

Abstract
An array of human cancers, including hepatocellular carcinoma (HCC), overexpress the oncogene Astrocyte elevated gene-1 (AEG-1). It is now firmly established that AEG-1 is a key driver of carcinogenesis, and enhanced expression of AEG-1 is a marker of poor prognosis in cancer patients. In-depth studies have revealed that AEG-1 positively regulates different hallmarks of HCC progression including growth and proliferation, angiogenesis, invasion, migration, metastasis and resistance to therapeutic intervention. By interacting with a plethora of proteins as well as mRNAs, AEG-1 regulates gene expression at transcriptional, post-transcriptional, and translational levels, and modulates numerous pro-tumorigenic and tumor-suppressive signal transduction pathways. Even though extensive research over the last two decades using various in vitro and in vivo models has established the pivotal role of AEG-1 in HCC, effective targeting of AEG-1 as a therapeutic intervention for HCC is yet to be achieved in the clinic. Targeted delivery of AEG-1 small interfering ribonucleic acid (siRNA) has demonstrated desired therapeutic effects in mouse models of HCC. Peptidomimetic inhibitors based on protein-protein interaction studies has also been developed recently. Continuous unraveling of novel mechanisms in the regulation of HCC by AEG-1 will generate valuable knowledge facilitating development of specific AEG-1 inhibitory strategies. The present review describes the current status of AEG-1 in HCC gleaned from patient-focused and bench-top studies as well as transgenic and knockout mouse models. We also address the challenges that need to be overcome and discuss future perspectives on this exciting molecule to transform it from bench to bedside.
1. Introduction
Liver cancer is the fourth most lethal tumor globally with a 5-year survival of 18% (Bray et al., 2018; Villanueva, 2019). Hepatocellular carcinoma (HCC), arising from primary hepatocytes, constitutes over 80% of all liver cancers (El-Serag, 2011; Robertson, Srivastava, Rajasekaran, et al., 2015). HCC is generally a consequence of chronic liver disease in which there is injury and inflammation to the liver for decades leading to hepatic fibrosis, cirrhosis and ultimately HCC. The majority of HCC cases develop owing to viral hepatitis, and globally, Hepatitis B virus (HBV) and Hepatitis C virus (HCV) infections are considered as the most significant HCC risk factors (Dimitroulis et al., 2017; Donato, Boffetta, & Puoti, 1998; Sarkar, 2013). Heavy alcohol consumption for a long time may lead to chronic liver disease and HCC development. The increasing numbers of nonalcoholic fatty liver disease (NAFLD), as a consequence of metabolic syndrome and obesity, is currently considered a major risk factor for HCC (Anstee, Reeves, Kotsiliti, Govaere, & Heikenwalder, 2019; Friedman, Neuschwander-Tetri, Rinella, & Sanyal, 2018; Rajesh & Sarkar, 2020; Villanueva, 2019). Contamination of food and medicine may also lead to the development of HCC. For instance, aflatoxins, frequent contaminants of many staple cereals and oil-seeds, may give rise to serious public health jeopardy including the causation of HCC (Kew, 2013). Aristolochic acid (AA), found in traditional Chinese herbal medicines, can also increase the risk of HCC development (Ng et al., 2017). Other factors that can cause HCC are hemochromatosis, smoking, coinfection with the human immunodeficiency virus (HIV) with either HBV or HCV, α1-antitrypsin deficiency, autoimmune hepatitis, porphyrias, and Wilson disease (Sarkar, 2013; Villanueva, 2019; Yang et al., 2019).
More than 50% of HCC cases are diagnosed in China; and among those patients, 90% population are infected with HBV. It is expected that the etiologic landscape of HCC might be changed after universal HBV vaccination and a wide range of application of direct-acting antiviral agents against HCV infection. Indeed, HBV vaccination reduces the incidence of HCC (Chang et al., 2009). However, there are still a large number of individuals who remain to be vaccinated, and therefore, they are at risk of developing HCC (Yuen et al., 2018). Antiviral therapies reduce the incidence of HCC although they do not completely eliminate the risk (Liaw et al., 2004). HCC is generally regarded as rare cancer in Western countries, however, the incidence of HCC is rising in Western countries owing to HCV infection, alcohol abuse, and NAFLD. In HCV-infected patients who show a sustained virologic response to the interferon-based treatment schedule, the risk of HCC is decreased from 6.2% to 1.5%, as compared with patients who do not show a response (Morgan et al., 2013). Nevertheless, the increase in incidence and mortality of HCC is projected to continue in the coming decades because of the pandemic of obesity which shows a direct link to HCC by causing NAFLD (Anstee et al., 2019). Indeed, promoting a healthy lifestyle, such as decreased alcohol consumption coupled with diet and exercise facilitating prevention of metabolic syndrome, significantly reduces the risk of HCC development (Forner, Reig, & Bruix, 2018; Li, Park, et al., 2014). Intriguingly, coffee, statins, metformin, and aspirin have been shown to be protective against formation of HCC in various studies conducted around the world (Bravi, Tavani, Bosetti, Boffetta, & La Vecchia, 2017; Sahasrabuddhe et al., 2012; Singh, Singh, Singh, Murad, & Sanchez, 2013; Zhou et al., 2016). Although protective effects of these agents against HCC have not been confirmed yet in randomized controlled trials (RCTs), coffee use is presently recommended by the European Association for the Study of the Liver (EASL) 2018 clinical practice guidelines for HCC (European Association for the Study of the Liver, Electronic address: easloffice@easloffice.eu, & European Association for the Study of the Liver, 2018).
Pathologically, HCC is generally a compact tumor with little stroma and a central necrotic core because of hypoxia. Pathological analysis identifies several types of HCC according to macroscopic and microscopic characteristics. Macroscopically, HCC can be classified into three types, nodular, massive, and infiltrative type (Kojiro, 2005). The nodular form is the most common HCC type and is characterized by single or multiple nodular neoplasms that are well-circumscribed. Massive HCCs consist of a large mass that almost completely replaces one of the lobes of the liver. The infiltrative type of HCC is not circumscribed and is characterized by diffuse infiltration of the liver, with potential to infiltrate into the portal or hepatic vein. HCCs can be divided into two types based on their microscopic appearances, such as well-differentiated and poorly differentiated HCCs. In well-differentiated type, HCC cells are like hepatocytes and form trabeculae, cords, and nests, whereas in poorly differentiated type, tumor cells are pleomorphic, anaplastic, and giant (Kojiro, 2005).
The diagnosis of HCC is of great interest to physicians evaluating patients with liver cirrhosis (Talwalkar & Gores, 2004). HCC can be diagnosed with the use of imaging techniques in cirrhotic patients as a result of the vascular shift that occurs during the malignant transformation of hepatocytes (Villanueva, 2019). The vascular shift converts into a characteristic pattern of hyperenhancement in the arterial phase and washout in venous or delayed phases on contrast-enhanced computed tomography (CT) or magnetic resonance imaging (MRI). That pattern has a sensitivity between 66% and 82% and a specificity greater than 90% for the diagnosis of HCC in cirrhotic patients having nodules larger than 1cm in diameter. For patients having nodules with an inconclusive pattern on imaging, and for patients without cirrhosis, the diagnosis should depend on biopsy (Roberts et al., 2018; Villanueva, 2019).
Surveillance for HCC aims to decrease disease-related mortality (Ayuso et al., 2018). Biannual ultrasonography of the abdomen is the recommended method for surveillance, with or without measurement of serum levels of HCC marker alpha-fetoprotein. Biannual ultrasonography allows an early diagnosis when effective therapies are feasible (Forner et al., 2018). However, the performance of ultrasound surveillance is problematic in obese patients (Villanueva, 2019). Cirrhotic patients with nodules that are less than 1cm in diameter should undergo ultrasound surveillance every 3 to 4 months and be considered for a return to conventional surveillance if the nodule is stable in size after 12 months (European Association for the Study of the Liver, Electronic address: easloffice@easloffice.eu, & European Association for the Study of the Liver, 2018).
After the diagnosis of HCC, it is important to determine if the tumor has spread, and if so, how far, which is determined by staging of HCC. The staging process helps describe how much cancer is in the body, how serious the cancer is, and how to treat cancer effectively. The majority of HCC patients have concomitant liver complications, especially cirrhosis who markedly compromises liver function, and as such the risk vs. benefit ratio should be carefully determined before the commencement of the treatment schedule. Indeed, the complex ecosystem of HCC calls for a multidisciplinary approach for the management of the disease with expertise in hepatology, surgical procedure, pathology, bioengineering, chemotherapy, oncology, radiology, and specialized nursing. Thus, the application of interdisciplinary science may enhance the survival of HCC patients. For the adequate estimation of the survival of HCC patients, any staging system must quantify the tumor burden as well as the amount of liver dysfunction and performance status. The Barcelona Clinic Liver Cancer (BCLC) algorithm introduced in 1999 is the most widely applied staging system, and this system measured all the components for the adequate estimation of survival of HCC patients (Ayuso et al., 2018; Forner et al., 2018; Llovet, Bru, & Bruix, 1999). Other staging systems do exist, but their application is restricted to certain geographic locations (Villanueva, 2019). The BCLC system is endorsed in clinical practice guidelines and is also applied for the clinical trial design in HCC (European Association for the Study of the Liver, Electronic address: easloffice@easloffice.eu, & European Association for the Study of the Liver, 2018; Marrero et al., 2018). This staging system divides patients to be in one of five stages and recommends treatment suggestions for each stage (Fig. 1).
Fig. 1.

Current treatment recommendations in different stages of HCC. The staging system is based on the BCLC algorithm introduced in 1999. The lenvatinib clinical trial did not include patients with 50% or higher occupation of the liver with tumors or invasion of the bile duct or main portal vein. None of the second-line therapies were tested in patients after lenvatinib therapy.
HCC is a highly aggressive malignancy and hypervascular in nature with rapid growth and early vascular invasion; therefore, the occurrence and loss of life run in parallel in the case of HCC. Clinical management options for HCC patients include surgical resection, tumor ablation with radiofrequency, liver transplantation, transarterial therapies, and systemic therapies. The majority of HCC patients are diagnosed with advanced symptoms that are not manageable by surgical resection or transplantation (Robertson, Srivastava, Rajasekaran, et al., 2015; Villanueva, 2019). For advanced HCC, multi-kinase inhibitor sorafenib has been the treatment of choice for more than a decade and recently other tyrosine kinase inhibitors (TKIs), such as lenvatinib, regorafenib, and cabozantinib, and ramucirumab, a monoclonal antibody blocking VEGF2R signaling, have been approved either as first line therapy or following sorafenib treatment (Abou-Alfa et al., 2018; Bruix et al., 2017; Kudo et al., 2018; Llovet et al., 2008; Zhu et al., 2019). In recent years, immunotherapy has come into limelight as a promising approach for HCC which includes immune checkpoint blockers/monoclonal antibodies against the programmed cell death protein 1 (PD-1), PD-1 ligand (PD-L1) and cytotoxic T lymphocyte antigen-4 (CTLA-4) such as nivolumab, pembrolizumab, MED14736, ipilimumab and tremelimumab (Johnston & Khakoo, 2019). PD-1 inhibitor Nivolumab showed efficacy in ~20% HCC patients of all etiologies with significantly improved survival benefits compared to TKIs and nivolumab and pembrolizumab have been approved for HCC treatment as a second line therapy following sorafenib (El-Khoueiry et al., 2017). Even with existence of HCC biomarkers, such as alpha-fetoprotein and glypican-3 (GPC-3), as well as newly discovered gene signatures, early detection and surveillance are still suboptimal contributing to continuing increase in global incidence and mortality of HCC (Liu et al., 2020; Yang et al., 2019). This scenario is further complicated by the fact that HCC is inherently resistant to conventional chemo- and radiotherapy and approved therapeutics for the treatment of HCC increase the survival of patients for only a few months as compared to placebo. This morbid scenario calls for identification of unique molecules driving HCC development and progression leading to the discovery of novel molecular medicine that can mitigate the suffering of these patients.
2. The molecular biology of HCC
Recent data have identified several molecular abnormalities, including somatic DNA alterations, in HCC. Mutations in various proto-oncogenes, such as insulin-like growth factor 2 (IGF2), CTNNB1 (encoding β-catenin), c-Myc, and cyclin D1, and tumor suppressor genes, such as p53, p73, retinoblastoma (Rb), adenomatous polyposis coli (APC), deleted in liver cancer 1 (DLC1), deleted in liver cancer 2 (DLC2), phosphatase and tensin homolog (PTEN), suppressor of cytokine signaling 1 (SOCS1), glutathione S-transferase pi 1 (GSTP1), hepatocellular carcinoma suppressor 1 (HCCS1)/VPS53, SMAD family member 2 and 4 (SMAD2/4), and axis inhibition protein 1 (AXIN1), have been identified in HCC (Boyault et al., 2007; Mann et al., 2007; Teufel et al., 2007). A plethora of non-coding RNAs, including miRNA and long non-coding RNAs, has been implicated in HCC pathogenesis (Manna & Sarkar, 2020). Comprehensive studies combining exome sequencing, transcriptome analysis, and genomic characterization of HCCs have shown that HCC is heterogeneous at the molecular level (Cancer Genome Atlas Research Network, Electronic address: wheeler@bcm.edu, & Cancer Genome Atlas Research Network, 2017; Hoshida et al., 2009; Schulze et al., 2015). One validated analysis has identified six robust sub-groups of HCC, designated G1–G6, that have characteristic genetic and clinical features (Amaddeo et al., 2015; Boyault et al., 2007). Mutations in the telomerase reverse transcriptase (TERT) promoter (occurring ~60% of patients with HCC), CTNNB1 (27–40%) and p53 (21–31%,) genes in HCC are the most common (Calderaro et al., 2017; Cancer Genome Atlas Research Network, Electronic address: wheeler@bcm.edu, & Cancer Genome Atlas Research Network, 2017). Specific etiologies of HCC are associated with particular genetic alterations (Calderaro et al., 2017). For instance, TERT promoter and p53 mutations are the most frequent genetic events in HBV-associated HCC, whereas CTNNB1 mutations are strongly associated with alcohol-related HCC. Besides, p53 mutations are linked with decreased survival (Amaddeo et al., 2015; Cancer Genome Atlas Research Network, Electronic address: wheeler@bcm.edu, & Cancer Genome Atlas Research Network, 2017). IL-6/Janus kinase (JNK)/signal transducer and activator of transcription (STAT) pathway activation without TERT, CTNNB1, or pP53 pathway alterations is frequently seen in the steatohepatitic subtype of HCC (Calderaro et al., 2017). These integrated analyses indicate the molecular diversity of HCC, and different etiologies with distinct mechanisms are involved in hepatocarcinogenesis.
Next-generation sequencing studies in HCC patients treated with systemic therapies are beginning to provide insights into the various cell signaling pathways and the disease control processes in response to specific classes of systemic therapies. For instance, in HCC patients treated with immune checkpoint inhibitors, activating mutations in the Wnt/β-catenin signaling pathway have been linked with a lower disease control rate and survival (Harding et al., 2019; Yang et al., 2019). Several abnormal signaling pathways play important role in liver carcinogenesis, and these aberrant signals may provide a source of novel molecular targets for effective therapies. The major signaling cascades activated in HCC are (A) MAPK/ERK pathway, (B) PI3K/Akt/mTOR pathway, (C) NF-κB pathway, and (D) Wnt/β-catenin signaling pathway.
3. A brief history of AEG-1
It has been almost two decades since the publication of the first discovery and cloning of AEG-1 (Su et al., 2002). AEG-1 was identified as an HIV- and tumor necrosis factor alpha (TNF-α)-inducible gene in primary human fetal astrocytes (PHFAs) with the protein primarily localizing at endoplasmic reticulum (ER) (Kang et al., 2005; Su et al., 2002). Since then, a comprehensive body of data on AEG-1 has elucidated diverse functional aspects of this molecule, ranging from its role in cancer biology to fundamental biological processes, such as inflammation and lipid metabolism, and the molecular mechanisms by which it exerts these functions. Initially, in vivo studies confirmed that as a cell membrane protein AEG-1 facilitates metastasis of breast cancer cells to the lung and was named metadherin, and the GenBank symbol for AEG-1 is MTDH (which stands for metadherin), for its participation in tumor metastasis and adhesion (Brown & Ruoslahti, 2004). Rodent (e.g., rat/mouse) AEG-1 was also cloned subsequently as an ER/nuclear envelop protein and as a tight junction protein, and was named lysine-rich CEACAM-1 co-isolated protein (LYRIC) (Britt et al., 2004; Sutherland, Lam, Briers, Lamond, & Bickmore, 2004). It was documented that AEG-1 is overexpressed in different cancer cell lines, such as melanoma, breast cancer and malignant glioma, compared to their normal counterparts (Kang et al., 2005). Indeed, a large set of studies currently indicates that AEG-1 is overexpressed in all types of cancers analyzed to date and it is an essential component critical to the onset and progression of cancer (Emdad et al., 2016; Sarkar & Fisher, 2013; Yoo, Emdad, et al., 2011). AEG-1 expression positively correlates with tumor progression, especially in the metastatic stage, and in vivo studies using nude mice and metastatic models with various cancer cell lines and transgenic and knockout mouse models point out that AEG-1 overexpression induces an aggressive, angiogenic, and metastatic phenotype, and AEG-1 knockdown or knockout markedly hampers tumor initiation, growth, and metastasis (Emdad et al., 2009; Hu et al., 2009; Hu et al., 2014; Robertson et al., 2014; Robertson et al., 2018; Shen et al., 2020; Wan, Hu, et al., 2014; Wan, Lu, et al., 2014; Yoo, Emdad, et al., 2009).
