

Abstract
Background:
Perfluorooctanoic acid (PFOA) is an environmental contaminant associated with adverse metabolic outcomes in developmentally exposed human populations and mouse models. Hexafluoropropylene oxide-dimer acid (HFPO-DA, commonly called GenX) has replaced PFOA in many industrial applications in the U.S. and Europe and has been measured in global water systems from <1 to 9350 ng/L HFPO-DA. Health effects data for GenX are lacking.
Objective:
Determine the effects of gestational exposure to GenX on offspring weight gain trajectory, adult metabolic health, liver pathology and key adipose gene pathways in male and female CD-1 mice.
Methods:
Daily oral doses of GenX (0.2, 1.0, 2.0 mg/kg), PFOA (0.1, 1.0 mg/kg), or vehicle control were administered to pregnant mice (gestation days 1.5–17.5). Offspring were fed a high- or low-fat diet (HFD or LFD) at weaning until necropsy at 6 or 18 weeks, and metabolic endpoints were measured over time. PFOA and GenX serum and urine concentrations, weight gain, serum lipid parameters, body mass composition, glucose tolerance, white adipose tissue gene expression, and liver histopathology were evaluated.
Results:
Prenatal exposure to GenX led to its accumulation in the serum and urine of 5-day old pups (P = 0.007, P < 0.001), which was undetectable by weaning. By 18 weeks of age, male mice fed LFD in the 2.0 mg/kg GenX group displayed increased weight gain (P < 0.05), fat mass (P = 0.016), hepatocellular microvesicular fatty change (P = 0.015), and insulin sensitivity (P = 0.014) in comparison to control males fed LFD. Female mice fed HFD had a significant increase in hepatocyte single cell necrosis in 1.0 mg/kg GenX group (P = 0.022) and 1.0 mg/kg PFOA group (P = 0.003) compared to control HFD females. Both sexes were affected by gestational GenX exposure; however, the observed phenotype varied between sex with males displaying more characteristics of metabolic disease and females exhibiting liver damage in response to the gestational exposure.
Conclusions:
Prenatal exposure to 1 mg/kg GenX and 1 mg/kg PFOA induces adverse metabolic outcomes in adult mice that are diet- and sex-dependent. GenX also accumulated in pup serum, suggesting that placental and potentially lactational transfer are important exposure routes for GenX.
Keywords: PFOA, HFPO-DA, GenX, Gestational exposure, Metabolism, PFAS, Microvesicular fatty change
1. Introduction
Prevalence of obesity in the U.S. population is on the rise, and recent data from the National Health and Nutrition Examination Survey found that 42.2% of Americans were classified as obese in the years 2017 and 2018 [1]. Increasing rates of obesity are concerning as obesity can lead to other adverse health outcomes, one of which is metabolic syndrome (MetS).
Metabolic syndrome describes a cluster of conditions that tend to co-occur and indicate overall poor metabolic health. In humans, these conditions include increased waist circumference, decreased high density lipoprotein C (HDL), increased triglycerides, elevated fasting glucose, and high blood pressure [2]. The prevalence of both MetS and obesity have increased in the US population at a rate that exceeds the effects of genetic predisposition and lifestyle alone, suggesting a role for environmental exposures [3]. Furthermore, an individual’s risk for developing obesity or MetS is influenced by their in utero environment. This phenomenon, called the Developmental Origins of Adult Health and Disease, explains how certain in utero conditions, such as maternal nutrition, can influence later life metabolic health [4–7]. Previous studies of humans and animals have demonstrated an association between gestational perfluorooctanoic acid (PFOA) exposure and adverse metabolic outcomes in exposed offspring [8].
PFOA is an environmental contaminant belonging to the family of chemicals known as per- and polyfluoroalkyl substances (PFAS). PFOA is a synthetic chemical containing a fully fluorinated eight-carbon backbone [9]. PFOA works as a surfactant, making it widely used in industrial processes and consumer products such as stain or water-resistant fabrics, food packaging, and cleaning products. This wide variety of uses, along with its high degree of chemical stability, has made PFOA a globally ubiquitous environmental contaminant [10–13]. In addition to its environmental persistence, PFOA is biologically persistent with a half-life of ~3.5 years in human serum [14]. Collaborative efforts by manufacturers and the U.S. Environmental Protection Agency (EPA) have led to reductions in the use of long-chain perfluoroalkyl acids, but these restrictions have also created the need for PFAS alternatives [15]. While levels of PFOA have declined in the US population since the introduction of the voluntary US EPA PFOA Stewardship Program, which aimed to first reduce and then eliminate production of PFOA [16], there is an increasing need to evaluate the toxicological effects of emerging replacement PFAS.
Hexafluoropropylene oxide-dimer acid (HFPO-DA) is a PFOA substitute and a public health concern due to its presence as an environmental pollutant. HFPO-DA and its ammonium salt are commonly referred to as “GenX chemicals” or simply “GenX”, though the term “GenX” technically refers to the trade name for the PFOA-free technology used in fluoropolymer manufacturing. For simplicity, here we use GenX and HFPO-DA interchangeably. GenX has been detected in the Cape Fear River watershed (North Carolina, USA) [17], residential soils in China [18], and in drinking water near a fluoropolymer manufacturing plant in the Netherlands [19] at levels between <1 and 9350 ng/L [20], which suggests that GenX contamination is a widespread, global concern. Despite widespread pollution in the environment, the human health effects of GenX exposure are not well studied including the potential for GenX to exert adverse effects during critical windows of in utero development.
Epidemiologic studies have shown associations between prenatal PFOA exposure and low birth weight as well as elevated body mass index, weight gain, and adiposity in children [21–25]. In addition to impacting metabolic outcomes in infants and children, an association between prenatal PFOA exposure and increased risk for obesity has been reported in young adult women [26]. Increased visceral fat mass in children has also been associated with serum levels of several PFAS [27]. The ability of prenatal PFOA exposure to disrupt metabolic disease outcomes in offspring has also been demonstrated in mice. Developmental exposure in CD-1 mice to 5–10 mg PFOA/kg body weight/day (mg/kg/day) caused low birth weight and reduced early life weight gain in offspring [8,28], while developmental exposure to lower doses (0.01, 0.1, 0.3 mg/kg/day PFOA) caused increased weight gain, insulin, and leptin in adult offspring [8,29].
The potential for prenatal PFOA exposure to disrupt developmental programming of metabolic homeostasis has been explored in both epidemiologic and in vivo studies, yet little is known regarding the metabolic health risks posed by the emerging PFOA replacement, GenX. One study in rats reported prenatal exposure to 125 mg/kg/day GenX caused decreased weight in female offspring from the perinatal period through puberty [30], and in mice it enhanced gestational weight gain [31], indicating that GenX may also induce adverse metabolic programming effects. These studies suggest altered metabolic programming as a mechanism as opposed to direct effects to target organs, as GenX has an estimated half-life of <24 h (vs ~3 weeks for PFOA) in mice and therefore would not be present in offspring that were only exposed in utero [32]. These initial findings of GenX-induced metabolic alterations, in addition to community-based exposure concerns, indicate a critical gap in knowledge regarding the potential lifelong consequences of developmental exposure to replacement PFAS, such as GenX, on metabolism.
In this study we compared the effects of developmental exposure to GenX or PFOA on metabolism in male and female CD-1 mouse offspring that were subsequently exposed to high or low fat diets by analyzing weight gain, serum lipid profile, body mass composition, glucose tolerance, liver pathology, and white adipose tissue gene expression.
2. Methods
2.1. Animals
Naïve female CD-1 mice from the National Institute of Environmental Health Sciences (NIEHS) mouse colony were bred in-house overnight and the next morning copulatory plug-positive animals were designated as gestational day (GD) 0.5, removed from the cage, weighed, and assigned to experimenter-blinded color-coded treatment groups. Plug-positive mice were distributed across color-coded groups to ensure baseline body weights were similar across all groups [range of 20–30 grams (g) on GD 0.5]. Animals were maintained on a 12/12hr light/dark schedule, with NIH-31 diet (Zeigler, PA, USA) and reverse osmosis deionized (RODI) water was provided ad libitum to dams during gestation and through weaning (PND 22). At weaning, offspring of all litters (50:50 by sex and number, when possible) were randomly assigned to either a high fat diet (HFD, 60% kcal fat diet; 5.21 kcal/g diet; Product #D12492, Research Diets) or control low fat diet (LFD, 10% kcal fat diet; 3.8 kcal/g diet; Product #D12450B, Research Diets, NJ, USA; Table S1). Diets and RODI water were provided ad libitum postweaning. Same sex offspring were housed together, and enrichment was added to cages. Animal care and use were in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals (https://olaw.nih.gov/policies-laws/phs-policy.htm). All animal procedures were approved by the NIEHS Animal Care and Use Committee (NTPL ASP#2018–0022).
2.2. Chemicals
Perfluorooctanoic acid ammonium salt, CAS# 3825-26-1 was purchased from Millipore Sigma (Darmstadt, Germany) and GenX (Ammonium 2,3,3,3-tetrafluoro-2-(heptafluoropropoxy) propanoate, CAS# 62,037-80-3) was purchased from SynQuest Laboratories (Florida, USA). Dosing solutions were prepared every 3–4 days in RODI water at concentrations allowing for a daily oral gavage administered volume of 0.01 mL dosing solution per g mouse body weight (mL/g).
2.3. Experimental design
Plug-positive dams (target of n = 10 per group) were assigned to one of the following six groups on GD 0.5: 0.1 or 1.0 mg/kg/day PFOA; 0.2, 1.0, or 2.0 mg/kg/day GenX; or vehicle control (VC, RODI water only). Sample size (# dams per treatment group) was determined a priori from data in Blake et al. [31]. Experimenters and technicians were blinded to the identity of the treatment groups throughout the experiments. PFOA doses were selected based on past studies [8,33]: 1.0 mg/kg/day was a critical dose used to inform the current human lifetime health advisory level for PFOA in drinking water of 70 parts per trillion (ppt [34]) and developmental exposure to 0.1 mg/kg/day had metabolic and developmental effects in mice in previous studies [8,29]. GenX doses were selected based on previous work that indicated a lowest observed adverse effect level (LOAEL) of 2 mg/kg/day for adverse maternal and placental outcomes [31]. Dams were weighed daily throughout gestation and were exposed to treatments via daily oral gavage from GD 1.5 to GD 17.5; doses were adjusted daily based on the previous day’s weight. Dams were monitored every few hours on the early morning of GD 18.5 to determine the approximate time of parturition. If a litter was present upon the start of the light cycle on GD 18.5, the litter age was considered postnatal day 0.5 (PND 0.5). At PND 5.5, litters were standardized to 10 pups, equalizing the sex distribution to 5 male and 5 female pups when possible.
On PND 22, necropsy of dams and one pup per sex per litter was performed and the remaining pups were weaned and assigned to HFD or LFD (Table S1). Offspring received assigned diets from PND 22 (3 weeks of age) to 18 weeks of age. These diets were matched in protein source and content, vitamin, and mineral levels [35,36]. From weaning until necropsy, offspring were co-housed with 2–5 animals per cage of the same sex and from the same treatment group, and at 12 weeks of age some males were individually housed due to fighting. Offspring were weighed weekly and numerous inlife outcomes were recorded as described below. A timeline demonstrating the experimental design is shown in Fig. 1A.
Fig. 1. Study design and measured levels of PFOA and GenX in the serum and urine of developmentally exposed offspring at postnatal day (PND) 5.5.
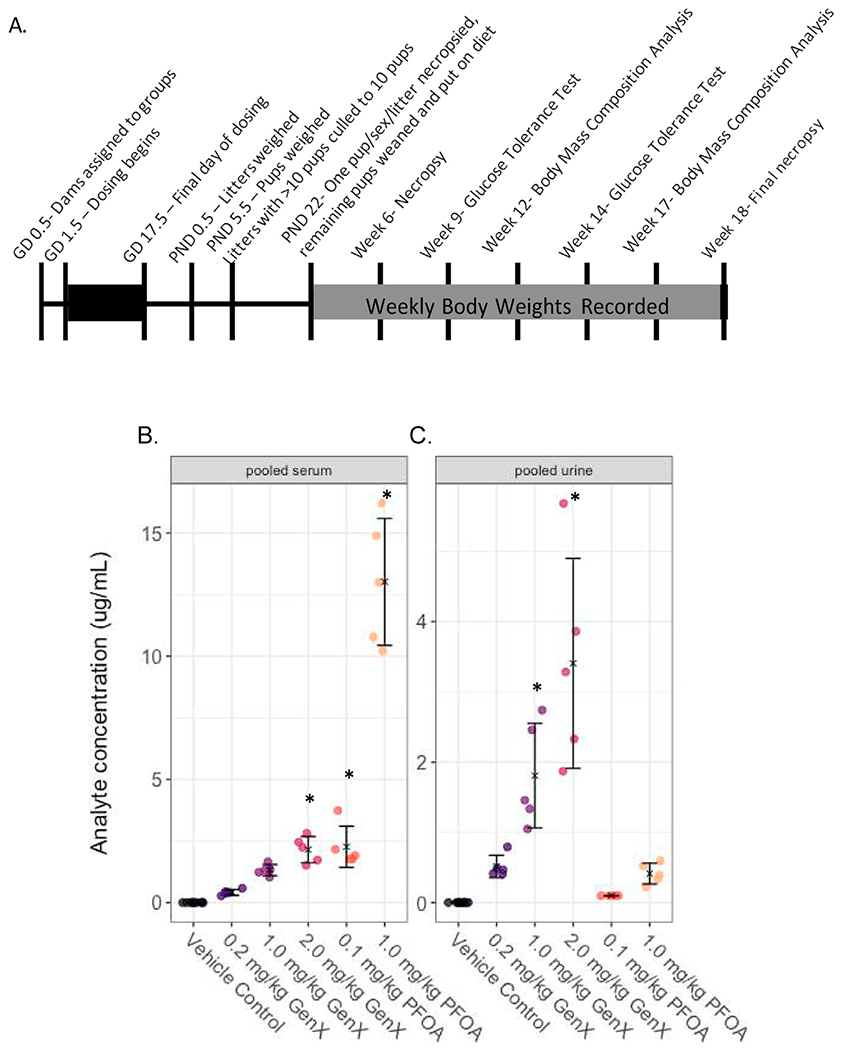
(A) The study design encompassed about 22 weeks from start to finish. (B) PFOA or GenX concentrations in samples obtained from pooling serum from littermates (μg analyte/mL serum) and (C) PFOA or GenX concentration in samples obtained from pooling urine from littermates (μg analyte/mL urine) were determined by high performance liquid chromatography tandem mass spectrometry. Treatment group mean values are denoted with an “X” flanked above and below by error bars showing standard deviation and individual data points are shown as circles (N = 5 litters per group). Note: Vehicle control (VC) samples were quantified for PFOA and GenX. Vehicle control samples were run against a low concentration calibration curve with a limit of detection of 5 ng/mL. GenX and PFOA experimental samples were run against higher calibrations curve with a LOD of 100 ng/mL. All vehicle control samples were below the LOD of 5 ng/mL for both PFOA and GenX and all 0.1 mg/kg PFOA samples were below the LOD of 100 ng/mL for PFOA. Sufficient serum and urine sample quantities were achieved by pooling across pups within the same litter. Statistical comparisons across all treatment groups for serum and urine samples are shown in Supplemental Table 3.
