

Abstract
Astrocytes are a type of glial cell that are activated in the brain tissue of patients with Alzheimer’s disease to induce the accumulation of amyloid (Aβ). We previously found that a combination of low-dose gemfibrozil (GFB, a drug approved to treat high cholesterol) and retinoic acid (RA, a vitamin A derivative) induces lysosomal biogenesis through peroxisome proliferator-activated receptor α (PPARα)-mediated transcription of the gene encoding transcription factor EB (TFEB), a master regulator of lysosomal biogenesis and autophagy. Here, we found that the same combination (GFB-RA) enhanced the uptake of Aβ from the extracellular space and its subsequent degradation in astrocytes through a PPARα-dependent pathway. GFB-RA stimulated the abundance of both low-density lipoprotein receptor (LDLR) and TFEB in astrocytes through PPARα. LDLR was critical for Aβ uptake, whereas TFEB was critical for its degradation. GFB-RA treatment also increased autophagic flux and lysosomal activity in astrocytes. Consistent with these effects and in a manner dependent on astroglial PPARα, oral administration of GFB-RA switched astroglial activation to a neuroprotective state, lowered Aβ burden in the brain and improved spatial learning and memory in the 5XFAD mouse model of Alzheimer’s disease. These findings uncover a new function of PPARα in stimulating astroglial uptake and degradation of Aβ and suggest possible repurposing of GFB-RA combination therapy for AD.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease with classic memory impairment and cognitive disorder. Pathological hallmarks of AD are the presence of senile plaques (SPs) containing fibrillar β-amyloid (Aβ40/42) and neurofibrillary tangles (NFTs), originating from increased phosphorylation of Tau, in the cortex and hippocampus of brain (1, 2). The abnormal accumulation of Aβ and formation NFTs induces neuro-inflammation and subsequent neuronal loss, which is the primary cause of AD (3). Aggregate prone Aβ40/42 fragments are generated by the sequential activity of β- and γ-secretase on amyloid precursor protein (APP), whereas the action of α-secretase produces soluble APPα (sAPPα) fragments that are not prone to aggregation (4, 5). The α-secretase is mainly associated to the plasma membrane, whereas majority of β-secretase is present in the endosomal-lysosomal compartments (6, 7). The processing of APP could happen in either secretory pathway or endosomal-lysosomal pathway. Newly synthesized APP could be either be delivered to plasma membrane where it is processed mainly by α-secretase (secretory pathway), or occasionally the APPs are recycled back into endosomes by endocytosis, where it could be processed by β- and γ-secretase (endosomal-lysosomal pathway) producing Aβ fragments (8). Under normal conditions, further cleavage by other proteases (mainly cathepsin B) in the lysosomes degrade the Aβ fragments into even smaller non-toxic fragments, which are recycled or expunged from the cell (9). Also both in vitro and in vivo conditions, extracellular Aβ could also be endocytosed and degraded in the lysosomes (10). Decline in lysosomal function due to ageing or other pathological condition may result in abnormal accumulation of Aβ fragments inside the lysosome and increase the lysosomal load. This may lead to rupture of lysosomal membrane, which not only releases the toxic Aβ into the cytosol but also triggers lysosomal membrane permeability that ultimately induces necrotic or apoptotic cell death (11). Therefore, it is imperative that enhanced lysosomal function could be a possible therapeutic mechanism of Aβ clearance in AD.
We have demonstrated that gemfibrozil (GFB), an agonist of peroxisome proliferator-activated receptor α (PPARα), either alone or in conjunction with all-trans-retinoic acid (RA), enhances TFEB expression and increases lysosomal biogenesis in mouse brain cells (12). Here, we examined whether GFB-RA could stimulate the uptake of Aβ in astrocytes and found that activation of PPARα by GFB-RA stimulated astroglial uptake of Aβ through low-density lipoprotein receptor (LDLR) and degradation of Aβ through TFEB. Our in vivo results also revealed that oral GFB-RA stimulated lysosomal biogenesis, reduced plaque load and improved cognitive functions in 5XFAD mouse model of AD via astroglial PPARα. These results highlight the importance PPARα-dependent astroglial plaque-clearance machinery in reducing one of the AD pathologies.
Results
Gemfibrozil (GFB) and retinoic acid (RA) treatment enhances Aβ uptake in mouse primary astrocytes.
As mentioned earlier, lysosomal activity is crucial for the clearance of Aβ in AD brain and earlier we have shown that a combination of GFB and RA (GFB-RA) enhances lysosomal biogenesis (12). Here, we investigated the effect of GFB-RA on the uptake of extracellular Aβ by mouse primary astrocytes. We performed both a quantitative in vitro assay and a qualitative microscopic analysis to measure the alterations in the levels of Aβ taken up by the cells.
For the former, cells were treated with GFB-RA and further incubated with FAM-Aβ(1–42) for various time points from 15 min to 8 hours. The signal intensity of Aβ was first normalized to that of Hoechst, to account for the variability in cell number in each well, if any. Then the normalized Aβ signals of GFB-RA–treated samples were compared to their DMSO-treated counterparts and percentage change in the Aβ signal was calculated for each time point. After 2 hours of incubation in Aβ containing media, the amount of Aβ inside the GFB-RA–treated cells were ~60% more compared to the DMSO-treated cells. At 4 hours, the Aβ signal in treated cells were about ~80% higher than the control (Fig 1A). However, additional incubation up to 8 hours did not yield any further increase in the Aβ content in treated cells. To confirm these results, we compared Aβ fluorescence from media and cells. While we found a peak of Aβ fluorescence in astrocytes at 4 hours of incubation (Fig. 1B), Aβ fluorescence gradually decreased in serum reaching the nadir at 4 hours (Fig. 1C). To further understand whether Aβ uptake was happening in astrocytes or any contaminating microglia, after 4 hours of treatment with FAM-Aβ(1–42), cells were stained for GFAP (a marker of astrocytes) and Iba1 (a marker of microglia). These data suggested that the uptake of FAM-Aβ(1–42) took place in astrocytes, not microglia (fig. S1, A and B). Therefore, for further uptake assays by astrocytes, the 4-hour time point of Aβ incubation was selected.
Figure 1. Combination of gemfibrozil and retinoic acid (GFB-RA) enhances Aβ uptake in mouse primary astrocytes.
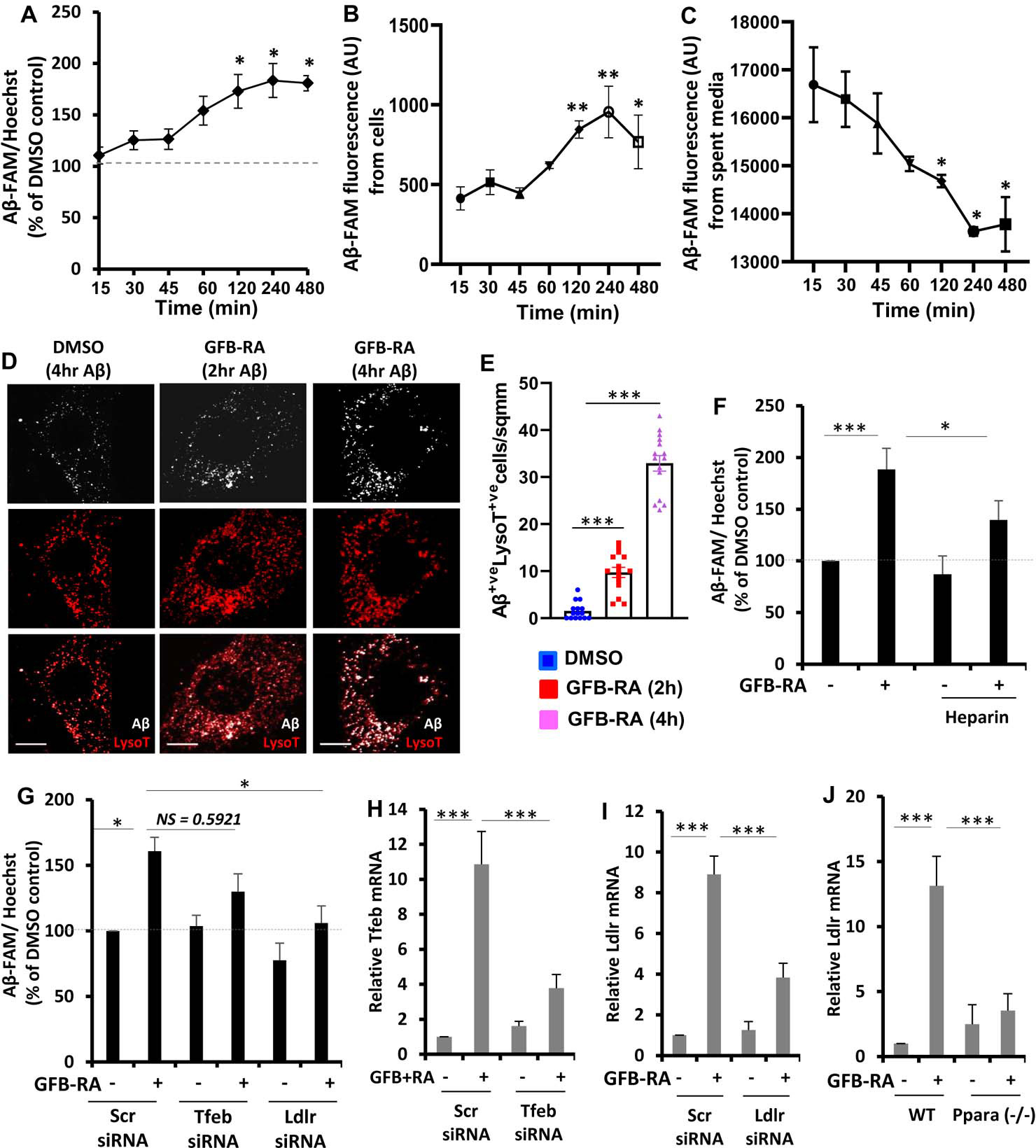
(A) Aβ uptake assay in mouse primary astrocytes were cultured for 24 hours with GFB-RA [GFB (10 μM) + RA (0.2 μM)], followed by 500 nM FAM-tagged Aβ1–42 for a further 15 min to 8 hours. Data are shown as a percent change compared to DMSO-treated controls. (B and C) The fluorescence of FAM-Aβ was monitored over time in the primary astrocytes and media described in (A). (D and E) Mouse primary astrocytes treated with GFB-RA were incubated with 500nM HF-Aβ1–42 and 75 nM Lysotracker Red and observed under microscope. Scale bar = 20 μm. Aβ+Lyso-T+ puncta were counted from 15 astrocytes from three independent experiments. (F) Mouse primary astrocytes were treated with DMSO or GFB-RA, followed by treatment with diluent of heparin (100 μg/ml) and further incubated in 500 nM FAM-Aβ for 4 hours. Aβ uptake assay was performed, and data is shown as percentage change with respect to untreated control. (G) Mouse primary astrocytes were transfected with scrambled siRNA, Tfeb siRNA or Ldlr siRNA, treated with GFB-RA, followed by incubation in 500 nM FAM-Aβ for 4 hours. Data from Aβ uptake assay is represented as percentage change with respect to DMSO and scrambled siRNA controls. (H and I) Real-time PCR was performed to measure the efficacy of siRNA silencing of Tfeb and Ldlr. (J) Wild-type (WT) and Ppara−/− astrocytes were treated with GFB-RA for 4 hours followed by monitoring the mRNA expression of LDLR by real-time PCR. All data are mean ± SEM of three independent experiments, each in duplicate. *P<0.05, ***P<0.001, and NS denotes not significant by one-way ANOVA followed by Dunnett’s multiple comparison test.
Fluorescence microscopy was performed by incubating the cells with HF-647-tagged Aβ(1–42) for 2 and 4 hours followed by incubation with LysoTracker Red. We observed increased punctate signal of HF-647-Aβ in both 2 and 4 hours in GFB-RA treated cells compared to DMSO control. Furthermore, the Aβ signal co-localized with the LysoTracker signal, indicating that the Aβ taken up by the cells were residing in the acidic vesicles inside the cell (late endosomes or lysosomes) (Fig. 1, D and E). Because the patterns of Aβ signal and LysoTracker signal were expected to be similar, we incubated cells separately with LysoTracker and HF-647-Aβ and tested all channels for any bleed through signals. As expected, only LysoTracker showed slight signal overlap between CY2 and CY3 channels, but there was no significant bleed through signal in any other channel for HF-647-Aβ apart from its true signal in the CY5 channel (fig. S2).
