

Abstract
Background
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome is a genetically heterogeneous disorder caused by mitochondrial DNA (mtDNA) mutations in the MT-TL1 gene. The pathophysiology of neurological manifestations is still unclear, but neuronal hyperexcitability and neuron–astrocyte uncoupling have been suggested. Glutamatergic neurotransmission is linked to glucose oxidation and mitochondrial metabolism in astrocytes and neurons. Given the relevance of neuron–astrocyte metabolic coupling and astrocyte function regulating energetic metabolism, we aimed to assess glutamate and glutamine CSF levels in MELAS patients.
Methods
This prospective observational case–control study determined glutamate and glutamine CSF levels in patients with MELAS syndrome and compared them with controls. The plasma and CSF levels of the remaining amino acids and lactate were also determined.
Results
Nine adult patients with MELAS syndrome (66.7% females mean age 35.8 ± 3.2 years) and 19 controls (63.2% females mean age 42.7 ± 3.8 years) were included. The CSF glutamate levels were significantly higher in patients with MELAS than in controls (18.48 ± 1.34 vs. 5.31 ± 1.09 μmol/L, p < 0.001). Significantly lower glutamine concentrations in patients with MELAS than controls were shown in CSF (336.31 ± 12.92 vs. 407.06 ± 15.74 μmol/L, p = 0.017). Moreover, the CSF levels of alanine, the branched-chain amino acids (BCAAs) and lactate were significantly higher in patients with MELAS.
Conclusions
Our results suggest the glutamate–glutamine cycle is altered probably due to metabolic imbalance, and as a result, the lactate–alanine and BCAA–glutamate cycles are upregulated. These findings might have therapeutic implications in MELAS syndrome.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00415-021-10942-7.
Keywords: Mitochondrial disease, MELAS, Glutamate, Glutamine, Branched-chain amino acids
Introduction
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome is a genetically heterogeneous disorder. The most common mutation leading to MELAS syndrome is the m.3243A > G in the mitochondrial DNA (mtDNA) gene MT-TL1, which encodes mitochondrial transfer RNA (tRNA) Leu (UUR) and accounts for 80% of cases [1–3]. The dysfunction of the OXPHOS system, due to the resulting decrease in intramitochondrial protein synthesis, leads to insufficient energy production to maintain cellular homeostasis [4].
In the central nervous system, the inability to generate sufficient ATP can produce acute or subacute cortical neurological deficits that mimic ischemic damage with typical abnormalities on magnetic resonance imaging (MRI). The lesions have a predilection for the posterior areas of the brain and are not limited to the vascular territories [5]. Prodromal symptoms such as migraine-like headaches, mental changes, and focal onset seizures are strongly associated with stroke-like episodes [6, 7]. The mechanisms behind this connection and the precise pathophysiology are still unclear, but neuronal hyperexcitability and neuron–astrocyte uncoupling have been previously suggested [8, 9].
The importance of the neuronal and astrocytic compartments for understanding the development of seizures in patients with mitochondrial disease has been demonstrated in a model for mitochondrial epilepsy (including neuropathological studies on patients with the m.3243A > G mutation). This study emphasizes the deficiency of astrocytic glutamine synthetase, the importance of the GABA–glutamate–glutamine cycle regulating GABAergic-mediated inhibition, and the loss of an inhibitory interneuron population creating a disinhibited neuronal network for seizure generation [10].
Glutamatergic neurotransmission is intricately linked to glucose oxidation and mitochondrial metabolism in astrocytes and neurons [11–13]. Given the relevance of this tightly coupled unit for energy metabolism [14, 15], we hypothesis that mitochondrial dysfunction in MELAS syndrome could contribute to neuron–astrocyte metabolic uncoupling [8, 9], altering the glutamate–glutamine cycle [10], and this might be reflected in the interstitial fluid, and therefore, in the CSF.
This study aimed to assess glutamate and glutamine levels in CSF in patients diagnosed with MELAS syndrome and controls. In addition, we also evaluated the plasma and CSF levels of the remaining amino acids and lactate.
Materials and methods
This study was approved by the local ethics committee of University Hospital 12 de Octubre (Madrid, Spain; approval number: 20/032) and was performed in accordance with the Declaration of Helsinki from April 2015 to January 2017. All participants provided a written informed consent.
We designed a prospective observational case–control study. Patient’s inclusion criteria were as follows: (1) adult patients harboring a MELAS-related pathogenic mtDNA mutation and fulfilling the complete diagnostic criteria for the MELAS phenotype 1; and (2) patients who were stable (i.e. with no acute stroke-like episodes) for at least 6 months before being included in the study. Controls were subjects who received a lumbar puncture (LP) to obtain CSF for diagnostic purposes other than mitochondrial or inherited metabolic conditions, and absence of meningitis or encephalitis.
Patients with MELAS were recruited from the Spanish National Reference Center for Inborn Errors of Metabolism (CSUR) at University Hospital 12 de Octubre in Madrid, Spain. All patients underwent a comprehensive evaluation at the time of inclusion, which included a standardized medical and neurologic history and examination by a neurologist and an internist with expertise in hereditary metabolic and mitochondrial disorders. The diagnosis was based on the clinical characteristics, and all of them were genetically confirmed. Sex- and age-matched control subjects were recruited at the same institution (ratio 1:2).
Nine adult patients with MELAS syndrome and 19 control subjects were included in this prospective observational case–control study. Final diagnoses in subjects used as controls were demyelinating disease (n = 6), cranial neuropathy (n = 5), primary SNC tumor (n = 2), ischemic stroke (n = 1), alcohol-related cerebellar degeneration (n = 1), postinfectious cerebellar ataxia (n = 1), neurosarcoidosis (n = 1), transient headache and neurologic deficits with CSF lymphocytosis (HaNDL) (n = 1), and spinal muscular atrophy (n = 1).
Measurement of lactate and amino acids
CSF samples were collected by LP in the L4-5 interspaces, which was performed by a neurologist between 9 a.m. and 12 a.m. The patients and controls were not fasting, and dietary intake of protein was not controlled.
Samples for lactate determination were immediately kept on ice after blood extraction. Routine biochemical CSF parameters and CSF and plasma amino acids were immediately analyzed. Lactate levels were measured using a spectrophotometric method. CSF and plasma amino acids were assayed by high-performance liquid chromatography with a methodology fulfilling the National Accreditation Body (ENAC, ISO 15189). After collection, samples were deproteinized with 50% sulfosalicylic acid. Amino acids of the deproteinized samples were separated on an ion-exchange column and quantified (µmol/L) by a postcolumn reaction with ninhydrin at 570 nm and 440 nm using a Biochrom 30 + amino acid analyzer (Biochrom, Cambridge, UK).
