

Abstract
Serum‐ and glucocorticoid‐inducible kinase 3 (SGK3) is a downstream mediator of PI3K, which is essential for maintaining the functional integrity of podocytes. However, little is known about the role of SGK3 in podocyte function. Herein, we demonstrated that SGK3 contributes to the maintenance of podocyte integrity. Conditionally immortalized mouse podocyte cells (MPCs) were treated with puromycin aminonucleoside (PAN). PAN treatment inhibited the activity of SGK3 and the expression of podocin. Short hairpin RNA (shRNA)‐mediated knockdown of SGK3 also reduced podocin expression in the absence of PAN. Adriamycin (ADR)‐treated mice developed proteinuria and had decreased renal glomerular SGK3 expression in comparison to control mice. Consistent with a role for SGK3 in the ADR effect, SGK3 knockout (KO) mice had markedly reduced kidney podocin expression and significantly elevated proteinuria compared with wild‐type mice. Electron microscopy revealed that SGK3 KO mice displayed partial effacement of podocyte foot processes. Further, a SGK3 target protein, glycogen synthase kinase‐3 (GSK3), was discovered to be dramatically activated in PAN and SGK3 shRNA‐treated MPCs and in SGK3 KO mice. Taken together, these data strongly suggest that SGK3 plays a significant role in regulating podocyte function, likely by controlling the expression and activity of GSK3.—Peng, L.‐Q., Zhao, H., Liu, S., Yuan, Y.‐P., Yuan, C.‐Y., Mwamunyi, M.‐J., Pearce, D., Yao, L.‐J. Lack of serum‐ and glucocorticoid‐inducible kinase 3 leads to podocyte dysfunction. FASEB J. 32, 576–587 (2018). www.fasebj.org
Keywords: cell viability, podocin, proteinuria
ABBREVIATIONS
- ADR
adriamycin
- BUN
blood urea nitrogen
- CD2AP
CD2‐associated protein
- eGFP
enhanced green fluorescent protein
- GSK3
glycogen synthase kinase‐3
- KO
knockout
- MPC
mouse podocyte cell
- PAN
puromycin aminonucleoside
- PAS
periodic acid‐Schiff
- p‐GSK3
phospho‐glycogen synthase kinase‐3
- p‐SGK3
phosphorylation‐serum‐and glucocorticoid‐inducible kinase 3
- MTT
methylthiazoletetrazolium
- SGK
serum‐ and glucocorticoid‐inducible kinase
- shRNA
short hairpin RNA
- WT
wild type
Serum and glucocorticoid‐inducible kinase 3 (SGK3), a member of the SGK family of serine/threonine kinases, is activated by PI3K and phosphoinositide‐dependent kinase 1 and plays an important role in cell growth, proliferation, and migration (1, 2). SGK3 is widely expressed in mammalian kidney cells and regulates the activity of a broad range of enzymes via phosphorylation. SGK3 also upregulates a diversity of transporters (1, 3–7), the Na+/K+ ATPase (8), and channels (1, 7, 9, 10), including Ca2+ channels (9, 11) and voltage‐gated K+ channels (1, 12, 13). Additionally, SGK3 shares significant homology with AKT kinase family members, which have been shown to be involved in many cellular processes that affect podocyte injury and integrity (14). However, the biologic role of SGK3 in podocytopathy remains to be elucidated.
Podocytes, also called glomerular visceral epithelial cells, are highly specialized, terminally differentiated cells that serve as a size and charge barrier to prevent renal disorders (15). Podocin is a member of the band‐7‐stomatin protein family of lipid raft‐associated proteins, which interacts with the adapter protein CD2‐associated protein (CD2AP). Podocin functions as a supporting protein in the maintenance of the podocyte process and slit diaphragm integrity (16). CD2AP functions as a gatekeeper for reorganization of the podocyte microfilament system and consequent proteinuria (17). Accumulating evidence has demonstrated that podocin is regulated by the PI3K/AKT pathway (18, 19), and abnormal expression of podocin is associated with glomerular deterioration and proteinuria (19–21). The role of SGK3 in regulating podocin expression in podocytes is still unknown.
Podocyte injury typically presents as an effacement of podocyte foot processes and a reduction in podocyte number. Desmin, as a characteristic protein of myogenic cells, is expressed at low levels in podocytes under normal conditions. Damaged podocytes undergo phenotypic transformations and increase desman expression (20). Thus, in this study we investigated the expression of podocin and desman in an adriamycin (ADR) nephritis model and in SGK3 knockout (KO) mice. We also examined the involvement of SGK3 in podocin and desmin expression during puromycin aminonucleoside (PAN)‐induced podocyte injury in vitro.
MATERIALS AND METHODS
Reagents and antibodies
Polyclonal rabbit anti‐SGK3 antibody for immunoblotting and polyclonal rabbit anti‐phospho‐GSK‐3α/β (Ser21/9) antibody were purchased from Cell Signaling Technology (Beverly, MA, USA). Polyclonal rabbit anti‐phospho‐SGK3 (Ser486) and monoclonal mouse anti‐glycogen synthase kinase‐3 (GSK3) (α/β) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Polyclonal rabbit anti‐SGK3 antibody for immunohistochemistry was purchased from Abnova (Taipei, Taiwan, China). Polyclonal rabbit anti‐desmin (I‐448), anti‐podocin, and anti‐CD2AP antibodies were purchased from Bioworld Technology (Nanjing, China). Monoclonal mouse anti‐GAPDH and horseradish peroxidase‐conjugated anti‐rabbit and anti‐mouse secondary antibodies were purchased from Beyotime Biotechnology (Shanghai, China). Cocktail and phosphatase inhibitors were purchased from Roche Applied Science (Indianapolis, IN, USA). A ThGV118 vector digested with HpaI/XhoI was obtained from Shanghai Genechem (Shanghai, China).
Animals
All animal experiments were conducted in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animas and were approved by local authorities.
ADR‐induced nephritis mouse model
Nephritis models could be induced by injecting ADR or PAN intravenously to investigate proteinuria. Most rats show good reactivity to these agents. Only BALB/C mice have been shown to be susceptible to ADR, and no mouse model of PAN nephritis has been developed (21, 22). Thus, this model was established by injecting ADR intravenously into BALB/C mice. Male BALB/C mice (20–25 g, 7–8 wk old) were purchased from the Animal Experiment Center of Wuhan University (Wuhan, China). The mice were kept in a specific pathogen‐free environment and were randomly assigned to 2 groups. A single injection of ADR (10.5 mg/kg) (D1515; Sigma‐Aldrich, St. Louis, MO, USA) was used to induce nephritis after 1 wk of adaptive breeding (ADR group; n = 7). The mice in the control group were given an equivalent volume of PBS (control group; n = 7). On d 7 and 14, spot urine was collected for analysis of urinary total protein and urinary creatinine levels. On d 14, the mice were euthanized under anesthesia, and blood samples and kidneys were collected. The harvested kidneys were halved: one half was immediately frozen in liquid nitrogen and stored at ‐80°C for immunoblotting analysis and RT‐PCR analysis, and the other half was fixed in 10% neutral buffered formalin for immunohistochemical analysis.
Generation of SGK3 KO mice
Generation and basic properties of the SGK3 KO mice (8–10 wk old) were previously described (23). The mice were compared with the wild‐type (WT) mice and genotyped by PCR from tail DNA using SGK3‐ and neoR‐specific primers as previously described (23, 24). Spot urine, plasma, and kidney were harvested from 8‐wk‐old male SGK3 KO mice (n = 6) and WT mice (n = 6) for subsequent experiments.
Urine and serum biochemistry
Total protein levels in urine were determined using a Urinary Total Protein Determination Kit (2115–717; Ningbo Elejech Biologic Technology, Zhejiang, China), and urinary creatinine levels were measured using a Creatinine Determination Kit (2046–717; Ningbo Elejech Biologic Technology). Serum levels of creatine, albumin, and blood urea nitrogen (BUN) were determined using the respective kits (Ningbo Elejech Biologic Technology).
