 5 to 7 per group). (M) NASH diet–fed WT mice were allowed to develop liver fibrosis and then administered weekly subcutaneous injections of siCtrl or siJag1. (N) Liver Jag1 and Notch target gene expression, (O) expression of markers of liver HSC activation, and (P) Sirius red staining (n = 5 to 6 per group). *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to the indicated controls by two-tailed t tests (two groups). Data are presented as means ± SEM.
ASOs may target multiple liver cell types, as well as potentially other tissues, such as muscle and WAT (fig. S7, E and F). To overcome this potential limitation, we developed a hepatocyte-specific Jagged1 inhibitor, N-acetylgalactosamine (GalNAc)–modified siJag1 (hence, siJag1). As above, we treated WT mice midway through the 16-week NASH diet–feeding paradigm with siJag1 or siCtrl (Fig. 6G). siJag1 treatment decreased liver Jag1 expression, leading to reduced Jagged1 protein staining in hepatocytes (Fig. 6, H to J), without effects on body or liver/WAT weight (fig. S7, G and H), serum ALT (fig. S7I), or Jag1 expression in other tissues (fig. S7, J and K). Nevertheless, siJag1 reduced liver Spp1 expression, markers of HSC activation, and liver collagen content (Fig. 6, K and L, and fig. S7L). We next pushed this experimental paradigm to see whether siJag1 could reverse established NASH diet–induced fibrosis (Fig. 6M). Even with delayed “treatment,” siJag1 reduced liver Jag1, Notch targets, and fibrogenic gene expression (Fig. 6, N and O) as well as liver fibrosis (Fig. 6P). These data provide proof-of-concept evidence for hepatocyte Jagged1 as a therapeutic target for NASH.
DISCUSSION
Hepatocyte Notch activation is associated with liver fibrosis (24, 37), but before this work, how hepatocyte Notch received signals and from where the signals were sent were unknown. This was an important knowledge gap not only in understanding the pathophysiology of NASH/fibrosis but also in design of therapeutic strategies to reduce the excess hepatocyte Notch signaling in the obese liver. Here, we identified an inter-hepatocyte Jagged1-Notch circuit necessary and sufficient for NASH-induced liver fibrosis. This finding is note-worthy because <5% of hepatocytes are Jagged1+ in healthy liver, but as Jagged1+ hepatocyte number increases in NASH, Notch activity follows. Although this may seem intuitive, these data represent an unusual description of Notch ligand-receptor signaling between the same cell type in the developed organism. For example, as bipotential hepatoblasts are faced with a cell fate decision in embryogenesis, cells that receive appropriate ligand stimulation—also from Jagged1 but derived from the portal mesenchyme—become “Notch-active” and proceed down a cholangiocyte lineage (16, 18). These cells simultaneously repress their own ligand expression, leading to low Notch activity in neighboring hepatoblasts, which then directs differentiation to hepatocytes. This sets the quiescent Notch tone for normal hepatocytes. So, what goes awry in obesity? Our loss-of-function mouse studies suggest that the proximal hit is aberrant hepatocyte Jag1 expression, which precedes and is necessary for liver Notch activity in NASH. Because JAG1 correlates with liver Notch activity in longitudinal biopsies from patients, we posit that this signaling circuit is inappropriately activated in human NASH as well. Pathologic consequence also appears conserved, as increased Jag1 tracks with increased NAS and liver fibrosis in longitudinal data from the PIVENS trial as well as in cross-sectional studies (38, 39).
The finding of our work, of an inter-hepatocyte Notch signaling circuit, does not abrogate prior data showing abundant Jagged1 in liver NPC populations. Basal Jag1 expression in lean mice is primarily in NPC. Although we find increased hepatocyte, but not overall NPC Jag1 expression with NASH diet feeding, that does not exclude the possibility that specific NPC populations may also show increased Jag1. This may synergistically contribute to hepatocyte Notch activity. We also find that increased hepatocyte Jag1 expression was sufficient to induce (or exacerbate) liver fibrosis but only in obese mice. This suggests an additional hit, such as a simultaneous increase in expression of Notch receptors or other ligands, or a necessity for the presence of liver injury or inflammation (40–44). Along these lines, we used Hes/Hey expression as indicators of Notch activity, but each individual target is neither sensitive nor specific for Notch. For instance, Hes/Hey can be regulated by other transcriptional factors, such as cyclic adenosine monophosphate (cAMP)–responsive transcription factor (CREB) (45) and hypoxia-inducible factor 1 (HIF1) (46). By corollary, even in the presence of Notch signaling, Hes/Hey genes may not be simultaneously changed because of individual sensitivity to NICD signal intensity and duration, variable mRNA and protein half-lives, or oscillation of target gene expression (47–51).
