

ABSTRACT
Cathelicidin-WA (CWA) is a novel cathelicidin peptide isolated from snakes that has been suggested to exert anti-inflammatory effects. The aim of our study was to investigate whether cathelicidin-WA (CWA) could protect the heart from diabetic cardiomyopathy (DCM). Streptozotocin (STZ) injection was used to establish a mouse model of DCM. CWA peptide (2 mg/kg or 8 mg/kg) was continuously administered to the mice from 10 weeks to 16 weeks after STZ injection. The mice in the DCM group exhibited cardiac dysfunction, while 8 mg/kg CWA ameliorated this cardiac dysfunction. Cardiac fibrosis, inflammation, and oxidative stress as well as cardiomyocyte apoptosis in the DCM mice were decreased by treatment with 8 mg/kg CWA. We isolated neonatal rat cardiomyocytes and stimulated the cells with high glucose to establish an in vitro model of myocyte cell injury. Consistently, CWA inhibited high glucose-induced cell death, inflammation and oxidative stress in the myocytes. Moreover, CWA reduced the formation of the NLR family pyrin domain-containing 3 (NRLP3) inflammasome by regulating thioredoxin-interacting protein expression and p65 activation. NLRP3 overexpression inhibited the beneficial effects of CWA on the heart during DCM and on high glucose-induced myocyte injury. In summary, CWA attenuates cardiac injury and preserves cardiac function during DCM by targeting the NLRP3 pathway.
Abbreviations: AAV9: Adeno associated virus; AGE: Advanced Glycation End products; CWA: Cathelicidin-WA; DCM: diabetic cardiomyopathy; Gpx: glutathione peroxidase; HG: high glucose; IL: Interleukin; NLR: Family Pyrin Domain Containing 3 (NRLP3); TXNIP: Thioredoxin interacting protein; LVEF: left ventricular ejection fraction; MDA: Malondialdehyde; MnSOD: manganese superoxide dismutase; NADPH: Nicotinamide adenine dinucleotide phosphate; NAC: N-acetyl-cysteine; NRCMs: Neonatal rat cardiomyocytes; ROS: reactive oxygen species; STZ: Streptozotocin; TNFa: tumor necrosis factor a.
KEYWORDS: Cathelicidin-WA, diabetic cardiomyopathy, NLRP3, thioredoxin interacting protein, NF-κB p65
Introduction
Diabetes-induced cardiac dysfunction is called diabetic cardiomyopathy (DCM) when the cause of cardiac dysfunction is not coronary heart disease, hypertension or other diseases [1,2]. Cardiac hypertrophy, fibrosis, inflammation, and apoptosis are pathological manifestations of diabetic cardiomyopathy, and the molecular mechanisms involve mitochondrial dysfunction and oxidative stress [3,4]. A variety of factors are important for the initiation and progression of diabetic cardiomyopathy, but the specific molecular mechanisms have not yet been fully elucidated. There is currently no specific drug available for the treatment of DCM in the clinic, and most patients inevitably develop heart failure. Therefore, it is critical to explore the pathogenesis of DCM and to identify new treatment methods.
NLR family pyrin domain-containing 3 (NLRP3) is an immune-related inflammatory molecule, and studies have confirmed that NLRP3 is closely associated with the development of DCM [5]. NF-κB regulates the transcriptional activation of NLRP3 and provides an initiation signal for the formation of NLRP3-containing inflammatory complexes at the onset of inflammation [6]. The dual-acting thioredoxin interaction/inhibitory protein thioredoxin-interacting protein (TXNIP) binds to NLRP3 and provides a second signal for its oligomerization [7,8]. During the pathogenesis of DCM, high glucose can lead to an increase in reactive oxygen species, which activate NF-κB, promote the expression of TXNIP, and ultimately induce the activation of NLRP3, thus promoting the occurrence of cellular inflammation and apoptosis [8,9]. Many studies have suggested that NLRP3-containing inflammatory complexes may be therapeutic targets for DCM and other cardiovascular diseases [5,10,11].
Cathelicidin-WA (CWA) is an antimicrobial peptide extracted from the genus Agaricus. Studies have shown that CWA exerts multiple anti-inflammatory effects [12–14]. Wu W showed that CWA could protect macrophages from lipopolysaccharide (LPS)-induced inflammatory injury [12]. Recently, Yi H also showed that CWA protected against postweaning diarrhea by inhibiting inflammation [14]. Moreover, cathelicidin family members were found to inhibit heart injury during myocardial ischaemia/reperfusion [15] and myocardial infarction [16]. These studies suggest that CWA may play a functional role in cardiovascular disease. We established a DCM model by injecting STZ into mice and simultaneously administered CWA to the mice to investigate whether CWA affects the development of DCM.
Animals and animal model
This study was approved by the Animal Care and Use Committee of our medical university and was performed in accordance with our institutional guidelines and the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH publication, revised 2011). The animals were housed as previously described [17]. The Institute of Laboratory Animal Science provided us with 8- to 10-week-old male C57BL/6 mice (Beijing, China). The mice were injected with STZ (S0130, Sigma, Germany, dissolved in 0.1 mol/L citrate buffer, pH 4.5) to establish the DCM model (intraperitoneal injection, 50 mg/kg for 5 days) [18]. The mice in the control group were administered the same volume of vehicle (0.1 mol/L citrate buffer, pH 4.5). Fasting blood glucose levels ≥16.6 mmol/L in three independent measurements were used to define diabetes. The CWA peptide was synthesized and purified (≥95%) by GL Biochem (Shanghai, China), and it was verified by using an Agilent 1200 Series high-performance liquid chromatograph (Agilent Technologies, CA, USA). The mice were also administered CWA (2 or 8 mg/kg, every other day; Shanghai, China) via intraperitoneal injection for 4 weeks, namely, from weeks 12–16 after the final STZ injection. Adeno-associated virus (AAV) 9-NLRP3 was used to overexpress NRLP3 in the hearts of the mice 10 weeks after the final STZ injection. Then, the mice were administered CWA (8 mg/kg, 4 weeks) from weeks 12 to 16 after the final STZ injection.
CWA peptide
CWA (KFFRKLKKSVKKRAKEFFKKPRVIGVSIPF) was synthesized by GL Biochem (Shanghai, China). Peptides were purified at greater than 95% purity via semi-preparative HPLC and characterized by analytical HPLC (Agilent 121 Technologies, CA, USA). The molecular weight of CWA was confirmed using a Thermo Finnigan LCQ ion trap mass spectrometer (Thermo Finnigan, CA, USA). CWA was prepared in saline before injection.
