

Abstract

Direct photocatalyzed hydrogen atom transfer (d-HAT) can be considered a method of choice for the elaboration of aliphatic C–H bonds. In this manifold, a photocatalyst (PCHAT) exploits the energy of a photon to trigger the homolytic cleavage of such bonds in organic compounds. Selective C–H bond elaboration may be achieved by a judicious choice of the hydrogen abstractor (key parameters are the electronic character and the molecular structure), as well as reaction additives. Different are the classes of PCsHAT available, including aromatic ketones, xanthene dyes (Eosin Y), polyoxometalates, uranyl salts, a metal-oxo porphyrin and a tris(amino)cyclopropenium radical dication. The processes (mainly C–C bond formation) are in most cases carried out under mild conditions with the help of visible light. The aim of this review is to offer a comprehensive survey of the synthetic applications of photocatalyzed d-HAT.
1. Introduction
The selective manipulation of C–H bonds (especially C(sp3)–H bonds) represents a remarkable challenge in synthetic campaigns because organic molecules contain many of these bonds of different nature. The acidity of hydrogens in the proximity of electron-withdrawing groups has been largely exploited for the smooth generation of enolates, versatile nucleophiles to forge C–C bonds.1,2 Apart from this fundamental reactivity pattern, aliphatic C–H bonds have been referred to as “unfunctional groups”3 due to their lack of reactivity and the more articulated strategies needed for their functionalization.4 Accordingly, the vast majority of C–H bond activation strategies in organic synthesis rely on the use of activating or directing groups, either to enable a particular reaction pathway or to improve selectivity as well as efficiency.5 Even though the use of temporary directing groups, that is functions that are reversibly bound to the substrate to drive selectivity, has been proposed,6 the direct aliphatic C–H bond elaboration in organic molecules still remains the unfound Holy Grail in chemistry.3,7−12 Notably, this is an intense area of research, because it is a godsend for late-stage functionalization13−17 and in function-oriented synthesis18 thanks to the innate atom-economy related to the direct elaboration of C–H bonds. Moreover, the selective activation of these bonds in structurally complex molecules is of immense value in medicinal chemistry,14 where small changes in a given structure may have a profound impact on its biological activity and in natural product synthesis.19,20
To address this challenging task, different metal-based strategies4,19,21−24 have been devised: in particular, Fe-,25,26 Cu-,27 Mn-,28−30 Co-,31,32 Rh-,33 Ir-,34,35 Ru-,36−38 and Pd-based39−43 catalysts have been successfully tested. Within this frame, one of the most appealing concepts consists in the homolytic cleavage of the C–H bond via a hydrogen atom transfer (HAT) event.44−46 This consists in the concerted movement of an electron and a proton (H• ≡ H+ + e–) from the substrate, aka hydrogen donor, to an accepting species (a hydrogen abstractor); all in a single kinetic step (Scheme 1).
Scheme 1. Homolytic Cleavage of a C–H Bond via a Hydrogen Atom Transfer Step.

The factors that rule this chemistry (and in general the approaches devoted to design a selective HAT) may be tentatively classified as depending on the substrate or on the hydrogen abstractor structures as well as “medium dependent”, as summarized in Scheme 2.
Scheme 2. Factors Affecting C–H Bond Cleavage.
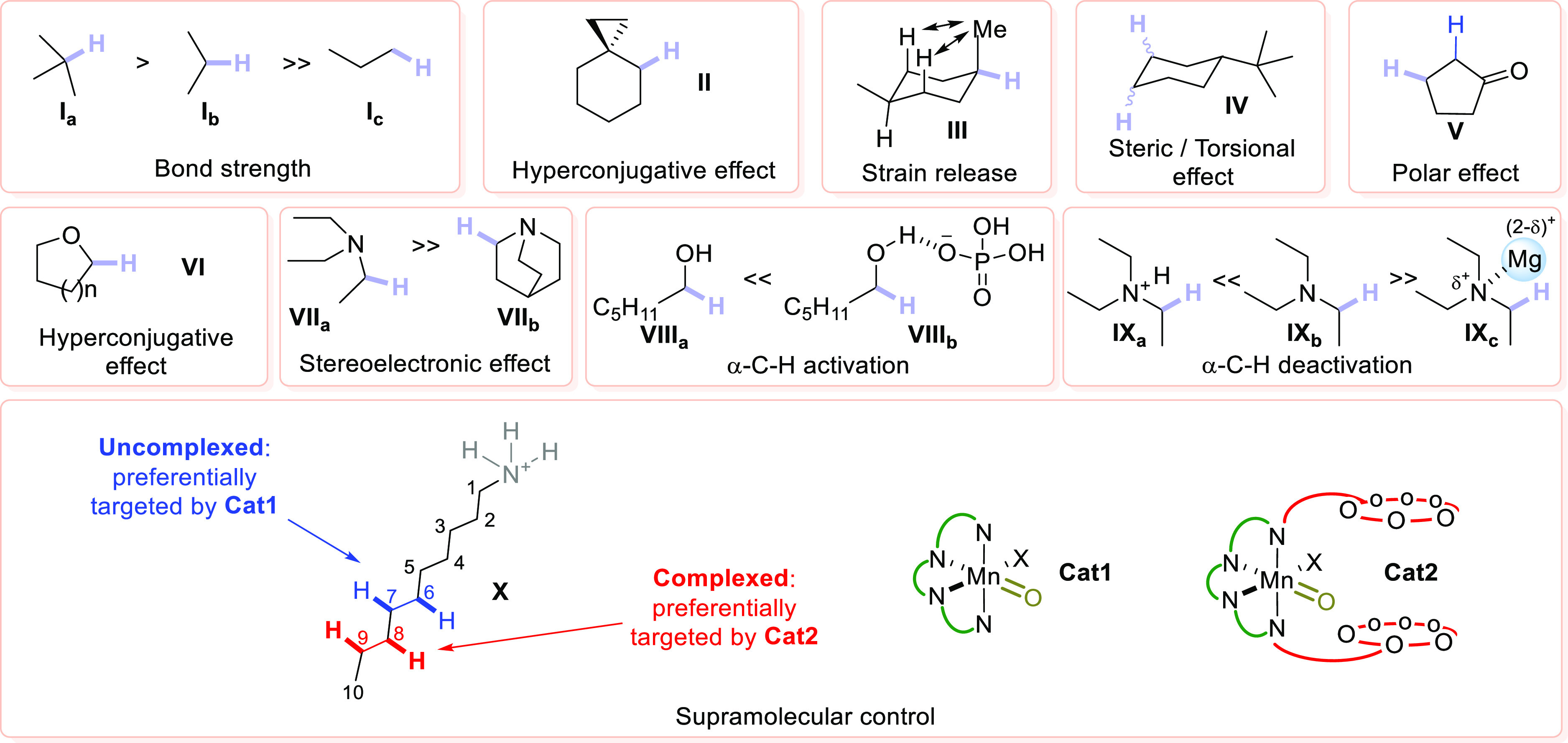
Different factors operating in the selective HAT-based C–H functionalization in organic compounds. In violet the reactive site where the hydrogen is preferentially cleaved during the functionalization (for the explanation of the effects, see the text).
One of the main effects belonging to the former class is the bond dissociation energy (BDE) of the C–H bond to be cleaved. In other words, the lower the BDE and the more stable the generated radical, the easier the bond to break (Scheme 3). However, this is just a rule of thumb and applies only under certain conditions. More often, other factors must be carefully evaluated to account for the difference in selectivity observed in the derivatization of a certain substrate (Scheme 2).47−52
Scheme 3. Bond Dissociation Energies (BDEs) in kcal/mol of Representative Compounds.
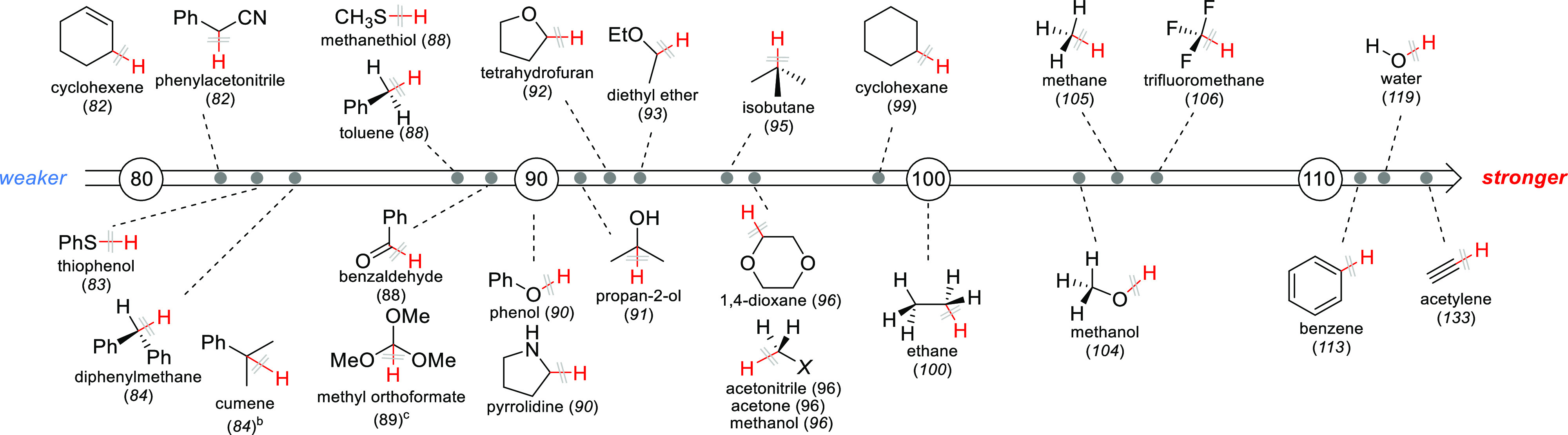
Bond dissociation energies (BDEs) in kcal/mol of the X–H bonds (in red) in representative compounds. Values taken from ref (53) except where otherwise noted.
Value taken from ref (54).
Value taken from ref (55).
Taking the case of hydrocarbons as reference, the relative stability of the generated organoradical drives the cleavage of a tertiary C–H bond (e.g., in compound Ia) over that of a secondary (as in Ib) or a primary C–H bond (in Ic). Also, hyperconjugation and conjugation play an important role in steering the selectivity of HAT. For example, hyperconjugative effects may operate in the case of a hydrocarbon containing a cyclopropyl ring (II). In this case, the overlap between the C–C σ bonding orbital of the three membered ring and the neighboring C–H σ* antibonding orbital directs the C–H activation to the vicinal position (Scheme 2).56−59 In some instances, the strain release connected with the cleavage of a C–H bond is another driving force for a selective process. Indeed, this factor is rather significant for six-membered carbocyclic structures (see the case of compound III). In substituted cyclohexanes, the hydrogen abstraction from the equatorial C–H bond that is adjacent to a bulkier axial group leads to the release of 1,3-diaxial strain thanks to the planarization of the incipient carbon centered radical in the HAT transition state.56,60−76
While depending also on the nature/identity of the hydrogen abstractor, steric hindrance plays an important role as well, as known for the reactivity of tert-butylcyclohexane IV.77 This is another nice example of a “substrate-dependent” effect where the geometry of the molecule forces the activation toward precise reaction sites (e.g., positions 3 and 4) due to simple steric/torsional effects.75,77
Turning to substrates containing heteroatoms (e.g., O, N, etc.), the presence of polar functionalities may influence neighboring C–H bonds and their reactivity. Notably, the polarity match (or mismatch) between the character of the C–H bond to be cleaved and the hydrogen abstractor is another factor that hugely affects HAT. For example, an electrophilic species (e.g., an alkoxy radical or a related derivative) strongly prefers to abstract an hydridic (nucleophilic) C–H bond rather than an electron-poor (electrophilic) one of similar strength, a feature that is directly linked to the electronic character of the accessible radical intermediate.78 This allows the use of solvents having (rather labile) electrophilic C–H bonds (e.g., acetonitrile, acetone, Scheme 3), when an electrophilic hydrogen abstractor operates in the elaboration of nucleophilic C–H bonds.45,79−82
The polarity of C–H bonds in the substrate is thus influenced by the presence of electron-withdrawing or electron-donating groups.56,60−66 This is apparent in the case of cyclopentanone V, where the more labile electrophilic α-hydrogens are not activated by an electrophilic hydrogen abstractor since a partial positive charge on the incipient C-centered radical makes the corresponding transition state unfavorable. As a result, the β-C–H cleavage occurs instead.83
Another common “substrate-depending” effect governing HAT is that exerted by electron-donating functionalities, notably oxygen and nitrogen atoms. The donation of the nonbonding electrons by these atoms activates vicinal C–H bonds through hyperconjugation. Typical is the case of ethers (e.g., VI), acetals, alcohols, amines, and amides, where the heteroatom causes the decrease of the BDE values of the vicinal C–H bond via hyperconjugation and stabilizes the corresponding radical intermediate (see Scheme 2). Furthermore, the presence of heteroatoms may influence the selectivity via stereoelectronic effects,56,60−66,84 which allows rationalization of the different reactivities of open chain vs cyclic derivatives. By considering the cleavage of the α-to-N C–H bond in amines VIIa and VIIb as a representative example, the hydrogen abstraction in VIIa is more effective than in VIIb. In fact, the process is more efficient when the bond being broken can be eclipsed with the heteroatom lone pair, not a favorable situation in VIIb due to the rigidity of the molecular scaffold.
Alternatively, “medium-dependent” effects (Scheme 2) can tune the reactivity pattern in chemical transformations occurring via HAT, again altering the reactivity of substrates containing heteroatoms through the activation or deactivation of the α-C–H bonds. Indeed, the solvent itself may function as hydrogen bond donor or acceptor due to its acidity or basicity;85−89 albeit, the presence of additives with peculiar acid/base properties may have a similar role.90−106 The activation effect is well illustrated by the selective C–H functionalization occurring in alcohol VIIIa. Tetrabutylammonium dihydrogen phosphate forms a complex with the substrate via hydrogen bonding with the alcoholic O–H bond, thus increasing the n−σ* delocalization of the oxygen lone pair and making the α-to-O C–H bond more prone to a HAT process.90 On the other hand, a deactivation effect can be induced by an acidic solvent (e.g., a fluorinated alcohol) or by the addition of a Lewis or a Brønsted acid.56,60−66,107−110 Accordingly, both in the protonated form (IXa) and in the complexed form (IXc) the α-to-N C–H bond of triethylamine (IXb) is less prone to be cleaved due to the reduced availability of the nitrogen lone pair, and in some instances the selectivity is shifted to the β- (or, in general terms, remote) C–H bonds.111
Quite recently, supramolecular chemistry has been exploited to induce chemoselectivity in HAT-based processes.112−114 The ammonium salt X is functionalized by a Mn-oxo complex (Cat1) preferentially at C6 and C7. By employing a more sophisticated catalyst (Cat2) bearing two crown ether moieties able to complex the ammonium salt, it was possible to shift the functionalization toward C8 and C9.
Upon suitable conditions, a moiety embedded in the molecular scaffold can be activated and triggers the hydrogen abstraction at a specific site in an intramolecular fashion thus inducing a remote activation of a C–H bond (r-HAT).41,115−122 Typically, such site-selectivity is granted by the formation of a favorable six-membered cyclic transition state, which results in the occurrence of a 1,5-HAT step, despite the fact that the 1,n-HAT mode (n ≥ 6) may compete in some cases.123
Given the above, a synthetic route based on HAT has to be judiciously planned, starting from the choice of the proper hydrogen abstractor X• (Scheme 4).124 Thermodynamics-wise, the newly formed X–H bond has to be stronger than the C–H bond to cleave to provide the driving force for the overall process, despite BDE not being the only parameter to be considered. As shown in Scheme 3, the BDE of α-to-heteroatom C–H bonds is mostly comprised between 85 and 95 kcal/mol, while primary and secondary C–H bonds in aliphatic hydrocarbons are quite strong (ca. 100 kcal/mol) calling for a highly reactive species (X•) to promote the cleavage event. Different hydrogen abstractors are known to have a radical or a radical-like character, including alkoxyl,122,125−127 aminoxyl,62,128 amidyl,129,130 and sulfonamidyl,131 azidyl,132 iodanyl,133 thiyl,134−138 and even C-centered116,139−142 radicals or halogen atoms,143,144 amine radical cations,90,117,145−147N-ammonium ylides,148 dioxiranes,59,70,149,150 or metal–oxo complexes.29,151 These hydrogen abstractors may be thermally or photochemically generated.
Scheme 4. Common Hydrogen Abstractors Used in Synthetic Planning.