Histochemistry and immunohistochemistry
Kidneys were fixed in 10% formalin neutral fixative overnight after removal. Kidneys were processed, embedded in paraffin, and sliced to make 5‐μm‐thick sections that were stained with periodic acid‐Schiff (PAS) reagent for histopathology. For immunohistochemical analysis, all slides were depar‐affinized in xylene, dehydrated using a graded ethanol series, and rehydrated. Antigen retrieval was performed by incubating slides in a citrate buffer solution for 2 min in a 95°C water bath. Slides were cooled at room temperature for 20 min and washed 3 times with PBS. Endogenous peroxidase activity was blocked with 3% H2O2 in methanol for 30 min. Nonspecific binding was blocked using 10% normal gat serum for 30 min. The slides were washed with PBS and incubated with polyclonal rabbit anti‐SGK3 at 4°C overnight, followed by incubation with polyperoxidase‐anti‐rabbit IgG secondary antibody (OriGene Technologies, Rockville, MD, USA) at room temperature for 30 min. Staining was then performed using a DAB Kit (Fuzhou Maixin Biotech, Fujian, China). Histologic examination was performed via light microscopy. Positive expression for SGK3 was determined semi‐quantitatively using Image Pro Plus software. SGK3 expression intensity was assessed by estimating the area of the objects and the medium pixel intensity per object as the integrated optical density. Incidental light was measured without a slide and was used as the light level for each acquired image. All glomeruli for a single section were counted, and the final data corresponded to the average of 7 measurements for each group.
Transmission electron microscopy
For transmission electron microscopy, kidney cortical tissues were cut into small pieces (1 mm3), fixed with 2.5% glutaraldehyde, and embedded in Epon Resin (Polysciences, Warrington, PA, USA). Conventional electron micrographs were obtained using a Tecnai G2 12 microscope (FEI, Hillsboro, OR, USA) operated at 60 kV. The podocyte foot process density was estimated by dividing the total length of glomerular basement membrane by the total number of foot processes present in each micrograph.
Generation of recombinant lentiviruses harboring SGK3 short hairpin RNAs
Synthesized sense and antisense oligos representing short hairpin RNAs (shRNAs) for SGK3 were annealed using a touchdown protocol on an Applied Biosystems 2720 thermal cycler at 90°C for 15 min and then allowed to cool to room temperature. The annealed shRNAs were ligated with the GV118 vector tagged with enhanced green fluorescent protein (eGFP) and digested with HpaI/XhoI using T4 DNA ligase. The ligated DNA was transformed into competent DH5a Escherichia coli. Ampicillin‐resistant colonies were selected and grown in LB broth for 16 h. Positive recombinants were identified by DNA sequencing. Recombinant lentiviruses were generated by cotransfection of plasmids harboring the shRNAs and a mixture of packaging plasmids pHelper 1.0 and pHelper 2.0 into human embryonic kidney‐293T packaging cells. Viral supernatants were harvested 48 h after transfection and concentrated. For determination of the viral titer, a 10‐fold dilution series of viruses was made and used to infect fresh human embryonic kidney‐293T cells. A viral titer of 4 × 108 TU/ml was routinely observed by visualizing cells for eGFP fluorescence.
Cell culture and PAN treatment
Conditional immortalized mouse podocyte cells (MPCs) (Mei‐Lian Biotechnology, Shanghai, China) were cultured under growth‐permissive conditions on rat collagen type I‐coated plastic dishes (Corning, Palo Alto, CA, USA) at 33°C under 5% CO2 and 95% air in RPMI 1640 medium (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA), 10 U/ml mouse recombinant γ‐IFN, 100 U/ml penicillin, and 100 μg/ml streptomycin (MP Biomedical, Santa Ana, CA, USA). After growing to 50–60% confluency, cells were transferred to 37°C under 5% CO2 and 95% air γ‐IFN‐free medium to induce differentiation. Cells between 10 and 14 d were used for all experiments. Before the experiments, cells were subcultured to 70–80% confluence in 6‐well plates. Then MPCs were cultured under different concentrations of PAN (0, 25, and 50 μg/ml) (Sigma‐Aldrich) for 24 h to induce podocyte injury. All experiments were performed in triplicate.
Recombinant DNA transduction
MPCs were seeded to 50–60% confluence in antibiotic‐free medium and grown for 24 h. They were then transfected with pMO/Flag/mSGK3‐S486D (Flag‐epitope at the N terminus of mouse SGK3), pCMV/Flag/mGSK3p, or the empty vector using lipofectamine, according to the manufacturer's instructions (Thermo Fisher Scientific). For lentivirus transfection, cells were individually infected with 30 multiplicity of infection of Lentivirus‐SGK3‐RNAi and negative control (con049), the medium was refreshed 14 h after transfection, and 48 h later the cell lysates were prepared for real‐time PCR and immunoblot analysis.
Cell viability assay
Cell viability was measured with a methylthiazoletetrazolium (MTT) detection kit (Amresco, Solon, OH, USA) according to the manufacturer's instructions. This assay identifies living cells and is based on the cellular conversion of a tetrazolium salt into a formazan product, a chromophore, which can be quantified by absorbance at 570 nm using a microplate reader (BioTek, Winooski, VT, USA).
Immunoblotting analysis
Incubated cells were trypsinized and washed twice in cold phosphate‐buffered saline. After washing, the cells and kidney tissues were lysed by adding lysis buffer containing 150 mM NaCl, 50 mM Tris‐HCl (pH 7.4), 1% NP‐40, 0.02% SDS, 0.5% sodium deoxycholate, 1 mM PMSF, 1 mM cocktail, and 1 mM phosphatase inhibitor. The suspension was centrifuged at 1600 g at 4°C for 15 min. Protein concentration was determined using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific) using bovine serum albumin as a standard. The samples were separated by electrophoresis on a 10% SDS‐polyacrylamide gel. After electrophoresis, the proteins were transferred to a PVDF membrane (Millipore, Bedford, MA, USA). The membranes were blocked with blocking buffer (5% milk in 1 × PBS plus 0.1% Tween) for 2 h at room temperature and incubated with the primary antibody overnight at 4°C, followed by incubation with horseradish peroxidase‐conjugated secondary antibody for 1 h at room temperature. Immunoreactive proteins were detected by ECL (Millipore, Germany). GAPDH was used as a loading control. All experiments were performed in triplicate, and the blots of phosphor‐SGK3 (p‐SGK3) and SGK3, phosphor‐GSK3 (p‐GSK3), and GSK3 were taken from the same membrane. The intensity of the bands was quantified using ImageJ (National Institutes of Health, Bethesda, MD, USA) and normalized to the band intensity of GAPDH.
Isolation of RNA, reverse transcription, and real‐time quantitative PCR
Incubated cells were washed twice with PBS, and total RNA was extracted with Trizol reagent (Thermo Fisher Scientific) according to the manufacturer's protocol and then dissolved in RNase‐free water. cDNA was synthesized with oligo dT primer in a 20‐μl reaction from 2 μg of total RNA using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer's protocol. cDNA (1 μl) was added to Takara SYBR Premix Ex Taq II (RR820L; Takara Bio, Kusatsu, Japan) and subjected to PCR amplification (1 cycle at 94°C for 3 min, 40 cycles at 94°C for 30 s, 58°C for 30 s, and 72°C for 25 s, and extension at 72°C for 5 min) in a CFX96 Touch Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA, USA). PCR was conducted in triplicate for each sample. The PCR results for each sample were normalized using GAPDH mRNA as an internal control. The ΔΔCt technique was used to calculate cDNA content in each sample. The primers for SGK3, podocin, and desmin were designed based on the GenBank (National Center for Tiotechnology Information, Bethesda, MD, USA; https://www.ncbi.nlm.nih.gov/genbank/) accession numbers (SGK3: NM_133220; desmin: NM_010043). The sequences of the primers were as follows: SGK3 (forward) ACACCAGAGTACCTTGCACCT, (reverse) AGCCCGTACAGCATCTCATA; podocin (forward) AGTCTAGCCCATGTGTCCAA, (reverse) CAGGTCACTGCATCTAAGGC; desmin (forward) ATGTGAAGATGGCCTTGGAT, (reverse) GCTGGTTTCTCGGAAGTTGA; GAPDH (forward) GTCAAGCTCATTTCCTGGTATG, (reverse) CCTCTCTTGCTCAGTGTCCTT.
