

Abstract
T cells able to control neoplasia or chronic infections display a signature gene expression profile similar or identical to that of central memory T cells. These cells have qualities of self-renewal and a plasticity that allow them to repeatedly undergo activation (growth, proliferation, and differentiation), followed by quiescence. It is these qualities that define the ability of T cells to establish an equilibrium with chronic infectious agents, and also preserve the ability of T cells to be re-activated (by checkpoint therapy) in response to malignant cancers. Here we describe distinctions between the forms of inhibition mediated by tumors and persistent viruses, we review the properties of T cells associated with long-term immunity, and we identify the transcription factor, FOXO1, as the control point for a program of gene expression that allows CD8+ T cells to undergo serial reactivation and self-renewal.
Introduction
Cytotoxic T cells specific for an infectious intracellular pathogen (or neoplasia) rapidly diverge toward three major differentiation states characterized as memory precursor cells, that ultimately become central memory cells, short-lived effector cells, and tissue resident memory cells [1–5]. Between the extremes of central memory and cytotoxic effector cells there exists a continuum or at least multiple cell-types in which the differentiated function of cytotoxic T cells is present at the expense of a retained potential for self-renewal and serial reactivation [6,7]. The appearance and maintenance of these cell populations are strongly influenced by the course and disposition of an infection. With rapid clearance of the recognized foreign agent, the effector cells slowly contract and leave the long-lived memory population in a state of prepared responsiveness—an acute primary response followed by a state of immunity. In those instances where neutralizing antibodies are absent or not protective [8•], a secondary infection provokes a rapid expansion of the memory population that once again diverges into effector and long-term memory cells—a secondary or anamnestic response.
When the immune response does not clear the intracellular threat, be it an infection or neoplastic growth, the differentiation of CD8+ cytotoxic T cells is less well delineated. In addition to effector cells and a fate resembling memory cells, antigen-specific CD8+ T cells may enter yet another state of differentiation characterized by reduced responsiveness that has been termed exhaustion [9]. The importance of this state in the progression of cancer or the persistence of intracellular infections is that T cells which can be effectively reactivated to clear or restrain the inciting principle are those that retain characteristics of central memory [10••,11,12••,13••]. Highly differentiated effector cells do not expand further upon reencounter with antigen [14,15], whereas exhausted T cells, though they continue to possess the potential to mount a response [16], are attenuated in their capacity for dispatching an infection or a tumor mass [9]. In this review, we will highlight the state of T cells present in the face of chronic antigen exposure, and the variable ability of such T cells to continue to mount an effective immune response. This topic has important implications for the failure of immune mechanisms to contain or clear chronic viral infections and neoplastic growth.
Feedback control, evolved under pressure of chronic infections, is disadvantageous to cancer immunosurveillance
Long-lived animal hosts and their parasitic viruses are often characterized as engaging in an arms race, where each is selected to evolve the means to survive and procreate at the expense of the other, an example of Van Valen’s ‘Red Queen Hypothesis’ [17]. But this is not quite accurate. The evolution of a virus (and other infectious agents) is based on a generation time that is orders of magnitude shorter than that of its host. Furthermore, viruses are selected to replicate and be transmitted within and between a limited number of host species, and thus they can evolve specifically targeted mechanisms of virulence. Importantly, they are constrained to accomplish transmission before rendering their host unable to spread an infection. In contrast, the host is co-evolving with a plethora of diverse parasitic agents, each displaying distinct sets of virulence mechanisms. So, in addition to a long generation time, the need to simultaneously resist hundreds of different parasitic agents most probably means that host immune mechanisms evolve on a different time scale when compared with the world of parasites. We posit that this arms race is almost entirely one-sided where the parasites, such as persistent viruses, define their niche in a relatively static population of hosts.
This is not the case with neoplasia. With the exception of a few cancers found in Tasmanian devils, domestic canines, and bivalve species [18,19], cancers themselves are not infectious. The emergence and ‘success’ of one cancer is not inherited by the next. Rather, neoplasia is a ‘new formation’ that is selected for unrestrained growth, without selective pressure to be transmitted or keep its host alive; one cancer resembles another only through convergent evolution [20]. Embedded within this somatic evolution, cancer cells appear to be selected individually and as a population to frustrate mechanisms of immunity that can impede their growth. As such, the forms of immune attenuation and negative feedback control that occur as a result of persistent virus infections and that which occurs coincident with the growth of malignant tumors are based on distinct evolutionary pressures. The former we presume to be advantageous to the long-term survival of the host, the latter favors unrestrained, metastatic tumor growth.
Feedback attenuation displayed by CD8+ T cells is proportional to T cell antigen receptor (TCR) signaling, and it involves expression of receptors connected to inhibitory signaling pathways that activate, for example, tyrosine phosphatases that block signaling cascades based on tyrosine phosphorylation [21]. The prototype for this feedback pathway is initiated by the inhibitory receptor PDCD1 (PD-1) that is expressed following TCR-mediated signaling on subsets of both CD4+ and CD8+ T cells, and is also expressed by B cells and myeloid cells [22,23]. This type of control applied to CD8+ T cells would appear to allow a burst of cytotoxic T cell activity, followed by attenuation that limits immunopathology. It is consistent with an evolutionary acknowledgement by pathogen-susceptible hosts that all-out resistance is futile, if not life-threatening, and often unnecessary in that co-evolved, persistent, intracellular infectious agents are not selected to cause premature demise of their hosts [24–26]. It may be a manifestation of the concept of tolerance—the ability of a host to accommodate infectious agents without disease [27].
We speculate that most of the mechanisms we associate with acquired immunity were most strongly selected for resistance to infectious agents. Although immunodeficient mice and human beings can be found to exhibit higher rates of cancer compared with their wildtype brethren, this is most clearly seen under the influence of carcinogens [28]. Furthermore, completely immunodeficient mice housed under standard conditions may experience increased inflammation associated with opportunistic infections, a condition known to favor oncogenesis [29]. We assert that an animal with a severe congenital immunodeficiency in the wild is likely to die of an infection long before metastatic cancer takes hold. The importance of this is that mechanisms of immunity, but also mechanisms of control, have been most strongly selected for relative resistance to infectious agents counterbalanced by regulation that minimizes immunopathology associated with persistent or latent infections. From this we deduce that the immune reaction that arises in response to a cancerous growth is subject to these same regulatory mechanisms, even though they may be disadvantageous in the presence of a life-threatening malignancy. We propose that the vertebrate immune system, although displaying the plasticity to recognize neoplastic growth, has been selected to anticipate a latent or chronic infection, not a rapidly malignant, lethal cancer.
