

Abstract
Capsid allosteric modulators (CAMs) inhibit the encapsidation of hepatitis B virus (HBV) pregenomic RNA (pgRNA), which contains a pathogen‐associated molecular pattern motif. However, the effect of CAMs on the innate immune response of HBV‐infected hepatocytes remains unclear, and we examined this effect in this study. Administration of a CAM compound, BAY41‐4109 (BAY41), to HBV‐infected primary human hepatocytes (PHHs) did not change the total cytoplasmic pgRNA levels but significantly reduced intracapsid pgRNA levels, suggesting that BAY41 increased extracapsid pgRNA levels in the cytoplasm. BAY41 alone did not change the intracellular interferon (IFN)–stimulated gene (ISG) expression levels. However, BAY41 enhanced antiviral ISG induction by IFN‐α in HBV‐infected PHHs but did not change ISG induction by IFN‐α in uninfected PHHs. Compared with BAY41 or IFN‐α alone, coadministration of BAY41 and IFN‐α significantly suppressed extracellular HBV‐DNA levels. HBV‐infected human liver–chimeric mice were treated with vehicle, BAY41, pegylated IFN‐α (pegIFN‐α), or BAY41 and pegIFN‐α together. Compared with the vehicle control, pegIFN‐α highly up‐regulated intrahepatic ISG expression levels, but BAY41 alone did not change these levels. The combination of BAY41 and pegIFN‐α further enhanced intrahepatic antiviral ISG expression, which was up‐regulated by pegIFNα. The serum HBV‐DNA levels in mice treated with the combination of BAY41 and pegIFN‐α were the lowest observed in all the groups. Conclusion: CAMs enhance the host IFN response when combined with exogenous IFN‐α, likely due to increased cytoplasmic extracapsid pgRNA.
Abbreviations
- BAY41
BAY41‐4109
- CAM
capsid allosteric modulator
- cccDNA
covalently closed circular DNA
- CHB
chronic hepatitis B
- DAPI
4′,6‐diamidino‐2‐phenylindole
- dpi
days post infection
- GEq
genome equivalents
- HBc
hepatitis B core
- HBeAg
hepatitis B e antigen
- HBsAg
hepatitis B surface antigen
- HBV
hepatitis B virus
- IFN
interferon
- IRF
interferon regulatory factor
- ISG
interferon‐stimulated gene
- mRNA
messenger RNA
- N.D.
not detected
- NTCP
sodium‐taurocholate cotransporting polypeptide
- NUC
nucleos(t)ide analog
- OAS1
2'‐5'‐oligoadenylate synthetase 1
- PCR
polymerase chain reaction
- pegIFN‐α
pegylated IFNα
- pgRNA
pregenomic RNA
- PHH
primary human hepatocyte
- Poly (I:C)
polyinosinic‐polycytidylic acid
- RIG‐I
retinoic acid–inducible gene‐I
- TFV
tenofovir
Hepatitis B virus (HBV) is a global health concern. The World Health Organization estimates that 887,000 people living with HBV died in 2015, and most of these deaths occurred due to cirrhosis or hepatocellular carcinoma.( 1 ) The goal of chronic hepatitis B (CHB) treatment is to prevent the development of cirrhosis or hepatocellular carcinoma, and the ideal goal of antiviral therapy is to eliminate viral covalently closed circular DNA (cccDNA) and genome‐integrated viral DNA from the host liver. However, it is widely acknowledged that the realistic goal of therapy is the persistent suppression of viral replication and protein expression, as indicated by the sustained loss of hepatitis B surface antigen (HBsAg), which is considered a functional cure.( 2 , 3 ) The currently available drugs for treating CHB are nucleos(t)ide analogs (NUCs) and interferons (IFNs). Pegylated IFN‐α (pegIFN‐α) is more effective than NUCs in lowering HBsAg levels( 2 ); however, the annual rates of HBsAg loss due to treatment with NUCs and pegIFN‐α are reported to be 1%‐3% and 3%‐4%, respectively,( 4 ) which are insufficient for achieving functional cures. New drugs with different mechanisms of action and more effective combination therapies need to be developed. To date, new therapeutic targets in the viral life cycle and host immune system have been studied, and some novel antiviral agents are in clinical trials.( 5 , 6 )
Capsid allosteric modulators or capsid assembly modulators (CAMs) are anti‐HBV drugs currently in development that are expected to be introduced into clinical practice in the near future.( 5 , 6 , 7 ) CAMs inhibit the encapsidation of viral pregenomic RNA (pgRNA) by producing empty capsids( 8 ) or by misdirecting HBV core (HBc) proteins toward aberrant aggregation and degradation.( 9 , 10 ) The inhibition of pgRNA encapsidation by CAMs leads to reduction in extracellular HBV pgRNA levels, as previously described in in vitro ( 11 , 12 ) and in vivo ( 13 ) studies. On the other hand, the decrease in encapsidated pgRNA by CAMs may result in an increase in the amount of nonencapsidated pgRNA in the cells. HBV pgRNA contains a pathogen‐associated molecular pattern (PAMP) motif recognized by retinoic acid–inducible gene‐I (RIG‐I),( 14 ) and the host IFN response is transiently induced early after HBV infection.( 14 , 15 ) On the other hand, HBV barely induces the host IFN response in the context of chronic infection.( 16 , 17 ) CAMs may cause some changes in the intracellular immune response against pgRNA by inhibiting the encapsidation of pgRNA. Klumpp et al.( 13 ) previously reported that intrahepatic interferon‐stimulated gene (ISG) expression did not differ between livers of HBV‐infected human liver–chimeric uPA/SCID mice treated with CAM and pegIFN‐α for 42 days and those treated with pegIFN‐α alone for 42 days. However, there has been minimal discussion about the effect of CAM treatment on the intracellular IFN response in HBV‐infected hepatocytes, especially in the early phase of treatment. In the present study, we aimed to elucidate the effect of CAM treatment on intracellular HBV pgRNA and host IFN responses at the early phase of treatment.
Materials and Methods
Generation of Human Liver–Chimeric Mice
TK‐NOG‐based human liver‐chimeric mice( 18 ) were generated and provided by the Central Institute for Experimental Animals, Kawasaki, Japan. Serum human albumin levels were measured as described previously,( 19 ) and the human liver replacement rate was estimated according to a formula described in a previous report( 20 ) (Supporting Materials and Methods).
Isolation and Culture of Primary Human Hepatocytes From Human Liver–Chimeric Mice
Primary human hepatocytes (PHHs) were isolated from human liver–chimeric mice with estimated human liver–replacement rates of greater than 70% using a two‐step collagenase‐pronase liver perfusion method, as previously described.( 19 ) The PHHs were seeded on type I collagen‐coated plates (AGC Techno Glass Co., Ltd., Shizuoka, Japan) and cultured with medium (Supporting Materials and Methods), which was changed every 5 days. All of the experiments were initiated within 7 days after the isolation of the cells from the murine livers.
Poly (I:C) Transfection
Low molecular weight poly (I:C) (InvivoGen, San Diego, CA) was transfected using Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific, Waltham, MA) and Opti‐MEM (Thermo Fisher Scientific), according to the manufacturer’s protocol.
HBV Inoculum Preparation and HBV Infection
In in vitro experiments, HBV derived from the culture supernatants of Hep38.7‐tet cells that produce HBV serotype ayw ( 21 , 22 ) was used as the inoculum. The culture supernatants were collected 4 times at 7‐day intervals, passed through a 0.45‐μm filter (Merck Millipore, Billerica, MA), and concentrated by 30‐fold using 8% PEG8000 (Promega, Madison, WI), 0.1 M NaCl, and 0.01 M 4‐(2‐hydroxyethyl)‐1‐piperazine ethanesulfonic acid. PHHs were cultured with the HBV inoculum for 24 hours in the presence of 4% PEG8000. After incubation with the indicated genome equivalents (GEq)/cell HBV inoculum, the cells were washed twice.