4. Localization and sequence motifs of AEG-1
The initial discovery of AEG-1 led to an array of research on its biological functions and biochemical characteristics, and although there are still some disputed issues, a number of features of AEG-1 have been agreed upon with unequivocal consensus. AEG-1 gene is located on human chromosome 8q22 and contains 12 exons and 11 introns. In humans, AEG-1 is a lysine-rich highly basic protein having 582 amino acid (a.a.) residues, and the a.a. sequences are highly conserved among vertebrates (Emdad et al., 2007; Kang et al., 2005; Yoo, Emdad, et al., 2011). The three-dimensional structure of full AEG-1 protein has not been elucidated and as such the functional domains of this protein have not been precisely defined, although the structure of the region of AEG-1 with which it interacts with staphylococcal nuclease and tudor domain containing 1 (SND1) has been resolved (Guo et al., 2014). Intracellular localization of AEG-1 has been studied extensively to understand its biological functions showing variable results. It seems that the localization of AEG-1 in cells depends on the cell type examined and the imaging techniques employed. In various experimental procedures, AEG-1 can be detectable in the cytoplasm/ER as well as in the nucleus by immunohistochemical (IHC) and immunofluorescent (IF) staining of cultured cells or sectioned specimens of tissue samples (Emdad et al., 2006; Emdad et al., 2007; Kang et al., 2005; Li et al., 2008; Sutherland et al., 2004). Additionally, AEG-1 can be found on the cell membrane in rat liver as well as in mouse breast cancer cells (Britt et al., 2004; Brown & Ruoslahti, 2004), and in contrast to that, a green fluorescent protein (GFP)-fused AEG-1 shows a stronger signal in the nucleus and nucleolus (Thirkettle, Girling, et al., 2009; Thirkettle, Mills, Whitaker, & Neal, 2009).
These apparently discrepant findings can be explained by unique sequence motifs present in AEG-1 (Fig. 2). AEG-1 has a transmembrane domain (TMD) between 50 and 77 a.a. residues which allows it to anchor on ER membrane, the predominant site of its localization, as well as on cell membrane, which is mainly found in aggressive, metastatic cells (Alexia et al., 2013; Brown & Ruoslahti, 2004; Hsu, Reid, Hoffman, Sarkar, & Nicchitta, 2018; Kang et al., 2005). Three nuclear localization sequences (NLS) are present in the lysine-rich regions of AEG-1 between 79 and 91, 432–451 and 561–580 a.a. residues (Emdad et al., 2007; Sutherland et al., 2004; Thirkettle, Girling, et al., 2009). By utilizing the GFP-fusion system, NLS 1 and 3 and their flanking regions were determined to be able to target AEG-1 to nucleus and nucleolus (Thirkettle, Girling, et al., 2009). In benign human tissues, including prostate, thyroid and lung, as well as in primary mouse hepatocytes, AEG-1 is predominantly located in the nucleus, while in cancer cells and tissues it is located mainly in the cytoplasm (Srivastava et al., 2014; Thirkettle, Girling, et al., 2009). It has been suggested that nuclear AEG-1 is a sumoylated protein that undergoes mono-ubiquitination in its NLS2 motif facilitating its translocation out of the nucleus and increased stability in the cytoplasm (Srivastava et al., 2014; Thirkettle, Girling, et al., 2009). In a later study, it was documented that K486 and K491 of AEG-1, which lie in the extended NLS2 region, undergo mono-ubiquitination, and an E3 ubiquitin ligase, TOPORS, was implicated to mediate this reaction (Luxton et al., 2014). This post-translational modification might explain why AEG-1 of predicted 64kD molecular weight shows bands between 70 and 80kD when detected by antibodies raised against various AEG-1 immunogenic fragments. Nonetheless, the biological significance of post-translational modification of AEG-1 in normal body function as well as in the pathophysiology of various diseases including HCC remains to be elucidated. On the other hand, it has also been shown that when stimulated by TNFα AEG-1 translocates to the nucleus from the cytoplasm interacting with p65 subunit of NF-κB and CREB-binding protein (CBP) thereby augmenting NF-κB transcriptional activity (Emdad et al., 2006; Sarkar et al., 2008). Additionally, a number of clinical studies have detected increasing nuclear staining for AEG-1 with progression of cancer, although the significance of this finding has not been studied (Sarkar & Fisher, 2013). Thus, the regulation of AEG-1 localization and the mechanism of its shuttling among different intracellular compartments still requires clarification. Apart from these localization signals, a lung-homing domain has been identified in mouse AEG-1, which corresponds to 381–443 a.a. residues of human AEG-1, facilitating adhesion of breast cancer cells to lung endothelium (Brown & Ruoslahti, 2004; Sarkar et al., 2009). AEG-1 lacks any DNA-binding domains or motifs but it has an LXXLL motif present in its N-terminus (21–25 a.a. residues) with which AEG-1 interacts with the transcription factor retinoid X receptor (RXR) and negatively regulates its activity (Srivastava et al., 2014). The importance of this motif in regulating AEG-1 function will be described in later section.
Fig. 2.

Cartoon showing important structural features of human AEG-1 mediating its function. Please see text for details. TMD: transmembrane domain; NSL: nuclear localization signal; LHD: lung-homing domain. K63-linked polyubiquitin interaction region is required for interaction with upstream ubiquitinated activators of NF-κB, such as RIP1 and TRAF2. Numbers indicate amino acids (a.a.).
5. Mechanism of AEG-1 overexpression in cancer
A plethora of mechanisms contribute to overexpression of AEG-1 in cancer (Fig. 3). Gains and amplifications of chromosome 8q are frequently seen in a variety of cancers, including HCC (Knuutila et al., 1998; Zimonjic, Keck, Thorgeirsson, & Popescu, 1999). Gain of chromosome 8q22, which contains AEG-1 gene, was detected in poor-prognosis breast cancer, and Q-RT-PCR and fluorescence in-situ hybridization (FISH) confirmed AEG-1 gene amplification in breast cancer patients (Hu et al., 2009). Overexpression of AEG-1 is also linked with increased copy numbers of it, primarily due to gains of large regions of chromosome 8q in HCC (Wang et al., 2013; Yoo, Emdad, et al., 2009). Ha-ras was reported to be upstream of AEG-1, activating PI3K/Akt signaling and increasing binding of c-Myc to key E-box elements in the AEG-1 promoter, thus promoting AEG-1 transcription in transformed astrocytes (Lee, Su, Emdad, Sarkar, & Fisher, 2006). The observation that AEG-1 is under transcriptional control of three potent oncogenic drivers, Ras, Akt and c-Myc, helps explain why AEG-1 plays a pivotal role in cancer development and progression. AEG-1 synergized with Ha-ras to augment soft agar colony formation by immortal melanocytes and AEG-1 siRNA suppressed Ha-ras-mediated colony formation by transformed astrocytes thereby placing AEG-1 as a key nodal point in Ha-ras-mediated oncogenesis (Kang et al., 2005; Lee et al., 2006). Since PI3K/Akt is activated and c-Myc is overexpressed in human HCC, they may also regulate AEG-1 transcription in HCC (Sarkar, 2013). Indeed, c-Myc is also located in chromosome 8q and is co-amplified with AEG-1 in HCC patients, and a transgenic mouse with hepatocyte-specific AEG-1 and c-Myc overexpression developed highly aggressive HCC with lung metastasis demonstrating functional cooperation between these two molecules (Srivastava, Siddiq, et al., 2015). AEG-1 expression is induced by lipopolysaccharide (LPS) and inflammatory cytokines, such as IL-1β and TNFα, via activation of NF-κB, a mechanism which might contribute to AEG-1 overexpression in cancers generated as a consequence of chronic inflammation, such as HCC (Khuda et al., 2009; Srivastava et al., 2017; Vartak-Sharma, Gelman, Joshi, Borgamann, & Ghorpade, 2014). Post-transcriptionally, AEG-1 is controlled by multiple tumor-suppressor miRNAs, such as miR-375, miR-136, miR-302c, miR-466 and miR-30a-5p, which are downregulated in several cancers, including HCC (He et al., 2012; Jia et al., 2018; Li, Dai, Ou, Zuo, & Liu, 2016; Malayaperumal, Sriramulu, Jothimani, Banerjee, & Pathak, 2020; Zhao et al., 2014; Zhu et al., 2014). Several long non-coding RNAs (lncRNAs) have been implicated to upregulate AEG-1 by acting as a sponge for specific AEG-1-targeting miRNAs (Han, Fu, Zeng, Yin, & Li, 2020; Lu et al., 2018; Zhang et al., 2017). In malignant glioma cells, lncRNA human histocompatibility leukocyte antigen (HLA) complex P5 (HCP5) promotes malignant phenotype by upregulating Runt-related transcription factor 1 (RUNX1) via sponging miR-139, and RUNX1 in turn upregulates AEG-1 transcription by directly binding to its promoter (Teng et al., 2016). LINC01638 interacts with c-Myc protecting it from speckle type BTB/POZ protein (SPOP)-mediated ubiquitination and degradation with subsequent upregulation of AEG-1 and Twist1 promoting epithelial-mesenchymal transition (EMT) in triple-negative breast cancer cells (Luo et al., 2018). As mentioned earlier, post-translationally mono-ubiquitination rendered increased stabilization of cytoplasmic AEG-1 in cancer cells (Thirkettle, Girling, et al., 2009). It was documented that cytoplasmic polyadenylation element-binding protein 1 (CPEB1) binds to AEG-1 mRNA and increases its translation in glioblastoma cells (Kochanek & Wells, 2013). On the other hand, in HCC cells CPEB3, which functions as a tumor suppressor, binds to 3′-untranslated region of AEG-1 mRNA and inhibits its translation (Zhang et al., 2020). Thus, all potential mechanisms of gene regulation confer AEG-1 overexpression in cancer.
Fig. 3.
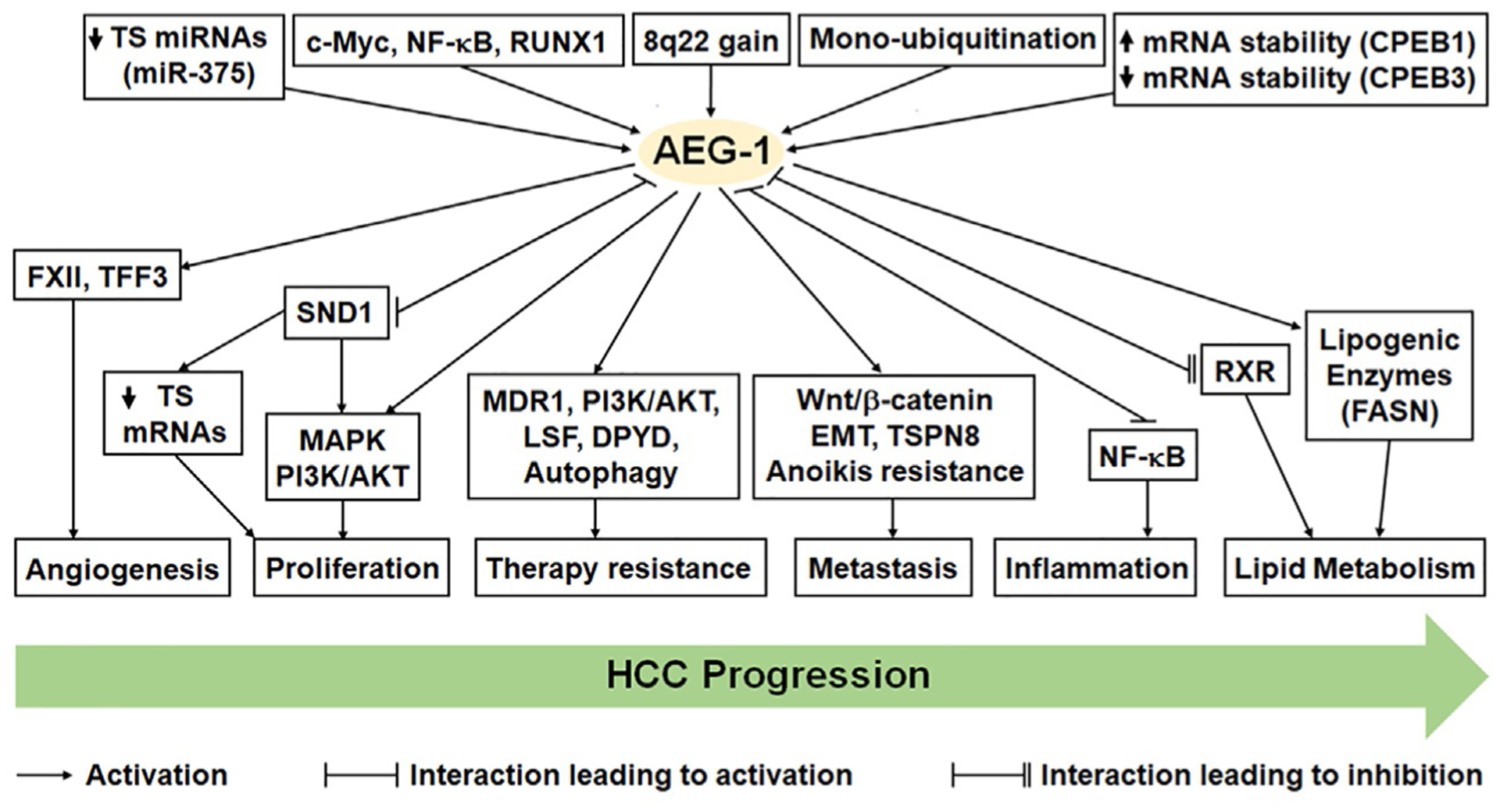
Schematic presentation of the mechanisms of AEG-1 regulation and AEG-1 downstream events in HCC. For details, please see the main text.
6. AEG-1: Clinicopathologic findings in HCC
As indicated earlier, AEG-1 functions as an oncogene in all types of cancers, and in this review, we focus on its role in HCC as evidenced from extensive research efforts over the last decade or so. IHC analysis of tissue microarray (TMA) containing 86 primary HCC, 23 metastatic HCC and 9 normal adjacent liver samples showed little to no AEG-1 staining in normal liver samples, while 93.58% of HCC samples showed variable AEG-1 levels which progressively increased with the stages I-IV and from well-differentiated to poorly differentiated (P < 0.0001) (Yoo, Emdad, et al., 2009). In a separate cohort, including 132 samples in various stages such as normal liver (n = 10), cirrhotic tissue (n = 13), low-grade dysplastic nodules (n = 10), high-grade dysplastic nodules (n = 8), and hepatocellular carcinoma (n = 91), AEG-1 mRNA expression was analyzed from Affymetrix microarray data. In HCV-HCC AEG-1 levels were significantly higher compared to normal and cirrhotic liver with mean up-regulation of 1.7-fold (P = 0.04) and 1.65-fold (P < 0.001), respectively (Yoo, Emdad, et al., 2009). Genomic amplification of AEG-1 was identified in 26% of HCC patients by DNA copy gain analysis which was also demonstrated in additional studies (Wang et al., 2013; Yoo, Emdad, et al., 2009).
Multiple clinicopathologic studies have established AEG-1 as a prognostic marker for HCC. IHC analysis in 323 HCC patients demonstrated AEG-1 expression in 54.2% patients (Zhu et al., 2011). AEG-1 levels were associated with microvascular invasion (P < 0.001), pathologic satellites (P = 0.007), tumor differentiation (P = 0.002), and TNM stage (P = 0.001). The 1-, 3-, 5-year overall survival (OS) in high AEG-1-expressing group were significantly lower than those in low AEG-1-expressing group (83.0% vs. 89.7%, 52.0% vs. 75.3%, 37.4% vs. 66.9%, respectively); and the 1-, 3-, 5-year cumulative recurrence rates were markedly higher in high AEG-1-expressing group than those in low AEG-1-expressing group (32.4% vs. 16.8%, 61.2% vs. 38.2%, 70.7% vs. 47.8%, respectively). AEG-1 was identified as an independent prognostic factor for both OS (HR = 1.870, P < 0.001) and recurrence (HR = 1.695, P < 0.001) by univariate and multivariate analyses (Zhu et al., 2011). Similar univariate and multivariate analyses with Cox regression following IHC of 85 HCC samples revealed that tumor size (HR, 2.285, 95% CI, P = 0.015), microvascular invasion (HR, 6.754, 95% CI, P = 0.008), and AEG-1 expression (HR, 4.756, 95% CI, P = 0.003) were independent prognostic factors for OS, and tumor size (HR, 2.245, 95% CI, P = 0.005) and AEG-1 expression (HR, 1.916, 95% CI, P = 0.038) served as prognostic factors for disease-free survival (DFS) (Jung et al., 2015). The cumulative 5-year survival and recurrence rates were 89.2% and 50.0% in low AEG-1-expressing group and 24.5% and 82.4% in high AEG-1-expressing group, respectively (Jung et al., 2015). A recent study incorporating IHC in HCC TMA, TCGA database analysis and meta-analysis of published data further confirmed the utility of AEG-1 as an HCC prognostic marker (He et al., 2017). AEG-1 has been identified as a prognostic marker for HBV-associated HCC and its levels correlated with the American Joint Committee on Cancer (AJCC, 7th edition) stage (P = 0.020), T classification (P = 0.007), N classification (P = 0.044), vascular invasion (P = 0.006) and histological differentiation (P = 0.020) in the HBV-HCC patients (Gong et al., 2012). In addition, patients with high AEG-1 levels had shorter survival times compared to those with low AEG-1 levels (P = 0.001) (Gong et al., 2012).
A known diagnostic marker for HCC is glypican-3 (GPC-3). The diagnostic value of AEG-1 and GPC-3 was analyzed by IHC on HCC, adjacent nontumor tissue (ANT) and dysplastic nodules (DN) (Cao, Sharma, Imam, & Yu, 2019). Compared to ANT and DN, in HCC both AEG-1 and GPC-3 levels were higher showing 92% and 54% positivity, respectively. Alone, AEG-1 showed high sensitivity but low specificity and accuracy, while GPC-3 showed high specificity but low sensitivity and accuracy. However, combination of both augmented the sensitivity, specificity and accuracy to 94.6%, 89.5%, and 90.5%, respectively, suggesting that combined AEG-1 and GPC-3 staining might facilitate early diagnosis of HCC (Cao et al., 2019).
These clinical observations were substantiated in a transgenic mouse with hepatocyte-specific overexpression of AEG-1 (Alb/AEG-1) that generated highly aggressive metastatic HCC in diethylnitrosamine (DEN)-induced HCC model (Srivastava et al., 2012). As a corollary, AEG-1 knockout mice, either total or conditional, were highly resistant to DEN/phenobarbital (PB)-induced hepatocarcinogenesis and metastasis (Robertson et al., 2014, 2018).
