

Abstract
BACKGROUND
Chronic hepatitis C virus (HCV) infection induces profound alterations in the cytokine and chemokine signatures in peripheral blood. Clearance of HCV by antivirals results in host immune modification, which may interfere with immune-mediated cancer surveillance. Identifying HCV patients who remain at risk of hepatocellular carcinoma (HCC) following HCV eradication remains an unmet need. We hypothesized that antiviral therapy-induced immune reconstruction may be relevant to HCC development.
AIM
To investigate the impact of differential dynamics of cytokine expression on the development of HCC following successful antiviral therapy.
METHODS
One hundred treatment-naïve HCV patients with advanced fibrosis (F3/4) treated with direct-acting antivirals (DAAs) or peginterferon/ribavirin who achieved sustained virologic response [SVR, defined as undetectable HCV RNA throughout 12 wk (SVR12) for the DAA group or 24 wk (SVR24) for the interferon group after completion of antiviral therapy] were enrolled since 2003. The primary endpoint was the development of new-onset HCC. Standard HCC surveillance (abdominal ultrasound and α-fetoprotein) was performed every six months during the follow-up. Overall, 64 serum cytokines were detected by the multiplex immunoassay at baseline and 24 wk after end-of-treatment.
RESULTS
HCC developed in 12 of the 97 patients over 459 person-years after HCV eradication. In univariate analysis, the Fibrosis-4 index (FIB-4), hemoglobin A1c (HbA1c), the dynamics of tumor necrosis factor-α (TNF-α), and TNF-like weak inducer of apoptosis (TWEAK) after antiviral therapy were significant HCC predictors. The multivariate Cox regression model showed that ΔTNF-α (≤ -5.7 pg/mL) was the most important risk factor for HCC (HR = 11.54, 95%CI: 2.27-58.72, P = 0.003 in overall cases; HR = 9.98, 95%CI: 1.88-52.87, P = 0.007 in the interferon group). An HCC predictive model comprising FIB-4, HbA1c, ΔTNF-α, and ΔTWEAK had excellent performance, with 3-, 5-, 10-, and 13-year areas under the curve of 0.882, 0.864, 0.903, and 1.000, respectively. The 5-year accumulative risks of HCC were 0%, 16.9%, and 40.0% in the low-, intermediate-, and high-risk groups, respectively.
CONCLUSION
Downregulation of serum TNF-α significantly increases the risk of HCC after HCV eradication. A predictive model consisting of cytokine kinetics could ameliorate personalized HCC surveillance strategies for post-SVR HCV patients.
Keywords: Hepatitis C virus, Hepatocellular carcinoma, Sustained virologic response, Tumor necrosis factor-α, Tumor necrosis factor-like weak inducer of apoptosis, Cytokine
Core Tip: Successful hepatitis C virus (HCV) eradication does not eliminate hepatocellular carcinoma (HCC) development. Clearance of HCV by antiviral agents results in host immune modification, which might interfere with immune-mediated cancer surveillance. We attempted to identify immune biomarkers to predict HCC occurrence after antiviral therapy. The dynamics of serum tumor necrosis factor-α (TNF-α) and TNF-like weak inducer of apoptosis were associated with HCC occurrence after HCV clearance. We established a predictive model to assess the risk of HCC among HCV patients after HCV eradication. Our findings provide a clue for the pathogenesis of hepatocarcinogenesis and a strategy for HCC surveillance based on risk stratification.
INTRODUCTION
Chronic hepatitis C virus (HCV) infection is a major cause of liver cirrhosis and hepatocellular carcinoma (HCC). As HCV treatment evolves from an interferon (IFN)-based regimen to a therapy based on direct-acting antiviral agents (DAAs), it yields a sustained virologic response (SVR) rate of more than 97% in chronic hepatitis C patients[1,2]. However, successful antiviral therapy does not eliminate HCC development. In a meta-analysis of observational studies, IFN therapy decreased the risk of HCC by 76% in patients with bridging fibrosis or cirrhosis who achieved SVR[3]. Recent studies have reported that HCC occurrence and recurrence rates are potentially increased in HCV patients treated with DAAs[4-6]. This concern remains controversial due to the heterogeneous cohorts, variations in the inclusion criteria, and short duration of follow-up.
Persistent inflammation is a hallmark of chronic hepatic injury. HCV infection induces endogenous type I and III IFN activation, which activates natural killer (NK) cells[7] and leads to the expression of IFN-stimulated genes (ISGs)[8]. It causes profound alterations in the cytokine and chemokine signature in peripheral blood. HCV-specific CD8+ T cells play a central role in viral clearance. Chronic HCV infection is characterized by impaired HCV-specific CD8+ T cell responses resulting from viral escape and T cell exhaustion[9]. IFN-based therapy failed to recover the function of HCV-specific CD8+ T cells. This result suggested that the damage to CD8+ T cells might be permanent even after virus elimination[10]. In contrast, the combination of deleobuvir and faldaprevir resulted in the downregulation of programmed death-1, which led to rapid restoration of HCV-specific CD8+ T cell functions[11]. DAA-mediated HCV clearance is correlated with mitigation of the IFN-α-induced immune response, followed by the downregulation of CXCL10 and CXCL11 and normalization of the phenotype and function of NK cells[12].
It is unclear whether host immunological modification after viral eradication influences the development of HCC. Although DAAs are the first choice for HCV clearance, they are not sufficient to abolish hepatic inflammation. Long-term inflammatory responses may change the liver microenvironment and cause irreversible hepatocyte damage. A rapid decline in HCV viral load induced by DAAs results in the reconstitution of immune surveillance[4]. HCV eradication during DAA treatment is accompanied by downregulation of type II and III IFN, their receptors, and downstream ISGs[13], which may affect the antitumor activity of immune cells. IFNs have immunomodulatory properties that regulate various immune cells to inhibit tumor proliferation and angiogenesis. Unlike IFNs, DAAs have neither antiproliferative nor antiangiogenic properties, which may allow the proliferation of malignant cells.
The identification of HCV patients who maintain a high risk of HCC following successful antiviral therapy remains an unmet need. Hepatocarcinogenesis despite HCV clearance is still unclear. First, this study aimed to investigate the impact of differential cytokine expression profiles on the development of HCC among chronic hepatitis C patients with advanced fibrosis who achieved SVR. Second, we attempted to identify immune biomarkers to predict the risk of HCC after successful antiviral therapy.
MATERIALS AND METHODS
Subjects
One hundred treatment-naïve chronic hepatitis C patients with advanced fibrosis treated with either pegylated IFN/ribavirin or IFN-free DAA who achieved SVR were recruited from Kaohsiung Medical University Hospital since 2003. Patients were required to satisfy any one of the following criteria to be diagnosed with advanced fibrosis (F3/4): Fibrosis-4 (FIB-4) index > 3.25[14], transient elastography (Fibroscan) > 9.1 kPa, or acoustic radiation force impulse elastography > 1.81 m/s. The exclusion criteria were coinfection with hepatitis B, hepatitis D or human immunodeficiency virus; history of liver transplantation; prior presence of HCC; decompensated liver cirrhosis; malignancy; alcoholism; primary biliary cholangitis; α1-antitrypsin deficiency; autoimmune hepatitis; renal function impairment; and psychiatric conditions.
