

Abstract
CNS inflammation is a key factor in Alzheimer’s Disease (AD), but its relation to pathological Aβ, tau, and APOE4 is poorly understood, particularly prior to the onset of cognitive symptoms. To better characterize early relationships between inflammation, APOE4, and AD pathology, we assessed correlations between cerebrospinal fluid (CSF) inflammatory markers and brain levels of Aβ and tau in cognitively normal older adults.
Each participant received a lumbar puncture to collect and quantify CSF levels of TNFα, IL-6, IL-8, and IL-10, a T1-weighted MRI, and PET scanning with [18F]flortaucipir (FTP; n = 57), which binds to tau tangles and/or [18F] florbetapir (FBP; n = 58), which binds to Aβ. Parallel voxelwise regressions assessed relationships between each CSF inflammatory marker and FTP and FBP SUVR, as well as APOE4*CSF inflammation interactions.
Unexpectedly, we detected significant negative associations between regional Aβ and tau PET uptake and CSF inflammatory markers. For Aβ PET, we detected negative associations with CSF IL-6 and IL-8 in regions known to show early accumulation of Aβ (i.e. lateral and medial frontal lobes). For tau PET, negative relationships were observed with CSF TNFα and IL-8, predominantly in regions known to exhibit early tau accumulation (i.e. medial temporal lobe). In subsequent analyses, significant interactions between APOE4 status and IL-8 on Aβ and tau PET levels were observed in spatially distinct regions from those showing CSF–Aβ/tau relationships.
Results from the current cross-sectional study support previous findings that neuroinflammation may be protective against AD pathology at a given stage of the disease, and extend these findings to a cognitively normal aging population. This study provides new insight into a dynamic relationship between neuroinflammation and AD pathology and may have implications for whom and when neuroinflammatory therapies may be appropriate.
Keywords: Glial activation, Amyloid-β, Tau, Neuroinflammation, PET imaging, Cytokine, Chemokine
1. Introduction
Along with amyloid-β (Aβ) plaques and hyperphosphorylated tau tangles, the hallmark pathological features of Alzheimer’s disease (AD), inflammation of the CNS has become increasingly recognized as a key factor in AD. Post-mortem and cerebrospinal fluid (CSF) markers of activated microglia and astrocytes are elevated in AD patients (Hopperton et al., 2018; Nordengen et al., 2019; Taipa et al., 2018). Activated glial cells are often colocalized with amyloid and tau in post-mortem tissue and AD models (Nilson et al., 2017; Stalder et al., 1999), as well as in in vivo positron emission tomography (PET) studies targeting the translocator protein (TSPO; Dani et al., 2018; Fan et al., 2017b; Hamelin et al., 2016; Knezevic et al., 2018; Parbo et al., 2017), which is a marker of activated glial cells. Furthermore, CSF markers of activated microglia are positively associated with CSF levels of phosphorylated tau (p-tau) (Janelidze et al., 2018), and preclinical data suggests microglia can drive pathological spreading of tau (Maphis et al., 2015). However, despite accumulating evidence that glial activation actively contributes to AD pathology at some point during the course of disease progression, there is also evidence suggesting that glial activation may be protective at certain disease stages (Fan et al., 2017b; Hamelin et al., 2016). These combined studies indicate that the involvement of neuroinflammation in AD is complex and dynamic, emphasizing the need for further research into inflammation at different timepoints.
One of the most commonly used methods to assess in vivo levels of glial activation is quantification of CSF inflammatory cytokines, such as IL-6, IL-8, TNFα, and IL-10, which are dysregulated in AD (Brosseron et al., 2014; Buchhave et al., 2010; Lue et al., 2001; Tarkowski et al., 2003). In response to glial activation, pro-inflammatory cytokines IL-6, IL-8, and TNFα are robustly released (Ehrlich et al., 1998; Olson and Miller, 2004), while increases in anti-inflammatory IL-10 occur secondarily (Burmeister and Marriott, 2018). In the brains of AD patients, elevated IL-6, IL-8, and TNFα levels are observed post-mortem (Ashutosh et al., 2011; Wood et al., 1993; Zhao et al., 2003) and are found in close proximity to amyloid plaques (Bauer et al., 1991; Dickson, 1997; Hüll et al., 1996; Xia et al., 1997). In vitro studies show that Aβ and tau exposure can increase release of IL-6, IL-8, and TNFα (Kovac et al., 2011; Lue et al., 2001). This body of work highlights the close relationship between inflammatory factors and AD pathology, but a definitive directional relationship is yet to be established. Generally, inflammatory processes are often thought to be deleterious and contribute to pathological aging and AD. This is supported by studies showing that IL-6 and TNFα induce expression of the amyloid precursor protein (Del Bo et al., 1995; Sommer et al., 2009). Two recent studies showed positive associations between several CSF inflammatory markers and levels of AD-related proteins from CSF (e.g. p-tau) in asymptomatic older adults (Bettcher et al., 2018) and in those with mild cognitive impairment and clinical AD (Brosseron et al., 2018). Positive correlations with CSF inflammatory markers were predominantly seen for p-tau, but Bettcher et al. did observe a counterintuitive positive association between CSF levels of IL-8 and Aβ42, which was suggested to reflect an initial increase in CSF Aβ42 prior to the decrease resulting from brain Aβ accumulation. Conversely, another study reported negative associations between CSF levels of TNFα and CSF p-tau (Tarkowski et al., 1999). The cumulative evidence suggests that while inflammation may contribute to and exacerbate pathological processes in aging and AD, there may also be circumstances in which inflammatory responses are protective. Therefore, a better understanding of associations between CSF inflammatory levels and early regional accumulation of amyloid and tau measured with positron emission tomography (PET) could help elucidate the role of neuroinflammation in aging and AD.
An additional factor, apolipoprotein E (APOE), is poised at the nexus of inflammation and AD pathology. The E4 variant of the APOE4 gene represents the highest genetic risk factor for sporadic AD (Colder et al., 1993). It has been linked to increased deposition and reduced clearance of Aβ aggregates (Kloske and Wilcock, 2020; Ohm et al., 1995), enhanced tau pathology in a mouse tauopathy model (Shi et al., 2017) and in human subjects (Ohm et al., 1995; Therriault et al., 2020), and elevated release of pro-inflammatory cytokines such as IL-6, IL-8, and TNFα (Fan et al., 2017a; Gale et al., 2014; Ophir et al., 2005). While it is possible APOE4 exerts these effects through independent pathways affecting levels of Aβ, tau and inflammatory cytokine, it is also likely that these pathways interact.
In order to assess early relationships between neuroinflammation and amyloid and tau levels, we conducted a study in cognitively normal older adults, comparing CSF inflammatory markers to brain PET amyloid and tau levels. Based on a large body of literature suggesting neuroinflammation contributes to AD pathology, we hypothesized that early neuroinflammation would be associated with increased brain deposition of Aβ and tau, and that this association would be stronger in APOE4 carriers.
2. Materials and methods
2.1. Participants
Seventy-three cognitively normal older adult participants (aged 49 – 85) were recruited through the Knight Alzheimer’s Disease Research Center at Washington University at St. Louis. All study procedures were approved by the local Institutional Review Board and performed in accordance with the 1964 Declaration of Helsinki. Informed consent was obtained from all participants before study enrollment. Clinical diagnosis for each participant was based on standard criteria incorporated in the Uniform Data Set (Morris et al., 2006). Inclusion in the study was restricted to participants who 1) demonstrated normal cognition, as determined by clinician assessment, a global Clinical Dementia Rating score of 0, and neuropsychological testing within a normal range; 2) received a lumbar puncture to collect cerebrospinal fluid (CSF); 3) received a T1-weighted MRI; and 4) received PET scanning with [18F] florbetapir (FBP; n = 58), which binds to Aβ plaques and /or [18F]flortaucipir (FTP; n = 57), which binds to tau tangles. All PET imaging sessions were conducted within one year of lumbar puncture (mean time difference ± SD = 4.59 ± 4.7 months).