In in vivo experiments, the serum of a patient with CHB (genotype C, 9.29 log copies/mL) was used. The experiments were approved by the institutional review board for Clinical Research at Osaka University Hospital (12050). Written informed consent was obtained from the patient. The patient’s serum was diluted 100‐fold with saline. Human liver–chimeric mice with estimated human liver–replacement rates of greater than 40% were intravenously injected with 100 μL of diluted serum. All of the mice were male. The median age of the mice was 24 weeks, and the mean serum human albumin level at the time of infection was 3.75 mg/mL. Blood samples were collected from the external jugular vein, and serum HBV‐DNA levels were measured.
Compounds
BAY41‐4109 (BAY41), tenofovir (TFV), IFN‐α (Sumiferon), and pegIFN‐α2a solution (0.09 mg/mL; PEGASYS) were purchased from Shanghai Haoyuan Chemexpress Co., Ltd. (Shanghai, China), Fujifilm Wako Pure Chemical Corp. (Osaka, Japan), Sumitomo Dainippon Pharma Co., Ltd. (Osaka, Japan), and Chugai Pharmaceutical Co., Ltd. (Tokyo, Japan), respectively. A CAM‐N tool compound (CAM‐N) was provided by Janssen Pharmaceutica NV (Beerse, Belgium). In the in vitro study, the compounds were diluted to the target concentrations with DMSO and culture media. In the in vivo study, BAY41 was administered with 0.5 wt%/vol% Methyl Cellulose 400 Solution (Fujifilm Wako Pure Chemical Corp.), and pegIFN‐α2a solution was diluted 36 times in phosphate‐buffered saline before injection.
PHH Viability
In vitro, PHH viability was evaluated using WST‐8 assays with cell count reagent kit (07553; Nacalai Tesque, Kyoto, Japan). The absorbance was measured at 450 nm with VARIOSKAN LUX (Thermo Fisher Scientific) according to the manufacturer’s instructions. PHH viability was also evaluated by analyzing the confluency of PHHs with Incucyte S1 (Sartorius, Göttingen, Germany).
Treatment of HBV‐Infected Human Liver–Chimeric Mice
HBV‐infected human liver–schimeric mice with serum HBV‐DNA levels of greater than 4.1 log IU/mL 10 weeks following infection were included in the treatment experiment. The mice were assigned to four treatment groups based on their HBV‐DNA levels at the beginning of the treatment. The mice were administered 100 μL of 0.5 wt%/vol% methyl cellulose (vehicle control), 40 mg/kg BAY41 twice daily by oral gavage, 25 μg/kg pegIFN‐α (2.5 µg/mL) twice weekly by subcutaneous injection, or a combination of BAY41 and pegIFN‐α for 14 days. Blood samples were collected on days 7 and 14. At the end of the treatment, the mice were sacrificed, and the livers were collected for RNA and DNA extraction.
Measurement of HBsAg, Hepatitis B e Antigen, and HBV‐DNA Levels
The HBsAg levels were measured in in vitro samples and in 50‐fold dilutions of in vivo serum samples using an chemiluminescent enzyme immunoassay (CLEIA; LUMIPULSE Presto HBsAg‐HQ; Fujirebio, Tokyo, Japan; lower limit of detection = 0.005 IU/mL). The hepatitis B e antigen (HBeAg) levels were measured in in vitro samples using CLEIA (LUMIPULSE Presto HBeAg, Fujirebio; lower limit of detection = 1.0 cutoff index). The HBV‐DNA levels were measured using the cobas 6800/8800 system HBV (Roche Diagnostics, Tokyo, Japan; lower limit of detection = 1.0 log IU/mL). The samples were diluted 10‐fold (in vitro experiments) or 100‐fold (in vivo experiments) for analysis.
Fluorescence Immunostaining
PHHs were seeded on type I collagen‐coated chamber slides (Corning Inc., Corning, NY). Fluorescence immunostaining was performed as previously described.( 23 ) The anti‐HBc antibody clone 7B2( 23 ) was used as the primary antibody, and goat anti‐mouse immunoglobulin G (IgG) conjugated with Alexa Fluor 594 (Cell Signaling Technology, Danvers, MA) was used as the secondary antibody to detect intracellular HBc protein. The cell nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI), and the stained cells were analyzed with EVOS FL Auto 2 (Invitrogen, Thermo Fisher Scientific).
Immunohistochemistry
The liver tissue of human liver chimeric mice was fixed with 4% (vol/vol) phosphate‐buffered formalin. Paraffin‐embedded sections were immunostained with anti‐human albumin antibodies (A80‐129 A; Bethyl Laboratories Inc., Montgomery, TX) and anti‐HBV core antigen antibodies (B0586; Dako, Agilent, Santa Clara, CA).
Western Blot
The western blot procedures were performed as previously described.( 24 ) Anti‐β‐actin monoclonal antibody (Sigma‐Aldrich, St. Louis, MO) and anti‐HBc antibody clone 7B2( 23 ) were used to detect human β‐actin and HBc protein, respectively. Horseradish peroxidase–conjugated anti‐mouse IgG (Cytiva, Tokyo, Japan) was used as the secondary antibody. HBc protein levels were calculated using ImageJ software( 25 ) and normalized to β‐actin protein levels in each sample. The results are presented as relative levels with the levels of each control set to 1.
RNA Extraction and Reverse‐Transcription Quantitative Real‐Time Polymerase Chain Reaction
Total RNA was extracted from PHHs and liver tissues using the RNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. For the analysis of the pgRNA levels, the prepared RNA was treated with DNase using the RNase‐Free DNase Set (Qiagen). The RNA was reverse‐transcribed using ReverTraAce qPCR RT Master Mix (Toyobo, Tokyo, Japan) and subjected to quantitative real‐time polymerase chain reaction (PCR) using the QuantStudio 6 Flex Standard Real‐Time System (Applied Biosystems, Thermo Fisher Scientific). The mRNA expression levels of specific genes were quantified using the TaqMan Gene Expression Assays (Thermo Fisher Scientific) listed in Supporting Table S1. The primer sets listed in Supporting Table S2 were used for detection of Genotype D and Genotype C pgRNA. The expression levels of all the target genes were normalized to the quantified expression levels of human β‐actin messenger RNA (mRNA). All results are presented as relative levels with the levels of each control set to 1 unless otherwise indicated in the figure legends.
Extraction of Cytoplasmic Total pgRNA and Intracapsid pgRNA and Quantification by Reverse‐Transcription Quantitative Real‐Time PCR
To analyze the cytoplasmic total HBV pgRNA and intracapsid pgRNA, the protocol reported by Jeong et al.( 26 ) was modified. The cells were treated with a lysis buffer composed of 0.05 M Tris/HCl, 0.01 M ethylene diamine tetraacetic acid and 1% NP‐40. The lysates were centrifuged at 4°C for 15 minutes at 13,500 rpm, and the supernatants were collected and considered the cell cytoplasm lysate containing HBV capsids. The lysates were divided into two portions of equal amounts. One portion was subjected to total RNA extraction using the mirVana PARIS RNA and Native Protein Purification Kit (Invitrogen) according to the manufacturer’s protocol. The other portion was treated with recombinant DNase I (Takara Bio, Shiga, Japan), RNase (Roche, Basel, Switzerland), and 5 mM CaCl2 and MgCl2 for 3 hours at 37°C to degrade the extracapsid nucleic acids; intracapsid RNA was then extracted using the same kit. The extracted RNA was treated with DNase, reverse‐transcribed, and subjected to quantitative real‐time PCR as described previously. The intracapsid pgRNA levels per cell were normalized to the cytoplasmic total β‐actin mRNA levels, which was measured using a sample that was initially divided to extract the total RNA of the cell.