Recently, photocatalysis has emerged as a powerful synthetic platform in organic chemistry because it allows taming the tremendous amount of energy associated with light to build molecular complexity. It relies on the use of chemical species, namely photocatalysts (PCs), that can convert light into chemical energy for substrate activation.152−180
This methodology has been used to trigger HAT and, in particular, all the reports that appeared so far can be classified in two approaches: indirect hydrogen atom transfer (i-HAT) and direct hydrogen atom transfer (d-HAT, Scheme 5).
Scheme 5. Photocatalyzed Indirect Hydrogen Atom Transfer (i-HAT) vs Direct Hydrogen Atom Transfer (d-HAT).

In the former case, the PC (PCSET) takes care of absorbing light and, once in the excited state, generates the hydrogen abstractor (X•, a radical or radical ion species) via a single-electron transfer (SET) step (Scheme 5, left).181−184 In the d-HAT process, the PC (PCHAT) triggers directly the HAT when in the excited state (Scheme 5, right).181−185 In other words, PC*HAT coincides with X•.
A common structural motif to the vast majoriy of PCsHAT currently known is the presence of an oxo group (Z=O), which acquires a peculiar O-centered radical character in the reactive excited state. The structure of the excited PC*HAT strictly resembles electrophilic alkoxyl radicals (Scheme 5) behaving as excellent hydrogen abstractors to cleave a C–H bond in the chosen substrate. This leads to the formation of the (protonated) reduced form of the PC (PC•-H). At each catalytic cycle, the spent PC must be recovered back to its original state, so that it can promote over and over again the process, according to the definition of “photocatalyst” offered by the IUPAC: “Catalyst able to produce, upon absorption of light, chemical transformations of the reaction partners. The excited state of the photocatalyst repeatedly interacts with the reaction partners forming reaction intermediates and regenerates itself after each cycle of such interactions”.186 The actual mechanism of the PC restoration depends on the synthetic application and can involve a back-HAT step or a sequential electron/proton transfer (ET/PT) mechanism toward a chemical species (Y, Scheme 5) present in the reaction mixture (e.g., a sacrificial hydrogen acceptor) or transiently formed during the process, also dictating the overall redox balance of the synthetic transformation.187
Depending on the X-element carrying the oxo moiety, PCsHAT can be grouped within different families (Figure 1). These comprise the class of carbonyl derivatives (Z = C),188,189 encompassing simple (aromatic) ketones and aldehydes,190−192 α-diketones,193 α-ketoacids,194 and (anthra)quinones,195−197 as well as the xanthene dye Eosin Y.198,199 On the other hand, inorganic derivatives including the decatungstate anion [W10O32]4– (Z = W)64,200−205 and the uranyl cation [UO2]2+ (Z = U)206,207 as well as antimony oxo porphyrin complexes (Z = Sb)208 have been likewise proposed as PCsHAT. A notable exception of PCHAT lacking the oxo group is known, namely the electrogenerated tris(amino)cyclopropenium (TAC) radical dication.209
Figure 1.
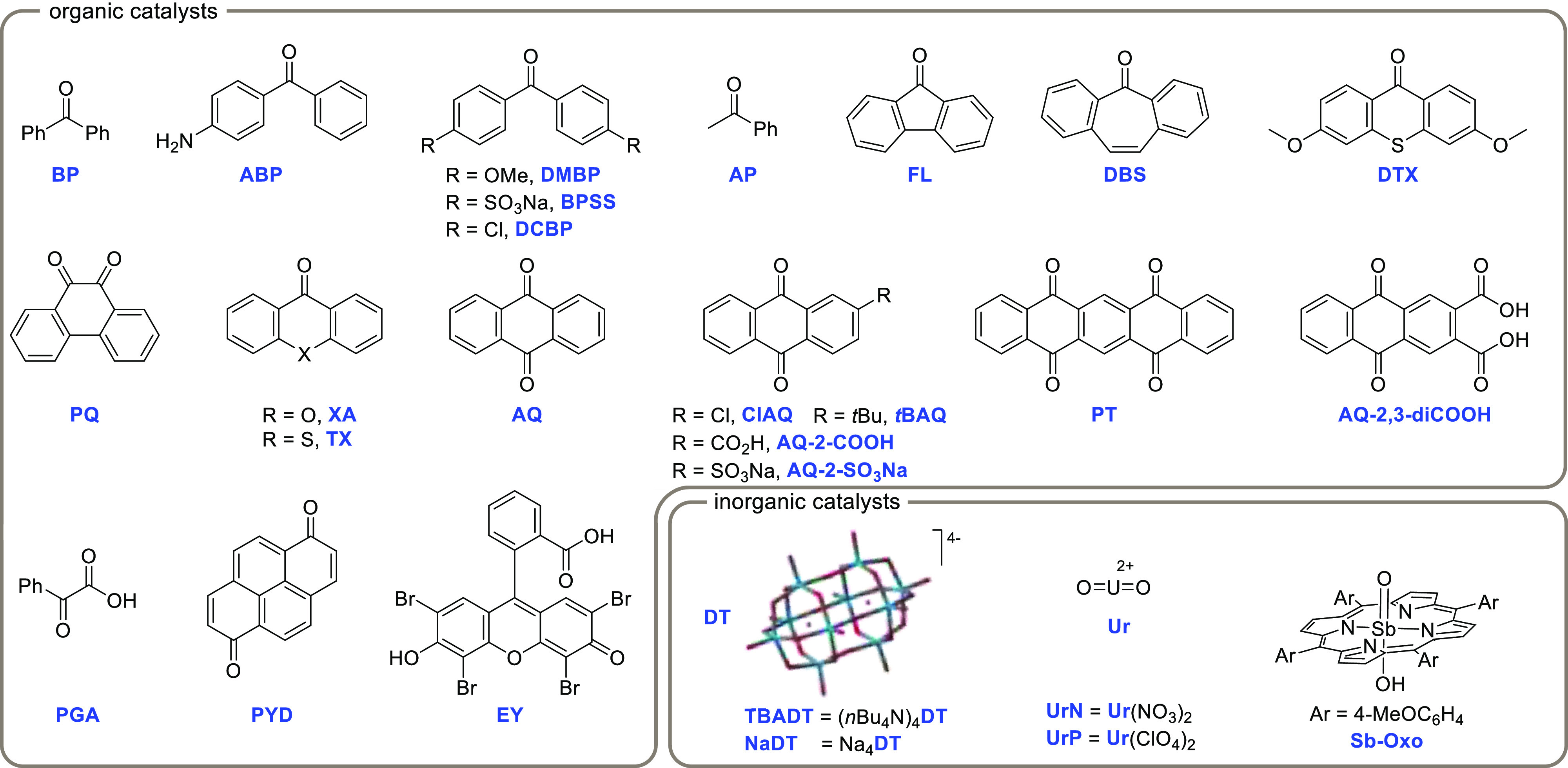
Main photocatalysts (PCsHAT) used in photocatalyzed HAT: BP, benzophenone; ABP, aminobenzophenone; DMBP, 4,4′-dimethoxybenzophenone; BPSS, disodium benzophenonedisulfonate; DCBP, 4,4′-dichlorobenzophenone; AP, acetophenone; FL, 9-fluorenone; DBS, dibenzosuberenone; DTX, 3,6-dimethoxy-9H-thioxanthen-9-one; PQ, 9,10-phenanthrenequinone; XA, xanthone; TX, thioxanthone; AQ, anthraquinone; ClAQ, 2-chloroanthraquinone; tBAQ, 2-tert-butylanthraquinone; AQ-2-COOH, anthraquinone-2-carboxylic acid; AQ-2-SO3Na, anthraquinone-2-sulfonic acid sodium salt; PT, 5,7,12,14-pentacenetetrone; AQ-2,3-diCOOH, anthraquinone-2,3-dicarboxylic acid; PGA, phenylglyoxylic acid; PYD, 1,6-pyrenedione; EY, Eosin Y; DT, decatungstate anion (TBADT, tetrabutylammonium decatungstate; NaDT, sodium decatungstate); Ur, uranyl cation (UrN, uranyl nitrate hexahydrate; UrP, uranyl perchlorate); Sb-Oxo, antimony-oxo tetra-(p-methoxyphenyl)porphyrin.
Apart from structural similarities, the behavior of excited PCsHAT featuring an oxo moiety shows many analogies. Thus, the capability of carbonyl derivatives, especially (aromatic) ketones, to act as hydrogen abstractor has been known for a long time.296−301 In particular, the photochemistry of these compounds is dominated by the triplet excited state and this is due to the very efficient intersystem crossing (ISC) from the first-formed singlet state. These triplet states have a lifetime in the microsecond range302 (Table 1) and, depending on the structure of the carbonyl derivative, may have a nπ* or a ππ* character (the former typically accountable for HAT reactivity).296,303−309 In carbonyl derivatives, the PC•-H form is a stable, long-living ketyl radical species (Scheme 5),310 featuring a very weak O–H bond (the calculated BDE of the O–H bond of the ketyl radical derived from acetone is ca. 16 kcal/mol).311 Accordingly, the restoration of the carbonyl group is the driving force for the recovery of the spent photocatalyst. However, a common drawback when using ketones as PCsHAT is that ketyl radical may dimerize in solution to form pinacols thus decreasing the efficiency of the reaction.312 A particular case is that of the excited carbonyl of EY that has some phenoxyl radical character.220
Table 1. Photophysical Properties of Selected PCsHAT.
| Photocatalyst | λmax (nm) | λuse (nm) | ΦISC | τT (μs) |
|---|---|---|---|---|
| AP | 243, 279, 315 (THF or MeCN)253 | 19 W CFL224 | ∼1 (computational)221 | 6.6 (H2O/MeCN 9:1)210 |
| ABP | 337 (i-PrOH),254 303 (C6H12),254 308 (CCl4)254 | 365, UV LED225 | 0.82 (C6H12)211 | 3.6211 |
| AQ | 325, 370 (DCM)255 | 360,226 390, 427227 | 0.95 (MeCN)212 | 1.62 (MeCN)212 |
| BP | 347, 415256 | 350,228 Hg lamp,229−232 350 Rayonet,233 366 Rayonet,234 365 LED lamp,235 18 W CFL,236 400 nm LED237 | 1 (MeCN)213 | 19.6 (MeCN)213 |
| BPSS | 330214 | 315, 366238 sunlight239 | 1 (H2O)214 | 352 (H2O)214 |
| ClAQ | 256, 265, 274, 325 (MeOH)257 | 365 LED lamp,240 Xe lamp241 | 1150 (CF3CH2OH at 77 K)215 | |
| DMBP | 339258 | 365 UV fluorescent lamp242 | 3.4–17.6 (CH2Cl2/CHCl3 1:1 at 77 K)216 | |
| DBS | 251, 306, 347 (C6H12),259 253, 316, 326 (EtOAc),259 253, 306, 347 (MeCN),259 255, 305, 348 (i-PrOH),259 255, 306, 349 (MeOH)259 | CFL243 | 980 (C6H12, Ar),217 890 (EtOH, Ar),217 740 (MeOH, Ar),217 650 (C6H6, Ar)217 | |
| PQ | 268, 324, 500 (CHCl3),260 319, 312, 503 (C6H6)261 | Blue LED strip244 | 1222 | 2.2218 |
| PGA | 350 (dioxane)262 | household lamps245 | ∼1194 | 8194 |
| DTX | 354 (MeCN)219 | 365 LED lamp246,247 | 0.93219 | 862 (MeCN)219 |
| EY | 541 (DMF)263 | Blue LED,248,249 460 Blue LED,250 470 blue LED,251 520 Green LED,250 Xe lamp (400 nm cutoff)252 | 0.32223 | ∼21220 |
| FL | 382 (C6H12),287 380 (C6H6),287 377 (MeCN),287 379 (EtOH)287 | Xe lamp,271 CFL lamp272 | 0.97 (MeCN)213 | 70 (MeCN)213 |
| NaDT/TBADT | 260, 323 (H2O),273 272, 321288 | BLB lamp,273 310 Multilamp reactor,274 366 LED,275 Xe lamp,271 Solar light,276 390 LED277 | 0.5 (MeCN)264 | 50 (MeCN)264 |
| PT | 270, 343 (CHCl3)289 | 365, 425,278 390, 427,279,280 455281 | ||
| PYD | 427 (MeCN/THF 1:1),290 266, 277, 398, 424, 448 (ether)291 | Blue LED282 | ||
| Sb-Oxo | 440265 | 405, 455265 | 8 (MeCN/H2O 95:5)265 | |
| TX | 380219 | 405 LED271 | 0.99 (MeCN),213 0.76267 | 28 (MeCN),213,266 760 ns (CD3CN),219 45 (MeCN)267 |
| UrN | 414292−294 | 456,283 Blue LED284 | 1270 | 400 (80 K, MeOH)268 |
| XA | 338295 | 365 LED,285 CFL272,286 | 1 (MeCN)213 | 4.8–8.3 (MeCN)213,269 |
Another deeply studied family of PCsHAT is that of inorganic polyoxometalate (POM) derivatives. The first reports describing a HAT reactivity upon excitation of these metal–oxygen clusters in the presence of organic substrates (mostly alcohols) dates back to the 1980s.313−317 It was soon realized that tungsten-based POMs, in particular DT, outperform all the other known POMs in terms of HAT reactivity,200,318 offering a catalytic tool for the elaboration of C–H bonds.319−330 A common occurrence in photocatalytic systems based on DT is the observation of a typical blue color of the PC•-H form ([W10O32]5–, either protonated or not)331 that accumulates in solution.332 The studies of DT-based systems by means of time-resolved spectroscopic techniques333−339 concluded that the state responsible for the HAT reactivity is a relaxed excited state (“wO” having a lifetime of 55 ns in acetonitrile),333 probably of triplet multiplicity,340 not directly accessible upon excitation (a so-called dark state).341,342 Theoretical simulations supported these experimental spectroscopic works.264,343,344
Turning to the uranyl cation, despite the very weak absorption in the blue region of the spectrum (ε ∼ 10 M–1 cm–1 at λ = 423 nm), visible light irradiation can be adopted to trigger its photochemistry. This transition has been proposed to populate a long-lived (μs range lifetime) state which contains an extremely reactive oxyl radical, well explaining the HAT reactivity.207,270,345−349
A partial oxyl radical character of the triplet excited state has been likewise postulated to be the active species in the hydrogen abstraction operated by Sb-Oxo.265 In the latter case, SbV (a high-valent oxidation state element of the p-block) is used in the dihydroxo form that contains two hydroxyl groups in the axial positions. Upon treatment with a base, one of the two hydroxy groups turns into the desired oxo moiety; the excited state of the so-generated oxo species was exploited for hydrogen abstraction.265,350 Even in this case the lifetime of the triplet involved is in the microsecond range (Table 1).
It is important to stress, however, that PCsHAT may be engaged in photocatalytic processes different from HAT (mainly electron transfer, but energy transfer may not be excluded).185,265,309,351 Accordingly, the real mechanism has to be checked carefully to ascertain if a HAT process is involved rather than an electron transfer followed by proton transfer or even a proton-coupled electron transfer (PCET) mechanism.162
In view of the above, the aim of the present review is to offer an overview of the synthetic applications based on photocatalyzed direct HAT (d-HAT), wherein the excited PCHAT triggers the HAT step. On the other hand, examples dealing with a photocatalyzed indirect HAT (i-HAT)181−184 or remote HAT (r-HAT)183 as well as the activation of C–H bonds via a PCET mechanism352 will be excluded. The threshold that we used throughout the entire work to consider an approach photocatalytic is 20 mol % of catalyst loading. We then considered photocatalytic HAT reactions where the generated radical must be incorporated in the desired compound, so the photogeneration of a thermally active redox agent will not be treated here.290,353−355 Photoinitiated processes wherein the light-absorbing species undergoes degradation during the process have been likewise excluded.356−358
Similarly, the adoption of a d-HAT strategy in polymerizations will not be mentioned; however, the reader is invited to refer to seminal works in the field.359−364 Thus, synthetic applications are preferentially treated here avoiding (when possible) the works simply devoted to mechanistic purposes and where the PCHAT tested gave a very low yield or a very low reagent consumption.
The following sections have been organized based on the bond being formed during the transformation and found in the final product, while different types of transformations (e.g., dehydrogenation and fragmentation reactions) have been reported in the final part of the review. Thus, under the section “formation of a C(sp3)–C(sp2) bond”, examples wherein a C(sp3)-centered radical (formed from the photocatalyzed homolysis of a C(sp3)–H bond via a HAT step) will be attached to a C(sp2) atom in the final product will be described. Moreover, despite the fact that most of the examples reported in this review deal with the functionalization of C–H bonds, the elaboration of P–H, Si–H, and S–H bonds via a photocatalyzed d-HAT step has been mentioned, for the sake of comprehensiveness.
All the schemes have been color-coded so that the bond activated via HAT has been reported in violet, while the bond formed has been highlighted in red.
2. Formation of C–C Bonds
2.1. Formation of C(sp3)–C(sp3) Bonds via Addition onto C=C Bonds
A typical reactivity mode that can be exploited to forge a C(sp3)–C(sp3) bond is the radical addition of nucleophilic radicals onto Michael acceptors. In this scenario, the C-centered radical generated via photocatalyzed HAT is trapped by an electrophilic olefin and the resulting radical adduct is quenched via back hydrogen atom transfer (or sequential electron/proton transfer) from the reduced form of the photocatalyst (PC•–H; see Scheme 5), thus closing the photocatalytic cycle. This is a very reliable and general protocol as also demonstrated by the vast amount of hydrogen donors that can be profitably employed, with notable examples including alcohols, ethers, dioxolanes, sulfides, amides, nitriles, as well as simple hydrocarbons such as toluenes, allylated derivatives, and even (cyclo)alkanes.
2.1.1. Oxygen- and Sulfur-Containing Compounds as Hydrogen Donors
Oxygenated derivatives have been the elective substrates for this reactivity manifold since the earliest reports on photocatalyzed HAT; this is because of the low BDE of the α-to-O C–H bond and the relatively stable α-oxy radical generated. In particular, to the best of our knowledge, alcohols are the first hydrogen donors ever investigated,365 and the earliest preparative example appeared in 1957 dealing with the photoaddition of isopropanol onto maleic acid 6.1 to give terebic acid 6.2 (Scheme 6).366 In those days, aromatic ketones (e.g., BP) were the elective class of PCsHAT to perform the reaction under UV light coming from a Hg lamp.366 Due to the low cost of isopropanol, this was used as the reaction medium. The mechanism is depicted in Scheme 6 (lower part) and, as mentioned in the introduction to this section, it is quite common in all cases wherein a radical Michael addition takes place. Thus, the excited PCHAT abstracts a hydrogen atom from the hydrogen donor and the resulting radical adds onto the olefin to give the radical adduct 6.3. Back hydrogen atom transfer from the reduced form of the PC to 6.3 yielded the hydroxy acid 6.4 (and 6.2 from it by spontaneous lactonization) with the concomitant regeneration of PCHAT.
Scheme 6. Photoaddition of Isopropanol onto Maleic Acid.

