

Abstract
The floral microbiome is of significant relevance to plant reproduction and crop productivity. While plant genotype is key to floral microbiome assembly, whether and how genotypic variation in floral traits and plant-level mutualistic and antagonistic interactions at the rhizosphere and phyllosphere influence the microbiome in the anthosphere remain little known. Using a factorial field experiment that manipulated biotic interactions belowground (mycorrhizae treatments) and aboveground (herbivory treatments) in three strawberry genotypes, we assessed how genotypic variation in flower abundance and size and plant-level biotic interactions influence the bidirectional relationships between floral volatile organic compounds (VOCs) and the floral microbiome using structural equation modeling. We found that plant genotype played a stronger role, overall, in shaping the floral microbiome than biotic interactions with mycorrhizae and herbivores. Genotypic variation in flower abundance and size influenced the emission of floral VOCs, especially terpenes (e.g. α- and β-pinene, ocimene isomers) and benzenoids (e.g. p-anisaldehyde, benzaldehyde), which in turn affected floral bacterial and fungal communities. While the effects of biotic interactions on floral traits including VOCs were weak, mycorrhizae treatments (mycorrhizae and herbivory + mycorrhizae) affected the fungal community composition in flowers. These findings improve our understanding of the mechanisms by which plant genotype influences floral microbiome assembly and provide the first evidence that biotic interactions in the rhizosphere and phyllosphere can influence the floral microbiome, and offer important insights into agricultural microbiomes.
Keywords: Horticultural plant reproductive biology
Introduction
The floral microbiome that consists of thousands or more taxa of bacteria and fungi mediates processes that are essential for plant reproduction and crop productivity [1–5]. While we are just beginning to understand the principles and functions of microbiome assembly in flowers [2, 3], studies have revealed that the floral microbiome is governed by plant genotype [1], and it overlaps with the leaf microbiome harboring a subset of the microbiota hosted by the root microbiome following a source–sink gradient [5]. Thus, plant genotypic variation in floral traits and species interactions that dictate the microbiomes in the rhizosphere (roots) and phyllosphere (leaves) are expected to influence the microbiome in the anthosphere (flowers).
Floral traits can influence the floral microbiome via distinct mechanisms. For instance, flower size is hypothesized to affect the niche availability for microbiota [6]. Flower abundance, on the other hand, can influence floral microbiome assembly by governing the source pool of colonizing microbes [1, 7] and by enhancing pollinator-mediated microbial dispersal [1]. In particular, floral volatile organic compounds (VOCs) have been shown to mediate pollinator foraging [8], which can potentially influence microbial dispersal [3]. In addition to the pollinator-mediated effect of floral VOCs on the floral microbiome, VOCs are known for their antimicrobial properties (e.g. terpenes, benzenoids, aliphatics) [9–12] and their roles as carbon sources (e.g. methanol) [9], and thus can potentially reduce pathogenic microbes [10, 13], modulate microbe–microbe interactions [14] and thereby affect microbial communities [12]. Microbiota, in turn, can influence plant VOCs by directly contributing to VOC emissions [3] or indirectly by metabolizing plant VOCs and/or affecting plant physiology that can influence plant VOC emissions [9, 10, 13]. Thus, the effect of microbiota on floral VOCs is determined, in part, by the quantity and quality of plant metabolites [9] in the anthosphere. Such bidirectional relationships between floral VOCs and floral microbiome are likely to be influenced by biotic interactions experienced by the whole plant. For instance, plant interactions with rhizosphere mutualists (e.g. mycorrhizae) [15] and phyllosphere antagonists (e.g. herbivores) [16, 17] may affect the source–sink dynamics of microbes that colonize the anthosphere and/or affect floral traits including VOCs [13, 15], which can influence floral microbiome assembly. Despite the connections between whole-plant biotic interactions, floral VOCs and the floral microbiome, no study has evaluated the relative importance of genotypic variation in floral traits and plant-level interactions in shaping the floral microbiome, nor in affecting the bidirectional relationships between floral VOCs and floral microbiome.
Here we leveraged a factorial field experiment [18] that manipulated biotic interactions belowground (mycorrhizae treatments) and aboveground (herbivory treatments) in three day-neutral strawberry cultivars (Fragaria ×ananassa “Seascape”, “Tribute” and “Wasatch”; Fig. 1). As clonal plants of each cultivar were used in this study, we referred to cultivars as genotypes. We first examined how epiphytic bacterial and fungal communities in flowers responded to plant genotype, mycorrhizae treatments and herbivory treatments. We then assessed how plant genotypes varied in floral traits (flower abundance, size and VOCs) that may shape the floral microbiome. Lastly, we used structural equation modeling to link genotypic variation in flower abundance and size and biotic treatments to the bidirectional relationships between floral VOCs and floral microbiome (bacterial and fungal α- and β-diversity).
Figure 1.
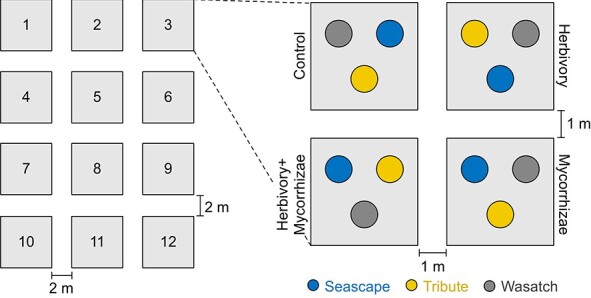
Experimental design. This factorial, potted strawberry experiment used split plot with randomized complete block design. The four treatments (control, herbivory, mycorrhizae, and herbivory + mycorrhizae) were grouped into and randomized within blocks (N = 1–12). The three strawberry genotypes (Seascape, Tribute, and Wasatch) were assigned to and completely randomized within each treatment. Floral microbiome samples were collected from 93 plants flowering on August 16, 2019, which represented a subset of 144 total plants in the experiment [18].
Results
Stronger effects of plant genotype than treatment on the floral microbiome
Strawberry genotypes influenced the α- and β-diversity of floral bacterial and fungal communities (Fig. 2 and Supplementary Table 1). Bacterial Shannon diversity differed among genotypes depending upon herbivory treatments, as revealed by a general linear mixed model (LMM, genotype–herbivory interaction, F = 7.06, df = 2, P = 0.002). For instance, bacterial Shannon diversity was higher in Tribute than Seascape in herbivory-absent treatments (least-squares mean, LS-mean, contrast: control, χ2 = 6.16, df = 1, P = 0.013; mycorrhizae, χ2 = 5.46, P = 0.019; Fig. 2a), but was similar in herbivory-present treatments (herbivory, χ2 = 2.47, P = 0.12; herbivory + mycorrhizae, χ2 = 0.90, P = 0.34). In addition, Tribute had lower bacterial Shannon diversity than Wasatch but only in herbivory + mycorrhizae treatment (χ2 = 4.74, df = 1, P = 0.030; Fig. 2a). Within genotypes, bacterial Shannon diversity was influenced by treatments, especially for Tribute (control > herbivory, χ2 = 7.22, df = 1, P = 0.007; control > herbivory + mycorrhizae, χ2 = 8.93, P = 0.003) and Seascape (herbivory > mycorrhizae, χ2 = 5.45, P = 0.020), but not for Wasatch (Fig. 2a).