2.4. Offspring serum and urine dosimetry
When the number of pups/litter was standardized at PND 5.5, urine and trunk blood was collected (following swift decapitation) from excess pups within a litter. Pup serum (PND 5.5 litter-pooled samples and PND 22 individual pup serum) and urine (PND 5.5 litter-pooled samples) were analyzed for PFOA and GenX concentrations using methods similar to those previously reported [31,37–40]. Briefly, serum or urine samples (25 μL) were spiked with 13C4 PFOA (Wellington Laboratories) or 13C3 HFPO-DA (Wellington Laboratories) as an internal standard in 0.1 M formic acid denaturation buffer, followed by a protein crash step using ice-cold acetonitrile. Samples were then vortex-mixed and centrifuged at 10,000×g for 5 min. Extract supernatants were separated using a Waters ACQUITY UPLC (Waters Corporation) fitted with a Waters ACQUITY UPLC BEH C18 Column (130 Å; 1.7 μm; 2.1 mm × 50 mm).
Detection was performed using a Waters Quattro Premier XE tandem quadrupole mass spectrometer in negative ionization mode. Isotope dilution mass spectrometry was used to quantify PFOA and GenX levels in the vehicle control (quantitative range 5–100 ng/mL; 7-point curve) and dosed animals (quantitative range 200–20,000 ng/mL; 9-point curve). Vehicle control and dosed animal samples were quantified for both PFOA and GenX using respective isotope labeled chemicals and calibration curves.
2.5. Glucose tolerance test
Intraperitoneal glucose tolerance tests were conducted at Week 9 (PND 54–58) and Week 14 (PND 96–100). Animals were weighed and then fasted for 5 h (Week 9) or 5.5 h (Week 14) prior to the test. Female mice were checked for estrous stage prior to fasting so that all females were tested during diestrus. Sterile glucose solutions were prepared prior to each round of testing (D-(+)-Glucose, Sigma, St. Louis, MO #G7021). Baseline glucose levels were determined using an Accu-Chek (Roche Diagnostics, Switzerland) glucometer and test strips, and glucose was administered at 1 g glucose/kg body weight through intraperitoneal injection. Blood glucose was measured at 20, 40, 60, 120, and 180 min post-injection using blood from tail vein collection. Animals were monitored until glucose levels returned to normal ranges. Area under the curve (AUC) was calculated for each animal using the trapezoidal method [41].
2.6. Body mass composition analysis
Body mass composition of mice was evaluated at Week 12 (PND 82–86) and Week 17 (PND 117–121). Mice were gently restrained, weighed, and then fat, lean and fluid mass were measured by time domain Nuclear Magnetic Resonance (TD-NMR) using an LF90 Minispec Body Mass Analyzer (Bruker, San Jose, CA), and then returned to their cages.
2.7. Necropsy
Necropsies were performed at PND 22, Week 6 (PND 42 ± 2), and Week 18 (PND 126 ± 2). On PND 22, body weights were recorded for the dam, one male pup, and one female pup per litter. Offspring were humanely euthanized via swift decapitation, liver weights were recorded, and trunk blood and liver were collected. A portion of the left lateral lobe was fixed in 10% neutral buffered formalin (NBF) for histological analysis. Dam uteri were collected and kept in cold phosphate buffered saline and implantation sites were counted using a light microscope (Leica Z16 APO macroscope). At Week 6, males (N = 8–12) and females (N = 8–10) from each diet and treatment group were weighed and euthanized. The same procedures were followed as described for PND 22 offspring necropsy.
At Week 18, the remaining mice were weighed and fasted for 5.5–6 h prior to sacrifice to allow for collection of serum for fasted glucose and insulin levels. Females were checked for stage of estrous so that all female mice were euthanized in diestrus. Vaginal lavage was conducted using 1 X phosphate buffered saline and wet preps were read immediately. Mice were euthanized by swift decapitation and trunk blood was collected. Males (N = 8–11) and females (N = 7–11) were collected from each diet and treatment group. The liver was removed and weighed, and a portion of the left lateral lobe was fixed in 10% NBF. White adipose tissue from the reproductive fat pads was collected and snap frozen. Frozen tissues were stored at −80 °C for future analysis.
2.8. Lipid panels and liver enzymes
Blood was collected at necropsy into Becton Dickinson Vacutainer serum separating tubes (ref # 367981) and serum prepared according to manufacturer’s directions. Samples of serum (10 μL) were analyzed for cholesterol (total cholesterol, triglycerides, LDL, and HDL), liver enzymes (ALP, ALT, and AST), and glucose using an AU480 clinical chemistry analyzer (Beckman Coulter Inc., Brea, CA, USA). Reagents and calibrators used to measure alkaline phosphatase (ALP), alanine aminotransferase (ALT), aspartate aminotransferase (AST), glucose (Glu), triglyceride (Trig), high density lipoprotein (HDL), and cholesterol (Chol), were purchased from Beckman Coulter Inc. (Melville, NY, USA). Reagents used to measure low density lipoprotein (LDL) were purchased from Diazyme Laboratories (Poway, CA, USA). Lipid panel measurements were conducted on serum samples collected at PND 22, Week 6, and Week 18. Liver enzymes were measured for Week 18 samples only.
2.9. Insulin
Fasting insulin was measured in serum collected at week 18 necropsy using the mouse metabolic immunoassay kit from Meso Scale Discovery (Catalog number: K15124C, Meso Scale Diagnostics, Rockville, MD). Each 25 μL sample was run in duplicate. Samples with a coefficient of variation (CV) greater than 20% were re-run two additional times for a total of three runs. If the CV across the three runs combined still exceeded 20%, the animal was excluded from the analysis (N = 2 mice, both in the male HFD GenX 0.2 mg/kg group). Means of samples from the same animal run in duplicate on a plate were considered a biological replicate in subsequent analyses. Quantitative insulin check index (QUICKI) scores were calculated for each animal [42].
2.10. Histopathology
Livers from offspring were fixed in 10% NBF, trimmed, processed into paraffin blocks, cut as 5 μM sections, mounted onto glass slides, and then stained with hematoxylin and eosin (H&E). Slides were evaluated by two board-certified veterinary pathologists (S.E. and J.H.) and reviewed by a Pathology Working Group with four additional board-certified pathologists to confirm final diagnoses. Severity grading was either none/minimal, mild, moderate or severe. Hepatocyte single cell necrosis and mixed cell infiltrate was graded as none/minimal (<2 foci), mild (2–7 foci), moderate (8–20 foci or 2–4 foci of >4 cell-layer thick periportal aggregates), or severe (20+ foci or >4 foci of >4 cell-layer thick periportal aggregates). Microvesicular fatty change was graded as none/minimal (<10% parenchyma affected), mild (multifocal random or centrilobular hepatocytes, >10% and <30% parenchyma affected with mild to moderate vacuoles or >30% affected with minimal vacuoles), moderate (centrilobular hepatocytes affected, >30% and <70% parenchyma affected with moderate to severe vacuoles or near diffuse pattern, >70% with mild vacuoles), or severe (near diffuse pattern, >70% hepatocytes affected with severe vacuoles). Macrovesicular fatty change was graded as none/minimal (no macrovesicular vacuolation), mild (multifocal random or periportal, <30% parenchyma affected), moderate (periportal hepatocytes affected, >30% and <70% parenchyma affected), or severe (near diffuse pattern, >70% hepatocytes affected). Digital images were captured as screen shots from H&E slides scanned on the Aperio ScanScope AT2 instrument (Leica Biosystems Inc., Buffalo Grove, IL) using ImageScope software, version 12.3 (Aperio). Formatting of images for publication was completed using Adobe Photoshop CC version 19.1.0 (Adobe Systems Inc., San Jose, CA).
2.11. Quantitative RT-PCR (RT-qPCR) analysis
White adipose tissue (WAT) was collected at the week 18 necropsy from the intra-abdominal perigonadal depots. Tissue from the four heaviest animals in each treatment group was selected to capture the effects in animals most sensitive to weight gain. A range in sensitivity was expected due to the genetic variation in the outbred CD-1 mice used in the study. RNA was isolated by homogenizing WAT with an automated ceramic bead homogenizer FastPrep-24 5G (MP Biomedicals, Irvine, CA) in Trizol (Thermofisher Scientific, PA, USA) for 2 cycles of 4 m/s for 30sec. RNA was purified using a Qiagen RNeasy kit (Qiagen, MD, USA) and genomic DNA was removed using on-the-column DNase I digestion (Qiagen, MD, USA) and quantified with Nanodrop 2000 spectrophotometer (ThermoFisher Scientific, PA, USA). RNA (1 mg) was transcribed into cDNA using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, USA). cDNA (25 ng) was amplified in triplicate using a QuantStudio 7 Flex Real Time PCR System (Applied Biosystems) and Power SYBR Green 2 × Mastermix (Applied Biosystems, Foster City, Canada). Primer sets for all genes were pre-designed assays (IDT DNA, Redwood City, CA, USA) and are shown in Table S2. Mean Ct values were normalized to Rpl19 (60S ribosomal protein L19; housekeeping gene) and relative fold change mRNA levels were calculated by using ΔΔ CT method [43].
2.12. Statistical analysis
Data were analyzed in R version 1.2.5019 (R Foundation for Statistical Computing; Vienna, Austria). Sample sizes for each endpoint are reported in the accompanying figure legends or tables. A threshold of P < 0.05 was used for determining statistical significance unless otherwise noted. For analyses of data obtained by a single measurement at a single time point (e.g. offspring serum and urine dosimetry, litter outcomes at PND 0.5, offspring liver weights, serum lipids, and body mass composition), data were analyzed for main effect of treatment group using analysis of variance and the lme4 (Bates et al., 2014) and lmerTest packages (Kuznetsova et al., 20,117). Simultaneous tests for general linear hypotheses were corrected for multiple comparisons of means between all groups using Tukey contrasts in the package multcomp (Hothorn et al., 2009) or by Dunnett’s test when comparing treatment groups to the vehicle control group in the same package. Jonckheere-Terpstra’s trend test in the package clinfun was used to determine dose-response trends in QUICKI scores [44]. Mixed effects models were used in analyses of data when multiple pups per litter were sampled or when individual pups were measured repeatedly for the same outcome over time (e.g. PND 5.5 pup weights and postweaning weight gain). Analyses of data collected after diet groups were assigned (PND 22 and older) were stratified a priori by offspring sex and diet group.
Average PND 0.5 pup weights were calculated as the litter weight divided by the number of pups in the litter. Offspring weights measured after PND 0.5 were obtained from individual pups. Multiple individual pups per litter were weighed for each litter at PND 5.5, therefore pup weights were analyzed using mixed effect models and included a priori fixed effects of treatment group and litter size and a random effects term for the dam (litter) using the lme4 package. Point estimates and 95% confidence intervals were determined from the final model using the Wald method. Offspring weights measured from PND 22 and later did not require a random effects term for the dam (litter), as only one pup per sex per litter was measured at these time points.
Postnatal weight gain analyses were stratified by diet type and sex, then analyzed using mixed effect models and included a priori fixed effects of treatment group and offspring age and a random effects term to account for repeated measures of the same individual using the lme4 package. Point estimates and 95% confidence intervals were determined from the final model using the Wald method.
Liver pathology scoring was analyzed using pairwise Fisher’s exact tests between control and treatment groups, with post-hoc correction of P values using the Holm method [45]. This analysis was done using the RVaidememoire package [46].
RT-qPCR data was analyzed using the delta delta Ct (ΔΔ Ct) method [43]. Delta Ct (Δ Ct) was calculated by normalizing to the Rpl19 housekeeping gene and mean Δ Ct was calculated for each gene in each animal. Mice were stratified by sex and diet, and within these sex and diet groups a one-way ANOVA with Dunnett’s post hoc test was used to compare treatment groups to the vehicle using GraphPad Prism (version 8.4.2).
3. Results
3.1. Internal dosimetry
To gain an understanding of the persistence of gestational exposures in the offspring, PFOA and GenX concentrations were measured using HPLC mass spectrometry in pooled serum or pooled urine from pups within a dose group when culled at PND 5.5. Quantified levels of both chemicals fell below the limits of detection of 5 ng/mL for the VC group. However, PFOA and GenX were detectable in serum and urine from respective treatment groups (Fig. 1). At PND 5.5, mean serum levels of PFOA in pups exposed prenatally to 0.1 mg/kg PFOA were 2.3 μg/mL and in pups exposed to 1.0 mg/kg PFOA were 13.0 μg/mL (Table S3). The pups exposed to 0.2, 1.0, and 2.0 mg/kg GenX had mean serum concentrations of 0.42 μg/mL, 1.32 μg/mL, and 2.15 μg/mL, respectively (Table S3). Serum measurements of GenX demonstrated a different slope than the administered dose. The serum concentration increased 3-fold between the 0.2 mg/kg GenX and 1.0 mg/kg GenX groups, which was a 5-fold increase in dose. The serum concentration increased 0.6-fold between the 1.0 mg/kg and 2.0 mg/kg GenX group (a 2-fold increase in dose); therefore, the observed relationship between increasing dose and serum accumulation was not parallel (Fig. 1B).
