

ABSTRACT
Dbf4-Dependent Kinase (DDK) has a well-established essential role at origins of DNA replication, where it phosphorylates and activates the replicative MCM helicase. It also acts in the response to mutagens and in DNA repair as well as in key steps during meiosis. Recent studies have indicated that, in addition to the MCM helicase, DDK phosphorylates several substrates during the elongation stage of DNA replication or upon replication stress. However, these activities of DDK are not essential for viability. Dbf4-Dependent Kinase is also emerging as a key factor in the regulation of genome-wide origin firing and in replication-coupled chromatin assembly. In this review, we summarize recent progress in our understanding of the diverse roles of DDK.
KEYWORDS: Dbf4-dependent Kinase, cyclin dependent Kinase, origins of DNA replication, replication forks, replication stress
Introduction
Dbf4-Dependent Kinase (DDK) is responsible for a critical step in the activation of the replicative helicase MCM and for the initiation of DNA replication at multiple origins throughout the eukaryotic genomes. This role of DDK has received significant attention and has been discussed in detail in several major reviews [1,2]. However, in recent years, we have witnessed the accumulation of evidence for multiple additional roles of this kinase. In this article, we briefly review the recent advances in our understanding of the mechanism of action of DDK during the initiation of DNA replication. We then focus on the potential role of DDK in the genome-wide control of origin activity and during the elongation stage of DNA replication.
DDK is built up of the Cdc7p kinase and its Dbf4p regulatory subunit [2]. Functionally related kinases have been identified in fission yeast (Hsk1p/Dfp1p) and vertebrates (CDC7/ASK) [3]. Orthologs of CDC7 and DBF4 have also been found in plants and Drosophila and were shown to complement the loss of the corresponding genes in budding or fission yeasts [4,5]. Hence, the role of DDK as a major regulator of DNA replication is conserved amongst various species. At the same time, in different species there are variations in the specific functions of DDK. In this review we stipulate if the discussed data has been obtained in studies with budding or fission yeasts or in vertebrate cells.
DDK is also known to act in Meiosis I. During this stage DDK collaborates with the polo-kinase Cdc5p and is involved in the recruitment of the Monopolin complex to the kinetochores and in the destruction of the cohesin rings and the synaptonemal complex [6–9]. In mitosis DDK associates with Rtt107p to control the activity of Mus81-Mms4 resolvase [10] and the degradation of the cohesin acetyltransferase Eco1p [11]. The meiotic and mitotic roles of DDK are not central to this review and will not be discussed in detail here. For recent reviews we recommend [6,12,13].
DDK and the activation of origins of DNA replication
Studies in S. cerevisiae have revealed that DDK, in tight coordination with the Cyclin Dependent Kinase (CDK) and other kinases, phosphorylates several S/T residues on the subunits of the MCM replicative helicase [1,2,14] (Figure 1a). In the G1 phase, the hexameric MCM helicase is loaded on specific DNA sequences (referred to as origins or Autonomously Replicating Sequences (ARS)) that are already occupied by the Origin Recognition Complex (ORC). The phosphorylation of the MCM proteins by CDK and DDK is required for the subsequent addition of Cdc45p-Sld3p and the GINS (Go Ichi Ni San) proteins to the MCM-occupied origins, for the activation of the MCM helicase and for the initiation of DNA replication (Figure 1a). The specificity of MCM phosphorylation by DDK is determined by DDK-docking sites on both Mcm2p and Mcm4p [15–17]. However, it had demonstrated that the sole essential role of DDK is to phosphorylate the N-terminal S/T-rich domain of Mcm4p and to relieve the inhibitory activity of this domain [18,19] (Figure 1a). The phosphorylation of other sites on the Mcm4p and the other MCM proteins is dispensable for viability and their precise role is not entirely understood.
Figure 1.
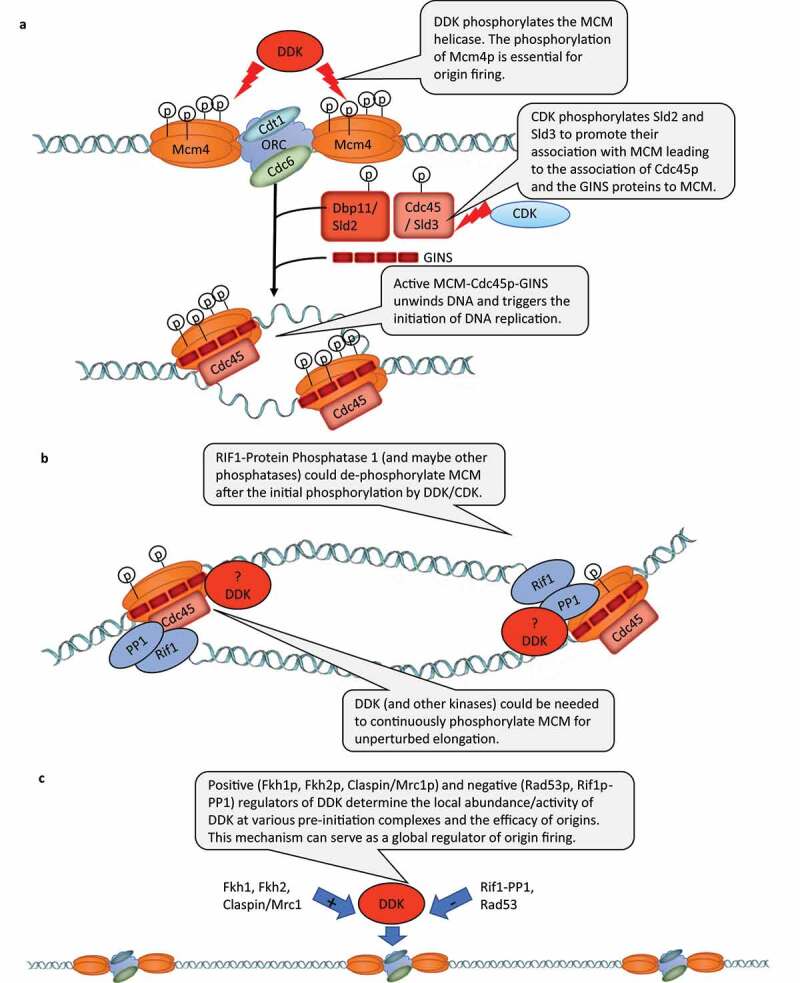
A) Essential role of DDK in the phosphorylation of MCM4 (and other MCM proteins) at pre-initiation complexes. b) RIF1-Protein Phosphatase 1 (and possibly other phosphatases) could de-phosphorylate MCM proteins after the initiation of DNA replication. Positive and negative regulators of DDK control the locus-specific activity of DDK and serve in the global control of origin firing
Speech bubbles are included in all figures to pinpoint the known function of DDK in the described processes.