DNA Extraction and HBV cccDNA Level Quantification by Quantitative Real‐Time PCR
Total DNA was prepared from PHHs and liver tissues of human liver–chimeric mice using a Biamp DNA Mini Kit (Qiagen) and DNeasy Blood and Tissue Kit (Qiagen), respectively, according to the manufacturer’s protocols. To remove single‐stranded DNA and double‐stranded linear DNA, 165 ng of the total DNA from each PHH sample and 500 ng of the total DNA from each liver tissue sample were digested overnight at 37°C with 10 U of Plasmid‐Safe ATP‐Dependent DNase (Lucigen Corp., Middleton, WI). The digested DNA was purified and precipitated using sodium acetate, glycogen, and ethanol. The primer sets listed in Supporting Table S2 were used for detection of Genotype D and Genotype C cccDNA. The quantitative real‐time PCR protocols were previously described.( 19 ) The levels of cccDNA were normalized to the quantified expression levels of human RNase P using the TaqMan Copy Number Reference Assay (Applied Biosystems).
Statistical Analysis
The data are presented as the mean ± SEM. The differences between two groups in the in vitro and in vivo studies were determined using Student t test. For comparisons between multiple groups in the in vitro and in vivo studies, analysis of variance was performed to detect an overall difference among the groups, followed by the Tukey‐Kramer test. A value of P < 0.05 was considered to indicate statistical significance.
Results
ISG Expression Is Not Induced in PHHs by HBV Infection
PHHs were inoculated with 100 or 1,000 GEq/cell HBV (Fig. 1A). At 0.5 days post infection (dpi) (12 hours after infection), fluorescence immunostaining showed that the percentages of PHHs positive for the HBc protein were 27.3% ± 2.5% and 73.2% ± 3.0%, respectively, and that the HBc positive rates did not differ between 0.5 and 20 dpi (Fig. 1B). Both the intracellular HBc expression levels and the intracellular total pgRNA levels significantly increased from 0.5 dpi to 20 dpi (Fig. 1C,D). There were no differences in the gene‐expression levels of ISGs with anti‐HBV activities( 27 ) between the HBV‐infected and uninfected PHHs at either 0.5 dpi or 20 dpi (Fig. 1E). Next, we examined whether HBV‐infected PHHs have the ability to induce antiviral ISG expression in response to poly (I:C), an agonist of RIG‐I and toll‐like receptor (TLR) 3. The antiviral ISGs in the HBV‐infected PHHs were robustly induced in response to poly (I:C) transfection (Fig. 1F). The induction of ISGs by poly (I:C) transfection was more potent in PHHs compared with HepG2 cells transfected with sodium‐taurocholate cotransporting polypeptide (HepG2‐hNTCP C4 cells) (Fig. 1F).
FIG. 1.
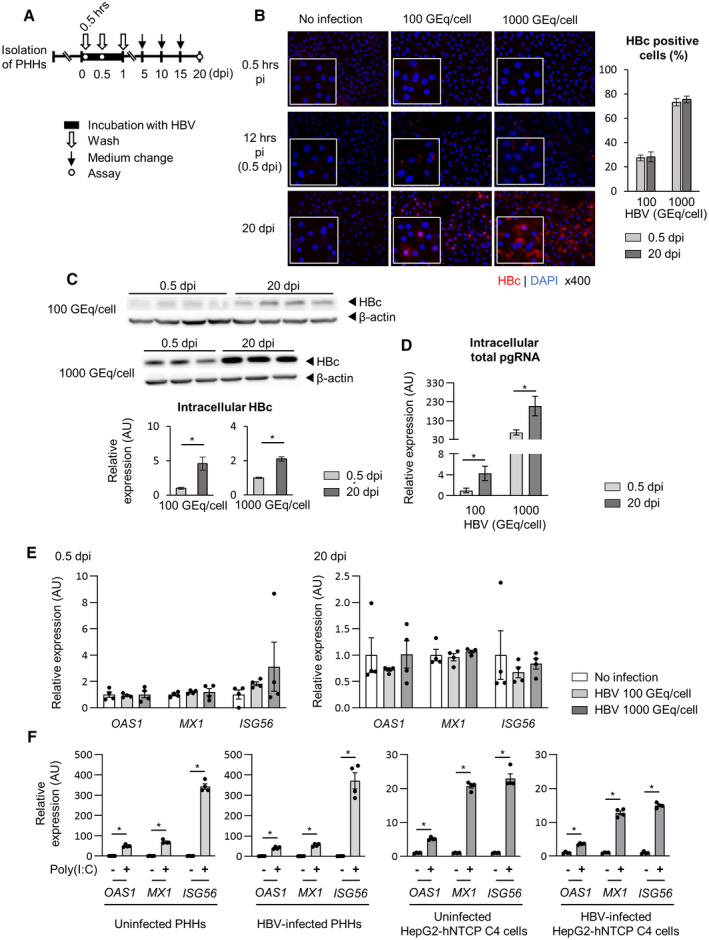
Intracellular ISG expression is not altered by HBV infection. (A‐E) PHHs isolated from human liver–chimeric mice were inoculated with HBV (100 GEq/cell or 1,000 GEq/cell), and the HBc‐positive cell ratio, intracellular pgRNA levels, and antiviral ISG expression levels were evaluated at indicated times. Each samples were analyzed after washing. (A) Schematic of the experimental procedure. (B) Representative image of fluorescence immunostaining (red, HBc; blue, nucleus). The HBc‐positive cell ratio was calculated based on the average of four fields of view. (C) Western blot of intracellular HBc protein and its quantifications (n = 3‐4). (D) Intracellular total pgRNA levels (n = 4). (E) The mRNA expression levels of antiviral ISGs (n = 4). Dot plot shows individual values. (F) Uninfected PHHs or HBV‐infected PHHs at 20 dpi were transfected with 0.01 μg/mL Poly (I:C). Two days later, the mRNA expression levels of antiviral ISGs were measured (n = 4). For reference, uninfected HepG2‐derived NTCP‐expressing cells (HepG2‐hNTCP C4 cells) or HBV‐infected (5,000 GEq/cell) HepG2‐hNTCP C4 cells at 8 dpi were transfected with 0.1 μg/ml Poly (I:C). Two days later, the mRNA expression levels of antiviral ISGs were measured (n = 4). Dot plot shows individual values. The data are shown as the mean ± SEM. *P < 0.05.
CAM Reduces the Intracellular pgRNA Levels in HBV‐Infected PHHs
BAY41 is a CAM classified as a heteroaryldihydropyrimidine that misdirects HBc proteins toward aberrant assembly into core multimers and leads to their intracellular depletion.( 9 , 10 , 28 ) PHHs were inoculated with HBV and administered BAY41 or TFV beginning on 20 dpi (Fig. 2A), given that we previously reported that it takes approximately 20 days for HBsAg levels in the supernatant to plateau after HBV inoculation to PHHs.( 19 ) To examine the cytotoxicity of BAY41 or TFV, WST assay and cell confluences of BAY41‐ and TFV‐treated PHHs were examined. There was no difference in cell viability assessed by WST assay or cell confluences between treated and untreated PHHs (Fig. 2B and Supporting Fig. S1). Intracellular HBc protein levels and pgRNA levels were significantly reduced by BAY41 but not by TFV (Fig. 2C,D). On the other hand, both BAY41 and TFV reduced HBV‐DNA levels, but not HBsAg and HBeAg levels, in the culture supernatants (Fig. 2E).
FIG. 2.