Aβ could be taken up through micropinocytosis assisted by heparan sulfate proteoglycans (HSPGs) (13). Therefore, to elucidate the mechanism of GFB-RA–mediated enhancement of Aβ uptake, we performed Aβ uptake assay in the presence of Heparin (inhibitor of HSPGs) first. Cells treated with GFB-RA in the presence of heparin showed ~40% increase in Aβ uptake compared to ~80% in those without heparin (Fig. 1F). Although, this reduction in the uptake level is statistically significant, but still there was about 40% uptake even in the presence of heparin, which indicates that other factors may also be responsible for the uptake process. We transfected the cells with Tfeb siRNA and observed slight decrease (not statistically significant, P=0.59) in the uptake level of Aβ in Tfeb siRNA-transfected cells compared to scrambled siRNA-transfected cells (Fig 1, G and H). These data further enforced the idea that neither HSPGs nor TFEB alone is responsible for GFB-RA–mediated enhanced uptake of Aβ. Reports suggest that lipoprotein receptors like LDLR and LRP1 also facilitate the internalization of Aβ in glial cells (14). Interestingly, there are reports that hepatic expression of LDLR is induced by fenofibrate (FF) via a PPARα-dependent mechanism involving Akt phosphorylation and transcriptional activation of SREBP2 (15, 16). Therefore, we further transfected cells with Ldlr siRNA and observed that the effect of GFB-RA on Aβ uptake was attenuated in the absence of LDLR (Fig. 1, G and I). Moreover, treatment with GFB-RA induced the expression of Ldlr in WT, but not in Ppara−/−, astrocytes (Fig. 1J) suggesting that GFB-RA stimulates the expression of Ldlr in astrocytes through PPARα. Together, these data indicate that GFB-RA promotes the uptake of Aβ in astrocytes via LDLR-mediated endocytosis.
GFB-RA treatment enhances the degradation of Aβ in mouse primary astrocytes.
Because we have seen colocalization of Aβ with the LysoTracker, a lysosomal dye (Fig. 1D), it is imperative that there will be degradation of Aβ inside the lysosome, provided there is proper functioning of the organelle. We wanted to observe, whether induction of TFEB (and subsequent induction of lysosomal genes and lysosomal biogenesis) could accelerate the process of degradation Aβ in the lysosome. We deployed the same in vitro assay for intracellular Aβ content, but this time, after incubation with Aβ for 4 hours, the cells were allowed to grow for various time points (from 15 min to 8 hours) in Aβ-free media. The normalized Aβ signal (normalized to Hoechst signal) for DMSO-treated cells and (GFB-RA)-treated cells were compared to their respective counterparts which were not allowed to grow in Aβ free media (termed as “0 min wash”). As expected, the basal level of lysosomal processing of Aβ caused reduction in signal intensity of intracellular Aβ by ~20% within 6 to 8 hours compared to 0’ wash cells (Fig. 2A). On the other hand, cells treated with GFB-RA showed an accelerated clearance rate, with a reduction of signal by ~40% within 6 to 8 hours (Fig. 2A). The data showed optimal degradation at 6 hours; hence that time point was used for further degradation assays (termed as “6 hour wash”). We also visualized reduced puncta of HF-647-Aβ after 6 hours of wash under the microscope (Fig. 2B). To determine whether the loss of Aβ signal was due to lysosomal processing, we incubated the cells with bafilomycin A1 (BafA1) that inhibits lysosomal acidification, thereby reducing its activity. The presence of BafA1 arrested the accelerated loss of Aβ as observed in GFB-RA treated cells, and rate of Aβ degradation was almost similar in both DMSO and GFB-RA treated cells, in presence of BafA1 (Fig. 2C). Furthermore, transfection of cells with Tfeb siRNA also attenuated the GFB-RA mediated accelerated lysosomal degradation of Aβ (Fig. 2D). Collectively, these data indicates that, GFB-RA–mediated induction of lysosomal biogenesis could accelerate the process of lysosomal Aβ degradation.
Figure 2. GFB-RA treatment enhances Aβ degradation in mouse primary astrocytes.

(A) Mouse primary astrocytes were treated for 24 hours with GFB-RA followed by incubation with 500nM FAM-tagged Aβ1–42 for 4 hours and allowed to grow in Aβ-free media for different time periods. Aβ degradation assay was performed as described in methods section. Data was represented a percentage change compared to unwashed control. (B) Mouse primary astrocytes treated with GFB-RA were incubated with 500 nM HF-Aβ1–42, washed for 4 or 6 hours, further incubated with 75 nM Lysotracker Red, and observed under microscope. Scale bar = 20μm. (C) Mouse primary astrocytes were treated with GFB and RA for 24 hours, followed by treatment with 100 nM Bafilomycin A1 for 45 min, followed by incubation with 500 nM FAM-Aβ, washed in Aβ free media for 6 hours and degradation assay was performed. Data is represented as percentage change with respect to unwashed controls. (D) Aβ degradation assay was done in mouse primary astrocytes which were either transfected with scrambled siRNA or Tfeb siRNA, prior to treatment with DMSO or GFB-RA. Data were compared to DMSO-treated, scrambled siRNA transfected controls. All data are mean ± SEM of three independent experiments run in duplicate. *P<0.05 and NS denotes not significant by one-way ANOVA followed by Dunnett’s multiple comparison test.
Role of PPARα and PPARβ in (GFB-RA)-mediated Aβ uptake and degradation:
Our previous results show that PPARα plays a key role in mediating the transcriptional activation of TFEB and subsequent enhancement in lysosomal biogenesis (12). Here, we tested whether the absence of PPARα and PPARβ affected the regulation of Aβ uptake and degradation in mouse primary astrocytes. Therefore, cells isolated from WT, Ppara−/− and Pparb−/− mice were treated with GFB-RA and further incubated with FAM-Aβ(1–42) (for in vitro assay) and HF-647-Aβ along with LysoTracker Red (for microscopy). As before, the Aβ signals were normalized to Hoechst signal to account for any variability in cell number.
The Aβ uptake assay, after 4 hours of incubation with Aβ, showed prominent increase in the Aβ content (measured by FAM-Aβ signal intensity) inside the cell, in both WT and Pparb−/−, but not in Ppara−/−, astrocytes (Fig. 3A). The signal intensity for all cells was compared to DMSO-treated WT controls. Although there was a slight increase in the levels of Aβ in Ppara−/− cells treated with GFB-RA (~20%), it was not significant compared to the ~80% and ~70% increase in GFB-RA–treated WT and Pparb−/− astrocytes, respectively (Fig. 3A). To assess the role of PPARs in Aβ degradation, the cells from WT and both knockout animals were treated, incubated with Aβ and further allowed to grow in Aβ-free media for 6 hours. We observed ~60% reduction in Aβ levels both in WT and Pparb−/− astrocytes, but only 30–35% loss in signal in case of Ppara−/− astrocytes (Fig. 3B).
Figure 3. Role of PPARα and PPARβ in Aβ uptake and degradation in mouse primary astrocytes.
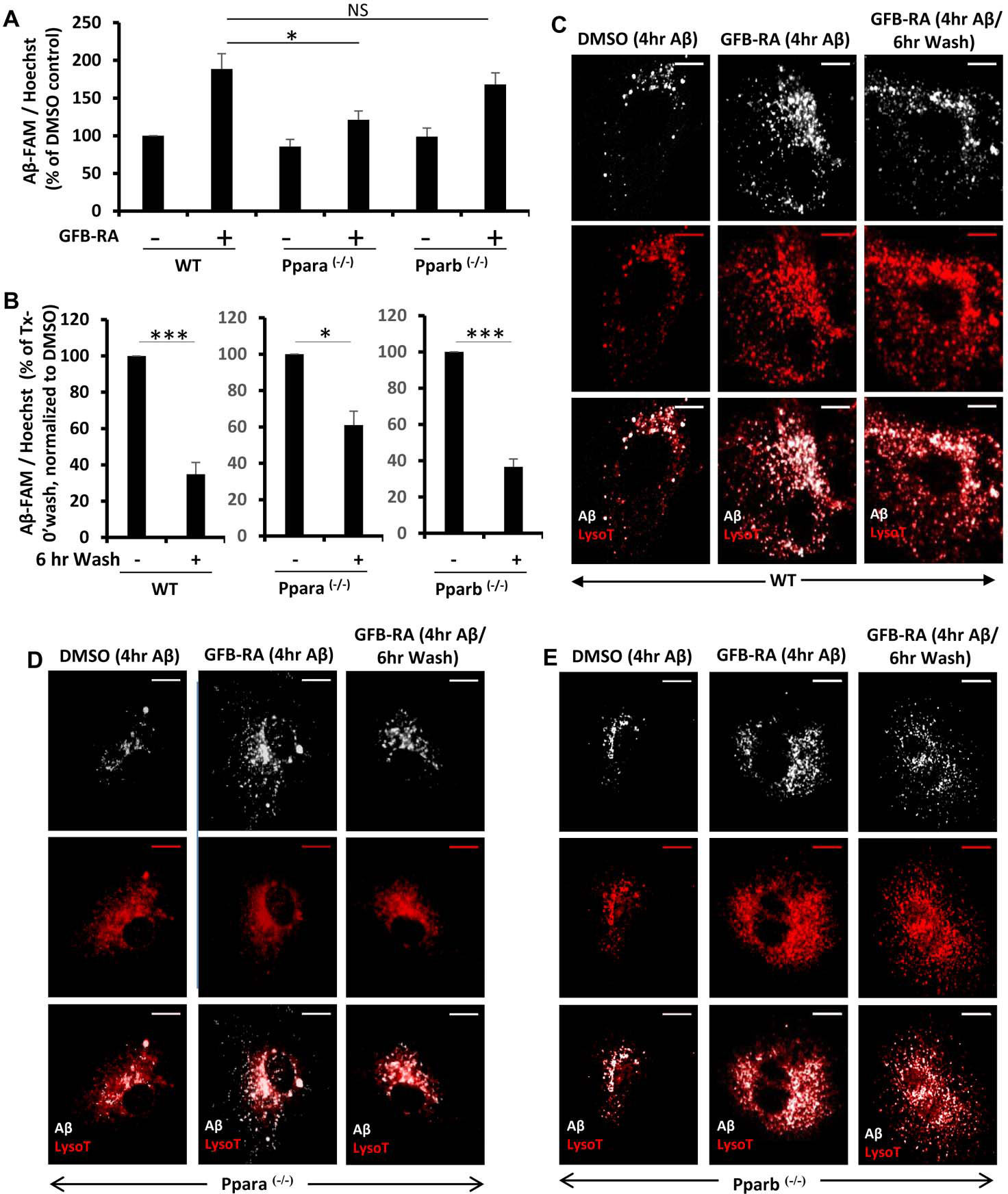
(A) Mouse primary astrocytes isolated from WT, Ppara−/− and Pparb−/− animals were treated with GFB-RA [GFB (10 μM) + RA (0.2 μM)] or DMSO, followed by incubation with 500 nM FAM-Aβ and subjected to Aβ uptake assay. Data were compared to DMSO-treated WT control and represented as percent change. (B) Mouse primary astrocytes isolated from WT, Ppara−/− and Pparb−/− animals were, treated with GFB-RA, followed by incubation with 500 nM FAM-Aβ for 4 hours, washed in Aβ free media for 6 hours, and subjected to an Aβ degradation assay. All data are representative of the mean ± SEM of three independent experiments run in duplicate. *P<0.05, ***P<0.001, and NS denotes not significant by one-way ANOVA followed by Dunnett’s multiple comparison test. (C to E) Mouse primary astrocytes isolated from WT (C), Ppara−/− (D) and Pparb−/− (E) animals were isolated, treated with DMSO, followed by incubation with 500 nM HF-647-Aβ for 4 hours and 75 nM Lysotracker for 45 min (first panel), treated with GFB-RA, followed by incubation with 500 nM HF-647-Aβ for 4 hours and 75 nM Lysotracker for 45 min (second panel), treated with GFB-RA, followed by incubation with 500 nM HF-647-Aβ for 4 hours and washed in Aβ-free media for 6 hours and incubated in 75 nM Lysotracker for 45 min (third panel) and observed under microscope. Scale bar = 20 μm. Results are representative of three independent experiments run in duplicate.
Furthermore, the observations from microscopy, also revealed reduced signal intensity of both Aβ and LysoTracker in Ppara−/− astrocytes compared to either Pparb−/− or WT astrocytes (Fig. 3, C, D and E). This is in agreement with our previous finding that the absence of PPARα abrogates the (GFB-RA)-mediated enhancement of lysosomal biogenesis (12) as well as attenuates the expression of Ldlr (Fig. 1J), a key component of Aβ uptake. Microscopic analysis also revealed reduced puncta of HF-647-Aβ in WT and Pparb−/− astrocytes after the 6-hour wash, but not a significant change in Ppara−/− astrocytes (Fig. 3, C, D, and E). Collectively, these data indicate that PPARα has a dual role - by regulating the expression of LDLR, it could facilitate the uptake Aβ and by enhancing lysosomal biogenesis via TFEB, it induces accelerated degradation of Aβ in the lysosomes.
GFB-RA treatment increases lysosomal activity and autophagic flux.