The mechanistic underpinnings of inappropriate Jag1 expression are potentially instructive to NASH pathophysiology. We find that hepatocyte Jag1 in mice was dependent on intact hepatocyte TLR4–NF-κB signaling (fig. S8), presumably triggered by intestinal dysbiosis and a dysfunctional intestinal barrier, permitting increased access of gut microbiota–derived products such as LPS to vulnerable hepatocytes (6). However, it is likely that other stimuli exacerbate increased hepatocyte Jag1, including fatty acid species also shown to activate TLR4 (52, 53). Another clue to this may be the preferential Jagged1 staining in pericentral hepatocytes in mice and patients with NASH, consistent with higher hepatocyte NF-κB activity in pericentral hepatocytes after injury (54). Oxygen, nutrient, and hormone gradient determine zonal distribution, and hypoxia is associated with the progression of NASH (55, 56) and NF-κB activity (57–59). However, whether Jag1 is regulated independently by hypoxia or other stimuli in NASH progression requires formal testing.
Jagged1 zonal distribution in NASH may also shed light on observed zonation preferences in fibrosis pathogenesis. Pericentral steatosis is the most common pattern in adults with NASH, with the severity of steatosis closely related to pericentral fibrosis stage (60, 61). Consistently, CV-associated HSCs were shown to preferentially contribute to liver fibrosis in rodent models (62). Whether this is due to local Jagged1-Notch signaling requires further study, but Notch inhibition has been shown to reduce HSC activity in vitro (63), and an NPC Notch activation state has been identified from single-cell RNA-sequencing studies in human cirrhosis (64). These data may suggest that the same proximal signal—increased hepatocyte Jagged1—may synergistically increase liver fibrosis both indirectly (via hepatocyte Notch-derived Spp1) and directly (HSC Notch activation). Although we found evidence of the former in this study, whether this leads to direct HSC Notch activation as well is an area of active research, as is the repercussions of successful Jagged1-Notch signaling to the signal-sending cell. After receptor binding, Jagged1 may be degraded or recycled back to the membrane to amplify hepatocyte and NPC Notch receptor activation (65, 66). Determinants of this decision tree are unclear, but Jagged1 can be cleaved by the same γ-secretase complex that mediates receptor cleavage, releasing the Jagged1 intracellular domain (JICD), which may act as a transcriptional factor, or modulate cross-talk with other signaling pathways during development (67, 68). Whether these signals exist in developed liver is not known.
NAFLD is the most common chronic liver disease and is now the second leading reason for liver transplantation. We developed two different therapeutic approaches to block Jagged1-induced Notch signaling; both had strong efficacy to reduce liver fibrosis without substantial impact on liver injury, consistent with phenotypes from hepatocyte Notch loss-of-function mice (24). Potential for Jag1 inhibitor–based toxicity is an important consideration because loss of Jagged1 during development is a cause of Alagille syndrome, which, in liver, manifests as bile duct paucity and cholestatic liver disease. However, we observe no biliary phenotypes with genetic or pharmacologic Jagged1 manipulation in adult mice. In retrospect, these data were entirely predictable because the defect in Alagille syndrome is due to loss of Jagged1 in the developing portal mesenchyme (16).
There are several limitations of this study. As discussed above, we focused on inter-hepatocyte Jagged1-Notch signaling to determine liver fibrosis, but this does not exclude the possibility of Jagged1 from a specific NPC population that may have synergistic effects. In addition, although we did not see differences in overall liver immune cell number, it is possible that increased hepatocyte Notch activity may drive expansion of specific immune cell populations. Last, as the intervention studies were conducted in rodent models, translation to patients with NASH and fibrosis requires formal testing. However, when combined with results from our analysis of patient-derived liver biopsies, these data suggest that hepatocyte-targeted Jagged1 inhibitors warrant exploration for treatment of NASH-induced fibrosis.
MATERIALS AND METHODS
Study design
The objectives of this study were to determine which Notch ligand, expressed by which liver cell type, tracks with aberrant Notch activity in NASH; to test the role of this Notch ligand in a mouse model of NASH-induced fibrosis using genetic gain- (L-Jag1TG) and loss-of-function (L-Jag1KO) mice; to determine molecular regulation of this ligand in liver; and to test whether pharmacologic inhibitors (Jag1 ASO and siJag1) could reduce NASH-induced fibrosis in mice.
