

Abstract
Purpose:
Tumor cells are dependent on the glutathione and thioredoxin antioxidant pathways to survive oxidative stress. Since the essential amino acid methionine is converted to glutathione, we hypothesized that methionine restriction (MR) would deplete glutathione and render tumors dependent on the thioredoxin pathway and its rate-limiting enzyme thioredoxin reductase (TXNRD).
Methods:
Triple (ER/PR/HER2)-negative breast cancer (TNBC) cells were treated with control or MR media and the effects on reactive oxygen species (ROS) and antioxidant signaling were examined. To determine the role of TXNRD in MR-induced cell death, TXNRD1 was inhibited by RNAi or the pan-TXNRD inhibitor auranofin, an antirheumatic agent. Metastatic and PDX TNBC mouse models were utilized to evaluate in vivo antitumor activity.
Results:
MR rapidly and transiently increased ROS, depleted glutathione and decreased the ratio of reduced glutathione/oxidized glutathione in TNBC cells. TXNRD1 mRNA and protein levels were induced by MR via a ROS-dependent mechanism mediated by the transcriptional regulators NRF2 and ATF4. MR dramatically sensitized TNBC cells to TXNRD1 silencing and the TXNRD inhibitor auranofin, as determined by crystal violet staining and caspase activity; these effects were suppressed by the antioxidant N-acetylcysteine. H-Ras-transformed MCF-10A cells, but not untransformed MCF-10A cells, were highly sensitive to the combination of auranofin and MR. Furthermore, dietary MR induced TXNRD1 expression in mammary tumors and enhanced the antitumor effects of auranofin in metastatic and PDX TNBC murine models.
Conclusion:
MR exposes a vulnerability of TNBC cells to the TXNRD inhibitor auranofin by increasing expression of its molecular target and creating a dependency on the thioredoxin pathway.
Keywords: methionine, cancer, nutrition, glutathione, thioredoxin, auranofin, oxidative stress
Introduction
Reactive oxygen species (ROS) are byproducts of aerobic metabolism that are buffered by antioxidant systems to prevent damage to cellular macromolecules [1]. Tumor cells have elevated ROS levels due to oncogenic alterations that fuel their rapid growth [2]. Transformed cells are dependent on the two principal nuclear factor erythroid 2-related factor 2 (NRF2)-regulated antioxidant pathways, the glutathione and thioredoxin pathways, to maintain redox homeostasis and prevent ROS-dependent cell death. Reduced glutathione and thioredoxin are abundant thiol-containing proteins that scavenge free radicals via thiol-disulfide exchange reactions to generate oxidized disulfides, which are in turn converted to their reduced dithiols by NADPH-dependent glutathione and thioredoxin reductases (TXNRD), respectively [3, 4]. There are two TXNRD isoforms, a cytosolic (TXNRD1) and a mitochondrial (TXNRD2) isoform [4]. Tumor cells survive disruption of either single antioxidant pathway, but combined inhibition of both the glutathione and thioredoxin pathways is synthetic lethal [5–8]. Deletion of the glutathione reductase gene in lung tumors exposes a synthetic lethal vulnerability to the pan-TXNRD inhibitor auranofin, an FDA-approved drug for Rheumatoid arthritis [8, 9]. Moreover, pharmacologic inhibition of glutathione biosynthesis with buthionine sulfoximine (BSO) in combination with auranofin resulted in ROS-dependent, synergistic cell death across a broad spectrum of cancer cell lines [6]. These studies suggest that the synergistic effects of combined glutathione and thioredoxin inhibition can be exploited therapeutically.
As an alternative to pharmacologic inhibition, dietary restriction of the essential amino acid methionine mimics BSO by depleting cellular glutathione levels and decreasing the reduced glutathione (GSH)/ oxidized glutathione (GSSG) ratio, thereby inducing ROS transiently [10–15]. These effects of methionine restriction (MR) reflect its conversion in the methionine cycle to homocysteine, which is subsequently transsulfurated to cysteine, a critical substrate for glutathione biosynthesis [16]. MR activates NRF2, which restores redox homeostasis by inducing expression of antioxidant genes such as glutathione S-transferase and TXNRD1 [12, 17]. Additionally, many tumors are dependent on methionine for cell growth and survival, and MR has emerged as a promising metabolic therapy in cancer [10, 14, 18–21]. We postulated that glutathione depletion by MR would render tumor cells dependent on the thioredoxin pathway by NRF2-dependent induction of thioredoxin reductase, thereby exposing a targetable vulnerability. Consistent with this idea, we observed in an unbiased proteomics screen that TXNRD1 protein levels were increased in TNBC cells by MR [22].
Here we report that MR rapidly and transiently increases ROS levels and depletes glutathione in TNBC cells. MR induces TXNRD1 by a ROS-dependent mechanism mediated by the master transcriptional regulators NRF2 and ATF4. Moreover, MR exposes a ROS-dependent vulnerability of TNBC cells to TXNRD1 inhibition. Human breast epithelial cells transformed by H-Ras, but not untransformed cells, are highly sensitive to the combination of auranofin and MR. Additionally, dietary MR induced TXNRD1 expression in mammary tumors and enhanced the antitumor effects of auranofin in metastatic and PDX TNBC models. Collectively, our findings indicate that MR exposes a druggable vulnerability of TNBC cells to TXNRD1 inhibition.
Material and Methods
Cell culture
MDA-MB-468, GILM2 and GILM2-mCherry cells were cultured as described [23, 24]. Cell lines were authenticated by STR analyses and tested for mycoplasma. MCF-10A cells expressing H-RasV12 or vector were grown as described [25]. NRF2- and ATF4-deficient mouse embryonic fibroblasts (MEFs) were kindly provided by Nobunao Wakabayashi (Fred Hutchinson Cancer Research Center) [26] and Craig Thompson (Memorial Sloan Kettering Cancer Center) [27], respectively. For MR experiments, complete medium (100 μM methionine) was formulated by supplementing RPMI 1640 with additional nutrients to closely match the original media for each cell line. MR medium (3% methionine unless otherwise specified) was formulated in the same way with reduced L-methionine (0.45 mg/L). Auranofin, BSO and N-acetylcysteine (NAC) were purchased (Sigma-Aldrich).
ROS assay
ROS levels were determined using the DCFDA Cellular ROS Assay kit (Abcam) according to the manufacturer’s instructions. Cells were seeded in 96-well plates (2.5 × 103 cells/well). The next day, DCFDA was added, the media was replaced with control or MR media, and cells were incubated for 0–72 hours. ROS levels were measured using a fluorescence microplate reader with excitation/emission at 495 nm/529 nm, corrected for background fluorescence and normalized to cell confluency using crystal violet staining of the cells cultured under the same conditions in a parallel experiment. ROS levels were expressed as the percentage of t=0.
Reduced glutathione (GSH)/oxidized glutathione (GSSG) assay
The ratio of GSH/GSSG was determined using the Quantification kit for GSSG and GSH (Sigma-Aldrich) according to the manufacturer’s protocol. Cells were seeded in 100 mm plates (5 × 106 cells/plate). The next day, the media was replaced with control or MR media, and cells were incubated for 0–72 hours. Cells (1 × 106) were lysed, and the GSSG concentration was determined by masking GSH and measuring the absorption at 412 nm after adding DNTB. The GSH concentration was determined by subtracting the GSSG concentration from the total glutathione concentration.
Real-time PCR
cDNA was synthesized from total RNA using the iScript™ cDNA Synthesis Kit (Bio-Rad) and the following primers (Integrated DNA Technologies): TXNRD1 (forward 5-GCTTTCACGTACTGGGTCCA-3, reverse 5-TGCACAGACAGGGTGGATTC-3) and GAPDH (5-GAAGGTGAAGGTCGGAGTC-3, reverse 5-GAAGATGGTGATGGGATTTC-3). PCR reactions were performed using iQ™ SYBR Green supermix (Bio-Rad) and the reaction products were detected using a CFX96 Real Time PCR system (Bio-Rad). RNA levels were normalized to control (GAPDH) RNA levels using a comparative Ct method.
Immunoblotting
Proteins were immunoblotted as described [25]. Abs against TXNRD1, TXNRD2, ATF4, p-NRF2 (all from Cell Signaling Technology) and β-actin (Sigma-Aldrich) were used.
Thioredoxin reductase assay
TXNRD activity was measured using the Thioredoxin Reductase Assay kit (Sigma-Aldrich) according to the manufacturer’s protocol. Cells were seeded in 100 mm plates (5 × 106 cells/well). The next day, the media was replaced with control or MR media, and the cells were incubated for 0–72 hours. Cells were then lysed with RIPA buffer as described [25]. Lysates were loaded into a 96-well plate (10 μg/well). TXNRD activity was measured as the NADPH-dependent reduction of DTNB to TNB by colorimetric assay at 412 nm using a TXNRD inhibitor to control for non-specific activity.