Statistical analysis
All studied variables were expressed as the mean (± mean standard error) unless otherwise indicated. All variables were tested for normality of data distribution using the Shapiro–Wilk normality test. Paired t test was used to compare normally distributed continuous variables, Mann–Whitney U test to compare the two groups for non-normally distributed continuous variables, and χ2 tests for categorical variables. Spearman's correlation analyses were performed for the lactate, glutamine, and glutamate CSF levels and (i) heteroplasmy levels, (ii) disease duration (from MELAS syndrome diagnosis), and (iii) JMDRS (Japanese mitochondrial disease rating scale). Statistical significance was set at a p value < 0.05, and all analyses were performed with STATA, version 14.2 (StataCorp LP, College Station, TX).
Data availability statement
Further anonymized data can be made available to qualified investigators on request to the corresponding author.
Results
Demographics
The main clinical/diagnostic characteristics of the patients with MELAS who completed the study are shown in Table 1. Nine patients (six females; mean age 35.8 ± 3.2 years) and 19 controls (twelve females; mean age 42.7 ± 3.8 years) were included. There were no differences in age or sex (p = 0.252 and p = 0.856, respectively) between patients and controls. The mean age at the diagnosis of the first stroke-like episode, therefore, fulfilling the complete diagnostic criteria of MELAS syndrome, was 28.1 ± 3.6 years, and the number of stroke-like episodes was 2.7 ± 0.2. The mean age at genetic diagnosis was 26.8 ± 3.2 years. The duration of the disease from the MELAS syndrome diagnosis, was 7.6 ± 2.05 years. All patients with MELAS syndrome had hearing impairment, and four were diabetic (two on treatment with oral therapy and two patients with oral therapy and insulin replacement, glycosylated hemoglobin (HbA1c) mean 7.85 ± 0.41). All of them had epilepsy (mean number of antiepileptic drugs 2.45 ± 0.32), and four had cardiac involvement (three with mild left ventricular hypertrophy and one with left posterior fascicular block). Eight patients had myopathy: all with exercise intolerance, six with symmetrical weakness of proximal lower limbs, and two with mild external ophthalmoparesis. All were ambulant with no assistive devices. Seven subjects had the m.3243A > G mutation, one the m.3258 T > C mutation, and another the m.3271 T > C mutation in the MT-TL1 gene. Mutations were found in the leukocyte DNA of all probands, heteroplasmy ranging from 25 to 50% (n = 7, mean 37 ± 3.4). Heteroplasmy in uroepithelial cells from urine samples ranged from 33 to 95% (n = 9, mean 77 ± 7.1). The Japanese mitochondrial disease rating scale (JMDRS) was 17.89 (± 2.81) out of 80 points, the Mini-Mental State Examination (MMSE) score was 24.11 ± 1.84 out of 30 points, and the Interview for Deterioration of Daily Living in Dementia (IDDD) score was 53 ± 4.98 out of 99 points.
Table 1.
Clinical/diagnostic characteristics of the patients with MELAS síndrome
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
|
|---|---|---|---|---|---|---|---|---|---|
| Age at genetic diagnosis (years) | 30 | 21 | 43 | 33 | 35 | 25 | 21 | 22 | 11 |
| Age at first stroke | 38 | 21 | 43 | 36 | 37 | 25 | 22 | 22 | 9 |
| Age at baseline | 41 | 26 | 49 | 38 | 46 | 36 | 42 | 24 | 22 |
| Sex (Male/Female) | M | M | F | F | F | M | F | F | F |
| Stroke-like episodes (no) | 3 | 2 | 1 | 1 | 2 | 3 | 2 | 2 | 6 |
| Epilepsy | + | + | + | + | + | + | + | + | + |
| JMDRS | 9 | 18 | 17 | 22 | 14 | 20 | 25 | 30 | 6 |
| Dementia | – | + | + | + | – | – | + | + | + |
| MMSE | 30 | 22 | 28 | 23 | 26 | 30 | 22 | 12 | 24 |
| IDDD | 41 | 59 | 48 | 60 | 40 | 39 | 65 | 83 | 42 |
| Hearing impairment | + | + | + | + | + | + | + | + | + |
| Myopathy | + | + | + | + | + | + | + | + | – |
| Cardiomyopathy | – | – | + | + | + | – | – | – | – |
| Diabetes | + | – | + | – | + | – | – | – | + |
| Lactic acidosis | + | – | + | + | + | + | + | + | + |
|
Mitochondrial DNA mutation |
m.3243A > G | m.3243A > G | m.3243A > G | m.3243A > G | m.3243A > G | m.3243A > G | m.3258 T > C | m.3271 T > C | m.3243A > G |
| Heteroplasmy in peripheral blood leucocytes (%) | 25 | 43 | 29 | 37 | 32 | ND | ND | 45 | 50 |
| Heteroplasmy in uroepithelial cells (%) | 95 | 94 | 70 | 58 | 68 | 92 | 33 | 90 | 90 |
| Treatment | |||||||||
| Arginine (g/d) | 14 | 14 | 14 | 14 | 14 | 14 | 14 | 14 | 14 |
| L-Citrulline (g/d) | – | 6 | 2 | 3 | – | 6 | – | 3 | 7.5 |
| Coenzyme Q10 (mg/d) | 200 | 300 | 300 | 300 | – | 300 | 300 | 300 | 300 |
| Carnitine (g/d) | 1 | 1 | 2 | 2 | – | 1 | 2 | – | 2 |
| Antiseizure medication |
LEV LCS |
LTG LCS CLB |
LEV LCS |
LCS BRI CLB |
LEV LCS |
LEV LCS CLB |
LTG CLB PER |
LEV LCS PER CLZ |
LEV LCS |
ND not done, IDDD interview for deterioration of daily living in dementia, MMSE mini-mental state examination, JMDRS Japanese mitochondrial disease rating scale, BRI brivaracetam, CLB clobazam, CLZ clonazepam, LEV levetiracetam, LCS lacosamide, LTG lamotrigine, PER perampanel
Among controls, two were diabetic, one had a memory disorder (Korsakoff syndrome), one had epilepsy, and three had migraine. None of the controls had myopathy or hearing impairment. The main clinical characteristics of the controls are shown in Supplement 1.
