

Abstract
Cerebrospinal fluid (CSF) synaptosomal-associated protein 25 (SNAP-25) and neurogranin (Ng) are recently described biomarkers for pre- and postsynaptic integrity known to be elevated in symptomatic Alzheimer disease (AD). Their relationship with Apolipoprotein E (APOE) ε4 carrier status, the major genetic risk factor for AD, remains unclear. In this study, CSF SNAP-25 and Ng were compared in cognitively normal APOE ε4 carriers and noncarriers (n = 274, mean age 65 ± 9.0 years, 39% APOE ε4 carriers, 58% female). CSF SNAP-25, not CSF Ng, was specifically elevated in APOE ε4 carriers versus noncarriers (5.95 ± 1.72 pg/mL, 4.44 ± 1.40 pg/mL, p < 0.0001), even after adjusting for age, sex, years of education, and amyloid status (p < 0.0001). CSF total tau (t-tau), phosphorylated-tau-181 (ptau181), and neurofilament light chain (NfL) also did not vary by APOE ε4 status. Our findings suggest APOE ε4 carriers have amyloid-related and amyloid-independent presynaptic disruption as reflected by elevated CSF SNAP-25 levels. In contrast, postsynaptic disruption as reflected by elevations in CSF neurogranin is related to amyloid status.
Keywords: APOE, SNAP-25, Neurogranin, Synapse, Biomarker, CSF
1. Background
Apolipoprotein E (APOE) genotype is the major genetic risk factor for Alzheimer disease (AD) and is thought to modify both amyloid- (Fleisher et al., 2013; Kok et al., 2009; Liu et al., 2017; Morris et al., 2010), and tau-related pathology (Shi et al., 2017; Shi et al., 2019). APOE genotype has also been implicated in a variety of neurodegenerative disorders including α-synucleinopathies such as Parkinson disease and Lewy Body dementia (Li et al., 2004; Zhao et al., 2020), Huntington disease (Panegyres et al., 2006), frontotemporal dementia (Agosta et al., 2009), and chronic traumatic encephalopathy (McKee et al., 2009). The mechanism by which APOE genotype affects these diverse disorders remains unclear. However, multiple studies have highlighted amyloid-independent toxicity through synapse related pathways (Dumanis et al., 2009; Love et al., 2006; Nwabuisi-Heath et al., 2014; Tannenberg et al., 2006; Wang et al., 2005; Zhao et al., 2020).
The APOE ε4 allele has been implicated in both presynaptic and postsynaptic dysfunction. This includes reductions of key presynaptic proteins (Tannenberg et al., 2006) and disruptions of presynaptic vesicular release and glutamine-to-glutamate production (Dumanis et al., 2013). Postsynaptic effects include disruptions of reelin-mediated long-term potentiation and plasticity (Weeber et al., 2002) and reductions in dendritic spine density and complexity (Dumanis et al., 2009; Jain et al., 2013; Wang et al., 2005) that may be further amplified in the presence of amyloid plaques (Holtzman et al., 20 0 0). However, the relationship between APOE genotype and pre- or postsynaptic dysfunction in cognitively normal older adults remains unclear.
Two recent cerebrospinal fluid (CSF) biomarkers have emerged for assessing synaptic integrity in humans: synaptosomal-associated protein 25 (SNAP-25) and neurogranin (Ng). SNAP-25 is a component of the presynaptic SNARE complex, which is essential for vesicular trafficking (Shin, 2014). Ng is expressed in postsynaptic dendritic spines (Chang et al., 1997). Both CSF SNAP-25 (Brinkmalm et al., 2014; Zhang et al., 2018), and Ng concentrations (De Vos A, et al., 2015; Kester et al., 2015; Kvartsberg et al., 2015a; Kvartsberg et al., 2015b; Portelius et al., 2015; Tarawneh et al., 2016; Thorsell et al., 2010) are elevated in individuals with AD dementia.
In this study, we compared levels of CSF SNAP-25 and Ng in cognitively normal individuals as a function of APOE ε4 status. We adjusted for the effects of age, sex, years of education and amyloid status. Additionally, we evaluated levels of CSF total tau (t-tau), tau phosphorylated at position 181 (ptau181), and neurofilament light chain (NfL). Finally, we replicated our major finding in an independent cohort.
2. Materials and methods
2.1. Participants
The primary cohort consisted of participants enrolled at the Knight Alzheimer Disease Research Center (Knight ADRC) at Washington University in St Louis. Inclusion criteria were the following: participants who were cognitively normal (Clinical Dementia Rating [CDR] 0; Morris, 1993), had APOE genotype data, and had undergone analysis of CSF SNAP-25 and/or Ng. Methods for recruitment and assessment have previously been described (Morris et al., 2019). This study was approved by the Washington University Institutional Review Board and each participant provided signed informed consent.
2.2. Genetic analyses
DNA samples were collected at enrollment and genotyped using either an Illumina 610 or Omniexpress chip, as previously described (Cruchaga et al., 2013). APOE ε4 carriers were defined by the presence of at least one ε4 allele (ε2/ε4, ε3/ε4, or ε4/ε4) in contrast to APOE ε4 noncarriers (ε2/ε2, ε2/ε3, or ε3/ε3).
2.3. CSF acquisition and processing
Participants underwent CSF collection as previously described (Fagan et al., 2006). Briefly, CSF was collected at 8 AM after overnight fasting in a polypropylene tube via gravity drip using an atraumatic Sprotte 22 gauge spinal needle. Samples were gently inverted and centrifuged at low speed to pellet any cellular debris. CSF was then aliquoted into 500 μL volumes in polypropylene tubes and stored at −80°C until the time of assay.
CSF Aβ42, t- tau, and ptau181 were measured with corresponding Elecsys immunoassays on the Roche cobas e601 analyzer (Schindler et al., 2018). Amyloid status was established per previously published cutoffs for CSF ptau181/Aβ42 (Schindler et al., 2018), with individuals with a CSF ptau181/Aβ42 ratio ≤0.0198 categorized as amyloid-negative and individuals with ptau181/Aβ42 > 0.0198 categorized as amyloid-positive.
CSF SNAP-25 and Ng were measured via the microparticle-based immunoassay, Single Molecule Counting Erenna system (EMD Millipore, Burlington MA) system, with antibodies developed in the laboratory of Dr. Jack Ladenson at Washington University. CSF NfL was measured with an immunoassay kit manufactured by Uman Diagnostics (UmanDiagnostics, Umeå, Sweden).
2.4. PET image acquisition and processing
Amyloid positron emission tomography (PET) images were acquired on a subset of participants per previously described methods (Mintun et al., 2006; Su et al., 2015; Su et al., 2018; Su et al., 2019) using either [11C] Pittsburgh Compound B (PiB) or florbetapir (18F-AV-45). Standard uptake value ratios (SUVR) were calculated for the 30–60 minute postinjection window for PiB and 50–70 minutes for 18F-AV-45. Raw PET data were then processed using a PET Unified Pipeline (github.com/ysu001/PUP). FreeSurfer 5.3 was employed for region of interest (ROI) segmentation. For each region, a tissue mask was generated based on segmentation, and partial volume correction performed (Su et al., 2015). SUVRs, also known as regional target-to-reference intensity ratios, were evaluated for each region using the cerebral cortex as the reference region. The partial volume corrected SUVR derived from cortical regions was used as a summary value for each PET imaging modality. To standardize across PiB and 18 F-AV-45, SUVRs were converted to centiloids (Klunk et al., 2004; Su et al., 2018).