Figure 2.

Stronger effects of genotype than treatment on the floral microbiome. a, Bacterial Shannon diversity (least-squares means) differed among strawberry genotypes contingent upon treatments, after controlling for all other factors in a general linear mixed model (LMM, detailed statistics in Supplementary Table 1). Significant least-squares mean contrasts are denoted: *P < 0.05; **P < 0.01. Error bars represent 1 SE. b, The least-squares means of fungal Shannon diversity (power-transformed, power parameter = 2) were similar among genotypes and treatments (LMM, detailed statistics in Supplementary Table 1). The least-squares mean contrasts showed that fungal Shannon diversity only differed in Wasatch between mycorrhizae and herbivory + mycorrhizae treatments. c and d, The constrained principal coordinates analyses (cPCoAs) revealed significant effects of genotype but not treatment on the bacterial and fungal community compositions (detailed statistics in Supplementary Table 1).
Figure 3.
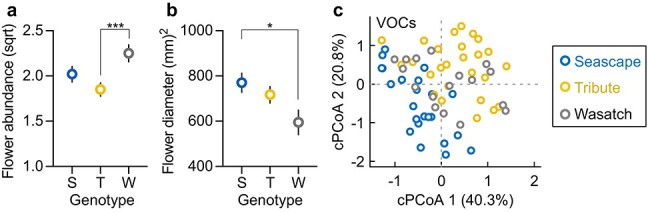
Strawberry genotypes differ in floral phenotype. a and b, The least-squares means of square-root transformed flower abundance and power-transformed flower diameter (power parameter = 2) are plotted for each genotype, after controlling for all other factors in general linear mixed models (LMMs; detailed statistics in Supplementary Table 2). Significant contrasts of least-squares means are denoted: *P < 0.05; ***P < 0.001. Error bars represent 1 SE. c, The constrained principal coordinates analysis (cPCoA) revealed significant differences in floral volatile organic compounds (VOCs) among genotypes (detailed statistics in Supplementary Table 3).
Similar to α-diversity, the β-diversity of floral bacterial communities was influenced by plant genotype (Fig. 2c and Supplementary Table 1). The permutational multivariate analysis of variance (PERMANOVA) of Bray–Curtis dissimilarity identified genotype as the primary source of variation in bacterial community composition (4% of variation, F = 1.70, df = 2, P = 0.035; Supplementary Table 1), which was driven by bacterial community differences between Tribute and Wasatch (pairwise PERMANOVA, F = 2.53, df = 1, P = 0.016) but not other pairs (Seascape vs. Tribute, F = 1.08, P = 0.32; Seascape vs. Wasatch, F = 1.32, P = 0.22). Supporting the PERMANOVA results, the constrained principal coordinates analysis (cPCoA) of bacterial communities revealed significant separation between Tribute and Wasatch (pairwise cPCoA, F = 2.48, df = 1, P = 0.016; Fig. 2c), with Seascape being intermediate between them but not different from either (Seascape vs. Tribute, F = 1.07, P = 0.33; Seascape vs. Wasatch, F = 1.31, P = 0.23).
For floral fungal communities, although Shannon diversity was similar among genotypes (LMM, F = 0.34, df = 2, P = 0.71; Fig. 2b), community composition varied significantly with genotype (PERMANOVA, 3% of variation, F = 1.32, df = 2, P = 0.045; cPCoA, 3%, F = 1.31, P = 0.050; Fig. 2d and Supplementary Table 1). Albeit subtler than bacterial communities (Fig. 2c, d), fungal community separation was also primarily between Tribute and Wasatch (pairwise cPCoA, F = 1.42, df = 1, P = 0.067; Fig. 2c) and less so between other genotypes (Seascape vs. Wasatch, F = 1.32, P = 0.10; Seascape vs. Tribute, F = 0.96, P = 0.50).
Plant genotypes differ in flower abundance, size and VOCs
Given the significant effect of genotype on the floral microbiome, we then examined genotypic variation in aspects of floral phenotype (e.g. flower abundance, size and VOCs) potentially important in governing floral microbiome assembly. The LMM revealed that flower abundance differed among genotypes (F = 6.06, df = 2, P = 0.004) but not among treatments (herbivory treatments, F = 1.36, df = 1, P = 0.25; mycorrhizae treatments, F = 0.11, P = 0.74; Supplementary Table 2). In particular, Wasatch produced more flowers than Tribute (LS-mean contrast, χ2 = 12.1, df = 1, P = 0.001; Fig. 3a), with Seascape being intermediate (Wasatch vs. Seascape, χ2 = 3.3, P = 0.068; Tribute vs. Seascape, χ2 = 2.5, P = 0.12). Similar to flower abundance, flower size (estimated by flower diameter) also differed among genotypes (LMM, F = 3.34, df = 2, P = 0.041; Supplementary Table 2), with Seascape having the largest and Wasatch the smallest (Fig. 3b). Yet, there was no significant trade-off between flower abundance and flower size (Pearson’s correlation, t = −0.78, df = 78, P = 0.44) across genotypes.
Figure 4.

Structural equation models (SEMs) linking floral phenotype and treatment to floral bacterial microbiome. a, Hypothesized effects of flower abundance and size as well as treatments on floral volatile organic compounds (VOCs) and bacterial microbiome. The floral bacterial microbiome was characterized using α-diversity (Shannon diversity) and β-diversity (the first two axes of constrained principal coordinates analysis, cPCoA1 and cPCoA2). VOCs were evaluated at three levels (see Materials and methods): individual VOCs, major classes of VOCs, and VOC profile (VOC cPCoA1 and cPCoA2). The bidirectional links between VOCs and bacterial microbiome reflected potential reciprocal influence on each other. Treatments were coded using the control treatment as the reference level. SEMs were fitted based on overall genotypic variation using lavaan [19]. b-g, SEMs that contained notable positive (solid black) and negative (dashed black) links between VOCs and bacterial microbiome, as well as other variables, were presented (see SEM details in Supplementary Table 4): †P < 0.10; *P < 0.05; **P < 0.01; ***P < 0.001. Grey links indicate P > 0.10. Link width and the numbers adjacent to links indicate standardized path coefficients.