At the PND 5.5 timepoint, GenX was detectable in the urine of all GenX exposed pups (Fig. 1C). The pups exposed to 0.2, 1.0, and 2.0 mg/kg GenX had mean urine concentrations of 0.51, 1.81, and 3.4 μg/mL, respectively (Table S3). For the PFOA exposed animals, 0.02 μg/mL was detected in the urine of the 0.1 mg/kg group, and 0.42 μg/mL in the 1.0 mg/kg group, suggesting a larger percentage of administered dose was excreted in urine in the higher dose group (Table S3). Serum levels of both PFOA exposed groups and the 2.0 mg/kg GenX group were significantly higher than the VC, and the urine levels of 1.0 and 2.0 mg/kg GenX were significantly different from VC (Table S4). Other significant differences that were present between dose groups are shown in Table S4. At the PND 22 timepoint, there was no detectable GenX in the blood of any of the GenX exposed pups. For the 0.1 mg/kg PFOA group, the mean serum concentration regardless of pup sex was approximately 0.3 μg/mL, and for the 1.0 mg/kg PFOA group, the serum concentration was approximately 4.0 μg/mL (Table S5).
3.2. Early life weight gain
To minimize stress on dams and pups, pup birth weight at PND 0.5 was determined by weighing the litter as a unit and dividing by the number of pups. Using this approach, there were no treatment-related differences in average pup weight at PND 0.5 (Table S6). Pup body weight at PND 5.5 was determined by weighing each pup individually. After adjusting for litter size and including a random effects term (dam), mixed effect models estimated a 6.3% and 6.7% reduction in pup weight for GenX 1.0 mg/kg and 2.0 mg/kg dose groups at PND 5.5 compared to VC, respectively (Fig. 2, Table S6).
Fig. 2. Mixed model estimates of pup body weight (g) at PND 5.5.
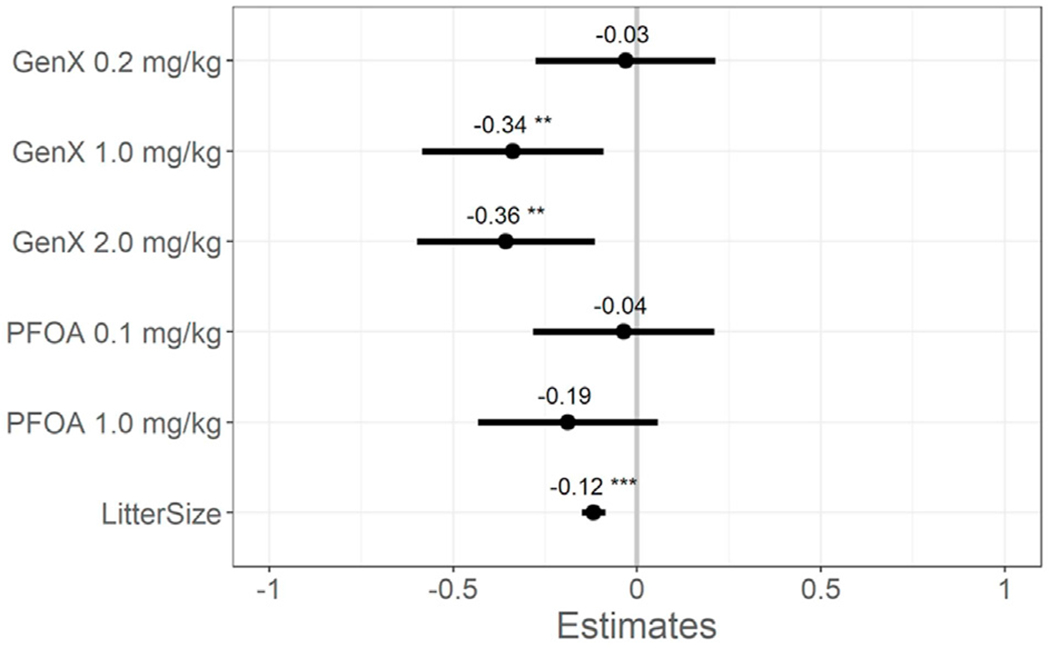
Effect estimates are centered around the vehicle control group (y = 0) and show the model point estimate for each main effect (dose group and litter size) with 95% confidence intervals (CIs). *P < 0.05. **P < 0.01. ***P < 0.001. Beta estimate 95% confidence intervals do not overlap zero (mixed-effect model adjusting a priori for litter size as a fixed effect and the dam as a random effect, vehicle control as reference group). Adjusted estimates and 95% CIs are shown in Table S6. N = 4–19 offspring per litter with 9–10 total litters per group.
3.3. Body and relative liver weights
Individual offspring body and liver weights were measured at necropsy on PND 22, Week 6 (PND 42 ± 2), and Week 18 (PND 126 ± 2).
3.3.1. PND 22
Prior to assigning offspring to specific diets, body weights in each group were determined. There were no significant differences in offspring body weights in the treatment groups compared to VC on PND 22 (Tables S7 and S8). There were no statistically significant changes in relative liver weights (liver weight as a percent of total body weight) in females, though the relative liver weight remained increased by 7.0% in the 1.0 mg/kg PFOA group compared to VC (P = 0.1; Table S7). Relative liver weight was significantly increased only in males exposed to 1.0 mg/kg PFOA by 11.6% compared to VC males (P = 0.02; Table S8).
3.3.2. Week 6
When assessed at six weeks of age, the female VC mice fed LFD weighed an average of 25.5 g, compared to 28.0 g for those fed HFD, but no significant treatment related changes in body weight were detected at this time point (Table S9). At this same age, males fed LFD demonstrated a 9.1% increase in body weight in the 2.0 mg/kg GenX group compared to VC and enlarged liver weight (average body weight 34.2 g; P = 0.036; Table S10). There were no significant treatment-related differences in average body weight for males fed HFD (Table S10). However, there was an 18% increase in relative liver weight for male offspring fed LFD in the 1.0 mg/kg GenX group compared to VC (P = 0.002) and a 13.6% increase in relative liver weight for male offspring fed LFD in the 1.0 mg/kg PFOA (P = 0.036) group compared to VC (Table S10).
3.3.3. Week 18
Females fed HFD exhibited substantial weight gain in the VC group by 18 weeks compared with VC females fed LFD (LFD = 35.5 g vs. HFD = 54.2 g; Table S11). In females, there was a 31.4% increase in relative liver weight in the 1.0 mg/kg PFOA HFD group compared to VC HFD (P = 0.011; Table S11). For males fed LFD, there was an 18.2% increase in body weight in the 2.0 mg/kg GenX group compared to VC (P = 0.026), a doubling in weight gain effect seen at week 6 necropsy, and a 17.6% increase in body weights of the 0.1 mg/kg PFOA group compared to VC (P = 0.046; Table S12). There was also a 16.0% increase in body weight in the males fed LFD in the 1.0 mg/kg PFOA group compared to VC; however, this did not reach statistical significance (P = 0.078; Table S12). There were no significant differences in body weight or relative liver weight across treatment groups in LFD females or HFD males at Week 18 (Table S11 & Table S12).
3.3.4. Weight Gain over Time (PND 22-Week 18)
Postnatal weight gain was analyzed using repeated measures mixed effect model estimates over time by diet and sex (Fig. 3). This revealed two important observations; first, female and male mice on LFD had considerably different patterns of weight gain following exposure to PFOA or GenX (Fig. 3A and C). Specifically, females exposed to the lower dose of PFOA were more sensitive to weight gain, while males exposed to the higher dose of GenX showed increased weight gain. Females fed LFD and exposed to 0.1 mg/kg/day PFOA gained more weight than VC females (17.16 g compared to 15.46 g in controls; Fig. 3C), but this difference did not reach statistical significance. However, male mice fed LFD and exposed to 2.0 mg/kg/day GenX gained on average 24.6 g from PND 22 to Week 18 (P < 0.05; Fig. 3A & Table S13); a 22.4% increase compared to the 20.1 g gained by VC mice fed LFD. There were no other significant changes in weight gain for males fed LFD, though the males in the 1.0 mg/kg PFOA group gained on average 23.4 g, which was a 16.4% increase compared to VC males (Table S13). Second, while the developmental exposures to PFOA and GenX exhibited a visual trend for dose-dependent weight changes over time in males fed LFD (GenX: 0.2 < 1.0 < 2.0 mg/kg/d; PFOA: 0.1 < 1.0 mg/kg/d; Fig. 3A), there were no significant changes due to chemical exposure on body weight of the male or female mice fed HFD (Fig. 3B and D; Tables S11 and 12).
Fig. 3. Mixed model estimates of weight gain (g) from PND 22 to 18 weeks of age.
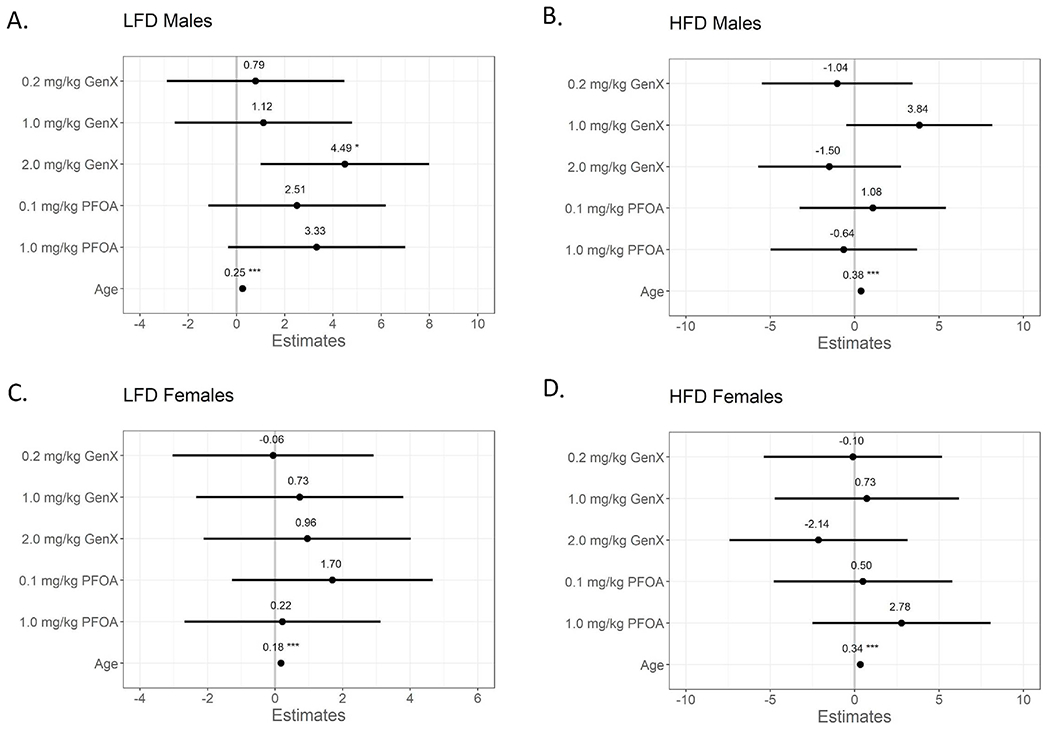
(A) Low fat diet (LFD) male offspring, (B) high fat diet (HFD) male offspring, (C) LFD female offspring, and (D) HFD female offspring. Effect estimates are centered around the vehicle control group (y = 0) and show the model point estimate for each main effect (dose group) with 95% confidence intervals (CIs). *P < 0.05. **P < 0.01. ***P < 0.001. Beta estimate 95% confidence intervals do not overlap zero (mixed-effect model adjusting a priori for repeated measures of the same animals over time, vehicle control as reference group). Adjusted estimates and 95% CIs are shown in Table S13. N = 7–11 mice per group.
3.4. Serum lipids
3.4.1. PND 22
At PND 22, serum triglycerides were significantly reduced in all female treatment groups compared to VC females (P < 0.001; Table 1), even though the GenX-exposed pups had no measurable circulating exposure remaining (Table S5). The decrease in serum triglycerides in females relative to VC was similar across the GenX dose groups: 42.3% for 0.2 mg/kg GenX, 45.5% for 1.0 mg/kg GenX, 43.5% for 2.0 mg/kg GenX. For PFOA-exposed females, triglycerides were decreased by 34.8% (0.1 mg/kg PFOA), and 52.8% (1.0 mg/kg PFOA; Table 1). In the females, there were no differences in total cholesterol (TC), LDL, or HDLdue to exposures; however, the HDL to LDL ratio was increased by 16.9% in the GenX 2.0 mg/kg group (P = 0.008; Table 1). In the males, serum triglycerides were reduced by 34.2% in 1.0 mg/kg GenX (P = 0.023), 38.9% in 2.0 mg/kg GenX, (P = 0.006), and 40.7% (P = 0.005) in 1.0 mg/kg PFOA groups relative to VC males (Table 1, bottom). In the males, there was a 21.3% increase in LDL cholesterol in the 1.0 mg/kg PFOA group compared to VC males (P = 0.047; Table 1). Although mean TC levels were increased 15–19% due to some PFOA and GenX exposures in males, the considerable variability in the endpoint obscured the ability to detect statistical significance and no significant differences in TC, HDL, or the HDL:LDL ratio were noted (Table 1).
Table 1.