The enhancement of lysosomal degradation of Aβ led us to investigate the markers for lysosomal activity and autophagy. Cathepsin B (CtsB) and cathepsin D (CtsD) are two important cathepsins involved in the degradation of Aβ fragments in the lysosomes. Our data indicates an increase in the activity both the cathepsins upon treatment with GFB-RA (fig. S3, A and B). The protein levels of both cathepsins were also found to increase by about 2 to 3-fold in astrocytes treated with GFB-RA (fig. S3, C and D). However, silencing of TFEB by siRNA abrogated the effect of GFB-RA on cathepsins (fig. S3, A and B). This is in accordance with the findings that CtsB and CtsD are direct targets of TFEB (17) and enhancement of TFEB activity subsequently induces the levels and activity of cathepsins as well.
It has been reported that deficiency in autophagy or blockage of autophagic pathway, results in abnormal accumulation of Aβ in autophagic vacuoles inside the cell and that this unwanted buildup of Aβ is one of the main causes for Aβ-induced neurotoxicity (8). Therefore, we observed the changes in autophagic flux in GFB-RA–treated cells by monitoring the levels of LC3 (LC3-I/LC3-II) and p62/SQSTM1. GFB-RA treatment increased the levels of LC3-II, the phosphatidylethanolamine conjugated form of LC3-I (fig. S3, E and F). The conversion of LC3-I to LC3-II is a hallmark of autophagy induction. We further blocked lysosomal activity by using BafA1 and observed further accumulation of LC3-II (fig. S3, E and F). In accordance of previous studies, we also observed reduced levels of p62 in conditions where there is accumulation of LC3-II, further enforcing the enhancement of autophagic flux (fig. S3, E and F).
Oral administration of GFB-RA induces lysosomal biogenesis in vivo in the hippocampus of 5XFAD mouse model of AD.
Next, we investigated the status of lysosomal biogenesis and autophagy in the hippocampus of 5XFAD mice and examined if oral GFB-RA treatment could normalize and/or upregulate these processes in the brain of 5XFAD mice. Western blot analysis of hippocampal homogenates (Fig. 4A) followed by densitometric scanning (Fig. 4, B to G) showed significant decrease in TFEB (Fig. 4B), LAMP2 (Fig. 4C) and TPP1 (Fig. 4D) in the hippocampus of 5XFAD mice as compared to non-Tg mice. Immunofluorescence analysis (Fig. 4H–I) followed by cell counting (Fig. 4, J and K) also indicated decrease in TFEB (Fig. 4H & J) and LAMP2 (Fig. 4I & K) in the hippocampus of 5XFAD mice as compared to non-Tg mice. In contrast, we noticed significant upregulation of p62 (Fig. 4E), LC3-I (Fig. 4F) and LC3-II (Fig. 4G) in the hippocampus of 5XFAD mice in comparison with non-Tg mice. Together, these results indicate downregulation of lysosomal biogenesis and autophagy in the hippocampus of 5XFAD mice. However, GFB-RA treatment significantly increased the protein levels of TFEB (Fig. 4, A and B), LAMP2 (Fig. 4, A and C) and TPP1 (Fig. 4, A and D) in the hippocampus of 5XFAD mice. The increase of TFEB (Fig. 4, H and J) and LAMP2 (Fig. 4, I and K) by GFB-RA treatment in the hippocampus of 5XFAD mice was also confirmed by immunofluorescence analysis. It was found that GFB-RA treatment increased the level of TFEB and LAMP2 in both astrocytes (Fig. 4, H to K) and neurons (fig. S4, A to D) in the hippocampus of 5XFAD mice. On the other hand, GFB-RA treatment decreased the levels of p62 (Fig. 4E), LC3-I (Fig. 4F) and LC3-II (Fig. 4G) in the hippocampus of 5XFAD mice. These results suggest that oral GFB-RA treatment is capable of increasing lysosomal biogenesis and autophagy in the hippocampus of 5XFAD mice.
Figure 4. Oral administration of the combination of gemfibrozil and retinoic acid (GFB-RA) activates lysosomal biogenesis in vivo in the hippocampus of 5XFAD mice.
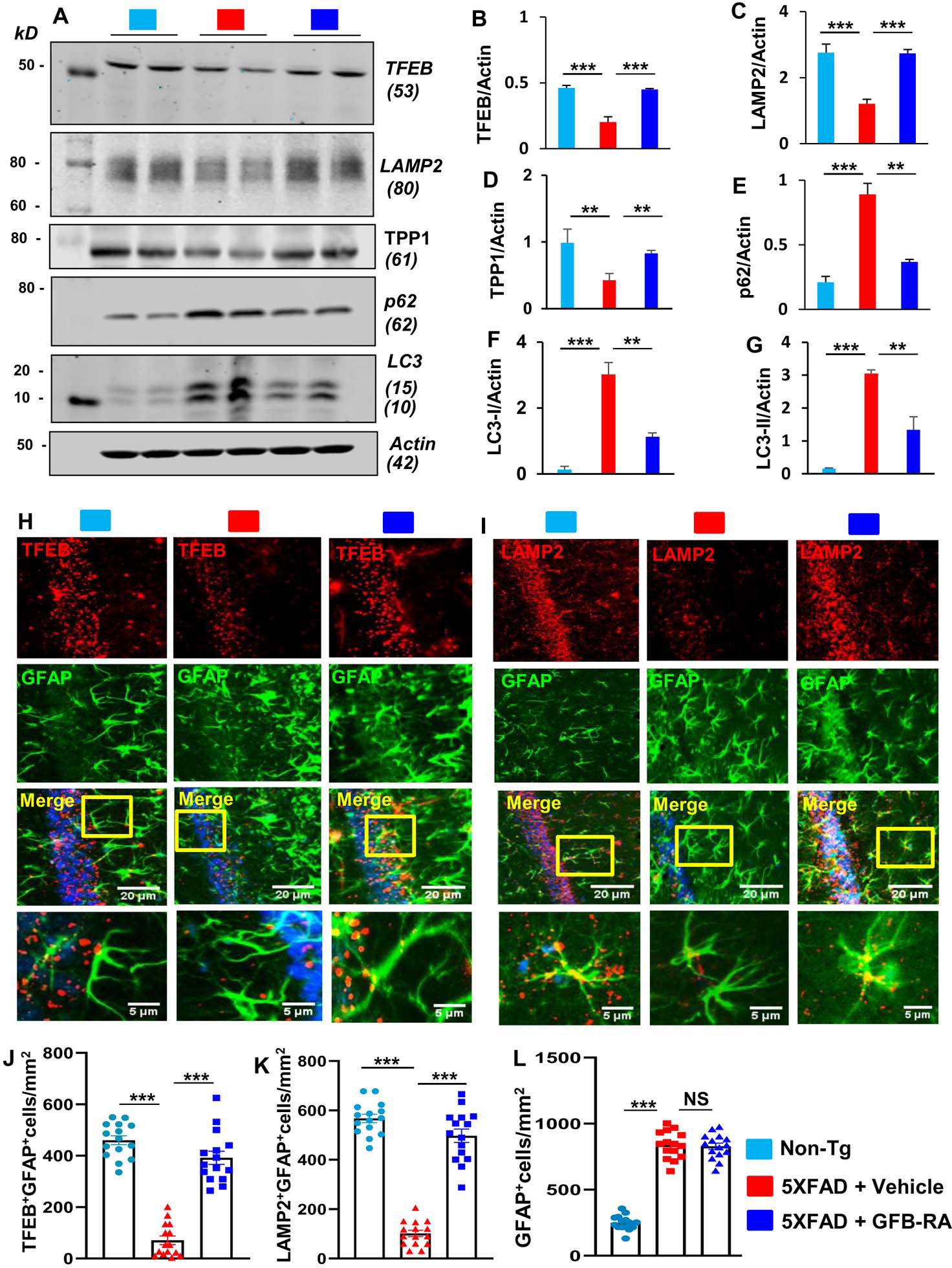
(A to G) Six-month-old 5XFAD mice (n=5/group in two independent experiments) were orally administered with GFB-RA [the combination of gemfibrozil (8 mg/Kg/day) and retinoic acid (150 IU/day)] for two months. The control group of 5XFAD mice received 0.5% methylcellulose as vehicle. Label color code: bottom right. Hippocampal protein homogenates from these mice were then subjected to Western blotting for protein expression of TFEB, LAMP2, TPP1, p62, and LC3 (A). Densitometry analysis of each (B to G) was measured with ImageJ, normalized to that of actin, and presented as relative to control. Data are mean ± SEM of five mice per group. (H to L) Hippocampal sections of mice described in (A) were double-labeled for either TFEB and GFAP (H) or LAMP2 and GFAP (I). Label color code: bottom right; scale bars as marked. TFEB+GFAP+ (J), LAMP2+GFAP+ (K) and GFAP+ (L) cells were counted in one section (3 images per section) of each of five mice per group. *P<0.05, **P<0.01, ***P<0.001, and NS denotes not significant by one-way ANOVA followed by Tukey’s multiple comparison test.
Although GFB-RA upregulated both TFEB and LAMP2 in astrocytes, GFB-RA treatment remained unable to modulate the number of astrocytes in the hippocampus of 5XFAD mice (Fig. 4L). This is important because astroglial activation plays an important role in the pathogenesis of different neurodegenerative disorders including AD. The 5XFAD mice also exhibit glial inflammation marked by activated glial cells surrounding plaques starting at 2 months that increases further with age and plaque deposition (18). Therefore, to monitor astroglial inflammation, next, we checked the expression of a proinflammatory marker, iNOS in the hippocampus by double labeling iNOS and GFAP. As described before (19, 20), marked increase in iNOS was seen in the hippocampus of 5XFAD mice as compared to non-Tg mice (fig. S5, A and B). However, (GFB-RA)-treated 5XFAD mice had decreased expression of iNOS in hippocampus relative to the vehicle group (fig. S5, A and B). In contrast, GFAP immunoreactivity did not change with GFB-RA treatment (Fig. 4L and fig. S5, A and B).
GRB-RA treatment reduces amyloid beta (Aβ) plaque load in the hippocampus of 5XFAD mice.
The hippocampus is the most affected region of the brain in 5XFAD mouse model of AD. These mice harboring five familial mutations linked to AD in APP and PS1 exhibit amyloid deposition starting from two months of age (18). Since astrocytes in the CNS are the main cell types to express and secrete insulin-degrading enzyme (IDE), one of the major proteases for the degradation of Aβ, we examined the effect of GFB-RA on the status of IDE in hippocampal astrocytes of 5XFAD mice. As expected, we observed marked loss of IDE in hippocampus of 5XFAD mice as compared to non-Tg mice (fig. S6, A and B). However, GFB-RA treatment increased the level of IDE in hippocampal astrocytes of 5XFAD mice (Fig. S6A–B). Future studies may be carried out to find out whether cultured astrocytes could release IDE in response to GFB-RA for the degradation of Aβ. In addition to increasing IDE, since GFB-RA treatment also increased lysosomal biogenesis and autophagy in the hippocampus of 5XFAD mice, we evaluated the cerebral plaque status by double labeling hippocampal sections with Aβ-specific monoclonal antibody 6E10 and thioflavin-S (Thio-S), a classic amyloid-binding dye that detects the β-pleated sheet of the amyloid plaques. As expected, we found marked increase in Thio-S positive and Aβ-immunoreactive plaques throughout the cortex and hippocampus of 5XFAD mice as comparted to non-Tg mice (Fig. 5A). However, oral GFB-RA treatment markedly reduced the plaque load in 5XFAD mice (Fig. 5A). Counting of 82e1+ and 82e1+Thio-S+ plaques (Fig. 5B) as well as quantification of Thio-S staining (Fig. 5, C to F) also revealed a lower number of plaques (Fig. 5B) and Thio-S puncta (Fig. 5C), a decline in plaque size (Fig. 5D), a decrease in Thio-S positive area (Fig. 5E), and reduction in Thio-S fluorescence along the perimeter (Fig. 5F) in the hippocampus of GFB-RA–treated 5XFAD mice relative to the vehicle-treated 5XFAD mice. To further confirm, we also performed immunoblot analysis with antibodies 82e1 (Fig. 5, G to J) and 6e10 (Fig. 5, K to N) and found remarkable increase in the Aβ levels (Fig. 5, G, H, K, and L) in the hippocampal homogenates from 5XFAD mice compared with non-Tg mice. However, a significant reduction in Aβ levels was seen in the hippocampus of (GFB-RA)-treated 5XFAD mice when compared to vehicle treated 5XFAD mice (Fig. 5, G, H, K, and L). In contrast, GFB-RA treatment remained unable to alter the level of either C-terminal fragments (CTFs) (Fig. 5, I and M) or amyloid precursor protein (APP) (Fig. 5, J and N), indicating that GFB-RA does not modulate the formation of Aβ plaques in the hippocampus of 5XFAD mice. Together, these results demonstrate that oral feeding of GFB-RA decreases Aβ plaque load in the hippocampus of 5XFAD mice.
Figure 5. Oral administration of GFB-RA decreases amyloid beta (Aβ) plaque pathology in 5XFAD mouse model of AD.