Adenoviral vector construction and viral delivery protocol
Vigene Bioscience (Jinan, China) provided recombinant AAV9 expressing mouse NLRP3 (AAV9-NLRP3) and control AAV9-LacZ. Myocardial injection of AAV9 was performed 10 weeks after the final STZ injection [19]; the virus was injected into a total of 3 sections, including the left ventricular apex, anterior wall, and lateral wall. For each mouse, 1 × 1011 vp was administered in a total volume of 50 μL with a 29-gauge syringe after exposure of the hearts and removal of the pericardium.
Echocardiography and hemodynamic evaluation
Echocardiography and hemodynamic measurements were performed as described in our previous study [20]. Briefly, a MyLab 30CV ultrasound (Esaote SpA, Genoa, Italy) with a 10-MHz linear array ultrasound transducer was used to measure the echocardiographic parameters. A Millar catheter transducer (SPR-839; Millar Instruments, Houston, TX) and PVAN data analysis software were used to measure the hemodynamic parameters.
Cardiac morphology and histomorphometric analysis
For all the pathological staining, including picrosirius red (PSR) staining, immunohistochemical staining, and TUNEL staining, the steps were performed according to a previous study [21]. For the immunohistochemical staining, a heating method was used to retrieve the antigen. After blocked with 10% goat serum, heart sections were incubated with different primary antibodies: anti-CD45, anti-F480 (Abcam), anti-4-hydroxynonenal (4-HNE) (Abcam) and then incubated with horseradish peroxidase-labeled secondary antibody and detected by DAB assay.
Neonatal rat cardiomyocyte (NRCM) culture and treatment
One- to three-day-old SD rats were used to isolate NRCMs according to the steps described in a previous study [18]. NRCMs were identified with anti–sarcomeric α-actin (Abcam), anti-titin, and anti–troponin T (Abcam) antibodies. CWA (5, 10, 20, and 40 μg/ml) was used to treat the NRCMs for 24 h. High glucose (HG; 33 mM glucose, 24 h) was used to induce cardiomyocyte injury. Mannitol (27.5 mM) and normal glucose (NG; 5.5 mM glucose) were added to the control cells. Cell counting kit-8 assays (CCK8, Dojindo, Tokyo, Japan) were used to detect cell viability. N-acetyl-cysteine (NAC) (2 mM, 12 h, Sigma, St. Louis, MO, USA) was used to scavenge reactive oxygen species (ROS). BAY-117082 (10 μM, 12 h, Sigma) was used to inhibit NF-κB. Ad-NLRP3 (MOI = 50) was used to overexpress NLRP3, and NLRP3 siRNA (purchased from Suzhou Ribo Life Science Co., Ltd.) was used to knockdown NLRP3. All the in vitro experiments were repeated three independent times.
Oxidative stress
A 2ʹ,7ʹ-dichlorodihydrofluorescein diacetate fluorescent probe (Beyotime, Haimen, China) was used to measure ROS. In brief, cells were incubated with 1 × 10−5 mol/L DCFH-DA in a 37°C incubator for 30 min. After washed, a fluorescence microplate reader was used to detect the ROS level at 488 nm excitation wavelength (Synergy HT, Biotek, Vermont, USA). Manganese superoxide dismutase (MnSOD) activity, glutathione peroxidase (Gpx) activity, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity and lipid peroxidation-induced malondialdehyde (MDA) contents were measured with commercial kits (Beyotime, Haimen, China) in fresh mouse hearts (80–120 mg) or cardiomyocytes according to the manufacturer’s instructions.
ELISA
The proinflammatory cytokine levels in the heart tissues from diabetic mice were measured with commercially available enzyme-linked immunosorbent assay (ELISA) kits to quantify the TNF-α, IL-1 IL-6 and IL-18 levels according to the manufacturer’s protocol (Biolegend, USA). Briefly, tissue samples were lysed. The lysates were then incubated on the corresponding plate. After the plate was washed, the antibodies were added and incubated for 1 h at room temperature. The plate was washed, and HRP-conjugated IgG was added and incubated at room temperature for 1 h. The plate was washed, 150 μl of the substrate (1 μl H2O2 with 0.5 mg/ml ABTS in citrate buffer) was added, and the optical density of each well was measured at 405 nm by an ELISA reader (Synergy HT, Biotek, Vermont, USA).
Caspase-1 activity assay
Caspase-1 activity was measured using a caspase-1 activity assay kit (Beyotime, China) according to the manufacturer’s instructions. This assay kit is based on the ability of caspase-1 to cleave the substrate acetyl-Tyr-Val-Ala-Asp p p-nitroanilide (Ac-YVAD pNA) to produce yellow p-nitroaniline (pNA). Then, the absorption at 405 nm was measured by an ELISA reader (Synergy HT, Biotek, Vermont, USA).
Quantitative real-time PCR and western blot analysis
Total RNA was isolated and purified as described in our previous study [20]. The SuperScript first-strand complementary DNA system (Invitrogen) was used for reverse transcription. A LightCycler 480 SYBR Green Master Mix system (Roche) was used for quantification. GAPDH was used as the reference gene. Primers used was listed in Table 1.
Table 1.
Primer sequences used for RT-PCR
| mRNA | Forward | Reverse |
|---|---|---|
| Collagen I M | AGGCTTCAGTGGTTTGGATG | CACCAACAGCACCATCGTTA |
| Collagen III M | AAGGCTGCAAGATGGATGCT | GTGCTTACGTGGGACAGTCA |
| CTGF M | AGGGCCTCTTCTGCGATTTC | CTTTGGAAGGACTCACCGCT |
| TGFβM | ATCCTGTCCAAACTAAGGCTCG | ACCTCTTTAGCATAGTAGTCCGC |
| IL-1 M | CCGTGGACCTTCCAGGATGA | GGGAACGTCACACACCAGCA |
| IL-6 M | AGTTGCCTTCTTGGGACTGA | TCCACGATTTCCCAGAGAAC |
| TNFα M | ACTGAACTTCGGGGTGATCGGT | TGGTTTGCTACGACGTGGGCTA |
| GAPDH M | ACTCCACTCACGGCAAATTC | TCTCCATGGTGGTGAAGACA |
| IL-1 R | GGGATGATGACGACCTGCTAG | ACCACTTGTTGGCTTATGTTCTG |
| IL-6 R | GTTGCCTTCTTGGGACTGATG | ATACTGGTCTGTTGTGGGTGGT |
| TNFαR | AGCATGATCCGAGATGTGGAA | TAGACAGAAGAGCGTGGTGGC |
| GAPDH R | GACATGCCGCCTGGAGAAAC | AGCCCAGGATGCCCTTTAGT |
Sequences are listed 5ʹ–3ʹ. M, mouse; R, rat.
