

Abstract
Rheumatoid arthritis (RA) patients have almost twice the risk of heart failure (HF) of patients without RA, even when adjusting for presence of ischemic heart disease. Moreover, RA patients remain at two-fold higher risk of mortality from HF compared to non-RA patients. These observations suggest that RA specific inflammatory pathways are significant contributors to this increased risk of HF. We summarize the epidemiology of HF in RA patients, the differences in myocardial structure or function between RA vs non-RA patients without clinical signs of HF, and data on the role of systemic and local inflammation in RA HF pathophysiology. We also discuss the impact of subduing inflammation thorough the use of RA disease modifying therapies (DMARDs) on HF and myocardial structure and function, emphasizing gaps in literature and areas needing further research.
Introduction
Rheumatoid arthritis (RA) is a systemic inflammatory disease affecting approximately 0.5-1% of the population. RA patients have almost twice the risk of heart failure (HF) of patients without RA, even when adjusting for conventional cardiovascular (CV) risk factors and coronary artery disease (CAD)1,2. This observation suggests that RA specific immune/inflammatory pathways are significant contributors to this increased risk of HF. This review will: 1) summarize the epidemiology of clinical HF in RA; 2) in RA patients without clinical HF, delineate differences in myocardial structure and function compared to non-RA patients; 3) examine data in RA patients supporting the pathophysiologic roles of systemic and local inflammation in driving HF and subclinical myocardial dysfunction; 4) review available data on the effect of RA disease modifying therapies (DMARDs) on HF and subclinical myocardial structure and function in RA and 5) discuss future areas for additional research.
Epidemiology of Heart Failure in Patients with RA
RA patients are at an almost 50% higher risk for incident CV disease (CVD) than non-RA patients (pooled Relative Risk (RR) of 1.48)3. In table 1, we summarize the incidence rates of HF specifically, revealing hazard ratios (HR) in RA vs non-RA of 1.21 to 1.872,4–6. While several of the more recent epidemiologic studies7–9 suggest declining overall CV event rates, and CV associated mortality rates, in RA patients diagnosed after 2000 vs those diagnosed prior to 2000, these studies did not include or clearly distinguish HF as an outcome. HF associated mortality is also increased two-fold, and time to onset of HF is shorter, in RA vs non-RA groups1,5,10 (Table 1). However despite the higher incidence of HF, RA patients were less likely to report orthopnea and paroxysmal nocturnal dyspnea than non-RA controls 10. These data raise the possibility that RA associated HF may be underdiagnosed and that aggressive screening for abnormalities in myocardial structure and function, while RA patients are still without HF symptoms, could present an opportunity for early intervention and prevention of HF.
Table 1.
Epidemiologic Studies of HF in RA Patients vs Non-RA Patients
| Incidence of HF in RA | |||||
|---|---|---|---|---|---|
| Study | Design | N (RA vs. non-RA) | HR/OR (95% CI) RA vs. non-RA | Incidence (95% CI or p-value) RA vs. non-RA | Statistical Adjustments |
| Wolfe et al, 200312 Wolfe et al,200411 |
Retrospective longitudinal cohort study | 9093 vs 2479 RA vs. OA 13171 vs 2568 RA vs. OA |
OR 1.43 (1.28-1.59) RA vs. OA Not reported |
Not reported 3.9% (3.4-4.3) vs. 2.3% (1.6-3.3) RA vs. OA |
Demographics, CV risk factors |
| Crowson et al,20051 | Retrospective longitudinal cohort study | 575 vs 583 | Not reported | 36.3% vs. 20.4% p<0.001 | Sex, CV risk factors, alcohol use |
| Nicola et al, 20052, 2006 4 | Retrospective longitudinal cohort study | 575 vs 583; 603 vs 603 | HR 1.87 (1.47–2.39) | 37.1% vs. 27.7% p<0.001 | Age, sex, CV risk factors, CAD |
| Mantel et al, 20175 | Prospective cohort study | 12,943 vs 113,884 | Overall HF: HR 1.22 (1.09-1.37) Ischemic HF: HR 1.27 (1.07-1.51) Non-ischemic HF: HR 1.22 (1.04-1.42) | Rates per 1000 person-years: Overall HF: 5.8 vs 3.1; Ischemic HF: 3.5 vs 1.9; Non-ischemic HF: 2.7 vs 1.4 | Age, sex |
| Ahlers et al, 20206 | Prospective cohort study | 9889 vs 9889 | Cumulative HR (HFpEF and HFrEF): 1.21 (1.03-1.42) | HFpEF: 64% vs 62% p=0.67 | Age, sex, race, CAD, CV meds |
| HF Mortality in RA vs non-RA | |||||
| Study | Design | N (RA vs. non-RA) | HR/OR (95% CI) RA vs. non-RA | Incidence (95% CI or p-value) RA vs. non-RA | Statistical Adjustments |
| Nicola et al, 20064 | Retrospective longitudinal cohort study | 603 vs 603 | 39.0 vs. 29.2 per person-years p<0.001 | Age, sex, calendar year | |
| Davis et al, 200810 | Prospective cohort study | 103 vs 852 | HR 1.89 (1.26-2.84) | 35% vs. 19% | Age, sex, calendar year, CV meds, CAD |
| Ahlers et al, 20206 | Prospective EHR study | 323 vs 443 | HR 1.68 (1.45-1.95) | 22.6% vs 14.6% p=0.006 | Age, sex, race |
BMI= Body Mass Index; CAD= Coronary Artery Disease; CI= Confidence Interval; CV= Cardiovascular; FH=Family History; HF=Heart Failure; HFpEF=HF with Preserved Ejection Fraction; HFrEF= HF with Reduced Ejection Fraction; HR=Hazard Ratio; HTN= Hypertension; OA= Osteoarthritis; OR= Odds Ratio; RA= Rheumatoid Arthritis
The etiology of the increased risk of HF in RA has not been well delineated. While higher rates of CAD pose a large risk for HF in RA4, the relative contribution of CAD to HF is attenuated in RA compared to non-RA patients (HR 3.25 [95% CI 2.35-4.51] vs HR 4.94 [95% CI 3.30-7.38], respectively)1 . Likewise, the attributable risk of HF due to conventional CV risk factors was only 54% in RA, compared to 77% in non-RA patients (p<0.01)1. This suggests that CAD and CV risk factors cannot solely account for the increased risk of HF in RA. Of note, most cohort studies1,2,4,6,11,12 in Table 1 do not distinguish ischemic vs. non-ischemic HF. However, Mantel et al5 recently found similar hazard ratios in RA vs non-RA patients for incident ischemic and non-ischemic HF of 1.27 (95% CI 1.07-1.51) and 1.22 (95% CI 1.04-1.42), respectively. Taken together these data suggest that a significant proportion of RA patients develop HF independently of CAD.
HF comprises a heterogeneous group of disorders that may be primarily cardiac in nature or secondary to systemic disease. HF can be stratified by left ventricular (LV) ejection fraction (EF), as reduced (EF < 40%), midrange (EF 40-49%), or preserved (EF ≥ 50%)13. HF with reduced EF (HFrEF) is characterized by systolic dysfunction, often with chamber dilation and eccentric remodeling, and is most commonly associated with ischemia, hypertension and valvular disease. In contrast, HFpEF (previously ‘diastolic HF’) is characterized by normal EF and LV volumes, but concentric remodeling or LV hypertrophy14. HFpEF is commonly associated with systemic proinflammatory states such as obesity, aging and diabetes 14. With regard to RA, Davis et al10 reported that in patients with clinical HF, the mean EF in RA patients was higher than that of non-RA patients (50% vs 43%, respectively, p=0.007) and the RA group was twice as likely to have preserved EF (OR 1.90 [95% CI 0.98–3.67]). Schau et al15 reported that of 38 RA patients with clinical HF, nearly all (n=36, 95%) had a diastolic phenotype with preserved EF. These observations suggest that RA may be added to the list of chronic inflammatory states that predispose to HFpEF.
Due to the retrospective nature of most of the HF studies in RA, limited data are available on the relationship of RA associated factors to the risk of developing HF, particularly HFpEF. However, in the Mantel5 study, non-ischemic HF was associated more potently than ischemic HF with erythrocyte sedimentation rate (ESR) > 40 and with RA disease activity score with 28 joints (DAS28) > 5.1 (HR 3.03 [95% CI 1.69-2.73] for ESR>40; HR 3.35 [95% CI 1.84-6.09] for DAS28>5.1). Moreover, rheumatoid factor (RF)-positive RA patients had a 40% higher risk of incident HF than RF-negative patients. Investigators at the Mayo Clinic2,4,16 also observed an elevated risk of HF with RF positivity (HR 1.6 [95% CI 1.0-2.5]), as well as with elevated ESR (HR 2.1[ 95% CI 1.2-3.5]), and extraarticular disease (HR 3.1 [95% CI 1.9-5.1]). Taken together, these data suggest that rheumatoid inflammation represents an independent risk factor for incident HF, and perhaps more strongly for the HFpEF phenotype.
Traditional diagnostic and prognostic CVD biomarkers include N-terminal pro B-type natriuretic peptide (NT-proBNP), BNP and troponin. BNP, released with atrial contraction, has long been heralded as a biomarker for predicting systolic, decompensated HF risk and all-cause mortality17. And a gradient increase in cumulative incidence of CV death per every unit increase of troponin was noted in a large population of stable CAD patients in the general population18. However, diagnostic/prognostic biomarkers for HF in RA patients are understudied. An association between NT-pro BNP level and all-cause mortality in RA (HR 2.36 [95% CI 1.42-3.94]) was reported, but HF associated mortality was not separately identified19. The elevated mortality of HF in RA adds urgency to the identification of sensitive measures to detect early myocardial dysfunction in patients with RA.
Measures of Myocardial Structure and Function in RA Patients without Clinical HF
It is useful to examine whether echocardiographic parameters, known to predict the development of clinical HF, are overrepresented in RA patients without clinical HF compared to non-RA patients.
LV Structure
In the general population, values of LV mass above defined cut-offs have been linked to an increased risk of composite CV endpoints, including HF20 . In RA patients without symptoms of HF, LV mass has been compared to non-RA controls in cross-sectional transthoracic echocardiographic (TTE) studies, summarized and analyzed in two meta-analyses (Table 2). In these meta-analyses comprising 25 and 16 individual studies21,22, respectively, higher mean differences in LV mass index (LVMI) of +6.2 g/m2 and +0.47 g/m2, respectively, were reported in the RA compared to non-RA groups. In contrast, two more recent TTE studies reported lower average LVMI23 in the RA group, or no significant difference in LVMI24 between groups.