2.2. Cerebrospinal fluid biomarkers
A Meso Scale Discovery (MSD) multiplex assay was used to determine CSF levels of tumor necrosis factor alpha (TNFα) and interleukin-6 (IL-6), IL-8, and IL-10 (catalog no. K15049G, MSD, Rockville, MD). An MSD multiplex assay was used to determine CSF levels of Aβ42 (catalog no. K15200E, MSD, Rockville, MD). An enzyme-linked immunosorbent assay was used to determine CSF levels of phosphorylated tau (pT181) (catalog no. 81581, Innotest, Fujirebio US, Inc., Malvern, PA).
2.3. APOE genotyping
APOE genotyping was performed as described in Nation et al. (2019). Participants with at least one copy of the E4 allele were considered APOE4 carriers. There were no E4 homozygous carriers.
2.4. Image acquisition
2.4.1. MRI
Structural T1-weighted MR images were acquired either on a Siemens Biograph mMR (n = 59) or a Siemens 3 T TrioTim (n = 14) using a magnetization prepared rapid acquisition gradient echo (MPRAGE) sequence with the following parameters: repetition time/echo time 2300/2.95 ms, in-plane resolution 240 × 256 mm, 176 slices, 1.2 mm slick thickness.
2.4.2. PET
[18F]florbetapir (FBP) PET scans were acquired on a Siemens Biograph mMR scanner. Scans were initiated with IV injection of 9.97 ± 0.70 mCi FBP and 4 × 5-minute frames were acquired from 50 to 70 min post-injection. [18F]flortaucipr (FTP) PET scans were acquired on a Siemens Biograph mMR scanner. Scans were initiated with IV injection of 8.89 ± 0.77 mCi FTP and 6 × 5-minute frames were acquired from 75 to 105 min post-injection.
2.5. Data processing
All data processing was performed using an automated in-house processing pipeline as detailed below.
2.5.1. T1 MRI
A sample-specific MR template was created using the Advanced Normalization Tools (ANTs) package template building tool (Avants et al., 2011, 2010). Each participants’ T1 image was processed with FreeSurfer v6.0.0 (https://surfer.nmr.mgh.harvard.edu) to generate subject-specific regions of interest (ROIs).
2.5.2. PET
Dynamic FBP and FTP images were motion corrected by aligning each frame to an average image. Motion-corrected PET frames were then averaged, coregistered to the T1 image, moved into template space, and smoothed with an 8 mm Gaussian kernel. To create reference regions, bilateral cerebellar gray and white matter labels from FreeSurfer were combined and eroded by 1 voxel to reduce partial volume effects (PVE). For FBP data, whole cerebellum was used as a reference region. For FTP data, inferior cerebellar gray matter was used as a reference region (Baker et al., 2017). Cerebellar segmentation was performed with SUIT (http://www.diedrichsenlab.org/imaging/suit.htm), and dorsal regions were removed from the cerebellar ROI. Average PET signal was extracted from reference ROIs in native PET space. Parametric FBP and FTP SUVR images were created by dividing PET signal in each voxel by the average signal in the respective reference regions. To characterize general levels of PET binding, composite ROIs were created. For FBP, the composite ROI consisted of frontal, parietal, lateral temporal, and cingulate cortices (Landau and Jagust, 2015). For FTP, the composite ROIs consisted of Braak I-II and Braak III-IV regions (Baker et al., 2017; Schöll et al., 2016).
2.6. Statistical analyses
Differences in subject characteristics between APOE4 carriers and non-carriers were assessed with independent samples t-tests.
To determine associations between brain levels of Aβ/tau and CSF inflammatory markers, we performed parallel voxelwise linear regressions between FBP/FTP SUVR images and CSF TNFα, IL-6, IL-8, and IL-10 using Statistical Parametric Mapping (SPM12, Wellcome Trust Center for Neuroimaging). All analyses were spatially restricted with a gray matter mask, obtained by segmentation of the MRI template. Age, sex, and APOE4 status were included as covariates. Statistical significance was determined with a voxelwise threshold of p < 0.001, uncorrected, with a cluster size threshold of k > 100 voxels. To assess the effect of APOE4 status on relationships between Aβ/tau and CSF inflammation, we conducted parallel voxelwise linear regressions identical to those described above, with the addition of an APOE4*CSF inflammation interaction term.
Secondary voxelwise analyses were conducted to assess whether Aβ/tau relationships with CSF inflammation existed independently of the other pathology, and to determine if PET – CSF relationships were influenced by gray matter volume. Therefore, average FBP/FTP SUVR were extracted from significant clusters in all voxelwise partial correlations. In order to reduce the number of ROIs for multiple comparisons, bilateral clusters and clusters within the same anatomic region (e.g. left and right inferior parietal lobe [IPL]) were combined into one ROI when more than 4 clusters were present. To correct for potential confounding effects of atrophy, average gray matter volume was extracted from each ROI by moving the ROI from template space into subject space and multiplying with the gray matter probability from SPM segmentation. Partial correlations were performed between FBP SUVR and CSF inflammatory markers including CSF p-tau and gray matter volume as covariates, and between FTP SUVR and CSF inflammatory markers including CSF Aβ42 and gray matter volume as covariates.
The FTP tracer exhibits high uptake in the choroid plexus (CP), a structure adjacent to the hippocampus, which may reflect off-target binding (Marquié et al., 2017). To correct for this, we employed a regression-based approach (Lee et al., 2018; Wang et al., 2016) for regions showing significant tau – CSF inflammation relationships that were directly adjacent to the CP. CP SUVR was extracted using the FreeSurfer CP segmentation as a mask. Partial correlations correcting for CP SUVR, age, sex, and APOE4 status were performed between FTP SUVR in CP-adjacent regions and CSF inflammatory markers. Statistical analyses were performed with SPSS.
3. Results
3.1. Participants
Demographic and study outcome variables for all participants are displayed in Table 1. APOE4 carriers exhibited higher composite FBP SUVR (p = 0.023) and lower CSF Aβ42 levels (p = 0.011) compared to non-carriers, but no significant differences in any other variable.
Table 1.
Subject characteristics. Mean ± SD or frequency (%) are displayed for each variable.
| Participant characteristics | Whole sample (n = 73) | APOE4 carriers (n = 24) | APOE4 non-carriers (n = 49) |
|---|---|---|---|
| Age | 67.1 ± 7.9 | 65.5 ± 8.5 | 67.8 ± 7.6 |
| Sex | 46F (63%) | 14 (58%) | 32 (65%) |
| Education (yrs) | 16.9 ± 2.6 | 16.8 ± 2.5 | 16.9 ± 2.6 |
| FBP composite SUVR (n = 58)* | 0.96 ± 0.1 | 1.01 ± 0.2 | 0.94 ± 0.1 |
| Braak I-II FTP SUVR (n = 57) | 1.17 ± 0.1 | 1.20 ± 0.2 | 1.01 ± 0.1 |
| Braak III-IV FTP SUVR (n = 57) | 1.14 ± 0.1 | 1.16 ± 0.1 | 1.14 ± 0.1 |
| CSF TNFα (n = 72) | 0.23 ± 0.1 | 0.20 ± 0.1 | 0.24 ± 0.1 |
| CSF IL-8 | 39.5 ± 10 | 40.0 ± 8.5 | 39.3 ± 11 |
| CSF IL-6 | 1.14 ± 0.6 | 1.06 ± 0.5 | 1.19 ± 0.7 |
| CSF IL-10 (n = 72) | 0.08 ± 0.0 | 0.08 ± 0.04 | 0.08 ± 0.04 |
| CSF Aβ42 (n = 72)* | 286 ± 120 | 236 ± 102 | 310 ± 110 |
| CSF p-tau (n = 71) | 49.3 ± 19 | 54.5 ± 21 | 46.9 ± 18 |
Significant difference between APOE4 carriers and non-carriers at p < 0.05 Data available for all subjects unless specified in parentheses.