Total proteins were extracted as described in our previous study [20]. A 10% SDS gel was used to separate the proteins. The following primary antibodies were used: MnSOD, gp91, TXNIP, total (T) and phosphorylated (P) p65, NLRP3, ASC, c-caspase-1 (Abcam), IL-1, and GAPDH (Santa Cruz). Image Lab 5.2.1 software was used for quantification. GAPDH was used as the reference protein.
Statistical analysis
All the data are expressed as the mean ± SEM. One-way ANOVA followed by a post hoc Tukey’s test was used to compare differences between groups. Differences between two groups were analyzed by unpaired, two‐sided Student’s t-tests. A P value less than 0.05 was considered statistically significant.
Results
CWA suppresses cardiac dysfunction in the hearts of mice with DCM
Sixteen weeks after the final STZ injection, blood glucose and body weight were measured in each group. Blood glucose was significantly higher in the mice with DCM than in the control mice, and body weights were significantly lower in the mice with DCM than in the control mice. These data indicated the successful establishment of our model. Both blood glucose and body weight were altered in the CWA (2 and 8 mg/kg) group compared to the vehicle-STZ group (Figure 1(a,b)). This result indicates that CWA may not affect glucose metabolism. The mice in the DCM group showed impaired cardiac systolic function and increased ventricular diameters (Figure 1(c,h)). Treatment with both 2 mg/kg CWA and 8 mg/kg CWA enhanced the left ventricular (LV) fractional shortening (LVFS) and the maximum rate of LV pressure rise (dp/dtmax) (Figure 1(c,h)). However, 8 mg/kg CWA treatment further enhanced the LV improved ejection fraction (LVEF) and maximum rate of LV pressure decay (dp/dtmin) and reduced the LV end diastolic diameter (LVEDd) (Figure 1(c,h)). Thus, the 8 mg/kg CWA treatment was chosen for further studies.
Figure 1.
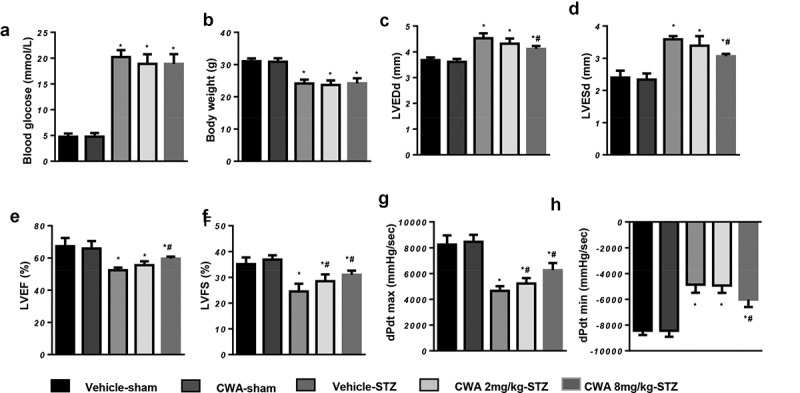
Cardiac function of mouse hearts
Mice were injected with STZ to establish the DCM model and then treated with CWA (2 mg/kg and 8 mg/kg) from 12–16 weeks after the final STZ injection. a. Blood glucose of the mice at 16 weeks after the final STZ injection in the indicated groups (n = 15). b. Body weight of the mice at 16 weeks after the final STZ injection (n = 15). c-f. Echocardiographic measurements of the mice at 16 weeks after the final STZ injection in the indicated groups (n = 10). g and h. Hemodynamic measurements in mouse hearts in the indicated groups (n = 10). *P < 0.05 vs. vehicle/sham; #P < 0.05 vs. vehicle/STZ.
CWA suppresses fibrosis in the hearts of mice with DCM
Interstitial and vascular collagen deposition in the hearts of mice with DCM were increased significantly. As shown in Figure 2a, LV collagen volume was much higher in the vehicle-treated mouse hearts, while CWA treatment reduced collagen deposition in the hearts of mice with DCM (Figure 2(a,b)). We also detected the transcriptional and protein levels of fibrosis markers, including collagen I, collagen III, transforming factor β (TGFβ) and Connective tissue growth factor (CTGF). CWA reduced both the transcriptional and protein levels of these fibrosis markers in the hearts of mice with DCM (Figure 2(c,e)).
Figure 2.
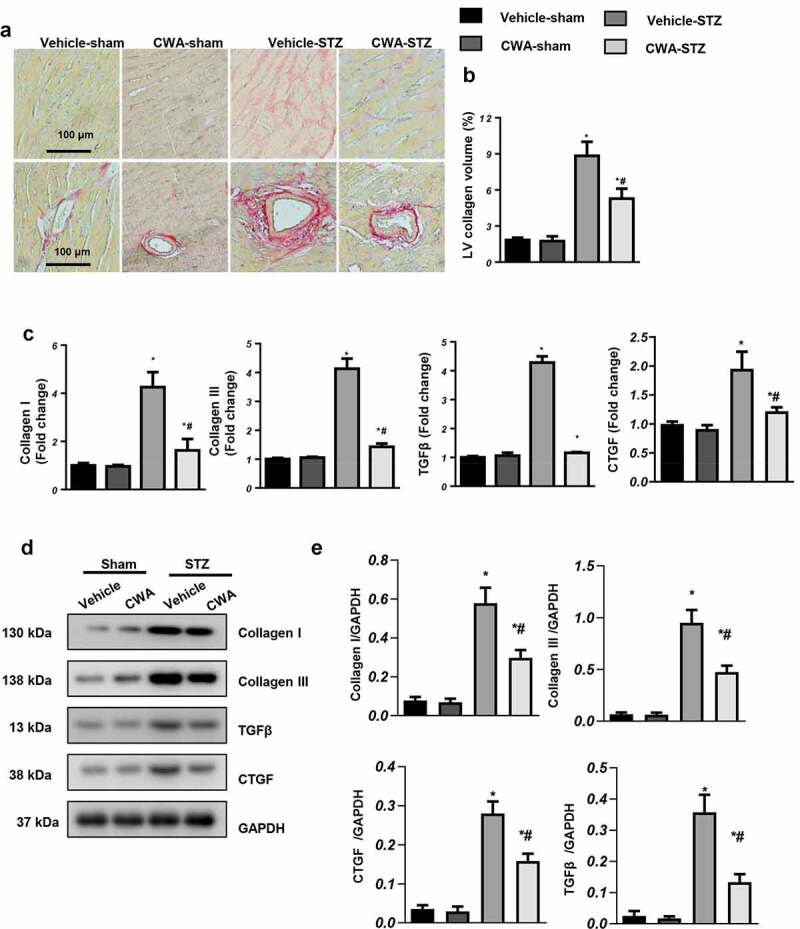
Cardiac fibrosis and inflammation in mice
Mice were injected with STZ to establish the DCM model and then treated with CWA (2 mg/kg and 8 mg/kg) from 12 to 16 weeks after the final STZ injection. a and b. Representative image of picrosirius red (PSR) staining and quantification results of collagen volume in the mouse hearts in the indicated groups (n = 6). c-e. Transcription (C) and protein levels (d, e) of fibrosis markers (n = 6). *P < 0.05 vs. vehicle/sham; #P < 0.05 vs. vehicle/STZ.