Offspring serum lipid levels at PND 22.
| Triglycerides (mg/dL) | Cholesterol (mg/dL) | HDL (mg/dL) | LDL (mg/dL) | HDL:LDL | ||
|---|---|---|---|---|---|---|
| Female | Vehicle | 194.4 ± 44.8 | 107.1 ± 13.5 | 62.5 ± 8.1 | 21.1 ± 2.7 | 3.0 ± 0.3 |
| GenX 0.2 mg/kg | 112.2 ± 30.8 * | 106.8 ± 12.3 | 64.1 ± 7.1 | 19.1 ± 2.4 | 3.4 ± 0.3 | |
| GenX 1.0 mg/kg | 106 ± 26.5 * | 104.2 ± 20.7 | 59.6 ± 10.4 | 18.7 ± 3.4 | 3.2 ± 0.2 | |
| GenX 2.0 mg/kg | 109.9 ± 26.8 * | 112.0 ± 12.1 | 66.7 ± 7.7 | 19.3 ± 2.0 | 3.5 ± 0.4 * | |
| PFOA 0.1 mg/kg | 126.8 ± 50.6 * | 100.5 ± 18.6 | 58.4 ± 11.2 | 18.5 ± 2.3 | 3.1 ± 0.4 | |
| PFOA 1.0 mg/kg | 91.8 ± 21.0 * | 117.3 ± 12.0 | 70.2 ± 6.6 | 23.1 ± 3.1 | 3.1 ± 0.3 | |
| Male | Vehicle | 174.6 ± 67.2 | 105.3 ± 16.4 | 64.5 ± 9.2 | 18.8 ± 2.7 | 3.5 ± 0.4 |
| GenX 0.2 mg/kg | 124.0 ± 38.0 | 121.4 ± 11.1 | 71.4 ± 7.6 | 21.2 ± 2.9 | 3.4 ± 0.3 | |
| GenX 1.0 mg/kg | 114.9 ± 43.5 * | 125.5 ± 30.2 | 68.3 ± 13.3 | 22.4 ± 4.9 | 3.1 ± 0.4 | |
| GenX 2.0 mg/kg | 106.6 ± 27.6 * | 111.0 ± 10.4 | 67.4 ± 6.3 | 18.6 ± 1.8 | 3.6 ± 0.3 | |
| PFOA 0.1 mg/kg | 132.7 ± 34.9 | 107.9 ± 17.5 | 63.0 ± 8.4 | 19.4 ± 2.3 | 3.3 ± 0.3 | |
| PFOA 1.0 mg/kg | 103.6 ± 24.6 * | 124.3 ± 11.5 | 74.4 ± 8.4 | 22.8 ± 3.5 * | 3.3 ± 0.3 |
Note: Data presented as mean ± SD, N = 7–10.
P < 0.05 relative to vehicle control (one-way ANOVA, Dunnett’s post hoc test).
Abbr: HDL = high density lipoprotein; LDL = low density lipoprotein.
3.4.2. Week 6
By week 6 of age, GenX significantly affected TC in males fed LFD. Relative to LFD VC males, TC was increased by 37% in LFD males exposed to 0.2 mg/kg GenX (P = 0.005), 44% in LFD males exposed to 1.0 mg/kg GenX (P < 0.001), and 29% in LFD males exposed to 2.0 mg/kg GenX (P = 0.022; Table S14). Males fed LFD in the 1.0 mg/kg GenX group also had a 21% increase in HDL (P = 0.084) and a 41% increase in LDL (P = 0.071) relative to VC males fed LFD, though these results were not statistically significant (Table S14). Males fed LFD in the 1.0 mg/kg PFOA group exhibited a 20% decrease in HDL:LDL ratio relative to VC males fed, but these effects did not reach statistical significance (P = 0.06; Table S14). HFD males in the 1.0 mg/kg PFOA group had a 19.3% decrease in the ratio of HDL to LDL (P = 0.012) relative to VC males fed HFD (Table S14). Triglyceride levels in males exposed to either PFOA or GenX had returned to VC levels by 6 weeks of age.
At 6 weeks of age, LFD females exhibited increased HDL:LDL ratios of 22.5% for the 0.2 mg/kg GenX group (P = 0.029), and 22.0% for the 0.1 mg/kg PFOA group (P = 0.043; Table S15). Although not detected in males, the decrease in triglyceride levels persisted in PFOA and GenX-exposed LFD females; triglycerides were decreased 42.1% in the 1.0 mg/kg GenX (P = 0.002), 30.4% in the 0.1 mg/kg PFOA (P = 0.038), and 34.2% in the 1.0 mg/kg PFOA (P = 0.012) groups, compared to LFD VC females (Table S15). TC was increased by 20.5% in females fed LFD in the 2.0 mg/kg GenX group (P = 0.057) compared to VC (Table S15). There were no significant differences due to treatment in lipid panels for females on the HFD.
3.4.3. Week 18
Effects on blood lipids due to developmental exposure to PFOA or GenX were diminished by 18 weeks of age compared to effects noted earlier in life. At Week 18, males fed LFD following exposure to 1.0 mg/kg GenX had a 20.8% increase in HDL (P = 0.04) and a persistent 30.3% elevation in TC (P = 0.04), relative to LFD vehicle males (Table S16). There were no differences in the male HFD groups or LFD groups in other serum lipid levels compared to their respective VC groups. Females fed HFD in the 1.0 mg/kg PFOA had a 65.8% increase in LDL (P = 0.01), resulting in a 33.6% decrease in HDL:LDL ratio (p < 0.001) relative to HFD VC females (Table S17).
3.5. Glucose metabolism
3.5.1. Week 18 Fasted serum measurements
Serum glucose and insulin levels were measured in fasted male and female mice at 18 weeks of age (Table 2). Fasting glucose levels were slightly elevated due to the HFD in VC mice relative to LFD VC mice (especially in males), but there was no significant effect of developmental PFOA or GenX exposure on fasting glucose levels in male or female offspring (Table 2). However, in male offspring, GenX had a substantial effect on fasting insulin levels compared to VC for both diet conditions. On HFD, 1.0 mg/kg GenX exposure caused more than a 4-fold increase in insulin levels compared to HFD VC males (P = 0.004) and on LFD, 2.0 mg/kg GenX exposures induced more than a 7-fold increase in insulin compared to LFD VC males (P = 0.02; Table 2). Within the LFD males exposed to GenX, there was a dose-dependent increase in serum insulin (P < 0.001, Jonckheere’s trend test; Table 2). Insulin levels in LFD males exposed to PFOA also increased but did not reach significance when compared by ANOVA. However, PFOA also caused a dose-dependent increase in insulin levels (P < 0.001, Jonckheere’s trend test; Table 2). As such, the QUICKI score was decreased in males fed the LFD by 42.2% in the 2.0 mg/kg GenX (P = 0.01) and by 43.1% in the 1.0 mg/kg PFOA (P = 0.02) groups and these changes were driven by elevated serum insulin (Table 2). Males in the 1.0 mg/kg GenX group fed LFD also had a 33.9% decrease in QUICKI, though this decrease did not reach statistical significance (P = 0.085; Table 2).
Table 2.
Serum glucose and insulin of fasted offspring at Week 18.
| Treatment | Glucose (mg/dL) | Insulin (pg/mL) | QUICKI | ||
|---|---|---|---|---|---|
| Female | LFD | Vehicle | 167.8 ± 28.0 | 602.8 ± 374.1 | 0.760 ± 0.170 |
| GenX 0.2 mg/kg | 159.9 ± 18.7 | 686.9 ± 598.6 | 0.741 ± 0.139 | ||
| GenX 1.0 mg/kg | 162.0 ± 27.1 | 543.0 ± 383.7 | 0.790 ± 0.176 | ||
| GenX 2.0 mg/kg | 158.4 ± 56.0 | 373.9 ± 271.8 | 1.174 ± 0.850 | ||
| PFOA 0.1 mg/kg | 180.4 ± 14.9 | 411.1 ± 307.5 | 0.910 ± 0.300 | ||
| PFOA 1.0 mg/kg | 155.3 ± 19.1 | 701.8 ± 846.9 | 0.857 ± 0.279 | ||
| HFD | Vehicle | 188.0 ± 19.0 | 1539.3 ± 1067.1 | 0.573 ± 0.127 | |
| GenX 0.2 mg/kg | 153.1 ± 16.9 | 903.0 ± 619.1 | 0.676 ± 0.128 | ||
| GenX 1.0 mg/kg | 183.0 ± 22.3 | 2876.7 ± 2614.2 | 0.547 ± 0.165 | ||
| GenX 2.0 mg/kg | 172.8 ± 36.4 | 829.8 ± 581.0 | 0.713 ± 0.231 | ||
| PFOA 0.1 mg/kg | 165.6 ± 17.1 | 653.0 ± 689.9 | 0.808 ± 0.229 | ||
| PFOA 1.0 mg/kg | 203.1 ± 50.4 | 6162.4 ± 7279.9 * | 0.577 ± 0.315 | ||
| Male | LFD | Vehicle | 179.7 ± 26.5 | 933.5 ± 791.3 | 0.802 ± 0.473 |
| GenX 0.2 mg/kg | 190.4 ± 32.3 | 1043.9 ± 515.6† | 0.600 ± 0.094 | ||
| GenX 1.0 mg/kg | 192.4 ± 31.1 | 2506.5 ± 1588.8† | 0.530 ± 0.187 | ||
| GenX 2.0 mg/kg | 181.0 ± 22.9 | 6597.5 ± 8869.8*,† | 0.463 ± 0.103 * | ||
| PFOA 0.1 mg/kg | 190.0 ± 29.3 | 2048.4 ± 1301.0† | 0.587 ± 0.281 | ||
| PFOA 1.0 mg/kg | 250.2 ± 199.0 | 4271.2 ± 3391.1† | 0.456 ± 0.114 * | ||
| HFD | Vehicle | 255.6 ± 113.6 | 4775.8 ± 1899.4 | 0.407 ± 0.044 | |
| GenX 0.2 mg/kg | 251.0 ± 137.8 | 7282.4 ± 7409.6 | 0.407 ± 0.078 | ||
| GenX 1.0 mg/kg | 252.6 ± 118.0 | 20027.8 ± 18986.2 * | 0.354 ± 0.074 | ||
| GenX 2.0 mg/kg | 196.2 ± 46.1 | 6135.3 ± 4838.6 | 0.423 ± 0.063 | ||
| PFOA 0.1 mg/kg | 208.4 ± 93.4 | 4452.9 ± 3456.6 | 0.444 ± 0.064 | ||
| PFOA 1.0 mg/kg | 198.1 ± 36.6 | 4954.8 ± 2416.4 | 0.421 ± 0.045 |
Note: Data presented as mean ± SD, N = 7–11.
P < 0.05 relative to vehicle control (one-way ANOVA, Dunnett’s post hoc test) within diet.
P < 0.001, Jonckheere’s trend test for a dose response across the GenX or PFOA doses.
Abbr: QUICKI = Quantitative Insulin Sensitivity Check Index.
3.5.2. Glucose Tolerance
Male and female offspring were also challenged by a glucose injection at 9 and 14 weeks of age, and blood glucose levels were measured at numerous timepoints thereafter. There were no significant differences in glucose tolerance when measured as area under the curve at 9 or 14 weeks across the treatment groups relative to respective same-sex and same-diet VC groups (Tables S18–19).
3.6. Body composition
Body composition was measured to determine if fat mass was increasing over time due to PFOA or GenX exposure. There were no statistically significant differences in body mass composition measurements across treatment groups within sex or within diet vs. VC groups at Week 12 (Tables S20–21). There were also no significant body mass composition changes in males fed HFD, or females fed either HFD or LFD at Week 17 (Tables S22–23; Fig. 4B, C, D). However, by Week 17, males fed LFD and exposed to 2.0 mg/kg GenX exhibited an 18.5% increase in body weight (P = 0.027; Fig. 4A), a 94.3% increase in grams of body fat (P = 0.011), a 66.6% increase in percent fat (P = 0.016; Fig. 4E), a 92.6% increase in the fat to lean ratio (P = 0.010; Fig. 4F), and a 12.3% decrease in percent lean mass (P = 0.017) compared to VC males fed LFD (Table S23).
Fig. 4. Body composition parameters at Week 17.
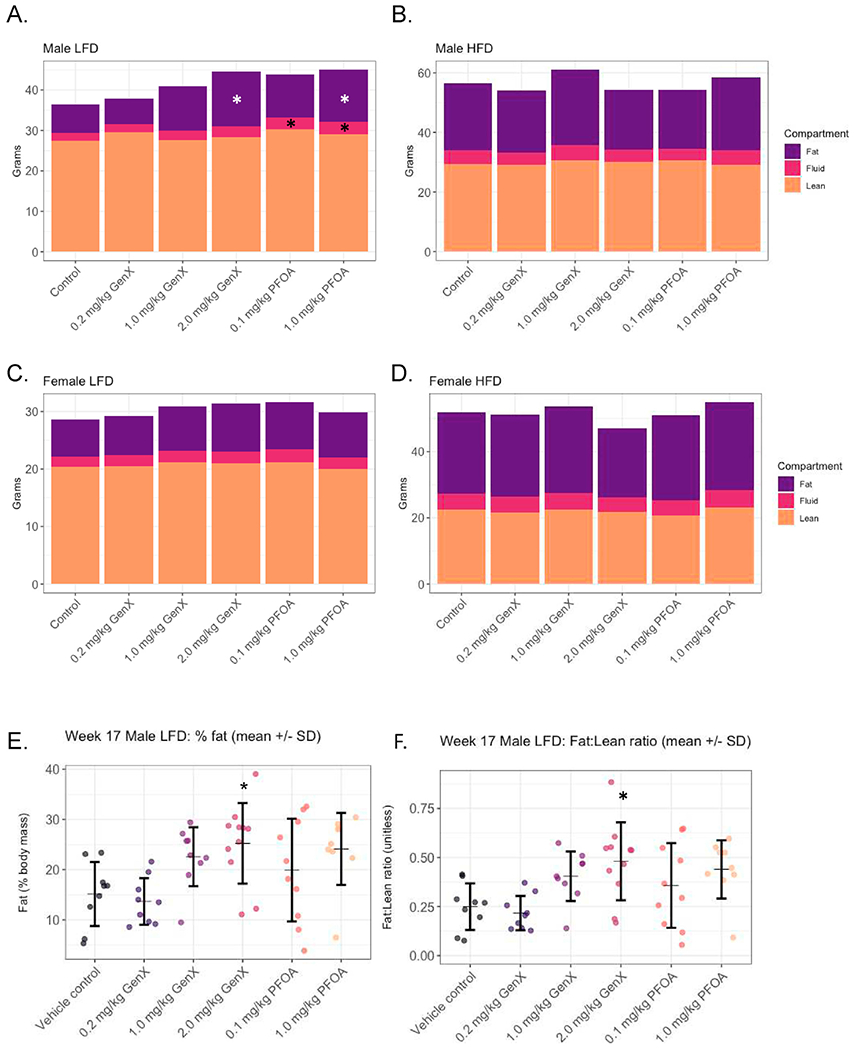
(A) Low fat diet (LFD) male offspring, (B) high fat diet (HFD) male offspring, (C) LFD female offspring, and (D) HFD female offspring grams of fat mass, fluid mass, and lean mass. Bars represent mean grams of mass per group calculated using LF90 Minispec Body Mass Analyzer. Additional data for males on LFD are shown in E) Relative fat mass expressed as percent total body mass, and F) Ratio of fat mass to lean mass. Circles represent individual data points within a treatment group (N = 7–11 mice/group) and horizontal dashes represent the group mean flanked by the standard deviation above and below in black error bars. *P < 0.05.