Plasma and CSF results: The CSF glutamate levels were significantly higher in patients with MELAS than in controls (18.48 ± 1.34 vs. 5.31 ± 1.09 μmol/L, p < 0.001) (Table 2, Fig. 1). In contrast, the plasma levels of glutamate were significantly lower in patients with MELAS than in controls (22.67 ± 3.37 vs. 45.05 ± 5.68 μmol/L, p = 0.017) (Table 3). Significant differences in the levels of glutamine were also detected, with lower glutamine concentrations in patients with MELAS than in the control group, both in CSF (336.31 ± 12.92 vs. 407.06 ± 15.74 μmol/L, p = 0.017) (Table 2, Fig. 1) and in plasma (393.67 ± 25.19 vs. 517.37 ± 23.25 μmol/L, p = 0.005) (Table 3).
Table 2.
CSF lactate and amino acid results in MELAS patients and control
| Analyte (μmol/l) | MELAS (n = 9) | Controls (n = 19) | p value |
|---|---|---|---|
| Lactate [1.1–2.4] (× 103) | 5.04 ± 0.46 | 1.68 ± 0.15 | < 0.001* |
| Alanine [12.6–49.0] | 62.30 ± 6.83 | 26.58 ± 1.38 | < 0.001* |
| Aminobutirate [0.6–5.6] | 2.41 ± 0.64 | 2.81 ± 0.34 | 0.209 |
| Arginine [11–28] | 37.83 ± 3.07 | 18.03 ± 1.12 | < 0.001* |
| Citrulline [0.7–4.6] | 2.81 ± 0.58 | 0.66 ± 0.05 | 0.001* |
| Glutamate [0.7–8.9] | 18.48 ± 1.34 | 5.31 ± 1.09 | < 0.001* |
| Glutamine [335–567] | 336.31 ± 12.92 | 407.06 ± 15.74 | 0.017* |
| Glycine [3–9.6] | 4.74 ± 0.52 | 3.69 ± 0.37 | 0.085 |
| Histidine [7.6–13.6] | 12.66 ± 0.82 | 9.85 ± 0.46 | 0.011* |
| Isoleucine [2.3–7.2] | 14.23 ± 1.42 | 8.83 ± 0.33 | < 0.001* |
| Leucine [6.0–18.6] | 24.44 ± 3.33 | 15.52 ± 1.14 | 0.002* |
| Lysine [14.7–36.0] | 24.17 ± 3.11 | 23.86 ± 1.07 | 0.522 |
| Methionine [1.0–4.5] | 3.03 ± 0.33 | 2.51 ± 0.13 | 0.191 |
| Ornithine [2.6–12.8] | 5.22 ± 0.73 | 3.14 ± 0.24 | 0.005 |
| Phenylalanine [4.1–12.0] | 9.57 ± 0.90 | 8.15 ± 0.42 | 0.258 |
| Serine [16–37] | 22.88 ± 1.51 | 25.16 ± 1.11 | 0.192 |
| Taurine [3.3–7.7] | 8.43 ± 0.56 | 5.78 ± 0.41 | 0.004* |
| Threonine [17–42] | 25.4 ± 3.71 | 33.81 ± 2.54 | 0.052 |
| Tyrosine [4.0–13.0] | 9.14 ± 1.26 | 8.57 ± 0.69 | 0.883 |
| Valine [7.7–25] | 26.61 ± 5.88 | 15.46 ± 1.47 | 0.032* |
Data are shown as mean ± SE and expressed as μmol/l. Normal range for each metabolite is shown in brackets
*Significant result (p < 0.05)
Fig. 1.

CSF levels of glutamate (A), glutamine (B), lactate (C), alanine (D) in MELAS patients and controls. Colored symbols correspond to individual subjects according to Table 1. Horizontal lines represent mean values for each group. Statistical Significances were assessed by Mann–Whitney U tests. Parallel dotted lines correspond to our laboratory reference ranges in healthy adults
Table 3.
Plasma lactate and amino acid results in MELAS patients and controls
| Analyte (μmol/l) | MELAS (n = 9) | Controls (n = 19) | p value |
|---|---|---|---|
| Lactate [0.5–2.2] (× 103) | 3.37 ± 0.52 | 1.22 ± 0.10 | 0.003* |
| Alanine [200–480] | 492.78 ± 22.43 | 451.00 ± 29.16 | 0.147 |
| Aminobutirate [7–40] | 13.67 ± 1.67 | 26.79 ± 2.90 | 0.003* |
| Arginine [30–120] | 142.56 ± 41.93 | 59.26 ± 5.17 | 0.002* |
| Asparagine [20–125] | 46.11 ± 3.49 | 63.37 ± 4.63 | 0.021* |
| Cystine [15–50] | 37.56 ± 2.59 | 55.53 ± 3.10 | 0.002* |
| Citrulline [10–50] | 64.11 ± 15.87 | 31.79 ± 3.00 | 0.089 |
| Glutamate [15–80] | 22.67 ± 3.37 | 45.05 ± 5.68 | 0.017* |
| Glutamine [350–650] | 393.67 ± 25.19 | 517.37 ± 23.25 | 0.005* |
| Glycine [130–315] | 132.33 ± 9.86 | 230.95 ± 12.55 | < 0.001* |
| Histidine [45–110] | 72.89 ± 4.00 | 76.37 ± 3.10 | 0.712 |
| Hydroxyproline [5–50] | 10.89 ± 0.86 | 13.42 ± 1.18 | 0.167 |
| Isoleucine [30–85] | 76.11 ± 3.36 | 69.37 ± 4.61 | 0.389 |
| Leucine [70–150] | 140.89 ± 6.53 | 135.89 ± 9.41 | 0.941 |
| Lysine [100–240] | 148.33 ± 12.59 | 220.95 ± 11.96 | 0.002* |
| Methionine [15–45] | 17.67 ± 2.07 | 26.21 ± 2.40 | 0.013* |
| Ornithine [30–100] | 152.89 ± 32.41 | 104.16 ± 8.80 | 0.201 |
| Phenylalanine [40–80] | 50.22 ± 2.74 | 58.58 ± 3.23 | 0.099 |
| Phosphoethanolamine [30–100] | 24.67 ± 1.39 | 48.10 ± 9.59 | 0.120 |
| Phosphoserine [3–10] | 4.00 ± 0.41 | 6.47 ± 0.50 | 0.004* |
| Proline [80–300] | 165.44 ± 30.70 | 181.26 ± 11.01 | 0.258 |
| Serine [75–180] | 83.67 ± 6.37 | 132.63 ± 6.27 | < 0.001* |
| Taurine [25–110] | 50.78 ± 7.39 | 51.52 ± 4.23 | 0.824 |
| Threonine [50–200] | 85.33 ± 1.74 | 168.63 ± 12.19 | < 0.001* |
| Tryptophan [20–70] | 28.78 ± 2.45 | 33.68 ± 2.16 | 0.160 |
| Tyrosine [35–80] | 56.11 ± 4.35 | 61.26 ± 3.68 | 0.389 |
| Valine [150–300] | 251.56 ± 13.69 | 251.47 ± 14.83 | 0.941 |
Data are shown as mean ± SE and expressed as μmol/l. Normal range for each metabolite is shown in brackets
*Significant result (p < 0.05)
The CSF lactate levels and plasma lactate levels were significantly higher in patients with MELAS than in controls (5.04 ± 0.46 vs. 1.68 ± 0.15 mmol/L, p < 0.001, and 3.37 ± 0.52 vs. 1.22 ± 0.10 mmol/L, p = 0.003, respectively) (Tables 2 and 3).