The same reaction was later on replicated on a gram scale by using the SOLFIN (SOLar synthesis of FINe chemicals) apparatus as solar light concentrator placed in Almeria (Spain).239 In this case, BPSS was used in the role of PCHAT, which was synthesized by sulfonation of parent BP (the sulfonation took place both at the 3- and 4-positions of the aromatic rings). The reason was that the thus-obtained PCHAT was easily removed at the end of the reaction by extraction with water. Thus, ca. 14 g of terebic acid (6.2) was isolated in 75% yield upon 14 h solar light irradiation of an isopropanol/water 1:1 solution of 6.1 in the presence of 10 mol % BPSS.239 This PCHAT has been likewise used to trigger the addition of alcohols (isopropanol, ethanol, and methanol) onto α,β-unsaturated aldehydes for the preparation of γ-lactols and γ-lactones upon treatment of the crude lactols with bromine.238 When maleic or fumaric acids were converted to the corresponding chiral (−)-menthyl diesters, the BP (19 mol %) photocatalyzed addition of isopropanol gave the acyclic diaterebic acid ester (63% yield) with a modest degree (8%) of diastereoselectivity.367
Alcohols (in particular, methanol) were used to functionalize carbohydrate enones, such as hex-2-enopyranosid-4-ulose 7.1, to form branched-chain monosaccharides (Scheme 7).233,368−370 Irradiation of this α-enone in MeOH in the presence of BP afforded 1,4-ketoalcohol 7.2 in 66% yield. Interestingly, the incorporation of the alcohol took place from the less-hindered side of the enone in a complete stereo- and regioselective fashion.369
Scheme 7. Photocatalyzed Functionalization of an α-Enone.

Isopropanol was likewise used for the derivatization of 1,3-dioxin-4-ones having a (−)-menthone moiety embedded as chiral auxiliary in the 2-position (8.1, Scheme 8). The resulting 1,5-dioxaspiro[5.5]undecane-2-one (8.2) was formed, however, in a poor yield (<30% by using 15 mol % BP).371
Scheme 8. Photocatalyzed Synthesis of 1,5-Dioxaspiro[5.5]undecane-2-ones.

On the other hand, the adoption of TBADT allowed the activation of isopropanol even by using a low amount of the PCHAT (2–4 mol %) in the reaction with acrylonitrile (72% yield).81,372
The addition of isopropanol onto a Michael acceptor (e.g., furanone 9.1, Scheme 9) was likewise carried out under flow conditions373 by using either an LED-driven microchip reactor,374 a continuous-flow photoreactor with parallel capillaries,375 or a multimicrocapillary flow reactor.242 In all cases, the adduct 9.2 was formed in a less than 10 min irradiation. Of note, DMBP was found to be the best PCHAT among the several aromatic ketones tested.242
Scheme 9. Photocatalyzed Functionalization of Furanones under Flow Conditions.

Cyclic alcohols have been rarely used, but the methine hydrogen atom in cyclohexanol (10.1, Scheme 10) was selectively abstracted by the excited state of PT and the radical formed was engaged in an allylation reaction to give homoallyl alcohol 10.3.289
Scheme 10. Synthesis of Homoallyl Alcohols.

The lability of the C–H bonds in position 2- in (2-substituted) 1,3-dioxolanes has been exploited for the generation of dioxolan-2-yl radicals, which moiety was used to formally introduce a masked formyl group. The reaction was initially tested on α-enones similar to 7.1 by using 1,3-dioxolane as the solvent.233 Later on, the process was extended to other enones, such as 1-phenyl-2-propen-1-one or chalcone 11.1 (Scheme 11).226 In that case, AQ was adopted as a visible-light-absorbing photoorganocatalyst (POC)153 and, despite the long reaction time needed (ca. 60 h), the final adduct 11.2 was isolated in 85% yield. The same reaction described in Scheme 11 was also performed in a 3D-printed, chemically resistant, nonswelling, and UV–vis transparent postfunctionalized flow reactor by using ABP as an immobilized PCHAT; however, an unsatisfactory yield (13%) was reported.225
Scheme 11. Photocatalyzed Incorporation of a Masked Formyl Group.

The BP-photocatalyzed addition of 1,3-dioxolane onto 5-alkoxymethyl-2(5H)-furanone was used as the key step for the preparation of a bis-tetrahydrofuranyl ligand for HIV protease inhibitor UIC-94017 (TMC-114).376BPSS was likewise used as PCHAT to promote the radical addition of 1,3-dioxolane onto α,β-unsaturated aldehydes to give 1,4-monoprotected succinaldehydes upon solar light exposure.239 The radical 1,3-dioxolanylation of alkenoic acids was also performed by using DTX (10 mol %) as POC.247 Shifting to a metal-based PC, the adoption of UrN allowed the hydrogen activation in 1,3-dioxolane for the addition onto Michael acceptors.283
Along the same line, 2-alkyl-1,3-dioxolanes were exploited as hydrogen donors for the (formal) incorporation of a ketone moiety, but they had to be used as cosolvents. In such a way, 1,4-monoprotected ketoaldehydes were obtained upon radical alkylation of α,β-unsaturated aldehydes.238
TBADT was the elective PCHAT for the activation of the methylene hydrogens in substituted 1,3-benzodioxoles (e.g., 12.1) to give the corresponding 2-substituted derivatives by reaction with various Michael acceptors377 or with styrene378 (in the latter case in the presence of a disulfide cocatalyst). When the process was carried out on the β-substituted cyclic enone 12.2 (Scheme 12) in the presence of a chiral organocatalyst (i.e., carbazole derivative (S,S)-12.3), an enantioselective radical conjugate addition took place with formation of 12.4 in 99% yield. Notably, the latter product was formed with e.e. 88% and contains two quaternary carbon stereocenters.379 The reaction is based on an electron relay mechanism. In fact, the carbazole moiety is oxidized by an intramolecular electron transfer with the unstable radical cation formed by radical addition onto the chiral iminium ion intermediate, thus functioning as an electron donor.
Scheme 12. Dual-Catalytic Asymmetric Formation of Quaternary Carbons.

Another class of widely used oxygen-based hydrogen donors is that of cyclic ethers, wherein the HAT step occurs at the labile α-to-O C–H bonds. It is perhaps important to stress here that cyclic ethers cannot be easily activated otherwise; in fact, they are routinely used as inert solvents. Scheme 13 collects some representative examples concerning the derivatization of butendioate esters 13.1. TBADT enabled the facile cleavage of the C–H bond adjacent to the oxygen atom both in 1,4-dioxane 13.2(276) and in oxetane 13.4.380 In the former case, sunlight was effectively used to irradiate the solution poured in a glass vessel placed on a window ledge. Despite the long time required (4 days), the reaction did not make use of any external source of artificial energy.276 The same process was performed upon UV light irradiation under flow conditions in a shorter period.381
Scheme 13. Cyclic Ethers as H-Donors in the Functionalization of Butendioate Esters.

The generation of radicals from tetrahydrofuran (14.1) is useful to compare different PCsHAT in their role and to stress the versatility of the photocatalyzed HAT process (Scheme 14). Thus, 14.1 may be photoactivated by having recourse to several PCs, including aromatic ketones such as TX and FL under visible light LED irradiation,271TBADT under solar simulated conditions,271,382 as well as with Sb-Oxo,265UrN,283PYD,282EY,220 and ClAQ.240 In all cases, satisfactory yields of adducts 14.3, 14.5, 14.7, and 14.9 were obtained.
Scheme 14. Different PCsHAT for the Photocatalyzed Cleavage of the C–H Bond in THF.

The photocatalyzed addition of THF was also applied to quinones 15.1a–d (4-benzylidene-2,6-di-tert-butylcyclohexa-2,5-dien-1-ones, Scheme 15) under blue LED irradiation by using UrN (5 mol %) to give 2,6-di-tert-butyl-4-[phenyl(tetrahydrofuran-2-yl)methyl]phenols 15.2a–d.383
Scheme 15. Photocatalyzed Addition of THF onto Cyclohexa-2,5-dien-1-ones.

In rare instances, the activation of the C–H bond was applied to cyclic carbonates (16.1a,b, Scheme 16), where the presence of the carbonyl group did not hamper the C–H cleavage in these substrates. The importance of using TBADT is evident in this case, since the same process promoted by aromatic ketones gave no products 16.3a,b.271 Introducing a methyl group in carbonate 16.1b drove the cleavage to the most labile C–H bond present.
Scheme 16. TBADT-Mediated Derivatization of Cyclic Carbonates.

Apart from the case of carbonates, other carbonyl-containing derivatives (ketones, esters, lactones, etc.) can be used as hydrogen donors. However, it is important to remember that in these cases the selectivity of the HAT step is shifted toward remote positions due to the mismatched polarity (Scheme 2). A typical case is the photoactivation of cyclopentanones 17.1a,b (Scheme 17a). Despite the lability (and the acidity) of the α-C–H bonds with respect to β-C–H bonds in compound 17.1a, the former are left untouched under the action of TBADT and a selective β-C–H to C–C bond conversion occurred.83 A more favorable polar HAT transition state has been invoked in this case to rationalize the observed regioselectivity.83b The presence of a methyl group in compound 17.1b made the methine hydrogen sufficiently labile to allow the preparation of compound 17.3b as the sole product. Notably, the combination of polar and steric effects may direct the selective C–H cleavage in cycloalkanones and lactones. As an example, compound 17.4 underwent a selective β-C–H cleavage since both the hydrogen abstraction from the α-C–H and the γ-C–H bonds is prevented by polar and steric effects, respectively (Scheme 17b).384 In particular cases, the regioselective cleavage may be induced even in open chain esters (e.g., 17.7) exploiting the lability of the methine hydrogen of the isopropyl group and taking advantage of the bulkiness of the tBu group that prevents any other competitive C–H cleavage (Scheme 17c).384
Scheme 17. Selective C–H Cleavage in Ketones and Esters.

Sparse examples have been reported involving the use of sulfides (mainly cyclic derivatives) as hydrogen donors. Thus, tetrahydrothiophene 18.1 (or thioxane) was allylated at the C–H bond adjacent to the S-atom by reaction with allyl sulfone 18.2 (Scheme 18).289 Despite the easy oxidizability of these sulfides, the adoption of PT avoided any competitive electron transfer reaction. Additionally, both ClAQ(240) and EY(220) were likewise effective PCsHAT to trigger the C–H to C–C bond conversion in tetrahydrothiophene.
Scheme 18. Allylation of Tetrahydrothiophene.

2.1.2. Nitrogen-Containing Compounds as Hydrogen Donors
A widely used class of nitrogen-containing hydrogen donors is that of amides (often used as the solvent) and carbamates; albeit, often the PCHAT has to be used in a (super)stoichiometric amount for their activation.157 Nevertheless, catalytic amounts of TBADT smoothly promoted the C–H functionalization in amides and carbamates, used only in a 4 equiv excess (Scheme 19). The C–H bonds adjacent to the nitrogen atom in protected pyrrolidine 19.1 were sufficiently labile to be cleaved under photocatalyzed conditions to afford nitrile 19.3 (Scheme 19a), while excess 19.1 could be recovered during the purification.385 The reaction was found to be effective even under sunlight exposure.276 A similar C–H activation has been reported by using ClAQ (10 mol %)240 and PT (5 mol %)289 as PCsHAT.
Scheme 19. Amides, Carbamates, and Amines as H-Donors.