2.5. Statistical analyses
Testing between subgroups was compared using unpaired t-tests for continuous variables and chi-square testing for categorical variables. Secondary validation for multiple comparisons was performed by calculating a false discovery rate. Analysis for covariance were implemented using the Matlab function LinearModel.fit between CSF SNAP-25 or Ng and APOE ε4 status, amyloid status, age, gender (female), and years of education.
2.6. Data availability policy
Data are available to qualified investigators upon request to the Knight ADRC (https://knightadrc.wustl.edu/Research/ResourceRequest.htm)
2.7. Replication cohort
For replication of the major finding, data were also obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early AD. For up-to-date information, see www.adni-info.org. Inclusion criteria were identical to the primary cohort: participants who were cognitively normal (CDR 0), had available APOE genotype data, and had undergone analysis of CSF SNAP-25 and/or Ng using consistent assay lot number.
3. Results
3.1. Participant characteristics
The Knight ADRC cohort consisted of 274 participants who met inclusion criteria. The characteristics of the cohort, grouped by either amyloid status based on CSF ptau181/Aβ42 or APOE ε4 carrier status, are shown in Table 1. There was no significant difference in years of education or gender by either amyloid status or APOE ε4 carrier status. Amyloid-positive individuals tended to be older and were more likely to carry an APOE ε4 allele (p < 0.0001). As expected, the individuals categorized as amyloid-positive by CSF ptau181/Aβ42 had significantly higher PET centiloid values. APOE ε4 carriers also had a higher average PET centiloid values (p = 0.0004).
Table 1.
Knight Alzheimer Disease Research Center (ADRC) participant characteristics
| Group | All | Amyloid | Amyloid+ | p |
|---|---|---|---|---|
| N | 274 | 191 | 83 | |
| Age (years ± SD) | 65 ± 9.0 | 62.3 ± 8.3 | 71.2 ± 7.4 | < 0.0001 |
| Education (years ± SD) | 16.0 ± 2.50 | 16.1 ± 2.41 | 15.8 ± 2.70 | N.S. |
| Sex (n, % Female) | 159, 58% | 116, 61% | 43, 52% | N.S. |
| Race (n, %) | ||||
| Asian | 1, <1% | 1, < 1% | 0, 0% | N.S. |
| Black | 23, 8% | 19, 10% | 4, 5% | N.S. |
| Non-Hispanic White | 250, 91% | 171, 90% | 79, 95% | N.S. |
| APOE ε4 statusa (n, %) | 107, 39% | 56, 29% | 51, 61% | < 0.0001 |
| PET Centiloid (mean ± SD) | 13.3 ± 28.3 | −2.14 ± 5.16 | 46.4 ± 29.4 | < 0.0001 |
| CSF Aβ42 (pg/mL) | 1330 ± 620 | 1600 ± 540 | 730 ± 260 | < 0.0001 |
| CSF t-tau (pg/mL) | 229 ± 106 | 188 ± 64.1 | 321 ± 124 | < 0.0001 |
| CSF ptau181 (pg/mL) | 21.2 ± 11.8 | 16.5 ± 5.71 | 32.1 ± 14.6 | < 0.0001 |
| CSF SNAP-25 (pg/mL) | 5.03 ± 1.72 | 4.66 ± 1.56 | 5.9 ± 1.76 | < 0.0001 |
| CSF Ng (pg/mL) | 2080 ± 1120 | 1770 ± 880 | 2790 ± 1290 | < 0.0001 |
| CSF NfL (pg/mL) | 1390 ± 690 | 1230 ± 620 | 1750 ± 690 | < 0.0001 |
| Group | APOE ε4− | APOE ε4+ | p | |
| n | 167 | 107 | ||
| Age (years ± SD) | 64.9 ± 8.5 | 65.2 ± 9.78 | N.S. | |
| Education (years ± SD) | 16 ± 2.5 | 16 ± 2.58 | N.S. | |
| Sex (n, % Female) | 101, 60% | 58, 54% | N.S. | |
| Race (n, %) | ||||
| Asian | 1, 1% | 0, 0% | N.S. | |
| Black | 15, 9% | 8, 7% | N.S. | |
| Non-Hispanic White | 151, 90% | 99, 93% | N.S. | |
| Amyloid statusa (n, %) | 32, 19% | 51, 48% | < 0.0001 | |
| PET Centiloid (mean ± SD)b | 7.82 ± 26.0 | 22.9 ± 30.0 | 0.0004 | |
| CSF Aβ42 (pg/mL) | 1460 ± 630 | 1130 ± 550 | < 0.0001 | |
| CSF t-tau (pg/mL) | 216 ± 100 | 249 ± 109 | 0.01 | |
| CSF ptau181 (pg/mL) | 19.8 ± 11 | 23.5 ± 12 | 0.01 | |
| CSF SNAP-25 (pg/mL) | 4.44 ± 1.4 | 5.95 ± 1.72 | < 0.0001 | |
| CSF Ng (pg/mL) | 1970 ± 1070 | 2250 ± 1190 | 0.04 | |
| CSF NfL (pg/mL) | 1340 ± 680 | 1470 ± 690 | N.S. |
Demographic comparison of amyloid-positive and amyloid-negativea groups, and APOE ε4 carriers and noncarriers. P values reflect between group comparisons using unpaired t-tests for continuous variables and chi-square tests for categorical variables. All participants are CDR 0.
Amyloid-negative if CSF ptau181/Aβ42 < 0.0198; Amyloid-positive if CSF ptau181/Aβ42 ≥0.0198 (Schindler et al., 2018).
Average across the subset of participants who had PET Centiloid data; all participants underwent CSF testing.
3.2. Differences in CSF biomarkers by amyloid status or APOE ε4 carrier status
Concentrations of CSF biomarkers including Aβ42, t-tau, ptau181, SNAP-25, Ng, and NfL were examined as a function of amyloid status and or APOE ε4 carrier status. All six CSF biomarkers were significantly different between amyloid-positive individuals and amyloid-negative individuals (p < 0.0001). When grouped by APOE ε4 carrier status, significant group differences in Aβ42 (p < 0.0001), t-tau (p = 0.01), and ptau181 (p = 0.01) were observed (Table 1). Significant elevations in SNAP-25 (p < 0.0001) and Ng (p = 0.04) were also observed, although the difference in Ng did not survive after correction for multiple comparisons. Finally, no significant group difference in NfL was observed between APOE ε4 carriers and noncarriers.
3.3. CSF biomarkers in different APOE allele genotypes
CSF SNAP-25 and Ng as a function of APOE genotype were evaluated (Fig. 1). Presence of the APOE ε4 allele was associated with higher CSF SNAP-25 levels (Fig. 1A). In contrast, Ng levels did not vary consistently by APOE genotype (Fig. 1B). Similarly, no consistent relationship was observed between APOE ε4 carrier status and CSF t-tau, ptau181, or NfL (Fig. S1 A–C).
Fig. 1.
