

Abstract
Cryptosporidiosis is caused by infection of the small intestine by Cryptosporidium parasites, resulting in severe diarrhea, dehydration, malabsorption and potentially death. The only FDA-approved therapeutic is only partially effective in young children and ineffective for immunocompromised patients. Triazolopyridine MMV665917 is a previously reported anti-Cryptosporidium screening hit with in vivo efficacy but suffers from modest inhibition of the hERG ion channel which could portend cardiotoxicity. Herein, we describe our initial development of structure-activity relationships of this novel lead series with a particular focus on optimization of the piperazine-urea linker. We have discovered that piperazine-acetamide is a superior linker resulting in identification of SLU-2633 which has an EC50 of 0.17 μM, an improved projected margin versus hERG, a prolonged pharmacokinetic exposure in small intestine, and oral efficacy in vivo with minimal systemic exposure. SLU-2633 represents a significant advancement towards the identification of a new effective and safe treatment for cryptosporidiosis.
Keywords: cryptosporidium; cryptosporidiosis; anticryptosporidial; antiparasitic; diarrhea; triazolopyridazine; [1,2,4]triazolo[4,3-b]pyridazine; hERG
Graphical Abstract

Introduction
Cryptosporidiosis is a disease caused by the apicomplexan protozoan pathogen Cryptosporidium. Although diarrhea is its primary symptom, Cryptosporidium infection is strongly associated with malnutrition, stunted growth and delayed cognitive development.1 Cryptosporidium is the third most common pathogen causing life-threatening diarrhea in children under one year of age.2, 3 It is mostly found in developing countries where access to clean water and healthy food is difficult. It is also the main cause of waterborne diarrhea in the United States for which an infectious agent is identified, usually through contaminated recreational and drinking water.4 In addition to its strong association with mortality in young children, cryptosporidiosis is a main cause of diarrhea in immunocompromised patients.5–7 One study concluded that Cryptosporidium causes between 2.9 and 4.7 million cases of diarrhea annually for children under 2 years of age in seven Sub-Saharan and South-Asian countries, potentially resulting in ~220,000 deaths.8 Another study recently estimated that acute and chronic effects of cryptosporidiosis in children under the age of 5 led to ~12.9 million disability-adjusted life-years lost globally in 2016.9
Out of twenty-seven (27) described Cryptosporidium species, C. parvum and C. hominis are primarily responsible for human disease.1 The disease is mainly transmitted through water and food, by direct person-to-person contact, and by bathing in swimming pools through ingesting water contaminated with feces of infected persons or animals containing Cryptosporidium oocysts. The parasite infects epithelial cells of the small intestine and replicates within an extra-cytoplasmic parasitophorous vacuole, but the mechanism of diarrhea remains poorly understood.10
The only FDA-approved drug for treating cryptosporidiosis is nitazoxanide (NTZ). For immunocompetent adults, NTZ reduces the duration of diarrhea by approximately one day. However, it is currently not recommended for use in infants less than 12 months of age, is just over 50% effective for older children, and is equivalent to a placebo for immunocompromised patients, such as those with AIDS.11–13 It has only modest potency in suppressing parasite growth in vitro (EC50 3.8 μM) and fails to effectively kill existing parasites, which could enable relapse.14 Nitazoxanide potently inhibits the pyruvate-ferredoxin oxidoreductase (PFOR) iron:sulfur metabolism of other anaerobic intestinal protozoa, e.g. Entamoeba histolytica and Giardia lamblia, but the Cryptosporidium PFOR is atypical and nitazoxanide’s anticryptosporidial activity appears to be due to one or more of the off-target effects that appear responsible for its reported anti-viral and anti-cancer activity.15 Clearly, more effective therapeutics need to be identified.
Towards this end, we recently reported the discovery of triazolopyridazine MMV665917 (1) as a novel anti-Cryptosporidium lead compound (Figure 1).14 As a screening hit, 1, with a modest potency in vitro (EC50 2.1 μM), is remarkable with oral efficacy having been demonstrated against C. parvum in several mouse models and a neonatal dairy calf model, and against C. hominis in a gnotobiotic piglet model.14, 16, 17 The antiparasitic effect of 1 occurs during the late-stages of asexual C. parvum growth and it also blocks sexual development.18, 19 Based on stage-specific in vitro assays, its antiparasitic mode-of-action is distinct from that of other major anticryptosporidial lead series, including reported tRNA-synthetase inhibitors, calcium-dependent protein kinase 1 inhibitors, a benzoxaborole believed to target mRNA processing, and phosphatidyl-inositol-4-kinase (PI4K) inhibitors.18–20 However, 1 also modestly (IC50 ~ 10 μM) inhibits the human Ether-a-go-go-Related Gene (hERG), a K+ ion channel required for cardiac muscle repolarization for which inhibitors may cause cardiac arrythmias;21–23 thus, 1 has a narrow therapeutic index between anti-Cryptosporidium efficacy and potential cardiotoxicity.
Figure 1.

Lead Compounds MMV665917 (1) and D-28 (2) and medicinal chemistry strategy (3) focused on optimization of the Linker, X, and Aryl Tail.
In our previous work, we conducted a preliminary structure-activity relationship (SAR) study with commercially available analogs that identified 2 as a modestly more potent analog than 1 with an EC50 of 0.55 μM.14 In that previous work, SAR was limited to only a few replacements for the heteroaryl head group and no replacements for the piperazinyl linker while urea and carboxamide tail groups were more extensively profiled but yielded no further improvements in potency than 2. The work described herein significantly extends this SAR through chemical synthesis in order to identify analogs of 1 with improved anti-cryptosporidial efficacy. Furthermore, we examine inhibition of the hERG channel to improve the therapeutic index of triazolopyradizines. We report on our initial structure-activity relationship (SAR) studies on the piperazine urea linker and have identified piperazine-acetamides as a promising lead series with an improved efficacy and safety profile over the original piperazine-urea hit.
Results and Discussion
In our previous work, dichlorophenyl urea 2 was found to be about 4-fold more potent than MMV665917.14 Thus, we focused on synthesizing analogues of 2 as our lead compound. Based on this previous work, we surmised that the piperazine serves primarily as a scaffolding linker whose role is proper positioning of the triazolopyridazine head group and the dichlorophenyl aryl tail group (Fig. 1). We elected to first survey potential linker replacements for the piperazine by systematically interrogating the role of each carbon and nitrogen atom of the linker and evaluating the other sized-rings and the overall linker length.
Synthesis of Piperazine and Diazepine “N-linked” Analogs.
Piperazine- and diazepine-based analogs were prepared as outlined in Scheme 1. Heteroaryl headgroup 6-chloro-[1,2,4]triazolo[4,3-b]pyridazine (4) was prepared from 3,6-dichloropyridazine in a manner similar to previous methods using hydrazine followed by condensation with formic acid.24, 25 Displacement of the chloride of 4 was accomplished with amines 5a-e by heating in EtOH or NMP in a microwave reactor to give Boc-protected amines 6a-e.26 The Boc-protected amine intermediates were deprotected using HCl in dioxane to yield the HCl salt of the amines. The resultant amines 7a-e were then coupled with the aryl acetic acids using EDCI and HOBt to furnish aryl acetamides 8, 9a-b, and 12a-d.27 Urea analogs 10a-c were synthesized using aryl isocyanates and diisopropylethylamine. Urea 10a was further substituted through alkylation with sodium hydride and iodomethane to furnish methylated urea 11.28 Arylmethanesulfonamide 13 was prepared by treatment of amine 7c with dichlorobenzylsulfonyl chloride.
Scheme 1. Synthesis of Piperazine and Diazepine “N-linked” Analogs.
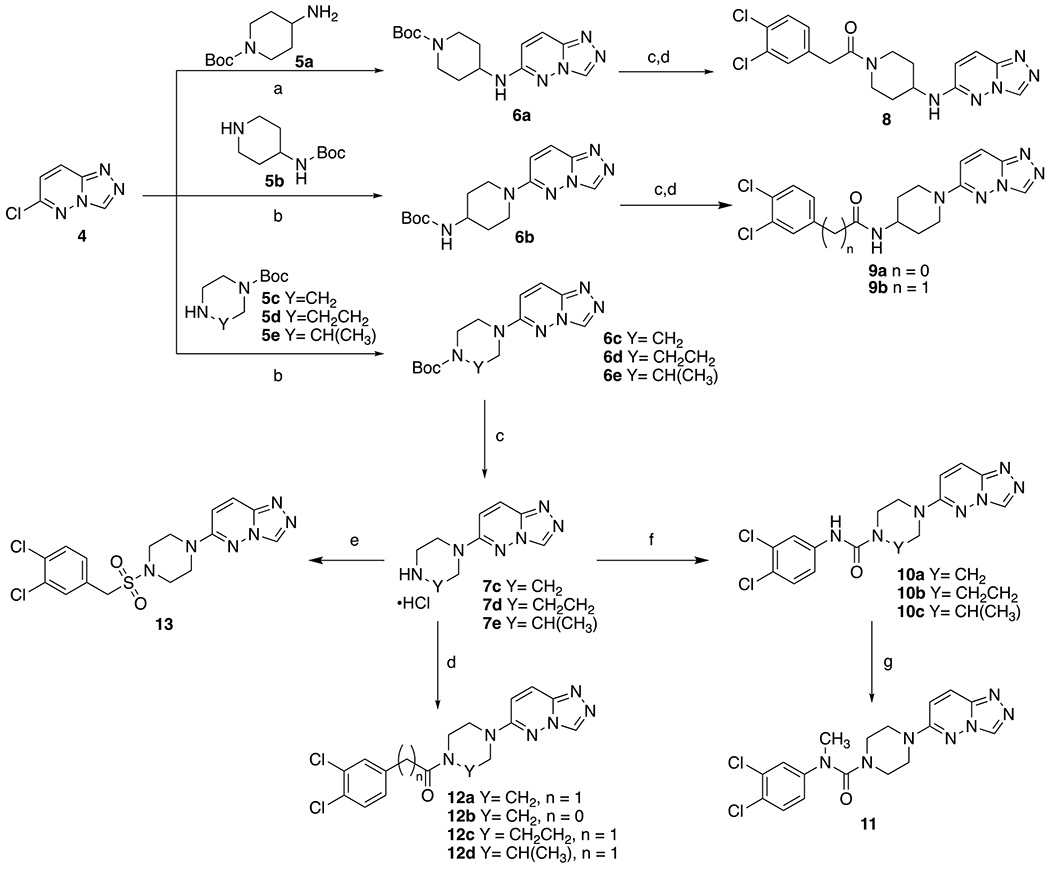
Reagents and conditions: (a) DIEA, EtOH, 100 °C, 2h, 80-88%; (b) DIEA, NMP, 120 °C, 8h, 80-88%; (c) 4M HCl in dioxane, 2h, 89-98%; (d) EDCI/HOBt or HBTU, DIEA, 3,4-Cl2Ph(CH2)nCO2H, DMF, 30-85%; (e) DIEA, 3,4-dichlorobenzylsulfonyl chloride, DMF, 15%; (f) DIEA, 3,4-dichloroisocyanate, DMF, 10-45%; (g) NaH, THF, CH3I, 0 °C to rt, 22%.
Synthesis of Piperidine “C-linked” Analogs.
Piperidine-based analogs were prepared as outlined in Scheme 2. Boc-protected vinyl-piperidine intermediate 14 was synthesized by Suzuki coupling of chloropyrazolopyridazine 4 and the Boc-protected boronic ester of the vinyl-piperidine. A portion of the olefin 14 was reduced by catalytic hydrogenation to produce piperidine 15. 14 and 15 were deprotected to produce the HCl salts of piperidines 16 and 17, which were used to synthesize the final compounds 18a,b and 19a,b as aryl acetamides and aryl ureas.
Scheme 2. Synthesis of Piperidine “C-linked” Analogs.
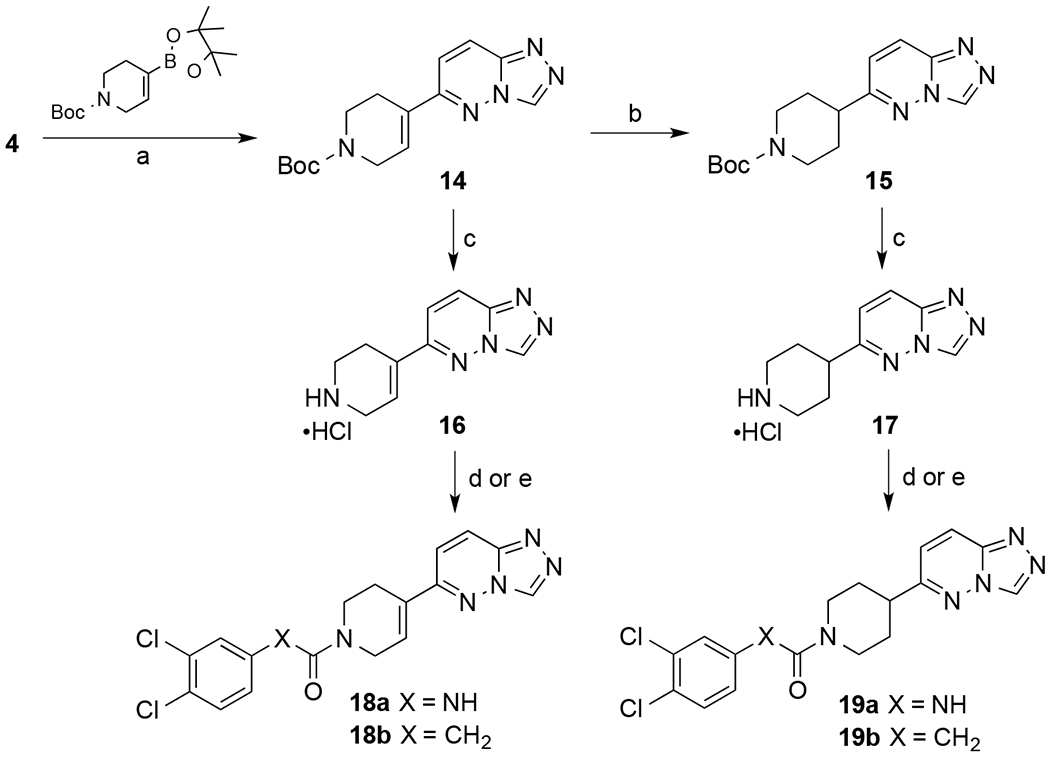
Reagents and conditions: (a) Cs2CO3, Pd((t-butyl)3P)2, toluene: EtOH: H2O (9:3:1), 120 °C, overnight, 30-35% (b) 10% Pd/C, 60 psi, CH3OH, 16 h, 80%, (c) 4M HCl in dioxane 89-98%, (d) DIEA,3,4-dichloroisocyanate, DMF, 10-45% (e) DIEA, EDCI, HOBT, 3,4-dichlorophenylacetic acid, DMF, 30-85%
Synthesis of Aryl Acetamide Derivatives.
Exploration of substitution of the acetamide methylene as oxo and α,α-difluoro moieties is illustrated in Scheme 3. The synthesis began with oxidation of 3,4-dichlorophenone 20 with selenium (IV) oxide to furnish acid 21. Carboxylic acid 21 was then coupled with amine 7c under our standard amide coupling conditions to give oxo analog 22. Ester 21b was obtained by refluxing carboxylic acid 21a with SOCl2 in ethanol. The α-carbonyl of intermediate 21b was difluorinated using 4-tert-butyl-2,6-dimethylphenylsulfur trifluoride to give α,α-difluoro ester 23.28, 29 Hydrolysis of compound 23 followed by amide coupling with amine 7c furnished α,α-difluoro analog 25. Commercially available cyclopropyl carboxylic acid 26 was transformed to the corresponding amide 27 under the same amide coupling conditions.
Scheme 3. Synthesis of α-substituted 3,4-dichlorophenylacetamide derivatives.

