

Abstract
A chiral dimer of an organic semiconductor was assembled from octaniline (octamer of polyaniline) conjugated to DNA. Facile reconfiguration between the monomer and dimer of octaniline–DNA was achieved. The geometry of the dimer and the exciton coupling between octaniline molecules in the assembly was studied both experimentally and theoretically. The octaniline dimer was readily switched between different electronic states by protonic doping and exhibited a Davydov splitting comparable to those previously reported for DNA–dye systems employing dyes with strong transition dipoles. This approach provides a possible platform for studying the fundamental properties of organic semiconductors with DNA-templated assemblies, which serve as candidates for artificial light-harvesting systems and excitonic devices.
Keywords: organic semiconductor, exciton delocalization, DNA conjugation, circular dichroism, proton doping
Introduction
The judicious placement of chromophores1,2 and coherent exciton delocalization3,4 in natural photosynthetic systems has inspired the development of artificial energy transfer systems5,6 and quantum excitonic devices.7 DNA nanotechnology offers the advantage of constructing excitonic devices using chemistry by rational design.8,9 Covalent bonding to DNA enhances control of placement and the number of dye molecules within a DNA template, which offers the ability to tune dye proximity and tailor exciton delocalization. A number of studies have demonstrated exciton delocalization in DNA-templated dye aggregates within linear duplex (dsDNA) structures.10−16 Several studies have examined dye aggregates assembled using three-armed DNA junctions, which have focused on either light-harvesting applications or excimer behavior.17,18 Both mobile and immobile DNA Holliday junction19,20 scaffolds have been used to organize several types of aggregates including dimers, trimers, and tetramers.21−23 Exciton delocalization has been observed in dimers templated via DX tiles and compared to a dsDNA templated dimer.24−26 In this work, we apply this strategy to the study of an organic semiconductor related to polyaniline.
Conductive polymers, such as poly(phenylene vinylene),27,28 polyaniline29,30 and polythiophene,31 were introduced into DNA nanostructures in recent years. The delocalization of electrons along the quasi-one-dimensional backbone of oligomers in conductive polymers provides a platform for constructing molecular electronic and excitonic devices with DNA as the template. Assemblies incorporating a discrete number of conductive polymer molecules would allow exploration of their fundamental properties and advance the construction of functional devices with such materials. However, unlike the dye aggregates discussed above, only isolated molecules or uncontrolled aggregates of conductive polymers have been templated by DNA nanostructures, due to the hydrophobicity of these molecules.27,29−31
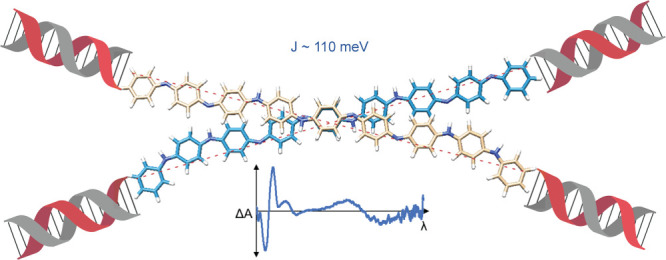
Here, we report an example of an octaniline dimer formed by an octaniline–DNA conjugate in a controlled manner. The dimer formed by octaniline molecules exists as a monodispersed assembly in aqueous solution with a chiral configuration. Moreover, by addition of a surfactant or dopant, the octaniline dimer can be readily switched between different monomeric states and electronic states. Large Davydov splitting was observed in this chiral dimer in the DNA-templated dye aggregate system based on our theoretical fitting,32 indicating strong exciton exchange between octaniline molecules in the dimer (Figure 1). The size of octaniline molecule employed in this study (∼4 nm) serves as a great advantage compared to small dye molecules (usually smaller than 2 nm), given that the diameter of the DNA duplex is 2 nm. Interhelical bridging of octaniline molecules can be used for constructing complex networks for long-range exciton delocalization, eventually on bigger DNA nanostructures such as DNA origami.
Figure 1.

Octaniline–DNA assemblies and the different states in response to environmental change. Three different octaniline–DNA constructs were synthesized and prepared (top): IS, I0, and II. Construct I0 spontaneously assembles into a chiral dimer in aqueous solution, and the dimeric state is switched between two different monomer constructs by adding a triggering DNA strand or a surfactant (middle). The octaniline molecules can exist in different electronic states, emeraldine base and emeraldine salt, which both show strong exciton exchange as a result of electronic coupling in the dimer (bottom).