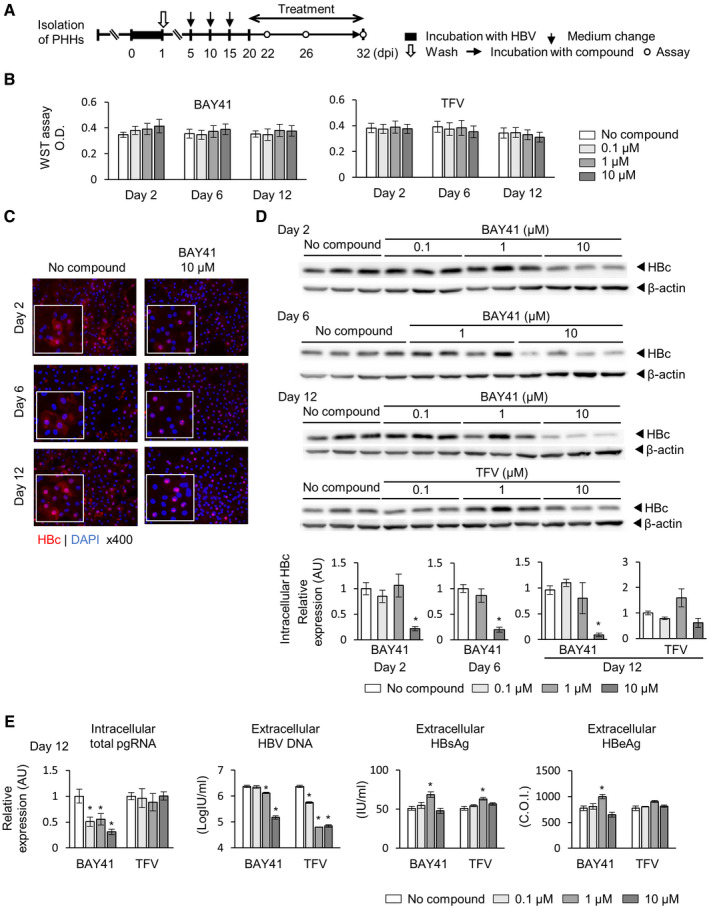
BAY41 reduces intracellular HBc and pgRNA levels. Cells were incubated with HBV inoculum (1,000 GEq/cell) for 24 hours and then treated with BAY41 or TFV beginning on 20 dpi. (A) Schematic of the experimental procedure. (B) WST assay at indicated days after treatment (n = 6). (C) Representative image of fluorescence immunostaining (red, HBc; blue, nucleus). (D) Western blot of intracellular HBc protein after indicated days after treatment, and its quantifications (n = 3‐4). (E) Intracellular total pgRNA, HBV DNA, HBsAg, and HBeAg levels in the culture supernatants at the end of the 12‐day treatment (n = 4). Each value was compared with that in the untreated control. The data are shown as the mean ± SEM. *P < 0.05.
CAM Reduces Intracapsid pgRNA Levels per Cell Without Inducing the Host IFN Response
Next, we examined the effect of a CAM on intracellular HBV pgRNA. BAY41 was administered to HBV‐infected PHHs beginning 20 dpi for 2, 6 or 12 days, and intracellular pgRNA levels were evaluated (Fig. 3A). Twelve‐day administration of BAY41 reduced the intracellular total pgRNA levels (Fig. 3B). On the other hand, 2‐day administration of BAY41 did not reduce the intracellular total pgRNA levels or the cytoplasmic total pgRNA levels, but did reduce the intracapsid pgRNA levels per cell (Fig. 3B,C), suggesting that extracapsid pgRNA levels increased in the cell cytoplasm at this time point with BAY41 treatment. The decrease in HBc expression per cell assessed by western blot (Fig. 2D) and the decrease in intracapsid pgRNA levels per cell (Fig. 3C) were comparable. When considering that BAY41 inhibits encapsidation of pgRNA by misdirecting HBc proteins toward aberrant assembly and by depleting HBc proteins,( 9 , 10 , 28 ) the decrease in intracapsid pgRNA levels detected in PHHs may represent a stronger effect of HBc depletion rather than HBc abnormal assemblies. Given that pgRNA contains a PAMP motif recognized by RIG‐I,( 14 ) we further examined the effect of CAM treatment on the host IFN response. The administration of BAY41 did not change the gene‐expression levels of type I and type III IFNs, RIG‐I, interferon regulatory factor 7 (IRF7), and antiviral ISGs in HBV‐infected PHHs (Fig. 3D).
FIG. 3.
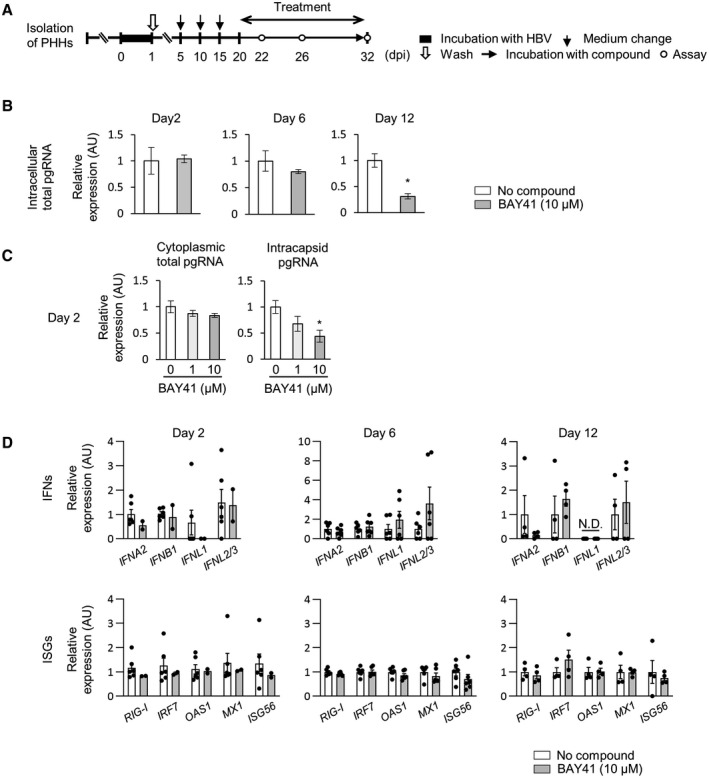
BAY41 reduces intracapsid pgRNA levels per cell without inducing the host IFN response. (A‐D) Cells were incubated with HBV inoculum (1,000 GEq/cell) for 24 hours and treated with BAY41 for 2, 6, or 12 days beginning at 20 dpi. (A) Schematic of the experimental procedure. (B) Intracellular total pgRNA levels in untreated PHHs or PHHs treated with BAY41 over time (n = 4‐6). (C) Cytoplasmic total pgRNA and intracapsid pgRNA levels per cell after 2 days of treatment with BAY41 (n = 4). The cytoplasmic total pgRNA levels and intracapsid pgRNA levels were normalized to the cytoplasmic β‐actin mRNA level. (D) The mRNA expression levels of IFNs and antiviral ISGs in untreated PHHs and PHHs treated with BAY41 monotherapy (n = 4‐6). The results are shown as relative levels with the levels of each data of untreated PHHs set to 1. Dot plot shows individual values. The data are shown as the mean ± SEM. *P < 0.05. Abbreviation: N.D., not detected.