Statistical analysis
All experiments were performed in triplicate, and all values were expressed as mean ± se. SPSS 13.0 was used for data analysis. Differences between groups were analyzed using an unpaired Student's t test for 2 groups, or a 1‐way ANOVA with Dunnett's posttest for more than 2 groups. A value of P < 0.05 was considered statistically significant. Analyses were performed using GraphPad InStat software (GraphPad Software, La Jolla, CA, USA).
RESULTS
PAN treatment induces podocyte injury and decreases SGK3 activity in cultured mouse podocytes
Many studies have reported that PAN treatment can result in enhanced apoptosis, increased desmin expression, and decreased expression levels of slit diaphragm proteins in cultured podocytes, such as synaptopodin, nephrin, CD2AP, and podocin (25, 26). The PAN cell model was constructed to study the activity of SGK3 during podocyte injury (27). First, an MTT assay was performed to observe the effect of PAN on podocyte viability. The optical density at a wavelength of 570 nm decreased by PAN treatment in a dose‐dependent manner, suggesting that PAN negatively affected podocyte viability (Fig. 1 A ). Additionally, in PAN‐treated podocytes, there was a significant dose‐dependent decreased expression of podocin and increased expression of desmin (Fig. 1 B) and a significant decrease in CD2AP protein expression (Fig. 1 C). These results suggest that a podocyte injury cell model was successfully generated via PAN application. The protein expression ratio of p‐SGK3 to total SGK3 was used to indicate the activity status of SGK3 (2). There was a progressive decrease of the p‐SGK3 to SGK3 ratio in PAN‐treated podocytes (Fig. 1 D). This observed decrease in SGK3 activity in PAN‐treated MPCs suggested that SGK3 may be involved in podocyte injury.
Figure 1.
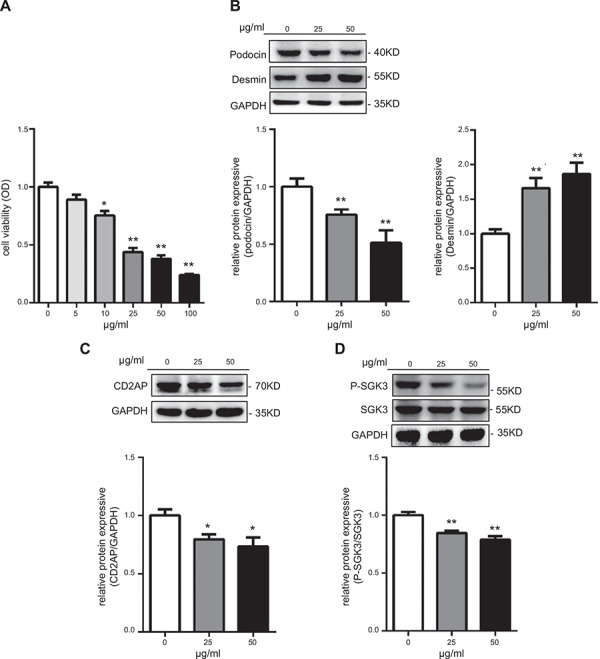
PAN treatment induced podocyte injury and decreased phosphorylation levels of SGK3 in cultured mouse podocytes. Immortalized MPCs were seeded and exposed for 24 h to PAN (0, 25, and 50 μg/ml). A) Cytotoxic effects of PAN were determined by MTT. B) Top: immunoblot analysis for podocin and desmin expression; GAPDH was used for normalization. Bottom: quantification of podocin and desmin by densitometry. C) Top: immunoblot analysis for CD2AP expression; GAPDH was used for normalization. Bottom: quantification of CD2AP by densitometry. D) Top: immunoblot analysis for p‐SGK3 and SGK3 expression. Bottom: quantification of p‐SGK3/SGK3 by densitometry. Data are means ± se from 3 independent experiments. *P < 0.05 vs. 0 PAN‐treated group, **P < 0.01 vs. 0 PAN‐treated group.
SGK3 shRNA transfection results in podocyte damage
To explore the ability of SGK3 to directly affect podocyte function, a recombinant lentivirus harboring SGK3 shRNAs was constructed to down‐regulate SGK3 expression in podocytes. The expression of exogenous eGFP‐labeled shRNA was detected via microscopy 48 h after transfection, revealing a 64% transfection efficiency (Fig. 2 A ). Moreover, SGK3 shRNA reduced SGK3 mRNA levels by ~51% (Fig. 2 B), which corresponded well to the degree of knockdown in SGK3 protein expression (Fig. 2 C). MTT assays revealed that the podocyte viability decreased significantly in the SGK3 shRNA‐treated group compared with the control group (Fig. 2 D). Examining the expression level of desmin and podocin in SGK3 down‐regulated MPCs revealed that SGK3 knockdown significantly increased both mRNA and protein levels of desmin, whereas podocin expression levels were repressed in cultured MPCs (Fig. 2 E, F). Moreover, the protein expression level of CD2AP was decreased dramatically in the SGK3 shRNA‐treated group (Fig. 2 F). These findings strongly support the idea that SGK3 down‐regulation could directly affect podocyte viability and function.
Figure 2.
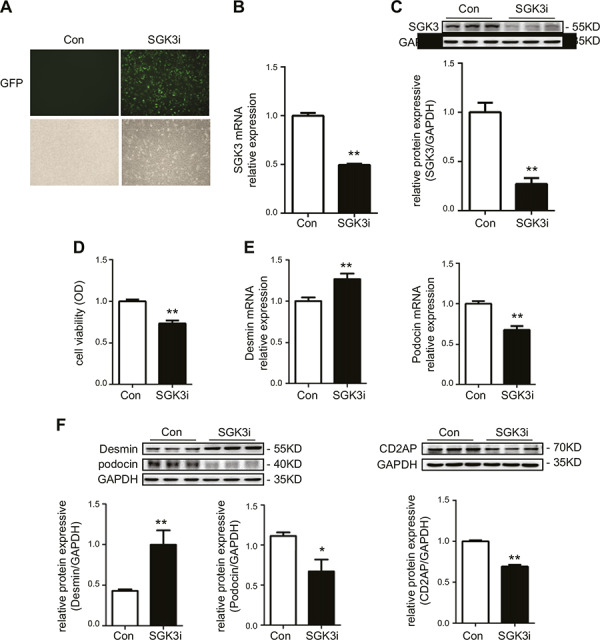
SGK3 knockdown results in podocyte injury in vitro. MPCs were infected with SGK3‐eGFP shRNA. A). Immunofluorescence revealed the effect of transfection with SGK3 shRNA. B) The SGK3 mRNA expression level was determined by real‐time PCR. C) Top: immunoblot analysis for SGK3 expression; GAPDH was used for normalization. Bottom: quantification of SGK3 by densitometry. D) Podocyte viability was determined by MTT. E) The mRNA expression levels of podocin and desmin were detected using real‐time PCR. F) Top: immunoblot analysis for desmin, podocin, and CD2AP expression. Bottom: quantification of desmin, podocin, and CD2AP by densitometry. OD, optical density. Data are means ± se of 3 independent experiments; 3 wells were repeated for each sample. *P < 0.05 vs. control group, **P < 0.01 vs. control group.