At week 17, males fed LFD in the 1.0 mg/kg PFOA group also had an 18.0% increase in body weight (P = 0.05; Fig. 4A), an 84.3% increase in body fat (P = 0.04), a 59.3% increase in percent fat (P = 0.05; Fig. 4E), and a 76.1% increase in fat to lean ratio (P = 0.06; Fig. 4F), compared to VC males fed LFD (Table S23). Males fed LFD and in the 0.1 mg/kg PFOA group had a 16.2% increase in body weight (P = 0.07; Fig. 4A, Table S23).
Of note, some of the treated groups retained more body fluids. Compared to VC males on the same diet, LFD males had a 35.0% increase in grams of fluid in the 2.0 mg/kg GenX group (P = 0.059), a 40.0% increase in the 0.1 mg/kg PFOA group (P = 0.044), and 50.0% increase in the 1.0 mg/kg PFOA group (P = 0.009; Table S23). The percent fluid was also increased in the LFD males in the 0.1 mg/kg PFOA group by 20.8% (P = 0.0827), and in the 1.0 mg/kg PFOA group by 28.5% (P = 0.011) compared to VC males fed LFD (Table S23).
3.7. Liver histopathology and liver enzymes
Liver histopathology was assessed in mice sacrificed at the 18-week timepoint, approximately 4.5 months after the gestational exposure to PFOA or GenX had ended. Representative pictures of the primary diagnoses affected by treatments are portrayed in Fig. 5, with accompanying data shown in Tables S24 and S25. For the HFD mice, there was a significant increase in hepatocyte single cell necrosis in the female 1.0 mg/kg GenX group (P = 0.022) and 1.0 mg/kg PFOA group (P = 0.003) compared to VC (VC liver shown in Fig. 5A; Table S25). The hepatocyte single cell necrosis was characterized by multifocal, single or small clusters of individual swollen, necrotic hepatocytes with associated pyknotic and karyorrhectic debris and neutrophilic inflammation (Fig. 5B). There was also an increase in microvesicular fatty change in LFD mice (shown in Fig. 5C and D), with a significant increase in the male 2.0 mg/kg GenX group (P = 0.015) compared to VC (Table S24). Microvesicular fatty change was characterized by expansion of the hepatocellular cytoplasm with multiple, small, discrete, clear lipid-type vacuoles and was often present in a diffuse pattern when severe but was sometimes only present in centrilobular areas (Fig. 5C and D). Among the LFD group within the 18 Week timepoint, there was a notably low incidence (0%) of microvesicular fatty change within the control group and, as such, all treatment groups appear to have an increased incidence relative to this control group. Although diagnoses in livers of exposed mice were made, no statistically significant differences due to treatment were observed for macrovesicular fatty changes (shown in Fig. 5E) or mixed cell infiltrates (Shown in Fig. 5F; Tables S24–25).
Fig. 5. Examples of liver histopathology in mice at the 18-week timepoint.
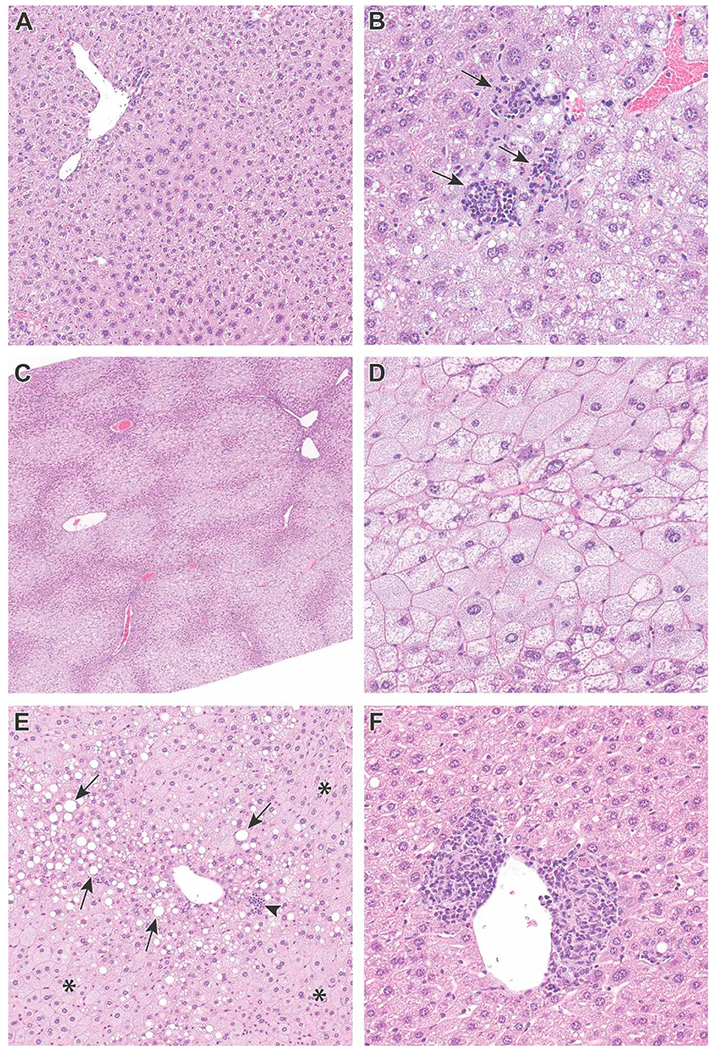
(A) Liver from control male mouse fed LFD (20x). (B) Example of multifocal single cell hepatocyte necrosis in a liver from a 1.0 mg/kg/day GenX exposed male mouse fed HFD (40x). There are three foci of single cell necrosis with associated inflammatory cells (arrows). (C) Example of microvesicular fatty change in a liver from a 2.0 mg/kg/day GenX-exposed male mouse fed LFD (4x). Note the centrilobular and periportal bridging pattern (paler regions). (D) Higher magnification of panel C shows hepatocytes filled with numerous small lipid vacuoles giving a “foamy” appearance (40x). (E) Example of periportal macrovesicular fatty change from a female mouse gestationally exposed to 0.2 mg/kg/day GenX and fed HFD (20x). The affected hepatocytes contain large, well-defined rounded vacuoles, that displace the nucleus and cytoplasm to the periphery (arrows). Microvesicular fatty change (asterisk) and single cell necrosis (arrowhead) are also present. (F) Example of perivascular mixed cell infiltrates in a liver from a female mouse gestationally exposed to 0.2 mg/kg/day GenX and fed HFD (10x). Hematoxylin and eosin. LFD = low fat diet; HFD = high fat diet.
Liver enzymes were also measured at the 18-week timepoint and data are shown in Tables S26–S27. There were no differences between treatment groups and vehicle for aspartate aminotransferase (AST), alanine transaminase (ALT), or alkaline phosphatase (ALP) for males fed HFD (Table S27). ALP was decreased by 32.5% in males fed LFD in the 0.2 mg/kg GenX group compared to vehicle (P = 0.03; Table S27). For HFD females, AST, ALT, and ALP were increased in the 1.0 mg/kg PFOA group by 22.8%, 129.9%, and 42.8% respectively (Table S26). For females fed HFD, ALT and ALP were increased in the 2.0 mg/kg GenX group by 142.8% and 39.3% respectively (Table S26). There were no differences in liver enzymes between treatment groups and vehicle for females fed LFD.
3.8. RT-qPCR analysis of adipose tissues
RT-qPCR analysis was performed on intra-abdominal perigonadal WAT collected at the week 18 necropsy. Sex-specific differentially expressed gene patterns were observed. WAT of GenX-exposed males fed LFD exhibited decreased expression of genes involved in metabolic processes, including acetyl-CoA carboxylase alpha and beta (Acaca, Acacb), adiponectin (Adipoq), fatty acid synthase (Fasn), insulin receptor (Insr), insulin receptor substrate 1 (Irs1), peroxisome proliferator-activated receptor gamma (Pparg), retinoid X receptor alpha (Rxra), and sterol regulatory element-binding factor 1 (Srebf1; Fig. 6). WAT of females fed LFD had significantly increased expression of the estrogen related receptor gamma (Esrrg) in all PFOA and GenX dose groups relative to VC females and elevated Pparg expression in the GenX 1.0 mg/kg dose group (Fig. 6). The glucose transporter 4 (Glut4) was significantly increased (2.4-fold) in the WAT of PFOA 1.0 mg/kg females fed LFD. There were no consistent trends in gene expression for WAT of animals on the HFD (Fig. 6). However, WAT of GenX-exposed males showed a 4-fold increase of estrogen receptor alpha (Esr1) expression while on the HFD, when compared to the HFD VC (adjusted P = 0.099; Fig. 6).
Fig. 6. Heat map of white adipose tissue (WAT) mRNAs altered by PFOA or GenX at 18 weeks of age.
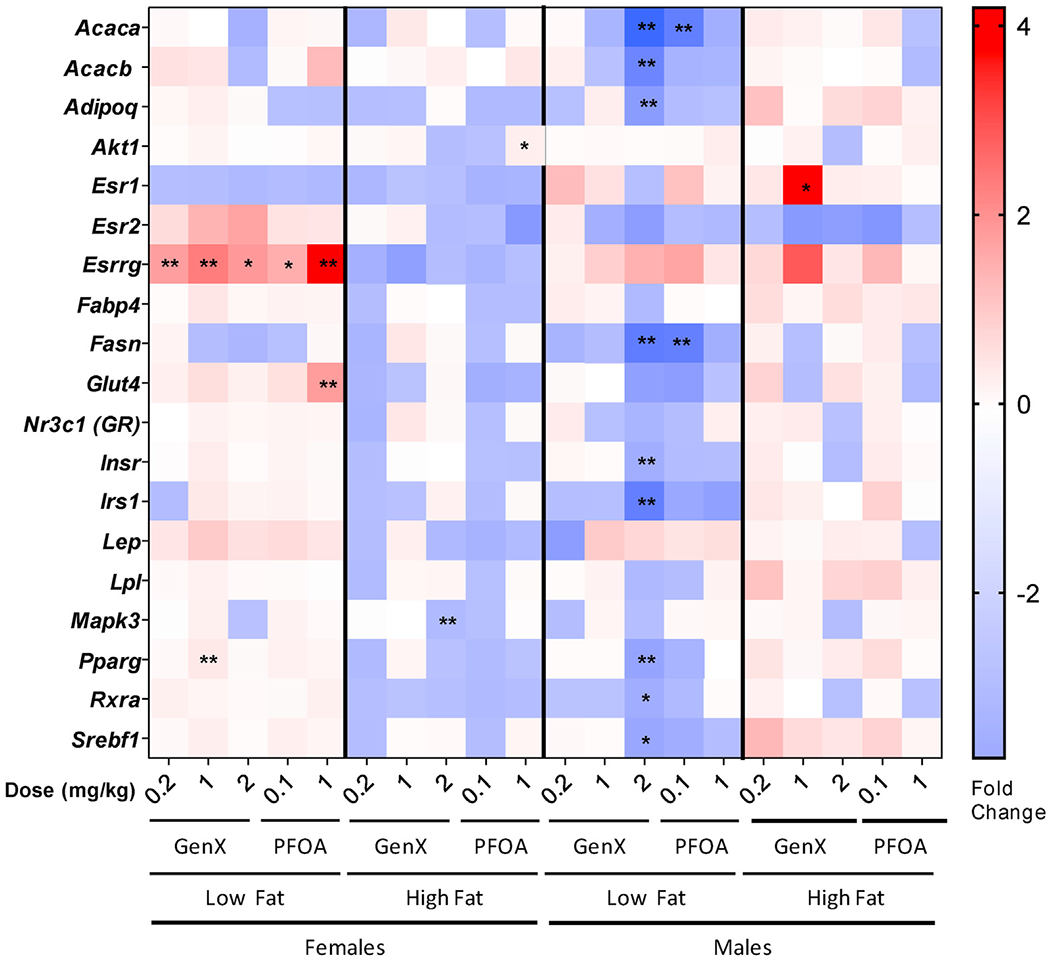
Messenger RNA expression was quantified by RT-qPCR using WAT from 18-week old mice prenatally exposed to either PFOA or GenX. Relative gene expression data are represented as mean fold change over VC in heatmap format, with red indicating an increase expression up to a 4-fold change and blue indicating a decrease in expression down to a 4-fold change, N = 4. Low fat and high fat denote the different diets. ANOVA with multiple comparisons Dunnett’s post hoc was performed on mean dCt values with statistical significance of *P < 0.1 and **P < 0.05.
3.9. Effect of diet
The above results were focused on the effect of the exposures within the diet groups. However, we also observed expected effects of HFD compared to LFD within vehicle control animals. HFD significantly increased mean body weights of both VC male and female mice compared with LFD VC mice at 18 weeks (P < 0.001; Tables S11–12). VC mice fed HFD exhibited altered lipid panels (e.g. increased TC, HDL, and HDL:LDL ratio and decreased triglycerides) relative to VC mice fed LFD of the same sex, a trend that persisted across observed timepoints (Tables S14–17). Although not yet apparent at the 9-week timepoint, glucose tolerance AUC increased in response to HFD at the 14-week timepoint for both VC males and females (P < 0.01; Tables S19–20). The body mass composition analysis at both 12 and 17 weeks indicated that the increase in mean body weight in response to HFD was due mainly to increased fat mass (P < 0.001), but there were also increases in fluid mass in both sexes in response to HFD (P < 0.001; Tables S20–23). Finally, patterns in gene expression in white adipose tissue were also shifted in response to HFD and appeared to differ by sex (Fig. 6).
4. Discussion
In this study we demonstrated for the first time that prenatal exposure to GenX, a replacement chemical for PFOA and an emerging contaminant of water systems across the globe [17–19], induces adverse, sex-specific, phenotypes in mouse offspring fed a typical fixed-formula chow. More specifically, male offspring exhibited adverse metabolic phenotypes when gestationally exposed to GenX and fed LFD, and females had evidence of liver damage at the histological and functional level. Both sexes showed some evidence of both types of phenotypes, but in adulthood the varied sex-specific manifestations were evident. Our previous work suggested that prenatal exposure to low levels of PFOA led to metabolic alterations in adult female mice, but lacked evaluation of both sexes [8] and was conducted on a variable-formula diet, meaning that phytoestrogen content of the diet could have varied between our previous work and present study. In this study, we have identified several latent adverse outcomes associated with prenatal exposure to PFOA and its contemporary replacement, GenX, in mice.