Negative feedback and the regulation of ‘exhaustion’
A manifestation of negative feedback control in response to persistent infectious agents is ultimately a state of attenuated responsiveness termed exhaustion. Exhaustion in CD8+ T cells was first observed in the context of a chronic lymphocytic choriomeningitis virus (LCMV) infection in mice [30–32], and it has been observed in both mice and human beings subject to several chronic infectious agents as well as the persistent inflammation present in autoimmune pathologies or in the context of cancer [33–41]. Exhaustion has also been reported to occur in CD4+ T cells, although not necessarily characterized by the expression of negative feedback receptors such as PDCD1 [42], and yet, CD4+ T cells play an essential role in the viral-immune equipoise characterizing both chronic and latent infections [43]. Exhausted CD8+ T cells are characterized by the sequential loss of (presumably pathogenic) effector function, and particularly, impairment in IL-2, TNF, and IFNγ production [41]. This altered state is also marked by the sustained induction of inhibitory receptors that, in addition to PDCD1, include LAG3 (Lymphocyte Activation Gene-3), HAVCR2 (TIM3) and TIGIT (T cell immunoglobulin and ITIM domain) [44,45].
Programs of gene expression associated with CD8+ T cell differentiation
Transcriptional profiling and network analysis of individual T cells have revealed the different programs of gene expression that correlate with memory, effector function, and exhaustion [9]. The transcription factors that appear to be necessary for long-lived memory cell formation and maintenance are: FOXO1, EOMES, and TCF7 (Tcf-1), whereas the alternative CD8+ T cell fate, short-lived effector cells, requires TBX21 (TBET), PRDM1 (Blimp-1), and ID2 (reviewed in Ref. [3]). These differences in memory and effector T cells appear to arise with an initial asymmetric division followed by the dynamics of metabolism, proliferation and survival determining the subsequent make-up of the T cell response [46–48]. An interesting concept to emerge, and one that we will discuss below is that most of the highly expressed genes in memory cells are commonly expressed by naïve T cells and many by hematopoietic stem cells. A conclusion is that there are common programs of gene expression found in cells endowed with the capacity for self-renewal [3].
The transcriptional profile of exhausted T cells differs from that of effector and memory cells and extends beyond genes encoding inhibitory feedback circuitry. The profile includes genes encoding transcription factors, metabolic pathways, and signaling intermediates, as well as chemokines, cytokines and their respective receptors [44,49]. Some of the major transcription factors implicated in the T cell exhaustion include EOMES and TBX21, TCF7, TOX, PRDM1, NFAT, FOXO1, FOXP1, BATF, IRF4 and VHL [50–57]. Most recently, the transcription factor TOX was shown to be required for important aspects of an exhausted phenotype, although one report provided evidence that functional exhaustion could take place without the presence of TOX [58••,59•,60•,61••,62•]. Interestingly, transcription factors that are key to T cell exhaustion are also important for T effector and memory formation, but they are utilized distinctly in the context of exhaustion. In chronic infection, the expression of TBX21 and EOMES appears to guide the formation of specific subsets within the T exhausted population, such that elevated expression of TBX21 is associated with a non-terminally exhausted, progenitor, subset (TBEThi PDCD1int EOMESlo, while, contrary to its role in T cell memory, high expression of EOMES is associated with a terminally exhausted T cell subset (EOMEShi PDCD1hi).
Perhaps most importantly for this discussion, TCF7, characterized for its essential role in the induction and maintenance of memory T cells, similarly plays a key role in sustaining long-lived T cells labeled as ‘exhausted’. It is this subpopulation, TCF7+ PDCD1int HAVCR2lo, that is credited with mediating a therapeutic response to ‘checkpoint’ immunotherapy [10••,12••,13••,63••]. The ability of this subset to revive a functional response when inhibitory signals are blocked is further evidenced by the ability of these TCF7+ T cells to clear a viral infection upon transfer to a naïve host [64,65••,66]. scRNA-sequencing analyses reveal that during a chronic infection TCF7 overrides T effector differentiation and skews the differentiation to exhausted T cells via the upregulation of MYB which is a known regulator of BCL2 and EOMES (for persistence of exhaustion phenotype) [67]. We emphasize that PDCD1 is induced under a variety of circumstances in CD4+ and CD8+ T cells, and alone, it does not signify unresponsive, exhausted T cells.
The origin of responsive TCF7-expressing T cells depends on FOXO1
Studies to date consistently reveal TCF7 to be a nexus of signaling and transcription necessary for T cell survival and self-renewal—and this transcends species (at least applies to human beings and mice) and the inciting chronic immune stimulus [67–69]. In addition to its role in late-stage T cell differentiation, survival and function, TCF7 is important, along with its paralog, LEF, for differentiation and transition through several stages of early T cell development [70]. It also functions to promote the differentiation of precursors to all innate lymphoid cells (ILCs) (reviewed in Refs. [69,71]). The means by which TCF7 accomplishes these varying roles depends upon transcriptional context, but also the many splice forms known for Tcf7 transcripts. These variously include the N-terminal β-catenin interacting domain mediating Wnt signaling, a 30 amino acid domain with intrinsic HDAC activity [72], and a C-terminal high-mobility-group (HMG) box DNA binding domain that allows TCF7 to directly mediate transcriptional activity [73]. There is presently little understanding of the splice forms that are important for TCF7 activity in mature T cell differentiation, although reports show its role in establishing a memory precursor cell requires WNT signaling and β-catenin [74–76,77•,78].
In its role as a central transcription factor in memory and as a linchpin describing CD8+ T cells able to respond to checkpoint therapy, RNA expression studies show that Tcf7 is often coordinately expressed with Il7r (encoding IL7 receptor α-chain, CD127), Sell (encoding L-selectin, CD62L), Ccr7 (encoding CCR7) and in opposition to the expression of Klrg1, Havcr2, Cx3cr1 (encoding fractalkine receptor) and effector molecules such as Gzmb (encoding granzyme B) [7,69]. This is a program of gene expression that is directly controlled by the forkhead ‘O’ family member FOXO1.
TCF7 is expressed at high levels in naïve CD8+ T cells—either wildtype or those deleted for Foxo1, but with antigen-induced T cell activation, Tcf7 expression is rapidly lost [79], possibly through the action of inflammatory cytokines [80]. In LCMV gp33-specific T cells it is only reestablished in a minor subset of responding cells that can be observed starting around day five post-activation; however, as described above, the dichotomy of memory and effector precursors may be established at the very first asymmetric cell division, one where MYC and FOXO1 are segregated into effector and memory precursors, respectively [47]. At the peak of the T cell response, about day seven, the memory precursor population is characterized as TCF7+ IL7R+ KLRG1− HAVCR2− GZMB−, but with the deletion of Foxo1, this subset is completely absent [79,81••]. Without Foxo1, TCF7 expression is never again reestablished, and the CD8+ T cell response consists entirely of effector cells that lack the ability to undergo a secondary expansion, that is, an anamnestic response [81••,82,83,84••]. In fact, the characteristic properties of memory cells generated after an acute infection required continuous expression of FOXO1, as late Foxo1 deletion using tamoxifen-induced CRE recombinase expression in Foxo1f/f T cells, resulted in a reversion to effector phenotype: the gain of KLRG1 and GZMB and loss of BCL2, self-renewal potential, and an ability to proliferate in response to reinfection. Under conditions of chronic or latent virus infection, where we presume T cells continued to receive antigen-mediated signals, deletion of Foxo1 caused a greatly accelerated loss of memory potential [85•,86•].