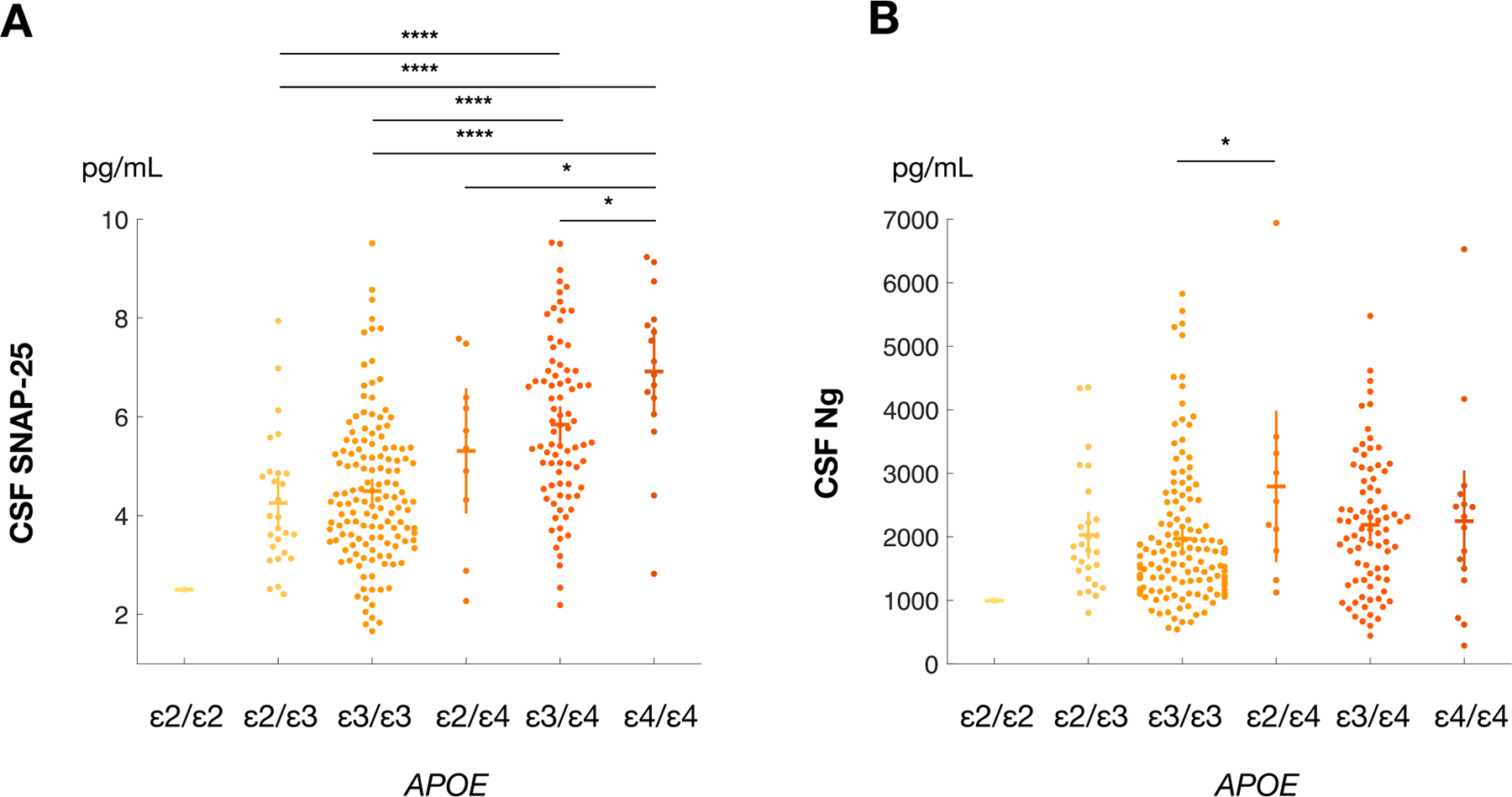
CSF SNAP-25 and Ng by APOE Allele Genotype. Figure 1. Levels of CSF SNAP-25 (A) and Ng (B) by APOE genotype. CSF SNAP-25 was significantly elevated in APOE ε4 allele carriers compared to noncarriers. A similar relationship was not observed for CSF Ng; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001
3.4. CSF SNAP-25 and Ng by amyloid status
We next determined whether elevations in either CSF SNAP-25 or Ng were present when controlling for amyloid status in our cohort of cognitively normal participants (Fig. 2). Amyloid-positive individuals had higher CSF SNAP-25 levels (p < 0.0001; Fig. 2A). Among amyloid-negative individuals, APOE ε4 carriers had higher SNAP-25 levels (p < 0.0001); among amyloid-positive individuals, APOE ε4 carriers also had higher SNAP-25 levels (p < 0.05; Fig. 2C). In contrast, while amyloid-positive individuals had higher CSF Ng levels (Fig. 2B), there was no difference between APOE ε4 noncarriers or carriers after controlling for amyloid status (Fig. 2D).
Fig. 2.
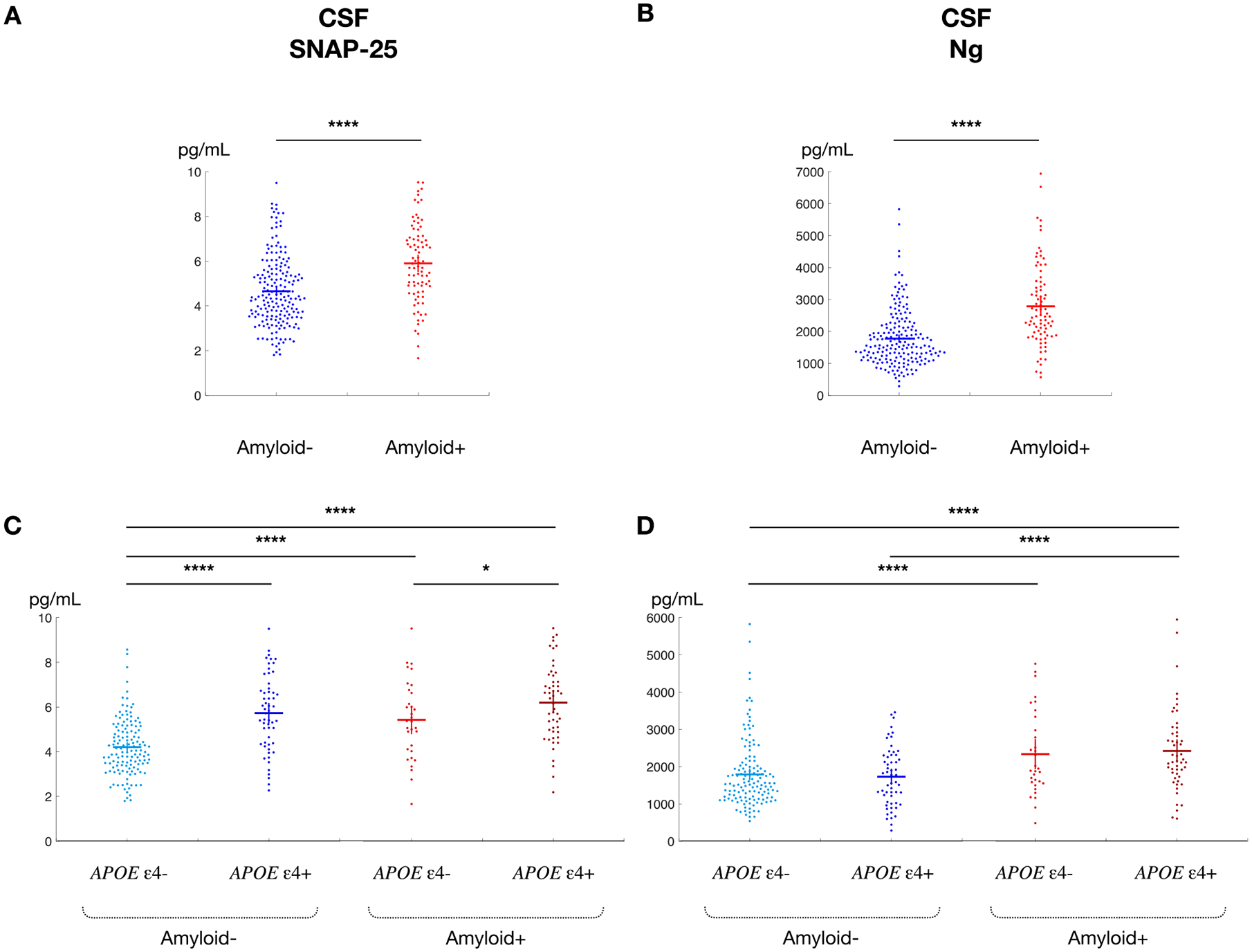
CSF SNAP-25 and Ng by amyloid status. Figure 2. Unadjusted CSF SNAP-25 and Ng as a function of amyloid status and APOE ε4 carrier status with confidence intervals. CSF SNAP-25 (A) and Ng (B) are significantly higher in cognitively normal participants who are amyloid-positive compared to amyloid-negativea. Even after adjusting for amyloid status, CSF SNAP-25 (C) is greater in APOE ε4 carriers than APOE ε4 noncarriers. CSF Ng (D) does not vary by APOE ε4 carrier status. aAmyloid-negative if CSF ptau181/Aβ42 < 0.0198; Amyloid-positive if CSF ptau181/Aβ42 ≥ 0.0198 (36) *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
3.5. Modeling CSF biomarker as function of amyloid status and APOE ε4 carrier status
All previous group comparisons were between unadjusted values for each CSF biomarker. Linear modeling was next used to examine the relationship between either CSF SNAP-25 or Ng and APOE ε4 carrier status, amyloid status, participant’s age, sex, and years of education (Table 2). Linear modeling revealed that both APOE ε4 carrier status (p < 0.0001) and amyloid status (p = 0.004) significant determinants of CSF SNAP-25 levels. In contrast, CSF Ng levels were affected by amyloid status (p < 0.0001) and age (p = 0.003), but not by APOE ε4 carrier status.
Table 2.
Effects of amyloid status and APOE ε4 status on CSF SNAP-25 and Ng levels
| Overall Model | SNAP-25 | Ng | ||||||
|---|---|---|---|---|---|---|---|---|
| F-statistic | p | F-statistic | p | |||||
| 17.3 | < 0.0001 | 13.6 | < 0.0001 | |||||
| Estimate | Standard error | tStat | p | Estimate | Standard Error | tStat | p | |
| APOE ε4 carrier | 1.30 | 0.20 | 6.5 | < 0.0001 | 55.70 | 133 | 0.42 | N.S. |
| Amyloid positive | 0.69 | 0.24 | 2.9 | 0.004 | 784 | 158 | 4.95 | < 0.0001 |
| Age (years) | 0.012 | 0.012 | 0.98 | 0.33 | 23.9 | 7.87 | 3.04 | 0.003 |