In this study we report three key novel findings [1]: GenX persists in offspring after birth, suggesting both transplacental and lactational transfer [2], prenatal PFOA or GenX exposure induces adverse metabolic outcomes in adult male offspring, and [3] prenatal PFOA or GenX induces liver damage in female and male offspring.
GenX was selected as a replacement chemical for PFOA due in part to its more rapid elimination from the body. In mice, GenX has a half-life of about 20 h, whereas PFOA has a half-life of 17–19 days [47,48]. GenX similarly has a shorter estimated serum half-life in humans of approximately 81 hours, while the human serum half-life of PFOA is approximately 3 years (Registration Dossier - ECHA (europa.eu); study 002) [47,49,50]. A shorter half-life is thought to impart a more favorable toxicological profile through quick elimination and, presumably, fewer adverse health outcomes. Our recent work has shown that GenX and PFOA are transplacentally transferred to mouse embryos under the same exposure conditions used in this study [31]. We have also shown that milk is a substantial contributor to PFOA exposure in mouse offspring from exposed dams [51], though lactational transfer has not previously been shown for GenX. Based on the previously reported half-life of approximately 20 h for GenX in mice, we did not expect to detect GenX in pups at PND 5.5 [31]. However, we observed GenX in both the serum and the urine of pups at PND 5.5, six days (approximately 7.2 half-lives) after the last dose was administered to the dams on the morning of GD 17.5. These findings suggest altered elimination kinetics in the maternal:fetal unit (significantly slower than anticipated) and/or support the hypothesis that like PFOA, GenX is transferred to the pups via lactation, and a combination of the load of GenX obtained as an embryo and continual dosing through the milk results in the presence of GenX in the pups at PND 5.5 [52]. This finding is concerning for human health, as PFOA and other PFAS have been shown to be excreted via lactation in humans, resulting in increased serum PFAS in children with extended months of breastfeeding [53,54]. Future studies should confirm the presence of GenX in milk for both mice and human cohorts to better understand the risk profile of this compound. Multiple studies have reported the effects of several legacy PFAS on lactation duration/nursing insufficiency in women [55–57], but GenX exposure/excretion data in women does not exist. As little is known about the transport and excretion of GenX in the adult body, further evaluation of the half-life of GenX in offspring may be needed. The possibility exists that the newborn/infant and adult vary in their ability to metabolize and/or eliminate this chemical.
A major concern within the field of PFAS research is consistent reports of PFAS on reduced birth weight. Low weight at birth and in early life has been associated with the development of metabolic syndrome later in life in humans and mouse models [5,58,59]. The Navigation Guide, a standardized method for conducting robust systematic reviews, was applied to both mouse and human cohort studies and concluded that prenatal exposure to PFOA results in reduced birth weight in both mice and humans [23,60,61]. In this study, PFOA doses were chosen (high dose of 1 mg/kg/day) that were not previously associated with a significant decrease in birth weight [8,60]. We did not find significant reductions in PND 0.5 litter weight in any dose group when litter weight was averaged (PFOA or GenX). However, at PND 5.5 we found that individual pup body weight was significantly reduced in the 1.0 mg/kg GenX and 2.0mg/kg GenX groups. This result was apparent when we statistically controlled for litter size, which is a critical consideration due to the inverse relationship between litter size and pup weight in rodents [62]. These findings are consistent with reduced pup weights previously reported for higher doses of PFOA [60] and GenX [37,63]. In the report, CD-1 mice exposed to 5 mg/kg/day HFPO-DA (GenX) from preconception through weaning showed more severe effects than herein, resulting in reduced pup weight from PND 1–21, and more severe effects in male offspring [63]. Another study has found that prenatal exposure to HFPO-DA results in altered expression of genes involved in glucose metabolism in offspring both directly after birth and at PND20 [64]. These findings, along with the increases in MetS-associated phenotypes seen in these groups later in life, suggest developmental exposure to GenX results in lifelong alterations in metabolic function, making metabolic disease a concern for both GenX and PFOA exposures [5]. Metabolic programming in response to chemical exposure has previously been documented with bisphenol A(BPA) and phthalates through various proposed mechanisms including epigenetic regulation [65–68].
Several environmental contaminants have been classified as obesogens due to their ability to disrupt adipocyte function and contribute to the development of obesity [69]. Animal models have previously been used to understand the effect of prenatal exposure to obesogens, such as BPA, on metabolic outcomes [70]. Prenatal exposure to BPA in CD-1 mice induced increased weight gain, reduced glucose tolerance, increased abdominal fat mass, and increased insulin levels in male offspring [71]. Recent work evaluating the effect of ozone exposure during gestation on offspring in rats found that programming of metabolic pathways in the fetal liver is impaired in males, but not females [72]. Increased sensitivity in males compared to females has also been well documented in toxicology studies [73–75]. Here we similarly observed increased susceptibility of male mouse offspring to a suite of adverse metabolic outcomes following developmental PFOA and especially GenX exposures.
In LFD males exposed to 2.0 mg/kg GenX, we observed consistent alterations in metabolic endpoints that worsened with time. Weight gain was increased in this group from PND 22 to age 18 weeks relative to controls, suggesting that a prenatal exposure to GenX sensitizes male offspring to higher weight gain across their lifespan. Additionally, males fed LFD in the 2.0 mg/kg GenX group also showed significant alterations in body mass composition compared to LFD male controls, indicated by an increased ratio of fat:lean mass. Taken together, these findings suggest that excess weight gain in GenX-exposed males was driven by increased body fat mass, unrelated to the increase in liver weight. An additional contributor to the excess weight gain in the GenX- and PFOA-exposed males, which is a novel finding in both this study and our previous work, is excess fluid retention [31]. Blake et al. [31] reported fluid retention in the abdomen of pregnant dams at term necropsy and here we found male offspring with the same exposures exhibited fluid retention measured by magnetic resonance imaging. This consistent finding deserves further study and we hypothesize that this may indicate hypertension onset in both cases, another commonly reported endpoint in humans with metabolic disease. A growing body of research has indicated circulating biomarkers in the hypertensive pregnant dam may be passed to the developing fetus and affect birthweight and offspring health [76–78].
There is a strong literature base demonstrating excess weight gain in children and young adults developmentally exposed to PFAS [21,22,24–26] and a recent report confirms associations between visceral fat depot size and developmental PFAS exposures [27]. In addition to increased weight gain and greater fat mass, male mice in our study exposed to GenX and fed LFD exhibited further signs of persistent metabolic disease as adults, including elevated serum cholesterol and insulin levels. Total cholesterol was increased in all GenX groups of males fed LFD at Week 6, relative to LFD vehicle control males. Cholesterol and HDL remained elevated in LFD male mice exposed to GenX (1 mg/kg) at 18 weeks of age compared to LFD VC mice. Additionally, male mice fed LFD and exposed to 2.0 mg/kg GenX or 1.0 mg/kg PFOA were insulin resistant (according to QUICKI calculation) in comparison to LFD VC males. Increased serum insulin has been reported previously in female mice prenatally exposed to PFOA [8], and cholesterol is a reported target of PFAS chemicals in rodent models and human populations [32]. Serum triglycerides, another indicator of overall metabolic health, were significantly decreased in weanling mice exposed to all PFOA and GenX doses compared to appropriate VC, but this shift was transient and attenuated over time, possibly indicating adaptation by the mice. PFOA- and GenX-exposed LFD female mice at 6 weeks still had significantly reduced triglyceride levels, but by 18 weeks of age, that effect was gone. Glucose tolerance tests did not reveal any differences in the ability to process glucose which could be due to the large variation in weight of the animals within the groups, a common occurrence in outbred strains such as CD-1. Our previous work using transmission electron microscopy demonstrated GenX induced depletion of glycogen stores in the liver of pregnant mice [31], and recent work reported newborn rat pups exposed in utero to GenX as “hypoglycemic at birth” due to observed decrease in serum glucose in neonatal rats prenatally exposed to GenX relative to controls [64]. Therefore, even though glucose tolerance was not altered, effects of emerging PFAS on insulin sensitivity should be evaluated in future studies by using an inbred strain (though the strain would need to have a comparable sensitivity to PFOA and GenX), or conducting experiments in mice at older ages previously shown to have altered glucose sensitivity in response to PFOA [8].
An important health effect associated with PFAS exposure in humans that has been recapitulated in both in vitro and in vivo experimental models is fatty liver, or steatosis [32]. Blake et al. [31] recently reported significant fatty liver effects of PFOA and GenX in sectioned livers of adult-dosed pregnant CD-1 mice, and this has been previously reported in livers of male CD-1 mice exposed in utero to 1 mg/kg PFOA [79]. Although GenX was no longer present in the pups at weaning in the present study, there was a significant increase in hepatic microvesicular fatty change in males (LFD, 2 mg/kg GenX) and an increase in individual hepatocyte necrosis in females (HFD, 1 mg/kg GenX and 1.0 mg/kg PFOA) in the 18 week LFD groups compared to VC. Non-alcoholic fatty liver disease is associated with risk of MetS in humans, and known to alter metabolic processes in the liver [80,81]. Taken together, these progressive, persistent, and sometimes dose-dependent physiological changes suggest that developmental exposure to PFOA or GenX enhanced metabolic disease in male mice and led to liver damage in female and male mice. We hypothesize that those effects were programmed early in development as the chemical exposure ceased shortly after birth.
We did not observe increased weight gain, altered body mass composition, elevated cholesterol, or insulin resistance in GenX-exposed males fed HFD. It is possible the effects of long-term HFD on these endpoints masked our ability to detect treatment-specific effects. In many cases, the effects of PFAS on adverse metabolic and liver outcomes mirrored those of HFD alone [82]. We hypothesize that in our study, the effect of the HFD contributed more to the alteration of these phenotypes than the PFAS exposure, making it difficult to distinguish the PFAS related changes from diet related changes when combined. Another study has shown that prolonged exposure to legacy PFAS in adult mice led to altered triglyceride and cholesterol content in the liver, which was exacerbated by a high fat, high carb diet [83]. While this confirms the importance of diet in long-term adult exposure studies on metabolic outcomes, the evidence for role of diet following a prenatal exposure of PFAS is still unclear. The interaction between dietary fats and developmental exposure to PFAS, including emerging compounds like GenX, should be investigated in future studies to determine if diet later in life influences susceptibility to or exacerbation of adverse metabolic outcomes.
Gene expression data from WAT of exposed mice in these studies supports the hypothesis that prenatal exposure to GenX induces a suite of latent metabolic outcomes and endocrine system disorder. Male mice fed LFD in the 2.0 mg/kg GenX group had reductions in Acaca, Acacb, Adipoq, Fasn, Insr, Irs1, Pparg, Rxra, and Srebfl mRNA expression, relative to LFD VC males. Decreases in Adipoq [84], Insr [85], Pparg [86], and Irs1 [87] gene expression have been associated with altered glucose metabolism. Decreased protein levels of INSR [88], ADIPOQ [89], and IRS1 [87] in mice are associated with insulin resistance and increased insulin levels in the blood. In our study, we report a ~7-fold increase in blood insulin over vehicle control in the GenX-exposed group. Based on these findings and the consistency of other studies reporting similar findings related to insulin sensitivity following PFOA exposure in mice and children [8,90](64), it may be that the serum insulin increases (highest in the same dose group where we observed the most dramatic WAT gene changes) affected feedback mechanisms in the WAT. These pathways should be interrogated in future studies to further understand how related proteins are altered.
Furthermore, our qPCR results corroborate the serum lipid data for the 2.0 mg/kg GenX group fed LFD, which also exhibited increased TC and HDL levels. Decreased WAT expression of Insr [91], Rxra [92], and Srebf1 [93] are all associated with increased circulating concentrations of cholesterol. Additionally, decreased Pparg [94], Insr [95], Acacb [96], and Irs1 [87] are linked to dyslipidemia, which again is consistent with the serum lipid data for this group. Finally, the sex-specific increase in expression of estrogen-regulated and related genes in WAT of female offspring exposed to PFOA and GenX deserves further research.
Currently, the North Carolina state health goal of 140 ppt in drinking water for GenX relies on a Reference Dose (RfD) derived from a no-observed-adverse-effect-level (NOAEL) of 0.1 mg/kg/day based on the critical effect of liver single-cell necrosis in adult male mice (0/10 mice, 0%) after a 28-day subchronic exposure [97,98]. The incidence of liver single-cell necrosis was 40% (4/10 mice) in the 3 mg/kg/day GenX dose group [97]. In our study, we observed a liver single-cell necrosis incidence of 38.9% (7/18 mice, males and females combined) in offspring exposed for 16 days in utero to 0.2 mg/kg/day GenX, nearly double the incidence in controls (3/17 mice, 17.6%; Supplemental Table 24). Given the regulatory significance of effects like liver single-cell necrosis, future studies should evaluate low-dose effects using larger sample sizes for increased statistical power.
In conclusion, we have identified several adverse outcomes associated with metabolic disease that were caused by prenatal exposure to GenX and/or PFOA. These consistent metabolic effects were most prominent in male mice exposed to the PFOA replacement, GenX. Liver effects were predominantly observed in female mice exposed to GenX or PFOA. We have shown that GenX persists in offspring after birth, suggesting transplacental and lactational transfer. These exposure routes are human relevant and suggest that prenatal exposure to GenX may cause metabolic disease even when it cannot be measured in the serum or urine of the exposed individuals. We have also shown that GenX exposure results in adverse, sex-specific phenotypes. In male mice, we found that prenatal GenX exposure led to increased weight gain, fat mass, retained water weight, fatty liver changes, and decreased insulin sensitivity at exposures as low as 1 mg/kg. At the 1 and 2 mg/kg doses of GenX, female mice displayed signs of liver damage including hepatocyte single cell necrosis, and elevated ALP and ALT. This evidence suggests that GenX, a PFOA-replacement chemical that is of high concern due to its widespread environmental contamination, causes adverse metabolic health effects that may occur at lower internal doses than PFOA-exposed groups, and may be unique compared to those effects caused by PFOA in some cases. GenX may not be a safe alternative for PFOA.