Control of Tcf7 transcription and alternate exon usage is likely to be complex, and this may be a focus of future studies. We originally looked at the Tcf7 gene from CD4+ T cells by analyzing regions of open chromatin, chromosome marks indicative of poised and active enhancers, and binding by FOXO1 by chromatin immunoprecipitation and genomic sequencing (ChIP-Seq) [81••]. Subsequently, we have carried out similar studies on CD8+ LCMV-specific T cells, before, and 12 days post infection by LCMV-Armstrong (data not shown). The results were similar to that of CD4+ T cells in that within a 60 kb region that includes the body of the Tcf7 gene and a 30 kb region upstream of the transcriptional start site, we found six distinct regions of open chromatin as detected by ATAC-Seq [87]. Each of these sites was flanked by chromosome marks of H3K27 acetylation (H3K27Ac) indicative of active enhancers [88], and each of these sites was bound by FOXO1. FOXO1 also bound to a 7th site characterized by H3K4 trimethylation (H3K4me3) located at the transcriptional start site (TSS), presumably the promoter [89,90]. Although these results are correlative, the requirement for FOXO1 in expression post-activation, and the identification of FOXO1 binding to nearby enhancers is consistent with a direct role for FOXO1 in post-activation Tcf7 regulation; however, we note that other gene elements located much further away may play a role in Tcf7 transcriptional regulation. For example, loss of Foxo1 did not affect TCF7 expression in naïve T cells, yet the Tcf7 proximal sites bound by FOXO1 were identical between naïve T cells and antigen-specific T cells from day 12 post-infection; however, a FOXO1 binding site co-localized with a site of open chromatin 280 kb downstream of the TSS appears to be inaccessible in Foxo1-null T cells (data not shown). Without a systematic mutation of these gene elements, alone and in combination, we are presently unable to more exactly identify the mechanism of Tcf7 regulation by FOXO1.
In addition to Tcf7 regulation, FOXO1 controls several genes that are coordinately found to be expressed in T cells associated with response to checkpoint therapy. These include Il7r, Sell, and Ccr7 [91–94]. In particular, FOXO1 is required for the expression of Il7r that encodes the alpha-chain of the receptor for IL7, an important factor in the viability of memory T cells [95,96]. FOXO1 binds to an enhancer 3.5 kb upstream of the Il7r TSS [92] in order to displace FOXP1 acting as a transcription repressor [97]. From these studies, FOXO1 emerges as the upstream control point for the program of gene expression that is essential for differentiation and survival of T cells able to control viral or neoplastic parasites.
FOXO1 regulation and the physiology of cell renewal
The role played by FOXO family transcription factors in survival, plasticity and self-renewal has been described previously [98–101]. In particular, FOXO transcription factors have a central role in the ability of metazoans to establish pluripotency and characteristics of stem cells. They were first described as essential to the downstream signaling module necessary for an extended life-span in nematodes or flies subject to diminished insulin-like growth factor signaling [102–104]. More recently, a FOXO ortholog has been found to be key to the establishment of self-renewal in all three stem cell lineages in hydra, an immortal cnidarian genus diverged from bilateral phyla on the order of 600 million years ago [105]. Closer to home, FOXO1 was found to regulate pluripotency in human embryonic stem cells by binding and activating the promotors of two essential ‘Yamanaka factors’, OCT4 (POU5F1) and SOX2 [106,107]. In addition, genome-wide association studies have revealed FOXO1 and FOXO3 to be the most prominent among a small number of genes associated with increased age at death or age at natural menopause [108–114]. As such, it is perhaps not surprising that FOXO factors are prominent in controlling survival and longevity in T cells [115], and a possibility is that the acquired immune system has coopted this ancient pathway for maintaining self-renewal properties as a means of accommodating life-long parasitic infections. We speculate that, while chronic pathogens accommodate FOXO1-expressing long-lived CD8 T cells (selected to maintain host viability), there may be tumors that have evolved mechanisms to impede the activity of FOXO1 in T cells.
To understand the program of gene expression consistent with tumor immunity or control of latent or persistent viral infections, a future task will be to describe how FOXO transcription factors are themselves regulated, and how they regulate downstream gene targets. This challenge is highlighted by the observation that multiple studies using single cell transcriptional profiling for genes that characterize T cells responsive to check-point therapy have identified a common program of gene expression that has not included Foxo1 [11,12••,13••]—even though it is clearly controlling key aspects of the definitive program of gene expression. This is likely due to the fact that FOXO transcription factors, although regulated at the level of gene expression [116], are prominently regulated by most known post-transcriptional mechanisms.
Foxo1 mRNA amounts are altered by multiple micro RNAs in different cell types [117–119] including CD8+ T cells via miR-150 [120]. In addition, FOXO factors are potently regulated by post-translational modifications, and most prominent are the inactivating serine-threonine phosphorylations mediated by AKT (but probably not SGK1 [121]). Two pathways converge to activate AKT: PI3K (Phosphoinositide 3-kinase) activation of PDPK1 (3-Phosphoinositide-Dependent Protein Kinase 1, PDK1) that phosphorylates AKT at Thr308 [122], and the mTORC2 complex that phosphorylates AKT at Ser473 [123] (Figure 1). Loss of mTORC2 signaling enhances CD8+ memory cell generation [124], presumably by preventing the activation of AKT and thus the inactivation of FOXO1. The outcome of phosphorylation of FOXO1 by AKT is its association with 14-3-3, exclusion from the nucleus, and degradation through ubiquitination [125,126]. AKT phosphorylation can also be directly countered by phosphatase 2A [127]. The control of PI3K and mTORC2 signaling in T cells, especially under conditions of chronic infection or neoplasia is complex and has not been studied in detail [128]; however, PI3K delta syndrome is a primary immunodeficiency resulting from a gain-of-function in PI3K-delta, in principle leading to the increased constitutive phosphorylation of FOXO transcription factors among other possibilities. The phenotypic effects of this congenital mutation are varied between affected individuals, even within the same family, but they often include severe and recurrent α, β, and γ herpes family viral infections [129].
Figure 1.