CWA inhibits inflammation, apoptosis and oxidative stress in the hearts of mice with DCM
Inflammation is another main feature of DCM. We detected inflammatory cell infiltration by immunohistochemistry. As shown in Figure 3a, CWA decreased the numbers of F4/80-labeled macrophages and CD45-labeled leukocytes in the hearts of mice with DCM (Figure 3(a)–c). CWA also decreased the transcription level and the release of proinflammatory factors, such as tumor necrosis factor α (TNFα), interleukin (IL)-1, IL-6, and IL-18 (Figure 3(a–c)).
Figure 3.
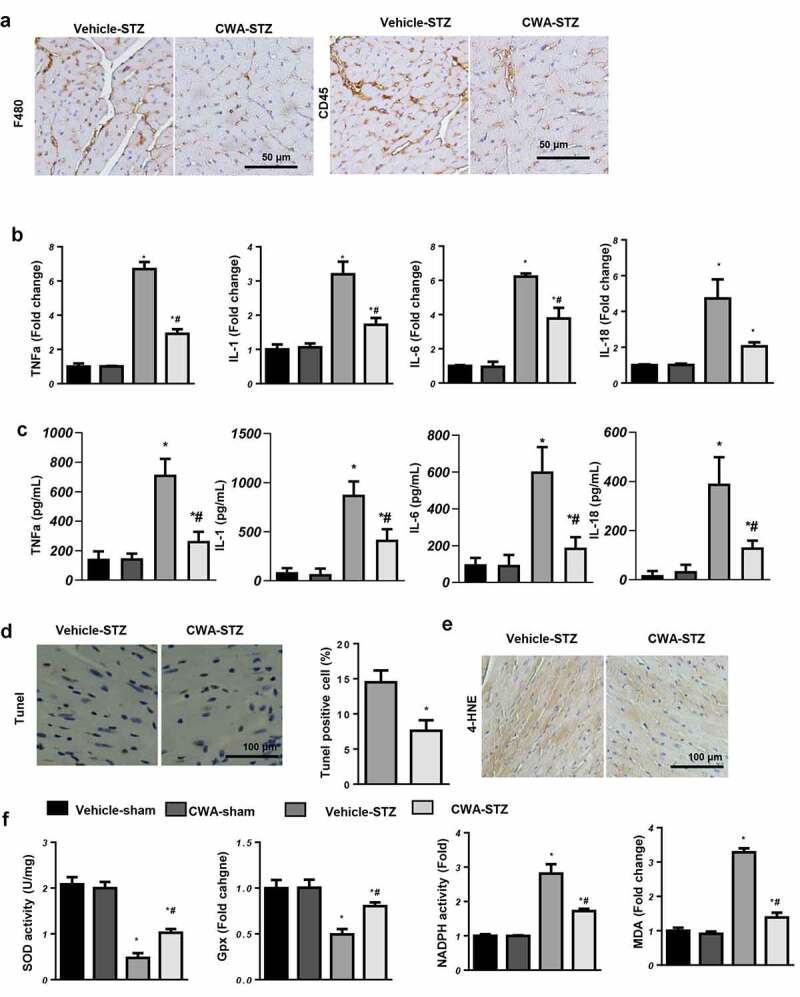
Cardiac apoptosis and oxidative stress in mice
a. Representative image of immunohistochemical staining of F4/80 and CD45 (n = 6). b and c. Transcription and protein levels of inflammation markers (n = 6). d. TUNEL staining and quantification 16 weeks after the final STZ injection in the indicated groups (n = 6). e. 4-HNE staining in the mouse hearts in the indicated groups (n = 6). f. SOD, Gpx, NADPH oxidase activity and MDA levels in the mouse hearts in the indicated groups (n = 6). *P < 0.05 vs. vehicle/sham; #P < 0.05 vs. vehicle/STZ.
Cardiomyocyte apoptosis was detected by TUNEL staining. As the results show, the numbers of TUNEL-positive apoptotic cells were lower in the CWA-treated mouse hearts than in the vehicle-treated mouse hearts (vehicle-STZ group: 14.5%, CWA-STZ group 7.6%) (Figure 3(d)). Oxidative stress occurs due to insufficient antioxidant enzymes to offset the excess reactive oxygen species, resulting in an redox imbalance. Sixteen weeks after the final STZ injection, the level of 4-HNE, an intermediate metabolite of lipid peroxidation, was reduced in the CWA-STZ group compared with the vehicle-STZ group (Figure 3(e)). The activity of antioxidant enzymes, such as MnSOD and Gpx, was enhanced in the CWA-STZ group, while the NADPH oxidase and MDA levels were reduced in the CWA-STZ group compared with the vehicle-STZ group (Figure 3(f)).
CWA attenuates HG-induced cardiomyocyte injury
NRCMs were treated with 0, 5, 10, 20, or 40 μg/ml CWA. Cell viability was not significantly different between the CWA groups and the control group (Figure 4(a)). As the exposure time increased, the cell viability in the HG group gradually decreased, and CWA prevented this HG-induced decrease in cell viability (Figure 4(b)). The cell viability in the 40 μg/ml CWA group was much higher than that in the other CWA groups (Figure 4(b)). Thus, 40 μg/ml CWA was chosen for the subsequent experiments. The inflammatory response was increased in HG-stimulated cells, as determined by increased mRNA levels of IL-1, IL-6, IL-18, and TNF-α in the HG groups (Figure 4(c)). CWA inhibited the inflammatory response, as evidenced by decreased proinflammatory cytokine levels (Figure 4(c)). CWA also reduced the oxidative stress induced by HG, as the ROS levels and NADPH oxidase activity decreased sharply in the CWA-treated cells, while the SOD and Gpx activities were markedly increased (Figure 4(d,e)). NF-κB, a molecule that is well-known to trigger inflammation and oxidative stress, was assessed by staining. CWA reduced the nuclear translocation of NF-κB in NRCMs exposed to HG (Figure 4(f)).
Figure 4.