The floral VOC profile consisted of 22 identified VOCs: 10 terpenes (α-pinene, β-pinene, 6-methyl-5-hepten-2-one, d-limonene, ocimene isomers, geranyl acetate, dihydro-β-ionone, β-ionone, α-muurolene and α-farnesene), seven benzenoids (anisole, benzaldehyde, benzyl alcohol, methyl salicylate, p-anisaldehyde, 3-hexen-1-ol benzoate and benzyl benzoate), three aliphatics (trans-2-hexen-1-al, (E)-3-hexen-1-ol and cis-3-hexenyl acetate), one (sulfur) S-containing compound (benzothiazole) and one C5 branched-chain compound (hexenyl tiglate). Plant genotype explained the largest source of variation in VOCs (PERMANOVA, 12%, F = 4.47, df = 2, P = 0.001; cPCoA, 11%, F = 3.81, P = 0.001; Fig. 3c and Supplementary Table 3), with the first two axes of VOC cPCoA (cPCoA1 and cPCoA2; Fig. 3c) driven primarily by variation in benzaldehyde and cis-3-hexenyl acetate, respectively (Supplementary Table 3). Pairwise PERMANOVAs and cPCoAs further revealed that all three genotypes produced significantly different VOC profiles from each other (all P < 0.05). The random forest classification identified the specific VOCs that differed between genotypes, that is, α-pinene, β-pinene, geranyl acetate and benzaldehyde between Seascape and Tribute (out-of-bag error, OOB = 6.12%), α-pinene and benzaldehyde between Seascape and Wasatch (OOB = 17.5%), and α-pinene, ocimene isomers and benzyl alcohol between Tribute and Wasatch (OOB = 9.3%).
SEMs linking floral phenotype and treatment to the floral microbiome
Given the significant genotypic differences in both floral phenotype and microbiome, we then used SEMs to link genotypic variation in flower abundance, size and VOCs, as well as treatments, to the α- and β-diversity of floral bacterial and fungal communities (Figs. 4a and 5a). The SEMs focused on VOCs at three different levels: (1) individual VOCs that differed between genotypes as identified by the random forest classification (see above), and that predicted the α- or β-diversity of bacterial and fungal communities based on backward predictor selection (see Materials and methods; Supplementary Tables 4 and 5); (2) the major classes of VOCs (terpenes, aliphatics, benzenoids, S-containing compounds and C5 branched-chain compounds); and (3) VOC profile (VOC cPCoA1 and cPCoA2; Fig. 3c). The SEMs were examined in parallel considering overall genotypic (within- and between-genotype) variation using lavaan [19] (Figs. 4 and 5), and within-genotype variation alone using piecewiseSEM [20] (with genotype as a random effect; Supplementary Figs. 1 and 2). This approach was used to confirm whether the detected relationships based on overall genotypic variation were also supported within genotypes.
Figure 5.

Structural equation models (SEMs) linking floral phenotype and treatment to floral fungal microbiome. a, Hypothesized effects of flower abundance and size as well as treatments on floral volatile organic compounds (VOCs) and fungal microbiome. The floral fungal microbiome was characterized using α-diversity (Shannon diversity) and β-diversity (the first two axes of constrained principal coordinates analysis, cPCoA1 and cPCoA2). VOCs were evaluated at three levels (see Materials and methods): individual VOCs, major classes of VOCs, and VOC profile (VOC cPCoA1 and cPCoA2). The bidirectional links between VOCs and fungal microbiome reflected potential reciprocal influence on each other. Treatments were coded using the control treatment as the reference level. SEMs were fitted based on overall genotypic variation using lavaan [19]. b-g, SEMs that contained notable positive (solid black) and negative (dashed black) links between VOCs and fungal microbiome, as well as other variables, were presented (see SEM details in Supplementary Table 5): †P < 0.10; *P < 0.05; **P < 0.01; ***P < 0.001. Grey links indicate P > 0.10. Link width and the numbers adjacent to links indicate standardized path coefficients.
The SEMs based on overall genotypic variation revealed that α-pinene and β-pinene were negatively linked with bacterial Shannon diversity (r = −0.21, P = 0.050 and r = −0.26, P = 0.017, respectively; Fig. 4b,c and Supplementary Table 4). Bacterial ASVs associated with the emission of α-pinene and β-pinene (Supplementary Table 6) were common residents of the floral microbiome in cultivated strawberry [1] and wild strawberries [5], including, for instance, Hymenobacter, Massilia, Deinococcus and nectar taxa (Acinetobacter, Arthrobacter, Methylobacterium, Nocardioides, Sphingomonas and Streptomyces) [21]. While treatments and flower abundance showed no notable effects (P > 0.10), flower size influenced bacterial Shannon diversity indirectly via enhancing the emission of α-pinene (r = 0.41, P < 0.001; Fig. 4b) and β-pinene (r = 0.41, P < 0.001; Fig. 4c). Different from α-pinene and β-pinene, other VOCs, major VOC classes and VOC profile (VOC cPCoA1 and cPCoA2) were not linked with bacterial Shannon diversity (Supplementary Table 4). For fungal Shannon diversity, only S-containing compound (benzothiazole) showed a positive relationship (r = 0.31, P = 0.039; Fig. 5b). Eight fungal ASVs that were commonly found in strawberry flowers [1] were identified as associated with the emission of the S-containing compound (Kendall’s correlation coefficient, P < 0.05; Supplementary Table 6), including those positively associated with the S-containing compound such as Cystofilobasidium macerans (yeast found in both flowers [1] and pollinators [22]), Phaeosphaeria caricicola (strains of this genus often pathogenic [23]) and Ramularia pratensis (some strains with biocontrol activities [24]), as well as those negatively associated with the S-containing compound such as pathogenic Blumeria graminis [25]. These detected relationships based on overall genotypic variation were also supported by the SEMs that examined within-genotype variation (Supplementary Fig. 1b, c; Supplementary Fig. 2b).