Activation of SGK3 mostly reverses PAN‐induced podocyte dysfunction
To further confirm the role of SGK3 on PAN‐induced podocyte injury in vitro, cultured MPCs were transfected with a eukaryotic expression vector encoding the constitutively activated S486D mutant of SGK3 (pMO/Flag/mSGK3‐S486D) and then treated with 25 μg/ml PAN for 24 h. SGK3 expression significantly increased in the SGK3‐S486D transfection group compared with the empty plasmid transfection group (Fig. 3 A ). Consistent with the PAN model, PAN stimulation resulted in a significantly decreased podocyte viability (Fig. 3 B), elevated expression of desmin (Fig. 3 C), and a subsequent significant reduction in podocin (Fig. 3 C) and CD2AP expression (Fig. 3 D). SGK3 phosphorylation was impaired by PAN treatment, thus reducing its activity, which supported the hypothesis that SGK3 inactivation is involved in PAN‐induced podocyte damage. Consequently, transfection of MPCs with SGK3‐S486D was performed to cause persistent activation of SGK3. In comparison to the PAN‐/S486D‐group, the cell viability and expression level of podocin was increased in the PAN–/S486D+ group (Fig. 3 B, C). Simultaneously, in the PAN+/S486D+ group, the cell viability and the protein expression level of podocin were remarkably higher, whereas the protein expression level of desmin was lower, than in the PAN+/S486D– group. More meaningful was that the cell viability and protein levels of desmin and podocin in the PAN+/S486D+ group were similar to physiologic conditions. The change in expression level of CD2AP was not significant in the PAN – / S486D+ group compared with the PAN– /S486D– group (Fig. 3 D). Overall, the enhanced activation of SGK3 by SGK3‐S486D plasmid transfection could reverse the decreased cell viability and expression of podocin and increase the expression of desmin in PAN‐induced MPCs (Fig. 3 B, C). These data indicate that SGK3 could ameliorate PAN‐induced podocyte dysfunction.
Figure 3.
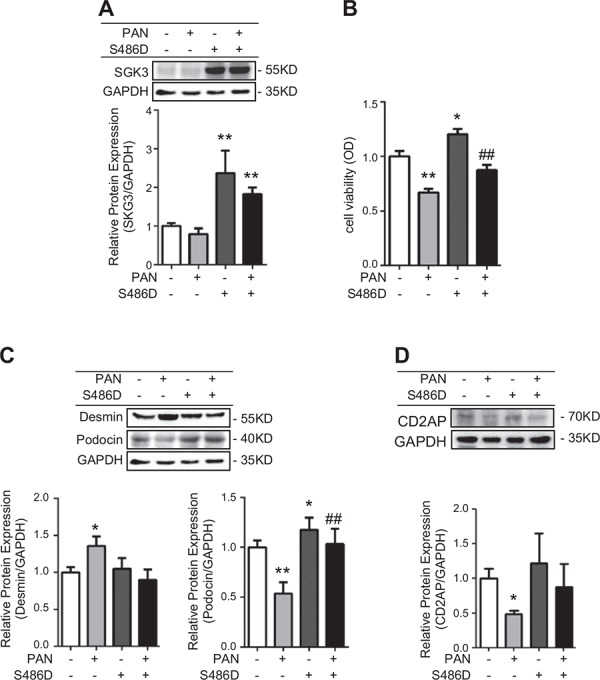
SGK3 activation mostly reversed PAN‐induced podocyte injury. The SGK3‐S486D plasmid was transiently transfected into cultured MPCs, and then the podocytes were exposed for 24 h to 0 and 25 μg/ml PAN. A) Top: immunoblot analysis for SGK3 expression; GAPDH was used for normalization. Bottom: quantification of SGK3 by densitometry. B) Podocyte viability was determined by MTT. C) Top: immunoblot analysis for desmin and podocin expression. Bottom: quantification of desmin and podocin by densitometry. D) Top: immunoblot analysis for CD2AP expression. Bottom: quantification of CD2AP by densitometry. OD, optical density. Data are means ± se of in 3 independent experiments. *P < 0.05 vs. PAN–/S486D– group, **P < 0.01 vs. PAN‐/S486D– group, ## P < 0.01 vs. PAN+/S486D– group.
Expression of SGK3 is decreased in the ADR nephritis mouse model
To assess whether SGK3 associates with podocyte dysfunction in vivo, an ADR nephritis mouse model was established by a single injection of ADR (10.5 mg/kg body weight) to the mice. Consistent with previous reports, ADR injection induced a marked proteinuria by 14 d (Fig. 4 A ) (28, 29). Meanwhile, there were some differences in the plasma urea nitrogen, creatinine, and albumin levels between the ADR‐treated and control mice, but these differences were not statistically significant (Fig. 4 A). PAS staining revealed that mice with ADR‐induced nephritis developed significant segmental obliteration of capillary lumina by the matrix and hyalinosis lesions (arrow) and protein casts in the renal tubules (arrowheads) as compared with normal mice (Fig. 4 B). In addition, desmin expression was dramatically increased and podocin was decreased in the kidneys of ADR‐treated mice (Fig. 4 C). These data suggest that a successful ADR nephritis mouse model had been established. Accompanied with the podocyte injury found in ADR‐induced mice, immunohistochemistry analysis revealed that SGK3 was abundantly expressed in the glomerulus rather than in the proximal tubules of BALB/C mice kidney, and ADR administration to mice resulted in decreased SGK3 expression in the glomerulus (Fig. 4 D). Taken together, these results suggest that SGK3 is involved in ADR‐induced podocyte dysfunction in vivo.
Figure 4.
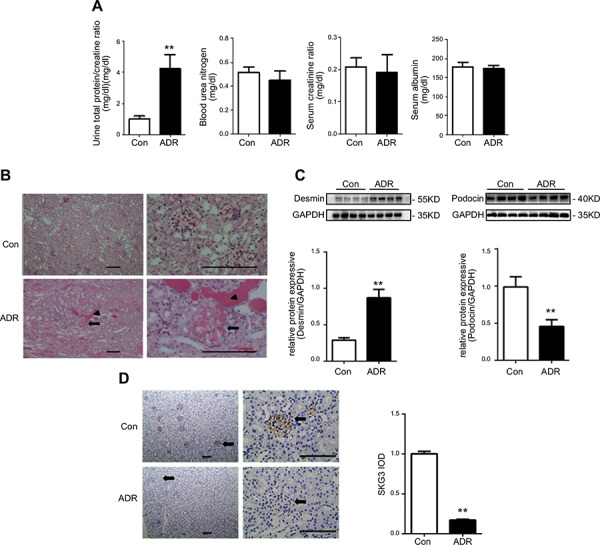
SGK3 protein expression was inhibited in mice with ADR nephritis. An intravenous injection of ADR (10.5 mg/kg) was used to induce nephritis. A) Plasma BUN, creatinine, albumin, and the urinary albumin/creatinine ratio were measured in mice after treatment with ADR. B) PAS staining demonstrates differences between control and ADR treated mice. Representative images from 7 mice in each group are shown. Arrows indicate sclerotic glomeruli. Arrowheads indicate tubular casts and dilatation. C) Top: immunoblot analysis for desmin and podocin expression; GAPDH was used for normalization. Bottom: quantification of desmin and podocin by densitometry. D) Left: histochemistry staining of SGK3 was performed on 5‐μm paraffin‐embedded kidney sections. Representative images from 7 mice in each group are shown. Arrows indicate glomeruli. Brown staining represents expression of SGK3. Right: computerized morphometric quantification of SGK3 staining in glomeruli in control and ADR mice. Data are given as mean ± se (n = 7/group). **P < 0.01 vs. control group. Scale bars, 100 μm.
The SGK3 KO mouse displays proteinuria and podocyte dysfunction
To further determine the direct role of SGK3 on podocyte function in vivo, SGK3 KO mice were generated as previously reported (23, 24). Spot urine of SGK3 KO and WT mice was harvested for proteinuria measurement. SGK3 KO mice had higher urinary protein levels than WT mice, whereas the plasma levels of BUN, creatinine, and albumin were not different between SGK3 knockout and WT mice (Fig. 5 A ). Observing kidney ultrastructure via electron microscopy revealed that the foot processes of podocytes, which line the outer surface of the glomerular basement membrane, displayed an apparent foot process effacement in the SGK3 KO mice when compared with WT mice (Fig. 5 B). To further confirm the podocyte injury seen in the SGK3 KO mice, kidney tissues were investigated using antibodies against desmin and podocin. This analysis revealed that the relative desmin expression level was significantly increased and that podocin was decreased in SGK3 KO mice (Fig. 5 C). These results provide evidence that SGK3 is essential for maintaining podocyte function.
Figure 5.