3.2. Associations between Aβ, tau, and neuroinflammation
3.2.1. Aβ is negatively correlated with CSF inflammatory markers
Contrary to our hypothesis, Aβ PET signal was negatively correlated with CSF markers of neuroinflammation. For IL-6, significant negative IL-6 – FBP SUVR associations were observed in several cortical regions, including prefrontal cortex (PFC), supramarginal gyrus (SMG), and precentral/postcentral gyri (Fig. 1A). For IL-8, significant negative IL-8 – FBP SUVR relationships were observed mainly in frontal regions, e.g. dorsolateral prefrontal cortex (dlPFC), inferior frontal gyrus (IFG), and in insula and inferior parietal lobe (IPL; Fig. 1B). There were no significant negative correlations between TNFα or IL-10 and Aβ PET, and no significant positive correlations for any CSF inflammatory marker and Aβ PET.
Fig. 1.
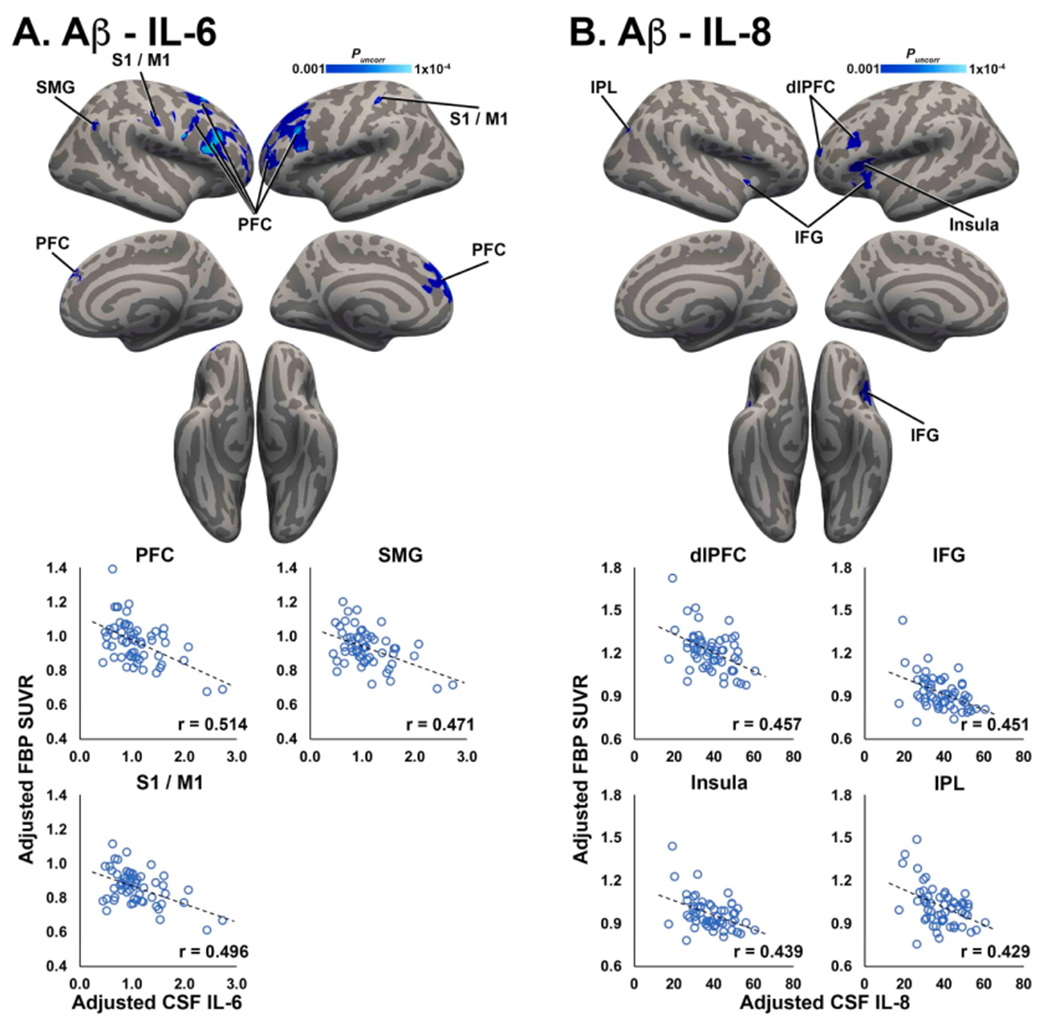
Aβ PET is negatively correlated with inflammatory markers. Clusters shown in blue colorscale depict significant negative correlations between FBP SUVR and CSF IL-6 (A) and IL-8 (B). For visualization, average FBP SUVR was extracted from significant clusters and plotted against CSF IL-6/IL-8, after correcting for age, sex, and APOE4 status. PFC: prefrontal cortex; SMG: supramarginal gyrus; S1/M1: primary somatosensoiy/motor cortex; IFG: inferior frontal gyrus. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In secondary analyses including CSF p-tau and gray matter volume as covariates to control for possible effects of tau pathology and gray matter atrophy, negative associations between IL and 6 and FBP SUVR observed in the primary analysis remained highly significant (r’s < −0.484, p’s < 0.001). Negative correlations between IL-8 and FBP SUVR also remained highly significant (r’s < −0.449, p’s < 0.001), indicating that associations between tau PET levels and pro-inflammatory cytokines were not influenced by CSF p-tau or atrophy.
3.2.2. Tau is negatively correlated with CSF inflammatory markers
Similar to observed relationships between Aβ PET uptake and CSF inflammatory markers, tau PET signal was negatively correlated with CSF markers of neuroinflammation. For TNFα, significant negative associations with FTP SUVR were observed in several cortical and subcortical structures, including bilateral medial temporal lobe (MTL; hippocampus and amygdala), entorhinal cortex (EC), inferior/middle temporal gyri (ITG/MTG; Fig. 2A). For IL-8, significant negative associations with FTP SUVR were observed in the bilateral hippocampus (Fig. 2B). There were no significant negative correlations between IL-6 or IL-10 and tau PET, and no significant positive correlations for any CSF inflammatory marker and tau PET.
Fig. 2.