Results and Discussion
Octaniline–DNA Construct Design and Synthesis
The octaniline–DNA conjugates were prepared following our previously reported synthesis, and two different asymmetric octaniline–DNA strands were prepared for this study (Figure S1).29 Given the hydrophobic nature of the octaniline molecule, three different constructs were designed in an attempt to control the number of octaniline molecules in the assemblies by tuning the composition, as shown in Figure 1. The IS construct consists of an octaniline–DNA single-strand and the long complementary strand with polyT in the middle, which was designed to form a linear duplex monomer by isolating octaniline molecules from each other. In contrast, the I0 construct was designed with an octaniline–DNA single strand and two short complementary strands, which leaves the octaniline molecules exposed to the environment to further assemble into larger structures. Furthermore, in order to bring two octaniline molecules proximate to one another by hybridization, we also prepared the II construct that contains an octaniline molecule in each complementary strand.
The self-assembly of the I0 construct into a dimer is most interesting of all three designs. Being “wrapped” by polyT from the complementary strand, the octaniline construct IS remains as a monomer at neutral pH. As characterized by non-denaturing gel electrophoresis, the IS construct migrates along with similarly sized linear DNA duplexes (Figure S2), whereas in the same gel, the I0 construct has much lower mobility and migrates in the same way as a double-sized linear DNA duplex, which suggests that I0 exists as (I0)2 under native conditions (Figure S2). We found that the dimerization of I0 is driven by the aggregation of octaniline molecules, as the dimer dissociates into monomers upon being treated with sodium dodecyl sulfate (SDS), a condition that can consistently prevent aggregation of such molecules. The monomeric state was further confirmed by a size-titration experiment using non-denaturing SDS gel (Figure S3). However, the II construct appears as multiple bands in both non-denaturing and SDS gels, suggesting that there are oligomers formed during assembly through cross-reaction and then further aggregation into larger structures (Figure S4). Although II is the most intuitive dimer among the three designs, the (I0)2 construct that spontaneously formed from the I0 construct is the monodispersed dimer assembly that is ideal for our study. Thus, we focused on using (I0)2 as a model dimer for further studies, with IS as an important control construct.
Structural and Optical Characterization of the Dimer Construct
The observation of distinctly different Ferguson plots of (I0)2 compared to similarly sized DNA linear duplex suggests that the dimer exists in a branched geometry (Figure 2), which is similar to the behavior of a DNA immobile four-arm junction.20 In contrast, the Ferguson plots of compositional monomer IS are almost identical to that of similarly sized linear DNA duplex. To investigate the dissociated monomer further, Ferguson plots were also used to study the I0 monomer in SDS gels. Surprisingly, the slope suggests that the dissociated monomer I0 maintained a nonlinear shape, much like a similarly sized DNA four-arm junction (Figure S5).
Figure 2.

Characterization of octaniline–DNA chiral dimer and two different monomers. (a) Schematic depiction of octaniline–DNA chiral dimer and two monomers. (b) Circular dichroism of dimeric and monomeric octaniline–DNA constructs. (c) Ferguson plots used to reveal the shape of different constructs, in comparison to similarly sized linear DNA duplexes.
One challenge with studies of octaniline is that the absorbance is broad (an inherent feature of bandgap-forming materials). As a result, detailed spectral features are more difficult to discern accurately. For example, dimeric constructs may show an obscured change or shift in the electronic absorbance spectrum compared to the monomer, so exciton delocalization may not be readily apparent in the absorbance data. However, we found that the circular dichroism (CD) spectra were useful to identify these interactions (Figure 2b).
CD spectroscopy has been used previously to characterize some polyaniline materials in the presence of various chiral dopants33−36 and also proved useful in this study. It has been used widely to characterize excitonic interactions37,38 as illustrated in Figure 3. The interaction of transition dipoles may result in a bisignate signal with red- and blue-shifted peaks. Thus, splitting that is not visible in UV–vis spectra can be observed in CD spectra since coupled peaks display opposite polarity. These coupled peaks then partially cancel, resulting in a characteristic bisignate signal.39
Figure 3.