Table 2.
Left Ventricular Structural and Functional Parameters in RA vs. Non-RA Patients Without HF
| Left Ventricular Mass Index (LVMI) | ||||
|---|---|---|---|---|
| TTE Studies | Design | N (RA vs non-RA) | RA vs non-RA(95% CI or p-value) | Statistical Adjustments |
| Aslam et al, 201321 | Meta-analysis; cross-sectional | 1614 vs 4222 | Mean Difference in LVMI: +6.2 g/m2 (1.08-11.33) | None |
| Rudominer et al, 200930 | Cross-sectional | 89 | RA status and LVMI: OR (95% CI) 3.24(1.05, 5.42), β 0.177; p=0.004 | Age, BMI, HTN |
| Myasoedova et al, 201323 | Cross-sectional | 200 vs 600 | LVMI (SD): 84.6± 15.9 g/m2 vs 91.7± 22.2 g/m2 (p<0.001) | CV risk factors and comorbidities |
| Corrao et al, 201522 | Meta-analysis of case-control studies; cross-sectional | 401 vs 383 | Mean Difference in LVMI: +0.47 g/m2 (0.32-0.62) | None |
| Midtbo et al, 201724 | Cross-sectional | 119 vs 46 | LVMI g/m2.7 (SD):Active RA vs Remission RA vs non-RA: 34.5 (12.1) vs 33.2 (10.2) vs 31.1 (8.1) no significant differences among 3 groups | None |
| Davis et al, 201730 | Prospective longitudinal cohort | 160 vs 1391 | Mean Difference in LVMI per year:−0.0004% (−8.917, −0.199) | Age, sex, CV risk factors |
| CMR Studies | Design | N (RA vs non-RA) | RA vs non-RA (95% CI or p-value) | Statistical Adjustments |
| Giles et al, 201025 | Cross-sectional | 75 vs 225 | Mean Difference in LVMI:−14.7 g/m2 (p<0.001) | Demographics, CV risk factors |
| Ahlers et al,20206 | Cross-sectional | 59 vs 56 | LVMI (SD): 44 g/m2 vs 42 g/m2 (p=0.19) | None |
| Bissell et al, 202026 | Cross-sectional | 76 vs 26 | Mean Difference in LVMI: −4.558 g/m2 (p<0.001) | Age, sex, CV risk factors |
| Plein et al, 202027 | Cross sectional: RA vs controls Prospective:RA only |
81 vs 30 | RA vs non-RA: Mean LVM (g) (95% CI): 78.2 (74.0-82.6) vs 92.9 (84.8-101.7); p<0.01 RA only: Mean LVM (g) (95% CI) at baseline vs 1 year after treatment: 78.2 (73.7-82.9) vs 81.4 (76.3-86.9); p=0.01 |
Age, sex, SBP, smoking |
| Systolic Strain | ||||
| TTE Studies | Design | N (RA vs non-RA) | RA vs non-RA (95% CI or p-value) | Statistical Adjustments |
| Fine et al, 201434 | Cross-sectional | 59 vs 59 | Systolic Strain (SD):−15.7±3.2% vs −18.1±2.4% (p<0.001) | None |
| Cioffi et al, 201735 | Prospective Cohort | 209 vs 52 | Systolic Strain (SD):−18.4± 3.4% vs −19.9± 2.6% (p<0.005) | None |
| Midtbo et al, 201724 | Cross-sectional | 78 vs 46 | Global Longitudinal Strain (SD): Active vs Remission RA: −18.9 % (3.1) vs −20.6 % (3.5) p=0.02 No significant differences between active RA vs controls, or remission RA vs controls | None |
| Lo Gullo et al, 201839 | Cross-sectional | 41 vs 58 | Systolic Strain (SD): −18.13 ± 1.36% vs −23.25 ± 1.80% (p<0.001) | None |
| CMR Studies | Design | N (RA vs non-RA) | RA vs non-RA (95% CI or p-value) | Statistical Adjustments |
| Ntusi et al, 201936 | Cross-sectional | 69 vs 63 | Mid Short Axis Circumferential Strain Rate Without CVRFs: −17.4 ± 1.3 vs. −19.2 ± 1.0 With CVRFs: −16.8 ± 1.1 vs. −18.2 ± 1.2 | None |
| Yokoe et al,202079 | Cross-sectional | 80 vs 20 | Systolic Strain (95% CI): −16.5 (−14.0 to −18.6) vs. −18.2 (−16.2 to −19.6); p<0.055 | None |
| Diastolic Function | ||||
| TTE Studies | Design | N (RA vs non-RA) | RA vs non-RA (95% CI or p-value) | Statistical Adjustments |
| Aslam et al, 201321 | Meta-analysis; Cross sectional | 1614 vs 4222 | Mean Differences: ● LAD: +0.09 cm (0.01-0.17) ● IVRT: +9.67 ms (5.78-13.56) ● E/A ratio: −0.17 (−0.25, −0.09) ● DT: +6.38 msec (−2.76, 15.51) | None |
| Davis et al, 201730 | Prospective longitudinal cohort | 160 vs 1391 | Mean Differences (annualized rate of change): ● LAVI: +0.251 (p<0.001) ● E/A ratio: −0.307 (p<0.001) ● E/e’ ratio: −0.038 (p=0.16) ● DT: −0.009 (p=0.90) | Age, sex, HTN, obesity, diabetes, CAD, smoking |
CAD=Coronary Artery Disease; CVRFs= Cardiovascular Risk Factors; DBP= Diastolic Blood Pressure; DT= Deceleration Time; E/A ratio= Ratio between peak early (E) and late (A) velocity of mitral flow; E/e’ ratio= Ratio between peak early (E) velocity of mitral flow and peak early diatolic velocities of lateral/septal mitral annulus (averaged); IVRT= Isovolumetric Relaxation Time; LAD= Left Atrial Dimension; LAVI=Left Atrial Ventricular Index; LVMI=Left Ventricular Mass Index; SBP=Systolic Blood Pressure; SD= Standard Deviation; TTE= Transthoracic Echocardiography
Other studies have utilized cardiac magnetic resonance (CMR) imaging to measure LV mass in RA. In three cross-sectional CMR studies of RA patients vs non-RA controls25–27, all without clinical HF, RA patients had lower LVMI (differences of −14.7 g/m2, −4.558 g/m2 and −14.7 g, respectively), while a fourth CMR study 6 showed no significant group difference (Table 2). CMR is considered the gold standard for assessing LV mass and volumes28 because of its high spatial and temporal resolution that is not limited by body habitus or ventricular geometry and thus, the ventricles can be imaged in their entirety without having to make geometrical assumptions. Yang et al29showed that adequate visualization of LV wall segments could be obtained in 97% of patients using CMR vs only 38% with TTE29. Thus, observed differences in LVMI may be attributable to technology. Other considerations include lack of statistical adjustment for potential confounders in the TTE meta-analyses, and differences among all studies in levels of severity or duration of RA. Indeed, positive associations of CRP and RA disease duration with LVMI, and current corticosteroid use with lower LVMI, have been reported27.
Additional insight might be gained from investigating differential rates of change in LVMI in RA vs non-RA groups over time. However, in a prospective observational TTE study30 in RA patients without clinical HF, while LVMI in both RA and non-RA groups declined significantly over 4-5 years, rates between groups were not statistically different. In the Plein27 CMR study, early untreated RA patients had a lower mean LVM at baseline than non-RA controls, but after a year of treatment, mean LVM increased in the RA group from 78.2g to 81.4g (p=0.01). These CMR studies suggest that RA itself may be associated with a decline in LVM, perhaps similar to the sarcopenia seen in peripheral muscle in RA, and that treatment of RA may facilitate re-gain of some muscle mass. However, proof of this hypothesis will require longer followup with carefully performed sequential MRI or TTE, and adjustment for treatment effect and CV risk factors. Until then, cross-sectional studies reporting associations of lower or higher LV mass with RA therapies are difficult to interpret.
Other descriptions of LV geometry31 such as concentric remodeling (normal LVMI and relative wall thickness (RWT)>0.42 cm), concentric hypertrophy (increased LVMI and RWT>0.42 cm), and eccentric hypertrophy (increased LVMI and RWT ≤0.42 cm), have been used to categorize phenotypes of LV remodeling. Descriptions of LV geometry in RA vs non-RA patients have been reported in three TTE studies. Rudominer et al32 observed that of 16 RA patients without clinical HF but with LV hypertrophy, 15 had eccentric hypertrophy. In contrast, Myasoedova et al23 reported that among individuals without HF, concentric remodeling was more prevalent in the RA compared to non-RA group (44% vs 19.2%; p<0.001). Cioffi et al33 also reported a significantly higher prevalence of concentric geometry in RA vs non-RA (47% vs 10%; p<0.001) groups without HF. Thus, the evidence currently points to a concentric geometry phenotype in the RA patients without clinical HF which would be in keeping with the presumed non-ischemic nature of RA associated HFpEF.
LV Function
Systolic Function.
In RA vs non-RA individuals without clinical CVD, the conventional measure of systolic function, EF, does not differ significantly by either TTE 21,24,34,35or CMR36. However, systolic strain, assessed by speckle tracking echocardiography or by tagging in CMR, is a more sensitive predictor of systolic dysfunction and of CV clinical endpoints including mortality37 in general population studies. While EF reflects change in LV volume only, systolic strain is an assessment of myocardial deformation during systole coupled to LV volume. GLS is reported as a negative value, reflecting shortening of the LV axis during contraction; a more negative value reflects greater contraction with normal values in the −15.9% to −22.1% range38. Systolic strain has been examined in RA patients without clinical HF (Table 2). All three TTE studies34,35,39and one CMR study36 reported lower GLS (i.e., less negative, worse function) in RA vs non-RA patients. In an RA cohort without clinical CVD35, low GLS predicted future CV hospitalizations for CHF, MI, limb ischemia, or atrial fibrillation (HR 4.50[95% CI 1.40-13.70]].
Diastolic Function.
