

Abstract
Although pulmonary arterial hypertension (PAH) leads to right ventricle (RV) hypertrophy and structural remodeling, the relative contributions of changes in myocardial geometric and mechanical properties to systolic and diastolic chamber dysfunction and their time courses remain unknown. Using measurements of RV hemodynamic and morphological changes over 10 wk in a male rat model of PAH and a mathematical model of RV mechanics, we discriminated the contributions of RV geometric remodeling and alterations of myocardial material properties to changes in systolic and diastolic chamber function. Significant and rapid RV hypertrophic wall thickening was sufficient to stabilize ejection fraction in response to increased pulmonary arterial pressure by week 4 without significant changes in systolic myofilament activation. After week 4, RV end-diastolic pressure increased significantly with no corresponding changes in end-diastolic volume. Significant RV diastolic chamber stiffening by week 5 was not explained by RV hypertrophy. Instead, model analysis showed that the increases in RV end-diastolic chamber stiffness were entirely attributable to increased resting myocardial material stiffness that was not associated with significant myocardial fibrosis or changes in myocardial collagen content or type. These findings suggest that whereas systolic volume in this model of RV pressure overload is stabilized by early RV hypertrophy, diastolic dilation is prevented by subsequent resting myocardial stiffening.
NEW & NOTEWORTHY Using a novel combination of hemodynamic and morphological measurements over 10 wk in a male rat model of PAH and a mathematical model of RV mechanics, we found that compensated systolic function was almost entirely explained by RV hypertrophy, but subsequently altered RV end-diastolic mechanics were primarily explained by passive myocardial stiffening that was not associated with significant collagen extracellular matrix accumulation.
Keywords: diastolic function, mathematical modeling, pulmonary arterial hypertension, sugen-hypoxia, systolic function
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a progressive vasculopathy characterized by structural remodeling of the pulmonary arteries. Clinically, PAH manifests as sustained elevated resting mean pulmonary arterial pressures of 20 mmHg or greater (1). The chronically elevated blood pressure in the pulmonary vasculature induces right ventricular pressure overload that stimulates chamber remodeling. Although remodeling of the right ventricle (RV) can be sufficient to compensate and maintain cardiac output, PAH commonly leads to RV failure and premature death (2).
RV systolic function was initially considered to be a good prognostic indicator of patient outcomes, but more recently, increases in RV diastolic stiffness have been shown to be a marker of diastolic dysfunction (3) and associated with disease severity (2, 4) and reduced survival (5, 6) in patients with PAH. Previous studies in animal models of RV pressure overload have shown myocyte hypertrophy (7–9), altered myocardial contractility (10, 11), fibrosis (12, 13), titin dephosphorylation (14), and passive myocardial stiffening (15), or combinations of these remodeling responses (16–21). But how these processes contribute to the time courses of altered systolic and diastolic RV function during PAH progression has not been studied. In particular, we still lack an understanding of how the sequences of right ventricular hypertrophy and changes in systolic and diastolic myocardial properties contribute to the time courses of altered systolic and diastolic RV chamber function. Therefore, we designed a longitudinal study of RV remodeling and chamber mechanics at six timepoints over 10 wk in the sugen-hypoxia male rat model of PAH.
The relative effects of changes in myocardial material properties versus RV hypertrophy to altered systolic and diastolic chamber mechanics also remain poorly understood (18). We used a computational model we developed earlier (18) to discriminate the contributions of altered chamber geometry and altered myocardial material properties to changes in end-systolic and end-diastolic right ventricular pressure-volume relations measured in vivo during the 10-wk study period. The analysis showed significant changes in RV chamber mechanics, in which the relative contributions of RV hypertrophy and remodeled myocardial material properties were distinct and different between diastole and systole.
MATERIALS AND METHODS
The sugen-hypoxia (SuHx) animal model of PAH was chosen for this study as it develops vascular lesions resembling those found at autopsy in patients with PAH, followed by RV remodeling (22). Animal care, housing, and feed were approved by the Institutional Animal Care and Use Committees at the University of Illinois at Chicago and the University of California San Diego. Hemodynamic measurements were taken by the same surgeon at both institutions using the same equipment in animals from the same vendor. Male Sprague–Dawley rats (7 wk old) weighing 216 ± 9.6 g (Charles River, Wilmington, MA) were randomized into control (n = 22) and SuHx (n = 52) groups. Pulmonary arterial hypertension was induced in rats by a single subcutaneous injection (20 mg/kg) of the vascular endothelial growth factor receptor antagonist sugen (SU5416, S8442 MilliporeSigma, CAS Number 204005-46-9, PubChem Substance ID 24278606 Sigma-Aldrich, MO) followed by exposure to chronic hypoxia (10% O2) for 3 wk. The animals were then returned to normoxia. The severity of the disease was augmented by the time (in wk) elapsed after returning to normoxia (up to 10-wk postinjection). Timepoints included in the study were 3-, 4-, 5-, 6-, 8-, and 10-wk postinjections. Age-matched normotensive control animals kept in normoxia throughout the study comprised the control group.
In-Vivo Hemodynamic Measurements
Right ventricular blood pressure and volume were measured in all the animals during an open-chest surgery, as previously described by Vélez-Rendón et al. (18). Briefly, the animals were administered 2.5% isoflurane (MWI Veterinary Supply, Cat. No. 502017) mixed with oxygen (100% O2) continuously via a nose cone. After a tracheotomy, animals were given isoflurane with concentrations monitored via calibrated vaporizer, supplied with a respiratory rate of 50 breaths/min and a tidal volume determined by the animal’s weight. A rectal probe (MicroTherma 2, ThermoWorks, UT) and a 12-lead electrocardiogram continuously monitored the body temperature and heart rate throughout the procedure.
A thoracotomy was performed to expose the heart and great vessels. With a 21-gauge needle the RV chamber was punctured apically to insert a 1.9-Fr admittance catheter (Transonic Scisense, ON, Canada), the gold-standard method for measuring ventricular blood pressure and volume (10, 23–25). Right ventricular pressure-volume (P-V) loops were taken during steady state and during occlusion of the inferior vena cava to generate RV P-V relations during changes in preload. When possible, occlusion of the inferior vena cava was repeated after steady-state measurements. In a subgroup of control and SuHx animals, after concluding RV hemodynamic measurements, left ventricular P-V measurements were taken to confirm systemic normotension. P-V time series were collected using the LabChart software (version 8 Pro, ADInstruments, Inc., Colorado Springs, CO) and analyzed off-line using a custom-written MATLAB code (version R2018b, The MathWorks, Natick, MA). A Savitzky–Golay finite impulse response convolution filter, implemented with sgolay function, was used to smooth pressure and volume time series using a third-order polynomial fit to approximate 15-timepoint windows using least-squares comparisons.
Steady-state P-V measurements immediately preceding caval occlusion, ranging from 80 to 200 heartbeats, were separated into single heartbeats, aligned within the cardiac cycle, and averaged to generate a representative P-V loop. The end-systolic (ES) P-V point was identified as the timepoint in the loop with the maximum pressure-to-volume ratio. The end-diastolic (ED) P-V point was identified as the pressure and volume at the first time following isovolumic relaxation when the volume was at a maximum (i.e., dV/dt ∼0). With the heart rate, ES and ED volumes were used to compute stroke volume (SV), ejection fraction (EF), and cardiac output (CO). Total pulmonary vascular resistance (tPVR) was determined by dividing RV ES pressure by CO. By fitting a linear function through the ES P-V points during caval occlusion, end-systolic elastance (Ees) was determined. End-diastolic elastance (Eed), on the other hand, was computed by fitting a nonlinear exponential function (18) through the ED P-V points during caval occlusion and evaluated at ED steady-state volume (as illustrated in Fig. 3, A and B). Effective arterial elastance (Ea) was calculated as the ratio of ES pressure to SV (26) and ventricular-vascular coupling was calculated as the ratio of Ees to Ea per animal. Maximum and minimum pressure rate changes (dP/dtmax and dP/dtmin) were found by identifying the largest local maxima and smallest local minima of the time derivative of pressure measurements, respectively. To avoid extrapolating volumes to pressures below zero, the unloaded volume (V0) was set to the RV volume measured during caval occlusions with the lowest end-diastolic pressure (18).
Figure 3.
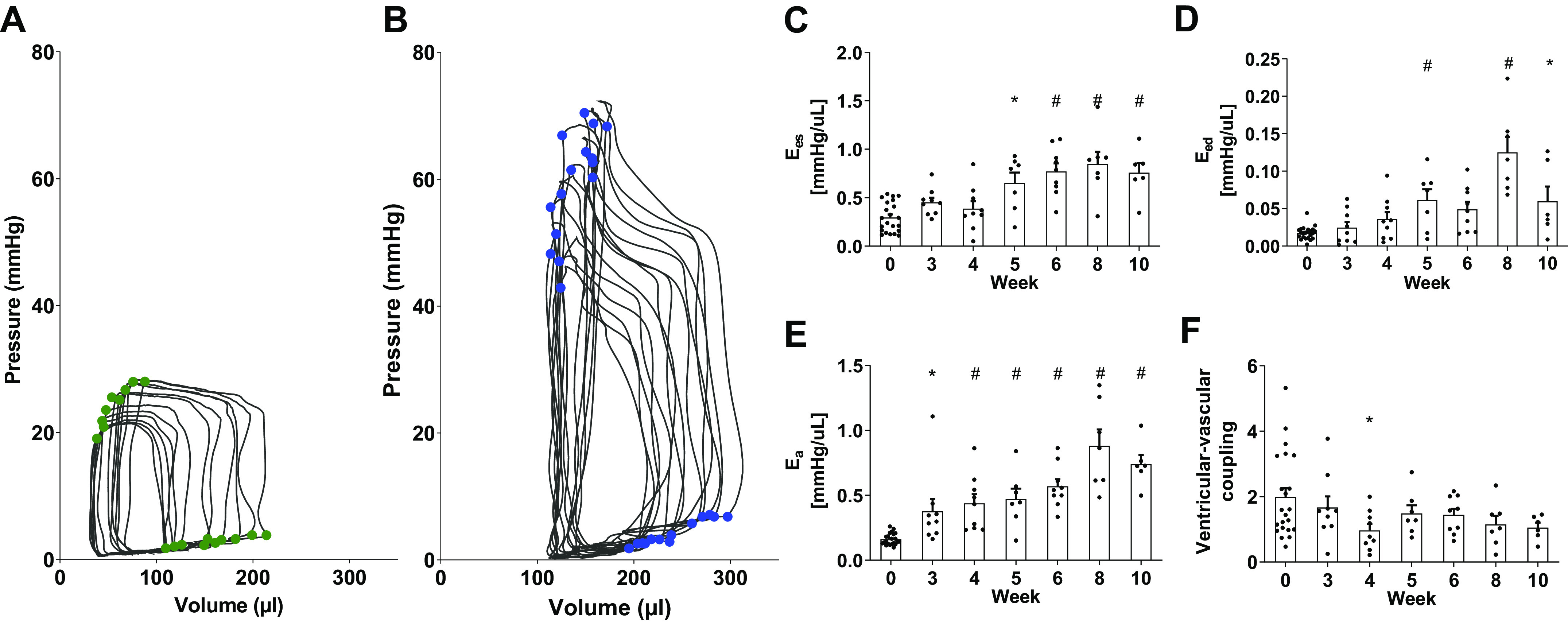
Pressure-volume (P-V) during caval occlusion to determine end-systolic (ES) and end-diastolic (ED) P-V relations for a control (A, green) and a 6-wk SuHx (B, blue) animal. ES elastance Ees (C), ED elastance Eed (D), arterial elastance Ea (E), and ventricular-vascular coupling Ees/Ea (F). Data are shown as means ± SE, *P < 0.05 and #P < 0.01 compared with the control group. SuHx, sugen-hypoxia.