| Female sex | −0.24 | 0.19 | −1.24 | 0.22 | 62.2 | 129 | 0.48 | N.S. |
| Education (years) | −0.06 | 0.038 | −1.58 | 0.12 | −14.2 | 25.2 | −0.56 | N.S. |
| Intercept | 4.89 | 1.14 | 4.3 | < 0.0001 | 394 | 762 | 0.52 | N.S. |
CSF SNAP-25 and CSF Ng as a function of APOE ε4 carrier status and amyloid statusa, adjusting for age, sex, and years of education. The estimate reflects the pg/mL change in biomarker levels associated with a unit change in the predictor values.
Amyloid-negative if CSF ptau181/Aβ42 < 0.0198; Amyloid-positive if CSF ptau181/Aβ42 ≥ 0.0198 (Schindler et al., 2018).
Identical models for CSF t-tau and ptau181 demonstrated only a clear relationship with amyloid status (p < 0.0001) and age (p < 0.0001) (Table S1). For CSF NfL, only age (p < 0.0001) and female sex (p < 0.0001) were significant determinants; no significant effect of amyloid status or APOE ε4 carrier status was observed (Table S2).
Finally, modeling was repeated using an independent cohort from the ADNI dataset (n = 57, mean age 76 ± 5.3 years, 21% APOE ε4 carriers, 40% female). In to contrast our Knight ADRC dataset, participants were older with a lower percentage of cognitively normal individuals who were APOE ε4 carriers and female participants (Table S3). The relationship between CSF SNAP-25, Ng, APOE ε4 carrier status, amyloid status, age, sex, and years of education was evaluated in the ADNI cohort using the same models as applied to the Knight ADRC cohort (Table 3). As before, CSF SNAP-25 levels were significantly higher in APOE ε4 carriers (p = 0.03). In this smaller cohort, CSF Ng levels were not significantly associated with any of the predictors. In summary, two independently collected datasets both reveal that CSF SNAP-25 levels are higher in cognitively normal APOE ε4 carriers, even after accounting for possible confounds.
Table 3.
Effects of Amyloid Status and APOE ε4 status on CSF SNAP-25 and Ng levels (ADNI)
| Overall model | SNAP-25 | Ng | ||||||
|---|---|---|---|---|---|---|---|---|
| F-statistic | p | F-statistic | p | |||||
| 2.39 | 0.05 | 1.93 | 0.105 | |||||
| Estimate | Standard Error | tStat | p | Estimate | Standard Error | tStat | p | |
| APOE ε4 carrier | 0.99 | 0.44 | 2.27 | 0.03 | 164.25 | 312.99 | 0.52 | N.S. |
| Amyloid positive | 0.17 | 0.36 | 0.48 | 0.64 | 423.9 | 258.14 | 1.64 | N.S. |
| Age (years) | −0.01 | 0.03 | −0.16 | 0.87 | 16.26 | 22.91 | 0.71 | N.S. |
| Female Sex | 0.68 | 0.38 | 1.78 | 0.08 | 437.14 | 272.9 | 1.6 | N.S. |
| Education (years) | 0 | 0.06 | −0.07 | 0.94 | 7.61 | 41.88 | 0.18 | N.S. |
| Intercept | 3.61 | 2.76 | 1.31 | 0.2 | 23.35 | 1973.57 | 0.01 | N.S. |
CSF SNAP-25 and CSF Ng as a function of APOE ε4 carrier status and amyloid statusa, accounting for confounds of age, sex, and years of education for the ADNI dataset. The estimate reflects the pg/mL change in biomarker levels associated with a unit change in the predictor values.