LV diastolic dysfunction (DD) is a characteristic finding in HFpEF and is manifested by increased myocardial stiffness, impaired relaxation and impaired systolic reserve40. DD is assessed by Doppler echocardiography by measurement of transmitral blood flow velocities in early (E) and late (A) diastole, septal and/or lateral mitral valve annular velocities (e’), and tricuspid regurgitant jet velocity41. Twenty-five case-control studies of DD in RA patients vs non-RA controls, all without clinical HF, were analyzed in a meta-analysis 21 (Table 2). DD (≥ 2 abnormal diastolic parameters) was reported in 26-36% of the RA vs 15-21.7% of the non-RA group. In the prospective TTE study of Davis et al30 comparing RA (n=160) vs. non-RA (n=1391) patients without HF, more rapid decreases in E/A, E/e,’ and deceleration time [DT]), and a more rapid increase in left atrial volume index (LAVI), all reflecting decline in diastolic function, occurred in the RA group (in contrast to no difference in rate of change in LVMI). Whether these changes in diastolic function herald the onset of HFpEF in RA is as yet unknown.
Biomarkers of Myocardial Dysfunction.
There are few reports of CVD biomarkers in RA patients without clinical HF. BNP, as a screening tool for asymptomatic DD in RA patients, had low positive predictive value (25%), sensitivity of only 40%, and specificity of 89%42. BNP and troponin levels may both be confounded by systemic inflammation. In fact, although there are no reports in RA evaluating associations of troponin T or I levels with subclinical LV remodeling, RA patients were reported to have higher levels of high sensitivity troponin I (cTn-I) than non-RA, and DAS28-CRP was independently associated with cTN-I levels in RA patients43. The paucity of work and potentially limited utility of conventional CV biomarkers in detection of subclinical LV dysfunction in RA underscores the need to incorporate novel biomarker studies into prospective studies of the natural history of LV remodeling in RA patients.
Pathophysiologic Roles of Systemic and Local Inflammation in HF and Subclinical Myocardial Remodeling in RA
There is substantial evidence of an association of RA characteristics, such as RA duration, disease activity and seropositivity44–46, and baseline biomarkers of inflammation (interleukin-6 [IL-6] and CRP), with both baseline and longitudinal changes in LV structure and function30,44–47 (see Table 3). However, investigations of specific molecular mechanisms that drive these changes in RA are few. In this section, we consider the following: 1) what is the body of evidence suggesting that circulating inflammatory molecules critical to synovial inflammation and joint destruction also cause LV dysfunction in RA; 2) can local (myocardial) inflammation be demonstrated and does it contribute to LV dysfunction in RA; 3) does endothelial dysfunction occur locally in the RA myocardium, is it associated with systemic and/or local myocardial inflammation, and does it contribute to LV remodeling and dysfunction. We also represent these hypotheses in graphic form in Figure 1.
Table 3.
Associations of Inflammatory Biomarkers and RA Characteristics with HF incidence (RA vs non-RA) and with Subclinical LV Structure/Function (RA without HF)
| Incidence in RA vs non-RA | |||||
|---|---|---|---|---|---|
| Study | Design | N (RA vs non-RA) | Biomarkers HR/OR (95%CI) | RA Characteristics HR/OR (95%CI) | Statistical Adjustments |
| Nicola et al, 20052 | Retrospective longitudinal cohort study | 575 vs 583 | None | RF+ RA vs. non-RA: HR 2.59 (1.95-3.43) | Age, sex, CV risk factors, CAD |
| Mantel et al, 20175 | Prospective cohort study | 12,943 vs. 113,884 | ESR≥40 vs ESR≤40 in non-ischemic HF: HR 3.03 (1.69-2.73); ESR≥40 vs ESR≤40 in ischemic HF: HR 2.41 (1.15-5.08) |
DAS28≥5.1 vs. DAS28≤5.1 in non-ischemic HF: HR 3.35 (1.84-6.09); DAS28≥5.1 vs. DAS28≤5.1 in ischemic HF: HR 2.68 (1.24-5.78) |
None |
| Ahlers et al, 20206 | Prospective cohort study | 9889 vs. 9889 | CRP and HFpEF: OR 1.24 (1.11-1.38) CRP and HFrEF: OR 1.17 (1.03-1.33) | Not reported | Age, sex, race, CAD, and CV Medications, DMARDs |
| Myocardial Measures (LVMI) in RA Patients without HF | |||||
| Study | Design | N (RA Only) | Biomarkers HR/OR (95%CI) | RA Characteristics HR/OR (95%CI) | Statistical Adjustments |
| Rudominer et al, 200932 | Cross-sectional TTE | 89 | No significant associations | No significant associations | Age, BMI, HTN |
| Giles et al, 201025 | Cross-sectional CMR | 75 | No significant associations between LVMI and CRP, IL-6 | LVMI associated with: bDMARDs (β −5.75, p<0.05) and CCP (β −0.46; p<0.05) |
Age, sex, BSA, SBP, DMARDs, smoking |
| Myasoedova et al, 201333 | Cross-sectional TTE | 200 | No significant associations between LVMI and CRP, IL-6, TNF | LVMI associated with glucocorticoid use (β −0.082, p=0.002) | None |
| Ntusi et al,201557 | Cross sectional CMR | 39 | Not reported | DAS28 and ECV: ρ 0.61; p<0.001 | None |
| Bissell et al, 202026 | Cross-sectional CMR | 76 | Not reported | LVMI not associated with DAS28, ACPA, HAQ-DI, RA duration | Age, Gender, CV risk factors, ACPA |
| Diastolic Function in RA Patients without HF | |||||
| Study | Design | N (RA Only) | Biomarkers HR/OR (95%CI) | RA Characteristics HR/OR (95%CI) | Statistical Adjustments |
| Di Franco et al, 200046 | Cross-sectional TTE | 32 | Not reported | RA duration and E/A ratio: r=0.40 (p=0.01) | None |
| Arslan et al, 200645 | Cross-sectional TTE | 52 | Not reported | RA duration and -E/A: r=0.40 (p=0.004) | None |
| Udayakumar et al, 200747 | Cross-sectional TTE | 45 | Not reported | RA duration and -E/A: r=−0.56 (p=0.001); | None |
| Liang et al,201044 | Cross-sectional TTE | 244 | Median IL-6 (IQR) for DD: OR 1.2 (1.01-1.4) | RA duration median (IQR) and DD: OR 3.3 (1.8-5.9) | Age, sex, and CV risk factors |
| Davis et al, 201730 | Prospective longitudinal cohort (5 year changes) |
160 | CRP and -E/A: r=−0.16 (p=0.047); IL-6 and E’: r=0.19 (p=0.02) | Significant associations of A velocity with glucocorticoid use (p=0.04), E/e’ ratio with pt global score (p=0.005) and RAPID 3 score (p=0.02). | None |
| Systolic Function in RA Patients without HF | |||||
| Study | Design | N (RA Only) | Biomarkers HR/OR (95%CI) | RA Characteristics HR/OR (95%CI) | Statistical Adjustment |
| Fine et al,201434 | Cross-sectional TTE | 87 | No significant associations between ESR and systolic longitudinal strain | Longitudinal strain and -Corticosteroid: β 1.84; p=0.062 -Methotrexate: β 1.46; p=0.054 | Age, gender, SBP, BMI, HR, LVMI |
| Cioffi et al,201735 | Prospective Cohort TTE | 209 | No significant associations of CRP with GCS/GLS | No association of RA duration, RF/CCP, CDAI, corticosteroid with GCS/GLS | None |
| Midtbo et al, 201733 | Cross-sectional TTE | 78 | Not reported | DAS28 and GLS: β 0.21; p=0.02 | Age, sex, BMI, SBP, and LVEF |
| Lo Gullo et al, 201839 | Cross-sectional TTE | 41 | Not reported | DAS28 and GLS: β 8.075; p<0.0001 DAS28 and GCS: β 7.214; p=0.002 | Age, BMI, CRP, ESR, SBP, DBP, others |
| Ntusi et al, 201936 | Cross-sectional CMR | 69 | CRP circumferential strain rate: β 0.02 (0.01;0.04); p=0.06 | ---- | Age, CV risk factors, aortic distensibility |
ACPA=Anti-citrullinated protein/peptide antibodies; bDMARD= biologic Disease Modifying Anti-Rheumatic Drugs ;BSA= Body Surface Area; CCP= Cyclic Citrullinated Protein; CDAI= Clinical Disease Activity Index; CMR= Cardiac Magnetic Resonance; CRP= C-Reactive Protein; DAS28= Disease Activity Score in 28 joints; DBP= Diastolic Blood Pressure; DD= Diastolic Dysfunction; DMARD= Disease Modifying Anti-Rheumatic Drugs; ESR: Erythrocyte Sedimentation Rate; GCS= Global Circumferential Strain; GLS= Global Longitudinal Strain; HAQ-DI= Health Assessment Questionnaire Disability Index; HR=Heart Rate; IL-6= Interleukin-6; IQR= Interquartile Range; LVEF= Left Ventricular Ejection Fraction; RAPID-3= Routine Assessment of Patient Index Data 3; RF=Rheumatoid Factor
Figure.