Reagents and conditions: (a) SeO2, pyridine, 110 °C, overnight, 95%; (b) 7c, HBTU, DIEA, DMF; (c) SOCl2, EtOH, reflux, 80 °C, 85%; (d) 4-tert-butyl-2,6-dimethylphenylsulfur trifluoride, HF-Py, DCM, RT to 45 °C, 98% crude yield; (e) LiOH, MeOH, then HCl, 98% crude yield.
Additional aryl acetamide derivatives were synthesized to provide a preliminary SAR on the aromatic ring (Scheme 4). Aryl acetic acids 28 were coupled with 7c through the previously described amide coupling procedures to give final compounds 29a-j.
Scheme 4. Synthesis of Arylacetamide Analogs.
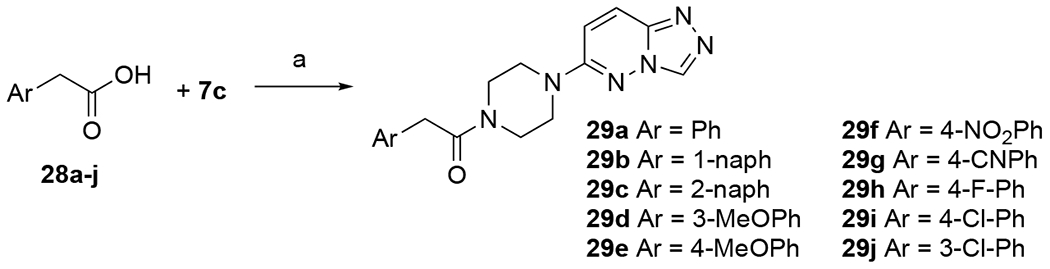
Reagents and conditions: (a) EDCI, HOBt, TEA, DMF, rt overnight (14-55%).
In vitro Cryptosporidium parvum-infected HCT-8 Cell Assay.
Anticryptosporidial activity of newly synthesized compounds was measured in vitro using a previously established high-content microscopy assay that measures asexual development of C. parvum within the colon carcinoma cell line HCT-8.30 HCT-8 cell monolayers were grown to near confluence in clear-bottomed microtiter plates and infected with C. parvum oocysts. Test compounds were added after allowing 3 hours for host cell invasion, followed by incubation for 48 hours, and then staining for parasite and host cells and enumeration by fluorescence microscopy. Results are shown in Tables 1 and 2.
Table 1.
Linker SAR.
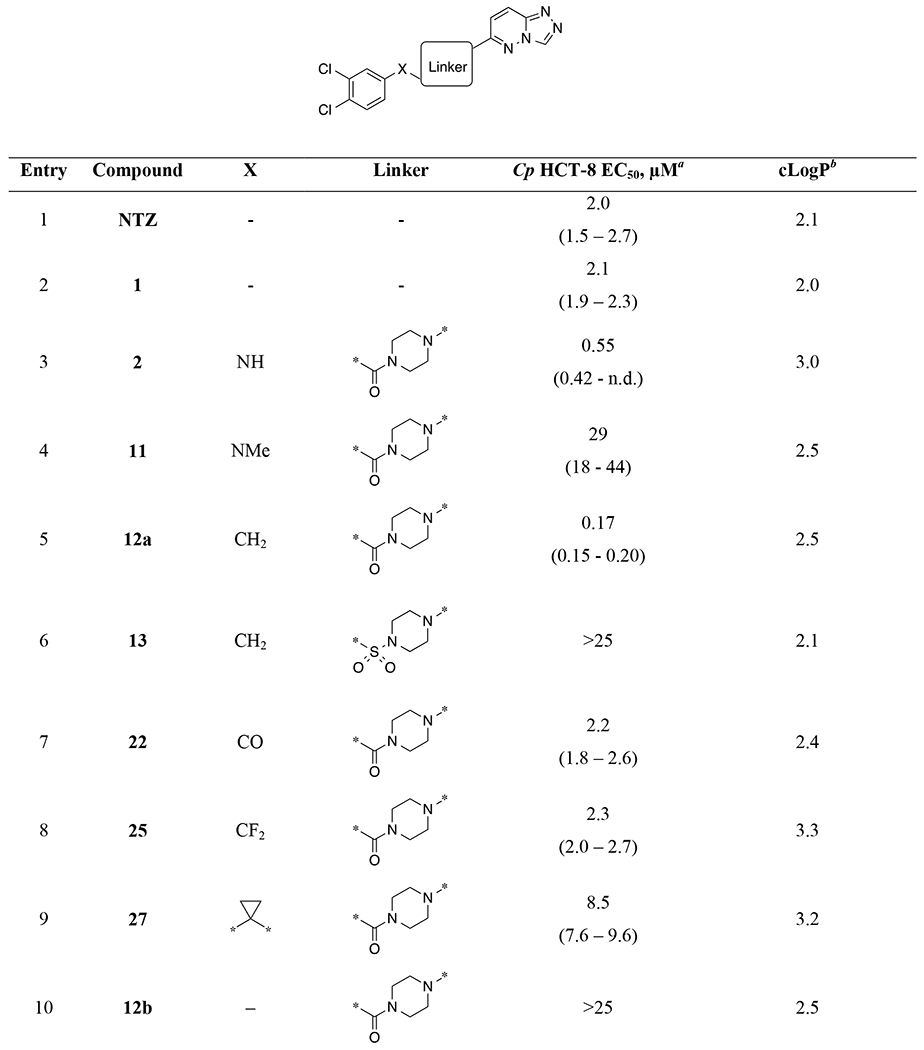
|

|
EC50 values and 95% confidence intervals are calculated from average potency in the Cp HCT-8 assay from at least two 9-point dose-response experiments (n=8 technical replicates per dose). n.d. = not determined.
ClogP represents an estimate of lipophilicity and is the log of the partition coefficient as calculated in CDD Vault using the ChemAxon fragment approach (www.collaborativedrug.com).
Table 2.
Arylacetamide SAR.
| ||||
|---|---|---|---|---|
| Entry | Compound | R | Cp HCT-8 EC50, μMa | cLogPb |
| 1 | 29a | H | 22 (20 – 24) | 1.3 |
| 2 | 29b |
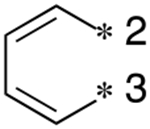
|
17.1 (13 - 23) | 2.30 |
| 3 | 29c |

|
14 (11 – 19) | 2.30 |
| 4 | 29d | 3-OMe | 18 (16 – 20) | 1.20 |
| 5 | 29e | 4-OMe | 17 (15 – 19) | 1.20 |
| 6 | 29f | 4-NO2 | 2.5 (2.0 – 2.9) | 1.20 |
| 7 | 29g | 4-CN | 11 (9.3 – 14) | 1.20 |
| 8 | 29h | 4-F | 0.86 (0.47 – 1.3) | 1.50 |
| 9 | 29i | 4-Cl | 0.66 (0.44 – 0.91) | 1.90 |
| 10 | 29j | 3-Cl | 3.5 (3.0 – 4.1) | 1.90 |
EC50 values and 95% confidence intervals are calculated from average potency in the Cp HCT-8 assay from at least two 9-point dose-response experiments (n=8 technical replicates per dose).
ClogP was calculated in CDD Vault using the ChemAxon fragment approach (www.collaborativedrug.com).
Piperazine and Diazepine “N-linked” SAR.
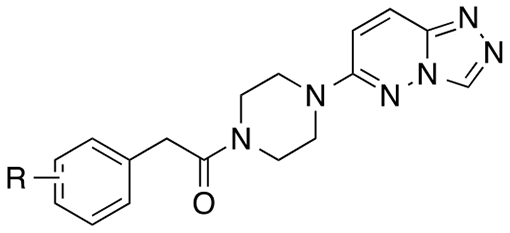
Keeping the 3,4-dichlorophenyl and the triazolopyridazine constant, we systematically replaced the central linker and X (Table 1). For reference, NTZ and original hit 1 are included (entries 1 and 2). Urea piperazine 2 was used as a comparator lead compound for new analogs (entry 3). Substitution of the NH of the urea with a methyl group (11; entry 4) led to a dramatic loss of potency. In contrast, replacement of the NH of the urea with a methylene resulted in arylacetamide 12a (EC50 = 0.17 μM; entry 5) with a modest 3-fold improvement in potency relative to 2 and >10-fold relative to NTZ and 1. In contrast, the corresponding sulfonamide 13 led to dramatic loss of potency (entry 6). Replacement of X as a carbonyl or difluoromethylene led to 4-fold potency losses (entries 7-8). Substitution as a fused cyclopropyl resulted in a 15-fold potency loss (entry 9).
Truncation to the directly linked aryl amide 12b led to >100-fold loss of potency relative to 12a (entry 10). Likewise, similar losses of potency were observed when maintaining linker length but shifting the amide linkage towards the phenyl ring in analog 9a (entry 11) and by extension of the amide using 4-aminopiperidine as a linker (9b and 8; entries 12-13). Expansion of the piperazine ring by addition of a methyl group (10c and 12d) or as the 7-membered diazepine ring system (10b and 12c) resulted in >50-fold losses of potency, with greater losses for ureas than for acetamides (entries 14-17).
Piperidine “C-linked” SAR.
A set of piperidine analogs of urea 2 and acetamide 12a utilizing a piperidine (“C-linked”) to the triazolopyridazine present a diverging SAR (Table 1, entries 18-21). Within this set of analogs, the unsaturated piperidine urea 18a was the most potent (EC50 = 0.58 μM) and equipotent to its N-linked counterpart 2. In contrast, unsaturated piperidine acetamide 18b as well as saturated piperidine urea 19a and acetamide 19b resulted in rather unexpected 10- to 30-fold losses in potency relative to 2.
Arylacetamides SAR.
The SAR in Table 1 suggests that the piperazine-acetamide linker is optimal and provides the precisely required geometry to present the triazolopyridazine head group and the aryl tail group. Small variations in linkers were not well tolerated despite similar lipophilicities (cLogP). To further extend this finding and confirm that 3,4-dichloro substitution of the aryl acetamide is preferred, we conducted a preliminary SAR of the aryl tail group on the acetamide scaffold. Table 2 shows the SAR of the substituents on the aryl tail group of the acetamide series. These substituents could influence activity through electronic, lipophilic and steric effects. The simple unadorned phenyl analog 29a (entry 1) is only weakly potent (EC50 22 μM). 1- and 2-napthyl variants (entries 2-3) have similar potencies. Electron donating groups 3- and 4-methoxy (entries 4-5) do not affect potency relative to hydrogen (entry 1). Electron withdrawing groups 4-nitro and 4-cyano (entries 6-7) show marginal improvement in potency, 8-fold and 2-fold, respectively.
Substitution patterns for monohalogenation were investigated as well (entries 8-10). 4-F 29h and 4-Cl 29i are 18- and 25-fold more potent than H, respectively (EC50 1.2 and 0.88 μM; entries 8-9 vs. entry 1). Moving the chlorine to the 3-position (29j; entry 10) was 4-fold less potent than in the 4-position (EC50 3.6 μM). It is interesting to note that combination of 3-Cl and 4-Cl in 12a gave an unpredicted synergistic improvement in potency relative to the singly substituted chloro analogs (Table 1, entry 4 vs. Table 2 entries 9 and 10).
Compound 12a, along with 1 and 2, were tested for inhibition of Cryptosporidium hominis-infected HCT-8 cells in a manner analogous to that for C. parvum. Potencies against C. hominis for this class of compounds is comparable to that for C. parvum with compounds 1, 2, and 12a having EC50 values of ~6, ~0.56, and ~0.2 μM (data not shown).
Rate-of-Action Studies.
The biomolecular target of these triazolopyridazines is not known. Prior phenotypic studies indicate that they affect parasites during late-stage merogeny and the development of gametocytes.14, 19 Further, 1 (MMV665917) rapidly eliminates C. parvum in the HCT-8 culture system, in contrast to NTZ, which blocks Cryptosporidium replication without eliminating the parasite.14 To determine if triazolopyridazines generally kill Cryptosporidium rapidly and further evaluate our new acetamide lead compound 12a (SLU-2633), we compared the in vitro rate of elimination by NTZ to that by 12a and 2 using a method analogous to an antibacterial time-kill curve assay (Figure 2). NTZ was cytotoxic at concentrations above the EC90 and blocked C. parvum growth without eliminating the parasite from HCT-8 cultures. In contrast, parasite numbers declined exponentially in the presence of both triazolopyridazines with the rate of parasite elimination maximized at concentrations at or above six times the EC90. Based on the estimated decay rate for 12a, 99.9% of parasites would be eliminated within 98 hours of exposure at this concentration.
Figure 2.
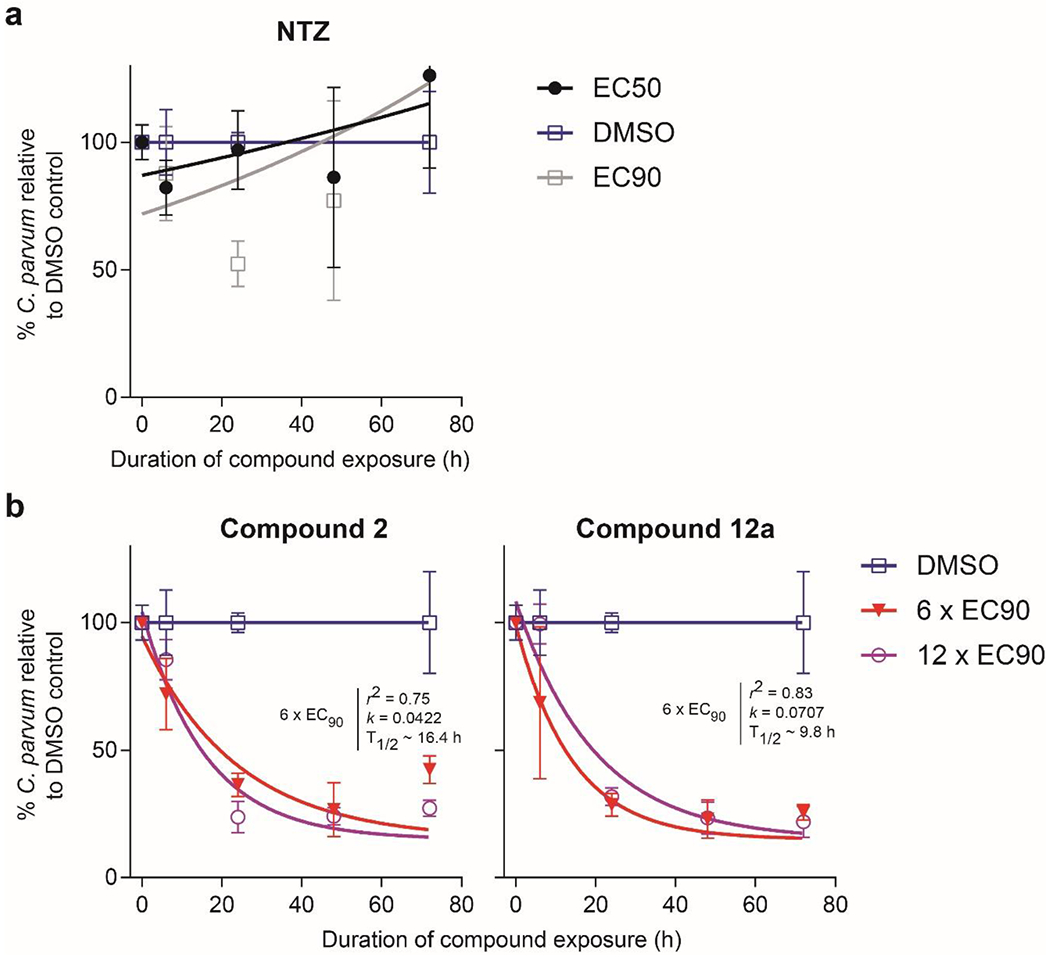
C. parvum time-kill curves. (a) Nitazoxanide (NTZ) or (b) the specified triazolopyridazine was added at multiples of the EC90 24 hours after infection of HCT-8 cell monolayers, followed by assaying the numbers of parasites and host cells at the indicated time points. One-phase exponential decay curves were fit to assay data after normalizing to the DMSO control at each time point. The lowest concentration of compound that maximized the rate of elimination is shown, along with the highest non-toxic concentration tested. Data points are the means and SDs for four culture wells per time point.