Amyloid-negative if CSF ptau181/Aβ42 < 0.0198; Amyloid-positive if CSF ptau181/Aβ42 > 0.0198.
4. Discussion
This study investigated CSF levels of the presynaptic marker SNAP-25 and the postsynaptic marker Ng in cognitively normal, older individuals. Presynaptic SNAP-25, but not postsynaptic Ng, was specifically elevated in the CSF of APOE ε4 carriers even after adjusting for age, sex, years of education, and amyloid status. The elevation of SNAP-25 but not Ng in APOE ε4 carriers may indicate selective presynaptic damage in APOE ε4 carriers; alternatively, Ng (or the Ng assay used in this study) may simply not be as sensitive to APOE ε4-related changes. CSF levels of t-tau, ptau181, and NfL also did not vary by APOE ε4 carrier status. These results are the first to demonstrate a relationship between CSF SNAP-25 elevation and APOE ε4 carrier status in cognitively normal older individuals without biomarker evidence of brain amyloidosis, and extend earlier reports of elevated CSF SNAP-25 levels in APOE ε4 carriers with early symptomatic AD (equivalent to mild cognitive impairment (MCI) due to AD and mild AD dementia (Galasko et al., 2019; Sutphen et al., 2018; Tible et al., 2020; Wang Q et al., 2018; Wang S et al., 2018; Zhang et al., 2018).
Previous work examining CSF SNAP-25 and Ng levels report that levels reach their maximum in individuals with early symptomatic AD, and then decline with progression to AD dementia (Sutphen et al., 2018). However, after accounting for APOE ε4 carrier status, differences between the cognitively normal and early symptomatic AD groups were present for Ng but not SNAP-25. This suggests that differences in SNAP-25 were related to APOE ε4 carrier status rather than diagnosis. Wang S et al. (2018) also demonstrated significantly higher levels of CSF SNAP-25 in APOE ε4 carriers compared to noncarriers with MCI, but no significant relationship was observed for participants who were cognitively normal or who had dementia due to AD. Both studies relied on the ADNI dataset, which includes a sizeable number of participants with MCI or AD dementia, but comparatively fewer cognitively normal elderly participants. Cognitively normal APOE ε4 carriers are particularly under-represented in the ADNI cohort, but are well represented in the Knight ADRC cohort, explaining why our current findings were not previously observed. More recently, Galasko et al. (2019) reported elevations in CSF SNAP-25 and Ng in AD dementia compared to cognitively normal individuals, but did not specifically evaluate the effect of APOE genotype in cognitively normal individuals. Finally, Tible et al. (2020) also reported CSF SNAP-25 and Ng elevations in APOE ε4 carriers with AD and non-AD dementia, but again, the effects of APOE genotype in cognitively normal individuals were not evaluated.
A number of studies have demonstrated that APOE ε4 is associated with synaptic dysfunction. Neuropathologic analyses of human brain from normal APOE ε4 carriers demonstrate decreased protein levels of synaptic markers (Love et al., 2006). APOE ε4 targeted replacement (TR) mice exhibit progressive loss of dendritic arbors and lower levels of excitatory synaptic activity (Dumanis et al., 2009; Klein et al., 2010). APOE ε4 has been shown to interfere with endosome recycling and glutamate receptor function via effects on Reelin signaling (Chen et al., 2010). Isogenic iPSC-derived human neurons expressing APOE ε4 exhibit early synaptic maturation and reduced expression of a number of genes, most of which are associated with synaptic function (Lin YT et al., 2018). Thus, most of the previously described mechanisms have been restricted to the postsynaptic compartment.
The mechanism(s) underlying possible presynaptic dysfunction in cognitively normal APOE ε4 carriers remains unclear. In animal models, the APOE ε4 allele has been associated with decreased presynaptic protein levels in response to environmental factors (Levi et al., 2003). Subsequent studies using transgenic mice expressing human APOE (ApoE4-TR, ApoE3-TR, and ApoE2-TR) reveal disruptions in vesicular release of several neurotransmitters (Dolejší et al., 2016; Dumanis et al., 2013). ApoE4-TR mice demonstrated impaired glutaminase activity resulting in a net decrease in glutamate present in the nerve terminals not observed in ApoE2-TR or ApoE3-TR mice (Dumanis et al., 2013). More recent work reveals inhibition of hippocampal ACh release from cholinergic nerve terminals in ApoE4-TR mice in a choline acetyltransferase-independent manner (Dolejší et al., 2016). Human studies are more limited, with reports of decreased presynaptic protein levels in APOE ε4 carriers (Tannenberg et al., 2006).
There are several limitations of this study. It remains unclear whether in vivo APOE ε4 mediated disruptions in presynaptic glutamate or acetylcholine are associated with increases in interstitial or CSF SNAP-25 levels. Furthermore, the specific pathological change reflected by elevated CSF SNAP-25 levels in cognitively normal, amyloid-negative APOE ε4 carriers remains unclear. It is possible that changes in SNAP-25 levels in APOE ε4 carriers are not specific to the presynaptic compartment and instead reflect global synaptic dysfunction or loss that is not reflected in the levels of other CSF synaptic markers. This uncertainty further extends to the neuroanatomical localization for elevated SNAP-25 levels. It remains unclear whether elevations in SNAP-25 reflect a localized or more cortically distributed phenomena. Additional studies are needed to better characterize the source of CSF SNAP-25 and associated neuropathologic and neuroanatomic changes at the synaptic level in cognitively normal, amyloid-negative APOE ε4 carriers.
This study also duplicates a significant association of age and sex with CSF NfL levels (Khalil et al., 2020). No clear association between NfL and APOE ε4 or amyloid status was observed in our study, also as previously reported (Bos et al., 2019). Previous studies exploring NfL in healthy adults reported no change in association of CSF NfL with risk of early symptomatic AD after adjustment for APOE status (Kern et al., 2019). Further NfL elevations observed in dementia associated with Parkinson’s disease (Lin YS et al., 2018) support NfL as a sensitive global marker of cumulative neural injury due to multiple etiologies rather than a highly specific marker of AD-related pathology.
5. Conclusions
Increased CSF SNAP-25 levels in cognitively normal APOE ε4 carriers, even those without brain amyloidosis, suggest that APOE ε4 may be associated with presynaptic dysfunction unrelated to amyloid. This difference is not seen with Ng, a postsynaptic marker, or another marker of neuronal injury, NfL. Differences in the longitudinal change of SNAP-25 in relation to APOE ε4 status remains unknown. It is also unclear if APOE ε4 carriers under 50 years old also have significant elevations in CSF SNAP-25. Further studies are needed to further discern the mechanism by which APOE ε4 modulates SNAP-25 levels and presynaptic dysfunction.
Supplementary Material
Acknowledgements
We thank all participants at the Knight Alzheimer Disease Research Center for their role in the sample provision and data collection. This work was made possible due to by the generous support of Barnes-Jewish Hospital, the Knight Alzheimer Disease Research Center, the Hope Center for Neurological Disorders, the Paula and Rodger O. Riney Fund, the Daniel J. Brennan MD Fund, and the Fred Simmons and Olga Mohan Fund.