Crystal violet cell survival assay
Cells were seeded on 6-well plates (3 × 105 cells/well). The next day, the cells were washed with PBS, and complete or MR media was added. After 48 hours, cells were treated with vehicle, auranofin (1 μM) or NAC (2 mM) for an additional 24 hours. Surviving cells were stained with crystal violet as described [28, 29].
Caspase-3/7 activity assay
Caspase-3/7 activity was measured in cell lysates using the Caspase-Glo 3/7 Assay System (Promega) according to the manufacturer’s instructions. Briefly, cells were seeded in 96-well plates (2.5 × 103 cells/well). The next day, the media was replaced with control or MR media, the cells were incubated for 72 hours and treated with vehicle or auranofin (1 μM) for the final 24 hours. Caspase-3/7 activity was measured by luminescence normalized to cell confluence as described above and expressed as fold activity compared to vehicle-treated cells.
TXNRD1 siRNA experiments
ON-TARGETplus siRNAs targeting the sequences of human TXNRD1 and non-silencing control siRNA were purchased from Dharmacon/Horizon Discovery. Cells were transfected with siRNAs using Lipofectamine RNAiMAX Reagent (Invitrogen) according to the manufacturer’s instructions.
Mouse model of metastatic TNBC
GILM2-mCherry cells (1 × 106) were resuspended in Matrigel (BD Bioscience) and injected bilaterally into the ducts of the 4th mammary glands of 4–5 week-old female NOD scid IL2 receptor γ chain knockout (NSG) mice (Jackson Laboratory) as described [23]. Mice were randomized into four groups (10 mice/group): (1) control 15% protein diet (Teklad TD.01084) plus vehicle (PBS i.p. daily for two weeks), (2) control diet plus auranofin (10 mg/kg i.p. daily for two weeks), (3) an isocaloric 15% protein methionine-free diet (Teklad TD.140119) plus vehicle, or (4) an isocaloric 15% protein methionine-free diet plus auranofin (10 mg/kg i.p. daily for two weeks). Mice were placed on their diets 0.5 weeks before beginning treatment 2.5 weeks after tumor cell inoculation. The diets were continued throughout the 2-week treatment period. Mammary tumors were measured with calipers and tumor volume was calculated as described [28]. At autopsy, mCherry-fluorescent metastases were visualized in isolated whole lungs using a Leica MZ10F fluorescent stereomicroscope; tumor burden was scored using NIH ImageJ analysis as described [28]. All animal experiments were approved by the University of Wisconsin-Madison IACUC.
PDX TNBC Model
Female NSG mice with PDX TNBC model TM00098 were purchased from Jackson Laboratory. When tumors were 10 mm in diameter, they were resected, minced into 2 mm pieces and transplanted bilaterally into the 4th mammary fat pads of 4–5 week-old female NSG mice (Jackson Laboratory). Mice were randomized into the same four treatment groups (10 mice per group) as in the metastatic model. The diets were started 0.5 weeks before beginning treatment 8 weeks after tumor transplantation and were continued throughout the 3-week treatment period. Tumor volume was measured with calipers [28].
Tumor apoptosis assay
The percentage of active caspase-3-positive tumor cells was determined by immunohistochemistry using a cleaved caspase-3 Ab (Cell Signaling) as described [28].
Statistics
Statistical significance was determined by ANOVA with posttests using GraphPad Prism software.
Results
MR transiently initiates oxidative stress and induces TXNRD
Methionine regulates redox homeostasis via its conversion to homocysteine, which is subsequently transsulfurated to cysteine, a critical precursor of the antioxidant glutathione [16]. Consistent with this role, MR rapidly increased ROS levels in GILM2 and MDA-MB-468 TNBC cells within 3–6 hours (Fig. 1A and 1B). These effects were accompanied by a correspondingly rapid depletion in total glutathione levels, a decrease in the GSH/GSSG ratio and an increase in the NADP/NADPH ratio within 3–6 hours, indicating oxidative stress (Fig. 1A and 1B and Fig. S1A and S1B). These rapid redox alterations were transient. Within 24 hours of MR, ROS levels, GSH/GSSG and NADP/NADPH ratios returned to baseline. Prolonged MR reduced ROS levels below baseline and modestly increased glutathione levels consistent with prior reports [30, 31]. The increased glutathione levels may reflect increased cystine import by the glutamate-cystine antiporter xCT since both subunits of this antiporter (xCT and CD98hc) were induced by MR (Fig. S2).
Fig. 1. Methionine restriction transiently initiates oxidative stress and induces TXNRD1 and TXNRD2.
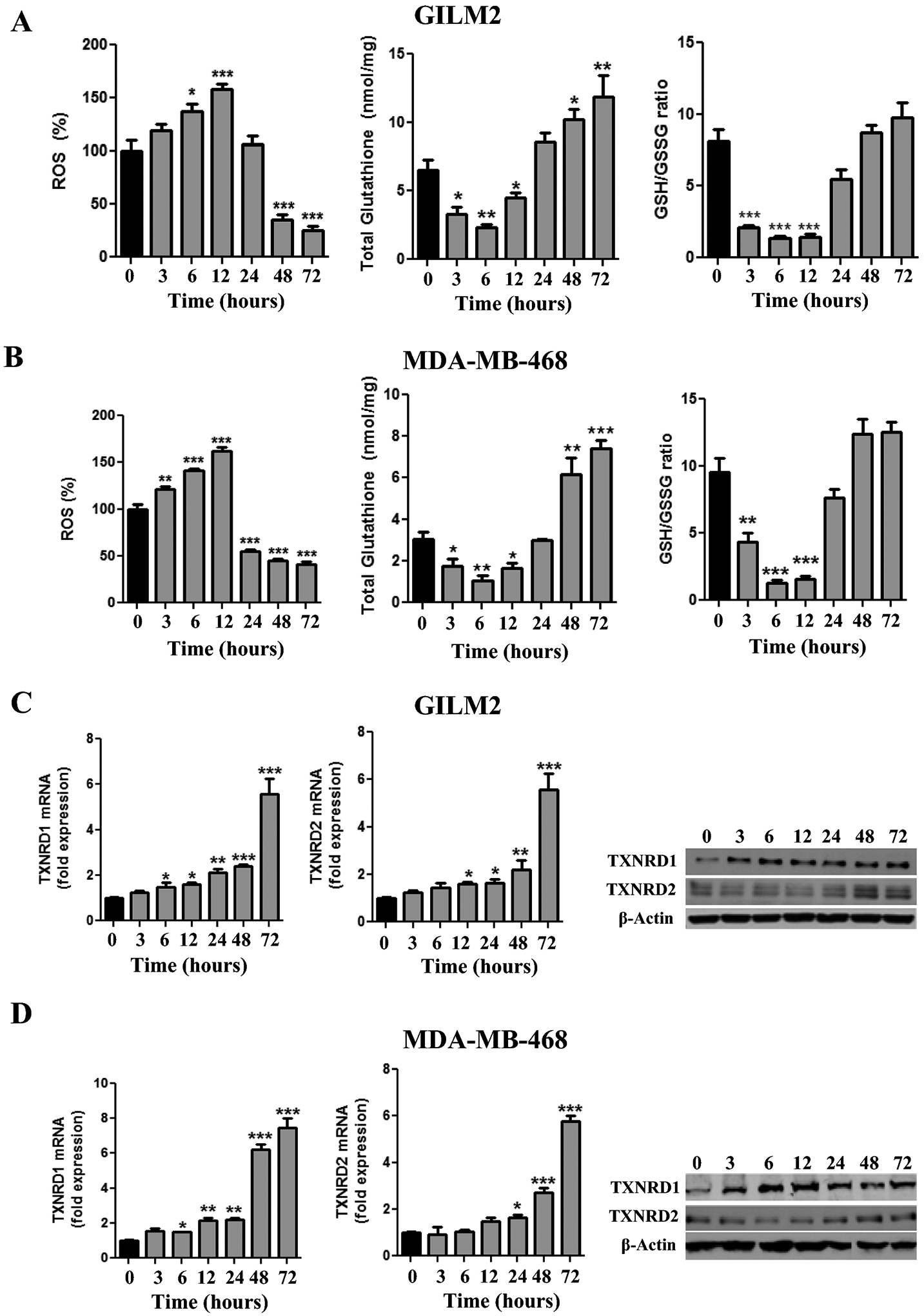
A and B, GILM2 (A) and MDA-MB-468 (B) TNBC cells were grown in control or MR media for the indicated number of hours. ROS levels (expressed as percentage at time t=0), total glutathione levels, and GSH/GSSG ratios were determined (mean ± SEM, n = 3). C and D, GILM2 (C) and MDA-MB-468 (D) were grown in control or MR media for the indicated number of hours. mRNA levels of TXNRD1 (left panel) and TXNRD2 (middle panel) in TNBC cells were determined by real-time PCR and were normalized to expression in control media (mean ± SEM, n = 3). Right Panel: Immunoblot of TXNRD1 and TXNRD2 protein levels in TNBC cells grown for the indicated number of hours in MR media. In all panels, *, P < 0.05, **, P < 0.01, ***, P < 0.001 vs. t = 0
Having previously identified TXNRD1 as an MR-regulated protein [22], we examined the effects of MR on TXNRD1 and TXNRD2 mRNA and protein levels. MR increased TXNRD1 mRNA levels within 6 hours and TXNRD2 mRNA levels within 24 hours in GILM2 and MDA-MB-468 cells, with maximal induction of both transcripts at 72 hours (Fig. 1C and 1D). TXNRD1 and TXNRD2 protein levels showed a similar increase in response to MR (Fig. 1C and 1D). These changes were accompanied by an increase in TXNRD activity (Fig. S1A and S1B). Hence, MR rapidly and transiently initiates oxidative stress and depletes glutathione, which is followed by induction of TXNRD1 and TXNRD2 expression and TXNRD activity.