As an alternative to typical Michael acceptors, the α-amidoalkyl radical formed from dimethylformamide 19.4 in the presence of TBADT was trapped by vinyl pyridine 19.5 to give adduct 19.6 in a very high yield (82%, Scheme 19b).386 It is important to note that in the latter case no C–H cleavage of the formyl hydrogen competed (see further section 2.5).
A related C–H functionalization of carbamates to perform a Giese-type alkylation was carried out by combining the action of a POC (BP, 20% mol) with a catalytic amount of Cu(OAc)2 (2% mol) under UV-A irradiation. In this case, the copper species prevents the otherwise feasible polymerization of Michael acceptors, such as unsubstituted acrylates, acrylonitrile, or methyl vinyl ketone.387
Even EY may be used for the selective, photocatalyzed addition of acetamide onto benzylidenemalononitrile.220 When using UrN, the C–H cleavage in 19.4 was not selective since competitive hydrogen abstraction from the C(sp2)–H bond took place (ca. 1/3 ratio).283 When N-methylacetamide was subjected to a hydrogen abstraction reaction by using DCBP (20 mol %), the resulting α-amidoalkyl radical was trapped by β-phenyl allyl sulfone to give the corresponding allylated derivative.388
The TBADT-photocatalyzed addition of tertiary amides (e.g., 19.7, Scheme 19c) onto vinyl sulfones under flow conditions was selected for the easy preparation of γ-aminopropylsulfones (19.9).389 The latter conditions allowed scale-up of the process with a substrate concentration up to 0.5 M.389
In rare instances, an amine functioned as the hydrogen donor. Indeed, the electron-donor capability of such substrate may engage an electron transfer rather than a hydrogen atom transfer reaction (this is a typical case when using aromatic ketones).185,351 However, EY was able to functionalize amine 19.10 via enantioselective addition onto α,β-unsaturated N-acyl-3,5-dimethylpyrazole 19.11 (Scheme 19d).390 The asymmetric Giese-type addition of the photogenerated α-amino radical was promoted by the presence of the chiral rhodium Lewis acid catalyst Λ-RhS. As a result, adduct 19.12 was formed in a modest yield but with a high e.e..390
Similarly, in one instance a primary amine was derivatized under photocatalyzed conditions (Scheme 20). Thus, the visible light irradiation of a mixture of amine 20.1, a styrene (20.2a–d), and a catalytic amount of EY caused the C–H cleavage of the methine hydrogen in 20.1, finally affording the hoped-for 2-methyl-4-arylbutan-2-amine derivatives 20.3a–d.391 Apart from the mildness of the reaction conditions, this is an important example dealing with the derivatization of vinyl aromatics.391
Scheme 20. Photocatalyzed C(sp3)–H Alkylation of Amines.

A particular case in the activation of the C–H bond in amines is depicted in Scheme 21 and deals with the introduction of a trifluoromethyl group, which is known to improve the pharmacokinetic properties of drugs.392 This challenging C(sp3)–C(sp3) bond formation was made possible by merging NaDT chemistry with copper catalysis and made use of the Togni’s reagent 21.2 as the trifluoromethylating agent.393 The adopted acidic conditions here caused the formation of the ammonium salt of pyrrolidine 21.1, thus deactivating the C–H bonds adjacent to the nitrogen atom (Scheme 2). Overall, the strategy is based on the addition of the photocatalyzed C-centered radical onto a CuII–CF3 species. The hoped-for trifluoromethylated product 21.3 (trifluoroacetate salt) was then obtained in 68% isolated yield as a single regioisomer. The same procedure enabled the trifluoromethylation of benzylic C–H bonds and of biologically valuable compounds such as lidocaine, prilocaine, celecoxib, and torsemide. Mechanistic studies are consistent with the involvement of a “Cu-CF3 complex”.393
Scheme 21. Metallaphotoredox Strategy for the Trifluoromethylation of Amines.

Other nitrogen-containing hydrogen donors have been reported, such as aliphatic nitriles and alkylpyridines, where the influence of the heteroatom is not so important as in the previous cases; albeit, it still has a role in directing the C–H cleavage event. A representative case is that of adiponitrile 22.1 (Scheme 22a). The electron-withdrawing effect of the cyano group hampers the cleavage of α-C–H but not of β-C–H bonds. Thus, the TBADT-photocatalyzed reaction between 22.1 and dimethyl maleate 22.2 easily gave tetrafunctionalized adduct 22.3 in a satisfying yield.394 Related reactions involve the photocatalyzed addition of 4-methylpentanenitrile to phenyl vinyl sulfone (under flow conditions)381 or to a vinylpyridine.386
Scheme 22. Regioselective Photocatalyzed C–H Cleavage in (a) Aliphatic Nitriles and (b) Alkylpyridines.

The same site-selective C–H to C–C bond conversion in nitriles took place when DT was incorporated within the pores of a copper-based metal organic framework (MOF) ([Cu4(BPY)6Cl2(W10O32)]·3H2O; DT-BPY, BPY = 4,4′-bipyridine). This is a rare case where the HAT process is carried out under heterogeneous conditions. The new PCHAT showed high catalytic efficiency, high stability, and good recyclability, allowing use of a lower excess of the aliphatic nitrile substrate (only 5 equiv), thus improving the sustainability of the process (Scheme 22a).395
The C–H activation in alkylpyridines is interesting, since the labile benzylic hydrogens are not involved in the process, at variance with the alkylbenzene counterparts (see also section 2.1.3). This is well exemplified by the case of 22.4, wherein the methine hydrogen was selectively cleaved and the resulting tertiary radical was then trapped by ketone 22.5 to give adduct 22.6 (Scheme 22b).396 In the last case, preference of the excited PCHAT to abstract the less acidic (or, in other words, the less electrophilic) hydrogen atom in the investigated alkylpyridine was observed.
2.1.3. Hydrocarbons as Hydrogen Donors
In hydrocarbons, it is possible to find quite labile hydrogens that can be easily cleaved under photocatalytic conditions. Hydrocarbons displaying labile benzylic397 and allylic C–H bonds can be easily cleaved at these sites under photocatalytic conditions. In fact, the BDEs of the most labile C–H bonds in toluene and cyclohexene are 88 and 82 kcal/mol, respectively (see Scheme 3).
The main problem here is the high stability of the radical formed and its reluctancy to react with the reaction partner (e.g., a C=C bond) to forge a C(sp3)–C(sp3) bond. This probably explains why very few processes involving these substrates have been reported. Simple alkylaromatics have been derivatized by using TBADT to perform valuable benzylations. However, only easily reducible olefins, including fumaronitrile, maleic anhydride, and substituted maleic imides, gave good results.398 The same conjugate radical additions were carried out in a mesoscale flow photoreactor by adopting a water-cooled 500 W medium-pressure Hg-vapor lamp as the light source. The use of this apparatus led to a marked increase of the STY (space time yield) and a reduction of the irradiation time compared with the same processes developed under batch conditions.381 Other PCsHAT were likewise useful for this C–H activation strategy, as collected in Scheme 23. Thus, the benzylic position in toluene 23.1 was functionalized under EY photocatalysis despite heating at 60 °C being required (Scheme 23a).220 The activation of allylic hydrogens was also attempted by using DT, albeit not on a preparative scale.200
Scheme 23. Functionalization of (a) Benzylic and (b,c) Allylic Hydrogens.

More recently, cyclohexene has been used as the hydrogen donor for the preparation of allylated derivatives 23.5 and 23.7 (Schemes 23b,c). Both EY(220) and ClAQ(240) were employed in the functionalization of very good Michael acceptors 23.2 and 23.6.
The most challenging reaction for the construction of C(sp3)–C(sp3) bonds is related to the functionalization of (cyclo)alkanes,124 due to the high BDE of the C–H bonds involved (ca. 100 kcal/mol, see Scheme 3). Early photocatalytic experiments made use of a high amount of the PCHAT to pursue this issue,157 but the use of DT allowed performing a real photocatalyzed process with only a few mol % loading of the PCHAT. Simple symmetric cycloalkanes were the preferred substrates.81,274,399 As shown in Scheme 24a, cyclohexane 24.1a easily gave access to the corresponding cycloalkyl radical that was in turn trapped by dinitrile 24.2 to give 24.3 through a C–C bond formation step.274
Scheme 24. Photocatalyzed C–H Cleavage in (Cyclo)alkanes.

Similarly, various 5- to 12-membered cycloalkanes were used to functionalize conjugated enones (24.6)400 even with the help of a chiral spiro phosphoric acid (24.7) to promote an asymmetric C–H functionalization (Scheme 24b).401 A chiral phosphoric acid similar to 24.7 was likewise adopted as a chiral proton-transfer shuttle in the cycloalkane addition onto α-substituted acrylates (e.g., N-acyl dehydroalanine benzyl esters) used as Michael acceptors for the smooth forging of enantioenriched α-stereogenic esters.402
Methylene norbornanone was alkylated in a good yield by using cyclohexane as the hydrogen donor under UrN photocatalysis.283 Similarly, the activation of nonacidic C(sp3)–H bonds in cyclohexane (or adamantane) was carried out upon UV light irradiation (ClAQ as the PCHAT) by using 1,1-bis(phenylsulfonyl)ethylene as the radical trap.240 The allylation of alkanes has been performed by means of the PT-photocatalyzed addition of cyclohexane, cyclododecane, or adamantane onto 1,2-bis(phenylsulfonyl)-2-propene as the allyl source.289
In rare instances, the reaction was applied to substituted cycloalkanes. Thus, the presence of a tBu group in compound 24.1b exerted a profound effect in steering the hydrogen abstraction process. In fact, the bulkiness of the tBu group completely shielded the hydrogens in positions 1- and 2-, allowing the selective C–H cleavage in positions 3- and 4- (Scheme 24a). The bulkiness of the PCHAT and the radical trap 24.2 further helped in reducing the number of possible isomers formed, with only cis-3-substituted 24.4 and trans-4-substituted 24.5 formed in an overall 70% yield.77 Interestingly, in the latter case, when BP (1 equiv) was used in place of TBADT, the same product distribution was roughly observed.77 However, when alkane 24.1b reacted with acrylonitrile (TBADT as the PCHAT), a more complex mixture resulted.77
Open-chain alkanes were poorly investigated. A rare case is that reported in Scheme 24c. Despite the fact that compound 24.9 has five different types of hydrogen atoms, only the methine C–H position was effectively cleaved, and the reaction with maleate 24.10 led to diester 24.11 as the sole product.372 Even in this case, the bulkiness of the tBu group helped in the regioselective cleavage of the C–H bond.
The activation of methane (BDE = 105 kcal/mol) was proved to be feasible by adopting DT photocatalysis. The process required specifically optimized conditions, namely the adoption of flow conditions and application of a high pressure (45 bar), to allow the correct mixing of the reagents (Scheme 25). Unfortunately, the C–H bond was so reluctant to undergo cleavage that acetonitrile competed in the HAT event. Accordingly, during the alkylation of dinitriles 25.1a–c, a deuterated acetonitrile/water 7:1 mixture was mandatory to obtain a decent yield of methylated derivatives 25.2a–c.403 The functionalization of ethane and propane was likewise carried out under milder conditions, with no need of deuterated solvents.403
Scheme 25. Photocatalyzed Derivatization of Methane under Flow Conditions.

The addition of an alkyl radical onto an electron-poor olefin may ultimately lead to difunctionalization of the double bond thanks to a dual-catalytic approach, as shown in Scheme 26. TBADT was again used as PCHAT to generate an alkyl radical from cyclohexane. In this case, however, the adduct radical formed by addition of the cyclohexyl radical onto acrylate ester 26.2 was intercepted by a Ni0 catalyst to form the alkyl-NiI intermediate 26.3. Oxidative addition of selected aryl derivatives 26.1a–d onto 26.3 led to ester 26.4 in variable amounts depending on the leaving group X on the aromatic ring, with the bromine atom being the best choice. This approach showed a broad substrate scope since it may be applicable to several functionalized tertiary, secondary, and primary alkyl radicals.404 A related approach was likewise devised by combining a POC (BP) with the same [Ni(dtbbpy)Br2] catalyst.405
Scheme 26. Photocatalyzed Three-Component Difunctionalization of Alkenes.

Another recent example where HAT catalysis was merged with metal catalysis involved a Pd-catalyzed allylic alkylation. In this strategy, the alkyl radical was trapped by a Michael acceptor and the resulting adduct radical was reduced and the resulting carbanion interacted with in situ formed π-allylpalladium species that finally released the desired allylation product.406 The approach developed was then used for the concise synthesis of (±)-mesembrine.406
As a final note to this section, it is worth highlighting that the addition of photogenerated radicals onto olefins different from Michael acceptors (e.g., electron-rich C=C bonds or captodative olefins) intended for the formation of C(sp3)–C(sp3) bonds has only a few precedents in the literature. These processes, however, took place only in the presence of high PCHAT loadings or showed a low conversion of the starting materials.157,200,325
A representative example is shown in Scheme 27 where ethylene 27.1 was alkylated under TBADT photocatalysis to give 27.2 as the major product.325 Such examples have not been presented in detail here due to their limited synthetic significance.
Scheme 27. Photocatalyzed Derivatization of Electron-Rich Olefins.

2.2. Formation of C(sp3)–C(sp3) Bonds via Addition onto C=X (X = N, O) Bonds
The formation of C(sp3)–C(sp3) bonds can be realized also via the addition of a photogenerated radical onto a C=X (X = N, O) double bond. In order to promote reactivity, the N-atom typically bears an electron-withdrawing S-based substituent, either S(=O)2R or S(=O)R. Thus, N-tosylimines have been reported to act as excellent radical traps in TBADT-triggered alkylations with alkanes, ethers, and DMF. As reported in Scheme 28a, cyclohexane 28.1 (10 equiv) underwent addition onto the C=N bond of 28.2 to give the hydroalkylated adduct 28.3 in 85% yield in the presence of TBADT (2 mol %) upon irradiation with 400 nm LEDs (16 h). The occurrence of a chain mechanism (at least in part), however, could not be excluded.407 Similarly, chiral N-sulfinyl imines were smoothly alkylated by adamantane scaffolds in the presence of a catalytic amount of PT (5 mol %) upon irradiation with 390 nm LEDs. Notably, this strategy allowed the enantioselective synthesis of the saxagliptin core, containing an adamantyl-glycine motif.279 In another instance, a dual-catalytic system based on PT and a chiral Cu-based complex containing a bisoxazoline (BOX) ligand allowed the regio- and stereoselective functionalization of benzylic, allylic, and even unactivated hydrocarbons with an imine derivative. As shown in Scheme 28b, toluene 28.6 (10 equiv) reacted with 28.7 to give product 28.9 in an excellent yield (93%) and enantioselectivity (e.e. 93%) in the presence of PT (2 mol %) and Cu(BF4)2 (10 mol %) and chiral BOX ligand 28.8 (11 mol %).281
Scheme 28. Photocatalyzed Addition of Hydrocarbons onto Imine Derivatives.

Very recently, the preparation of amines has been realized via a multicomponent carbonyl alkylative amination strategy. The protocol was promoted by TBADT (2 mol %) and comprised of N-arylamines, aldehydes, and hydrocarbons as starting materials. Slightly different conditions were required depending on the nature of the amine, being either an aniline or a diphenylamine. As shown in Scheme 29, the process involved the in situ formation of an iminium ion (29.6+), which acted as the trap of the photogenerated radical. When adopting cyclohexane 29.1, benzaldehyde 29.2, and anilines 29.3a–c, secondary amines 29.4a–c were obtained in good yields upon irradiation at 390 nm for 24 h, only requiring acetic acid (0.5 equiv) as an additive.408
Scheme 29. Multicomponent Synthesis of Secondary Amines.

The challenging addition of photogenerated intermediates onto C=O bonds has been realized only in a few instances. One notable example involved a strategy comprised of TBADT and a CrIII salt, where the role of the latter was to promote the formation of an organochromium compound via interception of the photogenerated radical. Indeed, this approach has been exploited to trigger the alkylation, aminomethylation, and oxymethylation of both aliphatic and aromatic aldehydes. Thus, N,N-dimethylacetamide 30.1 reacted with aldehydes 30.2a–c to give 1,2-aminoalcohol derivatives 30.3a–c in the presence of TBADT (10 mol %) and CrCl3 (3 equiv) upon irradiation with 390 nm LEDs for 48 h (Scheme 30).409
Scheme 30. Photocatalyzed Functionalization of C=O Bonds.