Safety pharmacology profiling.
Host cell cytotoxicity was assessed for all compounds during dose-response testing by exposure of HCT-8 cells for 48 hours to concentrations up to 25 μM, followed by staining cell nuclei and measuring the effect on cell numbers using high-density microscopy. None of the compounds tested affected the number of host cells under these conditions. The 50% cytotoxic concentration (CC50) of three compounds of high interest was determined in an additional toxicity assay using a colorimetric assay to measure NADPH or NADH produced by dehydrogenases in metabolically active cells.31 Assays were performed using the human colon carcinoma cell line HCT-8, which is the same cell line used to assay Cryptosporidium growth. The CC50 of all four compounds was >100 μM (Table 3), giving selectivity indices of over 400-fold for 12a.
Table 3.
In vitro Safety Profiling Data
| HCT-8 | hERG bindinga | hERG-CHO automated patch clamp, % inhibitionb |
|||
|---|---|---|---|---|---|
| Compound | CC50, μM | %inh, 10 μM | 1 μM | 10 μM | 100 μM |
| 1 | >100 | 8 | 12 | 48 | 88 |
| 2 | >100 | 56 | 41 | 87 | 94 |
| 12a | >100 | 11 | 19 | 67 | 94 |
| 18a | - | 56 | - | - | - |
| 18b | - | 9 | - | - | - |
| 29a | - | 4 | - | - | - |
| 29i | - | 2 | - | - | - |
Compound binding was calculated as a % inhibition of the binding of [3H]-dofetilide to the human potassium channel hERG (Eurofins, Saint Charles, MO, USA);
Functional hERG cell-based electrophysiology automated patch clamp assay; Compounds were evaluated in duplicate with average % inhibition of tail current reported at each concentration.33 (Eurofins, Saint Charles, MO, USA)
To assess whether lead compound 12a inhibits the activity of CYP enzymes which could produce undesirable drug-drug interactions, we profiled it for inhibition against a panel of CYP enzymes at 10 μM (1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 3A4). Most CYPs were not inhibited more than 10% at this concentration, although modest inhibition was observed for 2C9 and 2C19, 31% and 44%, respectively.
As an early probe to identify any off-target pharmacology that would raise potential safety concerns, we contracted Eurofins Discovery Services (St. Charles, MO) to conduct off-target safety profiling for 12a in their Safety47 assay panel.32 12a was tested in a dose response up to 30 μM in functional cell assays against 47 diverse host off-target proteins including many enzymes, GPCRs, ion channels, and transporters. Many of these assays were run in two modes to detect both agonist and antagonist activities (78 assays total). 12a had EC50 values >30 μM for 45 of the 47 targets (76 of 78 assays; Supplementary Table S1). The only host targets inhibited at <30 μM were human β−2 adrenergic receptor (ADRB2; antagonist, EC50 12 μM, with a 55% maximum response) and the hERG ion channel (antagonist, EC50 7.7 μM with a 79% max response). Overall, these results indicate that these compounds are highly selective for Cryptosporidium versus human host targets commonly associated with potential for toxicity.
Affinity for the hERG channel.
A potential safety concern with this series is its potential for binding the hERG channel given the modest hERG affinity reported for the MMV665917 (1) lead reported as having an IC50 of ~10 μM.21 To address this, we evaluated eight compounds in this series for hERG binding in a [3H]-dofetilide competitive binding assay (Table 3). Most compounds showed modest or no inhibition of binding of dofetilide at 10 μM in this assay. Only the dichlorophenyl urea 2, and C-linked analog 18a, also a urea, inhibited binding in the 50% range.
Given the discrepancy with what had been previously observed with 1 in functional hERG assays, we then tested 1, 2, and 12a in a cell-based hERG patch clamp electrophysiology assay at concentrations of 1, 10 and 100 μM to determine functional relevance (Table 3).33, 34 This assay is conducted in the absence of serum binding proteins. At 100 μM, all three compounds inhibited in the ~90% range. At 10 μM, dichlorophenyl urea 2 remained a strong inhibitor while 1 and 12a were modest inhibitors (48% and 67%, respectively). At 1 μM, only compound 2 inhibited above 40% and new lead compound 12a inhibited only 19%.
While new lead 12a did not reduce functional inhibition on the hERG channel at 10 μM relative to hit 1, it does exhibit a superior estimated safety margin. The general consensus in the field is that the margin of hERG IC50 (patch-clamp) versus Cmax (free; i.e., corrected for protein binding) ratio should be greater than 30-fold.23, 35–37 To determine our progress towards an improved margin, we elected to determine margins between hERG IC50 (patch-clamp) versus the Cp EC90 corrected for protein binding since the EC90 represents a concentration at which we see parasite killing within 24 hours. Plasma protein binding for lead 12a was found to be moderately high (92.4 and 97.8% in mouse and human plasma, respectively). For 1 and 12a, the Cp EC90 values corrected for murine protein binding are 2.8 μM and 0.036 μM, respectively. The protein-free hERG IC50 values are ~10 μM and ~8 μM, respectively. Thus, the hERG IC50 versus Cp EC90(free) ratios for 1 and 12a are ~4-fold and >200-fold, respectively. Note that fetal bovine serum is used in the CpHCT-8 assay, so we used the more conservative lower PPB value (mouse) in the calculation to approximate Cp EC90(free) value for 12a. Even if we do not correct for any protein plasma binding and just use EC50 values, 12a would have a margin of >40-fold.
In vitro profiling of pharmacologic properties.
Compound 12a and six other compounds were profiled for kinetic solubility in aqueous solution at pH 7.4 (Table 4). The compounds exhibit a range of solubilities from 1.8 μM to >200 μM. Lead 12a has a modest solubility in this assay of 18 μM. Seven compounds were profiled for metabolic stability in mouse and human liver microsomes (MLM and HLM, respectively; Table 4). The compounds are generally quite metabolically stable in both MLM and HLM with half-lives greater than 30 min. Some compounds were tested in an assay with cells (MDCK) overexpressing the human p-glycoprotein (Pgp) multi-drug resistance (MDR) transporter to assess their potential for efflux from mammalian cells. All exhibited an efflux ratio significantly > 1 suggesting they are Pgp substrates.
Table 4.
In vitro ADME Profiling Data
| Entry | Cmpd | Aq. Sol. pH 7.4, μM | MLM t1/2, min | HLM t1/2, min | MDCK-MDR Efflux Ratio |
|---|---|---|---|---|---|
| 1 | 1 | 200 | >30 | >30 | 8.9 |
| 2 | 2 | 1.8 | >30 | >30 | 12 |
| 3 | 12a | 18 | >30 | >30 | 5.8 |
| 4 | 18a | 59 | - | >30 | 6.5 |
| 5 | 19a | - | - | >30 | 6.6 |
| 6 | 19b | - | - | 7.8 | 9.8 |
| 7 | 29i | 10 | >30 | >30 | 3 |
In vivo pharmacokinetic properties of compound 12a.
Although the observed in vitro efflux of the compounds could act to limit their cellular uptake and accumulation and in vivo efficacy,38 it also may be expected to limit the efficiency of oral absorption to the systemic compartment. We and others39 speculate this could be a beneficial property to treat an infection confined mostly to intestinal enterocytes since high systemic distribution has also been shown to be unnecessary for efficacy and may only increase the possibility of side effects.40–42 Furthermore, due to the locally high concentrations of the drug that would occur in the intestinal lumen following oral dosing, higher exposure of the drug to parasite-infected intestinal cells might be sustained for hours even in the presence of ongoing cellular efflux, albeit with the caveat that the period during which this high concentration gradient exists could be shortened by increased intestinal motility associated with diarrhea.43
To evaluate the pharmacokinetics of 12a in mice, we performed studies in which a single dose was administered to a group of healthy CD1 mice by either the intravenous or oral route. Plasma and intestinal (jejunum) tissue samples were then collected at multiple time points for compound quantitation. Importantly, the dissected intestinal samples were opened and carefully rinsed to remove luminal contents. The results showed that after oral dosing, the compound’s exposure (AUC) and maximum obtained concentration (Cmax) were dramatically higher in the intestinal tissue than in plasma (Figure 3 and Table 5). This is also reflected by the very low observed oral bioavailability (F), indicating little orally administered compound is distributed to the systemic plasma compartment. Comparison of the total compound levels in the jejunum and plasma to the EC90 (0.47 μM) and 6X multiple of the EC90 (2.8 μM) from the Cp-infected HCT-9 assay suggest that anti-parasitic activity might be sustained in the former for up to 12 hours even with this low dose that was administered orally.
Figure 3. Oral Pharmacokinetic Profile of 12a in mice.

Compound concentrations of 12a in plasma (solid circles) and jejunum (solid squares) post oral dosing (10 mg/kg) are plotted as a function of time.
Table 5.
Mouse Pharmacokinetic Data for 12a
| Dose | Tissue | AUC, h*ng/mL | Cmax, ng/mL | t1/2, h | CL, mL/hr/kg | Vd, mL/kg | F, % |
|---|---|---|---|---|---|---|---|
| IV, 1 mg/kg | plasma | 50,000 | 17,000 | 2.2 | 19 | 62 | - |
| jejunum | 55,000 | 15,000 | 5.4 | 42 | 130 | - | |
|
| |||||||
| PO, 10 mg/kg | plasma | 5,200 | 1,500 | - | - | - | 1 |
| jejunum | 240,000 | 36,000 | - | - | - | - | |
It should be noted that a tissue lysate mixes interstitial and intracellular fluids, and we typically cannot know how the drug is partitioned between the extracellular and intracellular compartments.44 We also cannot accurately determine the free (unbound) concentration in the lysate since its protein composition is different from plasma. Thus, the precise unbound concentration at the site of action, presumably within the parasite which itself is located within the parasitophorous vacuole in the enterocyte, cannot be readily determined accurately. However, these data clearly indicate that the drug concentration in the local environment (intestinal tissue) greatly exceeds that observed in the plasma over the full 24-hour time course.
Compound 12a is orally efficacious in NSG mouse infection model.
We tested the efficacy of 12a in a previously described NOD SCID gamma (NSG) mouse model of C. parvum infection.14 Most of the established mouse models used for Cryptosporidium drug efficacy trials simulate an acute C. parvum infection that is self-resolving.42, 45–47 NSG mice, on the other hand, become persistently infected after oral C. parvum challenge, and thus, enable tests of the efficacy of compounds for clearance of established infections and monitoring for relapse following the completion of treatment. Infection was established by oral gavage of C. parvum on day 0 followed by administration of 50 mg/kg of 12a twice daily by oral gavage on days 7, 8, 9, and 10 of infection (Figure 4a). As measured by qPCR, parasite shedding was well-established prior to treatment (D7 values, Figure 4b). Parasite shedding was reduced by over 99% when measured the morning after completing treatment (D11) and continued to fall during the 10 day follow up period; no parasites were detected in 3 of 4 mice by ten days after completing treatment.
Figure 4. Efficacy of 12a in immunocompromised mice.

(a) Experimental scheme. Nod SCID gamma (NSG) mice were infected with C. parvum, followed by oral 12a administration twice daily. Mice were monitored for recrudescent infection for ten days after completing treatment. (b) C. parvum detected in the feces by qPCR on the indicated days. Data are the mean and SEM for each condition. Dots indicate individual mice. Negative PCR results are shown as 0.1 oocysts/mg feces, which is the PCR assay’s limit of detection (n=4 mice per group; note that one mouse in the vehicle control group was euthanized because of weight loss prior to day 21). * indicates p < 0.04 vs. pre-treatment measurement by one-way ANOVA with Dunnett’s multiple comparisons test.
Conclusions
In summary, we have taken initial steps in the optimization of triazolopyridazine hit compound 1 by systematic evaluation of the piperazinyl urea linker. The optimal linker identified in this study is piperazinyl-acetamide which led to the discovery of SLU-2633 (12a). This compound is 12-fold more potent than the hit 1 with an EC50 of 0.17 μM (0.15 to 0.20) and EC90 of 0.47 μM (0.20 to 0.72). A preliminary SAR on the terminal aryl ring revealed the preference for 3,4-dichlorosubstitution over mono-substituted halogens and other electron withdrawing and electron donating groups. Compound 12a exhibits fast-killing anticryptosporidial properties, in contrast to the parasitistatic agent NTZ. 12a is non-cytotoxic in HCT-8 cells and exhibits a remarkably clean profile against panels of CYPs and more than 40 other human off-targets of concern for the development of a safe drug. While 12a does not exhibit reduced effect on the hERG channel relative to hit 1, it does exhibit a significantly improved safety margin in vitro due to its improved potency against the parasite. In vitro ADME profiling of 12a shows good metabolic stability in liver microsomes but also that it and related compounds are efflux pump substrates. This is consistent with in vivo PK studies described here which show oral dosing of 12a provides very high exposures in the jejunum, the principal site of parasite infection, while having very low systemic exposure (F = 1%). An oral efficacy experiment in the NSG mouse chronic infection model revealed that 12a provides 99% reduction in parasite shedding after 4 days of treatment at 50 mg/kg BID and three of the four mice remained parasite-free for 10 days post-treatment. Thus, 12a represents a significant progression in the optimization of triazolopyridazines towards the identification of a novel treatment for cryptosporidiosis. Future work includes efforts towards developing SAR and optimizing the triazolopyridazine head and 3,4-dichlorophenyl tail groups.
Experimental Section
General.
Commercially available reagents and solvents were used without further purification unless stated otherwise. Reactions that were heated under microwave irradiation were conducted using a Biotage Inititiator+ Robot8 microwave. HPLC and LC-MS analyses were performed on an Agilent 1100 HPLC/MSD electrospray mass spectrometer in positive ion mode with scan range was 100-1000d. Preparative normal phase chromatography was performed on a CombiFlash Rf+ (Teledyne Isco) with SiliaFlash F60 40-63 μm (230-400 mesh) silica gel (SiliCycle Inc.). Preparative reverse phase HPLC was performed on a CombiFlash Rf+ (Teledyne Isco) equipped with RediSep Rf Gold pre-packed C18 cartridges and an acetonitrile/water/0.05% TFA gradient. NMR spectra were recorded on a Bruker 400 MHz and 700 MHz spectrometer. The signal of the deuterated solvent was used as internal reference. Chemical shifts (δ) are given in ppm and are referenced to residual not fully deuterated solvent signal. Coupling constants (J) are given in Hz. HRMS spectra were recorded on an ABSciex 5600+ instrument. All final compounds were purified to ≥95% as determined by HPLC UV absorbance unless noted otherwise.
3-Chloro-6-hydrazinylpyridazine hydrochloride.
To a microwave vial was added 3,6-dichloropyridazine (1.00 g, 7.00 mmol) and hydrazine monohydrate (0.38 mL, 7.70 mmol). The mixture was dissolved in 10 mL of ethanol and microwaved for 2 h at 100 °C. The crude reaction mixture was extracted three times with dichloromethane. The organic layer was dried with sodium sulfate and concentrated in vacuo to give the crude product (1.69 g). 1H NMR (400 MHz, DMSO-d6) δ ppm 7.08 (d, J = 9.2, 1H), 7.42 (d, J = 9.3, 1H), 8.05 (s, 4H), 8.17 - 8.23 (m, 1H). LC-MS: m/z 145.0 (MH)+, 147.0.
6-Chloro-[1,2,4]triazolo[4,3-b]pyridazine (4).
To a microwave vial was added 3-chloro-6-hydrazinopyridazine (3.24 g, 22.5 mmol). The starting material was dissolved in 15 mL formic acid, sealed, the heated to 100 °C in the microwave reactor for 2 h. The reaction mixture was neutralized with satd NaHCO3. The aqueous layer was then extracted three times with dichloromethane. The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo to give 1.54 g of the crude title compound. 1H NMR (400 MHz, D2O) δ ppm 7.43 - 7.49 (m, 1H), 8.21 - 8.27 (m, 1H), 9.30 - 9.34 (m, 1H). LC-MS m/z 155.0 (MH)+, 157.0.
tert-butyl 4-([1,2,4]triazolo[4,3-b]pyridazin-6-ylamino)piperidine-1-carboxylate (6a).