CAM Enhances ISG Induction by Exogenous IFN in HBV‐Infected PHHs
IFNα induces RIG‐I and IRF7, which plays an important role in the positive feedback regulation of the RIG‐I pathway.( 29 ) We assessed whether coadministration of BAY41 enhances the innate IFN response in HBV‐infected PHHs under administration of IFNα (Fig. 4A). Although 6‐day or 12‐day administration of IFN‐α reduced intracellular total pgRNA levels in HBV‐infected PHHs, 2‐day administration of IFN‐α did not reduce these levels (Fig. 4B). When compared with the administration of IFN‐α alone for 2 days, coadministration of BAY41 with IFN‐α for 2 days did not reduce intracellular total pgRNA levels or cytoplasmic total pgRNA levels, but did reduce the intracapsid pgRNA levels (Fig. 4B,C). IFN‐α administration alone for 12 hours induced ISGs in HBV‐infected PHHs, but there was no significant difference in ISG levels between cells treated with IFN‐α alone and cells cotreated with BAY41 with IFN‐α at this time point (Fig. 4D). Interestingly, coadministration of BAY41 with IFN‐α for 2 days up‐regulated the expression of IFN‐α‐induced ISGs in HBV‐infected PHHs compared with IFN‐α administration alone for 2 days, whereas treatment with BAY41 alone for 2 days did not induce ISGs or IFNs (Fig. 4D). The enhancement of ISG and IFN inductions by BAY41 in HBV‐infected PHHs was also detected at day 6 (Fig. 4D). In contrast, 2‐day or 6‐day coadministration of BAY41 and IFN‐α did not change the IFNα‐induced expression of IFNs and ISGs in uninfected PHHs compared with 2‐day or 6‐day IFN‐α administration alone, respectively (Fig. 4E,F). Twelve‐day coadministration of BAY41 and IFN‐α also failed to up‐regulate the IFN‐α‐induced expression of IFNs and ISGs in HBV‐infected PHHs (Fig. 4D), and the intracellular total pgRNA levels were more suppressed in these cells compared with HBV‐infected PHHs treated for 2 or 6 days (Fig. 4B). Similar to BAY41, CAM‐N, another type of CAM that inhibits pgRNA encapsidation and induces the formation of morphologically intact empty capsids,( 12 ) also enhanced IFN‐α‐induced ISG expression (Supporting Fig. S2).
FIG. 4.
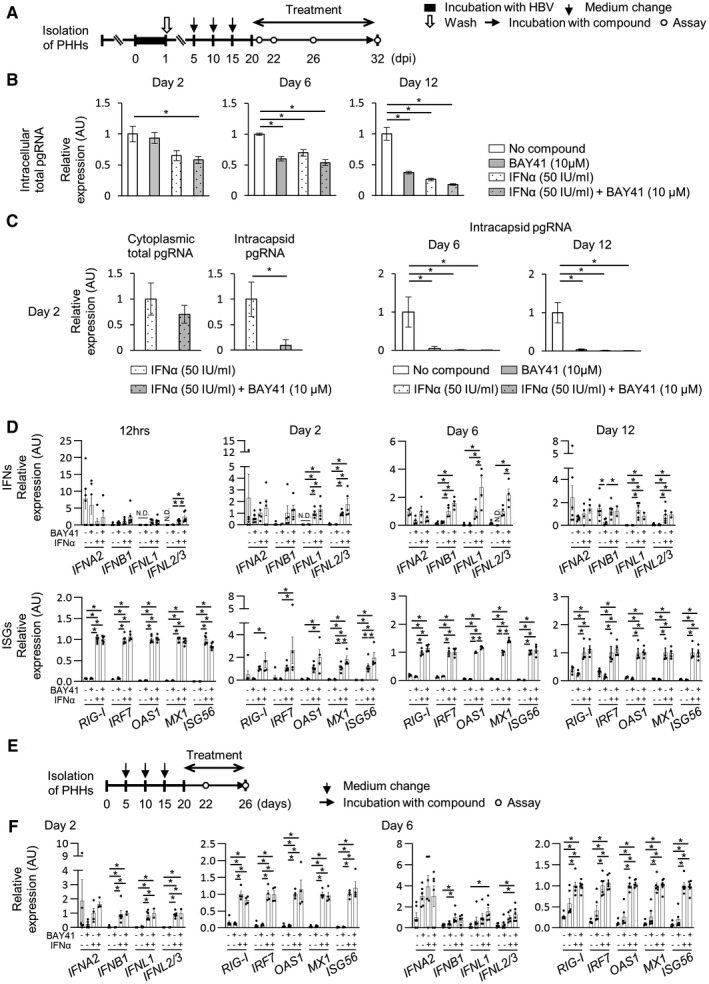
BAY41 enhances the antiviral ISG expression induced by IFN‐α in HBV‐infected PHHs. (A‐D) The cells were incubated with HBV inoculum (1,000 GEq/cell) for 24 hours and treated with BAY41 alone, IFN‐α alone, or in combination with BAY41 for indicated days beginning on 20 dpi. (A) Schematic of the experimental procedure. (B) Intracellular total pgRNA levels (n = 4‐6). (C) Cytoplasmic total pgRNA and intracapsid pgRNA levels (n = 4‐5). Total cytoplasmic pgRNA and intracapsid pgRNA levels in each sample were normalized to the cytoplasmic β‐actin mRNA level. (D) The mRNA expression levels of IFNs and ISGs in untreated PHHs or PHHs treated with BAY41 (10 μM) alone, IFN‐α (50 IU/ml) alone, or in combination with BAY41 (n = 4‐6). The results are shown as relative levels with the levels of each data of PHHs treated with IFN‐α alone set to 1. Dot plot shows individual values. (E,F) Uninfected PHHs were treated with BAY41 (10 μM) alone, IFN‐α (50 IU/ml) alone, or in combination with BAY41 and IFN‐α for 2 or 6 days beginning at 20 days after seeding. Same lot of PHHs as those in Fig. 4D were used. (E) Schematic of the experimental procedure. (F) The mRNA expression levels of the ISGs in uninfected PHHs after the treatment (n = 4‐6). The results are presented as relative levels, and the levels of each datapoint of PHHs treated with IFNα alone is set to 1. Dot plot shows individual values. The data are shown as the mean ± SEM. *P < 0.05.
CAM in Combination With IFN‐α Robustly Suppresses Extracellular HBV‐DNA Levels
To address the antiviral effect of the combination of BAY41 and IFN‐α, HBV‐infected PHHs were treated with BAY41 alone (BAY41 mono), IFN‐α alone (IFN mono), or the combination of IFN‐α and BAY41 (IFN/BAY41 combo) for 2 days or 12 days beginning on 20 dpi (Fig. 5A). The cytotoxicity was assessed by WST assay and cell confluences. The results showed no change in cell viability as assessed by WST assay or cell confluence in response to any treatment compared with the untreated control over time (Fig. 5B and Supporting Fig. S1). After 2 days of treatment, no significant differences in extracellular HBsAg levels and intracellular cccDNA levels were noted among all of the groups (Fig. 5C). Extracellular HBV‐DNA levels were significantly reduced by the BAY41 mono and IFN/BAY41 combo treatments compared with the untreated control; however, no significant differences were noted between the levels observed after these two treatments. After 12 days of treatment, no significant differences in the intracellular cccDNA levels were noted among all of the groups. The extracellular HBV‐DNA levels were significantly reduced in the IFN/BAY41 combo group compared with the BAY41 mono and the IFN mono groups. The HBsAg and HBeAg levels in the culture supernatants were significantly reduced by the IFN mono and the IFN/BAY41 combo treatments; however, no significant difference was noted between the levels observed after these two treatments (Fig. 5C).
FIG. 5.