In aggressive cancers AEG-1 is detected on the cell membrane giving rise to the hypothesis that autoantibody against AEG-1 might serve as a marker for advanced disease. The lung-homing domain (a.a. 381–443) of human AEG-1 was used as the antigen to detect anti-AEG-1 antibody in sera from 483 different cancer patients, including 98 breast cancer, 96 HCC, 88 colorectal cancer, 51 lung cancer and 88 gastric cancer, by ELISA (Chen et al., 2012). At titers of ≥1:50 anti-AEG-1 antibody was detected in 49% of these patients, including 45% breast cancer, 50% HCC, 49% colorectal cancer, 45% lung cancer and 49% gastric cancer patients, with none of 230 normal individuals displaying positivity (P < 0.01) (Chen et al., 2012). Even though AEG-1 is an established diagnostic/prognostic marker for cancer, its use is limited by the availability of tumor biopsy samples and as such anti-AEG-1 antibody might serve as an important surrogate for AEG-1. However, since the original publication these findings have not been replicated by other studies thereby requiring validation in a large cohort of patients.
7. Cellular signaling affected by AEG-1 in HCC
Interest in the effect of AEG-1 on the malignant phenotype of HCC cells is emerging as a new hotspot of research in the field of cancer biology. As a multifunctional protein, AEG-1 significantly alters a diverse array of signaling networks and effector molecules involved in tumor progression, and HCC is not an exception. In the context of HCC, AEG-1 is a strong activator of multiple pro-tumorigenic signal transduction pathways including NF-κB, PI3K/Akt/mTOR, Wnt/β-catenin, and MAPK/ERK (Fig. 3). However, there is still a need to explore the network of AEG-1 in the complex HCC ecosystem. The present status of downstream mediators of AEG-1 in HCC is discussed below.
7.1. Activation of NF-κB by AEG-1: Promotion of inflammation
Majority of documented risk factors of HCC, such as HBV or HCV infection, alcoholic liver disease and NAFLD, cause long-term liver inflammation and cirrhotic damage leading to HCC (El-Serag, 2011; Forner et al., 2018). Chronic inflammation in HCC is characterized by sustained expression of cytokines and recruitment of various immune cells to the liver. Activated inflammatory cells release free radicals, e.g., reactive oxygen species (ROS), which can cause DNA damage, lead to gene mutations, and ultimately cancer formation (Budhu & Wang, 2006; Karin, 2006; Leonardi et al., 2012). NF-κB is a key transcriptional regulator of the inflammatory response and plays an essential role in regulating inflammatory signaling in the liver (Pikarsky et al., 2004; Taniguchi & Karin, 2018). HCC develops as a consequence of chronic inflammation and as such NF-κB activation is a frequent and early event in human HCC of viral and non-viral etiologies (Hosel et al., 2009; Kim, Lee, & Jung, 2010; Liu et al., 2002; Lu et al., 2010; Tai, Tsai, Chang, et al., 2000; Tai, Tsai, Chen, et al., 2000). Increased levels of LPS, resulting in the activation of NF-κB in the liver, are detected in patients with advanced liver diseases, and fatty acids also activate NF-κB in NAFLD patients (Schwabe, Seki, & Brenner, 2006; Shi et al., 2006). Interestingly, while NF-κB induces AEG-1 expression, the first signaling pathway that was found to be activated by AEG-1 was NF-κB (Emdad et al., 2006; Sarkar et al., 2008). It was documented that upon TNF-α treatment AEG-1 translocates to the nucleus where it interacts with the p65 subunit of NF-κB and CREB-binding protein (CBP) and functions as a bridging factor between NF-κB and basal transcriptional machinery promoting NF-κB-induced transcription (Emdad et al., 2006; Sarkar et al., 2008). Subsequently, it was shown that AEG-1, anchored on ER membrane, associates with upstream ubiquitinated activators of NF-κB, such as RIP1 and TRAF2, facilitating their accumulation and as a consequence NF-κB activation (Alexia et al., 2013). AEG-1 is directly phosphorylated by IKKβ at serine 298 which is essential for IκBα degradation and NF-κB activation (Krishnan et al., 2015). Thus AEG-1 functions in multiple steps in NF-κB activation pathway and as such it is fundamentally required for inflammation which has been clearly demonstrated in AEG-1-deficient mouse models (Robertson et al., 2014, 2018). LPS-induced NF-κB activation is markedly abrogated in AEG-1−/− hepatocytes and macrophages vs. WT (Robertson et al., 2014). While 16-month-old WT mice showed signs of aging-associated inflammation no such changes were observed in AEG-1−/− littermates, and infiltration of macrophages was observed in aged WT liver and spleen but not in AEG-1−/− (Robertson et al., 2014). Indeed, AEG-1−/− mice lived longer than WT littermates and showed profound resistance to DEN-induced activation of oncogenic IL-6/STAT3 signaling and development of HCC (Robertson et al., 2014, 2015). Communication between tumor cells and tumor microenvironment is necessary for HCC development, and it has been shown that NF-κB activation in hepatocytes and macrophages is required for inflammation-induced HCC (Haybaeck et al., 2009; He & Karin, 2011). In a follow-up study it was documented that hepatocyte-specific AEG-1 deficiency (AEG-1ΔHEP) led to only an attenuation (and not complete abrogation), while myeloid-specific AEG-1 deficiency (AEG-1ΔMAC) led to complete abrogation of DEN-induced HCC, indicating that AEG-1 plays a key role in initial macrophage activation that is crucial for hepatocyte transformation (Robertson et al., 2018). AEG-1 deficiency made macrophages anergic so that they did not respond to polarization stimuli and their functional activity was markedly hampered (Robertson et al., 2018). It should be noted that AEG-1-induced inflammation has been attributed to regulate other inflammatory cancers, such as gastric cancer (Li, Wang, et al., 2014). AEG-1 plays a seminal role in contributing to the inflammatory component of NASH, a precursor to HCC, and other inflammatory conditions, such as diabetic kidney disease, rheumatoid arthritis, and HIV-1-associated neuroinflammation (Hong, Wang, & Shi, 2017; Liu et al., 2019; Srivastava et al., 2017; Vartak-Sharma et al., 2014).
7.2. Activation of Wnt/β-catenin pathway by AEG-1
Oncogenic Wnt/β-catenin pathway can be activated by multiple mechanisms in HCC, the most common being mutations in CTNNB1 in 30% cases or AXIN1/2 in 5–10% cases (Rebouissou et al., 2016; Satoh et al., 2000). Our endeavors to identify AEG-1-downstream genes by comparing global gene expression between control and AEG-1-overexpressed HCC cells first identified significant modulation of genes belonging to Wnt/β-catenin pathway by AEG-1 (Yoo, Emdad, et al., 2009). We documented that AEG-1 can activate Wnt/β-catenin pathway by multiple ways: (A) AEG-1 increases expression of lymphoid enhancer-binding factor 1 (LEF1), a transcription factor activated by Wnt signaling, and LEF1-regulated genes, such as c-Myc. (B) AEG-1 downregulates expression of negative regulators of the Wnt pathways, like APC and C-terminal-binding protein 2 (CTBP2). (C) AEG-1 activates ERK42/44 which phosphorylates and inactivates glycogen synthase kinase 3 beta (GSK3β) resulting in nuclear translocation of β-catenin (Yoo, Emdad, et al., 2009). Subsequent studies showed that AEG-1 knockdown abrogated nuclear translocation of β-catenin which was associated with decrease in EMT in HCC cells (Zhu et al., 2011). We showed that AEG-1 forms a complex with LEF1 and β-catenin, and AEG-1-mediated activation of Wnt/β-catenin pathway facilitated maintenance of glioma stem-like cells and their self-renewal (Hu et al., 2017). Using Co-immunoprecipitation (co-IP) and mass spectrometry, protein arginine methyltransferase 5 (PRMT5) was identified as an interacting partner of AEG-1, and PRMT5 inhibition abrogated AEG-1-induced increases in proliferation and migration of HCC cells (Zhu, Peng, et al., 2020). It was documented that PRMT5 and β-catenin competitively bind to the same domains of AEG-1, so that AEG-1 can sequester PRMT5 in the cytoplasm, allowing β-catenin to translocate to the nucleus and regulate gene expression (Zhu, Peng, et al., 2020). Altogether, these findings indicate that similar to NF-κB, AEG-1 can also activate Wnt/β-catenin by multiple ways contributing not only to HCC but other cancers as well.
7.3. Activation of PI3K/Akt pathway by AEG-1
Phosphatase and Tensin homolog (PTEN) is a negative regulator of the oncogenic PI3K/Akt pathway and acts by dephosphorylating phosphatidylinositol 3,4,5-triphosphate (PIP3), generated by PI3K. PTEN inactivation, by a variety of mechanisms including loss-of-function mutation and gene deletion, resulting in PI3K/Akt activation, is observed in ~40% HCC patients (Hu et al., 2003). Hepatocyte-specific Pten knockout mouse develops NASH with increase in SREBP-1c and lipogenic genes and eventually HCC (Horie et al., 2004). Conversely, liver-specific Akt2 knockout inhibited hepatic triglyceride (TG) accumulation and a hepatocyte-specific Pik3ca transgenic mouse developed steatosis and HCC (Kudo et al., 2011; Leavens, Easton, Shulman, Previs, & Birnbaum, 2009). While activation of PI3K/Akt pathway induces AEG-1, AEG-1, in turn, activates this pathway which mediates AEG-1-mediated protection from serum starvation-induced apoptosis as well as anoikis resistance in multiple cell types including HCC (Lee et al., 2006; Lee et al., 2008; Zhou et al., 2014). Mechanistically, it was demonstrated that AEG-1 interacts with Akt2 resulting in prolonged stabilization of Akt S474 phosphorylation and activation of downstream signaling in glioma cells (Hu et al., 2014). Although it remains to be seen whether similar interaction also happens in HCC cells, AEG-1-mediated activation of PI3/Akt signaling has also been demonstrated in Alb/AEG-1 hepatocytes (Srivastava et al., 2012).
7.4. Activation of MAPK/ERK pathway by AEG-1
In most cancers, MAPK pathway is activated by RAS mutations, which, however, are rare in HCC (Yea et al., 2008). Increased phosphorylation of MAPK/ERK and its upstream kinase MAP2K/MEK1/2 has been observed in HCC tissues compared to the adjacent normal liver (Huynh et al., 2003). It is postulated that the main mechanisms underlying MAPK activation in HCC involves ligand overexpression and aberrant epigenetic regulation (Newell et al., 2009). Overexpression of AEG-1 markedly activates MAPK/ERK as well as p38 MAPK and inhibition of either pathway significantly inhibited AEG-1-induced cell proliferation of human HCC cells (Yoo, Emdad, et al., 2009). Similar finding has also been observed in Alb/AEG-1 hepatocytes with concomitant increased activation of EGFR, an upstream activator of MAPK/ERK signaling (Srivastava et al., 2012; Srivastava, Siddiq, et al., 2015). Proteomic analysis of conditioned media (CM) from WT and Alb/AEG-1 hepatocytes identified upregulation of several components of the complement pathway, most notably Factor XII (FXII) by AEG-1, and knocking down FXII showed decreased activation of EGFR and consequently MAPK/ERK (Srivastava et al., 2012). These observations indicate that ligand overexpression is one mechanism by which AEG-1 activates MAPK/ERK signaling. This hypothesis is supported by the observation that AEG-1−/− primary mouse hepatocytes responded to EGF treatment, with activation of EGFR and MAPK/ERK, to the same level compared to WT hepatocytes (Robertson et al., 2014), indicating that AEG-1 is not required for normal activation of MAPK/ERK but its overexpression results in production of aberrant ligands, such as FXII, activating MAPK/ERK pathway. Activation of MAPK/ERK results in activation of the transcription factor AP-1 and it was documented that AEG-1 knockdown results in marked inhibition of AP-1 DNA binding in prostate cancer cells (Kikuno et al., 2007).
8. Cooperation/interaction of AEG-1 with other oncogenes/proteins to promote HCC
As a scaffold protein, AEG-1 interacts with many different proteins and protein complexes to mediate its action. Some of these interactions, leading to activation of specific oncogenic signaling pathways, have been described in the previous section. Here we highlight additional seminal interactions which helps AEG-1 execute its functions.
8.1. AEG-1 co-operates with c-Myc to promote hepatocarcinogenesis
In HCC, c-Myc is often overexpressed and functions as a driver oncogene for the induction and maintenance of the neoplastic state as indicated by the studies involving c-Myc overexpression mouse models (Coulouarn et al., 2006; Murakami et al., 1993; Shachaf et al., 2004). AEG-1 is transcriptionally regulated by c-Myc, and can control c-Myc expression by a variety of mechanisms, such as by activation of Wnt/β-catenin pathway and by interacting with and inhibiting a transcriptional repressor PLZF thereby facilitating c-Myc-mediated transcription (Lee et al., 2006; Thirkettle, Mills, et al., 2009; Yoo, Emdad, et al., 2009). Both AEG-1 and c-Myc genes are located at human chromosome 8q and co-amplified in HCC patients. A transgenic mouse with hepatocyte-specific overexpression of both AEG-1 and c-Myc (Alb/AEG-1/c-Myc) developed highly aggressive, metastatic HCC, both spontaneously and following DEN exposure, as compared with transgenic mice having overexpression of either oncogene alone (Srivastava, Siddiq, et al., 2015). Alb/AEG-1/c-Myc hepatocytes showed strong and sustained activation of various pro-survival signaling pathways as well as positively altered EMT. Indeed, in in vitro assays Alb/AEG-1/c-Myc hepatocytes manifested the cancer hallmarks, such as proliferation, invasion and chemoresistance, at a significantly higher levels compared to Alb/AEG-1 or Alb/c-Myc hepatocytes. RNA sequencing (RNA-Seq) analysis indicated that livers of Alb/AEG-1/c-Myc mice showed a distinct gene signature mimicking human HCC. Compared to the single transgenics, robust upregulation of various ncRNAs, e.g., Rian, Mirg, and Meg3, was observed only in the double transgenic mice. Knocking down these ncRNAs considerably reduced proliferation and invasion by Alb/AEG-1/c-Myc hepatocytes, suggesting that these ncRNAs mediated AEG-1- and c-Myc-induced survival advantages. These studies unraveled a novel cooperative oncogenic effect of AEG-1 and c-Myc in HCC and Alb/AEG-1/c-Myc mouse model might serve as a valuable tool to evaluate novel therapeutic strategies targeting HCC (Srivastava, Siddiq, et al., 2015).
8.2. AEG-1-SND1: A key interaction mediating AEG-1 function
Although many intracellular proteins that interact with AEG-1 have been identified, the most representative protein binding with high affinity is SND1 that provides interesting insights into the mechanism of action of AEG-1 (Blanco et al., 2011; Wan, Lu, et al., 2014; Yoo, Santhekadur, et al., 2011). Two independent approaches, namely yeast two-hybrid screening using a human liver cDNA library, and co-immunoprecipitation (Co-IP) followed by mass spectrometry, identified SND1 as the protein which most strongly interacts with AEG-1 (Yoo, Santhekadur, et al., 2011). A similar strategy also identified AEG-1-SND1 interaction in breast cancer cells (Blanco et al., 2011). SND1, also known as p100 coactivator or Tudor staphylococcal nuclease (Tudor-SN), is a multifunctional protein regulating a variety of cellular processes like transcription (Leverson et al., 1998; Wang et al., 2010), RNA splicing (Gao et al., 2012), and RNA metabolism (Gao et al., 2010). SND1 can be found both in the nucleus and cytoplasm. It facilitates transcription as a co-activator and mRNA splicing through interaction with the spliceosome machinery in the nucleus (Yang et al., 2007). In the cytoplasm, it acts as a nuclease in the RNA-induced silencing complex (RISC) in which small RNAs (e.g., siRNAs or miRNAs) are complexed with ribonucleoproteins to carry out RNAi-mediated gene silencing (Caudy et al., 2003). It was documented that AEG-1 interacts with SND1 in the cytoplasm and both AEG-1 and SND1 are required for optimum RISC activity (Yoo, Santhekadur, et al., 2011). It has been demonstrated that. Increased RISC activity, granted by AEG-1 or SND1, was found to result in increased degradation of tumor-suppressor mRNAs, which are targets of oncogenic miRNAs, including the mRNA of the tumor suppressor PTEN, a target of miRNA-221 which is overexpressed in HCC (Yoo, Santhekadur, et al., 2011). Interestingly, SND1 is highly expressed in HCC, SND1 overexpression increased and SND1 knockdown abrogated growth of human HCC xenografts in nude mice and a transgenic mouse with hepatocyte-specific overexpression of SND1 (Alb/SND1) developed spontaneous and augmented DEN-induced HCC. (Jariwala et al., 2017; Yoo, Santhekadur, et al., 2011). SND1 promoted expansion of tumor-initiating cells (TICs) in Alb/SND1 mice (Jariwala et al., 2017). A selective SND1 inhibitor, 3′,5′-deoxythymidine bisphosphate (pdTp), inhibited AEG-1-induced increased proliferation of human HCC cells, and effectively reduced tumor burden in human xenograft models of subcutaneous or orthotopic HCC (Jariwala et al., 2017; Yoo, Santhekadur, et al., 2011). SND1 regulates HCC angiogenesis by the activation of NF-κB and miR-221 inducing angiogenic factors like CXCL16 (Santhekadur, Das, et al., 2012). Using a variety of mouse models, a key role of AEG-1 in expansion of TICs in breast cancer was elucidated, facilitating metastasis, and it was documented that AEG-1 exerted its effect by interacting and stabilizing SND1 (Wan, Lu, et al., 2014). Under steady-state condition SND1 levels did not differ between WT and AEG-1 knocked-down cells. However, upon induction of DNA replication stress, a common type of stress during tumor development, half-life of SND1 protein was significantly reduced in AEG-1 knocked-down cells compared to control indicating that AEG-1-SND1 interaction is required for survival under stressful condition, e.g., during tumor initiation (Wan, Lu, et al., 2014). Similarly, overexpression of AEG-1 showed increased stabilization of SND1 upon heat shock (Guo et al., 2014). AEG-1 mutants, which failed to interact with SND1, lost their tumor-initiating potential (Guo et al., 2014; Wan, Lu, et al., 2014). The importance of SND1 in AEG-1-mediated oncogenesis has been shown in additional cancers (He et al., 2020), and collectively, these studies show a seminal role of AEG-1-SND1 interaction in carcinogenesis.