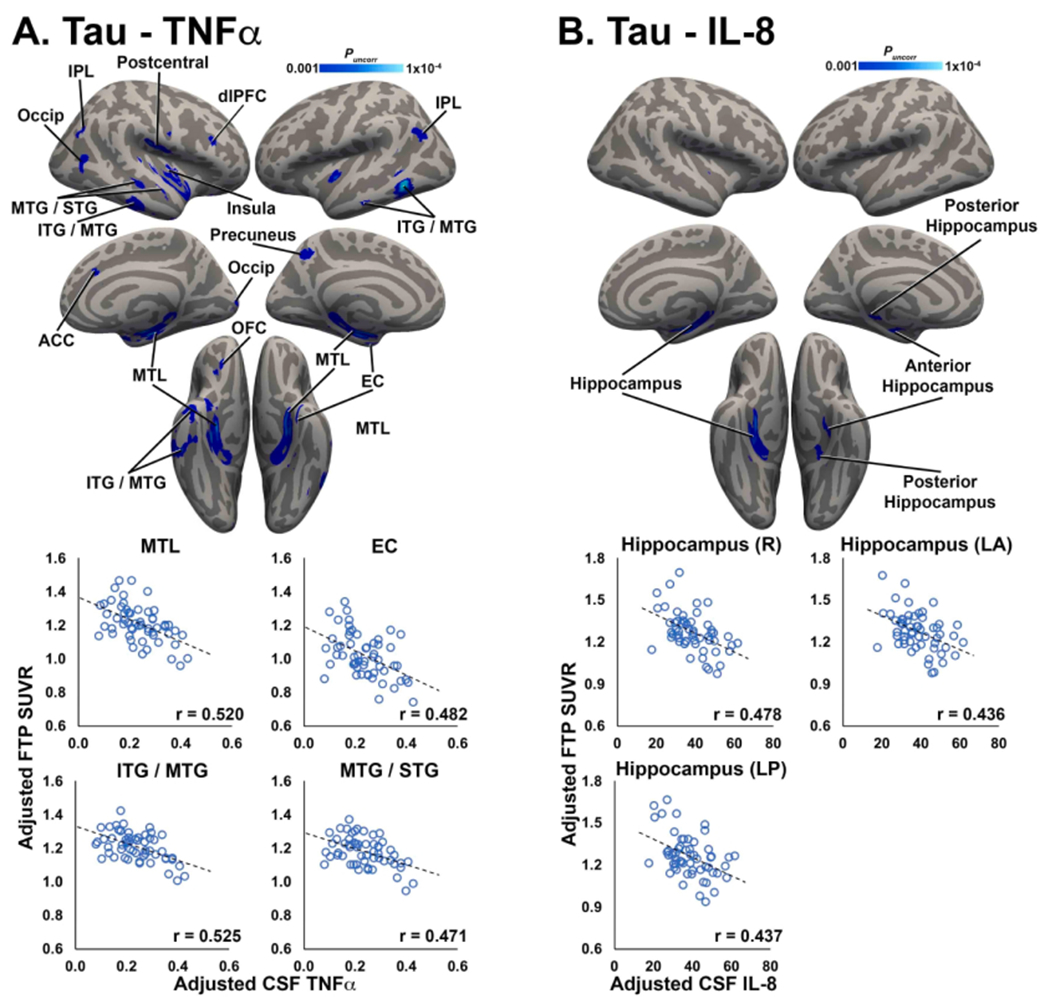
Tau PET is negatively correlated with CSF inflammatory markers. Clusters shown in blue colorscale depict significant negative correlations between FTP SUVR and CSF TNFα (A) and IL-8 (B). For visualization, average FTP SUVR was extracted from significant clusters and plotted against CSF TNFα/IL-8, after correcting for age, sex, and APOE4 status. MTL: medial temporal lobe; EC: entorhinal cortex; ITG/MTG/STG: inferior/middle/superior temporal gyrus; IPL: inferior parietal lobule; dlPFC: dorsolateral prefrontal cortex; ACC: anterior cingulate cortex; OFC: orbitofrontal cortex; R: right; LA: left anterior; LP: left posterior. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In secondary analyses including CSF Aβ42 and gray matter volume as covariates to control for potential effects of amyloid pathology and gray matter atrophy, negative associations between TNFα and FTP SUVR remained highly significant across all ROIs (r’s < −0.408, p’s < 0.003). Negative associations between IL and 8 and FTP SUVR also remained significant when correcting for CSF Aβ42 and gray matter volume (r’s < −0.392, p’s < 0.005), indicating that associations between tau PET levels and pro-inflammatoiy cytokines were independent of CSF levels of Aβ42 and gray matter atrophy.
3.2.3. Effects of choroid plexus FTP binding on FTP – CSF associations
Voxelwise regressions revealed negative associations between TNFα/IL-8 and FTP SUVR in several regions directly adjacent to choroid plexus (CP; Fig. 2). For TNFα, when FTP SUVR from CP was corrected for, correlations between TNFα and MTL FTP SUVR remained significant (r = −0.287, p = 0.039), but the correlation between TNFα and thalamic FTP SUVR was no longer significant (r = − 0.139, p = 0.327), indicating that FTP binding in the CP may have influenced the association between TNFα and FTP SUVR in the thalamus, but likely not the MTL. For IL-8, when CP SUVR was corrected for, correlations between IL-8 and FTP SUVR were no longer significant for the right hippocampus (r = −0.213, p = 0.125), left anterior hippocampus (r = −0.191, p = 0.171), and left posterior hippocampus (r = −0.127, p = 0.364). This suggests that correlations between IL-8 and FTP SUVR in these regions may have been influenced by FTP binding in the CP.
3.3. Interactions between APOE4 and neuroinflammation on Aβ and tau levels
3.3.1. Negative relationship between IL and 8 and Aβ PET is enhanced in APOE4 carriers
Significant interactions between APOE4 and IL-8 on Aβ PET signal were detected in the SMG and IPL (Fig. 3A). In this interaction, APOE4 carriers displayed a steeper negative relationship between IL and 8 and FBP SUVR than APOE4 non-carriers. There were no significant APOE4*CSF inflammation interactions on FBP SUVR for TNFα, IL-6, or IL-10.
Fig. 3.