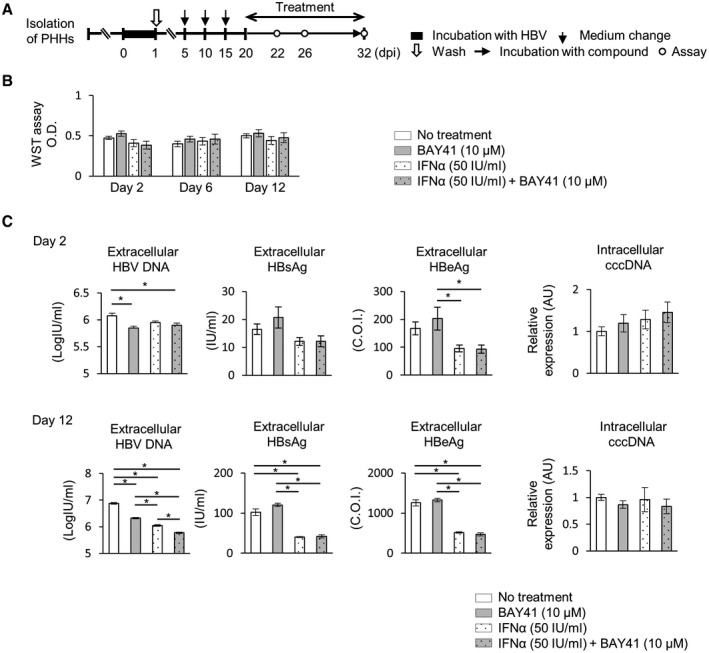
The combination of BAY41 and IFN‐α reduces HBV‐DNA levels more effectively than BAY41 or IFN‐α monotherapy. (A‐C) HBV‐infected PHHs were treated with BAY41 alone, IFN‐α alone, or their combination for indicated days. (A) Schematic of the experimental procedure. (B) WST assay at indicated days after treatment (n = 6). Each value was compared to that in the untreated control. (C) cccDNA levels and HBV‐DNA, HBsAg, and HBeAg levels in the culture supernatants after 2 days or 12 days of treatment (n = 4). The data are shown as the mean ± SEM. *P < 0.05.
CAM Enhances Intrahepatic ISG Expression During IFN Treatment in HBV‐Infected Human Liver–Chimeric Mice
Eighteen human liver–chimeric mice were infected with HBV. On 70 dpi, mice were assigned to four treatment groups: the vehicle control group (vehicle, n = 4), BAY41 monotherapy group (BAY41 mono, n = 5), pegIFN‐α monotherapy group (pegIFN mono, n = 4), and combination therapy group (pegIFN/BAY41 combo, n = 5). After 14 days of treatment, the mice were sacrificed for analysis (Fig. 6A). The mean serum HBV‐DNA level was 5.5 ± 0.04 log IU/ml (mean ± SEM) at the start of treatment (Fig. 6B). Although BAY41 mono exerted no significant suppressive effect on serum HBV‐DNA levels, pegIFN‐α mono significantly reduced serum HBV‐DNA levels. PegIFN/BAY41 combo exerted the strongest suppressive effect on the serum HBV‐DNA levels among all of the treatments (Fig. 6C). Although no significant differences in IFN‐α, IFN‐β, and IFN‐λ levels were noted at this time point, intrahepatic RIG‐I, IRF7, and antiviral ISG expression was up‐regulated in the pegIFN‐α mono group compared with the vehicle and BAY41 mono groups and further enhanced in the pegIFN‐α/BAY41 combo group (Fig. 6D). Intrahepatic pgRNA, cccDNA, and HBc levels were not different among all of the groups at the end of the 14‐day treatment (Fig. 6E,F).
FIG. 6.
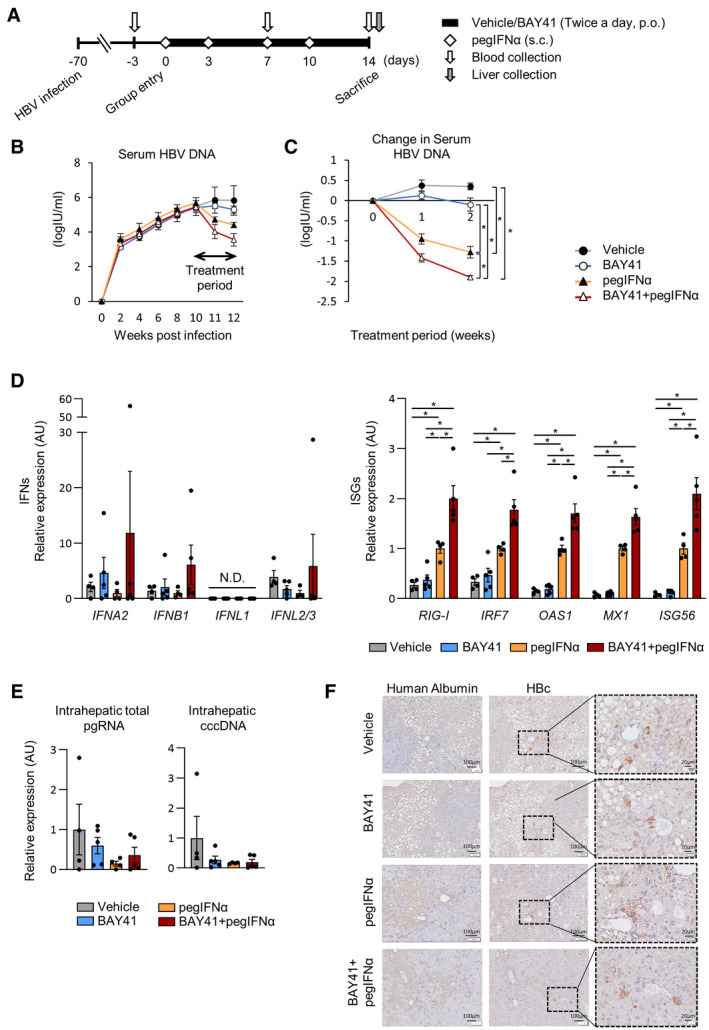
BAY41 enhances the intrahepatic ISG expression induced by pegIFN‐α in HBV‐infected human liver–chimeric mice, and BAY41 and pegIFN‐α combination therapy reduces serum HBV‐DNA levels more effectively than BAY41 or pegIFN‐α alone. HBV‐infected mice were treated with vehicle (n = 4), BAY41 (40 mg/kg, twice a day, p.o.) (n = 5), pegIFN‐α (25 mg/kg, twice per week, s.c.) (n = 4), or the combination of BAY41 and pegIFNα (n = 5) for 14 days. (A) Schematic of the experimental procedure. (B) Serum HBV‐DNA levels from inoculation to the end of the treatment. (C) Change in the serum HBV‐DNA levels over the treatment period. (D) Analysis of the intrahepatic mRNA expression levels of human‐specific IFN and ISGs. The results are presented as relative levels with the levels of each datapoint of mice treated with pegIFN‐α alone set to 1. Dot plot shows individual values. (E) Intrahepatic pgRNA and cccDNA levels at the end of the treatment. Dot plot shows individual values. (F) Representative image of HBc immunohistochemistry. The data are shown as the mean ± SEM. *P < 0.05. Abbreviations: p.o., per os; s.c., subcutaneous administration.
Discussion
In the present study, treatment with CAM alone altered intracellular posttranscriptional HBV‐RNA kinetics but failed to promote the intracellular IFN response. However, CAM enhanced intrahepatic ISG induction by IFN‐α in both HBV‐infected PHHs and the livers of human liver–chimeric mice. These findings indicate how CAMs influence innate immune responses in HBV‐infected hepatocytes during treatment with IFN‐α.