All data presented here have been replicated in independent cohorts of mice, or in at least three biological replicates for in vitro experiments, with staining data quantitatively analyzed as indicated in the figure. Statistical analysis was performed by two-tailed t tests (two groups) or one-way analysis of variance (ANOVA) followed by post hoc t tests (multiple groups). We based sample size predictions on data from hepatocyte Notch gain- and loss-of-function mice and, with a power of 0.8 and P < 0.05, calculated a sample size necessary of between five and eight mice per group. We used pre-experiment weight matching to eliminate this potential confounding variable; thereafter, animals were randomly assigned to control and experimental groups, and all data were analyzed in a blinded fashion without exclusion of any animal. Human samples were obtained from a completed, randomized control trial (PIVENS) or cross-sectional analysis in patients with suspected NASH, for which randomization was not applicable (25, 26). Group assignments and other demographic information were blinded to the investigators until all data were obtained. Data collection methods are detailed in the figures, figure legends, and Materials and Methods.
Liver biopsies
PIVENS trial
We analyzed gene expression of liver biopsy samples from the PIVENS trial, a NASH treatment trial (NCT00063622) sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases and conducted by the NASH CRN. Gene expression was compared to change in NAS.
Cross-sectional analysis in patients with suspected NASH
We analyzed liver gene expression in individuals who underwent liver biopsy as standard of care for suspected NASH due to the presence of persistent elevations in liver enzymes or due to severe obesity. The protocol was approved by the Ethical Committee of the Fondazione IRCCS of Milan, and each patient signed a written informed consent. For statistical analysis, comparisons were made by fitting data to generalized linear models, unadjusted (univariate analyses), or considering as independent variables: age, gender, body mass index (BMI), presence of impaired fasting glucose, or type 2 diabetes (T2D). Hepatic HES1 and JAG1 mRNA expression were normalized to ACTB expression and natural log–transformed before analyses to ensure a normal distribution.
Animals
Jag1flox/flox, Jag1teto, rtTAflox/flox, and Tlr4flox/flox mice have been described (12, 69–71). To generate hepatocyte-specific KO mice, we transduced 8- to 10-week-old mice carrying floxed alleles with 1.5 × 1011 genome copies of AAV8-Tbg-CRE (AV-8-PV1091, Addgene) or AAV8-Tbg-GFP (AV-8-PV0146) as a control by tail vein injection. We allowed AAV8-Tbg-CRE–transduced Jag1teto/−/rtTA flox/− and rtTAflox/− control mice ad libitum access to doxycycline (1 g/liter; Sigma-Aldrich) in drinking water to induce hepatocyte-specific Jag1 overexpression. Mice were weaned to standard chow (PicoLab Rodent Diet 20) for all experiments and then started on 60% HFD (Research Diets) or a NASH-provoking diet enriched in saturated fat, sucrose, and cholesterol (Teklad) (27) with fructose-containing drinking water (23.1 g of fructose and 18.9 g of glucose dissolved in 1 liter of water and then filter-sterilized) at 8 weeks of age, unless otherwise noted. The animals were housed three to five per cage in standard cages at 22°C in a 12-hour light/12-hour dark cycle and monitored for overall well-being and signs of distress with BW measured weekly. Upon completion of each study, the mice were weighed and euthanized, and blood was collected by cardiac puncture. We removed and weighed perigonadal adipose tissues and livers, which were split for fixation and RNA and protein isolation and frozen for later analyses. All animal experiments were conducted in accordance per guidelines of the Columbia University Institutional Animal Care and Utilization Committee.
Plasmids
The p65 expression construct (72) was obtained from Addgene (no. 23255). We generated the Jag1 promoter-luciferase vector by inserting 2.8 kb upstream of the coding sequence, including the NF-κB binding site (mouse: −2351 GGGAGTCCC −2343) (33) into the pGL4.10 plasmid backbone. We inserted the mouse Jag1 coding region [amplified by polymerase chain reaction (PCR) from a liver cDNA library] into a pLive vector (MIR5420, Mirus) containing a mouse minimal albumin promoter using the In-Fusion HD Cloning Plus Kit (638909, Clontech). Plasmids were dissolved in TransIT-EE Delivery Solution (MIR5340, Mirus), and then 20 μg per mouse was hydrodynamically injected into the tail vein following the manufacturer’s instructions.