Treatment
In the IFN group, the patients were subcutaneously administered peginterferon α-2a (180 μg/wk) plus weight-based ribavirin (1200 mg/d for weights ≥ 75 kg or 1000 mg/d for weights < 75 kg) for 24 to 48 wk depending on the HCV genotype. In the DAA group, the physician selected IFN-free DAA regimens for 12 to 24 wk that were discreetly based on the HCV international treatment guidelines (The Asian Pacific Association for the Study of the Liver, European Association for the Study of the Liver and American Association for The Study of Liver Diseases).
Outcome assessment
SVR was defined as undetectable HCV RNA throughout 12 wk (SVR12) for the DAA group or 24 wk (SVR24) for the IFN group after completion of antiviral therapy[15,16]. The primary endpoint was the occurrence of new-onset HCC. Standard HCC surveillance [abdominal ultrasound and α-fetoprotein (AFP) every six months] was performed during the follow-up[17]. HCC development within six months of initiation of antiviral treatment was excluded. Proof of HCC was directly linked to the National Cancer Registration of Taiwan in Health and Welfare Data Science Center (Taiwan). This study was approved by the Institutional Review Board of Kaohsiung Medical University Hospital (No. KMUHIRB-E(I)-20180307 & KMUHIRB-G(II)-20170020). Written informed consent was acquired from all participants.
HCV genotyping and quantification
Anti-HCV antibodies were identified by a third-generation commercially available enzyme-linked immunosorbent assay (Abbott Laboratories, Chicago, IL, United States). HCV RNA was quantified by real-time polymerase chain reaction assay with a lower limit of detection of 12 IU/mL (RealTime HCV; Abbott Molecular, Des Plaines IL, United States)[18]. HCV genotypes were determined using a commercial kit (Abbott RealTime HCV Genotype II; Abbott Molecular, Des Plaines, IL, United States).
Cytokine measurement
Serum samples were collected from the participants at baseline and SVR24. In total, 64 serum cytokines and chemokines (Supplementary Tables 1-3) were measured by the magnetic bead multiplex immunoassay according to the manufacturer’s instructions (Merck KGaA, Darmstadt, Germany)[19,20]. In brief, a calibration curve based on 1:3 dilutions of the highest standard was used for quantification. Beads were premixed and put into wells containing diluted serum and reagents. After fixation of the antigen on the capture antibody linked with the microspheres, a biotinylated detection antibody was added. The concentration of the analyte was quantified based on the bead color and the intensity of the fluorescent signal using the multiplex Luminex-200 (Luminex Corporation, Austin, TX, United States). All samples were analyzed in duplicate.
Statistical analysis
Student’s t test and the Mann–Whitney U test were performed to compare the continuous variables. The chi-square (χ2) test with Yates correction or Fisher’s exact test was used to assess the categorical variables. Differences in the cumulative incidence of HCC between groups were analyzed by Kaplan-Meier survival analysis and the log-rank test. The risk factors for HCC were evaluated using multivariate Cox regression analysis. In conjunction with receiver operating characteristic area (ROC) analysis[21], the optimum cutoff value to distinguish between the risk strata was calculated by the Youden index[22]. The performance of biomarkers to predict the risk of HCC was calculated by time-dependent ROC curve analysis. The area under the ROC area (AUROC) was assessed by the timeROC package of R software (http://www.r-project.org). The statistical power for the comparison of survival curves between two groups under the Cox proportional hazards model was calculated by the powerSurvEpi package of R software. A two-tailed P value < 0.05 was considered statistically significant. The statistical analysis was conducted by the Statistic Packages for Social Science Program (SPSS v19.0 for Windows, SPSS Inc., United States). The statistical methods of this study were reviewed by Dr. Tsai PC from Kaohsiung Medical University.
RESULTS
Baseline demographics
The baseline demographics of the study subjects are shown in Table 1. There were no significant differences in age, sex, HCV genotype, FIB-4 index, or AFP levels between the DAA and IFN groups. HCV RNA was significantly higher in the DAA group than in the IFN group. Aspartate aminotransferase, alanine aminotransferase and hemoglobin A1c (HbA1c) were significantly elevated in the IFN group compared to the DAA group.
Table 1.
Baseline demographics of study subjects
|
Group
|
Total
|
DAA
|
IFN
|
P
value (DAA vs IFN)
|
| n | 100 | 50 | 50 | |
| Age (yr) | 63.8 ± 7.2 | 64.9 ± 7.9 | 62.6 ± 6.3 | 0.100 |
| Sex, n (%) | ||||
| Female | 66 (66.0) | 38 (76.0) | 28 (56.0) | 0.057 |
| Male | 34 (34.0) | 12 (24.0) | 22 (44.0) | |
| HCV genotype, n (%) | ||||
| 1 | 65 (65.0) | 37 (74.0) | 28 (56.0) | 0.098 |
| 2 | 23 (23.0) | 10 (20.0) | 13 (26.0) | |
| Mixed | 12 (12.0) | 3 (6.0) | 9 (18.0) | |
| HCV RNA (log IU/mL) | 2.47 ± 0.89 | 2.67 ± 0.84 | 2.28 ± 0.91 | 0.027 |
| FIB-4 | 6.14 ± 3.28 | 6.55 ± 3.69 | 5.73 ± 2.80 | 0.213 |
| AFP (ng/mL) | 26.3 ± 56.7 | 28.8 ± 74.0 | 23.6 ± 29.3 | 0.662 |
| Platelet (k/μL) | 119.8 ± 34.7 | 115.2 ± 35.9 | 124.4 ± 33.2 | 0.186 |
| AST (IU/L) | 136.9 ± 79.9 | 115.2 ± 64.7 | 158.7 ± 87.9 | 0.006 |
| ALT (IU/L) | 177.5 ± 138.4 | 127.9 ± 83.9 | 227.0 ± 163.2 | 2.7 × 10-4 |
| γ-GT (IU/L) | 67.6 ± 48.8 | 57.2 ± 42.6 | 76.7 ± 52.4 | 0.053 |
| Cholesterol (mg/dL) | 161.6 ± 34.8 | 158.6 ± 37.1 | 165.2 ± 32.0 | 0.388 |
| Triglyceride (mg/dL) | 97.7 ± 41.4 | 99.2 ± 40.5 | 96.0 ± 42.8 | 0.722 |
| HDL (mg/dL) | 47.0 ± 13.3 | 49.0 ± 11.6 | 44.1 ± 15.1 | 0.107 |
| LDL (mg/dL) | 90.6 ± 26.4 | 86.0 ± 24.4 | 97.2 ± 28.1 | 0.068 |
| Cr (mg/dL) | 0.79 ± 0.24 | 0.77 ± 0.27 | 0.82 ± 0.20 | 0.222 |
| HbA1c (%) | 5.8 ± 1.2 | 5.5 ± 0.7 | 6.0 ± 1.5 | 0.016 |
| BMI (kg/m2) | 24.4 ± 4.7 | 24.0 ± 6.0 | 24.8 ± 2.9 | 0.381 |
DAA: Direct-acting antiviral agent; IFN: Interferon; HCV: Hepatitis C virus; FIB-4: Fibrosis-4 index; AFP: Alpha-fetoprotein; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; γ-GT: γ-glutamyltransferase; HDL: High-density lipoprotein; LDL: Low-density lipoprotein; Cr: Creatinine; HbA1c: Hemoglobin A1c; BMI: Body mass index.