Funding:
This work was supported by National Institute on Aging (NIA) grants R03AG050921 (SES, PI) and K23AG053426 (SES, PI). Data from the Knight ADRC cohort was funded by NIA grants P50 AG05681 (JCM, PI), P01 AG03991 (JCM, PI), and P01 AG026276 (JCM, PI).
In addition, data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Health-care; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Abbreviations:
- Aβ
amyloid-β
- AD
Alzheimer’s disease
- ADNI
Alzheimer’s Disease Neuroimaging Initiative
- APOE
Apolipoprotein E
- CDR
clinical dementia rating
- CSF
cerebrospinal fluid
- Knight ADRC
Knight Alzheimer Disease Research Center
- MCI
mild cognitive impairment (early symptomatic AD)
- NfL
neurofilament light chain
- Ng
neurogranin
- NS
not significant
- PiB
Pittsburgh compound B
- ptau181
tau phosphorylated at 181
- ROI
region of interest
- SNAP-25
synaptosomal-associated protein 25
- SUVR
standardized uptake value ratio
- t-tau
total tau
- TR
targeted replacement (transgenic)
Footnotes
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Author AMF is a member of the scientific advisory boards for Roche Diagnostics, Genentech and AbbVie and also consults for Araclon/Grifols, DiademRes, DiamiR and Otsuka Pharmaceuticals.
All remaining authors report no conflict of interest.
Credit author statement
Omar Butt: Conceptualization, Methodology, Software, Visualization, Formal Analysis, Writing-Original Draft, Validation Justin Long: Writing-Original Draft Rachel Henson: Investigation, Data Curation Elizabeth Herries: Investigation, Data Curation Courtney Sutphen: Investigation, Visualization Anne Fagan: Project administration, Resources, Funding acquisition Carlos Cruchaga: Resources Jack Ladenson: Resources David Holtzman: Writing-Original Draft John Morris: Resources, Funding acquisition Beau Ances: Conceptualization, Writing-Original Draft, Resources, Supervision Suzanne Schindler: Conceptualization, Methodology, Software, Formal analysis, Validation, Data Curation, Writing-Original Draft, Funding acquisition, Project Administration, Supervision.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.neurobiolaging.2021.02.008.
References
- Agosta F, Vossel KA, Miller BL, Migliaccio R, Bonasera SJ, Filippi M, Boxer AL, Karydas A, Possin KL, Gorno-Tempini ML, 2009. Apolipoprotein E epsilon4 is associated with disease-specific effects on brain atrophy in Alzheimer’s disease and frontotemporal dementia. Proc Natl Acad Sci U S A 106 (6), 2018–2022. doi: 10.1073/pnas.0812697106, Epub 2009 Jan 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos I, Vos S, Verhey F, Scheltens P, Teunissen C, Engelborghs S, Sleegers K, Frisoni G, Blin O, Richardson JC, Bordet R, Tsolaki M, Popp J, Peyratout G, Martinez-Lage P, Tainta M, Lleó A, Johannsen P, Freund-Levi Y, Frölich L, Vandenberghe R, Westwood S, Dobricic V, Barkhof F, Legido-Quigley C, Bertram L, Lovestone S, Streffer J, Andreasson U, Blennow K, Zetterberg H, Visser PJ, 2019. Cerebrospinal fluid biomarkers of neurodegeneration, synaptic integrity, and astroglial activation across the clinical Alzheimer’s disease spectrum. Alzheimers Dement 15 (5), 644–654. doi: 10.1016/j.jalz.2019.01.004, Epub 2019 Mar 8. [DOI] [PubMed] [Google Scholar]
- Brinkmalm A, Brinkmalm G, Honer WG, Frölich L, Hausner L, Minthon L, Hansson O, Wallin A, Zetterberg H, Blennow K, Öhrfelt A, 2014. SNAP-25 is a promising novel cerebrospinal fluid biomarker for synapse degeneration in Alzheimer’s disease. Mol Neurodegener 9, 53. doi: 10.1186/1750-1326-9-53, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JW, Schumacher E, Coulter PM 2nd, Vinters HV, Watson JB, 1997. Dendritic translocation of RC3/neurogranin mRNA in normal aging, Alzheimer disease and fronto-temporal dementia. J Neuropathol Exp Neurol 56 (10), 1105–1118. doi: 10.1097/00005072-199710000-00004, [DOI] [PubMed] [Google Scholar]
- Chen Y, Durakoglugil MS, Xian X, Herz J, 2010. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A. 107 (26), 12011–12016. doi: 10.1073/pnas.0914984107, Epub 2010 Jun 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM, Benitez BA, Jeng AT, Skorupa T, Carrell D, Bertelsen S, Bailey M, McKean D, Shulman JM, De Jager PL, Chibnik L, Bennett DA, Arnold SE, Harold D, Sims R, Gerrish A, Williams J, Van Deerlin VM, Lee VM, Shaw LM, Trojanowski JQ, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD, Peskind ER, Galasko D, Fagan AM, Holtzman DM, Morris JC GERAD Consortium; Alzheimer’s Disease Neuroimaging Initiative (ADNI); Alzheimer Disease Genetic Consortium (ADGC), Goate AM, 2013. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron. 78 (2), 256–268. doi: 10.1016/j.neuron.2013.02.026, Epub 2013 Apr 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos A, Jacobs D, Struyfs H, Fransen E, Andersson K, Portelius E, Andreasson U, De Surgeloose D, Hernalsteen D, Sleegers K, Robberecht C, Van Broeck-hoven C, Zetterberg H, Blennow K, Engelborghs S, Vanmechelen E, 2015. C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer’s disease. Alzheimers Dement 11 (12), 1461–1469. doi: 10.1016/j.jalz.2015.05.012, Epub 2015 Jun 16. [DOI] [PubMed] [Google Scholar]
- Dolejší E, Liraz O, Rudajev V, Zimčík P, Doležal V, Michaelson DM, 2016. Apolipoprotein E4 reduces evoked hippocampal acetylcholine release in adult mice. J Neurochem 136 (3), 503–509. doi: 10.1111/jnc.13417, Epub 2015 Nov 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumanis SB, DiBattista AM, Miessau M, Moussa CE, Rebeck GW, 2013. APOE genotype affects the pre-synaptic compartment of glutamatergic nerve terminals. J Neurochem 124 (1), 4–14. doi: 10.1111/j.1471-4159.2012.07908.x, Epub 2012 Sep 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, Weeber EJ, Turner RS, Xu B, Rebeck GW, Hoe HS, 2009. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci 29 (48), 15317–15322. doi: 10.1523/JNEUROSCI.4026-09.2009, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM, 2006. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol 59 (3), 512–519. doi: 10.1002/ana.20730, [DOI] [PubMed] [Google Scholar]
- Fleisher AS, Chen K, Liu X, Ayutyanont N, Roontiva A, Thiyyagura P, Protas H, Joshi AD, Sabbagh M, Sadowsky CH, Sperling RA, Clark CM, Mintun MA, Pontecorvo MJ, Coleman RE, Doraiswamy PM, Johnson KA, Carpenter AP, Skovron-sky DM, Reiman EM, 2013. Apolipoprotein E ε4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease. Neurobiol Aging 34 (1), 1–12. doi: 10.1016/j.neurobiolaging.2012.04.017, Epub 2012 May 24. [DOI] [PubMed] [Google Scholar]