MR induces TXNRD1 by a ROS-, NRF2- and ATF4-dependent mechanism
Since the induction of TXNRD1 corresponded temporally with ROS initiation by MR, we chose to focus on this TXNRD isoform. To identify the underlying mechanisms, we examined the potential role of the transcription factors NRF2 and ATF4, master regulators of the antioxidant and integrated stress response pathways, respectively [2, 32]. Pretreatment with the antioxidant NAC attenuated the induction of ATF4, p-NRF2 and TXNRD1 by MR in GILM2 and MDA-MB-468 cells, indicating that these events are ROS-dependent (Fig. 2A). Deletion of ATF4 or NRF2 in MEFs diminished the induction of TXNRD1 expression (Fig. 2B) and TXNRD activity (Fig. 2C) by MR, although basal levels of TXNRD1 expression/TXNRD activity were not suppressed. Consistent with prior reports [33, 34], deletion of NRF2 also completely inhibited ATF4 induction by MR (Fig. 2B), indicating that ATF4 induction by MR is mediated by NRF2. These findings indicate that MR induces TXNRD1/TXNRD activity by a ROS-, ATF4- and NRF2-dependent mechanism.
Fig. 2. Methionine restriction induces TXNRD1 by a ROS-, NRF2- and ATF4-dependent mechanism.
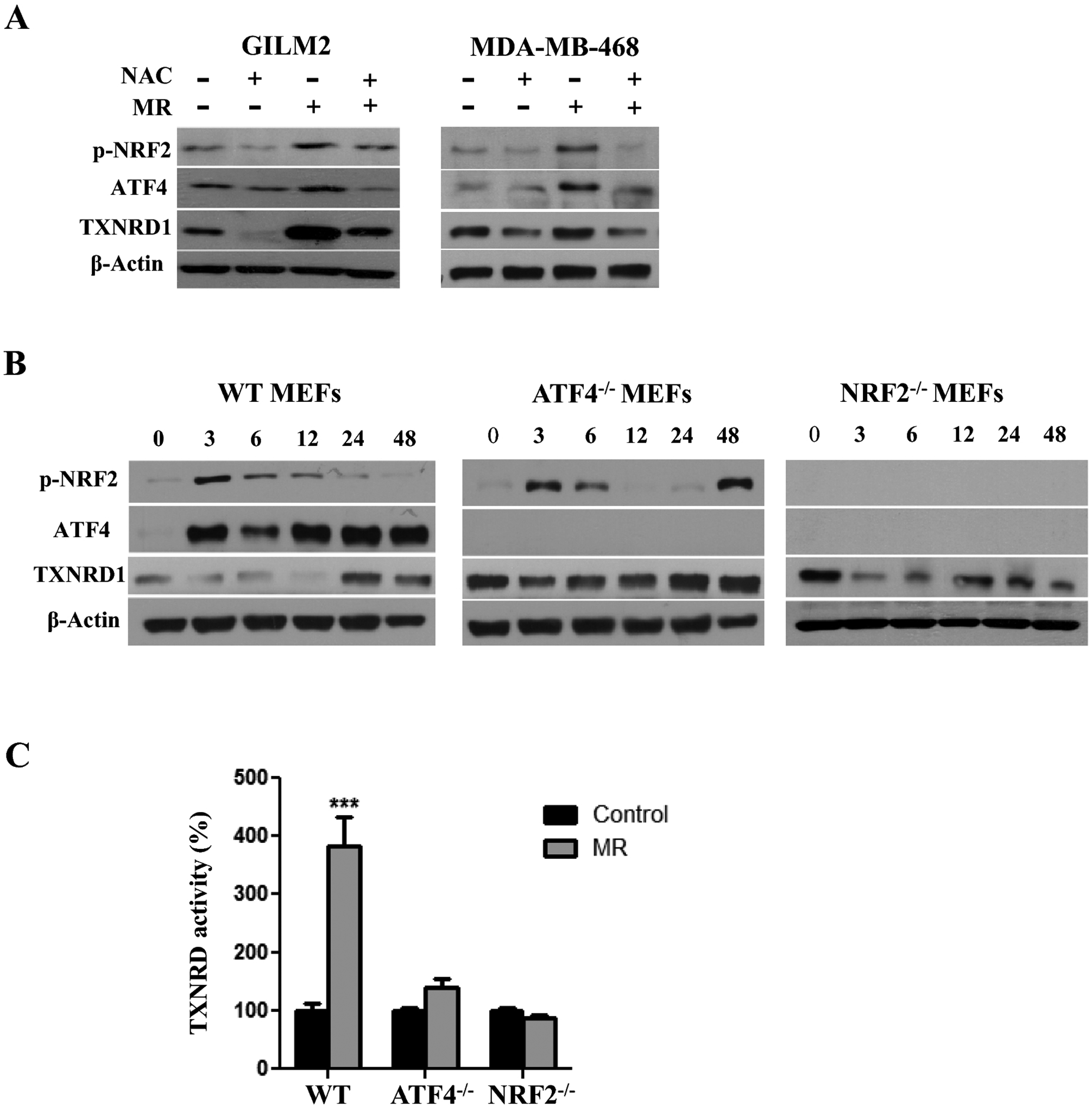
A, Immunoblot of p-NRF2, ATF4 and TXNRD1 expression in GILM2 (left) and MDA-MB-468 (right) cells cultured in control or MR media for 24 hours in the presence or absence of NAC (2 mM). B, Immunoblot of p-NRF2, ATF4 and TXNRD1 expression in WT, ATF4−/− and NRF2−/− MEFs grown in MR media for the indicated number of hours. C, TXNRD activity (expressed as percentage of time t=0) was determined in the indicated MEFs cultured in control or MR media for 48 hours (mean ± SEM, n = 3). ***, P < 0.001 vs. control
MR exposes a redox vulnerability to the TXNRD inhibitor auranofin
Given the robust induction of TXNRD activity by MR, we hypothesized that MR renders TNBC cells dependent on the thioredoxin antioxidant pathway for survival, exposing a vulnerability to the pan-TXNRD inhibitor auranofin [9]. Treatment of GILM2 and MDA-MB-468 cells with MR or auranofin alone resulted in modest or no cytotoxicity (Fig. 3A and 3B). In contrast, MR dramatically sensitized TNBC cells to auranofin, resulting in the virtual eradication of all TNBC cells (Fig. 3A and 3B). The interaction between MR and auranofin was synergistic (Fig. S3). The combination of MR and auranofin also enhanced caspase-3/7 activity much more robustly than either treatment alone (Fig. 3A and 3B, right panels). Similar results were observed with the methionine-degrading enzyme methioninase (Fig. S4). Unlike auranofin, BSO, an inhibitor of glutathione biosynthesis [35], did not enhance MR-induced cell death (Fig. S5). Moreover, the cytotoxicity of the combination of MR and auranofin was dependent on oxidative stress: NAC attenuated cell death and ROS induction in response to MR and auranofin (Fig. 3C and 3D). These results indicate that MR exposes a specific vulnerability of TNBC cells to auranofin, which together result in lethal oxidative stress.
Fig. 3. Methionine restriction sensitizes TNBC cells to auranofin by an ROS-dependent mechanism.