Cumulative probability of HCC development
HCC developed in 12 (IFN group n = 11, DAA group n = 1) of the 97 patients over 459 person-years of follow-up. Three patients were excluded because HCC occurred within six months of initiation of the antiviral treatment. The mean follow-up time was 7.46 years [interquartile range (IQR) = 3.65-12.23] in the IFN group and 1.84 years (IQR = 1.19-2.43) in the DAA group. The annual incidence of HCC was 2.95% in the IFN group and 1.16% in the DAA group. The Kaplan-Meier survival analysis showed no statistical significance in the accumulative probability of HCC between the IFN and DAA groups (log-rank P value = 0.712) (Figure 1).
Figure 1.
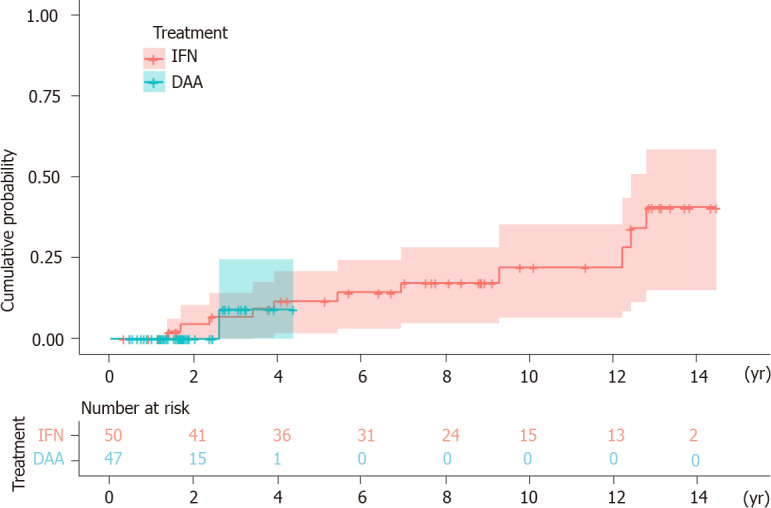
Kaplan-Meier survival analysis. HCC: Hepatocellular carcinoma; IFN: Interferon; DAA: Direct-acting antiviral agent.
Cytokines associated with HCC development
In total, 64 cytokines were used to analyze the relationship with HCC (Supplementary Table 1). Seven of the sixty-four cytokines were excluded from subsequent analysis because more than 80% of the samples were below the limit of detection. Members of the tumor necrosis factor (TNF) superfamily, including TNF-α and TNF-like weak inducer of apoptosis (TWEAK), were associated with the development of HCC. The baseline TNF-α level was significantly elevated in the HCC group compared to the non-HCC group (22.22 ± 4.33 vs 12.53 ± 1.03 pg/mL, P = 0.036). There was no significant difference in the posttreatment TNF-α levels between the HCC and non-HCC groups. The change in TNF-α levels (ΔTNF-α) before and after treatment significantly differed between the HCC and non-HCC groups (-15.11 ± 3.93 vs -3.78 ± 1.16 pg/mL, P = 8.2 × 10-4) (Figure 2A). The baseline TWEAK expression was similar in both groups. The posttreatment TWEAK level was significantly lower in the HCC group than in the non-HCC group (434.82 ± 84.18 vs 660.65 ± 30.34 pg/mL, P = 0.018). ΔTWEAK showed a reciprocal change between the HCC and non-HCC groups and achieved statistical significance (-71.78 ± 54.56 vs 142.81 ± 27.69 pg/mL, P = 3.6 × 10-3) (Figure 2B).
Figure 2.

Cytokine expression between the hepatocellular carcinoma and non-hepatocellular carcinoma groups among chronic hepatitis C patients with advanced fibrosis. A: Tumor necrosis factor-α; B: Tumor necrosis factor-like weak inducer of apoptosis. Δ = (posttreatment cytokine level) - (pretreatment cytokine level). The bar represents the means ± SE. The P value was tested by the Mann–Whitney U test. HCC: Hepatocellular carcinoma; TNF-α: Tumor necrosis factor-α; TWEAK: TNF-like weak inducer of apoptosis; IFN: Interferon; Pre-Tx: Pretreatment; Post-Tx: Posttreatment.
Among the HCV patients treated with pegIFN/ribavirin, the baseline TNF-α level was significantly higher in the HCC group than in the non-HCC group (22.55 ± 4.72 vs 9.13 ± 7.79 pg/mL, P = 0.017). The posttreatment TNF-α concentration was comparable between the HCC and non-HCC groups. ΔTNF-α levels significantly declined in HCC compared to non-HCC patients (-15.86 ± 4.22 vs -4.56 ± 1.85 pg/mL, P = 0.007) (Supplementary Figure 1A). The dynamic change in TWEAK did not show significant variations between the HCC and non-HCC groups (Supplementary Figure 1B).
Cox regression analysis of the relationship between the differentially expressed cytokines and HCC
In univariate Cox regression analysis, FIB-4 (≥ 9 vs < 9, crude HR = 4.04, 95%CI: 1.27-12.86, P = 0.018), HbA1c (≥ 7 vs < 7%, crude HR = 5.38, 95%CI: 1.38-20.99, P = 0.015), pretreatment TNF-α (≥ 18 vs < 18 pg/mL, crude HR = 5.15, 95%CI: 1.57-16.87, P = 0.007), ΔTNF-α (≤ -5.7 vs > -5.7 pg/mL, crude HR = 11.07, 95%CI: 2.27-53.87, P = 0.003), and ΔTWEAK (≤ -70 vs > -70 pg/mL, crude HR = 4.01, 95%CI: 1.20-13.40, P = 0.024) were significant predictors of HCC. Multivariate stepwise Cox regression analysis revealed that ΔTNF-α was the only independent risk factor for HCC (HR = 11.54, 95%CI: 2.27-58.72, P = 0.003) (Table 2).
Table 2.