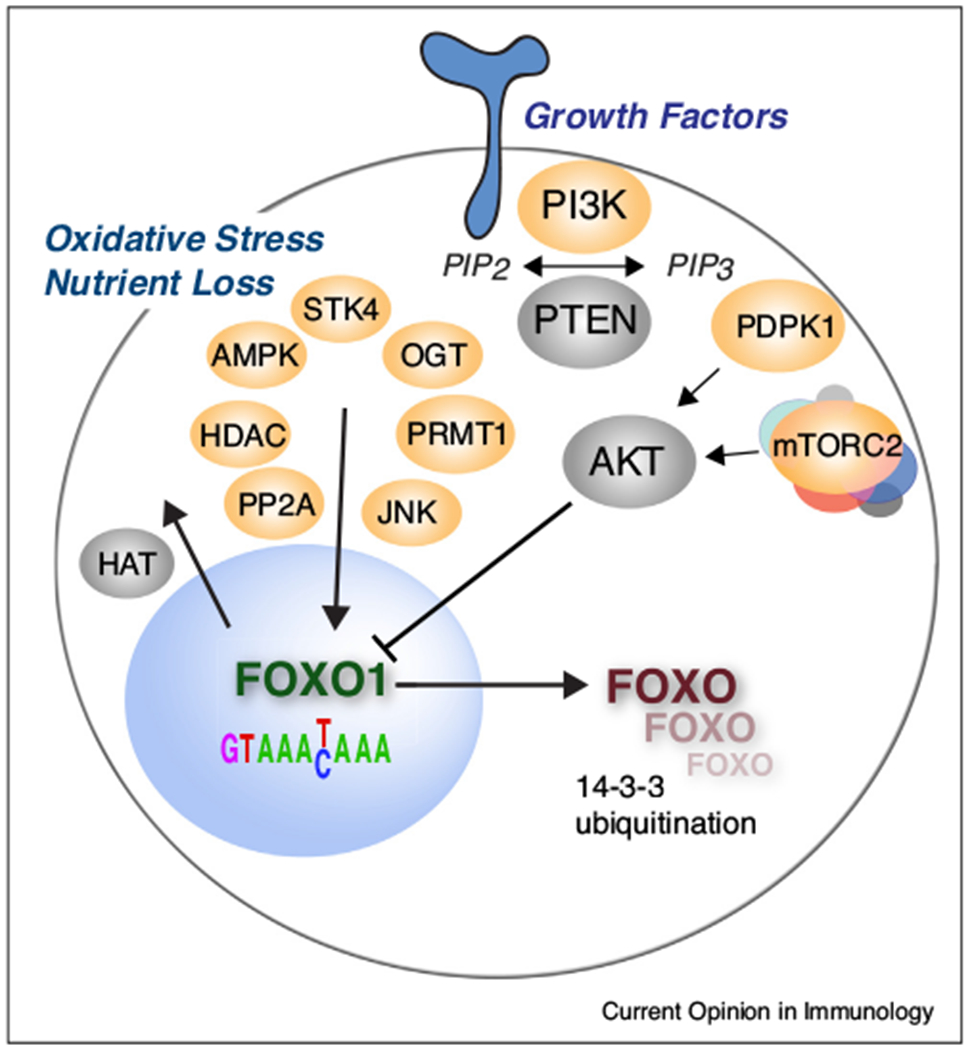
Opposing influences on FOXO1 cellular localization and transcriptional activity. PI3K, Phosphoinositide 3-kinase; PTEN, Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase; PDPK1, 3-Phosphoinositide-Dependent Protein Kinase 1 (PDK1); mTORC2, mTOR complex 2 (consisting of 7 components); AKT, (Protein Kinase B); PP2A, Protein phosphatase 2A; HDAC, Histone deacetylase; AMPK, 5′ adenosine monophosphate-activated protein kinase; STK4, Serine/Threonine Kinase 4 (MST1); OGT, O-Linked N-Acetylglucosamine (GlcNAc) Transferase; PRMT1, protein arginine methyltransferase; JNK, Jun N-terminal protein kinase.
Other serine-threonine phosphorylation sites on FOXO1 oppose AKT-mediated nuclear exclusion. 5′ adenosine monophosphate-activated protein kinase (AMPK) responds to reduced energy levels and activates FOXO transcription factors indirectly, and through direct phosphorylation [130–132]. Jun N-terminal protein kinase (JNK), responding to oxidative stress, phosphorylates FOXO1, and this enhances its nuclear localization, possibly by dissociation from 14-3-3 [133]. The biological importance of activating serine-threonine FOXO1 phosphorylations is exemplified by familial deficiencies in STK4 (MST1). These congenital deficiencies, resulting from consanguineal marriages, arose from STK4 nonsense mutations that were found to be associated with several combined skin and respiratory infections and multiple herpes virus family infections. Patients experienced a progressive loss of naive CD4 T cells and central memory T cells that correlated with cellular abnormalities including a loss of FOXO1 expression and its downstream targets, most notably IL7R. Further studies showed that Stk4 loss-of-function mice showed a very similar phenotype [134,135].
Additional FOXO1 post-translational modifications mediated by oxidative stress or restricted nutrients include acetylation regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs) [136], glycosylation catalyzed by O-Linked N-Acetylglucosamine (GlcNAc) Transferase (OGT) [137], methylation carried out by protein arginine methyltransferase (PRMT) [138], and ubiquitination at multiple sites [100,139–141] (Figure 1). These modifications also affect nuclear versus cytoplasmic localization, and in addition, stability, turnover, and transcriptional activity.
A large caveat is that most of the studies characterizing signaling pathways important for FOXO1 regulation have not been carried out in T cells, but rather in other differentiated cell types especially those that regulate metabolism such as liver, fat and muscle. We do not understand the progression of FOXO1 posttranslational modifications in detail in any T cell subset, and certainly not in T cells found under conditions of chronic virus infection or neoplasia. On top of this, all of these signaling modules, and especially those involving mTOR are entangled such that drawing linear pathways is mainly uninformative. The means by which FOXO1 regulates physiology in each differentiated cell-type may need to be studied from a systems analysis approach that would begin by correlating FOXO protein modifications, intracellular localization, chromatin binding and gene expression in T cell subsets (or individual cells) at different times subsequent to viral infection or tumor growth. If the goal is to understand the biology underlying long-term stem-like activity and a continuing capacity for immune activation, a reasonable approach is to compare wildtype and Foxo1-deficient cells.
Acknowledgements
We wish to posthumously credit our valued colleague, Arnaud Delpoux, whose conversations and ideas made this review possible. Funding of this work came from National Institutes of Health [grant numbers R01AI131081 and R01AI073885] to SMH. NM has been supported by the Tata Institute for Genetics and Society.