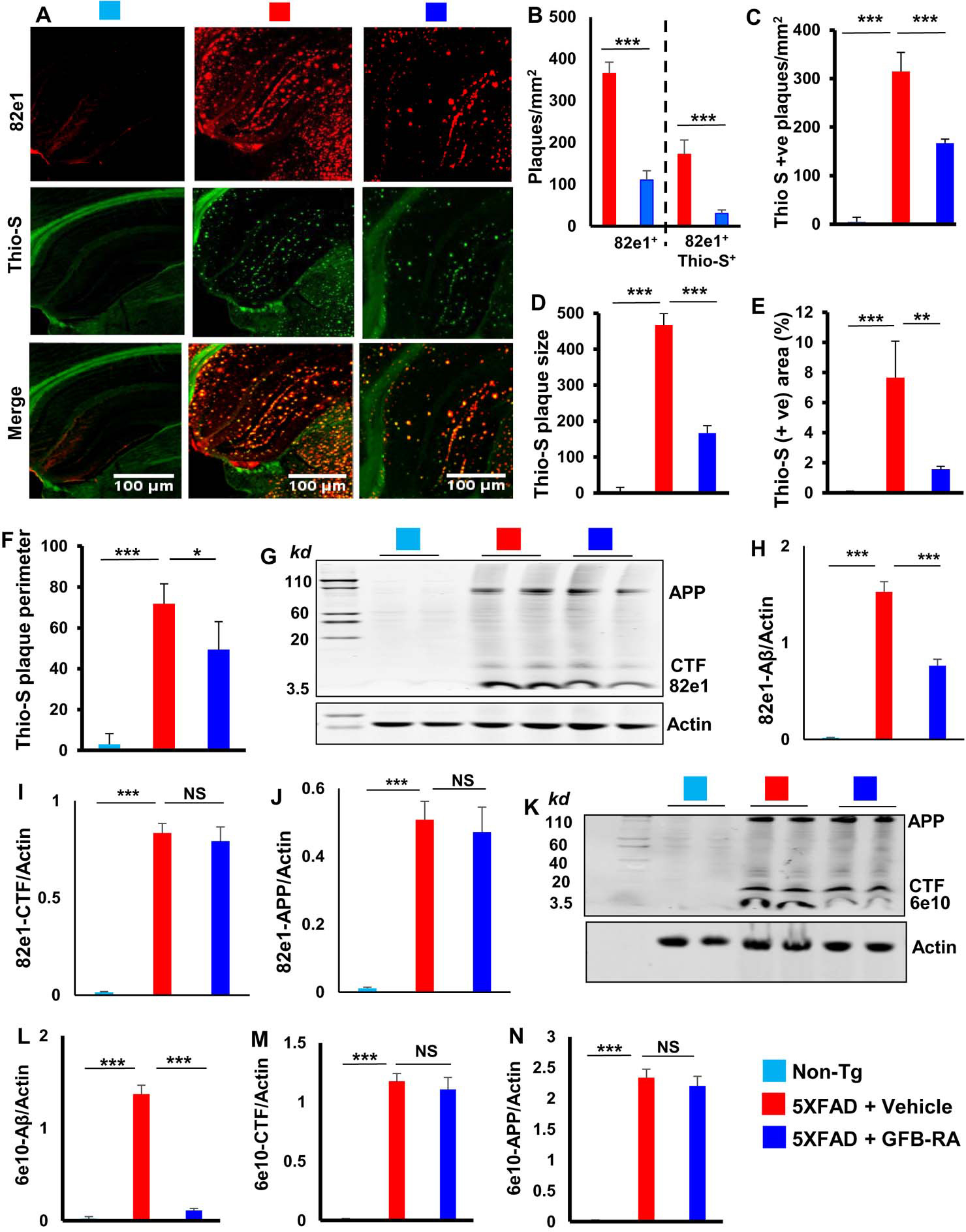
(A to F) Six-month old 5XFAD mice (n=5/group in two independent experiments) were orally administered with GFB-RA [the combination of gemfibrozil (8 mg/Kg/day) and retinoic acid (150 IU/day)] or vehicle (0.5% methylcellulose) for two months. Label color code: bottom right. Hippocampal sections from these mice were immunohistochemically assessed for Aβ plaques using 82E1 antibody (red) and counterstained with Thioflavin-S (Thio-S; green) followed by counting of 82e+ and 82e+Thio-S+ plaques in one section (3 images per section) of each of five mice per group (A and B; scale bars as marked). Quantification of Thio-S+ Aβ plaques were further evaluated for (C) Thio-S+ plaque counts, (D) average plaque size, (E) total area fraction representing Thio-S+ area as percentage of total area of cortex and hippocampus, and (F) perimeter of Thio-S+ plaques. Quantification was done using imageJ. (G to N) Hippocampal homogenates from mice described in (A) were immunoblotted with 82e1 (G) and 6e10 (K) 6e10 antibodies. Actin was used as loading control. Densitometric quantifications of 82e1-Aβ (H), 82e1-CTF (I), 82e1-APP (J), 6e10-Aβ (L), 6e10-CTF (M), and 6e10-APP (N) and normalized to actin. Data are mean ± SEM of 5 mice per group. *P<0.05, **P<0.01, ***P<0.001, and NS denotes not significant by one-way ANOVA followed by Tukey’s multiple comparison test.
GFB-RA treatment improves cognitive deficits in 5XFAD mice.
The final goal of neuroprotection in cognitive disorders like AD is to protect and/or improve memory. Since the major functions of the hippocampus are to generate and organize long-term memory and spatial learning, we examined whether oral GFB-RA protected memory and learning in 5XFAD mice. Expectedly, 5XFAD mice took much longer to find the reward hole (Fig. 6A) with greater latency (Fig. 6B) and higher errors (Fig. 6C) in the Barnes maze in comparison with non-Tg mice. However, oral GFB-RA markedly enhanced the performance of 5XFAD mice on Barnes maze (Fig. 6, A to C). Similarly, on the T-maze, a context-dependent hippocampal behavior test, 5XFAD mice also had fewer positive turns (Fig. 6D) and a higher number of negative turns (Fig. 6E) than non-Tg mice. However, upon oral GFB-RA treatment, 5XFAD mice exhibited significant improvement in number of successful positive turns with fewer errors (Fig. 6, D and E).
Figure 6. Oral administration of GFB-RA increases spatial learning and memory in 5XFAD mice.

(A to E) Six-month-old 5XFAD mice (n=5/group in two independent experiments) were orally administered with GFB-RA, the combination of gemfibrozil (8mg/Kg/day) and retinoic acid (150 IU/day). The control group received 0.5% methylcellulose as vehicle. Label color code: bottom right. After 2 months of treatment, spatial learning and memory was examined by the Barnes maze [(A), representative heat maps; (B), latency to goal box; and (C), number of errors made prior to reaching goal box] and the T maze [(D), positive turns; and (E), negative turns]. Data are mean ± SEM of five mice per group. ***P<0.001 by one-way ANOVA followed by Tukey’s multiple comparison test.
GFB-RA reduces Aβ plaque load in the hippocampus of 5XFAD mice via astrocytic PPARα:
Given that GFB-RA increased the uptake and degradation of Aβ in astrocytes in a manner dependent on PPARα, we investigated whether GFB-RA–mediated reduction in plaque burden in 5XFAD mice was dependent on PPARα specifically in astrocytes. Therefore, using Cre-Lox system, we generated 5XFAD-PparaΔAstro mice, 5XFAD mice lacking PPARα in astrocytes (fig. S7A). To validate PPARα knockdown from astrocytes, DG sections from non-Tg, 5XFAD and 5XFAD-PparaΔAstro mice were double-labeled for PPARα and GFAP. Although there was significant decrease in the expression of PPARα in both astrocyte and non-astrocyte cell populations in the hippocampus of 5XFAD mice as compared to non-Tg mice, we observed a complete absence of astrocytic PPARα in 5XFAD-PparaΔAstro mice (fig. S7, B and C).
Because GFB-RA induced the expression of LDLR in astrocytes via PPARα (Fig. 1J) to mediate the uptake of Aβ (Fig. 1G), we examined whether GFB-RA also upregulated LDLR in vivo in the hippocampus in a manner dependent on astroglial PPARα. While in non-Tg mice, LDLR was expressed by neurons as evident from the widespread presence of LDLR in hippocampal track (fig. S8, A to B), in 5XFAD mice, LDLR was mainly expressed by astrocytes (fig. S8, A and B). Treatment of 5XFAD mice with GFB-RA led to the restoration of LDLR abundance in hippocampal track and a marked increase in LDLR abundance in astrocytes (fig. S8, A and B). However, GFB-RA remained unable to upregulate LDLR expression in astrocytes in the hippocampus of 5XFAD-PparaΔAstro mice (fig. S8, A and B). Given that GFB-RA treatment increased TFEB (Fig. 4J) and LAMP2 (Fig. 4K) in astrocytes without modulating the number of astrocytes (Fig. 4L), we examined whether GFB-RA was associated with the switching of astrocytic properties in the hippocampus of 5XFAD mice. Whereas C3-expressing “A1” reactive astrocytes generate various molecules that are destructive to neurons and myelin, S100A10-expressing “A2” astrocytes are protective of neurons and reduce tissue damage in the CNS (21). As expected, greater number of A1 astrocytes (fig. S9, A and C) and very few A2 astrocytes (fig. S9, B and D) were seen in the hippocampus of 5XFAD mice as compared to non-Tg mice. However, this trend was reversed by oral GFB-RA treatment (fig. S9, A to D). On the other hand, GFB-RA remained unable to shift A1 astrocytes to A2 ones in the hippocampus of 5XFAD-PparaΔAstro mice (fig. S9, A to D). These results suggest that GFB-RA switches astroglial reactive state from A1 to A2 and upregulates astroglial LDLR in the hippocampus of 5XFAD mice in a manner dependent on astroglial PPARα.
Next, to examine the role of astrocytic PPARα in lysosomal biogenesis, we monitored the levels of LAMP2 and TFEB in hippocampal homogenates of (GFB-RA)-treated and untreated 5XFAD and 5XFAD-PparaΔAstro mice. Our results indicate that (GFB-RA)-treated 5XFAD mice exhibited significantly higher levels of LAMP2 (Fig. 7, A and B) and TFEB (Fig. 7, C and D) as compared to vehicle treatment. However, GFB-RA treatment remained unable to upregulate the level of LAMP2 (Fig. 7, A and B) and TFEB (Fig. 7, C and D) in the hippocampus of 5XFAD-PparaΔAstro mice, indicating an important role of astroglial PPARα in (GFB-RA)-mediated lysosomal biogenesis in the hippocampus of 5XFAD mice.
Figure 7. GFB-RA treatment mitigates Aβ plaque load in 5XFAD mice via astrocytic PPARα.