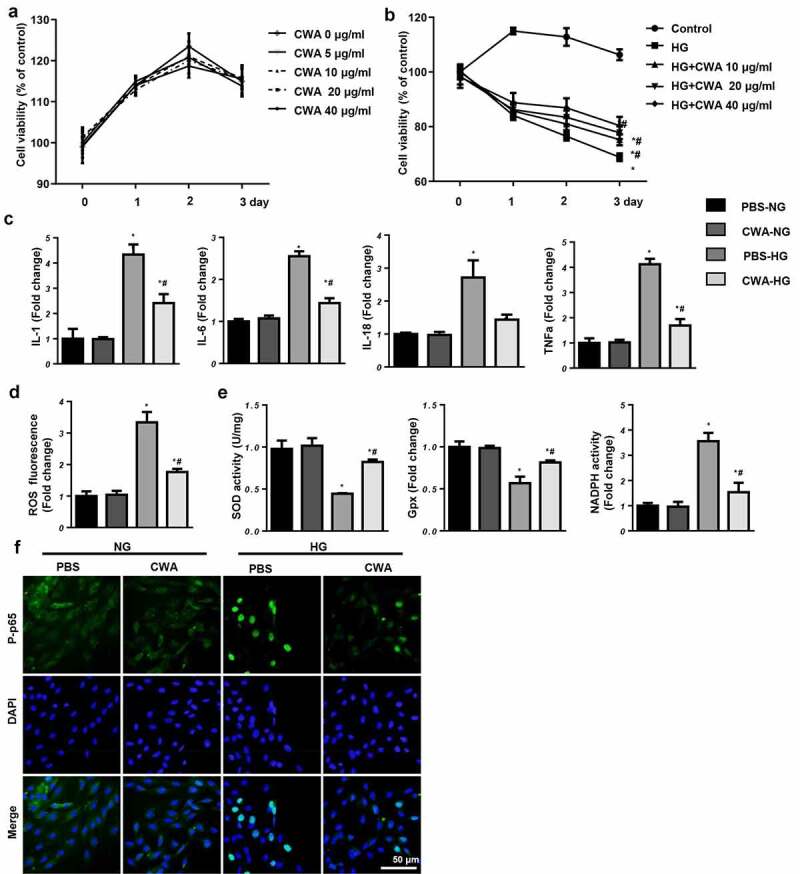
Effects of CWA in vitro
a. NRCMs were treated with CWA (0, 5, 10, 20, and 40 μg/ml). Cell viability in each group (n = 6). b. NRCMs were cultured with CWA (10, 20, and 40 μg/ml) and exposed to 33.3 mM glucose. Cell viability in each group (n = 6). c-f. NRCMs were cultured with CWA (40 μg/ml) and exposed to 33.3 mM glucose. c. IL-1, IL-6, IL-18, and TNFα mRNA levels (n = 6). d. ROS levels in each group (n = 6). e. SOD, Gpx, and NADPH oxidase activities in each group (n = 6). f. Immunofluorescent staining of p-p65 in each group (n = 6). *P < 0.05 vs. PBS/NG; #P < 0.05 vs. PBS/HG. All the in vitro experiments were performed 3 independent times.
CWA inhibits the NLRP3 inflammasome
We then examined whether CWA affects NLRP3 inflammasome signaling. We observed that TXNIP and P-p65 were increased both in the hearts of mice with DCM and in myocytes exposed to HG. NLRP3 inflammasome components, including NLRP3, ASC1, cleaved (c-) caspase1, and IL-1, were also increased in the hearts of mice with DCM and in HG-stimulated myocytes (Figure 5(a,d)). CWA inhibited the increased expression of TXNIP, P-p65, and NLRP3 in the hearts of mice with DCM (Figure 5(a,b)). Furthermore, the increased levels of TXNIP, P-p65, and NLRP3 in HG-stimulated myocytes were also inhibited by CWA (Figure 5(d,e)). Cleaved-caspase1, as a regulator of the expression of the inflammatory cytokines IL-1 and IL-18, was also inactivated by CWA treatment in both treated mouse hearts and cells (Figure 5(c,f)). CWA further reduced the release of IL-18 by CWA-treated cells exposed to HG (Figure 5g).
Figure 5.
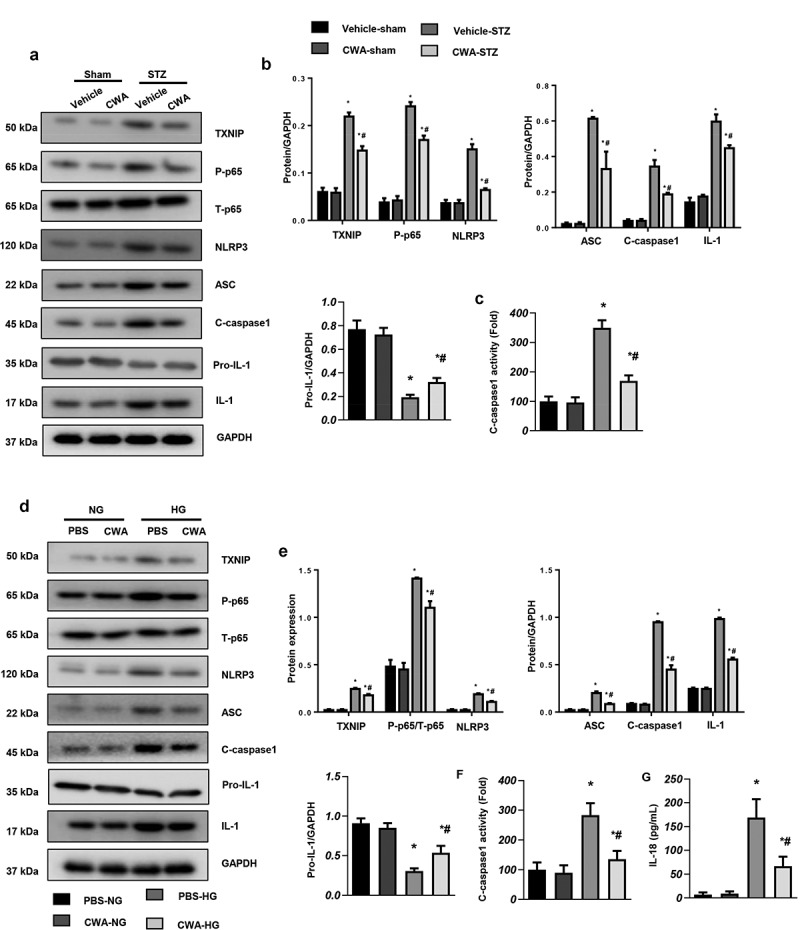
Effects of CWA on the NLRP3 inflammasome
a and b. Protein expression of NLRP3 signaling factors in the hearts of mice with DCM (n = 6). C. The activity of c-caspase1 in the hearts of mice with DCM (n = 6). *P < 0.05 vs. vehicle/sham; #P < 0.05 vs. vehicle/STZ. d and e. Protein expression of NLRP3 signaling factors in myocytes stimulated with HG (n = 6). f. The activity of c-caspase1 in myocytes (n = 6). g. IL-18 release by myocytes was measured by ELISA (n = 6). *P < 0.05 vs. PBS/NG; #P < 0.05 vs. PBS/HG. All the in vitro experiments were performed 3 independent times.