Factors associated with the onset of hepatocellular carcinoma: Univariate and multivariate Cox regression models
| Variables |
Univariate Cox regression
|
Multivariate Cox regression
|
||
|
Crude HR (95%CI)
|
P
value
|
Adjusted HR (95%CI)
|
Adjusted P value
|
|
| Age (yr) | 1.07 (0.98-1.18) | 0.149 | - | - |
| Sex (male vs female) | 3.19 (0.96-10.65) | 0.059 | - | - |
| HCV genotype | 1.00 (0.47-2.13) | 0.995 | - | - |
| HCV RNA (log IU/mL) | 0.68 (0.37-1.25) | 0.213 | - | - |
| FIB-4 | 1.13 (0.98-1.31) | 0.089 | - | - |
| FIB-4 (≥ 9 vs < 9) | 4.04 (1.27-12.86) | 0.018 | - | - |
| Platelet (k/μL) | 0.99 (0.98-1.01) | 0.408 | - | - |
| AFP (ng/mL) | 1.00 (1.00-1.01) | 0.308 | - | - |
| HbA1c (%) | 1.28 (1.01-1.62) | 0.041 | - | - |
| HbA1c (≥ 7 vs < 7%) | 5.38 (1.38-20.99) | 0.015 | - | - |
| BMI (kg/m2) | 1.00 (0.83-1.20) | 0.993 | - | - |
| Treatment (DAA vs IFN) | 0.66 (0.07-6.15) | 0.713 | - | - |
| TNF-α (pg/mL) | ||||
| Pre-Tx TNF-α ≥ 18 | 5.15 (1.57-16.87) | 0.007 | - | - |
| Post-Tx TNF-α ≥ 6 | 0.79 (0.25-2.46) | 0.683 | - | - |
| ΔTNF-α ≤ -5.7 | 11.07 (2.27-53.87) | 0.003 | 11.54 (2.27-58.72) | 0.003 |
| TWEAK (pg/mL) | ||||
| Pre-Tx TWEAK ≥ 500 | 2.18 (0.64-7.39) | 0.213 | - | - |
| Post-Tx TWEAK ≥ 600 | 0.80 (0.20-3.11) | 0.744 | - | - |
| ΔTWEAK ≤ -70 | 4.01 (1.20-13.40) | 0.024 | - | - |
The forward stepwise multivariate Cox regression model was adjusted by age, sex, hepatitis C virus (HCV) genotypes, HCV RNA, Fibrosis-4 index (FIB-4), platelet, alpha-fetoprotein, hemoglobin A1c, body mass index, treatment, tumor necrosis factor-α and tumor necrosis factor-like weak inducer of apoptosis (pretreatment, posttreatment, Δ). The cut-off value for each cytokine and FIB-4 was determined by Youden index of receiver operating characteristic curve. Δ = (posttreatment cytokine level) - (pretreatment cytokine level). DAA: Direct-acting antiviral agent; IFN: Interferon; HCV: Hepatitis C virus; FIB-4: Fibrosis-4 index; AFP: Alpha-fetoprotein; HbA1c: Hemoglobin A1c; BMI: Body mass index; HCV: Hepatitis C virus; TNF-α: tumor necrosis factor-α; TWEAK: TNF-like weak inducer of apoptosis; Pre-Tx: Pretreatment; Post-Tx: Posttreatment; HR: Hazard ratio; CI: Confidence interval.
Among the HCV patients treated with pegIFN/ribavirin, univariate Cox regression showed that the significant predictors of HCC included sex, FIB-4 (≥ 9 vs < 9), HbA1c, baseline TNF-α (≥ 18 vs < 18 pg/mL) and ΔTNF-α (≥ -5.7 vs < -5.7 pg/mL). The association between ΔTWEAK (≤ -70 vs > -70 pg/mL) and HCC was borderline statistically significant. Stepwise multivariate Cox regression revealed that the ΔTNF-α level was the only independent risk factor for HCC in the IFN group (HR = 9.98, 95%CI: 1.88-52.87, P = 0.007 (Supplementary Table 2).
Subgroup analysis for the association between TNF-α and HCC
Since age and diabetes mellitus were important risk factors for HCC, the subjects were further stratified by age and HbA1c. The high- and low-risk groups were dichotomized based on ΔTNF-α with a cutoff value of -5.7 pg/mL. The multivariate Cox regression analysis revealed that the high-risk group (ΔTNF-α ≤ -5.7 pg/mL) had an 11-fold cumulative probability of HCC compared to that of the low-risk group (HR = 11.02, 95%CI: 1.86-65.17, P = 0.008) among HCV patients with HbA1c less than 7%. In the younger population (age < 65 years old), the HCC risk was borderline significant between the high- and low-risk groups (HR = 8.51, 95%CI: 0.78-92.86, P = 0.079). Among patients with both HbA1c < 7% and age below 65 years old, the high-risk group had a 20-fold cumulative probability of HCC in comparison with the low-risk group (HR = 19.99, 95%CI: 0.90-443.91, P = 0.058) (Figure 3). The level of ΔTNF-α did not influence the development of HCC in either the patients aged ≥ 65 years old or with HbA1c ≥ 7% (Supplementary Figure 2).
Figure 3.

Multivariate Cox regression analysis of tumor necrosis factor-α associated with hepatocellular carcinoma in subgroups. Comparison of the cumulative probability of hepatocellular carcinoma development divided by Δtumor necrosis factor-α with a cutoff value of -5.7 pg/mL in patients with (A) hemoglobin A1c (HbA1c) < 7%, (B) age < 65 years old and (C) HbA1c < 7% and age < 65 years old. The P value was adjusted by age, sex, Fibrosis-4 index, and HbA1c. HR: Hazard ratio; CI: Confidence interval; HCC: Hepatocellular carcinoma; TNF-α: Tumor necrosis factor-α; HbA1c: Hemoglobin A1c.
HCC predictive model
Based on previous analyses, the FIB-4 index, HbA1c, ΔTNF-α, and ΔTWEAK were selected as parameters to predict the risk of HCC. The HCC predictive model was as follows: Score = 4 × FIB-4 (≥ 9, yes = 1, no = 0) + 5 × HbA1c (≥ 7, yes = 1, no = 0) + 11 × ΔTNF (≤ -5.7, yes = 1, no = 0) + 4 × ΔTWEAK (≤ -70, yes = 1, no = 0).
The weighting coefficient for each parameter was derived from the crude hazard ratio of the univariate Cox proportional hazards model. The performance of this HCC predictive model was assessed by time-dependent ROC curve analysis. In overall cases, the 3-year, 5-year, 10-year, and 13-year areas under the ROC curve (AUCs) were 0.882, 0.864, 0.903, and 1.000, respectively (Figure 4A). In the IFN group, the 3-year, 5-year, 10-year, and 13-year areas under the ROC curve (AUCs) were 0.782, 0.802, 0.870, and 1.000, respectively (Figure 4B).