8.3. AEG-1 regulates metabolic functions by interacting with RXR
RXR is a ligand-dependent transcription factor that functions as a key regulator of cell growth, differentiation, metabolism and development (Lefebvre, Benomar, & Staels, 2010). RXR heterodimerizes with one third of the 48 human nuclear receptor superfamily members, including retinoic acid receptor (RAR), thyroid hormone receptor (TR), vitamin D receptor (VDR), Liver X Receptor (LXR), Peroxisome Proliferator Activated Receptor (PPAR) and Farnesoid X Receptor (FXR), and regulates corresponding ligand-dependent gene transcription. Cholesterol metabolites, fatty acid derivatives and bile acids serve as endogenous ligands for LXR, PPAR and FXR, respectively, which play important role in regulating lipid metabolism (Lefebvre et al., 2010). In the absence of ligand, RXR heterodimers interact with co-repressors that maintain histones in a deacetylated state and inhibit transcription. Upon ligand binding there is a conformational change so that the co-repressors are replaced by co-activators inducing histone acetylation and transcriptional activation. The co-activators harbor a unique LXXLL motif through which they interact with the transcription factors (Heery, Kalkhoven, Hoare, & Parker, 1997). Interestingly AEG-1 also harbors an LXXLL motif and yeast two-hybrid assay using the region of AEG-1 harboring the LXXLL motif identified RXR as its interacting partner (Srivastava et al., 2014). We documented that in the nucleus, interaction of AEG-1 with RXR blocks co-activator recruitment thereby abrogating retinoic acid-, thyroid hormone and fatty acid-mediated gene transcription (Srivastava et al., 2014, 2017, Srivastava, Robertson, et al., 2015). Knocking down AEG-1 markedly augmented retinoic acid-mediated killing and this concept was used to develop and evaluate a therapeutic protocol in mouse models (Rajasekaran, Srivastava, et al., 2015; Srivastava et al., 2014). Non-thyroidal illness syndrome (NTIS), characterized by low serum 3,5,3′-triiodothyronine (T3) with normal l-thyroxine (T4) levels, is associated with malignancy and decreased activity of type I 5′-deiodinase (DIO1), which converts T4 to T3, contributes to NTIS (Warner & Beckett, 2010). T3 binds to TR/RXR heterodimer and regulate transcription of target genes, including DIO1. It was demonstrated that AEG-1 overexpression repressed and AEG-1 knockdown induced DIO1 expression (Srivastava, Robertson, et al., 2015). An inverse correlation was observed between AEG-1 and DIO1 levels in human HCC patients. Low T3 with normal T4 was observed in the sera of HCC patients and Alb/AEG-1 mice (Srivastava, Robertson, et al., 2015). Altogether these observations suggested that AEG-1 might play a role in NTIS associated with HCC and other cancers.
The RXR inhibitory activity allows AEG-1 to profoundly regulate lipid metabolism. AEG-1−/− mice are significantly leaner with prominently less body fat compared to the WT (AEG-1+/+) littermates (Robertson, Srivastava, Siddiq, et al., 2015). When fed high fat and cholesterol diet (HFD), WT mice rapidly gained weight while AEG-1−/− did not gain weight at all even thought their food intake was similar. AEG-1−/− mice showed decreased fat absorption from the intestines because of increased activity of LXR and PPARα (Robertson, Srivastava, Siddiq, et al., 2015). In enterocytes, activation of LXR inhibits cholesterol absorption by downregulating cholesterol transporter Npc1l1 and upregulating cholesterol efflux proteins, Abca1, Abcg5 and Abcg8. Activation of PPARα in the enterocytes promotes β-oxidation of absorbed fatty acids (FA) thereby downregulating fatty acid absorption into the circulation. Thus, increased activity of LXR and PPARα in AEG-1−/− enterocytes impaired overall fat absorption contributing to leanness (Robertson, Srivastava, Siddiq, et al., 2015). On the contrary, Alb/AEG-1 mice developed spontaneous NASH and AEG-1ΔHEP mice were protected from HFD-induced NASH (Srivastava et al., 2017). One underlying mechanism of increased steatosis in Alb/AEG-1 mice is inhibition of PPARα-mediated FA β-oxidation allowing accumulation of fat in the liver (Srivastava et al., 2017). Thus AEG-1-RXR interaction has profound implication in regulating metabolism as well as functions of vitamins and hormones, especially in the liver.
9. AEG-1 binds to RNA: Regulation of translation
Several RNA interactome screen identified AEG-1 as a selective ER mRNA-binding protein (Baltz et al., 2012; Castello et al., 2012; Chen, Jagannathan, Reid, Zheng, & Nicchitta, 2011; Kwon et al., 2013). In a recent study it was confirmed that AEG-1 is an ER-resident integral membrane RNA-binding protein (RBP) (Hsu et al., 2018). Analysis of AEG-1 RNA interactome by HITS-CLIP and PAR-CLIP methods revealed an enrichment for endomembrane organelle-encoding transcripts, most prominently those encoding ER-resident proteins as well as integral membrane protein-coding RNAs (Hsu et al., 2018). Secretory and cytosolic protein encoding mRNAs were also represented in the AEG-1 RNA interactome, with the latter category enriched in genes functioning in mRNA localization, translational regulation and RNA quality control. AEG-1 does not have a consensus RNA-binding domain and deletion mapping analysis identified the central disordered region of AEG-1, comprised of a.a. 138–350 to bind to RNA (Hsu et al., 2018). These findings corelate nicely with several of our previous findings. We showed that overexpression of AEG-1 increases protein levels, and not mRNA levels, of multidrug resistance gene 1 (MDR1) contributing to chemoresistance, FXII contributing to angiogenesis, and fatty acid synthase (FASN) contributing to de novo lipogenesis, hence NASH (Srivastava et al., 2012, 2017; Yoo, Chen, et al., 2010). All these three proteins are endomembrane or secreted and we documented that AEG-1 facilitates association of all three mRNAs with polysomes resulting in increased translation (Srivastava et al., 2012, 2017; Yoo, Chen, et al., 2010). It should be noted that in addition to FASN, AEG-1 bound mRNAs also code for additional fatty acid synthesizing enzymes, and in Gene Ontology (GO) analysis of AEG-1 bound mRNAs encoding endomembrane proteins, lipid metabolism-associated proteins came out to be the most significant category (Hsu et al., 2018). Thus AEG-1 promotes NASH by translational upregulation of enzymes of de novo lipogenesis, inhibition of PPARα-mediated FA β-oxidation and stimulation of inflammation by activating NF-κB (Fig. 4). A separate study also identified AEG-1 as an RBP in endometrial cancer cells by RNA immunoprecipitation followed by microarray (Meng et al., 2012). However, the RNA interactome was not characterized and it was documented that protein levels of two AEG-1-interacting mRNAs, PDCD11 and KDM6A, were increased in AEG-1 knockdown cells, and the consequence of this observation was not studied (Meng et al., 2012).
Fig. 4.

Mechanisms by which AEG-1 induces NASH. AEG-1 binds to RXR using LXXLL motif which inhibits PPARα and decreases fatty acid β-oxidation. AEG-1 binds to specific mRNAs increasing translation of lipogenic enzymes thus increasing de novo lipogenesis. These two events lead to increased steatosis. AEG-1 activates NF-κB by multiple mechanisms. It binds to p65 subunit of NF-κB and functions as a bridging factor between NF-κB and basal transcription machinery. It interacts with upstream ubiquitinated molecules of NF-κB pathway such as TRAF2 and RIP1. It is directly phosphorylated by IKKβ which is necessary for subsequent IKKβ-mediated phosphorylation of IκBα leading to its proteasomal degradation and translocation of NF-κB to the nucleus. NF-κB activation leads to increased inflammation. Thus AEG-1 increases both steatotic and inflammatory components of NASH. Image created using tools from Biorender.
10. AEG-1 and hallmarks of cancer: Specific examples
By its pleiotropic action AEG-1 has the ability to augment all hallmarks of cancer as described by Hanahan and Weinberg (2011) leading to tumor initiation and progression. Here we highlight unique aspects of AEG-1 regulating some of these hallmarks with particular focus on HCC.
10.1. AEG-1 and resistance to therapy
Resistance developed after the commencement of treatment is a major obstacle that an oncologist confronts during cancer treatment. AEG-1 over-expression in cancer plays a crucial role in conferring resistance to chemotherapy (chemoresistance). Response to various chemotherapeutics, e.g., doxorubicin (Dox), cisplatin, 5-fluorouracil (5-FU), is markedly blunted due to higher expression of AEG-1 in neoplastic tissues (Meng, Thiel, & Leslie, 2013). Chemotherapy may be administered to HCC patients whose cancer cannot be removed by surgery, has not responded to local therapies such as ablation or embolization, or when targeted therapy is no longer helpful. Doxorubicin (Dox) is a common anticancer drug used for the treatment of HCC, but the clinical efficacy of Dox is not profound thus emphasizing an inherent resistance of HCC cells to Dox (Kato et al., 2001). ABC transporters are ATP-dependent efflux pumps with broad drug specificity overexpression of which contribute to chemoresistance. AEG-1 overexpression resulted in marked upregulation of MDR1 (ABCB1) protein by increased translation resulting in increased efflux of Dox and promotion of Dox-resistance in HCC cells (Yoo, Chen, et al., 2010). While, AEG-1 can directly bind to MDR1 mRNA and increase its translation (Hsu et al., 2018), it was documented that inhibition of PI3K/Akt pathway could also inhibit AEG-1-mediated polysome association of MDR1 mRNA (Yoo, Chen, et al., 2010), indicating that AEG-1 modulates MDR1 translation by multiple ways. AEG-1 overexpression increased phosphorylation of eukaryotic translation initiation factor 4G (eIF4G), but not mTOR-sensitive eIF4E and 4E-BP, and interestingly this activation was not blocked by PI3K/Akt inhibitor, indicating that AEG-1 can stimulate the translational machinery in a PI3K/Akt/mTOR-independent pathway (Yoo, Chen, et al., 2010). In-depth protein-protein interaction studies need to be carried out to elucidate the underlying mechanism of this phenomenon. Resistance to 5-FU, another chemotherapeutic used to treat HCC, can also be conferred by AEG-1 (Yoo, Gredler, et al., 2009). Gene expression analysis of AEG-1-overexpressed human HCC cells identified upregulation of several genes, e.g., drug-metabolizing enzymes like dihydropyrimidine dehydrogenase (DPYD) and the transcription factor late SV40 factor (LSF/TFCP2), by AEG-1 (Yoo, Emdad, et al., 2009). LSF transcriptionally regulates thymidylate synthase (TS), the substrate for 5-FU, and upregulation of LSF by AEG-1 induced TS (Yoo, Gredler, et al., 2009). Thus, the dual inhibitory effects produced by AEG-1, i.e., increase in the 5-FU catabolizing enzyme DPYD, and increase in 5-FU substrate TS, resulted in significant resistance to 5-FU. A lentivirus expressing AEG-1 short hair-pin RNA in combination with Dox or 5-FU dramatically inhibited growth of aggressive human HCC, compared to either agent alone, in nude mice xenograft experiments (Yoo, Chen, et al., 2010; Yoo, Gredler, et al., 2009). Subsequent studies identified overexpression of LSF in HCC and unraveled its role as an oncogene via transcriptional regulation of osteopontin, resulting in activation of hepatocyte growth factor receptor c-Met, and that of MMP-9, and a small molecule inhibitor of LSF abrogated HCC in an endogenous HCC model (Grant et al., 2012; Rajasekaran, Siddiq, et al., 2015; Santhekadur, Gredler, et al., 2012; Yoo, Emdad, et al., 2010, Yoo, Gredler, et al., 2011). Interestingly, studies using breast cancer cells showed resistance to broad-spectrum chemotherapeutics conferred by AEG-1 that involves upregulation of aldehyde dehydrogenase 3 family, member A1 (ALDH3A1) and c-Met (Hu et al., 2009).
Another mechanism by which AEG-1 causes chemoresistance is by induction of protective autophagy (Bhutia et al., 2010). Autophagy is an evolutionarily conserved process that can be observed in all cells, maintaining cellular homeostasis by removing cellular contents through the lysosomal compartment for degradation (Das, Banerjee, & Mandal, 2020). Overexpression of AEG-1 induced protective autophagy in multiple cell types by activating AMPK/mTOR pathway leading to increased expression of the autophagy regulator ATG5, and inhibition of AEG-1-induced autophagy in cancer cells enhanced sensitivity to chemotherapeutic agents (Bhutia et al., 2010). However, the mechanism by which AEG-1 induces an energy-deprived state leading to activation of AMPK remains to be determined. Altogether these studies suggest that localized inhibition of AEG-1 might be a good strategy to re-sensitize HCC with chemotherapy treatment.
Like chemoresistance, failure of radiotherapy is a major clinical challenge owing to the radio-resistance in the course of treatment. It has been reported that Aurora-A confers radio-resistance in HCC by upregulating the NF-κB signaling pathway (Shen et al., 2019). As AEG-1 also activates NF-κB, AEG-1 may likely have a role in conferring radio-resistance in HCC, which has been shown for other cancers (Zhao et al., 2012).
10.2. AEG-1 and senescence
Senescence is a potential anti-cancer mechanism and is characterized by a senescence-associated secretory phenotype (Collado & Serrano, 2010). Insulin-like growth factor binding protein-7 (IGFBP7) is a secreted protein that induces senescence in cancer cells and functions as a tumor suppressor in HCC (Akiel et al., 2017; Chen, Yoo, et al., 2011). IGFBP7 was identified as the most robustly downregulated gene by AEG-1 in HCC (Yoo, Emdad, et al., 2009) and forced overexpression of IGFBP7 in AEG-1-overexpressing HCC cells inhibited in vitro growth and induced senescence, and suppressed in vivo tumor growth in nude mice (Chen, Yoo, et al., 2011). These findings suggest that AEG-1 protects from senescence by downregulating IGFBP7. Primary hepatocytes, mouse and human, do not divide in vitro and starts becoming senescent after 96 h. It was documented that lentivirus-mediated overexpression of AEG-1 markedly protected human hepatocytes from induction of senescence and Alb/AEG-1 hepatocytes showed a significant reduction and delay in senescence compared to WT hepatocytes (Srivastava et al., 2012; Srivastava, Siddiq, et al., 2015). Senescence is associated with activation of DNA damage response (DDR), leading to activation of ATM and ATR, their downstream kinases CHK1 and CHK2 leading to p53 phosphorylation and increase in p53 and p21 levels, which was markedly dampened in Alb/AEG-1 hepatocytes compared to WT (Srivastava et al., 2012; Srivastava, Siddiq, et al., 2015). DNA-damaging agent DEN induces DDR in hepatocytes leading to senescence and/or apoptosis which functions as a protective mechanism. If the hepatocytes still survive following DEN treatment the ensuing DNA damage mutagenizes and transforms them. AEG-1ΔHEP mice are protected from DEN-induced HCC, and it was demonstrated that DEN-induced DDR is significantly augmented in AEG-1−/− hepatocytes leading to senescence and apoptosis compared to their WT counterparts (Robertson et al., 2018). AEG-1 can interact with MDM2 resulting in stabilization of MDM2 protein precluding p53 activation (Ding et al., 2019). However, the mechanism by which AEG-1 interferes with the initial steps of DDR remains to be elucidated.
10.3. AEG-1 promotes angiogenesis
Angiogenesis is a fundamental requirement for the development of any solid tumor (Folkman, 1971; Kerbel, 2000). AEG-1-overexpressing human HCC cells generated highly vascular tumors in nude mice associated with increased expression of angiogenic factors, such as VEGF, placental growth factor (PIGF) and FGFα (Yoo, Emdad, et al., 2009). Stable overexpression of AEG-1 in normal immortal cloned rat embryo fibroblast (CREF) transformed them and allowed them to form highly vascular tumors characterized by enhanced expression of biomarkers of angiogenesis, e.g., angiopoietin-1, Tie 2 and HIF-1α (Emdad et al., 2009). In vitro analysis documented that activation of PI3K/Akt pathway plays a role in regulating angiogenesis by AEG-1 (Emdad et al., 2009). Similarly, conditioned media (CM) from Alb/AEG-1 hepatocytes, but not from WT hepatocytes, induced a marked angiogenic response in human umbilical vein endothelial cells (HUVEC) differentiation assay and chicken chorioallantoic membrane (CAM) assay (Srivastava et al., 2012). Further studies documented that trefoil factor 3 (TFF3) and FXII, secreted from Alb/AEG-1 hepatocytes, played a seminal role in mediating AEG-1-induced angiogenesis, especially proliferation and differentiation of endothelial cells (Srivastava et al., 2012). Both hypoxia and glucose deprivation, stimulators of angiogenesis, induced AEG-1 by HIF-1α activation and ROS generation, respectively, and the induced AEG-1 then protected from stress and supported survival in glioma cells (Noch, Bookland, & Khalili, 2011).
10.4. AEG-1 promotes metastasis
AEG-1 was cloned as a metastasis-promoting gene for breast cancer and since then extensive research has confirmed the pivotal role of AEG-1 in facilitating invasion and metastasis of many types of cancer including HCC (Brown & Ruoslahti, 2004; Emdad et al., 2009; Hu et al., 2009; Kikuno et al., 2007; Srivastava et al., 2012; Srivastava, Siddiq, et al., 2015; Yoo, Emdad, et al., 2009). The importance of AEG-1 in metastasis is emphasized by its inclusion in MammaPrint, the only US Food and Drug Administration (FDA)-approved individualized metastasis risk assessment assay for breast cancer patients. The predictive diagnostic test MammaPrint includes 70 genes including AEG-1 for assessment. Gradually higher expression of AEG-1 is detected with the progression of different cancers, especially overexpression of AEG-1 can be observed in the metastatic stage of the disease (Sarkar & Fisher, 2013).