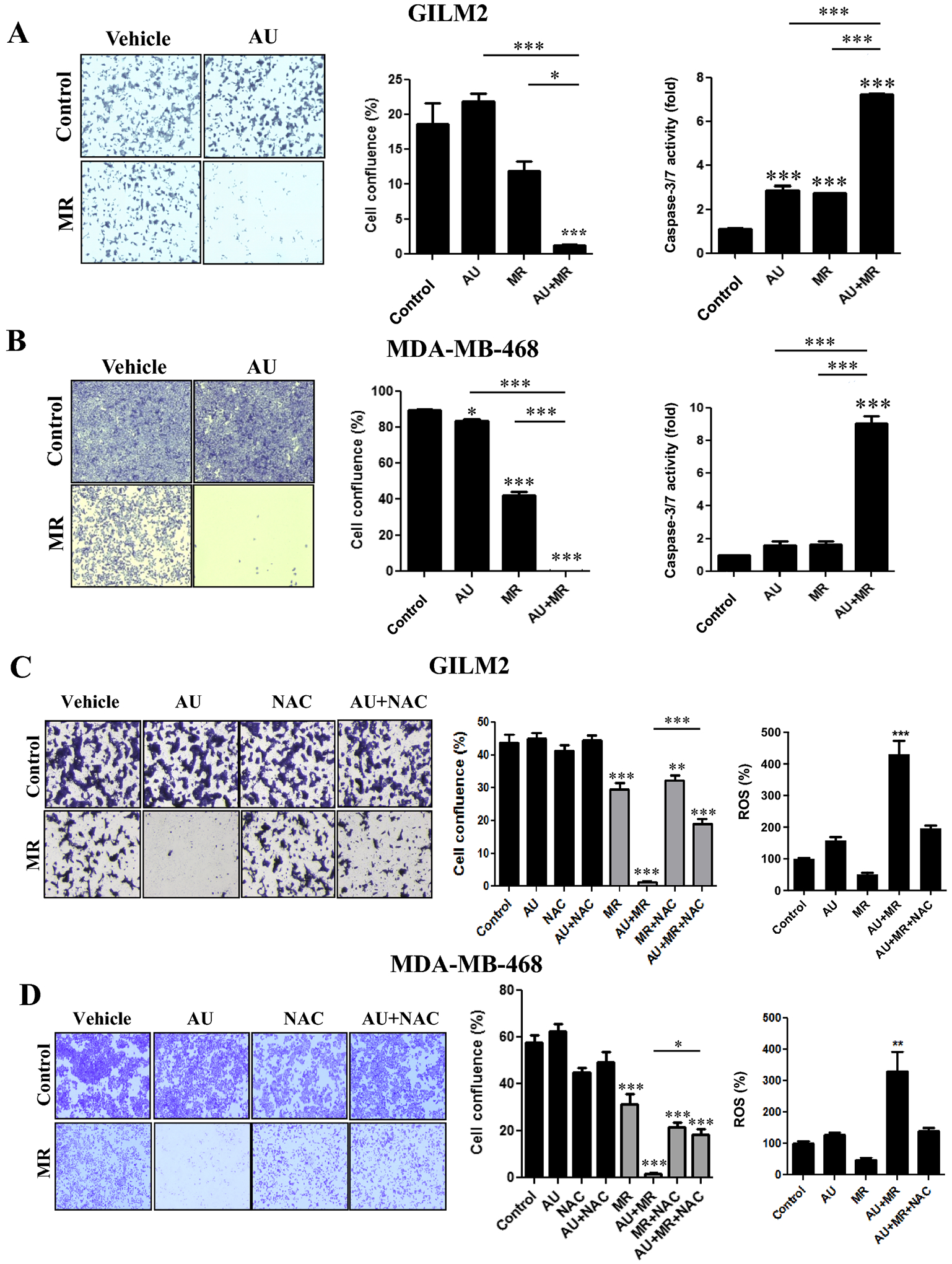
A and B, Crystal violet cell survival assay of GILM2 (A) and MDA-MB-468 (B) TNBC cells grown in control or MR media for 72 hours and treated with vehicle or auranofin (1 μM) for the final 24 hours. Left panel: representative images. Middle panel: quantification of percentage confluence performed by counting cells in 3 fields of each well (mean ± SEM, n = 3). Right panel: GILM2 (A) and MDA-MB-468 (B) TNBC cells grown in control or MR media for 72 hours, treated with vehicle or auranofin (1 μM) for the final 24 hours, and caspase-3/7 activity was measured (expressed as fold control, mean ± SEM, n=3). C and D, Crystal violet cell survival assay of GILM2 (C) and MDA-MB-468 (D) TNBC cells cultured in control or MR media for 72 hours in presence or absence of NAC (2 mM) and treated with vehicle or auranofin (1 μM) for the final 24 hours. Left panel: representative images. Middle panel: quantification of percentage confluence performed by counting cells in 3 fields of each well (mean ± SEM, n = 3). Right panel: GILM2 (C) and MDA-MB-468 (D) cells were grown in control or MR media for 72 hours, treated with vehicle or auranofin (1 μM) for the final 24 hours, and ROS levels (expressed as percentage control) were determined (mean ± SEM, n = 3). In all panels, *, P < 0.05, **, P < 0.01, ***, P < 0.001 vs. control or the indicated comparison
Transformed breast epithelial cells are more sensitive to MR and auranofin
To assess whether the combination of MR and auranofin is selectively cytotoxic against transformed cells, MCF-10A cells transformed by H-RasV12 and isogenic untransformed MCF-10A cells expressing vector were grown in different methionine concentrations (0–100 μM). Although TXNRD1 protein levels were increased in both MCF-10A-Ras and MCF-10A-Vector cells grown in MR media (10 μM methionine or less), the fold induction (from 100 μM to 3 μM) was much more robust in transformed MCF-10A-Ras cells (Fig. 4A). Consistent with our results in TNBC cells (Fig. 3), MR alone had a moderate effect on cell viability in MCF-10A-RasV12 cells and auranofin alone had no effect, while the combination resulted in enhanced cytotoxicity and caspase-3/-7 activity (Fig. 4B). In contrast, MCF-10A-Vector cells were largely resistant to the combination of MR and auranofin (Fig. 4C). Hence, transformed breast epithelial cells are selectively vulnerable to the combination of MR and auranofin.
Fig. 4. Transformed breast epithelial cells are more sensitive to the combination of methionine restriction and auranofin.
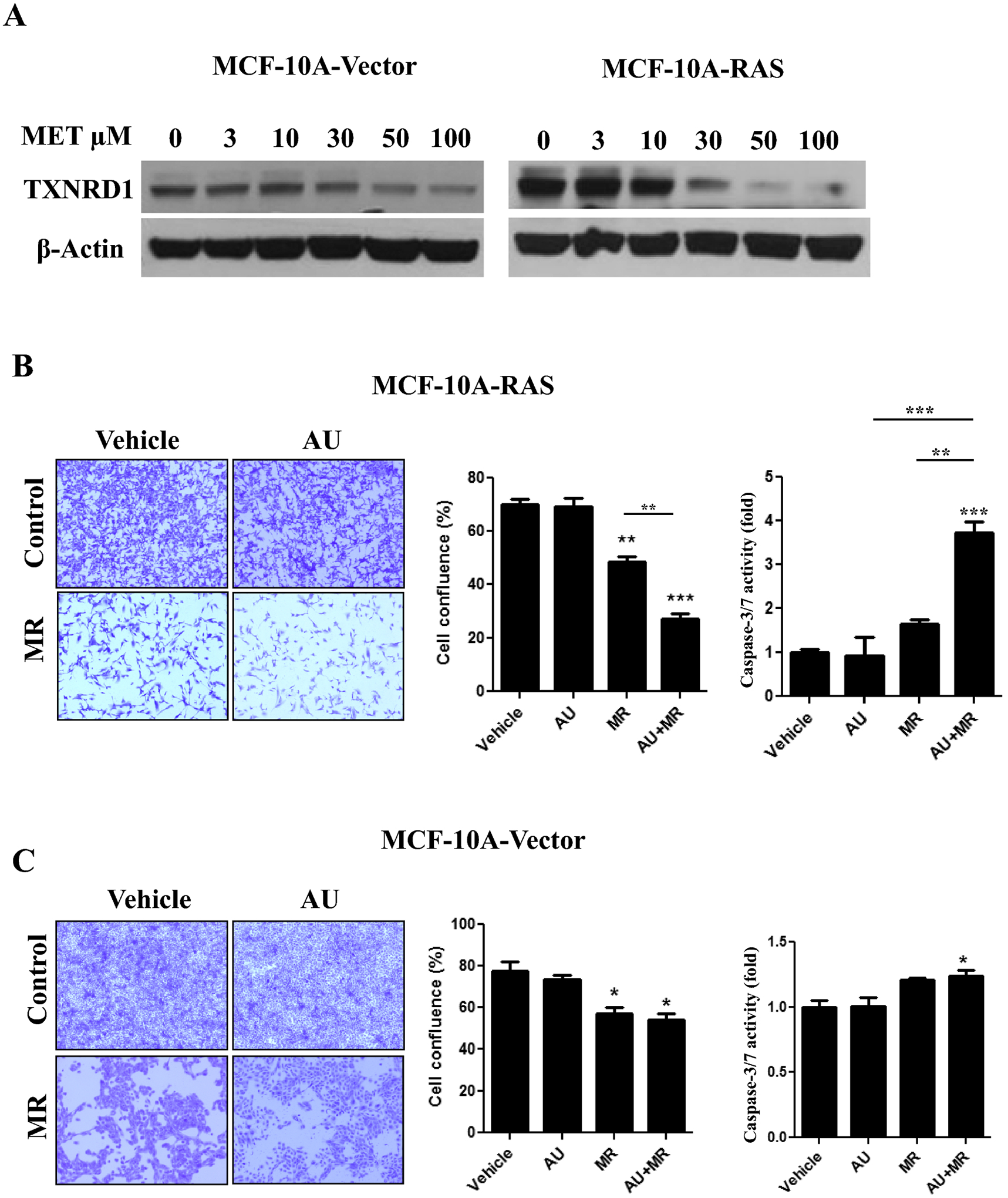
A, Immunoblot of TXNRD1 expression in MCF-10A breast epithelial cells stably expressing vector or H-RasV12 cultured in different concentrations of methionine (0–100 μM) for 72 hours. B and C, Crystal violet cell survival assay of MCF-10A-Ras (B) and MCF-10A-Vector (C) cells grown in control or MR media for 72 hours and treated with vehicle or auranofin (1 μM) for the final 24 hours. Left panel: representative images. Middle panel: quantification of percentage confluence performed by counting cells in 3 fields of each well (mean ± SEM, n = 3). Right panel: MCF-10A-Ras (B) and MCF-10A-Vector (C) cells were grown in control or MR media for 72 hours, treated with vehicle or auranofin (1 μM) for the final 24 hours, and caspase-3/7 activity was determined (expressed as fold control, mean ± SEM, n=3). In all panels, *, P < 0.05, **, P < 0.01, ***, P < 0.001 vs. vehicle control or the indicated comparison
Silencing TXNRD1 sensitizes TNBC cells to MR