Figure 4.

Time-dependent receiver operating characteristic curve analysis for the hepatocellular carcinoma predictive model. IFN: Interferon; AUC: Area under the curve.
Kaplan-Meier analysis for HCV patients stratified by risk scores
To classify the predictive score according to the risk of HCC, the patients were further stratified into low- (score = 0-7), intermediate- (score = 8-15), and high-risk groups (score > 15). In the high-risk group, the 3-year, 5-year, and 10-year cumulative risks of HCC were 20.0%, 40.0%, and 60.0%, respectively. In the intermediate-risk group, the 3-year, 5-year, and 10-year cumulative probabilities of HCC were 11.4%, 16.9%, and 31.0%, respectively. In contrast, none of the low-risk patients had HCC within 14 years of follow-up after successful viral eradication among the overall cases (log-rank P value = 6.8 × 10-6) (Figure 5A). Likewise, the Kaplan-Meier survival analysis revealed a significant difference in the cumulative probability of HCC among the IFN group stratified by the risk scores (log-rank P value = 9.6 × 10-5) (Figure 5B).
Figure 5.

Kaplan-Meier survival analysis for chronic hepatitis C patients stratified by the risk scores. IFN: Interferon.
DISCUSSION
This study revealed that there was no significant difference in the risk of HCC between the DAA and IFN groups after successful antiviral therapy. Downregulation of TNF-α and TWEAK increased the risk of hepatic carcinogenesis. ΔTNF-α was identified as an independent predictor of new-onset HCC among HCV patients with SVR. The effect of TNF-α was more prominent in young adults with normoglycemia. An HCC predictive model comprising FIB-4, HbA1c, ΔTNF-α, and ΔTWEAK had excellent performance, with 3-, 5-, 10-, and 13-year areas under the curve of 0.882, 0.864, 0.903, and 1.000, respectively. The 5-year accumulative risk of HCC was 0.0%, 16.9%, and 40.0% in the low-, intermediate-, and high-risk groups, respectively. These findings remained statistically significant among the HCV patients treated with pegIFN/ribavirin. Because there was only one HCC in the DAA group, the role of TNF-α in HCC should be further verified in this population. The HCC risk could be modified by the pre-existing host background and adjusted by the immune signatures after viral eradication. This predictive model helps clinicians adopt appropriate surveillance strategies for chronic hepatitis C patients following successful antiviral therapy according to the risk of HCC.
Our study showed that elevation of pretreatment TNF-α levels raised the possibility of new-onset HCC. Consistent with our study, Tarhuni et al[23] found that HCV-related cirrhotic patients carrying TNF-α 308 G>A had higher basal TNF-α production and exhibited a higher risk of HCC. Elevated basal TNF-α indicates sustained hepatic inflammation accompanied by persistent liver damage, which is susceptible to carcinogenesis. A systematic review showed that TNF-α was one of the strongest host genetic predictors for HCC in HCV-infected patients[24]. These findings suggested that the immune background was affected before antiviral therapy.
Interestingly, the abrupt decline in TNF-α levels after successful antiviral therapy increased the risk of HCC in our study. This implies a potential modification of the immune milieu by antiviral therapy that may trigger HCC development. Stimulation of the immune system effectively protects tissues from malignant cell invasion. Both cytotoxic T lymphocytes and NK cells are potent effectors in immune surveillance. TNF-α mediates the immune response against tumor cells by creating a microenvironment toward immunogenic activation rather than suppression[25]. Suppression of TNF signaling enables tumor cells to evade attack by cytotoxic T lymphocytes and attenuate in vivo antitumor responses[26]. Antiviral therapy may disrupt the balance from TNF-α activation to inhibition in immune surveillance. Alternatively, Debes et al[27] found that HCV patients with early-onset or recurrent HCC within 18 mo maintained stable or even higher TNF-α levels after DAA therapy. This implied that those patients might exhibit precarcinogenic or ongoing carcinogenic activity induced by TNF-α in response to occult HCC.
Both TNF-α and TWEAK belong to members of the TNF superfamily. These cytokines are mainly produced by macrophages, monocytes, and lymphocytes. TWEAK is a multifunctional cytokine that regulates a variety of cellular activities, including cell proliferation, differentiation, apoptosis, inflammation, and angiogenesis, via the fibroblast growth factor-inducible 14 receptor[28]. TWEAK appears to attenuate the innate response switch to adaptive immunity[29]. In addition, TWEAK is a weak inducer of apoptosis and also participates in tissue repair[30]. In chronic liver injury and repair, TWEAK appears to initiate liver progenitor cell expansion and ductal proliferation[31]. Hyperstimulation of inflammatory cells simultaneously results in excessive matrix deposition by activated hepatic stellate cells and myofibroblasts via the lymphotoxin-β signaling pathway[32]. Our study showed that posttreatment TWEAK expression was upregulated in the non-HCC group. Viral clearance alleviates the inflammatory status in the liver. It provides a microenvironment to facilitate the reconstruction of hepatocytes aided by TWEAK, which may further delay HCC development.
Our study confirms the consensus that DAA treatment does not markedly increase the risk of HCC compared to IFN treatment[33]. Most evidence has shown a decline in HCC risk regardless of whether SVR was achieved by IFN alone, DAA-only, or combined regimens[34]. However, successful antiviral therapy cannot eliminate the risk of HCC. The standard surveillance strategy (ultrasound and AFP every six months) advocates for all HCV patients. However, interindividual variations in HCC risk raise the question of whether the recommendations for HCC screening should be adjusted. Age, male sex, diabetes mellitus, and advanced fibrosis are well-known independent predictors of HCC after viral eradication[35-37]. In the absence of diabetes mellitus and old age (> 65 years old), the presence of ΔTNF-α ≤ -5.7 pg/mL increased the risk of HCC in patients with advanced fibrosis by 20-fold after HCV clearance. According to this HCC predictive model, patients with scores exceeding 15 should be closely monitored, since the 5-year cumulative risk of HCC reaches up to 40.0%. Nevertheless, none of the HCC cases had been identified over 14 years of follow-up in patients with a score of less than 7. The surveillance intervals may be extended among HCV patients achieving SVR in the absence of concurrent risk factors. In the post-DAA era, the risk model-based algorithm provides a cost-effective surveillance strategy for HCC.
The advancement of high-throughput technology makes early HCC detection more feasible. Currently, integrating multiomics data for HCC screening is also frequently observed[38]. The GALAD score consists of clinical factors (sex, age) and biomarkers (AFP, AFP-L3, and Des-carboxyprothrombin) that have an excellent performance to predict HCC, with an AUROC up to 0.97[39]. Using a miRNA panel (miR-22, miR-199a-3p) with AFP provided high diagnostic accuracy (AUROC = 0.988) for the early detection of HCC in HCV patients[39]. The methylation pattern of circulating cell-free DNA (APC, SFRP1, GSTP1, and RASSF1A) has demonstrated sufficient detection value to distinguish HCC patients from healthy controls[40]. Nevertheless, a majority of studies collected a cohort with a small sample size, and the analytical methods varied even in the same testing platform. These factors have limited the clinical application of these biomarkers.
