

Abstract
Airborne particulate matter (PM) is associated with an increased risk for cardiovascular diseases. Although the goal of thermal remediation is to eliminate organic wastes through combustion, when incomplete combustion occurs, organics chemisorb to transition metals to generate PM-containing environmentally persistent free radicals (EPFRs). Similar EPFR species have been detected in PM found in diesel and gasoline exhaust, woodsmoke, and urban air. Prior in vivo studies demonstrated that EPFRs reduce cardiac function secondary to elevations in pulmonary arterial pressures. In vitro studies showed that EPFRs increase ROS and cytokines in pulmonary epithelial cells. We thus hypothesized that EPFR inhalation would promote lung inflammation and oxidative stress, leading to systemic inflammation, vascular endothelial injury, and a decline in vascular function. Mice were exposed to EPFRs for either 4 h or for 4 h/day for 10 days and lung and vascular function were assessed. After a 4-h exposure, plasma nitric oxide (NO) was reduced while endothelin-1 (ET-1) was increased, however lung function was not altered. After 10 day, plasma NO and ET-1 levels were again altered and lung tidal volume was reduced. These time course studies suggested the vasculature may be an early target of injury. To test this hypothesis, an intermediate time point of 3 days was selected. Though the mice exhibited no marked inflammation in either the lung or the blood, we did note significantly reduced endothelial function concurrent with a reduction in lung tidal volume and an elevation in annexin V protein levels in the lung. Although vascular dysfunction was not dependent upon inflammation, it may be associated with an injury at the air-blood interface. Gene expression analysis suggested roles for oxidative stress and aryl hydrocarbon receptor (Ahr) signaling. Studies probing the relationship between pulmonary oxidative stress and AhR signaling at the air-blood interface with vascular dysfunction seem warranted.
NEW & NOTEWORTHY Particulate matter (PM) resulting from the combustion of organic matter is known to contribute to cardiopulmonary disease. Despite hypotheses that cardiovascular dysfunction occurring after PM exposures is secondary to lung or systemic inflammation, these studies investigating exposures to PM-containing environmentally persistent free radicals (EPFRs) demonstrate that cardiovascular dysfunction precedes pulmonary inflammation. The cardiopulmonary health consequences of EPFRs have yet to be thoroughly evaluated, especially in healthy, adult mice. Our data suggest the vasculature as a direct target of PM exposure, and our studies aimed to elucidate the mechanisms contributing to EPFR-induced vascular dysfunction.
Keywords: cardiovascular, free radicals, inhalation, particulate matter
INTRODUCTION
Particulate matter (PM) air pollution forms in the air as solid particles or liquid droplets and from sources ranging from dust and dirt to soot and smoke. The detrimental health impacts associated with PM are well known, with studies indicating correlations between PM concentration and cardiopulmonary diseases (1–6). Although airborne PM is commonly linked with cardiopulmonary morbidity and mortality, the specific mechanisms underlying the development of PM-related cardiopulmonary diseases are not well characterized. Nevertheless, as PM levels increase, the incidence of asthma increases (7, 8) and a decrease in lung function has been demonstrated (9). Moreover, for every 5 µg/m3 that PM is increased over a year’s time, coronary events increase by 13%, and for every 10 µg/m3 PM increase, hypertension increases by 13% (10). In addition, ∼200,000 deaths each year in the United States alone are attributed to PM exposure (11). Studies from the Multi-Ethnic Study of Atherosclerosis (MESA) have also revealed the association between PM exposure and cardiovascular diseases. Higher PM levels were associated with elevations in systolic blood and pulse pressures (12). Even children exposed to elevated PM levels exhibited increased pulmonary arterial pressures, suggesting that PM exposure has important consequences in both adults and children (13). Important to studies presented here, PM exposures were shown to induce endothelial dysfunction, measured as a decreased flow-mediated dilation in the brachial artery of human subjects (14). These data suggest that in humans, the endothelium may be a particular locus of injury.
PM resulting from thermal remediation of hazardous organic wastes and contaminated soils contains environmentally persistent free radicals (EPFRs). EPFRs are a result of incomplete combustion occurring when oxygen-poor cool pockets form within the flame and on the outer edges of the flame where the temperatures are not as hot. In these regions, organic pollutants chemisorb to transition metals in the PM, producing EPFRs (15, 16). These EPFRs are unique pollutant-particle systems that redox cycle, resulting in the generation of reactive oxygen species (ROS) in soils and air for extended periods of time, ranging from days to weeks (17, 18), and conferring a toxicity that is greater than that of the particle or pollutant alone (19). The redox activity of EPFRs is maintained in biological fluids and tissues (19). Inhaled EPFRs elicit detrimental effects on the cardiopulmonary system (20), resulting in elevations in vascular pulmonary resistance (21) and reductions in pulmonary function (22–24). It should be noted that although our studies emphasize EPFRs produced through thermal remediation processes, similar quinoid radicals have been identified in PM from a variety of sources associated with combustion processes, including diesel and gasoline exhaust particles, woodsmoke, and urban air PM (25–27).
Despite epidemiologic data regarding PM exposures per se, there remains a significant gap in literature linking EPFR content of PM exposures with impacts on human health. This lack of data likely stems from the fact that although PM levels are monitored and regulated, their EPFR content is not, perhaps due to the necessity of expensive instrumentation, for example, electron paramagnetic resonance (EPR), for their assessment.
Early PM research focused on the pulmonary system and pulmonary/systemic inflammation as a cause of PM-associated cardiovascular dysfunction (28–30), as the pulmonary system is the first line of defense against PM and PM is typically removed from the body via inflammatory cells. However, numerous studies are now emphasizing the cardiovascular system and vasculature as important early targets of PM (31–33). The effects that EPFRs exert on vascular endothelial function are of concern and any expected detrimental effects to the vasculature after EPFR exposure need to be mechanistically dissected.
Oxidative stress is inevitably an underlying factor in the development of cardiopulmonary diseases associated with PM (34) and presumably EPFR exposure. Increased inflammatory responses such as increased cytokines and activation of the aryl hydrocarbon receptor (AhR) have been documented in in vitro studies (35). In addition, although AhR mediates the toxic effects of hydrocarbons as well as PM (36), it has recently been shown to facilitate normal immune and cardiovascular physiological processes (34), potentially illuminating new signaling pathways for cardiopulmonary disease initiation as well as molecular targets for therapy.
Whole body animal exposures that mimic real-world human exposures, coupled to EPR verification of EPFR levels within PM are needed to fully understand the risks posed by EPFR inhalation and their resulting health effects, particularly with respect to their impacts on the pulmonary and vasculature systems. Elucidating the mechanisms and driving forces behind EPFR-associated cardiopulmonary disease events is necessary for improving human health outcomes through either targeted therapies or exposure mitigation strategies. Building upon our prior findings that inhalation of EFPRs, but not non-EFPR controls, reduces cardiac function via increased pulmonary arterial pressures (21), we hypothesized that inhaled EPFRs induce cardiopulmonary inflammation and oxidative stress responses, which in turn promote vascular dysfunction. To test this hypothesis, we conducted time- and dose-response studies of EPFR inhalation in mice. Measures of EPFR-mediated inflammation and oxidative stress in the lungs and cardiovascular system were coupled to assessments of lung and vascular function, as well as gene expression studies of oxidative stress compared with AhR signaling pathways, so as to tease apart cause-effect relationships.
MATERIALS AND METHODS
Inhalation Exposure System
A whole body inhalation system was developed, using a custom-designed 18-L Plexiglas chamber containing 16 individual compartments, enabling simultaneous exposure of separated mice to the same EPFR concentration for hours at a time. EPFR PM aerosols were generated by a dry powder dispersion technique, using a Venturi disperser (37). Desired EPFR concentration was achieved by adjusting the initial quantity of EPFR particles used. No aerosol neutralizer was used (38, 39). The flow rate of filtered air for controls and for diluting the EPFRs was ∼4 L/min. Total mass concentration in the chamber was determined gravimetrically and monitored real time via a TSI DustTrak II Model 8530.
Generation and Characterization of Combustion-Derived EPFRs
The generation and characterization of the EPFR particles used in these studies has been previously described (16). The EPFRs were generated with copper as the transition metal and dichlorobenzene (DCB) as the organic substrate, with heating at 230°C. Hence, EPFRs used for studies presented here are denoted as DCB230. Particles were characterized for size via transmission electron microscopy (TEM). Briefly, DCB230 particles were collected during a mock exposure where they were aerosolized into the inhalation chamber in the same manner and at the same flow rate as used for mouse exposures. Aerosolized particles were collected onto a precoated carbon Formvar copper grid, glued to a 25-mm polycarbonate filter with Loctite Superglue. Sampling took place for either 2.5 or 5 min. Characterization of the aerosolized particles was performed by TEM using the JEOL JEM-1010 microscope. The PM particle size after aerosolization was determined to be <0.2 µm in diameter, well within the ultrafine range.
Mice and Inhalation Dosimetry
All animal exposures and protocols were approved by the LSU Institutional Animal Care and Use Committee (Protocol No. 15–044 and 18–074) and adhered to practices recommended in the Guide for the Care and Use of Laboratory Animals (8th ed.). Our initial dosing paradigm was chosen based on PM2.5 levels reported in polluted environments. For example, a recent report noted that PM2.5 concentrations in several cities of China (Beijing, Baoding, Langfang) often reach 0.8 mg/m3 (40, 41). Male C57BL/6 mice were obtained from Jackson Laboratory (Bar Harbor, ME) at 8–10 wk of age, were acclimated to the room for 1 wk before testing, were allowed ad libitum access to food and water and were maintained on a regular 12-h:12-h light/dark cycle. At 10–12 wk of age, mice were exposed to 0.5 mg/m3 or 1.5 mg/m3 DCB230 for 4 h (single exposure) or for 4 h a day for either 3 or 10 consecutive days. Mice exposed to filtered air (FA) were used as controls for each of the experiments and were monitored along with the DCB230-exposed mice. After each day’s exposures, the mice were returned to FA cage banks for the remainder of the 24-h period. Thus, average daily exposures were 0.083 or 0.250 mg/m3/h.
With respect to the dosing paradigm, we considered two additional variables: 1) the concentration of EPFRs (i.e., free radicals) generated in relation to that known associated with urban PM and 2) pulmonary deposition expected for these exposures. First, ambient air PM contains a wide concentration range of EPFRs. Thus, EPFR concentration can be expressed as a function of its particulate surface reactivity. Recent analysis of EPFR concentrations in Memphis, TN demonstrated that radical concentration in airborne PM ranges from 1.1e17–3.7e19 radicals/g (42). Using the NAAQS 24 h compliance concentration for PM2.5 of 35 μg/m3 and the EPFR concentration range of 1.1e17–3.7e19 radicals/g, the calculated EPFR/m3 concentration in Memphis would be 3.9e12–1.3e15 EPFR/m3. Studies presented here are exposing mice at a lower EPFR concentration, on the order of 1.8e13–5.3e13 EPFR/m3.
With respect to pulmonary deposition, our dosing rationale is supported by our calculations of the estimated cumulative pulmonary deposited doses, which we calculated using the formula: pulmonary deposition = deposition fraction × minute ventilation × aerosol concentration × exposure duration (PD = F × V × C × T). F is the deposition fraction (10%); V represents minute ventilation based on body weight; C equals the mass concentration (mg/m3); and T equals the exposure duration (minutes). We used a mouse minute volume of 24 mL/min (≈ 20 g mouse). Estimated pulmonary doses in EPFR -exposed mice were thus 0.26, 3.17, 0.77 and 9.85 µg, for the low and high exposure concentrations and at 4 h and 10 days, respectively. These estimated pulmonary doses were used as surrogates for internal doses. This allows for comparison with levels of ultrafine particles found in real-world human exposure scenarios, including concentrations greater than 0.1 µg/m3 measured in four Californian cities, and exposure concentrations of 10 µg/m3 used in asthmatic human studies (43, 44).