Footnotes
Conflict of interest statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Lefrancois L, Obar JJ: Once a killer, always a killer: from cytotoxic T cell to memory cell. Immunol Rev 2010, 235:206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Youngblood B, Hale JS, Ahmed R: T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology 2013, 139:277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gray SM, Kaech SM, Staron MM: The interface between transcriptional and epigenetic control of effector and memory CD8(+) T-cell differentiation. Immunol Rev 2014, 261:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosato PC, Beura LK, Masopust D: Tissue resident memory T cells and viral immunity. Curr Opin Virol 2016, 22:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Omilusik KD, Goldrath AW: Remembering to remember: T cell memory maintenance and plasticity. Curr Opin Immunol 2019, 58:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaech SM, Cui W: Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 2012, 12:749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin MD, Badovinac VP: Defining memory CD8 T cell. Front Immunol 2018, 9:2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.•.Zinkernagel RM, Hengartner H: On immunity against infections and vaccines: credo 2004. Scand J Immunol 2004, 60:9–13. [DOI] [PubMed] [Google Scholar]; Although this perspective paper is old, it makes an important point concerning the immunology of viral infections. That is, for many or most acute viral infections, neutralizing antibodies are protective for a secondary infection, thus making the reactivation of CD8 T cells difficult to measure and possibly irrelevant to immunity. To examine anamnestic T cell responses, we have to use mice lacking B cells, carry out an adoptive transfer, or rechallenge mice with a second recombinant virus expressing the original T cell epitope.
- 9.Kahan SM, Wherry EJ, Zajac AJ: T cell exhaustion during persistent viral infections. Virology 2015, 479-480:180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.••.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH et al. Defining CD8(+) T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537:417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]; This was the first work showing the gene expression associated with T cells that responded to ‘checkpoint’ therapy.
- 11.Miron M, Kumar BV, Meng W, Granot T, Carpenter DJ, Senda T, Chen D, Rosenfeld AM, Zhang B, Lerner H et al. Human lymph nodes maintain TCF-1hi memory T cells with high functional potential and clonal diversity throughout life. J Immunol 2018, 201:2132–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.••.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen jH, Frederick DT, Barzily-Rokni M et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 2018, 175:998–1013.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using signal cell sequencing, this massive study defines the T cell subset associated with a successful response to check-point therapy in patients with melanoma. This subset is identified by the expression of TCF7 and other genes that constitute targets of FOXO1.
- 13.••.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S et al. Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 2019, 50:195–211.e10. [DOI] [PubMed] [Google Scholar]; Studies in human beings and mice show that response to checkpoint therapy relies on the proliferation of a self-renewing, stem-like population that expresses TCF7.
- 14.Voehringer D, Blaser C, Brawand P, Raulet DH, Hanke T, Pircher H: Viral infections induce abundant numbers of senescent CD8 T cells. J Immunol 2001, 167:4838–4843. [DOI] [PubMed] [Google Scholar]
- 15.Hikono H, Kohlmeier JE, Takamura S, Wittmer ST, Roberts AD, Woodland DL: Activation phenotype, rather than central- or effector-memory phenotype, predicts the recall efficacy of memory CD8+ T cells. J Exp Med 2007, 204:1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Utzschneider DT, Alfei F, Roelli P, Barras D, Chennupati V, Darbre S, Delorenzi M, Pinschewer DD, Zehn D: High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J Exp Med 2016. 213:1819–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Valen L: A new evolutionary law. Evol Theory 1973, 1:1–30. [Google Scholar]
- 18.Ujvari B, Gatenby RA, Thomas F: The evolutionary ecology of transmissible cancers. Infect Genet Evol 2016, 39:293–303. [DOI] [PubMed] [Google Scholar]
- 19.Metzger MJ, Villalba A, Carballal MJ, Iglesias D, Sherry J, Reinisch C, Muttray AF, Baldwin SA, Goff SP: Widespread transmission of independent cancer lineages within multiple bivalve species. Nature 2016, 534:705–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fortunato A, Boddy A, Mallo D, Aktipis A, Maley CC, Pepper JW:Natural selection in cancer biology: from molecular snowflakes to trait hallmarks. Cold Spring Harb Perspect Med 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okazaki T, Honjo T: The PD-1-PD-L pathway in immunological tolerance. Trends Immunol 2006, 27:195–201. [DOI] [PubMed] [Google Scholar]
- 22.Iwai Y, Okazaki T, Nishimura H, Kawasaki A, Yagita H, Honjo T: Microanatomical localization of PD-1 in human tonsils. Immunol Lett 2002, 83:215–220. [DOI] [PubMed] [Google Scholar]
- 23.Jin HT, Ahmed R, Okazaki T: Role of PD-1 in regulating T-cell immunity. Curr Top Microbiol Immunol 2011, 350:17–37. [DOI] [PubMed] [Google Scholar]
- 24.Anderson RM, May RM: Population biology of infectious diseases: part I. Nature 1979, 280:361–367. [DOI] [PubMed] [Google Scholar]
- 25.May RM, Anderson RM: Population biology of infectious diseases: part II. Nature 1979, 280:455–461. [DOI] [PubMed] [Google Scholar]
- 26.Klenerman P, Oxenius A: T cell responses to cytomegalovirus. Nat Rev Immunol 2016, 16:367–377. [DOI] [PubMed] [Google Scholar]
- 27.Ayres JS, Schneider DS: Tolerance of infections. Annu Rev Immunol 2012, 30:271–294. [DOI] [PubMed] [Google Scholar]
- 28.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD: IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001,410:1107–1111. [DOI] [PubMed] [Google Scholar]
- 29.Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA: Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 2013, 13:759–771. [DOI] [PubMed] [Google Scholar]
- 30.Moskophidis D, Lechner F, Pircher H, Zinkernagel RM: Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362:758–761. [DOI] [PubMed] [Google Scholar]
- 31.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R: Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 1998, 188:2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gallimore A, Glithero A, Godkin A, Tissot AC, Plückthun A, Elliott T, Hengartner H, Zinkernagel R: Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med 1998, 187:1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zinkernagel RM, Planz O, Ehl S, Battegay M, Odermatt B, Klenerman P, Hengartner H: General and specific immunosuppression caused by antiviral T-cell responses. Immunol Rev 1999, 168:305–315. [DOI] [PubMed] [Google Scholar]
- 34.Radziewicz H, Ibegbu CC, Fernandez ML, Workowski KA, Obideen K, Wehbi M, Hanson HL, Steinberg JP, Masopust D, Wherry EJ et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol 2007, 81:2545–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El-Far M, Halwani R, Said E, Trautmann L, Doroudchi M, Janbazian L, Fonseca S, van Grevenynghe J, Yassine-Diab B, Sekaly RP, Haddad EK: T-cell exhaustion in HIV infection. Curr HIV/AIDS Rep 2008, 5:13–19. [DOI] [PubMed] [Google Scholar]
- 36.Wykes MN, Horne-Debets JM, Leow CY, Karunarathne DS: Malaria drives T cells to exhaustion. Front Microbiol 2014, 5:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schietinger A, Greenberg PD: Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol 2014, 35:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thorp EB, Stehlik C, Ansari MJ: T-cell exhaustion in allograft rejection and tolerance. Curr Opin Organ Transplant 2015, 20:37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG: T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 2015, 523:612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wieland D, Kemming J, Schuch A, Emmerich F, Knolle P, Neumann-Haefelin C, Held W, Zehn D, Hofmann M, Thimme R: TCF1+ hepatitis C virus-specific CD8+ T cells are maintained after cessation of chronic antigen stimulation. Nat Commun 2017, 8:15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLane LM, Abdel-Hakeem MS, Wherry EJ: CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol 2019, 37:457–495. [DOI] [PubMed] [Google Scholar]
- 42.Han S, Asoyan A, Rabenstein H, Nakano N, Obst R: Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc Natl Acad Sci USA 2010, 107:20453–20458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walton S, Mandaric S, Oxenius A: CD4 T cell responses in latent and chronic viral infections. Front Immunol 2013, 4:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R: Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27:670–684. [DOI] [PubMed] [Google Scholar]