The β-diversity of floral bacterial communities was linked with a different set of VOCs relative to the α-diversity (Fig. 4), as revealed by the SEMs based on overall genotypic variation. Specifically, p-anisaldehyde was positively linked with the first axis of bacterial community composition (cPCoA1, r = 0.24, P = 0.019; Fig. 4d), which was dominated by Lactobacillus micheneri (found in both flowers [1] and pollinators [1, 26]) and Methylobacterium and Acinetobacter nectaris (nectar taxa [21]) on the positive axis and Delftia (common taxa in strawberry flowers [1]) on the negative axis (Fig. 2c; Supplementary Table 1). Moreover, ocimene isomers (r = −0.32, P = 0.029; Fig. 4e), aliphatics (r = −0.18, P = 0.069; Fig. 4f) and the C5 branched-chain compound (hexenyl tiglate, r = −0.28, P = 0.023; Fig. 4g) were negatively linked with the cPCoA2 of bacterial communities, which was dominated by Delftia, Lactobacillus micheneri, Methylobacterium and Acinetobacter nectaris on the positive axis and Phaseolibacter (common taxa in strawberry flowers [1]) and Sphingomonas and Rosenbergiella (nectar taxa [21]) on the negative axis (Fig. 2c; Supplementary Table 1). The cPCoA2 of bacterial communities was also influenced by flower size indirectly via the enhanced emission of VOCs (ocimene isomers, r = 0.28, P = 0.021, Fig. 4e; aliphatics, r = 0.29, P = 0.008, Fig. 4f), but was not influenced by flower abundance or treatments (Fig. 4). These relationships based on overall genotypic variation were also detected within genotypes (Supplementary Fig. 1).
The β-diversity of fungal communities, on the other hand, were influenced by treatments and flower abundance (Fig. 5c–f), as revealed by the SEMs based on overall genotypic variation. Relative to the control, mycorrhizae treatment positively affected the cPCoA1 of fungal communities (all P < 0.10; Fig. 5c, e, f and Supplementary Table 5), which was dominated by Pseudopithomyces chartarum (potentially pathogenic [25]) and Candelaria concolor (lichenized fungal taxon [27]) on the positive axis and pathogenic Alternaria alternata [28] and Cladosporium delicatulum [25] on the negative axis (Fig. 2d; Supplementary Table 1). In addition, herbivory + mycorrhizae treatment positively affected the cPCoA2 of fungal communities (r = 0.48, P = 0.003; Fig. 5g), which was dominated by C. concolor and A. alternata on the positive axis and Rhodotorula graminis (nectar yeast [21]) and Naganishia randhawae (=Cryptococcus randhawai [29], yeast also found in strawberry flowers [1]) on the negative axis (Fig. 2d; Supplementary Table 1). In addition to treatments, flower abundance affected the cPCoA1 of fungal communities indirectly via the reduced emission of VOCs per unit flower dry weight (Fig. 5c–f), including benzenoids (r = −0.24, P = 0.014; Fig. 5e) such as benzaldehyde (r = −0.21, P = 0.024; Fig. 5c) and VOC profile (VOC cPCoA1, r = −0.26, P = 0.027; Fig. 5f) that also primarily reflected the variation in benzaldehyde (Fig. 3c), as well as ocimene isomers (r = −0.24, P = 0.053; Fig. 5d). For the cPCoA2 of fungal communities, flower size exhibited an indirect effect via the enhanced emission of terpenes (r = 0.39, P < 0.001; Fig. 5g). While most of these relationships were also detected within genotypes (Supplementary Fig. 2), the links between fungal cPCoA1 and benzaldehyde (and related VOCs including benzenoids and VOC cPCoA1 that was dominated by benzaldehyde; Fig. 5c, e, f) as well as mycorrhizae treatment showed otherwise, indicating the role of among-genotype variation in driving some of the relationships.
Discussion
Our results demonstrated that strawberry genotype played a stronger role, overall, in shaping the floral microbiome than whole-plant biotic interactions both belowground (with mycorrhizae) and aboveground (with herbivores). In particular, we found that genotypic variation in flower abundance and size influenced floral VOCs, especially terpenes and benzenoids, which in turn affected the floral bacterial and fungal communities. While the effects of biotic interactions on floral phenotype including VOCs were weak (Figs. 3–5), mycorrhizae treatments (mycorrhizae and herbivory + mycorrhizae) directly affected the fungal communities in flowers relative to the control (Fig. 5). By linking genotypic variation in floral traits and biotic interactions to floral bacterial and fungal communities, our study revealed the mechanisms by which plant genotype influences floral microbiome assembly [1] and also provided the first evidence that plant-level biotic interactions at the rhizosphere and phyllosphere can influence the anthosphere microbiome.
In line with previous studies on the floral microbiome [1], genotypic variation in flower abundance influenced the floral microbiome. Flower abundance has been shown to govern floral microbiome assembly both directly by influencing the source pool of microbes that colonize individual flowers [1, 7] and indirectly by influencing pollinator visits [30] that can mediate microbial dispersal [1]. Our results showed that flower abundance affected fungal α- and β-diversity via the bidirectional links between VOCs and the fungal communities among genotypes but not within genotypes. In addition to flower abundance, flower size that is hypothesized to influence the floral microbiome has, nevertheless, rarely been tested [6]. In this study, we found that flower size affected bacterial and fungal communities via the bidirectional links between VOCs and the floral microbiome, which was supported by SEMs based on overall genotypic variation and within-genotype variation as well. While the causation of these relationships awaits experimental validation, it provides a new insight into the mechanisms by which flower abundance and size drive microbiome assembly.
Our results provided evidence for the hypothesized bidirectional relationships [9] between plant VOCs and microbiome in the anthosphere. Specifically, monoterpenes, namely α- and β-pinene and ocimene isomers contributed most to the variation in floral VOC profile and floral bacterial communities. Bacterial Shannon diversity was negatively associated with α- and β-pinene, whereas bacterial community composition was associated with p-anisaldehyde, ocimene isomers, aliphatics and hexenyl tiglate (the only C5 branched-chain compound in this study). These bidirectional relationships were supported by overall genotypic variation and within-genotype variation. On the other hand, fungal Shannon diversity was positively linked with benzothiazole, the only S-containing compound detected in strawberry volatile collections here, whereas fungal community composition was associated with terpenes (especially ocimene isomers) and benzenoids (especially benzaldehyde). Yet, the bidirectional relationship between benzaldehyde and fungal community composition was only supported among genotypes. To further verify that the relationship is not caused by unmeasured genotypic differences, experimental investigation that directly tests the reciprocal effects is needed. Overall, despite the known antimicrobial effects of floral VOCs such as terpenes, benzenoids, aliphatics and S-containing compounds [9–12], our results detected microbial taxa both negatively and those positively associated with the emission of these VOCs. This finding suggests the potential dual roles of floral VOCs as antimicrobial agents and carbon sources for some microbial taxa [9, 10] and/or their role in mediating complex microbe–microbe interactions [14] where the suppression of some taxa can potentially increase other taxa via direct and indirect interactions through the microbial network [1]. While how dynamic the relationships are between floral VOCs and the floral microbiome remains an open question, our results are expected to hold despite the time interval between VOC and microbiome collections, due to stable VOC composition (i.e. relative abundances of individual VOCs) among these genotypes detected over a longer time period (4 weeks) in this field experiment [18]. Taken together, our findings highlight the needs for future research on unraveling the specific mechanisms that underlie the relationships between floral VOCs and the floral microbiome as well as their temporal dynamics.