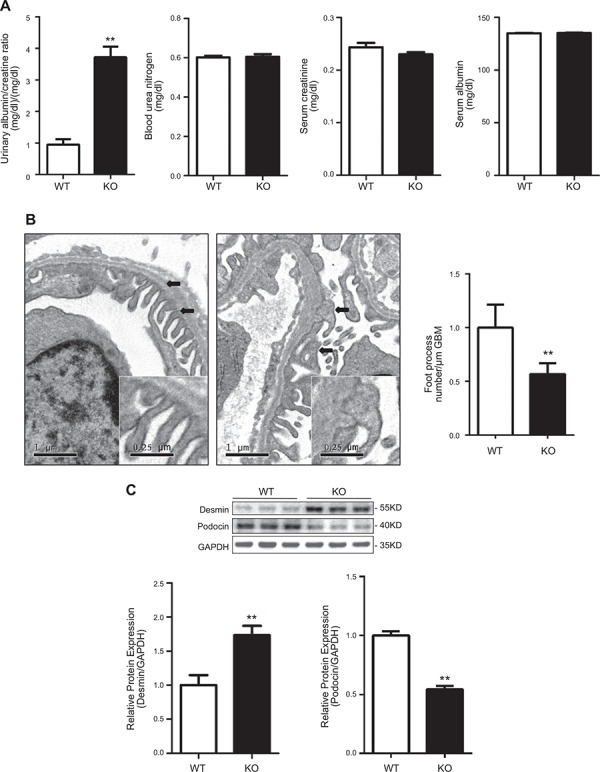
SGK3 KO mice exhibit a subtle but significant increase in albuminuria with fusion of the foot processes. A) Plasma urea nitrogen, creatinine, albumin, and the urinary albumin/creatinine ratio were measured in SGK3 KO and WT mice. B) Left: electron microscopy was performed to assess ultrastructural changes in podocyte morphology. Representative images are shown from 4 mice in each group. Black arrows indicate the change in podocyte foot process morphology. Right: absolute count of the number of foot processes per unit length of glomerular basement membrane on electron micrographs of kidney specimens. C) Top: Immunoblot analysis for desmin and podocin expression; GAPDH was used for normalization. Bottom: quantification of desmin and podocin by densitometry. GBM, glomerular basement membrane. Data are given as means ± se (n = 6/group). **P < 0.01 vs. SGK3 WT group.
p‐GSK3 regulated by SGK3 is related to podocyte injury in vivo and in vitro
GSK3 is a Ser/Thr kinase that is inhibited by phosphorylation of an N‐terminal Ser residue (Ser21 in GSK3a and Ser9 in GSK3β) and is involved in a plethora of functions, ranging from the glycogen metabolism to transcriptional regulation (30, 31). GSK3 activity is related to the formation of proteinuria, and GSK3 can be phosphorylated by SGK3 (32). To further explore the mechanism by which SGK3 maintains the normal expression and function of podocin, the protein expression levels of p‐GSK3 and GSK3 were measured in the podocyte injury model in vitro and in the SGK3 KO mice. The p‐GSK3/GSK3 ratio declined in correlation to the variation of SGK3 in PAN‐treated MPCs (Fig. 6 A ). The same trend was observed for GSK3 when the transcription level of SGK3 was interrupted using shRNA (Fig. 6 B). In addition, the reduced ratio p‐GSK3/GSK3 observed in PAN‐treated MPCs could be reversed by the addition of activated SGK3 (Fig. 6 C). Furthermore, investigating the GSK3 activity in SGK3 KO mice revealed that the phosphorylation levels of GSK3 decreased significantly in SGK3 KO mice (Fig. 6 D). Several studies have revealed that GSK3β mediates podocyte injury in vitro and in vivo (32–34). To further confirm the role of GSK3 on podocyte function, cultured MPCs were transfected with a GSK3β‐flag plasmid. GSK3β overexpression significantly decreased the protein expression of podocin and podocyte viability (Fig. 6 E‐G). These results suggest that GSK3 may participate in SGK3‐mediated podocyte injury. Combining the present observation that GSK3 activation was regulated by SGK3 with previous reports reveal that increased GSK3 activity could result in proteinuria (35). This suggests that SGK3 could maintain normal expression and function of podocin in podocyte, which involves GSK3 phosphorylation.
Figure 6.
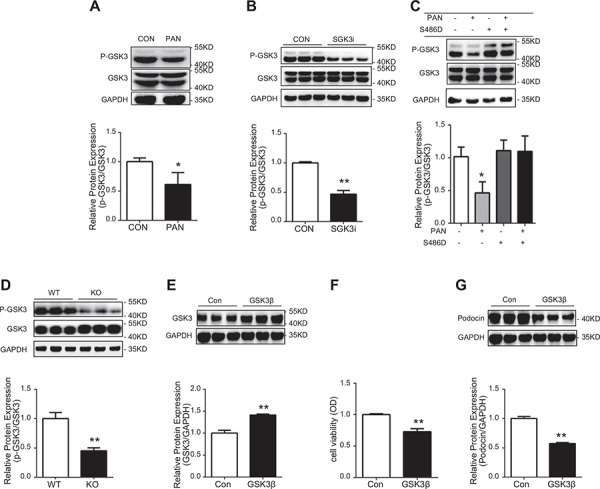
P‐GSK3 regulated by SGK3 is related to the podocyte injury in vivo and in vitro. Top: immunoblot analysis for p‐GSK3 and GSK3 expression was performed. Bottom: quantification of p‐GSK3/GSK3 by densitometry. A) PAN treatment. *P < 0.05 vs. control group. B) SGK3 shRNA transfection. **P < 0.01 vs. control group. C) PAN treatment with S486D transfection *P < 0.05 vs PAN–/S486D– group. D) SGK3 KO mice. **P < 0.01 vs. SGK3 WT group. E) Top: immunoblot analysis for GSK3β expression; GAPDH was used for normalization. Bottom: quantification of GSK3β by densitometry. F) Podocyte viability was determined by MTT. G) Top: immunoblot analysis for podocin expression; GAPDH was used for normalization. Bottom: quantification of podocin by densitometry. OD, optical density. Data are means ± se of in 3 independent experiments. **P < 0.01 vs. control group.
DISCUSSION
The SGK family plays a pivotal role in cell survival, proliferation, migration, and sodium balance (1), although the interplay between SGK3 and podocyte injury is unknown. The present study demonstrates that SGK3 deficiency leads to podocyte injury both in vivo and in vitro and that the glomerular expression of SGK3 was inhibited by ADR‐induced nephrosis. Furthermore, increased SGK3 activity promoted podocyte homeostasis, as evidenced by a reverse in the decreased podocyte viability and expression of podocin induced by PAN treatment. These results indicate that SGK3 plays an important role in maintaining podocyte normal function and that increasing SGK3 activity may provide a new therapeutic strategy for patients with chronic progressive renal diseases.
Podocytes are one of the key components of the renal filtration barrier, and podocyte injury plays an important role in proteinuria formation. Canaud et al. (36) reported that the mTOR/Akt2 pathway is a novel factor for influencing podocyte survival, and Huber et al. demonstrated that the slit diaphragm proteins nephrin, CD2AP, and podocin could initiate PI3K/AKT‐dependent signal transduction in glomerular podocytes (18); thus, AKT activation may contribute to the synthesis and/or maintenance of an intact glomerular basement membrane (37). SGK3 is a downstream target protein of PI3K and can be phosphorylated immediately by PI3K (1, 2). Consistent with these results, this study explored the concentration‐dependent inhibition of SGK3 activity in in vitro PAN‐treated MPCs. PAN caused decreased expression of podocin, which could be mostly reversed by SGK3 activation. These results verified the protective role of SGK3 in podocyte function. SGK3 has high structural homology to the AKT family, which suggests that it may play a similar role to AKT in influencing podocyte survival, although this remains to be investigated.