To a microwave vial with a stirring bar was added 6-Chloro-[1,2,4]triazolo[4,3-b]pyridazine 4 (400 mg, 2.59 mmol) and 4-amino-1-boc-piperidine (1700 mg, 8.50 mmol). The mixture was then dissolved in ethanol, after which the vial was sealed, the heated to 120 °C in the microwave reactor for 8h. The mixture was then concentrated under vacuum and extracted three times with ethyl acetate (30 mL) and the combined organic layers were washed with water. The organic fractions were then dried over Na2SO4, filtered, and concentrated under vacuum. The yellowish oily compound was then used for the next step (200 mg, 24%). LCMS m/z 319.1 (MH)+.
tert-butyl (1-([1,2,4]triazolo[4,3-b]pyridazin-6-yl)piperidin-4-yl)carbamate (6b).
To a microwave vial with a stirring bar was added 6-Chloro-[1,2,4]triazolo[4,3-b]pyridazine 4 (1200 mg, 7.76 mmol) and 4-(N-boc)-piperidine (1690 mg, 8.73 mmol). The mixture was then dissolved in ethanol, after which the vial was sealed, the heated to 120 °C in the microwave reactor for 8h. The mixture was then concentrated under vacuum and extracted three times with ethyl acetate (30 mL) and the combined organic layers were washed with water. The organic fractions were then dried over Na2SO4, filtered, and concentrated under vacuum. The white compound was then used for the next step (940 mg, 38%). LCMS m/z 319.1 (MH)+.
tert-Butyl 4-([1,2,4]triazolo[4,3-b]pyridazin-6-yl)piperazine-1-carboxylate (6c).
To a microwave vial was added 6-chloro-[1,2,4]triazolo[4,3-b]pyridazine 4 (1.54 g, 9.96 mmol), N-Boc-piperazine (2.04 g, 11.0 mmol), and DIEA (2.22 mL, 13.0 mmol). The starting materials were dissolved in ethanol. The vial was sealed and heated to 100 °C in the microwave reactor for 2 hours. Ethanol was removed in vacuo, water was added and the crude product was extracted with dichloromethane three times. The organic layer was dried over sodium sulfate, filtered, and concentrated in vacuo to give 2.16 g of crude 6c. 1H NMR (400 MHz, CDCl3) δ ppm 1.50 (s, 9H), 3.52 - 3.63 (m, 8H), 6.94 (d, J = 10.2, 1H) 7.91 (d, J = 10.2, 1H), 8.79 (s, 1H). LC-MS: m/z 305.1 (MH)+.
tert-butyl 4-([1,2,4]triazolo[4,3-b]pyridazin-6-yl)-1,4-diazepane-1-carboxylate (6d).
To a microwave vial with a stirring bar was added 6-Chloro-[1,2,4]triazolo[4,3-b]pyridazine 4 (200 mg, 1.29 mmol) and homopiperazine (285 mg, 1.42 mmol). The mixture was then dissolved in NMP, after which the vial was sealed, the heated to 120 °C in the microwave reactor for 8h. The mixture was then extracted three times with ethyl acetate (30 mL) and the combined organic layers were washed with water. The organic fractions were then dried over Na2SO4, filtered, and concentrated under vacuum. The brown oily compound was then used for the next step (380 mg, 92%). LCMS m/z 319.1 (MH)+.
tert-butyl 4-([1,2,4]triazolo[4,3-b]pyridazin-6-yl)-2-methylpiperazine-1-carboxylate (6e).
To a microwave vial with a stirring bar was added 6-Chloro-[1,2,4]triazolo[4,3-b]pyridazine 4 (200 mg, 1.29 mmol) and tert-butyl 2-methylpiperazine-1-carboxylate (0.29 mL, 1.42 mmol). The mixture was then dissolved in NMP, after which the vial was sealed, the heated to 160 °C in the microwave reactor for 8h. The mixture was then extracted three times with ethyl acetate (30 mL) and the combined organic layers were washed with water. The organic fractions were then dried over Na2SO4, filtered, and concentrated under vacuum. The brown oily compound was then used for the next step (392 mg, 95%). LCMS m/z 319 (MH)+.
6-(Piperazin-1-yl)-[1,2,4]triazolo[4,3-b]pyridazine hydrochloride (7c).
To a round-bottomed flask was added 6c (2.16 g, 7.10 mmol) and excess 4M HCl in dioxane (12 mL). The reaction mixture was stirred at room temperature for 2 h. Dioxane was removed in vacuo, and the crude product was rinsed with DCM. The DCM was removed in vacuo, then the crude product was rinsed with diethyl ether, which was subsequently removed in vacuo. The crude product was rinsed again with diethyl ether. The diethyl ether was decanted, giving 1.76 g of 7c. 1H NMR (400 MHz, D2O) δ 3.38-3.44 (m,4H), 3.91-3.96 (m, 4H), 7.54-7.60 (m, 1H), 8.08-8.13 (m, 1H), 9.17-9.20 (m, 1H). LC-MS: m/z 205.1 (MH)+.
General procedure Boc group removal (7a-b, 7d-e, 16-17).
Boc-protected amines were suspended in 4M HCl in dioxane (10 mL) in separate round bottom flasks with stirring bars. The mixtures were allowed to stir for 2 h after which they were concentrated and washed with Et2O.
N-(piperidin-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-6-amine hydrochloride (7a).
Brown oily solid (449 mg, 98 %). LC-MS m/z 219 (MH)+.
N-(piperidin-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-6-amine hydrochloride (7b).
White solid (200 mg, 95%). LC-MS m/z 219 (MH)+.
6-(1,4-diazepan-1-yl)-[1,2,4]triazolo[4,3-b]pyridazine hydrochloride (7d).
Yellow crystalline solid (440 mg, 97%). LC-MS m/z 219 (MH)+.
6-(3-methylpiperazin-1-yl)-[1,2,4]triazolo[4,3-b]pyridazine hydrochloride (7e).
Brown solid (458 mg, 99%). LC-MS m/z 219 (MH)+.
6-(1,2,3,6-tetrahydropyridin-4-yl)-[1,2,4]triazolo[4,3-b]pyridazine hydrochloride (16)
Light yellow cream solid (100 mg, 99%). LC-MS m/z 202 (MH)+.
6-(piperidin-4-yl)-[1,2,4]triazolo[4,3-b]pyridazine hydrochloride (17).
Light brown solid (150 mg, 84 %). LC-MS m/z 204 (MH)+.
2-(3,4-dichlorophenyl)-1-[4-({[1,2,4]triazolo[4,3-b]pyridazin-6-yl}amino)piperidin-1-yl]ethan-1-one (8).
A suspension of N-(piperidin-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-6-amine hydrochloride (100 mg, 0.393 mmol) and 3,4-dichlorophenylacetic acid (121 mg, 0.590 mmol) was treated with TEA (50.0 μL, 0.393 mmol) , EDCI (90 mg, 0.390 mmol) and HOBT (53 mg, 0.390 mmol) in 5 mL of DMF was stirred at room temp. After 2 h, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30 mL) and the combined organic layers were washed with aq. NaHCO3. The extraction was done three times. The organic fraction was then dried over Na2SO4, filtered, and concentrated under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide title compound 8 as light yellow solid (38 mg, 0.0941 mmol, 24%). HPLC purity 98%. LCMS m/z 405 (MH)+. 1H NMR (400 MHz, DMSO-d6) δ 9.18 (s, 1H), 7.91 - 7.97 (m, 1H), 7.54 (d, J = 18.34, 2H), 7.15 - 7.28 (m, 1H), 6.74 - 6.88 (m, 1H), 4.53 - 5.27 (br. s, 1H), 4.16 - 4.29 (m, 1H), 4.04 - 4.14 (m, 1H), 3.43 (s, 2H), 3.18 - 3.30 (m, 1H), 3.05 - 3.18 (m, 1H), 2.81 - 2.96 (m, 1H), 1.94 - 2.06 (m, 1H), 1.76 - 1.89 (m, 1H), 1.37 - 1.51 (m, 1H), 1.21 - 1.35 (m, 1H). HRMS (ESI) m/z: [M + H]+ Calcd for C18H18Cl2N6O 405.0992; found 405.0986.
3,4-dichloro-N-(1-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperidin-4-yl)benzamide (9a)
A suspension of 1-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperidin-4-amine hydrochloride (50.0 mg, 0.200 mmol) and 3,4-dichlorobenzoic acid (75.0 mg, 0.390 mmol) was treated with TEA (82.0 μL, 0.590 mmol) , EDCI (75.3 mg, 0.390 mmol) and HOBT (53.1 mg, 0.390 mmol) at room temp. After 2 h, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30 mL) and the combined organic layers were washed with aq. NaHCO3. The extraction was done three times. The organic fraction was then dried over Na2SO4, filtered, and concentrated under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide title compound 9a as white solid (21 mg, 0.054 mmol, 28%). HPLC purity 96%. LCMS m/z 391(MH)+. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.22 (s, 1H) , 8.48 (d, J = 7.09, 1H), 8.04 - 8.21 (m, 2H), 7.67 - 7.90 (m, 2H), 7.43 (d, J = 10.03, 1H), 3.95 - 4.31 (m, 3H), 3.13 (t, J = 12.10, 2H), 1.91 (d, J = 11.49, 2H), 1.60 (q, J = 10.43, 2H). HRMS (ESI) m/z: [M + H]+ Calcd for C17H16Cl2N6O 391.0836; found 391.0829.
2-(3,4-dichlorophenyl)-N-(1-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperidin-4-yl)acetamide (9b)
A suspension of 1-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperidin-4-amine hydrochloride (50.0 mg, 0.200 mmol) and 3,4-dichlorophenylacetic acid (60.5 mg, 0.300 mmol) was treated with TEA (82.0 μL, 0.590 mmol) , EDCI (43.0 mg, 0.220 mmol) and HOBT (30.3 mg, 0.220 mmol) at room temp. After 2 h, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30 mL) and the combined organic layers were washed with aq. NaHCO3. The extraction was done three times. The organic fraction was then dried over Na2SO4, filtered, and concentrated under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide the title compound 9b as yellow solid (27 mg, 0.067 mmol, 30%). HPLC purity 96%. LCMS m/z 405 (MH)+. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.57 (s, 1H), 8.31 (s, 1H), 7.57 (d, J = 8.31, 1H), 7.52 (d, J = 2.20, 1H), 7.40 (d, J = 9.29, 1H), 7.17 - 7.30 (m, 1H), 4.00 - 4.21 (m, 2H), 3.44 - 3.73 (m, 2H), 3.02 - 3.25 (m, 2H), 2.56 - 2.94 (m, 1H), 1.88 - 2.07 (m, 2H), 1.39 - 1.79 (m, 2H). 13C NMR (176 MHz, DMSO-d6) δ ppm 168.42, 154.96, 138.21, 137.47, 130.85, 130.49, 130.16, 129.28, 128.90, 123.97, 115.11, 45.62, 44.29, 40.95, 30.52. HRMS (ESI) m/z: [M + H]+ Calcd for C18H18Cl2N6O 405.0992; found 405.0988.
4-([1,2,4]triazolo[4,3-b]pyridazin-6-yl)-N-(3,4-dichlorophenyl)piperazine-1-carboxamide (10a).
A suspension of 7c (100 mg, 0.420 mmol) and 1,2-dichloro-4-isocyanatobenzene (148 mg, 1.41 mmol) was treated with DIEA (50 μL, 0.290 mmol) in DMF (4 mL) was stirred for overnight at room temp. The crude mixture was then concentrated under vacuum and purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide title compound as yellowish crystalline solid (13 mg, 0.034 mmol, 17%), HPLC purity 97%. LCMS m/z 392 (MH)+. 1H NMR (400 MHz, DMSO-d6) δ 9.24 (s, 1H), 8.93 (s, 1H), 8.13 (d, J = 10.03, 1H), 7.86 (d, J = 1.71, 1H), 7.46 - 7.52 (m, 2H), 7.43 (d, J = 10.27, 1H), 3.61 (s, 8H).
N-(3,4-dichlorophenyl)-4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}-1,4-diazepane-1-carboxamide (10b).
A suspension of the intermediate 7d (220 mg, 0.864 mmol) and 3,4-dichlorophenyl isocyanate (243 mg, 1.30 mmol) was treated with DIEA (1.00 mL, 0.00600 mmol at room temperature. After reaction period of 2 hours, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30mL) and wash with NaHCO3.The extraction was done three times. The organic fraction was then dried over Na2SO4 under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→5% CH3CN/H2O) as to provide title compound 10b as white solid (54 mg, 0.13 mmol, 25%). HPLC purity 97%. LCMS m/z 406 (MH)+. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.12 (d, J = 0.73, 1H), 8.63 (s, 1H), 8.04 (dd, J = 10.27, 0.73, 1H), 7.71 - 7.81 (m, 1H), 7.35 - 7.44 (m, 2H), 7.29 (d, J = 10.03, 1H), 3.77 - 3.86 (m, 2H), 3.65 - 3.74 (m, 2H), 3.49 (t, J = 5.87, 2H), 2.54 (s, 2H), 1.82 - 1.97 (m, 2H). HRMS (ESI) m/z: [M + H]+ Calcd for C17H17Cl2N7O 406.0945; found 406.0938.
2-(3,4-dichlorophenyl)-1-(2-methyl-4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethan-1-one (10c).
A suspension of 7e (100.0 mg, 0.390 mmol) and 3,4-dichlorophenylacetic acid (120.0 mg, 0.210 mmol) was treated with DIPEA (50 μL, 0.590 mmol), and HBTU (224 mg, 0.590 mmol) in 4 mL was stirred at room temp. After 2 h, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30 mL) and the combined organic layers were washed with aq. NaHCO3. The extraction was done three times. The organic fraction was then dried over Na2SO4, filtered, and concentrated under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide 10c as light yellow solid (42 mg, 0.10 mmol, 27%). HPLC purity 97%. LCMS m/z 405 (MH)+. 1H NMR (400 MHz, CDCl3) δ ppm 1.09 (d, J = 6.36, 3H), 2.82 - 3.13 (m, 2H), 3.17 - 3.28 (m, 1H), 3.38 - 3.51 (m, 1H), 3.80 (br. s., 2H), 3.86 - 4.12 (m, 2H), 4.13 - 4.30 (m, 1H), 4.32 - 4.72 (m, 1H), 7.17 - 7.29 (m, 1H), 7.34 - 7.43 (m, 1H), 7.56 (d, J = 8.31, 2H), 8.10 (d, J = 10.03, 1H), 9.21 (s, 1H). HRMS (ESI) (ESI) m/z: [M + H]+ Calcd for C18H18Cl2N6O 405.0992; found 405.0986.
N-(3,4-dichlorophenyl)-N-methyl-4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazine-1-carboxamide (11).
100.0 mg (0.256 mmol) of urea 2 was weighed into a dried flask and dissolved with 5 mL of THF. 12.3 mg (0.513 mmol) of NaH was added and stirred with the flask in an ice bath. The reaction mixture allowed to stir for about 15 minutes after which was added 17.5 μL (0.583 mmol) of methyl iodide and allowed to stir overnight. the mixture was then concentrated under vacuum and extracted using ethyl acetate (30 mL). The extraction was done three times. The organic fraction was then dried over Na2SO4, filtered, and concentrated under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide title compound 11 as yellowish solid (22 mg, 0.054 mmol, 22%), HPLC purity 97%. LCMS m/z 406 (MH)+. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.21 (s, 1H), 8.09 (d, J = 10.27, 1H), 7.45 - 7.59 (m, 2H), 7.32 (d, J = 10.27, 1H), 7.15 (dd, J = 8.80, 2.45, 1H), 3.43 - 3.53 (m, 4H), 3.15 (s, 2H), 2.54 (s, 4H). HRMS (ESI) m/z: [M + H]+ Calcd for C17H17Cl2N7O 406.0945; found 406.0940.