- 45.Legat A, Speiser DE, Pircher H, Zehn D, Fuertes Marraco SA: Inhibitory receptor expression depends more dominantly on differentiation and activation than “exhaustion” of human CD8 T cells. Front Immunol 2013, 4:455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science 2007, 315:1687–1691. [DOI] [PubMed] [Google Scholar]
- 47.Verbist KC, Guy CS, Milasta S, Liedmann S, Kamihski MM, Wang R, Green DR: Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature 2016, 532:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kakaradov B, Arsenio J, Widjaja CE, He Z, Aigner S, Metz PJ, Yu B, Wehrens EJ, Lopez J, Kim SH et al. Early transcriptional and epigenetic regulation of CD8+ T cell differentiation revealed by single-cell RNA sequencing. Nat Immunol 2017, 18:422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ: Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 2012, 37:1130–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agnellini P, Wolint P, Rehr M, Cahenzli J, Karrer U, Oxenius A: Impaired NFAT nuclear translocation results in split exhaustion of virus-specific CD8+ T cell functions during chronic viral infection. Proc Natl Acad Sci U S A 2007, 104:4565–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, Julg B, Jesneck JL, Brosnahan K, Imam S et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med 2010, 16:1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 2015, 42:265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, Intlekofer AM, Boss JM, Reiner SL, Weinmann AS, Wherry EJ: Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol 2011, 12:663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff Ek, Robertson EJ, Lauer GM, Reiner SL, Wherry EJ: Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012, 338:1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, Johnson RS, Goldrath AW: Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol 2013, 14:1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, Cui G, Li MO, Kaech SM: The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity 2014, 41:802–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stephen TL, Rutkowski MR, Allegrezza MJ, Perales-Puchalt A, Tesone AJ, Svoronos N, Nguyen JM, Sarmin F, Borowsky ME, Tchou J, Conejo-Garcia JR: Transforming growth factor β-mediated suppression of antitumor T cells requires FoxP1 transcription factor expression. Immunity 2014, 41:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.••.Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, López-Moyado IF, Georges RO, Zhang W, Onodera A et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc Natl Acad Sci U S A 2019, 116:12410–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]; A series of papers showed that the transcription factor, TOX, is important or essential in the establishment of exhaustion. This particular study shows that TCR signaling induces NFAT, and even in the absence of its partner AP1 factors (FOS-JUN), there occurs the induction of Nr4a family members, Tox, and Tox2. Deletion of both Tox and Tox2 or all three Nr4a paralogs facilitated a CAR-T response and survival of tumor-bearing mice. This was the only study to analyze the genetics of all the Tox and Nr4a paralogs in the induction of exhaustion.
- 59.•.Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571:270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that deletion of Tox was sufficient to diminish the expression of inhibitory receptors, for example, PD1, and yet Tox-deleted T cells remained dysfunctional. The authors assert that this uncouples exhaustion from inhibitory receptor expression. A possible complication is the potential for redundancy in between the two Tox paralogs: Tox and Tox2.
- 60.•.Wang X, He Q, Shen H, Xia A, Tian W, Yu W, Sun B: TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J Hepatol 2019, 71:731–741. [DOI] [PubMed] [Google Scholar]; This study purports to show that TOX acts via endocytic recycling of PD1 between the cell surface and the endosome. The contention is that TOX acts directly to enhance cell surface expression of PD1, and not necessarily as a transcription factor.
- 61.••.Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, von Hoesslin M, Cullen JG, Fan Y et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019, 571:265–269. [DOI] [PubMed] [Google Scholar]; Here Tox was studied in chronic viral infections, and an important point was that the DNA binding domain is important for TOX function. Thus, TOX most likely acts as a transcription factor to promote exhaustion. Consistent with the selective advantage of the exhaustion response, T cells with a Tox DNA binding domain deletion showed increased effector function and caused more immunopathology; yet, these T cells exhibited a massive decline in the self-renewing subpopulation expressing TCF7.
- 62.•.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571:211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]; TOX was shown here to be essential for exhaustion, and participate in the regulation of its own expression, an example of positive-feedback control (as opposed to feed-forward control that is independent of the parameter in question, in this case PD1 expression). They showed that strong expression of TOX results in a more permanent commitment to the exhaustion state.
- 63.••.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1-CD8+ tumor-infiltrating T cells. Immunity 2019, 50:181–194.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]; Experiments demonstrated the requirement for TCF7 in T cells able to carry out an anti-tumor response upon treatment with checkpoint therapy.
- 64.Jin X, Bauer DE, Tuttleton SE, Lewin S, Gettie A, Blanchard J, Irwin CE, Safrit JT, Mittler J, Weinberger L et al. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med 1999, 189:991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.••.Utzschneider DT, Legat A, Fuertes Marraco SA, Carrie L, Luescher I, Speiser DE, Zehn D: T cells maintain an exhausted phenotype after antigen withdrawal and population reexpansion. Nat Immunol 2013, 14:603–610. [DOI] [PubMed] [Google Scholar]; This study established the concept that PD1+ T cells from a chronic viral infection, that is, exhausted, are able to contain a subsequent response measured by adoptive transfer. The authors propose that T cells in the ‘exhausted’ state are still able to limit viral replication, and yet could not cause excessive immunopathology.
- 66.Zehn D, Utzschneider DT, Thimme R: Immune-surveillance through exhausted effector T-cells. Curr Opin Virol 2016, 16:49–54. [DOI] [PubMed] [Google Scholar]
- 67.Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C et al. TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity 2019, 51:840–855.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kratchmarov R, Magun AM, Reiner SL: TCF1 expression marks self-renewing human CD8+ T cells. Blood Adv 2018, 2:1685–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raghu D, Xue HH, Mielke LA: Control of lymphocyte fate, infection, and tumor immunity by TCF-1. Trends Immunol 2019, 40:1149–1162. [DOI] [PubMed] [Google Scholar]
- 70.Rothenberg EV: Transcriptional drivers of the T-cell lineage program. Curr Opin Immunol 2012, 24:132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.De Obaldia ME, Bhandoola A: Transcriptional regulation of innate and adaptive lymphocyte lineages. Annu Rev Immunol 2015, 33:607–642. [DOI] [PubMed] [Google Scholar]
- 72.Xing S, Li F, Zeng Z, Zhao Y, Yu S, Shan Q, Li Y, Phillips FC, Maina PK, Qi HH et al. Tcf1 and Lef1 transcription factors establish CD8(+)T cell identity through intrinsic HDAC activity. Nat Immunol 2016, 17:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oosterwegel MA, van de Wetering ML, Holstege FC, Prosser HM, Owen MJ, Clevers HC: TCF-1, a T cell-specific transcription factor of the HMG box family, interacts with sequence motifs in the TCR beta and TCR delta enhancers. Int Immunol 1991, 3:1189–1192. [DOI] [PubMed] [Google Scholar]
- 74.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med 2009, 15:808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao DM, Yu S, Zhou X, Haring JS, Held W, Badovinac VP, Harty JT, Xue HH: Constitutive activation of Wnt signaling favors generation of memory CD8 T cells. J Immunol 2010, 184:1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH: Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 2010, 33:229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.•.Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, Held W: Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8T cell memory. Proc Natl Acad Sci U S A 2010, 107:9777–9782. [DOI] [PMC free article] [PubMed] [Google Scholar]; The essential role of TCF7 in the formation of T cell memory was established with this study.