For animals with repeated steady-state measurements, ED and ES pressures and volumes, SV, EF, CO, tPVR, dP/dtmax, and dP/dtmin of each steady-state measurement were averaged per animal. For animals in which multiple caval occlusions were performed, occlusions were averaged. By normalizing RV volumes by V0, the pressure-volume curves were averaged across occlusions. P-V loops for a single animal were then averaged and interpolated over the measured volume ranges. Volumes were then unnormalized by multiplying by the mean minimum volume. Hemodynamic measurements obtained from animals at the University of Illinois at Chicago were compared with data obtained at the University of California San Diego. Since no significant differences were found, the data from both institutions were pooled.
Cardiac Magnetic Resonance Imaging
To examine how the open-chest preparation may have affected ventricular volumes in this study, we performed noninvasive cardiac magnetic resonance imaging (cMRI) in two week 4 SuHx and one age-matched control rats in a 11.7-T preclinical scanner (Bruker Biospec, Billerica, MA). Rats were anesthetized using 1%–2% isoflurane in 100% O2. Body temperature was regulated at 37°C using an external warm air flow while electrocardiogram (EKG) and respiration were continuously monitored. Sixteen EKG-gated short-axis images per slice were acquired at each of five to eight slices spanning the biventricular length. Image analysis was performed blinded to the treatment and hemodynamics of the animal. Endocardial contours were drawn manually on short-axis slices using ImageJ (NIH, Bethesda, MD). Right ventricular ED and ES volumes were calculated from the short-axis images with largest and smallest RV chamber volumes using the volumetric summation of disks method (27). The cardiac MRI parameters were slice thickness (1.50 mm), image resolution (200 × 200 microns), imaging matrix (250 × 250 pixels), field of view (50 × 50 mm), and frame acquisition duration of 312 ms.
Tissue-Level Measurements
Immediately following in vivo hemodynamic measurements, animals were exsanguinated, and the heart was flushed with ice-cold phosphate-buffered saline (pH 7.4), consisting of 0.137 M NaCl, 0.0027 M KCl, 0.01 M Na2HPO4, 0.0018 M KH2HPO4, and 5,000 U/mL heparin solution (USP, MWI Veterinary Supply, Cat. No. 054255). The heart was excised, and the RV free wall was isolated and weighed. RV thickness was measured at three locations near the mid region of the free wall using a precision gauge (1010Z, Starrett, Athol, MA, with a range of 0–0.375″ and accuracy of ±0.001″) and averaged per animal. Left ventricular (LV) and septal weights were also measured and used to compute septal volume and Fulton Index (i.e., the ratio of RV mass to the sum of LV and septal masses). Tissue samples from the mid region of the RV free wall were taken for histological analysis. Samples were fixed in 10% formalin (Thermo Fisher Scientific, MA, Cat. No. SF100-4) for 48 h, stored in 70% ethanol, then paraffin-embedded and sectioned parallel to myofiber direction. The samples were stained with Masson’s Trichrome to identify collagen (blue), myocytes (red), and nuclei (black). Imaging the samples at ×40 magnification (Evos FL Auto Fluorescent Microscope, Thermo Fisher Scientific, MA), the collagen to myocyte area fractions were determined by converting bright-field images from the red-green-blue (RGB) color scale to the hue saturation value (HSV) scale to screen for the different tissue constituents.
Myocardial Collagen Quantification
A hydroxyproline assay (QuickZyme Total Collagen Assay, QuickZyme BioSciences, Lei, NL) was used to measure total RV collagen content. Free wall tissue samples weighing 5–15 mg were prepared and assayed according to the manufacturer’s recommended protocol. Hydroxyproline residues were quantified using a microplate reader (Tecan M200, Tecan LifeSciences, CH). Absorbances were calibrated with a linear collagen standard curve, adjusted for sample dilution and expressed as total collagen content.
Collagen types I and III contents were quantified using enzyme-linked immunosorbent assay kits (Rat Collagen type I ELISA Kit, NBP2-75823, Collagen type III ELISA Kit, NBP2-81205, Novus Biologicals, CO). Tissue samples were isolated in cold 1× PBS at a 1:9 wt/vol ratio and diluted 1:100. Collagen I and III concentrations were measured at 450-nm wavelength (Tecan F200 Microplate Reader, Tecan LifeSciences, CH). Absorbances were calibrated with collagen I and collagen III standard curves ranging from 0 to 100 ng/mL and expressed as the ratio of collagen I to collagen III.
Model of Right Ventricular Biomechanics
Right ventricular pressure, myocardial wall mechanics, and morphology were analyzed with a biomechanics model described previously (18) to estimate the contributions of altered RV geometry versus changes in intrinsic myocardial material properties to measured changes in RV P-V relations during the progression of hypertensive remodeling. Briefly, the RV was modeled as a fraction Kr of a sphere of radius R and thickness h. The volume enclosed by the outside of the sphere was calculated as the sum of the RV chamber (Vrv), RV free-wall (Vw), and septal (Vsw) volumes. Based on the control-group rats RV wall mass to thickness ratio, the fraction Kr of the sphere occupied by the RV was 0.55. The stress-free reference geometry of the model was calculated using V0. The left ventricle and the effects of left ventricular pressure on stresses and strains in the RV free wall were not included.
Since the ratio of RV radius to wall thickness is typically greater than 5:1 (18), even in models of RV hypertrophy, a thin-walled assumption (28, 29) was used to relate RV blood pressure to myocardial circumferential wall stress T by Laplace’s Law with midwall radius r = R − h/2 and wall thickness h. Circumferential stress T was assumed to equal the passive fiber stress (Tp) as a function of midwall sarcomere length l at ED, plus an additional active fiber stress (Ta) as a function of l and calcium-dependent myofilament activation k3 at ES. Tp was modeled as an exponential function of stretch ratio λ = l/lR, where lR is the slack sarcomere length (set to 1.85 µm) (30), and k1 and k2 are parameters of the exponential material law.
| (1) |
The active fiber stress Ta was modeled as a saturation equation (31, 32) with Ta as a function of l and k3,
| (2) |
where l0 is the constant sarcomere length at which zero active isometric tension is developed (set to 1.6 μm; 31, 33), C is set to 4.35 μM (31), Tmax is the constant maximal isometric tension achieved at normal activation (set to 20,000 mmHg; 31), and the exponent B (set to 11; 31) modulates the shape of peak isometric tension-length relation.
The free wall outer radius R of the RV model was calculated so that the volume enclosed by the outer surface of the partial sphere matched the sum of the measured Vrv, Vw, and Vsw. A myocardial density of 1.053 g/mL (34) was used to compute Vw for each rat from the measured RV free wall mass. Vsw for each rat was computed from the measured total LV (septal plus free wall) mass using a ratio of septal to free wall mass of 0.45 (based on measurements in a subset of 2 control and 9 SuHx rats). The wall thickness h was then calculated from Vw using R and Kr. The resting material parameters k1 and k2 were chosen to give the best fit of model-predicted ED volumes to measured ED volumes, as a function of ED pressure. The myofilament activation material parameter k3 was chosen to fit model-predicted ES pressures to measured ES pressures as a function of ES volume in each animal. The model-generated pressure-volume curves were used to partition the contributions of altered RV volume (measured ED/ES volume), geometry (RV, LV and septum mass, and V0), and material properties (k1, k2, and k3) to the differences in end-systolic or end-diastolic pressures between control and hypertensive rats. The fraction Kr was held constant. Fitted-model parameters were then used to predict and analyze resting and active sarcomere length-stress relations for each animal. Further details on model implementation are included in the Supplemental Data (Supplemental Figs. S2–S4; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.14780016).
Statistical Analysis
Descriptive statistics were performed using JMP Pro Statistical software (version 14, SAS Institute Inc., NC) for group comparisons of hemodynamic and morphological measurements and derived model results. For normally distributed data, one-factor ANOVA were used to test for differences in treatment groups followed by the Dunnett’s post hoc test. Otherwise, the nonparametric Wilcoxon–Kruskal–Wallis statistic was used followed by the Dunnett’s post hoc test. Model predictions of the rat-specific sarcomere mechanics were analyzed using a general linear model with mixed factors to compare intergroup and interlevel effects. For a given parameter, the statistical model was where µ is the mean, αi is the treatment effect i (control, weeks 3–10), τj is the fiber stress or sarcomere length, ατ is the interaction term, γ is the subject variability [independent N(0,σ2)], and is the error term. Fixed factors of this statistical model were fiber stress for ES-derived data, sarcomere length for ED-derived data, and treatment group for ES- and ED-derived data, and the random factor was animals. Statistical significance was determined at a level of P < 0.05. Errors in the model fits were quantified via root mean square errors (RMSE). Graphs were generated using GraphPad Prism software (version 8.4.3, GraphPad Software, CA).
RESULTS
Hemodynamic and morphological measurements were obtained from 22 control and 52 SuHx rats, of which 5 SuHx-induced animals died during surgeries before hemodynamic measurements were completed. No rats died before the terminal procedure. Initially, control animals at ages corresponding to each SuHx timepoint were compared, but since no changes in hemodynamics were found, these animals were aggregated into a single group for the purposes of presentation and statistical analysis. The group sizes for hemodynamic measures at each disease stage were n = 22 (control), n = 9 (week 3), n = 9 (week 4), n = 7 (week 5), n = 9 (week 6), n = 7 (week 8), and n = 6 (week 10).
Ventricular Hemodynamics and Morphology
In SuHx-treated rats, RV ES pressure, RV ED pressure, tPVR (Fig. 1, A–C), and RV mass (Fig. 2A) had all increased significantly (P < 0.05) and more than doubled compared with control by 5-wk postinduction, and these increases were all sustained through week 10. Immediately after hypoxia (week 3), ES volume (P < 0.01, Fig. 1D) and wall thickness (p < 0.01, Fig. 2D) were both significantly higher than control. By week 4, EF was significantly lower than control (from 66 ± 10% to 40 ± 6%, P < 0.01, Fig. 1H), but thereafter, ejection fraction stabilized, coinciding with significant increases in peak positive and negative dP/dt by week 5 compared with controls (P < 0.05, Fig. 1, F and I). Despite the significant increase in RV ED pressure by week 5, ED volume (Fig. 1E) remained unchanged.