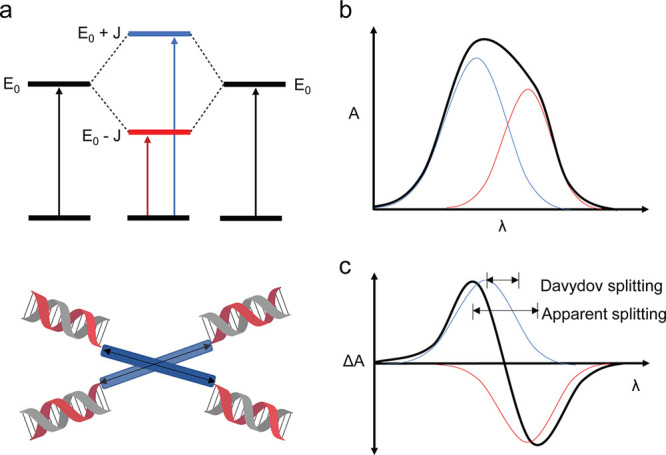
Coupling of octaniline chromophores. (a) Two aggregated octaniline moieties interact, resulting in a couplet and affording two broad, unresolved peaks in the electronic absorption spectrum (b). The coupled peaks are out of phase in the CD spectrum (c), affording a bisignate curve.39
The CD spectrum of (I0)2 indicates the presence of asymmetry (Figure 2b). Besides the characteristic CD signal from the B-form DNA duplex, there is a sharp peak around 330 nm and a broad peak around 620 nm in the CD spectrum of the dimer, and the bisignate CD curves correlate very well to the UV–vis spectrum of octaniline (Figure S6). The CD spectrum of the octaniline is quite strong given its relatively weaker extinction compared to the signal from DNA bases (which are also far more preponderant). Interestingly, the shape of the signal arising from the octaniline dimer is opposite to that of B-form DNA, suggesting that the dimer may adopt a left-handed asymmetric orientation. For both compositional and dissociated monomers, there are weak to nonexistent CD signals from octaniline, which further indicates that the CD signal in the dimer is the result of electronic coupling between octaniline molecules (Figure 2b). We also compared the UV–vis spectrum of (I0)2 to that of IS and dissociated I0, and in both cases, there is a blue shift in the dimer aggregates (Figure S7). Although chiral, the blue shift in the UV–vis spectrum indicates that the octaniline dimer has an H-like configuration; thus the transition between the ground state and the lower energy state in Davydov splitting was restricted.
Theoretical Spectral Modeling
The spectral theoretical analysis of the octaniline dimer was performed by taking advantage of the isothermal strand-displacement (Figure S8) experiment; we used the data collected from the in situ generated (I0)2 construct from a “toehold” version of IS (Figure 4a). Vibronic shoulders in the monomer absorbance peaks arise from the presence of a dominant vibronic mode or bands of modes. These modes can strongly influence absorbance peak positions in chromophore aggregates for which the vibronic energy of the dominant modes is comparable to the exciton exchange energy. A simple Frenkel Hamiltonian, neglecting vibronic effects due to the absence of prominent vibronic shoulders on the octaniline absorbance peaks, presented in the SI, is employed in the analysis presented here. Another consequence of the coupling between the electronic and vibrational degrees of freedom is to give width to the absorbance peaks. This effect is accounted for here by convolving the discrete absorbance spectrum calculated from the Frenkel Hamiltonian with a Gaussian line shape whose width is taken to match that of the absorbance data. The phenomenological parameters of the Frenkel model are extracted by simultaneous fitting of the absorbance and CD spectra. The extracted parameters are given in Table S1, and a comparison between the model and experimental spectra is given in Figure 4b.
Figure 4.

Octaniline–DNA dimer construct generated by strand displacement and the theoretical analysis. (a) Octaniline–DNA compositional monomer spontaneously assembled into the chiral dimer by adding triggering DNA strand. (b) Absorbance and CD spectral theoretical fits. Experimental data sets are shown as black lines, while theoretical fits are given as red curves.