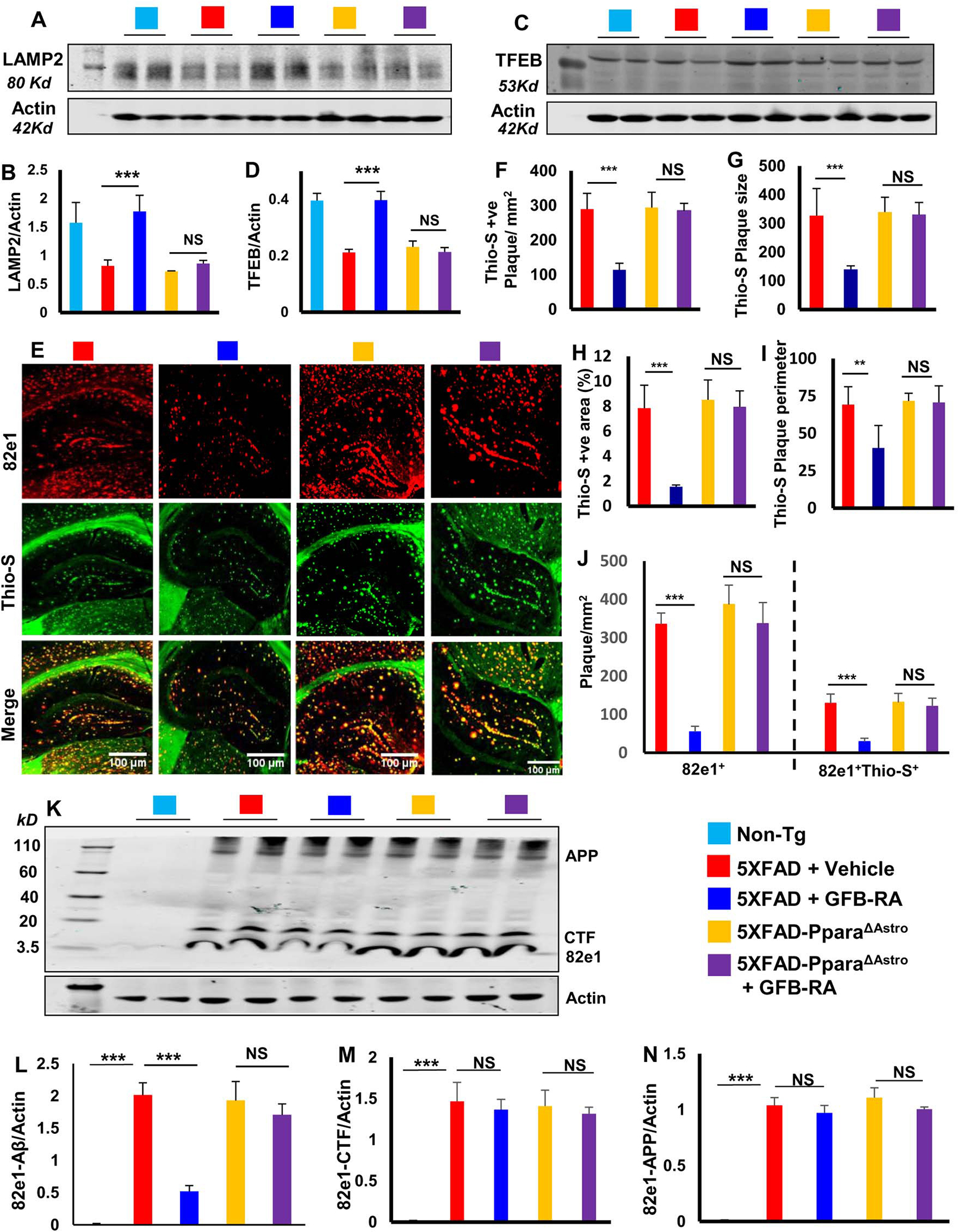
(A to D) Immunoblotting and analysis of TFEB and LAMP2 protein levels in hippocampal homogenates from 6-month-old 5XFAD (n=6/group in two independent experiments) and 5XFAD-PparaΔAstro mice (n=7/group in two independent experiments) orally administered with GFB-RA (8 mg/Kg/day gemfibrozil and 150 IU/day retinoic acid) or vehicle (0.5% methylcellulose) for two months. Label color code: bottom right. (E to J) Imaging of Thio-S (green) and 82e1 (red) staining on hippocampal sections from all groups described in (A to D), analyzed for density (F), size (G), area (as percentage of total area in cortex and hippocampus; H) and perimeter (I) of Thio-S-positive plaques, as well as (J) number of 82e+ and 82e+Thio-S+ plaques in one section (3 images per section) of each of five mice per group. Scale bars as marked. (K to N) Immunoblotting and analysis of Aβ plaques in the above-described tissues using 82e1, quantified relative to actin. Data are mean ± SEM of five mice per group. *P<0.05, **P<0.01, ***P<0.001, and NS denotes not significant by one-way ANOVA followed by Tukey’s multiple comparison test.
We then assessed plaque load and found that GFB-RA treatment significantly lowered the Thio- S+ amyloid plaques in the hippocampus and cortex of 5XFAD mice but was unable to do so in 5XFAD-PparaΔAstro (Fig. 7E). Quantification of Thio-S staining (Fig. 7, F to I) and counting of 82e1+ and 82e1+Thio-S+ puncta (Fig. 7J) revealed that GFB-RA treatment reduced the number (Fig. 7, F and J), size (Fig. 7G), area (Fig. 7H), and perimeter (Fig. 7I) of plaques in 5XFAD, but not 5XFAD-PparaΔAstro, mice. Immunoblot analyses with 82e1 antibody also confirmed decrease in Aβ plaques by GFB-RA in 5XFAD, but not 5XFAD-PparaΔAstro, mice (Fig. 7, K and L). However, GFB-RA treatment did not change the level of CTF (Fig. 7M) and APP (Fig. 7N) in either 5XFAD or 5XFAD-PparaΔAstro mice. These results suggest that oral administration of GFB-RA reduces amyloid plaque in 5XFAD mice in a manner dependent on astroglial PPARα.
GFB-RA improves learning and memory in 5XFAD mice via astrocytic PPARα.
Lastly, we investigated whether the effect of GFB-RA on learning and memory in 5XFAD mice was also dependent on astrocytic PPARα. As expected, on Barnes maze (Fig. 8A), GFB-RA-treated 5XFAD mice displayed increased spatial behaviors, as indicated by latency (Fig. 8B) and errors (Fig. 8C) when compared with vehicle-treated 5XFAD mice. In contrast, GFB-RA treatment remained unable to improve the performance of 5XFAD-PparaΔAstro mice on the Barnes maze (Fig. 8, A to C). Similarly, GFB-RA treatment increased the number of positive turns (Fig. 8D) and reduced the number of errors 5XFAD, but not 5XFAD-PparaΔAstro, mice made on the T maze (Fig. 8E). These results indicate an essential role of astrocytic PPARα in GFB-RA–mediated improvement in spatial memory and learning in 5XFAD mice. We further tested these mice for novel object recognition, which is a commonly used behavioral tool for investigating various aspects of short-term memory in mice. Similar to effects seen using the Barnes maze and the T maze, GFB-RA treatment also enhanced the short-term memory of 5XFAD mice as evidenced by the preferential index (Fig. 8F). However, we did not observe any improvement in preferential index of 5XFAD-PPARαΔAstro mice upon GFB-RA treatment. Inability of GFB-RA to modulate general locomotor activities such as total distance traveled (fig. S10, A and B) and velocity (fig. S10 and C) in either 5XFAD mice or 5XFAD-PparaΔAstro mice suggests that GFB-RA–mediated improvement in cognitive performance is due to increased cognitive behavior, not any effect on general locomotor activities. Together, our results demonstrate that GFB-RA improves cognitive performance of 5XFAD mice via astrocytic PPARα.
Figure 8. GFB-RA improves memory in 5XFAD mice via astrocytic PPARα.
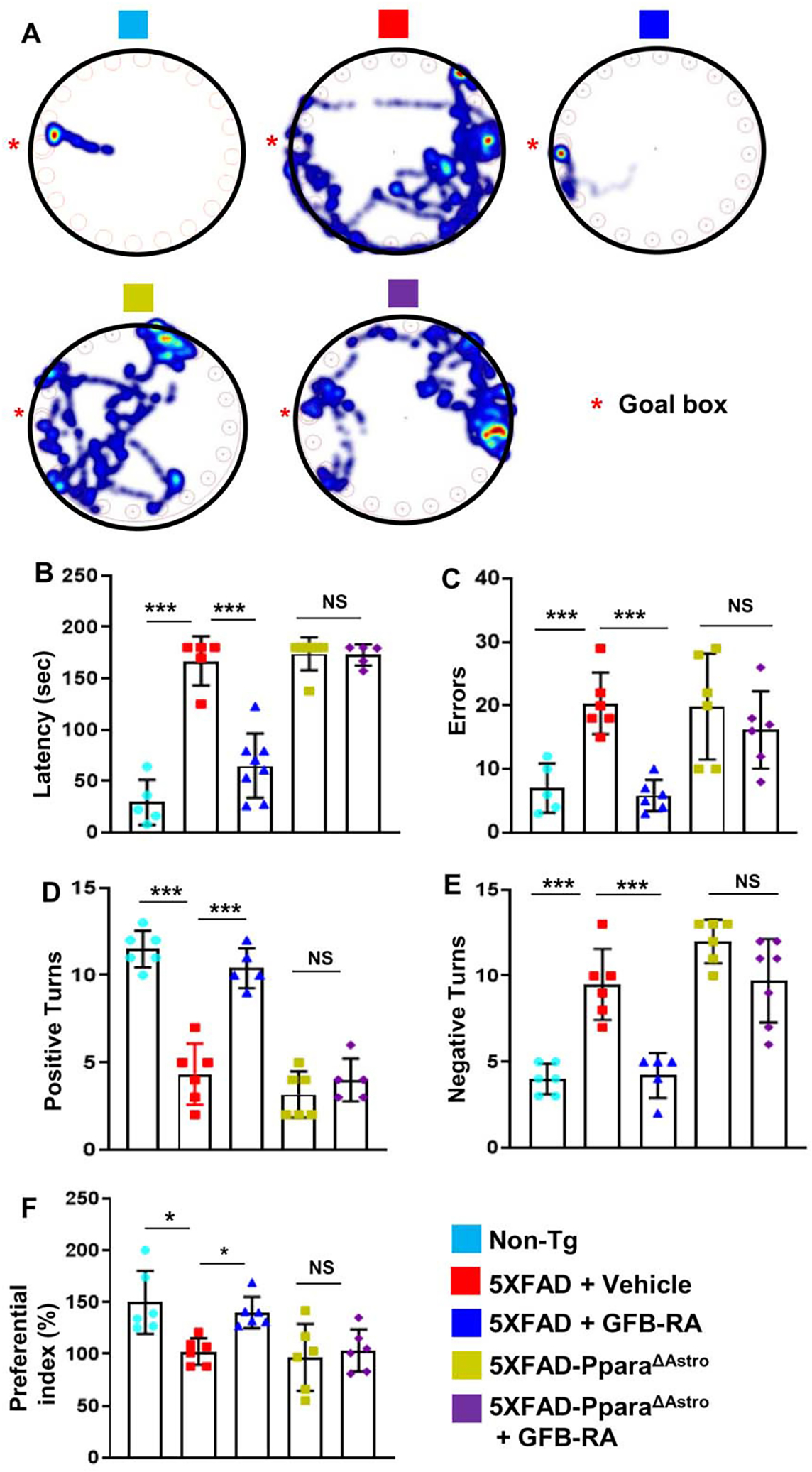
(A to F) Six-month-old 5XFAD mice (n=6/group in two independent experiments) and 5XFAD-PparaΔAstro mice (n=7/group in two independent experiments) were orally administered with GFB-RA [the combination of gemfibrozil (8 mg/Kg/day) and retinoic acid (150 IU/day)] or vehicle (0.5% methylcellulose). Label color code: bottom right. After 60 days of treatment, behavioral tests [Barnes maze (A to C), T maze (D and E), and novel object recognition (F)] were performed to evaluate memory improvement in 5XFAD mice (n=6/group) and 5XFAD-PparaΔAstro mice (n=7/group). Data are mean ± SEM of 6 to 7 mice per group. *P<0.05, ***P<0.001, and NS denotes not significant by one-way ANOVA followed by Tukey’s multiple comparison test.
Discussion
Effective clearance of Aβ from the brain parenchyma is thought to reduce the development and progression of AD. In AD brain, while neurons die, glial cells like astrocytes and microglia do not die. Although microglia are known to play an important role in Aβ clearance by a variety of phagocytic and digestive mechanisms, very few cells in the brain are microglia. On the other hand, astrocytes are the major cell type in the brain. Therefore, it makes sense to utilize astrocytes for the clearance of cerebral Aβ load. However, drugs for stimulating astroglial plaque clearance are poorly understood. Gemfibrozil is a FDA-approved lipid-lowering drug with a nice track record and retinoic acid, a vitamin A derivative, regulates the expression of a wide variety of target genes (22, 23). We have seen that a combination of gemfibrozil (GFB) and retinoic acid (RA), at low doses, induces TFEB and upregulates lysosomal biogenesis in astrocytes (12). Because lysosomes are critically involved in the metabolism of Aβ (24, 25), we examined the effect of combination of GFB and RA (GFB-RA) on the uptake and degradation of Aβ1–42 and found that it increased both in cultured astrocytes. However, at present, we do not know whether this effect is specific for Aβ or GFB-RA could stimulate the uptake and degradation of other peptides or proteins as well in astrocytes. Many drugs exhibit therapeutic effect in cell culture models, however, very few show efficacy in vivo in the brain. We previously reported that after oral administration, GFB enters the brain of mice (26). In this current study, oral administration of GFB-RA upregulated lysosomal biogenesis and autophagy in vivo in the hippocampus of 5XFAD mice, ultimately leading to reduction of plaque load and protection of memory and learning. During the course of the study, we did not observe any drugrelated side effects (such as hair loss, weight loss, diarrhea, and infections) in any of the mice used. These results suggest that oral GFB-RA may be considered to stimulate autophagy in the hippocampus, increase cerebral plaque clearance and protect memory in AD.