Figure 1.
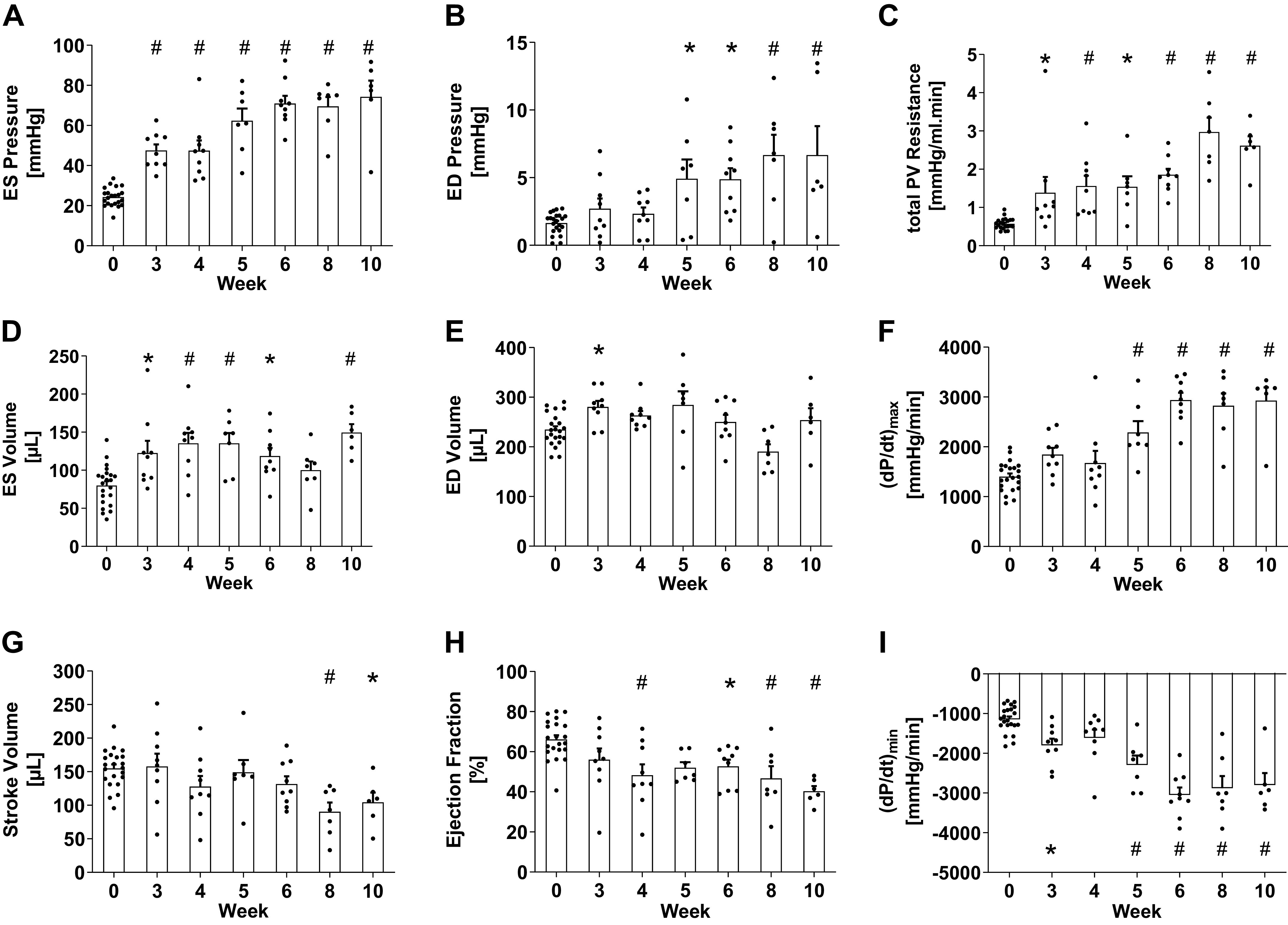
In vivo hemodynamic measurements in SuHx rats through the 10-wk time course of the study showed significant increases by week 3 in end-systolic (ES) right ventricular pressure (A) and total pulmonary vascular resistance (C), and significant increases by week 5 in end-diastolic (ED) pressure (B) and peak positive time derivative of right ventricular pressure (F). Although ES volume (D) increased significantly, ED volume (E) was preserved throughout the study apart from a small but significant increase at week 3, and stroke volume (G) was only significantly lower than control after week 6. Mean ejection fraction (H) decreased significantly by week 4 but did not change after week 4 (P > 0.05, comparison not shown). The magnitude of peak negative time derivative of pressure (I) increased significantly at week 3 and after week 4. Data are shown as means ± SE, *P < 0.05 and #P < 0.01 compared with the control group. SuHx, sugen-hypoxia.
Figure 2.
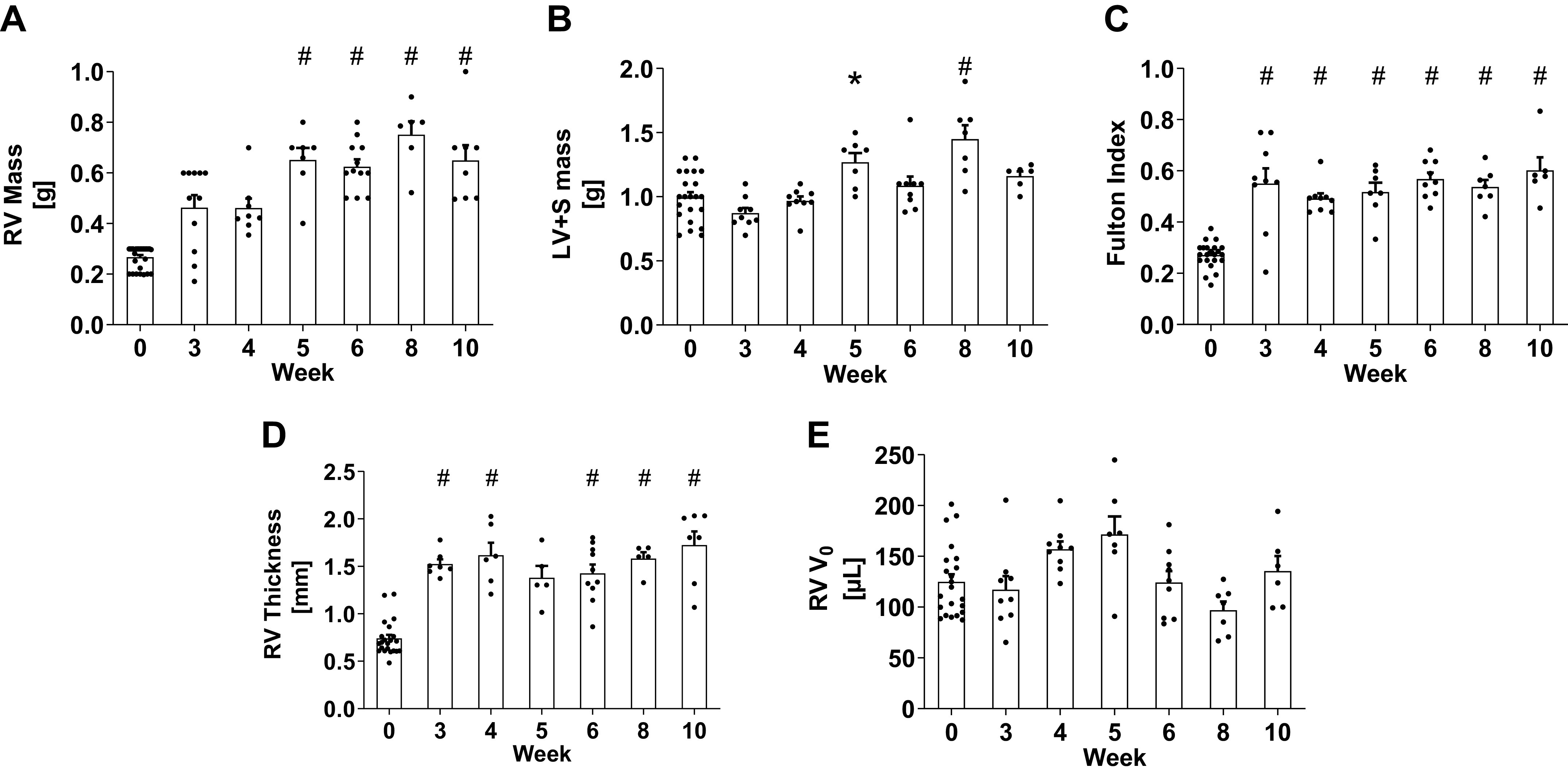
Masses of right ventricle (A), left ventricle plus septum (B), and their mass ratio (C), and right ventricular wall thickness (D) and estimated V0 (E). Data are shown as means ± SE, *P < 0.05 and #P < 0.01 compared with the control group. LV, left ventricle; S, septum; RV, right ventricle; V0, unloaded volume.
No changes in LV end-systolic pressure (from 91 ± 9 mmHg, n = 8 to 87 ± 7 mmHg, n = 12, P = 0.77) or in LV ejection fraction (from 76 ± 3%, n = 8 to 70 ± 5%, n = 12, P = 0.38) were found in the SuHx animals compared with control animals. The sum of LV and septum masses increased in SuHx weeks 5 and 8 (Fig. 2B) compared with control animals but remained unchanged at all other timepoints (P > 0.05).
Mean pressure-volume measurements during caval occlusion showed significant changes in SuHx rats from control rats in the end-diastolic and end-systolic pressure-volume relations after week 4 (Fig. 3 and Supplemental Fig. S1). End-systolic elastance Ees (Fig. 3C) significantly increased at week 5 compared with control and at all timepoints thereafter (P < 0.003). End-diastolic elastance Eed (Fig. 3D) was significantly larger than control at weeks 5, 8, and 10 (P < 0.05). Arterial elastance Ea (Fig. 3E) increased significantly compared with control by week 3 (P < 0.05) and continued to increase until week 8. Ventricular-vascular coupling Ees/Ea (Fig. 3F) remained unchanged.
RV volumes of individual rats obtained noninvasively via cMRI were compared with the respective group’s distribution. Although end-diastolic and end-systolic volumes were smaller in the cMRI measures, volumes were within two standard deviations of the invasive group means (Table 1) and the differences between control and SuHx RV volumes were consistent. cMRI-derived ejection fractions were 54% and 44% for the SuHx animals compared with the group mean of 48 ± 16% and 72% in the control animal compared with the group mean of 66 ± 10%. Similarly, cMRI-derived stroke volumes were 127 and 118 μL in the SuHx rats compared with group mean of 128 ± 48 μL and 129 μL in the control animal compared with the group mean of 155 ± 28 μL (Table 1).
Table 1.