Overexpression of AEG-1 in human HCC cells markedly increased in vitro invasion and induced lung metastasis in in vivo xenograft assays (Yoo, Emdad, et al., 2009). As a corollary, the most robust phenotype observed upon knockdown of AEG-1 in HCC cells was in vitro invasion (Yoo, Emdad, et al., 2009). Similarly knocking down AEG-1 in human HCC cells resulted in reduced pulmonary and abdominal metastases in nude mice and was associated with alterations in EMT markers, namely, down-regulation of N-cadherin and snail, upregulation of E-cadherin, and increased cytoplasmic localization of β-catenin (Zhu et al., 2011). Under naı¨ve condition, Alb/AEG-1 mice did not develop spontaneous HCC, Alb/c-Myc mice developed spontaneous HCC without distant metastasis, and Alb/AEG-1/c-Myc mice developed highly aggressive HCC with frank lung metastasis, emphasizing the role of AEG-1 in promoting metastasis (Srivastava, Siddiq, et al., 2015). DEN-induced carcinogenesis was significantly accelerated in all three groups with most markedly pronounced effect with lung metastasis in Alb/AEG-1/c-Myc mice.AEG-1 or c-Myc alone was able to induce EMT signaling pathways in hepatocytes, but activation of these pathways was sustained Alb/AEG-1/c-Myc hepatocytes (Srivastava, Siddiq, et al., 2015). Tetraspanin 8 (TSPN8), a cell surface protein known to play a role in metastasis was found to be robustly increased in AEG-1-overexpressing human HCC cells, and TSPN8 knockdown markedly inhibited AEG-1-induced in vitro invasion and intrahepatic metastasis in an orthotopic xenograft model, suggesting a potential role of TSPN8 in AEG-1-induced metastasis of HCC cells (Akiel et al., 2016; Yoo, Emdad, et al., 2009). Resistance to anoikis is an important component for survival of cancer cells in circulation to establish metastasis (Kim, Koo, Sung, Yun, & Kim, 2012).AEG-1 expression was found to be enhanced in HCC cells grown in suspension culture and AEG-1 could promote anoikis resistance of detached HCC cells (Zhu, Liu, et al., 2020). A role of AEG-1-induced autophagy in conferring anoikis resistance was suggested and inhibition of autophagy prevented AEG-1-induced metastasis of HCC xenografts to liver and lungs of nude mice (Zhu, Liu, et al., 2020). A potential role of protein kinase RNA-like ER kinase (PERK)-eIF2α-ATF4-CHOP signaling axis was implicated in regulating AEG-1-induced autophagy in HCC cells (Zhu, Liu, et al., 2020). In a separate study it was shown that elevated AEG-1 expression promoted anoikis resistance and orientation chemotaxis of HCC cells toward human pulmonary microvascular endothelial cells (HPMEC) through the activation of the PI3K/Akt signaling pathway and the metastasis-associated chemokine receptor CXCR4, respectively (Zhou et al., 2014). It was documented that HPMEC secreted CXCL12, the ligand for CXCR4, and CXCR4 antagonist AMD3100 reduced AEG-1-induced orientation chemotaxis (Zhou et al., 2014). A role of AEG-1-PRMT5 interaction leading to activation of Wnt/β-catenin pathway and promoting AEG-1-induced metastasis of HCC cells has been described (Zhu, Peng, et al., 2020). Collectively, these results suggest that AEG-1 positively regulates HCC cell metastasis by multiple mechanisms.
11. AEG-1 targeting as a potential therapy for HCC
There is no effective therapy for advanced-stage HCC, and most of the FDA-approved drugs increase patient survival for only ~3 months compared to placebo in non-resectable HCC patients (Abou-Alfa et al., 2018; Bruix et al., 2017; Kudo et al., 2018; Llovet et al., 2008; Rimassa et al., 2018; Zhu et al., 2019). HCC develops on a cirrhotic background with compromised liver function which markedly interferes with the ability of the liver to detoxify drugs. Hence drug-induced toxicity is a major limiting factor for treating HCC patients, and relatively non-toxic gene- and immune-based therapies are being increasingly appreciated as the treatment of choice (Reghupaty & Sarkar, 2019). Mouse model studies have clearly established AEG-1 as a key driver of advanced AEG-1 and as such targeting AEG-1 should be a viable and effective strategy for treating HCC. As yet no small molecule inhibitor for AEG-1 exists. However, RNA-interference (RNAi) strategy to inhibit AEG-1 can be effective in HCC. Indeed, nanoparticle-delivered siRNA targeting oncogenes has shown strong efficacy in inhibiting HCC in pre-clinical models as well as in phase I trials (Bogorad et al., 2014; Dudek et al., 2014; Tabernero et al., 2013). We have developed hepatocyte-targeted nanoplexes by conjugating polyamidoamine (PAMAM) dendrimers with polyethylene glycol (PEG) and galactose lactobionic acid (PAMAM-PEG-Gal) which were complexed with AEG-1 siRNA (PAMAM-AEG-1si) (Rajasekaran, Srivastava, et al., 2015; Srivastava et al., 2017). PAMAM complexes and compacts siRNA, PEG reduces charge density and increases circulation half-life, and lactobionic acid binds to asialoglycoprotein receptors overexpressed selectively in hepatocytes thus allowing targeted uptake (Rajasekaran, Srivastava, et al., 2015). AEG-1 overexpression inhibits anti-tumorigenic activity of all-trans retinoic acid (ATRA) (Srivastava et al., 2014). In a nude mice orthotopic human HCC xenograft assay, we documented that ATRA alone had no effect on tumor growth, PAMAM-AEG-1si alone significantly inhibited tumor growth, and the combination of PAMAM-AEG-1si and ATRA completely eliminated the tumors without exerting any toxicity (Rajasekaran, Srivastava, et al., 2015). In a separate study, we showed that PAMAM-AEG-1si could effectively protect C57BL/6 mice from HFD-induced NASH development (Srivastava et al., 2017). Thus PAMAM-AEG-1si could be a therapeutic strategy for HCC as well as a preventive strategy by inhibiting NASH and thereby preventing NASH to progress into HCC. AEG-1 knockdown markedly augmented anti-HCC activity of Dox and 5-FU, indicating that PAMAM-AEG-1si can be combined with standard chemotherapy or TKIs. AEG-1 plays a profound role in regulating macrophage function and inflammation, and as such PAMAM-AEG-1si can also be combined with anti-inflammatory strategies and immunotherapy. In-depth studies using endogenous mouse models of HCC need to be performed to evaluate these strategies for their potential transition to the clinics. A recent study described therapeutic efficacy of locked nucleic acid (LNA)-modified AEG-1 antisense oligonucleotide (ASO) in inhibiting primary tumor growth and attenuating metastasis of syngeneic breast, colorectal and lung tumors in C57BL/6 mice (Shen et al., 2020).
Very recently the interaction between AEG-1 and SND1 was exploited to design a peptide that can inhibit AEG-1-SND1 interaction (Li et al., 2021). AEG-1-interacting domains of SND1 were used as bait in a phage display screening to identify a 12 a.a. peptide which could disrupt AEG-1-SND1 interaction in vivo, induce SND1 degradation, and inhibit growth of human breast cancer xenografts (Li et al., 2021). This is an exciting development which needs to be evaluated in other cancers and opens up modification of this strategy for generating clinically relevant peptidomimetics or small molecule inhibitors.
12. Challenges and future perspectives
Over the years, our knowledge regarding AEG-1 has expanded through extensive research since its discovery. Nevertheless, a lot of challenges, unanswered questions, and areas remain that need to be addressed to comprehensively understand structural and functional aspects of AEG-1. AEG-1 is a unique molecule that has no homolog. Apart from the LXXLL motif, AEG-1 does not have any known structural domains or motifs to assign it to a specific molecular class, e.g., AEG-1 exerts many of its important functions by RNA binding but it does not have any classical RNA-binding motif (Hsu et al., 2018). Having a drosophila or worm model would have simplified research on AEG-1. However, AEG-1 is present only in vertebrates creating another level of challenge. Understanding of AEG-1 function has been gleaned by fishing expeditions, such as identification of AEG-1 protein and RNA interactomes, analysis of global gene or protein expression changes following AEG-1 overexpression or knockdown/knockout, and systematically analyzing phenotypes after modulating AEG-1 levels. The availability of total and conditional knockout and tissue-specific transgenic mice has provided novel insights into the consequences of AEG-1 modulation in vivo, e.g., identification of AEG-1’s role in promoting NASH (Srivastava et al., 2017). One field of study that needs further investigation is the mechanism of AEG-1-mediated metastatic process. The lung-homing domain of AEG-1 interacts with an unknown receptor in endothelial cells facilitating pro-metastatic function of AEG-1 (Brown & Ruoslahti, 2004). What is the nature of this receptor? Can an inhibitor of the receptor be developed to block AEG-1-induced metastasis? Does the lung-homing domain also mediate AEG-1-induced metastasis to other organs, such as liver? Structural resolution of AEG-1-SND1 interaction regions has provided granular insight into the key residues required for this interaction (Guo et al., 2014). However, lack of structural information of the complete AEG-1 protein hinders understanding of other pivotal molecular interactions. A peptide has been identified interfering AEG-1-SND1 interaction (Li et al., 2021) and resolution of crystal structure of full AEG-1 protein will help develop additional peptidomimetic inhibitors using a fragment-based drug discovery approach and may pave the way for developing a small molecule inhibitor of AEG-1. The mechanism of regulation of autophagy by AEG-1 at different stages of hepatocarcinogenesis also warrants further investigation and may shed light on AEG-1-induced metastatic process. A relatively understudied area is AEG-1’s role in regulating the immune system. With elucidation of the role of AEG-1 in regulating macrophage function (Robertson et al., 2018), it remains to be seen whether AEG-1 has any role in regulating cellular and humoral immune, in physiology as well as in disease, such as cancer.
Another important area that needs further clarification is the intracellular localization of AEG-1. To be precise, what is the function of AEG-1 at different cellular compartments? AEG-1 is detected mainly in the nucleus of normal hepatocytes (Srivastava et al., 2014). However, in HCC cells it is localized primarily in the cytoplasm, indicating that in hepatocytes, AEG-1 may promote unique functions that are different from its role in HCC cells. In the nucleus AEG-1 modulates the functions of transcription factors, but it is also detected in nucleolus the functional significance of which still remains to be determined. In vivo, AEG-1 preferentially inhibits PPARα, even though AEG-1 interacts with RXR and as such it should inhibit other RXR heterodimer partners. What determines this preference? Does AEG-1 compete with PPARα-specific co-activators, such as PGC1α, specifically and/or strongly compared to other co-activators? ER membrane-anchored AEG-1 binds to and promotes translation of specific subsets of mRNAs. Is there any specific motif or RNA secondary structure that determines the specificity? These unanswered questions, unexplored areas and inherent challenges create a wide scope and multiple avenues for future studies on AEG-1.
13. Conclusions
HCC is a highly aggressive cancer that develops chemoresistant and radioresistant phenotype quickly. Virtually, no therapy exists for the advanced stage of HCC. Therefore, the need to develop novel diagnostic techniques as well as therapeutic interventions is paramount to save the lives of HCC patients. At least, the development of new modalities of treatment that provides lasting disease-free survival benefit to the patients is needed. Our extensive studies in HCC cell lines and different in vivo HCC models confirm that AEG-1 is required for HCC progression. Even though in mouse model AEG-1 does not have the potency, like c-Myc, to function as a stand-alone oncogene to transform hepatocytes, it is clear that AEG-1 inhibition can block initiation and progression of HCC and help overcome therapy resistance. Nanoparticle-mediated targeted delivery for in vivo inhibition of AEG-1 is a clinically relevant strategy in the context of HCC as the payload delivery after intravenous administration is the highest for liver compared to other organs. This approach of targeted delivery of AEG-1 siRNA to HCC cells markedly inhibited their growth as xenografts (Rajasekaran, Srivastava, et al., 2015). Considering that inhibition of AEG-1-induced inflammation also blocks HCC initiation (Rajasekaran, Srivastava, et al., 2015), it would be interesting to see whether targeting AEG-1 by siRNA in macrophages shows the same efficacy as targeting it in HCC cells, and whether combination of these two approaches exert a synergistic effect. Testing combination of AEG-1 inhibition with immunomodulators, commonly used chemotherapy and targeted small molecule inhibitors will help develop therapeutic protocols having significant impact on HCC patient outcome and survival. Coming years beacons exciting mechanistic and translational research to harness the potential of AEG-1 in HCC patient management.
Acknowledgments
The present study was supported in part by The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant 1R01DK107451-01A1 (D.S.), National Cancer Institute (NCI) Grants 1R01CA230561-01A1 (D.S.), 1R01CA240004-01 (D.S.) and 1R01CA244993-01 (D.S. and P.B.F.), and Department of Defense (DOD) Grant CA170048 (D.S.).
List of abbreviations
- AA
aristolochic acid
- a.a.
amino acid
- Abca1
ATP-binding cassette transporter member 1
- Abcg5
ATP-binding cassette subfamily G member 5
- Abcg8
ATP-binding cassette subfamily G member 8
- AEG-1
astrocyte elevated gene-1
- AEG-1ΔHEP
hepatocyte-specific AEG-1 knockout mice
- AEG-1ΔMAC
myeloid-specific AEG-1 knockout mice
- AEG-1−/−
germline knockout of AEG-1 in mice
- Akt
protein kinase B
- Akt2
serine/threonine-protein kinase (or Akt kinase) 2
- Alb/AEG-1
A transgenic mouse with hepatocyte-specific overexpression of AEG-1
- Alb/c-Myc
A transgenic mouse with hepatocyte-specific overexpression of c-Myc
- Alb/SND1
A transgenic mouse with hepatocyte-specific overexpression of SND1
- Alb/AEG-1/c-Myc
A transgenic mouse with hepatocyte-specific overexpression of both AEG-1 and c-Myc
- ALDH3A1
aldehyde dehydrogenase 3 family, member A1
- AMPK
5′-adenosine monophosphate-activated protein kinase
- ANT
adjacent nontumor tissue
- AP-1
activator protein-1
- APC
adenomatous polyposis coli
- ASO
antisense oligonucleotide
- ATF4
activating transcription factor 4
- ATG5
autophagy related protein 5
- ATM
ataxia telangiectasia mutated kinase
- ATR
ataxia telangiectasia and Rad3-related protein kinase
- ATRA
all-trans retinoic acid
- ATP
adenosine triphosphate
- AXIN1
axis inhibition protein 1
- BCLC
Barcelona clinic liver cancer
- CAM
chorioallantoic membrane
- cAMP
cyclic adenosine monophosphate
- CBP
CREB-binding protein
- cDNA
complementary DNA
- CEACAM-1
carcinoembryonic antigen-related cell adhesion molecule 1
- C/EBP
CCAAT/enhancer-binding protein
- CHK1
checkpoint kinase 1
- CHK2
checkpoint kinase 2
- CHOP
C/EBP homologous protein
- CM
conditioned media
- c-Met
tyrosine-protein kinase Met
- co-IP
co-immunoprecipitation
- CPEB1
cytoplasmic polyadenylation element-binding protein 1
- CPEB3
cytoplasmic polyadenylation element-binding protein 3
- CREB
cAMP-response element-binding protein
- CREF
cloned rat embryo fibroblast
- CT
computed tomography
- CTBP2
C-terminal-binding protein 2
- CTLA-4
Cytotoxic T lymphocyte antigen-4
- CTNNB1
gene encoding β-catenin
- CXCL12
C-X-C motif chemokine ligand 12
- CXCL16
C-X-C motif chemokine ligand 16
- CXCR4
C-X-C chemokine receptor type 4
- DDR
DNA damage response
- DEN
diethylnitrosamine
- DFS
disease-free survival
- DIO1
type I 5′-deiodinase
- DLC1
deleted in liver cancer 1
- DLC2
deleted in liver cancer 2
- DN
dysplastic nodules
- DNA
deoxyribonucleic acid
- Dox
doxorubicin
- DPYD
dihydropyrimidine dehydrogenase
- EASL
European association for the study of the liver
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- eIF2α
eukaryotic initiation factor 2 sub-unit α
- eIF4E
eukaryotic translation initiation factor 4E
- eIF4E-BP
eukaryotic initiation factor 4E binding protein
- eIF4G
eukaryotic translation initiation factor 4G
- ELISA
enzyme-linked immunosorbent assay
- EMT
epithelial-mesenchymal transition
- ER
endoplasmic reticulum
- ERK
extracellular-signal-regulated kinase
- FA
fatty acids
- FASN
fatty acid synthase
- FDA
Food and Drug Administration
- FGFα
fibroblast growth factor-α
- FISH
fluorescence in-situ hybridization
- 5-FU
5-fluorouracil
- FXII
factor XII
- FXR
farnesoid X receptor
- Gal
galactose lactobionic acid
- GFP
green fluorescent protein
- GO
gene ontology
- GPC-3
Glypican-3
- GSK3β
glycogen synthase kinase 3 beta
- GSTP1
glutathione S-transferase pi 1
- Ha-ras
Harvey rat sarcoma viral oncogene homolog
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCCS1
hepatocellular carcinoma suppressor 1
- HCP5
HLA complex P5
- HCV
hepatitis C virus
- HFD
high fat and cholesterol diet
- HIF-1α
hypoxia-inducible factor 1-alpha
- HITS-CLIP
high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation
- HIV
human immunodeficiency virus
- HLA
histocompatibility leukocyte antigen
- HPMEC
human pulmonary microvascular endothelial cell
- HUVEC
human umbilical vein endothelial cell
- IGF2
insulin-like growth factor 2
- IHC
immunohistochemical staining
- IF
immunofluorescent staining
- IκBα
nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
- IKKβ
inhibitor of NF-κB kinase subunit beta
- IL-1β