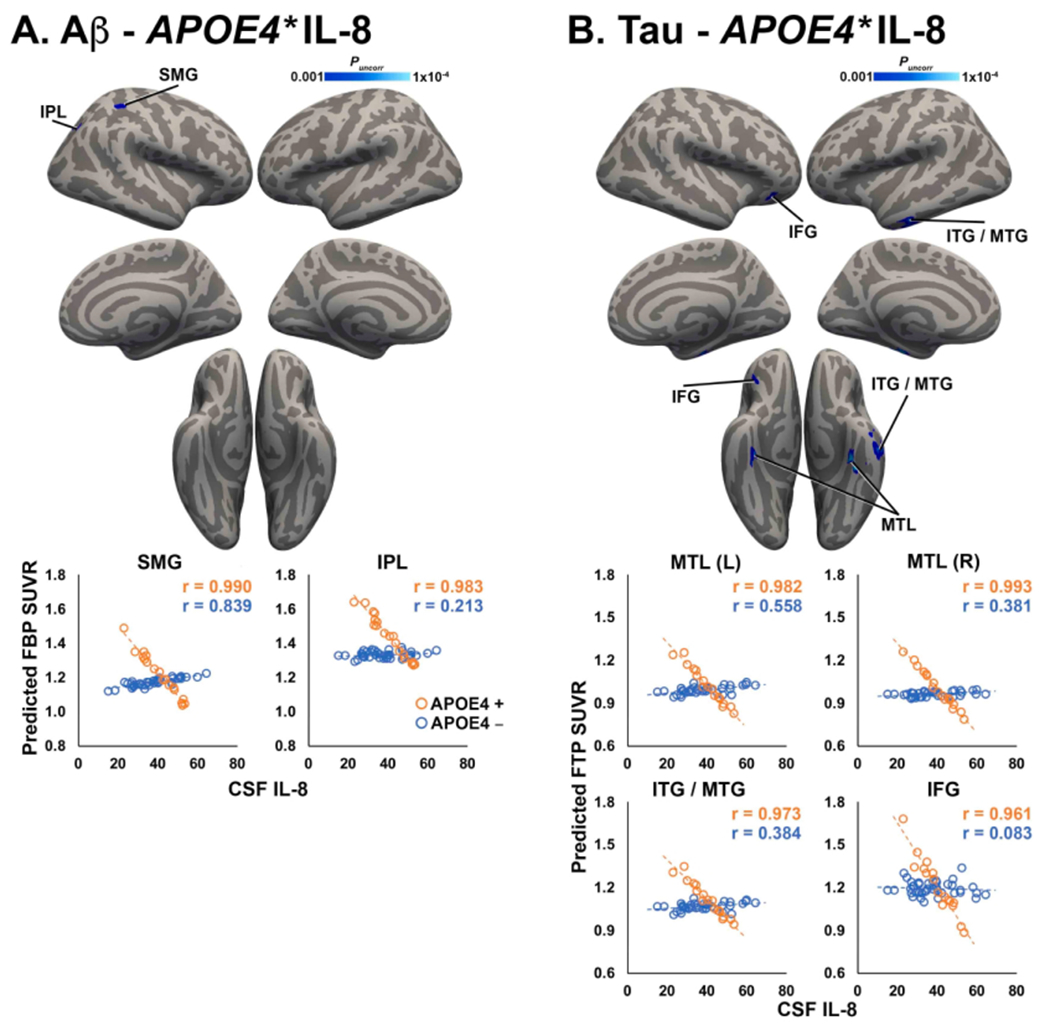
Significant interactions between APOE4 and IL-8 on Aβ and tau PET. Clusters shown in blue colorscale depict significant interactions between APOE4 status and IL-8 on FBP SUVR (A) and FTP SUVR (B). For visualization, average FBP/FTP SUVR was extracted from significant clusters and plotted against CSF IL-8, after correcting for age, sex, and APOE4 status. APOE4 carriers are shown in orange, non-carriers are shown in blue. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3.2. Negative relationship between IL and 8 and tau PET is enhanced in APOE4 carriers
Significant interactions between APOE4 and IL-8 on tau PET signal were detected in bilateral MTL (including parahippocampal gyrus, entorhinal cortex, and fusiform gyrus), ITG/MTG, and IFG (Fig. 3B). In this interaction, APOE4 carriers displayed a much steeper negative relationship between IL-8 and FTP SUVR than APOE4 non-carriers. There were no significant APOE4*CSF inflammation interactions on FTP SUVR for TNFα, IL-6, or IL-10.
4. Discussion
In the current study, we unexpectedly observed negative relationships between brain PET levels of both Aβ and tau and CSF inflammation markers, such that higher levels of inflammation were associated with lower AD pathology. Furthermore, we observed APOE4*IL-8 interactions on brain Aβ and tau PET signal, such that negative Aβ/tau – CSF relationships were stronger in APOE4 carriers. These combined results suggest that early neuroinflammation, prior to the onset of cognitive symptoms, may be an adaptive mechanism to prevent accumulation of Aβ and tau proteins.
Our finding of inverse relationships between Aβ/tau levels and CSF inflammatory markers is in line with previous longitudinal studies that utilized TSPO PET. In one study, a mixed sample of prodromal AD patients (CDR = 0.5) and AD dementia patients (CDR > 0.5) was followed for 2 years (Hamelin et al., 2016). Patients exhibiting slow cognitive decline longitudinally showed higher glial activation compared to patients with faster cognitive decline, indicating that glial activation might play a protective role in early clinical progression of AD. Another study reported that patients with mild cognitive impairment (MCI) exhibited decreased glial activation over the course of ~14 months that was associated with increased amyloid load (Fan et al., 2017b), suggesting that early glial activation may reflect an adaptive mechanism to prevent against AD pathology. Results from the current study are in agreement with these previous outcomes in MCI and prodromal AD patients, and further add evidence suggesting that early glial activation, as reflected by enhanced CSF inflammatory cytokines, may be protective against accumulation of Aβ and tau in a cognitively normal aging population.
Significant negative correlations were observed between IL-6/IL-8 and Aβ levels, particularly in frontal regions, which are known to demonstrate early Aβ accumulation in aging and AD (Gonneaud et al., 2017; Mattsson et al., 2019). The negative direction of inflammation – Aβ relationships is consistent with a potential protective role for neuroinflammation prior to the onset of cognitive symptoms. Our result suggesting IL-6 may be protective against increased Aβ levels in cognitive normal older adults is consistent with evidence that IL-6 suppresses amyloid deposition in an AD mouse model (Chakrabarty et al., 2010). Though there are relatively few studies directly investigating the effects of IL-8 on amyloid burden, previous results have indicated neuroprotective effects of IL-8 (Araujo and Cotman, 1993), and attenuation of Aβ-induced apoptosis after administration of IL-8 (Ashutosh et al., 2011). Furthermore, a well-known action of IL-8 is the chemotactic recruitment of neutrophils across the blood–brain barrier (BBB). Although some recent evidence suggests that neutrophils may play a deleterious role in AD (Zenaro et al., 2015), other studies have found that specific neutrophil proteins may inhibit accumulation of AD pathology (Stock et al., 2018). Additionally, IL-8 is known to have pro-angiogenic actions (Koch et al., 1992), which could potentially contribute to increased Aβ clearance across the BBB. The specific mechanisms underlying IL-6/IL-8 relationships with Aβ are still poorly understood and require further investigation. There were no significant correlations between Aβ PET and TNFα or IL-10. This could be due to a smaller effect size, as voxelwise correlations between FBP SUVR and TNFα/IL-10 showed similar spatial patterns to those with IL-6/IL-8 when significance thresholds were lowered (data not shown). Longitudinal studies are needed to elucidate the dynamic effects of inflammatory cytokine release and their downstream effects on Aβ deposition.
We observed significant negative associations between tau levels and both TNFα and IL-8 that were independent of CSF amyloid and atrophy, particularly in regions known to exhibit early accumulation of tau in aging and AD, i.e. MTL and inferior temporal lobe (Lowe et al., 2018; Pontecorvo et al., 2017). Mechanistic evidence consistent with this finding suggests a suppressive role for TNFα on AD pathology. For example, in a triple-transgenic AD mouse model (3xTg-AD), knocking out both TNFα receptors, TNF-R1 and TNF-R2, reduced the phagocytic ability of microglia and exacerbated both Aβ and tau pathology over time (Montgomery et al., 2011). A study in AD patients reported negative associations between CSF levels of TNFα and p-tau similar to those in the current study (Tarkowski et al., 1999), suggesting a potential neuroprotective role for TNFα. Regarding the observed correlations between IL-8 and tau levels, there is little direct evidence linking IL-8 to tau. Because TNFα signaling can itself induce the release of IL-8 from astrocytes (Aloisi et al., 1992) and microglia (Ehrlich et al., 1998), and the spatial patterns of TNFα – tau relationships was largely overlapping with that of IL-8 – tau relationships (Fig. 2), it is possible that the results with IL-8 were an indirect result influenced by TNFα levels. Additionally, after covarying for the effect of choroid plexus binding, IL-8 – tau relationships were no longer significant, indicating that they could also have been an artifact of FTP binding in the CP. There were also no significant correlations between tau PET and IL-6 or IL-10. Because IL-6 has been reported to induce tau phosphorylation, an initial step in accumulation of pathological tau (Quintanilla et al., 2004), any AD pathology-suppressing effects related to pro-inflammatory actions of IL-6 could potentially be balanced by its tau phosphorylating effects. Further studies are needed to better understand the relationships of pro-inflammatory factors and tau levels in the context of aging and AD.