DDK and RIF1-Protein Phosphatase 1
An interesting addition to our understanding of the roles of DDK in DNA replication have been provided by studies on RIF1 [20]. Rif1p had been described as a protein that binds to Rap1p and counteracts its gene silencing activity [21]. Later studies in S. pombe and S. cerevisiae indicated that the deletion of RIF1 generated hyperphosphorylated Mcm4p and that Rif1p interacted with the protein phosphatase Glc7p [22–24]. Mutations that interfere with the Glc7p-Rif1p interaction in cells with defective CDC7 or DBF4 produced a normal phenotype arguably because of the reduced RIF1-dependent dephosphorylation of MCM [23,24]. These results suggested that during early S phase Rif1p negatively impacts the firing of late origins by counteracting the phosphorylation of MCM (and possibly other targets) and by balancing out the activity of DDK [25] (Figure 1b). However, it is not known if at the repressed origins Glc7p-Rif1p counteracts the essential phosphorylation of Mcm4p by DDK or some other function of the kinase.
Similar effects of DDK and RIF1 had been observed in vertebrate cells. The immuno-depletion of RIF1 from Xenopus extracts increased the DDK-dependent phosphorylation of chromatin-bound MCM and the rate of replication initiation [26]. In HeLa cells, the suppression of RIF1 lead to a speedy transition through S-phase and to a substantial deficiency in blocking spurious replication initiation [26]. Importantly, the use of inhibitors for DDK, ATR/Chk1 and Protein Phosphatase 1 (PP1) demonstrated that PP1-dependent MCM dephosphorylation occurs on the elongating CMG helicase, but with different kinetics and at different S/T residues compared to MCM complexes at licensed origins [26] (Figure 1b). In addition, fork stability seemed to be dependent on the continuous phosphorylation of MCM within the CMG (Cdc45/MCM/GINS) complex and that both DDK and ATR/Chk1 were shown to be the contributing kinases [26]. Another study demonstrated that the loss of ETAA1 (an activator of ATR/Chk1) or RIF1 reduce the sensitivity to inhibition of DDK during late S phase, thus reiterating that both DDK and ATR counter the activity of RIF1-PP1 [27]. In agreement, the inhibition of DDK alone did not reverse the excessive nascent DNA resection in cells lacking RIF1 suggesting that RIF1-PP1 balances the activity of other kinases involved in DNA repair [28]. Finally, a recent study showed that in vertebrate cells RIF1 is enriched at stalled replication forks and that cells without RIF1 excessively degrade reversed forks [29].
It is apparent that in addition to its roles in negatively controlling the firing of origins in vertebrates, RIF1-PP1 also acts during the elongation stage of DNA replication and could be a protecting factor for stalled forks. However, the detailed mechanisms of interplay between RIF1-PP1 and DDK (and other kinases) during elongation remain to be elucidated.
DDK and the global control of origin firing in S. cerevisiae
Recent studies have indicated that DDK is not a simple molecular “on” switch for origins, but also acts as a global regulator of origin activity. Earlier, a link between the transcription factors Fkh1p and Fkh2p and the timing of origin firing has been observed [30]. In 2017, it was reported that Dbf4p is specifically enriched at early origins through its interactions with Fkh1p and Fkh2p and that the fusion of the Fkh1p DNA-binding domain to Dbf4p restores the FKH1-dependent origin firing [31] (Figure 1c). The authors also indicated that Dbf4p directly interacts with Sld3p and promotes the recruitment of downstream limiting factors to these FKH1-dependent origins. Another paper demonstrated that in G1 and G2 phases of the cell cycle the Rad53p kinase phosphorylates Sld3p and Dbf4p at the same sites as in S-phase [32]. This activity had a negative effect on origin firing and was required to prevent spurious initiation at the G1/S transition and re-replication in G2/M [32]. It is not clear if only the kinase activity of Rad53p is necessary for the inhibition of DDK and Sld3p outside of S-phase. For example, it has been shown that in a reconstituted in vitro system that Rad53p reduces the association of DDK to the MCM hexamer, but this function was not dependent on the Rad53 kinase activity [33]. Hence, steric inhibition or a sequestration of DDK by Rad53p is possible [25], but it remains unknown if this activity of Rad53p works outside S-phase. Importantly, the above papers indicated that the abundance of DDK at various loci can be positively or negatively regulated by Fkh1p, Fkh2p, Rad53p, and possibly other factors (Figure 1c). In this regard, a recent study unveiled how the differential abundance of DDK at origins in S-phase can regulate the genome-wide regulation of the initiation of DNA replication [34]. The authors demonstrated that multiple intermediate Cdc45p/GINS complexes are formed on MCM in vivo and that the activity of DDK determines the efficiency of assembly of these intermediate complexes [34]. In a second, less efficient step, some of these complexes form the functional CMG complex that is poised to initiate DNA replication [34].
The above studies suggest that the control of DDK levels over various loci and/or the balancing of DDK activity by protein phosphatases contribute to the control of the efficiency/timing of origin firing throughout the genome (Figure 1c). It is not clear if and how this novel role of DDK is related to its essential function as a triggering kinase at the origins. For example, it is not clear if the essential phosphorylation of Mcm4p, the phosphorylation of other MCM proteins or the phosphorylation of other substrates are central to this role of DDK.
DDK and “replication stress
Replication forks encounter various impediments to elongation in the form of DNA damage, tightly bound proteins, unusual DNA structures or a deficiency of dNTPs. All these are jointly referred to as “replication stress” and are known to activate a conserved checkpoint pathway involving a sensor kinase (Mec1p/Rad3p/ATR), Mrc1p/Claspin and an effector kinase (Rad53p/Cds1p/Chk1p) [3].
In S. cerevisiae DDK is required for the activation of the effector kinase and could play a direct role in the response to certain forms of replication stress [25]. For example, it has been demonstrated that hypomorphic alleles of CDC7 drastically reduce the rate of UV-induced or chemically induced mutagenesis [35–37]. Conversely, the overexpression of CDC7 increases the levels of induced mutagenesis [38]. Interestingly, increased DDK expression in human cancers was also correlated to increased chemoresistance and higher mutation frequencies [39]. These studies suggested that DDK may be required for some form of error-prone DNA repair or translesion error-prone DNA replication. In agreement, other studies in budding yeasts have presented evidence that the activation of the checkpoint by exposure to mutagens or dNTP depletion leads to the phosphorylation and the suppression of DDK and Sld3p activity by the effector Rad53p kinase and to the repression of the late-firing origins [40,41] (Figure 1c). The suppression of DDK is also mediated by an additional non-canonical interaction between Dbf4p and Rad53p [42].