interleukin 1 beta
- IL-6
interleukin 6
- IGFBP7
insulin-like growth factor binding protein-7
- JNK
Janus kinase
- kD
kilodalton
- KDM6A
A gene encoding protein called lysine-specific demethylase 6A
- LEF1
lymphoid enhancer-binding factor 1
- LNA
locked nucleic acid
- lncRNA
long non-coding RNA
- LPS
lipopolysaccharide
- LSF
late SV40 factor
- LXR
liver X receptor
- LYRIC
lysine-rich CEACAM-1 co-isolated protein
- MAP2K
mitogen-activated protein kinase kinase
- MAPK
mitogen-activated protein kinase
- MDM2
mouse double minute 2 homolog
- MDR1
multidrug resistance gene 1
- MEK1/2
MAPK/ERK Kinase 1 and 2
- miR
MicroRNA
- miRNA
MicroRNA
- MMP-9
matrix metalloproteinase-9
- MRI
magnetic resonance imaging
- mRNA
messenger RNA
- MTDH
metadherin
- mTOR
mechanistic target of rapamycin
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- ncRNA
non-coding RNA
- NF-κB
nuclear factor kappa B
- NLS
nuclear localization sequences
- NPC1L1
niemann-Pick C1-Like 1
- NTIS
non-thyroidal illness syndrome
- OS
overall survival
- PAMAM
polyamidoamine
- PAMAM-AEG-1si
PAMAM-PEG-Gal complexed with AEG-1 siRNA
- PAMAM-PEG-Gal
conjugation of PAMAM dendrimers with PEG and Gal
- PAR-CLIP
photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation
- PB
phenobarbital
- PD-1
programmed cell death protein 1
- PDCD11
programmed cell death 11
- PD-L1
PD-1 ligand
- pdTp
3′,5′-deoxythymidine bisphosphate
- PEG
polyethylene glycol
- PERK
protein kinase RNA-like ER kinase
- PGC1α
peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PHFA
primary human fetal astrocyte
- Pik3ca
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subuni alpha
- PIP3
phosphatidylinositol 3,4,5-triphosphate
- PI3K
phosphoinositide 3-kinase
- PIGF
placental growth factor
- PLZF
promyelocytic leukemia zinc finger
- POZ
pox virus and zinc finger protein
- PPAR
peroxisome proliferator-activated receptor
- PPARα
peroxisome proliferator-activated receptor alpha
- PRMT5
protein arginine methyltransferase 5
- PTEN
phosphatase and tensin homolog
- Q-RT-PCR
quantitative reverse transcription polymerase chain reaction
- RAR
retinoic acid receptor
- RAS
rat sarcoma
- Rb
retinoblastoma
- RBP
RNA-binding protein
- RCT
randomized controlled trial
- RIP1
receptor interacting protein 1
- RNA
ribonucleic acid
- RNAi
RNA interference
- RNA-Seq
RNA sequencing
- ROS
reactive oxygen species
- RUNX1
runt-related transcription factor 1
- RXR
retinoid X receptor
- siRNA
small interfering RNA
- SMAD2/4
SMAD family member 2 and 4
- SND1
staphylococcal nuclease and tudor domain containing 1
- SOCS1
suppressor of cytokine signaling 1
- SPOP
speckle type BTB/POZ protein
- SREBP-1c
sterol regulatory element-binding protein 1c
- STAT
signal transducer and activator of transcription
- STAT3
signal transducer and activator of transcription 3
- T3
3,5,3′-triiodothyronine
- T4
l-thyroxine
- TCGA
the cancer genome atlas
- TERT
telomerase reverse transcriptase
- TFF3
trefoil factor 3
- TG
triglyceride
- TIC
tumor initiating cell
- Tie 2
angiopoietin-1 receptor
- TKI
tyrosine kinase inhibitors
- TMA
tissue microarray
- TMD
transmembrane domain
- TNF-α
tumor necrosis factor alpha
- TNM
tumor-nodes-metastasis
- TOP1
DNA topoisomerase 1
- TOPORS
TOP1 binding arginine/serine rich protein, E3 ubiquitin ligase
- TR
thyroid hormone receptor
- TRAF2
TNF receptor associated factor 2
- TS
thymidylate synthase
- TSPN8
tetraspanin 8
- Tudor-SN
tudor staphylococcal nuclease
- VDR
vitamin D receptor
- VEGF
vascular endothelial growth factor
- WT
wild-type
References
- Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY, et al. (2018). Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. The New England Journal of Medicine, 379(1), 54–63. 10.1056/NEJMoa1717002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiel M, Guo C, Li X, Rajasekaran D, Mendoza RG, Robertson CL, et al. (2017). IGFBP7 deletion promotes hepatocellular carcinoma. Cancer Research, 77(15), 4014–4025. 10.1158/0008-5472.CAN-16-2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiel MA, Santhekadur PK, Mendoza RG, Siddiq A, Fisher PB, & Sarkar D (2016). Tetraspanin 8 mediates AEG-1-induced invasion and metastasis in hepatocellular carcinoma cells. FEBS Letters, 590(16), 2700–2708. 10.1002/1873-3468.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexia C, Poalas K, Carvalho G, Zemirli N, Dwyer J, Dubois SM, et al. (2013). The endoplasmic reticulum acts as a platform for ubiquitylated components of nuclear factor kappaB signaling. Science Signaling, 6(291), ra79. 10.1126/scisignal.2004496. [DOI] [PubMed] [Google Scholar]
- Amaddeo G, Cao Q, Ladeiro Y, Imbeaud S, Nault JC, Jaoui D, et al. (2015). Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut, 64(5), 820–829. 10.1136/gutjnl-2013-306228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstee QM, Reeves HL, Kotsiliti E, Govaere O, & Heikenwalder M (2019). From NASH to HCC: Current concepts and future challenges. Nature Reviews. Gastroenterology & Hepatology, 16(7), 411–428. 10.1038/s41575-019-0145-7. [DOI] [PubMed] [Google Scholar]
- Ayuso C, Rimola J, Vilana R, Burrel M, Darnell A, Garcia-Criado A, et al. (2018). Diagnosis and staging of hepatocellular carcinoma (HCC): Current guidelines. European Journal of Radiology, 101, 72–81. 10.1016/j.ejrad.2018.01.025. [DOI] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, et al. (2012). The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Molecular Cell, 46(5), 674–690. 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Bhutia SK, Kegelman TP, Das SK, Azab B, Su ZZ, Lee SG, et al. (2010). Astrocyte elevated gene-1 induces protective autophagy. Proceedings of the National Academy of Sciences of the United States of America, 107(51), 22243–22248. 10.1073/pnas.1009479107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco MA, Aleckovic M, Hua Y, Li T, Wei Y, Xu Z, et al. (2011). Identification of staphylococcal nuclease domain-containing 1 (SND1) as a Metadherin-interacting protein with metastasis-promoting functions. The Journal of Biological Chemistry, 286(22), 19982–19992. 10.1074/jbc.M111.240077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogorad RL, Yin H, Zeigerer A, Nonaka H, Ruda VM, Zerial M, et al. (2014). Nanoparticle-formulated siRNA targeting integrins inhibits hepatocellular carcinoma progression in mice. Nature Communications, 5, 3869. 10.1038/ncomms4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, Jeannot E, et al. (2007). Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology, 45(1), 42–52. 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- Bravi F, Tavani A, Bosetti C, Boffetta P, & La Vecchia C (2017). Coffee and the risk of hepatocellular carcinoma and chronic liver disease: A systematic review and meta-analysis of prospective studies. European Journal of Cancer Prevention, 26(5), 368–377. 10.1097/CEJ.0000000000000252. [DOI] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, & Jemal A (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 68(6), 394–424. 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Britt DE, Yang DF, Yang DQ, Flanagan D, Callanan H, Lim YP, et al. (2004). Identification of a novel protein, LYRIC, localized to tight junctions of polarized epithelial cells. Experimental Cell Research, 300(1), 134–148. 10.1016/j.yexcr.2004.06.026. [DOI] [PubMed] [Google Scholar]
- Brown DM, & Ruoslahti E (2004). Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell, 5(4), 365–374. 10.1016/s1535-6108(04)00079-0. [DOI] [PubMed] [Google Scholar]
- Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, et al. (2017). Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet, 389(10064), 56–66. 10.1016/S0140-6736(16)32453-9. [DOI] [PubMed] [Google Scholar]
- Budhu A, & Wang XW (2006). The role of cytokines in hepatocellular carcinoma. Journal of Leukocyte Biology, 80(6), 1197–1213. 10.1189/jlb.0506297. [DOI] [PubMed] [Google Scholar]
- Calderaro J, Couchy G, Imbeaud S, Amaddeo G, Letouze E, Blanc JF, et al. (2017). Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. Journal of Hepatology, 67(4), 727–738. 10.1016/j.jhep.2017.05.014. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, Electronic address: wheeler@bcm.edu, & Cancer Genome Atlas Research Network. (2017). Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell, 169(7), 1327–1341.e23. 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Sharma M, Imam R, & Yu J (2019). Study on diagnostic values of astrocyte elevated gene 1 (AEG-1) and glypican 3 (GPC-3) in hepatocellular carcinoma. American Journal of Clinical Pathology, 152(5), 647–655. 10.1093/ajcp/aqz086. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, et al. (2012). Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell, 149(6), 1393–1406. 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Caudy AA, Ketting RF, Hammond SM, Denli AM, Bathoorn AM, Tops BB, et al. (2003). A micrococcal nuclease homologue in RNAi effector complexes. Nature, 425(6956), 411–414. 10.1038/nature01956. [DOI] [PubMed] [Google Scholar]
- Chang MH, You SL, Chen CJ, Liu CJ, Lee CM, Lin SM, et al. (2009). Decreased incidence of hepatocellular carcinoma in hepatitis B vaccinees: A 20-year follow-up study. Journal of the National Cancer Institute, 101(19), 1348–1355. 10.1093/jnci/djp288. [DOI] [PubMed] [Google Scholar]
- Chen X, Dong K, Long M, Lin F, Wang X, Wei J, et al. (2012). Serum anti-AEG-1 auto-antibody is a potential novel biomarker for malignant tumors. Oncology Letters, 4(2), 319–323. 10.3892/ol.2012.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Jagannathan S, Reid DW, Zheng T, & Nicchitta CV (2011). Hierarchical regulation of mRNA partitioning between the cytoplasm and the endoplasmic reticulum of mammalian cells. Molecular Biology of the Cell, 22(14), 2646–2658. 10.1091/mbc.E11-03-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Yoo BK, Santhekadur PK, Gredler R, Bhutia SK, Das SK, et al. (2011). Insulin-like growth factor-binding protein-7 functions as a potential tumor suppressor in hepatocellular carcinoma. Clinical Cancer Research, 17(21), 6693–6701. 10.1158/1078-0432.CCR-10-2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, & Serrano M (2010). Senescence in tumours: Evidence from mice and humans. Nature Reviews. Cancer, 10(1), 51–57. 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulouarn C, Gomez-Quiroz LE, Lee JS, Kaposi-Novak P, Conner EA, Goldina TA, et al. (2006). Oncogene-specific gene expression signatures at preneoplastic stage in mice define distinct mechanisms of hepatocarcinogenesis. Hepatology, 44(4), 1003–1011. 10.1002/hep.21293. [DOI] [PubMed] [Google Scholar]
- Das CK, Banerjee I, & Mandal M (2020). Pro-survival autophagy: An emerging candidate of tumor progression through maintaining hallmarks of cancer. Seminars in Cancer Biology, 66, 59–74. 10.1016/j.semcancer.2019.08.020. [DOI] [PubMed] [Google Scholar]
- Dimitroulis D, Damaskos C, Valsami S, Davakis S, Garmpis N, Spartalis E, et al. (2017). From diagnosis to treatment of hepatocellular carcinoma: An epidemic problem for both developed and developing world. World Journal of Gastroenterology, 23(29), 5282–5294. 10.3748/wjg.v23.i29.5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Zhang Z, Jin X, Chen P, Lv F, Liu D, et al. (2019). Interaction with AEG-1 and MDM2 is associated with glioma development and progression and correlates with poor prognosis. Cell Cycle, 18(2), 143–155. 10.1080/15384101.2018.1557489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato F, Boffetta P, & Puoti M (1998). A meta-analysis of epidemiological studies on the combined effect of hepatitis B and C virus infections in causing hepatocellular carcinoma. International Journal of Cancer, 75(3), 347–354. 10.1002/(sici)1097-0215(19980130)75:3<347::aid-ijc4>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Dudek H, Wong DH, Arvan R, Shah A, Wortham K, Ying B, et al. (2014). Knockdown of beta-catenin with dicer-substrate siRNAs reduces liver tumor burden in vivo. Molecular Therapy, 22(1), 92–101. 10.1038/mt.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. (2017). Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet, 389(10088), 2492–2502. 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Serag HB (2011). Hepatocellular carcinoma. The New England Journal of Medicine, 365(12), 1118–1127. 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- Emdad L, Das SK, Hu B, Kegelman T, Kang DC, Lee SG, et al. (2016). AEG-1/MTDH/LYRIC: A promiscuous protein partner critical in cancer, obesity, and CNS diseases. Advances in Cancer Research, 131, 97–132. 10.1016/bs.acr.2016.05.002. [DOI] [PubMed] [Google Scholar]
- Emdad L, Lee SG, Su ZZ, Jeon HY, Boukerche H, Sarkar D, et al. (2009). Astrocyte elevated gene-1 (AEG-1) functions as an oncogene and regulates angiogenesis. Proceedings of the National Academy of Sciences of the United States of America, 106(50), 21300–21305. 10.1073/pnas.0910936106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emdad L, Sarkar D, Su ZZ, Lee SG, Kang DC, Bruce JN, et al. (2007). Astrocyte elevated gene-1: Recent insights into a novel gene involved in tumor progression, metastasis and neurodegeneration. Pharmacology & Therapeutics, 114(2), 155–170. 10.1016/j.pharmthera.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emdad L, Sarkar D, Su ZZ, Randolph A, Boukerche H, Valerie K, et al. (2006). Activation of the nuclear factor kappaB pathway by astrocyte elevated gene-1: Implications for tumor progression and metastasis. Cancer Research, 66(3), 1509–1516. 10.1158/0008-5472.CAN-05-3029. [DOI] [PubMed] [Google Scholar]
- European Association for the Study of the Liver, Electronic address: easloffice@easloffice.eu, & European Association for the Study of the Liver. (2018). EASL clinical practice guidelines: Management of hepatocellular carcinoma. Journal of Hepatology, 69(1), 182–236. 10.1016/j.jhep.2018.03.019. [DOI] [PubMed] [Google Scholar]
- Folkman J (1971). Tumor angiogenesis: Therapeutic implications. The New England Journal of Medicine, 285(21), 1182–1186. 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Forner A, Reig M, & Bruix J (2018). Hepatocellular carcinoma. Lancet, 391(10127), 1301–1314. 10.1016/S0140-6736(18)30010-2. [DOI] [PubMed] [Google Scholar]
- Friedman SL, Neuschwander-Tetri BA, Rinella M, & Sanyal AJ (2018). Mechanisms of NAFLD development and therapeutic strategies. Nature Medicine, 24(7), 908–922. 10.1038/s41591-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Ge L, Shao J, Su C, Zhao H, Saarikettu J, et al. (2010). Tudor-SN interacts with and co-localizes with G3BP in stress granules under stress conditions. FEBS Letters, 584(16), 3525–3532. 10.1016/j.febslet.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Zhao X, Zhu Y, He J, Shao J, Su C, et al. (2012). Tudor staphylococcal nuclease (Tudor-SN) participates in small ribonucleoprotein (snRNP) assembly via interacting with symmetrically dimethylated Sm proteins. The Journal of Biological Chemistry, 287(22), 18130–18141. 10.1074/jbc.M111.311852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Liu W, You N, Wang T, Wang X, Lu P, et al. (2012). Prognostic significance of metadherin overexpression in hepatitis B virus-related hepatocellular carcinoma. Oncology Reports, 27(6), 2073–2079. 10.3892/or.2012.1749. [DOI] [PubMed] [Google Scholar]
- Grant TJ, Bishop JA, Christadore LM, Barot G, Chin HG, Woodson S, et al. (2012). Antiproliferative small-molecule inhibitors of transcription factor LSF reveal oncogene addiction to LSF in hepatocellular carcinoma. Proceedings of the National Academy of Sciences of the United States of America, 109(12), 4503–4508. 10.1073/pnas.1121601109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Wan L, Zheng A, Stanevich V, Wei Y, Satyshur KA, et al. (2014). Structural insights into the tumor-promoting function of the MTDH-SND1 complex. Cell Reports, 8(6), 1704–1713. 10.1016/j.celrep.2014.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Fu Y, Zeng N, Yin J, & Li Q (2020). LncRNA FAM83H-AS1 promotes triple-negative breast cancer progression by regulating the miR-136–5p/metadherin axis. Aging (Albany NY), 12(4), 3594–3616. 10.18632/aging.102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, & Weinberg RA (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Harding JJ, Nandakumar S, Armenia J, Khalil DN, Albano M, Ly M, et al. (2019). Prospective genotyping of hepatocellular carcinoma: Clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clinical Cancer Research, 25(7), 2116–2126. 10.1158/1078-0432.CCR-18-2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, et al. (2009). A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell, 16(4), 295–308. 10.1016/j.ccr.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XX, Chang Y, Meng FY, Wang MY, Xie QH, Tang F, et al. (2012). MicroRNA-375 targets AEG-1 in hepatocellular carcinoma and suppresses liver cancer cell growth in vitro and in vivo. Oncogene, 31(28), 3357–3369. 10.1038/onc.2011.500. [DOI] [PubMed] [Google Scholar]
- He R, Gao L, Ma J, Peng Z, Zhou S, Yang L, et al. (2017). The essential role of MTDH in the progression of HCC: A study with immunohistochemistry, TCGA, meta-analysis and in vitro investigation. American Journal of Translational Research, 9(4), 1561–1579. [PMC free article] [PubMed] [Google Scholar]