To examine the role of TXNRD1 in sensitizing cells to MR, TNBC cells were transfected with a scrambled siRNA (siC) or two different siRNAs targeting human TXNRD1 (si1 and si2). Both siRNAs targeting TXNRD1 potently reduced protein levels of TXNRD1 compared to the scrambled control siRNA (Fig. 5A). Moreover, each siRNA targeting TXNRD1 dramatically reduced TXNRD activity in TNBC cells (Fig. 5B), suggesting that TXNRD1 is the principal isoform contributing to TXNRD activity in these cells. Although silencing TXNRD1 in both TNBC cell lines had little effect on cell viability (Fig. 5C) or caspase-3/7 activity (Fig. 5D), each siRNA targeting TXNRD1 potentiated the cytotoxic effects and enhanced caspase-3/7 activity by MR compared to the scrambled control siRNA. These results indicate that TXNRD1 is the principal isoform mediating the redox vulnerability to MR.
Fig. 5. Silencing TXNRD1 sensitizes TNBC cells to methionine restriction.
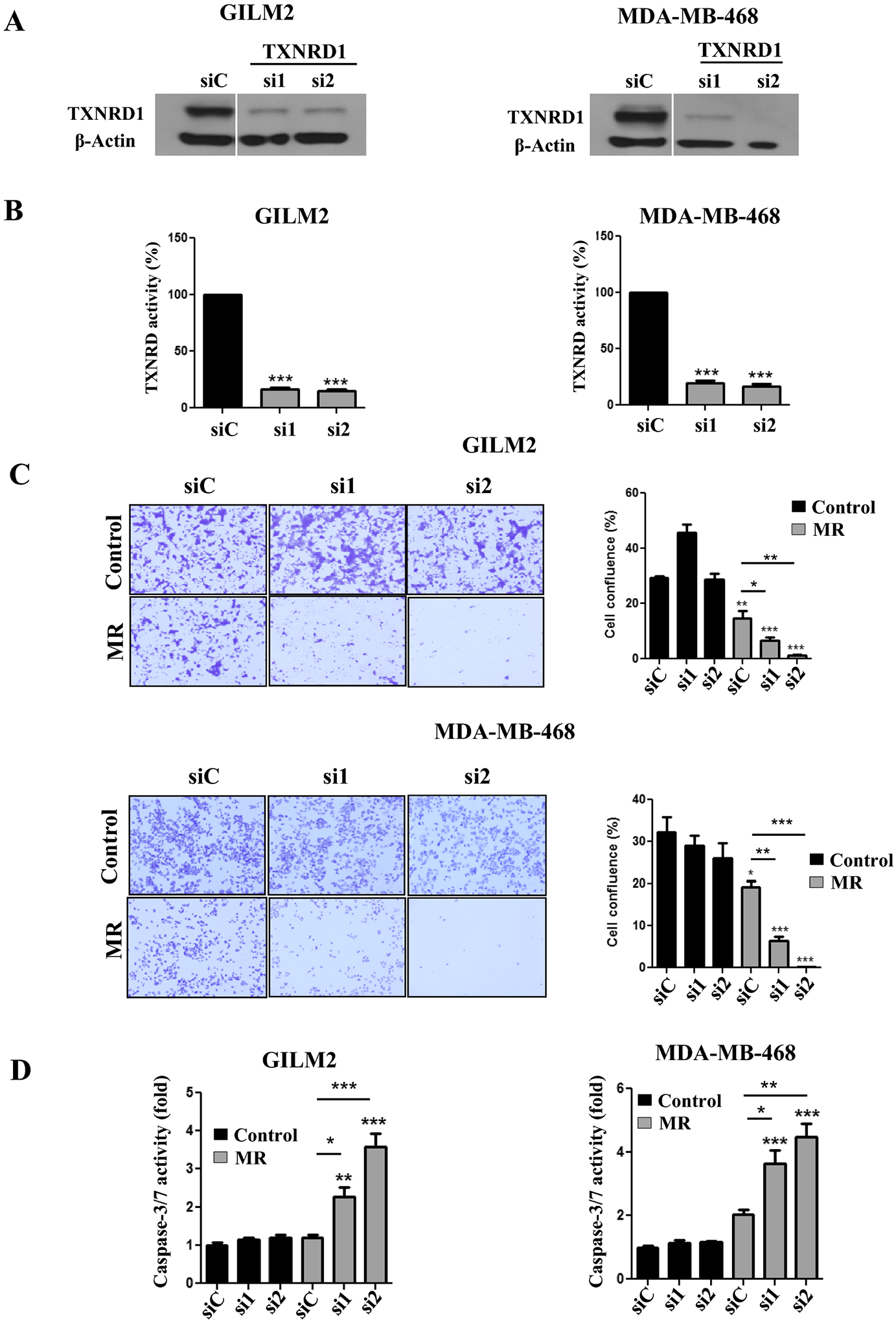
A, Immunoblot analysis of TXNRD1 expression in GILM2 (left) and MDA-MB-468 (right) cells transfected with a scrambled control siRNA (siC) or one of two different siRNAs targeting TXNRD1 (si1 and si2) and grown in control media for 72 hours. B, TXNRD activity in GILM2 (left) and MDA-MB-468 (right) cells transfected with a scrambled control siRNA or one of two different siRNAs targeting TXNRD1 (si1 and si2) and grown in control media for 72 hours. TXNRD activity is presented as the percentage activity in TNBC cells treated with the scrambled control siRNA (mean ± SEM, n = 3). C, Crystal violet cell survival assay of GILM2 and MDAMB-468 cells transfected with a scrambled control siRNA or one of two different siRNAs targeting TXNRD1 (si1 and si2) and cultured in control or MR media for 72 hours. Left panel: representative images. Right panel: quantification of percentage confluence performed by counting cells in 3 fields of each well (mean ± SEM, n = 3). D, GILM2 (left) and MDA-MB-468 (right) were transfected with a scrambled control siRNA or one of two different siRNAs targeting TXNRD1 (si2 and si2), grown in control or MR media for 48 hours, and caspase-3/7 activity was measured (expressed as fold activity of control cells, mean ± SEM, n=3). In all panels, *, P < 0.05, **, P < 0.01, ***, P < 0.001 vs. control or the indicated comparison
Dietary MR augments the antitumor effects of auranofin in TNBC mouse models
To examine the antitumor activity of MR, auranofin and the combination in vivo, we placed female NSG mice with transplanted GILM2-mCherry mammary tumors on a control or methionine-free (MR) diet and treated them with vehicle or auranofin (10 mg/kg daily). Two interventions (MR alone and MR plus auranofin) inhibited mammary tumor growth compared to vehicle-treated mice on a control diet, although the combination of MR plus auranofin was more effective than MR alone or auranofin alone at the completion of the study (Fig. 6A). Both MR and MR plus auranofin resulted in modest weight loss (Fig. S6A). Mammary tumors from mice on MR expressed higher protein levels of TXNRD1 and TXNRD2 than those from mice on the control diet (Fig. 6B), providing in vivo evidence of thioredoxin pathway activation by dietary MR. To evaluate the impact of treatment on lung metastases in vivo, mCherry-fluorescent metastatic lesions in the lungs were identified at autopsy. MR had a modest effect on lung metastases, but the combination of MR and auranofin was more effective at reducing lung metastases (Fig. 6C). Notably, the combination of MR and auranofin was more effective at suppressing lung metastases than primary mammary tumor growth. Moreover, the combination of MR and auranofin robustly induced apoptosis in mammary tumors and lung metastases as determined by active caspase-3 immunohistochemistry (Fig. 6D). The combination of dietary MR and auranofin was also more effective at inhibiting mammary tumor growth (Fig. 6E) and inducing tumor cell apoptosis (Fig. 6F) than either treatment alone in a TNBC PDX model. MR alone and MR in combination with auranofin resulted in modest weight loss (Fig. S6B). Immunohistochemistry of PDX tumors confirmed their triple-negative expression status (Fig. S7). Our results indicate that dietary MR induces TXNRD expression and enhances the activity of auranofin in vivo, with more robust activity against lung metastases than mammary tumors.