2-(3,4-Dichlorophenyl)-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethan-1-one (12a; SLU-2633).
A suspension of 7c (103.8 mg, 0.43 mmol) and 3,4-dichlorophenylacetic acid (127.8 mg, 0.62 mmol) was treated with DIEA (50 μL, 0.29 mmol) and HBTU (129.8 mg, 0.34 mmol) at room temp. After 2 h, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30 mL) and the combined organic layers were washed with aq. NaHCO3. The extraction was done three times. The organic fraction was then dried over Na2SO4, filtered, and concentrated under vacuum. The crude compound was purified by silica gel chromatography with 0-30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) to provide the title compound 12a as a yellowish solid (20 mg, 0.051 mmol, 12%). HPLC purity 98%. LCMS m/z 391 (M+1). 1H NMR (400 MHz, CDCl3) δ 8.80 (d, J = 0.7, 1H), 7.91 - 7.96 (m, 1H), 7.35 - 7.40 (m, 1H), 7.07 - 7.12 (m, 1H), 6.99 - 7.03 (m, 1H), 6.92 (d, J = 10.0, 1H), 3.76 (s, 4H), 3.61 - 3.65 (m, 2H), 3.53 - 3.58 (m, 2H), 3.48 - 3.52 (m, 2H). 13C NMR (176 MHz, DMSO-d6) δ 168.5, 155.4, 141.9, 138.5, 137.2, 131.6, 130.7, 130.3, 130.0, 129.1, 124.3, 115.2, 45.5, 45.2, 44.5, 40.8, 38.1. HRMS (ESI) m/z: [M + H]+ Calcd for C17H16Cl2N6O 391.0841 ; found 391.0827.
1-(3,4-dichlorobenzoyl)-4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazine (12b).
A suspension of 7c (50.0 mg, 0.210 mmol) and 3,4-dichlorobenzoic acid (39.7 mg, 0.210 mmol) was treated with TEA (90 μL, 0.65 mmol), EDCI (45.4, 0.24 mmol) and HOBT (32.0 mg, 0.24 mmol) at room temp. After 2 h, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30 mL) and the combined organic layers were washed with aq. NaHCO3. The extraction was done three times. The organic fraction was then dried over Na2SO4, filtered, and concentrated under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide 12b as yellowish crystalline solid (13 mg, 0.034 mmol, 17%), HPLC purity 97%. LCMS m/z 377 (MH)+. 1H NMR (400 MHz, CDCl3) δ ppm 8.83 (br. s., 1H), 8.10 (d, J = 9.78, 1H), 7.49 - 7.60 (m, 2H), 7.30 (dd, J = 8.07, 1.96, 1H), 7.06 (d, J = 9.78, 1H), 3.65 (br. s., 8H). HRMS (ESI) m/z: [M + H]+ Calcd for C16H14Cl2N6O 377.0679; found 377.0672.
2-(3,4-dichlorophenyl)-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}-1,4-diazepan-1-yl)ethan-1-one (12c).
A suspension of amine 7d (220 mg, 0.89 mmol) and 3,4-dichlorophenylacetic acid, (266 mg, 1.20 mmol) in DMF (5 mL) was treated with DIEA (0.30 mL, 0.30 mmol) and HBTU (328 mg, 0.86 mmol) at room temperature. After 2 h, the mixture was concentrated under vacuum and extracted with ethyl acetate (30 mL) and the combined organic layers were washed with NaHCO3. The extraction was done three times. The organic fractions were then dried over Na2SO4, filtered and concentrated under vacuum. The crude compound was purified by silica gel chromatography (0 to 30% methanol/ethyl acetate) and further purified by reverse-phase HPLC (5 to 95% CH3CN/H2O) to furnish 12c as a yellowish sticky solid (68 mg, 0.17 mmol, 43%). LCMS m/z 405 (MH)+. HPLC purity 97%. 1H NMR (700 MHz, DMSO-d6) δ 9.16 (s, 1H), 8.04 (d, J = 10.72, 1H), 7.38 - 7.42 (m, 2H), 7.36 (d, J = 8.58, 1H), 7.30 (d, J = 2.14, 1H)*, 7.25 (dd, J = 10.01, 16.44, 2H)*, 7.10 (dd, J = 1.79, 8.22, 1H)*, 7.01 (dd, J = 1.79, 8.22, 1H), 3.79 - 3.84 (m, 2H), 3.65 - 3.76 (m, 4H), 3.56 (t, J = 6.07, 2H), 3.41 - 3.47 (m, 2H), 1.79 (quint d, J = 5.89, 11.48, 2H). 13C NMR (176 MHz, DMSO-d6) δ 169.6*, 169.3, 154.0*, 153.8, 141.8*, 141.8, 138.4*, 138.3, 137.5*, 137.3, 131.5*, 131.4, 130.7, 130.2*, 130.2, 129.8, 129.8*, 129.1, 124.5*, 124.3, 114.3*, 114.1, 48.8*, 48.0, 47.5, 46.9*, 46.6, 45.3, 44.6, 38.2, 37.9*. *indicates presumed ~1:1 rotamer peaks observed in NMR. HRMS (ESI) m/z: [M + H]+ Calcd for C18H18Cl2N6O 405.0992; found 405.0986.
2-(3,4-dichlorophenyl)-1-(2-methyl-4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethan-1-one (12d).
A suspension of amine 7e (100 mg, 0.39 mmol) and 3,4-dichlorophenylacetic acid, (120 mg, 0.59 mmol) in DMF (5 mL) was treated with DIEA (50 μL, 0.29 mmol) and HBTU (129.8 mg, 0.34 mmol) at room temperature. After 2 h, the mixture was concentrated under vacuum and extracted with ethyl acetate (30 mL) and the combined organic layers were washed with NaHCO3. The extraction was done three times. The organic fractions were then dried over Na2SO4, filtered and concentrated under vacuum. The crude compound was purified by silica gel chromatography (0 to 30% methanol/ethyl acetate) and further purified by reverse-phase HPLC (5 to 95% CH3CN/H2O) to furnish 12d as a white solid (42 mg, 0.10 mmol, 27%). LCMS m/z 405 (MH)+. HPLC purity 97%. 1H NMR (400 MHz, DMSO-d6) δ 9.21 (s, 1H), 8.10 (d, J = 10.03, 1H), 7.56 (d, J = 8.31, 2H), 7.34 - 7.43 (m, 1H), 7.17 - 7.29 (m, 1H), 4.32 - 4.72 (m, 1H), 4.13 - 4.30 (m, 1H), 3.86- 4.12 (m, 2H), 3.80 (br. s., 2H) , 3.38 - 3.51 (m, 1H), 3.17 - 3.28 (m, 1H), 2.82 - 3.13 (m, 2H), 1.09 (d, J = 6.36, 3H). HRMS (ESI) m/z: [M + H]+ Calcd for C18H18Cl2N6O 405.0992; found 405.0986.
1-[(3,4-dichlorophenyl)methanesulfonyl]-4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazine (13).
A suspension of 7c (110 mg, 0.460 mmol), 3,4-dichlorophenylmethylsulfonyl chloride (120 mg, 0.49 mmol) were treated with DIEA (0.100 mL, 0.560 mmol) and dissolved in 4 mL of DMF and placed in an ice bath. After 3hr, the mixture was then concentrated and purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O as to provide title compound 13 as white solid (20 mg, 0.047 mmol, 11%), HPLC purity 98%. LCMS m/z 427 (MH)+. 1H NMR (400 MHz, CDCl3) δ ppm 8.84 (s, 1H), 8.19 (d, J = 10.03, 1H), 7.43 - 7.55 (m, 2H), 7.25 (d, J = 1.96, 1H), 7.06 (d, J = 10.27, 1H), 4.20 (s, 2H), 3.56 - 3.68 (m, 4H), 3.28 - 3.38 (m, 4H).. HRMS (ESI) m/z: [M + H]+ Calcd for C16H16Cl2N6O2S 427.0511; found 427.0500.
tert-butyl 4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}-1,2,3,6-tetrahydropyridine-1-carboxylate (14).
6-chloro-[1,2,4]triazolo[4,3-b]pyridazine (700 mg, 4.53 mmol), 3,6-dihydro-2H-pyridine-1-N-Boc-boronic acid, pinacol ester (1.82 mg, 5.89 mmol), and Cs2CO3 (4.43 mg, 13.6 mmol) were weighed into a round bottom flask and dissolved in a solvent mixture of toluene, water and ethanol in the ratio 9:3:1 respectively. The reaction mixture was vacuum and purged with nitrogen for 20 mins., after which the reaction was allowed to reflux for 18 h, after which the reaction mixture was then allowed to cool and filtered. The reaction mixture was purified using EtOAc and hexane (light brown solid, 3.80 g, 33%).
tert-butyl 4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperidine-1-carboxylate (15).
Hydrogenation reaction vessel was charged with tert-butyl 4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}-1,2,3,6-tetrahydropyridine-1-carboxylate 14 (789 mg, 2.55 mmol) and Pd/C(130 mg, 10% mol) was dissolved in ~100 mL of EtOH:MeOH (50:50). The reaction mixture was hydrogenated by a pressure of 60 psi of H2(g) for 48h. The reaction mixture was filtered and purified by flash chromatography using normal phase with EtOAc and hexane (brown oil, 750 mg, 97%). LCMS m/z 304 (MH)+.
N-(3,4-dichlorophenyl)-4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}cyclohex-3-ene-1-carboxamide (18a).
A suspension of 16 (100 mg, 0.50 mmol) and 3,4-dichlorophenyisocyanate (120 mg, 0.64 mmol) in DMF was treated with DIEA (100 μL, 0.50 mmol) at 0 °C and allowed to room temperature.. After reaction period of 2 hours, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30mL) and wash with NaHCO3.The extraction was done three times. The organic fraction was then dried over Na2SO4 under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide the title compound 18a as white solid (20 mg, 0.051 mmol, 10%), HPLC purity 98%. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.61 (s, 1H), 8.89 (s, 1H), 8.33 (d, J = 9.78, 1H), 7.75 - 7.93 (m, 2H), 7.50 (d, J = 1.47, 2H), 7.03 (br. s., 1H), 4.29 (d, J = 3.18, 2H), 3.70 (t, J = 5.62, 2H), 2.67 (br. s., 2H). 13C NMR (176 MHz, DMSO-d6) δ ppm 158.4, 158.2, 154.4, 153.2, 140.8, 139.3, 131.4, 130.8, 130.5, 130.2, 123.8, 123.0, 120.5, 119.4, 119.1, 44.1, 24.6. HRMS (ESI) m/z: [M + H]+ Calcd for C17H14Cl2N6O 389.0684; found 389.0674.
2-(3,4-dichlorophenyl)-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}-1,2,3,6-tetrahydropyridin-1-yl)ethan-1-one (18b).
A suspension of 16 (171 mg, 0.719 mmol) and 3,4-dichlorophenyl acetic acid (220 mg, 1.07 mmol) was treated with DIEA (0.500 mL, mmol) and HBTU (200 mg, 0.528 mmol) at room temp. After 2 h, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30 mL) and the combined organic layers were washed with aq. NaHCO3. The extraction was done three times. The organic fraction was then dried over Na2SO4, filtered, and concentrated under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide title compound 18b as brownish solid (46 mg, 0.12 mmol, 17%), HPLC purity 96%, LCMS m/z 388(MH)+. 1H NMR (400 MHz, DMSO-d6) δ 9.60 (br. s., 1H), 8.32 (br. s., 2H), 7.79 (br. s., 2H), 7.46 - 7.64 (m, 3H), 7.24 (br. s., 2H), 6.98 (br. s., 2H), 4.17 4.43 (m, 3H), 3.64 - 3.94 (m, 6H), 2.64 (br. s., 4H). HRMS (ESI) m/z: [M + H]+ Calcd for C18H15Cl2N5O 388.0727; found 388.0722.
N-(3,4-dichlorophenyl)-4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperidine-1-carboxamide (19a).
A suspension of the 17 (40.0 mg, 0.710 mmol) and 3,4-dichlorophenyl isocyanate (51.4 mg, 0.25 mmol) was treated with DIEA (50 μL, 0.29 mmol at room temperature. After reaction period of 2 hours, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30mL) and wash with NaHCO3.The extraction was done three times. The organic fraction was then dried over Na2SO4 under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide title compound 19a as white deliquescent solid (30 mg, 0.077 mmol, 46%), HPLC purity 95%. LCMS m/z 390 (MH)+. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.58 (s, 1H), 8.83 (s, 1H), 8.31 (d, J = 9.54, 1H), 7.86 (s, 1H), 7.23 - 7.57 (m, 2H), 4.25 (d, J = 12.96, 2H), 3.07 - 3.19 (m, 2H), 2.97 (t, J = 11.86, 2H), 1.99 (d, J = 11.49, 2H), 1.57 - 1.77 (m, 1H). HRMS (ESI) m/z: [M + H]+ Calcd for C17H16Cl2N6O 391.0836; found 391.0827.
2-(3,4-dichlorophenyl)-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperidin-1-yl)ethan-1-one (19b).
A suspension of 17 (40.0 mg, 0.710 mmol) and 3,4-dichlorophenyl acetic acid (51.4 mg, 0.25 mmol) was treated with DIEA (0.500 mL, 0.29 mmol) and HBTU (63.4 mg, 0.17 mmol) at room temp. After 2 h, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30 mL) and the combined organic layers were washed with aq. NaHCO3. The extraction was done three times. The organic fraction was then dried over Na2SO4, filtered, and concentrated under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide title compound 19b as white solid (34 mg, 0.087 mmol, 60%), HPLC purity 95%, LCMS m/z 391.0(M+1), 1H NMR (400 MHz, DMSO-d6) δ ppm 9.57 (s, 1H), 8.32 (d, J = 9.78, 1H), 7.57 (d, J = 8.31, 1H), 7.52 (d, J = 1.71, 1H), 7.41 (d, J = 9.54, 1H), 7.24 (dd, J = 8.19, 1.83, 1H), 4.50 (d, J = 12.96, 2H), 4.10 (d, J = 13.20, 2H), 3.06 - 3.26 (m, 1H), 2.73 (t, J = 11.49, 1H), 1.98 (d, J = 12.47, 2H), 1.48 - 1.78 (m, 1H), 1.17 - 1.33 (m, 2H). HRMS (ESI) m/z: [M + H]+ Calcd for C18H17Cl2N5O 390.0883; found 390.0873.
3,4-dichlorophenylglyoxylic acid (21a).
To a round bottom flask, with a stirring bar was added 3,4-dichloroacetophenone (100 mg, 0.53 mmol) and selenium dioxide (88 mg, 0.79 mmol), then 5 mL of anhydrous pyridine was quickly added. The reaction mixture was then stirred and heated under a nitrogen atmosphere for 110°C for 1 h. The temperature was lowered to 90°C for another 4h. The reaction mixture was allowed overnight, after which was filtered and concentrated, 20 mL of 2N NaOH was added and later, 30mL of 2N HCl was added. The product was filtered from the mixture. The residue was dissolved in EtOAc and concentrated under vacuum. Crude product (white to pinkish solid, 299 mg, 99%) was used for the next reaction.
Ethyl-3,4-dichlorophenylglyoxyloate (21b).
50 mL round bottom flask was charged with 3,4-dichlorophenylglyoxylic acid (400 mg, 1.83 mmol) and which dissolved with 30 mL of EtOH. The reaction vessel was cooled to 0°C in water bath. Followed by addition of 2 mL of thionyl chloride (27 mmol). The reaction vessel was then refluxed at 80°C for overnight. The reaction mixture was concentrated under vacuum and extracted using EtOAc and water and washed with NaHCO3. The organic extract was then dried using NaSO4, filtered and concentrated to give the title compound as a yellow oily compound (457 mg, 85%).
1-(3,4-dichlorophenyl)-2-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethane-1,2-dione (22).