CWA targets NLRP3 in myocytes
To further confirm the functional role of CWA in NF-κB and NLRP3 signaling, NRCMs were incubated with BAY-117082 to inhibit p65 activity. NRCMs were also treated with NAC to eliminate ROS. Both BAY-117082 and NAC inhibited the negative effects of HG, as determine by the reduced ROS levels, increased cell viability and decreased inflammatory cytokine levels in the BAY-117082 and NAC groups. However, when the cells were treated with both NAC and CWA, CWA did not enhance the protective effects of NAC (Figure 6(a–c)). Interestingly, when the cells were treated with both BAY-117082 and CWA, CWA enhanced the protective effects of BAY-117082 (Figure 6(a,c)). These data indicate that CWA targets pathways other than NF-κB.
Figure 6.
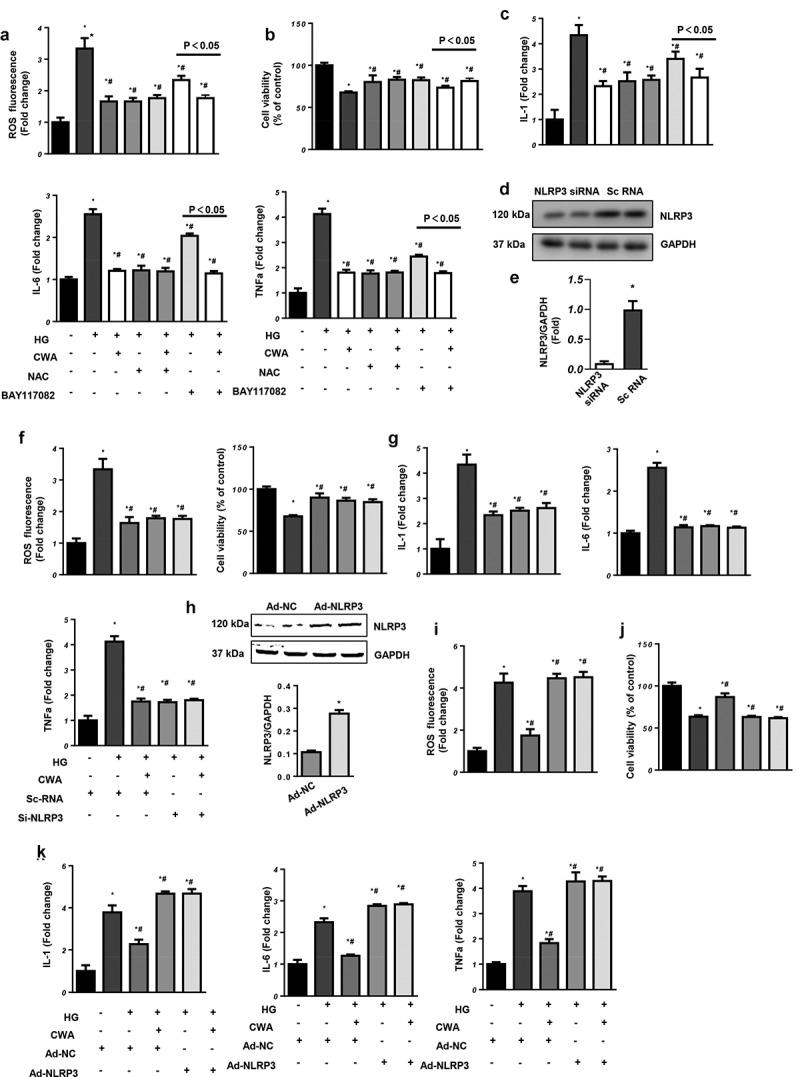
CWA targets NLRP3
a-c. NRCMs were treated with CWA, NAC (2 mM), and BAY-117082 (10 μM) and exposed to 33.3 mM glucose. a. ROS levels in the indicated groups (n = 6). b. Cell viability in the indicated groups (n = 6). c. Transcription levels of inflammatory markers (n = 6).d-g. NRCMs were transfected with NLRP3-specific siRNA or CWA and exposed to 33.3 mM glucose. d. Protein expression of NLRP3 in cells transfected with NLRP3-specific siRNA (n = 6, *P < 0.05 vs. scRNA). e. ROS levels in the indicated groups (n = 6). f. Cell viability in the indicated groups (n = 6). g. Transcription levels of inflammatory markers (n = 6). *P < 0.05 vs. scRNA/NG; #P < 0.05 vs. scRNA/HG. h-k. NRCMs were transfected with Ad-NRLP3, treated with CWA (40 μg/ml) and exposed to 33.3 mM glucose. h. Protein expression of NLRP3 in cells transfected with Ad-NRLP3 (n = 6, *P < 0.05 vs. Ad-NC). i. ROS levels in the indicated groups (n = 6). j. Cell viability in the indicated groups (n = 6). k. Transcription levels of inflammatory markers (n = 6). *P < 0.05 vs. Ad-NC/NG; #P < 0.05 vs. Ad-NC/HG. All the in vitro experiments were performed 3 independent times.
We then verified the effect of CWA on NLRP3. NRCMs were transfected with NLRP3-specific siRNA to silence NLRP3 (Figure 6(d)). NLRP3 knockdown protected the cells from oxidative and inflammatory injury, while CWA did not enhance the protective effects of NLRP3 silencing (Figure 6(d,g)). When we overexpressed NLRP3 by adenovirus transfection (Figure 6(h)), NLRP3 overexpression counteracted the protective effects of CWA (Figure 6(h,k)). These results indicate that NLRP3 is the target of CWA in cardiomyocytes.
NLRP3 inhibits the protective effects of CWA in mice
To confirm the effect of CWA on NLRP3 in vivo, mice with DCM were administered AAV9-NLRP3 to overexpress NLRP3 in the heart and then treated with CWA (8 mg/kg) for 4 weeks. The protein level of NLRP3 in the mouse hearts was increased at 1, 2, 4, and 6 weeks after AAV9-NLRP3 injection (Figure 7(a)). Reduced cardiac function was observed in the mice with DCM and with overexpressed NLRP3, and CWA treatment did not exert protective effects on cardiac dysfunction in the mice with DCM (Figure 7(b)). NLRP3 overexpression induced severe cardiac fibrosis (Figure 7(c,e)) and inflammation (Figure 7(f,g)). CWA treatment could not ameliorate these changes. Moreover, cardiomyocyte apoptosis (Figure 7(h)) and oxidative stress (Figure 7(i,j)) were also increased in the AAV9-NLRP3-STZ group, and CWA treatment did not inhibit the NLRP3 overexpression-induced apoptosis or oxidative stress observed in the mice with DCM. Taken together, CWA exerts cardioprotective effects via NLRP3 regulation.