Replication stress in vertebrates also leads to the phosphorylation of DBF4 by the effector ATR/Chk kinases, however DDK remains active and could be involved in fork protection and reactivation [43,44]. Several recent studies have provided significant mechanistic details about functions of DDK in these organisms. In vertebrates, DDK is directly involved in the regulation of Trans-Lesion DNA synthesis [45]. It has been shown that in UV light treated cells DDK phosphorylates RAD18 and stimulates its association with DNA polymerase η. In turn, RAD18 acts as a chaperone to recruit Pol η to PCNA, which is ubiquitinated at the stalled fork (Figure 2a). Another study has indicated that DDK promotes the resection of newly synthesized DNA in human cells treated with Hydroxyurea [46]. The treatment with Hydroxyurea is known to induce replication fork stalling and the generation of extended stretches of ssDNA [46]. This effect was likely due to the phosphorylation of the EXO1 nuclease, which is required for nascent strand degradation [46] (Figure 2a). It has not been explicitly shown if DDK is recruited to the stalled fork to phosphorylate RAD18 and EXO1 or if they are phosphorylated elsewhere in the nucleoplasm. However, a third study clearly demonstrated that DDK co-localizes with stalled replication forks [47]. DDK was also necessary for the slowing down of fork progression upon replication stress and for the stability of the slowed/paused forks and for their subsequent restarting [47]. These effects were attributed to the DDK-mediated phosphorylation of MRE11, another exonuclease involved in the resection of DNA at stalled forks [47](Figure 2a).
Figure 2.
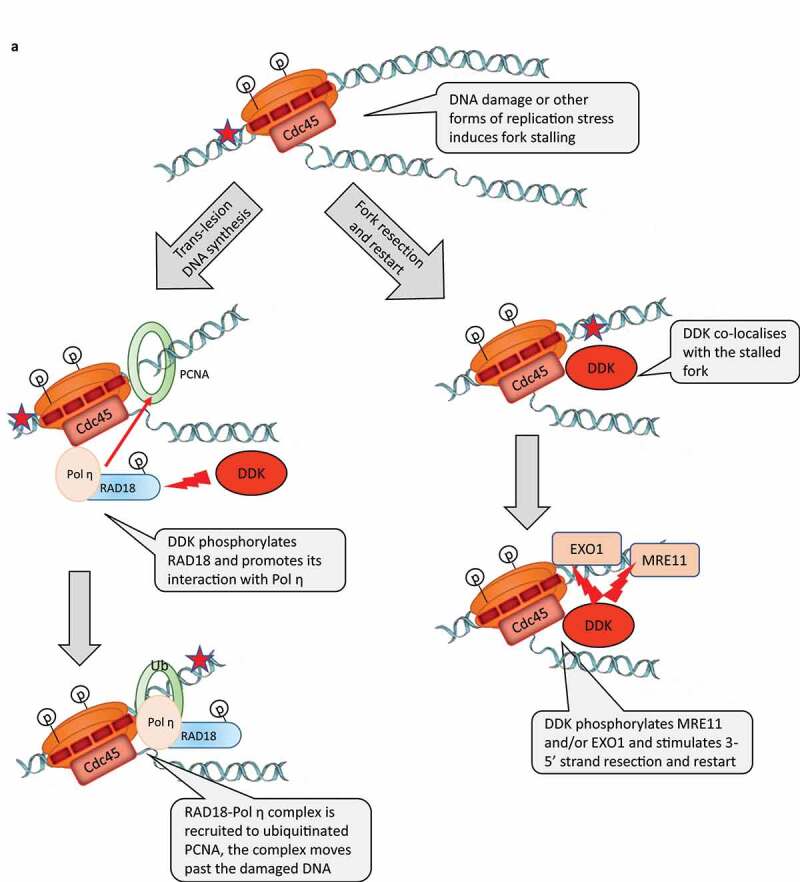
A) DDK participates in the response to DNA damage. Left-hand column: role of DDK in trans-lesion DNA synthesis. Right-hand panel: role of DDK in strand resection and restart of stalled forks. Programmed stalling of replication forks at the RFB loci of S. cerevisiae. DDK participates the replication-coupled reassembly of chromatin
Speech bubbles are included in all figures to pinpoint the known function of DDK in the described processes.
Another study performed in human cells and Xenopus extracts reiterated that DDK has multiple roles during the elongation stage of DNA replication [44]. Of significance, most of the experiments in this paper were performed without exposure to mutagens. As expected, the highly specific inhibition of DDK affected the firing of late origins that are normally inhibited by ATR/Chk [44]. In addition, the inhibition of DDK induced spontaneous stalling of early-firing forks and DDK was required to stabilize and restart these arrested forks [44]. Furthermore, the authors identified multiple DDK targets as effectors in fork stabilization. Similar to its effect on Mcm4p, the phosphorylation by DDK alleviated an auto-inhibitory effect of intrinsically disordered regions in these substrates [44]. Many of the effects of the inhibition of DDK could be reversed by the inhibition of the ATR/Chk kinases [44] suggesting that the balance between the activities of these two kinases is a critical regulator of both origin firing and fork progression in vertebrates. Further support to the significance of DDK in post-initiation events in DNA replication came from studies in embryonic stem cells, where replication stress is suppressed by the high abundance of the MYBL2 protein [48]. In these cells the loss of MYBL2, ATM or MRE11 activity lead to a significant reduction in fork speed, increased fork stalling and elevated origin firing [48]. Interestingly, the concomitant inhibition of CDC7 reversed the effect on replication fork speed and replication fork stability without any significant effect on the overall replication rates [48].
It is noteworthy that that CDC7 and DBF4 are overexpressed in a variety of cancer cells, where dealing with replication stress is critical [49]. Even more, the suppression of DDK by newly developed drugs has been identified as a promising anti-cancer strategy [50,51]. For this and other good reasons the detailed mechanistic description of these post-initiation roles of DDK will undoubtedly be a focus of future studies. Apart from the identification of relevant DDK substrates, it will be interesting to address these issues in the context of other kinases and phosphatases. As discussed earlier, the activity of DDK is countered by the Protein Phosphatase 1 [25]. It will be interesting to test if the inhibition of PP1 or other phosphatases could abolish the requirement of DDK for fork stability, fork speed, and for the resumption of elongation upon various forms of replication stress.