A suspension of the 7c (100 mg, 0.39 mmol) and 3,4-dichlorophenylacetic acid, (136 mg, 0.62 mmol) was treated with DIEA (50 μL, 0.29 mmol) and HBTU (129.8 mg, 0.34 mmol) at room temperature. After reaction period of 2 hours, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30mL) and wash with NaHCO3.The extraction was done three times. The organic fraction was then dried over Na2SO4 under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) to provide the title compound 22 as white solid (32 mg, 0.079 mmol, 35%), HPLC purity 95%. 1H NMR (400 MHz, DMSO-d6) ppm 9.25 (s, 1H), 8.08 - 8.18(m, 3H), 7.91 (d, J = 0.98, 1H), 7.40 (d, J = 10.27, 1H), 3.77 (d, J = 9.29, 4H), 3.45 - 3.61 (m, 4H). HRMS (ESI) m/z: [M + H]+ Calcd for C17H14Cl2N6O2 405.0628; found 405.0621.
2-(3,4-dichlorophenyl)-2,2-difluoroacetic acid (24).
To a plastic tube was then dried by flashing N2(g) through and charged with 6 mL of anhydrous DCM, followed by addition of ethyl-3,4-dichlorophenylglyoxyloate (457 mg, 1.85 mmol) and addition of 4-tert-butyl-2,6-dimethylphenylsulfur trifluoride (694.7 mg, 2.77 mmol) and allowed to str for about 5 minutes. ~44 μL of HF-Py was added lastly. The reaction mixture was stirred for ~10 minutes after which the temperature was increased to 45 °C for 3 hours after which it was allowed to stir at room temperature for 16 h. The reaction was quenched with NaHCO3 and extracted with EtOAc and water. The crude brown oil compound was then subjected to hydrolysis (brown solid compound, 460 mg, crude yield 98%).
2-(3,4-dichlorophenyl)-2,2-difluoro-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethan-1-one (25).
A suspension of the 7c (304 mg, 1.18 mmol) and 3,4-dichlorophenyl,-2,2-difluoroacetic acid 24 (300 mg, 1.24 mmol) was treated with DIEA (0.21 mL, 1.23 mmol) and HBTU (480 mg, 1.26 mmol) at room temperature. After reaction period of 2 hours, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30mL) and wash with NaHCO3.The extraction was done three times. The organic fraction was then dried over Na2SO4 under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide the title compound 25 as white solid (64 mg, 0.15 mmol, 13%), HPLC purity 95%, LCMS m/z 427 (MH)+. 19F NMR (376 MHz, DMSO-d6) δ ppm −93.22 (s, 1 F). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.24 (d, J = 0.49, 1H), 8.09 - 8.19(m, 2H), 7.91 (d, J = 0.73, 2H), 7.39 (d, J = 10.03, 1H), 3.77 (d, J = 9.78, 4H), 3.45 - 3.61 (m, 4H). HRMS (ESI) m/z: [M + H]+ Calcd for 427.0647; found 427.0643.
1-[1-(3,4-dichlorophenyl)cyclopropanecarbonyl]-4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazine (27).
A suspension of the 7c (100 mg, 0.39 mmol) and 3,4-dichlorophenylacetic acid, (143 mg, 0.62 mmol) was treated with DIEA (50 μL, 0.29 mmol) and HBTU (129.8 mg, 0.34 mmol) at room temperature. After reaction period of 2 hours, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30mL) and wash with NaHCO3.The extraction was done three times. The organic fraction was then dried over Na2SO4 under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as the title compound 27 as yellow solid (93 mg, 0.22 mmol, 57%), HPLC purity 97%, LCMS m/z 417 (MH)+. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.20 (s, 1H), 8.09 (d, J = 10.27, 1H), 7.56 (d, J = 8.31, 1H), 7.38 (d, J = 2.20, 1H), 7.32 (d, J = 10.27, 1H), 7.20 (dd, J = 8.56, 2.20, 1H), 3.55 (br. s., 8H), 1.24 - 1.42 (m, 4H). HRMS (ESI) m/z: [M + H]+ Calcd for 417.0992; found 417.0986.
1-(4-([1,2,4]triazolo[4,3-b]pyridazin-6-yl)piperazin-1-yl)-2-phenylethanone (29a):
A suspension of 7c 100 mg ( 0.39 mmol) and phenylacetic acid 107 mg (0.79 mmol) was treated with DIEA 7.0 μL (0.39 mmol) and HBTU (179 mg (0.47 mmol) and at room temp. After 2 h, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30 mL) and the combined organic layers were washed with aq. NaHCO3. The extraction was done three times. The organic fraction was then dried over Na2SO4, filtered, and concentrated under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O)) to provide the title compound 29a as a light-yellow solid (18.00 mg, 0.056 mmol, 14%). HPLC purity 95%. LCMS m/z 323 (MH)+. 1H NMR (400 MHz, CDCl3) δ ppm 8.77 (s, 1H), 7.91 (d, J = 10.27, 1H), 7.19 - 7.45 (m, 5H), 6.94 (d, J = 10.03, 1H), 3.81 (s, 2H), 3.50 - 3.69 (m, 4H), 3.30 - 3.48 (m, 4H). HRMS (ESI) m/z: [M + H]+ Calcd for C17H17Cl2N7O 323.1615; found 323.1609.
2-(naphthalen-1-yl)-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethan-1-one (29b).
Purchased from Life Chemicals (www.lifechemical.com; catalog F5123-0045).
2-(naphthalen-2-yl)-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethan-1-one (29c).
A suspension of the 7c (100 mg, 0.39 mmol) and 2-naphthylacetic acid, (155 mg, 0.92 mmol) was treated with DIEA (50 μL, 0.29 mmol) and HBTU (129.8 mg, 0.34 mmol) at room temperature. After reaction period of 2 hours, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30mL) and wash with NaHCO3.The extraction was done three times. The organic fraction was then dried over Na2SO4 under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide title compound 29c as white solid (73 mg, 0.20 mmol, 47%), HPLC purity 97%, LCMS m/z 373 (MH)+. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.22 (s, 1H), 8.09 (d, J = 10.03, 1H), 7.86 (d, J = 8.31, 3H), 7.76 (s, 1H), 7.48 (s, 2H), 7.37 (d, J = 10.03, 2H), 3.96 (s, 2H), 3.62 - 3.73 (m, 4H), 3.47 - 3.58 (m, 4H). HRMS (ESI) m/z: [M + H]+ Calcd for C21H20N6O 373.1771; found 373.1767.
2-(3-methoxyphenyl)-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethan-1-one (29d).
3-Methoxy-phenylacetic acid (100 mg, 0.6 mmol) was dissolved in 10 mL of DMF. To the solution was added 7c (147 mg, 0.72 mmol), HOBt (162 mg, 1.2 mmol), EDCI (230 mg, 1.2 mmol), and TEA (0.25 mL, 1.8 mmol). The reaction mixture was allowed to stir overnight at room temperature. The DMF was removed in vacuo and the remaining solid was extracted 3 times with ethyl acetate, washing 2 times with water and once with brine. The organic layer was dried with Na2SO3, filtered, and concentrated. The remaining oil was purified via Reverse Phase Flash Chromatography (5→95% CH3CN/H2O) to give 29d as a yellow solid (102 mg, 0.29 mmol, 48%), HPLC purity 95%, LCMs m/z 353 (MH)+. 1H NMR (400 MHz, DMSO-d6) δ ppm 3.49 (d, J = 18.8, 4H), 3.62 (br.s., 4H), 3.67 - 3.76 (m, 6H), 6.87 (d, J = 8.2, 2H), 7.16 (d, J = 7.8, 2H), 7.37 (d, J = 10.0, 1H), 8.10 (d, J = 10.0, 1H), 9.22 (s, 1H). HRMS (ESI) m/z: [M + H]+ Calcd for 353.1721; found 353.1714.
2-(4-methoxyphenyl)-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethan-1-one (29e).
4-Methoxy-phenylacetic acid (100 mg, 0.6 mmol) was dissolved in 10 mL of DMF. To the solution was added 7c (147 mg, 0.72 mmol), HOBt (162 mg, 1.2 mmol), EDCI (230 mg, 1.2 mmol), and TEA (0.25 mL, 1.8 mmol). The reaction mixture was allowed to stir overnight at room temperature. The DMF was removed in vacuo and the remaining solid was extracted 3 times with ethyl acetate, washing 2 times with water and once with brine. The organic layer was dried with Na2SO3, filtered, and concentrated. The remaining oil was purified via Reverse Phase Flash Chromatography (5→95% CH3CN/H2O) to give 29e as a yellow solid (114 mg, 0.32 mmol, 54%), HPLC purity 95%, LCMs m/z 353 (MH)+. 1H NMR (400 MHz, DMSO-d6) δ ppm 3.49 (d, J = 18.8, 4H), 3.62 (br.s., 4H), 3.67 - 3.76 (m, 6H), 6.87 (d, J = 8.2, 2H), 7.16 (d, J = 7.8, 2H), 7.37 (d, J = 10.0, 1H), 8.10 (d, J = 10.0, 1H), 9.22 (s, 1H). HRMS (ESI) m/z: [M + H]+ Calcd for 353.1721; found 353.1714.
2-(4-nitrophenyl)-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethan-1-one (29f).
A suspension of the 7c (100 mg, 0.39 mmol) and 4-nitrophenylacetic acid, (151 mg, 0.83 mmol) was treated with DIEA (50 μL, 0.29 mmol) and HBTU (129.8 mg, 0.34 mmol) at room temperature. After reaction period of 2 hours, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30mL) and wash with NaHCO3.The extraction was done three times. The organic fraction was then dried over Na2SO4 under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide the title compound 29f as white solid (20 mg, 0.054 mmol, 14%), HPLC 97%, LCMS m/z 367 (MH)+. 1H NMR (700 MHz, DMSO-d6) δ ppm 9.23 (s, 1H), 8.16 - 8.22 (m, 2H), 8.08 - 8.14 (m, 1H), 7.48 - 7.57 (m, 1H), 7.34 - 7.44 (m, 1H), 5.75 (s, 1H), 3.98 (s, 2H), 3.51 - 3.75 (m, 8H). HRMS (ESI) m/z: [M + H]+ Calcd for C17H17N7O3 368.1466; found 368.1462.
4-[2-oxo-2-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethyl]benzonitrile (29g).
A suspension of the intermediate 7c synthesized in step 4 (70 mg, 0.29 mmol) and 4-cyanophenylacetic acid, (93.7 mg, 0.58 mmol) was treated with DIEA (47 μL, 0.29 mmol) and HBTU (104 mg, 0.27 mmol) at room temperature. After reaction period of 2 hours, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30mL) and wash with NaHCO3.The extraction was done three times. The organic fraction was then dried over Na2SO4 under vacuum. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide the title compound 29g as light-yellow solid (25 mg, 0.072 mmol, 26%), HPLC purity 95%, LCMS m/z 348 (MH)+. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.17 - 9.29 (m, 1H), 8.01 - 8.17 (m, 1H), 7.77 (s, 2H), 7.42 - 7.48 (m, 2H), 7.36 - 7.42 (m, 1H), 3.92 (s, 2H), 3.51 - 3.72 (m, 8H). HRMS (ESI) m/z: [M + H]+ Calcd for C18H17N7O 348.1568; found 348.1556.
2-(4-fluorophenyl)-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethan-1-one (29h).
A suspension of intermediate 7c (103.8mg,0.43mmol) and 4-fluorophenylacetic acid (127.8 mg, 0.62 mmol) was treated with DIEA (0.05mL) and HBTU (129.8mg) at room temperature. After reaction period of 2 hours, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30mL) and wash with NaHCO3.The extraction was done three times. The organic fraction was then dried over Na2SO4 under vacuum. The LCMS was taken. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide the title compound 29h as yellowish solid (50 mg, 0.088 mmol, 35%), HPLC purity 98%, LCMS m/z 341.1 (MH)+. 1H NMR (400 MHz, CDCl3) δ ppm 8.79 (s, 1H), 7.92 (d, J = 10.03, 1H), 7.22 - 7.26 (m, 2H), 7.05 (t, J = 8.68, 2H), 6.91 (d, J = 10.27, 1H), 3.79 - 3.84 (m, 2H), 3.77 (s, 2H), 3.59 - 3.66 (m, 2H), 3.51 - 3.57 (m, 2H), 3.44 (d, J = 5.62, 2H). 13C NMR (176 MHz, DMSO-d6) δ 169.2, 161.7, 160.3, 155.3, 141.8, 138.5, 132.0, 131.1, 124.3, 115.2, 115.1, 115.0, 45.4, 45.1, 44.6, 40.7, 38.5. HRMS (ESI) m/z: [M + H]+ Calcd for C17H17FN6O 134.1521; found 341.1513.
2-(4-chlorophenyl)-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethan-1-one (29i).
A suspension of intermediate 7c (103.0mg, 0.43mmol) and 4-chlorophenylacetic acid(109.9mg,0.64mmol) was treated with DIEA (0.05mL) and HBTU (106mg) at room temperature. After reaction period of 2 hour, the mixture was then concentrated under vacuum and extracted using ethyl acetate (30mL) and wash with NaHCO3.The extraction was done three times. The organic fraction was then dried over Na2SO4 under vacuum. The LCMS was taken. The crude compound was purified by silica gel chromatography with 0→30% methanol/ethyl acetate and further purified by reverse-phase HPLC (5→95% CH3CN/H2O) as to provide the title compound 29i as yellowish white solid (30 mg, 0.084 mmol, 18%), HPLC purity 98%. LCMS m/z 357 (MH)+. 1H NMR (400 MHz, CDCl3) δ 8.79 (d, J = 0.73, 1H), 7.92 (dd, J = 0.61, 10.15, 1H), 7.29 - 7.36 (m, 2H), 7.18 - 7.24 (m, 2H), 6.91 (d, J = 10.03, 1H), 3.78 - 3.84 (m, 2H), 3.77 (s, 2H), 3.59 - 3.64 (m, 2H), 3.51 - 3.57 (m, 2H), 3.40 - 3.48 (m, 2H). HRMS (ESI) m/z: [M + H]+ Calcd for C17H17ClN6O 357.1230; found 357.1221.
2-(3-chlorophenyl)-1-(4-{[1,2,4]triazolo[4,3-b]pyridazin-6-yl}piperazin-1-yl)ethan-1-one (29j).
3-chloro-phenylacetic acid (100 mg, 0.63 mmol) was dissolved in 10 mL of DMF. To the solution was added 7c (155 mg, 0.72 mmol), HOBt (172 mg, 1.3 mmol), EDCI (244 mg, 1.3 mmol), and TEA (0.26 mL, 1.9 mmol). The reaction mixture was allowed to stir overnight at room temperature. The DMF was removed in vacuo and the remaining solid was extracted 3 times with ethyl acetate, washing 2 times with water and once with brine. The organic layer was dried with Na2SO3, filtered, and concentrated. The remaining oil was purified via Reverse Phase Flash Chromatography (5→95% CH3CN/H2O) to give 29j as a yellow solid (132 mg, 0.34 mmol, 55%), HPLC purity 95%, LCMS m/z 357 (MH)+, 359. 1H NMR (400 MHz, DMSO-d6) δ 3.55 (d, J = 5.4, 4H), 3.65 (d, J = 18.1, 4H), 3.81 (s, 2H), 7.18-7.24 (m, 1H), 7.29-7.35 (m, 3H), 7.40 (s, 1H), 8.08 - 8.14 (m, 1H), 9.23 (s, 1H). HRMS (ESI) m/z: [M + H]+ Calcd for 357.1225; found 357.1221.
Microsome Stability Assay.