Even though the sample size was limited in this pilot study, the statistical power was sufficient to be reliable. In overall cases, the statistical power of the association between ΔTNF-α and HCC was 99.9% to reject the null hypothesis at a P value < 0.05 under a hazard ratio of 11.54. In the IFN group, the statistical power of ΔTNF-α on HCC risk was 94.6% to reject the null hypothesis at a P value < 0.05 under a hazard ratio of 9.98. There are several limitations to this study. Although expensive IFN-free DAAs have been on the market since 2014, the National Health Insurance in Taiwan has reimbursed DAAs for HCV patients with advanced fibrosis since 2017. The follow-up time in most HCV patients treated with DAAs was less than 3 years. Owing to the small sample size and short follow-up time of the DAA group, a larger study cohort is necessary to validate the performance of this predictive model. IFN may induce distinct host immune alterations in comparison with DAA. As only one HCC case was identified in the DAA group throughout the follow-up period, it was unable to compare the diversity of cytokine profiles regarding HCC between the IFN and DAA groups. This predictive model was restricted to HCV patients with advanced fibrosis following successful antiviral therapy. Additionally, the optimal cutoff value should be further verified in other populations. The parameters of this predictive model were composed of serum cytokines involving the TNF superfamily. Host inflammation elicited by other etiologies may interfere with the predictive accuracy. Serum cytokines may not reflect the microenvironment within hepatocytes. To interpret this HCC predictive model, more care should be given to HCV patients presenting coinfection with other viruses, inflammatory disease, or malignancies.
CONCLUSION
This study revealed that downregulation of TNF-α increases the risk of HCC among HCV patients following successful antiviral therapy. Inhibition of TNF-α may attenuate host immune surveillance against tumor cells. Our findings provide a clue for the pathogenesis of hepatocarcinogenesis and a strategy for HCC surveillance based on risk stratification. With the development of high-throughput molecular technology, it is believed that more novel biomarkers will be applied in the early detection of HCC in the future.
ARTICLE HIGHLIGHTS
Research background
Successful hepatitis C virus (HCV) eradication cannot eliminate hepatocellular carcinoma (HCC) development. Chronic HCV infection induces profound alterations in cytokine and chemokine signatures. Clearance of HCV results in host immune modification, which may interfere with immune-mediated cancer surveillance.
Research motivation
The mechanism of hepatocarcinogenesis despite HCV clearance is still unclear.
Research objectives
To investigate the impact of differential cytokine expression on the development of HCC following HCV eradication.
Research methods
One hundred treatment-naïve HCV patients with advanced fibrosis who received antiviral therapy and achieved sustained virologic response (SVR) were enrolled. The primary endpoint was the development of new-onset HCC. In total, 64 serum cytokines were detected by the multiplex immunoassay at baseline and 24 wk after end-of-treatment.
Research results
The dynamics of serum tumor necrosis factor-α (TNF-α) and TNF-like weak inducer of apoptosis (TWEAK) were associated with HCC occurrence after HCV clearance. Multivariate Cox regression analysis showed that ΔTNF-α ≤ -5.7 pg/mL was an independent risk factor for HCC. An HCC predictive model comprising the Fibrosis-4 index, hemoglobin A1c, ΔTNF-α, and ΔTWEAK had excellent performance in stratifying the risk of HCC among HCV patients with SVR.
Research conclusions
Downregulation of serum TNF-α significantly increased the risk of HCC after HCV eradication.
Research perspectives
Our findings provide a clue for the pathogenesis of hepatocarcinogenesis and a strategy for HCC surveillance based on risk stratification.
Footnotes
Institutional review board statement: This study was reviewed and approved by the Institutional Review Board of Kaohsiung Medical University Hospital [No. KMUHIRB-E(I)-20180307&KMUHIRB-G(II)-20170020].
Informed consent statement: All study participants provided informed written consent prior to study enrollment.
Conflict-of-interest statement: The authors declare that they have no competing interests.
STROBE statement: The authors have read the STROBE Statement - checklist of items, and the manuscript was prepared and revised according to the STROBE Statement -checklist of items.
Provenance and peer review: Unsolicited article; Externally peer reviewed.
Peer-review model: Single blind
Peer-review started: September 8, 2021
First decision: October 16, 2021
Article in press: December 23, 2021
Specialty type: Gastroenterology and hepatology
Country/Territory of origin: Taiwan
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Emran TB, Sergi CM S-Editor: Yan JP L-Editor: A P-Editor: Yan JP
Contributor Information
Ming-Ying Lu, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Ming-Lun Yeh, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan; School of Medicine and Hepatitis Research Center, College of Medicine, Center for Cancer Research and Center for Liquid Biopsy, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
Ching-I Huang, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan; School of Medicine and Hepatitis Research Center, College of Medicine, Center for Cancer Research and Center for Liquid Biopsy, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
Shu-Chi Wang, Department of Medical Laboratory Science and Biotechnology, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
Yi-Shan Tsai, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Pei-Chien Tsai, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Yu-Min Ko, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Ching-Chih Lin, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Kuan-Yu Chen, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Yu-Ju Wei, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Po-Yao Hsu, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Cheng-Ting Hsu, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Tyng-Yuan Jang, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Ta-Wei Liu, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Po-Cheng Liang, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Ming-Yen Hsieh, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Zu-Yau Lin, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan; School of Medicine and Hepatitis Research Center, College of Medicine, Center for Cancer Research and Center for Liquid Biopsy, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
Shinn-Cherng Chen, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan; School of Medicine and Hepatitis Research Center, College of Medicine, Center for Cancer Research and Center for Liquid Biopsy, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
Chung-Feng Huang, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan; School of Medicine and Hepatitis Research Center, College of Medicine, Center for Cancer Research and Center for Liquid Biopsy, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
Jee-Fu Huang, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan; School of Medicine and Hepatitis Research Center, College of Medicine, Center for Cancer Research and Center for Liquid Biopsy, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
Chia-Yen Dai, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan; School of Medicine and Hepatitis Research Center, College of Medicine, Center for Cancer Research and Center for Liquid Biopsy, Kaohsiung Medical University, Kaohsiung 80708, Taiwan; Health Management Center, Department of Community Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
Wan-Long Chuang, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan; School of Medicine and Hepatitis Research Center, College of Medicine, Center for Cancer Research and Center for Liquid Biopsy, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
Ming-Lung Yu, Hepatitis Center and Hepatobiliary Division, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan; School of Medicine and Hepatitis Research Center, College of Medicine, Center for Cancer Research and Center for Liquid Biopsy, Kaohsiung Medical University, Kaohsiung 80708, Taiwan; Institute of Biomedical Sciences, National Sun Yat-Sen University, Kaohsiung 80708, Taiwan. fish6069@gmail.com.