2.3. Formation of C(sp3)–C(sp2) Bonds
This section describes the formation of C(sp3)–C(sp2) bonds between a photocatalytically generated C(sp3)-centered radical and suitable reaction partners, which include alkynes via an addition process or vinyl/aryl derivatives via a (formal) substitution or cross-dehydrogenative coupling reaction. Likewise, the addition onto carbon monoxide (CO) or carbon dioxide (CO2) will be reported here.
Seminal works in the field focused on the addition of cycloalkyl radicals, obtained from the corresponding hydrocarbons, onto electron-poor alkynes in the presence of aromatic carbonyls. Although these PCsHAT were routinely adopted stoichiometrically, the reaction was demonstrated to work smoothly also in the presence of a catalytic amount of BP. Thus, methyl propiolate 31.2 was functionalized by cyclopentane (31.1, used as the solvent) to give an E/Z mixture of vinylcycloalkanes 31.3 (Scheme 31) in a very good yield. BP loading could be lowered to 9 mol % without affecting the reaction yield; albeit, a longer irradiation time was required in the latter case.228 Notably, the employed aromatic ketone could be supported onto a solid material (a polystyrene matrix or silica), rendering the PCHAT potentially recyclable. This heterogeneous variant has been shown to work to some extent under natural sunlight irradiation.410,411 When applied to alcohols as substrates and dimethyl acetylenedicarboxylate as radical trap, this (heterogeneous) methodology opened the way to the generation of α-hydroxyalkyl radicals and to the preparation of γ-butenolides from them.412 Very recently, an analogous strategy based on the use of chloroalkynes (and, in selected cases, terminal alkynes) has been reported. DCBP (15 mol %) was used as the PCHAT, while the substrate scope included alcohols, ethers, amides, and even alkanes. Furthermore, when applied to THF, this process could be performed on the gram scale, without any significant yield decrease. Mechanistic studies revealed that this process occurred with a quantum yield >1, indicating the involvement of a radical chain mechanism.413 Similarly, the functionalization of chloroalkynes to give functionalized vinyl chlorides has been likewise carried out in the presence of EY(220) and TBADT,277 respectively.
Scheme 31. Photocatalyzed Addition of Cycloalkanes onto Alkynes.

Another option to forge a C(sp3)–C(sp2) bond is to intercept the photogenerated radical with an olefinic reaction partner containing a suitable radicofugal group. Thus, the alkenylation of ethers and amides with a library of vinyl sulfones smoothly occurred in the presence of DCBP (20 mol %) upon irradiation with CFL bulbs, wherein the loss of a sulfonyl radical occurred during the process. Thus, 2-pyrrolidone 32.1 (used as the solvent) reacted with sulfones 32.2a–c to give the expected alkenylated amides 32.3a–c in good yields and with a marked preference for the formation of the E-isomer (Scheme 32).388
Scheme 32. Photocatalyzed Alkenylation of Amides.

The dehydrogenative coupling between alkanes and aryl alkenes is also possible and has been recently realized thanks to a dual-catalytic strategy. This approach relies on the synergistic combination of TBADT photocatalysis with cobaloxime-mediated hydrogen-evolution cross-coupling. The Co-catalyst is responsible for intercepting the radical adduct formed upon addition of the photogenerated radical onto the olefin and then undergoes a photoinduced β-hydride elimination, restoring the original double bond. As depicted in Scheme 33, a series of cycloalkanes (33.1a–d) was alkenylated by styrene 33.2 (10 equiv) in the presence of TBADT (4 mol %), Co(dmgH)(dmgH2)Cl2 (1 mol %; dmgH2 and dmgH: dimethylglyoxime and its monoanion), and 2,6-lutidine (10 mol %) as the ligand to deliver adducts 33.3a–d in good yields with complete regio- and stereoselectivity. The reaction took place in acetonitrile at 60 °C upon irradiation with a 370 nm LED and could be applied in the late-stage alkenylation of natural products, including steroid derivatives.414
Scheme 33. Dual-Catalyzed Dehydrogenative (E)-Alkenylation of Cycloalkanes.

Turning to arylation reactions, the preparation of alkylated pyrimidines was realized through the coupling of saturated heterocycles (including oxygen-, nitrogen-, and sulfur-based derivatives) with sulfonylated pyrimidines in the presence of BP (10 mol %). As an example, 5-membered heterocycles 34.1a,b were arylated by 34.2 to give pyrimidine derivatives 34.3a,b in good yields upon irradiation with a medium-pressure Hg lamp via an ipso-substitution process (Scheme 34).415 Similarly, EY (2 mol %) was employed to promote the arylation of THF at the 2-position upon reaction with 2-phenylsulfonylbenzothiazole.220 In a related instance, the 4-pyridination of cumene at the benzylic position was performed in the presence of a catalytic amount of BP (10 mol %). In the process, the photogenerated radical added onto 4-cyanopyridine, while the desired product was formed upon loss of HCN from the initially formed adduct.229 In a very recent report, N-aminopyridinium salts have been likewise used as radical traps for photogenerated C-centered radicals (AQ as the photocatalyst) and enabled the site-selective C–H pyridylation of unactivated alkanes. Notably, this protocol could be adopted for the late-stage site-selective functionalization of biorelevant compounds.416
Scheme 34. Photocatalyzed Arylation of Five-Membered Heterocycles.

The merging of HAT photocatalysis with Ni-catalysis opened new avenues on the route toward the arylation of (strong) aliphatic C–H bonds, allowing adoption of aromatic halides (mostly, bromides) as coupling partners. In particular, the Ni-based cocatalyst was responsible for activating the C(sp2)–Br bond and intercepting the photogenerated radical. This chemistry was successfully combined with different classes of PCsHAT, including DT(417) and aromatic carbonyls.418,419 Thus, cyclohexane 35.1 was functionalized by (hetero)aryl bromides 35.2a,b to deliver cross-coupled products 35.3a–b in very good yields upon irradiation with a 390 nm LED (Scheme 35). Of note, this protocol could be applied to the manipulation of natural products (see the case of 35.5).417 Very recently, a dual-catalytic strategy based on DCBP and a Ni-based complex enabled the construction of C(sp3)–C(sp2) bonds via the acylation of methylbenzenes with N-acylsuccinimides.420
Scheme 35. Arylation of Strong C–H Bonds via a TBADT/Nickel Dual-Catalyzed Strategy.

Another opportunity for C(sp3)–C(sp2) bond construction is represented by photocatalytic cross-dehydrogenative couplings (CDC)421 between aliphatic H-donors and (hetero)arenes. These processes require the adoption of oxidative conditions to remove the extra electrons, and this has been realized either by having recourse to a chemical oxidant or through electrochemical means. As for the former case, DT photocatalysis has been successfully exploited to trigger the functionalization of alkanes, ethers, and amides with heteroarenes in the presence of a persulfate salt. As an example, this Minisci-type reaction allowed the functionalization of quinaldine (36.2) with DMF (36.1) to give 36.3 as the only product (73% isolated yield) in the presence of TBADT (4 mol %) and K2S2O8 (2 equiv) upon irradiation with simulated solar light (Scheme 36).422 More recently, a similar strategy has been applied to the preparation of 2-alkylated benzothiazoles under chemical oxidant-free photoelectrochemical conditions.423
Scheme 36. Photocatalyzed Cross-Dehydrogenative Coupling between Amides and Heteroarenes.

A different cross-dehydrogenative coupling encompassing the merging between HAT photocatalysis and electrochemistry allowed the regioselective functionalization of ethers with isoquinolines. This strategy, tagged “electrophotocatalysis”, was based on the use of a trisaminocyclopropenium ion (TAC+), which was electrochemically converted to the stable TAC•2+ species via one-electron oxidation. The latter species then underwent excitation and, once in the excited state, triggered the desired HAT from the chosen ether. Thus, adducts 37.3a–d have been prepared by reaction between THF (37.1) and substituted isoquinolines 37.2a–d in the presence of TAC+ (perchlorate salt; 1 mol %) upon application of a constant potential (Ecell = 1.5 V) under irradiation with a CFL (Scheme 37).209
Scheme 37. Electrophotocatalytic Arylation of Ethers Mediated by the Trisaminocyclopropenium (TAC+) Ion.

Carbon monoxide (CO) is an excellent radical trap and has been frequently exploited to get access to valuable acyl radicals.424−426 Seminal examples of this chemistry within photocatalytic applications date back to the early 90s, when aromatic carbonyls where adopted to promote the carbonylation of alkanes (mainly, cyclohexane) to afford cyclohexanecarboxaldehyde either under high CO pressure (20–80 atm)427 or in the presence of metal carbonyl complexes based on Ir, Rh, or Ru.428 Around the same period, an 8% formation of cyclohexanecarboxaldehyde from cyclohexane and CO (1 atm) in the presence of TBADT was reported.326 More recently, the functionalization of alkanes with electron-poor olefins under photocatalytic conditions mediated by TBADT was realized in the presence of CO. This allowed the preparation of unsymmetrical ketones in an atom-economical fashion in an overall multicomponent process, where the photogenerated alkyl radical was trapped by CO to form a C(sp3)–C(sp2) bond and then by the chosen electron-poor olefin. Thus, upon irradiation with a Xe lamp equipped with a Pyrex filter, 5- to 7-membered cycloalkanes 38.1a–c reacted with dibutyl maleate 38.3 under an atmosphere of CO (38.2, 80 atm) in the presence of TBADT (4 mol %) to afford ketones 38.4a–c in good isolated yields (Scheme 38).429 Later, the same protocol was applied to the regioselective β-acylation of cyclopentanone in the role of H-donor,83 as well as to the preparation of acyl hydrazides using diisopropyl azodicarboxylate (DIAD) in place of electron-poor olefins.430
Scheme 38. Three-Component Photocatalyzed Synthesis of Unsymmetrical Ketones.

In one instance, the carboxylation of the allylic position in simple alkenes by CO2 has been realized in the presence of 3,6-diphenylxanthone and a Cu-based complex. The process has been proposed to occur through a sequence involving two independent steps, where activation of the allylic C–H bond was promoted by the excited carbonyl, while the copper complex operated the desired carboxylation, also restoring the initial ketone. Indeed, it was demonstrated that both the xanthone derivative and the copper complex behaved catalytically in the overall process.285
2.4. Formation of C(sp3)–C(sp) Bonds
The HAT-photocatalyzed C(sp3)–H to C(sp3)–C(sp) bond conversion can be related to two different families of processes, namely the introduction of an alkynyl or a cyano group. As for the first instance, the photogenerated radicals have been trapped by suitable alkynylating agents, namely alkynes substituted with a convenient radicofugal group. Indeed, only a handful of examples of this chemistry have been reported, which are based on the use of bromoalkynes,413 alkynylbenziodoxolones,237 or alkynylsulfones.277,388 Either the aromatic ketone DCBP(237,388,413) or DT(277) was used as the PCsHAT. As an example, THF (39.1, used as the solvent) reacted with bromoalkynes 39.2a–c in the presence of DCBP (15 mol %) and KOAc (1.5 equiv) to give alkynes 39.3a–c in good isolated yield, independently from the electronic character of the aromatic substituent in 39.2a–c (Scheme 39). In the process, the formation of a vinyl bromide intermediate initially takes place, which then undergoes HBr elimination to give the desired alkynylated product aided by the employed base (KOAc).413
Scheme 39. Photocatalyzed Alkynylation of Ethers by Bromoalkynes.

Turning to cyanation processes, an excellent option to intercept the photogenerated radical is using tosyl cyanide, which allows introduction of the desired cyano group via displacement of the sulfonyl moiety. Thus, BP can be successfully adopted to trigger this transformation in a variety of substrates, including ethers, alkanes, and nitrogen-containing substrates; however, only in the latter case it behaves as a real PCHAT. Thus, protected nitrogen-heterocycles 40.1a–c were cyanated by tosyl cyanide 40.2 (2 equiv) to give 40.3a–c in good to excellent yield in the presence of BP (20 mol %) and 2,6-di-tert-butylpyridine (4 equiv; functioning as an acid scavenger) upon irradiation with a medium-pressure Hg lamp (Scheme 40). Worthy of notice is the example related to 40.1c, wherein the functionalization of the α-to-N position occurred chemoselectively.230 Similarly, EY (2 mol %) has been adopted in the C–H to C–CN conversion in 1,4-dioxane in the presence of tosyl cyanide.220 A different strategy is based on the use of a seven-coordinated (chiral) Ur salen complex (2 mol %), which was used for the cyanation of a variety of (substituted) N,N-dimethylanilines under oxidative conditions (H2O2) in the presence of NaCN and AcOH.431
Scheme 40. BP-Photocatalyzed Cyanation of Nitrogen-Containing Heterocycles.

2.5. Formation of C(sp2)–C(sp3) Bonds
This section gathers examples enabling the formation of a C(sp2)–C(sp3) bond via the intermediacy of a photogenerated C(sp2)-hybridized radical. Specifically, either aldehydes or formamides (see also section 2.1.2) can be exploited as H-donors in the formation of acyl and carbamoyl radicals, respectively. Seminal works in the area involved the use of BP to trigger the acylation of enones (mainly carbohydrate enones, see also Scheme 7 for the analogous C(sp3)–C(sp3) bond formation)233 and α,β-unsaturated esters or acids234 with aldehydes. An interesting example is reported in Scheme 41, showing the hydroacylation of crotonic acid 41.2 with acetaldehyde 41.1 to give 4-oxoalkanoic acid 41.3 (60% yield) in the presence of BP (10 mol %) upon irradiation at 366 nm for 24 h.234
Scheme 41. Photocatalyzed Acetylation of Crotonic Acid.

More recently, given their excellent reactivity as H-donors, aldehydes have been adopted as substrates in combination with a plethora of PCsHAT to perform the hydroacylation of a huge variety of unsaturated systems. One of the most studied systems involves the functionalization of electron-poor olefins (α,β-unsaturated esters, ketones, and nitriles, as well as vinyl sulfones) triggered by TBADT.382,432,433 As an example, 2-cyclohexenone 42.1 was smoothly acylated by both hydrocinnamaldehyde 42.2a and p-anisaldehyde 42.2b to give interesting 1,4-diketones 42.3a,b in a good yield (Scheme 42). Thus, the optimal TBADT loading was 2 mol %, but contrary to aliphatic aldehydes, a slight excess of 42.1 (1.2 equiv), a longer irradiation time (30 vs 24 h), and an increased light intensity were required in the preparation of 42.3b.(432,433) Of note, these reactions have been demonstrated to occur under natural sunlight irradiation by simply exposing the reaction vessel containing the mixture on a window ledge for a few days. In the acylation of dimethyl maleate with heptanal, it was possible to increase the concentration of the starting materials up to 0.5 M, therefore reducing the amount of solvent needed and bringing about important ecological advantages.276
Scheme 42. Cyclohexenone Acylation by Addition of Aliphatic (Upper Part) and Aromatic (Lower Part) Aldehydes.

The environmental performance of these acylations was further ameliorated making use of continuous flow conditions.381 The adoption of this operation mode also allowed design of multistep procedures, wherein the photocatalytic C(sp2)–C(sp3) bond formation was followed by additional thermal steps on the resulting acylated derivatives.434,435
An elegant one-pot protocol comprised of two distinct photochemical steps was adopted for the preparation of homoallyl ketones starting from cyclopentanones and electron-poor olefins. The sequence involved an initial Norrish type-I photoinduced fragmentation of the 5-membered ring to give a 4-pentenal derivative followed by the TBADT-photocatalyzed hydroacylation of an electron-poor olefin. Thus, cyclopentanone 43.1 underwent ring opening via photoinduced cleavage of the C1–C2 bond to give aldehyde 43.2. Next, TBADT (4 mol %) and the chosen electron-poor olefins (e.g., 43.3a,b) were added to the crude mixture, finally affording the desired adducts (43.4a,b) in a good yield upon irradiation for an additional 24 h (Scheme 43).436
Scheme 43. Preparation of Homoallyl Ketones from Cyclopentanones via a Two-Step Photochemical Norrish Type-I Cleavage/Photocatalyzed Hydroacylation Sequence.

Apart from TBADT, other PCsHAT have been recently reported to promote the hydroacylation of electron-poor olefins under visible light irradiation, including EY,220UrN,283 and Sb-Oxo.(265)Scheme 44 gathers selected examples describing the hydroacylation of benzylidene malononitrile 44.1. Thus, EY and UrN allowed the hydroacylation of 44.1 with hexanal 44.2a and heptanal 44.2b to give the corresponding adducts 44.3a and 44.3b in 84 and 93% isolated yield, respectively (Schemes 44a,b).220,283 On the other hand, the preparation of 44.3b in the presence of Sb-Oxo proceeded with a partial conversion of the starting materials; however, an almost quantitative yield based on remaining starting material (99% brsm) was observed (Scheme 44c).265
Scheme 44. Hydroacylation of Benzylidene Malononitrile Triggered by Different PCsHAT.