ASO and siRNA
Control (5′-GGCCAATACGCCGTCA-3′) and Jag1 ASO (5′-GCGA-TACTGAGATGGC-3′) were synthesized by Ionis Pharmaceuticals, diluted in saline before injection, and administered by intraperitoneal injection to male C57BL/6J mice at a dose of 25 mg/kg BW weekly for 10 weeks. GalNAc-modified Jag1 [sense: 5′-cscsaGfgGfcUfuG-fuAfgUfuUfcUfuUfaAfs(NHC6)(GalNAc3)-3′, antisense: 5′-UfsUf-saAfaGfaAfaCfuAfcAfaGfcCfcsdTsdT-3′] and control small interfering RNA (siRNA) [sense: 5′-usasuAfuCfgUfaCfgUfaCfcGfuCfgUfaUfs(N-HC6)(GalNAc3)-3′, antisense: 5′-AfsUfsaCfgAfcGfgUfaCfgUfaCf-gAfusdTsdT-3′] were provided by Sanofi and administered by subcutaneous injection to male C57BL/6J mice at a dose of 15 mg/kg BW weekly for 8 weeks. dA, dG, dT, dC: DNA nucleotides; Af, Gf, Uf, Cf: 2′-fluoro nucleotides; a, g, u, c: 2′-O-methyl nucleotides; s: phosphorothioate.
Cell isolation
Primary hepatocytes, NPCs, and HSCs were isolated as previously described (24, 73). Briefly, we anesthetized mice and digested livers by perfusion of EGTA buffer and collagenase buffer through the inferior vena cava, purified hepatocytes with Percoll, and concentrated the remaining NPC or HSC by Nycodenz density centrifugation.
Immunofluorescence and Sirius red staining
We fixed mouse livers overnight in 4% paraformaldehyde and then dehydrated in 30% sucrose for another 24 hours. Liver tissue was then embedded in Tissue-Tek O.C.T. compound (VWR, 25608–930) before frozen sectioning. For immunostaining of human liver biopsies, paraffinized slides were deparaffinized and rehydrated as described (23). Before immunofluorescence staining, slides were air-dried at room temperature (RT) (for the frozen section) or deparaffinized (for the paraffin section), and then antigens were retrieved using HistoVT One (Nacalai, 06380–05). Sections were blocked in 3% bovine serum albumin (BSA) and 10% donkey serum in phosphate-buffered saline–Tween 20 (PBST) for 1 hour at RT and then incubated with primary antibody {hJagged1 (AF1277, R&D Systems, 1:50), mJagged1 (AF599, R&D Systems, 1:100), HNF4α [3113s, Cell Signaling Technology (CST), 1:500], GS (ab49873, Abcam, 1:500), CD45 (BD Bio-sciences, 550539, 1:100), CK19 (Abcam, ab52625, 1:100), and TLR4 (Thermo Fisher Scientific, 48–2300, 1:500)} overnight at 4°C. After secondary antibody [donkey anti-rabbit Alexa Fluor 555 conjugate [(31572, Thermo Fisher Scientific, 1:500), donkey anti-goat Alexa Fluor 647 (A32849, Thermo Fisher Scientific, 1:500), and donkey anti-rat Alex Fluor 488 (A21208, Thermo Fisher Scientific, 1:500)] incubation, we used 4′,6-diamidino-2-phenylindole (DAPI) mounting medium to counterstain nuclei and then took images with a Zeiss light microscope coupled with an AxioCam camera (MR3; Carl Zeiss) or Aperio Versa 8 fluorescence scanner (Leica Biosystems). We counted HNF4α+/Jagged1− and HNF4α+/Jagged1+ hepatocytes in pictures of 15 to 20 nonoverlapping and randomly chosen fields per section and quantitated Jagged1-positive staining area using the HALO platform. Liver paraffin sections were deparaffinized and rehydrated to distilled water, and collagen content was detected by Sirius red staining (214901, Polysciences) per the manufacturer’s protocol and quantitated with ImageJ [National Institutes of Health (NIH)].
RNA extraction, qPCR, and chromatin immunoprecipitation
We extracted RNA with TRIzol (Thermo Fisher Scientific), reverse-transcribed to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems), and performed quantitative PCR with a CFX96 real-time PCR detection system (Bio-Rad). For chromatin immunoprecipitation (ChIP) assays, we homogenized 100 mg of liver, fixed and fragmented DNA, and then immune-precipitated with control immunoglobulin G (IgG) or p65 antibodies (8242, CST) as per SimpleChIP Plus Enzymatic Chromatin IP Kit (CST) instructions followed by PCR with Jag1 promoter-specific primers (forward: 5′-GAGCAGTAGTTCCCCATT-3′; reverse: 5′-GGGTTCCCAGGCACAGAA-3′).