Fig. 6. Dietary methionine restriction augments the antitumor effects of auranofin in a metastatic TNBC mouse model.
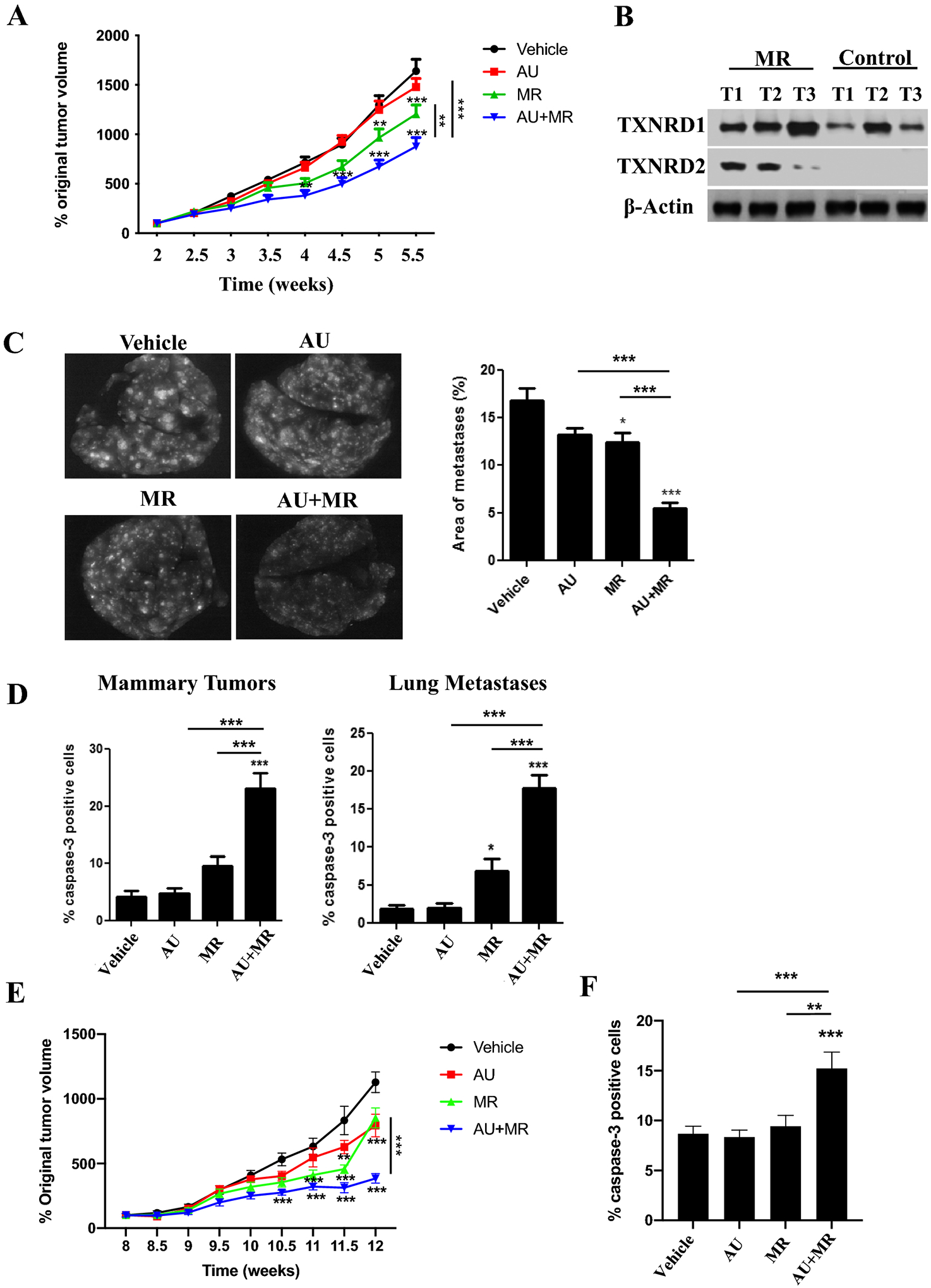
Female NSG mice with GILM2-mCherry mammary tumors (two per mouse, A-D) or PDX TNBC tumors (E-F) were randomly assigned to one of four treatment groups (10 mice per group): control diet plus vehicle, control diet plus auranofin (10 mg/kg daily), MR diet plus vehicle, or MR diet plus auranofin (10 mg/kg daily). The diets were started 2.5 (GILM2) or 8 (PDX) weeks after tumor injection and were continued throughout the treatment period. A, Percentage original GILM2 mammary tumor volume at 2.5 weeks in each group (mean ± SEM, n = 10 mice per group). B, Immunoblot of TXNRD1 and TXNRD2 expression in GILM2 mammary tumors from mice on the control or MR diet. C, Representative images of resected whole lungs at autopsy visualized by fluorescence microscopy in GILM2 model. The percentage of the surface area occupied by fluorescent lung metastases (mean ± SEM, n = 10 mice per group) is indicated. D, The percentage active caspase-3-positive tumor cells in GILM2 mammary tumors (left panel) or GILM2 lung metastases (right panel) after treatment (mean ± SEM, n = 3 tumors per group). E, Percentage original PDX tumor volume at 8 weeks in each group (mean ± SEM, n = 10 mice per group). F, The percentage active caspase-3-positive tumor cells in primary PDX mammary tumors. In all panels, *, P < 0.05, **, P < 0.01, ***, P < 0.001 vs. vehicle-treated mice or the indicated comparison
Discussion
We have demonstrated that MR exposes a vulnerability of TNBC cells to TXNRD1 inhibition by ROS-dependent induction of TXNRD1. MR rapidly initiates oxidative stress by depleting glutathione and reducing the GSH/GSSG ratio, consistent with prior reports [11–15]. Glutathione depletion by MR likely reflects diminished levels of homocysteine [10], which is converted to cysteine, a critical precursor of glutathione [16]. This transient oxidative stress produced by MR results in the induction of TXNRD1, TXNRD2 and TXNRD activity. MR induces TXNRD1 by a ROS-dependent mechanism mediated by NRF2 and ATF4, master transcriptional regulators of the antioxidant and integrated stress responses, respectively [2, 32]. The TXNRD1 gene is an established transcriptional target of NRF2, which binds to an antioxidant response element in the TXNRD1 promoter to regulate its expression [17]. Although both TXNRD1 and TXNRD2 are induced by MR and inhibited by auranofin, TXNRD1 is the principal isoform responsible for the observed synergy with MR because silencing TXNRD1 mimics the effects of auranofin. Moreover, silencing TXNRD1 reduced TXNRD activity, indicating that TXNRD1 is the major isoform contributing to TXNRD enzymatic activity in these TNBC cells. TXNRD1, but not TXNRD2, acts cooperatively with the cystine-glutamate antiporter xCT to rescue cells from glutathione deficiency [36], providing additional evidence that TXNRD isoforms perform at least partly non-redundant functions. Intriguingly, TXNRD1 expression in node-negative breast cancer correlates with poor metastasis-free survival, pointing to TXNRD1 as an attractive therapeutic target [37]. Indeed, selective TXNRD1 inhibitors were recently shown to have antitumor activity in preclinical models and may be less toxic than auranofin [38]. Notably, the synergistic cytotoxicity of combined MR and TXNRD1 inhibition is mediated by the resultant oxidative stress: NAC attenuates this effect. Additionally, our observation that non-transformed breast epithelial cells are resistant to the combination of MR and auranofin suggests that this redox vulnerability may be intrinsic to the transformed phenotype. Our findings underscore that MR creates a tumor dependency on TXNRD1 to maintain redox homeostasis that can be exploited therapeutically.