Inhalation Methods
C57BL/6 male mice were placed into an 18-L Plexiglas chamber containing 16 individual compartments, allowing simultaneous exposure of multiple animals to the same concentration of EPFRs, while also keeping the animals separate. Thus, there was no chance that the animals would cuddle or lick one another during exposures and all of the animals were exposed to the same concentration of EPFRs. Male mice were chosen for these experiments because previous studies showed that they were more susceptible than females to effects of ultrafine PM inhalation (45–47). EPFR PM aerosols were generated by a dry powder dispersion technique, using a Venturi disperser (37). Desired EPFR concentration was achieved by modifying the initial quantity of EPFR particles used and by adjusting air flows. No aerosol neutralizer was used (38, 39). Venturi air flow (17–35 L/min) enabled the particles to be aerosolized directly from the EPFR particle container into the top of the inhalation chamber. Additional dilution air (0–10 L/min) created an airstream mixture inside of the inhalation chamber, and thus contributed to particle distribution within the chamber. At the bottom of the chamber, the exhaust flow (2–4 L/min) consistently removed aerosolized particles and allowed for chamber air exchanges. To monitor for aerosol concentration stability, total mass concentration in the chamber was displayed in real time, every second of the 4-h exposure, via a TSI DustTrak II Model 8530 (Shoreview, MN). Off-line determination of total mass concentration was measured gravimetrically by sampling and collecting the aerosols on glass fiber filters (Millipore Sigma, Burlington, MA) that were weighed, before and after sampling, on a Sartorius MC5 microbalance (Sartorius, Goettingen, Germany). EPFR air samples were collected throughout the daily exposure using a Sensidyne Gilian BDX-II air sampling pump (Sensidyne, St-Petersburg, FL). Testing of the EPFR dynamic exposure system showed that the EPFR aerosol sampled close to the wall of the Plexiglas chamber was within 93% of the targeted mass concentration of 1.5 mg/m3 and the EPFR aerosol sampled near the center of the chamber was within 120% of this targeted mass concentration (Table 1). Thus, the distribution of particles within the exposure chamber was most likely uniform, ±20% of the targeted mass concentration. Because the inhalation chamber allowed for 16 mice to be exposed at one time, at each of 4-h or 10-day time points, 16 mice were exposed to filtered air, to 0.5 mg/m3 DCB230 or to 1.5 mg/m3 DCB230, for a total of 48 mice used for each of two time points. At the end of each exposure, DCB230-exposed animals were returned to their cages where they breathed HEPA-filtered, clean air (FA). When experiments were completed, mice were euthanized by pneumothorax under anesthesia and tissues and blood were collected. Eight mice from each group were used for pulmonary function measures, collection of bronchoalveolar lavage fluid (BALF), lung histopathology, and gene expression in the lung. The remaining eight mice in each group were used for evaluation of vessel reactivity, plasma biomarkers, and vascular gene expression.
Table 1.
Two distinct mass concentrations were obtained for the exposures to DCB230 particles
| DCB230, 4 h |
DCB230, 4 h |
DCB230, 3 days |
DCB230, 10 days |
DCB230, 10 days |
||||
|---|---|---|---|---|---|---|---|---|
| Measured Parameters | Filtered Air, 4 h |
0.5 mg/m3 | 1.5 mg/m3 | Filtered Air, 3 days |
1.5 mg/m3 | Filtered Air, 10 days |
0.5 mg/m3 | 1.5 mg/m3 |
| Mass concentration, mg/m3* | 0.45 | 1.34 | 1.79 ± 0.7 | 0.55 ± 0.1 | 1.71 ± 0.8 | |||
| Temperature, °C | 26.4 ± 0.6 | 25.9 ± 0.8 | 25.6 ± 1.0 | 24.9 ± 1.1 | 25.3 ± 1.0 | 25.4 ± 0.7 | 26.1 ± 0.7 | 26.2 ± 0.9 |
| Relative humidity, % | 15.6 ± 15.2 | 9.1 ± 11.6 | 7.5 ± 9.1 | 50.5 ± 14.0 | 27.8 ± 8.8 | 51.3 ± 9.1 | 25.8 ± 3.4 | 20.4 ± 3.9 |
Values are means ± SD. Data were recorded daily and represent exposure parameters inside the chambers. *Average mass concentration was determined gravimetrically. For the gravimetric analysis of mass concentration, only one measure was collected daily; hence, no standard deviation is indicated for the 4-h exposures.
Once data from the first experiment were integrated, a follow-up study was conducted to examine toxicological responses occurring between 1 and 10 days. As such, 16 mice were exposed to 1.5 mg/m3 DCB230 and 16 to filtered air for 4 h a day for 3 consecutive days. After the final exposure, mice were euthanized and blood and tissues were collected. Eight mice from each group were used for pulmonary function assessments and for collection of BALF. Eight of the animals were used for measures of vascular function (i.e., vessel reactivity) and eight were used for flow cytometry analysis of inflammatoric cell populations.
Plasma Endothelin-1
Enzyme-linked immunosorbent assays (ELISAs) were performed on plasma samples from DCB230-exposed and FA control mice. Plasma samples exhibiting hemolysis were excluded from analysis. Plasma endothelin-1 (ET-1) was determined with commercially available ELISA kits (Abcam, Cambridge, MA). Plasma levels were assessed on 100 µL of plasma, and all assays were carried out in accordance with manufacturer’s directions. The minimal detectable dose (MDD) for the ET-1 ELISA was 0.41 pg/mL with an intra-assay/interassay precision of 8.1% and 9.8% coefficient of variation (CV), respectively.
Plasma Nitric Oxide
Plasma from the 4-h and 10-day DCB230-exposed and air control animals was aliquoted, treated with a mix of potassium ferricyanide and N-ethylmaleimide in PBS+NP40 at a ratio of 1:6 (mix:plasma) and frozen at −80°C until analysis of nitric oxide (NO). NO levels were determined by measuring levels of its stable metabolite, nitrite, with an NO analyzer (Sievers, Boulder, CO). Plasma samples exhibiting hemolysis were excluded from analysis. The procedure was carried out according to the protocol provided by Sievers (GE Analytical Instruments, Maintenance and Operation Manual). Briefly, sodium iodide in acetic acid at 1% wt/vol was used as the reducing agent to generate a calibration curve. The calibration curve included seven concentrations ranging from 10 nM–10 µM. Plasma samples from controls and treated animals were analyzed in an alternating order; a control was injected with each group of samples to ensure accuracy, and the reaction vessel was cleaned and prepped when the solution turned yellow from saturation.
Bronchoalveolar Lavage Fluid Cytology
Bronchoalveolar lavage fluid (BALF) was harvested as described previously (48). Briefly, mice were euthanized with an intraperitoneal injection of Beuthanasia-D (Schering-Plough, NJ; 0.1 mL/10 g of body wt) before exsanguination via cardiac puncture. The diaphragm was cut and the thoracic cavity opened to expose the trachea, which was cannulated using a 19-gauge needle. Silk suture was used to hold the needle in place. The lungs were then lavaged twice with 0.5 mL phosphate-buffered saline passed through the cannula. Pooled BALF was placed on ice immediately. BALF was further processed and analyzed if more than 200 µL were recovered. Modified Wright’s-stained cytocentrifuge slide preparations were used and 300-cell differential counts were performed. The percentages of neutrophils, lymphocytes, and macrophages were determined, and binucleated macrophages per eight high-power fields were also recorded, all by a “blinded” veterinary anatomic pathologist. Note, however, that samples were excluded from analysis if the pathologist deemed the samples of insufficient cellularity for assessing differential cell counts or if the sample was contaminated with blood. Before lung fixation, a small piece of lung was cut and quick-frozen at −80°C for subsequent protein analysis; another small piece of lung was placed in RNALater for RNA extraction and gene expression analysis. The remaining lungs were pressure-fixed (25 cm) with buffered formalin (10%) administered by intratracheal instillation for histopathology assessment.
Lung Function-Tidal Volume
Whole body plethysmography was used to assess lung function. After the final day of exposure for each time point, mice were placed into individual chambers of the plethysmograph (Buxco) and their baseline tidal and minute volumes were measured, as previously described (45, 47). Responses were recorded for 5 min and were averaged for each mouse.
Flow Cytometry
For animals exposed to EPFRs for 3 days, both circulating and lung populations of lymphocytes, neutrophils, dendritic cells, and monocytes/macrophages were determined at the time of euthanasia. Flow cytometry analysis was used to detect and quantify each cell type. Whole blood was collected and mixed well, and 100 µL was added to Falcon tubes (Corning, Corning, NY) incubated with 2 µL of Fc block (BD Biosciences, San Jose, CA) for 5 min. The left lung of each animal was rinsed with 1× PBS/0.5% BSA and kept on ice until processing. The lungs were minced thoroughly with a sterile razor blade in a petri dish on ice and then placed in a collagenase (5 mg/mL)/DNase (2 mg/mL) solution (Roche Life Science) for digestion at 37°C for 1 h. Following incubation, an 18-gauge needle with syringe was used to further disrupt the tissue and the fluid was filtered through a 40-μm filter (BD Biosciences) into a 50-mL conical tube. Lung cell samples were centrifuged at 300 g for 5 min at 4°C, and the supernatant, discarded. Fc block (2 µL) was added to each cell pellet, vortexed, and incubated for 5 min before being transferred to Falcon tubes. All samples, both blood and lung cells, were treated identically for the rest of the staining procedure as follows: AKC lysing buffer (1 mL; Invitrogen, Carlsbad, CA) was added to each tube, and the tubes were vortexed and incubated at room temperature for 5 min to lyse any remaining erythrocytes. PBS (1×; 1 mL) was added to each of the tubes before centrifugation at 300 g for 5 min at 4°C. The supernatant was discarded and the remaining pellets were washed with cold PBS. Anti-mouse MHCII -APC, Ly-6G-FITC, CD11c-PE, and CD11b-PerCP-Cy5.5 were added to the tubes at concentrations supplied by the manufacturer (BD Biosciences), vortexed and incubated for 30 min at room temperature in the dark. Tubes were then centrifuged, the supernatant was decanted, and the cell pellets were washed with PBS before a final centrifugation. The remaining pellet was resuspended in PBS and stored at 4°C until FACS analysis. Finally, flow cytometry was conducted by a blinded technician. Cell populations were determined as follows: lymphocytes: MHCII+ low forward scatter cells comprised B cells and MHCII negative low forward scatter cells comprised the T-cell population based on MHCII versus forward scatter plot; neutrophils: Ly6G labeled cells based on Ly6G versus forward scatter; dendritic cells: coexpressed population of MHCII and CD11c on dual parameter plot; and monocytes/macrophages: CD11b staining of intermediate forward scatter population in the lung based on CD11b versus forward scatter plot. However, in peripheral blood monocytes were MHCII+ intermediate forward scatter cells based on MHCII versus forward scatter due to lack of CD11b staining in peripheral blood. The percent of positive staining cells above the negative control was determined for each individual mouse and the mean values for each group calculated.