EY has been further tested for the asymmetric synthesis of 1,4-dicarbonyls. This strategy encompassed a dual-catalytic system, which also involved the use of a chiral rhodium catalyst responsible for coordinating the chosen electron-poor olefin (an unsaturated N-acylpyrazole) and driving the radical addition step (for a related example, see Scheme 19d).390
Very recently, the formation of a C(sp2)–C(sp3) bond by the EY-photocatalyzed reaction of aldehydes with N-(hetero)arylsulfonyl propiolamides has been proposed. This transformation led to the preparation of the isothiazolidin-3-one 1,1-dioxide core and proceeded through a cascade involving addition of the photogenerated radical onto the C≡C triple bond of propiolamide, Smiles rearrangement, and 5-endo-trig cyclization.437 As an example, N-heterocycles 45.3a–c have been prepared from aldehydes 45.1a–c and amide 45.2 upon irradiation with blue light for 48 h in the presence of EY (4 mol %, Scheme 45). The preparation of 45.3c has been successfully realized on a gram scale in 83% yield, and of note, some of the synthesized compounds may have potential anticancer activity.437
Scheme 45. Photocatalyzed Preparation of Isothiazolidin-3-one 1,1-Dioxides.

Apart from the use of electron-poor olefins, (hetero)aromatic alkenes can be used as well as radical traps for the photogenerated acyl radicals, with the driving force for the process being the formation of a stabilized benzyl radical. Thus, different combinations of the PCHAT and alkenes have been adopted, including vinylpyridines,386 α-trifluoromethyl aryl alkenes (in this case, an aliphatic trifluoromethylalkene has been used as well),438 and aryl alkenes.378,439Scheme 46 gathers the case of benzaldehyde 46.1, that has been adopted for the functionalization of 2-vinylpyridine 46.2a, CF3-substituted alkene 46.2b, styrene 46.2c (in all cases TBADT as the PCHAT), and p-fluorostyrene 46.2d (EY as the PCHAT).
Scheme 46. Photocatalyzed Hydroacylation of Vinyl (Hetero)aromatics.

Finally, N-tosyl imines (for a related example, see Scheme 28a)407 and dehydroalanine derivatives440 have been likewise adopted as acyl radical traps under TBADT-mediated photocatalytic conditions.
Along the same line, a dual-catalytic strategy comprised of TBADT and a Ni-based cocatalyst allowed the asymmetric acyl-carbamoylation of alkenes starting from aldehydes and a carbamoyl chloride incorporating a C=C double bond. In this case, the Ni-based cocatalyst intercepted the photogenerated acyl radical and triggered the activation of the carbamoyl chloride, supervising the sequence of steps leading to the formation of the final product. Thus, butanal 47.1 reacted with aryl carbamic chlorides 47.2a–c in the presence of TBADT (5 mol %), Ni(OTf)2 (10 mol %), ligand 47.4 (12 mol %), and K3PO4 (1.1 equiv). The process took place in MeCN upon irradiation with a 390 nm LED for 9 h, delivering oxindoles 47.3a–c in good yields and excellent enantioselectivity (Scheme 47).441
Scheme 47. Asymmetric Acyl-Carbamoylation of Alkenes.

A similar reaction system, comprised of TBADT and a Ni-based cocatalyst, enabled the cross-coupling between acyl radicals, photogenerated from aldehydes, and (fluorinated) α-bromoacetates. As reported in Scheme 48, 1,3-dicarbonyl derivatives 48.3a–c were readily accessed upon reaction between p-anisaldehyde 48.1 and esters 48.2a–c.442
Scheme 48. Synthesis of 1,3-Dicarbonyl Derivatives via a Dual-Catalytic Strategy.

As mentioned above, apart from aldehydes, formamides can be likewise adopted as H-donors for the generation of carbamoyl radicals. These, in turn, are interesting intermediates for C(sp2)–C(sp3) bond formation campaigns.157 Thus, only a handful of PCsHAT have been employed to trigger this reactivity, including TBADT(381,382,385,386,389) and UrN.283 In the latter case, however, this chemistry represented a minor pathway with respect to the preferred α-to-N C–H cleavage (see also section 2.1.2).283
On one hand, TBADT has been shown to cleave chemoselectively the C(=O)–H bond in primary and secondary formamides, while a completely different reactivity has been observed with tertiary formamides, (e.g., DMF, see Scheme 19b).385 As shown in Scheme 49, methyl crotonate 49.2 has been successfully carbamoylated by formamide 49.1a and N-methyl formamide 49.1b in the presence of TBADT (2 mol %) upon irradiation with phosphor-coated lamps centered at 310 nm. Indeed, the former H-donor led to the formation of product 49.3a in a higher yield (76%) than the latter (49.3b, 46% yield). The carbamoylation of electron-poor olefins has been successfully implemented under continuous flow conditions, delivering the desired products in shorter reaction times and increased productivity.381,389 More recently, the same reactivity has been likewise applied to the carbamoylation of vinylpyridines386 and styrenes378 (in the latter case, in the presence of a disulfide cocatalyst). In sharp contrast, UrN enabled the functionalization of the C(=O)–H bond even when using DMF as substrate, being a competitive path to the usual α-to-N functionalization.283
Scheme 49. TBADT-Photocatalyzed Carbamoylation of Electron-Poor Olefins.

2.6. Formation of C(sp2)–C(sp2) Bonds
As in the previous section, the formation of C(sp2)–C(sp2) bonds made use of aldehydes or formamides as radical precursors. The photogenerated radicals, however, are here used in the addition onto C≡C triple bonds or in (formal) substitution reactions or cross-dehydrogenative couplings with substituted arenes or alkenes.
Early examples described the acylation of quinones in the presence of BP,231,232 in what has been tagged as “photo-Friedel–Crafts acylation”.232 Albeit the reaction proceeded to some extent also in the absence of BP, its addition in catalytic quantities allowed an improvement of the product yield. As depicted in Scheme 50, 1,4-naphthoquinone 50.2 reacted with ortho-substituted benzaldehydes 50.1a–c to afford hydroquinones 50.3a–c in a good yield upon irradiation with a high-pressure mercury lamp for 5 days in the presence of BP (6 mol %).231
Scheme 50. Photocatalyzed Acylation of 1,4-Naphthoquinone.

More recently, the construction of the 3-acyl-4-arylcoumarin scaffold has been realized upon photocatalyzed addition of aldehydes (e.g., 51.1a–c) onto an aromatic ynoate (51.2) triggered by an anthraquinone derivative (Scheme 51). Thus, the photogenerated acyl radical added regioselectively onto the α-position of the ynoate to give the vinyl radical 51.4•, which cyclized onto the tethered aromatic ring followed by rearomatization to give coumarins 51.3a–c in good yields. The reaction was promoted under visible light irradiation by adopting tBAQ (10 mol %) and benzoyl peroxide (BPO) as a stoichiometric oxidant. A possible role of the carboxyl radical resulting from BPO decomposition in the desired C–H cleavage has been proposed. Of note, the biological activity of some of the synthesized coumarins has been studied, suggesting their efficacy as potential candidates for new therapeutics.443
Scheme 51. Photocatalyzed Acylation of Ynoates on the Route to Coumarin Scaffolds.

Very recently, AQ (1 mol %) has been used as the PCHAT to promote the C(=O)–H pyridylation of a small library of aldehydes and formamide with N-aminopyridinium salts. As an example, Scheme 52 depicts the reaction between aldehydes 52.1a,b and 52.2, which took place upon visible light irradiation for 48 h, to deliver acylated pyridines 52.3a,b. In the process, AQ triggered the formation of acyl radicals 52.4•, which underwent addition onto the pyridinium derivative in a regioselective fashion and ultimately gave the desired products upon deprotonation and release of sulfonamidyl radical 52.5• via N–N bond cleavage. Indeed, the possible involvement of sulfonamidyl radical 52.5• in a chain pathway has been likewise proposed.416
Scheme 52. Photocatalyzed Preparation of Pyridyl Ketones.

Another opportunity for C(sp2)–C(sp2) bond formation is the cross-coupling between aldehydes and aromatics. Most of the reported routes rely on the adoption of a dual-catalytic strategy comprised of a PCHAT (TBADT227,442 or an aromatic ketone227) and a transition-metal cocatalyst (based on Ni441 or Pd227), responsible for intercepting the photogenerated acyl radical and activating the aromatic derivative. Scheme 53a describes the arylation of cyclohexanecarboxaldehyde 53.1 with 4-bromotoluene 53.2. Interestingly, two different PCsHAT have been used in the preparation of 53.3, either TBADT (2 mol %) or AQ (10 mol %), in the presence of the same cocatalyst, viz. Pd(OAc)2 (5 mol %) complexed with the Xantphos ligand (5 mol %). Notably, while the former system required UV light irradiation (390 nm), the latter worked upon visible light exposure (427 nm). Apart from aryl bromides, aryl iodides and triflates have been demonstrated to be competent substrates as well. One example also demonstrated that N-methyl formamide 53.4 could be used instead of aldehydes, enabling the carbamoylation of aryl bromide 53.5 to give benzamide 53.6 (Scheme 53b).227 Very recently, a dual-catalytic approach based on PQ and a Pd-based cocatalyst enabled the acylation of arenes via a double C–H activation strategy. The process took place under visible light irradiation in the presence of Ag2O as the terminal oxidant.444 In another instance, the net-oxidative cross-coupling between heptanal and quinaldine in the presence of TBADT (4 mol %) and K2S2O8 (2 equiv) has been described (see Scheme 36 for a related process).422 Also in this case, the aldehyde could be substituted with N-methyl formamide as the H-donor to unlock quinaldine carbamoylation.422
Scheme 53. Photocatalyzed Preparation of Aromatic Ketones and Benzamides.

Apart from aromatic derivatives, the formation of C(sp2)–C(sp2) bonds starting from aldehydes and alkenes has been reported. Only a couple of examples of this chemistry are available in the literature, both making use of dual-catalytic strategies. The first one enabled the cross-dehydrogenative coupling between aldehydes and alkenes, based on the use of TBADT and a Co-based cocatalyst. Thus, adopting the same protocol described in Scheme 33, hexanal 54.1 reacted with styrene 54.2 to give the α,β-unsaturated ketone 54.3 in a good yield (Scheme 54). Notably, the process occurred in a stereoselective fashion, affording exclusively the E-isomer.414 In one instance, the alkenylation of cyclohexanecarboxaldehyde with β-bromostyrene has been likewise realized through the merging of TBADT photocatalysis with Pd-catalysis (see also Scheme 53).227
Scheme 54. Dual-Catalytic Cross-Dehydrogenative Coupling between Aldehydes and Aryl Alkenes.

2.7. Formation of C(sp2)–C(sp) Bonds
Very recently, an example of C(sp2)–C(sp) bond formation has been reported, making use of a SOMOphilic alkynylation protocol. As shown in Scheme 55, heptanal 55.1 reacted with alkynyl sulfone 55.2 to give ynone 55.3 in a good isolated yield (67%) upon irradiation with a 390 nm LED in the presence of TBADT (2 mol %).277
Scheme 55. TBADT-Photocatalyzed Alkynylation of Aldehydes.

2.8. Functionalization of Carbon Nanostructures
Fullerene is one of the most investigated nanomaterials nowadays, and this is mainly due to its versatility in supramolecular chemistry,445,446 in technological applications,447,448 as well as in medicinal chemistry and nanotechnology.449 One of the most convenient ways to edit the fullerene structure, taking advantage of the high number of unsaturations, is by radical addition, wherein fullerene works as a radical sponge.450 However, the challenging aspect of this chemistry stems from the tendency of C60 to give a complex mixture of multiadducts. This reactivity could be tamed by having recourse to DT-photocatalyzed HAT;451−453 albeit, only in one case the PCHAT could be used in a 20 mol % loading. Therein, the functionalization of fullerene C60 with acyl radicals generated from aromatic, α,β unsaturated and aliphatic aldehydes was reported.454 The mechanism is based on a fast radical–radical anion coupling between the acyl radical and the fullerene radical anion generated concomitantly with the recovery of the PCHAT. Intriguingly, it was found that when a pivaloyl or phenylacetyl radical was generated (for both intermediates the decarbonylation rate is >105 s–1), CO loss occurred already at 10 °C. Notably, the decarbonylation step could be suppressed and the corresponding acylated fullerenes were synthesized with great selectivity by decreasing the temperature down to −40 °C. In fact, when pivalaldehyde (56.1a) or phenylacetaldehyde (56.1b) was subjected to the optimized reaction conditions at 10 °C, a mixture of 56.3 and 56.4 was obtained in the ratio 1:1.8 or 1:2, respectively. However, by decreasing the temperature, the ratio was completely reversed to 12:1 for 56.1a, while for 56.1b the decarbonylation product was not even observed (Scheme 56).
Scheme 56. Functionalization of Fullerene-C60 via Addition of Photogenerated Acyl Radicals.

In another instance, the DT-photocatalyzed PEGylation of carbon nanotubes was reported.455,456 In particular, when a solution of single-walled carbon nanotubes was irradiated in the presence of TBADT in a PEG400/MeCN solution, the grafting occurred smoothly in 48 h, albeit with modest efficiency (20 wt %, 1 PEG400 chain every 120 carbon atoms). Interestingly, it was shown that sunlight could be used as an inexpensive light source and afforded the PEGylated carbon nanotubes in a comparable yield.455,456
3. Formation of C–Y Bonds (Y ≠ C)
Besides the formation of C–C bonds, photocatalyzed HAT has been successfully employed to forge C–Y bonds. Specifically, this section gathers synthetic examples dealing with the formation of C–N, C–O, C–S, C–F, and C–Cl bonds. Finally, the adoption of d-HAT for the deuteration of organic molecules is presented.
3.1. Formation of C–N Bonds
Diisopropyl azodicarboxylate (DIAD) is one of the most widely adopted unsaturated compounds to forge C–N bonds via photocatalyzed HAT. Thus, its remarkable electrophilic character makes DIAD an excellent trap for nucleophilic radicals such as alkyl, acyl, α-oxyalkyl, and α,α-dioxyalkyl. Upon addition of these nucleophilic radicals onto the N=N double bond, the corresponding N-centered radical is generated, which is typically entrusted for the recovery of the spent PCHAT.
In one instance, DIAD was exploited for the synthesis of hydrazides, as shown in Scheme 57.430 Therein, TBADT was used for the functionalization of (cyclo)alkanes (e.g., cyclohexane 57.1), ethers, and acetals via homolytic C–H bond cleavage. The photogenerated C-centered radicals were trapped by DIAD 57.2 to deliver the corresponding hydrazides (e.g., 57.3) in good to excellent yields. Interestingly, aldehydes were competent substrates as well, allowing the synthesis of acyl hydrazides through the formation of a C(sp2)–N bond.430
Scheme 57. TBADT-Photocatalyzed Synthesis of Hydrazides.

The synthesis of hydrazides, including adduct 57.3, was also reported under continuous-flow conditions. In such case, the photochemical reactor (V = 50 mL) consisted of a water-cooled 500 W medium-pressure Hg-vapor lamp wrapped in FEP (fluorinated ethylene propylene) tubing. When a MeCN solution of THF (58.1), DIAD, and TBADT (0.4 mol %) was flown through the tubing, the labile α-to-O C(sp3)–H bond of 58.1 was cleaved, thus delivering the corresponding α-oxyalkyl radical and, ultimately, product 58.3 in 73% yield upon reaction with 58.2 (Scheme 58). Interestingly, this reaction showed an excellent process mass intensity (PMI, i.e. the ratio of the total mass of materials to the mass of isolated product) of 9.36 kg kg–1 and a specific productivity (SP, i.e. mmol of product formed with respect to the energy consumed) of 22 mol W–1 h–1.381
Scheme 58. TBADT-Photocatalyzed Formation of a C–N Bond in Flow.