Western blots
We homogenized liver in radioimmunoprecipitation assay (RIPA) buffer with protease and phosphatase inhibitors. We quantified protein and loaded equal amounts for polyacrylamide gel electrophoresis. After blocking as described, we applied primary antibodies for Jagged1 (AF599, R&D Systems, 1:700 or AF1277, R&D Systems, 1:250), p65 (8242, CST, 1:1000), actin (ab5694, Abcam, 1:2000), or tubulin (ab18207, Abcam, 1:2000) and secondary antibodies and quantitated band intensities with either Quantity One (Bio-Rad) or ImageJ.
Statistical analysis
We expressed results as means ± SEM. Shapiro-Wilk test was used to assess normality. Differences between two groups were calculated using a two-tailed Student’s t test if the data followed a normal distribution; otherwise, nonparametric Mann-Whitney U tests were used. Analyses involving multiple groups were performed using one-way ANOVA followed by post hoc t tests. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments:
We thank A. Flete, T. Kolar, Z. Wang, and J. Liang for excellent technical support and members of the Pajvani laboratory for insightful discussion. We also thank K. Loomes (Penn), R. Adams (Max Planck), and J. Kitajewski (UIC) for sharing mouse strains, as well as the NASH CRN investigators and the Ancillary Studies Committee for providing clinical samples and relevant data from the PIVENS trial.
Funding:
This work was supported by NIH DK103818 (U.B.P.), NIH DK119767 (U.B.P.), NIH DK116620 (I.T.), MyFIRST AIRC grant no.16888 (L.V.), Ricerca Finalizzata Ministero della Salute RF-2016-02364358 (L.V.), European Union (EU) Programme Horizon 2020 grant agreement no. 777377 (L.V.), a Sanofi iAward (U.B.P.), ALF Liver Scholar Award (X.W.), NRF grant no. 2020R1C1C1004015 (K.K.), and a Russell Berrie Foundation Fellowship in Diabetes Research (J.Y.).
Footnotes
SUPPLEMENTARY MATERIALS
Competing interests: L.V., speaking for MSD, Gilead, AlfaSigma, and AbbVie; consulting for Gilead, Pfizer, AstraZeneca, Novo Nordisk, Intercept Pharmaceuticals, Diatech Pharmacogenetics, and IONIS; and research for Gilead. I.T., consulting for Akero Therapeutics Inc. and research with Takeda Pharmaceutical Company. J.E.L., consulting for Mirum Pharmaceuticals, Prosciento, Gannex, Surrozen, Intercept, Novo Nordisk, and Pfizer; unpaid Scientific Advisory Board for Thetis Pharmaceuticals; and research with Genfit. U.B.P., consulting for Jannsen Pharmaceuticals and Casma Therapeutics and research with Takeda Pharmaceutical Company. All other authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. Primary data are available in data file S1.
REFERENCES AND NOTES
- 1.Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, Zelber-Sagi S, Wong VW-S, Dufour J-F, Schattenberg JM, Kawaguchi T, Arrese M, Valenti L, Shiha G, Tiribelli C, Yki-Järvinen H, Fan J-G, Grønbæk H, Yilmaz Y, Cortez-Pinto H, Oliveira CP, Bedossa P, Adams LA, Zheng M-H, Fouad Y, Chan W-K, Mendez-Sanchez N, Ahn SH, Castera L, Bugianesi E, Ratziu V, George J, A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol 73, 202–209 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Eslam M, Sanyal AJ, George J; International Consensus Panel, MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158, 1999–2014.e1 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Rockey DC, Bell PD, Hill JA, Fibrosis––A common pathway to organ injury and failure. N. Engl. J. Med 372, 1138–1149 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Michelotti GA, Machado MV, Diehl AM, NAFLD, NASH, and liver cancer. Nat. Rev. Gastroenterol. Hepatol 10, 656–665 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Wiest R, Albillos A, Trauner M, Bajaj JS, Jalan R, Targeting the gut-liver axis in liver disease. J. Hepatol 67, 1084–1103 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Brandl K, Schnabl B, Intestinal microbiota and nonalcoholic steatohepatitis. Curr. Opin. Gastroenterol 33, 128–133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuster S, Cabrera D, Arrese M, Feldstein AE, Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol 15, 349–364 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Luedde T, Schwabe RF, NF-κB in the liver—Linking injury, fibrosis, and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol 8, 108–118 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M, From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol 16, 411–428 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Couso JP, Martinez Arias A, Notch is required for wingless signaling in the epidermis of Drosophila. Cell 79, 259–272 (1994). [DOI] [PubMed] [Google Scholar]