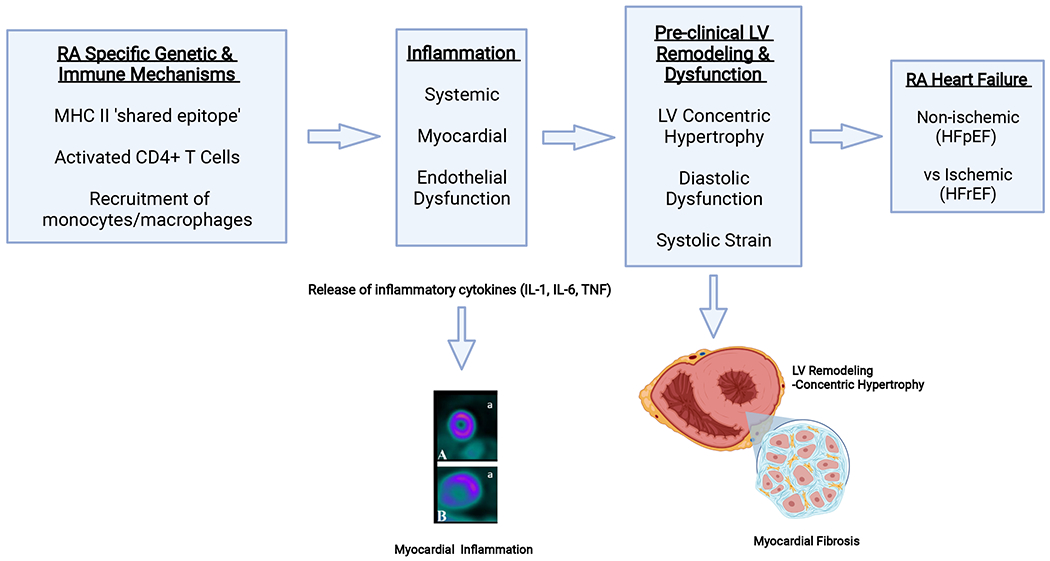
Mechanisms Driving LV Dysfunction and HF in RA Patients
Antigen presenting cells expressing major histocompatibility complex II (MHC II) alleles that encode RA specific ‘shared epitopes’ interact with and select CD4+ T cells. Activated antigen-specific CD4+ T cells interact, in turn, with circulating monocytes and/or tissue macrophages to induce release of key inflammatory cytokines (IL-1, IL-6, TNF). These cytokines, possibly coupled with APCA-containing immune complexes, induce myocardial inflammation with subsequent endothelial damage and microvascular dysfunction. In addition, inflammatory cytokines activate local myofibroblasts that promote myocardial fibrosis and LV remodeling (concentric hypertrophy) that results in diastolic dysfunction and/or systolic strain. These changes in turn may lead to clinical HF with preserved EF (HFpEF). Since RA also promotes accelerated atherosclerosis through similar mechanisms, ischemic damage to the myocardium may also contribute to HF resulting in a reduced EF (HFrEF).
Systemic inflammation and LV structure/function in RA
Several inflammatory molecules that play a key role in RA synovitis and joint destruction, such as tumor necrosis factor (TNF), IL-1, IL-6, and matrix metalloproteases (MMPs), have also been implicated in the pathogenesis of HF in the general population. Inflammatory cytokines critical to HF pathophysiology can be broadly categorized into those implicated in negative LV inotropy (TNF, IL-1, IL-6, IL-18)48 or in LV remodeling (TNF, TIMP-1, MMP-3, MMP-9, MCP-1, IL-8, IL-17)49,50.
Given the limited number of studies in RA patients and in experimental RA models, we focus on TNF, IL-1 and MMPs. Higher levels of TNF have been reported in both blood and myocardia of patients with HF in the general population compared to those without HF.51 Animal studies further support a direct role for TNF in HF pathophysiology. Infusion of TNF causes acute hemodynamic collapse and inflammatory infiltrates in the LV which are reversed with cessation of infusion52. In mice with cardiac-restricted overexpression of a human TNF transgene, depression of LV function, LV dilatation, marked myocardial inflammation, and ultimately myocardial fibrosis, HF and death were observed50.
IL-1 levels are also elevated in patients with chronic HF53. IL-1 acutely depresses myocyte contractility, due in part to impairments in cytoplasmic calcium handling and β-adrenergic receptor signaling53. At the histologic level, IL-1 is implicated in cardiac myocyte hypertrophy via NF-kB, JAK/STAT and PI3K pathways and ultimately in myocardial fibrosis53. Although evidence is lacking in RA, it can be hypothesized that TNF and IL-1, circulating in high levels in RA patients, bathe the myocardium and engage cognate receptors on myocardial cells, inducing the types of deleterious effects outlined above.
Myocardial inflammation and LV structure/function in RA
Just as the RA synovium becomes infiltrated with inflammatory/immune cells including monocytes/macrophages, T cells and B cells, there has been interest in whether a similar process occurs in RA myocardia. In early autopsy studies54, higher prevalences of inflammatory cell infiltrates and myofiber degeneration were reported in RA vs non-RA hearts. However, there is almost no modern literature on histopathology of RA hearts. Moreover, given the risks involved in endomyocardial biopsy and its potential diagnostic inaccuracy due to sampling error (as myocarditis tends to be patchy), the research field has turned to non-invasive cardiac imaging, including CMR and cardiac PET/CT scanning, as alternative methods to identifying myocardial inflammation and fibrosis.
Several studies in RA patients have utilized CMR with late gadolinium enhancement (LGE) to identify myocardial fibrosis/inflammation (Table 3). In RA patients without clinical CVD, Kobayashi et al55,56 reported a prevalence of LGE of up to 38.9%. Moreover, LGE was associated with DAS28 in a multivariable model adjusted for CV risk factors. Ntusi et al57reported a significantly higher prevalence of LGE in RA vs non RA patients without HF (46% vs. 0%, respectively) on CMR, and confirmed an association of LGE with disease activity in the RA patients. Moreover, they demonstrated moderate correlation between DAS28-CRP and LV extracellular volume (ECV) estimation, a quantitative measure that is postulated to reflect the extent of myocardial fibrosis (R = 0.61, p<0.001). A limitation to the interpretation of LGE is that it can represent inflammation, edema, necrosis or fibrosis or any combination thereof. Use of 18-fluorodeoxyglucose positron emission tomography with computed tomography (FDG PET-CT) has emerged as a potentially more specific method for detecting myocardial inflammation. That myocardial FDG uptake reflects inflammation is supported by studies demonstrating accumulation of 18FDG in monocyte/macrophages in post-MI mice58, and high association (R2=0.92) between localization of CD68+ macrophages and 18F-FDG signaling in autoimmune myocarditis rat models59. Cardiac FDG PET-CT scans require careful pre-scan preparation with a very low carbohydrate diet to downregulate glucose receptors on cardiomyocytes, thus hypothetically isolating inflammatory cells as the only residual glucose-receptor expressing cells. In the only myocardial PET/CT study in RA60, nearly 40% (46/119) of patients without clinical CVD had visually detected myocardial FDG uptake. Using a quantitative software package and scans from healthy controls, a cut-off value for elevated FDG uptake was derived. Using this metric, 18% of RA patients had significantly elevated mean myocardial standardized uptake values (SUV), and myocardial SUV was correlated with RA disease activity (p=0.005) in multivariable analyses. The weight of evidence suggests that subclinical myocarditis and/or fibrosis may be present in a significant proportion of RA patients without clinically evident CVD.
Mechanisms by which inflammatory myocarditis is initiated and/or propagated in RA are unknown. Antibodies to proteins that have a post-translational modification called citrullination, termed ACPA (anti-citrullinated protein antibody) are highly specific for RA 61. Data from autopsied RA hearts indicate higher levels of myocardial citrullination in RA compared to control hearts62. It is possible that antibodies are generated in RA not just to synovial, but also to cardiac-specific, citrullinated antigens, triggering an autoimmune response within the heart. In RA patients without clinical CVD, levels of seroreactivity against citrullinated fibrinogen and citrullinated vimentin correlated with higher LVMI (p<0.05)63. These putative immune complexes may lead to the local myocardial inflammation and remodeling, but these conjectures will require further investigation and confirmation.
Myocardial endothelial (microvascular) dysfunction and LV structure/function in RA.
Another potential mechanism of HF in RA is inflammation-induced endothelial dysfunction, leading to impaired vasodilation of the microvasculature and decreased perfusion of the surrounding territory64. This is also thought to be a mechanism underpinning the enhanced risk of HFpEF in mildly inflammatory conditions such as obesity and diabetes 65. Indeed, in RA, studies utilizing diverse methodologies –e.g., brachial artery reactivity, laser Doppler imaging, peripheral arterial tonometry - have demonstrated microvascular (defined by arteries smaller than 500 mm) dysfunction in RA patients and its association with disease activity, circulating cytokines, and future atherosclerosis64. However, few studies have directly investigated myocardial microvascular function in RA. The intra-myocardial arterioles and capillaries of the heart comprise 75% of the resistance in the coronary circulation; thus, dysfunction in these vessels can lead to ischemia even in the absence of significant CAD65. Microvascular disease is quantified by myocardial flow reserve (MFR; also called coronary flow reserve) – i.e., the ratio of myocardial blood flow at peak vasodilatory stress to blood flow at rest. In the absence of significant CAD, this ratio is thought to represent the vasodilatory reserve of the microvascular circulation. MFR cut-offs of < 1.5 or < 2.0 have been suggested to represent microvascular dysfunction65,66. In the general population, impaired MFR has been linked with subclinical DD and with HFpEF66. Decreased nitric oxide bioavailability resulting from microvascular dysfunction has been suggested as a mechanism leading to concentric LV modeling and myocardial stiffness66.
There are few investigations of myocardial microvascular perfusion in RA patients without clinical CVD. Using TTE techniques, investigators67,68 reported significantly lower MFRs in RA patients without HF compared to controls. Using cardiac PET-CT, Recio-Mayoral et al69 also reported lower (impaired) MFR in RA and SLE patients vs controls (p<0.001), and MFR correlated inversely with disease activity (r= −0.65; p<0.001). Microvascular dysfunction is a well documented complication of diabetes mellitus (DM); Liao et al70 reported similar rates of impaired MFR in RA and DM patients (54% and 64%, respectively), and MFR < 2 was significantly associated with all-cause mortality (HR 2.43 [95% CI1.40-4.22]). Amigues et al 71 reported a mean MFR < 2.5 in 29%, and mean MFR < 2.0 in 12%, of RA patients without clinical CVD. In multivariable analyses, TNF inhibitor (TNFi) use was associated with higher (better) MFR (p= 0.023), while lower (worse) MFR was associated with higher IL-6 levels and higher LVMI, suggesting a relationship of depressed MFR with inflammation and myocardial remodeling. Longitudinal studies examining the potential role of microvascular disease in the development of clinical HF in RA are needed.
In summary, RA-specific autoimmune mechanisms that trigger release of inflammatory cytokines may lead to local activation of macrophages and myofibroblasts in the myocardium, subsequent myocardial inflammation, endothelial damage, decreased perfusion, and ultimately, LV remodeling and clinical HF. However, much work is needed to confirm these events and elucidate causative molecular pathways.