Finally, we observed interactions between APOE4 status and CSF IL-8 concentrations on both Aβ and tau PET uptake. The direction of this interaction was such that negative Aβ/tau – IL-8 relationships were significantly steeper in APOE4 carriers compared to non-carriers. Although the APOE4 allele represents the greatest genetic risk factor for late onset AD, several studies have shown that middle-aged APOE4 carriers exhibit paradoxically enhanced cognitive performance (Alexander et al., 2007; Zokaei et al., 2017), indicating that APOE4 may impart early beneficial effects prior to its later harmful effects. The APOE4 interactions observed in the current study could be due to enhanced levels of cytokines, as APOE4 expression induces greater release of inflammatory cytokines in transfected cells and in vivo in humans (Fan et al., 2017a; Gale et al., 2014; Theendakara et al., 2016). However, there were no significant differences in CSF inflammatory cytokines between APOE4 carriers and non-carriers in the current study. Alternatively, the APOE4*IL-8 interactions could be related to a recent discovery linking APOE4 to BBB dysfunction (Montagne et al., 2020). The increased BBB leakage demonstrated by APOE4 carriers prior to cognitive impairment could potentially interact with the chemotactic properties of IL-8 to allow for greater infiltration of immune cells like neutrophils that target AD pathology. This interpretation is supported by a preclinical study showing that a disease-modifying agent enhanced infiltration of neutrophils and reduced amyloid and tau levels (Gabbita et al., 2015). Additionally, the APOE4*IL-8 interactions could be associated with pro-angiogenic actions of IL-8 (Koch et al., 1992), either independently of or complementary to immune cell recruitment. Interestingly, the regions showing significant APOE4*IL-8 interactions were spatially distinct from those demonstrating IL-8 – Aβ/tau correlations (Figs. 1B and 2B), suggesting that APOE4 may exert effects in a regionally distinct manner. This observation could be related to the documented influence of APOE4 on regional brain metabolism (Jagust et al., 2012), as the regions exhibiting APOE4 interactions (e.g. lateral parietal lobe, MTL) are hubs in the default mode network, which is marked by a high metabolic rate (Raichle et al., 2001). Further research is needed to understand the cellular mechanisms underlying relationships between APOE4, IL-8, and amyloid and tau pathology.
The current study has several limitations. Recent investigations have emphasized variability in the FTP PET signal, particularly in individuals with low levels of brain Aβ (Baker et al., 2019; Lowe et al., 2020), and we cannot exclude the possibility that noise in the FTP PET signal unrelated to tau protein in our sample of predominantly amyloid- participants could have masked results. However, the majority of CSF-FTP PET correlations were observed in lateral and medial temporal areas. FTP PET signal in these areas has been shown to correspond well with tau burden in post mortem samples (Lowe et al., 2020). Regardless, these results will need to be confirmed in future studies. Additionally, due to several factors, including the cross-sectional nature of the study and the inclusion of only cognitively normal participants, it is impossible to determine whether the observed relationships between CSF inflammatory markers and brain Aβ and tau levels have any influence on the development of cognitive impairment and clinical AD. Longitudinal studies are necessary to elucidate the influence of inflammation in cognitively normal individuals on the development of future cognitive decline.
In conclusion, the current study provides evidence of negative relationships between neuroinflammation and Aβ and tau accumulation in a cognitively normal aging population. Our study, along with several lines of previous research, suggest that the relationship between neuroinflammation and proteins associated with AD pathology may be dynamic and differ across stages of aging and dementia severity. Future studies are needed to carefully delineate these complex interactions and to better understand when and for whom neuroinflammatory therapies may be appropriate.
Funding
This work was supported by National Institute on Aging grants P01AG03991, P50AG05681, P01AG026276 (JC Morris PI), P01AG052350 (B Zlokovic), and R01AG054617 (J Pa, PI).
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Alexander DM, Williams LM, Gatt JM, Dobson-Stone C, Kuan SA, Todd EG, Schofield PR, Cooper NJ, Gordon E, 2007. The contribution of apolipoprotein E alleles on cognitive performance and dynamic neural activity over six decades. Biol. Psychol 75, 229–238. [DOI] [PubMed] [Google Scholar]
- Aloisi F, Carè A, Borsellino G, Gallo P, Rosa S, Bassani A, Cabibbo A, Testa U, Levi G, Peschle C, 1992. Production of hemolymphopoietic cytokines (IL-6, IL-8, colony-stimulating factors) by normal human astrocytes in response to IL-1 beta and tumor necrosis factor-alpha. J. Immunol 149, 2358–2366. [PubMed] [Google Scholar]
- Araujo DM, Cotman CW, 1993. Trophic effects of interleukin-4, -7 and -8 on hippocampal neuronal cultures: potential involvement of glial-derived factors. Brain Res. 600, 49–55. [DOI] [PubMed] [Google Scholar]
- Ashutosh KW, Cotter R, Borgmann K, Wu L, Persidsky R, Sakhuja N, Ghorpade A, 2011. CXCL8 protects human neurons from amyloid-β-induced neurotoxicity: relevance to Alzheimer’s disease. Biochem. Biophys. Res. Commun 412, 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Tustison NJ, Song G, Cook PA, Klein A, Gee JC, 2011. A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage 54, 2033–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Yushkevich P, Pluta J, Minkoff D, Korczykowski M, Detre J, Gee JC, 2010. The optimal template effect in hippocampus studies of diseased populations. Neuroimage 49, 2457–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SL, Harrison TM, Maass A, La Joie R, Jagust WJ, 2019. Effect of off-target binding on. J. Nucl. Med 60, 1444–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SL, Maass A, Jagust WJ, 2017. Considerations and code for partial volume correcting [Data Brief 15, 648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer J, Strauss S, Schreiter-Gasser U, Ganter U, Schlegel P, Witt I, Yolk B, Berger M, 1991. Interleukin-6 and alpha-2-maeroglobulin indicate an acute-phase state in Alzheimer’s disease cortices. FEBS Lett. 285, 111–114. [DOI] [PubMed] [Google Scholar]
- Bettcher BM, Johnson SC, Fitch R, Casaletto KB, Heffernan KS, Asthana S, Zetterberg H, Blennow K, Carlsson CM, Neuhaus J, Bendlin BB, Kramer JH, 2018. Cerebrospinal fluid and plasma levels of inflammation differentially relate to CNS markers of Alzheimer’s disease pathology and neuronal damage. J. Alzheimers Dis 62, 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosseron F, Krauthausen M, Kummer M, Heneka MT, 2014. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol. Neurobiol 50, 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosseron F, Traschütz A, Widmann CN, Kummer MP, Tacik P, Santarelli F, Jessen F, Heneka MT, 2018. Characterization and clinical use of inflammatory cerebrospinal fluid protein markers in Alzheimer’s disease. Alzheimers Res. Ther 10, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchhave P, Zetterberg H, Blennow K, Minthon L, Janciauskiene S, Hansson O, 2010. Soluble TNF receptors are associated with Aβ metabolism and conversion to dementia in subjects with mild cognitive impairment. Neurobiol. Aging 31, 1877–1884. [DOI] [PubMed] [Google Scholar]