Metabolic stability was assessed using human and mouse liver microsomes (Sekisui Xenotech, Kansas City, MO). Pooled, mixed-gender liver microsome preparations were diluted to a final cytochrome P450 content of 0.25 μM in 0.1 M potassium phosphate buffer+ 3.3 mM MgCl2, pH 7.5 and kept on ice. Drug (2 μM final concentration) was added to the microsome suspension and warmed to 37oC with mixing. The assay was started by the addition of NADPH (1.2 mM final concentration). Samples were removed (50 μL) at 0 min, 5 min, 10 min, 20 min, 30 min and added to 3 volumes of acetonitrile containing internal standard (5 ng/mL Enalapril). Samples were centrifuged at 3000 rpm for 5 minutes and 100 μL supernatant transferred to a 96-well plate. Both drug and internal standard concentrations were determined by LC/MS/MS. Analyte/internal standard peak area ratio was used to plot analyte disappearance over time, and half-lives and other metabolic parameters were calculated using the embedded functions within Graphpad Prism.
Plasma Protein Binding Assay.
Plasma protein binding was performed using equilibrium dialysis (HTDialysis, Gales Ferry, CT). Briefly, dialysis membrane (MW cutoff 12-14K) was rehydrated and the assay block assembled. Pooled human or mouse plasma (Innovative Research, Novi, MI) spiked with test compound (10 μM final concentration) was added to one side of the dialysis membrane and phosphate buffered saline (PBS, pH 7.4, Gibco) added to the other. The sample wells were covered with sealing film and the apparatus placed in a 5% CO2 cell culture incubator at 37oC with rotational mixing for 6 hrs. At the conclusions of the incubation, plasma- containing and plasma-free sides of the dialysis membrane were sampled, adjusted for differences in matrix with neat plasma or buffer, and protein precipitated with 3 volumes of acetonitrile containing internal standard (5 ng/mL Enalapril). Samples were centrifuged at 300 rpm for 5 minutes and 100 μL supernatant transferred to a 96-well plate. Both analyte and internal standard concentration was determined by LC/MS/MS. Analyte/internal standard peak area ratio was used to calculate fraction of unbound (fu) compound using the following equation:
Caco-2 Permeability Assay.
Caco-2 cells (ATCC, Manassas, VA) were plated on 96-well transwell plates (0.4 μm, Corning, Corning NY) at 100,000/cm2 and the cells incubated at 37°C/5% CO2 for 21 days with media changed every 2-3 days. Prior to the start of the assay, cells were washed with pre-warmed Hank’s Buffered Salt Solution (HBSS, pH7.4, Gibco) 3x with the final wash left in the well, and the cells allowed to equilibrate for 30 mins at 37°C. Apparent permeability was measured in both directions using 10 μM drug after 2 hours. Buffer from both the donor and receiver compartments was added to 3 volumes of acetonitrile containing internal standard (5 ng/mL Enalapril). Samples were centrifuged and analyzed as above and the apparent permeability (Papp) and efflux ratio calculated using the following equations.
Vr is the volume of the receiver, dC/dt the change of analyte concentration over time, A is the membrane surface area, and C0 the initial compound concentration.
MDR1 Transporter Assay.
Canine MDR1 knockout, human MDR1 knockin MDCKII cells were obtained from Sigma-Aldrich. The cells were plated on 96-well transwell plates (0.4 μm, Corning, Corning NY) at 100,000 cell/cm2 and incubated at 37°C/5%CO2 for 3 days. Prior to the start of the assay, cells were washed with pre-warmed Hank’s Buffered Salt Solution (HBSS, pH7.4, Gibco) 3x with the final wash left in the well, and the cells were allowed to equilibrate for 30 mins at 37°C. 10 μM test compound was added and the apparent permeability (Papp) was measured in both directions after 2 hours. The efflux ratio was calculated using the above equations described for Caco-2 permeability assay.
Mouse PK study.
Both intravenous and oral pharmacokinetic studies were conducted using 6-8-week-old male CD-1 mice (Charles River Labs). Prior to each study, they were housed in a temperature- and light-controlled environment for 5 days. All mice had free access to standard feed and water prior to and throughout the study. All procedures were performed in accordance with the Institutional Animal Care and Use Committee of Saint Louis University.
Intravenous dosing solution was prepared the day of the study in 60% Saline/40% Formal Glycerol to a final concentration of 0.24 mg/mL, which resulted in a clear solution. Drug was administered into the tail vein (3 mice/timepoint) at 1 mg/kg. At the given time point (0.08, 0.17, 0.5, 1, 2, 4, 6 hr), the mouse was euthanized by CO2 exposure and samples collected.
Oral administration of drug (2.4 mg/mL, 10 mg/kg) was performed with a solution of 0.5% Tween 80/0.5% Carboxymethyl cellulose in water. Drug was given by mouth using a stainless-steel feeding needle (3 in x 2.9 mm ball). At the given time point (0.15, 0.5, 1, 2, 4, 8, 24 hr), the mouse was euthanized by CO2 exposure and samples collected.
In both studies, blood was obtained by cardiac puncture into sodium heparin tube (vacutainer) and placed on ice. The blood was separated by centrifugation and the plasma stored at −80°C until analysis. Plasma proteins were denatured by the addition of 3 volumes of ice-cold acetonitrile containing internal standard (5 ng/ml Enalapril). Plasma samples were then vortexed and centrifuged at 12,000xg for 10 minutes. Supernatants were then transferred to a 96-well sample plates for analysis.
The intestinal tissue was obtained by dissection. Briefly, after exposing the intestine, the entire length of the intestine was removed and the middle 4 cm excised. The segment was sliced along the longitudinal axis, and the intestinal contents thoroughly rinsed away with sterile water. The tissue stored at −80°C until analysis.
Intestinal tissue was macerated in a bead beater in PBS, pH 7.2. Tissue mascerates were centrifuged at 12,000g for 10 minutes. The supernate was then treated with 3 volumes of ice-cold acetonitrile containing internal standard (5 ng/ml Enalapril) followed by centrifugation. The final supernate was then transferred to a 96-well sample plate for analysis.
Both plasma and lung samples were analyzed by liquid chromatography-tandem mass spectrometry (LC/MS/MS), using compound spiked into control plasma as a standard. Noncompartmental analysis was performed using Phoenix WinNonlin version 8.2 (Certara, Clayton, MO).
Antiparasitic activity.
Anticryptosporidial activity was measured using a previously described high-content microscopy assay, a human ileocecal adenocarcinoma (HCT-8) cell infection model and C. parvum Iowa isolate oocysts (Bunch Grass Farms).30 Oocysts treated were treated to induce excystation (10 mM HCL ( 10 min, 37° C) followed by 2 mM sodium taurocholate in PBS (10 min, 16 °C)) and used to infect nearly confluent HCT-8 cell monolayers grown in 384-well clear-bottomed plates. Compounds were added after 3 hours incubation, followed by incubation for an additional 45 hours (37°C, 5% CO2). Wells were then washed 3 times with PBS containing 111 mM D-galactose, fixed with 4% formaldehyde, permeabilized with 0.25% Triton X-100, blocked with 4% bovine serum albumin (BSA), and stained for fluorescence microscopy using fluorescein-labeled Vicia villosa lectin (Vector Laboratories) to label the parasitophorous vacuoles and Hoechst 33258 (Anaspec) to label the cell nuclei. A Nikon Eclipse Ti2000 epifluorescence microscope with an automated stage was programmed to focus on the center of each well and take a 3×3 composite image using an EXi blue fluorescence microscopy camera (QImaging) with a 20X objective (NA = 0.45). Nuclei and parasite images were separately exported as .tif files, and parasites and host cells were counted using custom NIH ImageJ macros. Dose-response curves were plotted and half maximal effective concentrations (EC50) and 90% effective concentrations (EC90) were calculated using GraphPad Prism software version 6.01.
Parasite time-kill curve assay.
A modification of the growth inhibition assay was used to determine compound rate-of-action.14 C. parvum was grown in 384-well plate HCT-8 cell cultures for 24 hours, followed by addition of compounds at the EC50 or multiples of the EC90. Culture wells were fixed at 24 hours (the time of compound addition) or the indicated time intervals thereafter, followed by staining of host nuclei and parasites and imaging as above. A separate 384-well plate was used for each time interval. Parasite numbers were normalized to host nuclei numbers and expressed as % parasite per nuclei, and then parasite decay curves were fit by expressing parasite numbers as the % of DMSO control for each time point and using GraphPad Prism.
Host cell toxicity.
Host cell toxicity was measured using HCT-8 Cells and the CellTiter AQueous assay kit (Promega) following the manufacturer’s instructions. Increasing concentrations of compounds were added to 95% confluent HCT-8 cell monolayers, followed by incubation for 48 hours (37°C, 5% CO2). Cell numbers were expressed as the percent of vehicle control (DMSO). GraphPad Prism software version 6.01 was used to plot graphs and calculate the concentration that inhibited 50% of host cell proliferation compared to DMSO control (CC50).
In vivo NSG mouse assay.
All mouse efficacy studies were performed at University of Vermont in compliance with animal care guidelines and were approved by the University of Vermont Institutional Animal Care and Use Committee. Normal flora NOD SCID gamma mice (NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ) were purchased from Jackson Laboratory (Maine, USA) and housed for one week for acclimatization. Mice were then infected by oral gavage at four-to-five weeks of age with 105 C. parvum Iowa isolate oocysts (n=4 mice per experimental group). From days seven to ten following infection, mice were treated by oral gavage with the indicated compounds. Previously prepared aliquots of compound in DMSO were thawed on the day of administration, thoroughly mixed, and diluted in 1% hydoxymethylcellulose/PBS to a total volume of 100 μL and final concentration 5% DMSO. Oocyst shedding in feces was measured using a previously validated qPCR assay.
PAINS Evaluation.
Compounds were filtered for PAINS substructures using the PAINS (Pan-assay interference compounds) filters available at http://fafdrugs4.mti.univ-paris-diderot.fr. There were no compounds identified as PAINS.
Supplementary Material
Acknowledgments.
The authors would like to thank Michael Armbruster for HRMS sample analysis, Michael Prinsen for bioanalysis of samples for in vitro and in vivo PK experiments, and Kirtika Chatri for assistance with in vitro dose-response assays.
Funding.
Research reported in this publication was supported by a grant to CDH, MJM and DG from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01 AI143951.
Abbreviations.
- ADME
absorption, distribution, metabolism and elimination
- CC50
50% cytotoxic concentration
- Cp
Cryptosporidium parvum
- EC50
50% efficacious concentration
- SI
Selectivity Index
- MLM
mouse liver microsomes
- NTZ
nitazoxanide
- PK
pharmacokinetics
Footnotes
Declarations of interest: EO, TJS, EES, CDH and MJM are inventors on a patent application filed that includes some of the compounds described herein.
References
- 1.Checkley W; White AC Jr.; Jaganath D; Arrowood MJ; Chalmers RM; Chen XM; Fayer R; Griffiths JK; Guerrant RL; Hedstrom L; Huston CD; Kotloff KL; Kang G; Mead JR; Miller M; Petri WA Jr.; Priest JW; Roos DS; Striepen B; Thompson RC; Ward HD; Van Voorhis WA; Xiao L; Zhu G; Houpt ER, A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium. Lancet Infect. Dis 2015, 15 (1), 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu J; Platts-Mills JA; Juma J; Kabir F; Nkeze J; Okoi C; Operario DJ; Uddin J; Ahmed S; Alonso PL; Antonio M; Becker SM; Blackwelder WC; Breiman RF; Faruque AG; Fields B; Gratz J; Haque R; Hossain A; Hossain MJ; Jarju S; Qamar F; Iqbal NT; Kwambana B; Mandomando I; McMurry TL; Ochieng C; Ochieng JB; Ochieng M; Onyango C; Panchalingam S; Kalam A; Aziz F; Qureshi S; Ramamurthy T; Roberts JH; Saha D; Sow SO; Stroup SE; Sur D; Tamboura B; Taniuchi M; Tennant SM; Toema D; Wu YK; Zaidi A; Nataro JP; Kotloff KL; Levine MM; Houpt ER, Use of quantitative molecular diagnostic methods to identify causes of diarrhoea in children: a reanalysis of the GEMS case-control study. Lancet 2016, 388 (10051), 1291–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kotloff KL; Nataro JP; Blackwelder WC; Nasrin D; Farag TH; Panchalingam S; Wu Y; Sow SO; Sur D; Breiman RF; Faruque AS; Zaidi AK; Saha D; Alonso PL; Tamboura B; Sanogo D; Onwuchekwa U; Manna B; Ramamurthy T; Kanungo S; Ochieng JB; Omore R; Oundo JO; Hossain A; Das SK; Ahmed S; Qureshi S; Quadri F; Adegbola RA; Antonio M; Hossain MJ; Akinsola A; Mandomando I; Nhampossa T; Acacio S; Biswas K; O’Reilly CE; Mintz ED; Berkeley LY; Muhsen K; Sommerfelt H; Robins-Browne RM; Levine MM, Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 2013, 382 (9888), 209–222. [DOI] [PubMed] [Google Scholar]
- 4.Hlavsa MC; Cikesh BL; Roberts VA; Kahler AM; Vigar M; Hilborn ED; Wade TJ; Roellig DM; Murphy JL; Xiao L; Yates KM; Kunz JM; Arduino MJ; Reddy SC; Fullerton KE; Cooley LA; Beach MJ; Hill VR; Yoder JS, Outbreaks associated with treated recreational water - United States, 2000–2014. MMWR Morb. Mortal Wkly. Rep 2018, 67 (19), 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerrant DI; Moore SR; Lima AA; Patrick PD; Schorling JB; Guerrant RL, Association of early childhood diarrhea and cryptosporidiosis with impaired physical fitness and cognitive function four-seven years later in a poor urban community in northeast Brazil. Am. J. Trop. Med. Hyg 1999, 61 (5), 707–713. [DOI] [PubMed] [Google Scholar]
- 6.Navin TR; Weber R; Vugia DJ; Rimland D; Roberts JM; Addiss DG; Visvesvara GS; Wahlquist SP; Hogan SE; Gallagher LE; Juranek DD; Schwartz DA; Wilcox CM; Stewart JM; Thompson SE 3rd; Bryan RT, Declining CD4+ T-lymphocyte counts are associated with increased risk of enteric parasitosis and chronic diarrhea: results of a 3-year longitudinal study. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol 1999, 20 (2), 154–159. [DOI] [PubMed] [Google Scholar]
- 7.Malebranche R; Arnoux E; Guerin JM; Pierre GD; Laroche AC; Pean-Guichard C; Elie R; Morisset PH; Spira T; Mandeville R; Seemayer T; Dupuy J-M, Acquired immunodeficiency syndrome with severe gastrointestinal manifestations in Haiti. Lancet 1983, 2 (8355), 873–878. [DOI] [PubMed] [Google Scholar]