Mechanisms for stimulating astroglial plaque clearance are poorly understood. PPARα is a transcription factor that regulates fatty acid catabolism in the liver. Although hippocampus does not catabolize fat, recently we have demonstrated that PPARα is constitutively expressed in hippocampal neurons and astrocytes (27–29). Uptake of Aβ by astrocytes is the first step in this clearance process. Interestingly, GFB-RA stimulated the uptake of Aβ in astrocytes isolated from WT and Pparb−/−, but not Ppara−/−, mice, indicating an important role of PPARα in the uptake of Aβ. Prior to this study, we reported that PPARα is capable of regulating Tfeb at the transcriptional level (12). However, siRNA knockdown of TFEB did not abrogate GFB-RA–mediated uptake of Aβ in astrocytes, ruling out a role of TFEB in this process. On the other hand, knockdown of LDLR in astrocytes attenuated (GFB-RA)-mediated enhancement of Aβ uptake, indicating the involvement of LDLR in the uptake of Aβ. Previous studies showed that overexpression of Ldlr inhibited Aβ deposition and enhanced clearance of extracellular Aβ (30). The effect could be mediated with or without the involvement of Apolipoprotein E (ApoE), one of the strongest genetic risk factors for AD (15, 16, 30). Furthermore, Ldlr overexpression has been also been shown to facilitate the rate of brain-to-blood transport of cerebral Aβ, thereby enhancing removal of pathologic Aβ from the brain (31). Moreover, when Ldlr was deleted in 5XFAD mice, there was evidence of increased amyloid beta deposition (32). Here, we have seen that GFB-RA increased the mRNA expression of Ldlr in astrocytes isolated from WT, but not Ppara−/−, mice. Together, these results suggest that GFB-RA increases the uptake of Aβ in astrocytes via PPARα – LDLR, but not PPARα – TFEB, pathway.
The co-localization of HF-647-Aβ signal and LysoTracker observed under microscope showed that internalized Aβ indeed ended up in the lysosomes. Accordingly, similar to the uptake of Aβ, we found that GFB-RA stimulated the degradation of Aβ in astrocytes isolated from WT and Pparb−/−, but not Ppara−/−, mice, suggesting a role of PPARα in the degradation of Aβ as well. Although we did not see an involvement of TFEB in the uptake of Aβ, siRNA knockdown of TFEB significantly decreased the degradation of Aβ in (GFB-RA)-treated astrocytes, indicating that GFB-RA needs TFEB, a target of PPARα, to enhance the degradation of Aβ in astrocytes. Upon activation, TFEB is known to stimulate the autophagy machinery including the cathepsins and it has been also shown that increased activity of cathepsins results in effective degradation of Aβ and reduction in Aβ plaques (33). Consistent to the upregulation of TFEB, GFB-RA increased activity and levels of both cathepsin B (CtsB) and cathepsin D (CtsD). Moreover, enhanced autophagy results in lysosomal degradation of Aβ (34). Microtubule associated protein 1 (MAP1) light chain 3 (MAP-LC3 or simply LC3) exists as a free soluble form (LC3-I), which is covalently conjugated to phosphatidylethanolamine (LC3-II) by the enzymatic action of Atg4 (35, 36). Signals leading to the induction of autophagy trigger the conversion. LC3-II remains bound to the autophagosome membrane and is essential for the de novo production of autophagic vacuole (37, 38). Monitoring the changes in the levels of LC3-I/II is considered to be a simple and effective way to monitor autophagy induction. However, mere increase in the levels of LC3-II does not necessarily indicate complete autophagy. LC3-II itself is degraded in the later stages of autophagic degradation, which makes the interpretation of LC3 immunoblot results more complex. Therefore, monitoring LC3-I/II levels both in presence of activators and inhibitors of autophagy has been proposed to be a better way to interpret the data (39, 40). Another marker for autophagy, p62, also known as sequestsome1 (SQSTM1), which delivers LC3-II to the autophagosome and majority of p62, is degraded in the early stages of autophagosome formation (39–42). The expected negative correlation of p62 and LC3 in astrocytes upon GFB-RA treatment indicates increased autophagic flux. Therefore, it appears that GFB-RA increases the degradation of Aβ in astrocytes via “PPARα – TFEB – autophagy” pathway.
TFEB overexpression led to induction in lysosomal biogenesis and eventually resulted in enhanced uptake and clearance of Aβ from the interstitial fluids by astrocytes and enhanced processing of APP by neurons thus reducing Aβ production (24, 25). While these studies underscore the importance of astrocytic clearance of Aβ in AD like conditions, drug-mediated enhancement of astroglial plaque clearance in vivo has not been described so far. Since (GFB-RA)-mediated increased uptake and degradation of Aβ in astrocytes depended on PPARα, to delineate the role of astroglial plaque clearance machinery in vivo in 5XFAD mice, we prepared 5XFAD mice that lacked PPARα in GFAP-positive astrocytes. Whereas oral GFB-RA increased lysosomal biogenesis and autophagy to clear Aβ plaques from the hippocampus and improve spatial memory and learning in 5XFAD mice, the same treatment remained unable to upregulate lysosomal biogenesis and autophagy, reduce Aβ plaques and increase spatial memory and learning in 5XFAD mice lacking PPARα in astrocytes. These results highlight an active role of PPARα-dependent plaque clearance machinery in reducing cerebral plaque load. Although being in the liver, PPARα controls fatty acid metabolism, recent studies have described that PPARα is involved in hippocampal plasticity via transcription of cAMP response element-binding protein (CREB) (27, 29) and that PPARα is also involved in non-amyloidogenic processing of APP (43). Here, we describe another new function of PPARα in stimulating astroglial uptake and degradation of Aβ.
In summary, we delineate that GFB-RA stimulates astroglial uptake of Aβ via PPARα - LDLR pathway and increases Aβ degradation in astrocytes via PPARα – TFEB pathway, thus fueling interest in understanding the crosstalk among the lipid metabolism, lysosomal biogenesis and astroglial Aβ processing. Finally, we demonstrate that oral GFB-RA switches astroglial activation state from a neurotoxic A1-type to a neuroprotective A2-type state, augments astroglial LDLR, stimulates lysosomal biogenesis and autophagy, lowers plaque load, and improve spatial memory and learning in 5XFAD mouse model of AD dependent upon astrocytic PPARα. While from academic standpoint, these results highlight the role of PPARα-dependent astroglial plaque clearance machinery, from a therapeutic angle, these results suggest that low-dose GFB-RA might be repurposed as a treatment for reducing plaque burden and improving cognition in AD patients.
Materials and Methods
Reagents:
Cell culture materials (DMEM/F-12, L-glutamine, Hank’s balanced salt solution, 0.05% trypsin, and antibiotic-antimycotic) were purchased from Mediatech. Fetal bovine serum (FBS) was obtained from Atlas Biologicals. Aspirin and all molecular biology-grade chemicals were obtained from Sigma. FAM-tagged Aβ(1–42) and HF-647-tagged Aβ(1–42) were obtained from Anaspec. Primary antibodies, their sources and concentrations used are listed in the supplement (table S1). Alexa-fluor antibodies used in immunostaining were obtained from Jackson ImmunoResearch and IR-dye-labeled reagents used for immunoblotting were from Li-Cor Biosciences.
Isolation of Primary Mouse Astroglia:
Astroglia were isolated from mixed glial cultures as described (44, 45) according to the procedure of Giulian and Baker (46). Briefly, on day 9, the mixed glial cultures were washed three times with Dulbecco’s modified Eagle’s medium/F-12 and subjected to shaking at 240 rpm for 2 h at 37°C on a rotary shaker to remove microglia. After 2 days, the shaking was repeated for 24 h for the removal of oligodendroglia and to ensure the complete removal of all non-astroglial cells. In several studies (12, 28, 47, 48), we have seen that the attached cells express glial fibrillary acidic protein (GFAP) and that these cells are 96–98% pure astrocytes. These cells were seeded onto new plates for further studies.
Quantitative Real-Time PCR:
The mRNA quantification was performed using the ABI-Prism7700 sequence detection system using SYBR Select master mix as described earlier (49, 50). The mRNA expression of the targeted genes was normalized to the level of Gapdh mRNA and data was processed by the ABI Sequence Detection System 1.6 software.
Immunoblotting:
Western blotting was conducted as described earlier (51, 52) with modifications. Briefly, cells were scraped in 1X RIPA buffer, protein was measured using Bradford reagent and sodium dodecyl sulfate (SDS) buffer was added and electrophoresed on NuPAGE® Novex® 4–12% Bis-Tris gels (Invitrogen) and proteins transferred onto a nitrocellulose membrane (Bio-Rad) using the Thermo-Pierce Fast Semi-Dry Blotter. The membrane was then washed for 15 min in TBS plus Tween 20 (TBST) and blocked for 1 hour in TBST containing BSA. Next, membranes were incubated overnight at 4°C under shaking conditions with primary antibodies listed in Table S1. The next day, membranes were washed in TBST for 1 hour, incubated in secondary antibodies (all 1:10,000; Jackson ImmunoResearch) for 1 hour at room temperature, washed for one more hour and visualized under the Odyssey® Infrared Imaging System (Li-COR, Lincoln, NE).
For hippocampal tissue, the weight of the tissue was measured followed by homogenization in RIPA buffer containing protease and phosphatase inhibitors. Homogenates were centrifuged at 15,000 rpm for 30 mins followed by measuring protein in supernatants using the Bradford method (Bio-Rad). Total protein (30μg) was mixed with 2X SDS sample buffer, boiled for 5 min and electrophoresed on SDS-Polyacrylamide gels using the Tris/Glycine/SDS PAGE buffer. All primary antibodies are listed in table S1.
Densitometric analysis:
Protein blots were analyzed using ImageJ (NIH, Bethesda, MD) and bands were normalized to their respective β-actin loading controls. Immunofluorescence quantification data are representative of the average fold change with respect to control for at least 25 different images per condition from three independent set of experiments.
Amyloid beta uptake assay:
Mouse primary astrocytes were plated in black 96-well plates. After appropriate treatment, the wells were incubated at 37°C with 500nM FAM-tagged Aβ(1–42) for appropriate time-points. Finally, the Aβ-containing medium was removed and wells were gently washed with normal media, followed by quenching of extracellular Aβ with 100 μl 0.2% trypan blue in PBS for 2mins. After aspiration the fluorescence was measured Ex./Em. of 485/535 in Victor X2 microplate reader (Perkin Elmer). The wells were further incubated with 100 μl of 50μg/ml Hoechst 33342 dye in PBS for 30 mins and fluorescence was measured Ex./Em. of 360/465nm (24). The Aβ fluorescence was normalized to Hoechst fluorescence to account for cell number variability if any.
Amyloid beta degradation assay:
Mouse primary astrocytes were plated, treated and then incubated for 4 h with FAM-tagged Aβ(1–42). After incubation, Aβ containing media was removed and after a single gentle wash, the plates were incubated with normal media at 37°C for different time points. The measurement of Aβ and Hoechst fluorescence was measure as mentioned above. When we assayed for Aβ degradation in WT, Ppara−/− and Pparb−/− astrocytes, we calculated the degradation the following way. Because we had three different cell types that responded differentially to GFB-RA treatment in terms of Aβ uptake, for proper assessment of degradation, levels of Aβ post 6h wash were compared with the fold change in Aβ prior to wash (0’ wash), individually, for each type of GFB-RA treated cells.
1st order derivation: Aβ signal normalized to Hoechst signal = Aβnorm (for all conditions). 2nd order derivation: Aβnorm (Tx, 0’ wash) normalized to Aβnorm (DMSO, 0’ wash) = Aβfold (Tx, 0’ wash); and Aβnorm (Tx, 6h wash) normalized to Aβnorm (DMSO, 6h wash) = Aβfold (Tx, 6h wash). 3rd order derivation: {Aβfold (Tx, 6h wash) / Aβfold (Tx, 0’ wash)}* 100 = % change. This third order derivation of the Aβ signal allowed us to compare between the net reduction in Aβ content in the cell compared to the net uptake of Aβ by the same cells prior to wash.
Immunocytochemistry for Aβ uptake/degradation:
Mouse primary astrocytes were cultured on square coverslips placed in 6 well plates. After treatment cells were incubated with 500nM of HF-647-tagged Aβ(1–42). For degradation study, the cells were further allowed to grow in normal media, after removal of Aβ containing media. After incubation, cells were further incubated in media containing 75nM LysoTracker Red DND99 for 30mins. The cells were then washed, fixed on glass slides and observed under BX41 fluorescence microscope (24).
Cathepsin assay:
Mouse primary astrocytes were cultured, treated and lysed in 100 mm sodium acetate, pH 5.5, with 2.5 mm EDTA, 0.01% Triton X-100, and 2.5 mm DTT. For Cathepsin B assay, the supernatant was incubated for 30mins at pH 6.0 with 100 μm Z-Arg-Arg-AMC. 7-amino-4-methylcoumarin, AMC was used as standard. The fluorescence was measured at Ex./Em. of 355/460nm in Victor X2 microplate reader. For Cathepsin D assay, the supernatant was incubated 10 μm substrate 7-methoxycoumarin-4-acetyl-(Mca)-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys-2,4 nitrophenyl (Dnp)-d-Arg-NH2 at pH 4.0 for 30 min. Mca-Pro-Leu-OH was used as standard. The fluorescence was measured at Ex./Em. of 320/420 nm. The fluorescence readings of the samples were compared to the respective standard to measure the amount of product obtained. Cathepsin activity (in Units) was calculated per mg of cell extract, considering 1 unit of enzyme activity released 1 nmole of product per hour at 37°C (53, 54).