Vessel Reactivity
After completion of the 3-day exposures, mice were euthanized and their aortas were carefully harvested and placed in oxygenated, cold Krebs-Henseleit solution, pH 7.4, to preserve physiological responses. The vessels were then cleaned and analyzed in a blinded manner. First, aortas were gently trimmed of connective tissue under a microscope and were cut into rings for analysis of endothelial function via wire myography (DMT, Ann Arbor, MI). The myograph chamber was filled with warm Krebs–Henseleit solution, oxygenated, and kept at 37°C for the duration of the experiment. Under a microscope, vessels were carefully attached by thin, titanium wires to the jaws of the myograph. Once in place and secured, a tension of 9.8 mN was set for each vessel and the vessel was allowed to equilibrate. The physiological salt solution was replaced every 15 min to ensure that the vessels remained viable. After the vessels had equilibrated, phenylephrine (PE) was applied at a concentration that was previously determined via creating a phenylephrine curve to constrict the vessels to 80% of their maximum contraction. Note that for quality control purposes, vessels were deemed viable and acceptable for analysis if they exhibited an ability to contract in the presence of phenylephrine. The response to PE was allowed to plateau; then, to measure endothelium-dependent vasodilation, increasing concentrations of acetylcholine (ACh) were added to the chamber as the decreasing tension was recorded. Vessels were washed thoroughly afterward, allowed to equilibrate back to 9.8 mN, and again were treated with PE. After the tension had plateaued, increasing doses of sodium nitroprusside (SNP) were added to ensure that all of the vessels fully relaxed, thus testing whether any lack of relaxation after ACh treatment was endothelium-dependent and not caused by a direct effect on vascular smooth muscle.
Gene Expression Analysis
In a blinded manner, aortas and lungs stored frozen in RNA-later were processed using the Quick-RNA MicroPrep Kit (Qiagen, Hilden, Germany) and according to the manufacturer’s instructions. RNA was validated for quantity and purity using a NanoDrop (manufacturer) spectrophotometer and only samples with an A260/A280 greater than 1.85 were used to prepare cDNA for RT-PCR analysis. mRNAs analyzed included those associated with oxidative stress/antioxidant response element signaling [glutathione peroxidase-2 (Gpx2), heme oxygenase-1 (Hmox1), NAD(P)H quinone dehydrogenase 1 (Nqo1), glutamate-cysteine ligase (Gclc), nitric oxide synthase-2 (Nos2), AhR signaling (aryl hydrocarbon receptor repressor (Ahrr), cytochrome P450 1B1 (Cyp1b1)], and endothelial function (endothelin-1 (Edn1), endocan or endothelial cell specific molecule-1 (Esm1), intracellular adhesion molecule-1 (Icam)). See Supplemental Table S1 (All Supplemental material available at https://doi.org/10.6084/m9.figshare.14755377.v3) for a list of primers used for studies. Gene expression was calculated using the ΔΔCt method. ΔCt data were calculated using an average of reference housekeeping genes, and the fold change was calculated using the 2−ΔΔCt method.
RT2 Profiler PCR Array
In the aortas and lungs of the 3-day-exposure mice, the expression of 84 mouse endothelial cell biology genes (Cat. No. PAMM-015Z) was analyzed using a RT2 PCR array (Qiagen) per the manufacturer’s instructions. After DNAse treatment, total RNA (0.5 μg) was reverse-transcribed using the RT2 First Strand Kit (Qiagen 330404), and cDNA was diluted with RNase-free water to a volume of 111 μL. Converted cDNA sample (100 μL) was mixed with RT2 SYBR Green qPCR Master mix (Qiagen 330523). Equal aliquots of 25 μL were added onto the corresponding wells of the PCR Array plate which contained the pre-dispensed, gene-specific primer sets, and PCR was performed as per the cycling conditions recommended for Applied Biosystems model 7300 real-time cyclers. Gene expression and fold change was calculated using the ΔΔCt method. ΔCt data were calculated using an average of reference housekeeping genes, and the fold change was calculated using the 2−ΔΔCt method, with the web-based PCR Array data analysis software.
Western Blot Analysis
Lung tissues collected from mice exposed to filtered air, 0.5 or 1.5 mg/m3 DB230 for 10 days were snap frozen and stored at −80°C until Western blot analysis could be performed. Tissues were homogenized in RIPA lysis buffer, total protein was determined and 15 µg of protein from each sample were analyzed using polyacrylamide gel electrophoresis on 10% Invitrogen gels. Electrophoresis was run at 120 V for 1.5 h before transferring the proteins to PVDF membranes at 80 V for 2 h. Blots were blocked with 5% nonfat milk in 0.1% TBST for 1 h before overnight incubation at 4°C with annexin V antibody (1:500) diluted in 5% nonfat milk in 0.1% TBST. Blots were washed three times in 0.1% TBST for 15 min each time before the addition of secondary anti-rabbit antibody (1:4,000) diluted in 5% nonfat milk in 0.1% TBST and incubation for 1 h at room temperature. Membranes were again washed as before and ECL substrate was added to image the blots on a ChemiDoc. β-Actin was used to ensure that equal amounts of protein were loaded in each well and Bio-Rad Image Lab software was used to compute the ratio of annexin to β-actin in each lane.
Immunohistochemistry
Lung tissues from mice exposed to filtered air, 0.5 and 1.5 mg/m3 DCB230 (n = 8/group) were paraffin embedded and sectioned before staining. Sections were immunostained for the endothelial protein factor VIII in the Louisiana Animal Disease Diagnostic Laboratory. A control tissue (lung from an untreated mouse) was also stained using the same protocol. Briefly, 4-μm sections of fixed tissues were mounted on positively charged Superfrost Plus slides (VWR, Radnor PA) and subjected to IHC using the automated BOND-MAX and the Polymer Refine Detection kit (Leica Biosystems, Buffalo Grove, IL). Following automated deparaffinization, heat-induced epitope retrieval (HIER) was performed using a ready-to-use citrate-based buffer (pH 6.0; Leica Biosystems) at 100°C for 20 min before incubation with anti-Factor VIII (rabbit polyclonal, diluted 1:2,000; A0082; Dako; Carpinteria, CA). Sections were incubated with the primary antibody for 30 min at room temperature, followed by a polymer-labeled goat anti-rabbit IgG coupled with horseradish peroxidase (Leica Biosystems) for 8 min at room temperature. 3,3′-diaminobenzidine tetrahydrochloride (DAB) was used as the chromogen (10 min), and counterstaining was performed with hematoxylin. Slides were mounted with a permanent mounting medium (Micromount, Leica Biosystems). Positive staining was determined using the analyze function in ImageJ software (https://imagej.nih.gov/nih-image/), and lung staining of tissues from each group was assessed as a percent of the control.
Statistics
One-way ANOVA was used to determine significance between individual data points for the 4-h and 10-day studies. For the 3-day study, a Student’s t test was used to determine significance between the DCB230 and FA mice. Finally, for the semiquantitative assessments of lung histology, the nonparametric Kruskill–Wallis one-way ANOVA by ranks was used to determine differences between treatment groups. Data in most figures are expressed as means ± SE and in all cases, P < 0.05 was accepted as significant. All statistical analyses with the exception of RT2 Profiler data were conducted using GraphPad Prism (La Jolla, CA).
RESULTS
Characterization of DCB230
DCB230 particles were visualized via TEM for size determination. Individual particles measured ∼0.02 µm, but the particles formed aggregates that measured from 0.1 µm to 0.2 µm (Fig. 1A). The particles generated a broad singlet with some splitting and an average EPR g value of 2.00558 (Fig. 1B). The average radical content of the particles used for exposures was determined to be 3.5e16 spins/g.
Figure 1.
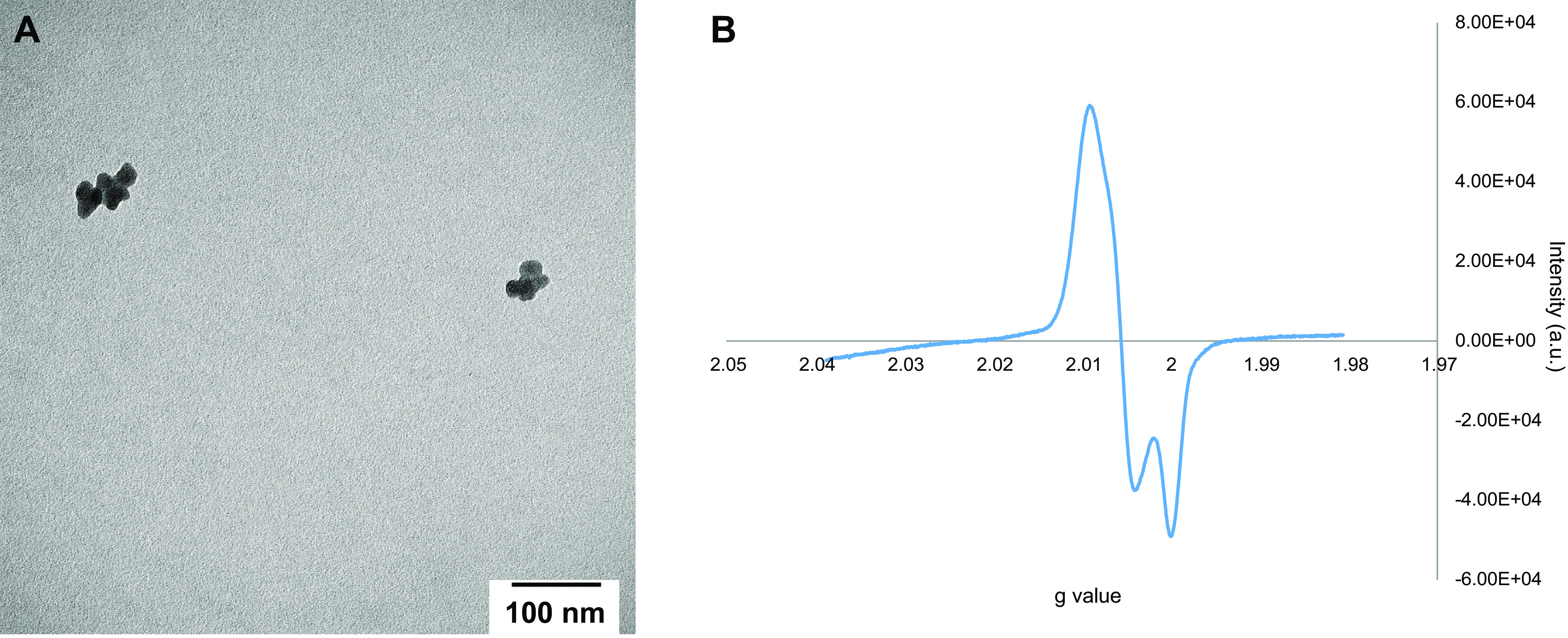
Transmission electron microscopy (TEM) of aerosolized DCB230 particles. A: DCB230 particles were aerosolized and collected on precoated carbon Formvar copper grids for 5 min and characterized for shape, agglomeration, and structure using TEM. Individual particles were rounded and below the 20 nm range, classifying them as ultrafine particulate matter, but formed agglomerates which were around 100–200 nm, and still within the ultrafine range. B: representative EPR spectrum measuring g value, which on average, was 2.00558.
4-H and 10-Day Exposures
Following the 4-h and 10-day exposures to DCB230 at either 0.5 mg/m3 or 1.5 mg/m3, plasma collected from the DCB230 and air control mice was analyzed for ET-1 and NO concentrations. In both the 4-h and 10-day animals, mice exposed to DCB230 at 1.5 mg/m3 had significantly elevated ET-1 (Fig. 2A) and significantly reduced NO compared to their air control counterparts (Fig. 2B). Together, these data indicated the possibility of vascular dysfunction and vascular injury in response to EPFR-containing particulate matter. However, mice exposed to DCB230 at the lower dose for either 4 h or 10 days showed no significant alterations in plasma ET-1 or NO levels.
Figure 2.

DCB230 exposure for only 4 h significantly altered plasma levels of endothelial biomarkers. After one 4-h exposure to DCB230 at 1.5 mg/m3, plasma endothelin-1 (ET-1) (A) was significantly increased and nitric oxide (NO) (B) was significantly reduced in the DCB230-exposed animals compared to air controls. There also was a significant increase in ET-1 (A) and decrease in NO (B) in plasma of mice exposed to 1.5 mg/m3 DCB230 for 4 h a day for 10 consecutive days. Note that ET-1 was measured using ELISA and NO was determined via an NO analyzer (n = 7–15/group, *P ≤ 0.05).