RV chamber volumes, ejection fractions, and stroke volumes
| Control |
SuHx Week 4 |
||||
|---|---|---|---|---|---|
| Invasive | cMRI | Invasive | cMRI 1 | cMRI 2 | |
| n | 22 | 1 | 9 | 1 | 1 |
| RV ED volume, μL | 234 ± 32 | 180 | 263 ± 27 | 236 | 266 |
| RV ES volume, μL | 80 ± 26 | 51 | 135 ± 41 | 109 | 148 |
| RV stroke volume, μL | 155 ± 28 | 129 | 128 ± 48 | 127 | 118 |
| RV ejection fraction, % | 66 ± 10 | 72 | 48 ± 16 | 54 | 44 |
Values are means ± SD and measured by invasive catheterization for control and sugen-hypoxia (SuHx) week 4 groups compared with those in individual animals measured using cardiac magnetic resonance imaging (cMRI). ED, end-diastolic; ES, end-systolic; RV, right ventricular.
Model Analysis of Pressure-Volume Relations
When accounting for measured changes in RV geometry and optimizing myocardial material properties, the biomechanics models closely predicted end-systolic pressures as a function of volume with an average RMSE of 5.7 ± 0.5 mmHg (9% of peak ES pressure) and end-diastolic volumes as a function of pressure with an average RMSE of 18 ± 1 µL (7% of peak ED volume, Supplemental Fig. S5).
The model-predicted relative contributions of volume, geometry, and material property changes to measured SuHx group pressures compared with the control group pressures are summarized in Fig. 4. Models with measured geometries and control ES material properties were sufficient to explain most or all of the significant increases in ES elastance that were measured in SuHx rats after week 4, except at week 5, where there was also a significant contribution of increased myofilament activation. In contrast, the significant increase in ED elastances were not explained by RV geometric remodeling. Hence, the increased ED chamber elastance in SuHx rats after week 5 were explained almost entirely by increased passive myocardial material stiffness. Although increased RV ES volume alone (green bars in Fig. 4A) was sufficient to generate all of the increased RV ES pressure at weeks 3 and 4, at weeks 5 and 6, there was also a significant, transient contribution of increased myofilament activation to RV ES pressure (blue bars). After week 5, significant contributions of geometry (RV hypertrophy, black bars) explained all or nearly all the measured increases in end-systolic pressure generation. From the governing equations, the effects of altered RV geometry on computed wall mechanics are due to changes in the wall thickness to radius ratio in the model (Supplemental Table S1).
Figure 4.
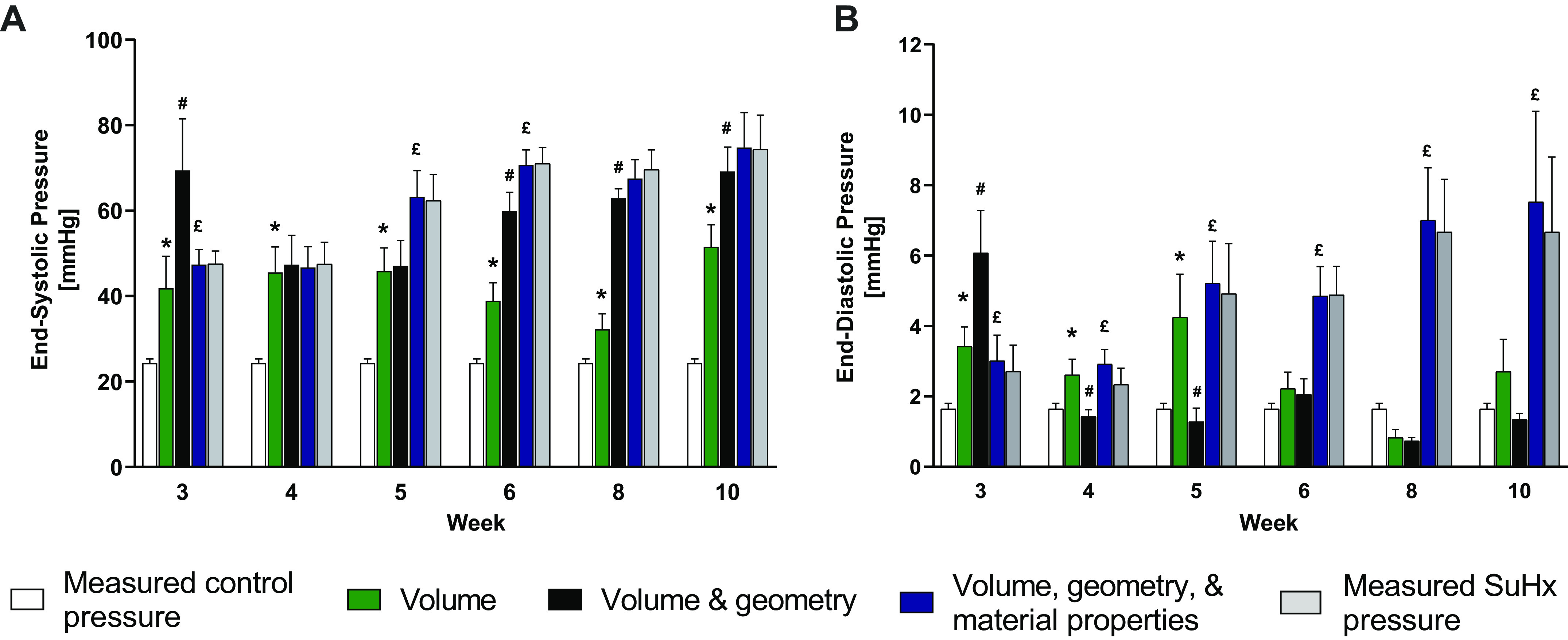
Measurements of control (white bars) and SuHx (gray bars) RV pressure for end systole (A) and end diastole (B) compared with model predictions of pressure using SuHx volume with control geometry and material properties (green); SuHx volume and geometries with control material properties (black); and SuHx volume, geometries, and material properties (blue). Green bars with * indicate a significant contribution of RV volume to increased ES or ED pressure compared with control (white bars). Black bars with # indicate a significant contribution of RV hypertrophy to increased ES (A) or ED (B) pressure compared with the contributions of volume alone (green bars). Blue bars with £ indicate a significant contribution of altered myofilament activation (A) or myocardial resting stiffness (B) to increased ES or ED pressure compared with volume and geometry (black bars). Data are shown as means ± SE, P < 0.05. ED, end-diastolic; ES, end-systolic; RV, right ventricle; SuHx, sugen-hypoxia.
The same analysis for end diastole (Fig. 4B) showed that the significant increases in RV ED pressure from control in week 5–10 rats were initially explained by increased RV ED volumes (at week 5) but subsequently were due almost entirely to increased passive myocardial stiffness (blue bars) with no significant contributions of altered RV volume (green bars) or geometry (black bars).
A graphical comparison of mean RV P-V relations computed with the optimized models of SuHx rats with mean model-computed control curves (green) and mean computed P-V relations accounting for measured RV geometries in SuHx rats with unchanged material properties (black) and measured RV geometries in SuHx rats with altered material properties (blue) is illustrated in Supplemental Fig. S2.
Myocardial Wall Stresses
Right ventricular systolic active fiber stress increased from a mean of 14.6 kPa at control to 25.8 kPa at week 3 and did not change significantly thereafter (Table 2). Had the RV not remodeled in SuHx rats, the increase in ES pressure would have more than doubled fiber stresses by week 3 (from 14.6 to 31.4 kPa) and more than tripled by week 10 (from 14.6 to 48.8 kPa). RV geometric remodeling caused active fiber stress in weeks 5–10 SuHx rats to be about half as high as it would have been without hypertrophy (26.7–29.8 kPa vs. 41.8–48.8 kPa). End-diastolic RV free wall fiber stress did not increase from the control mean (1.05 kPa) until week 5 (2.10 kPa) and continued to increase further to 2.6 kPa by weeks 8 and 10. Had the RV not remodeled, the increase in ED pressure would have more than tripled passive wall stresses by week 5 (from 1.05 to 3.61 kPa) and increased fivefold by weeks 8 and 10. Hence, although RV anatomic and material property remodeling were not sufficient to completely normalize ED and ES fiber stresses, they did offset the majority of the increases that would otherwise have occurred.
Table 2.
Model predictions of fiber strain and stress
|
Week
|
|||||||
|---|---|---|---|---|---|---|---|
| Control | 3 | 4 | 5 | 6 | 8 | 10 | |
| Control geometry and control mechanics | |||||||
| ED fiber strain | 1.05 | 1.07 | 1.06 | 1.08 | 1.08 | 1.09 | 1.09 |
| ED fiber stress, kPa | 1.12 | 1.91 | 1.50 | 3.61 | 3.58 | 5.00 | 5.00 |
| ES active fiber strain | 0.98 | 1.01 | 1.01 | 1.03 | 1.03 | 1.03 | 1.04 |
| ES active fiber stress, kPa | 14.6 | 31.4 | 29.7 | 41.8 | 46.9 | 46.1 | 48.8 |
| SuHx geometry and control mechanics | |||||||
| ED fiber strain | 1.06 | 1.05 | 1.07 | 1.07 | 1.08 | 1.08 | |
| ED fiber stress, kPa | 1.21 | 1.03 | 2.31 | 2.20 | 2.93 | 2.93 | |
| ES active fiber strain | 0.99 | 0.99 | 1.00 | 1.01 | 1.01 | 1.01 | |
| ES active fiber stress, kPa | 19.4 | 19.9 | 26.7 | 29.7 | 27.6 | 29.8 | |
| SuHx geometry and SuHx mechanics | |||||||
| ED fiber strain | 1.06 | 1.04 | 1.03 | 1.04 | 1.02 | 1.04 | |
| ED fiber stress, kPa | 1.21 | 0.99 | 2.10 | 2.04 | 2.56 | 2.64 | |
| ES active fiber strain | 1.00 | 1.00 | 0.99 | 1.00 | 1.00 | 1.00 | |
| ES active fiber stress, kPa | 25.8 | 21.5 | 26.3 | 27.7 | 25.6 | 27.1 | |
Values are based on measurements of control and sugen-hypoxia (SuHx) animal geometry, end-diastolic (ED) pressure for passive stress, and end-systolic (ES) pressure for active stress.
Myocardial Passive and Active Material Properties
The adjustable material parameters (k1 and k2 for end diastole and k3 for end systole) of the models that best fitted the ED and ES P-V relations (blue error bars in Supplemental Fig. S1) were measured in each rat for each group, shown in Supplemental Fig. S3. Mean passive fiber stress-sarcomere length relations of each group (Fig. 5) computed from the fitted rat-specific values of k1 and k2 were stiffer than control relations by week 5 and the stiffest at week 8. Sarcomere lengths at matched fiber stresses were significantly lower (P < 0.05) than control values at weeks 5, 8, and 10 by two-factor ANOVA. There were no significant differences between groups in the computed active stress-sarcomere length relations (Fig. 5), except in the SuHx week 5 group (Fig. 5C). A two-factor ANOVA of model-computed systolic fiber stress showed a significant interaction effect between experimental group and sarcomere length (P < 0.01). A post hoc comparison showed that the slope of the active stress-sarcomere length relation was significantly greater than control only at week 5 (P < 0.01).