DDK and stalled replication forks
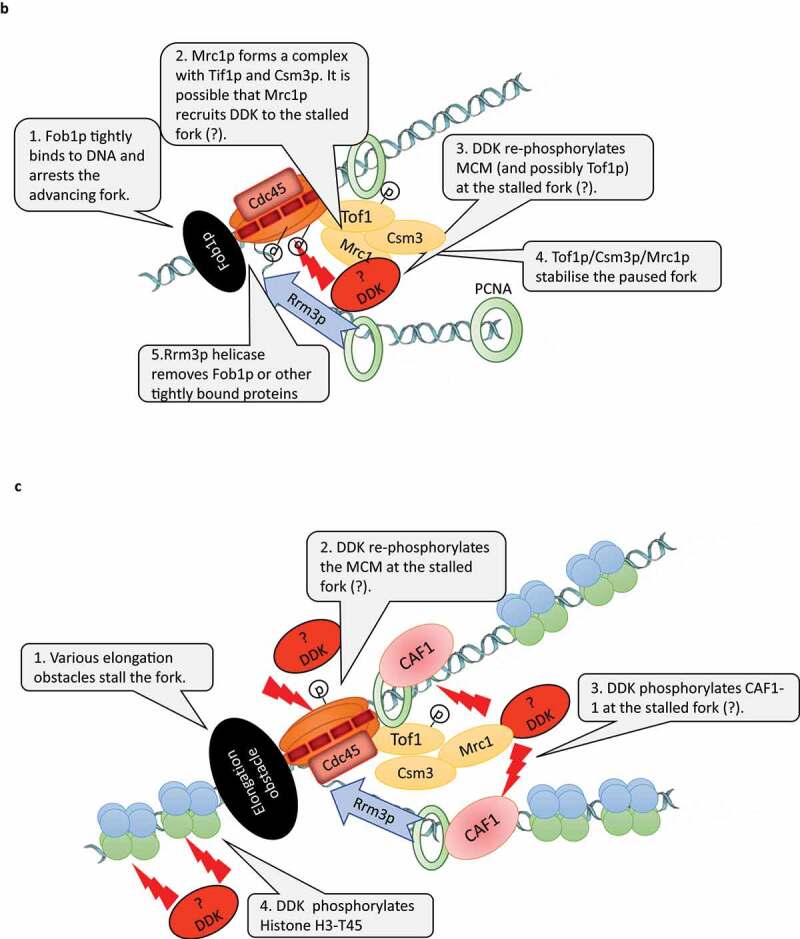
The replication forks in the rRNA gene cluster of S. cerevisiae undergo a programmed arrest to prevent collisions with the RNA polymerase I that is transcribing in the opposite direction [52]. The pausing is produced by the tight binding of the Fob1p protein to the RFB (Replication Fork Barriers) sites between the rRNA gene repeats (Figure 2b). Upon pausing, a Fork Stabilization Complex (FSC) is formed by Mrc1p and the Tof1p/Csm3p dimer [53] (Figure 2b). FSC inhibits the helicase activity of MCM and bridges the DNA helicase and DNA polymerase to prevent fork distortions and collapse. It had demonstrated that in addition to the stabilization of the paused forks, Tof1p is also needed to counteract the activity of the Rrm3p helicase [54]. Rrm3p directly binds to the replication clamp (PCNA) and is believed to facilitate the rescue of the arrested forks by displacing the Fob1p protein from the RFB site via its 5ʹ-3ʹ helicase activity [55]. Similar Mrc1p/Tof1p/Csm3p complexes are observed on chromatin upon the treatment of cells with hydroxyurea [56] while the deletion of RRM3 leads to fork pausing at numerous loci outside of the rRNA gene cluster [57–59]. It seems that the mechanisms of fork stabilization and rescue at the rRNA gene clusters also operates at other difficult to replicate sites throughout the genome.
Figure 2.
Continued
Recently, it was found that the phosphorylation of MCM by DDK is required for the programmed fork arrest and for the formation of FSC at the RFB sites [53] (Figure 2b). Tof1p must also be phosphorylated for the formation of FSC, but it is not clear if DDK or another kinase fulfills this function [53]. Still, DDK seems to play a major role in the stabilization and processing of paused replication forks at the RFB sites. It remains to be established if the phosphorylation of MCM by DDK at the time of initiation is a prerequisite for the proper fork arrest or, alternatively, DDK is required to re-phosphorylate MCM (and possibly Tof1p) upon the pausing of the fork. The later possibility is fueled by two studies suggesting that the continuous re-phosphorylation of MCM by DDK is necessary for efficient elongation (Figure 1b). It is known that the phosphorylation of MCM2 at S164 and S170 allows for the opening of the MCM ring and its loading onto DNA [60]. Interestingly, MCM2 with S164E/S170E phosphor-mimicry substitutions reduced the helicase activity of MCM while increasing its DNA binding [61]. The mutant MCM2-S164E/S170E protein had a dominant-negative effect and increased the sensitivity of the cells to exposure to mutagens [61]. So, the inability to dephosphorylate these Serines acts as an impediment to the helicase function of MCM and to the proper processing of DNA damage [61]. More recent studies in vertebrates suggested that MCM is gradually dephosphorylated upon the progression of the forks [26,44]. If indeed MCM needs to be re-phosphorylated upon fork arrest, it will be interesting to establish which components of the fork act as DDK recruiters and if other kinases are involved. Furthermore, the downstream effects of DDK at paused forks needs to be addressed by mechanistic studies.
Mrc1p/claspin and DDK
Mrc1p (called claspin in S. pombe and vertebrates) is required for the efficient progression of S phase in both budding yeast and in vertebrates and associates with the elongating forks in S. cerevisiae [62,63]. In a defined in vitro replication system from budding yeasts Mrc1p stimulates fork progression [64]. As mentioned, at arrested forks in the rRNA gene cluster Mrc1p/Tof1p/Csm3p form the Fork Stabilization Complex [53,56]. Hence, Mrc1p needs to be considered as a bona fide elongation factor. On the other hand, several lines of evidence suggest that DDK is involved in post-initiation events mediated by Mrc1p/Tof1p/Csm3p. In S. cerevisiae, the binding of Mrc1p to chromatin during the mitotic S phase is dependent on Dbf4p [65]. In addition, during the pre-meiotic S phase Tof1p/Csm3p recruits DDK to newly synthesized DNA. Later on, DDK phosphorylates key substrates to enable meiotic recombination [66]. In S. pombe DDK is required for the activation of Cds1 (the homologue of the effector kinase Rad53p) and the concomitant phosphorylation of claspin, both of which are induced by replication fork blockages and mediated by Rad3p (the S. pombe homologue of Mec1p) [67]. Remarkably, the deletions of the CDS1 and MRC1 genes in S. pombe abolished the requirement for DDK for viability and for the initiation of DNA replication [68].
Mrc1p/claspin physically interacts with multiple replication factors and could have additional roles in DNA replication [3]. A key study in vertebrates demonstrated that MRC1/claspin plays a fundamental role in the initiation of DNA replication [69]. It turned out that claspin recruits DDK to origins via an interaction with CDC7 and that this interaction is necessary and sufficient for the phosphorylation of MCM and for the firing of origins693. The CDC7-binding domain of claspin was shown to be involved in an inhibitory intramolecular interaction, which is masking the DNA-binding domain and a PIP (PCNA Interacting Peptide) in claspin [69]. The DDK-dependent phosphorylation of claspin abolished this intramolecular interaction and is likely to contribute to the loading of claspin to the replication fork [69]. Another paper from Masai lab showed that DDK participates in the Mec1p/ATR-Mrc1p/Claspin-Rad53p/Chk1 pathway by phosphorylating the Chk1-binding-domain of Claspin [70]. This domain is also phosphorylated by Casein Kinase 1 [71]. It remains to be established how the two kinases cooperate in the response to replication stress.