As discussed above, clear indications
of coherent exciton exchange
giving rise to exciton delocalization have been observed. To predict
the configuration that the DNA organized octaniline dimer aggregates
adopt and to quantify the exciton exchange energy of these dimers,
data analysis was performed by simultaneously fitting the absorbance
data and CD data for the I0 construct used in the isothermal strand-displacement
experiment (Table S1). 3D molecular models
were generated using the quasi-1D model of the octaniline molecule
and assuming that the aggregates adopt a single conformation rather
than an ensemble of conformations. As shown in Figure 5, the intermolecular distance between octaniline
molecules was predicted to be 0.53 nm with an inter-backbone angle
of 34°. Taking the octaniline length l to be
4 nm, the distance between the ends is given by 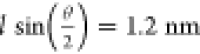 ,
which can be used as a good approximation
for the in-solution dimer assembly given that duplex DNA is 2 nm wide.
An exciton exchange energy of 110 meV was obtained. This is comparable
to the exciton exchange energy exhibited between dye pairs in DNA-assembled
dye aggregates employing dyes with strong transition dipoles.32
,
which can be used as a good approximation
for the in-solution dimer assembly given that duplex DNA is 2 nm wide.
An exciton exchange energy of 110 meV was obtained. This is comparable
to the exciton exchange energy exhibited between dye pairs in DNA-assembled
dye aggregates employing dyes with strong transition dipoles.32
Figure 5.
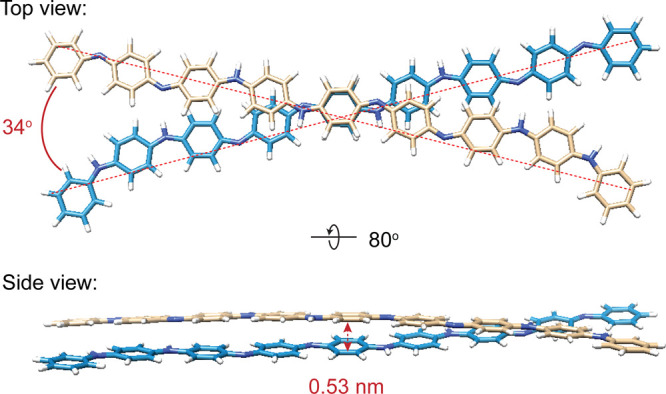
Molecular models of the octaniline dimer. Top view shows the oblique angle, in degrees, as an angle between vectors of the two octaniline molecules. Side view shows the intermolecule distance from center to center. The position and orientation of the long axes of the octaniline molecules are from the theoretical fitting, but the rotation around the long axis of both molecules was arbitrarily chosen.
Proton Doping of Octaniline Dimer
So far, our discussion has involved the dimer with the octaniline molecules in emeraldine base form. However, the most interesting property of octaniline is the transition from emeraldine base to emeraldine salt, which is the conductive form.40 The dimer can be readily protonated around pH 4.5 in solution, similar to the monomer as shown in a previous study.29 Despite the octaniline molecules being positively charged after proton doping, the construct remains a dimer as indicated by gel electrophoresis at lower pH (Figure S9). The CD signal of the emeraldine salt dimer shifted to longer wavelengths (Figure 6), which follows the same trend as the UV–vis spectra. We attribute this dramatic change to a greater delocalization of electrons in the backbone of octaniline in the emeraldine salt state upon proton doping. Although the longer wavelength part of the spectrum is beyond our instrument measurement range, the CD curve in the shorter wavelength region (360–480 nm) indicates strong exciton exchange between octaniline molecules at emeraldine salt state. The bisignate CD curve also suggests that the emeraldine salt dimer exists in the same chirality compared to its emeraldine base state. It is worth mentioning that, because of the facile switch between emeraldine base and emeraldine salt states, the octaniline dimer covers a broad range of absorbance including almost the entire UV–vis spectrum and reaches the near-infrared region (Figure 6, Figure S10). This feature potentiates construction of an artificial photosynthesis system with the ability to adapt to environmental changes, such as pH.
Figure 6.

Proton doping of octaniline–DNA dimer construct. Octaniline chiral dimer undergoes proton doping at a lower pH. The emeraldine base (blue) and emeraldine salt (green) states of octaniline dimer were confirmed by circular dichroism spectra.