Tidal volume was significantly reduced only in mice that had been exposed to 1.5 mg/m3 of DCB230 for 10-days (Fig. 3, A and B). In contrast, the BALF of mice exposed to 1.5 mg/m3 of DCB230 in the 4-h and 10-day groups showed no alterations in inflammatory cell counts (%neutrophils, lymphocytes or macrophages; Supplemental Table S2) or binucleated macrophages, although the latter were slightly elevated in the 10-day treatment group (Supplemental Fig. S3). Again, mice exposed to 0.5 mg/m3 did not display any significant differences in tidal volume or binucleated macrophages. In addition, histological sections were analyzed blindly by a board-certified veterinary anatomic pathologist revealed no significant impact of DCB230 at either dose on lung structure (Supplemental Fig. S4, A–C) nor levels of alveolar inflammatory cells (Supplemental Fig. S4D).
Figure 3.

Tidal volume was significantly reduced and binucleated macrophages trended higher after 10 days exposure to DCB230. Tidal volume was determined via whole body plethysmography. No change was noted in tidal volume at the 4-h time point (A), but a significant decrease was observed for the mice exposed for 10 days to 1.5 mg/m3 DCB230 (B). Data represent n = 8/group. *P ≤ 0.05. C: BALF was collected from mice from each of the three groups and microscopically assessed for the presence of binucleated macrophages. Data shown are representative stained images of BALF macrophages from each treatment group. Although there was an increase in binucleated macrophages after exposure to both DCB230 concentrations, the levels did not reach significance. No increase was noted in any of the mice exposed for 4 h (n = 7 or 8/group, P ≤ 0.05).
Gene expression changes in AhR-related genes, antioxidant response signaling pathways and endothelial genes were analyzed in the lungs of mice exposed to DCB230, at both low and high concentrations, compared with FA controls (n = 8/group) for the 4-h and-10 day time points (Fig. 4, A and B). At the 4-h time point, the aryl hydrocarbon related genes AhRr (low: −1.5, high: −1.6) and Cyp1b1 (low: −1.3) were downregulated compared to filtered-air controls for both concentrations of DCB230. Nqo1 was downregulated by a fold change of −1.4 at the low DCB230 concentration; Hmox1 (low: 1.3, high:1.3) and Gpx2 (high: 1.4) were upregulated, and Nos2 was downregulated (low: −1.6, high: −1.3) at both DCB230 concentrations. Esm1, a gene associated with endothelial function, was downregulated at both concentrations of DCB230 by −2.4 and −1.8-fold, respectively. For the 10-day exposure group, antioxidant response genes Hmox1 and Gclc were both down regulated by a fold change of 1.2 at the low concentration of DCB230. The AhR associated gene Cyp1b1 (1.3) was up-regulated at the higher concentration of DCB230, and endothelial genes, End1 (1.3), Esm (1.5) and Icam (1.2), were all upregulated at the 1.5 mg/m3 concentration of DCB230. A summary of this data is shown as a heat map in Fig. 4. This heat map for the 4-h and 10-day time points clearly shows that while there were small, yet statistically significant changes in lung gene expression early on, the majority of these changes resolved by 10 days, and only endothelial- and AhR related-changes persisted.
Figure 4.

Heat map representing lung gene expression data from 4 h and 10 day studies. Heat maps were generated to show significant changes in lung gene expression. Genes were grouped according to function and color coded according to the degree in which they were altered. Shades of red and green indicate increases and decreases in the gene versus control mice, respectively, and the brighter the color, the greater the change. Black represents no significant change in gene expression and the scale on the right denotes the range of values for each color (n = 8/group, P ≤ 0.05).
3-Day Exposure
Once the results from the 4-h and 10-day studies were evaluated, an intermediate time point between 4 h and 10 days was selected (i.e., 3-days) and only the high dose (1.5 mg/m3) of DCB230 was used. The same measurements were completed for these animals and again, ET-1 was significantly increased (7.3 pg/mL vs. 9.5 pg/mL) whereas NO was reduced (142 nM vs. 126 nM) in DCB230-exposed mice, indicating vascular dysfunction (Fig. 5, A and B).
Figure 5.

Biomarkers of endothelial function after 3 days exposure to DCB230. Mice were exposed to DCB230 (1.5 mg/m3) or filtered air for 4 h daily for 3 consecutive days before euthanasia and blood collection. Plasma ET-1 levels were measured via ELISA and plasma NO concentrations were determined using an NO analyzer. Similar to the other time points, mice exposed to DCB230 demonstrated a significant increase in ET-1 (A), whereas NO was significantly reduced (B), suggesting endothelial dysfunction after only 3 days of exposure (n = 7 or 8/group, *P ≤ 0.05).
In addition to measuring the plasma biomarkers ET-1 and NO, we also determined the functional responses of the endothelium to EPFRs in eight mice. Aortas isolated from mice exposed for 3 days were evaluated for their ability to relax after vasoconstriction with phenylephrine (PE). The aortas from mice exposed to DCB230 exhibited diminished acetylcholine-stimulated vasodilation compared to mice exposed to filtered air (Fig. 6A). No difference between groups was observed when relaxation was induced using the NO donor sodium nitroprusside, indicating that the diminished vasodilation is endothelium-dependent (Fig. 6B). In addition, there was no difference in PE-induced vasoconstriction between the FA and EPFR groups (Fig. 6B). These functional measures confirmed the ET-1 and NO data, indicating that endothelial dysfunction is an early target of EPFR exposure.
Figure 6.

Endothelium-dependent vasorelaxation was reduced after DCB230 exposure for 3 days. After exposure of mice for 3 days to DCB230 compared with filtered air, aortas were extracted and analyzed using wire myography for their ability to vasorelax, a functional measure of endothelial health. A: after precontraction with phenylephrine, aortas from mice exposed to DCB230 exhibited significantly reduced relaxation in the presence of increasing concentrations of acetylcholine. B: maximal relaxation, calculated by estimation of the curve using GraphPad Prism, was also reduced (n = 8/group, *P < 0.05). C: pilot studies demonstrated no impact of DCB230 exposure on phenylephrine- (PE-) induced relaxation. D: sodium nitroprusside (SNP) induced near complete relaxation of vessels of mice exposed to either DCB230 or filtered air, suggesting no impact of EPFR inhalation on vascular smooth muscle.
With respect to lungs, a mild but significant reduction in lung tidal volumes was detected (Fig. 7A). However, no significant alterations in BALF cytology or bi-nucleated macrophages were measured (Supplemental Figs. S5 and S6). Flow cytometry revealed that inhalation of DCB230 only modestly altered inflammatory cell populations in the lung and blood (Fig. 7B). Specifically, between air control mice and mice exposed to DCB230, there was only a slight but significant increase in lung dendritic cells and in peripheral blood lymphocytes in the DCB230 group (dendritic cells: 0.47 versus 1.41% and lymphocytes: 67.3 versus 76.3%, respectively. Thus, although lung function was reduced, DCB230 exposure had no marked impact on pulmonary or systemic inflammation.
Figure 7.

Inhalation of EPFRs for 3 days reduced lung tidal volume but had only a modest impact on lung function. Mice were exposed to either filtered air or 1.5 mg/m3 DCB230 for 4 h/day for 3 days. Tidal volumes were measured using whole body plethysmography (A) and inflammatory cell populations in the lungs and peripheral blood were determined using flow cytometry (B). *Significance compared with filtered air (lungs: n = 7 or 8/group; peripheral blood: n = 4–8/group).
Gene expression in the aortas and lungs of mice exposed for 3 days also was measured via qRT-PCR (Fig. 7, n = 5–8/group). We observed a significant and robust decrease in the antioxidant response genes Nqo1 (−8.6) and Gpx2 (−8.0) in the aortas of the 3-day, DCB230-exposed mice compared to FA control mice, while in lungs from mice exposed to DCB230, we noted an increase in Hmox (2.8) expression. In addition to finding differences in antioxidant response genes, the endothelial marker Esm1 was expressed at a significantly lower level of –1.5 in the lungs of DCB230 mice compared to FA controls and Edn1 was also down regulated by –1.4. These data are represented as a heat map in Fig. 8A and show that after the 3-day exposures there were robust changes in expression of antioxidant response genes in the aorta, and in Hmox1, Esm1, and Edn1 in the lung.
Figure 8.

Heat maps representing lung and aorta gene expression data from the 3-day study. Heat maps were generated for the 3-day gene expression data for both the lung and aorta tissues. A: as with the 4-h and 10 day heat maps, the genes were grouped according to function and shaded to reflect change. Red and green indicate increases and decreases in fold change, respectively, and the brighter the color, the greater the change. Black represents no significant change in gene expression and the scale on the right of the heat maps describes the range of values for each color (n = 5–8/group, P ≤ 0.05). B: RT2 Profiler PCR Array was also used to determine the expression of 84 endothelial cell biology genes in the aortas and lungs of the 3 day mice and a heat map for the analysis was generated for any significant fold changes (aortas: n = 4/group; lungs: n = 8/group, P ≤ 0.05).
To further elucidate the pathways mediating the cardiovascular damage resulting from DCB230 exposure, an RT2 Profiler Array for endothelial cell biology-related genes was also performed on the lungs and aortas from mice exposed to DCB230 or FA for 3 days (Fig. 8B). In the lung, only four genes were significantly altered. Two immune-related genes Cxcl1 (3.3) and Cxcl2 (1.6) were significantly increased and two vascular response genes Agtr1a (−1.5), and Edn (−1.7) were decreased versus FA control mice. In the aorta, however, 12 genes were altered after a 3-day exposure to DCB230. The majority of alterations were noted in genes related to coagulation and platelet activation. F2rl1 (−5.5), Fn1 (−2.0), Pf4 (−2.4), Procr (−1.6), Tfpi (−1.7), and Thbd (−1.6) were all significantly decreased in the aorta of mice exposed to DCB230 versus FA controls. Immune and cell adhesion genes were also significantly decreased in the aortas of the DCB230 mice, where decreased fold changes were identified in IL7 (−2.4), Pdgfra (−1.7), and Icam (−1.9). In the aortas, a 2.3-fold increase in expression was noted for the angiogenesis gene Vegfa, and an increased fold change of 2.0 was identified for Agtr1a in the aortas. Anxa5, a gene associated with apoptosis, was also increased 1.6-fold in mice exposed to DCB230 versus FA controls. Finally, Ingenuity Pathway Analysis (IPA) revealed that in aortic tissues, networks of genes dysregulated by 3 days of exposures to EPFRs were associated with blood pressure disorder, artery occlusion, and atherosclerosis lesioning (Supplemental Fig. S7). Also, dysregulated genes were related to function, including vascuologenesis, vascular morphology, and blood cell adhesion.
Expression of Lung Proteins Detected by Western Blot and Immunohistochemistry
Our findings for elevated vascular Anxa5 mRNA levels were particularly intriguing. Given a lack of remaining vascular tissue for confirming these findings, we used both lung homogenates and lung tissue cross sections from the mice exposed to EPFRs for 10 days to conduct both Western blot and immunohistochemical analyses, as lungs contain a wealth of vascular tissue. As shown in Fig. 9A, annexin V protein levels were elevated in the lungs by ∼50% after DCB230 exposure. With respect to FVIII staining, both arterial and capillary endothelial cells stained strongly for FVIII, and visual inspection of the tissues revealed that the endothelial monolayer remained intact (Fig. 9B). Magnification (×100) of these sections highlighting staining in the alveoli likewise revealed no significant impact on the capillary endothelial cells (Supplemental Fig. S8). Furthermore, its expression was unchanged, as image analysis revealed no change in color density (Fig. 9B).