By initially depleting glutathione levels, MR mimics the effects of the BSO, an inhibitor of glutamate cysteine ligase [35], the rate-limiting enzyme in glutathione synthesis. Like BSO, MR alone is unable to robustly induce apoptosis in cancer cells, but each acts synergistically with TXNRD inhibition to initiate apoptosis and inhibit tumor growth in vivo [5–7]. Notably, BSO does not enhance cell death induction by MR, while recombinant methioninase also synergizes with TXNRD1 inhibition. Our findings are consistent with multiple studies demonstrating synthetic lethality of combined glutathione and thioredoxin pathway inhibition [5–8]. However, MR has several potential advantages over BSO. BSO is rapidly cleared in vivo with a t1/2 of 37 minutes, requiring continuous intravenous infusion for robust glutathione depletion, and BSO has been associated with significant toxicity (leukopenia and thrombocytopenia) and deaths when used in combination with melphalan [39, 40]. In contrast, MR is well tolerated by patients with advanced solid tumors [41–43]. Indeed, we initiated two clinical trials of MR to examine its tumor and metabolic effects in newly diagnosed TNBC (NCT03186937) and its activity in combination with a novel TRAIL agonist in patients with metastatic TNBC (NCT03733119). Moreover, auranofin is currently being investigated in multiple clinical trials in cancer (clinicaltrials.gov). These findings point to the synergistic combination of MR and TXNRD1 inhibition as a novel therapeutic intervention to target the glutathione and thioredoxin pathways to modulate oxidative stress.
We have also demonstrated that dietary MR induces the expression of TXNRD1 and TXNRD2 in mammary tumors and enhances the antitumor effects of auranofin in murine TNBC models. Although the combination of MR and auranofin inhibited growth of both mammary tumors and lung metastases in the metastatic model by activating apoptosis, the effects on lung metastases were more robust. These findings are consistent with recent results suggesting that melanoma circulating tumor cells and metastatic tumors have reduced GSH/GSSG and increased ROS levels compared to primary subcutaneous tumors and are more susceptible to inhibitors of the folate pathway, which deplete NADPH and enhance oxidative stress [44]. Conversely, antioxidants enhance tumor progression and metastases in multiple preclinical models and increase cancer risk in some chemoprevention trials [45–48]. Hence, precision medicine-based approaches to enhance oxidative stress (rather than attenuating it) may be particularly effective against advanced solid tumors. Collectively, our results provide critical mouse model data for a clinical trial of MR in combination with TXNRD1 inhibition to target this redox vulnerability.
Our results substantially expand our “metabolic priming” strategy by MR. We have demonstrated that MR primes breast cancer cells to respond to proapoptotic TRAIL receptor-2 (DR5) agonists by inducing cell surface expression of TRAIL receptor-2 [22]. Additionally, MR induces the expression of MAT2A, the enzyme that converts methionine to the universal methyl-donor S-adenosylmethionine [49–51]. We have demonstrated that MR primes cancer stem cells to respond to MAT2A inhibition [51]. Moreover, dietary MR induces MAT2A in orthotopic mammary tumors in mice, and the combination of MR and the MAT2A inhibitor cycloleucine is more effective than either alone at suppressing tumor burden in a murine TNBC model [51]. Similar results pointing to enhanced methionine cycle activity and dependence on MAT2A in cancer stem cells/tumor-initiating cells were recently reported by others [52]. MR primes tumors to respond to TXNRD1 inhibition by ROS- and NRF2/ATF4-dependent induction of this enzyme. Methionine restricted breast tumor cells, but not non-transformed breast epithelial cells, become addicted to TXNRD1 activity for survival, rendering them exquisitely sensitive to auranofin. Given the demonstrated safety of both MR and auranofin [9, 41–43], these preclinical findings may be readily translated into a clinical trial.
Supplementary Material
Acknowledgments
We are indebted to Drs. Nobunao Wakabayashi and Craig Thompson for providing MEFs, Dr. Vadim Pokrovsky for providing methioninase, and to Dr. Mark Burkard, Dr. Dudley Lamming and members of the Cryns lab for their critical reading of the manuscript.
Funding
This work was supported by grants from the Breast Cancer Research Foundation (VLC), V Foundation for Cancer Research (VLC), Wisconsin Partnership Program (VLC), and P30CA14520 core facility support.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Data availability
The data from this study are available from the corresponding author upon request. Additional data are available in Supplementary Data.
References
- 1.Schieber M, Chandel NS. (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24:R453–R462. 10.1016/j.cub.2014.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorrini C, Harris IS, Mak TW. (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12:931–947. 10.1038/nrd4002 [DOI] [PubMed] [Google Scholar]
- 3.Bansal A, Simon MC. (2018) Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol 217:2291–2298. 10.1083/jcb.201804161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J, Li X, Han X, Liu R, Fang J. (2017) Targeting the thioredoxin system for cancer therapy. Trends Pharmacol Sci 38:794–808. 10.1016/j.tips.2017.06.001 [DOI] [PubMed] [Google Scholar]
- 5.Fath MA, Ahmad IM, Smith CJ, Spence J, Spitz DR. (2011) Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clin Cancer Res 17:6206–17. 10.1158/1078-0432.CCR-11-0736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, et al. (2015) Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27:211–22. 10.1016/j.ccell.2014.11.019 [DOI] [PubMed] [Google Scholar]
- 7.Mandal PK, Schneider M, Kolle P, Kuhlencordt P, Forster H, Beck H, et al. (2010) Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res 70:9505–9514. 10.1158/0008-5472.CAN-10-1509 [DOI] [PubMed] [Google Scholar]
- 8.Yan X, Zhang X, Wang L, Zhang R, Pu X, Wu S, et al. (2019) Inhibition of thioredoxin/thioredoxin reductase induces synthetic lethality in lung cancers with compromised glutathione homeostasis. Cancer Res 79:125–32. 10.1158/0008-5472.CAN-18-1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roder C, Thomson MJ. (2015) Auranofin: repurposing an old drug for a golden new age. Drugs R D 15:13–20. 10.1007/s40268-015-0083-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao X, Sanderson SM, Dai Z, Reid MA, Cooper DE, Lu M, et al. (2019) Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 572:397–401. 10.1038/s41586-019-1437-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghosh S, Forney LA, Wanders D, Stone KP, Gettys TW. (2017) An integrative analysis of tissue-specific transcriptomic and metabolomic responses to short-term dietary methionine restriction in mice. PLoS One 12:e0177513. 10.1371/journal.pone.0177513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin AH, Chen HW, Liu CT, Tsai CW, Lii CK. (2012) Activation of Nrf2 is required for up-regulation of the π class of glutathione S-transferase in rat primary hepatocytes with L-methionine starvation. J Agric Food Chem 60:6537–45. 10.1021/jf301567m [DOI] [PubMed] [Google Scholar]
- 13.Pettit AP, Jonsson WO, Bargoud AR, Mirek ET, Peelor FF 3rd, Wang Y, et al. (2017) Dietary methionine restriction regulates liver protein synthesis and gene expression independently of eukaryotic initiation factor 2 phosphorylation in mice. J Nutr 147:1031–40. 10.3945/jn.116.246710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poirson-Bichat F, Goncalves RA, Miccoli L, Dutrillaux B, Poupon MF. (2000) Methionine depletion enhances the antitumoral efficacy of cytotoxic agents in drug-resistant human tumor xenografts. Clin Cancer Res 6:643–653. [PubMed] [Google Scholar]
- 15.Tsai CW, Lin AH, Wang TS, Liu KL, Chen HW, Lii CK. (2010) Methionine restriction up-regulates the expression of the π class of glutathione S-transferase partially via the extracellular signal-regulated kinase-activator protein-1 signaling pathway initiated by glutathione depletion. Mol Nutr Food Res 54:841–850. 10.1002/mnfr.200900083 [DOI] [PubMed] [Google Scholar]
- 16.Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. (2013) Nat Rev Cancer 13:572–583. 10.1038/nrc3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakurai A, Nishimoto M, Himeno S, Imura N, Tsujimoto M, Kunimoto M, et al. (2005) Transcriptional regulation of thioredoxin reductase 1 expression by cadmium in vascular endothelial cells: role of NF-E2-related factor-2. J Cell Physiol 203:529–537. 10.1002/jcp.20246 [DOI] [PubMed] [Google Scholar]