Effect of RA DMARDs on HF and on subclinical measures of LV structure and function in RA
The association of inflammatory cytokines with LV remodeling in experimental models generated considerable interest in cytokine blockade as a therapy for HF. However, clinical trials of TNFi’s for treatment of moderate to severe HF in the general population were disappointing. In the RENEWAL trial72 there was neither significant benefit nor increased risk in all-cause-mortality or HF hospitalizations in etanercept vs placebo treated patients (RR 1.10 [95% CI 0.91–1.33; p=0.33]). In contrast, in the ATTACH trial 73, a higher risk of death and/or HF hospitalizations was reported in infliximab vs placebo treated patients (HR 2.84[95% CI 1.01-7.97; p=0.043]). An ensuing report of 38 cases of new onset HF in patients receiving etanercept or infliximab for conditions other than HF were reported, raising further concern74. As a result, the US Food and Drug Administration (FDA) issued a warning regarding use of TNFi’s in individuals with HF.
Consequently, no randomized clinical trials (RCT) of TNF inhibitors to treat HF in RA patients have been conducted. However, several observational studies of the association of TNFi’s with HF incidence or prevalence in RA have been published (see Table 4). In the prospective ‘RABBIT’ RA cohort study, a non-statistically significant difference in incident HF risk in TNFi vs conventional synthetic DMARD (csDMARD) treatment was reported (adjusted HR 1.66 [95% CI 0.67-4.1])75. In a retrospective cohort study of RA patients > 65 years, the hazard ratio (HR) for new HF hospitalizations in TNFi vs MTX users was also numerically elevated but not statistically significant (HR 1.61 [95% CI 0.75-3.49])76 . Using a combined Medicaid/Medicare database of over 10,000 RA patients, Solomon et al 77 reported no statistically significant difference in risk of incident HF in TNFi vs csDMARDs users (HR 0.84[95% CI 0.62-1.12]). Finally, lower rates of incident HF in TNFi vs csDMARD treated RA patients were observed in two studies11,78. Taken together, these studies suggest that TNFi’s may reduce, or at least not elevate, risk of HF in RA. While an RCT would provide more definitive evidence, it seems unlikely that such a trial will be forthcoming, given the number of patients and extended length of follow-up needed.
Table 4.
Effects of Anti-Cytokine Therapy on Incidence/Prevalence of Clinical HF, and on Subclinical LV Structure and Function, in RA patients
| Studies of Incident or Prevalent HF | Design | TNFi vs no Use (N) | Primary HF Outcomes | Adjustments |
|---|---|---|---|---|
| Wolfe et al, 200412 | Retrospective review of longitudinal survey | TNFi (ETN/IFX) vs no TNFi: 5832 vs 7339 | Adjusted Rates of HF: TNFi vs no TNFi: 2.8 vs 3.4-3.9 (p=0.03) IFX vs ETN vs no TNFi: 2.6 vs 2.9 vs 3.4-3.9 | Propensity score matched |
| Bernatsky et al, 200578 | Nested Case Control | TNFi (ETN/IFX) vs csDMARDs: 187 vs 3656 | Adjusted RR for HF hospitalization (95% CI): Any DMARDs vs No DMARDs: 0.7 (0.6-0.9) | Age, sex, cohort, ischemic heart disease, stroke, peripheral arterial disease, HTN, DM, HL, RA meds |
| Listing et al, 200875 | Prospective Cohort; RABBIT | TNFi (ADA/IFX/ETN) vs csDMARDS: 2,757 vs 1,491 | Prevalent HF adjusted HR (95% CI) for TNFi vs csDMARDs: 1.49 (0.70-3.18) Incident HF adjusted HR (95% CI) for TNFi vs csDMARDs: 1.66 (0.67-4.1) Worsening HF adjusted HR (95% CI) for TNFi vs csDMARDs: 1.18 (0.30-4.73) |
Age, male sex, CVD, BMI, DAS28, functional capacity Age, sex, CVD, BMI, functional capacity, disease activity at follow up Age, male sex, GC>10 mg/day |
| Setoguchi et al, 200876 | Retrospective Cohort study; Medicare | TNFi (ADA/IFX/ETN) vs MTX: 1,002 vs 5,593 | New HF hospitalization adjusted HR (95% CI) for TNFi vs MTX: -with previous HF: 1.50 (0.41-4.79) -without previous HF: 3.41 (0.73-16.05) -combined (HF or not): 1.61 (0.75-3.49) | Age, sex, race, CV comorbidities including CAD, other DMARDs, ESR, CRP, CKD, diabetes, HL |
| Solomon et al,201377 | Cohort study; Medicaid and Medicare | TNFi (ADA/IFX/ETN) vs csDMARDs: 11,587 vs 8,656 | HR (95% CI) new or recurrent HF hospitalizations: TNFi vs csDMARDs: 0.85 (0.63-1.14) | Propensity score matched |
| Studies of LV Structure and Function in Patients without HF | Design | DMARD Use (N) | Primary LV Structure/Function Outcomes | Statistical Adjustments |
| Kotyla et al, 201280 | Prospective Cohort Study: TTE | TNFi (IFX): 23 | Before and 1 yr after IFX: median EF: 58.5% vs 63%; p<0.05 | None |
| Santos et al, 201281 | Prospective Cohort Study: TTE | TNFi (IFX): 14 | Before and 2-hr after IFX: -CO: 7.04 ± 2.3 vs 6.12 ± 2.1 L/min; p<0.001 -SV: 91± 29.0 to 83 ± 28.8 mL/beat; p<0.001 | None |
| Daien et al, 201383 | Prospective Cohort Study; TTE | TNFi (ETN) vs csDMARDs: 28 vs 20 | Change in LVMI at 3 and 6 months: ETN: −6.3±7.6; −14.2±9.3 g/m2 csDMARD: −2.2±10.9; −2.7±10.2 g/m2 | None |
| Vizzardi et al, 2016 82 | Prospective Cohort Study; TTE | TNFi (ADA/IFX/ETN): 13 | Baseline vs one year after TNFi: No significant changes in EF or GLS | None |
| Giles et al, 201025 | Cross-sectional: CMR | Non-bDMARD (MTX) vs. bDMARDs (ETN, ADA, IFX, rituximab): 53 vs. 37 | Association of any bDMARD use with LVM: β −5.75; p<0.05 | Age, sex, BSA, SBP, and smoking |
| Plein et al, 202027 | RCT: CMR | TNFi (ETN) + MTX N=81 | Baseline vs one year after treatment: Geometric mean LVM (g) (95% CI): 78.2 (73.7-82.9) vs 81.4 (76.3-86.9); p=0.01 | None |
| Yokoe et al,2020 79 | Cross-sectional: CMR | csDMARDS or bDMARDS:80 | GCS and bDMARDs use: β 0.26 ; p= 0.021 | ACPA, SJC, SDAI, MMP-3 |
ADA=Adalimumab; CKD= Chronic Kidney Disease; CO= Cardiac Output; csDMARDs= Conventional Synthetic Disease Modifying Anti-Rheumatic Drugs; CVD= Cardiovascular Disease; DM= Diabetes Mellot GC= Glucocorticosteroids; HL= Hyperlipidemia; IFX= Infliximab; MMP-3= Matrix Metalloproteinase-3; MTX= Methotrexate; RCT= Randomized Controlled Trial; SJC=Swollen Joint Count; SDAI= Simplified Disease Activity Index; SV=Stroke Volume; TNFi= Tumor Necrosis Factor Inhibitor
In RA patients without HF, the effect of TNFi on measures of LV structure and function has been examined in small sample sizes and with variable outcomes, and which taken together do not offer a clear conclusion (Table 4). In a cross-sectional study, Giles et al25 reported an association of any biologic DMARD (bDMARD) use (most were receiving TNFi’s) with lower LVMI compared to no bDMARD use. In an RCT, Plein et al27 reported a modest increase in geometric mean LV mass after one year of treatment in 81 RA patients treated with ETN+ MTX. In a cross sectional study, Yokoe et al79 reported better global circumferential strain (GCS) in patients treated with bDMARD than with csDMARDs. Kotyla et al80 reported an increase in EF, and decrease in LVM, after 1 year of infliximab in 23 RA patients. Other small and/or very short duration echocardiographic studies are included in Table 481–83. In the absence of an RCT to discern the effects of TNFi’s on HF risk in RA, the American College of Rheumatology (ACR) 2021 guidelines for the treatment of RA continue to recommend non-TNFi biologics over TNFi in RA patients with HF, and switch from TNFi to non-TNFi DMARD if HF develops while on a TNFi.
Despite the success of the CANTOs RCT84 in which an IL-1 antagonist was shown to reduce nonfatal MI, stroke, and CV death in CAD patients in the general population, there is a dearth of studies of IL-1 inhibition in patients with HF. In a clinical trial of combined HFrEF and HFpEF patients in the general population, incidence of HF readmission or death at 24 weeks did not differ between anakinra vs placebo groups85. However, in experimental RA models, IL-1 blockade was associated with improvements in LVEF, LV dilatation and fractional shortening49. Since IL-1 inhibitors are only modestly efficacious for the treatment of RA, data on the effect of IL-1 on LV function in RA are also scant. In several short-term studies with small sample sizes, Ikonomidis et al 86 reported significant improvements in flow mediated dilation, MFR, and strain measures in RA patients.
Even fewer RA studies examine the impact of IL-6 blockade on HF or LV structure/function or cardiac biomarkers in patients with RA. In a post hoc analysis of RA patients receiving tocilizumab (TCZ) vs placebo, there were no statistically significant differences in decreases in troponin (hsTNT) or NT-proBNP levels between groups 87. However, Kobayashi et al88 reported a significant reduction in LVMI (p<0.001) after 52 weeks of tocilizumab treatment in RA patients, and a significant correlation between the change in CDAI with change in LVMI (p = −0.580; p=0.007). However, as noted previously, it is not clear which direction of change in LVMI (higher vs lower; increasing vs decreasing over time) is considered to be beneficial in RA HF pathophysiology and its natural progression.
In summary, the risk/benefit of TNFi’s in RA patients with co-morbid HF remains unclear and mandates further investigation. Likewise, insufficient data preclude conclusions about use of IL-1 or IL-6 inhibitors in clinical HF or to slow or prevent subclinical LV remodeling in RA patients.
As corticosteroids and NSAIDs are both well-recognized factors for triggering or worsening acute HF, current European Society of Cardiology (ESC) guidelines13 recommend against their use in patients in the general population with HF. Limited data are available in RA patients with HF, however, on the contribution of glucocorticoids and/or nonsteroidal anti-inflammatory drugs (NSAIDs) to the incidence or prevalence of HF, or to abnormal echocardiographic measures of LV structure/function (such as LVMI). In a prospective cohort study of RA patients by Mantel et al5, use of corticosteroids was strongly associated with non-ischemic HF (HR 3.12 [95% CI 1.30–7.44]) but not statistically significantly associated with ischemic HF or overall HF. However, in another prospective study6 of RA patients, the use of corticosteroids was not a significant risk factor for incident HFpEF (OR 0.99 [95% CI 0.64-1.54]).