The current evidence indicates that DDK is recruited to vertebrate origins via a docking site on Claspin [69] and to budding yeast origins via a docking site on Mcm4p [19]. In budding yeast Tof1p/Csm3p is involved in the recruitment of DDK to elongating forks [72]. As mentioned, it is also possible that DDK is recruited to paused forks by presently unknown “recruiters”. Given the recent advances in our understanding of the DDK-claspin interactions, it is important to establish if the Mrc1p/claspin-DDK association is employed in the detection of replication stress, in the suppression of late-firing origins and in the re-activation of stalled forks (Figure 2 b, c). It also remains to be resolved if Mrc1p and DDK in S. cerevisiae play a role in the initiation of DNA replication.
DDK and replication-coupled assembly of chromatin
The disassembly of nucleosomes and their reassembly behind the fork is a key parallel process during the replication of DNA in eukaryotes [73]. The proper recycling of old histones and the reassembly of nucleosomes is critical for the faithful transmission of epigenetic marks and for the rebuilding of the same type of chromatin after each passage of the fork. The deregulation of the disassembly/reassembly of chromatin can lead to altered epigenetic transmission and is best exemplified by the loss of the heterochromatin-mediated gene silencing at various loci in budding and fission yeasts as well as other eukaryotes [74]. While a role of DDK in the replication-coupled disassembly/reassembly of chromatin is yet to be detailed, certain evidence suggests that it could be directly or indirectly involved in these processes.
It is known that in addition to its effects on DNA replication, mutations in CDC7, DBF4 and other replication factors significantly reduce the heterochromatin-mediated gene silencing at the sub-telomeres and the mating-type loci in S. cerevisiae[75]. However, it is not clear if these effects are linked to defects in the silencing function of dormant origins (these are non-firing origins and act as silencers at the mating-type loci or as proto-silencers at the telomeres [76]) or by a deficiency in the elongation replisome. Recent evidence suggests that these effects could be linked to the replication-coupled assembly of chromatin (Figure 2c). DDK, as well CDK, have been shown to phosphorylate the largest subunit of Chromatin Assembly Factor 1 (CAF1) in both yeast and vertebrates [77,78]. CAF1 is known to directly binds to the replication clamp (PCNA). It is believed that via this association CAF1 travels behind the fork and assembles H3/H4 histone tetramers [73]. It is not clear if this histone chaperone contributes to the reassembly of the old dis-assembled H3/H4 histones or if it builds up tetramers from the H3/H4 dimers delivered from the cytoplasm [74]. In S. cerevisiae, the phosphorylation of CAF1 by CDK, but not DDK, is required for the loading of CAF1 to chromatin [78]. It is plausible that the phosphorylation of CAF1 by DDK has a post-initiation role that affects chromatin structure. In agreement, it has been recently shown that S→A mutations in the DDK and CDK target sites of CAF1 have a dominant-negative effect and lead to a substantial loss of gene silencing at multiple loci as well as to a prolonged S-phase and to increased sensitivity to mutagens [79]. Mutations in the CDK target sites only did not have this effect. Hence, its is possible that the observed defects in gene silencing in cdc7 and dbf4 mutants could be mediated by the phosphorylation of CAF1 and by the disrupted reassembly of chromatin behind the forks. Of notice, another major substrate of DDK, the MCM helicase, also plays a key role in the replication-coupled reassembly of chromatin. It has long been speculated that the Mcm2p subunit of the MCM helicase also acts as a histone chaperone. Recently, it has been demonstrated that in both human and budding yeast cells Mcm2p distributes the disassembled H3/H4 histone tetramers to the lagging strand [80,81] while DNA polymerase ε distributes them to the leading strand [82]. At present, it is not known if the phosphorylation of Mcm2p by DDK is necessary for its histone chaperone function. Another study in S. cerevisiae had demonstrated that upon replication stress DDK phosphorylates Histone H3-T45 [83]. The H3-T45 residue resides at the DNA entry/exit site of the nucleosome. For this reason, it has been suggested that its phosphorylation could facilitate the resumption of elongation at stalled forks [83]. The role of DDK at paused replication forks and the phosphorylation of Mcm2p and CAF1 remains enigmatic and needs to be addressed in future studies.
Concluding remarks
DDK has been discovered more than 30 years ago. Initially, most of the research on this kinase had been focused on its essential role in the initiation of DNA replication. Indeed, in many reviews and most textbooks the only discussed function of DDK is the phosphorylation of the MCM helicase at the pre-assembled initiation complexes. Later, studies have demonstrated that DDK works during mitosis and meiosis as well as in trans-lesion DNA synthesis. More recently we have witnessed several lines of solid evidence pointing to far more diverse roles of DDK at all stages of DNA replication. Many studies have also explored the potential of DDK as a drug target for cancer therapy. The continuation of the studies on the emerging roles of DDK in the global control of origin firing and upon replication stress will certainly provide valuable mechanistic background to these clinical studies. Its less-characterized involvement in the replication-coupled chromatin assembly will be of interest to the fields of chromatin biology and epigenetics. By all means, DDK can no longer be treated as a simple molecular switch at origins. In the near future we expect to achieve a far-better understanding of its non-essential functions.
Acknowledgments
This work was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada [RGPIN-2015-06727)].