While plant interactions with herbivores at the phyllosphere have been found to influence the leaf microbiome [16, 17], our results showed, for the first time, that plant interactions at the rhizosphere and phyllosphere can influence the floral microbiome. Such effects were detected on shaping the floral fungal communities. Specifically, the mycorrhizae treatment caused the deviation of fungal community composition from the control (Fig. 5) by reducing pathogenic taxa (e.g. A. alternata, Cladosporium delicatulum) in flowers, whereas the herbivory + mycorrhizae treatment influenced fungal community composition by reducing floral yeasts (e.g. R. graminis, Naganishia randhawae) and increasing pathogenic taxa (e.g. A. alternata). While the effect of mycorrhizae treatment occurred among genotypes, the effect of herbivory + mycorrhizae treatment was supported by SEMs based on overall genotypic variation and within-genotype variation as well. These effects of biotic interactions on the floral microbiome were, nevertheless, not via floral VOCs, although plant mutualists and antagonists have been shown previously to alter floral VOCs [15, 31, 32]. In this study, the mycorrhizae treatments and herbivory treatments exhibited weak effects on the VOC profile. Yet, we could not rule out the possibility that the effects of the biotic treatments on floral VOCs can be dynamic and thus attenuate over time, and plant genotypes and their floral VOCs might respond differently to natural mycorrhizal inocula compared to a single mycorrhizal inoculum used in this study. Whether the effects of mycorrhizae and herbivory + mycorrhizae treatments on floral fungal microbiome are caused by altered source pools of microbes from the rhizosphere and phyllosphere that can affect the source–sink dynamics or altered plant traits not measured here that can affect floral fungal taxa merits further investigation.
To conclude, plant genotype governs floral microbiome assembly via both direct and indirect mechanisms mediated by diverse floral traits, especially the bidirectional relationships between floral VOCs and bacterial and fungal communities. In addition to the intrinsic characteristics of plant genotypes, interactions with non-focal mutualists and antagonists at the rhizosphere and phyllosphere influence the anthosphere microbiome that can profoundly affect plant reproduction [1, 2, 4]. These findings should not be unique to strawberries but may be generalizable to other crops and wild plants, because the VOCs of important relevance to the floral microbiome are common among plant species [9–13], and mycorrhizal partners and herbivore pests are ubiquitous in natural and agricultural ecosystems [33, 34]. We emphasize that our findings are based on plants with pre-existing endophytic microbiomes; as such, the extent to which endophytic microbiomes influence the detected relationships remain to be determined. Nevertheless, our findings based on strawberries improve the understanding of microbiome assembly in flowers and offer important insights into agricultural microbiomes that can influence plant reproduction and crop productivity.
Materials and methods
Factorial field experiment
The factorial field experiment [18] was conducted during March–September, 2019, at the Oakland University Student Farm, Rochester, Michigan (42.66073°N, 83.19558°W). Three strawberry genotypes (“Seascape”, “Tribute” and “Wasatch”) with 48 bare-root nursery stock plants per genotype were included in the experiment. The fresh biomass was recorded for each bare-root plant (i.e. initial plant weight) to account for potential influence of variation in initial plant size on the experiment. These bare-root plants (N = 144 total) were grown in 1-gallon pots filled with 2:1:1 mixture of sphagnum peat moss (Lambert, Rivière-Ouelle, Québec, Canada), washed play sand (Kolorscape, Atlanta, GA) and Turface MVP (Turface, Buffalo Grove, IL) during March 10–16, 2019, and re-potted into 2-gallon white fabric pots (247Garden, Montebello, CA) filled with the same media during May 24–29, 2019. These plants were subjected to four treatments (control, herbivory, mycorrhizae, and herbivory + mycorrhizae). Within each treatment, a single plant from each genotype was randomly selected (Fig. 1). The four treatments were then grouped into and replicated across 12 blocks (Fig. 1). For mycorrhizae-present treatments (mycorrhizae and herbivory + mycorrhizae), plants were inoculated with Rhizophagus irregularis spores (Elite 91 Myco Jordan, Murietta, CA) upon initial potting by dusting roots with inocula and also filling the 1-gallon pots half full of media and adding a thin layer of 5 mL inocula at the point of contact with roots. For herbivory-present treatments (herbivory and herbivory + mycorrhizae), plants experienced both natural herbivory from Vanessa cardui caterpillars (Carolina Biological Supply, Charlotte, NC; with three to five 2nd and 3rd instar larvae feeding on a single leaf per plant for six days) and simulated herbivory (by clipping the distal half each leaf from half of the leaves per plant and then applying 1 mM jasmonic acid solution in water [35] to cover all upper leaf surface) during June 17–July 6, 2019. The combination of natural and simulated herbivory is commonly used [36] to ensure that plants received the stimulus of a live herbivore and associated cues (e.g. enzymes in caterpillar saliva) while standardizing for the leaf area damaged per unit time.
Floral traits and VOC collection
In this study, we recorded the number of open flowers produced over a two-week period (August 4–17, 2019) prior to and during the floral microbiome collection. We sampled floral volatile emissions eight weeks after herbivory treatments, and 5–15 days prior to floral microbiome collection (July 30–August 10, 2019) to minimize the potential influence of the activities of VOC measurements that can cause artificial microbial arrival and destructive flower collection on floral microbiome. Floral volatiles were collected from new fully expanded flowers between 11 AM and 3 PM on sunny to partly sunny days, with no to little chance of rain. During the 4-h sampling period each day, 16 floral and two blank samples were collected using a dynamic headspace sampling method [31], by enclosing flowers in 12 oz. polyethylene terephthalate cups with dome lids (Comfy Package, Rikkel Corp, NY) and pulling air through a semi-open system at a flow rate of 200 mL/min through volatile traps with 30 mg of HayeSep Q adsorbent (VAS, Rensselaer, NY). We used either a PVAS22 portable volatile assay system (VAS, Rensselaer, NY) or an Air Lite low-flow air sample pump (SKC, Eighty Four, PA), which were shown functionally equivalent in volatile collection during initial trials. Volatiles were analyzed on an Agilent 7890A gas chromatograph (GC) with an Agilent 5977B mass spectrometer (MS) and separated on a HP-5 column (30 m × 250 μm × 0.25 μm) with helium as a carrier gas. We measured flower diameter and dry weight for all flowers sampled. Floral VOC emissions (per hour) were standardized by flower dry weight.