Figure 9.
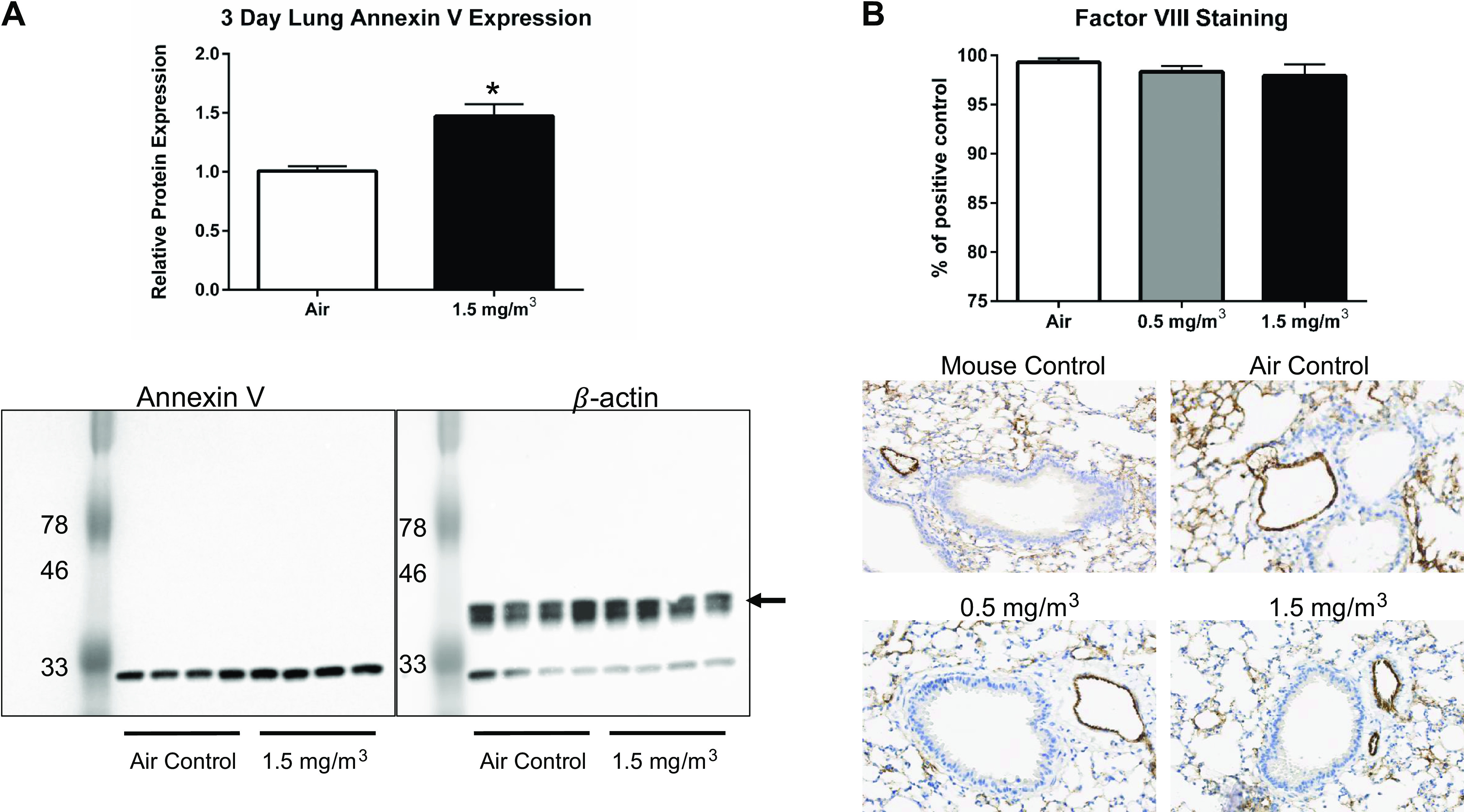
DCB230 exposure increased apoptosis marker Annexin V in the lung, but did not disrupt the integrity of the vascular endothelium. A: Annexin V protein levels were measured in lung tissue homogenates by Western blot analysis. The blot was probed first for Annexin V protein (left) and then for β-actin (right) as a housekeeping protein. Note the lower band in the right panel represents residual Annexin V staining. *P < 0.05. B: the endothelium of lung cross sections was visualized by immunohistochemical staining for factor VIII protein. Representative sections are shown for control mouse lung compared with the lungs of mice exposed to filtered air, 0.5 mg/m3 or 1.5 mg/m3 DCB230. Staining in images were quantified using Image J software and the data are expressed as a % of the control (normal mouse lung).
DISCUSSION
The health impacts of particulate matter (PM) have been well established over the last two decades, with cardiopulmonary diseases a primary concern (5, 49, 50). Studies have repeatedly demonstrated a correlation between PM concentration and the severity of cardiopulmonary events (49, 51, 52). Using laboratory-generated PM that mimic PM collected in urban areas near thermal remediation sites like waste incinerators, our studies aimed to establish the impact that EPFR-containing PM have on cardiovascular and pulmonary health. Exposure to 1.5 mg/m3 DCB230 for only 4 h or for 10 days significantly increased ET-1 and reduced NO, which is suggestive of vascular endothelial dysfunction (Fig. 2). Ideally, ET-1 and NO levels in the circulation are balanced to preserve vascular homeostasis and maintain vasorelaxation (53). Reductions in vascular function lead to an inability of the vessel to fully relax, ultimately increasing blood pressure and exacerbating the progression of cardiovascular disease (54). After 10 days of EPFR exposure, lung tidal volumes were reduced (Fig. 3). Moreover, lung inflammation, assessed as changes in inflammatory cell populations in BALF, was unremarkable through 10 days of treatment with either dose (Supplemental Fig. S2). Overall, these data suggest that mild alterations in pulmonary vessel capacities may precede pulmonary inflammatory responses.
Although the main objective of these studies was to test our hypothesis that lung inflammation and pulmonary function were drivers for EPFR-induced cardiovascular dysfunction, we used measures of gene expression across several pathways to begin to ascribe a molecular mechanism for the observed cardiovascular dysfunction. We began by measuring the expression of genes related to 1) oxidative stress (i.e., the antioxidant response element), 2) AhR activation and 3) endothelial function. The data showed slight, yet significant reductions in AhRR at the 4-h time point (Fig. 4). As AhRR regulates AhR signaling by interfering with the formation of the AhR/ARNT dimer complex necessary for AhR activation (55–57), it is possible that AhR was not normally regulated early in the exposure time course, which could lead to increased AhR activation and an exacerbated oxidative stress in the lungs as the exposures continued. Other genes associated with AhR signaling such as Cyp1b1 were significantly reduced in the lung tissue of mice after the 4-h exposure (Fig. 4). Two antioxidant-related genes: Hmox1 and Gpx2 (58, 59, 60, 61), which are involved in detoxification of heme oxygenase and scavenging hydroperoxides, respectively, were both upregulated at the 4-h time point. Increased expression of Hmox1 and Gpx2 was also reported in lung tissues of animals exposed to cigarette smoke, likely serving to protect the lung against inflammation and oxidative stress (61). However, Nqo1, which is responsible for the detoxification of quinones, was significantly reduced in the lungs at 4 h. Although Nqo1 is a component of the typical Nrf-2 signaling pathway and is known to be regulated through the antioxidant response element (ARE), AhR activation is also known to regulate the transcription of Nqo1 (62). We did not identify an increase in AhR expression, but the data suggest that AhR regulation was altered. Further investigation into canonical versus noncannonical AhR signaling using in vitro studies may be necessary to fully identify the mechanisms driving gene expression changes. With the longer 10-day exposures to EPFRs, antioxidant genes were no longer altered, the AhR-related gene Cyp1b1 was increased and endothelial biomarkers Edn1 and Esm1 were upregulated (Fig. 4). Increases in endocan suggest endothelial activation may also be a factor (63, 64).
We then chose to examine an intermediate time point (i.e., 3 days) to further clarify the cardiopulmonary gene expression changes we identified in our 4-h and 10-day studies and test whether the observed changes in gene expression and endothelial biomarkers were correlated with a reduction in vascular function assessed physiologically. Again, ET-1 was increased and NO was reduced in the circulation of DCB230-exposed mice versus FA controls. However, we also found reductions in endothelium-dependent vascular relaxation demonstrating that after only 3 days of exposure, the endothelium is sufficiently impacted such that vascular physiology is altered. Our findings for EPFR exposure support earlier reports of PM-induced endothelial dysfunction assessed in humans as a decrease in flow-mediated dilation in the brachial artery (14). Interestingly, at the 3-day time point, we also began to detect decreases in lung function, evidenced by decreases in lung tidal volumes (Fig. 7A). Given our findings of concurrent changes in lung and vascular function at this early time point, it begs the question of whether these are tightly associated, interdependent phenomena.
Peripheral blood and lung tissue collected from the 3-day exposed mice were also used to determine immune cell populations in DCB230-exposed mice versus the air controls. Differences in immune cells between DCB230 and FA control mice at this time point were modest. There was a slight, yet significant increase in dendritic cells in the lung tissue and an increased number of lymphocytes in the circulation of animals exposed to DCB230. Dendritic cells are critical in barrier tissues such as the lung and they induce the adaptive immune response when tissue damage and inflammation are present (65). Potentially, at a later time point, once the dendritic cells have had sufficient time to fully initiate the adaptive immune response, other lung cell populations such as lymphocytes would also increase in an attempt to manage inflammation and lung injury resulting from the PM inhalation.
To summarize the physiological findings, despite hypotheses that cardiovascular dysfunction occurring after PM exposures is secondary to lung or systemic inflammation, biomarkers for vascular dysfunction preceded pulmonary and systemic inflammation, but physiological reductions in lung and vascular function occurred within the same window of time during the exposure time-course. To further delineate a mechanism for these physiological findings, we conducted a more thorough analysis of gene expression changes in lung compared to vasculature at 3 days, a key time point where reductions in vasorelaxation begin to develop.
Beginning with our gene expression analyses directed toward AhR and oxidative stress signaling, gene expression changes observed at the 4-h time point were mostly preserved. The addition of RT2 Profiler data was used as an untargeted approach for identifying pathways beyond those we hypothesized as integral to EPFR-mediated injury. In these studies, we noted significant increases in both Cxcl1 and Cxcl2 in the lungs of mice exposed for 3 days, while the vascular genes Agtr1a and Edn1 were both reduced. Although we did not detect marked cellular inflammation in the lungs, we contemplated whether an injury at the air-blood interface would lead to these observations of increased chemokines and decreased endothelial biomarkers, especially since the apoptosis-related gene Anxa5 was significantly increased in the vasculature (Fig. 8B). Furthermore, in addition to being chemoattractants, Cxcl1 and Cxcl2 are known to promote angiogenesis and remodeling of vascular smooth muscle in the lung. Thus, increased expression of Cxcl1 and 2 potentially resulting from an underlying subclinical injury in the lung or pulmonary vascular tissue may signal vascular remodeling. In addition, expression of Pf4 and Fn1, which play a role in inhibiting angiogenesis, were reduced in the aorta. While in our prior DCB230 studies, an increase in pulmonary arterial resistance and pressure was identified after exposure to DCB230 (21), lung angiogenesis generally follows an acceleration in pulmonary arterial pressure as a way to compensate for reduced blood flow (66).