Data sharing statement
Technical appendix, statistical code, and dataset available from the corresponding author at fish6069@gmail.com. Participants gave informed consent for data sharing.
References
- 1.Yu ML. Hepatitis C treatment from "response-guided" to "resource-guided" therapy in the transition era from interferon-containing to interferon-free regimens. J Gastroenterol Hepatol. 2017;32:1436–1442. doi: 10.1111/jgh.13747. [DOI] [PubMed] [Google Scholar]
- 2.Falade-Nwulia O, Suarez-Cuervo C, Nelson DR, Fried MW, Segal JB, Sulkowski MS. Oral Direct-Acting Agent Therapy for Hepatitis C Virus Infection: A Systematic Review. Ann Intern Med. 2017;166:637–648. doi: 10.7326/M16-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan RL, Baack B, Smith BD, Yartel A, Pitasi M, Falck-Ytter Y. Eradication of hepatitis C virus infection and the development of hepatocellular carcinoma: a meta-analysis of observational studies. Ann Intern Med. 2013;158:329–337. doi: 10.7326/0003-4819-158-5-201303050-00005. [DOI] [PubMed] [Google Scholar]
- 4.Reig M, Mariño Z, Perelló C, Iñarrairaegui M, Ribeiro A, Lens S, Díaz A, Vilana R, Darnell A, Varela M, Sangro B, Calleja JL, Forns X, Bruix J. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J Hepatol. 2016;65:719–726. doi: 10.1016/j.jhep.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Meringer H, Shibolet O, Deutsch L. Hepatocellular carcinoma in the post-hepatitis C virus era: Should we change the paradigm? World J Gastroenterol. 2019;25:3929–3940. doi: 10.3748/wjg.v25.i29.3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hernáez-Alsina T, Caballol-Oliva B, Díaz-González Á, Guedes-Leal C, Reig M. Risk of recurrence of hepatocellular carcinoma in patients treated with interferon-free antivirals. Gastroenterol Hepatol. 2019;42:502–511. doi: 10.1016/j.gastrohep.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Ahlenstiel G, Titerence RH, Koh C, Edlich B, Feld JJ, Rotman Y, Ghany MG, Hoofnagle JH, Liang TJ, Heller T, Rehermann B. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon-alfa-dependent manner. Gastroenterology. 2010;138:325–35.e1. doi: 10.1053/j.gastro.2009.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, Filipowicz W, Heim MH. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A. 2008;105:7034–7039. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 10.Missale G, Pilli M, Zerbini A, Penna A, Ravanetti L, Barili V, Orlandini A, Molinari A, Fasano M, Santantonio T, Ferrari C. Lack of full CD8 functional restoration after antiviral treatment for acute and chronic hepatitis C virus infection. Gut. 2012;61:1076–1084. doi: 10.1136/gutjnl-2011-300515. [DOI] [PubMed] [Google Scholar]
- 11.Martin B, Hennecke N, Lohmann V, Kayser A, Neumann-Haefelin C, Kukolj G, Böcher WO, Thimme R. Restoration of HCV-specific CD8+ T cell function by interferon-free therapy. J Hepatol. 2014;61:538–543. doi: 10.1016/j.jhep.2014.05.043. [DOI] [PubMed] [Google Scholar]
- 12.Serti E, Chepa-Lotrea X, Kim YJ, Keane M, Fryzek N, Liang TJ, Ghany M, Rehermann B. Successful Interferon-Free Therapy of Chronic Hepatitis C Virus Infection Normalizes Natural Killer Cell Function. Gastroenterology. 2015;149:190–200.e2. doi: 10.1053/j.gastro.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meissner EG, Wu D, Osinusi A, Bon D, Virtaneva K, Sturdevant D, Porcella S, Wang H, Herrmann E, McHutchison J, Suffredini AF, Polis M, Hewitt S, Prokunina-Olsson L, Masur H, Fauci AS, Kottilil S. Endogenous intrahepatic IFNs and association with IFN-free HCV treatment outcome. J Clin Invest. 2014;124:3352–3363. doi: 10.1172/JCI75938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kayadibi H. Letter: changes in FIB-4 cut-off points for viral hepatitis. Aliment Pharmacol Ther. 2017;45:1007–1008. doi: 10.1111/apt.13956. [DOI] [PubMed] [Google Scholar]
- 15.Burgess SV, Hussaini T, Yoshida EM. Concordance of sustained virologic response at weeks 4, 12 and 24 post-treatment of hepatitis c in the era of new oral direct-acting antivirals: A concise review. Ann Hepatol. 2016;15:154–159. doi: 10.5604/16652681.1193693. [DOI] [PubMed] [Google Scholar]
- 16.Lin CP, Liang PC, Huang CI, Yeh ML, Hsu PY, Hsu CT, Wei YJ, Liu TW, Hsieh MY, Hou NJ, Jang TY, Lin YH, Wang CW, Lin ZY, Chen SC, Huang CF, Huang JF, Dai CY, Chuang WL, Yu ML. Concordance of SVR12, SVR24 and SVR durability in Taiwanese chronic hepatitis C patients with direct-acting antivirals. PLoS One. 2021;16:e0245479. doi: 10.1371/journal.pone.0245479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marrero JA, Kulik LM, Sirlin CB, Zhu AX, Finn RS, Abecassis MM, Roberts LR, Heimbach JK. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology. 2018;68:723–750. doi: 10.1002/hep.29913. [DOI] [PubMed] [Google Scholar]
- 18.Vermehren J, Yu ML, Monto A, Yao JD, Anderson C, Bertuzis R, Schneider G, Sarrazin C. Multi-center evaluation of the Abbott RealTime HCV assay for monitoring patients undergoing antiviral therapy for chronic hepatitis C. J Clin Virol. 2011;52:133–137. doi: 10.1016/j.jcv.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 19.McKay HS, Margolick JB, Martínez-Maza O, Lopez J, Phair J, Rappocciolo G, Denny TN, Magpantay LI, Jacobson LP, Bream JH. Multiplex assay reliability and long-term intra-individual variation of serologic inflammatory biomarkers. Cytokine. 2017;90:185–192. doi: 10.1016/j.cyto.2016.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dressen K, Hermann N, Manekeller S, Walgenbach-Bruenagel G, Schildberg FA, Hettwer K, Uhlig S, Kalff JC, Hartmann G, Holdenrieder S. Diagnostic Performance of a Novel Multiplex Immunoassay in Colorectal Cancer. Anticancer Res. 2017;37:2477–2486. doi: 10.21873/anticanres.11588. [DOI] [PubMed] [Google Scholar]
- 21.Zweig MH, Campbell G. Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin Chem. 1993;39:561–577. [PubMed] [Google Scholar]