Conclusion
In summary, we have controlled the association–dissociation of the amphiphilic octaniline–DNA conjugate using a variety of ways, such as tuning the composition, adding surfactant, and switching by strand displacement. The H-like dimer construct showed strong exciton coupling in both the emeraldine base and emeraldine salt states, and the theoretical analysis fits agree well with the data. Nevertheless, the octaniline–DNA single-strand and II construct also formed larger aggregates, and their optical properties are also similar to the dimer (Figures S11 and S12). Advancing the control of the number and geometry of octaniline molecules in such aggregates will be of great interest in the future. We envision harnessing exciton delocalization in aggregates so as to allow the development of light-harvesting systems, quantum computing systems, and metamaterials based on octaniline or other organic semiconductors templated by DNA nanostructures.
Methods
Materials
Commercially available reagents were purchased and used without purification. Spin filters (3K), 0.22 μm filters, and 4–7 pH strips were purchased from Millipore. Nucleoside phosphoramidite and propargyl-modified phosphoramidite for oligonucleotide synthesis were purchased from Chemgenes. DNA solid phase synthesis was carried on ABI394 synthesizer. Unmodified oligonucleotides were purchased from Integrated DNA Technologies and further purified. Ultraviolet–visible (UV–vis) spectra were collected using Nanodrop 2000. Circular dichroism spectra were collected using Jasco J-1500 circular dichroism spectrophotometer with a microcuvette.
Self-Assembly of DNA Constructs
Constructs were formed by mixing a stoichiometric quantity of each strand, as estimated by OD260. The mixtures were dried and dissolved in 1×TAE Mg (40 mM Tris-HCl, 20 mM acetic acid, 2 mM EDTA, and 12.5 mM magnesium acetate) buffer, pH ≈ 7.5. The samples were sealed in PCR tubes and were then incubated by the following the fast-annealing protocol: 20 min at 65 °C, 20 min at 45 °C, 30 min at 37 °C, and 30 min at room temperature.
Circular Dichroism Measurement
The CD spectra were collected using Jasco J-1500 circular dichroism spectrophotometer at room temperature. The octaniline–DNA samples were loaded in a microcuvette, which was formed by two parallel glass slides, and the path length was controlled by a spacer between them. The samples were held with two glass slides by the capillary effect. For samples without SDS (sodium dodecyl sulfate), a 1 mm spacer was used, and thus the path length was 1 mm. For samples with SDS, a 0.2 mm spacer was used, which made the path length 0.2 mm.
Acknowledgments
This work was supported by the following grants to N.C.S.: CCF-1526650 and CHE-1708776 (also to J.W.C.) from the National Science Foundation, MURI N00014-19-1-2506 from the Office of Naval Research (also to J.W.C.), DE-SC0007991 from the Department of Energy for DNA synthesis and partial salary support, and RGP0010/2017 from the Human Frontiers Science Program. Work at Boise State University (W.B.K. and B.Y.) was supported by the National Science Foundation (NSF) through the Integrated NSF Support Promoting Interdisciplinary Research and Education (INSPIRE) via Award No. 1648655.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.1c09143.
Description of octaniline–DNA conjugate synthesis, gel electrophoresis characterization of octaniline–DNA constructs, UV–vis and circular dichroism of octaniline–DNA constructs, and theoretical analysis of the octaniline dimer assembly (PDF)
Author Contributions
X.W., R.S., W.B.K., N.C.S., J.W.C., and B.Y. analyzed the data and wrote the manuscript. All authors have given approval to the final version of the manuscript. X.W. and R.S. carried out experiments. B.Y. carried out theoretical analysis. N.C.S., J.W.C., and B.Y. initiated and directed the project.
The authors declare no competing financial interest.