The CSF levels of alanine, taurine, and BCAAs (isoleucine, leucine, and valine) were significantly higher in patients with MELAS than in controls (p < 0.05, see Table 2, Figs. 1, 2). In contrast, the plasma levels of these amino acids showed no significant differences between the groups (Table 3).
Fig. 2.

CSF levels of leucine (A), isoleucine (B), taurine (C) and Valine (D) in MELAS patients and controls. Colored symbols correspond to individual subjects according to Table 1. Horizontal lines represent mean values for each group. Statistical significances were assessed by Mann–Whitney U tests. Parallel dotted lines correspond to our laboratory reference ranges in healthy adults
The arginine plasma levels were significantly higher (patients were taking dietary supplements, see Table 1) in patients with MELAS than in the control group (142.56 ± 41.93 vs. 59.26 ± 5.17, p = 002). The arginine, ornithine, citrulline, and histidine CSF levels were also significantly higher in patients with MELAS syndrome (Table 2).
Most amino acids (aminobutyrate, asparagine, cysteine, glycine, lysine, methionine, phosphoserine, serine, and threonine) showed significantly decreased levels in plasma. The rest of the amino acids did not show significant differences between the two groups (Table 3).
A significant inverse correlation was found between CSF glutamine and the JMDRS score (Spearman’s correlation coefficient: − 0.72, p = 0.03) (Fig. 3). A correlation was not found between JMDRS and the CSF levels of lactate or glutamate. No significant correlations were found between the CSF levels of glutamine, glutamate or lactate and (i) the heteroplasmy of the mtDNA mutation in uroepithelial cells or (ii) duration of disease (all p > 0.05).
Fig. 3.
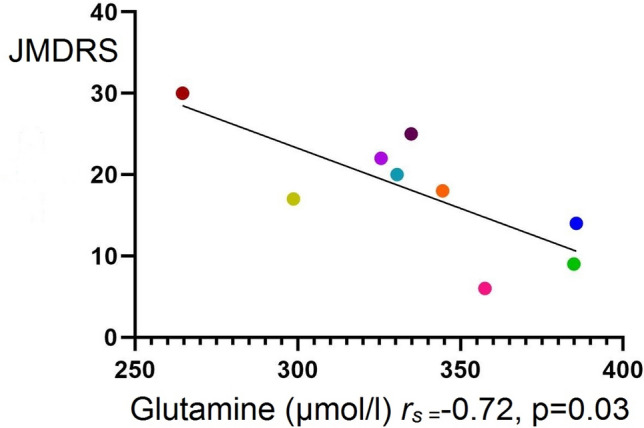
Spearman’s correlation coefficient between CSF glutamine and the JMDRS. JMDRS: Japanese mitochondrial disease rating scale, rs: Spearman’s correlation coefficient. Colored symbols correspond to individual subjects according to Table 1
Discussion
We found that patients diagnosed with MELAS syndrome showed significantly increased CSF glutamate and significantly decreased CSF glutamine compared with controls. Moreover, the CSF levels of alanine, taurine, and BCAAs were significantly higher in patients with MELAS than in controls, although the plasma levels of these amino acids did not present significant differences between the groups. To our best knowledge, no previous studies have systematically evaluated glutamine, glutamate and the other amino acids in CSF and plasma in patients diagnosed with MELAS syndrome during a stationary situation (i.e., no stroke-like episodes).
It has been previously reported in hybrid cell lines carrying the m.3243A > G mutation that impaired oxidative phosphorylation and decreased energy supply reduce glutamate transporter activity [16]. Astroglial glutamate excitatory amino acid transporters (EAAT2) require cellular ion gradients (supported by ATP-dependent activities) to maintain low extracellular glutamate, preserving synaptic signaling and preventing excitotoxicity [14]. In our study, all patients showed remarkably higher concentrations of CSF glutamate than controls (all also above the upper limit of normal range of our laboratory. Table 2), reflecting glutamate clearance could be affected in the CNS because of mitochondrial dysfunction. Since CSF amino acid concentrations are assumed to be similar to that of brain extracellular fluids [17], the increased CSF glutamate levels found in our study could be toxic and leading to neuronal damage [18, 19], thus contributing to cognitive impairment and dementia observed in MELAS patients [20, 21]. An imbalance between release and reuptake with an excess of glutamate in the synapses inducing cortical hyperexcitability, has been implicated in migraine pathophysiology [22, 23] and epileptic seizures [24, 25], which both are common manifestations in MELAS patients [6, 26]. In addition, neuronal hyperexcitability has been proposed as a cellular pathogenic basis of stroke-like episodes [8], and the astrocyte dysfunction and neuron–astrocyte uncoupling caused by ATP deficiency is considered to be one of the critical mechanisms [8, 9]. The taurine levels in CSF were also significantly higher in patients with MELAS syndrome. A possible explanation is that excitotoxic concentrations of glutamate may potentiate taurine release from glial cells as a neuroprotective mechanism to counteract glutamate-mediated neuronal damage [27].