Supplementary Material
Acknowledgements
This research was funded by NIEHS/DNTP (Z01-ES102785 [S.E.F.]). The authors acknowledge the excellent assistance of the NIEHS animal care team, especially Louise Harris (dosing). We also thank Debra King and Ralph Wilson for assistance in processing serum/clinical chemistry, and Beth Mahler for producing our pathology Fig. 5. We appreciate the time and constructive comments from Dr. Mimi Huang (DNTP) and Dr. Colette Miller (US EPA) that were submitted in NIEHS and EPA clearance of this manuscript, as well as the time and training of Dr. Keith Shockley in statistical methodology (helped train B.B.). The research presented was not performed or funded by EPA and was not subject to EPA’s quality system requirements. The views expressed are those of the author(s) and do not necessarily represent the views or the policies of the U.S. Environmental Protection Agency.
Abbreviations:
- PFOA
Perfluorooctanoic Acid
- HFPO-DA/GenX
Hexafluoropropylene oxide-dimer acid
- PFAS
Per- and Polyfluoroalkyl substance
- MetS
Metabolic Syndrome
- HDL
High Density Lipoprotein Cholesterol
- LDL
Low Density Lipoprotein Cholesterol
- CFR
Cape Fear River
- GD
Gestational Day
- PND
Postnatal Day
- AUC
Area Under the Curve
- CV
Coefficient of Variation
- QUICKI
Quantitative insulin-sensitivity check index
- VC
Vehicle Control
- TC
Total Cholesterol
Footnotes
Declaration of competing interest
The authors report no conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.emcon.2021.10.004.
References
- [1].Hales CM, Prevalence of obesity and severe obesity among adults, United States (360) (2020) 8, 2017–2018. [PubMed] [Google Scholar]
- [2].Samson Metabolic, Syndrome | elsevier enhanced reader, Internet, https://reader.elsevier.com/reader/sd/pii/S0889852913001084?token=0819074B4FE565598A26B58375F508F249F68E5434FFE2F07C09862A86B6CC924807EAFEAE5964BE12C50911EBA1F27B. (Accessed 21 January 2020). Available from.
- [3].Ogden CL, Carroll MD, Kit BK, Flegal KM, Prevalence of obesity and trends in body mass index among US children and adolescents, 1999-2010, J. Am. Med. Assoc 307 (5) (2012. Feb 1) 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bruce KD, Hanson MA, The developmental origins, mechanisms, and implications of metabolic syndrome, J. Nutr 140 (3) (2010. Mar 1) 648–652. [DOI] [PubMed] [Google Scholar]
- [5].Barker DJP, The developmental origins of chronic adult disease, Acta Paediatr Oslo Nor 93 (446) (2004. Dec) 26–33, 1992 Suppl. [DOI] [PubMed] [Google Scholar]
- [6].Desai M, Jellyman JK, Ross MG, Epigenomics, gestational programming and risk of metabolic syndrome, Int. J. Obes 39 (4) (2015. Apr) 633–641. [DOI] [PubMed] [Google Scholar]
- [7].de Rooij SR, Painter RC, Holleman F, Bossuyt PM, Roseboom TJ, The metabolic syndrome in adults prenatally exposed to the Dutch famine, Am. J. Clin. Nutr 86 (4) (2007. Oct) 1219–1224. [DOI] [PubMed] [Google Scholar]
- [8].Hines EP, White SS, Stanko JP, Gibbs-Flournoy EA, Lau C, Fenton SE, Phenotypic dichotomy following developmental exposure to perfluorooctanoic acid (PFOA) in female CD-1 mice: low doses induce elevated serum leptin and insulin, and overweight in mid-life, Mol. Cell. Endocrinol 304 (1–2) (2009. May 25) 97–105. [DOI] [PubMed] [Google Scholar]
- [9].Interstate Technology and Regulatory Council (ITRC), Naming conventions and physical and chemical properties of per-and polyfluoroalkyl substances (pfas) [Internet]. [cited 2020 Apr 30]. Available from: https://pfas-1.itrcweb.org/fact-sheets/.
- [10].Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Tully JS, Needham LL, Serum concentrations of 11 polyfluoroalkyl compounds in the u.s. population: data from the national health and nutrition examination survey (NHANES), Environ. Sci. Technol 41 (7) (2007. Apr 1) 2237–2242. [DOI] [PubMed] [Google Scholar]
- [11].Domingo JL, Nadal M, Human exposure to per- and polyfluoroalkyl substances (PFAS) through drinking water: a review of the recent scientific literature, Environ. Res 177 (2019. Oct) 108648. [DOI] [PubMed] [Google Scholar]
- [12].Hanssen L, Dudarev AA, Huber S, Odland Jø, Nieboer E, Sandanger TM, Partition of perfluoroalkyl substances (PFASs) in whole blood and plasma, assessed in maternal and umbilical cord samples from inhabitants of arctic Russia and Uzbekistan, Sci. Total Environ 447 (2013. Mar 1) 430–437. [DOI] [PubMed] [Google Scholar]
- [13].Olsen GW, Mair DC, Lange CC, Harrington LM, Church TR, Goldberg CL, et al. , Per- and polyfluoroalkyl substances (PFAS) in American Red Cross adult blood donors, 2000-2015, Environ. Res 157 (2017) 87–95. [DOI] [PubMed] [Google Scholar]
- [14].Olsen Geary W, Burris Jean M, Ehresman David J, Froehlich John W, Seacat Andrew M, Butenhoff John L, et al. , Half-life of serum elimination of Perfluorooctanesulfonate,Perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers, Environ. Health Perspect 115 (9) (2007. Sep 1) 1298–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].US EPA O, Risk Management for Per- and Polyfluoroalkyl Substances (PFAS) under TSCA, Internet, US EPA, 2015. Available from, https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/risk-management-and-polyfluoroalkyl-substances-pfas. (Accessed 7 April 2020). [Google Scholar]
- [16].Agency for Toxic Substances and Disease Registry (ATSDR), Toxicological profile for perfluoroalkyls, Internet, https://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=1117&tid=237. (Accessed 30 April 2020). Available from. [PubMed]
- [17].Strynar M, Dagnino S, McMahen R, Liang S, Lindstrom A, Andersen E, et al. , Identification of novel perfluoroalkyl ether carboxylic acids (PFECAs) and sulfonic acids (PFESAs) in natural waters using accurate mass time-of-flight mass spectrometry (TOFMS), Environ. Sci. Technol 49 (19) (2015. Oct 6) 11622–11630. [DOI] [PubMed] [Google Scholar]
- [18].Li J, He J, Niu Z, Zhang Y, Legacy per- and polyfluoroalkyl substances (PFASs) and alternatives (short-chain analogues, F-53B, GenX and FC-98) in residential soils of China: present implications of replacing legacy PFASs, Environ. Int 135 (2020. Feb 1) 105419. [DOI] [PubMed] [Google Scholar]
- [19].Brandsma SH, Koekkoek JC, van Velzen MJM, de Boer J, The PFOA substitute GenX detected in the environment near a fluoropolymer manufacturing plant in The Netherlands, Chemosphere 220 (2019. Apr 1) 493–500. [DOI] [PubMed] [Google Scholar]
- [20].Heydebreck F, Tang J, Xie Z, Ebinghaus R, Alternative and legacy perfluoroalkyl substances: differences between European and Chinese river/estuary systems, Environ. Sci. Technol 49 (14) (2015. Jul 21) 8386–8395. [DOI] [PubMed] [Google Scholar]
- [21].Braun JM, Chen A, Romano ME, Calafat AM, Webster GM, Yolton K, et al. , Prenatal perfluoroalkyl substance exposure and child adiposity at 8 years of age: the HOME study, Obes Silver Spring Md 24 (1) (2016. Jan) 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Høyer BB, Ramlau-Hansen CH, Vrijheid M, Valvi D, Pedersen HS, Zviezdai V, et al. , Anthropometry in 5- to 9-year-old Greenlandic and Ukrainian children in relation to prenatal exposure to perfluorinated alkyl substances, Environ. Health Perspect 123 (8) (2015. Aug) 841–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Johnson PI, Sutton P, Atchley DS, Koustas E, Lam J, Sen S, et al. , The navigation guide—evidence-based medicine meets environmental health: systematic review of human evidence for PFOA effects on fetal growth, Environ. Health Perspect 122 (10) (2014. Oct) 1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Manzano-Salgado CB, Casas M, Lopez-Espinosa M-J, Ballester F, Iñiguez C, Martinez D, et al. , Prenatal exposure to perfluoroalkyl substances and cardiometabolic risk in children from the Spanish INMA birth cohort study, Environ. Health Perspect 125 (9) (2017. 20), 097018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Starling AP, Adgate JL, Hamman RF, Kechris K, Calafat AM, Ye X, et al. , Perfluoroalkyl substances during pregnancy and offspring weight and adiposity at birth: examining mediation by maternal fasting glucose in the healthy start study, Internet], Environ. Health Perspect 125 (6) (2017. Jun 26). Available from, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5743451/. (Accessed 30 April 2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Halldorsson TI, Rytter D, Haug LS, Bech BH, Danielsen I, Becher G, et al. , Prenatal exposure to perfluorooctanoate and risk of overweight at 20 years of age: a prospective cohort study, Environ. Health Perspect 120 (5) (2012. May) 668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Janis JA, Rifas-Shiman SL, Seshasayee SM, Sagiv S, Calafat AM, Gold DR, et al. , Plasma concentrations of per- and polyfluoroalkyl substances and body composition from mid-childhood to early adolescence, Internet, J. Clin. Endocrinol. Metab (2021. Mar 19), 10.1210/clinem/dgab187(dgab187). Available from. (Accessed 25 March 2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wolf CJ, Fenton SE, Schmid JE, Calafat AM, Kuklenyik Z, Bryant XA, et al. , Developmental toxicity of perfluorooctanoic acid in the CD-1 mouse after cross-foster and restricted gestational exposures, Toxicol Sci Off J Soc Toxicol 95 (2) (2007. Feb) 462–473. [DOI] [PubMed] [Google Scholar]
- [29].van Esterik Jcj, Sales LB, Dollé MET, Håkansson H, Herlin M, Legler J, et al. , Programming of metabolic effects in C57BL/6JxFVB mice by in utero and lactational exposure to perfluorooctanoic acid, Arch. Toxicol 90 (3) (2016. Mar 1) 701–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Conley JM, Lambright CS, Evans N, Strynar MJ, McCord J, McIntyre BS, et al. , Adverse maternal, fetal, and postnatal effects of hexafluoropropylene oxide dimer acid (GenX) from oral gestational exposure in sprague-dawley rats, Internet, Environ. Health Perspect 127 (3) (2019. Mar 28). Available from, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6768323/. (Accessed 3 January 2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Blake BE, Cope HA, Hall SM, Keys RD, Mahler BW, McCord J, et al. , Evaluation of maternal, embryo, and placental effects in CD-1 mice following gestational exposure to perfluorooctanoic acid (PFOA) or hexafluoropropylene oxide dimer acid (HFPO-da or GenX), Environ. Health Perspect 128 (2) (2020. Feb), 27006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fenton SE, Ducatman A, Boobis A, DeWitt JC, Lau C, Ng C, et al. , Per- and polyfluoroalkyl substance toxicity and human health review: current state of knowledge and strategies for informing future research, Environ. Toxicol. Chem (2020. Oct 5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lau C, Thibodeaux JR, Hanson RG, Narotsky MG, Rogers JM, Lindstrom AB, et al. , Effects of perfluorooctanoic acid exposure during pregnancy in the mouse, Toxicol Sci Off J Soc Toxicol 90 (2) (2006. Apr) 510–518. [DOI] [PubMed] [Google Scholar]
- [34].UEJEDN., Drinking Water Health Advisory for Perfluorooctanoic Acid (PFOA), 2016, 103. [Google Scholar]
- [35].D12450B Formula, OpenSource diets - search formulas - research diets, Inc, Internet, https://researchdiets.com/formulas/d12450B. (Accessed 30 April 2020). Available from.
- [36].D12492 Formula, OpenSource diets - search formulas - research diets, Inc, Internet, https://researchdiets.com/formulas/d12492. (Accessed 30 April 2020). Available from.
- [37].Conley JM, Lambright CS, Evans N, Strynar MJ, McCord J, McIntyre BS, et al. , Adverse maternal, fetal, and postnatal effects of hexafluoropropylene oxide dimer acid (GenX) from oral gestational exposure in sprague-dawley rats, Environ. Health Perspect 127 (3) (2019), 37008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].McCord J, Newton S, Strynar M, Validation of quantitative measurements and semi-quantitative estimates of emerging perfluoroethercarboxylic acids (PFECAs) and hexfluoroprolyene oxide acids (HFPOAs), J. Chromatogr. A 1551 (2018. May 25) 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Reiner JL, Nakayama SF, Delinsky AD, Stanko JP, Fenton SE, Lindstrom AB, et al. , Analysis of PFOA in dosed CD1 mice: Part 1. Methods development for the analysis of tissues and fluids from pregnant and lactating mice and their pups, Reprod. Toxicol 27 (3) (2009. Jun 1) 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rushing BR, Hu Q, Franklin JN, McMahen R, Dagnino S, Higgins CP, et al. , Evaluation of the immunomodulatory effects of 2,3,3,3-tetrafluoro-2-(heptafluoropropoxy)-propanoate in C57BL/6 mice, Toxicol Sci Off J Soc Toxicol (2017. Jan 23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sakaguchi K, Takeda K, Maeda M, Ogawa W, Sato T, Okada S, et al. , Glucose area under the curve during oral glucose tolerance test as an index of glucose intolerance, Diabetol Int 7 (1) (2015. May 14) 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Katz A, Nambi SS, Mather K, Baron AD, Follmann DA, Sullivan G, et al. , Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans, J. Clin. Endocrinol. Metab 85 (7) (2000. Jul 1) 2402–2410. [DOI] [PubMed] [Google Scholar]
- [43].Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods San Diego Calif 25 (4) (2001. Dec) 402–408. [DOI] [PubMed] [Google Scholar]
- [44].Jonckheere AR, A distribution-free k-sample test against ordered alternatives, Biometrika 41 (1/2) (1954) 133–145. [Google Scholar]
- [45].Holm S, A simple sequentially rejective multiple test procedure, Scand.J. Stat 6 (2) (1979) 65–70. [Google Scholar]
- [46].RVAideMemoire, Internet, https://cran.r-project.org/web/packages/RVAideMemoire/RVAideMemoire.pdf. (Accessed 20 May 2020). Available from.