Funding Statement
This work was supported by the Canadian Network for Research and Innovation in Machining Technology, Natural Sciences, and Engineering Research Council of Canada [RGPIN-2015-06727)].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Bell SP, Labib K.. Chromosome duplication in saccharomyces cerevisiae. Genetics. 2016;203:1027–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Masai H, Matsumoto S, You Z, et al. Eukaryotic chromosome DNA replication: where, when, and how? Annu. Rev Biochem. 2010;79(1):89–130. [DOI] [PubMed] [Google Scholar]
- [3].Masai H, Yang CC, Matsumoto S. Mrc1/Claspin: a new role for regulation of origin firing. Curr Genet. 2017;63(5):813–818. [DOI] [PubMed] [Google Scholar]
- [4].Jouannic S, Champion JA, Segui-Simarro JM, et al. The protein kinases AtMAP3Kε1 and BnMAP3Kε1 are functional homologues of S. pombe cdc7p and may be involved in cell division. Plant J. Internet] 2001. [cited 2021 May 14]; 26:637–649. Available from;6: https://pubmed.ncbi.nlm.nih.gov/11489177/ [DOI] [PubMed] [Google Scholar]
- [5].Stephenson R, Hosler MR, Gavande NS, et al. Characterization of a Drosophila ortholog of the Cdc7 kinase a role for Cdc7 in endoreplication independent of chiffon. J Biol Chem. Internet] 2015. [cited 2021 May 14]; 290(3):1332–1347. Available from https://pubmed.ncbi.nlm.nih.gov/25451925/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gómez-Escoda B, Wu P-YJ. Roles of CDK and DDK in genome duplication and maintenance: meiotic singularities. 2017; [DOI] [PMC free article] [PubMed]
- [7].Challa K, Ghanim Fajish V, Shinohara M, et al. Meiosis-specific prophase-like pathway controls cleavage-independent release of cohesin by Wapl phosphorylation. PLoS Genet. Internet] 2019. [cited 2021 May 12]; 15(1):e1007851. Available from https://pubmed.ncbi.nlm.nih.gov/30605471/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Galander S, Barton RE, Borek WE, et al. Reductional meiosis i chromosome segregation is established by coordination of key meiotic kinases. Dev Cell. 2019;49(4):526–541.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Argunhan B, Leung W, Afshar N, et al. Fundamental cell cycle kinases collaborate to ensure timely destruction of the synaptonemal complex during meiosis. EMBO J. Internet] 2017. [cited 2021 May 12]; 36:2488–2509. Available from. ;(17):. https://pubmed.ncbi.nlm.nih.gov/28694245/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Princz LN, Wild P, Bittmann J, et al. Pfander B. Dbf4‐dependent kinase and the Rtt107 scaffold promote Mus81‐Mms4 resolvase activation during mitosis. EMBO J. Internet] 2017. [cited 2021 May 12]; 36:664–678. Available from. ;(5):. https://pubmed.ncbi.nlm.nih.gov/28096179/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Seoane AI, Morgan DO. Firing of replication origins frees Dbf4-Cdc7 to Target Eco1 for destruction. Curr Biol. 2017;27(18):2849–2855.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mackenzie AM, Lacefield S. CDK regulation of meiosis: lessons from S. cerevisiae and S. pombe [Internet]. Genes (Basel). 2020. [cited 2021 Jun 8];11(7):1–27. Available from: https://pubmed.ncbi.nlm.nih.gov/32610611/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fulcher LJ, Sapkota GP. Mitotic kinase anchoring proteins: the navigators of cell division [Internet]. Cell Cycle. 2020. [cited 2021 Jun 8];19(5):505–524. Available from: https://pubmed.ncbi.nlm.nih.gov/32048898/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lin YC, Prasanth SG. Replication initiation: implications in genome integrity. DNA Repair (Amst). 2021;103:103131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sheu YJ, Stillman B. Cdc7-Dbf4 Phosphorylates MCM proteins via a docking site-mediated mechanism to promote s phase progression. Mol Cell. 2006;24(1):101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ramer MD, Suman ES, Richter H, et al. Dbf4 and Cdc7 proteins promote DNA replication through interactions with distinct Mcm2-7 protein subunits. J Biol Chem. Internet] 2013. [cited 2021 May 18]; 288:14926–14935. Available from. ;(21):. https://pubmed.ncbi.nlm.nih.gov/23549044/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bruck I, Kaplan D. Dbf4-Cdc7 phosphorylation of Mcm2 is required for cell growth. J Biol Chem. Internet] 2009. [cited 2021 May 18]; 284:28823–28831. Available from. ;(42):. https://pubmed.ncbi.nlm.nih.gov/19692334/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sheu YJ, Kinney JB, Lengronne A, et al. Domain within the helicase subunit Mcm4 integrates multiple kinase signals to control DNA replication initiation and fork progression. Proc Natl Acad Sci U S AInternet] 2014. [cited 2021 May 18]; 11118:E1899.Available from:/pmc/articles/PMC4020090/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sheu YJ, Stillman B. The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature. 2010;463(7277):113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Alavi S, Ghadiri H, Dabirmanesh B, et al. G-quadruplex binding protein Rif1, a key regulator of replication timing [Internet]. J Biochem. 2021. [cited 2021 Jul 5];169(1):1–14. Available from: https://pubmed.ncbi.nlm.nih.gov/33169133/ [DOI] [PubMed] [Google Scholar]
- [21].Hardy CFJ, Sussel L, Shore D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. Internet] 1992. [cited 2021 Jun 3]; 6:801–814. Available from. ;(5):. https://pubmed.ncbi.nlm.nih.gov/1577274/ [DOI] [PubMed] [Google Scholar]
- [22].Hiraga SI, Alvino GM, Chang FJ, et al. Rif1 controls DNA replication by directing protein phosphatase 1 to reverse Cdc7- mediated phosphorylation of the MCM complex. Genes Dev. Internet] 2014. [cited 2021 May 18]; 28:372–383. Available from. ;(4):. https://pubmed.ncbi.nlm.nih.gov/24532715/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Davé A, Cooley C, Garg M, et al. Protein phosphatase 1 recruitment by Rif1 regulates DNA replication origin firing by counteracting DDK activity. Cell Rep. Internet] 2014. [cited 2021 May 18]; 7(1):53–61. Available from https://pubmed.ncbi.nlm.nih.gov/24656819/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mattarocci S, Shyian M, Lemmens L, et al. Rif1 Controls DNA replication timing in yeast through the PP1 phosphatase Glc7. Cell Rep. Internet] 2014. [cited 2021 May 18]; 71):62–69. Available from https://pubmed.ncbi.nlm.nih.gov/24685139/ [DOI] [PubMed] [Google Scholar]
- [25].Larasati DBP, Duncker B. Mechanisms Governing DDK Regulation of the Initiation of DNA Replication. Genes (Basel). Internet] 2016. [cited 2021 May 11]; 8. Available from. ;(1):3. http://www.ncbi.nlm.nih.gov/pubmed/28025497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Alver RC, Chadha GS, Gillespie PJ, et al. Reversal of DDK-mediated MCM phosphorylation by Rif1-PP1 regulates replication initiation and replisome stability independently of ATR/Chk1. Cell Rep. Internet] 2017. [cited 2021 May 27]; 18(10):2508–2520. Available from https://pubmed.ncbi.nlm.nih.gov/28273463/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rainey MD, Bennett D, O’Dea R, et al. ATR Restrains DNA synthesis and mitotic catastrophe in response to CDC7 inhibition. Cell Rep. Internet] 2020. [cited 2021 Jun 3]; 32(9): Available from.108096. https://pubmed.ncbi.nlm.nih.gov/32877678/ [DOI] [PubMed] [Google Scholar]