- 78.Sharma A, Chen Q, Nguyen T, Yu Q, Sen JM: T cell factor-1 and β-catenin control the development of memory-like CD8 thymocytes. J Immunol 2012, 188:3859–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Delpoux A, Lai CY, Hedrick SM, Doedens AL: FOXO1 opposition of CD8(+) T cell effector programming confers early memory properties and phenotypic diversity. Proc Natl Acad Sci U S A 2017, 114:E8865–E8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Danilo M, Chennupati V, Silva JG, Siegert S, Held W: Suppression of Tcf1 by inflammatory cytokines facilitates effector CD8 T cell differentiation. Cell Rep 2018, 22:2107–2117. [DOI] [PubMed] [Google Scholar]
- 81.••.Hess Michelini R, Doedens AL, Goldrath AW, Hedrick SM:Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med 2013, 210:1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study we established FOXO1 as essential to the formation of memory T cells capable of a secondary, anamnestic response to rechallenge with an acute virus. We also established the requirement for FOXO1 in the post-activation expression of TCF7.
- 82.Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA: Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity 2012, 36:374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tejera MM, Kim EH, Sullivan JA, Plisch EH, Suresh M: FoxO1 controls effector-to-memory transition and maintenance of functional CD8 T cell memory. J Immunol 2013, 191:187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.••.Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO: The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity 2013, 39:286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]; This report established FOXO1 control of Tcf7 as an essential control point for T cell memory formation.
- 85.•.Utzschneider DT, Delpoux A, Wieland D, Huang X, Lai CY, Hofmann M, Thimme R, Hedrick SM: Active maintenance of T cell memory in acute and chronic viral infection depends on continuous expression of FOXO1. Cell Rep 2018, 22:3454–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this study we compared the requirement for continuous FOXO1 expression in acute and chronic infections — post infection. Upon deletion of Foxo1, central memory T cells declined and differentiated into a phenotype reminiscent of effector cells. This decline was accelerated in a chronic infection in comparison with an acute infection.
- 86.•.Delpoux A, Michelini RH, Verma S, Lai CY, Omilusik KD, Utzschneider DT, Redwood AJ, Goldrath Aw, Benedict CA, Hedrick SM: Continuous activity of Foxo1 is required to prevent anergy and maintain the memory state of CD8+T cells. J Exp Med 2018, 215:575–594. [DOI] [PMC free article] [PubMed] [Google Scholar]; The need for memory T cells in the control of a chronic cytomegalovirus infection was examined by deleting Foxo1 at different times following infection. Results established that Foxo1 is continually required to maintain stem-like responsive T cells.
- 87.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ: Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 2013, 10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010, 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ:Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 2006, 442:91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pekowska A, Benoukraf T, Zacarias-Cabeza J, Belhocine M, Koch F, Holota H, Imbert J, Andrau JC, Ferrier P, Spicuglia S: H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J 2011, 30:4198–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fabre S, Carrette F, Chen J, Lang V, Semichon M, Denoyelle C, Lazar V, Cagnard N, Dubart-Kupperschmitt A, Mangeney M et al. FOXO1 regulates L-selectin and a network of human T cell homing molecules downstream of phosphatidylinositol 3-kinase. J Immunol 2008, 181:2980–2989. [DOI] [PubMed] [Google Scholar]
- 92.Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, Hedrick SM: Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol 2009, 10:176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gubbels Bupp MR, Edwards B, Guo C, Wei D, Chen G, Wong B, Masteller E, Peng SL: T cells require Foxo1 to populate the peripheral lymphoid organs. Eur J Immunol 2009, 39:2991–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ouyang W, Beckett O, Flavell RA, Li MO: An essential role of the forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity 2009, 30:358–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kallies A: Distinct regulation of effector and memory T-cell differentiation. Immunol Cell Biol 2008, 86:325–332. [DOI] [PubMed] [Google Scholar]
- 96.Colpitts SL, Dalton NM, Scott P: IL-7 receptor expression provides the potential for long-term survival of both CD62Lhigh central memory T cells and Th1 effector cells during Leishmania major infection. J Immunol 2009, 182:5702–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Feng X, Wang H, Takata H, Day TJ, Willen J, Hu H: Transcription factor Foxp1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nat Immunol 2011, 12:544–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Salih DA, Brunet A: FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol 2008, 20:126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Burgering BM, Medema RH: Decisions on life and death: FOXO forkhead transcription factors are in command when PKB/Akt is off duty. J Leukoc Biol 2003, 73:689–701. [DOI] [PubMed] [Google Scholar]
- 100.Eijkelenboom A, Burgering BM: FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol 2013, 14:83–97. [DOI] [PubMed] [Google Scholar]
- 101.Hand SC, Denlinger DL, Podrabsky JE, Roy R: Mechanisms of animal diapause: recent developments from nematodes, crustaceans, insects, and fish. Am J Physiol Regul Integr Comp Physiol 2016, 310:R1193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lin K, Dorman JB, Rodan A, Kenyon C: daf-16: an HNF-3/ forkhead family member that can function to double the lifespan of Caenorhabditis elegans. Science 1997, 278:1319–1322. [DOI] [PubMed] [Google Scholar]
- 103.Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G: The fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 1997, 389:994–999. [DOI] [PubMed] [Google Scholar]
- 104.Hwangbo DS, Gershman B, Tu MP, Palmer M, Tatar M: Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 2004, 429:562–566. [DOI] [PubMed] [Google Scholar]
- 105.Boehm AM, Khalturin K, Anton-Erxleben F, Hemmrich G, Klostermeier UC, Lopez-Quintero JA, Oberg HH, Puchert M, Rosenstiel P, Wittlieb J, Bosch TC: FoxO is a critical regulator of stem cell maintenance in immortal hydra. Proc Natl Acad Sci U S A 2012, 109:19697–19702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang X, Yalcin S, Lee DF, Yeh TY, Lee SM, Su J, Mungamuri SK, Rimmele P, Kennedy M, Sellers R et al. FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat Cell Biol 2011, 13:1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Takahashi K, Yamanaka S: A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol 2016, 17:183–193. [DOI] [PubMed] [Google Scholar]