Figure 5.
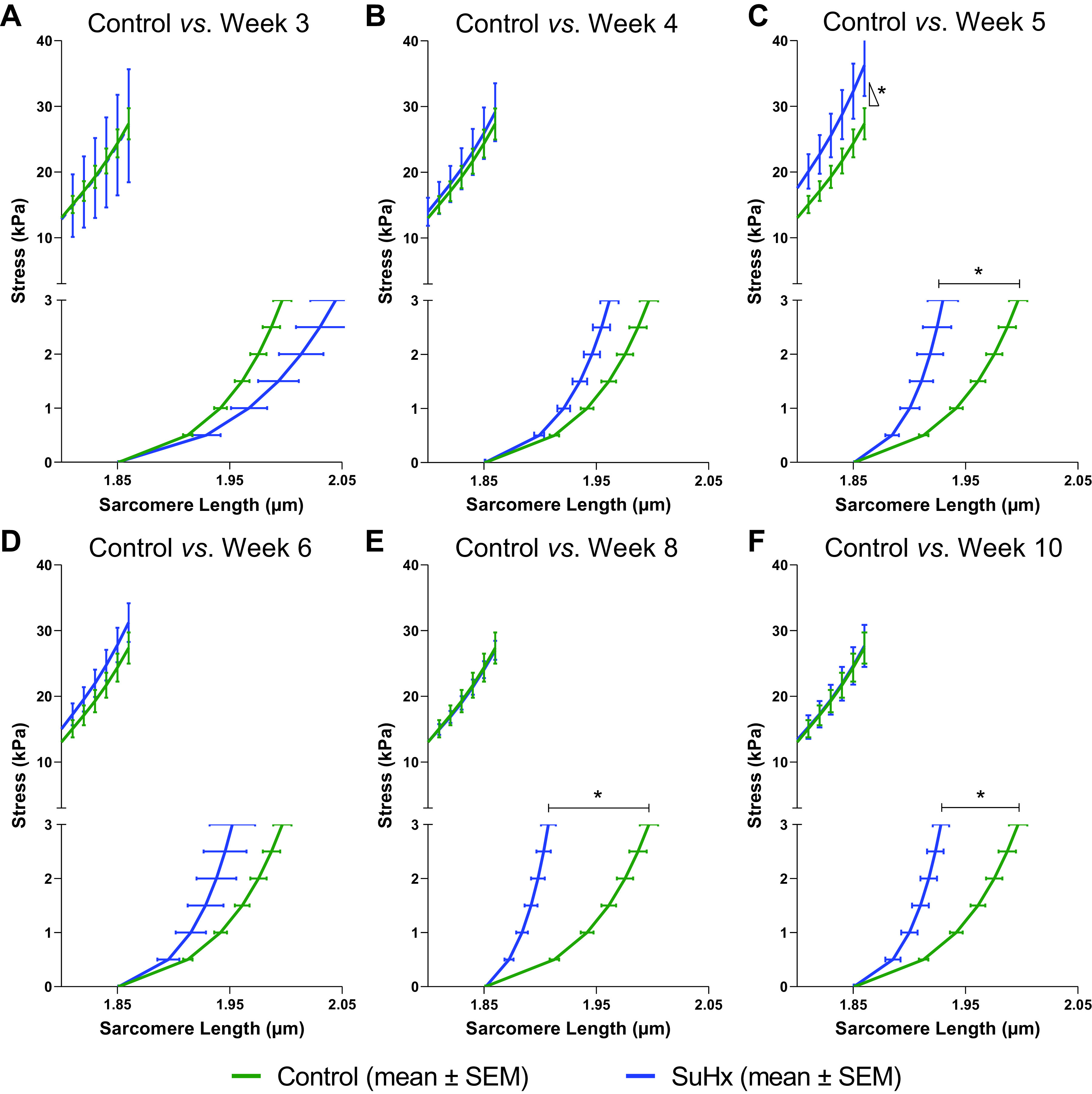
Model-predicted mean active end-systolic (top) and passive end-diastolic (bottom) fiber stress-sarcomere length relations for control (green) and SuHx (blue) rats at weeks 3 (A), 4 (B), 5 (C), 6 (D), 8 (E), and 10 (F). The slope of the active stress-sarcomere length relation was only significantly higher than control in week 5 (P < 0.05). Predicted mean passive stress-sarcomere length relations were significantly stiffer in SuHx than in control rats at weeks 5, 8, and 10 (*P < 0.05). SuHx, sugen-hypoxia.
Myocardial Collagen Quantification
Collagen to myocyte histological-area ratios remained unchanged in SuHx RV samples except for animals at week 6 (P = 0.04, Fig. 6, A and D–I). Hydroxyproline assays showed no significant changes in RV free wall collagen content (P > 0.16) when compared with the control group at all timepoints (Fig. 6B). There were no significant differences in RV collagen type I to III ratios between SuHx and control rats at any time (Fig. 6C).
Figure 6.
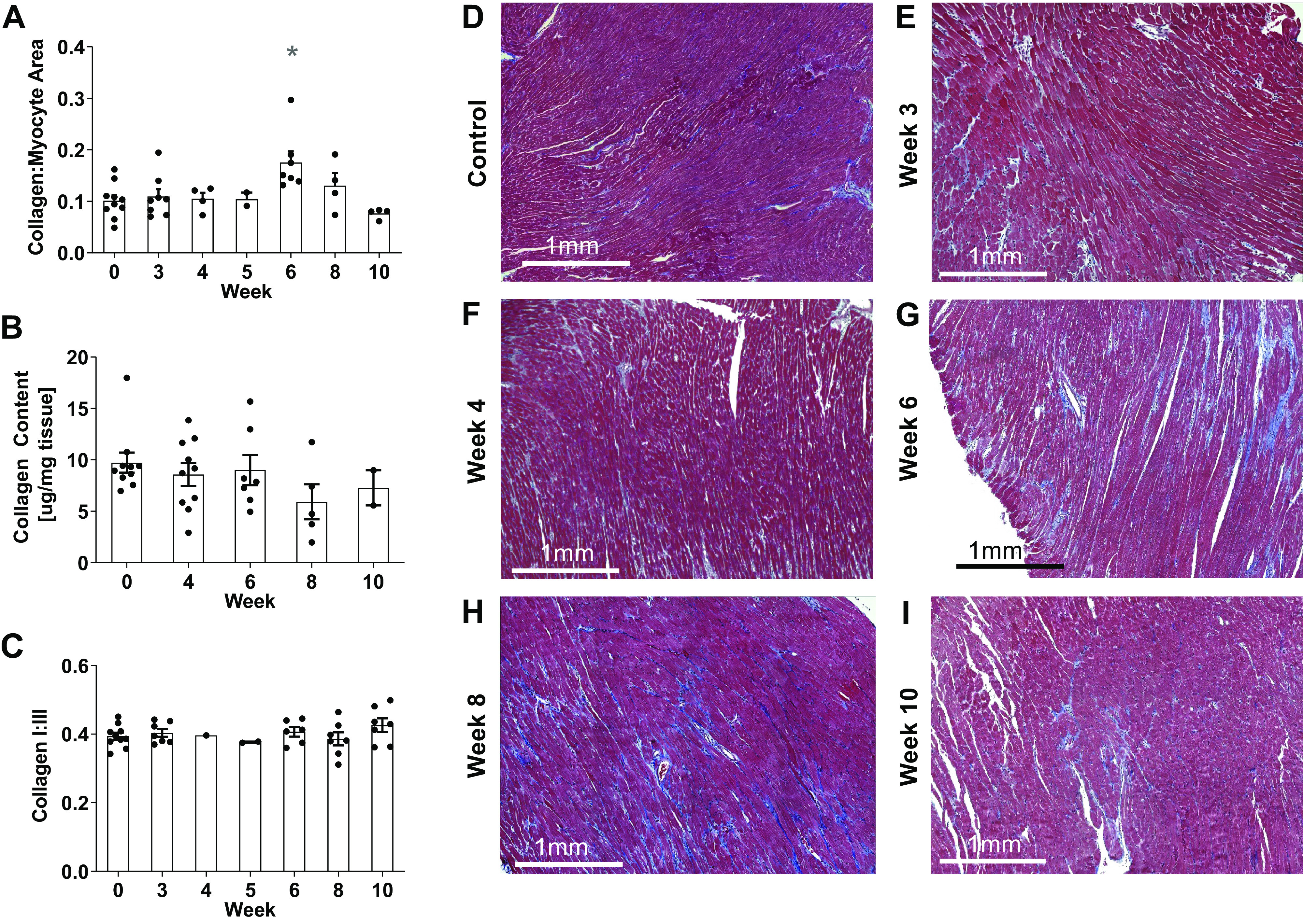
Histological collagen to myocyte area fraction (A) from Masson’s trichrome-stained RV sections in control (D) rats and SuHx rats at weeks 3 (E), 4 (F), 6 (G), 8 (H), and 10 (I) was only significantly different from control at week 6 (*P < 0.05). RV myocardial total collagen content (B) and collagen type I to III ratio (C) showed no significant changes throughout the study (P > 0.05). Data are shown as means ± SE compared with the control group. RV, right ventricle; SuHx, sugen-hypoxia.
DISCUSSION
In this study, we analyzed RV hemodynamic and morphological ventricular remodeling in the male SuHx rat model of PAH up to 10-wk postdisease induction. Initially, RV ejection fraction decreased in response to increased pulmonary arterial pressure but then stabilized after week 4. In contrast RV end-diastolic pressure increased after week 4 with no change in RV end-diastolic volume. We used computational models fitted to measured pressure-volume relations to discriminate the contributions of changes in measured RV geometry from those due to changes in resting and active RV myocardial material properties to altered systolic and diastolic RV chamber function. The analysis showed that right ventricular geometric remodeling was the major factor maintaining systolic function in response to pressure overload, whereas increased passive myocardial stiffness was the principal determinant of the increased end-diastolic chamber stiffness during PAH remodeling.
Systolic Function
In the SuHx-treated rats, total pulmonary vascular resistance and right ventricular end-systolic pressures were significantly larger than in control rats. At week 4, there was an initial decrease in ejection fraction with no significant changes in the ES P-V relation. Pressure-volume analysis explained this decrease by the increased end-systolic volume due to the observed increase in RV end-systolic pressure. Ejection fraction and end-systolic volumes stabilized after week 4 despite further substantial increases in RV ES pressures (from 48 ± 5 mmHg at week 4 to 74 ± 8 mmHg at week 10). Our model analysis showed that this preservation of systolic function was due to increasing Ees that was primarily attributable after week 5 to continued RV hypertrophy rather than recruitment of myofilament activation. From the model equations, this effect was due to the increase in RV wall thickness to radius ratio, corresponding to the observed increase in RV mass to volume ratio from 3.4 mg/μL at week 4 to 4.3 mg/μL by week 10. End-systolic active fiber stress almost doubled by week 3 and remained essentially unchanged thereafter; in contrast, if RV geometry had not remodeled, end-systolic fiber stress would have tripled by week 10 (Table 2). Therefore, the stabilized response of RV systolic function to pulmonary arterial pressure overload after week 4 was primarily due to RV hypertrophy with only transient changes in myofiber contractility in the 10-wk time course of this study.