Floral microbiome collection and sequencing
The floral microbiome was collected from individual strawberry plants that flowered on August 16, 2019 (N = 93 out of 144 plants). A single flower (without pedicel) per plant was collected into a sterile 15 mL centrifuge tube using ethanol-rinsed forceps. These flower samples were transported on dry ice to the University of Pittsburgh and stored at −20°C within 12 h. Following the previous protocol [1], epiphytic microbes were collected by sonicating flowers in 3 mL phosphate-buffered saline at 40 kHz for 10 min and vortexing for 3 min. The microbes were pelleted by centrifuging at 10000 x g for 5 min, and then subject to DNA extraction using Quick-DNA Fecal/Soil Microbe Kits (Zymo Research, Irvine, CA). Samples and one negative control (without flower but in the process of microbial isolation and DNA extraction) were sent to Argonne National Laboratory for bacterial (16S rRNA V5–V6 region, 799f–1115r primer pair) and fungal (ITS1f–ITS2) library preparation [1]. Because the negative control failed in library preparation, the 93 samples were sequenced on a 1/2 lane of Illumina MiSeq (paired-end 250 bp).
Microbial sequence analysis
Demultiplexed paired-end (PE) reads were used for detecting bacterial and fungal amplicon sequence variants (ASVs) using package DADA2 v1.14.0 [37] in R v3.6.2 [38] and QIIME 2 v2019.4 [39]. Following the previous pipeline [1] for bacterial ASV detection in DADA2, PE reads were trimmed and filtered [truncLen = c(245, 245), trimLeft = c(0, 10), maxN = 0, truncQ = 2], prior to specific variant identification that took into account sequence errors. The PE reads were then end joined (minOverlap = 20, maxMismatch = 4) for ASV detection. After chimera removal, ASVs were assigned with taxonomic identification based on the SILVA reference database (132 release NR 99) implemented in DADA2. For fungal ASV detection in DADA2, the PE reads were first screened to remove potential primer contaminations. Due to the low-quality end (~50 bp), the fungal reads were truncated at 200 bp during quality filtering [truncLen = c(200, 200), maxN = 0, truncQ = 2] to ensure the accuracy in ASV detection. This method provided a conservative estimate of fungal ASVs [1], due to the potential loss of information based on ITS length variation [37]. After chimera removal, fungal ASVs were assigned with taxonomic identification based on the UNITE reference database (v8.0 dynamic release) using QIIME 2.
Bacterial and fungal ASV tables were further filtered separately before conversion into microbial community matrices using package phyloseq [40]. First, we removed non-focal ASVs (Archaea, chloroplasts and mitochondria). Second, we filtered out samples of low reads (<200 reads). Third, we normalized per-sample reads to the same number (i.e. the median reads, 4114 and 10 377, bacterial and fungal data set, respectively) [1,5]. Lastly, we removed low-frequency ASVs (<0.001% of total observations). The final community matrices consisted of 1500 and 976 ASVs for bacteria (N = 86 samples) and fungi (N = 87), respectively.
Floral microbial α- and β-diversity
To evaluate how floral microbial α-diversity was influenced by plant genotype, herbivory treatments (present: herbivory, herbivory + mycorrhizae; absent: control, mycorrhizae), and mycorrhizae treatments (present: mycorrhizae, herbivory + mycorrhizae; absent: control, herbivory), we analyzed the bacterial and fungal communities separately using general linear mixed models (LMMs) in package lme4 [41]. The response variable (Shannon diversity) was calculated using package vegan [42], and power transformed to improve normality with the optimal power parameter determined using the Box–Cox method in package car [43] (i.e. power parameter = 1 and 2 for bacterial and fungal Shannon diversity, respectively). The predictors included genotype, herbivory treatments and mycorrhizae treatments as well as their two-way interactions and three-way interaction, with initial plant weight as a covariate. The split plot with randomized complete block design in this experiment (Fig. 1) required nested random effects (i.e. treatment plots nested within blocks). However, due to only a subset of the 144 samples (N = 86 and 87 for bacterial and fungal data sets, respectively) for the LMMs, nested random effects led to difficulties in model convergence and thus were not used. Instead, the LMMs considered the random effect of blocks alone. Statistical significance (type III sums of squares) and least-squares means (LS-means) of predictors were assessed using packages lmerTest [44] and emmeans [45].
Microbial β-diversity (Bray–Curtis dissimilarity) was evaluated using PERMANOVA and constrained principal coordinates analysis (cPCoA) in vegan for bacterial and fungal communities separately. To assess the significance of the main effects, PERMANOVA and cPCoA included genotype, herbivory treatments and mycorrhizae treatments, with initial plant weight as a covariate and block as the random effect. To assess the significance of two-way interactions, PERMANOVA and cPCoA included both the main effects and their two-way interaction terms. Similarly, to assess the significance of the three-way interaction, the main effects, two-way interactions and three-way interaction were included. Once significant genotypic effect was identified, we further conducted the same PERMANOVAs and cPCoAs between pairwise genotypes to detect which specific genotypes were divergent from each other in microbial community composition.
Floral phenotype analysis
To evaluate whether strawberry genotypes differed in flower abundance (sample size N = 93) and flower size (N = 80), we conducted LMMs with the response variables power transformed to improve normality (i.e. power parameter = 0.5, square-root transformation, for flower abundance, and 2 for flower diameter). The predictors included genotype, herbivory treatments and mycorrhizae treatments as well as their two-way interactions and three-way interaction, with initial plant weight as a covariate and block as the random effect.
To evaluate whether and how strawberry genotypes differed in VOC profile (sample size N = 66), we conducted PERMANOVA and cPCoA. The predictors included genotype, herbivory treatments and mycorrhizae treatments as well as their two-way interactions and three-way interaction, with covariates (i.e. initial plant weight, pump ID and average daily temperature during VOC collection) and random effect (block). The PERMANOVA and cPCoA were conducted as in the above-mentioned microbial analysis to assess the significance of the main effects and interaction terms. Once significant genotypic effect was detected, we further identified the specific VOCs that differed between genotypes using random forest (RF) classification in packages caret [46] and randomForest [47]. The RF classification models were run for the full data with 1000 trees, and the number of randomly selected variables (i.e. individual VOCs) at each split of a decision tree was optimized using 10-fold cross validation in caret. RF model performance was assessed using out-of-bag (OOB) error. The set of important, non-redundant VOCs (in the presence of potentially correlated VOCs) were selected using backward variable elimination with package varSelRF [48].