A similar approach was reported for the one-pot synthesis of hydroxamic acids from aldehydes.245 In detail, acyl radicals were generated via HAT by PGA (10 mol %) and trapped by DIAD. The resulting acylhydrazides (e.g., 59.3) were then converted in a one-pot fashion to the corresponding hydroxamic acids via nucleophilic acyl substitution with hydroxylamine. For example, propanal (59.1) was smoothly converted to hydroxamic acid 59.4 in 78% yield over two steps (Scheme 59).
Scheme 59. One-Pot Synthesis of Hydroxamic Acids from Aldehydes.

This approach was also extended to the synthesis of amides by employing primary and secondary amines in the place of hydroxylamine.457 The validity and the robustness of the protocol were demonstrated through the preparation of Moclobemide (∼30% yield over two steps), a drug used against depression and social anxiety, starting from 4-chlorobenzaldehyde.457
A conceptually different route for the formation of C–N bonds consists in the synthesis of organic azides. This approach is particularly appealing since these compounds are widely used in medicinal chemistry for biorthogonal labeling via the Cu-catalyzed Huisgen reaction. In one instance, photocatalyzed HAT has been used to introduce the N3 group on leucine in the synthesis of Manzacidin C.458,459 The first step of the synthetic route consisted in the TBADT-promoted activation of the methine site of the amino acid (60.1); ensuing quenching of the photogenerated radical with sulfonyl azide 60.2 readily afforded the hoped-for azide 60.3 in 49% yield (Scheme 60).
Scheme 60. Azidation of the Methine Site of Leucine.

Recently, the azidation of C(sp3)–H bonds via HAT has been achieved through an electrophotocatalytic strategy, where DMBP (5.5 mol %) was used as PCHAT and a Mn-salt as the cocatalyst.460 In detail, compound 61.1 was activated via HAT to afford the corresponding (stabilized) benzyl radical. In the meanwhile, the manganese ion (from MnF2) was complexed by the added ligand (1,10-phenantroline) and the azide anion to afford a MnIII/L–N3 complex after anodic oxidation. The interaction between the benzyl radical and the latter species afforded the hoped-for organic azide and the MnII ion, prone to start a new catalytic cycle. It is however important to mention that the reaction proceeded to a certain extent (>40%) even without the photocatalyst, suggesting that a competitive direct anodic oxidation of the N3– anion might be likewise responsible for the observed reactivity. Notably, the reaction could be run on a gram scale and product 61.3 was obtained in 71% yield (1.36 g, Scheme 61).
Scheme 61. Electrophotocatalytic Azidation via a DMBP/Mn Dual-Catalytic System.

Very recently, a different concept for the C–H to C–N bond conversion based on radical-polar crossover (RPC) has been disclosed. RPC opens the world of polar chemistry to radicals and enables a whole new range of synthetic possibilities for radical chemistry.461,462 More specifically, HAT was coupled with a subsequent chemical oxidation of the first-formed C-centered radical to afford a carbocation. The latter intermediate could be conveniently trapped with N-heteroaryl-based nucleophiles, thus establishing the targeted C–N bond.463 Thus, when an acetonitrile solution of tetrahydrofuran (62.1, 6 equiv), pyrazole (62.2), TBADT (5 mol %), and TBHP (tert-butyl hydroperoxide, 3 equiv) was irradiated with a UV LED (365 nm) for 16 h, product 62.3 was formed in 86% 1H NMR yield. The reaction was made more efficient by adopting a flow apparatus, which allowed reduction of reaction time to just 1 h giving a similar result (86% 1H NMR yield, 81% isolated yield), even though 18 equiv of 62.1 was required (Scheme 62). Notably, the reaction remained efficient when performed on a 10 mmol scale (80% yield) and the synthetic approach could be employed for the late-stage functionalization of biologically active molecules such as acetylated podophyllotoxin (62.4) and stanozolol (62.5).463
Scheme 62. C(sp3)–H Heteroarylation via Radical-Polar Crossover.

3.2. Formation of C–O Bonds
The formation of C–O bonds is a widely investigated transformation among those that can be carried out via photocatalyzed HAT. In these manifolds, the C-centered radical generated via HAT is typically intercepted by a sacrificial oxidant, such as molecular oxygen (O2). The first-formed oxygenated intermediate is a hydroperoxyl radical, which can be isolated as the corresponding hydroperoxide or in situ converted to alcohols, ketones, and even carboxylic acids. Besides its operational simplicity, this transformation has remarkable commercial and industrial implications. For example, when using cyclohexane as the starting material, a mixture of cyclohexanol and cyclohexanone is obtained, the so-called KA–oil (ketone–alcohol mixture).464 The latter has an immense value in modern industry, since it is used for the synthesis of adipic acid, in turn needed to produce Nylon-6,6.465
In view of the above, several research groups have designed photocatalytic systems able to perform this transformation in a sustainable way and carried out meticulous mechanistic studies. Most of them rely on the use of metal-oxo compounds (e.g., the DT or Ur ions) or POCs (e.g., aromatic ketones) under aerobic conditions. Homogeneous conditions are routinely adopted;201,314,472−476,333,338,466−476 albeit, heterogeneous systems have been reported as well.288,477,478
Hereby, the synthetic applications of this chemistry have been reviewed and classified according to the aimed-for product, namely hydroperoxides, alcohols, and ketones or carboxylic acids.
3.2.1. Synthesis of Hydroperoxides
A seminal report on the synthesis of hydroperoxides via photocatalyzed HAT was published in the 1980s and focused on the oxidation of isobutane relying on the use of polyoxometalates as PCsHAT.200,323 In such a case, an acetonitrile solution of the light alkane was irradiated in the presence of TBADT (4 mol %) to afford tert-butylhydroperoxide in a quantitative yield (at 55% conversion of isobutane) after 2 h of irradiation with a 1000 W Xe lamp.323 In another instance, the hydroperoxidation of benzylic C–H bonds by using EY has been reported.250 The irradiation of an acetonitrile solution of 63.1 (0.2 M) under an O2-atmosphere with a blue LED in the presence of the organic dye (2 mol %) delivered compound 63.2 in 80% isolated yield (Scheme 63). Notably, the same reaction was run on a gram scale and 1.18 g of 63.1 was smoothly converted into 0.95 g of 63.2 (68% yield) after 15 h of irradiation.250
Scheme 63. Photocatalyzed Benzylic Hydroperoxidation under Visible Light Irradiation.

3.2.2. Preparation of Alcohol and Ketone Mixtures
Under certain circumstances, hydroperoxyl radicals undergo spontaneous disproportionation to yield a mixture of the corresponding alcohol and ketone.479 This chemistry has been exploited in several instances, especially with cyclohexane as the substrate and TBADT as the PCHAT, to obtain a mixture of cyclohexanol and cyclohexanone.322,480−487
Thus, when a TBADT solution (2 × 10–4 M in CH2Cl2/C6H12/CH3CN 6:3:1) was irradiated at 325 nm with a 250 W Xe lamp (equipped with a grating monochromator), the formation of the KA–oil was observed with a quantum yield of 0.35.480 It is important to mention that a great influence of the oxygen pressure on the final product distribution was observed. In fact, while the formation of the alcohol was not affected when increasing O2 pressure from ∼0.03 to 1 atm, the percentage of ketone in the mixture increased considerably. Thus, while at 0.03 atm the alcohol/ketone ratio was found to be 7:3, at 1 atm the ratio was fully reversed (ca. 3:7).480 As stated by the authors, the distribution of the final products depends on a delicate interplay of the involved radical species. In particular, at low oxygen concentration, the reduced photocatalyst interacts with O2 to afford H2O2 and eventually OH•. Radical recombination of the latter intermediate with the photocatalytically generated cyclohexyl radical affords cyclohexanol. In such a scenario, the formation of the ketone is proposed to be a marginal phenomenon. Conversely, at high pressure of oxygen, different pathways are stimulated, such as radical trapping of cyclohexyl radical by triplet oxygen to get the corresponding hydroperoxyl intermediate. The decomposition of such an intermediate, as also mentioned above, leads to the formation of a mixture of ketone and alcohol through radical chain autoxidation, with a preference for the former.
3.2.3. Synthesis of Carbonyl Derivatives
As mentioned in the previous paragraph, when TBADT-photocatalyzed HAT is carried out under an O2-rich atmosphere, the selectivity can be diverted toward the formation of the C=O double bond. The direct, 4-electron oxidation of a CH2 group into ketones has been achieved on a preparative scale by combining the use of TBADT (5 mol %) and O2 with a flow technology (V = 5 mL; PFA tubing, øin = 750 μm).275 Thus, a MeCN/1 M HCl 2.5:1 solution of cyclohexane (64.1, ∼0.1 M) and molecular oxygen were pumped through a PFA tubing coiled around a 3D-printed PLA cylinder and the mixture was irradiated with UV LEDs (λ = 365 nm). As a result, a cyclohexanone/cyclohexanol mixture (64.2/64.2′ 9:1) was obtained in 90% overall yield from cyclohexane, outperforming the results obtained in batch. Interestingly, this methodology could be extended to natural scaffolds (64.3–64.5) and, for instance, allowed us to realize the oxidation of artemisinin to 9-artemisitone 64.3 with great efficiency and selectivity (59%, gram-scale) in a MeCN/DCM mixture (acid was omitted, Scheme 64).
Scheme 64. Molecular Oxygen as the Oxidant in the Selective CH2 to C=O Conversion.

In another instance, the use of NaDT for the remote oxidation of aliphatic amines was reported.111 Typically, protected amines undergo photocatalyzed HAT only at the α-to-N position. Building upon previous reports, however, it was envisaged that the protonation of amines could divert the HAT reactivity toward remote C–H bonds98 (see also the Introduction, Scheme 2). Thus, the use of flow technology enabled the conversion of pyrrolidine 65.1 to (protected) 3-pyrrolidinone 65.3 under mild conditions on a 5 g scale in 46% isolated yield in the presence of 1.5 equiv of sulfuric acid and molecular oxygen as the oxidant (Scheme 65). The initially formed ketoamine 65.2 was not stable enough for purification, so isolation was performed after derivatization, for example through Boc-protection to give 65.3. The reaction could also be conducted in batch by adopting hydrogen peroxide as the oxidant.111
Scheme 65. Gram-Scale Remote Oxidation of Pyrrolidine.

The direct oxidation of labile benzylic C–H bonds for the synthesis of carbonyl compounds was reported to take place under visible light. For example, UrN was used for the benzylic oxidation of indane and isochroman, respectively, to indanone and isochromanone in high yields (up to 87 and 80%) upon irradiation at 420 nm.488 Similarly, UrP was employed for the oxidation of toluene to benzaldehyde, with minor amounts of benzyl alcohol detected.466
Ketones can be smoothly prepared upon photooxidation of the corresponding alcohols via photocatalyzed HAT. Accordingly, TBADT was used to convert benzyl alcohols to the corresponding ketones in high yields on a 0.1 mmol scale.489,490 Molecular oxygen was used as the oxidant and was bubbled with a constant flow (20 mL min–1) in the solution. Thus, when an acetonitrile solution of 1-(4-methylphenyl)ethanol in the presence of 1 mol % of the PCHAT was irradiated for 90 min, 82% conversion to the corresponding acetophenone was observed (>99% yield). Notably, when the very same PCHAT was immobilized on TiO2, a competition between hydrogen atom transfer and single-electron transfer was observed and the conversion rate was halved (35% in 90 min).489 It is important to mention that, albeit homogeneous TBADT showed higher reaction rates, the heterogenized photocatalyst showed better chemoselectivity toward the α-to-O C–H bond when two distinguishable benzylic sites were present in the substrate, as demonstrated for 1-(4-ethylphenyl)ethanol.489
In another instance, TBADT immobilized on silica (10% w/w) promoted the oxygen-assisted chemoselective photooxidation of 1-pentanol or 3-pentanol to pentanal or 3-pentanone, respectively.491 When the alcohol (5 mM) was irradiated under O2 for 1 h at λ > 290 nm in the presence of the heterogeneous PCHAT (8 g L–1), 90% mass balance was observed. Of note, no overoxidation products were detected in this case, contrary to what was observed under the same conditions when using homogeneous TBADT. The performances of homogeneous and heterogeneous TBADT were then directly compared, and the role of the matrix (silica) was evoked to explain the differences in reactivity. Indeed, the oxidation of 1-pentanol occurred 4-times faster when the PCHAT was supported on SiO2 due to the positive effect of adsorption phenomena. Later on, this heterogeneous PCHAT was also used for the oxidation of diols with similar results in terms of chemoselectivity.492 Similarly, silica-heterogenized Ur has been used for the oxidation of alcohols.478
Intriguingly, a conceptually similar approach was reported for the oxidation of benzyl alcohols (and of aliphatic alcohols to some extent) by NaDT on a preparative scale.493 In detail, zirconia was used as a solid support for this PCHAT (∼19% w/w) and the transformation occurred by irradiating a 0.2 M solution of the starting alcohol with a 400 W high-pressure Hg lamp using a cutoff filter (λ > 320 nm) for less than 2 h in most cases, delivering the expected ketone in >90% yield. However, when benzyl alcohols bearing strongly electron-withdrawing substituents were used, longer irradiation times were required (up to 4 h). As an example, the oxidation of 1-phenylethanol (66.1) occurred smoothly on a 2 mmol scale to give the corresponding acetophenone (66.2) in 94% yield after only 1 h of irradiation (Scheme 66). Notably, the use of the corresponding homogeneous PCHAT led to decreased yields (68%). The photocatalyzed oxidation of 66.1 could be conducted on a 100 mmol scale in 3.25 h to deliver the expected ketone in 92% isolated yield. When the same methodology was applied to nonbranched benzylic alcohols, aldehydes were formed. Thus, benzyl alcohol was smoothly converted to benzaldehyde in 88% yield after 2 h of irradiation.
Scheme 66. Heterogenized NaDT-Photocatalyzed Oxidation of Benzylic Alcohols.

The photooxidation of benzyl alcohols to aldehydes under aerobic conditions has also been reported using water as a benign solvent.494 In particular, a tandem transformation was reported where this first step was triggered by AQN-2-SO3Na (10 mol %), followed by an asymmetric aldol reaction with ketones in the presence of a chiral pyrrolidine-based organocatalyst. Prolonged irradiation (48 h) and substoichiometric acetic acid (50 mol %) were needed to obtain full conversion of the starting materials. For example, (4-bromophenyl)methanol (67.1) was a competent substrate in the presence of cyclohexanone (67.2, 5 equiv), delivering product 67.3 in 71% yield with exquisite diastereo- and enantioselectivity. Benzyl alcohols bearing strong electron-donating substituents failed to give the expected products due to the diminished electrophilicity of the intermediate aldehyde (Scheme 67).
Scheme 67. One-Pot Asymmetric Aldol Reaction Starting from Benzyl Alcohols under Aerobic Conditions.

The same PCHAT was later used to promote the oxidation of both hydrocarbons and alcohols to the corresponding carbonyl compounds. For example, it was found that, upon irradiation with a 200 W white LED lamp, the water-soluble AQN-2-SO3Na (3 mol %) could be used to promote efficiently the oxidation of toluene to benzaldehyde (GC yield: 81%).495 It was observed that the transformation proceeded through the rate-determining formation of benzyl alcohol.495 In another instance, the use of EY as POC extended this transformation to primary alcohols, besides easily oxidizable benzyl alcohols, for the synthesis of quinazolinones in a one-pot fashion.252 As an example, when ethanol (68.1) was used as the substrate in DMSO in the presence of EY (1 mol %) and the mixture was irradiated for 72 h with a 50 W Xe lamp (cutoff > 400 nm) under O2 atmosphere, acetaldehyde was formed in situ and conveniently trapped by o-aminobenzamide 68.2 to give the expected product 68.3 in 66% yield (Scheme 68). Remarkably, this procedure was adopted for the synthesis of 2-alkylquinazolinones, including the sedative-hypnotic drugs methaqualone 68.4 and mecloqualone 68.5.252
Scheme 68. EY-Photocatalyzed Synthesis of Alkyl Quinazolinones via Oxidation of Primary Alcohols.