- 18.Hoffman RM, Erbe RW. (1976) High in vivo rates of methionine biosynthesis in transformed human and malignant rat cells auxotrophic for methionine. Proc Natl Acad Sci USA 73:1523–1527. 10.1073/pnas.73.5.1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coalson DW, Mecham JO, Stern PH, Hoffman RM. (1982) Reduced availability of endogenously synthesized methionine for S-adenosylmethionine formation in methionine-dependent cancer cells. Proc Natl Acad Sci USA 79:4248–4251. 10.1073/pnas.79.14.4248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stern PH, Hoffman RM. (1986) Enhanced in vitro selective toxicity of chemotherapeutic agents for human cancer cells based on a metabolic defect. J Natl Cancer Inst 76:629–639. 10.1093/jnci/76.4.629 [DOI] [PubMed] [Google Scholar]
- 21.Chaturvedi S, Hoffman RM, Bertino JR. (2018) Exploiting methionine restriction for cancer treatment. Biochem Pharmacol 154:170–173. 10.1016/j.bcp.2018.05.003 [DOI] [PubMed] [Google Scholar]
- 22.Strekalova E, Malin D, Good DM, Cryns VL. (2015) Methionine deprivation induces a targetable vulnerability in triple-negative breast cancer cells by enhancing TRAIL receptor-2 expression. Clin Cancer Res 21:2780–2791. 10.1158/1078-0432.CCR-14-2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malin D, Strekalova E, Petrovic V, Deal AM, Al Ahmad A, Adamo B, et al. (2014) αB-crystallin: a novel regulator of breast cancer metastasis to the brain. Clin Cancer Res 20:56–67. 10.1158/1078-0432.CCR-13-1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strekalova E, Malin D, Rajanala H, Cryns VL. (2017) Metformin sensitizes triple-negative breast cancer to proapoptotic TRAIL receptor agonists by suppressing XIAP expression. Breast Cancer Res Treat 163:435–447. 10.1007/s10549-017-4201-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moyano JV, Evans JR, Chen F, Lu M, Werner ME, Yehiely F, et al. (2006) αB-crystallin is a novel oncoprotein that predicts poor clinical outcome in breast cancer. J Clin Invest 116:261–70. 10.1172/JCI25888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin S, Wakabayashi N, Misra V, Biswal S, Lee GH, Agoston ES, et al. (2007) NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol Cell Biol 27:7188–97. 10.1128/MCB.00915-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye J, Palm W, Peng M, King B, Lindsten T, Li MO, et al. (2015) GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Dev 29:2331–2336. 10.1101/gad.269324.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malin D, Chen F, Schiller C, Koblinski J, Cryns VL. (2011) Enhanced metastasis suppression by targeting TRAIL receptor 2 in a murine model of triple-negative breast cancer. Clin Cancer Res 17:5005–5015. 10.1158/1078-0432.CCR-11-0099 [DOI] [PubMed] [Google Scholar]
- 29.Lu M, Strohecker A, Chen F, Kwan T, Bosman J, Jordan VC, et al. (2008) Aspirin sensitizes cancer cells to TRAIL-induced apoptosis by reducing survivin levels. ClinCancer Res 14:3168–3176. 10.1158/1078-0432.CCR-07-4362 [DOI] [PubMed] [Google Scholar]
- 30.Sanz A, Caro P, Ayala V, Portero-Otin M, Pamplona R, Barja G. (2006) Methionine restriction decreases mitochondrial oxygen radical generation and leak as well as oxidative damage to mitochondrial DNA and proteins. FASEB J 20:1064–73. 10.1096/fj.05-5568com [DOI] [PubMed] [Google Scholar]
- 31.Najim N, Podmore ID, McGown A, Estlin EJ. (2009) Methionine restriction reduces the chemosensitivity of central nervous system tumour cell lines. Anticancer Res 29:3103–3108. [PubMed] [Google Scholar]
- 32.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. (2016) The integrated stress response. EMBO Rep 17:1374–1395. 10.15252/embr.201642195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, et al. (2015) NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47:1475–1481. 10.1038/ng.3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyamoto N, Izumi H, Miyamoto R, Bin H, Kondo H, Tawara A, et al. (2011) Transcriptional regulation of activating transcription factor 4 under oxidative stress in retinal pigment epithelial ARPE-19/HPV-16 cells. Invest Ophthalmol Vis Sci 52:1226–1234. 10.1167/iovs.10-5775 [DOI] [PubMed] [Google Scholar]
- 35.Griffith OW, Meister A. (1979) Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J Biol Chem 254:7558–7560. [PubMed] [Google Scholar]
- 36.Mandal PK, Seiler A, Perisic T, Kolle P, Banjac Canak A, Forster H, et al. (2010) System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J Biol Chem 285:22244–22253. 10.1074/jbc.M110.121327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cadenas C, Franckenstein D, Schmidt M, Gehrmann M, Hermes M, Geppert B, et al. (2010) Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res 12:R44. 10.1186/bcr2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stafford WC, Peng X, Olofsson MH, Zhang X, Luci DK, Lu L, et al. (2018) Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Sci Transl Med 10(428). 10.1126/scitranslmed.aaf7444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson CP, Matthay KK, Perentesis JP, Neglia JP, Bailey HH, Villablanca JG, et al. (2015) Pilot study of intravenous melphalan combined with continuous infusion L-S,R-buthionine sulfoximine for children with recurrent neuroblastoma. Pediatr Blood Cancer 62:1739–1746. 10.1002/pbc.25594 [DOI] [PubMed] [Google Scholar]
- 40.Bailey HH, Ripple G, Tutsch KD, Arzoomanian RZ, Alberti D, Feierabend C, et al. (1997) Phase I study of continuous-infusion L-S,R-buthionine sulfoximine with intravenous melphalan. J Natl Cancer Inst 89:1789–1796. 10.1093/jnci/89.23.1789 [DOI] [PubMed] [Google Scholar]
- 41.Durando X, Thivat E, Farges MC, Cellarier E, D’Incan M, Demidem A, et al. (2008) Optimal methionine-free diet duration for nitrourea treatment: a Phase I clinical trial. Nutr Cancer 60:23–30. 10.1080/01635580701525877 [DOI] [PubMed] [Google Scholar]
- 42.Epner DE, Morrow S, Wilcox M, Houghton JL. (2002) Nutrient intake and nutritional indexes in adults with metastatic cancer on a phase I clinical trial of dietary methioninerestriction. Nutr Cancer 42:158–166. 10.1207/S15327914NC422_2 [DOI] [PubMed] [Google Scholar]
- 43.Thivat E, Farges MC, Bacin F, D’Incan M, Mouret-Reynier MA, Cellarier E, et al. (2009) Phase II trial of the association of a methionine-free diet with cystemustine therapy in melanoma and glioma. Anticancer Res 29:5235–5240. [PubMed] [Google Scholar]
- 44.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. (2015) Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527:186–91. 10.1038/nature15726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, Bergo MO. (2014) Antioxidants accelerate lung cancer progression in mice. Sci Transl Med 6:221ra15. 10.1126/scitranslmed.3007653 [DOI] [PubMed] [Google Scholar]
- 46.Wiel C, Le Gal K, Ibrahim MX, Jahangir CA, Kashif M, Yao H, et al. (2019) BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell 178:330–345 e22. 10.1016/j.cell.2019.06.005 [DOI] [PubMed] [Google Scholar]
- 47.α-Tocopherol BCCPSG. (1994) The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med 330:1029–1035. 10.1056/NEJM199404143301501 [DOI] [PubMed] [Google Scholar]
- 48.Klein EA, Thompson IM Jr., Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, et al. (2011) Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 306:1549–1556. 10.1001/jama.2011.1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martinez-Chantar ML, Latasa MU, Varela-Rey M, Lu SC, Garcia-Trevijano ER, Mato JM, et al. (2003) L-methionine availability regulates expression of the methionine adenosyltransferase 2A gene in human hepatocarcinoma cells: role of S-adenosylmethionine. J Biol Chem 278:19885–19890. 10.1074/jbc.M211554200 [DOI] [PubMed] [Google Scholar]
- 50.Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, et al. (2014) Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab 19:780–794. 10.1016/j.cmet.2014.03.017 [DOI] [PubMed] [Google Scholar]
- 51.Strekalova E, Malin D, Weisenhorn EMM, Russell JD, Hoelper D, Jain A, et al. (2019) S-adenosylmethionine biosynthesis is a targetable metabolic vulnerability of cancer stem cells. Breast Cancer Res Treat 175:39–50. 10.1007/s10549-019-05146-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Z, Yip LY, Lee JHJ, Wu Z, Chew HY, Chong PKW, et al. (2019) Methionine is a metabolic dependency of tumor-initiating cells. Nat Med 25:825–837. 10.1038/s41591-019-0423-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data from this study are available from the corresponding author upon request. Additional data are available in Supplementary Data.