- 11.Furukawa T, Mukherjee S, Bao ZZ, Morrow EM, Cepko CL, rax, Hes1, and notch1 promote the formation of Muller glia by postnatal retinal progenitor cells. Neuron 26, 383–394 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, Adams RH, The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137, 1124–1135 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Weng AP, Ferrando AA, Lee W, Morris JP IV, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC, Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Henrique D, Schweisguth F, Mechanisms of Notch signaling: A simple logic deployed in time and space. Development 146, dev172148 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Kopan R, Ilagan MXG, The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 137, 216–233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hofmann JJ, Zovein AC, Koh H, Radtke F, Weinmaster G, Iruela-Arispe ML, Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: Insights into Alagille syndrome. Development 137, 4061–4072 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams JM, Jafar-Nejad H, The roles of notch signaling in liver development and disease. Biomolecules 9, 608 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geisler F, Strazzabosco M, Emerging roles of Notch signaling in liver disease. Hepatology 61, 382–392 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont ME, Rand EB, Piccoli DA, Hood L, Spinner NB, Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat. Genet 16, 243–251 (1997). [DOI] [PubMed] [Google Scholar]
- 20.Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, Chandrasekharappa SC, Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat. Genet 16, 235–242 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Pajvani UB, Qiang L, Kangsamaksin T, Kitajewski J, Ginsberg HN, Accili D, Inhibition of Notch uncouples Akt activation from hepatic lipid accumulation by decreasing mTorc1 stability. Nat. Med 19, 1054–1060 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pajvani UB, Shawber CJ, Samuel VT, Birkenfeld AL, Shulman GI, Kitajewski J, Accili D, Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat. Med 17, 961–967 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valenti L, Mendoza RM, Rametta R, Maggioni M, Kitajewski C, Shawber CJ, Pajvani UB, Hepatic notch signaling correlates with insulin resistance and nonalcoholic fatty liver disease. Diabetes 62, 4052–4062 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, Meroni M, Graham MJ, Yates KP, Diehl AM, Schwabe RF, Tabas I, Valenti L, Lavine JE, Pajvani UB, Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med 10, eaat0344 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, Van Natta M, Clark J, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR; NASH CRN, Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med 362, 1675–1685 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanyal AJ, Brunt EM, Kleiner DE, Kowdley KV, Chalasani N, Lavine JE, Ratziu V, McCullough A, Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 54, 344–353 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, Masia R, Chung RT, Lefkowitch JH, Schwabe RF, Tabas I, Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab 24, 848–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brosnan ME, Brosnan JT, Hepatic glutamate metabolism: A tale of 2 hepatocytes. Am. J. Clin. Nutr 90, 857S–861S (2009). [DOI] [PubMed] [Google Scholar]
- 29.Gebhardt R, Mecke D, Heterogeneous distribution of glutamine synthetase among rat liver parenchymal cells in situ and in primary culture. EMBO J 2, 567–570 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Messeguer X, Escudero R, Farré D, Núñez O, Martínez J, Albà MM, PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18, 333–334 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Farré D, Roset R, Huerta M, Adsuara JE, Roselló L, Albà MM, Messeguer X, Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res 31, 3651–3653 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bash J, Zong W-X, Banga S, Rivera A, Ballard DW, Ron Y, Gélinas C, Rel/NF-κB can trigger the Notch signaling pathway by inducing the expression of Jagged1, a ligand for Notch receptors. EMBO J 18, 2803–2811 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnston DA, Dong B, Hughes CC, TNF induction of jagged-1 in endothelial cells is NFκB-dependent. Gene 435, 36–44 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto M, Taguchi Y, Ito-Kureha T, Semba K, Yamaguchi N, Inoue J.