SGK3 can inactivate GSK3 by phosphorylation (31), and the activation of GSK3 is closely related to proteinuria (32). Thus, SGK3 may be involved with GSK3‐related podocyte damage and proteinuria. In the described ADR‐induced nephritis model, ADR‐treated mice had significantly increased proteinuria in relation to the control group and presented progressive glomerular hyalinosis/sclerosis and podocyte enlargement and hyperplasia. The SGK3 level decreased in ADR‐treated mice compared with the control mice, and the decreased SGK3 expression was associated with increased proteinuria. These results suggest that SGK3 is involved in the pathophysiology of ADR nephritis. Moreover, in the present study, there were not higher serum levels of BUN and creatinine or the lower serum levels of albumin, suggesting that the renal function of the ADR mice was not altered even in mice that presented greater proteinuria than normal mice at the fourteenth day after ADR treatment. The normal plasma BUN, creatinine, and albumin levels from ADR mice are inconsistent with previous reports (21, 22, 38), which may be due to the different times used for harvesting the plasma.
As a member of the SGK family, the catalytic domains of SGK3 share 80% amino acid sequence identity with SGK1 (39), and these two proteins can complement each other in some respects (35). Garofalo reported that SGK3 KO mice exert notable hair follicle development insufficiencies (40), which was not observed in SGK1‐deficient mice (41–45); this finding helps to elucidate the different role of the SGK isoforms. Regarding the phenotype of SGKs in kidney, SGK1 is related to the formation of proteinuria and is up‐regulated in patients with diabetic nephropathy (46) or glomerular nephritis (47). However, SGK1 KO mice do not exhibit proteinuria. Unlike SGK1‐deficient mice, the results of this study revealed that SGK3 KO mice presented proteinuria and podocyte injury, supporting a role for SGK3 in maintaining podocyte function (41–45). Interestingly, the expression of GSK3 is partially mediated by SGK3, and mice expressing PKB/SGK‐resistant GSK3 can show proteinuria (48). A series of studies have demonstrated that blocking GSK3 can ameliorate proteinuria, podocyte injury, and glomerulosclerosis in several experimental glomerular diseases, including diabetic nephropathy (47) and lupus nephritis (48). Liang et al. (49) reported that GSK3 was related to the epithelial‐mesenchymal transition during podocyte damage, which supports the changes to desmin expression in our lentivirus and animal experiments. Moreover, GSK3 participates in podocyte injury and apoptosis (14, 33), and activation of GSK3 (50) could inhibit podocin expression (33, 48), all of which are consistent with the findings in this study, which suggests that inhibiting SGK3 activation could result in decreased mRNA and protein expression levels of podocin. Based on this information, SGK3 could be involved in regulating podocyte injury by affecting the transcriptional levels of podocin. Additional studies are required to address whether and how the SGK3/GSK3 pathway regulates podocin expression and if there are other SGK3 target proteins that may be important for podocyte function and viability (49).
In summary, our findings suggest a mechanistic basis for the deficiency of SGK3 in controlling cell viability and podocin expression in mouse podocytes. According to this view, GSK3, which is phosphorylated and thus inactivated by SGK3, may contribute to SGK3‐modulated podocyte dysfunction (shown schematically in Fig. 7 ). These data provide potential insight into the molecular mechanisms underlying PAN‐induced podocyte injury in vitro and ADR‐induced nephritis in vivo, suggesting a method for regulating podocin expression through the SGK3 signaling pathway.
Figure 7.
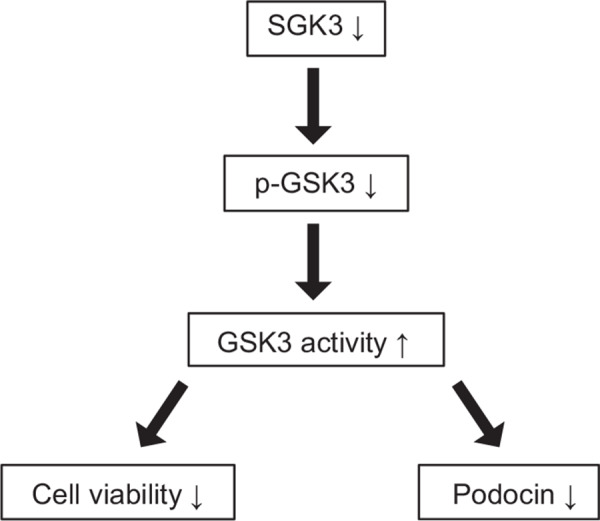
GSK3 activation is mediated by SGK3 deficiency in regulating podocyte dysfunction. Lack of SGK3 results in decreased phosphorylation of GSK3, thereby activating GSK3. Activation of GSK3 inhibits cell viability and podocin expression, which contributes to podocyte dysfunction.
AUTHOR CONTRIBUTIONS
H. Zhao, D. Pearce, and L.‐J. Yao designed the research; L.‐Q. Peng, S. Liu, Y.‐P. Yuan, and C.‐Y. Yuan performed the experiments and analyzed the data; and L.‐Q. Peng, M.‐J. Mwamunyi, and L.‐J. Yao wrote the paper.
ACKNOWLEDGMENTS
The authors thank Zi‐Fang Li (Union Hospital, Tongji Medical College, Huazhong University of Science and Technology) for help in revising the manuscript. This work was supported by the National Nature Science foundation of China (Grant 81370818). The authors declare no conflicts of interest.
Peng, L.‐Q. , Zhao, H. , Liu, S. , Yuan, Y.‐P. , Yuan, C.‐Y. , Mwamunyi, M.‐J. , Pearce, D. , Yao, L.‐J. Lack of serum‐ and glucocorticoid‐inducible kinase 3 leads to podocyte dysfunction. FASEB J. 32, 576–587 (2018). www.fasebj.org
REFERENCES
- 1. Lang, F. , Böhmer, C. , Palmada, M. , Seebohm, G. , Strutz‐Seebohm, N. , and Vallon, V. (2006) (Patho)physiological significance of the serum‐ and glucocorticoid‐inducible kinase isoforms. Physiol. Rev. 86, 1151–1178 [DOI] [PubMed] [Google Scholar]
- 2. Tessier, M. , and Woodgett, J. R. (2006) Role of the Phox homology domain and phosphorylation in activation of serum and glucocorticoid‐regulated kinase‐3. J. Biol. Chem. 281, 23978–23989 [DOI] [PubMed] [Google Scholar]
- 3. Ahmed, M. , Fezai, M. , Uzcategui, N. L. , Hosseinzadeh, Z. , and Lang, F. (2016) SGK3 sensitivity of voltage gated K+ channel Kv1.5 (KCNA5). Cell. Physiol. Biochem. 38, 359–367 [DOI] [PubMed] [Google Scholar]
- 4. Bhandaru, M. , Kempe, D. S. , Rotte, A , Capuano, P. , Pathare, G. , Sopjani, M. , Alesutan, I. , Tyan, L. , Huang, D. Y. , Siraskar, B. , Judenhofer, M. S. , Stange, G. , Pichler, B. J. , Biber, J. , Quintanilla‐Martinez, L. , Wagner, C. A. , Pearce, D. , Föller, M. , and Lang, F. (2011) Decreased bone density and increased phosphaturia in gene‐targeted mice lacking functional serum‐ and glucocorticoid‐inducible kinase 3. Kidney Int. 80, 61–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boehmer, C. , Palmada, M. , Rajamanickam, J. , Schniepp, R. , Amara, S. , and Lang, F. (2006) Post‐translational regulation of EAAT2 function by co‐expressed ubiquitin ligase Nedd4–2 is impacted by SGK kinases. J. Neurochem. 97, 911–921 [DOI] [PubMed] [Google Scholar]