- Galasko D, Xiao M, Xu D, Smirnov D, Salmon DP, Dewit N, Vanbrabant J, Jacobs D, Vanderstichele H, Vanmechelen, EAlzheimer’s Disease Neuroimaging Initiative (ADNI), Worley P, 2019. Synaptic biomarkers in CSF aid in diagnosis, correlate with cognition and predict progression in MCI and Alzheimer’s disease. Alzheimers Dement (N Y) 5, 871–882. doi: 10.1016/j.trci.2019.11.002, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM, 2000. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 97 (6), 2892–2897. doi: 10.1073/pnas.050004797, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Yoon SY, Leung L, Knoferle J, Huang Y, 2013. Cellular source-specific effects of apolipoprotein (apo) E4 on dendrite arborization and dendritic spine development. PLoS One 8 (3), e59478. doi: 10.1371/journal.pone.0059478, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern S, Syrjanen JA, Blennow K, Zetterberg H, Skoog I, Waern M, Hagen CE, van Harten AC, Knopman DS, Jack CR Jr, Petersen RC, Mielke MM, 2019. Association of cerebrospinal fluid neurofilament light protein with risk of mild cognitive impairment among individuals without cognitive impairment. JAMA Neurol 76 (2), 187–193. doi: 10.1001/jamaneurol.2018.3459, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kester MI, Teunissen CE, Crimmins DL, Herries EM, Ladenson JH, Scheltens P, van der Flier WM, Morris JC, Holtzman DM, Fagan AM, 2015. Neurogranin as a cerebrospinal fluid biomarker for synaptic loss in symptomatic Alzheimer disease. JAMA Neurol 72 (11), 1275–1280. doi: 10.1001/jamaneurol.2015.1867, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil M, Pirpamer L, Hofer E, Voortman MM, Barro C, Leppert D, Benkert P, Ropele S, Enzinger C, Fazekas F, Schmidt R, Kuhle J, 2020. Serum neurofilament light levels in normal aging and their association with morphologic brain changes. Nat Commun 11 (1), 812. doi: 10.1038/s41467-020-14612-6, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RC, Mace BE, Moore SD, Sullivan PM, 2010. Progressive loss of synaptic integrity in human apolipoprotein E4 targeted replacement mice and attenuation by apolipoprotein E2. Neuroscience 171 (4), 1265–1272. doi: 10.1016/j.neuroscience.2010.10.027, Epub 2010 Oct 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B, 2004. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 55 (3), 306–319. doi: 10.1002/ana.20009, [DOI] [PubMed] [Google Scholar]
- Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, Karhunen PJ, 2009. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol 65 (6), 650–657. doi: 10.1002/ana.21696, [DOI] [PubMed] [Google Scholar]
- Kvartsberg H, Duits FH, Ingelsson M, Andreasen N, Öhrfelt A, Andersson K, Brinkmalm G, Lannfelt L, Minthon L, Hansson O, Andreasson U, Teunissen CE, Scheltens P, Van der Flier WM, Zetterberg H, Portelius E, Blennow K, 2015a. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dement 11 (10), 1180–1190. doi: 10.1016/j.jalz.2014.10.009, Epub 2014 Dec 19. [DOI] [PubMed] [Google Scholar]
- Kvartsberg H, Portelius E, Andreasson U, Brinkmalm G, Hellwig K, Lelental N, Kornhuber J, Hansson O, Minthon L, Spitzer P, Maler JM, Zetterberg H, Blennow K, Lewczuk P, 2015b. Characterization of the postsynaptic protein neurogranin in paired cerebrospinal fluid and plasma samples from Alzheimer’s disease patients and healthy controls. Alzheimers Res Ther 7 (1), 40. doi: 10.1186/s13195-015-0124-3, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi O, Jongen-Relo AL, Feldon J, Roses AD, Michaelson DM, 2003. ApoE4 impairs hippocampal plasticity isoform-specifically and blocks the environmental stimulation of synaptogenesis and memory. Neurobiol Dis 13 (3), 273–282. doi: 10.1016/s0969-9961(03)00045-7, [DOI] [PubMed] [Google Scholar]
- Li YJ, Hauser MA, Scott WK, Martin ER, Booze MW, Qin XJ, Walter JW, Nance MA, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Goetz CG, Small GW, Mastaglia F, Haines JL, Pericak-Vance MA, Vance JM, 2004. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology 62 (11), 2005–2009. doi: 10.1212/01.wnl.0000128089.53030.ac, [DOI] [PubMed] [Google Scholar]
- Lin YS, Lee WJ, Wang SJ, Fuh JL, 2018. Levels of plasma neurofilament light chain and cognitive function in patients with Alzheimer or Parkinson disease. Sci Rep 8 (1), 17368. doi: 10.1038/s41598-018-35766-w, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, Rueda R, Kritskiy O, Abdurrob F, Peng Z, Milo B, Yu CJ, Elmsaouri S, Dey D, Ko T, Yankner BA, Tsai LH, 2018. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98 (6), 1141–1154. doi: 10.1016/j.neuron.2018.05.008, e7Epub 2018 May 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai CW, Bu G, 2017. ApoE4 accelerates early seeding of amyloid pathology. Neuron 96 (5), 1024–1032. doi: 10.1016/j.neuron.2017.11.013, e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love S, Siew LK, Dawbarn D, Wilcock GK, Ben-Shlomo Y, Allen SJ, 2006. Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol Aging 27 (6), 797–803. doi: 10.1016/j.neurobiolaging.2005.04.008, Epub 2005 Jun 23. [DOI] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA, 2009. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68 (7), 709–735. doi: 10.1097/NEN.0b013e3181a9d503, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC, 2006. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 67 (3), 446–452. doi: 10.1212/01.wnl.0000228230.26044.a4, [DOI] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, Mintun MA, 2010. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 67 (1), 122–131. doi: 10.1002/ana.21843, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Schindler SE, McCue LM, Moulder KL, Benzinger TLS, Cruchaga C, Fagan AM, Grant E, Gordon BA, Holtzman DM, Xiong C, 2019. Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol 76 (3), 264–273. doi: 10.1001/jamaneurol.2018.4249, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, 1993. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43 (11), 2412–2414. doi: 10.1212/wnl.43.11.2412-a, [DOI] [PubMed] [Google Scholar]
- Nwabuisi-Heath E, Rebeck GW, Ladu MJ, Yu C, 2014. ApoE4 delays dendritic spine formation during neuron development and accelerates loss of mature spines in vitro. ASN Neuro 6 (1), e00134. doi: 10.1042/AN20130043, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panegyres PK, Beilby J, Bulsara M, Toufexis K, Wong C, 2006. A study of potential interactive genetic factors in Huntington’s disease. Eur Neurol 55 (4), 189–192. doi: 10.1159/000093867, Epub 2006 Jun 13. [DOI] [PubMed] [Google Scholar]