- He A, He S, Huang C, Chen Z, Wu Y, Gong Y, et al. (2020). MTDH promotes metastasis of clear cell renal cell carcinoma by activating SND1-mediated ERK signaling and epithelial-mesenchymal transition. Aging (Albany NY), 12(2), 1465–1487. 10.18632/aging.102694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, & Karin M (2011). NF-kappaB and STAT3—Key players in liver inflammation and cancer. Cell Research, 21(1), 159–168. 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heery DM, Kalkhoven E, Hoare S, & Parker MG (1997). A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature, 387(6634), 733–736. 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- Hong R, Wang K, & Shi H (2017). Astrocyte elevated gene-1 promotes inflammation and invasion of fibroblast-like synoviocytes in rheumatoid arthritis. Tissue & Cell, 49(6), 672–679. 10.1016/j.tice.2017.09.005. [DOI] [PubMed] [Google Scholar]
- Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, et al. (2004). Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. The Journal of Clinical Investigation, 113(12), 1774–1783. 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosel M, Quasdorff M, Wiegmann K, Webb D, Zedler U, Broxtermann M, et al. (2009). Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology, 50(6), 1773–1782. 10.1002/hep.23226. [DOI] [PubMed] [Google Scholar]
- Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, et al. (2009). Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Research, 69(18), 7385–7392. 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JC, Reid DW, Hoffman AM, Sarkar D, & Nicchitta CV (2018). Oncoprotein AEG-1 is an endoplasmic reticulum RNA-binding protein whose interactome is enriched in organelle resident protein-encoding mRNAs. RNA, 24(5), 688–703. 10.1261/rna.063313.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Chong RA, Yang Q, Wei Y, Blanco MA, Li F, et al. (2009). MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell, 15(1), 9–20. 10.1016/j.ccr.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Emdad L, Bacolod MD, Kegelman TP, Shen XN, Alzubi MA, et al. (2014). Astrocyte elevated gene-1 interacts with Akt isoform 2 to control glioma growth, survival, and pathogenesis. Cancer Research, 74(24), 7321–7332. 10.1158/0008-5472.CAN-13-2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Emdad L, Kegelman TP, Shen XN, Das SK, Sarkar D, et al. (2017). Astrocyte elevated gene-1 regulates beta-catenin signaling to maintain glioma stem-like stemness and self-renewal. Molecular Cancer Research, 15(2), 225–233. 10.1158/1541-7786.MCR-16-0239. [DOI] [PubMed] [Google Scholar]
- Hu TH, Huang CC, Lin PR, Chang HW, Ger LP, Lin YW, et al. (2003). Expression and prognostic role of tumor suppressor gene PTEN/MMAC1/TEP1 in hepatocellular carcinoma. Cancer, 97(8), 1929–1940. 10.1002/cncr.11266. [DOI] [PubMed] [Google Scholar]
- Huynh H, Nguyen TT, Chow KH, Tan PH, Soo KC, & Tran E (2003). Over-expression of the mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK in hepatocellular carcinoma: Its role in tumor progression and apoptosis. BMC Gastroenterology, 3, 19. 10.1186/1471-230X-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jariwala N, Rajasekaran D, Mendoza RG, Shen XN, Siddiq A, Akiel MA, et al. (2017). Oncogenic role of SND1 in development and progression of hepatocellular carcinoma. Cancer Research, 77(12), 3306–3316. 10.1158/0008-5472.CAN-17-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia C, Tang D, Sun C, Yao L, Li F, Hu Y, et al. (2018). MicroRNA466 inhibits the aggressive behaviors of hepatocellular carcinoma by directly targeting metadherin. Oncology Reports, 40(6), 3890–3898. 10.3892/or.2018.6763. [DOI] [PubMed] [Google Scholar]
- Johnston MP, & Khakoo SI (2019). Immunotherapy for hepatocellular carcinoma: Current and future. World Journal of Gastroenterology, 25(24), 2977–2989. 10.3748/wjg.v25.i24.2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HI, Ahn T, Bae SH, Chung JC, Kim H, Chin S, et al. (2015). Astrocyte elevated gene-1 overexpression in hepatocellular carcinoma: An independent prognostic factor. Annals of Surgical Treatment and Research, 88(2), 77–85. 10.4174/astr.2015.88.2.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DC, Su ZZ, Sarkar D, Emdad L, Volsky DJ, & Fisher PB (2005). Cloning and characterization of HIV-1-inducible astrocyte elevated gene-1, AEG-1. Gene, 353(1), 8–15. 10.1016/j.gene.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Karin M (2006). Tracking the road from inflammation to cancer: The critical role of IkappaB kinase (IKK). Harvey Lectures, 102, 133–151. 10.1002/9780470593042.ch7. [DOI] [PubMed] [Google Scholar]
- Kato A, Miyazaki M, Ambiru S, Yoshitomi H, Ito H, Nakagawa K, et al. (2001). Multidrug resistance gene (MDR-1) expression as a useful prognostic factor in patients with human hepatocellular carcinoma after surgical resection. Journal of Surgical Oncology, 78(2), 110–115. 10.1002/jso.1129. [DOI] [PubMed] [Google Scholar]
- Kerbel RS (2000). Tumor angiogenesis: Past, present and the near future. Carcinogenesis, 21(3), 505–515. 10.1093/carcin/21.3.505. [DOI] [PubMed] [Google Scholar]
- Kew MC (2013). Aflatoxins as a cause of hepatocellular carcinoma. Journal of Gastrointestinal and Liver Diseases, 22(3), 305–310. [PubMed] [Google Scholar]
- Khuda II, Koide N, Noman AS, Dagvadorj J, Tumurkhuu G, Naiki Y, et al. (2009). Astrocyte elevated gene-1 (AEG-1) is induced by lipopolysaccharide as toll-like receptor 4 (TLR4) ligand and regulates TLR4 signalling. Immunology, 128(1 Suppl), e700–e706. 10.1111/j.1365-2567.2009.03063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, Tanaka Y, et al. (2007). Knockdown of astrocyte-elevated gene-1 inhibits prostate cancer progression through upregulation of FOXO3a activity. Oncogene, 26(55), 7647–7655. 10.1038/sj.onc.1210572. [DOI] [PubMed] [Google Scholar]
- Kim YN, Koo KH, Sung JY, Yun UJ, & Kim H (2012). Anoikis resistance: An essential prerequisite for tumor metastasis. International Journal of Cell Biology, 2012. 10.1155/2012/306879, 306879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Lee SH, & Jung G (2010). The hepatitis B viral X protein activates NF-kappaB signaling pathway through the up-regulation of TBK1. FEBS Letters, 584(3), 525–530. 10.1016/j.febslet.2009.11.091. [DOI] [PubMed] [Google Scholar]
- Knuutila S, Bjorkqvist AM, Autio K, Tarkkanen M, Wolf M, Monni O, et al. (1998). DNA copy number amplifications in human neoplasms: Review of comparative genomic hybridization studies. The American Journal of Pathology, 152(5), 1107–1123. [PMC free article] [PubMed] [Google Scholar]
- Kochanek DM, & Wells DG (2013). CPEB1 regulates the expression of MTDH/AEG-1 and glioblastoma cell migration. Molecular Cancer Research, 11(2), 149–160. 10.1158/1541-7786.MCR-12-0498. [DOI] [PubMed] [Google Scholar]
- Kojiro M (2005). Histopathology of liver cancers. Best Practice & Research. Clinical Gastroenterology, 19(1), 39–62. 10.1016/j.bpg.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Krishnan RK, Nolte H, Sun T, Kaur H, Sreenivasan K, Looso M, et al. (2015). Quantitative analysis of the TNF-alpha-induced phosphoproteome reveals AEG-1/MTDH/LYRIC as an IKKbeta substrate. Nature Communications, 6, 6658. 10.1038/ncomms7658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. (2018). Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet, 391(10126), 1163–1173. 10.1016/S0140-6736(18)30207-1. [DOI] [PubMed] [Google Scholar]
- Kudo Y, Tanaka Y, Tateishi K, Yamamoto K, Yamamoto S, Mohri D, et al. (2011). Altered composition of fatty acids exacerbates hepatotumorigenesis during activation of the phosphatidylinositol 3-kinase pathway. Journal of Hepatology, 55(6), 1400–1408. 10.1016/j.jhep.2011.03.025. [DOI] [PubMed] [Google Scholar]
- Kwon SC, Yi H, Eichelbaum K, Fohr S, Fischer B, You KT, et al. (2013). The RNA-binding protein repertoire of embryonic stem cells. Nature Structural & Molecular Biology, 20(9), 1122–1130. 10.1038/nsmb.2638. [DOI] [PubMed] [Google Scholar]
- Leavens KF, Easton RM, Shulman GI, Previs SF, & Birnbaum MJ (2009). Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metabolism, 10(5), 405–418. 10.1016/j.cmet.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SG, Su ZZ, Emdad L, Sarkar D, & Fisher PB (2006). Astrocyte elevated gene-1 (AEG-1) is a target gene of oncogenic Ha-ras requiring phosphatidylinositol 3-kinase and c-Myc. Proceedings of the National Academy of Sciences of the United States of America, 103(46), 17390–17395. 10.1073/pnas.0608386103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SG, Su ZZ, Emdad L, Sarkar D, Franke TF, & Fisher PB (2008). Astrocyte elevated gene-1 activates cell survival pathways through PI3K-Akt signaling. Oncogene, 27(8), 1114–1121. 10.1038/sj.onc.1210713. [DOI] [PubMed] [Google Scholar]
- Lefebvre P, Benomar Y, & Staels B (2010). Retinoid X receptors: Common heterodimerization partners with distinct functions. Trends in Endocrinology and Metabolism, 21(11), 676–683. 10.1016/j.tem.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Leonardi GC, Candido S, Cervello M, Nicolosi D, Raiti F, Travali S, et al. (2012). The tumor microenvironment in hepatocellular carcinoma (review). International Journal of Oncology, 40(6), 1733–1747. 10.3892/ijo.2012.1408. [DOI] [PubMed] [Google Scholar]
- Leverson JD, Koskinen PJ, Orrico FC, Rainio EM, Jalkanen KJ, Dash AB, et al. (1998). Pim-1 kinase and p100 cooperate to enhance c-Myb activity. Molecular Cell, 2(4), 417–425. 10.1016/s1097-2765(00)80141-0. [DOI] [PubMed] [Google Scholar]
- Li WF, Dai H, Ou Q, Zuo GQ, & Liu CA (2016). Overexpression of microRNA-30a-5p inhibits liver cancer cell proliferation and induces apoptosis by targeting MTDH/PTEN/AKT pathway. Tumour Biology, 37(5), 5885–5895. 10.1007/s13277-015-4456-1. [DOI] [PubMed] [Google Scholar]
- Li P, He Y, Chen T, Choy KY, Chow TS, Wong ILK, et al. (2021). Disruption of SND1-MTDH interaction by a high affinity peptide results in SND1 degradation and cytotoxicity to breast cancer cells in vitro and in vivo. Molecular Cancer Therapeutics, 20(1), 76–84. 10.1158/1535-7163.MCT-20-0130. [DOI] [PubMed] [Google Scholar]
- Li WQ, Park Y, McGlynn KA, Hollenbeck AR, Taylor PR, Goldstein AM, et al. (2014). Index-based dietary patterns and risk of incident hepatocellular carcinoma and mortality from chronic liver disease in a prospective study. Hepatology, 60(2), 588–597. 10.1002/hep.27160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Wang Z, Ye J, Zhang X, Wu H, Peng J, et al. (2014). Uncontrolled inflammation induced by AEG-1 promotes gastric cancer and poor prognosis. Cancer Research, 74(19), 5541–5552. 10.1158/0008-5472.CAN-14-0968. [DOI] [PubMed] [Google Scholar]
- Li J, Zhang N, Song LB, Liao WT, Jiang LL, Gong LY, et al. (2008). Astrocyte elevated gene-1 is a novel prognostic marker for breast cancer progression and overall patient survival. Clinical Cancer Research, 14(11), 3319–3326. 10.1158/1078-0432.CCR-07-4054. [DOI] [PubMed] [Google Scholar]
- Liaw YF, Sung JJ, Chow WC, Farrell G, Lee CZ, Yuen H, et al. (2004). Lamivudine for patients with chronic hepatitis B and advanced liver disease. The New England Journal of Medicine, 351(15), 1521–1531. 10.1056/NEJMoa033364. [DOI] [PubMed] [Google Scholar]
- Liu W, Chen X, Wang Y, Chen Y, Chen S, Gong W, et al. (2019). Micheliolide ameliorates diabetic kidney disease by inhibiting Mtdh-mediated renal inflammation in type 2 diabetic db/db mice. Pharmacological Research, 150, 104506. 10.1016/j.phrs.2019.104506. [DOI] [PubMed] [Google Scholar]
- Liu P, Kimmoun E, Legrand A, Sauvanet A, Degott C, Lardeux B, et al. (2002). Activation of NF-kappa B, AP-1 and STAT transcription factors is a frequent and early event in human hepatocellular carcinomas. Journal of Hepatology, 37(1), 63–71. 10.1016/s0168-8278(02)00064-8. [DOI] [PubMed] [Google Scholar]
- Liu J, Tang W, Budhu A, Forgues M, Hernandez MO, Candia J, et al. (2020). A viral exposure signature defines early onset of hepatocellular carcinoma. Cell, 182(2), 317–328.e10. 10.1016/j.cell.2020.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet JM, Bru C, & Bruix J (1999). Prognosis of hepatocellular carcinoma: The BCLC staging classification. Seminars in Liver Disease, 19(3), 329–338. 10.1055/s-2007-1007122. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. (2008). Sorafenib in advanced hepatocellular carcinoma. The New England Journal of Medicine, 359(4), 378–390. 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- Lu B, Guo H, Zhao J, Wang C, Wu G, Pang M, et al. (2010). Increased expression of iASPP, regulated by hepatitis B virus X protein-mediated NF-kappaB activation, in hepatocellular carcinoma. Gastroenterology, 139(6), 2183–2194.e5. 10.1053/j.gastro.2010.06.049. [DOI] [PubMed] [Google Scholar]
- Lu Q, Shan S, Li Y, Zhu D, Jin W, & Ren T (2018). Long noncoding RNA SNHG1 promotes non-small cell lung cancer progression by up-regulating MTDH via sponging miR-145–5p. The FASEB Journal, 32(7), 3957–3967. 10.1096/fj.201701237RR. [DOI] [PubMed] [Google Scholar]
- Luo L, Tang H, Ling L, Li N, Jia X, Zhang Z, et al. (2018). LINC01638 lncRNA activates MTDH-Twist1 signaling by preventing SPOP-mediated c-Myc degradation in triple-negative breast cancer. Oncogene, 37(47), 6166–6179. 10.1038/s41388-018-0396-8. [DOI] [PubMed] [Google Scholar]
- Luxton HJ, Barnouin K, Kelly G, Hanrahan S, Totty N, Neal DE, et al. (2014). Regulation of the localisation and function of the oncogene LYRIC/AEG-1 by ubiquitination at K486 and K491. Molecular Oncology, 8(3), 633–641. 10.1016/j.molonc.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malayaperumal S, Sriramulu S, Jothimani G, Banerjee A, & Pathak S (2020). A review on AEG-1 oncogene regulating MicroRNA expression in colon cancer progression. Endocrine, Metabolic & Immune Disorders Drug Targets, 21, 27–34. 10.2174/1871530320666200618104116. [DOI] [PubMed] [Google Scholar]
- Mann CD, Neal CP, Garcea G, Manson MM, Dennison AR, & Berry DP (2007). Prognostic molecular markers in hepatocellular carcinoma: A systematic review. European Journal of Cancer, 43(6), 979–992. 10.1016/j.ejca.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Manna D, & Sarkar D (2020). Non-coding RNAs: Regulating disease progression and therapy resistance in hepatocellular carcinoma. Cancers (Basel), 12(5), 1243. 10.3390/cancers12051243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrero JA, Kulik LM, Sirlin CB, Zhu AX, Finn RS, Abecassis MM, et al. (2018). Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American Association for the Study of Liver Diseases. Hepatology, 68(2), 723–750. 10.1002/hep.29913. [DOI] [PubMed] [Google Scholar]
- Meng X, Thiel KW, & Leslie KK (2013). Drug resistance mediated by AEG-1/MTDH/LYRIC. Advances in Cancer Research, 120, 135–157. 10.1016/B978-0-12-401676-7.00005-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Zhu D, Yang S, Wang X, Xiong Z, Zhang Y, et al. (2012). Cytoplasmic Metadherin (MTDH) provides survival advantage under conditions of stress by acting as RNA-binding protein. The Journal of Biological Chemistry, 287(7), 4485–4491. 10.1074/jbc.C111.291518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RL, Baack B, Smith BD, Yartel A, Pitasi M, & Falck-Ytter Y (2013). Eradication of hepatitis C virus infection and the development of hepatocellular carcinoma: A meta-analysis of observational studies. Annals of Internal Medicine, 158 (5 Pt 1), 329–337. 10.7326/0003-4819-158-5-201303050-00005. [DOI] [PubMed] [Google Scholar]
- Murakami H, Sanderson ND, Nagy P, Marino PA, Merlino G, & Thorgeirsson SS (1993). Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: Interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Research, 53(8), 1719–1723. [PubMed] [Google Scholar]
- Newell P, Toffanin S, Villanueva A, Chiang DY, Minguez B, Cabellos L, et al. (2009). Ras pathway activation in hepatocellular carcinoma and anti-tumoral effect of combined sorafenib and rapamycin in vivo. Journal of Hepatology, 51(4), 725–733. 10.1016/j.jhep.2009.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng AWT, Poon SL, Huang MN, Lim JQ, Boot A, Yu W, et al. (2017). Aristolochic acids and their derivatives are widely implicated in liver cancers in Taiwan and throughout Asia. Science Translational Medicine, 9(412), eaan6446. 10.1126/scitranslmed.aan6446. [DOI] [PubMed] [Google Scholar]
- Noch E, Bookland M, & Khalili K (2011). Astrocyte-elevated gene-1 (AEG-1) induction by hypoxia and glucose deprivation in glioblastoma. Cancer Biology & Therapy, 11(1), 32–39. 10.4161/cbt.11.1.13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. (2004). NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature, 431(7007), 461–466. 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Rajasekaran D, Siddiq A, Willoughby JL, Biagi JM, Christadore LM, Yunes SA, et al. (2015). Small molecule inhibitors of late SV40 factor (LSF) abrogate hepatocellular carcinoma (HCC): Evaluation using an endogenous HCC model. Oncotarget, 6(28), 26266–26277. 10.18632/oncotarget.4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran D, Srivastava J, Ebeid K, Gredler R, Akiel M, Jariwala N, et al. (2015). Combination of nanoparticle-delivered siRNA for astrocyte elevated gene-1 (AEG-1) and all-trans retinoic acid (ATRA): An effective therapeutic strategy for hepatocellular carcinoma (HCC). Bioconjugate Chemistry, 26(8), 1651–1661. 10.1021/acs.bioconjchem.5b00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh Y, & Sarkar D (2020). Molecular mechanisms regulating obesity-associated hepatocellular carcinoma. Cancers (Basel), 12(5), 1290. 10.3390/cancers12051290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebouissou S, Franconi A, Calderaro J, Letouze E, Imbeaud S, Pilati C, et al. (2016). Genotype-phenotype correlation of CTNNB1 mutations reveals different ss-catenin activity associated with liver tumor progression. Hepatology, 64(6), 2047–2061. 10.1002/hep.28638. [DOI] [PubMed] [Google Scholar]