Animals and oral GFB-RA treatment:
Animal maintenance and experiments were conducted according to NIH guidelines that were approved by Rush University Medical Center Institutional Animal Care and Use Committee (IACUC). B6SJL-Tg (APPSwFlLon, PSEN1*M146L*L286V) 6799 Vas/J) transgenic (5XFAD) mice were obtained from Jackson Laboratories (Bar Harbor, ME). To prepare 5XFAD mice lacking PPARα in astrocytes (5XFAD-PparaΔAstro), Gfap-cre mice were crossed with Ppara-flox mice to make PparaΔAstro mice. Then PparaΔAstro mice were crossed with 5XFAD mice to generate 5XFAD-PparaΔAstro mice. Six-month old male and female 5XFAD and 5XFAD-PparaΔAstro mice were orally administered with a combination of gemfibrozil (GFB) (8mg/kg bodyweight/day) and retinoic acid (RA) (150 IU) or vehicle (0.5% methylcellulose) via oral gavage for 60 days. Non-transgenic mice from the same background were used as a control.
Immunohistochemistry:
For immunohistochemistry, hemi brains incubated in 30% sucrose were washed thoroughly in PBS cryo-sectioned using a sliding microtome (American opticals 860) as described (20, 28, 43). Prior to staining, 40μm free floating hippocampal sections were washed thoroughly in PBS. The sections were blocked using 2% BSA in PBSTT (PBS+Triton X-100+Tween-20) for 1h. Next, the sections were incubated with primary antibody in 1% PBSTT at 4°C overnight. The following day, sections were washed in PBSTT and incubated with 488 or 647- conjugated secondary antibody (Jackson ImmunoResearch Laboratories) for 3 h at room temperature. Following washes in PBSTT, the sections were mounted on glass sides (12, 55). The samples were visualized under Olympus BX41 fluorescence microscope.
For thioflavin-S (Thio-S) and 82E1 amyloid colabeling, following primary and secondary antibody incubation for 82E1 antibody, sections were incubated in 0.05% Thio-S for 5 min. Next, the sections were washed in 50% ethanol and PBS followed by mounting on a glass slide (56). The samples were visualized under Olympus BX41 fluorescence microscope. All primary antibodies used are described in table S1.
Counting of Aβ plaques:
Amyloid plaques in hippocampus and cortex were scored blinded to the experimental conditions using the touch counting module of the Olympus Microsuite 5™ imaging software. Briefly, captured images were opened in the infinity image viewer window and the area of the entire image was measured by drawing a rectangular object around the image. After that, plaques were counted by touch counting. Both area of the image and counted signals were exported to excel and calculated as a unit of number of signals per square millimeter area. Plaque area, size and perimeter were calculated using NIH’s ImageJ software (56).
Barnes maze and T maze:
Maze experiments were performed as described by us (27, 57, 58). Briefly, for the Barnes maze, mice were trained for 2 consecutive days followed by examination on day 3. During training, the overnight food-deprived mouse was placed in the middle of the maze in a 10 cm high cylindrical start chamber. After 10 s, the start chamber was removed to allow the mouse to move around the maze to find out the color food chips in the baited tunnel. The session was ended when the mouse entered the baited tunnel. The tunnel was always located underneath the same hole (stable within the spatial environment). After each training session, maze and escape tunnel were thoroughly cleaned with a mild detergent to avoid instinctive odor avoidance due to mouse’s odor from the familiar object. On day 3, a video camera (Basler Gen I Cam - Basler acA 1300–60) connected to a Noldus computer system was placed above the maze and was illuminated with high wattage light that generated enough light and heat to motivate animals to enter into the escape tunnel. The performance was monitored by the video tracking system (Noldus System). Cognitive parameters were analyzed by measuring latency (duration before all four paws were on the floor of the escape box) and errors (incorrect responses before all four paws were on the floor of the escape box).
For the T-maze, mice were also habituated in the T-maze for two days under food-deprived conditions so that animals can eat food rewards at least five times during 10 minutes period of training. During each trial, mice were placed in the start point for 30 s and then forced to make a right arm turn which was always baited with color food chips. On entering the right arm, they were allowed to stay there for 30–45 s, then returned to the start point, held for 30 s and then allowed to make right turn again. As described above, after each training session, T-maze was thoroughly cleaned with a mild detergent. On day 3, mice were tested for making positive turns and negative turns. The reward side is always associated with a visual cue. The number of times the animal eats the food reward would be considered as a positive turn.
Novel object recognition task:
Novel object recognition task was performed to monitor the short-term memory as described by others (59) and us (27). Briefly, during training, mice were placed in the NORT testing apparatus comprised of a wooden floor square arena of 40 cm long and 40 cm wide, with walls 30 cm high. Two plastic toys (between 6 and 7 cm) that varied in color, shape, and texture were placed in specific locations in the environment 30 cm away from each other. The mice were able to explore freely the environment and objects for 15 min and then were placed back into their individual home cages. After 30 min, mice were placed back into the environment with two objects in the same locations, but now one of the familiar objects was replaced with a third novel object. The mice were then again allowed to explore freely both objects for 15 min. The objects were thoroughly cleaned with a mild detergent. A video camera (Basler Gen I Cam - Basler acA 1300–60) connected to a Noldus computer system was placed above the box. Each mouse was placed individually on the center of the NORT arena and the performance was monitored by the live video tracking system (Noldus System).
Open field test:
The Open field tests were performed as described earlier (60, 61) with slight modifications and used to assess spontaneous exploratory activity. Briefly, each mouse was allowed to freely explore an open field arena for 5 min. The testing apparatus was a classic open field (i.e., a wooden floor square arena, 40 × 40 cm, with walls 30 cm high). A video camera (Basler Gen I Cam - Basler acA 1300–60) connected to a Noldus computer system was placed above the box. Each mouse was placed individually on the center of the arena and the performance was monitored by the live video tracking system (Noldus System). Exploratory parameters in OFT that were analyzed include total distance traveled by the mice during 5 min of free exploration in the open field arena and the velocity with which this distance was covered.
Statistics:
Statistical analyses were performed using Graph Pad Prism 9.0.0.121. Data sets were analyzed by one-way ANOVA followed by Dunnett’s and Tukey’s multiple comparison tests or student’s t-test as described previously (20, 43). Data represented as mean ± SD or mean ± SEM from three independent experiments. A P level < 0.05 was considered statistically significant: * P < 0.05, ** P < 0.01 and *** P < 0.001.
Study ethics approval:
Mice were maintained and experiments conducted in accordance with National Institute of Health guidelines and were approved (protocol # 18–045) by the Rush University Medical Center Institutional Animal Care and Use Committee.
Supplementary Material
Funding:
This study was funded by the Zenith Fellows Award (ZEN-17– 438829) from Alzheimer’s Association and a grant from NIH (AG050431) to KP. Moreover, Dr. Pahan is the recipient of a Research Career Scientist Award (1IK6 BX004982) from the Department of Veterans Affairs.
Footnotes
Competing interests: KP and AG are inventors on patent application (20200297657) held/submitted by Rush University Medical Center that covers the stimulation of astroglial plaque clearance. The other authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes:
- 1.Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK, Co-occurrence of Alzheimer’s disease ss-amyloid and tau pathologies at synapses. Neurobiol Aging 31, 1145–1152; published online EpubJul (S0197–4580(08)00277–7 [pii] 10.1016/j.neurobiolaging.2008.07.021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li M, Chen L, Lee DH, Yu LC, Zhang Y, The role of intracellular amyloid beta in Alzheimer’s disease. Prog Neurobiol 83, 131–139 (2007); published online EpubOct (S0301–0082(07)00157–8 [pii] 10.1016/j.pneurobio.2007.08.002). [DOI] [PubMed] [Google Scholar]
- 3.Citron M, Teplow DB, Selkoe DJ, Generation of amyloid beta protein from its precursor is sequence specific. Neuron 14, 661–670 (1995); published online EpubMar (0896–6273(95)90323–2 [pii]). [DOI] [PubMed] [Google Scholar]
- 4.Okochi M, Eimer S, Bottcher A, Baumeister R, Romig H, Walter J, Capell A, Steiner H, Haass C, A loss of function mutant of the presenilin homologue SEL-12 undergoes aberrant endoproteolysis in Caenorhabditis elegans and increases abeta 42 generation in human cells. J Biol Chem 275, 40925–40932 (2000); published online EpubDec 29 ( 10.1074/jbc.M005254200M005254200 [pii]). [DOI] [PubMed] [Google Scholar]
- 5.Capell A, Steiner H, Willem M, Kaiser H, Meyer C, Walter J, Lammich S, Multhaup G, Haass C, Maturation and pro-peptide cleavage of beta-secretase. J Biol Chem 275, 30849–30854 (2000); published online EpubOct 6 ( 10.1074/jbc.M003202200M003202200 [pii]). [DOI] [PubMed] [Google Scholar]
- 6.Vetrivel KS, Zhang YW, Xu H, Thinakaran G, Pathological and physiological functions of presenilins. Mol Neurodegener 1, 4 (2006)1750–1326-1–4 [pii] 10.1186/1750-1326-1-4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vetrivel KS, Thinakaran G, Amyloidogenic processing of beta-amyloid precursor protein in intracellular compartments. Neurology 66, S69–73 (2006); published online EpubJan 24 (66/1_suppl_1/S69 [pii] 10.1212/01.wnl.0000192107.17175.39). [DOI] [PubMed] [Google Scholar]
- 8.Nixon RA, Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 120, 4081–4091 (2007); published online EpubDec 1 (120/23/4081 [pii] 10.1242/jcs.019265). [DOI] [PubMed] [Google Scholar]
- 9.Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J, Wang X, Yu G, Esposito L, Mucke L, Gan L, Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron 51, 703–714 (2006); published online EpubSep 21 (S0896–6273(06)00597–6 [pii] 10.1016/j.neuron.2006.07.027). [DOI] [PubMed] [Google Scholar]
- 10.Zhang L, Sheng R, Qin Z, The lysosome and neurodegenerative diseases. Acta Biochim Biophys Sin (Shanghai) 41, 437–445 (2009); published online EpubJun ( [DOI] [PubMed] [Google Scholar]
- 11.Ditaranto K, Tekirian TL, Yang AJ, Lysosomal membrane damage in soluble Abeta-mediated cell death in Alzheimer’s disease. Neurobiol Dis 8, 19–31 (2001); published online EpubFeb ( 10.1006/nbdi.2000.0364S0969-9961(00)90364-4 [pii]). [DOI] [PubMed] [Google Scholar]
- 12.Ghosh A, Jana M, Modi K, Gonzalez FJ, Sims KB, Berry-Kravis E, Pahan K, Activation of peroxisome proliferator-activated receptor alpha induces lysosomal biogenesis in brain cells: implications for lysosomal storage disorders. J Biol Chem 290, 10309–10324 (2015); published online EpubApr 17 (M114.610659 [pii] 10.1074/jbc.M114.610659). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Horssen J, Wesseling P, van den Heuvel LP, de Waal RM, Verbeek MM, Heparan sulphate proteoglycans in Alzheimer’s disease and amyloid-related disorders. Lancet Neurol 2, 482–492 (2003); published online EpubAug (S1474442203004848 [pii]). [DOI] [PubMed] [Google Scholar]