As for the association of corticosteroid use and abnormal measures of LV structure and function in RA patients without clinical HF, the data are somewhat conflicting. Each study focuses on a different outcome (LVMI23, systolic longitudinal strain35, diastolic function30,44) and reported either positive or negative association with corticosteroid use. None of the reviewed studies in RA patients specifically evaluated the association between NSAID use and myocardial measures or HF risk. Given the small number of studies and heterogeneity of findings in this area, clear conclusions are not possible but clinicians are wise to exercise caution in the use of these medications in RA patients with HF.
Future Directions
A keener awareness of the increased risk of HF in RA is needed, particularly given the reports of higher mortality in RA. Typical symptoms and physical exam findings of HF could be misinterpreted as RA-associated interstitial lung disease, and a normal EF on echocardiogram may be dismissed as normal before considering the possibility of HFpEF. The development of guidelines for screening RA patients to identify those at high risk for developing HF would be beneficial, but prospective imaging and biomarker data in RA are currently too scarce to inform guidelines. Davis et al 89 reported that a multi-cytokine immune response score discriminated between normal diastolic function and moderate to severe DD but this cell-based assay may be unwieldy to translate into clinical use. Longitudinal studies that delineate the natural history of pre-clinical echocardiographic findings to clinical HF, and that incorporate novel biomarker investigations, are critically needed in RA.
To supplement RA clinical studies, expanded investigation of in vitro HF models specific to RA should be pursued. A promising development in this regard, particularly given the limited availability of RA myocardial tissue, is the generation of engineered human cardiac tissue from an RA patient’s own induced pluripotent stem cells (iPSCs). Rim et al90 derived iPSCs from RA fibroblast-like synoviocytes, and Lee et al91 demonstrated successful differentiation of cardiomyocytes from those iPSCs. HF may also be investigated as a potential co-morbidity of the induction of experimental inflammatory arthritis. Zhou et al92 reported myocardial inflammation and fibrosis, upregulated gene expression of TNF, IL-6, IL-17 and MMP3 in cardiomyocytes and cardiac fibroblasts, and a decline in LV function, in mice with collagen-induced arthritis. Additional work in animal models with concurrent inflammatory arthritis and HF could aid in defining shared molecular pathways between the two processes.
A critical area for further study is investigation of the direct effect of cytokine inhibitors on parameters of LV structure and function in RA patients without clinical HF. If these studies were to indicate absence of a detrimental effect on LV function, then further study of the safety of these agents in RA patients with clinical HF could conceivably progress.
In conclusion, the morbidity and mortality burden of HF in RA patients is higher than in the general population and appears to be predominantly of the HFpEF phenotype. Substantial evidence supports a role for chronic inflammation in driving HF in RA. Whether DMARDs prevent or worsen HF and/or subclinical LV dysfunction in RA remain unclear.
Acknowledgments
This work was supported by NIH NIAMS R01 AR050026. Acknowledgement is provided at end of manuscript.
Sources of Funding:
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases under Award Number AR-050026 (JMB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
There are no relevant financial interests or relationships to disclose for any of the authors.
Disclosures:
None
References
- 1.Crowson CS, Nicola PJ, Kremers HM, et al. How much of the increased incidence of heart failure in rheumatoid arthritis is attributable to traditional cardiovascular risk factors and ischemic heart disease? Arthritis Rheum. 2005;52(10):3039–3044. doi: 10.1002/art.21349 [DOI] [PubMed] [Google Scholar]
- 2.Nicola PJ, Maradit-Kremers H, Roger VL, et al. The risk of congestive heart failure in rheumatoid arthritis: A population-based study over 46 years. Arthritis Rheum. 2005;52(2):412–420. doi: 10.1002/art.20855 [DOI] [PubMed] [Google Scholar]
- 3.Avina-Zubieta JA, Thomas J, Sadatsafavi M, Lehman AJ, Lacaille D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. 2012;71(9):1524–1529. doi: 10.1136/annrheumdis-2011-200726 [DOI] [PubMed] [Google Scholar]
- 4.Nicola PJ, Crowson CS, Maradit-Kremers H, et al. Contribution of congestive heart failure and ischemic heart disease to excess mortality in rheumatoid arthritis. Arthritis Rheum. 2006;54(1):60–67. doi: 10.1002/art.21560 [DOI] [PubMed] [Google Scholar]
- 5.Mantel Ä, Holmqvist M, Andersson DC, Lund LH, Askling J. Association Between Rheumatoid Arthritis and Risk of Ischemic and Nonischemic Heart Failure. Journal of the American College of Cardiology. 2017;69(10):1275–1285. doi: 10.1016/j.jacc.2016.12.033 [DOI] [PubMed] [Google Scholar]
- 6.Ahlers MJ, Lowery BD, Farber-Eger E, et al. Heart Failure Risk Associated With Rheumatoid Arthritis–Related Chronic Inflammation. JAHA. 2020;9(10). doi: 10.1161/JAHA.119.014661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Myasoedova E, Gabriel SE, Matteson EL, Davis JM, Therneau TM, Crowson CS. Decreased Cardiovascular Mortality in Patients with Incident Rheumatoid Arthritis (RA) in Recent Years: Dawn of a New Era in Cardiovascular Disease in RA? J Rheumatol. 2017;44(6):732–739. doi: 10.3899/jrheum.161154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Myasoedova E, Davis JM, Roger VL, Achenbach SJ, Crowson CS. Improved Incidence of Cardiovascular Disease in Patients With Incident Rheumatoid Arthritis in the 2000s: A Population-based Cohort Study. J Rheumatol. Published online February 15, 2021:jrheum.200842. doi: 10.3899/jrheum.200842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lacaille D, Avina-Zubieta JA, Sayre EC, Abrahamowicz M. Improvement in 5-year mortality in incident rheumatoid arthritis compared with the general population—closing the mortality gap. Ann Rheum Dis. 2017;76(6):1057–1063. doi: 10.1136/annrheumdis-2016-209562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis JM, Roger VL, Crowson CS, Kremers HM, Therneau TM, Gabriel SE. The presentation and outcome of heart failure in patients with rheumatoid arthritis differs from that in the general population. Arthritis Rheum. 2008;58(9):2603–2611. doi: 10.1002/art.23798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolfe F, Michaud K. Heart failure in rheumatoid arthritis: rates, predictors, and the effect of anti–tumor necrosis factor therapy. The American Journal of Medicine. 2004;116(5):305–311. doi: 10.1016/j.amjmed.2003.09.039 [DOI] [PubMed] [Google Scholar]
- 12.Wolfe F, Freundlich B, Straus WL. Increase in cardiovascular and cerebrovascular disease prevalence in rheumatoid arthritis. Published online 2020:6. [PubMed] [Google Scholar]
- 13.Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37(27):2129–2200. doi: 10.1093/eurheartj/ehw128 [DOI] [PubMed] [Google Scholar]
- 14.Redfield MM. Heart Failure with Preserved Ejection Fraction. Solomon CG, ed. N Engl J Med. 2016;375(19):1868–1877. doi: 10.1056/NEJMcp1511175 [DOI] [PubMed] [Google Scholar]
- 15.Schau T, Gottwald M, Arbach O, et al. Increased Prevalence of Diastolic Heart Failure in Patients with Rheumatoid Arthritis Correlates with Active Disease, but Not with Treatment Type. J Rheumatol. 2015;42(11):2029–2037. doi: 10.3899/jrheum.141647 [DOI] [PubMed] [Google Scholar]
- 16.Myasoedova E, Crowson CS, Nicola PJ, et al. The Influence of Rheumatoid Arthritis Disease Characteristics on Heart Failure. J Rheumatol. 2011;38(8):1601–1606. doi: 10.3899/jrheum.100979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khanam SS, Son J-W, Lee J-W, et al. Prognostic value of short-term follow-up BNP in hospitalized patients with heart failure. BMC Cardiovasc Disord. 2017;17(1):215. doi: 10.1186/s12872-017-0632-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Omland T, de Lemos JA, Sabatine MS, et al. A Sensitive Cardiac Troponin T Assay in Stable Coronary Artery Disease. N Engl J Med. 2009;361(26):2538–2547. doi: 10.1056/NEJMoa0805299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mirjafari H, Welsh P, Verstappen SMM, et al. N-terminal pro-brain-type natriuretic peptide (NT-pro-BNP) and mortality risk in early inflammatory polyarthritis: results from the Norfolk Arthritis Registry (NOAR). Ann Rheum Dis. 2014;73(4):684–690. doi: 10.1136/annrheumdis-2012-202848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lieb W, Gona P, Larson MG, et al. The Natural History of Left Ventricular Geometry in the Community. JACC: Cardiovascular Imaging. 2014;7(9):870–878. doi: 10.1016/j.jcmg.2014.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aslam F, Bandeali SJ, Khan NA, Alam M. Diastolic Dysfunction in Rheumatoid Arthritis: A Meta-Analysis and Systematic Review. Arthritis Care Res. 2013;65(4):534–543. doi: 10.1002/acr.21861 [DOI] [PubMed] [Google Scholar]
- 22.Corrao S, Argano C, Pistone G, Messina S, Calvo L, Perticone F. Rheumatoid arthritis affects left ventricular mass: Systematic review and meta-analysis. European Journal of Internal Medicine. 2015;26(4):259–267. doi: 10.1016/j.ejim.2015.02.008 [DOI] [PubMed] [Google Scholar]