-i., NF-κB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat. Commun 4, 2299 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Carpino G, Del Ben M, Pastori D, Carnevale R, Baratta F, Overi D, Francis H, Cardinale V, Onori P, Safarikia S, Cammisotto V, Alvaro D, Svegliati-Baroni G, Angelico F, Gaudio E, Violi F, Increased liver localization of lipopolysaccharides in human and experimental NAFLD. Hepatology 72, 470–485 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Zhang G, Ghosh S, Molecular mechanisms of NF-κB activation induced by bacterial lipopolysaccharide through Toll-like receptors. J. Endotoxin Res 6, 453–457 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Schwabe RF, Tabas I, Pajvani UB, Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 158, 1913–1928 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lefebvre P, Lalloyer F, Baugé E, Pawlak M, Gheeraert C, Dehondt H, Vanhoutte J, Woitrain E, Hennuyer N, Mazuy C, Bobowski-Gérard M, Zummo FP, Derudas B, Driessen A, Hubens G, Vonghia L, Kwanten WJ, Michielsen P, Vanwolleghem T, Eeckhoute J, Verrijken A, Van Gaal L, Francque S, Staels B, Interspecies NASH disease activity whole-genome profiling identifies a fibrogenic role of PPARα-regulated dermatopontin. JCI Insight 2, e92264 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, Murphy SK, Ashley-Koch AE, Choi SS, Michelotti GA, Hampton DD, Chen Y, Tillmann HL, Hauser MA, Abdelmalek MF, Diehl AM, Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 59, 471–482 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang YM, Noureddin M, Liu C, Ohashi K, Kim SY, Ramnath D, Powell EE, Sweet MJ, Roh YS, Hsin I-F, Deng N, Liu Z, Liang J, Mena E, Shouhed D, Schwabe RF, Jiang D, Lu SC, Noble PW, Seki E, Hyaluronan synthase 2–mediated hyaluronan production mediates Notch1 activation and liver fibrosis. Sci. Transl. Med 11, eaat9284 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aimaiti Y, Jin X, Wang W, Chen Z, Li D, TGF-β1 signaling regulates mouse hepatic stellate cell differentiation via the Jagged1/Notch pathway. Life Sci 192, 221–230 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Nakano Y, Nakao S, Sumiyoshi H, Mikami K, Tanno Y, Sueoka M, Kasahara D, Kimura H, Moro T, Kamiya A, Hozumi K, Inagaki Y, Identification of a novel α-fetoprotein-expressing cell population induced by the Jagged1/Notch2 signal in murine fibrotic liver. Hepatol. Commun 1, 215–229 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang G, Weng Z, Song J, Chen Y, Reversal effect of Jagged1 signaling inhibition on CCl4-induced hepatic fibrosis in rats. Oncotarget 8, 60778–60788 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitade M, Kaji K, Nishimura N, Seki K, Nakanishi K, Tsuji Y, Sato S, Saikawa S, Takaya H, Kawaratani H, Namisaki T, Moriya K, Mitoro A, Yoshiji H, Blocking development of liver fibrosis augments hepatic progenitor cell-derived liver regeneration in a mouse chronic liver injury model. Hepatol. Res 49, 1034–1045 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Herzig S, Hedrick S, Morantte I, Koo SH, Galimi F, Montminy M, CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR-γ. Nature 426, 190–193 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Chiu DK-C, Tse AP-W, Law C-T, Xu IM-J, Lee D, Chen M, Lai RK-H, Yuen VW-H, Cheu JW-S, Ho DW-H, Wong C-M, Zhang H, Ng IO-L, Wong CC-L, Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell Death Dis 10, 934 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nandagopal N, Santat LA, LeBon L, Sprinzak D, Bronner ME, Elowitz MB, Dynamic ligand discrimination in the notch signaling pathway. Cell 172, 869–880.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seymour PA, Collin CA, la Rosa Egeskov-Madsen A, Jørgensen MC, Shimojo H, Imayoshi I, de Lichtenberg KH, Kopan R, Kageyama R, Serup P, Jag1 modulates an oscillatory Dll1-Notch-Hes1 signaling module to coordinate growth and fate of pancreatic progenitors. Dev. Cell 52, 731–747.e8 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Kobayashi T, Kageyama R, Expression dynamics and functions of Hes factors in development and diseases. Curr. Top. Dev. Biol 110, 263–283 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Ilagan MXG, Lim S, Fulbright M, Piwnica-Worms D, Kopan R, Real-time imaging of notch activation with a luciferase complementation-based reporter. Sci. Signal 4, rs7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fryer CJ, White JB, Jones KA, Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 16, 509–520 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Kim JK, Fat uses a TOLL-road to connect inflammation and diabetes. Cell Metab 4, 417–419 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Lee JY, Sohn KH, Rhee SH, Hwang D, Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem 276, 16683–16689 (2001). [DOI] [PubMed] [Google Scholar]