- [28].Garzón J, Ursich S, Lopes M. Hiraga S Ichiro, donaldson AD. Human RIF1-Protein Phosphatase 1 prevents degradation and breakage of nascent DNA on Replication Stalling. Cell Rep. Internet] 2019. [cited 2021 May 27]; 27(9):2558–2566.e4. Available from https://pubmed.ncbi.nlm.nih.gov/31141682/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mukherjee C, Tripathi V, Manolika EM, et al. RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat CommunInternet] 2019. [cited 2021 May 27]; 101: Available from. https://pubmed.ncbi.nlm.nih.gov/31337767/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Knott SRV, Peace JM, Ostrow AZ, et al. Forkhead transcription factors establish origin timing and long-range clustering in Scerevisiae. Cell. 2012;148(1–2):99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fang D, Lengronne A, Shi D, et al. Dbf4 recruitment by forkhead transcription factors defines an upstream rate-limiting step in determining origin firing timing. Genes Dev. Internet] 2017. [cited 2021 May 12]; 31:2405–2415. Available from;23–24: https://pubmed.ncbi.nlm.nih.gov/29330352/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Johnson MC, Can G, Santos MM, et al. Checkpoint inhibition of origin firing prevents inappropriate replication outside of s-phase. Elife Internet] 2021. [cited 2021 May 13]; 10:1–16. Available from. https://pubmed.ncbi.nlm.nih.gov/33399537/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wahab SA, Remus D. Antagonistic control of ddk binding to licensed replication origins by mcm2 and rad53. Elife. 2020;9:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].De Jesús-Kim L, Friedman LJ, Lõoke M, et al. Ddk regulates replication initiation by controlling the multiplicity of cdc45-gins binding to mcm2-7. Elife Internet] 2021. [cited 2021 May 12]; 10:1–83. Available from https://pubmed.ncbi.nlm.nih.gov/33616038/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gd N, BJ K. cdc7-1 a temperature sensitive cell-cycle mutant which interferes with induced mutagenesis in Saccharomyces cerevisiae. Mol Gen Genet. Internet] 1982. [cited 2021 Sep 15]; 186:478–481. Available from.(4): https://pubmed.ncbi.nlm.nih.gov/6752657/ [DOI] [PubMed] [Google Scholar]
- [36].Ostroff RM, Sclafani RA. Cell cycle regulation of induced mutagenesis in yeast. Mutat Res - Fundam Mol Mech Mutagen. 1995;329(2):143–152. [DOI] [PubMed] [Google Scholar]
- [37].Re H, Rm O, Mb K, et al. Molecular genetic studies of the Cdc7 protein kinase and induced mutagenesis in yeast. Genetics. Internet] 1992. [cited 2021 Sep 15]; 132(1):53–62. Available from. https://pubmed.ncbi.nlm.nih.gov/1398063/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ra S,MP,JR, WL F. Differential regulation of the yeast CDC7 gene during mitosis and meiosis. Mol Cell Biol [Internet] 1988. [cited 2021 Sep 15]; 8:293–300. Available from: https://pubmed.ncbi.nlm.nih.gov/3275871/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].NK S, Bhutkar A, Lanning NJ, A B, NJ L, JP M, M W . DDK promotes tumor chemoresistance and survival via multiple pathways. NeoplasiaInternet] 2017. [cited 2021 Sep 15]; 19:439–450. Available from. ;5: https://pubmed.ncbi.nlm.nih.gov/28448802/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zegerman P, Diffley JFX. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature. Internet] 2010. [cited 2021 May 18]; 467(7314):474–478. Available from: https://pubmed.ncbi.nlm.nih.gov/20835227/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lopez-Mosqueda J, Maas NL, Jonsson ZO, et al. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature. Internet] 2010. [cited 2021 May 18]; 467(7314):479–483. Available from: https://pubmed.ncbi.nlm.nih.gov/20865002/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Matthews LA, Selvaratnam R, Jones DR, et al. A novel non-canonical forkhead-associated (FHA) domain-binding interface mediates the interaction between Rad53 and Dbf4 proteins. J Biol Chem. Internet] 2014. [cited 2021 May 18]; 289(5):2589–2599. Available from:https://pubmed.ncbi.nlm.nih.gov/24285546/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lee AYL, Chiba T, Truong LN, et al. is direct downstream target of ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) protein to regulate intra-S-phase checkpoint. J Biol Chem. 2012;287(4):2531–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jones MJK, Gelot C, Munk S, et al. Human DDK rescues stalled forks and counteracts checkpoint inhibition at unfired origins to complete DNA replication. Mol Cell. Internet] 2021. [cited 2021 May 12]; 81:426–441.e8. Available from;3: https://pubmed.ncbi.nlm.nih.gov/33545059/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Day TA, Palle K, Barkley LR, et al. Phosphorylated Rad18 directs DNA polymerase η to sites of stalled replication. J Cell Biol. 2010;191(5):953–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sasi NK, Coquel F, Lin YL, et al. Has a primary role in processing stalled replication forks to initiate downstream checkpoint signaling. Neoplasia (United States). Internet] 2018. [cited 2021 May 27]; 20(10):14926–14935. Available from:https://pubmed.ncbi.nlm.nih.gov/30157471/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rainey MD, Quinlan A, Cazzaniga C, et al. CDC7 kinase promotes MRE11 fork processing, modulating fork speed and chromosomal breakage. EMBO RepInternet] 2020. [cited 2021 May 12]; 21. Available from. ;8: https://pubmed.ncbi.nlm.nih.gov/32496651/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Blakemore D, Vilaplana‐Lopera N, Almaghrabi R, et al. MYBL2 and ATM suppress replication stress in pluripotent stem cells. EMBO RepInternet] 2021. [cited 2021 Jul 8]; 225. Available from: /pmc/articles/PMC8097389/: 10.15252/embr.202051120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yamada M, Masai H, Bartek J. Regulation and roles of Cdc7 kinase under replication stress [Internet]. Cell Cycle. 2014. [cited 2021 Jul 8];13(12):1859–1866. Available from: https://pubmed.ncbi.nlm.nih.gov/24841992/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang C, Vegna S, Jin H, C W, S V, H J, B B, C L, C R, RL de O, B M, J G, W W, et al . Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nat. 2019. cited 2021 Sep 15; 5747777: 268–272Available fromhttps://pubmed.ncbi.nlm.nih.gov/31578521/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rp M,JH, VE van der N,JR, Ja F, et al. A kinase inhibitor screen identifies a dual cdc7/CDK9 inhibitor to sensitise triple-negative breast cancer to EGFR-targeted therapy. Breast Cancer Res Internet] 2019. [cited 2021 Sep 15]; 21. Available from. https://pubmed.ncbi.nlm.nih.gov/31262335/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kaplan DL, Bastia D. Mechanisms of polar arrest of a replication fork: microReview. Mol Microbiol. 2009;72(2):279–285. [DOI] [PubMed] [Google Scholar]