Compensatory RV hypertrophy or increased contractility in response to increased afterload has been described in patients (35–37) and animal models (38, 39). In human-skinned cardiomyocytes isolated from patients with PAH, Rain et al. (2) measured greater maximal active tension development, but no change in myofilament calcium sensitivity or length-dependent activation compared with cells from patients suspected of PAH but confirmed with normal pulmonary pressures after right heart catheterization. This result may reflect a recruitment of myocyte contractility in compensated PAH but did not eliminate the possibility that RV hypertrophy or dysfunction in the control group may have depressed myocyte force generation. Hsu et al. (40) compared myocyte contractility in patients with severe systemic sclerosis-associated PAH and patients with less severe idiopathic PAH. Skinned myocytes from patients with idiopathic PAH had significantly greater maximal steady-state tension development than control myocytes, but those from patients with systemic sclerosis-associated PAH generated significantly lower maximal force. These findings are consistent with our observation of a transient increase in myofilament activation at week 5 and with the interpretation that RV myocytes can recruit myofilament force generation in response to RV pressure overload but may not be able to maintain this contractile upregulation indefinitely. Wang et al. (20) reported a similar time course of changes in RV systolic function over 8 wk in a sugen-hypoxia mouse model of PAH. They reported no changes in crossbridge kinetics or sarcomere length-dependent myofilament activation in skinned trabeculae from these animals. Although our model predictions of sarcomere length-dependent tension development did not fall significantly below control during the 10-wk study, it is possible that a downregulation of myocyte force development may occur later as seen in patients with more severe PAH (40). Although other studies have also reported decreased RV systolic function in small animal models of pulmonary hypertension, there have not been enough long-term studies in SuHx rats to conclude whether or when they decompensate and fail.
Over the time course in this model, essentially all the RV hypertrophy was explained by RV wall thickening. We saw the same pattern in the monocrotaline-treated rat model, which displayed comparable increases in wall thickness and mass after 4 wk as seen over the same time course in this study (18). Jayasekera et al. (41) observed RV dilatation in SuHx rats at 8 wk and also observed no significant decreases in ejection fraction (from 68 ± 5% to 62 ± 6%) and concluded that RV systolic function remained compensated. SuHx rats in the present study did exhibit a significant reduction in mean EF, but this stabilized by week 4 and never fell below the threshold of 35% that has been reported to correlate with significantly reduced survival in patients with PAH (42, 43).
Diastolic Function
Right ventricular end-diastolic pressures also rose and were significantly higher than control by week 5. However, this increase was not accompanied by any increase in end-diastolic volume due to corresponding increases in RV end-diastolic chamber stiffness. Our model analysis showed that chamber stiffening was not explained by geometric remodeling, but rather was due entirely to an increase in resting myocardial stiffness of up to threefold (Fig. 5). Some researchers have reported RV chamber dilation in 8-wk SuHx rats (41) and increased RV inner diameter in SuHx mice after 3 wk (19). In contrast, other groups using SuHx rats have reported no changes to RV end-diastolic volume in SuHx rats at 8 wk (44) or in SuHx mice at 4 wk (45). Thus, there is no consistent evidence that RV dilation in the first 10 wk is a phenotype of the male rodent sugen-hypoxia model of PAH. Although ED stiffening was sufficient to prevent RV dilation in this study, it may subsequently reverse, become maladaptive, and contribute to diastolic dysfunction or become insufficient permitting subsequent RV enlargement and decompensation.
Diastolic RV chamber stiffening has been observed in patients (2, 4, 6) and animal models (7, 12, 14, 44) of PAH and has been shown to be a prognostic indicator of diastolic dysfunction and PAH disease severity (2, 6). In skinned RV myocytes isolated from patients with PAH who had significantly increased RV diastolic stiffness with significant fibrosis and titin dephosphorylation but no changes in titin isoform composition, Rain et al. (2) reported increased passive sarcomere stiffness. The same group (14) measured increased passive trabecular stiffness in pulmonary artery banded rats that was reversed by incubating tissues in protein kinase A. Interestingly, they also found changes in collagen type I:III ratio but only in rats with severe RV systolic dysfunction. In another study in pulmonary artery banded rats, Baicu et al. (12) reported increased myocardial insoluble collagen, indicative of increased cross-linking. Extracellular matrix (ECM) remodeling in response to right (8, 45) and left (46) ventricular pressure overload and myocardial stiffness is modulated by many properties of the collagen matrix including fiber diameter (47), tortuosity (48, 49), and cross-linking (46, 50, 51). Although our findings are consistent with previous human and animal studies showing highly significant diastolic chamber and myocardial stiffening before the onset of severe systolic dysfunction in PAH, the extent to which stiffness is due to collagen matrix structural remodeling, titin isoform switching or phosphorylation or all three remains unclear.
In our SuHx rat model, we did see histological evidence of fibrosis but increased collagen area fraction only reached statistical significance at week 6. Moreover, there were no significant changes throughout the time course in total collagen content, collagen type I to III ratio, or enzymatic collagen cross-linking (data not shown). Consistent with the predictions of the present model analysis, our previous biaxial mechanical testing of RV myocardium from week 6 SuHx rats showed significantly increased passive biaxial stiffness (15). When the samples were decellularized, the remaining RV collagen ECM also had significantly higher circumferential stiffness than control decellularized myocardium. Taken together with the current results, these findings suggest that passive myocardial stiffening in the SuHx rat model of PAH is at least partly due to ECM remodeling, but the changes to the collagen ECM that cause myocardial stiffening may not be in the total collagen amount, type, or intramolecular cross-linking, but instead may be due to structural alterations in myocardial fiber architecture such as changes in coiled perimysial collagen fiber diameter or tortuosity (47–49).
Although RV hypertrophy did not contribute significantly to increased end-diastolic chamber stiffness, it was important for reducing end-diastolic stress in the free wall. Without the observed RV geometric remodeling, the model showed that RV ED fiber stress would have been substantially greater (Table 2). Hence, although changes in passive material properties were primarily responsible for preventing increased RV ED volumes during PAH, geometric remodeling acted primarily to limit increased fiber stress.
Animal Model of PAH
The male SuHx rat model was chosen as it closely mimics human pathology in vascular plexiform lesion formation and RV pressure overload and remodeling (22, 41, 52–54). Although a comprehensive longitudinal study of PAH in the SuHx model has not been studied before, our study is in agreement with previously reported hemodynamic and morphological measurements in SuHx-treated animals (22, 41, 52). It is well known clinically that PAH is two to four times more prevalent in females (55), and studies suggest that women may develop less systolic dysfunction and more concentric hypertrophy than males with equivalent pulmonary arterial pressures (56). Therefore, to avoid confounding effects of possible sex differences in RV remodeling between male and female SuHx rats, we only used males in this study. Interestingly, studies of sex differences in RV remodeling between male and female SuHx rats to date have only shown differences when the females were ovariectomized (57).
Limitations
Hemodynamic measurements were obtained in an open-chest pericardiectomized state, which negates the effects of intrathoracic pressures, transmural pressure gradients, and the boundary condition due to the absence of the pericardium (58). Noninvasive cardiac MRI measurements in an albeit very small subset of animals showed differences between control and SuHx animals in end-systolic and end-diastolic volume that were consistent with our invasive measurements. Stroke volumes and ejection fractions showed close agreement between our closed- and opened-chest measures (Table 1). In a previous larger cohort study of pressure-overloaded rats, Andersen et al. (7) reported CMR measures of ejection fraction (from 72 ± 1% to 52 ± 2%, 20% decrease) in agreement with the limited CMR (from 72% to 49%, 23% decrease) and extensive catheterization measures (from 66 ± 2 to 48 ± 6%, 18% decrease) of ejection fraction in this study. Furthermore, the changes in ejection fraction that we measured in this study are similar to those observed in patients with PAH by Rain et al. (2) (from 57 ± 5% to 36 ± 4%, 21% decrease) and others (43, 59, 60). Therefore, the differences due to the measurement technique were likely small compared with the differences due to the SuHx treatment.
The biomechanics model used a thin-walled simplifying approximation, which ignores transmural gradients of stress and strain. The radius to wall thickness ratio was 11.1 ± 0.4 in the control group animals, and although RV thickness increased due to pulmonary pressure overload, this ratio was greater than 6 in all treatment groups and suggested that the thin-walled assumption remained justified. To model end-systolic and end-diastolic pressure-volume relations incorporating RV geometry and myocardial passive and active properties, a spherical model of the RV was adapted from a biventricular model (18, 61). Although our mathematical model included the effects of septal hypertrophy, it did not account for trans-septal pressure gradients or indirect LV-RV interactions. In the SuHx male rats, we did not observe any significant changes in left ventricular systolic pressure or ejection fraction in the 10-wk study or changes in LV plus septal mass at weeks 3, 4, 6, and 10. These observations are in agreement with previous studies of SuHx mouse (20) and rat (22, 41) models of PAH. We did account for length-dependent myofilament activation and nonlinear stress-strain relations. Our constitutive models were parsimonious and only required two adjustable material parameters for end diastole and one for end systole. Thus, the risk of overfitting was low. The model analysis assumed time-varying elastance, so that changes in RV mechanics due to changes in anelastic properties such as viscoelasticity and the force-velocity relation were not accounted for, but these factors are not likely to be major contributors to altered RV mechanics in PAH. The model results also depended on estimating unloaded RV volumes. We used the minimum RV volumes measured during caval occlusions to estimate this volume, but these measurements were variable likely because RV shape is less cylindrical at low volumes, which may affect the accuracy of the admittance catheter measurements.
Although the changes in passive myocardial material properties during PAH that we report here are predictions of a biomechanics model, these predictions have been validated with ex vivo planar biaxial mechanical measurements in resting myocardium (15) from the same cohort animals. The threefold increase in myocardial stiffness measured in the circumferential direction of RV free wall samples at weeks 5 and 6 closely matched the changes in end-diastolic fiber stress-sarcomere length relations predicted by the model at these timepoints.