Supplementary Material
References
- Koepke J.; Hu X.; Muenke C.; Schulten K.; Michel H. The Crystal Structure of the Light-Harvesting Complex II (B800–850) from Rhodospirillum Molischianum. Structure 1996, 4 (5), 581–597. 10.1016/S0969-2126(96)00063-9. [DOI] [PubMed] [Google Scholar]
- Camara-Artigas A.; Blankenship R. E.; Allen J. P. J. P. R. The Structure of the FMO Protein from Chlorobium Tepidum at 2.2 Å Resolution. Photosynthesis Research 2003, 75 (1), 49–55. 10.1023/A:1022406703110. [DOI] [PubMed] [Google Scholar]
- Engel G. S.; Calhoun T. R.; Read E. L.; Ahn T.-K.; Mančal T.; Cheng Y.-C.; Blankenship R. E.; Fleming G. R. Evidence for Wavelike Energy Transfer through Quantum Coherence in Photosynthetic Systems. Nature 2007, 446 (7137), 782–786. 10.1038/nature05678. [DOI] [PubMed] [Google Scholar]
- Chenu A.; Scholes G. D. Coherence in Energy Transfer and Photosynthesis. Annu. Rev. Phys. Chem. 2015, 66 (1), 69–96. 10.1146/annurev-physchem-040214-121713. [DOI] [PubMed] [Google Scholar]
- Dutta P. K.; Varghese R.; Nangreave J.; Lin S.; Yan H.; Liu Y. DNA-Directed Artificial Light-Harvesting Antenna. J. Am. Chem. Soc. 2011, 133 (31), 11985–11993. 10.1021/ja1115138. [DOI] [PubMed] [Google Scholar]
- Dutta P. K.; Levenberg S.; Loskutov A.; Jun D.; Saer R.; Beatty J. T.; Lin S.; Liu Y.; Woodbury N. W.; Yan H. A DNA-Directed Light-Harvesting/Reaction Center System. J. Am. Chem. Soc. 2014, 136 (47), 16618–16625. 10.1021/ja509018g. [DOI] [PubMed] [Google Scholar]
- Yurke B.; Kuang W. Passive Linear Nanoscale Optical and Molecular Electronics Device Synthesis from Nanoparticles. Phys. Rev. A 2010, 81 (3), 033814. 10.1103/PhysRevA.81.033814. [DOI] [Google Scholar]
- Seeman N. C. DNA in a Material World. Nature 2003, 421 (6921), 427–431. 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]
- Yurke B.; Turberfield A. J.; Mills A. P.; Simmel F. C.; Neumann J. L. A DNA-Fuelled Molecular Machine Made of DNA. Nature 2000, 406 (6796), 605–608. 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- Asanuma H.; Shirasuka K.; Takarada T.; Kashida H.; Komiyama M. DNA-Dye Conjugates for Controllable H* Aggregation. J. Am. Chem. Soc. 2003, 125 (8), 2217–2223. 10.1021/ja021153k. [DOI] [PubMed] [Google Scholar]
- Haner R.; Samain F.; Malinovskii V. L. DNA-Assisted Self-Assembly of Pyrene Foldamers. Chem.—Eur. J. 2009, 15 (23), 5701–5708. 10.1002/chem.200900369. [DOI] [PubMed] [Google Scholar]
- Markova L. I.; Malinovskii V. L.; Patsenker L. D.; Haner R. J- vs. H-Type Assembly: Pentamethine Cyanine (Cy5) as a Near-IR Chiroptical Reporter. Chem. Commun. 2013, 49 (46), 5298–5300. 10.1039/c3cc42103a. [DOI] [PubMed] [Google Scholar]
- Cunningham P. D.; Kim Y. C.; Diaz S. A.; Buckhout-White S.; Mathur D.; Medintz I. L.; Melinger J. S. Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds. J. Phys. Chem. B 2018, 122 (19), 5020–5029. 10.1021/acs.jpcb.8b02134. [DOI] [PubMed] [Google Scholar]
- Kringle L.; Sawaya N. P. D.; Widom J.; Adams C.; Raymer M. G.; Aspuru-Guzik A.; Marcus A. H. Temperature-Dependent Conformations of Exciton-Coupled Cy3 Dimers in Double-Stranded DNA. J. Chem. Phys. 2018, 148 (8), 085101. 10.1063/1.5020084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham P. D.; Díaz S. A.; Yurke B.; Medintz I. L.; Melinger J. S. Delocalized Two-Exciton States in DNA Scaffolded Cyanine Dimers. J. Phys. Chem. B 2020, 124 (37), 8042–8049. 10.1021/acs.jpcb.0c06732. [DOI] [PubMed] [Google Scholar]