Figure 7.
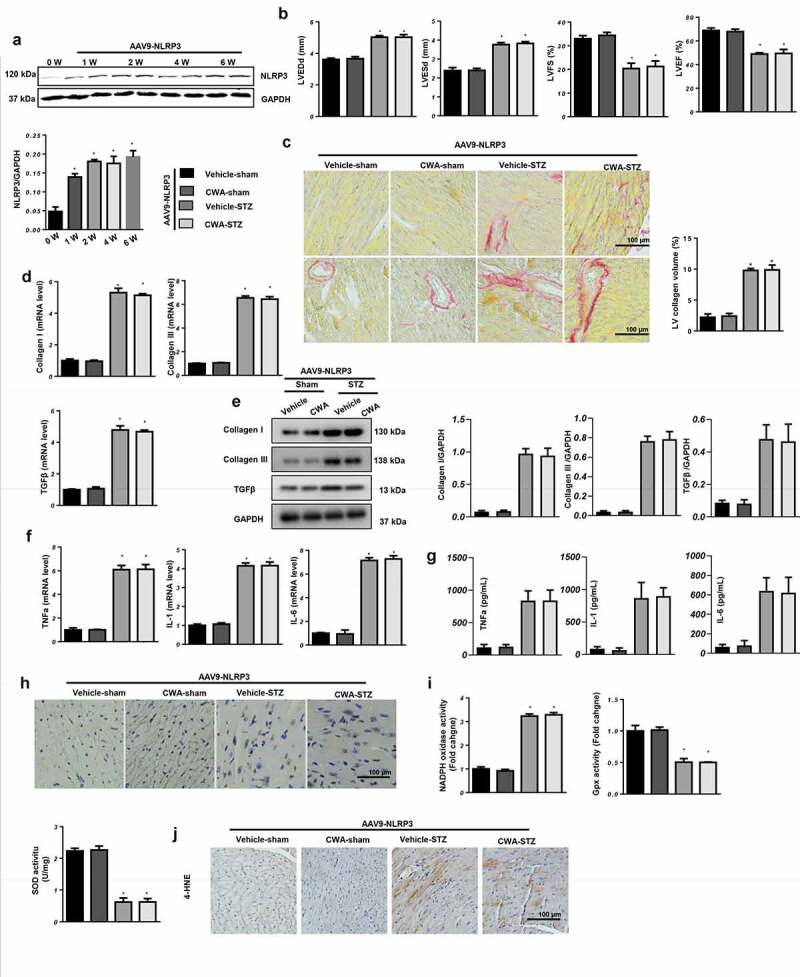
NLRP3 counteracts the cardioprotective effects of CWA
Mice were injected with AAV9-NLRP3 and treated with CWA (8 mg/kg, 4 weeks). a. NLRP3 expression in the mouse hearts at the indicated times after AAV9-NLRP3 injection (n = 6, *P < 0.05 vs. 0 W group). b. Echocardiographic parameters in the indicated groups (n = 10). c. Representative image of picrosirius red (PSR) staining and quantification results (n = 6). d and e. Transcription (D) and protein levels (E) of fibrosis markers (n = 6). f and g. Transcription levels (F) and release (G) of inflammatory markers (n = 6). H. Representative image of TUNEL staining (n = 6). i. SOD, Gpx, NADPH oxidase activities (n = 6). J. 4-HNE staining of mice hearts (n = 6). *P < 0.05 vs. vehicle/sham; #P < 0.05 vs. vehicle/STZ.
Discussion
In the progression of diabetes, metabolic disorders (hyperglycemia, hyperlipidemia, and hyperinsulinaemia) can lead to changes in various molecular pathways in cardiomyocytes, leading to myocardial cell dysfunction, injury, and death and subsequently to cardiac dysfunction [3,22]. Currently, the molecular mechanism underlying diabetic cardiomyopathy has not yet been fully elucidated. To date, DCM treatment has mainly focused on glucose regulation. However, cardiac injury during the progression of hyperglycemia, hyperlipidemia, and hyperinsulinaemia could not be restored with therapy targeting hypoglycemia alone. Therefore, it is important to identify a treatment method for DCM. The present study reveals a previously unrecognized biological function of CWA in DCM. We clearly showed that CWA acts as a negative regulator of cardiac fibrosis, inflammation, apoptosis and oxidative stress. Mechanistically, CWA inhibited the NLRP3 inflammasome, which inhibited the inflammatory response and oxidative stress, leading to improved cardiac function.
Hyperglycemia and induced ROS increase the accumulation of nonenzymatic advanced glycation end products (AGEs) in the cardiovascular system. The increased expression of AGE receptors (RAGEs) promotes AGE-mediated damage to the heart, which is an important mechanism underlying the occurrence of cardiovascular complications in diabetes patients [23,24]. Increased AGE levels also enhance the crosslinking of collagens, which inhibit collagenase degradation and promote cardiac fibrosis [25,26]. NF-κB signaling can also be activated by AGEs, subsequently activating inflammatory signaling [25]. In our study, NF-κB activation was suppressed by CWA treatment. CWA also reduced the production of ROS. This may be the mechanism by which CWA exerts its cardioprotective effects. When we eliminated ROS with NAC, CWA could no longer exert its synergistic protective effects. However, when cells were treated with an NF-κB inhibitor, CWA exerted synergistic protective effects. These results suggest that CWA exerts cardioprotective effects in part by regulating NF-κB.
NLRP3 is an immune-related inflammatory molecule, and studies have confirmed that NLRP3 is closely associated with the development of DCM [5]. ROS promote the activation of NLRP3 by activating p65 and increasing the level of TXNIP [6,7]. Activated NLRP3 binds to the ASC protein to form inflammasomes that activate pro-caspase-1 and promote caspase-1 cleavage and activation [27]. Activated caspase 1 hydrolyses the IL-1 precursor to form mature IL-1β and IL-18, which play important roles in inflammation [27]. We observed that CWA suppressed the expression of TXNIP and NLRP3, inhibited the activation of the inflammasome, and decreased the levels of IL-1β and IL-18. This NLRP3 pathway may be the target of CWA. When we overexpressed NLRP3 in cardiomyocytes, the protective effects of CWA were completely abrogated. NLRP3 knockdown in vitro completely mimicked the effects of CWA. We also confirmed the significance of NLRP3 in the functional role of CWA in vivo. Overexpression of NLRP3 in the heart abolished the cardioprotective effects of CWA. Thus, by eliminating ROS and inhibiting the NF-κB and TXNIP levels, CWA suppresses the activation and formation of the NLRP3 inflammasome, which protects the heart from inflammation and oxidative injury. Cathelicidins have been reported to suppress LPS-induced inflammation in macrophages via 1) binding to LPS; 2) perturbation of the MyD88 signaling pathway; 3) inhibition of NF-κB translocation [28]. Yi H also found that CWA-mediated suppression of IL-6 expression by directly affecting the TLR4 and MyD88 signaling pathways [14]. Thus we suppose that CWA may affect NLRP3 signaling by either binding to it or through the up-stream molecular located in the cell membrane.