- 6. Böhmer, C. , Sopjani, M. , Klaus, F. , Lindner, R. , Laufer, J. , Jeyaraj, S. , Lang, F. , and Palmada, M. (2010) The serum and glucocorticoid inducible kinases SGK1–3 stimulate the neutral amino acid transporter SLC6A19. Cell. Physiol. Biochem. 25, 723–732 [DOI] [PubMed] [Google Scholar]
- 7. Strutz‐Seebohm, N. , Seebohm, G. , Korniychuk, G. , Baltaev, R. , Ureche, O. , Striegel, M. , and Lang, F. (2006) Additive regulation of GluR1 by stargazin and serum‐ and glucocorticoid‐inducible kinase isoform SGK3. Pflugers Arch. 452, 276–282 [DOI] [PubMed] [Google Scholar]
- 8. Henke, G. , Setiawan, I. , Böhmer, C. , and Lang, F. (2002) Activation of Na+/K+‐ATPase by the serum and glucocorticoid‐dependent kinase isoforms. Kidney Blood Press. Res. 25, 370–374 [DOI] [PubMed] [Google Scholar]
- 9. Schmid, E. , Bhandaru, M. , Nurbaeva, M. K. , Yang, W. , Szteyn, K. , Russo, A. , Leibrock, C. , Tyan, L. , Pearce, D. , Shumilina, E. , and Lang, F. (2012) SGK3 regulates Ca(2+) entry and migration of dendritic cells. Cell. Physiol. Biochem. 30, 1423–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trepiccione, F. , and Capasso, G. (2011) SGK3: a novel regulator of renal phosphate transport? Kidney Int. 80, 13–15 [DOI] [PubMed] [Google Scholar]
- 11. Böhmer, C. , Palmada, M. , Kenngott, C. , Lindner, R. , Klaus, F. , Laufer, J. , and Lang, F. (2007) Regulation of the epithelial calcium channel TRPV6 by the serum and glucocorticoid‐inducible kinase isoforms SGK1 and SGK3. FEBS Lett. 581, 5586–5590 [DOI] [PubMed] [Google Scholar]
- 12. Pasham, V. , Rotte, A. , Bhandaru, M. , Eichenmüller, M. , Fröhlich, H. , Mack, A. F. , Bobbala, D. , Yang, W. , Pearce, D. , and Lang, F. (2011) Regulation of gastric acid secretion by the serum and glucocorticoid inducible kinase isoform SGK3. J. Gastroenterol. 46, 305–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maier, G. , Palmada, M. , Rajamanickam, J. , Shumilina, E. , Böhmer, C. , and Lang, F. (2006) Upregulation of HERG channels by the serum and glucocorticoid inducible kinase isoform SGK3. Cell. Physiol. Biochem. 18, 177–186 [DOI] [PubMed] [Google Scholar]
- 14. Qi, R. , and Sheng, Y. Y. (2016) CD2‐associated protein participates in podocyte apoptosis via PI3K/Akt signaling pathway. J. Recept. Signal Transduc. Res. 36, 288–291 [DOI] [PubMed] [Google Scholar]
- 15. Mundel, P. , and Shankland, S. J. (2002) Podocyte biology and response to injury. J. Am. Soc. Nephrol. 13, 3005–3015 [DOI] [PubMed] [Google Scholar]
- 16. Kang, H. G. , Paik, K. H. , Cho, H. Y. , Lee, B. H. , Ha, I. S. , Choi, Y. , and Cheong, H. I. (2010) Transcriptome analysis of the response of cultured murine podocytes to puromycin aminonucleoside. Nephron, Exp. Nephrol. 115, e1–e8 [DOI] [PubMed] [Google Scholar]
- 17. Yaddanapudi, S. , Altintas, M. M. , Kistler, A. D. , Fernandez, I. , Möller, C. C. , Wei, C. , Peev, V. , Flesche, J. B. , Forst, A. L. , Li, J. , Patrakka, J. , Xiao, Z. , Grahammer, F. , Schiffer, M. , Lohmüller, T. , Reinheckel, T. , Gu, C. , Huber, T. B. , Ju, W. , Bitzer, M. , Rastaldi, M. P. , Ruiz, P. , Tryggvason, K. , Shaw, A. S. , Faul, C. , Sever, S. , and Reiser, J. (2011) CD2AP in mouse and human podocytes controls a proteolytic program that regulates cytoskeletal structure and cellular survival. J. Clin. Invest. 121, 3965–3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huber, T. B. , Hartleben, B. , Kim, J. , Schmidts, M. , Schermer, B. , Keil, A. , Egger, L. , Lecha, R. L. , Borner, C. , Pavenstädt, H. , Shaw, A. S. , Walz, G. , and Benzing, T. (2003) Nephrin and CD2AP associate with phosphoinositide 3‐OH kinase and stimulate AKT‐dependent signaling. Mol. Cell. Biol. 23, 4917–4928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ha, T. S. , Hong, E. J. , and Han, G. D. (2015) Diabetic conditions downregulate the expression of CD2AP in podocytes via PI3‐K/Akt signalling. Diabetes Metab. Res. Rev. 31, 50–60 [DOI] [PubMed] [Google Scholar]
- 20. Zou, J. , Yaoita, E. , Watanabe, Y. , Yoshida, Y. , Nameta, M. , Li, H. , Qu, Z. , and Yamamoto, T. (2006) Upregulation of nestin, vimentin, and desmin in rat podocytes in response to injury. Virchows Arch. 448, 485–492 [DOI] [PubMed] [Google Scholar]
- 21. Fogo, A. B. (2003) Animal models of FSGS: lessons for pathogenesis and treatment. Semin. Nephrol. 23, 161–171 [DOI] [PubMed] [Google Scholar]
- 22. Fogo, A. B. (2015) Causes and pathogenesis of focal segmental glomerulosclerosis. Nat. Rev. Nephrol. 11, 76–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yao, L. J. , McCormick, J. A. , Wang, J. , Yang, K. Y. , Kidwai, A. , Colussi, G. L. , Boini, K. M. , Birnbaum, M. J. , Lang, F. , German, M. S. , and Pearce, D. (2011) Novel role for SGK3 in glucose homeostasis revealed in SGK3/Akt2 double‐null mice. Mol. Endocrinol. 25, 2106–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sandu, C. , Rexhepaj, R. , Grahammer, F. , McCormick, J. A. , Henke, G. , Palmada, M. , Nammi, S. , Lang, U. , Metzger, M. , Just, L. , Skutella, T. , Dawson, K. , Wang, J. , Pearce, D. , and Lang, F. (2005) Decreased intestinal glucose transport in the sgk3‐knockout mouse. Pflugers Arch. 451, 437–444 [DOI] [PubMed] [Google Scholar]
- 25. Zheng, C. X. , Chen, Z. H. , Zeng, C. H. , Qin, W. S. , Li, L. S. , and Liu, Z. H. (2008) Triptolide protects podocytes from puromycin aminonucleoside induced injury in vivo and in vitro. Kidney Int. 74, 596–612 [DOI] [PubMed] [Google Scholar]
- 26. Dong, N. , Meng, L. , Xue, R. , Yu, M. , Zhao, Z. , and Liu, X. (2017) Adrenomedullin ameliorates podocyte injury induced by puromycin aminonucleoside in vitro and in vivo through modulation of Rho GTPases. Int. Urol. Nephrol. 49, 1489–1506 [DOI] [PubMed] [Google Scholar]
- 27. Pippin, J. W. , Brinkkoetter, P. T. , Cormack‐Aboud, F. C. , Durvasula, R. V. , Hauser, P. V. , Kowalewska, J. , Krofft, R. D. , Logar, C. M. , Marshall, C. B. , Ohse, T. , and Shankland, S. J. (2009) Inducible rodent models of acquired podocyte diseases. Am. J. Physiol. Renal Physiol. 296, F213–F229 [DOI] [PubMed] [Google Scholar]
- 28. Yang, S. M. , Hua, K. F. , Lin, Y. C. , Chen, A. , Chang, J. M. , Kuoping Chao, L. , Ho, C. L. , and Ka, S. M. (2013) Citral is renoprotective for focal segmental glomerulosclerosis by inhibiting oxidative stress and apoptosis and activating Nrf2 pathway in mice. PLoS One 8, e74871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu, G. , Shi, Y. , Peng, X. , Liu, H. , Peng, Y. , and He, L. (2015) Astaxanthin attenuates adriamycin‐induced focal segmental glomerulosclerosis. Pharmacology 95, 193–200 [DOI] [PubMed] [Google Scholar]