- Mazuski R. J.; Díaz S. A.; Wood R. E.; Lloyd L. T.; Klein W. P.; Mathur D.; Melinger J. S.; Engel G. S.; Medintz I. L. Ultrafast Excitation Transfer in Cy5 DNA Photonic Wires Displays Dye Conjugation and Excitation Energy Dependency. J. Phys. Chem. Lett. 2020, 11 (10), 4163–4172. 10.1021/acs.jpclett.0c01020. [DOI] [PubMed] [Google Scholar]
- Probst M.; Langenegger S. M.; Haner R. A Modular LHC Built on the DNA Three-Way Junction. Chem. Commun. 2014, 50 (2), 159–161. 10.1039/C3CC47490A. [DOI] [PubMed] [Google Scholar]
- Probst M.; Wenger D.; Biner S. M.; Haner R. The DNA Three-Way Junction as a Mould for Tripartite Chromophore Assembly. Organic & Biomolecular Chemistry 2012, 10 (4), 755–759. 10.1039/C1OB06400B. [DOI] [PubMed] [Google Scholar]
- Seeman N. C. Nucleic-Acid Junctions and Lattices. J. Theor. Biol. 1982, 99 (2), 237–247. 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
- Kallenbach N. R.; Ma R. I.; Seeman N. C. An Immobile Nucleic-Acid Junction Constructed from Oligonucleotides. Nature 1983, 305 (5937), 829–831. 10.1038/305829a0. [DOI] [Google Scholar]
- Cannon B. L.; Kellis D. L.; Patten L. K.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Coherent Exciton Delocalization in a Two-State DNA-Templated Dye Aggregate System. J. Phys. Chem. A 2017, 121 (37), 6905–6916. 10.1021/acs.jpca.7b04344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B. L.; Patten L. K.; Kellis D. L.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates. J. Phys. Chem. A 2018, 122 (8), 2086–2095. 10.1021/acs.jpca.7b12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff J. S.; Davis P. H.; Christy A.; Kellis D. L.; Kandadai N.; Toa Z. S. D.; Scholes G. D.; Yurke B.; Knowlton W. B.; Pensack R. D. DNA-Templated Aggregates of Strongly Coupled Cyanine Dyes: Nonradiative Decay Governs Exciton Lifetimes. J. Phys. Chem. Lett. 2019, 10 (10), 2386–2392. 10.1021/acs.jpclett.9b00404. [DOI] [PubMed] [Google Scholar]
- Cannon B. L.; Patten L. K.; Kellis D. L.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates. J. Phys. Chem. A 2018, 122 (8), 2086–2095. 10.1021/acs.jpca.7b12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay M. S.; Roy S. K.; Huff J. S.; Mass O. A.; Turner D. B.; Wilson C. K.; Kellis D. L.; Terpetschnig E. A.; Lee J.; Davis P. H.; Yurke B.; Knowlton W. B.; Pensack R. D. Rotaxane Rings Promote Oblique Packing and Extended Lifetimes in DNA-Templated Molecular Dye Aggregates. Communications Chemistry 2021, 4 (1), 19. 10.1038/s42004-021-00456-8. [DOI] [PubMed] [Google Scholar]
- Hart S. M.; Chen W. J.; Banal J. L.; Bricker W. P.; Dodin A.; Markova L.; Vyborna Y.; Willard A. P.; Häner R.; Bathe M.; Schlau-Cohen G. S. Engineering Couplings for Exciton Transport Using Synthetic DNA Scaffolds. Chem. 2021, 7 (3), 752–773. 10.1016/j.chempr.2020.12.020. [DOI] [Google Scholar]
- Knudsen J. B.; Liu L.; Kodal A. L. B.; Madsen M.; Li Q.; Song J.; Woehrstein J. B.; Wickham S. F. J.; Strauss M. T.; Schueder F.; Vinther J.; Krissanaprasit A.; Gudnason D.; Smith A. A. A.; Ogaki R.; Zelikin A. N.; Besenbacher F.; Birkedal V.; Yin P.; Shih W. M.; et al. Routing of Individual Polymers in Designed Patterns. Nat. Nanotechnol. 2015, 10 (10), 892–898. 10.1038/nnano.2015.190. [DOI] [PubMed] [Google Scholar]