After clearance into astrocytes, glutamate is converted to glutamine by the astrocyte-specific glutamine synthetase, an ATP-dependent reaction. Finally, astrocytes release glutamine into the extracellular space where is used for synthesizing glutamate in excitatory or GABA in inhibitory neurons [12, 14]. Interestingly, we found that CSF glutamine levels are significantly and inversely associated with the JMDRS score, suggesting that decreased glutamine sensitively correlates with the severity of the disease in patients with MELAS. Notably, in postmortem human brain tissue of three patients with the m.3243A > G mutation, astrocytes showed a much lower level of glutamine synthetase than controls and a significant reduction of glutamine levels [10]. These results may explain, in part, the significantly lower glutamine levels in CSF observed in our patients. In addition, another mechanism may contribute to reducing glutamine levels not only in CSF but also in plasma. After conversion to α-ketoglutarate (αKG) and entry into the tricarboxylic acid (TCA) cycle, glutamine is rewired as a bioenergetic adaptation of human cells with mitochondrial DNA mutations [28]. This compensatory energy-generating anaplerotic mechanism and requirement of glutamine-glutamate-αKG oxidative metabolism for mtDNA mutant cell survival [28] could explain the significantly lower glutamine and glutamate plasma levels observed in our patients. Interestingly, low glutamine plasma levels and increased glutamine demand stimulate myofibrillar protein degradation for glutamine de novo synthesis [29]. This negative net muscle protein balance and upregulated glutamate metabolism shown in an animal model of mitochondrial myopathy [28] may contribute to the sarcopenia and muscle weakness described in MELAS patients. Except for arginine, whose levels were above the cutoff value (patients were taking dietary supplements), most amino acids (aminobutyrate, asparagine, cysteine, glycine, lysine, methionine, phosphoserine, serine, and threonine) showed significantly decreased levels in plasma. Moreover, most patients with mitochondrial disease may fulfill the criteria for malnutrition as the amount of ingested energy is slightly insufficient, contributing to breaking down muscle protein to meet their energy and protein requirements [30].
Arginine, citrulline, ornithine, and histidine CSF levels were also significantly higher in patients with MELAS syndrome. Except for arginine, the rest of the amino acids were in the normal range. In the case of citrulline and ornithine, higher levels in CSF could be explained because both are products of arginine metabolism [31]. However, we have no explanation for the higher histidine levels.
In MELAS patients, the BCAAs (valine, isoleucine, and leucine) CSF levels were significantly increased above the normal range. Extracellular levels of these essential amino acids together with glutamate were shown to be chronically elevated at baseline in human brain regions of seizure onset and increased significantly above baseline before spontaneous seizure [25]. Several explanations are delineated in this study, but we hypothesize that BCAAs could be increased because of the higher glutamate concentration at the synaptic cleft. Although astrocytes uptake most of the glutamate, neurons can also remove excess of extracellular glutamate through the EAAT3 transporter which expression is primarily neuronal and located with high densities at postsynaptic terminals [32]. It has been shown that this neuronal glutamate transporter is upregulated as a modulator of extracellular glutamate concentrations in response to recurrent seizures [33]. In neurons, we hypothesized that BCAA metabolism would afford a putative “glutamate buffering system” [34]. Glutamate transamination with the corresponding branched-chain α-ketoacid (in the case of leucine being α-ketoisocaproate) yields BCAAs and α-ketoglutarate. The leucine (as well as valine and isoleucine) formed in neurons is released back to astrocytes, where transamination with αKG produces glutamate, constituting a “BCAA-glutamate cycle” that could serve to shuttle nitrogen between neurons and astrocytes [34–36]. The αKG produced by transamination of glutamate inside the neuron could enter the TCA to support energy production [28], while glutamate shuttling into astrocytes could restore it to neurons as glutamine. Therefore, the increased BCAA levels observed in CSF of our MELAS patients could reflect an upregulation of these cycles between astrocytes and neurons, constituting a compensatory mechanism by which high (and potentially toxic) concentrations of glutamate are managed [34].
In all MELAS patients in this study, the lactate levels were higher in CSF than in plasma, reflecting higher production within the brain. Similar results have been reported in children with electron transport chain defects [37], but to date, they have not been assessed in a case series of adult MELAS patients. Although traditionally lactate has been considered a toxic waste product, the concept of lactate shuttling between astrocytes and neurons has emerged to include its function as a supplemental fuel [38]. When excitatory neurons release glutamate, they enhance glucose uptake and lactate production by astrocytes, which are taken up by neurons and oxidized to produce pyruvate (entering the TCA cycle) to sustain neuronal activity [11, 14, 38]. In addition, transamination of pyruvate and glutamate produces alanine and αKG. The resulting alanine transfers ammonia between glutamatergic neurons and astrocytes [39]. Interestingly, the CSF levels of alanine were significantly higher in patients with MELAS syndrome. The upregulated lactate–alanine shuttle, together with the upregulated BCAA–glutamate cycle, may indicate that the carbon skeleton of glutamate is oxidized by the TCA cycle in neurons as a bioenergetic adaptation [28]. This hypothesis may explain the presence of low glutamate production via TCA compared with high lactate synthesis by glycolysis demonstrated in a patient with MELAS using magnetic resonance spectroscopy (13C-MRS) with carbon 13-labeled glucose administration [40]. These data do not contradict the high glutamate levels found in CSF in our study as the concentration of glutamate in the cerebral extracellular fluid must be kept exceptionally low (typically µM). In contrast, the concentration of this amino acid in glutamate-containing vesicles may even exceed 100 mM [13]. Interestingly, in a brain slice model of mitochondrial epilepsy, a significant reduction in glutamine and glutamate reflects the downregulation of the astrocytic glutamate-glutamine cycle [10].
As noted above, impaired energy production by mutant mitochondria could disrupt astrocyte-neuron coupling, altering the glutamate (GABA)-glutamine cycle [10], responding adaptively to compensate for the metabolic imbalance. As the glutamate-glutamine cycle is an open cycle that actively interfaces with other pathways, inefficient ATP generation could force rewiring glutamine–glutamate as a substrate to replenish TCA cycle intermediates [13, 28]. Metabolism of the resulting ammonium in neurons would be fixed in amino acids such as alanine, valine, leucine, and isoleucine [13, 36, 39], upregulating these shuttles to be subsequently transferred to astrocytes (Supplement 2).