- [47].Sa G, Wj F, Mp M, Dl N, Rc B, Lw B, et al. , Absorption, distribution, metabolism, excretion, and kinetics of 2,3,3,3-tetrafluoro-2-(heptafluoropropoxy) propanoic acid ammonium salt following a single dose in rat, mouse, and cynomolgus monkey [Internet], Toxicology 340 (2016) [cited 2020 May 20]. Available from: https://pubmed.ncbi.nlm.nih.gov/26743852/. [DOI] [PubMed] [Google Scholar]
- [48].Lau C, Perfluoroalkyl acids: recent research highlights, Reprod. Toxicol 33 (4) (2012. Jul 1)405–409. [DOI] [PubMed] [Google Scholar]
- [49].Li Y, Fletcher T, Mucs D, Scott K, Lindh CH, Tallving P, et al. , Half-lives of PFOS, PFHxS and PFOA after end of exposure to contaminated drinking water, Occup. Environ. Med 75 (1) (2018) 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Charles River Laboratories Den Bosch BV, DuPont-C30031_516655: Determination of HFPO-DA in EDTA Human Plasma Samples, Wilmington, DE: The Chemours Company, 2017. [Google Scholar]
- [51].Fenton SE, Reiner JL, Nakayama SF, Delinsky AD, Stanko JP, Hines EP, et al. , Analysis of PFOA in dosed CD-1 mice. Part 2: disposition of PFOA in tissues and fluids from pregnant and lactating mice and their pups, Reprod Toxicol Elmsford N 27 (3–4) (2009. Jun) 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pizzurro DM, Seeley M, Kerper LE, Beck BD, Interspecies differences in perfluoroalkyl substances (PFAS) toxicokinetics and application to health-based criteria, Regul Toxicol Pharmacol RTP 106 (2019. Aug) 239–250. [DOI] [PubMed] [Google Scholar]
- [53].Mondal D, Weldon RH, Armstrong BG, Gibson LJ, Lopez-Espinosa M-J, Shin H-M, et al. , Breastfeeding: a potential excretion route for mothers and implications for infant exposure to perfluoroalkyl acids, Environ. Health Perspect 122 (2) (2014. Feb) 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kärrman A, Ericson I, van Bavel B, Darnerud PO, Aune M, Glynn A, et al. , Exposure of perfluorinated chemicals through lactation: levels of matched human milk and serum and a temporal trend, 1996–2004, in Sweden, Environ. Health Perspect 115 (2) (2007. Feb) 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Romano ME, Xu Y, Calafat AM, Yolton K, Chen A, Webster GM, et al. , Maternal serum perfluoroalkyl substances during pregnancy and duration of breastfeeding, Environ. Res 149 (2016) 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rosen EM, Brantsæter AL, Carroll R, Haug LS, Singer AB, Zhao S, et al. , Maternal plasma concentrations of per- and polyfluoroalkyl substances and breastfeeding duration in the Norwegian mother and child cohort, Environ Epidemiol Phila Pa 2 (3) (2018. Sep) e027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Timmermann CAG, Budtz-Jørgensen E, Petersen MS, Weihe P, Steuerwald U, Nielsen F, et al. , Shorter duration of breastfeeding at elevated exposures to perfluoroalkyl substances, Reprod Toxicol Elmsford N 68 (2017) 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Cn H, Dj B, The Thrifty Phenotype Hypothesis [Internet], Vol. 60, British medical bulletin, Br Med Bull, 2001. [cited 2020 May 20]. Available from: https://pubmed.ncbi.nlm.nih.gov/11809615/?from_term=barker+hypothesis&from_pos=6. [Google Scholar]
- [59].Beauchamp B, Ghosh S, Dysart M, Kanaan GN, Chu A, Blais A, et al. , Low birth weight is associated with adiposity, impaired skeletal muscle energetics, and weight loss resistance in mice, 2005, Int. J. Obes 39 (4) (2015. Apr) 702–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Koustas E, Lam J, Sutton P, Johnson PI, Atchley DS, Sen S, et al. , The navigation guide—evidence-based medicine meets environmental health: systematic review of nonhuman evidence for PFOA effects on fetal growth, Environ. Health Perspect 122 (10) (2014. Oct) 1015–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lam J, Koustas E, Sutton P, Johnson PI, Atchley DS, Sen S, et al. , The navigation guide—evidence-based medicine meets environmental health: integration of animal and human evidence for PFOA effects on fetal growth, Environ. Health Perspect 122 (10) (2014. Oct) 1040–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Epstein HT, The effect of litter size on weight gain in mice, J. Nutr 108 (1) (1978. Jan) 120–123. [DOI] [PubMed] [Google Scholar]
- [63].DuPont-18405-1037 EI du P de N and C. An Oral (Gavage) Reproduction Developmental Toxicity Screening Study of H-28548 in Mice. USEPA OPPTS 870.3550; OECD Test Guideline 421. Study conducted by WIL Research Laboratories, LLC, 2010. Dec 29. [Google Scholar]
- [64].Conley JM, Lambright CS, Evans N, McCord J, Strynar MJ, Hill D, et al. , Hexafluoropropylene oxide-dimer acid (HFPO-DA or GenX) alters maternal and fetal glucose and lipid metabolism and produces neonatal mortality, low birthweight, and hepatomegaly in the Sprague-Dawley rat, Environ. Int 146 (2020. Oct 27) 106204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Singh S, Li SS-L, Epigenetic effects of environmental chemicals bisphenol A and phthalates, Int. J. Mol. Sci 13 (8) (2012) 10143–10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Qin T, Zhang X, Guo T, Yang T, Gao Y, Hao W, et al. , Epigenetic alteration shaped by the environmental chemical bisphenol A, Front. Genet 11 (2020) 618966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Van Cauwenbergh O, Di Serafino A, Tytgat J, Soubry A, Transgenerational epigenetic effects from male exposure to endocrine-disrupting compounds: a systematic review on research in mammals, Clin. Epigenet 12 (1) (2020. May 12) 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kim S, Thapar I, Brooks BW, Epigenetic changes by per- and polyfluoroalkyl substances (PfAS), 1987, Environ Pollut Barking Essex 279 (2021. Mar 15), 116929. [DOI] [PubMed] [Google Scholar]
- [69].Darbre PD, Endocrine disruptors and obesity, Curr Obes Rep 6 (1) (2017) 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Egusquiza RJ, Blumberg B, Environmental Obesogens and Their Impact on Susceptibility to Obesity: New Mechanisms and Chemicals, Endocrinology [Internet], 2020. Mar 1 [cited 2020 Dec 11];161(3). Available from: https://academic.oup.com/endo/article/161/3/bqaa024/5739626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Angle BM, Do RP, Ponzi D, Stahlhut RW, Drury BE, Nagel SC, et al. , Metabolic Disruption in Male Mice Due to Fetal Exposure to Low but Not High Doses of Bisphenol A (BPA): Evidence for Effects on Body Weight, Food Intake, Adipocytes, Leptin, Adiponectin, Insulin and Glucose Regulation, Reprod Toxicol Elmsford N [Internet], 2013. Dec [cited 2020 Jun 4];42. Available from, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3886819/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Miller CN, Dye JA, Henriquez AR, Stewart EJ, Lavrich KS, Carswell GK, et al. , Ozone-induced fetal growth restriction in rats is associated with sexually dimorphic placental and fetal metabolic adaptation, Mol Metab 42 (2020. Dec) 101094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Mohar I, Stamper BD, Rademacher PM, White CC, Nelson SD, Kavanagh TJ, Acetaminophen-induced liver damage in mice is associated with gender-specific adduction of peroxiredoxin-6, Redox Biol 2 (2014) 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kratchman J, Wang B, Gray G, Which is most sensitive? Assessing responses of mice and rats in toxicity bioassays, J. Toxicol. Environ. Health 81 (7) (2018) 173–183. [DOI] [PubMed] [Google Scholar]
- [75].Siesky AM, Kamendulis LM, Klaunig JE, Hepatic effects of 2-butoxyethanol in rodents, Toxicol. Sci 70 (2) (2002. Dec 1) 252–260. [DOI] [PubMed] [Google Scholar]
- [76].Lin S, Leonard D, Co MAM, Mukhopadhyay D, Giri B, Perger L, et al. , Preeclampsia has an adverse impact on maternal and fetal health, Transl. Res 165 (4) (2015. Apr 1) 449–463. [DOI] [PubMed] [Google Scholar]
- [77].Jin J, Menon R, Placental exosomes: a proxy to understand pregnancy complications, Am. J. Reprod. Immunol 79 (5) (2018), e12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].West CA, Sasser JM, Baylis C, The enigma of continual plasma volume expansion in pregnancy: critical role of the renin-angiotensin-aldosterone system, Am. J. Physiol. Ren. Physiol 311 (6) (2016. Oct 5) F1125–F1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Filgo AJ, Quist EM, Hoenerhoff MJ, Brix AE, Kissling GE, Fenton SE, Perfluorooctanoic acid (PFOA)-induced liver lesions in two strains of mice following developmental exposures: PPARα is not required, Toxicol. Pathol 43 (4) (2015. Jun) 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Dietrich P, Hellerbrand C, Non-alcoholic fatty liver disease, obesity and the metabolic syndrome, Best Pract. Res. Clin. Gastroenterol 28 (4) (2014. Aug) 637–653. [DOI] [PubMed] [Google Scholar]
- [81].Samuel VT, Shulman GI, Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases, Cell Metabol. 27 (1) (2018. September) 22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Hariri N, Thibault L, High-fat diet-induced obesity in animal models, Nutr. Res. Rev 23 (2) (2010. Dec) 270–299. [DOI] [PubMed] [Google Scholar]
- [83].Pfohl M, Ingram L, Marques E, Auclair A, Barlock B, Jamwal R, et al. , Perfluorooctanesulfonic acid and perfluorohexanesulfonic acid alter the blood lipidome and the hepatic proteome in a murine model of diet-induced obesity, Toxicol. Sci 178 (2) (2020. Dec 1) 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, et al. , Disruption of adiponectin causes insulin resistance and neointimal formation, J. Biol. Chem 277 (29) (2002. Jul 19) 25863–25866. [DOI] [PubMed] [Google Scholar]
- [85].Kitamura T, Kahn CR, Accili D, Insulin receptor knockout mice, Annu. Rev. Physiol 65 (1) (2003. Mar 1) 313–332. [DOI] [PubMed] [Google Scholar]
- [86].Medina-Gomez G, Virtue S, Lelliott C, Boiani R, Campbell M, Christodoulides C, et al. , The link between nutritional status and insulin sensitivity is dependent on the adipocyte-specific peroxisome proliferator–activated receptor-γ2 isoform, Diabetes 54 (6) (2005. Jun 1) 1706–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Laustsen PG, Michael MD, Crute BE, Cohen SE, Ueki K, Kulkarni RN, et al. , Lipoatrophic diabetes in Irs1−/−/Irs3−/−double knockout mice, Genes Dev. 16 (24) (2002. Dec 15) 3213–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Tanabe K, Liu Z, Patel S, Doble BW, Li L, Cras-Méneur C, et al. , Genetic deficiency of glycogen synthase kinase-3β corrects diabetes in mouse models of insulin resistance, PLoS Biol. 6 (2) (2008. Feb 19) e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, et al. , Diet-induced insulin resistance in mice lacking adiponectin/ACRP30, Nat. Med 8 (7) (2002. Jul) 731–737. [DOI] [PubMed] [Google Scholar]
- [90].Li N, Liu Y, Papandonatos GD, Calafat AM, Eaton CB, Kelsey KT, et al. , Gestational and childhood exposure to per- and polyfluoroalkyl substances and cardiometabolic risk at age 12 years, Environ. Int 147 (2021. Feb 1), 106344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Miao J, Haas JT, Manthena P, Wang Y, Zhao E, Vaitheesvaran B, et al. , Hepatic insulin receptor deficiency impairs the SREBP-2 response to feeding and statins, J. Lipid Res 55 (4) (2014. Apr) 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wan Y-JY, An D, Cai Y, Repa JJ, Hung-Po Chen T, Flores M, et al. , Hepatocyte-specific mutation establishes retinoid X receptor α as a heterodimeric integrator of multiple physiological processes in the liver, Mol. Cell Biol 20 (12) (2000. Jun) 4436–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Shimano H, Shimomura I, Hammer RE, Herz J, Goldstein JL, Brown MS, et al. , Elevated levels of SREBP-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP-1 gene, J. Clin. Invest 100 (8) (1997. Oct 15) 2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Koutnikova H, , Cock T-A, Watanabe M, Houten SM, Champy M-F, Dierich A, et al. , Compensation by the muscle limits the metabolic consequences oflipodystrophy in PPAR gamma hypomorphic mice, Proc. Natl. Acad. Sci. U. S. A 100 (24) (2003. Nov 25) 14457–14462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Biddinger SB, Hernandez-Ono A, Rask-Madsen C, Haas JT, Alemán JO, Suzuki R, et al. , Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis, Cell Metabol. 7 (2) (2008. Feb 6) 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Diaz FJ, Meary A, Arranz MJ, Ruaão G, Windemuth A, de Leon J, Acetylcoenzyme A carboxylase α gene variations may be associated with the direct effects of some antipsychotics on triglyceride levels, Schizophr. Res 115 (2) (2009. Dec 1) 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Assessment UENC for E, DuPont-24459: a 28-day oral (gavage) toxicity study of H-28397 in mice with a 28-day recovery, Volume 1 [Internet]. 2009. [cited 2021 Oct 7]. Available from, https://hero.epa.gov/hero/index.cfm/reference/details/reference_id/4221051. [Google Scholar]
- [98].North Carolina Department of Environmental Quality and North Carolina Department of Health and Human Services, Secretaries’ Science Advisory Boars Review of the North Carolina Drinking Water Provisional Health Goal for GenX, 2018. Aug 29, pp. 1–27. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.