- 22.Schisterman EF, Perkins NJ, Liu A, Bondell H. Optimal cut-point and its corresponding Youden Index to discriminate individuals using pooled blood samples. Epidemiology. 2005;16:73–81. doi: 10.1097/01.ede.0000147512.81966.ba. [DOI] [PubMed] [Google Scholar]
- 23.Tarhuni A, Guyot E, Rufat P, Sutton A, Bourcier V, Grando V, Ganne-Carrié N, Ziol M, Charnaux N, Beaugrand M, Moreau R, Trinchet JC, Mansouri A, Nahon P. Impact of cytokine gene variants on the prediction and prognosis of hepatocellular carcinoma in patients with cirrhosis. J Hepatol. 2014;61:342–350. doi: 10.1016/j.jhep.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 24.Walker AJ, Peacock CJ, Pedergnana V STOP-HCV Consortium, Irving WL. Host genetic factors associated with hepatocellular carcinoma in patients with hepatitis C virus infection: A systematic review. J Viral Hepat. 2018;25:442–456. doi: 10.1111/jvh.12871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Showalter A, Limaye A, Oyer JL, Igarashi R, Kittipatarin C, Copik AJ, Khaled AR. Cytokines in immunogenic cell death: Applications for cancer immunotherapy. Cytokine. 2017;97:123–132. doi: 10.1016/j.cyto.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kearney CJ, Vervoort SJ, Hogg SJ, Ramsbottom KM, Freeman AJ, Lalaoui N, Pijpers L, Michie J, Brown KK, Knight DA, Sutton V, Beavis PA, Voskoboinik I, Darcy PK, Silke J, Trapani JA, Johnstone RW, Oliaro J. Tumor immune evasion arises through loss of TNF sensitivity. Sci Immunol. 2018;3 doi: 10.1126/sciimmunol.aar3451. [DOI] [PubMed] [Google Scholar]
- 27.Debes JD, van Tilborg M, Groothuismink ZMA, Hansen BE, Schulze Zur Wiesch J, von Felden J, de Knegt RJ, Boonstra A. Levels of Cytokines in Serum Associate With Development of Hepatocellular Carcinoma in Patients With HCV Infection Treated With Direct-Acting Antivirals. Gastroenterology. 2018;154:515–517.e3. doi: 10.1053/j.gastro.2017.10.035. [DOI] [PubMed] [Google Scholar]
- 28.Burkly LC, Michaelson JS, Hahm K, Jakubowski A, Zheng TS. TWEAKing tissue remodeling by a multifunctional cytokine: role of TWEAK/Fn14 pathway in health and disease. Cytokine. 2007;40:1–16. doi: 10.1016/j.cyto.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 29.Maecker H, Varfolomeev E, Kischkel F, Lawrence D, LeBlanc H, Lee W, Hurst S, Danilenko D, Li J, Filvaroff E, Yang B, Daniel D, Ashkenazi A. TWEAK attenuates the transition from innate to adaptive immunity. Cell. 2005;123:931–944. doi: 10.1016/j.cell.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 30.Shafritz DA. To TWEAK, or not to TWEAK: that is the question. Hepatology. 2010;52:13–15. doi: 10.1002/hep.23750. [DOI] [PubMed] [Google Scholar]
- 31.Jakubowski A, Ambrose C, Parr M, Lincecum JM, Wang MZ, Zheng TS, Browning B, Michaelson JS, Baetscher M, Wang B, Bissell DM, Burkly LC. TWEAK induces liver progenitor cell proliferation. J Clin Invest. 2005;115:2330–2340. doi: 10.1172/JCI23486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dwyer BJ, Olynyk JK, Ramm GA, Tirnitz-Parker JE. TWEAK and LTβ Signaling during Chronic Liver Disease. Front Immunol. 2014;5:39. doi: 10.3389/fimmu.2014.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li DK, Ren Y, Fierer DS, Rutledge S, Shaikh OS, Lo Re V 3rd, Simon T, Abou-Samra AB, Chung RT, Butt AA. The short-term incidence of hepatocellular carcinoma is not increased after hepatitis C treatment with direct-acting antivirals: An ERCHIVES study. Hepatology. 2018;67:2244–2253. doi: 10.1002/hep.29707. [DOI] [PubMed] [Google Scholar]
- 34.Gigi E, Lagopoulos VI, Bekiari E. Hepatocellular carcinoma occurrence in DAA-treated hepatitis C virus patients: Correlated or incidental? World J Hepatol. 2018;10:595–602. doi: 10.4254/wjh.v10.i9.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu ML, Huang CF, Yeh ML, Tsai PC, Huang CI, Hsieh MH, Hsieh MY, Lin ZY, Chen SC, Huang JF, Dai CY, Chuang WL. Time-Degenerative Factors and the Risk of Hepatocellular Carcinoma after Antiviral Therapy among Hepatitis C Virus Patients: A Model for Prioritization of Treatment. Clin Cancer Res. 2017;23:1690–1697. doi: 10.1158/1078-0432.CCR-16-0921. [DOI] [PubMed] [Google Scholar]
- 36.Huang CF, Yeh ML, Huang CY, Tsai PC, Ko YM, Chen KY, Lin ZY, Chen SC, Dai CY, Chuang WL, Huang JF, Yu ML. Pretreatment glucose status determines HCC development in HCV patients with mild liver disease after curative antiviral therapy. Medicine (Baltimore) 2016;95:e4157. doi: 10.1097/MD.0000000000004157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hajarizadeh B, Grebely J, Dore GJ. Epidemiology and natural history of HCV infection. Nat Rev Gastroenterol Hepatol. 2013;10:553–562. doi: 10.1038/nrgastro.2013.107. [DOI] [PubMed] [Google Scholar]
- 38.Liu XN, Cui DN, Li YF, Liu YH, Liu G, Liu L. Multiple "Omics" data-based biomarker screening for hepatocellular carcinoma diagnosis. World J Gastroenterol. 2019;25:4199–4212. doi: 10.3748/wjg.v25.i30.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson PJ, Pirrie SJ, Cox TF, Berhane S, Teng M, Palmer D, Morse J, Hull D, Patman G, Kagebayashi C, Hussain S, Graham J, Reeves H, Satomura S. The detection of hepatocellular carcinoma using a prospectively developed and validated model based on serological biomarkers. Cancer Epidemiol Biomarkers Prev. 2014;23:144–153. doi: 10.1158/1055-9965.EPI-13-0870. [DOI] [PubMed] [Google Scholar]
- 40.Huang ZH, Hu Y, Hua D, Wu YY, Song MX, Cheng ZH. Quantitative analysis of multiple methylated genes in plasma for the diagnosis and prognosis of hepatocellular carcinoma. Exp Mol Pathol. 2011;91:702–707. doi: 10.1016/j.yexmp.2011.08.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Technical appendix, statistical code, and dataset available from the corresponding author at fish6069@gmail.com. Participants gave informed consent for data sharing.