As all the analytical determinations were made while the MELAS patients were in a stationary situation (i.e., no stroke-like episodes), we hypothesize that this unstable, maladjusted balance could fail when the energetic demand increases, as occurs during epileptic seizures [7]. Upon metabolically already compromised neurons and astrocytes, these increased requirements may establish a vicious cycle producing a worsening energy failure leading to stroke-like episodes [7–9]. These lesions that mimic ischemic damage not confined to a particular vascular territory occur most frequently in the occipital cortex [2]. As there is a higher glia/neuron ratio in regions with larger average neuron sizes (i.e., prefrontal cortex) and a lower glia/neuron ratio in the occipital cortex [41], we hypothesized that the posterior region of brain cortex would be more prone to astrocyte–neuron uncoupling.
Several limitations of this study should be noted. (1) We did not measure the amino acids in CSF and plasma during fasting. We also did not control the participants’ diet; moreover, our patients were not drug-naïve and were heterogeneous regarding pharmacological treatment, such as antiseizure medication, oral L-arginine, cofactors, or vitamin supplementation. (2) The control group consisted of heterogeneous neurological patients, which might limit our findings. Concerning this aspect, the average CSF and plasma glutamate and glutamine levels in the controls were within the normal range. (3) We could not detect GABA in CSF, because it was below the limit of detection of the HPLC system used. Given the critical role of the GABA–glutamate–glutamine cycle in regulating GABAergic-mediated inhibition for understanding the development of seizures in patients with mitochondrial disease [9], measuring CSF GABA levels using a high-sensitivity analytical system could be of great interest. (4) We did not include patients harboring the m.3243A > G mtDNA mutation as they did not fulfill the diagnostic criteria for the complete MELAS phenotype, and the small sample size might limit the generalizability of our results. Future studies considering all these limitations are needed.
Conclusion
This study was designed to provide information on CSF glutamate and glutamine and subsequently on pathophysiological mechanisms in MELAS. The main finding is that patients with MELAS have decreased CSF glutamine and increased CSF glutamate, taurine, alanine, valine, leucine, and isoleucine compared with a group of controls. The glutamate–glutamine cycle could be amended adaptively to compensate for the metabolic imbalance. As a result, the lactate–alanine and BCAA–glutamate cycle would be upregulated. As human data on this topic are lacking, experimental investigations are required to assess the mechanisms described and their relevance to the pathogenesis of MELAS syndrome, particularly the role of astrocyte–neuron coupling. These data might have therapeutic implications for testing new pharmacological approaches. Since blood-to-brain glutamine transport can influence brain glutamine concentrations [42, 43], high-dose glutamine oral supplementation might help restore glutamine brain levels, improving the glutamate–glutamine cycle.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file2 Supplement 2 Proposed metabolic pathways altered in MELAS syndrome in the central nervous system: Glutamate (Glu) released from the presynaptic neuron is mainly taken up by astrocytic transporters (EAAT2) coupled to Na+/K+ ATPase activity. Once in the astrocyte, Glu is converted into glutamine (Gln) via the glutamine synthetase (GS) pathway (for which ATP is needed). Then, Gln is sent back to the neurons to regenerate Glu. In MELAS syndrome, inefficient ATP generation may alter Glu uptake leading to higher brain extracellular levels of Glu. Taurine (Tau) could be increased as a neuroprotective mechanism to counteract glutamate-mediated excitotoxicity. Impaired energy production by mutant mitochondria could disrupt astrocyte-neuron coupling, altering the glutamate-glutamine cycle. Neurons might switch to a glutamate-fueled mitochondrial metabolism. Glu must first be metabolized to α-ketoglutarate: 1) Glutamate transamination with the corresponding branched-chain α-ketoacid (in the case of leucine being α-ketoisocaproate) yields BCAAs and α-ketoglutarate. The leucine (as well as valine and isoleucine) formed in neurons is released back to astrocytes, where transamination with αKG produces glutamate, constituting a leucine (BCAAs)-glutamate cycle that could serve to shuttle nitrogen between neurons and astrocytes; 2) In addition, lactate (produced by astrocytes) is taken up by neurons and oxidized to produce pyruvate. Transamination of pyruvate and glutamate produces alanine (Ala) and αKG. The resulting lactate-alanine cycle could also serve to transfer ammonia between glutamatergic neurons and astrocytes. Downregulated pathways (glutamate-glutamine cycle) and lower glutamine CSF levels are highlighted in yellow; upregulated metabolic pathways (lactate-alanine cycle and leucine-glutamate cycle) and higher CSF levels of Ala, Glu, Leu, and Tau are highlighted in red. α-KG: alfa-ketoglutarate; Ala: Alanine; ATP: Adenosine triphosphate; EAAT2: Excitatory amino acid transporter 2; Lac: Lactate; Leu: Leucine; KIC: leucine α-ketoisocaproate; Gln: Glutamine; Glu: Glutamate; Pyr: Pyruvate; Tau: Taurine; TCA: tricarboxylic acid. (TIF 329 KB)
Acknowledgements
This research was supported by a grant from the Spanish Fundación Eugenio Rodríguez Pascual. The financial sponsors had no role in the analysis or the development of conclusions. The investigators are solely responsible for the content and the decision to submit the manuscript for publication.
Author contributions
MPG-M the main author and has taken part in: conceptualization, study design, data collection, statistical analysis, investigation about the problem, writing the original draft and validation of results and the final manuscript. MM-C has taken part in the conceptualization, the investigation, project administration, as well as supervision and validation of the work. AD has participated in amino acids analysis and methodology and resources and validation and supervision of this manuscript. MM has participated in the conceptualization (as she has been working in basic sciences in mitochondrial diseases in our center), methodology and supervision, and validation of our results and the manuscript. JA has participated in supervising the methodology, the validation of our data, and the manuscript's edition. He also helped us with the project administration. CD has participated in the conceptualization, methodology, supervision, validation, and editing of the final manuscript. AR has supported us with the project administration, supervision of data and results, and validation of the manuscript. EA has participated in data collection, validation of data, and visualization of results. MÁM has supported us during the project, especially during the investigation, in the project administration and supervising and validating our results. He had a contribution to the final view of the manuscript. JGA has led the conceptualization, supervision, and manuscript review and final editing. He has had an essential contribution in the investigation, methodology, project administration, validation of our results, and the final version.
Funding
This work was funded by Fundación Eugenio Rodríguez Pascual.
Declarations
Conflicts of interest
None.
Ethical approval
This study was approved by the local ethics committee of University Hospital 12 de Octubre (Madrid, Spain; approval number: 20/032) and was performed in accordance with the Declaration of Helsinki from April 2015 to January 2017.