Conclusions
We used a novel combination of structural and hemodynamic measurements of the RV in the male SuHx rat model of PAH with computational modeling of right ventricular mechanics to distinguish the contributions of right ventricular geometric remodeling from changes in intrinsic passive and active myocardial material properties to alterations in systolic and diastolic RV chamber function over a 10-wk time course. The analysis showed that systolic ventricular function was stabilized primarily by hypertrophic wall thickening with only transient changes in myofiber contractility, whereas significantly increased end-diastolic chamber stiffness was almost entirely attributable to increased resting myocardial stiffness that was not associated with altered myocardial collagen content or types. This previously unrecognized increase in right ventricular myocardial diastolic stiffness after RV hypertrophy has already stabilized systolic function may be a compensatory mechanism that prevents RV dilation, as observed in this study, but could eventually contribute to diastolic dysfunction.
SUPPLEMENTAL DATA
Supplemental Table S1 and Supplemental Figs. S1–S5: https://doi.org/10.6084/m9.figshare.14780016.
GRANTS
This work was funded by American Heart Association Grant 16SDG29670010 (to D. Valdez-Jasso); National Heart, Lung, and Blood Institute Grants 1R25HL145817-01 and 1R01HL155945-01 (to D. Valdez-Jasso) and 1T32HL105373 (to E. D. Kwan and E. Pursell); and National Science Foundation CAREER Award 2046259 (to D. Valdez-Jasso).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.V.-R., E.P., and D.V.-J. conceived and designed research; D.V.-R., E.D.K., H.M., M.P., E.P., J.S., and D.V.-J. performed experiments; E.D.K., D.V.-R, X.Z., H.M., M.P., J.S., and D.V.-J. analyzed data; E.D.K., D.V.-R, J.S., and D.V.-J. interpreted results of experiments; E.D.K. and X.Z. prepared figures; E.D.K. drafted manuscript; E.D.K., D.V.-R. and D.V.-J. edited and revised manuscript; E.D.K., D.V.-R., X.Z., H.M., M.P., E.P., J.S., and D.V.-R. approved final version of manuscript.
REFERENCES
- 1.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 53: 1801913, 2019. doi: 10.1183/13993003.01913-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rain S, Handoko ML, Trip P, Gan CTJ, Westerhof N, Stienen GJ, Paulus WJ, Ottenheijm CAC, Marcus JT, Dorfmüller P, Guignabert C, Humbert M, Macdonald P, Dos Remedios C, Postmus PE, Saripalli C, Hidalgo CG, Granzier HL, Vonk-Noordegraaf A, Van Der Velden J, De Man FS. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation 128: 2016–2025, 2013. doi: 10.1161/CIRCULATIONAHA.113.001873. [DOI] [PubMed] [Google Scholar]
- 3.Trip P, Rain S, Handoko ML, van der Bruggen C, Bogaard HJ, Marcus JT, Boonstra A, Westerhof N, Vonk-Noordegraaf A, de Man FS. Clinical relevance of right ventricular diastolic stiffness in pulmonary hypertension. Eur Respir J 45: 1603–1612, 2015. doi: 10.1183/09031936.00156714. [DOI] [PubMed] [Google Scholar]
- 4.Gan CT-J, Holverda S, Marcus JT, Paulus WJ, Marques KM, Bronzwaer JGF, Twisk JW, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Right ventricular diastolic dysfunction and the acute effects of sildenafil in pulmonary hypertension patients. Chest 132: 11–17, 2007. doi: 10.1378/chest.06-1263. [DOI] [PubMed] [Google Scholar]
- 5.Swift AJ, Capener D, Johns C, Hamilton N, Rothman A, Elliot C, Condliffe R, Charalampopoulos A, Rajaram S, Lawrie A, Campbell MJ, Wild JM, Kiely DG. Magnetic resonance imaging in the prognostic evaluation of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 196: 228–239, 2017. doi: 10.1164/rccm.201611-2365OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanderpool RR, Pinsky MR, Naeije R, Deible C, Kosaraju V, Bunner C, Mathier MA, Lacomis J, Champion HC, Simon MA. RV-pulmonary arterial coupling predicts outcome in patients referred for pulmonary hypertension. Heart 101: 37–43, 2015. doi: 10.1136/heartjnl-2014-306142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersen S, Schultz JG, Andersen A, Ringgaard S, Nielsen JM, Holmboe S, Vildbrad MD, De Man FS, Bogaard HJ, Vonk-Noordegraaf A, Nielsen-Kudsk JE. Effects of bisoprolol and losartan treatment in the hypertrophic and failing right heart. J Card Fail 20: 864–873, 2014. doi: 10.1016/j.cardfail.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Dias-Neto M, Luísa-Neves A, Pinho S, Gonçalves N, Mendes M, Eloy C, Lopes JM, Gonçalves D, Ferreira-Pinto M, Leite-Moreira AF, Henriques-Coelho T. Pathophysiology of infantile pulmonary arterial hypertension induced by monocrotaline. Pediatr Cardiol 36: 1000–1013, 2015. doi: 10.1007/s00246-015-1111-y. [DOI] [PubMed] [Google Scholar]
- 9.Neto-Neves EM, Frump AL, Vayl A, Kline JA, Lahm T. Isolated heart model demonstrates evidence of contractile and diastolic dysfunction in right ventricles from rats with sugen/hypoxia-induced pulmonary hypertension. Physiol Rep 5: e13438, 2017. doi: 10.14814/phy2.13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu S, Houston BA, Tampakakis E, Bacher AC, Rhodes PS, Mathai SC, Damico RL, Kolb TM, Hummers LK, Shah AA, McMahan Z, Corona-Villalobos CP, Zimmerman SL, Wigley FM, Hassoun PM, Kass DA, Tedford RJ. Right ventricular functional reserve in pulmonary arterial hypertension. Circulation 133: 2413–2422, 2016. doi: 10.1161/CIRCULATIONAHA.116.022082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabourin J, Boet A, Rucker-Martin C, Lambert M, Gomez AM, Benitah JP, Perros F, Humbert M, Antigny F. Ca2+ handling remodeling and STIM1L/Orai1/TRPC1/TRPC4 upregulation in monocrotaline-induced right ventricular hypertrophy. J Mol Cell Cardiol 118: 208–224, 2018. doi: 10.1016/j.yjmcc.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Baicu CF, Li J, Zhang Y, Kasiganesan H, Cooper IG, Zile MR, Bradshaw AD. Time course of right ventricular pressure-overload induced myocardial fibrosis: relationship to changes in fibroblast postsynthetic procollagen processing. Am J Physiol Heart Circ Physiol 303: H1128–H1134, 2012. doi: 10.1152/ajpheart.00482.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 120: 1951–1960, 2009. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 14.Rain S, Andersen S, Najafi A, Gammelgaard Schultz J, da Silva Gonçalves Bós D, Handoko ML, Bogaard H-J, Vonk-Noordegraaf A, Andersen A, van der Velden J, Ottenheijm CAC, de Man FS. Right ventricular myocardial stiffness in experimental pulmonary arterial hypertension. Circ Heart Fail 9: e002636, 2016. doi: 10.1161/CIRCHEARTFAILURE.115.002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vélez-Rendón D, Pursell ER, Shieh J, Valdez-Jasso D. Relative contributions of matrix and myocytes to biaxial mechanics of the right ventricle in pulmonary arterial hypertension. J Biomech Eng 141: 091011, 2019. doi: 10.1115/1.4044225. [DOI] [PubMed] [Google Scholar]
- 16.Hill MR, Simon MA, Valdez-Jasso D, Zhang W, Champion HC, Sacks MS. Structural and mechanical adaptations of right ventricle free wall myocardium to pressure overload. Ann Biomed Eng 42: 2451–2465, 2014. doi: 10.1007/s10439-014-1096-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Legchenko E, Chouvarine P, Borchert P, Fernandez-Gonzalez A, Snay E, Meier M, Maegel L, Mitsialis SA, Rog-Zielinska EA, Kourembanas S, Jonigk D, Hansmann G. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med 10: eaao0303, 2018. doi: 10.1126/scitranslmed.aao0303. [DOI] [PubMed] [Google Scholar]
- 18.Vélez-Rendón D, Zhang X, Gerringer J, Valdez-Jasso D. Compensated right ventricular function of the onset of pulmonary hypertension in a rat model depends on chamber remodeling and contractile augmentation. Pulm Circ 8: 2045894018800439, 2018. doi: 10.1177/2045894018800439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vitali SH, Hansmann G, Rose C, Fernandez-Gonzalez A, Scheid A, Mitsialis SA, Kourembanas S. The Sugen 5416/hypoxia mouse model of pulmonary hypertension revisited: long-term follow-up. Pulm Circ 4: 619–629, 2014. doi: 10.1086/678508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Patel JR, Schreier DA, Hacker TA, Moss RL, Chesler NC. Organ-level right ventricular dysfunction with preserved Frank-Starling mechanism in a mouse model of pulmonary arterial hypertension. J Appl Physiol (1985) 124: 1244–1253, 2018. doi: 10.1152/japplphysiol.00725.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zungu-Edmondson M, Shults NV, Wong CM, Suzuki YJ. Modulators of right ventricular apoptosis and contractility in a rat model of pulmonary hypertension. Cardiovasc Res 110: 30–39, 2016. doi: 10.1093/cvr/cvw014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121: 2747–2754, 2010. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 23.Lai N, Lu W, Wang J. Ca2+ and ion channels in hypoxia-mediated pulmonary hypertension. Int J Clin Exp Pathol 8: 1081–1092, 2015. [PMC free article] [PubMed] [Google Scholar]
- 24.Raghavan K, Porterfield JE, Kottam ATG, Feldman MD, Escobedo D, Valvano JW, Pearce JA. Electrical conductivity and permittivity of murine myocardium. IEEE Trans Biomed Eng 56: 2044–2053, 2009. doi: 10.1109/TBME.2009.2012401. [DOI] [PubMed] [Google Scholar]
- 25.Wei CL, Valvano JW, Feldman MD, Pearce JA. Nonlinear conductance-volume relationship for murine conductance catheter measurement system. IEEE Trans Biomed Eng 52: 1654–1661, 2005. doi: 10.1109/TBME.2005.856029. [DOI] [PubMed] [Google Scholar]
- 26.Vonk-Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J, Naeije R, Newman J, Oudiz RJ, Provencher S, Torbicki A, Voelkel NF, Hassoun PM. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol 62: D22–D33, 2013. doi: 10.1016/j.jacc.2013.10.027. [DOI] [PubMed] [Google Scholar]
- 27.Müller S, Schocke M, Hunold P, Pachinger O, Bartel T. Accurate, simplified and rapid three-dimensional echocardiographic volume quantifications: comparison of different algorithms in symmetric and asymmetric left ventricular geometry. Can J Cardiol 20: 1091–1096, 2004. [PubMed] [Google Scholar]