- 8.Sow SO; Muhsen K; Nasrin D; Blackwelder WC; Wu Y; Farag TH; Panchalingam S; Sur D; Zaidi AK; Faruque AS; Saha D; Adegbola R; Alonso PL; Breiman RF; Bassat Q; Tamboura B; Sanogo D; Onwuchekwa U; Manna B; Ramamurthy T; Kanungo S; Ahmed S; Qureshi S; Quadri F; Hossain A; Das SK; Antonio M; Hossain MJ; Mandomando I; Nhampossa T; Acacio S; Omore R; Oundo JO; Ochieng JB; Mintz ED; O’Reilly CE; Berkeley LY; Livio S; Tennant SM; Sommerfelt H; Nataro JP; Ziv-Baran T; Robins-Browne RM; Mishcherkin V; Zhang J; Liu J; Houpt ER; Kotloff KL; Levine MM, The burden of Cryptosporidium diarrheal disease among dhildren < 24 months of age in moderate/high mortality regions of Sub-Saharan Africa and South Asia, utilizing data from the Global Enteric Multicenter Study (GEMS). PLoS Negl. Trop. Dis 2016, 10 (5), e0004729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khalil IA; Troeger C; Rao PC; Blacker BF; Brown A; Brewer TG; Colombara DV; De Hostos EL; Engmann C; Guerrant RL; Haque R; Houpt ER; Kang G; Korpe PS; Kotloff KL; Lima AAM; Petri WA Jr.; Platts-Mills JA; Shoultz DA; Forouzanfar MH; Hay SI; Reiner RC Jr.; Mokdad AH, Morbidity, mortality, and long-term consequences associated with diarrhoea from Cryptosporidium infection in children younger than 5 years: a meta-analyses study. Lancet Glob. Health 2018, 6 (7), e758–e768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen XM; Keithly JS; Paya CV; LaRusso NF, Current concepts: Cryptosporidiosis. New Engl. J. Med 2002, 346 (22), 1723–1731. [DOI] [PubMed] [Google Scholar]
- 11.Abubakar I; Aliyu SH; Arumugam C; Usman NK; Hunter PR, Treatment of cryptosporidiosis in immunocompromised individuals: systematic review and meta-analysis. Br. J. Clin. Pharmacol 2007, 63 (4), 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amadi B; Mwiya M; Musuku J; Watuka A; Sianongo S; Ayoub A; Kelly P, Effect of nitazoxanide on morbidity and mortality in Zambian children with cryptosporidiosis: a randomised controlled trial. Lancet 2002, 360 (9343), 1375–1380. [DOI] [PubMed] [Google Scholar]
- 13.Amadi B; Mwiya M; Sianongo S; Payne L; Watuka A; Katubulushi M; Kelly P, High dose prolonged treatment with nitazoxanide is not effective for cryptosporidiosis in HIV positive Zambian children: a randomised controlled trial. BMC Infect. Dis 2009, 9, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jumani RS; Bessoff K; Love MS; Miller P; Stebbins EE; Teixeira JE; Campbell MA; Meyers MJ; Zambriski JA; Nunez V; Woods AK; McNamara CW; Huston CD, A novel piperazine-based drug lead for cryptosporidiosis from the Medicines for Malaria Venture Open-Access Malaria Box. Antimicrob. Agents Chemother 2018, 62 (4), e01505–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartelt LA; Bolick DT; Kolling GL; Stebbins E; Huston CD; Guerrant RL; Hoffman PS, Amixicile reduces severity of cryptosporidiosis but does not have in vitro activity against Cryptosporidium. Antimicrob. Agents Chemother 2018, 62 (12), e00718–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee S; Ginese M; Girouard D; Beamer G; Huston CD; Osbourn D; Griggs DW; Tzipori S, Piperazine-derivative MMV665917: An effective drug in the diarrheic piglet model of Cryptosporidium hominis. J. Infect. Dis 2019, 220 (2), 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stebbins E; Jumani RS; Klopfer C; Barlow J; Miller P; Campbell MA; Meyers MJ; Griggs DW; Huston CD, Clinical and microbiologic efficacy of the piperazine-based drug lead MMV665917 in the dairy calf cryptosporidiosis model. PLoS Negl. Trop. Dis 2018, 12 (1), e0006183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jumani RS; Hasan MM; Stebbins EE; Donnelly L; Miller P; Klopfer C; Bessoff K; Teixeira JE; Love MS; McNamara CW; Huston CD, A suite of phenotypic assays to ensure pipeline diversity when prioritizing drug-like Cryptosporidium growth inhibitors. Nat. Commun 2019, 10 (1), 1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Funkhouser-Jones LJ; Ravindran S; Sibley LD, Defining Stage-Specific Activity of Potent New Inhibitors of Cryptosporidium parvum Growth In Vitro. mBio 2020, 11 (2), e00052–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lunde CS; Stebbins EE; Jumani RS; Hasan MM; Miller P; Barlow J; Freund YR; Berry P; Stefanakis R; Gut J; Rosenthal PJ; Love MS; McNamara CW; Easom E; Plattner JJ; Jacobs RT; Huston CD, Identification of a potent benzoxaborole drug candidate for treating cryptosporidiosis. Nat. Commun 2019, 10 (1), 2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Voorhis WC; Adams JH; Adelfio R; Ahyong V; Akabas MH; Alano P; Alday A; Alemán Resto Y; Alsibaee A; Alzualde A; Andrews KT; Avery SV; Avery VM; Ayong L; Baker M; Baker S; Ben Mamoun C; Bhatia S; Bickle Q; Bounaadja L; Bowling T; Bosch J; Boucher LE; Boyom FF; Brea J; Brennan M; Burton A; Caffrey CR; Camarda G; Carrasquilla M; Carter D; Belen Cassera M; Chih-Chien Cheng K; Chindaudomsate W; Chubb A; Colon BL; Colón-López DD; Corbett Y; Crowther GJ; Cowan N; D’Alessandro S; Le Dang N; Delves M; DeRisi JL; Du AY; Duffy S; Abd El-Salam El-Sayed S; Ferdig MT; Fernández Robledo JA; Fidock DA; Florent I; Fokou PV; Galstian A; Gamo FJ; Gokool S; Gold B; Golub T; Goldgof GM; Guha R; Guiguemde WA; Gural N; Guy RK; Hansen MA; Hanson KK; Hemphill A; Hooft van Huijsduijnen R; Horii T; Horrocks P; Hughes TB; Huston C; Igarashi I; Ingram-Sieber K; Itoe MA; Jadhav A; Naranuntarat Jensen A; Jensen LT; Jiang RH; Kaiser A; Keiser J; Ketas T; Kicka S; Kim S; Kirk K; Kumar VP; Kyle DE; Lafuente MJ; Landfear S; Lee N; Lee S; Lehane AM; Li F; Little D; Liu L; Llinás M; Loza MI; Lubar A; Lucantoni L; Lucet I; Maes L; Mancama D; Mansour NR; March S; McGowan S; Medina Vera I; Meister S; Mercer L; Mestres J; Mfopa AN; Misra RN; Moon S; Moore JP; Morais Rodrigues da Costa F; Müller J; Muriana A; Nakazawa Hewitt S; Nare B; Nathan C; Narraidoo N; Nawaratna S; Ojo KK; Ortiz D; Panic G; Papadatos G; Parapini S; Patra K; Pham N; Prats S; Plouffe DM; Poulsen SA; Pradhan A; Quevedo C; Quinn RJ; Rice CA; Abdo Rizk M; Ruecker A; St Onge R; Salgado Ferreira R; Samra J; Robinett NG; Schlecht U; Schmitt M; Silva Villela F; Silvestrini F; Sinden R; Smith DA; Soldati T; Spitzmüller A; Stamm SM; Sullivan DJ; Sullivan W; Suresh S; Suzuki BM; Suzuki Y; Swamidass SJ; Taramelli D; Tchokouaha LR; Theron A; Thomas D; Tonissen KF; Townson S; Tripathi AK; Trofimov V; Udenze KO; Ullah I; Vallieres C; Vigil E; Vinetz JM; Voong Vinh P; Vu H; Watanabe NA; Weatherby K; White PM; Wilks AF; Winzeler EA; Wojcik E; Wree M; Wu W; Yokoyama N; Zollo PH; Abla N; Blasco B; Burrows J; Laleu B; Leroy D; Spangenberg T; Wells T; Willis PA, Open source drug discovery with the Malaria Box compound collection for neglected diseases and beyond. PLoS Pathog. 2016, 12 (7), e1005763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pearlstein R; Vaz R; Rampe D, Understanding the structure-activity relationship of the human ether-a-go-go-related gene cardiac K+ channel. A model for bad behavior. J. Med. Chem 2003, 46 (11), 2017–2022. [DOI] [PubMed] [Google Scholar]
- 23.Redfern WS; Carlsson L; Davis AS; Lynch WG; MacKenzie I; Palethorpe S; Siegl PKS; Strang I; Sullivan AT; Wallis R; Camm AJ; Hammond TG, Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res 2003, 58 (1), 32–45. [DOI] [PubMed] [Google Scholar]
- 24.Suikov SY; Kal’nits’kii MY; Lutsik OI; Plyushchakova LA; Bondarenko AV; Bogza SL; Popov AF, Derivatives of [1,2,4]triazolo[4.3-b]pyridazin-6-ylhydrazines as a new training set for predictive models of an octanol-water partition. Dopov. Nats. Akad. Nauk Ukr 2007, (7), 142–146. [Google Scholar]
- 25.Katrusiak A; Ratajczak-Sitarz M; Skierska U; Zinczenko W, Nucleophilic substitution and lipophilicity - Structure relations in methylazolopyridazines. Collect. Czech. Chem. Commun 2005, 70 (9), 1372–1386. [Google Scholar]
- 26.Bradbury RH; Hales NJ; Rabow AA; Walker GE; Acton DG; Andrews DM; Ballard P; Brooks NAN; Colclough N; Girdwood A; Hancox UJ; Jones O; Jude D; Loddick SA; Mortlock AA, Small-molecule androgen receptor downregulators as an approach to treatment of advanced prostate cancer. Bioorg. Med. Chem. Lett 2011, 21 (18), 5442–5445. [DOI] [PubMed] [Google Scholar]
- 27.Wang L.-x.; Zhou X.-b.; Xiao M.-l.; Jiang N; Liu F; Zhou W.-x.; Wang X.-k.; Zheng Z.-b.; Li S, Synthesis and biological evaluation of substituted 4-(thiophen-2-ylmethyl)-2H-phthalazin-1-ones as potent PARP-1 inhibitors. Bioorg. Med. Chem. Lett 2014, 24 (16), 3739–3743. [DOI] [PubMed] [Google Scholar]
- 28.Yang N; Zhang H; Yuan G, KI-catalyzed reactions of aryl hydrazines with α-oxocarboxylic acids in the presence of CO2: access to 1,3,4-oxadiazol-2(3H)-ones. Org. Chem. Front 2019, 6 (4), 532–536. [Google Scholar]
- 29.Umemoto T; Singh RP; Xu Y; Saito N, Discovery of 4-tert-butyl-2,6-dimethylphenylsulfur trifluoride as a deoxofluorinating agent with high thermal stability as well as unusual resistance to aqueous hydrolysis, and its diverse fluorination capabilities including deoxofluoro-arylsulfinylation with high stereoselectivity. J. Am. Chem. Soc 2010, 132 (51), 18199–18205. [DOI] [PubMed] [Google Scholar]
- 30.Bessoff K; Sateriale A; Lee KK; Huston CD, Drug repurposing screen reveals FDA-approved inhibitors of human HMG-CoA reductase and isoprenoid synthesis that block Cryptosporidium parvum growth. Antimicrob. Agents Chemother 2013, 57 (4), 1804–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berridge MV; Tan AS, Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys 1993, 303 (2), 474–482. [DOI] [PubMed] [Google Scholar]
- 32.Bowes J; Brown AJ; Hamon J; Jarolimek W; Sridhar A; Waldron G; Whitebread S, Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat. Rev. Drug. Discov 2012, 11 (12), 909–922. [DOI] [PubMed] [Google Scholar]
- 33.Mathes C, QPatch: the past, present and future of automated patch clamp. Expert Opin. Ther. Targets 2006, 10 (2), 319–327. [DOI] [PubMed] [Google Scholar]
- 34.Choi R; Hulverson MA; Huang W; Vidadala RSR; Whitman GR; Barrett LK; Schaefer DA; Betzer DP; Riggs MW; Doggett JS; Hemphill A; Ortega-Mora LM; McCloskey MC; Arnold SLM; Hackman RC; Marsh KC; Lynch JJ; Freiberg GM; Leroy BE; Kempf DJ; Choy RKM; de Hostos EL; Maly DJ; Fan E; Ojo KK; Van Voorhis WC, Bumped Kinase Inhibitors as therapy for apicomplexan parasitic diseases: lessons learned. Int. J. Parasitol 2020, 50 (5), 413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehmann DF; Eggleston WD; Wang DL, Validation and clinical utility of the hERG IC50:C-max ratio to determine the risk of drug-induced Torsades de Pointes: A meta-analysis. Pharmacotherapy 2018, 38 (3), 341–348. [DOI] [PubMed] [Google Scholar]
- 36.Leishman DJ; Abernathy MM; Wang EB, Revisiting the hERG safety margin after 20 years of routine hERG screening. J. Pharmacol. Toxicol. Methods 2020, 105, 106900. [DOI] [PubMed] [Google Scholar]
- 37.Ridder BJ; Leishman DJ; Bridgland-Taylor M; Samieegohar M; Han XM; Wu WW; Randolph A; Tran P; Sheng JS; Danker T; Lindqvist A; Konrad D; Hebeisen S; Polonchuk L; Gissinger E; Renganathan M; Koci B; Wei HY; Fan JS; Levesque P; Kwagh J; Imredy J; Zhai J; Rogers M; Humphries E; Kirby R; Stoelzle-Feix S; Brinkwirth N; Rotordam MG; Becker N; Friis S; Rapedius M; Goetze TA; Strassmaier T; Okeyo G; Kramer J; Kuryshev Y; Wu CY; Himmel H; Mirams GR; Strauss DG; Bardenet R; Li ZH, A systematic strategy for estimating hERG block potency and its implications in a new cardiac safety paradigm. Toxicol. Appl. Pharmacol 2020, 394, 114961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arnold SLM; Choi R; Hulverson MA; Whitman GR; McCloskey MC; Dorr CS; Vidadala RSR; Khatod M; Morada M; Barrett LK; Maly DJ; Yarlett N; Van Voorhis WC, P-glycoprotein-mediated efflux reduces the in vivo efficacy of a therapeutic targeting the gastrointestinal parasite Cryptosporidium. J. Infect. Dis 2019, 220 (7), 1188–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manjunatha UH; Chao AT; Leong FJ; Diagana TT, Cryptosporidiosis drug discovery: Opportunities and challenges. ACS Infect. Dis 2016, 2 (8), 530–537. [DOI] [PubMed] [Google Scholar]
- 40.Arnold SLM; Choi R; Hulverson MA; Schaefer DA; Vinayak S; Vidadala RSR; McCloskey MC; Whitman GR; Huang W; Barrett LK; Ojo KK; Fan E; Maly DJ; Riggs MW; Striepen B; Van Voorhis WC, Necessity of bumped kinase inhibitor gastrointestinal exposure in treating Cryptosporidium infection. J. Infect. Dis 2017, 216 (1), 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manjunatha UH; Vinayak S; Zambriski JA; Chao AT; Sy T; Noble CG; Bonamy GMC; Kondreddi RR; Zou B; Gedeck P; Brooks CF; Herbert GT; Sateriale A; Tandel J; Noh S; Lakshminarayana SB; Lim SH; Goodman LB; Bodenreider C; Feng G; Zhang L; Blasco F; Wagner J; Leong FJ; Striepen B; Diagana TT, A Cryptosporidium PI(4)K inhibitor is a drug candidate for cryptosporidiosis. Nature 2017, 546 (7658), 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gorla SK; McNair NN; Yang G; Gao S; Hu M; Jala VR; Haribabu B; Striepen B; Cuny GD; Mead JR; Hedstrom L, Validation of IMP dehydrogenase inhibitors in a mouse model of cryptosporidiosis. Antimicrob. Agents Chemother 2014, 58 (3), 1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin B-J; Thiagarajah JR; Verkman AS, Convective washout reduces the antidiarrheal efficacy of enterocyte surface–targeted antisecretory drugs. J. Gen. Physiol 2013, 141 (2), 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mouton JW; Theuretzbacher U; Craig WA; Tulkens PM; Derendorf H; Cars O, Tissue concentrations: do we ever learn? J. Antimicrob. Chemother 2008, 61 (2), 235–237. [DOI] [PubMed] [Google Scholar]
- 45.Campbell LD; Stewart JN; Mead JR, Susceptibility to Cryptosporidium parvum infections in cytokine- and chemokine-receptor knockout mice. J. Parasitol 2002, 88 (5), 1014–1016. [DOI] [PubMed] [Google Scholar]
- 46.Love MS; Beasley FC; Jumani RS; Wright TM; Chatterjee AK; Huston CD; Schultz PG; McNamara CW, A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Negl. Trop. Dis 2017, 11 (2), e0005373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.You X; Schinazi RF; Arrowood MJ; Lejkowski M; Juodawlkis AS; Mead JR, In-vitro activities of paromomycin and lasalocid evaluated in combination against Cryptosporidium parvum. J. Antimicrob. Chemother 1998, 41 (2), 293–296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.