- 30. Beurel, E. , Grieco, S. F. , and Jope, R. S. (2015) Glycogen synthase kinase‐3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther. 148, 114–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dai, F. , Yu, L. , He, H. , Chen, Y. , Yu, J. , Yang, Y. , Xu, Y. , Ling, W. , and Zhao, S. (2002) Human serum and glucocorticoid‐inducible kinase‐like kinase (SGKL) phosphorylates glycogen syntheses kinase 3 beta (GSK‐3beta) at serine‐9 through direct interaction. Biochem. Biophys. Res. Commun. 293, 1191–1196 [DOI] [PubMed] [Google Scholar]
- 32. Föller, M. , Kempe, D. S. , Boini, K. M. , Pathare, G. , Siraskar, B. , Capuano, P. , Alesutan, I. , Sopjani, M. , Stange, G. , Mohebbi, N. , Bhandaru, M. , Ackermann, T. F. , Judenhofer, M. S. , Pichler, B. J. , Biber, J. , Wagner, C. A. , and Lang, F. (2011) PKB/SGK‐resistant GSK3 enhances phosphaturia and calciuria. J. Am. Soc. Nephrol. 22, 873–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li, C. , Ge, Y. , Dworkin, L. , Peng, A. , and Gong, R. (2016) The β isoform of GSK3 mediates podocyte autonomous injury in proteinuric glomerulopathy. J. Pathol. 239, 23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paeng, J. , Chang, J. H. , Lee, S. H. , Nam, B. Y. , Kang, H. Y. , Kim, S. , Oh, H. J. , Park, J. T. , Han, S. H. , Yoo, T. H. , and Kang, S. W. (2014) Enhanced glycogen synthase kinase‐3beta activity mediates podocyte apoptosis under diabetic conditions. Apoptosis 19, 1678–1690 [DOI] [PubMed] [Google Scholar]
- 35. Grahammer, F. , Artunc, F. , Sandulache, D. , Rexhepaj, R. , Friedrich, B. , Risler, T. , McCormick, J. A. , Dawson, K. , Wang, J. , Pearce, D. , Wulff, P. , Kuhl, D. , and Lang, F. (2006) Renal function of gene‐targetedmice lacking both SGK1 and SGK3. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R945–R950 [DOI] [PubMed] [Google Scholar]
- 36. Canaud, G. , Bienaimé, F. , Viau, A. , Treins, C. , Baron, W. , Nguyen, C. , Burtin, M. , Berissi, S. , Giannakakis, K. , Muda, A. O. , Zschiedrich, S. , Huber, T. B. , Friedlander, G. , Legendre, C. , Pontoglio, M. , Pende, M. , and Terzi, F. (2013) AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat. Med. 19, 1288–1296 [DOI] [PubMed] [Google Scholar]
- 37. Li, X. , Chuang, P. Y. , D'Agati, V. D. , Dai, Y. , Yacoub, R. , Fu, J. , Xu, J. , Taku, O. , Premsrirut, P. K. , Holzman, L. B. , and He, J. C. (2015) Nephrin preserves podocyte viability and glomerular structure and function in adult kidneys. J. Am. Soc. Nephrol. 26, 2361–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang, Y. , Wang, Y. P. , Tay, Y. C. , and Harris, D. C. (2000) Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int. 58, 1797–1804 [DOI] [PubMed] [Google Scholar]
- 39. Kobayashi, T. , Deak, M. , Morrice, N. , and Cohen, P. (1999) Characterization of the structure and regulation of two novel isoforms of serum‐ and glucocorticoid‐induced protein kinase. Biochem. J. 344, 189–197 [PMC free article] [PubMed] [Google Scholar]
- 40. Garofalo, R. S. , Orena, S. J. , Rafidi, K. , Torchia, A. J. , Stock, J. L. , Hildebrandt, A. L. , Coskran, T. , Black, S. C. , Brees, D. J. , Wicks, J. R. , McNeish, J. D. , and Coleman, K. G. (2003) Severe diabetes, age‐dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J. Clin. Invest. 112, 197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fejes‐Tóth, G. , Frindt, G. , Náray‐Fejes‐Tóth, A. , and Palmer, L. G. (2008) Epithelial Na+ channel activation and processing in mice lacking SGK1. Am. J. Physiol. Renal Physiol. 294, F1298–F1305 [DOI] [PubMed] [Google Scholar]
- 42. Huang, D. Y. , Wulff, P. , Völkl, H. , Loffing, J. , Richter, K. , Kuhl, D. , Lang, F. , and Vallon, V. (2004) Impaired regulation of renal K+ elimination in the sgk1‐knockout mouse. J. Am. Soc. Nephrol. 15, 885–891 [DOI] [PubMed] [Google Scholar]
- 43. Huang, D. Y. , Boini, K. M. , Osswald, H. , Friedrich, B. , Artunc, F. , Ullrich, S. , Rajamanickam, J. , Palmada, M. , Wulff, P. , Kuhl, D. , Vallon, V. , and Lang, F. (2006) Resistance of mice lacking the serum‐ and glucocorticoid‐inducible kinase SGK1 against salt‐sensitive hypertension induced by a high‐fat diet. Am. J. Physiol. Renal Physiol. 291, F1264–F1273 [DOI] [PubMed] [Google Scholar]
- 44. Nasir, O. , Wang, K. , Föller, M. , Gu, S. , Bhandaru, M. , Ackermann, T. F. , Boini, K. M. , Mack, A. , Klingel, K. , Amato, R. , Perrotti, N. , Kuhl, D. , Behrens, J. , Stournaras, C. , and Lang, F. (2009) Relative resistance of SGK1 knockout mice against chemical carcinogenesis. IUBMB Life 61, 768–776 [DOI] [PubMed] [Google Scholar]
- 45. Sobiesiak, M. , Shumilina, E. , Lam, R. S. , Wölbing, F. , Matzner, N. , Kaesler, S. , Zemtsova, I. M. , Lupescu, A. , Zahir, N. , Kuhl, D. , Schaller, M. , Biedermann, T. , and Lang, F. (2009) Impaired mast cell activation in gene‐targeted mice lacking the serum‐ and glucocorticoid‐inducible kinase SGK1. J. Immunol. 183, 4395–4402 [DOI] [PubMed] [Google Scholar]
- 46. Kumar, J. M. , Brooks, D. P. , Olson, B. A. , and Laping, N. J. (1999) Sgk, a putative serine/threonine kinase, is differentially expressed in the kidney of diabetic mice and humans. J. Am. Soc. Nephrol. 10, 2488–2494 [DOI] [PubMed] [Google Scholar]
- 47. Friedrich, B. , Wärntges, S. , Klingel, K. , Sauter, M. , Kandolf, R. , Risler, T. , Müller, G. A. , Witzgall, R. , Kriz, W. , Gröne, H. J. , and Lang, F. (2002) Up‐regulation of the human serum and glucocorticoid‐dependent kinase 1 in glomerulonephritis. Kidney Blood Press. Res. 25, 303–307 [DOI] [PubMed] [Google Scholar]
- 48. Boini, K. M. , Amann, K. , Kempe, D. , Alessi, D. R. , and Lang, F. (2009) Proteinuria in mice expressing PKB/SGK‐resistant GSK3. Am. J. Physiol. Renal Physiol. 296, F153–F159 [DOI] [PubMed] [Google Scholar]
- 49. Liang, Y. , Jing, Z. , Deng, H. , Li, Z. , Zhuang, Z. , Wang, S. , and Wang, Y. (2015) Soluble epoxide hydrolase inhibition ameliorates proteinuria‐induced epithelial‐mesenchymal transition by regulating the PI3K‐Akt‐GSK‐3β signaling pathway. Biochem. Biophys. Res. Commun. 463, 70–75 [DOI] [PubMed] [Google Scholar]
- 50. Mariappan, M. M. , Prasad, S. , D'Silva, K. , Cedillo, E. , Sataranatarajan, K. , Barnes, J. L. , Choudhury, G. G. , and Kasinath, B. S. (2014) Activation of glycogen synthase kinase 3b ameliorates diabetes‐induced kidney injury. J. Biol. Chem. 289, 35363–35375 [DOI] [PMC free article] [PubMed] [Google Scholar]