Despite that our lung histology through 10 days failed to reveal overt lung injury (Supplemental Fig. S4), it remained possible that a cell type within the lung exhibited a subclinical injury and interacted with endothelial cells, perhaps at the air-blood interface, to produce vascular dysfunction even downstream of the lungs. In support of this assertion, tidal volumes at 3 days were significantly reduced after EPFR exposures and gene expression changes in lung tissues were also detected. In addition, injured alveolar epithelial cells have been shown to be a source for Cxcl1 and 2 expression and release (67). Although an intriguing finding was an elevation in Anxa5 mRNA levels in vascular tissues, using lung homogenates remaining from our 10-day exposure study, we assessed annexin V protein levels by Western blot analysis. We rationalized that since the lung is a rich source of vascular tissue, our findings of increased Anxa5 in aorta may be mirrored in our lung tissue samples. Moreover, other as yet unidentified cell types in the lungs may likewise undergo apoptosis. As is shown in Fig. 9A, annexin V protein was elevated by 50%. Because identifying the cell type injured early after EPFR exposure could inform future mechanistic studies, using lung tissue sections from the same study, we attempted to measure annexin V protein levels using immunohistochemistry. Unfortunately, we were unable to conclusively identify the cellular source for elevated annexin V, mainly because alveolar epithelial cells demonstrated a strong background staining. Thus, while the elevations in annexin V may reveal an important mechanistic detail about EPFR induced pathology, its cellular origin remains unclear.
As an alternative approach for identifying the injured cell type, we used immunohistochemistry for an endothelial biomarker to visually inspect whether the endothelial layer remained intact after EPFR inhalation. As an elevation in annexin V suggested apoptosis, endothelial apoptosis should result in a disrupted endothelial monolayer. We selected Factor VIII as our endothelial biomarker, because 1) FVIII is found tightly associated with von Willebrand Factor within Weibel–Palade bodies in endothelial cells (68) and many studies suggest that it is synthesized there (69); 2) it is released together with vWF upon endothelial activation (69); and 3) an immunohistochemistry (IHC) assay for its detection was readily available in our diagnostic laboratory. Remaining lung tissue sections from mice subjected to EPFR inhalation for 10-day were selected for examination, since no lung sections remained from our 3-day exposure study. Results were that both arterial and capillary endothelial cells stained strongly for FVIII, and its expression was unchanged (Fig. 9B). In addition, capillary endothelial cell staining within alveoli also showed similar staining patterns with and without EPFR exposure (Supplemental Fig. S8). Thus, flow cytometry approaches for sorting lung cells and probing for increases in apoptotic markers may be required for finally elucidating the locus of injury for EPFR exposures in the lung.
Although our current studies used aorta as a source for vascular tissue, it is likely that these changes are even more profound in vessels within the lung. Furthermore, Agtr1a and Vegfa expression were both increased in the aorta, suggesting that if there was an increase in vascular pressure after EPFR exposure, compensatory mechanisms may be stimulated to rectify this increased pressure through angio- or vasculogenesis.
Another interesting finding was that vascular genes known to inhibit coagulation were also reduced, suggesting a unregulated prothrombiotic environment. mRNA levels for thrombomodulin (Thbd), tissue factor pathway inhibitor (Tfpi) and anti-thrombotic protein C receptor (Procr) were all down regulated in the aorta of mice exposed to DCB230 versus FA. Thbd on the surface of endothelial cells binds thrombin to prevent excess free thrombin, and this complex in turn activates protein C (70). Under normal circumstances, activated protein C is bound by its receptor Procr to decrease clotting factors and thrombosis (70). In addition, Tfpi, which was also downregulated in the DCB230-exposed mice, normally functions to inhibit factor X and promote antithrombiotic events (70). Taken together, the reduced expression of these coagulation-regulating genes in the endothelium may suggest that endothelial dysfunction induced by EPFR exposure may culminate in a pro-thrombotic state.
To summarize our key findings, EPFR inhalation has an early impact on vascular function that does not seem to require pulmonary or systemic inflammation but may be tightly associated with lung injury and changes in gene expression. Other rodent studies also have identified the vascular endothelium as an early or even direct target for PM exposures, particularly those arising from diesel exhaust (71) and urban particulate matter (72). Our findings of cardiovascular dysfunction preceding pulmonary inflammation also are substantiated by literature reports of inhalation studies performed using similar doses and lengths of exposure (72, 73). In those studies, vascular effects occurred following exposures to inhaled ultrafine particles (<100 nm) without producing significant alterations in BALF cytology (72, 73). BALF inflammation is initiated by alveolar macrophage activation through phagocytosis (74), an optimal clearance mechanism for particles in a size range of 500 to 3,000 nm (75, 76). Below 300–260 nm, particles are not efficiently detected by alveolar macrophages and thus, can interact with epithelial cells (75, 76). The very small primary particle size (20–40 nm) of the EPFR particles used in our study (Fig. 1A) predicts a moderate-to-low alveolar macrophage uptake of these particles, and consequently, an unremarkable inflammatory reaction. Thus, the small size of the particles used in these studies may have been a contributing factor to their uptake, potentially due to their ability to cross the air-blood interface in the lungs and enter the circulation. Nanoparticles, classified as particles smaller than 100 nm, have been shown to transit into the circulation from the lungs (77, 78). Ultrafine particles, including titanium dioxide nanoparticles translocate from the lungs to extrapulmonary organs (79). The percentage of inhaled nanoparticles able to accomplish this transit however, remains heavily debated (80, 81). Thus, we are considering alternate mechanisms potentially involving interactions between cell types at the air-blood interface, i.e., within alveoli, where the ultrafine particles are likely deposited.
A limitation of our studies is that we did not compare the effects of EPFRs to those from other non-EPFR containing particle systems. Testing whether the direct impacts of EPFRs on vascular function are dependent upon their free radical content or are simply a response to the physical presence of particulate matter would be necessary for fully understanding the mechanism of action of EPFRs. However, in the experiments that led up to the current study, our colleagues showed that EPFRs but not amorphous silica of a similar size, introduced to the mice via nose-only inhalation increased pulmonary arterial pressures (21). We attempted to use amorphous silica as our negative control for these studies but were unable to do so, because the particles were not sufficiently dense for obtaining a stable dry particle aerosol with our whole body inhalation system. Nevertheless, our prior studies support that the EPFRs and not their non-EFPR controls exhibit vascular effects. Moreover, the in vivo studies presented here clearly demonstrate that antioxidant response element (ARE) activation occurred early after EPFR inhalation, supporting the view that EFPRs acted at least in part via an oxidant-mediated mechanism. Our prior studies demonstrated oxidative stress occurring in both the lung and heart for similar exposures (82). Furthermore, other investigators have reported similar findings from ozone exposures. Ozone, itself a reactive form of oxygen, is known to induce oxidative stress in lung tissues but is not expected to travel beyond the lung, due to its overt reactivity. In these studies, notable changes in biomarkers of oxidative stress occurred in the vasculature, while only mild pulmonary changes were present (83). In that same study, the investigators noted that phospholipid fatty acids were diminished. They hypothesized that byproducts of lipid oxidation could be the driving factor for the observed cardiovascular injury (83). Thus, although additional studies probing the link between 1) the specific EPFR content within particulate matter, 2) EPFR-induced redox cycling within tissues, and 3) the presentation of cardiovascular dysfunction are needed, data presented here supports that assertion that EPFR-induced oxidant injury in the lung may play a causal role in its mechanism of inhaled PM toxicity.
Another limitation of our study is that we used a whole body inhalation exposure system to expose mice to PM for 4 h a day and for 1, 3, and 10 consecutive days. For exposures to PM, with the lungs as a portal of entry, whole body inhalation is one of the most relevant and least stressful exposure methods for experimental animals (84, 85). Properly designed inhalation exposure systems simulate an aerosol distribution pattern similar to human exposures by rigorously modeling concentration and duration of exposure. Although nose-only inhalation exposures prevent PM ingestion via grooming, the exposures are more stressful to mice and are less suitable for longer-term studies, for example, the 3- and 10-day exposures conducted here. However, to minimize secondary ingestion via grooming, the mice were exposed in a custom-designed 18 L Plexiglas chamber containing 16 individual compartments, allowing for simultaneous exposure of separated mice. Although we cannot conclude that the effects that we observed were due solely to PM inhalation, we believe that by taking the additional precaution of having each mouse exposed in a separate compartment of the chamber minimized this possibility.
The studies presented here using DCB230 at two particulate concentrations and at three different time points are important for establishing a dose response between EPFR-containing PM and cardiopulmonary effects. They allowed the delineation of a physiological mechanism where vascular function was an early locus of injury. Although the vascular dysfunction was not dependent upon inflammation, it may be associated with an injury at the air-blood interface. Future studies are needed to focus on the development of a new combustion-generated PM system that better enables our ability to titer the free radical content while maintaining the mass density of the particle constant. With this design, we would be able to tease apart the specific effect of the free radical and its redox cycle on the cardiopulmonary system. It would further enhance our ability to assess the dose response for a given radical concentration and would allow us to begin to pinpoint the mechanisms by which cardiovascular injury is initiated by the inhalation of EPFRs.
SUPPLEMENTAL DATA
All Supplemental material: https://doi.org/10.6084/m9.figshare.14755377.v3.
GRANTS
This work was supported by Louisiana Governor’s Biotechnology Initiative Grant GBI-BOR#013 (to A. L. Penn); Louisiana State University Biomedical Collaborative Research Program (to A. Noel, A. L. Penn, K. J. Varner and T. R. Dugas); and National Institute of Environmental Health Sciences Grants R21ES03006202 (to K. J. Varner and T. R. Dugas), P42ES01364808A1 (to K. J. Varner and T. R. Dugas), and P42ES01364808A1 (to A. Noel).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.C.H., A.N., A.L.P., K.J.V., and T.R.D. conceived and designed research; A.C.H., A.N., B.S., Z.P., M.H.J., Y.-F.C., and K.L. performed experiments; A.C.H., A.N., Z.P., and D.B.P. analyzed data; A.C.H., A.N., and D.B.P. interpreted results of experiments; A.C.H. prepared figures; A.C.H. drafted manuscript; K.J.V., T.R.D., A.N., and A.P. edited and revised manuscript; A.C.H., A.N., B.S., Z.P., M.H.J., Y.-F.C., A.L.P., K.L., K.J.V., and T.R.D. approved final version of manuscript.