- [53].Bastia D, Srivastava P, Zaman S, et al. Phosphorylation of CMG helicase and tof1 is required for programmed fork arrest. Proc Natl Acad Sci U S A. 2016;113(26):E3639–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mohanty BK, Bairwa NK, Bastia D. The Tof1p-Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2006;103(4):897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Torres JZ. Local chromatin structure at the ribosomal DNA causes replication fork pausing and genome instability in the absence of the S. cerevisiae DNA helicase Rrm3p. Genes Dev. Internet] 2004. [cited 2021 Jun 22]; 18(5):498–503. Available from: https://pubmed.ncbi.nlm.nih.gov/15037547/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bando M, Katou Y, Komata M, et al. Tof1, and Mrc1 form a heterotrimeric mediator complex that associates with DNA replication forks. J Biol Chem. 2009;284(49):34355–34365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Doi G, Okada S, Yasukawa T, et al. Catalytically inactive Cas9 impairs DNA replication fork progression to induce focal genomic instability. Nucleic Acids Res. Internet] 2021. [cited 2021 Jun 22]; 49(2):954–968. Available from: https://pubmed.ncbi.nlm.nih.gov/33398345/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ivessa AS, Zhou JQ, Schulz VP, et al. Saccharomyces Rrm3p, a 5′ to 3′ DNA helicase that promotes replication fork progression through telomeric and subtelomeric DNA. Genes Dev. 2002;16:1383–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Azvolinsky A, Dunaway S, Torres JZ, et al. The S. cerevisiae Rrm3p DNA helicase moves with the replication fork and affects replication of all yeast chromosomes. Genes Dev. 2006;20(22):3104–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Li N, Zhai Y, Zhang Y, et al. Structure of the eukaryotic MCM complex at 3.8 Å. Nature. Internet] 2015. [cited 2021 May 18]; 524(7564):186–191. Available from: https://pubmed.ncbi.nlm.nih.gov/26222030/ [DOI] [PubMed] [Google Scholar]
- [61].Stead BE, Brandl CJ, Davey MJ. Phosphorylation of Mcm2 modulates Mcm2-7 activity and affects the cell’s response to DNA damage. Nucleic Acids Res. Internet] 2011. [cited 2021 May 18]; 39(16):6998–7008. Available from:https://pubmed.ncbi.nlm.nih.gov/21596784/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Szyjka SJ, Viggiani CJ, Aparicio OM. Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol Cell. Internet] 2005. [cited 2021 May 16]; 19(5):691–697. Available from: https://pubmed.ncbi.nlm.nih.gov/16137624/ [DOI] [PubMed] [Google Scholar]
- [63].Gambus A, Jones RC, Sanchez-Diaz A, et al. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol. 2006;8(4):358–366. [DOI] [PubMed] [Google Scholar]
- [64].Yeeles JTP, Janska A, Early A, et al. How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol Cell. 2017;65(1):105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Osborn AJ. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. Internet] 2003. [cited 2021 May 27]; 17(14):1755–1767. Available from: https://pubmed.ncbi.nlm.nih.gov/12865299/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Murakami H, Keeney S. Temporospatial coordination of meiotic DNA replication and recombination via DDK recruitment to replisomes. Cell. Internet] 2014. [cited 2021 Jun 4]; 158(4):861–873. Available from: https://pubmed.ncbi.nlm.nih.gov/25126790/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tanaka K, Mrc1 RP. channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat Cell Biol. Internet] 2001. [cited 2021 May 28]; 3(11):966–972. Available from: https://pubmed.ncbi.nlm.nih.gov/11715017/ [DOI] [PubMed] [Google Scholar]
- [68].Matsumoto S, Hayano M, Kanoh Y, et al. Multiple pathways can bypass the essential role of fission yeast Hsk1 kinase in DNA replication initiation. J Cell Biol. Internet] 2011. [cited 2021 May 28]; 195(3):387–401. Available from: https://pubmed.ncbi.nlm.nih.gov/22024164/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yang CC, Suzuki M, and Yamakawa S, et al. Claspin recruits Cdc7 kinase for initiation of DNA replication in human cells. Nat Commun. 2016;. 12(7): ;12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yang CC, Kato H, and Shindo M, et al. Cdc7 activates replication checkpoint by phosphorylating the chk1 binding domain of claspin in human cells. Elife. 2019. 8 ;e50796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Meng Z, Capalbo L, Glover DM, et al. Role for casein kinase 1 in the phosphorylation of claspin on critical residues necessary for the activation of Chk1. Mol Biol Cell. Internet] 2011. [cited 2021 May 27]; 22(16):2834–2847. Available from: https://pubmed.ncbi.nlm.nih.gov/21680713/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Murakami H, Keeney S. DDK links replication and recombination in meiosis [Internet]. Cell Cycle. 2014. [cited 2021 Jun 4];13(23):3621–3622. Available from: https://pubmed.ncbi.nlm.nih.gov/25483055/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Stewart-Morgan KR, Petryk N, Groth A. Chromatin replication and epigenetic cell memory [Internet]. Nat Cell Biol. 2020. [cited 2021 Jul 12];22(4):361–371. Available from: https://pubmed.ncbi.nlm.nih.gov/32231312/ [DOI] [PubMed] [Google Scholar]
- [74].Rowlands H, Dhavarasa P, Cheng A, et al. Forks on the run: can the stalling of DNA replication promote epigenetic changes? [Internet]. Front Genet. 2017. [cited 2021 May 11];8:86. Available from: www.frontiersin.org [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sauty SM, Shaban K, Yankulov K. Gene repression in S. cerevisiae—looking beyond Sir-dependent gene silencing [Internet]. Curr Genet. 2021. [cited 2021 Jun 26];67:3–17. Available from: https://pubmed.ncbi.nlm.nih.gov/33037902/ [DOI] [PubMed] [Google Scholar]
- [76].Rehman MA, Yankulov K. The dual role of autonomously replicating sequences as origins of replication and as silencers [Internet]. Curr Genet. 2009. [cited 2021 Jun 26];55(4):357–363. Available from: https://pubmed.ncbi.nlm.nih.gov/19633981/ [DOI] [PubMed] [Google Scholar]
- [77].Gérard A, Koundrioukoff S, Ramillon V, et al. The replication kinase Cdc7-Dbf4 promotes the interaction of the p150 subunit of chromatin assembly factor 1 with proliferating cell nuclear antigen. EMBO Rep. 2006;7(8):817–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jeffery DCB, Kakusho N, You Z, et al. CDC28 phosphorylates Cac1p and regulates the association of chromatin assembly factor i with chromatin. Cell Cycle. 2015;14(1):74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Rowlands H, Shaban K, Cheng A, et al. CAF-I reveals its role in cell cycle progression and differential regulation of gene silencing. Cell Cycle. Internet] 2019. [cited 2021 Jun 26]; 18(22):3223–3236. Available from: https://pubmed.ncbi.nlm.nih.gov/31564230/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gan H, Serra-Cardona A, Hua X, et al. The Mcm2-Ctf4-Polα axis facilitates parental histone H3-H4 transfer to lagging strands. Mol Cell. Internet] 2018. [cited 2021 Jun 26]; 72(1):140–151.e3. Available from: https://pubmed.ncbi.nlm.nih.gov/30244834/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Petryk N, Dalby M, Wenger A, et al. MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science. (80-) [Internet] 2018. [cited 2021 Jun 26]; 361(6409):1389–1392. Available from: https://pubmed.ncbi.nlm.nih.gov/30115746/ [DOI] [PubMed] [Google Scholar]
- [82].Yu C, Gan H, Serra-Cardona A, et al. A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science. (80-) [Internet] 2018. [cited 2021 Jun 26]; 361(6409):1386–1389. Available from: https://pubmed.ncbi.nlm.nih.gov/30115745/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Baker SP, Phillips J, Anderson S, et al. Histone H3 Thr 45 phosphorylation is a replication-associated post-translational modification in S. cerevisiae Nat Cell Biol. 2010;12(3):294–298 [DOI] [PMC free article] [PubMed] [Google Scholar]