- 54.Nilakantan H, Kuttippurathu L, Parrish A, Hoek JB, Vadigepalli R, In vivo zonal variation and liver cell-type–specific NF-κB localization after chronic adaptation to ethanol and following partial hepatectomy. PLOS ONE 10, e0140236 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mesarwi OA, Shin M-K, Bevans-Fonti S, Schlesinger C, Shaw J, Polotsky VY, Hepatocyte hypoxia inducible factor-1 mediates the development of liver fibrosis in a mouse model of nonalcoholic fatty liver disease. PLOS ONE 11, e0168572 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X, de Carvalho Ribeiro M, Iracheta-Vellve A, Lowe P, Ambade A, Satishchandran A, Bukong T, Catalano D, Kodys K, Szabo G, Macrophage-specific hypoxia-inducible factor-1α contributes to impaired autophagic flux in nonalcoholic steatohepatitis. Hepatology 69, 545–563 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Culver C, Sundqvist A, Mudie S, Melvin A, Xirodimas D, Rocha S, Mechanism of hypoxia-induced NF-κB. Mol. Cell. Biol 30, 4901–4921 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.D’Ignazio L, Rocha S, Hypoxia induced NF-κB. Cell 5, 10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savransky V, Nanayakkara A, Vivero A, Li J, Bevans S, Smith PL, Torbenson MS, Polotsky VY, Chronic intermittent hypoxia predisposes to liver injury. Hepatology 45, 1007–1013 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Chalasani N, Wilson L, Kleiner DE, Cummings OW, Brunt EM, Ünalp A; NASH Clinical Research Network, Relationship of steatosis grade and zonal location to histological features of steatohepatitis in adult patients with non-alcoholic fatty liver disease. J. Hepatol 48, 829–834 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yeh MM, Brunt EM, Pathological features of fatty liver disease. Gastroenterology 147, 754–764 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, Wallenborg K, Picelli S, Zagorska A, Pendem SV, Hudson TE, Wu MM, Budas GR, Breckenridge DG, Harrison EM, Mole DJ, Wigmore SJ, Ramachandran P, Ponting CP, Teichmann SA, Marioni JC, Henderson NC, Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep 29, 1832–1847.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen Y, Zheng S, Qi D, Zheng S, Guo J, Zhang S, Weng Z, Inhibition of Notch signaling by a γ-Secretase inhibitor attenuates hepatic fibrosis in rats. PLOS ONE 7, e46512 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA, Taylor RS, Efremova M, Vento-Tormo R, Carragher NO, Kendall TJ, Fallowfield JA, Harrison EM, Mole DJ, Wigmore SJ, Newsome PN, Weston CJ, Iredale JP, Tacke F, Pollard JW, Ponting CP, Marioni JC, Teichmann SA, Henderson NC, Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Antfolk D, Sjöqvist M, Cheng F, Isoniemi K, Duran CL, Rivero-Muller A, Antila C, Niemi R, Landor S, Bouten CVC, Bayless KJ, Eriksson JE, Sahlgren CM, Selective regulation of Notch ligands during angiogenesis is mediated by vimentin. Proc. Natl. Acad. Sci. U.S.A 114, E4574–E4581 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bray SJ, Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol 7, 678–689 (2006). [DOI] [PubMed] [Google Scholar]
- 67.Azimi M, Brown NL, Jagged1 protein processing in the developing mammalian lens. Biol. Open 8, bio041095 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Metrich M, Bezdek Pomey A, Berthonneche C, Sarre A, Nemir M, Pedrazzini T, Jagged1 intracellular domain-mediated inhibition of Notch1 signalling regulates cardiac homeostasis in the postnatal heart. Cardiovasc. Res 108, 74–86 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Loomes KM, Russo P, Ryan M, Nelson A, Underkoffler L, Glover C, Fu H, Gridley T, Kaestner KH, Oakey RJ, Bile duct proliferation in liver-specific Jag1 conditional knockout mice: Effects of gene dosage. Hepatology 45, 323–330 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Belteki G, Haigh J, Kabacs N, Haigh K, Sison K, Costantini F, Whitsett J, Quaggin SE, Nagy A, Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res 33, e51 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McAlees JW, Whitehead GS, Harley IT, Cappelletti M, Rewerts CL, Holdcroft AM, Divanovic S, Wills-Karp M, Finkelman FD, Karp CL, Cook DN, Distinct Tlr4-expressing cell compartments control neutrophilic and eosinophilic airway inflammation. Mucosal Immunol 8, 863–873 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen L.-f., Fischle W, Verdin E, Greene WC, Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293, 1653–1657 (2001). [DOI] [PubMed] [Google Scholar]
- 73.Mederacke I, Dapito DH, Affo S, Uchinami H, Schwabe RF, High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc 10, 305–315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. Primary data are available in data file S1.