- 108.Lunetta KL, D’Agostino RBS, Karasik D, Benjamin EJ, Guo CY, Govindaraju R, Kiel DP, Kelly-Hayes M, Massaro JM, Pencina MJ et al. Genetic correlates of longevity and selected age-related phenotypes: a genome-wide association study in the Framingham study. BMC Med Genet 2007, 8(Suppl. 1):S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li Y, Wang WJ, Cao H, Lu J, Wu C, Hu FY, Guo J, Zhao L, Yang F, Zhang YX et al. Genetic association of FOXO1A and FOXO3A with longevity trait in Han Chinese populations. Hum Mol Genet 2009, 18:4897–4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kenyon CJ: The genetics of ageing. Nature 2010, 464:504–512. [DOI] [PubMed] [Google Scholar]
- 111.Anselmi CV, Malovini A, Roncarati R, Novelli V, Villa F, Condorelli G, Bellazzi R, Puca AA: Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res 2009, 12:95–104. [DOI] [PubMed] [Google Scholar]
- 112.Flachsbart F, Caliebe A, Kleindorp R, Blanche H, von Eller-Eberstein H, Nikolaus S, Schreiber S, Nebel A: Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci U S A 2009, 106:2700–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zeng Y, Chen H, Ni T, Ruan R, Nie C, Liu X, Feng L, Zhang F, Lu J, Li J et al. Interaction between the FOXO1A-209 genotype and tea drinking is significantly associated with reduced mortality at advanced ages. Rejuvenation Res 2016, 19:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liang R, Ghaffari S: Stem cells seen through the FOXO lens: an evolving paradigm. Curr Top Dev Biol 2018, 127:23–47. [DOI] [PubMed] [Google Scholar]
- 115.Hedrick SM, Hess Michelini R, Doedens AL, Goldrath AW, Stone EL: FOXO transcription factors throughout T cell biology. Nat Rev Immunol 2012, 12:649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Welinder E, Mansson R, Mercer EM, Bryder D, Sigvardsson M, Murre C: The transcription factors E2A and HEB act in concert to induce the expression of FOXO1 in the common lymphoid progenitor. Proc Natl Acad Sci U S A 2011, 108:17402–17407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Saki N, Abroun S, Soleimani M, Hajizamani S, Shahjahani M, Kast RE, Mortazavi Y: Involvement of MicroRNA in T-cell differentiation and malignancy. Int J Hematol Oncol Stem Cell Res 2015, 9:33–49. [PMC free article] [PubMed] [Google Scholar]
- 118.Urbánek P, Klotz LO: Posttranscriptional regulation of FOXO expression: microRNAs and beyond. Br J Pharmacol 2017, 174:1514–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gagnon JD, Ansel KM: MicroRNA regulation of CD8+ T cell responses. Noncoding RNA Investig 2019, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ban YH, Oh SC, Seo SH, Kim SM, Choi IP, Greenberg PD, Chang J, Kim TD, Ha SJ: miR-150-mediated foxo1 regulation programs CD8(+) T cell differentiation. Cell Rep 2017, 20:2598–2611. [DOI] [PubMed] [Google Scholar]
- 121.Di Cristofano A: SGK1: The dark side of PI3K signaling. Curr Top Dev Biol 2017, 123:49–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P: Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase balpha. Curr Biol 1997, 7:261–269. [DOI] [PubMed] [Google Scholar]
- 123.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B: SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127:125–137. [DOI] [PubMed] [Google Scholar]
- 124.Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, Delgoffe GM, Powell JD: mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest 2015, 125:2090–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rena G, Prescott AR, Guo S, Cohen P, Unterman TG: Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in regulating 14-3-3 binding, transactivation and nuclear targetting. Biochem J 2001, 354:605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Plas DR, Thompson CB: Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem 2003, 278:12361–12366. [DOI] [PubMed] [Google Scholar]
- 127.Yan L, Lavin VA, Moser LR, Cui Q, Kanies C, Yang E: PP2A regulates the pro-apoptotic activity of FOXO1. J Biol Chem 2008, 283:7411–7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Myers DR, Wheeler B, Roose JP: mTOR and other effector kinase signals that impact T cell function and activity. Immunol Rev 2019, 291:134–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, Goodlad JR, Farmer G, Steele CL, Leahy TR et al. Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: a large patient cohort study. J Allergy Clin Immunol 2017, 139:597–606.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A: An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol 2007, 17:1646–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yun H, Park S, Kim MJ, Yang WK, Im DU, Yang KR, Hong J, Choe W, Kang I, Kim SS, Ha J: AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J 2014, 281:4421–4438. [DOI] [PubMed] [Google Scholar]
- 132.Yadav H, Devalaraja S, Chung ST, Rane SG: TGF-β1/Smad3 pathway targets PP2A-AMPK-FoxO1 signaling to regulate hepatic gluconeogenesis. J Biol Chem 2017, 292:3420–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Weng Q, Liu Z, Li B, Liu K, Wu W, Liu H: Oxidative stress induces mouse follicular granulosa cells apoptosis via JNK/FoxO1 pathway. PLoS One 2016, 11:e0167869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nehme NT, Schmid JP, Debeurme F, André-Schmutz I, Lim A, Nitschke P, Rieux-Laucat F, Lutz P, Picard C, Mahlaoui N et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood 2012, 119:3458–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Abdollahpour H, Appaswamy G, Kotlarz D, Diestelhorst J, Beier R, Schäffer AA, Gertz Em, Schambach A, Kreipe HH, Pfeifer D et al. The phenotype of human STK4 deficiency. Blood 2012, 119:3450–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Accili D, Arden KC: FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004, 117:421–426. [DOI] [PubMed] [Google Scholar]
- 137.Rahman MM, Stuchlick O, El-Karim EG, Stuart R, Kipreos ET, Wells L: Intracellular protein glycosylation modulates insulin mediated lifespan in C. elegans. Aging (Albany NY) 2010, 2:678–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K, Mukai H, Kasuya Y, Fukamizu A: Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell 2008, 32:221–231. [DOI] [PubMed] [Google Scholar]
- 139.Calnan DR, Brunet A: The FoxO code. Oncogene 2008, 27:2276–2288. [DOI] [PubMed] [Google Scholar]
- 140.Wang Z, Yu T, Huang P: Post-translational modifications of FOXO family proteins (review). Mol Med Rep 2016, 14:4931–4941. [DOI] [PubMed] [Google Scholar]
- 141.Brown AK, Webb AE: Regulation of FOXO factors in mammalian cells. Curr Top Dev Biol 2018, 127:165–192. [DOI] [PMC free article] [PubMed] [Google Scholar]