- 23.Myasoedova E, Davis JM, Crowson CS, et al. Brief Report: Rheumatoid Arthritis Is Associated With Left Ventricular Concentric Remodeling: Results of a Population-Based Cross-Sectional Study: Abnormal Left Ventricular Remodeling in RA. Arthritis & Rheumatism. 2013;65(7):1713–1718. doi: 10.1002/art.37949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Midtbø H, Semb AG, Matre K, Kvien TK, Gerdts E. Disease activity is associated with reduced left ventricular systolic myocardial function in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76(2):371–376. doi: 10.1136/annrheumdis-2016-209223 [DOI] [PubMed] [Google Scholar]
- 25.Giles JT, Malayeri AA, Fernandes V, et al. Left ventricular structure and function in patients with rheumatoid arthritis, as assessed by cardiac magnetic resonance imaging. Arthritis & Rheumatism. 2010;62(4):940–951. doi: 10.1002/art.27349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bissell LA, Erhayiem B, Hensor EMA, et al. Cardiovascular MRI evidence of reduced systolic function and reduced LV mass in rheumatoid arthritis: impact of disease phenotype. Int J Cardiovasc Imaging. 2020;36(3):491–501. doi: 10.1007/s10554-019-01714-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Plein S, Erhayiem B, Fent G, et al. Cardiovascular effects of biological versus conventional synthetic disease-modifying antirheumatic drug therapy in treatment-naïve, early rheumatoid arthritis. Ann Rheum Dis. Published online August 28, 2020:annrheumdis-2020–217653. doi: 10.1136/annrheumdis-2020-217653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keenan NG, Pennell DJ. CMR of Ventricular Function. Echocardiography. 2007;24(2):185–193. doi: 10.1111/j.1540-8175.2007.00375.x [DOI] [PubMed] [Google Scholar]
- 29.Yang PC, Kerr AB, Liu AC, et al. New real-time interactive cardiac magnetic resonance imaging system complements echocardiography. Journal of the American College of Cardiology. 1998;32(7):2049–2056. doi: 10.1016/S0735-1097(98)00462-8 [DOI] [PubMed] [Google Scholar]
- 30.Davis JM, Lin G, Oh JK, et al. Five-year changes in cardiac structure and function in patients with rheumatoid arthritis compared with the general population. International Journal of Cardiology. 2017;240:379–385. doi: 10.1016/j.ijcard.2017.03.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lang RM, Badano LP, Mor-Avi V, et al. Recommendations for Cardiac Chamber Quantification by Echocardiography in Adults: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Journal of the American Society of Echocardiography. 2015;28(1):1–39.e14. doi: 10.1016/j.echo.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 32.Rudominer RL, Roman MJ, Devereux RB, et al. Independent association of rheumatoid arthritis with increased left ventricular mass but not with reduced ejection fraction. Arthritis Rheum. 2009;60(1):22–29. doi: 10.1002/art.24148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cioffi G, Ognibeni F, Dalbeni A, et al. High prevalence of occult heart disease in normotensive patients with rheumatoid arthritis. Clin Cardiol. 2018;41(6):736–743. doi: 10.1002/clc.22926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fine NM, Crowson CS, Lin G, Oh JK, Villarraga HR, Gabriel SE. Evaluation of myocardial function in patients with rheumatoid arthritis using strain imaging by speckle-tracking echocardiography. Ann Rheum Dis. 2014;73(10):1833–1839. doi: 10.1136/annrheumdis-2013-203314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cioffi G, Viapiana O, Ognibeni F, et al. Prognostic Role of Subclinical Left Ventricular Systolic Dysfunction Evaluated by Speckle-Tracking Echocardiography in Rheumatoid Arthritis. Journal of the American Society of Echocardiography. 2017;30(6):602–611. doi: 10.1016/j.echo.2017.02.001 [DOI] [PubMed] [Google Scholar]
- 36.Ntusi NAB, Francis JM, Gumedze F, et al. Cardiovascular magnetic resonance characterization of myocardial and vascular function in rheumatoid arthritis patients. Hellenic Journal of Cardiology. 2019;60(1):28–35. doi: 10.1016/j.hjc.2018.01.008 [DOI] [PubMed] [Google Scholar]
- 37.Sengeløv M, Jørgensen PG, Jensen JS, et al. Global Longitudinal Strain Is a Superior Predictor of All-Cause Mortality in Heart Failure With Reduced Ejection Fraction. JACC: Cardiovascular Imaging. 2015;8(12):1351–1359. doi: 10.1016/j.jcmg.2015.07.013 [DOI] [PubMed] [Google Scholar]
- 38.Yingchoncharoen T, Agarwal S, Popović ZB, Marwick TH. Normal Ranges of Left Ventricular Strain: A Meta-Analysis. Journal of the American Society of Echocardiography. 2013;26(2):185–191. doi: 10.1016/j.echo.2012.10.008 [DOI] [PubMed] [Google Scholar]
- 39.Lo Gullo A, Rodríguez-Carrio J, Aragona CO, et al. Subclinical impairment of myocardial and endothelial functionality in very early psoriatic and rheumatoid arthritis patients: Association with vitamin D and inflammation. Atherosclerosis. 2018;271:214–222. doi: 10.1016/j.atherosclerosis.2018.03.004 [DOI] [PubMed] [Google Scholar]
- 40.Sharma K, Kass DA. Heart Failure With Preserved Ejection Fraction: Mechanisms, Clinical Features, and Therapies. Circ Res. 2014;115(1):79–96. doi: 10.1161/CIRCRESAHA.115.302922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagueh SF, Smiseth OA, Appleton CP, et al. Recommendations for the Evaluation of Left Ventricular Diastolic Function by Echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Journal of the American Society of Echocardiography. 2016;29(4):277–314. doi: 10.1016/j.echo.2016.01.011 [DOI] [PubMed] [Google Scholar]
- 42.Crowson CS, Myasoedova E, Davis JM, et al. Use of B-type natriuretic peptide as a screening tool for left ventricular diastolic dysfunction in rheumatoid arthritis patients without clinical cardiovascular disease. Arthritis Care Res. 2011;63(5):729–734. doi: 10.1002/acr.20425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avouac J, Meune C, Chenevier-Gobeaux C, et al. Inflammation and Disease Activity are Associated with High Circulating Cardiac Markers in Rheumatoid Arthritis Independently of Traditional Cardiovascular Risk Factors. J Rheumatol. 2014;41(2):248–255. doi: 10.3899/jrheum.130713 [DOI] [PubMed] [Google Scholar]
- 44.Liang KP, Myasoedova E, Crowson CS, et al. Increased prevalence of diastolic dysfunction in rheumatoid arthritis. Annals of the Rheumatic Diseases. 2010;69(9):1665–1670. doi: 10.1136/ard.2009.124362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arslan Ş, Bozkurt E, Ali Sari R, Erol MK. Diastolic function abnormalities in active rheumatoid arthritis evaluation by conventional Doppler and tissue Doppler: relation with duration of disease. Clin Rheumatol. 2006;25(3):294–299. doi: 10.1007/s10067-005-0014-3 [DOI] [PubMed] [Google Scholar]
- 46.Di Franco M Diastolic function abnormalities in rheumatoid arthritis. Evaluation by echo Doppler transmitral flow and pulmonary venous flow: relation with duration of disease. Annals of the Rheumatic Diseases. 2000;59(3):227–229. doi: 10.1136/ard.59.3.227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Udayakumar N, Venkatesan S, Rajendiran C. Diastolic function abnormalities in rheumatoid arthritis: relation with duration of disease. Singapore Med J. 2007;48(6):537–542. [PubMed] [Google Scholar]
- 48.Mann DL. Inflammatory Mediators and the Failing Heart: Past, Present, and the Foreseeable Future. Circulation Research. 2002;91(11):988–998. doi: 10.1161/01.RES.0000043825.01705.1B [DOI] [PubMed] [Google Scholar]
- 49.Hartman MHT, Groot HE, Leach IM, Karper JC, van der Harst P. Translational overview of cytokine inhibition in acute myocardial infarction and chronic heart failure. Trends in Cardiovascular Medicine. 2018;28(6):369–379. doi: 10.1016/j.tcm.2018.02.003 [DOI] [PubMed] [Google Scholar]
- 50.Sivasubramanian N, Coker ML, Kurrelmeyer KM, et al. Left Ventricular Remodeling in Transgenic Mice With Cardiac Restricted Overexpression of Tumor Necrosis Factor. Circulation. 2001;104(7):826–831. doi: 10.1161/hc3401.093154 [DOI] [PubMed] [Google Scholar]
- 51.Elevated Circulating Levels of Tumor Necrosis Factor in Severe Chronic Heart Failure | NEJM. Accessed January 1, 2020. [DOI] [PubMed]
- 52.Bozkurt B, Kribbs SB, Clubb FJ, et al. Pathophysiologically Relevant Concentrations of Tumor Necrosis Factor-␣ Promote Progressive Left Ventricular Dysfunction and Remodeling in Rats. :11. [DOI] [PubMed] [Google Scholar]
- 53.Buckley LF, Abbate A. Interleukin-1 blockade in cardiovascular diseases: a clinical update. European Heart Journal. 2018;39(22):2063–2069. doi: 10.1093/eurheartj/ehy128 [DOI] [PubMed] [Google Scholar]
- 54.Bonfiglio T Heart disease in patients with seropositive rheumatoid arthritis; a controlled autopsy study and review. Archives of Internal Medicine. 1969;124(6):714–719. doi: 10.1001/archinte.124.6.714 [DOI] [PubMed] [Google Scholar]
- 55.Kobayashi Y, Giles JT, Hirano M, et al. Assessment of myocardial abnormalities in rheumatoid arthritis using a comprehensive cardiac magnetic resonance approach: a pilot study. Arthritis Res Ther. 2010;12(5):R171. doi: 10.1186/ar3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kobayashi H, Kobayashi Y, Yokoe I, Akashi Y, Takei M, Giles JT. Magnetic Resonance Imaging-Detected Myocardial Inflammation and Fibrosis in Rheumatoid Arthritis: Associations With Disease Characteristics and N-Terminal Pro-Brain Natriuretic Peptide Levels: Myocardial Inflammation and Fibrosis in RA. Arthritis Care & Research. 2017;69(9):1304–1311. doi: 10.1002/acr.23138 [DOI] [PubMed] [Google Scholar]
- 57.Ntusi NAB, Piechnik SK, Francis JM, et al. Diffuse Myocardial Fibrosis and Inflammation in Rheumatoid Arthritis. JACC: Cardiovascular Imaging. 2015;8(5):526–536. doi: 10.1016/j.jcmg.2014.12.025 [DOI] [PubMed] [Google Scholar]