- 28.Lee LC, Zhihong Z, Hinson A, Guccione JM. Reduction in left ventricular wall stress and improvement in function in failing hearts using Algisyl-LVR. J Vis Exp (74): 50096, 2013. doi: 10.3791/50096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vonk-Noordegraaf A, Westerhof N. Describing right ventricular function. Eur Respir J 41: 1419–1423, 2013. doi: 10.1183/09031936.00160712. [DOI] [PubMed] [Google Scholar]
- 30.Ter Keurs HEDJ, Rijnsburger WH, Van Heuningen R, Nagelsmit MJ. Tension development and sarcomere length in rat cardiac trabeculae. Evidence of length-dependent activation. Circ Res 46: 703–714, 1980. doi: 10.1161/01.RES.46.5.703. [DOI] [PubMed] [Google Scholar]
- 31.Guccione JM, McCulloch AD. Mechanics of active contraction in cardiac muscle: part I—constitutive relations for fiber stress that describe deactivation. J Biomech Eng 115: 72–81, 1993. doi: 10.1115/1.2895473. [DOI] [PubMed] [Google Scholar]
- 32.Tözeren A. Continuum rheology of muscle contraction and its application to cardiac contractility. Biophys J 47: 303–309, 1985. doi: 10.1016/S0006-3495(85)83920-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Haynes P, Campbell KS, Wenk JF. Numerical evaluation of myofiber orientation and transmural contractile strength on left ventricular function. J Biomech Eng 137: 044502, 2015. doi: 10.1115/1.4028990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yipintsoi T, Scanlon PD, Bassingthwaighte JB. Density and water content of dog ventricular myocardium. Proc Soc Exp Biol Med 141: 1032–1035, 1972. doi: 10.3181/00379727-141-36927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naeije R, Manes A. The right ventricle in pulmonary arterial hypertension. Eur Respir Rev 23: 476–487, 2014. doi: 10.1183/09059180.00007414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van De Veerdonk MC, Bogaard HJ, Voelkel NF. The right ventricle and pulmonary hypertension. Heart Fail Rev 21: 259–271, 2016. doi: 10.1007/s10741-016-9526-y. [DOI] [PubMed] [Google Scholar]
- 37.Vonk Noordegraaf A, Chin KM, Haddad F, Hassoun PM, Hemnes AR, Hopkins SR, Kawut SM, Langleben D, Lumens J, Naeije R. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. Eur Respir J 53: 1801900, 2019. doi: 10.1183/13993003.01900-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Man FS, Handoko ML, van Ballegoij JJM, Schalij I, Bogaards SJP, Postmus PE, van der Velden J, Westerhof N, Paulus WJ, Vonk-Noordegraaf A. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail 5: 97–105, 2012. doi: 10.1161/CIRCHEARTFAILURE.111.964494. [DOI] [PubMed] [Google Scholar]
- 39.Karunanithi MK, Michniewicz J, Copeland SE, Feneley MP. Right ventricular preload recruitable stroke work, end-systolic pressure-volume, and dP/dtmax-end-diastolic volume relations compared as indexes of right ventricular contractile performance in conscious dogs. Circ Res 70: 1169–1179, 1992. doi: 10.1161/01.RES.70.6.1169. [DOI] [PubMed] [Google Scholar]
- 40.Hsu S, Kokkonen-Simon KM, Kirk JA, Kolb TM, Damico RL, Mathai SC, Mukherjee M, Shah AA, Wigley FM, Margulies KB, Hassoun PM, Halushka MK, Tedford RJ, Kass DA. Right ventricular myofilament functional differences in humans with systemic sclerosis-associated versus idiopathic pulmonary arterial hypertension. Circulation 137: 2360–2370, 2018. doi: 10.1161/CIRCULATIONAHA.117.033147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jayasekera G, Wilson KS, Buist H, Woodward R, Uckan A, Hughes C, Nilsen M, Church AC, Johnson MK, Gallagher L, Mullin J, MacLean MR, Holmes WM, Peacock AJ, Welsh DJ. Understanding longitudinal biventricular structural and functional changes in a pulmonary hypertension Sugen–hypoxia rat model by cardiac magnetic resonance imaging. Pulm Circ 10: 2045894019897513, 2020. doi: 10.1177/2045894019897513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brewis MJ, Bellofiore A, Vanderpool RR, Chesler NC, Johnson MK, Naeije R, Peacock AJ. Imaging right ventricular function to predict outcome in pulmonary arterial hypertension. Int J Cardiol 218: 206–211, 2016. doi: 10.1016/j.ijcard.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van De Veerdonk MC, Kind T, Marcus JT, Mauritz GJ, Heymans MW, Bogaard HJ, Boonstra A, Marques KMJ, Westerhof N, Vonk-Noordegraaf A. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol 58: 2511–2519, 2011. doi: 10.1016/j.jacc.2011.06.068. [DOI] [PubMed] [Google Scholar]
- 44.da Silva Gonçalves Bos D, Happé C, Schalij I, Pijacka W, Paton JFR, Guignabert C, Tu L, Thuillet R, Bogaard HJ, van Rossum AC, Vonk-Noordegraaf A, de Man FS, Handoko ML. Renal denervation reduces pulmonary vascular remodeling and right ventricular diastolic stiffness in experimental pulmonary hypertension. JACC Basic Transl Sci 2: 22–35, 2017. doi: 10.1016/j.jacbts.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Z, Schreier DA, Hacker TA, Chesler NC. Progressive right ventricular functional and structural changes in a mouse model of pulmonary arterial hypertension. Physiol Rep 1: e00184, 2013. doi: 10.1002/phy2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Norton GR, Tsotetsi J, Trifunovic B, Hartford C, Candy GP, Woodiwiss AJ. Myocardial stiffness is attributed to alterations in cross-linked collagen rather than total collagen or phenotypes in spontaneously hypertensive rats. Circulation 96: 1991–1998, 1997. doi: 10.1161/01.CIR.96.6.1991. [DOI] [PubMed] [Google Scholar]
- 47.MacKenna DA, Vaplon SM, McCulloch AD. Microstructural model of perimysial collagen fibers for resting myocardial mechanics during ventricular filling. Am J Physiol Heart Circ Physiol 273: H1576–H1586, 1997. doi: 10.1152/ajpheart.1997.273.3.h1576. [DOI] [PubMed] [Google Scholar]
- 48.MacKenna DA, Omens JH, Covell JW. Left ventricular perimysial collagen fibers uncoil rather than stretch during diastolic filling. Basic Res Cardiol 91: 111–122, 1996. doi: 10.1007/BF00799683. [DOI] [PubMed] [Google Scholar]
- 49.Robinson TF, Geraci MA, Sonnenblick EH, Factor SM. Coiled perimysial fibers of papillary muscle in rat heart: morphology, distribution, and changes in configuration. Circ Res 63: 577–592, 1988. doi: 10.1161/01.RES.63.3.577. [DOI] [PubMed] [Google Scholar]
- 50.Herrmann KL, McCulloch AD, Omens JH. Glycated collagen cross-linking alters cardiac mechanics in volume-overload hypertrophy. Am J Physiol Heart Circ Physiol 284: H1277–H1284, 2003. doi: 10.1152/ajpheart.00168.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herum KM, Lunde IG, Skrbic B, Louch WE, Hasic A, Boye S, Unger A, Brorson SH, Sjaastad I, Tønnessen T, Linke WA, Gomez MF, Christensen G. Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc Res 106: 217–226, 2015. doi: 10.1093/cvr/cvv002. [DOI] [PubMed] [Google Scholar]
- 52.Al-Husseini A, Wijesinghe DS, Farkas L, Kraskauskas D, Drake JI, Van Tassel B, Abbate A, Chalfant CE, Voelkel NF. Increased eicosanoid levels in the Sugen/chronic hypoxia model of severe pulmonary hypertension. PLoS One 10: e0120157, 2015. doi: 10.1371/journal.pone.0120157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dai Z, Zhu MM, Peng Y, Machireddy N, Evans CE, Machado R, Zhang X, Zhao YY. Therapeutic targeting of vascular remodeling and right heart failure in pulmonary arterial hypertension with a HIF-2a inhibitor. Am J Respir Crit Care Med 198: 1423–1434, 2018. doi: 10.1164/rccm.201710-2079OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drozd K, Ahmadi A, Deng Y, Jiang B, Petryk J, Thorn S, Stewart D, Beanlands R, DeKemp RA, DaSilva JN, Mielniczuk LM. Effects of an endothelin receptor antagonist, Macitentan, on right ventricular substrate utilization and function in a Sugen 5416/hypoxia rat model of severe pulmonary arterial hypertension. J Nucl Cardiol 24: 1979–1989, 2017. doi: 10.1007/s12350-016-0663-4. [DOI] [PubMed] [Google Scholar]
- 55.Guiot J, Parzibut G, Weber T, Davin L, Dulgheru R, Lancellotti P, Louis R, Vachiery JL. [Pulmonary arterial hypertensiony]. Rev Med Liege 74: 139–145, 2019. doi: 10.1161/circresaha.115.301146. [DOI] [PubMed] [Google Scholar]
- 56.Ventetuolo CE, Ouyang P, Bluemke DA, Tandri H, Barr RG, Bagiella E, Cappola AR, Bristow MR, Johnson C, Kronmal RA, Kizer JR, Lima JAC, Kawut SM. Sex hormones are associated with right ventricular structure and function: the MESA-right ventricle study. Am J Respir Crit Care Med 183: 659–667, 2011. doi: 10.1164/rccm.201007-1027OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lahm T, Frump AL, Albrecht ME, Fisher AJ, Cook TG, Jones TJ, Yakubov B, Whitson J, Fuchs RK, Liu A, Chesler NC, Brown MB. 17β-Estradiol mediates superior adaptation of right ventricular function to acute strenuous exercise in female rats with severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 311: L375–L388, 2016. doi: 10.1152/ajplung.00132.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoit BD, Ball N, Walsh RA. Invasive hemodynamics and force-frequency relationships in open- versus closed-chest mice. Am J Physiol Heart Circ Physiol 273: H2528–H2533, 1997. doi: 10.1152/ajpheart.1997.273.5.h2528. [DOI] [PubMed] [Google Scholar]
- 59.Li JH, Zhang H, Da Wang ZZ, Lu QQ, Li D, Lian TY, Lv ZC, Jiang X, Wu Y, Ye J, Zhao S, Yang Z. Acute iloprost inhalation improves right ventricle function in pulmonary artery hypertension: a cardiac magnetic resonance study. Front Pharmacol 9: 1550, 2019. doi: 10.3389/fphar.2018.01550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swift AJ, Rajaram S, Campbell MJ, Hurdman J, Thomas S, Capener D, Elliot C, Condliffe R, Wild JM, Kiely DG. Prognostic value of cardiovascular magnetic resonance imaging measurements corrected for age and sex in idiopathic pulmonary arterial hypertension. Circ Cardiovasc Imaging 7: 100–106, 2014. doi: 10.1161/CIRCIMAGING.113.000338. [DOI] [PubMed] [Google Scholar]
- 61.Lumens J, Delhaas T, Kirn B, Arts T. Three-wall segment (TriSeg) model describing mechanics and hemodynamics of ventricular interaction. Ann Biomed Eng 37: 2234–2255, 2009. doi: 10.1007/s10439-009-9774-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1 and Supplemental Figs. S1–S5: https://doi.org/10.6084/m9.figshare.14780016.