Informed consent
All participants provided a written informed consent.
References
- 1.Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol. 1984;16(4):481–488. doi: 10.1002/ana.410160409. [DOI] [PubMed] [Google Scholar]
- 2.El-Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. 2015;116(1–2):4–12. doi: 10.1016/j.ymgme.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Mariotti C, Savarese N, Suomalainen A, et al. Genotype to phenotype correlations in mitochondrial encephalomyopathies associated with the A3243G mutation of mitochondrial DNA. J Neurol. 1995;242:304–312. doi: 10.1007/BF00878873. [DOI] [PubMed] [Google Scholar]
- 4.Yasukawa T, Suzuki T, Ueda T, Ohta S, Watanabe K. Modification defect at anticodon wobble nucleotide of mitochondrial tRNAs(Leu)(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. J Biol Chem. 2000;275(6):4251–4257. doi: 10.1074/jbc.275.6.4251. [DOI] [PubMed] [Google Scholar]
- 5.Castillo M, Kwock L, Green C. MELAS syndrome: imaging and proton MR spectroscopic findings. AJNR Am J Neuroradiol. 1995;16(2):233–239. [PMC free article] [PubMed] [Google Scholar]
- 6.Yatsuga S, Povalko N, Nishioka J, et al. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820(5):619–624. doi: 10.1016/j.bbagen.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 7.Tzoulis C, Bindoff LA. Acute mitochondrial encephalopathy reflects neuronal energy failure irrespective of which genome the genetic defect affects. Brain. 2012;135(Pt 12):3627–3634. doi: 10.1093/brain/aws223. [DOI] [PubMed] [Google Scholar]
- 8.Iizuka T, Sakai F. Pathogenesis of stroke-like episodes in MELAS: analysis of neurovascular cellular mechanisms. Curr Neurovasc Res. 2005;2(1):29–45. doi: 10.2174/1567202052773544. [DOI] [PubMed] [Google Scholar]
- 9.Tetsuka S, Ogawa T, Hashimoto R, Kato H. Clinical features, pathogenesis, and management of stroke-like episodes due to MELAS. Metab Brain Dis. 2021 doi: 10.1007/s11011-021-00772-x. [DOI] [PubMed] [Google Scholar]
- 10.Chan F, Lax NZ, Voss CM, et al. The role of astrocytes in seizure generation: insights from a novel in vitro seizure model based on mitochondrial dysfunction. Brain. 2019;142(2):391–411. doi: 10.1093/brain/awy320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. 1994;91(22):10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKenna MC. The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res. 2007;85(15):3347–3358. doi: 10.1002/jnr.21444. [DOI] [PubMed] [Google Scholar]
- 13.Robinson MB, Lee ML, DaSilva S. Glutamate transporters and mitochondria: signaling, co-compartmentalization, functional coupling, and future directions. Neurochem Res. 2020;45(3):526–540. doi: 10.1007/s11064-020-02974-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benarroch EE. Astrocyte signaling and synaptic homeostasis: II: astrocyte-neuron interactions and clinical correlations. Neurology. 2016;87(7):726–735. doi: 10.1212/WNL.0000000000003019. [DOI] [PubMed] [Google Scholar]
- 15.Hertz L, Peng L, Dienel GA. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab. 2007;27(2):219–249. doi: 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- 16.DiFrancesco JC, Cooper JM, Lam A, et al. MELAS mitochondrial DNA mutation A3243G reduces glutamate transport in cybrids cell lines. Exp Neurol. 2008;212(1):152–156. doi: 10.1016/j.expneurol.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 17.Hawkins RA, O'Kane RL, Simpson IA, Viña JR. Structure of the blood-brain barrier and its role in the transport of amino acids. J Nutr. 2006;136(1 Suppl):218S–S226. doi: 10.1093/jn/136.1.218S. [DOI] [PubMed] [Google Scholar]
- 18.Rodríguez JJ, Olabarria M, Chvatal A, Verkhratsky A. Astroglia in dementia and Alzheimer's disease. Cell Death Differ. 2009;16(3):378–385. doi: 10.1038/cdd.2008.172. [DOI] [PubMed] [Google Scholar]
- 19.Olloquequi J, Cornejo-Córdova E, Verdaguer E, et al. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: therapeutic implications. J Psychopharmacol. 2018;32(3):265–275. doi: 10.1177/0269881118754680. [DOI] [PubMed] [Google Scholar]
- 20.Moore HL, Blain AP, Turnbull DM, Gorman GS. Systematic review of cognitive deficits in adult mitochondrial disease. Eur J Neurol. 2020;27(1):3–17. doi: 10.1111/ene.14068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finsterer J. Cognitive decline as a manifestation of mitochondrial disorders (mitochondrial dementia) J Neurol Sci. 2008;272(1–2):20–33. doi: 10.1016/j.jns.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 22.Zielman R, Wijnen JP, Webb A, et al. Cortical glutamate in migraine. Brain. 2017;140(7):1859–1871. doi: 10.1093/brain/awx130. [DOI] [PubMed] [Google Scholar]
- 23.Parker PD, Suryavanshi P, Melone M, et al. Non-canonical glutamate signaling in a genetic model of migraine with aura. Neuron. 2021;109(4):611–628.e8. doi: 10.1016/j.neuron.2020.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Çavuş I, Romanyshyn JC, Kennard JT, et al. Elevated basal glutamate and unchanged glutamine and GABA in refractory epilepsy: microdialysis study of 79 patients at the yale epilepsy surgery program. Ann Neurol. 2016;80(1):35–45. doi: 10.1002/ana.24673. [DOI] [PubMed] [Google Scholar]
- 25.Ong C, Damisah EC, Gruenbaum SE, et al. Increased branched-chain amino acids at baseline and hours before a spontaneous seizure in the human epileptic brain. Epilepsia. 2021;62(6):e88–e97. doi: 10.1111/epi.16920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo S, Esserlind AL, Andersson Z, et al. Prevalence of migraine in persons with the 3243A>G mutation in mitochondrial DNA. Eur J Neurol. 2016;23(1):175–181. doi: 10.1111/ene.12832. [DOI] [PubMed] [Google Scholar]
- 27.Saransaari P, Oja SS. Taurine and neural cell damage. Amino Acids. 2000;19(3–4):509–526. doi: 10.1007/s007260070003. [DOI] [PubMed] [Google Scholar]