- Reghupaty SC, & Sarkar D (2019). Current status of gene therapy in hepatocellular carcinoma. Cancers (Basel), 11(9), 1265. 10.3390/cancers11091265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimassa L, Assenat E, Peck-Radosavljevic M, Pracht M, Zagonel V, Mathurin P, et al. (2018). Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): A final analysis of a phase 3, randomised, placebo-controlled study. The Lancet Oncology, 19(5), 682–693. 10.1016/S1470-2045(18)30146-3. [DOI] [PubMed] [Google Scholar]
- Roberts LR, Sirlin CB, Zaiem F, Almasri J, Prokop LJ, Heimbach JK, et al. (2018). Imaging for the diagnosis of hepatocellular carcinoma: A systematic review and meta-analysis. Hepatology, 67(1), 401–421. 10.1002/hep.29487. [DOI] [PubMed] [Google Scholar]
- Robertson CL, Mendoza RG, Jariwala N, Dozmorov M, Mukhopadhyay ND, Subler MA, et al. (2018). Astrocyte elevated gene-1 regulates macrophage activation in hepatocellular carcinogenesis. Cancer Research, 78(22), 6436–6446. 10.1158/0008-5472.CAN-18-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CL, Srivastava J, Rajasekaran D, Gredler R, Akiel MA, Jariwala N, et al. (2015). The role of AEG-1 in the development of liver cancer. Hepatic Oncology, 2(3), 303–312. 10.2217/hep.15.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CL, Srivastava J, Siddiq A, Gredler R, Emdad L, Rajasekaran D, et al. (2014). Genetic deletion of AEG-1 prevents hepatocarcinogenesis. Cancer Research, 74(21), 6184–6193. 10.1158/0008-5472.CAN-14-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CL, Srivastava J, Siddiq A, Gredler R, Emdad L, Rajasekaran D, et al. (2015). Astrocyte elevated gene-1 (AEG-1) regulates lipid homeostasis. The Journal of Biological Chemistry, 290(29), 18227–18236. 10.1074/jbc.M115.661801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahasrabuddhe VV, Gunja MZ, Graubard BI, Trabert B, Schwartz LM, Park Y, et al. (2012). Nonsteroidal anti-inflammatory drug use, chronic liver disease, and hepatocellular carcinoma. Journal of the National Cancer Institute, 104(23), 1808–1814. 10.1093/jnci/djs452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhekadur PK, Das SK, Gredler R, Chen D, Srivastava J, Robertson C, et al. (2012). Multifunction protein staphylococcal nuclease domain containing 1 (SND1) promotes tumor angiogenesis in human hepatocellular carcinoma through novel pathway that involves nuclear factor kappaB and miR-221. The Journal of Biological Chemistry, 287(17), 13952–13958. 10.1074/jbc.M111.321646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhekadur PK, Gredler R, Chen D, Siddiq A, Shen XN, Das SK, et al. (2012). Late SV40 factor (LSF) enhances angiogenesis by transcriptionally up-regulating matrix metalloproteinase-9 (MMP-9). The Journal of Biological Chemistry, 287(5), 3425–3432. 10.1074/jbc.M111.298976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar D (2013). AEG-1/MTDH/LYRIC in liver cancer. Advances in Cancer Research, 120, 193–221. 10.1016/B978-0-12-401676-7.00007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar D, Emdad L, Lee SG, Yoo BK, Su ZZ, & Fisher PB (2009). Astrocyte elevated gene-1: Far more than just a gene regulated in astrocytes. Cancer Research, 69(22), 8529–8535. 10.1158/0008-5472.CAN-09-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar D, & Fisher PB (2013). AEG-1/MTDH/LYRIC: clinical significance. Advances in Cancer Research, 120, 39–74. 10.1016/B978-0-12-401676-7.00002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar D, Park ES, Emdad L, Lee SG, Su ZZ, & Fisher PB (2008). Molecular basis of nuclear factor-kappaB activation by astrocyte elevated gene-1. Cancer Research, 68(5), 1478–1484. 10.1158/0008-5472.CAN-07-6164. [DOI] [PubMed] [Google Scholar]
- Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, et al. (2000). AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nature Genetics, 24(3), 245–250. 10.1038/73448. [DOI] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, et al. (2015). Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nature Genetics, 47(5), 505–511. 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe RF, Seki E, & Brenner DA (2006). Toll-like receptor signaling in the liver. Gastroenterology, 130(6), 1886–1900. 10.1053/j.gastro.2006.01.038. [DOI] [PubMed] [Google Scholar]
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, et al. (2004). MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature, 431(7012), 1112–1117. 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- Shen ZT, Chen Y, Huang GC, Zhu XX, Wang R, & Chen LB (2019). Aurora-a confers radioresistance in human hepatocellular carcinoma by activating NF-kappaB signaling pathway. BMC Cancer, 19(1), 1075. 10.1186/s12885-019-6312-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M, Xie S, Rowicki M, Michel S, Wei Y, Hang X, et al. (2020). Therapeutic targeting of Metadherin suppresses colorectal and lung cancer progression and metastasis. Cancer Research, 81, 1014–1025. 10.1158/0008-5472.CAN-20-1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, & Flier JS (2006). TLR4 links innate immunity and fatty acid-induced insulin resistance. The Journal of Clinical Investigation, 116(11), 3015–3025. 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Singh PP, Singh AG, Murad MH, & Sanchez W (2013). Statins are associated with a reduced risk of hepatocellular cancer: A systematic review and meta-analysis. Gastroenterology, 144(2), 323–332. 10.1053/j.gastro.2012.10.005. [DOI] [PubMed] [Google Scholar]
- Srivastava J, Robertson CL, Ebeid K, Dozmorov M, Rajasekaran D, Mendoza R, et al. (2017). A novel role of astrocyte elevated gene-1 (AEG-1) in regulating nonalcoholic steatohepatitis (NASH). Hepatology, 66(2), 466–480. 10.1002/hep.29230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava J, Robertson CL, Gredler R, Siddiq A, Rajasekaran D, Akiel MA, et al. (2015). Astrocyte elevated gene-1 (AEG-1) contributes to non-thyroidal illness syndrome (NTIS) associated with hepatocellular carcinoma (HCC). The Journal of Biological Chemistry, 290(25), 15549–15558. 10.1074/jbc.M115.649707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava J, Robertson CL, Rajasekaran D, Gredler R, Siddiq A, Emdad L, et al. (2014). AEG-1 regulates retinoid X receptor and inhibits retinoid signaling. Cancer Research, 74(16), 4364–4377. 10.1158/0008-5472.CAN-14-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava J, Siddiq A, Emdad L, Santhekadur PK, Chen D, Gredler R, et al. (2012). Astrocyte elevated gene-1 promotes hepatocarcinogenesis: Novel insights from a mouse model. Hepatology, 56(5), 1782–1791. 10.1002/hep.25868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava J, Siddiq A, Gredler R, Shen XN, Rajasekaran D, Robertson CL, et al. (2015). Astrocyte elevated gene-1 and c-Myc cooperate to promote hepatocarcinogenesis in mice. Hepatology, 61(3), 915–929. 10.1002/hep.27339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su ZZ, Kang DC, Chen Y, Pekarskaya O, Chao W, Volsky DJ, et al. (2002). Identification and cloning of human astrocyte genes displaying elevated expression after infection with HIV-1 or exposure to HIV-1 envelope glycoprotein by rapid subtraction hybridization, RaSH. Oncogene, 21(22), 3592–3602. 10.1038/sj.onc.1205445. [DOI] [PubMed] [Google Scholar]
- Sutherland HG, Lam YW, Briers S, Lamond AI, & Bickmore WA (2004). 3D3/lyric: A novel transmembrane protein of the endoplasmic reticulum and nuclear envelope, which is also present in the nucleolus. Experimental Cell Research, 294(1), 94–105. 10.1016/j.yexcr.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, et al. (2013). First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discovery, 3(4), 406–417. 10.1158/2159-8290.CD-12-0429. [DOI] [PubMed] [Google Scholar]
- Tai DI, Tsai SL, Chang YH, Huang SN, Chen TC, Chang KS, et al. (2000). Constitutive activation of nuclear factor kappaB in hepatocellular carcinoma. Cancer, 89(11), 2274–2281. [PubMed] [Google Scholar]
- Tai DI, Tsai SL, Chen YM, Chuang YL, Peng CY, Sheen IS, et al. (2000). Activation of nuclear factor kappaB in hepatitis C virus infection: Implications for pathogenesis and hepatocarcinogenesis. Hepatology, 31(3), 656–664. 10.1002/hep.510310316. [DOI] [PubMed] [Google Scholar]
- Talwalkar JA, & Gores GJ (2004). Diagnosis and staging of hepatocellular carcinoma. Gastroenterology, 127(5 Suppl 1), S126–S132. 10.1053/j.gastro.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, & Karin M (2018). NF-kappaB, inflammation, immunity and cancer: Coming of age. Nature Reviews. Immunology, 18(5), 309–324. 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- Teng H, Wang P, Xue Y, Liu X, Ma J, Cai H, et al. (2016). Role of HCP5-miR-139-RUNX1 feedback loop in regulating malignant behavior of glioma cells. Molecular Therapy, 24(10), 1806–1822. 10.1038/mt.2016.103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Teufel A, Staib F, Kanzler S, Weinmann A, Schulze-Bergkamen H, & Galle PR (2007). Genetics of hepatocellular carcinoma. World Journal of Gastroenterology, 13(16), 2271–2282. 10.3748/wjg.v13.i16.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirkettle HJ, Girling J, Warren AY, Mills IG, Sahadevan K, Leung H, et al. (2009). LYRIC/AEG-1 is targeted to different subcellular compartments by ubiquitinylation and intrinsic nuclear localization signals. Clinical Cancer Research, 15(9), 3003–3013. 10.1158/1078-0432.CCR-08-2046. [DOI] [PubMed] [Google Scholar]
- Thirkettle HJ, Mills IG, Whitaker HC, & Neal DE (2009). Nuclear LYRIC/AEG-1 interacts with PLZF and relieves PLZF-mediated repression. Oncogene, 28(41), 3663–3670. 10.1038/onc.2009.223. [DOI] [PubMed] [Google Scholar]
- Vartak-Sharma N, Gelman BB, Joshi C, Borgamann K, & Ghorpade A (2014). Astrocyte elevated gene-1 is a novel modulator of HIV-1-associated neuroinflammation via regulation of nuclear factor-kappaB signaling and excitatory amino acid transporter-2 repression. The Journal of Biological Chemistry, 289(28), 19599–19612. 10.1074/jbc.M114.567644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva A (2019). Hepatocellular carcinoma. The New England Journal of Medicine, 380(15), 1450–1462. 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- Wan L, Hu G, Wei Y, Yuan M, Bronson RT, Yang Q, et al. (2014). Genetic ablation of metadherin inhibits autochthonous prostate cancer progression and metastasis. Cancer Research, 74(18), 5336–5347. 10.1158/0008-5472.CAN-14-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Lu X, Yuan S, Wei Y, Guo F, Shen M, et al. (2014). MTDH-SND1 interaction is crucial for expansion and activity of tumor-initiating cells in diverse oncogene- and carcinogen-induced mammary tumors. Cancer Cell, 26(1), 92–105. 10.1016/j.ccr.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Lim HY, Shi S, Lee J, Deng S, Xie T, et al. (2013). Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology, 58(2), 706–717. 10.1002/hep.26402. [DOI] [PubMed] [Google Scholar]
- Wang X, Liu X, Fang J, Lu Y, He J, Yao X, et al. (2010). Coactivator P100 protein enhances STAT6-dependent transcriptional activation but has no effect on STAT1-mediated gene transcription. The Anatomical Record: Advances in Integrative Anatomy and Evolutionary Biology, 293(6), 1010–1016. 10.1002/ar.21143. [DOI] [PubMed] [Google Scholar]
- Warner MH, & Beckett GJ (2010). Mechanisms behind the non-thyroidal illness syndrome: An update. The Journal of Endocrinology, 205(1), 1–13. 10.1677/JOE-09-0412. [DOI] [PubMed] [Google Scholar]
- Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, & Roberts LR (2019). A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nature Reviews. Gastroenterology & Hepatology, 16(10), 589–604. 10.1038/s41575-019-0186-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Valineva T, Hong J, Bu T, Yao Z, Jensen ON, et al. (2007). Transcriptional co-activator protein p100 interacts with snRNP proteins and facilitates the assembly of the spliceosome. Nucleic Acids Research, 35(13), 4485–4494. 10.1093/nar/gkm470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yea S, Narla G, Zhao X, Garg R, Tal-Kremer S, Hod E, et al. (2008). Ras promotes growth by alternative splicing-mediated inactivation of the KLF6 tumor suppressor in hepatocellular carcinoma. Gastroenterology, 134(5), 1521–1531. 10.1053/j.gastro.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BK, Chen D, Su ZZ, Gredler R, Yoo J, Shah K, et al. (2010). Molecular mechanism of chemoresistance by astrocyte elevated gene-1. Cancer Research, 70(8), 3249–3258. 10.1158/0008-5472.CAN-09-4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BK, Emdad L, Gredler R, Fuller C, Dumur CI, Jones KH, et al. (2010). Transcription factor late SV40 factor (LSF) functions as an oncogene in hepatocellular carcinoma. Proceedings of the National Academy of Sciences of the United States of America, 107(18), 8357–8362. 10.1073/pnas.1000374107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BK, Emdad L, Lee SG, Su ZZ, Santhekadur P, Chen D, et al. (2011). Astrocyte elevated gene-1 (AEG-1): A multifunctional regulator of normal and abnormal physiology. Pharmacology & Therapeutics, 130(1), 1–8. 10.1016/j.pharmthera.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BK, Emdad L, Su ZZ, Villanueva A, Chiang DY, Mukhopadhyay ND, et al. (2009). Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. The Journal of Clinical Investigation, 119(3), 465–477. 10.1172/JCI36460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BK, Gredler R, Chen D, Santhekadur PK, Fisher PB, & Sarkar D (2011). C-met activation through a novel pathway involving osteopontin mediates oncogenesis by the transcription factor LSF. Journal of Hepatology, 55(6), 1317–1324. 10.1016/j.jhep.2011.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BK, Gredler R, Vozhilla N, Su ZZ, Chen D, Forcier T, et al. (2009). Identification of genes conferring resistance to 5-fluorouracil. Proceedings of the National Academy of Sciences of the United States of America, 106(31), 12938–12943. 10.1073/pnas.0901451106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BK, Santhekadur PK, Gredler R, Chen D, Emdad L, Bhutia S, et al. (2011). Increased RNA-induced silencing complex (RISC) activity contributes to hepatocellular carcinoma. Hepatology, 53(5), 1538–1548. 10.1002/hep.24216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen MF, Chen DS, Dusheiko GM, Janssen HLA, Lau DTY, Locarnini SA, et al. (2018). Hepatitis B virus infection. Nature Reviews. Disease Primers, 4, 18035. 10.1038/nrdp.2018.35. [DOI] [PubMed] [Google Scholar]
- Zhang W, Bi Y, Li J, Peng F, Li H, Li C, et al. (2017). Long noncoding RNA FTX is upregulated in gliomas and promotes proliferation and invasion of glioma cells by negatively regulating miR-342–3p. Laboratory Investigation, 97(4), 447–457. 10.1038/labinvest.2016.152. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zou C, Qiu Z, E F, Li Q, Chen M, et al. (2020). CPEB3-mediated MTDH mRNA translational suppression restrains hepatocellular carcinoma progression. Cell Death & Disease, 11(9), 792. 10.1038/s41419-020-02984-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Moran MS, Yang Q, Liu Q, Yuan C, Hong S, et al. (2012). Metadherin regulates radioresistance in cervical cancer cells. Oncology Reports, 27(5), 1520–1526. 10.3892/or.2012.1692. [DOI] [PubMed] [Google Scholar]
- Zhao J, Wang W, Huang Y, Wu J, Chen M, Cui P, et al. (2014). HBx elevates oncoprotein AEG-1 expression to promote cell migration by downregulating miR-375 and miR-136 in malignant hepatocytes. DNA and Cell Biology, 33(10), 715–722. 10.1089/dna.2014.2376. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Deng H, Yan W, Luo M, Tu W, Xia Y, et al. (2014). AEG-1 promotes anoikis resistance and orientation chemotaxis in hepatocellular carcinoma cells. PLoS One, 9(6), e100372. 10.1371/journal.pone.0100372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YY, Zhu GQ, Liu T, Zheng JN, Cheng Z, Zou TT, et al. (2016). Systematic review with network meta-analysis: Antidiabetic medication and risk of hepatocellular carcinoma. Scientific Reports, 6, 33743. 10.1038/srep33743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Dai Z, Pan Q, Wang Z, Yang GH, Yu L, et al. (2011). Metadherin promotes hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clinical Cancer Research, 17(23), 7294–7302. 10.1158/1078-0432.CCR-11-1327. [DOI] [PubMed] [Google Scholar]
- Zhu AX, Kang YK, Yen CJ, Finn RS, Galle PR, Llovet JM, et al. (2019). Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased alpha-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology, 20(2), 282–296. 10.1016/S1470-2045(18)30937-9. [DOI] [PubMed] [Google Scholar]
- Zhu HD, Liu L, Deng H, Li ZB, Sheng JQ, He XX, et al. (2020). Astrocyte elevated gene 1 (AEG-1) promotes anoikis resistance and metastasis by inducing autophagy in hepatocellular carcinoma. Journal of Cellular Physiology, 235(6), 5084–5095. 10.1002/jcp.29377. [DOI] [PubMed] [Google Scholar]
- Zhu K, Pan Q, Jia LQ, Dai Z, Ke AW, Zeng HY, et al. (2014). MiR-302c inhibits tumor growth of hepatocellular carcinoma by suppressing the endothelial-mesenchymal transition of endothelial cells. Scientific Reports, 4, 5524. 10.1038/srep05524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Peng Y, Hu J, Zhan H, Yang L, Gao Q, et al. (2020). Metadherin-PRMT5 complex enhances the metastasis of hepatocellular carcinoma through the WNT-beta-catenin signaling pathway. Carcinogenesis, 41(2), 130–138. 10.1093/carcin/bgz065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimonjic DB, Keck CL, Thorgeirsson SS, & Popescu NC (1999). Novel recurrent genetic imbalances in human hepatocellular carcinoma cell lines identified by comparative genomic hybridization. Hepatology, 29(4), 1208–1214. 10.1002/hep.510290410. [DOI] [PubMed] [Google Scholar]