In summary, CWA ameliorates the development of DCM by regulating the ROS-induced NF-κB/TXNIP-NLRP3 pathway. CWA may be a new therapeutic strategy for the treatment of DCM.
Funding Statement
This work was supported by the National Natural Science Foundation of China (grant nos. 81172405 and 81572490), the Tianjin Science and Technology Committee (grant no. 18JCZDJC98600) and the Science and Technology fund of Tianjin Binhai New Area Health and Family Planning Commission (grant no. 2018BWKZ002).
Source of funding
This study was supported by the Youth Fund of the First Affiliated Hospital of Zhengzhou University (201,809,201).
Authorship contributions
Conducted experiments: Yuan Liu, Yawei Xu, Li Li
Performed data analysis: Yan Li
Wrote or contributed to the writing of the manuscript: Meng Peng, Haibo Yang
Disclosure statement
We confirm that there are no known conflicts of interest associated with this publication.
Data availability
The original data used to support the findings of this study are included in the article.
References
- [1].Marwick TH, Ritchie R, Shaw JE, et al. Implications of underlying mechanisms for the recognition and management of diabetic cardiomyopathy. J Am Coll Cardiol. 2018;71(3):339–351. [DOI] [PubMed] [Google Scholar]
- [2].Rubler S, Dlugash J, Yuceoglu YZ, et al. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30(6):595–602. [DOI] [PubMed] [Google Scholar]
- [3].Kain V, Halade GV.. Metabolic and biochemical stressors in diabetic cardiomyopathy. Front Cardiovasc Med. 2017;4:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Boudina S, Abel ED.. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11(1):31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Luo B, Liu HF, Liang Y, et al. NLRP3 inflammasome as a molecular marker in diabetic cardiomyopathy. Front Physiol. 2017;25(8):519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Liao PC, Chao LK, Chou J-C, et al. Lipopolysaccharide/adenosine triphosphate-mediated signal transduction in the regulation of NLRP3 protein expression and caspase-1-mediated interleukin-1beta secretion. Inflamm Res. 2013;62(1):89–96. [DOI] [PubMed] [Google Scholar]
- [7].Luo B, Li B, Wang W, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One. 2014;9(8):e104771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Franchi L,M-PR, Núñez G, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13(4):325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kumar S, Prasad S, Sitasawad SL. Multiple antioxidants improve cardiac complications and inhibit cardiac cell death in streptozotocin-induced diabetic rats. PLoS One. 2013;8(7):e67009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Martinez GJ, Robertson S, Barraclough J, et al. Colchicine acutely suppresses local cardiac production of inflammatory cytokines in patients with an acute coronary syndrome. J Am Heart Assoc. 2015;4(8):e002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Luo B, Li B, Wang W, et al. Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovasc Drugs Ther. 2014;28(1):33–43. [DOI] [PubMed] [Google Scholar]
- [12].Wu W, Wang S, Liu Q et al. Cathelicidin-WA attenuates LPS-induced inflammation and redox imbalance through activation of AMPK signaling. Free Radic Biol Med. 2018;129:338–353. [DOI] [PubMed] [Google Scholar]
- [13].Chen S, Lu Z, Wang F, et al. Cathelicidin-WA polarizes E. coli K88-induced M1 macrophage to M2-like macrophage in RAW264.7 cells. Int Immunopharmacol. 2018;54:52–59. [DOI] [PubMed] [Google Scholar]
- [14].Yi H, Zhang L, Gan Z, et al. High therapeutic efficacy of Cathelicidin-WA against postweaning diarrhea via inhibiting inflammation and enhancing epithelial barrier in the intestine. Sci Rep. 2016;6(1):25679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bei Y, Pan -L-L, Zhou Q, et al. Cathelicidin-related antimicrobial peptide protects against myocardial ischemia/reperfusion injury. BMC Med. 2019;17(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Klyachkin YM, Idris A, Rodell CB, et al. Cathelicidin Related Antimicrobial Peptide (CRAMP) enhances bone marrow cell retention and attenuates cardiac dysfunction in a mouse model of myocardial infarction. Stem Cell Rev. 2018;14(5):702–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yuan Y, Zong J, Zhou H, et al. Puerarin attenuates pressure overload-induced cardiac hypertrophy. J Cardiol. 2014;63(1):73–81. [DOI] [PubMed] [Google Scholar]
- [18].Gao L, Liu Y, Guo S, et al. LAZ3 protects cardiac remodeling in diabetic cardiomyopathy via regulating miR-21/PPARa signaling. Biochim Biophys Acta Mol Basis Dis. 2018;1864(10):3322–3338. [DOI] [PubMed] [Google Scholar]
- [19].Jiang DS, Wei X, Zhang X-F, et al. IRF8 suppresses pathological cardiac remodelling by inhibiting calcineurin signalling. Nat Commun. 2014;5(1):3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ma ZG, Zhang X, Yuan Y-P, et al. A77 1726 (leflunomide) blocks and reverses cardiac hypertrophy and fibrosis in mice. Clin Sci (Lond). 2018;132(6):685–699. [DOI] [PubMed] [Google Scholar]
- [21].Li F, Zong J, Zhang H, et al. Orientin reduces myocardial infarction size via eNOS/NO signaling and thus mitigates adverse cardiac remodeling. Front Pharmacol. 2017;8:926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia. 2018;61(1):21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Candido R, Forbes JM, Thomas MC, et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ Res. 2003;92(7):785–792. [DOI] [PubMed] [Google Scholar]
- [24].Bidasee KR, Zhang Y, Shao CH, et al. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes. 2004;53(2):463–473. [DOI] [PubMed] [Google Scholar]
- [25].Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. 2014;57(4):660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Norton GR, Candy G, Woodiwiss AJ. Aminoguanidine prevents the decreased myocardial compliance produced by streptozotocin-induced diabetes mellitus in rats. Circulation. 1996;93(10):1905–1912. [DOI] [PubMed] [Google Scholar]
- [27].Liu D, Zeng X, Li X, et al. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. 2017;113(1):5. [DOI] [PubMed] [Google Scholar]
- [28].Sun J, Furio L, Mecheri R, et al. Pancreatic beta-Cells limit autoimmune diabetes via an immunoregulatory antimicrobial peptide expressed under the influence of the gut microbiota. Immunity. 2015;43(2):304–317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The original data used to support the findings of this study are included in the article.