- 28.Chen Q, Kirk K, Shurubor YI, et al. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. 2018;27(5):1007–1025.e5. doi: 10.1016/j.cmet.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients. 2018;10(11):1564. doi: 10.3390/nu10111564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aubry E, Aeberhard C, Bally L, et al. Are patients affected by mitochondrial disorders at nutritional risk? Nutrition. 2018;47:56–62. doi: 10.1016/j.nut.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 31.Morris SM., Jr Arginine metabolism: boundaries of our knowledge. J Nutr. 2007;137(6 Suppl 2):1602S–1609S. doi: 10.1093/jn/137.6.1602S. [DOI] [PubMed] [Google Scholar]
- 32.Bjørn-Yoshimoto WE, Underhill SM. The importance of the excitatory amino acid transporter 3 (EAAT3) Neurochem Int. 2016;98:4–18. doi: 10.1016/j.neuint.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crino PB, Jin H, Shumate MD, Robinson MB, Coulter DA, Brooks-Kayal AR. Increased expression of the neuronal glutamate transporter (EAAT3/EAAC1) in hippocampal and neocortical epilepsy. Epilepsia. 2002;43(3):211–218. doi: 10.1046/j.1528-1157.2002.35001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yudkoff M. Interactions in the metabolism of glutamate and the branched-chain amino acids and ketoacids in the CNS. Neurochem Res. 2017;42(1):10–18. doi: 10.1007/s11064-016-2057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yudkoff M. Brain metabolism of branched-chain amino acids. Glia. 1997;21(1):92–98. doi: 10.1002/(sici)1098-1136(199709)21:1<92::aid-glia10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 36.Rothman DL, De Feyter HM, Maciejewski PK, Behar KL. Is there in vivo evidence for amino acid shuttles carrying ammonia from neurons to astrocytes? Neurochem Res. 2012;37(11):2597–2612. doi: 10.1007/s11064-012-0898-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hutchesson A, Preece MA, Gray G, Green A. Measurement of lactate in cerebrospinal fluid in investigation of inherited metabolic disease. Clin Chem. 1997;43(1):158–161. doi: 10.1093/clinchem/43.1.158. [DOI] [PubMed] [Google Scholar]
- 38.Magistretti PJ, Allaman I. Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci. 2018;19(4):235–249. doi: 10.1038/nrn.2018.19. [DOI] [PubMed] [Google Scholar]
- 39.Bak LK, Sickmann HM, Schousboe A, Waagepetersen HS. Activity of the lactate-alanine shuttle is independent of glutamate-glutamine cycle activity in cerebellar neuronal-astrocytic cultures. J Neurosci Res. 2005;79(1–2):88–96. doi: 10.1002/jnr.20319. [DOI] [PubMed] [Google Scholar]
- 40.Otsuki T, Kanamatsu T, Tsukada Y, Goto Y, Okamoto K, Watanabe H. Carbon 13-labeled magnetic resonance spectroscopy observation of cerebral glucose metabolism: metabolism in MELAS: case report. Arch Neurol. 2005;62(3):485–487. doi: 10.1001/archneur.62.3.485. [DOI] [PubMed] [Google Scholar]
- 41.Ribeiro PF, Ventura-Antunes L, Gabi M, et al. The human cerebral cortex is neither one nor many: neuronal distribution reveals two quantitatively different zones in the gray matter, three in the white matter, and explains local variations in cortical folding. Front Neuroanat. 2013;7:28. doi: 10.3389/fnana.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiang J, Ennis SR, Abdelkarim GE, Fujisawa M, Kawai N, Keep RF. Glutamine transport at the blood-brain and blood-cerebrospinal fluid barriers. Neurochem Int. 2003;43(4–5):279–288. doi: 10.1016/s0197-0186(03)00013-5. [DOI] [PubMed] [Google Scholar]
- 43.Bagga P, Behar KL, Mason GF, De Feyter HM, Rothman DL, Patel AB. Characterization of cerebral glutamine uptake from blood in the mouse brain: implications for metabolic modeling of 13C NMR data. J Cereb Blood Flow Metab. 2014;34(10):1666–1672. doi: 10.1038/jcbfm.2014.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file2 Supplement 2 Proposed metabolic pathways altered in MELAS syndrome in the central nervous system: Glutamate (Glu) released from the presynaptic neuron is mainly taken up by astrocytic transporters (EAAT2) coupled to Na+/K+ ATPase activity. Once in the astrocyte, Glu is converted into glutamine (Gln) via the glutamine synthetase (GS) pathway (for which ATP is needed). Then, Gln is sent back to the neurons to regenerate Glu. In MELAS syndrome, inefficient ATP generation may alter Glu uptake leading to higher brain extracellular levels of Glu. Taurine (Tau) could be increased as a neuroprotective mechanism to counteract glutamate-mediated excitotoxicity. Impaired energy production by mutant mitochondria could disrupt astrocyte-neuron coupling, altering the glutamate-glutamine cycle. Neurons might switch to a glutamate-fueled mitochondrial metabolism. Glu must first be metabolized to α-ketoglutarate: 1) Glutamate transamination with the corresponding branched-chain α-ketoacid (in the case of leucine being α-ketoisocaproate) yields BCAAs and α-ketoglutarate. The leucine (as well as valine and isoleucine) formed in neurons is released back to astrocytes, where transamination with αKG produces glutamate, constituting a leucine (BCAAs)-glutamate cycle that could serve to shuttle nitrogen between neurons and astrocytes; 2) In addition, lactate (produced by astrocytes) is taken up by neurons and oxidized to produce pyruvate. Transamination of pyruvate and glutamate produces alanine (Ala) and αKG. The resulting lactate-alanine cycle could also serve to transfer ammonia between glutamatergic neurons and astrocytes. Downregulated pathways (glutamate-glutamine cycle) and lower glutamine CSF levels are highlighted in yellow; upregulated metabolic pathways (lactate-alanine cycle and leucine-glutamate cycle) and higher CSF levels of Ala, Glu, Leu, and Tau are highlighted in red. α-KG: alfa-ketoglutarate; Ala: Alanine; ATP: Adenosine triphosphate; EAAT2: Excitatory amino acid transporter 2; Lac: Lactate; Leu: Leucine; KIC: leucine α-ketoisocaproate; Gln: Glutamine; Glu: Glutamate; Pyr: Pyruvate; Tau: Taurine; TCA: tricarboxylic acid. (TIF 329 KB)
Data Availability Statement
Further anonymized data can be made available to qualified investigators on request to the corresponding author.