- Portelius E, Zetterberg H, Skillbäck T, Törnqvist U, Andreasson U, Trojanowski JQ, Weiner MW, Shaw LM, Mattsson N, Blennow, KAlzheimer’s Disease Neuroimaging Initiative, 2015. Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer’s disease. Brain 138 (Pt 11), 3373–3385. doi: 10.1093/brain/awv267, Epub 2015 Sep 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler SE, Gray JD, Gordon BA, Xiong C, Batrla-Utermann R, Quan M, Wahl S, Benzinger TLS, Holtzman DM, Morris JC, Fagan AM, 2018. Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimers Dement 14 (11), 1460–1469. doi: 10.1016/j.jalz.2018.01.013, Epub 2018 Mar 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, Hoyle R, Holtzman DM, 2019. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med 216 (11), 2546–2561. doi: 10.1084/jem.20190980, Epub 2019 Oct 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM, Holtzman DM Alzheimer’s Disease Neuroimaging Initiative, 2017. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549 (7673), 523–527. doi: 10.1038/nature24016, Epub 2017 Sep 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin OH, 2014. Exocytosis and synaptic vesicle function. Compr Physiol 4 (1), 149–175. doi: 10.1002/cphy.c130021, [DOI] [PubMed] [Google Scholar]
- Su Y, Blazey TM, Snyder AZ, Raichle ME, Marcus DS, Ances BM, Bateman RJ, Cairns NJ, Aldea P, Cash L, Christensen JJ, Friedrichsen K, Hornbeck RC, Farrar AM, Owen CJ, Mayeux R, Brickman AM, Klunk W, Price JC, Thompson PM, Ghetti B, Saykin AJ, Sperling RA, Johnson KA, Schofield PR, Buckles V, Morris JC, Benzinger TLS, 2015. Dominantly Inherited Alzheimer Network, 2015. Partial volume correction in quantitative amyloid imaging. Neuroimage 107, 55–64. doi: 10.1016/j.neuroimage.2014.11.058, Epub 2014 Dec 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Flores S, Hornbeck RC, Speidel B, Vlassenko AG, Gordon BA, Koeppe RA, Klunk WE, Xiong C, Morris JC, Benzinger TLS, 2018. Utilizing the centiloid scale in cross-sectional and longitudinal PiB PET studies. Neuroimage Clin 19, 406–416. doi: 10.1016/j.nicl.2018.04.022, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Flores S, Wang G, Hornbeck RC, Speidel B, Joseph-Mathurin N, Vlassenko AG, Gordon BA, Koeppe RA, Klunk WE, Jack CR Jr, Farlow MR, Salloway S, Snider BJ, Berman SB, Roberson ED, Brosch J, Jimenez-Velazques I, van Dyck CH, Galasko D, Yuan SH, Jayadev S, Honig LS, Gauthier S, Hsiung GR, Masellis M, Brooks WS, Fulham M, Clarnette R, Masters CL, Wallon D, Hannequin D, Dubois B, Pariente J, Sanchez-Valle R, Mummery C, Ring-man JM, Bottlaender M, Klein G, Milosavljevic-Ristic S, McDade E, Xiong C, Morris JC, Bateman RJ, Benzinger TLS, 2019. Comparison of Pittsburgh compound B and florbetapir in cross-sectional and longitudinal studies. Alzheimers Dement (Amst) 11, 180–190. doi: 10.1016/j.dadm.2018.12.008, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutphen CL, McCue L, Herries EM, Xiong C, Ladenson JH, Holtzman DM, Fagan AM, ADNI, 2018. Longitudinal decreases in multiple cerebrospinal fluid biomarkers of neuronal injury in symptomatic late onset Alzheimer’s disease. Alzheimers Dement 14 (7), 869–879. doi: 10.1016/j.jalz.2018.01.012, Epub 2018 Mar 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannenberg RK, Scott HL, Tannenberg AE, Dodd PR, 2006. Selective loss of synaptic proteins in Alzheimer’s disease: evidence for an increased severity with APOE varepsilon4. Neurochem Int 49 (7), 631–639. doi: 10.1016/j.neuint.2006.05.004, Epub 2006 Jun 30. [DOI] [PubMed] [Google Scholar]
- Tarawneh R, D’Angelo G, Crimmins D, Herries E, Griest T, Fagan AM, Zipfel GJ, Ladenson JH, Morris JC, Holtzman DM, 2016. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer disease. JAMA Neurol 73 (5), 561–571. doi: 10.1001/jamaneurol.2016.0086, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsell A, Bjerke M, Gobom J, Brunhage E, Vanmechelen E, Andreasen N, Hansson O, Minthon L, Zetterberg H, Blennow K, 2010. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain Res 1362, 13–22. doi: 10.1016/j.brainres.2010.09.073, Epub 2010 Sep 25. [DOI] [PubMed] [Google Scholar]
- Tible M, Sandelius Å, Höglund K, Brinkmalm A, Cognat E, Dumurgier J, Zetterberg H, Hugon J, Paquet C, Blennow K, 2020. Dissection of synaptic pathways through the CSF biomarkers for predicting Alzheimer disease. Neurology 95 (8), e953–e961. doi: 10.1212/WNL.0000000000010131, Epub 2020 Jun 25. [DOI] [PubMed] [Google Scholar]
- Wang C, Wilson WA, Moore SD, Mace BE, Maeda N, Schmechel DE, Sullivan PM, 2005. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol Dis 18 (2), 390–398. doi: 10.1016/j.nbd.2004.10.013, [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhou W, Zhang J Alzheimer’s Disease Neuroimaging Initiative, 2018. Levels of cortisol in CSF are associated with SNAP-25 and tau pathology but not amyloid- β. Front Aging Neurosci 10, 383. doi: 10.3389/fnagi.2018.00383, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zhang J, Pan T for Alzheimer’s Disease Neuroimaging Initiative, 2018. APOE ε4 is associated with higher levels of CSF SNAP-25 in prodromal Alzheimer’s disease. Neurosci Lett. 685, 109–113. doi: 10.1016/j.neulet.2018.08.029, Epub 2018 Aug 23. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Beffert U, Jones C, Christian JM, Forster E, Sweatt JD, Herz J, 2002. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem 277 (42), 39944–39952. doi: 10.1074/jbc.M205147200, Epub 2002 Aug 7. [DOI] [PubMed] [Google Scholar]
- Zhang H, Therriault J, Kang MS, Ng KP, Pascoal TA, Rosa-Neto P, Gauthier S Alzheimer’s Disease Neuroimaging Initiative, 2018. Cerebrospinal fluid synaptosomal-associated protein 25 is a key player in synaptic degeneration in mild cognitive impairment and Alzheimer’s disease. Alzheimers Res Ther 10 (1), 80. doi: 10.1186/s13195-018-0407-6, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Attrebi ON, Ren Y, Qiao W, Sonustun B, Martens YA, Meneses AD, Li F, Shue F, Zheng J, Van Ingelgom AJ, Davis MD, Kurti A, Knight JA, Linares C, Chen Y, Delenclos M, Liu CC, Fryer JD, Asmann YW, McLean PJ, Dickson DW, Ross OA, Bu G, 2020. APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid. Sci Transl Med 12 (529), eaay1809. doi: 10.1126/scitranslmed.aay1809, [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available to qualified investigators upon request to the Knight ADRC (https://knightadrc.wustl.edu/Research/ResourceRequest.htm)