- Burmeister AR, Marriott I, 2018. The Interleukin-10 family of cytokines and their role in the CNS. Front. Cell. Neurosci 12, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Jansen-West K, Beccard A, Ceballos-Diaz C, Levites Y, Verbeeck C, Zubair AC, Dickson D, Golde TE, Das P, 2010. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 24, 548–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA, 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. [DOI] [PubMed] [Google Scholar]
- Dani M, Wood M, Mizoguchi R, Fan Z, Walker Z, Morgan R, Hinz R, Biju M, Kuruvilla T, Brooks DJ, Edison P, 2018. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 141, 2740–2754. [DOI] [PubMed] [Google Scholar]
- Del Bo R, Angeretti N, Lucca E, De Simoni MG, Forloni G, 1995. Reciprocal control of inflammatory cytokines, IL-1 and IL-6, and beta-amyloid production in cultures. Neurosci. Lett 188, 70–74. [DOI] [PubMed] [Google Scholar]
- Dickson DW, 1997. The pathogenesis of senile plaques. J. Neuropathol. Exp. Neurol 56, 321–339. [DOI] [PubMed] [Google Scholar]
- Ehrlich LC, Hu S, Sheng WS, Sutton RL, Rockswold GL, Peterson PK, Chao CC, 1998. Cytokine regulation of human microglial cell IL-8 production. J. Immunol 160, 1944–1948. [PubMed] [Google Scholar]
- Fan YY, Cai QL, Gao ZY, Lin X, Huang Q, Tang W, Liu JH, 2017a. APOE ε4 allele elevates the expressions of inflammatory factors and promotes Alzheimer’s disease progression: A comparative study based on Han and She populations in the Wenzhou area. Brain Res. Bull 132, 39–43. [DOI] [PubMed] [Google Scholar]
- Fan Z, Brooks DJ, Okello A, Edison P, 2017b. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 140, 792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbita SP, Johnson MF, Kobritz N, Eslami P, Poteshkina A, Varadarajan S, Turman J, Zemlan F, Harris-White ME, 2015. Oral TNFαc modulation alters neutrophil infiltration, improves cognition and diminishes tau and amyloid pathology in the 3xTgAD mouse model. PLoS ONE 10, e0137305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale SC, Gao L, Mikacenic C, Coyle SM, Rafaels N, Murray Dudenkov T, Madenspacher JH, Draper DW, Ge W, Aloor JJ, Azzam KM, Lai L, Blackshear PJ, Calvano SE, Barnes KC, Lowry SF, Corbett S, Wurfel MM, Fessler MB, 2014. APOε4 is associated with enhanced in vivo innate immune responses in human subjects. J. Allergy Clin. Immunol 134, 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonneaud J, Arenaza-Urquijo EM, Mézenge F, Landeau B, Gaubert M, Bejanin A, de Flores R, Wirth M, Tomadesso C, Poisnel G, Abbas A, Desgranges B, Chételat G, 2017. Increased florbetapir binding in the temporal neocortex from age 20 to 60 years. Neurology 89, 2438–2446. [DOI] [PubMed] [Google Scholar]
- Hamelin L, Lagarde J, Dorothée G, Leroy C, Labit M, Comley RA, de Souza LC, Corne H, Dauphinot L, Bertoux M, Dubois B, Gervais P, Colliot O, Potier MC , Bottlaender M, Sarazin M, team, C.I., 2016. Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain 139, 1252–1264. [DOI] [PubMed] [Google Scholar]
- Hopperton KE, Mohammad D, Trépanier MO, Giuliano V, Bazinet RP, 2018. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: a systematic review. Mol. Psychiatry 23, 177–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hïll M, Berger M, Volk B, Bauer J, 1996. Occurrence of interleukin-6 in cortical plaques of Alzheimer’s disease patients may precede transformation of diffuse into neuritic plaques. Ann. N. Y. Acad. Sci 777, 205–212. [DOI] [PubMed] [Google Scholar]
- Jagust WJ, Landau SM, Initiative A.s.D.N, 2012. Apolipoprotein E, not fibrillar β-amyloid, reduces cerebral glucose metabolism in normal aging. J. Neurosci 32, 18227–18233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janelidze S, Mattsson N, Stomrud E, Lindberg O, Palmqvist S, Zetterberg H, Blennow K, Hansson O, 2018. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology 91, e867–e877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloske CM, Wilcock DM, 2020. the important interface between apolipoprotein E and neuroinflammation in Alzheimer’s disease. Front. Immunol 11, 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knezevic D, Verhoeff NPL, Hafizi S, Strafella AP, Graff-Guerrero A, Rajji T, Pollock BG, Houle S, Rusjan PM, Mizrahi R, 2018. Imaging microglial activation and amyloid burden in amnestic mild cognitive impairment. J. Cereb. Blood Flow Metab 38, 1885–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM, 1992. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 258, 1798–1801. [DOI] [PubMed] [Google Scholar]
- Kovac A, Zilka N, Kazmerova Z, Cente M, Zilkova M, Novak M, 2011. Misfolded truncated protein τ induces innate immune response via MAPK pathway. J. Immunol 187, 2732–2739. [DOI] [PubMed] [Google Scholar]
- Landau S, Jagust W, 2015. Florbetapir processing methods, https://adni.bitbucket.io/reference/docs/UCBERKELEYAV45/ADNI_AV45_Methods_JagustLab_06.25.15.pdf. [Google Scholar]
- Lee CM, Jacobs HIL, Marquié M, Becker JA, Andrea NV, Jin DS, Schultz AP, Frosch MP, Gómez-Isla T, Sperling RA, Johnson KA, 2018. 18F-Flortaucipir binding in choroid plexus: related to race and hippocampus signal. J. Alzheimers Dis 62, 1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe VJ, Lundt ES, Albertson SM, Min HK, Fang P, Przybelski SA, Senjem ML, Schwarz CG, Kantarci K, Boeve B, Jones DT, Reichard RR, Tranovich JF , Hanna Al-Shaikh FS, Knopman DS, Jack CR, Dickson DW, Petersen RC, Murray ME, 2020. Tau-positron emission tomography correlates with neuropathology findings. Alzheimers Dement 16, 561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe VJ, Wiste HJ, Senjem ML, Weigand SD, Therneau TM, Boeve BF, Josephs KA, Fang P, Pandey MK, Murray ME, Kantarci K, Jones DT, Vemuri P, Graff-Radford J, Schwarz CG, Machulda MM, Mielke MM, Roberts RO, Knopman DS, Petersen RC, Jack CR, 2018. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 141, 271–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM, Brachova L, Yan SD, Walker DG, Shen Y, Rogers J, 2001. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 35, 72–79. [DOI] [PubMed] [Google Scholar]
- Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K, 2015. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138, 1738–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquié M, Verwer EE, Meltzer AC, Kim SJW, Agüero C, Gonzalez J, Makaretz SJ, Chong, Siao Tick M, Ramanan P, Amaral AC, Normandin MD, Vanderburg CR, Gomperts SN, Johnson KA, Frosch MP, Gómez-Isla T, 2017. Lessons learned about [F-18]-AV-1451 off-target binding from an autopsy-confirmed Parkinson’s case. Acta Neuropathol. Cornmun 5, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Palmqvist S, Stomrud E, Vogel J, Hansson O, 2019. Staging β-Amyloid pathology with amyloid positron emission tomography. JAMA Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, Pachicano M, Joe E, Nelson AR, D’Orazio LM, Buennagel DP, Harrington MG , Benzinger TLS, Fagan AM, Ringman JM, Schneider LS, Morris JC, Reiman EM, Caselli RJ, Chui HC, Tcw J, Chen Y, Pa J, Conti PS, Law M, Toga AW, Zlokovic BV, 2020. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 581, 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SL, Mastrangelo MA, Habib D, Narrow WC, Knowlden SA, Wright TW, Bowers WJ, 2011. Ablation of TNF-RI/RII expression in Alzheimer’s disease mice leads to an unexpected enhancement of pathology: implications for chronic pan-TNF-α suppressive therapeutic strategies in the brain. Am. J. Pathol 179, 2053–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Weintraub S, Chui HC, Cummings J, Decarli C, Ferris S, Foster NL, Galasko D, Graff-Radford N, Peskind ER, Beekly D, Ramos EM, Kukull WA, 2006. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis. Assoc. Disord 20, 210–216. [DOI] [PubMed] [Google Scholar]
- Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG, Benzinger TLS, Fagan AM, Ringman JM, Schneider LS, Morris JC, Chui HC, Law M, Toga AW, Zlokovic BV, 2019. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med 25, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilson AN, English KC, Gerson JE, Barton Whittle T, Nicolas Crain C, Xue J, Sengupta U, Castillo-Carranza DL, Zhang W, Gupta P, Kayed R, 2017. Tau oligomers associate with inflammation in the brain and retina of tauopathy mice and in neurodegenerative diseases. J. Alzheimers Dis 55, 1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordengen K, Kirsebom BE, Henjum K, Seines P, Gísladóttir B, Wettergreen M, Torsetnes SB, Grøntvedt GR, Waterloo KK, Aarsland D, Nilsson LNG, Fladby T, 2019. Glial activation and inflammation along the Alzheimer’s disease continuum. J. Neuroinflam 16, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohm TG, Kirca M, Bohl J, Scharnagl H, Gross W, März W, 1995. Apolipoprotein E polymorphism influences not only cerebral senile plaque load but also Alzheimer-type neurofibrillary tangle formation. Neuroscience 66, 583–587. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD, 2004. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol 173, 3916–3924. [DOI] [PubMed] [Google Scholar]
- Ophir G, Amariglio N, Jacob-Hirsch J, Elkon R, Rechavi G, Michaelson DM, 2005. Apolipoprotein E4 enhances brain inflammation by modulation of the NF-kappaB signaling cascade. Neurobiol. Dis 20, 709–718. [DOI] [PubMed] [Google Scholar]
- Parbo P, Ismail R, Hansen KV, Amidi A, Mårup FH, Gottrup H, Brændgaard H, Eriksson BO, Eskildsen SF, Lund TE, Tietze A, Edison P, Pavese N, Stokholm MG, Borghammer P, Hinz R, Aanerud J, Brooks DJ, 2017. Brain inflammation accompanies amyloid in the majority of mild cognitive impairment cases due to Alzheimer’s disease. Brain 140, 2002–2011. [DOI] [PubMed] [Google Scholar]
- Pontecorvo MJ, Devous MD, Navitsky M, Lu M, Salloway S, Schaerf FW, Jennings D, Arora AK, McGeehan A, Lim NC, Xiong H, Joshi AD, Siderowf A, Mintun MA, investigators,, F., A., ., A., 2017. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 140, 748–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Orellana DI, González-Billault C, Maccioni RB, 2004. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res 295, 245–257. [DOI] [PubMed] [Google Scholar]
- Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL, 2001. A default mode of brain function. Proc. Natl. Acad. Sci. USA 98, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöll M, Lockhart SN, Schonhaut DR, O’Neil JP, Janabi M, Ossenkoppele R, Baker SL, Vogel JW, Faria J, Schwimmer HD, Rabinovici GD, Jagust WJ, 2016. PET imaging of tau deposition in the aging human brain. Neuron 89, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM, Holtzman DM, Initiative,, A.s. D.N.„ 2017. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer G, Kralisch S, Lipfert J, Weise S, Krause K, Jessnitzer B, Lössner U, Blüher M, Stumvoll M, Fasshauer M, 2009. Amyloid precursor protein expression is induced by tumor necrosis factor alpha in 3T3-L1 adipocytes. J. Cell. Biochem 108, 1418–1422. [DOI] [PubMed] [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M, 1999. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am. J. Pathol 154, 1673–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock AJ, Kasus-Jacobi A, Pereira HA, 2018. The role of neutrophil granule proteins in neuroinflammation and Alzheimer’s disease. J Neuroinflam 15, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipa R, Ferreira V, Brochado P, Robinson A, Reis I, Marques F, Mann DM, Melo-Pires M, Sousa N, 2018. Inflammatory pathology markers (activated microglia and reactive astrocytes) in early and late onset Alzheimer disease: a post mortem study. Neuropathol. Appl. Neurobiol 44, 298–313. [DOI] [PubMed] [Google Scholar]
- Tarkowski E, Andreasen N, Tarkowski A, Blennow K, 2003. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 74, 1200–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski E, Blennow K, Wallin A, Tarkowski A, 1999. Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J. Clin. Immunol 19, 223–230. [DOI] [PubMed] [Google Scholar]
- Theendakara V, Peters-Libeu CA, Spilman P, Poksay KS, Bredesen DE, Rao RV, 2016. Direct transcriptional effects of Apolipoprotein E. J. Neurosci 36, 685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therriault J, Benedet AL, Pascoal TA, Mathotaarachchi S, Chamoun M, Savard M, Thomas E, Kang MS, Lussier F, Tissot C, Parsons M, Qureshi MNI, Vitali P, Massarweh G, Soucy JP, Rej S, Saha-Chaudhuri P, Gauthier S, Rosa-Neto P, 2020. Association of Apolipoprotein E ε4 with medial temporal tau independent of Amyloid-β. JAMA Neurol 77, 470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Benzinger TL, Su Y, Christensen J, Friedrichsen K, Aldea P, McConathy J, Cairns NJ, Fagan AM, Morris JC, Ances BM, 2016. Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between β-Amyloid and Tauopathy. JAMA Neurol 73, 1070–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JA, Wood PL, Ryan R, Graff-Radford NR, Pilapil C, Robitaille Y, Quirion R, 1993. Cytokine indices in Alzheimer’s temporal cortex: no changes in mature IL-1 beta or IL-1RA but increases in the associated acute phase proteins IL-6, alpha 2-macroglobulin and C-reactive protein. Brain Res 629, 245–252. [DOI] [PubMed] [Google Scholar]
- Xia M, Qin S, McNamara M, Mackay C, Hyman BT, 1997. Interleukin-8 receptor B immunoreactivity in brain and neuritic plaques of Alzheimer’s disease. Am. J. Pathol 150, 1267–1274. [PMC free article] [PubMed] [Google Scholar]
- Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, Turano E, Rossi B, Angiari S, Dusi S, Montresor A, Carlucci T, Nanì S, Tosadori G, Calciano L, Catalucci D, Berton G, Bonetti B, Constantin G, 2015. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med 21, 880–886. [DOI] [PubMed] [Google Scholar]
- Zhao M, Cribbs DH, Anderson AJ, Cummings BJ, Su JH, Wasserman AJ, Cotman CW, 2003. The induction of the TNFalpha death domain signaling pathway in Alzheimer’s disease brain. Neurochem. Res 28, 307–318. [DOI] [PubMed] [Google Scholar]
- Zokaei N, Giehl K, Sillence A, Neville MJ, Karpe F, Nobre AC, Husain M, 2017. Sex and APOE: A memory advantage in male APOE ε4 carriers in midlife. Cortex 88, 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]