PHHs were used in all of the in vitro experiments in the current study. Stable HBV‐transfected cells or sodium‐taurocholate cotransporting polypeptide (NTCP)–expressing cells are conventionally used as models of HBV infection. In HBV‐transfected cells, such as HepG2.2.15 or HepAD38 cells, the viral life cycle is not completed, and viral proteins or viral particles are produced primarily from the viral genome integrated in host DNA, not from cccDNA.( 30 ) NTCP‐expressing cells derived from HepG2 cells or HuH cells are permissive to viral infection, and the full viral life cycle is completed in these cells.( 30 ) However, such hepatoma‐derived cells are impaired in the production of IFN against viral infections.( 31 ) In our experiment, poly (I:C) more strongly induced antiviral ISGs in PHHs compared with HepG2‐hNTCP C4 cells (Fig. 1), indicating that RIG‐I and TLR3 in PHHs induce endogenous IFN responses more robustly than those in HepG2‐derived cells. Moreover, it has been reported that compared with PHHs, hepatoma‐derived cell lines induce weaker ISG expression in response to exogenous IFN.( 31 , 32 ) Based on these findings, PHHs are a suitable model to assess the IFN response in HBV‐infected cells.
We demonstrated that CAM treatment significantly reduced intracapsid pgRNA levels, but the cytoplasmic total pgRNA levels were not altered in the presence or absence of IFN‐α, suggesting that CAM treatment increases extracapsid pgRNA levels in the cytoplasm. The 5’‐ε stem loop of HBV pgRNA has been reported to be a PAMP recognized by host RIG‐I, leading to the IFN response( 14 ); however, in our study, the intracellular IFN response was not promoted in HBV‐infected PHHs treated with CAM monotherapy. This finding may be attributed to a suppressive effect of HBV infection on the host IFN response. HBV polymerase has been reported to inhibit the phosphorylation of IRF3 and IRF7, which are parts of the RIG‐I pathway.( 33 , 34 ) We showed that intracellular antiviral ISG expression was induced by IFN‐α and further enhanced by the coadministration of CAM. IRF7 is activated by IFN and plays a key role in promoting the RIG‐I pathway.( 29 ) We hypothesize that CAM augments the cytoplasmic nonencapsidated pgRNA levels and that the host IFN response is promoted when the RIG‐I pathway is activated by exogenous IFN‐α. However, we cannot exclude the possibility that the decrease in HBc or other viral components by CAM contributed to enhancing ISG induction by IFN. Mitra et al. reported that intranuclear HBc might inhibit ISG induction by IFN‐α.( 35 ) Further study of this issue will be needed.
In the present study, treatment with IFN for 6 days or 12 days reduced total intracellular pgRNA levels in HBV‐infected PHHs without changing cccDNA levels (Figs. 4B and 5C). In HBV‐infected human liver–chimeric mice, although the difference was not significant, the mean pgRNA level was lower in the INF‐treated mice than the untreated mice (Fig. 6E). IFN has been reported to suppress cccDNA transcription through histone hypoacetylation( 36 , 37 ) or the suppression of enhancer II.( 38 ) IFN‐induced ISGs, including ISG20 and 2'‐5'‐oligoadenylate synthetase 1 (OAS1), have been reported to suppress cccDNA transcription( 39 ) and promote pgRNA degradation.( 40 ) The suppression of pgRNA levels in the present study could have been caused by IFN‐mediated epigenetic suppression of cccDNA or ISG‐mediated HBV‐RNA suppression.
Klumpp et al.( 13 ) previously reported the results of a 42‐day treatment of HBV‐infected human liver–chimeric uPA/SCID mice with another CAM, NVR 3‐778. These authors reported that intrahepatic ISG expression was not enhanced by the combination of this CAM with pegIFN‐α at the end of the treatment. Inconsistent with that previous study, intrahepatic ISG expression was up‐regulated in the HBV‐infected human liver–chimeric mice treated with the combination of BAY41 and pegIFN‐α in the present study. The difference in the intrahepatic ISG induction may be due to the timing of liver sample collection. In the previous study, liver samples were collected at the end of the 42‐day treatment when the intrahepatic pgRNA levels were significantly reduced by pegIFN‐α. In our in vitro study, the intracellular pgRNA levels were reduced well after 12 days of treatment with IFN‐α alone and with the combination of IFN‐α and BAY41. At this time point, up‐regulation of the IFN response by the coadministration of BAY41 and IFN‐α was not observed. In our in vivo study, liver samples were collected after a relatively short treatment period, and intrahepatic pgRNA levels were not decreased by any treatments compared with the vehicle control. This short treatment period may show a stronger effect of CAM administration because the samples were collected while the pgRNA was still present in the cells. These results, together with a previous report by Klumpp et al.,( 13 ) suggest that two mechanisms may be involved in the antiviral effect of CAM IFN‐α cotreatment: an ISG‐independent pathway and an ISG‐dependent pathway.
In the experiment using PHHs, ISGs were enhanced, but HBV‐DNA levels were not further reduced after 2 days of BAY41 and IFN‐α combination therapy compared with those following IFN‐α monotherapy. On the other hand, after 12 days of treatment, ISGs were not enhanced, but HBV‐DNA levels were further reduced by combination therapy compared with those following treatment with IFN‐α alone (Figs. 4D and 5C). Although we cannot exclude the possibility that the decrease in HBV‐DNA levels detected after 12 days of treatment was mediated by the ISG‐independent pathway, it is also possible that the ISG‐dependent pathway takes longer to decrease HBV‐DNA levels. Furthermore, in in vitro and in vivo experiments, the decrease in HBV‐DNA levels mediated by BAY41 under IFN‐α treatment was not accomplished by a reduction in intracellular pgRNA levels (Figs. 4B and 6E). MxA, an ISG, was reported to inhibit pgRNA encapsidation.( 41 ) Combination treatment with BAY41 and IFN‐α may affect HBV replication without reducing pgRNA levels through the ISG‐dependent pathway.
In conclusion, CAMs enhance IFN‐induced antiviral ISG expression in HBV‐infected hepatocytes. Whether these effects could contribute to an improved functional cure rate remains to be demonstrated in clinical trials.
Supporting information
Fig S1
Fig S2
Table S1‐S2
Supplementary Material
Acknowledgment
The authors thank Janssen Pharmaceutica NV for providing a CAM‐N tool compound and Dr. Nao Yoneda at Central Institute for Experimental Animals for providing animals and technical advice. They also thank Professor Keiji Ueda for the advice on the experimental procedure and Ms. Miyuki Imai, Dr. Kumiko Shirai, and Dr. Satoshi Shigeno for supporting their experiments.
Supported by the Japan Agency for Medical Research and Development (JP19fk0310108 and JP20am0101121).
Potential conflict of interest: Dr. Hikita is on the speakers’ bureau for Chugai Pharmaceutical Co., Ltd. Dr. Takehara is on the speakers’ bureau for and received grants from Gilead Sciences and Chugai Pharmaceutical Co., Ltd.
References
Author names in bold designate shared co‐first authorship.
- 1. World Health Organization . Fact sheet, Hepatitis B 2019, July 18. https://www.who.int/news‐room/fact‐sheets/detail/hepatitis‐b. Accessed January 14, 2021.