SEMs linking floral phenotype to microbial α- and β-diversity
SEMs were conducted to link genotypic variation in flower abundance, size and VOCs, as well as treatments, to the α-diversity and β-diversity of floral bacterial and fungal communities (Figs. 4a and 5a). We focused on VOCs at three different levels: (1) individual VOCs that differed between genotypes as identified by the random forest classification (see above), and that predicted the α-diversity (Shannon diversity) or β-diversity (the first and second axis of cPCoA, cPCoA1 and cPCoA2) of bacterial and fungal communities based on backward predictor selection using packages caret and leaps [49]; (2) the major classes of VOCs (terpenes, aliphatics, benzenoids, S-containing compounds and C5 branched-chain compounds); and (3) VOC profile (VOC cPCoA1 and cPCoA2; Fig. 3c). For the SEMs, flower abundance and flower diameter were power transformed (i.e. power parameter = 0.5 and 2, respectively). For the categorical variables of treatments, we coded the control treatment as the reference level so that the effects of the other three treatments (herbivory, mycorrhizae, herbivory + mycorrhizae) were relative to the control. All the variables of VOCs were log transformed (i.e. power parameter = 0, log(x + 1)). For floral bacterial microbiome (Fig. 4a), a total of 41 SEMs were conducted (14 SEMs for Shannon diversity, 14 for cPCoA1 and 13 for cPCoA2, Supplementary Table 4). Similarly, for floral fungal microbiome (Fig. 5a), 41 SEMs were conducted (14 for α-diversity, 13 for cPCoA1 and 14 for cPCoA2, Supplementary Table 5). To reduce the number of models and model complexity, SEMs were only retained when notable paths (P < 0.10) were identified, and then re-fitted with only the notable paths present to estimate standardized path coefficients. The SEMs were fitted using package lavaan [19] based on overall genotypic (within- and among-genotype) variation. Model estimation used robust maximum likelihood with Satorra-Bentler scaled χ2 that can accommodate nonnormality in lavaan, and model fit was confirmed for each retained SEM (comparative fit index, CFI > 0.9; root mean squared error of approximation, RMSEA, the lower bound of 90% confidence interval < 0.05; standardized root mean squared residual, SRMR <0.1) [50]. With these retained SEMs (Supplementary Tables 4 and 5), to further confirm whether the detected relationships based on overall genotypic variation were also supported within genotypes, we re-fitted the retained SEMs (with only the notable paths present) based on within-genotype variation using package piecewiseSEM [20], where genotype was included as the random effect using package nlme [51]. The SEMs that contained notable links (P < 0.10) between VOCs and bacterial and fungal α- or β-diversity based on overall genotypic variation were presented in Figs. 4 and 5 (six for bacterial microbiome and six for fungal microbiome), and the same set of SEMs fitted based on within-genotype variation were presented in Supplementary Figs. 1 and 2.
Supplementary Material
Acknowledgements
This work was supported by the University of Pittsburgh Dietrich School of Arts and Sciences and the National Science Foundation (DEB 1452386 to TLA), Foundation for Food and Agricultural Research (FFAR 430876 to MAJ) and an Oakland University Provost Graduate Student Research Award (to RLW).
Author contributions
NW and TLA conceived the microbiome study and collected microbiome data. RLW and MAJ designed the factorial field experiment at Oakland University and collected floral trait data. NW performed data analyses and visualization. All authors contributed to writing.
Data availability
Microbiome sequence data are available from NCBI SRA (PRJNA793251).
Conflict of interest
The authors declare no conflict of interest.
Supplementary data
Supplementary data is available at Horticulture Research Journal online.
References
- 1. Wei N, Russell AL, Jarrett AR. et al. . Pollinators mediate floral microbial diversity and network under agrochemical disturbance. Mol Ecol. 2021;30:2235–47. [DOI] [PubMed] [Google Scholar]
- 2. Rebolleda-Gómez M, Forrester NJ, Russell AL.et al. . Gazing into the anthosphere: considering how microbes influence floral evolution. New Phytol. 2019;224:1012–20. [DOI] [PubMed] [Google Scholar]
- 3. Vannette RL. The floral microbiome: plant, pollinator, and microbial perspectives. Annu Rev Ecol Evol Syst. 2020;51:363–86. [Google Scholar]
- 4. Cullen NP, Fetters AM, Ashman T-L. Integrating microbes into pollination. Curr Opin Insect Sci. 2021;44:48–54. [DOI] [PubMed] [Google Scholar]
- 5. Wei N, Ashman T-L. The effects of host species and sexual dimorphism differ among root, leaf and flower microbiomes of wild strawberries in situ. Sci Rep. 2018;8:5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vannette RL, Hall G, Munkres I. Fungi are more dispersal-limited than bacteria among flowers. bioRxiv. 2020. 10.1101/2020.05.19.104968. [DOI] [Google Scholar]
- 7. Toju H, Vannette RL, Gauthier MPL.et al. . Priority effects can persist across floral generations in nectar microbial metacommunities. Oikos. 2018;127:345–52. [Google Scholar]
- 8. Rering CC, Beck JJ, Hall GW.et al. . Nectar-inhabiting microorganisms influence nectar volatile composition and attractiveness to a generalist pollinator. New Phytol. 2018;220:750–9. [DOI] [PubMed] [Google Scholar]
- 9. Farré-Armengol G, Filella I, Llusia J.et al. . Bidirectional interaction between phyllospheric microbiotas and plant volatile emissions. Trends Plant Sci. 2016;21:854–60. [DOI] [PubMed] [Google Scholar]
- 10. Hammerbacher A, Coutinho TA, Gershenzon J. Roles of plant volatiles in defence against microbial pathogens and microbial exploitation of volatiles. Plant Cell Environ. 2019;42:2827–43. [DOI] [PubMed] [Google Scholar]
- 11. Scala A, Allmann S, Mirabella R.et al. . Green leaf volatiles: a plant's multifunctional weapon against herbivores and pathogens. Int J Mol Sci. 2013;14:17781–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Junker RR, Tholl D. Volatile organic compound mediated interactions at the plant-microbe interface. J Chem Ecol. 2013;39:810–25. [DOI] [PubMed] [Google Scholar]
- 13. Sharifi R, Lee SM, Ryu CM. Microbe-induced plant volatiles. New Phytol. 2018;220:684–91. [DOI] [PubMed] [Google Scholar]
- 14. Weisskopf L, Schulz S, Garbeva P. Microbial volatile organic compounds in intra-kingdom and inter-kingdom interactions. Nat Rev Microbiol. 2021;19:391–404. [DOI] [PubMed] [Google Scholar]
- 15. Barber NA, Gorden NLS. How do belowground organisms influence plant-pollinator interactions? J Plant Ecol. 2015;8:1–11. [Google Scholar]
- 16. Humphrey PT, Whiteman NK. Insect herbivory reshapes a native leaf microbiome. Nat Ecol Evol. 2020;4:221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Perez JL, Victor French J, Summy KR.et al. . Fungal phyllosphere communities are altered by indirect interactions among trophic levels. Microb Ecol. 2009;57:766–74. [DOI] [PubMed] [Google Scholar]
- 18. Whyle RL, Trowbridge AM, Jamieson MA. Effects of mycorrhizal inoculation and herbivory on strawberry functional traits. (In Prep).