REFERENCES
- 1.Dockery DW. Epidemiologic evidence of cardiovascular effects of particulate air pollution. Environ Health Perspect 109, Suppl 4: 483–486, 2001. doi: 10.1289/ehp.01109s4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dockery DW, Stone PH. Cardiovascular risks from fine particulate air pollution. N Engl J Med 356: 511–513, 2007. doi: 10.1056/NEJMe068274. [DOI] [PubMed] [Google Scholar]
- 3.Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, Kaufman JD. Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med 356: 447–458, 2007. doi: 10.1056/NEJMoa054409. [DOI] [PubMed] [Google Scholar]
- 4.Poloniecki J, Atkinson R, de LA, Anderson H. Daily time series for cardiovascular hospital admissions and previous day's air pollution in London, UK. Occup Environ Med 54: 535–540, 1997. doi: 10.1136/oem.54.8.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pope CA 3rd, Dockery DW. Health effects of fine particulate air pollution: lines that connect. J Air Waste Manag Assoc 56: 709–742, 2006. doi: 10.1080/10473289.2006.10464485. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz J. Air pollution and blood markers of cardiovascular risk. Environ Health Perspect 109: 405–409, 2001. doi: 10.1289/ehp.01109s3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glad JA, Brink LL, Talbott EO, Lee PC, Xu X, Saul M, Rager J. The relationship of ambient ozone and PM(2.5) levels and asthma emergency department visits: possible influence of gender and ethnicity. Arch Environ Occup Health 67: 103–108, 2012. doi: 10.1080/19338244.2011.598888. [DOI] [PubMed] [Google Scholar]
- 8.Zhang YX, Liu Y, Xue Y, Yang LY, Song GD, Zhao L. Correlational study on atmospheric concentrations of fine particulate matter and children cough variant asthma. Eur Rev Med Pharmacol Sci 20: 2650–2654, 2016. [PubMed] [Google Scholar]
- 9.Amadeo B, Robert C, Rondeau V, Mounouchy MA, Cordeau L, Birembaux X, Citadelle E, Gotin J, Gouranton M, Marcin G, Laurac D, Raherison C. Impact of close-proximity air pollution on lung function in schoolchildren in the French West Indies. BMC Public Health 15: 45, 2015. doi: 10.1186/s12889-015-1382-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cesaroni G, Forastiere F, Stafoggia M, Andersen ZJ, Badaloni C, Beelen R, et al. Long term exposure to ambient air pollution and incidence of acute coronary events: prospective cohort study and meta-analysis in 11 European cohorts from the ESCAPE Project. BMJ 348: f7412, 2014. doi: 10.1136/bmj.f7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caiazzo F, Ashok A, Waitz IA, Yim SHL, Barrett SRH. Air pollution and early deaths in the United States. Part I: Quantifying the impact of major sectors in 2005. Atmos Environ 79: 198–208, 2013. doi: 10.1016/j.atmosenv.2013.05.081. [DOI] [Google Scholar]
- 12.Auchincloss AH, Diez Roux AV, Dvonch JT, Brown PL, Barr RG, Daviglus ML, Goff DC, Kaufman JD, O'Neill MS. Associations between recent exposure to ambient fine particulate matter and blood pressure in the Multi-Ethnic Study of Atherosclerosis (MESA). Environ Health Persp 116: 486–491, 2008. doi: 10.1289/ehp.10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calderón-Garcidueñas L, Vincent R, Mora-Tiscareño A, Franco-Lira M, Henríquez-Roldán C, Barragán-Mejía G, Garrido-García L, Camacho-Reyes L, Valencia-Salazar G, Paredes R, Romero L, Osnaya H, Villarreal-Calderón R, Torres-Jardón R, Hazucha MJ, Reed W. Elevated plasma endothelin-1 and pulmonary arterial pressure in children exposed to air pollution. Environ Health Perspect 115: 1248–1253, 2007. doi: 10.1289/ehp.9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishnan RM, Adar SD, Szpiro AA, Jorgensen NW, Van Hee VC, Barr RG, O'Neill MS, Herrington DM, Polak JF, Kaufman JD. Vascular responses to long- and short-term exposure to fine particulate matter: MESA Air (Multi-Ethnic Study of Atherosclerosis and Air Pollution). J Am Coll Cardiol 60: 2158–2166, 2012. doi: 10.1016/j.jacc.2012.08.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lomnicki S, Dellinger B. A detailed mechanism of the surface-mediated formation of PCDD/F from the oxidation of 2-chlorophenol on CuO/silica surface. J Phys Chem A 107: 4387–4395, 2003. doi: 10.1021/jp026045z. [DOI] [Google Scholar]
- 16.Lomnicki S, Truong H, Vejerano E, Dellinger B. Copper oxide-based model of persistent free radical formation on combustion-derived particulate matter. Environ Sci Technol 42: 4982–4988, 2008. doi: 10.1021/es071708h. [DOI] [PubMed] [Google Scholar]
- 17.Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, Wang M, Oberley T, Froines J, Nel A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect 111: 455–460, 2003. doi: 10.1289/ehp.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vejerano E, Lomnicki SM, Dellinger B. Formation and stabilization of combustion-generated, environmentally persistent radicals on Ni(II)O supported on a silica surface. Environ Sci Technol 46: 9406–9411, 2012. doi: 10.1021/es301136d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelley MA, Hebert VY, Thibeaux TM, Orchard MA, Hasan F, Cormier SA, Thevenot PT, Lomnicki SM, Varner KJ, Dellinger B, Latimer BM, Dugas TR. Model combustion-generated particulate matter containing persistent free radicals redox cycle to produce reactive oxygen species. Chem Res Toxicol 26: 1862–1871, 2013. doi: 10.1021/tx400227s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burn BR, Varner KJ. Environmentally persistent free radicals compromise left ventricular function during ischemia/reperfusion injury. Am J Physiol Heart Circ Physiol 308: H998–H1006, 2015. doi: 10.1152/ajpheart.00891.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahne S, Chuang GC, Pankey E, Kiruri L, Kadowitz PJ, Dellinger B, Varner KJ. Environmentally persistent free radicals decrease cardiac function and increase pulmonary artery pressure. Am J Physiol Heart Circ Physiol 303: H1135–H1142, 2012. doi: 10.1152/ajpheart.00545.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balakrishna S, Lomnicki S, McAvey KM, Cole RB, Dellinger B, Cormier SA. Environmentally persistent free radicals amplify ultrafine particle mediated cellular oxidative stress and cytotoxicity. Part Fibre Toxicol 6: 11, 2009. doi: 10.1186/1743-8977-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balakrishna S, Saravia J, Thevenot P, Ahlert T, Lominiki S, Dellinger B, Cormier SA. Environmentally persistent free radicals induce airway hyperresponsiveness in neonatal rat lungs. Part Fibre Toxicol 8: 11, 2011. doi: 10.1186/1743-8977-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fahmy B, Ding L, You D, Lomnicki S, Dellinger B, Cormier SA. In vitro and in vivo assessment of pulmonary risk associated with exposure to combustion generated fine particles. Environ Toxicol Pharmacol 29: 173–182, 2010. doi: 10.1016/j.etap.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dugas TR, Lomnicki S, Cormier SA, Dellinger B, Reams M. Addressing emerging risks: scientific and regulatory challenges associated with environmentally persistent free radicals. Int J Environ Res Public Health 13: 573, 2016. doi: 10.3390/ijerph13060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian LW, Koshland CP, Yano JK, Yachandra VK, Yu ITS, Lee SC, Lucas D. Carbon-centered free radicals in particulate matter emissions from wood and coal combustion. Energy Fuels 23: 2523–2526, 2009. doi: 10.1021/ef8010096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valavanidis A, Iopoulos N, Gotsis G, Fiotakis K. Persistent free radicals, heavy metals and PAHs generated in particulate soot emissions and residue ash from controlled combustion of common types of plastic. J Hazard Mater 156: 277–284, 2008. doi: 10.1016/j.jhazmat.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 28.Davel AP, Lemos M, Pastro LM, Pedro SC, de André PA, Hebeda C, Farsky SH, Saldiva PH, Rossoni LV. Endothelial dysfunction in the pulmonary artery induced by concentrated fine particulate matter exposure is associated with local but not systemic inflammation. Toxicology 295: 39–46, 2012. doi: 10.1016/j.tox.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 29.Farina F, Sancini G, Longhin E, Mantecca P, Camatini M, Palestini P. Milan PM1 induces adverse effects on mice lungs and cardiovascular system. Biomed Res Int 2013: 583513, 2013. doi: 10.1155/2013/583513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kampfrath T, Maiseyeu A, Ying Z, Shah Z, Deiuliis JA, Xu X, Kherada N, Brook RD, Reddy KM, Padture NP, Parthasarathy S, Chen LC, Moffatt-Bruce S, Sun Q, Morawietz H, Rajagopalan S. Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways. Circ Res 108: 716–726, 2011. doi: 10.1161/CIRCRESAHA.110.237560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hill BG, Rood B, Ribble A, Haberzettl P. Fine particulate matter (PM2.5) inhalation-induced alterations in the plasma lipidome as promoters of vascular inflammation and insulin resistance. Am J Physiol Heart Circ Physiol 320: H1836–H1850, 2021. doi: 10.1152/ajpheart.00881.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang Q, Sun M, Wang F, Ma Y, Lin L, Li T, Duan J, Sun Z. Short-term PM2.5 exposure and circulating von Willebrand factor level: a meta-analysis. Sci Total Environ 737: 140180, 2020. doi: 10.1016/j.scitotenv.2020.140180. [DOI] [PubMed] [Google Scholar]
- 33.Singh P, O'Toole TE, Conklin DJ, Hill BG, Haberzettl P. Endothelial progenitor cells as critical mediators of environmental air pollution-induced cardiovascular toxicity. Am J Physiol Heart Circ Physiol 320: H1440–H1455, 2021. doi: 10.1152/ajpheart.00804.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller MR. Oxidative stress and the cardiovascular effects of air pollution. Free Radic Biol Med 151: 69–87, 2020. doi: 10.1016/j.freeradbiomed.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harmon AC, Hebert VY, Cormier SA, Subramanian B, Reed JR, Backes WL, Dugas TR. Particulate matter containing environmentally persistent free radicals induces AhR-dependent cytokine and reactive oxygen species production in human bronchial epithelial cells. Plos One 13: e0205412, 2018. doi: 10.1371/journal.pone.0205412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawal AO. Air particulate matter induced oxidative stress and inflammation in cardiovascular disease and atherosclerosis: The role of Nrf2 and AhR-mediated pathways. Toxicol Lett 270: 88–95, 2017. doi: 10.1016/j.toxlet.2017.01.017. [DOI] [PubMed] [Google Scholar]
- 37.Noël A, Cloutier Y, Wilkinson KJ, Dion C, Hallé S, Maghni K, Tardif R, Truchon G. Generating nano-aerosols from TiO(2) (5 nm) nanoparticles showing different agglomeration states. Application to toxicological studies. J Occup Environ Hyg 10: 86–96, 2013. doi: 10.1080/15459624.2012.748340. [DOI] [PubMed] [Google Scholar]
- 38.Noël A, Charbonneau M, Cloutier Y, Tardif R, Truchon G. Rat pulmonary responses to inhaled nano-TiO(2): effect of primary particle size and agglomeration state. Part Fibre Toxicol 10: 48, 2013. doi: 10.1186/1743-8977-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noël A, Maghni K, Cloutier Y, Dion C, Wilkinson KJ, Hallé S, Tardif R, Truchon G. Effects of inhaled nano-TiO2 aerosols showing two distinct agglomeration states on rat lungs. Toxicol Lett 214: 109–119, 2012. doi: 10.1016/j.toxlet.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 40.Huang W, Wang L, Li J, Liu M, Xu H, Liu S, Chen J, Zhang Y, Morishita M, Bard RL, Harkema JR, Rajagopalan S, Brook RD. Short-term blood pressure responses to ambient fine particulate matter exposures at the extremes of global air pollution concentrations. Am J Hypertens 31: 590–599, 2018. doi: 10.1093/ajh/hpx216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao N, Yin Z, Liu F, Zhang M, Lv Y, Hao Z, Pan G, Zhang J. Environmentally persistent free radicals mediated removal of Cr(VI) from highly saline water by corn straw biochars. Bioresour Technol 260: 294–301, 2018. doi: 10.1016/j.biortech.2018.03.116. [DOI] [PubMed] [Google Scholar]