- 2. European Association for the Study of the Liver . EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol 2017;67:370‐398. [DOI] [PubMed] [Google Scholar]
- 3. Terrault NA, Lok ASF, McMahon BJ, Chang K‐M, Hwang JP, Jonas MM, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology 2018;67:1560‐1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Suk‐Fong LA. Hepatitis B treatment: what we know now and what remains to be researched. Hepatol Commun 2019;3:8‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fanning GC, Zoulim F, Hou J, Bertoletti A. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat Rev Drug Discov 2019;18:827‐844. [DOI] [PubMed] [Google Scholar]
- 6. Smolders EJ, Burger DM, Feld JJ, Kiser JJ. Review article: clinical pharmacology of current and investigational hepatitis B virus therapies. Aliment Pharmacol Ther 2020;51:231‐243. [DOI] [PubMed] [Google Scholar]
- 7. Zoulim F, Lenz O, Vandenbossche JJ, Talloen W, Verbinnen T, Moscalu I, et al. JNJ‐56136379, an HBV capsid assembly modulator, is well‐tolerated and has antiviral activity in a phase 1 study of patients with chronic infection. Gastroenterology 2020;159:521‐533.e9. [DOI] [PubMed] [Google Scholar]
- 8. Feld JJ, Colledge D, Sozzi V, Edwards R, Littlejohn M, Locarnini SA. The phenylpropenamide derivative AT‐130 blocks HBV replication at the level of viral RNA packaging. Antiviral Res 2007;76:168‐177. [DOI] [PubMed] [Google Scholar]
- 9. Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, et al. Inhibition of hepatitis B virus replication by drug‐induced depletion of nucleocapsids. Science 2003;299:893‐896. [DOI] [PubMed] [Google Scholar]
- 10. Stray SJ, Bourne CR, Punna S, Lewis WG, Finn MG, Zlotnick A. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc Natl Acad Sci U S A 2005;102:8138‐8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lam AM, Ren S, Espiritu C, Kelly M, Lau V, Zheng L, et al. HBV capsid assembly modulators, but not nucleoside analogs, inhibit the production of extracellular pregenomic RNA and spliced RNA variants. Antimicrob Agents Chemother 2017;61:e00680‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lahlali T, Berke JM, Vergauwen K, Foca A, Vandyck K, Pauwels F, et al. Novel potent capsid assembly modulators regulate multiple steps of the hepatitis B virus life‐cycle. Antimicrob Agents Chemother 2018;62:e835‐e918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klumpp K, Shimada T, Allweiss L, Volz T, Lütgehetmann M, Hartman G, et al. Efficacy of NVR 3–778, alone and in combination with pegylated interferon, vs entecavir In uPA/SCID mice with humanized livers and HBV infection. Gastroenterology 2018;154:652‐662.e8. [DOI] [PubMed] [Google Scholar]
- 14. Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, et al. The RNA sensor RIG‐I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 2015;42:123‐132. [DOI] [PubMed] [Google Scholar]
- 15. Luangsay S, Gruffaz M, Isorce N, Testoni B, Michelet M, Faure‐Dupuy S, et al. Early inhibition of hepatocyte innate responses by hepatitis B virus. J Hepatol 2015;63:1314‐1322. [DOI] [PubMed] [Google Scholar]
- 16. Lebossé F, Testoni B, Fresquet J, Facchetti F, Galmozzi E, Fournier M, et al. Intrahepatic innate immune response pathways are downregulated in untreated chronic hepatitis B. J Hepatol 2017;66:897‐909. [DOI] [PubMed] [Google Scholar]
- 17. Suslov A, Boldanova T, Wang X, Wieland S, Heim MH. Hepatitis B virus does not interfere with innate immune responses in the human liver. Gastroenterology 2018;154:1778‐1790. [DOI] [PubMed] [Google Scholar]
- 18. Suemizu H, Hasegawa M, Kawai K, Taniguchi K, Monnai M, Wakui M, et al. Establishment of a humanized model of liver using NOD/Shi‐scid IL2Rgnull mice. Biochem Biophys Res Commun 2008;377:248‐252. [DOI] [PubMed] [Google Scholar]
- 19. Nakabori T, Hikita H, Murai K, Nozaki Y, Kai Y, Makino Y, et al. Sodium taurocholate cotransporting polypeptide inhibition efficiently blocks hepatitis B virus spread in mice with a humanized liver. Sci Rep 2016;6:27782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hasegawa M, Kawai K, Mitsui T, Taniguchi K, Monnai M, Wakui M, et al. The reconstituted 'humanized liver' in TK‐NOG mice is mature and functional. Biochem Biophys Res Commun 2011;405:405‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, et al. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 1997;41:1715‐1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ogura N, Watashi K, Noguchi T, Wakita T. Formation of covalently closed circular DNA in Hep38.7‐Tet cells, a tetracycline inducible hepatitis B virus expression cell line. Biochem Biophys Res Commun 2014;452:315‐321. [DOI] [PubMed] [Google Scholar]
- 23. Murai K, Hikita H, Kai Y, Kondo Y, Fukuoka M, Fukutomi K, et al. Hepatitis C virus infection suppresses hepatitis B virus replication via the RIG‐I‐like helicase pathway. Sci Rep 2020;10:941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamai T, Hikita H, Fukuoka M, Fukutomi K, Murai K, Nakabori T, et al. SIRT1 enhances hepatitis virus B transcription independent of hepaticautophagy. Biochem Biophys Res Commun 2020;527:64‐70. [DOI] [PubMed] [Google Scholar]
- 25. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jeong JK, Yoon GS, Ryu WS. Evidence that the 5'‐end cap structure is essential for encapsidation of hepatitis B virus pregenomic RNA. J Virol 2000;74:5502‐5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tan G, Song H, Xu F, Cheng G. When hepatitis B virus meets interferons. Front Microbiol 2018;9:1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stray SJ, Zlotnick A. BAY 41‐4109 has multiple effects on hepatitis B virus capsid assembly. J Mol Recognit 2006;19:542‐548. [DOI] [PubMed] [Google Scholar]
- 29. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006;25:349‐360. [DOI] [PubMed] [Google Scholar]
- 30. Witt‐Kehati D, Bitton Alaluf M, Shlomai A. Advances and challenges in studying hepatitis B virus in vitro. Viruses 2016;8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Keskinen P, Nyqvist M, Sareneva T, Pirhonen J, Melen K, Julkunen I. Impaired antiviral response in human hepatoma cells. Virology 1999;263:364‐375. [DOI] [PubMed] [Google Scholar]
- 32. Shen F, Li Y, Wang Y, Sozzi V, Revill PA, Liu J, et al. Hepatitis B virus sensitivity to interferon‐alpha in hepatocytes is more associated with cellular interferon response than with viral genotype. Hepatology 2018;67:1237‐1252. [DOI] [PubMed] [Google Scholar]
- 33. Wang H, Ryu WS. Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog 2010;6:e1000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu S, Chen J, Wu M, Chen H, Kato N, Yuan Z. Hepatitis B virus polymerase inhibits RIG‐I‐ and toll‐like receptor 3‐mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J Gen Virol 2010;91(Pt 8):2080‐2090. [DOI] [PubMed] [Google Scholar]
- 35. Mitra B, Wang J, Kim ES, Mao R, Dong M, Liu Y, et al. Hepatitis B virus precore protein p22 inhibits alpha interferon signaling by blocking STAT nuclear translocation. J Virol 2019;93:e196‐e219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, et al. IFN‐alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest 2012;122:529‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu F, Campagna M, Qi Y, Zhao X, Guo F, Xu C, et al. Alpha‐interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccDNA minichromosomes. PLoS Pathog 2013;9:e1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nawa T, Ishida H, Tatsumi T, Li W, Shimizu S, Kodama T, et al. Interferon‐alpha suppresses hepatitis B virus enhancer II activity via the protein kinase C pathway. Virology 2012;432:452‐459. [DOI] [PubMed] [Google Scholar]
- 39. Imam H, Kim GW, Mir SA, Khan M, Siddiqui A. Interferon‐stimulated gene 20 (ISG20) selectively degrades N6‐methyladenosine modified Hepatitis B Virus transcripts. PLoS Pathog 2020;16:e1008338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park IH, Kwon YC, Ryu WS, Ahn BY. Inhibition of hepatitis B virus replication by ligand‐mediated activation of RNase L. Antiviral Res 2014;104:118‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li N, Zhang L, Chen L, Feng W, Xu Y, Chen F, et al. MxA inhibits hepatitis B virus replication by interaction with hepatitis B core antigen. Hepatology 2012;56:803‐811. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1‐S2
Supplementary Material