- 19. Rosseel Y. Lavaan: an R package for structural equation modeling. J Stat Softw. 2012;48:1–36. [Google Scholar]
- 20. Lefcheck JS, Freckleton R. piecewiseSEM: piecewise structural equation modelling in R for ecology, evolution, and systematics. Methods Ecol Evol. 2015;7:573–9. [Google Scholar]
- 21. Pozo MI, Lievens B, Jacquemyn H. Impact of microorganisms on nectar chemistry, pollinator attraction and plant fitness. In: Nectar: Production, Chemical Composition and Benefits to Animals and Plants. Nova Science Publishers: New York, NY, 2014,1–40. [Google Scholar]
- 22. Ye M-H, Fan S-H, Li X-Y.et al. . Microbiota dysbiosis in honeybee (Apis mellifera L.) larvae infected with brood diseases and foraging bees exposed to agrochemicals. R Soc Open Sci. 2021;8:201805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shoemaker RA, Babcock C. Phaeosphaeria. Can J Bot. 1989;67:1500–99. [Google Scholar]
- 24. Kernaghan G, Mayerhofer M, Griffin A. Fungal endophytes of wild and hybrid Vitis leaves and their potential for vineyard biocontrol. Can J Microbiol. 2017;63:583–95. [DOI] [PubMed] [Google Scholar]
- 25. Farr DF, Rossman AY Fungal Databases. U.S. National Fungus Collections, ARS, USDA. from https://nt.ars-grin.gov/fungaldatabases/. (Retrieved February 28, 2021). [Google Scholar]
- 26. Vuong HQ, McFrederick QS. Comparative genomics of wild bee and flower isolated Lactobacillus reveals potential adaptation to the bee host. Genome Biol Evol. 2019;11:2151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kirk P, Cannon P, Minter D.et al. . Ainsworth and Bisby's Dictionary of the Fungi. 10th ed. Wallingford, UK: CABI Publishing; 2008. [Google Scholar]
- 28. Nishikawa J, Nakashima C. Morphological and molecular characterization of the strawberry black leaf spot pathogen referred to as the strawberry pathotype of Alternaria alternata. Mycoscience. 2019;60:1–9. [Google Scholar]
- 29. Khan ZU, Ahmad S, Hagen F.et al. . Cryptococcus randhawai sp. nov., a novel anamorphic basidiomycetous yeast isolated from tree trunk hollow of Ficus religiosa (peepal tree) from New Delhi, India. Antonie Van Leeuwenhoek. 2010;97:253–9. [DOI] [PubMed] [Google Scholar]
- 30. Wei N, Kaczorowski RL, Arceo-Gómez G.et al. . Pollinators contribute to the maintenance of flowering plant diversity. Nature. 2021;597:688–92. [DOI] [PubMed] [Google Scholar]
- 31. Burkle LA, Runyon JB. Drought and leaf herbivory influence floral volatiles and pollinator attraction. Glob Change Biol. 2016;22:1644–54. [DOI] [PubMed] [Google Scholar]
- 32. Schiestl FP, Kirk H, Bigler L.et al. . Herbivory and floral signaling: phenotypic plasticity and tradeoffs between reproduction and indirect defense. New Phytol. 2014;203:257–66. [DOI] [PubMed] [Google Scholar]
- 33. Bernal JS, Medina RF. Agriculture sows pests: how crop domestication, host shifts, and agricultural intensification can create insect pests from herbivores. Curr Opin Insect Sci. 2018;26:76–81. [DOI] [PubMed] [Google Scholar]
- 34. Fester T, Sawers R. Progress and challenges in agricultural applications of arbuscular mycorrhizal fungi. Crit Rev Plant Sci. 2011;30:459–70. [Google Scholar]
- 35. Kleunen M, Ramponi G, Schmid B. Effects of herbivory simulated by clipping and jasmonic acid on Solidago canadensis. Basic Appl Ecol. 2004;5:173–81. [Google Scholar]
- 36. Waterman JM, Cazzonelli CI, Hartley SE.et al. . Simulated herbivory: the key to disentangling plant defence responses. Trends Ecol Evol. 2019;34:447–58. [DOI] [PubMed] [Google Scholar]
- 37. Callahan BJ, McMurdie PJ, Rosen MJ.et al. . DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. R Core Team . R: A Language and Environment for Statistical Computing. (R Foundation for Statistical Computing. https://www.R-project.org/, 2019). [Google Scholar]
- 39. Bolyen E, Rideout JR, Dillon MR.et al. . Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bates D, Machler M, Bolker BM.et al. . Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:1–48. [Google Scholar]
- 42. Oksanen J, Blanchet FG, Friendly M.et al. . Vegan: Community Ecology Package. R package version 2.5–5. https://CRAN.R-project.org/package=vegan (2019).
- 43. Fox J, Weisberg S. An R Companion to Applied Regression. 2nd ed. SAGE; 2011. [Google Scholar]
- 44. Kuznetsova A, Brockhoff PB, Christensen RHB. lmerTest package: tests in linear mixed effects models. J Stat Softw. 2017;82:1–26. [Google Scholar]
- 45. Lenth, R. Emmeans: Estimated Marginal Means, Aka Least-Squares Means. R package version 1.3.4. https://CRAN.R-project.org/package=emmeans (2019).
- 46. Kuhn, M. Caret: Classification and Regression Training. R package version 6.0–84. https://CRAN.R-project.org/package=caret (2019).
- 47. Liaw A, Wiener M. Classification and regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- 48. Diaz-Uriarte R. GeneSrF and varSelRF: a web-based tool and R package for gene selection and classification using random forest. BMC Bioinform. 2007;8:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miller, A. Leaps: Regression Subset Selection. R package version 3.1. https://CRAN.R-project.org/package=leaps (2020).
- 50. Kline RB. Principles and Practice of Structural Equation Modeling. 4th ed. Guilford Publications; 2015. [Google Scholar]
- 51. Pinheiro J, Bates D, DebRoy S.et al. . Nlme: Linear and Nonlinear Mixed Effects Models. R package version 3.1–152. https://CRAN.R-project.org/package=nlme (2021).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microbiome sequence data are available from NCBI SRA (PRJNA793251).