- 42.Oyana T, Lomnicki S, Guo CQ, Cormier SA. A scalable field study protocol and rationale for ambient air sampling: a spatial phytosampling for leaf data collection. Environ Sci Technol 51: 10663–10673, 2017. doi: 10.1021/acs.est.7b03643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chalupa DC, Morrow PE, Oberdörster G, Utell MJ, Frampton MW. Ultrafine particle deposition in subjects with asthma. Environ Health Perspect 112: 879–882, 2004. doi: 10.1289/ehp.6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu X, Venecek M, Kumar A, Hu JL, Tanrikulu S, Soon ST, Tran C, Fairley D, Kleeman MJ. Regional sources of airborne ultrafine particle number and mass concentrations in California. Atmos Chem Phys 19: 14677–14702, 2019. doi: 10.5194/acp-19-14677-2019. [DOI] [Google Scholar]
- 45.Noël A, Xiao R, Perveen Z, Zaman H, Le Donne V, Penn A. Sex-specific lung functional changes in adult mice exposed only to second-hand smoke in utero. Respir Res 18: 104, 2017. doi: 10.1186/s12931-017-0591-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woodward NC, Crow AL, Zhang Y, Epstein S, Hartiala J, Johnson R, Kocalis H, Saffari A, Sankaranarayanan I, Akbari O, Ramanathan G, Araujo JA, Finch CE, Bouret SG, Sioutas C, Morgan TE, Allayee H. Exposure to nanoscale particulate matter from gestation to adulthood impairs metabolic homeostasis in mice. Sci Rep 9: 1816, 2019. doi: 10.1038/s41598-018-37704-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao R, Perveen Z, Rouse RL, Le Donne V, Paulsen DB, Ambalavanan N, Penn AL. In utero exposure to second-hand smoke aggravates the response to ovalbumin in adult mice. Am J Respir Cell Mol Biol 49: 1102–1109, 2013. doi: 10.1165/rcmb.2013-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noël A, Xiao R, Perveen Z, Zaman HM, Rouse RL, Paulsen DB, Penn AL. Incomplete lung recovery following sub-acute inhalation of combustion-derived ultrafine particles in mice. Part Fibre Toxicol 13: 10, 2016. doi: 10.1186/s12989-016-0122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brook RD, Rajagopalan S, Pope CA 3rd, Brook JR, Bhatnagar A, Diez-Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, Peters A, Siscovick D, Smith SC Jr, Whitsel L, Kaufman JD; American Heart Association Council on Epidemiology and Prevention, Council on the Kidney in Cardiovascular Disease, and Council on Nutrition, Physical Activity and Metabolism. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 121: 2331–2378, 2010. doi: 10.1161/CIR.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- 50.Laden F, Neas LM, Dockery DW, Schwartz J. Association of fine particulate matter from different sources with daily mortality in six U.S. cities. Environ Health Perspect 108: 941–947, 2000. doi: 10.1289/ehp.00108941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones MR, Diez-Roux AV, O'Neill MS, Guallar E, Sharrett AR, Post W, Kaufman JD, Navas-Acien A. Ambient air pollution and racial/ethnic differences in carotid intima-media thickness in the Multi-Ethnic Study of Atherosclerosis (MESA). J Epidemiol Community Health 69: 1191–1198, 2015. doi: 10.1136/jech-2015-205588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaufman JD, Adar SD, Barr RG, Budoff M, Burke GL, Curl CL, Daviglus ML, Diez Roux AV, Gassett AJ, Jacobs DR Jr, Kronmal R, Larson TV, Navas-Acien A, Olives C, Sampson PD, Sheppard L, Siscovick DS, Stein JH, Szpiro AA, Watson KE. Association between air pollution and coronary artery calcification within six metropolitan areas in the USA (the Multi-Ethnic Study of Atherosclerosis and Air Pollution): a longitudinal cohort study. Lancet 388: 696–704, 2016. doi: 10.1016/S0140-6736(16)00378-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bourque SL, Davidge ST, Adams MA. The interaction between endothelin-1 and nitric oxide in the vasculature: new perspectives. Am J Physiol Regul Integr Comp Physiol 300: R1288–R1295, 2011. doi: 10.1152/ajpregu.00397.2010. [DOI] [PubMed] [Google Scholar]
- 54.Böhm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc Res 76: 8–18, 2007. doi: 10.1016/j.cardiores.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 55.Jackson DP, Joshi AD, Elferink CJ. Ah receptor pathway intricacies; signaling through diverse protein partners and DNA-motifs. Toxicol Res (Camb) 4: 1143–1158, 2015. doi: 10.1039/c4tx00236a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lamas B, Natividad JM, Sokol H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol 11: 1024–1038, 2018. doi: 10.1038/s41385-018-0019-2. [DOI] [PubMed] [Google Scholar]
- 57.Kerzee JK, Ramos KS. Constitutive and inducible expression of Cyp1a1 and Cyp1b1 in vascular smooth muscle cells: role of the Ahr bHLH/PAS transcription factor. Circ Res 89: 573–582, 2001. doi: 10.1161/hh1901.097083. [DOI] [PubMed] [Google Scholar]
- 58.Fredenburgh LE, Perrella MA, Mitsialis SA. The role of heme oxygenase-1 in pulmonary disease. Am J Respir Cell Mol Biol 36: 158–165, 2007. doi: 10.1165/rcmb.2006-0331TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sebastián VP, Salazar GA, Coronado-Arrázola I, Schultz BM, Vallejos OP, Berkowitz L, Álvarez-Lobos MM, Riedel CA, Kalergis AM, Bueno SM. Heme oxygenase-1 as a modulator of intestinal inflammation development and progression. Front Immunol 9: 1956, 2018. doi: 10.3389/fimmu.2018.01956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Day BJ. Catalase and glutathione peroxidase mimics. Biochem Pharmacol 77: 285–296, 2009. doi: 10.1016/j.bcp.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Singh A, Rangasamy T, Thimmulappa RK, Lee H, Osburn WO, Brigelius-Flohé R, Kensler TW, Yamamoto M, Biswal S. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am J Respir Cell Mol Biol 35: 639–650, 2006. [Erratum in Am J Respir Cell Mol Biol 36: 642, 2007]. doi: 10.1165/rcmb.2005-0325OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whitlock NC, Baek SJ. The anticancer effects of resveratrol: modulation of transcription factors. Nutr Cancer 64: 493–502, 2012. doi: 10.1080/01635581.2012.667862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kali A, Shetty KS. Endocan: a novel circulating proteoglycan. Indian J Pharmacol 46: 579–583, 2014. doi: 10.4103/0253-7613.144891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kechagia M, Papassotiriou I, Gourgoulianis KI. Endocan and the respiratory system: a review. Int J Chron Obstruct Pulmon Dis 11: 3179–3187, 2016. doi: 10.2147/COPD.S118692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cook PC, MacDonald AS. Dendritic cells in lung immunopathology. Semin Immunopathol 38: 449–460, 2016. doi: 10.1007/s00281-016-0571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frump AL, Bonnet S, de Jesus Perez VA, Lahm T. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 314: L443–L460, 2018. doi: 10.1152/ajplung.00374.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Manzer R, Wang J, Nishina K, McConville G, Mason RJ. Alveolar epithelial cells secrete chemokines in response to IL-1beta and lipopolysaccharide but not to ozone. Am J Respir Cell Mol Biol 34: 158–166, 2006. doi: 10.1165/rcmb.2005-0205OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Everett LA, Cleuren AC, Khoriaty RN, Ginsburg D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood 123: 3697–3705, 2014. doi: 10.1182/blood-2014-02-554501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turner NA, Moake JL. Factor VIII is synthesized in human endothelial cells, packaged in Weibel-Palade bodies and secreted bound to ULVWF strings. PloS One 10: e0140740, 2015. doi: 10.1371/journal.pone.0140740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ikezoe T. Thrombomodulin/activated protein C system in septic disseminated intravascular coagulation. J Intensive Care 3: 1, 2015. doi: 10.1186/s40560-014-0050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tabor CM, Shaw CA, Robertson S, Miller MR, Duffin R, Donaldson K, Newby DE, Hadoke PW. Platelet activation independent of pulmonary inflammation contributes to diesel exhaust particulate-induced promotion of arterial thrombosis. Part Fibre Toxicol 13: 6, 2016. doi: 10.1186/s12989-016-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Upadhyay S, Stoeger T, Harder V, Thomas RF, Schladweiler MC, Semmler-Behnke M, Takenaka S, Karg E, Reitmeir P, Bader M, Stampfl A, Kodavanti UP, Schulz H. Exposure to ultrafine carbon particles at levels below detectable pulmonary inflammation affects cardiovascular performance in spontaneously hypertensive rats. Part Fibre Toxicol 5: 19, 2008. doi: 10.1186/1743-8977-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nurkiewicz TR, Porter DW, Hubbs AF, Cumpston JL, Chen BT, Frazer DG, Castranova V. Nanoparticle inhalation augments particle-dependent systemic microvascular dysfunction. Part Fibre Toxicol 5: 1, 2008. doi: 10.1186/1743-8977-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morrissette N, Gold E, Aderem A. The macrophage–a cell for all seasons. Trends Cell Biol 9: 199–201, 1999. doi: 10.1016/S0962-8924(99)01540-8. [DOI] [PubMed] [Google Scholar]
- 75.Kreyling WG, Semmler M, Erbe F, Mayer P, Takenaka S, Schulz H, Oberdörster G, Ziesenis A. Translocation of ultrafine insoluble iridium particles from lung epithelium to extrapulmonary organs is size dependent but very low. J Toxicol Environ Health A 65: 1513–1530, 2002. doi: 10.1080/00984100290071649. [DOI] [PubMed] [Google Scholar]
- 76.Yang W, Peters JI, Williams RO 3rd.. Inhaled nanoparticles–a current review. Int J Pharm 356: 239–247, 2008. doi: 10.1016/j.ijpharm.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 77.Choi HS, Ashitate Y, Lee JH, Kim SH, Matsui A, Insin N, Bawendi MG, Semmler-Behnke M, Frangioni JV, Tsuda A. Rapid translocation of nanoparticles from the lung airspaces to the body. Nat Biotechnol 28: 1300–1303, 2010. doi: 10.1038/nbt.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raftis JB, Miller MR. Nanoparticle translocation and multi-organ toxicity: a particularly small problem. Nano Today 26: 8–12, 2019. doi: 10.1016/j.nantod.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gaté L, Disdier C, Cosnier F, Gagnaire F, Devoy J, Saba W, Brun E, Chalansonnet M, Mabondzo A. Biopersistence and translocation to extrapulmonary organs of titanium dioxide nanoparticles after subacute inhalation exposure to aerosol in adult and elderly rats. Toxicol Lett 265: 61–69, 2017. doi: 10.1016/j.toxlet.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 80.Mühlfeld C, Gehr P, Rothen-Rutishauser B. Translocation and cellular entering mechanisms of nanoparticles in the respiratory tract. Swiss Med Wkly 138: 387–391, 2008. [DOI] [PubMed] [Google Scholar]
- 81.Buckley A, Warren J, Hodgson A, Marczylo T, Ignatyev K, Guo C, Smith R. Slow lung clearance and limited translocation of four sizes of inhaled iridium nanoparticles. Part Fibre Toxicol 14: 5, 2017. doi: 10.1186/s12989-017-0185-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lord K, Moll D, Lindsey JK, Mahne S, Raman G, Dugas T, Cormier S, Troxlair D, Lomnicki S, Dellinger B, Varner K. Environmentally persistent free radicals decrease cardiac function before and after ischemia/reperfusion injury in vivo. J Recept Signal Transduct Res 31: 157–167, 2011. doi: 10.3109/10799893.2011.555767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kodavanti UP, Thomas R, Ledbetter AD, Schladweiler MC, Shannahan JH, Wallenborn JG, Lund AK, Campen MJ, Butler EO, Gottipolu RR, Nyska A, Richards JE, Andrews D, Jaskot RH, McKee J, Kotha SR, Patel RB, Parinandi NL. Vascular and cardiac impairments in rats inhaling ozone and diesel exhaust particles. Environ Health Perspect 119: 312–318, 2011. doi: 10.1289/ehp.1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pauluhn J. Overview of inhalation exposure techniques: strengths and weaknesses. Exp Toxicol Pathol 57: 111–128, 2005. doi: 10.1016/j.etp.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 85.Pauluhn J, Mohr U. Inhalation studies in laboratory animals–current concepts and alternatives. Toxicol Pathol 28: 734–753, 2000. doi: 10.1177/019262330002800514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All Supplemental material: https://doi.org/10.6084/m9.figshare.14755377.v3.