- 58.Lee WW, Marinelli B, van der Laan AM, et al. PET/MRI of Inflammation in Myocardial Infarction. Journal of the American College of Cardiology. 2012;59(2):153–163. doi: 10.1016/j.jacc.2011.08.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Werner RA, Wakabayashi H, Bauer J, et al. Longitudinal 18F-FDG PET imaging in a rat model of autoimmune myocarditis. European Heart Journal - Cardiovascular Imaging. 2019;20(4):467–474. doi: 10.1093/ehjci/jey119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Amigues I, Tugcu A, Russo C, et al. Myocardial Inflammation, Measured Using 18-Fluorodeoxyglucose Positron Emission Tomography With Computed Tomography, Is Associated With Disease Activity in Rheumatoid Arthritis. Arthritis Rheumatol. 2019;71(4):496–506. doi: 10.1002/art.40771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Willemze A, Trouw LA, Toes REM, Huizinga TWJ. The influence of ACPA status and characteristics on the course of RA. Nat Rev Rheumatol. 2012;8(3):144–152. doi: 10.1038/nrrheum.2011.204 [DOI] [PubMed] [Google Scholar]
- 62.Giles JT, Fert-Bober J, Park J, et al. Myocardial citrullination in rheumatoid arthritis: a correlative histopathologic study. Arthritis Res Ther. 2012;14(1):R39. doi: 10.1186/ar3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Geraldino-Pardilla L, Russo C, Sokolove J, et al. Association of anti-citrullinated protein or peptide antibodies with left ventricular structure and function in rheumatoid arthritis. Rheumatology. Published online December 19, 2016:kew436. doi: 10.1093/rheumatology/kew436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bordy R, Totoson P, Prati C, Marie C, Wendling D, Demougeot C. Microvascular endothelial dysfunction in rheumatoid arthritis. Nat Rev Rheumatol. 2018;14(7):404–420. doi: 10.1038/s41584-018-0022-8 [DOI] [PubMed] [Google Scholar]
- 65.Taqueti VR, Di Carli MF. Coronary Microvascular Disease Pathogenic Mechanisms and Therapeutic Options. Journal of the American College of Cardiology. 2018;72(21):2625–2641. doi: 10.1016/j.jacc.2018.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taqueti VR, Solomon SD, Shah AM, et al. Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. European Heart Journal. 2018;39(10):840–849. doi: 10.1093/eurheartj/ehx721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ciftci O, Yilmaz S, Topcu S, et al. Impaired coronary microvascular function and increased intima-media thickness in rheumatoid arthritis. Atherosclerosis. 2008;198(2):332–337. doi: 10.1016/j.atherosclerosis.2007.11.013 [DOI] [PubMed] [Google Scholar]
- 68.Turiel M, Atzeni F, Tomasoni L, et al. Non-invasive assessment of coronary flow reserve and ADMA levels: a case-control study of early rheumatoid arthritis patients. Rheumatology. 2009;48(7):834–839. doi: 10.1093/rheumatology/kep082 [DOI] [PubMed] [Google Scholar]
- 69.Recio-Mayoral A, Mason JC, Kaski JC, Rubens MB, Harari OA, Camici PG. Chronic inflammation and coronary microvascular dysfunction in patients without risk factors for coronary artery disease. European Heart Journal. 2009;30(15):1837–1843. doi: 10.1093/eurheartj/ehp205 [DOI] [PubMed] [Google Scholar]
- 70.Liao KP, Huang J, He Z, et al. Coronary microvascular dysfunction in rheumatoid arthritis compared to diabetes mellitus and association with all-cause mortality. Arthritis Care Res. Published online November 9, 2019:acr.24108. doi: 10.1002/acr.24108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amigues I, Russo C, Giles JT, et al. Myocardial Microvascular Dysfunction in Rheumatoid Arthritis: Quantitation by 13 N-Ammonia Positron Emission Tomography/Computed Tomography. Circ: Cardiovascular Imaging. 2019;12(1). doi: 10.1161/CIRCIMAGING.117.007495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mann DL, McMurray JJV, Packer M, et al. Targeted Anticytokine Therapy in Patients With Chronic Heart Failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation. 2004;109(13):1594–1602. doi: 10.1161/01.CIR.0000124490.27666.B2 [DOI] [PubMed] [Google Scholar]
- 73.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized, Double-Blind, Placebo-Controlled, Pilot Trial of Infliximab, a Chimeric Monoclonal Antibody to Tumor Necrosis Factor-α, in Patients With Moderate-to-Severe Heart Failure: Results of the Anti-TNF Therapy Against Congestive Heart failure (ATTACH) Trial. Circulation. 2003;107(25):3133–3140. doi: 10.1161/01.CIR.0000077913.60364.D2 [DOI] [PubMed] [Google Scholar]
- 74.Kwon HJ, Cot TR, Cuffe MS, Kramer JM, Braun MM. Case Reports of Heart Failure after Therapy with a Tumor Necrosis Factor Antagonist. Ann Intern Med. 2003;138(10):807. doi: 10.7326/0003-4819-138-10-200305200-00008 [DOI] [PubMed] [Google Scholar]
- 75.Listing J, Strangfeld A, Kekow J, et al. Does tumor necrosis factor α inhibition promote or prevent heart failure in patients with rheumatoid arthritis? Arthritis Rheum. 2008;58(3):667–677. doi: 10.1002/art.23281 [DOI] [PubMed] [Google Scholar]
- 76.Setoguchi S, Schneeweiss S, Avorn J, et al. Tumor necrosis factor-α antagonist use and heart failure in elderly patients with rheumatoid arthritis. American Heart Journal. 2008;156(2):336–341. doi: 10.1016/j.ahj.2008.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Solomon DH, Rassen JA, Kuriya B, et al. Heart failure risk among patients with rheumatoid arthritis starting a TNF antagonist. Ann Rheum Dis. 2013;72(11):1813–1818. doi: 10.1136/annrheumdis-2012-202136 [DOI] [PubMed] [Google Scholar]
- 78.Bernatsky S, Hudson M, Suissa S. Anti-rheumatic drug use and risk of hospitalization for congestive heart failure in rheumatoid arthritis. Rheumatology. 2005;44(5):677–680. doi: 10.1093/rheumatology/keh610 [DOI] [PubMed] [Google Scholar]
- 79.Yokoe I, Kobayashi H, Kobayashi Y, et al. Impact of biological treatment on left ventricular dysfunction determined by global circumferential, longitudinal and radial strain values using cardiac magnetic resonance imaging in patients with rheumatoid arthritis. Int J Rheum Dis. 2020;23(10):1363–1371. doi: 10.1111/1756-185X.13942 [DOI] [PubMed] [Google Scholar]
- 80.Kotyla PJ, Owczarek A, Rakoczy J, Lewicki M, Kucharz EJ, Emery P. Infliximab Treatment Increases Left Ventricular Ejection Fraction in Patients with Rheumatoid Arthritis: Assessment of Heart Function by Echocardiography, Endothelin 1, Interleukin 6, and NT-pro Brain Natriuretic Peptide. J Rheumatol. 2012;39(4):701–706. doi: 10.3899/jrheum.110751 [DOI] [PubMed] [Google Scholar]
- 81.Santos RC, Figueiredo VN, Martins LC, et al. Infliximab reduces cardiac output in rheumatoid arthritis patients without heart failure. Rev Assoc Med Bras:5. [PubMed] [Google Scholar]
- 82.Vizzardi E, Cavazzana I, Franceschini F, et al. Left ventricular function in rheumatoid arthritis during anti-TNF-α treatment: a speckle tracking prospective echocardiographic study. Monaldi Arch Chest Dis. 2016;84(1–2). doi: 10.4081/monaldi.2015.716 [DOI] [PubMed] [Google Scholar]
- 83.Daïen CI, Fesler P, du Cailar G, et al. Etanercept normalises left ventricular mass in patients with rheumatoid arthritis. Ann Rheum Dis. 2013;72(6):881–887. doi: 10.1136/annrheumdis-2012-201489 [DOI] [PubMed] [Google Scholar]
- 84.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377(12):1119–1131. doi: 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- 85.Van Tassell BW, Trankle CR, Canada JM, et al. IL-1 Blockade in Patients With Heart Failure With Preserved Ejection Fraction: Results From DHART2. Circ Heart Fail. 2018;11(8). doi: 10.1161/CIRCHEARTFAILURE.118.005036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ikonomidis I, Tzortzis S, Andreadou I, et al. Increased Benefit of Interleukin-1 Inhibition on Vascular Function, Myocardial Deformation, and Twisting in Patients With Coronary Artery Disease and Coexisting Rheumatoid Arthritis. Circ Cardiovasc Imaging. 2014;7(4):619–628. doi: 10.1161/CIRCIMAGING.113.001193 [DOI] [PubMed] [Google Scholar]
- 87.Welsh P, Tuckwell K, McInnes IB, Sattar N. Effect of IL-6 receptor blockade on high-sensitivity troponin T and NT-proBNP in rheumatoid arthritis. Atherosclerosis. 2016;254:167–171. doi: 10.1016/j.atherosclerosis.2016.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kobayashi H, Kobayashi Y, Giles JT, Yoneyama K, Nakajima Y, Takei M. Tocilizumab Treatment Increases Left Ventricular Ejection Fraction and Decreases Left Ventricular Mass Index in Patients with Rheumatoid Arthritis without Cardiac Symptoms: Assessed Using 3.0 Tesla Cardiac Magnetic Resonance Imaging. J Rheumatol. 2014;41(10):1916–1921. doi: 10.3899/jrheum.131540 [DOI] [PubMed] [Google Scholar]
- 89.Davis JM, Knutson KL, Strausbauch MA, et al. A signature of aberrant immune responsiveness identifies myocardial dysfunction in rheumatoid arthritis. Arthritis & Rheumatism. 2011;63(6):1497–1506. doi: 10.1002/art.30323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rim YA, Park N, Nam Y, Ju JH. Generation of Induced-pluripotent Stem Cells Using Fibroblast-like Synoviocytes Isolated from Joints of Rheumatoid Arthritis Patients. JoVE. 2016;(116):54072. doi: 10.3791/54072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee J, Jung SM, Ebert AD, et al. Generation of Functional Cardiomyocytes from the Synoviocytes of Patients with Rheumatoid Arthritis via Induced Pluripotent Stem Cells. Sci Rep. 2016;6(1):32669. doi: 10.1038/srep32669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou Z, Miao Z, Luo A, et al. Identifying a marked inflammation mediated cardiac dysfunction during the development of arthritis in collagen-induced arthritis mice. Clinical and Experimental Rheumatology. Published online 2020:10. [PubMed] [Google Scholar]