- 14.Basak JM, Verghese PB, Yoon H, Kim J, Holtzman DM, Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Abeta uptake and degradation by astrocytes. J Biol Chem 287, 13959–13971 (2012); published online EpubApr 20 ( 10.1074/jbc.M111.288746). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basak JM, Verghese PB, Yoon H, Kim J, Holtzman DM, Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Abeta uptake and degradation by astrocytes. J Biol Chem 287, 13959–13971; published online EpubApr 20 (M111.288746 [pii] 10.1074/jbc.M111.288746). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Basak JM, Kim J, Pyatkivskyy Y, Wildsmith KR, Jiang H, Parsadanian M, Patterson BW, Bateman RJ, Holtzman DM, Measurement of apolipoprotein E and amyloid beta clearance rates in the mouse brain using bolus stable isotope labeling. Mol Neurodegener 7, 14 (2012)#N/A [pii] 10.1186/1750-1326-7-14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carey KL, Paulus GLC, Wang L, Balce DR, Luo JW, Bergman P, Ferder IC, Kong L, Renaud N, Singh S, Kost-Alimova M, Nyfeler B, Lassen KG, Virgin HW, Xavier RJ, TFEB Transcriptional Responses Reveal Negative Feedback by BHLHE40 and BHLHE41. Cell Rep 33, 108371 (2020); published online EpubNov 10 ( 10.1016/j.celrep.2020.108371). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R, Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26, 10129–10140 (2006); published online EpubOct 4 ( 10.1523/JNEUROSCI.1202-06.2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Modi KK, Roy A, Brahmachari S, Rangasamy SB, Pahan K, Cinnamon and Its Metabolite Sodium Benzoate Attenuate the Activation of p21rac and Protect Memory and Learning in an Animal Model of Alzheimer’s Disease. PLoS One 10, e0130398 (2015) 10.1371/journal.pone.0130398). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rangasamy SB, Corbett GT, Roy A, Modi KK, Bennett DA, Mufson EJ, Ghosh S, Pahan K, Intranasal Delivery of NEMO-Binding Domain Peptide Prevents Memory Loss in a Mouse Model of Alzheimer’s Disease. J Alzheimers Dis 47, 385–402 (2015)JAD150040 [pii] 10.3233/JAD-150040). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhauser C, Volterra A, Carmignoto G, Agarwal A, Allen NJ, Araque A, Barbeito L, Barzilai A, Bergles DE, Bonvento G, Butt AM, Chen WT, Cohen-Salmon M, Cunningham C, Deneen B, De Strooper B, Diaz-Castro B, Farina C, Freeman M, Gallo V, Goldman JE, Goldman SA, Gotz M, Gutierrez A, Haydon PG, Heiland DH, Hol EM, Holt MG, Iino M, Kastanenka KV, Kettenmann H, Khakh BS, Koizumi S, Lee CJ, Liddelow SA, MacVicar BA, Magistretti P, Messing A, Mishra A, Molofsky AV, Murai KK, Norris CM, Okada S, Oliet SHR, Oliveira JF, Panatier A, Parpura V, Pekna M, Pekny M, Pellerin L, Perea G, Perez-Nievas BG, Pfrieger FW, Poskanzer KE, Quintana FJ, Ransohoff RM, Riquelme-Perez M, Robel S, Rose CR, Rothstein JD, Rouach N, Rowitch DH, Semyanov A, Sirko S, Sontheimer H, Swanson RA, Vitorica J, Wanner IB, Wood LB, Wu J, Zheng B, Zimmer ER, Zorec R, Sofroniew MV, Verkhratsky A, Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci 24, 312–325 (2021); published online EpubMar ( 10.1038/s41593-020-00783-4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim K, Kleinman HK, Lee HJ, Pahan K, Safety and potential efficacy of gemfibrozil as a supportive treatment for children with late infantile neuronal ceroid lipofuscinosis and other lipid storage disorders. Orphanet J Rare Dis 12, 113 (2017); published online EpubJun 17 ( 10.1186/s13023-017-0663-8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy A, Pahan K, Gemfibrozil, stretching arms beyond lipid lowering. Immunopharmacol Immunotoxicol 31, 339–351 (2009) 10.1080/08923970902785253). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Burchett JM, Schuler DR, Cirrito JR, Diwan A, Lee JM, Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J Neurosci 34, 9607–9620; published online EpubJul 16 (34/29/9607 [pii] 10.1523/JNEUROSCI.3788-13.2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Tripoli DL, Czerniewski L, Ballabio A, Cirrito JR, Diwan A, Lee JM, Neuronal-Targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing Abeta Generation and Amyloid Plaque Pathogenesis. J Neurosci 35, 12137–12151; published online EpubSep 2 (35/35/12137 [pii] 10.1523/JNEUROSCI.0705-15.2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dasgupta S, Roy A, Jana M, Hartley DM, Pahan K, Gemfibrozil ameliorates relapsing-remitting experimental autoimmune encephalomyelitis independent of peroxisome proliferator-activated receptor-alpha. Mol Pharmacol 72, 934–946 (2007); published online EpubOct (mol.106.033787 [pii] 10.1124/mol.106.033787). [DOI] [PubMed] [Google Scholar]
- 27.Roy A, Jana M, Corbett GT, Ramaswamy S, Kordower JH, Gonzalez FJ, Pahan K, Regulation of cyclic AMP response element binding and hippocampal plasticity-related genes by peroxisome proliferator-activated receptor alpha. Cell Rep 4, 724–737 (2013); published online EpubAug 29 (S2211–1247(13)00390–2 [pii] 10.1016/j.celrep.2013.07.028). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy A, Jana M, Kundu M, Corbett GT, Rangaswamy SB, Mishra RK, Luan CH, Gonzalez FJ, Pahan K, HMG-CoA Reductase Inhibitors Bind to PPARalpha to Upregulate Neurotrophin Expression in the Brain and Improve Memory in Mice. Cell Metab 22, 253–265 (2015); published online EpubAug 4 (S1550–4131(15)00265-X [pii] 10.1016/j.cmet.2015.05.022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roy A, Kundu M, Jana M, Mishra RK, Yung Y, Luan CH, Gonzalez FJ, Pahan K, Identification and characterization of PPARalpha ligands in the hippocampus. Nat Chem Biol 12, 1075–1083 (2016); published online EpubOct 17 (nchembio.2204 [pii] 10.1038/nchembio.2204). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM, Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron 64, 632–644 (2009); published online EpubDec 10 (S0896–6273(09)00896–4 [pii] 10.1016/j.neuron.2009.11.013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T, Paoletti AC, Kasper TR, DeMattos RB, Zlokovic BV, Holtzman DM, Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Abeta clearance in a mouse model of beta-amyloidosis. Proc Natl Acad Sci U S A 109, 15502–15507; published online EpubSep 18 (1206446109 [pii] 10.1073/pnas.1206446109). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katsouri L, Georgopoulos S, Lack of LDL receptor enhances amyloid deposition and decreases glial response in an Alzheimer’s disease mouse model. PLoS One 6, e21880 10.1371/journal.pone.0021880PONE-D-11-05741 [pii]). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bahr BA, Abai B, Gall CM, Vanderklish PW, Hoffman KB, Lynch G, Induction of beta-amyloid-containing polypeptides in hippocampus: evidence for a concomitant loss of synaptic proteins and interactions with an excitotoxin. Exp Neurol 129, 81–94 (1994); published online EpubSep (S0014–4886(84)71149–6 [pii] 10.1006/exnr.1994.1149). [DOI] [PubMed] [Google Scholar]
- 34.Nixon RA, Yang DS, Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb Perspect Biol 4, cshperspect.a008839 [pii] 10.1101/cshperspect.a008839). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanida I, Ueno T, Kominami E, Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem 279, 47704–47710 (2004); published online EpubNov 12 ( 10.1074/jbc.M407016200M407016200 [pii]). [DOI] [PubMed] [Google Scholar]
- 36.Tanida I, Ueno T, Kominami E, LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 36, 2503–2518 (2004); published online EpubDec ( 10.1016/j.biocel.2004.05.009S1357272504002110 [pii]). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scherz-Shouval R, Elazar Z, ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 17, 422–427 (2007); published online EpubSep (S0962–8924(07)00168–7 [pii] 10.1016/j.tcb.2007.07.009). [DOI] [PubMed] [Google Scholar]
- 38.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z, Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26, 1749–1760 (2007); published online EpubApr 4 (7601623 [pii] 10.1038/sj.emboj.7601623). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanida I, Yamaji T, Ueno T, Ishiura S, Kominami E, Hanada K, Consideration about negative controls for LC3 and expression vectors for four colored fluorescent protein-LC3 negative controls. Autophagy 4, 131–134 (2008); published online EpubJan (5233 [pii]). [DOI] [PubMed] [Google Scholar]
- 40.Mizushima N, Yoshimori T, How to interpret LC3 immunoblotting. Autophagy 3, 542–545 (2007); published online EpubNov-Dec (4600 [pii]). [DOI] [PubMed] [Google Scholar]
- 41.Tanida I, Ueno T, Kominami E, LC3 and Autophagy. Methods Mol Biol 445, 77–88 (2008) 10.1007/978-1-59745-157-4_4). [DOI] [PubMed] [Google Scholar]
- 42.Kuma A, Matsui M, Mizushima N, LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 3, 323–328 (2007); published online EpubJul-Aug (4012 [pii]). [DOI] [PubMed] [Google Scholar]
- 43.Corbett GT, Gonzalez FJ, Pahan K, Activation of peroxisome proliferator-activated receptor alpha stimulates ADAM10-mediated proteolysis of APP. Proc Natl Acad Sci U S A 112, 8445–8450 (2015); published online EpubJul 7 (1504890112 [pii] 10.1073/pnas.1504890112). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brahmachari S, Pahan K, Sodium benzoate, a food additive and a metabolite of cinnamon, modifies T cells at multiple steps and inhibits adoptive transfer of experimental allergic encephalomyelitis. J Immunol 179, 275–283 (2007); published online EpubJul 1 (179/1/275 [pii]). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saha RN, Pahan K, Differential regulation of Mn-superoxide dismutase in neurons and astroglia by HIV-1 gp120: Implications for HIV-associated dementia. Free Radic Biol Med 42, 1866–1878 (2007); published online EpubJun 15 (S0891–5849(07)00217–1 [pii] 10.1016/j.freeradbiomed.2007.03.022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giulian D, Baker TJ, Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci 6, 2163–2178 (1986); published online EpubAug ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brahmachari S, Fung YK, Pahan K, Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J Neurosci 26, 4930–4939 (2006); published online EpubMay 3 (26/18/4930 [pii] 10.1523/JNEUROSCI.5480-05.2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jana M, Jana A, Pal U, Pahan K, A simplified method for isolating highly purified neurons, oligodendrocytes, astrocytes, and microglia from the same human fetal brain tissue. Neurochem Res 32, 2015–2022 (2007); published online EpubDec ( 10.1007/s11064-007-9340-y). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kundu M, Roy A, Pahan K, Selective neutralization of IL-12 p40 monomer induces death in prostate cancer cells via IL-12-IFN-gamma. Proc Natl Acad Sci U S A 114, 11482–11487 (2017); published online EpubOct 24 ( 10.1073/pnas.1705536114). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mondal S, Kundu M, Jana M, Roy A, Rangasamy SB, Modi KK, Wallace J, Albalawi YA, Balabanov R, Pahan K, IL-12 p40 monomer is different from other IL-12 family members to selectively inhibit IL-12Rbeta1 internalization and suppress EAE. Proc Natl Acad Sci U S A 117, 21557–21567 (2020); published online EpubSep 1 ( 10.1073/pnas.2000653117). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corbett GT, Roy A, Pahan K, Gemfibrozil, a Lipid-Lowering Drug, Upregulates IL-1 Receptor Antagonist in Mouse Cortical Neurons: Implications for Neuronal Self-Defense. J Immunol 189, 1002–1013; published online EpubJul 15 (jimmunol.1102624 [pii] 10.4049/jimmunol.1102624). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saha RN, Liu X, Pahan K, Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. J Neuroimmune Pharmacol 1, 212–222 (2006); published online EpubSep ( 10.1007/s11481-006-9020-8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bond JS, Barrett AJ, Degradation of fructose-1,6-bisphosphate aldolase by cathepsin B. Biochem J 189, 17–25 (1980); published online EpubJul 1 ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barrett AJ, Fluorimetric assays for cathepsin B and cathepsin H with methylcoumarylamide substrates. Biochem J 187, 909–912 (1980); published online EpubJun 1 ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khasnavis S, Pahan K, Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J Neuroimmune Pharmacol 9, 569–581 (2014); published online EpubSep ( 10.1007/s11481-014-9552-2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chandra S, Pahan K, Gemfibrozil, a Lipid-Lowering Drug, Lowers Amyloid Plaque Pathology and Enhances Memory in a Mouse Model of Alzheimer’s Disease via Peroxisome Proliferator-Activated Receptor alpha. J Alzheimers Dis Rep 3, 149–168 (2019); published online EpubMay 18 ( 10.3233/ADR-190104). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Corbett GT, Roy A, Pahan K, Sodium phenylbutyrate enhances astrocytic neurotrophin synthesis via protein kinase C (PKC)-mediated activation of cAMP-response element-binding protein (CREB): implications for Alzheimer disease therapy. J Biol Chem 288, 8299–8312 (2013); published online EpubMar 22 (M112.426536 [pii] 10.1074/jbc.M112.426536). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patel D, Roy A, Kundu M, Jana M, Luan CH, Gonzalez FJ, Pahan K, Aspirin binds to PPARα to stimulate hippocampal plasticity and protect memory. Proc Natl Acad Sci U S A 115, E7408–E7417 (2018); published online Epub07 ( 10.1073/pnas.1802021115). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mansuy IM, Mayford M, Jacob B, Kandel ER, Bach ME, Restricted and regulated overexpression reveals calcineurin as a key component in the transition from short-term to long-term memory. Cell 92, 39–49 (1998); published online EpubJan ( 10.1016/s0092-8674(00)80897-1). [DOI] [PubMed] [Google Scholar]
- 60.Galeano P, Martino Adami PV, Do Carmo S, Blanco E, Rotondaro C, Capani F, Castano EM, Cuello AC, Morelli L, Longitudinal analysis of the behavioral phenotype in a novel transgenic rat model of early stages of Alzheimer’s disease. Front Behav Neurosci 8, 321 (2014) 10.3389/fnbeh.2014.00321). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen Y, Liang Z, Blanchard J, Dai CL, Sun S, Lee MH, Grundke-Iqbal I, Iqbal K, Liu F, Gong CX, A non-transgenic mouse model (icv-STZ mouse) of Alzheimer’s disease: similarities to and differences from the transgenic model (3xTg-AD mouse). Mol Neurobiol 47, 711–725 (2013); published online EpubApr ( 10.1007/s12035-012-8375-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.