- Wang X.; Li C.; Niu D.; Sha R.; Seeman N. C.; Canary J. W. Construction of a DNA Origami Based Molecular Electro-Optical Modulator. Nano Lett. 2018, 18 (3), 2112–2115. 10.1021/acs.nanolett.8b00332. [DOI] [PubMed] [Google Scholar]
- Wang X.; Sha R. J.; Kristiansen M.; Hernandez C.; Hao Y. D.; Mao C. D.; Canary J. W.; Seeman N. C. An Organic Semiconductor Organized into 3D DNA Arrays by “Bottom-Up” Rational Design. Angew. Chem., Int. Ed. 2017, 56 (23), 6445–6448. 10.1002/anie.201700462. [DOI] [PubMed] [Google Scholar]
- Wang Z. G.; Liu Q.; Ding B. Q. Shape-Controlled Nanofabrication of Conducting Polymer on Planar DNA Templates. Chem. Mater. 2014, 26 (11), 3364–3367. 10.1021/cm501445u. [DOI] [Google Scholar]
- Zessin J.; Fischer F.; Heerwig A.; Kick A.; Boye S.; Stamm M.; Kiriy A.; Mertig M. Tunable Fluorescence of a Semiconducting Polythiophene Positioned on DNA Origami. Nano Lett. 2017, 17 (8), 5163–5170. 10.1021/acs.nanolett.7b02623. [DOI] [PubMed] [Google Scholar]
- Mass O. A.; Wilson C. K.; Roy S. K.; Barclay M. S.; Patten L. K.; Terpetschnig E. A.; Lee J.; Pensack R. D.; Yurke B.; Knowlton W. B. Exciton Delocalization in Indolenine Squaraine Aggregates Templated by DNA Holliday Junction Scaffolds. J. Phys. Chem. B 2020, 124 (43), 9636–9647. 10.1021/acs.jpcb.0c06480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulikova T.; Porfireva A.; Evtugyn G.; Hianik T. Electrochemical DNA Sensors with Layered Polyaniline—DNA Coating for Detection of Specific DNA Interactions. Sensors 2019, 19 (3), 469. 10.3390/s19030469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Liang J.; Pan F.; Wang Z.; Zhang J.; Amin K.; Fang J.; Zou W.; Chen Y.; Shi X.; Wei Z. Macroscopic Helical Chirality and Self-Motion of Hierarchical Self-Assemblies Induced by Enantiomeric Small Molecules. Nat. Commun. 2018, 9 (1), 3808. 10.1038/s41467-018-06239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Yan J.; Hu X.; Liu T.; Sun C.; Xiao S.; Yuan J.; Chen P.; Zhou S. Conductive Polyaniline Helixes Self-Assembled in the Absence of Chiral Dopant. Chem. Commun. 2013, 49 (11), 1100–1102. 10.1039/c2cc38575a. [DOI] [PubMed] [Google Scholar]
- Wang Z.-G.; Zhan P.; Ding B. Self-Assembled Catalytic DNA Nanostructures for Synthesis of Para-Directed Polyaniline. ACS Nano 2013, 7 (2), 1591–1598. 10.1021/nn305424e. [DOI] [PubMed] [Google Scholar]
- Sutherland J. C.Measurement of the Circular Dichroism of Electronic Transitions. Comprehensive Chiroptical Spectroscopy: Instrumentation, Methodologies, and Theoretical Simulations, 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2012; Vol. 1, pp 35–63. [Google Scholar]
- Zahn S.; Canary J. W. Electron-Induced Inversion of Helical Chirality in Copper Complexes of N,N-Dialkylmethionines. Science 2000, 288 (5470), 1404–1407. 10.1126/science.288.5470.1404. [DOI] [PubMed] [Google Scholar]
- Berova N.; Bari L. D.; Pescitelli G. Application of Electronic Circular Dichroism in Configurational and Conformational Analysis of Organic Compounds. Chem. Soc. Rev. 2007, 36 (6), 914–931. 10.1039/b515476f. [DOI] [PubMed] [Google Scholar]
- Lu F. L.; Wudl F.; Nowak M.; Heeger A. J. Phenyl-Capped Octaaniline (COA): An Excellent Model for Polyaniline. J. Am. Chem. Soc. 1986, 108 (26), 8311–8313. 10.1021/ja00286a057. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.