3.2.4. Synthesis of Carboxylic Acids and Esters
Photocatalyzed HAT was likewise exploited for the oxidation of methyl aromatics to the corresponding benzoic acids.496 As an example, when 4-tert-butyltoluene 69.1 was reacted under aerobic conditions in the presence of ClAQ, a mixture of the corresponding benzoic and perbenzoic acids was obtained. It was found that the use of either a base or an acid could steer the selectivity toward the acid. Accordingly, when a 0.3 M ethyl acetate solution of 69.1 was irradiated in the presence of ClAQ (8 mol %), the corresponding benzoic acid 69.2 was isolated in 97% and 99% yields when using K2CO3 or TFA as an additive, respectively (Scheme 69). A stepwise mechanism was unveiled, where the alkyl aromatic is first converted to the aldehyde via a hydroperoxide intermediate, which is in turn hydrated and then transformed into the acid by a similar photooxidative process.496
Scheme 69. Anthraquinone-Photocatalyzed Benzylic Oxidation of Methylarenes.

Later on, this strategy was used for the direct aerobic photo-oxidative synthesis of aromatic methyl esters starting from methyl aromatics.241 In detail, when methanol was adopted as the reaction solvent, the transient aldehyde was converted to the corresponding hemiacetal and, eventually, to the methyl ester. For example, 69.1 was converted to 69.3 in 89% isolated yield by irradiating a solution containing AQ-2,3-diCOOH (10 mol %) for 24 h in methanol (Scheme 69). Unfortunately, this strategy could not be extended to the synthesis of other alkyl carboxylates, since other alcohols, such as ethanol or propanol, were found to undergo uncontrolled oxidation when used as solvents.
Intriguingly, when 69.1 was reacted in the presence of AQ-2-COOH (5 mol %) in pure aerated ethyl acetate without any additive, the perbenzoic acid 69.4 was formed predominantly (88% overall 1H NMR yield, perbenzoic:benzoic acid ratio 2.8:1). This product could be in turn used in a one-pot fashion to epoxidize 5-decene to give 69.5 in 86% isolated yield when MeCN was used as cosolvent (Scheme 69).497
3.3. Formation of C–S Bonds
Photocatalyzed HAT was also employed to forge C–S bonds via addition of the photogenerated C-centered radicals onto a weak S–S single bond (see also section 4.3). In one instance, PQ was chosen as visible light-triggered PCHAT (5 mol %) to promote the metal- and oxidant-free thioesterification of aldehydes.244 Thus, it was found that thiosulfonate 70.2 used in slight excess (1.2 equiv) was a suitable trap for the acyl radical generated via HAT from 70.1 (0.2 M) because of the high polarization of the S–SO2 bond (Scheme 70). Consequently, the generated sulfonyl radical accounted for the closure of the photocatalytic cycle, yielding product 70.3 in 95% yield after 14 h of irradiation under blue light. The presence of a base (Na2CO3, 0.5 equiv) was found crucial to neutralize the sulfinic acid developed during the reaction. Notably, this protocol could be extended to several structurally diverse aldehydes and thiosulfonate S-esters and could be adopted for the functionalization of complex biologically active molecules, such as probenecid and ursodeoxycholic aldehyde derivatives to give 70.4 and 70.5, respectively.244
Scheme 70. Photocatalyzed Thioesterification of Aldehydes.

Very recently, aldehydes functioned as substrates for a difluoromethylthiolation strategy triggered by photocatalyzed HAT (Scheme 71).498 Both aliphatic and aromatic aldehydes were activated by the excited state of TBADT (4 mol %) to form the corresponding acyl radicals that were in turn trapped by compound 71.2, used in excess. Indeed, the expected difluoromethylthiolated product could be isolated in good yields on a 0.2 mmol scale. As an example, 2-naphthaldehyde 71.1 was converted to thioester 71.3 in 86% yield. As reported in Scheme 71, this methodology was exploited to functionalize biologically relevant molecules, such as L-menthol (product 71.4) and dehydrocholic acid derivatives (product 71.5).
Scheme 71. Difluoromethylthiolation of Aldehydes.

A conceptually similar procedure was reported for the trifluoromethylthiolation of aromatic aldehydes.499 For example, TBADT (4 mol %) was used to cleave the formyl C–H bond in p-anisaldehyde (72.1) and the acyl radical was readily trapped by N-(trifluoromethylthio)phthalimide (72.2) to afford the expected product (72.3, Scheme 72). Notably, the protocol proved robust and enabled this transformation on a gram-scale (5 mmol), as well as the functionalization of fenbufen and ibuprofen to deliver products 72.4 and 72.5, respectively. It is perhaps important to mention that the authors found some limitations with substrates containing electron-withdrawing groups or substituents in the ortho position. They proposed the steric bulkiness of TBADT as the main reason for this diminished reactivity.499
Scheme 72. Trifluoromethylthiolation of Aldehydes.

Recently, a completely different strategy for the formation of C–S bonds has been disclosed. In this work, the authors made use of photocatalyzed HAT to generate C-centered radicals that were readily trapped by a SO2-surrogate (1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide), DABSO) to afford sulfonyl radicals. The latter intermediates were then exploited for a Ni-mediated enantioselective radical addition onto electrophilic olefins (Scheme 73).500 As an example, when adamantane (73.1) was irradiated in the presence of PT (5 mol %) under blue light (455 nm), the tertiary radical was generated. This intermediate was readily trapped by DABSO (73.3, 1.5 equiv) to forge the aimed-for C–S bond and afford a stable sulfonyl radical (e.g., 73.9•). In the meanwhile, an asymmetric Ni complex was formed upon coordination of NiII by chiral ligand 73.4; this Ni complex was responsible for imparting asymmetry on the following step via coordination of 73.2. Finally, the sulfonyl radical added onto 73.2 to eventually afford the expected product 73.5 (77% yield, e.e. 90%, r.r. > 50:1).500 This protocol was likewise amenable to activated benzylic C–H bonds and to functionalize celestolide (to give 73.6) and a synthetic intermediate of differin (73.7).
Scheme 73. Three-Component Asymmetric Sulfonylation via HAT.

3.4. Formation of C–F Bonds
3.4.1. Fluorination of C(sp3)–H Bonds
The introduction of a fluorine atom onto unactivated sites of organic molecules, such as strong aliphatic C(sp3)–H bonds, is a hot topic in medicinal and pharmaceutical sciences. In fact, fluorination makes a molecule more lipophilic, which results in ameliorated biological absorption and distribution. This transformation is of great importance in materials science too, where the incorporation of the halogen endows materials with a remarkable hydrophobicity.501 Photocatalyzed HAT has been widely used to promote this highly desirable transformation.502 This chemistry mainly relies on fluorinating agents containing a weak N–F bond (BDE = 61–63 kcal mol–1), such as N-fluorobenzenesulfonimide and Selectfluor.503
In one of the earliest examples of this chemistry, the use of aromatic ketones for the mono- and gem-difluorination of benzylic C(sp3)–H bonds in the presence of Selectfluor was reported.272 In particular, when an acetonitrile solution of 1-(tert-butyl)-4-methylbenzene (74.1, 0.08 M) was irradiated with a CFL lamp in the presence of FL (5 mol %) and 2 equiv of Selectfluor (74.2a), the monofluorination at the benzylic site occurred in 82% yield to give compound 74.3 in 6 h (Scheme 74). However, when the reaction was performed in the presence of XA (5 mol %) and Selectfluor II (74.2b, 3 equiv) as PCHAT and fluorinating agent, respectively, the gem-difluorinated product 74.4 was isolated in 72% yield after 16 h. From the mechanistic standpoint, the obtained organoradical was fluorinated by reaction with 74.2 to deliver the (di)fluorinated product and a N-centered radical cation, which is entrusted for the closure of the photocatalytic cycle.
Scheme 74. Diarylketone-Photocatalyzed Selective Benzylic Mono- and Difluorinations.

More recently, the above-mentioned procedure for the monofluorination of alkylaromatics was used to develop a one-pot, transition metal-free cross-dehydrogenative arylation of unactivated benzylic C–H bonds (Scheme 75).504 In particular, FL was used to trigger the halogenation step, while the so-formed benzyl fluorides (75.2) were used as electrophilic partners in a nucleophilic substitution with electron-rich (hetero)arenes (e.g. 75.3 to give 75.4).504
Scheme 75. One-Pot Arylation of Unactivated Benzylic C–H Bonds.

In a related example, the same monofluorination reaction was performed under flow conditions by circulating an acetonitrile solution of 76.1 through a transparent FEP tubing coiled around a glass cylinder (V = 28 mL, flow rate = 1 mL·min–1) containing a 105 W CFL.286 Under these conditions, it was found that XA (5 mol %) could perform the monofunctionalization of the benzylic site to get 76.3 in 89% yield with a residence time of 28 min (temperature of the system = 40 °C). This reaction manifold was adapted also to the fluorination of drugs, such as celestolide and ester-protected ibuprofen (Scheme 76). As for celestolide, milder conditions were required (flow rate = 3 mL min–1, tr = 9 min, temperature: 25 °C) to get the corresponding fluorinated compound in 88% yield and the reaction could be conveniently scaled up with comparable efficiency (87% yield). Contrarily, in the case of ibuprofen, “harsher” conditions were needed (flow rate = 1 mL min–1, tr = 28 min, temperature: 60 °C) to get the expected product in 80% yield, with the secondary benzylic C–H position being almost exclusively functionalized (>90% selectivity).
Scheme 76. Photocatalyzed Benzylic Fluorination under Flow Conditions.

In another case, the benzylic fluorination of alkyl aromatics was achieved in the presence of TBADT and NFSI (77.2) as the fluorinating agent.505 Thus, ibuprofen methyl ester 77.1 was smoothly functionalized at the less-hindered benzylic position in 75% yield. It is important to stress that, when a flow apparatus was adopted (V = 1.4 mL, FEP tubing wrapped around a blacklight blue lamp; λ = 365 nm), the reaction was significantly sped up, from 24 h required in batch to only 5 h (70% yield, Scheme 77). Hydrolysis of the fluorinated ester gave fluoroibuprofen, whose metabolic stability in human and rat microsomes was evaluated to be somewhat higher compared to the parent drug.
Scheme 77. TBADT-Photocatalyzed Benzylic Fluorination in Flow.

Similarly, DBS was used to promote the selective fluorination of phenylalanine-like residues in amino acids and short chain peptides at the benzylic site. In this case, Selectfluor was employed as the fluorinating agent while a 14 W CFL was used as the light source.243
Photocatalyzed HAT can be conveniently used also for the fluorination of strong aliphatic C–H bonds. In one case, the fluorination of cycloalkanes took place making use of aromatic ketones such as AP, which was effective in the C–H to C–F bond conversion upon irradiation with a commercially available CFL (Scheme 78). For example, the fluorination of cyclohexane, cycloheptane, and cyclooctane 78.1a–c occurred in 90%, 85%, and 82% 19F NMR yield, respectively.224
Scheme 78. Photocatalyzed Preparation of Fluorocycloalkanes.

Another convenient PCHAT that can be used for the fluorination of strong, unactivated C–H bonds is TBADT (Scheme 79).506 In particular, this polyoxometalate has been exploited for the fluorination of a wide array of compounds, including biologically valuable molecules such as (protected) amino acids and sclareolide (79.1). Thus, when an acetonitrile solution of 79.1 was irradiated for 17 h in the presence of NFSI (79.2, 1.2 equiv) and TBADT (2 mol %) at 365 nm (15 W black light bulbs), a mixture of the 8- and 9-fluorinated adducts was obtained (68% overall yield, ratio 79.3/79.4 5.8:1). Interestingly, the selectivity enabled by this system is analogous to that previously reported adopting manganese porphyrins in the presence of fluoride anion.507
Scheme 79. Photocatalytic Fluorination of Unactivated C–H Bonds.

In this context, a similar strategy was adopted for the functionalization of the methine site in leucine. Thus, NaDT was found useful for the selective fluorination of γ-leucine methyl ester (80.1), a key intermediate on the route to odanacatib, a potent inhibitor of cathepsin K. Intriguingly, this transformation was carried out under flow conditions and could be easily scaled up, to get ∼45 g of product (90% yield, Scheme 80).508
Scheme 80. TBADT-Photocatalyzed Fluorination of Leucine in Flow.

The same procedure was used to achieve the site-selective 18F-fluorination of unprotected amino acids273 and peptides,509 which has an intrinsic value for positron emission tomography (PET) imaging. Later on, the same fluorination concept was adopted for the labeling of ZJ-43 analogues, potent binders for PSMA (prostate specific membrane antigen), that are overexpressed in the case of prostate cancer. Parent ZJ-43 could not be efficiently labeled (<12%) with 18F; however, the addition of pendant ammonium groups was found to accelerate the functionalization (>46% yield).510
Quite recently, the UrN photocatalyst has been used for the fluorination of cycloalkanes under visible light irradiation. Thus, a deuterated acetonitrile solution of cyclooctane (81.1) was irradiated in the presence of UrN (1 mol %) and 1.5 equiv of NFSI (81.2) for 16 h under inert atmosphere to afford fluorocyclooctane (81.3) in 95% 1H NMR yield (Scheme 81).284
Scheme 81. Visible Light Fluorination of Cyclooctane.

3.4.2. Fluorination of C(sp2)–H Bonds
The fluorination of aldehydes via the activation of the formyl C(sp2)–H bond has been sparsely reported.511 Here, the capability of DT to generate acyl radicals from aldehydes (82.1) was exploited. These intermediates were in turn trapped by NFSI to get the corresponding acyl fluorides (82.3, Scheme 82). Since acyl fluorides were found to be unstable in several cases, they were treated in situ with benzylamine to get the corresponding amides (82.4). For example, when an acetonitrile solution of benzaldehyde 82.1 (0.3 M) was irradiated at 365 nm in the presence of NaDT (2.5 mol %) and NFSI (82.2, 1.2 equiv), the corresponding acyl fluoride 82.3 was formed in 79% 19F NMR yield. After irradiation, the addition of 2 equiv of benzylamine allowed isolation of benzamide 82.4 in 79% yield. This reactivity was extended to several aliphatic aldehydes as well. Notably, 82.4 could be obtained in 53% isolated yield starting from benzyl alcohol in the presence of a higher amount of NFSI (2.5 equiv) in a one-pot fashion.511
Scheme 82. Synthesis of Acyl Fluorides from Aldehydes.

3.5. Formation of C–Cl Bonds
Similarly to what was shown in the previous section for C–F bond formation, C–Cl bonds can also be formed via photocatalyzed HAT. In these cases, electrophilic reactants containing a weak N–Cl bond are exploited, with one typical example being N-chlorosuccinimide (NCS).
In one instance, the halogenation of α-to-boron C(sp3)–H bonds in benzyl N-methyliminodiacetyl (MIDA) boronates was reported.512 Thus, the authors started off by studying the bromination of said bonds and found an effective photochemical way to promote reactivity. In particular, the direct irradiation of a solution containing the boronate ester and N-bromosuccinimide (NBS) under inert atmosphere with a 13 W CFL led to the expected products. However, this protocol failed when replacing NBS with NCS to perform chlorination of C–H bonds; intriguingly, the authors found that the addition of 5 mol % of FL restored reactivity (Scheme 83). Upon mechanistic investigation, they found that FL acted as a photocatalyst for HAT. Interestingly, by replacing NCS with Selectfluor, the authors were able to achieve the fluorination of α-to-boron C(sp3)–H bonds.512
Scheme 83. Chlorination of Benzyl MIDA Boronates.

In another instance, a similar chlorine source was used for the chlorination of (+)-sclareolide (84.1) under flow conditions (Scheme 84).513 Interestingly, the authors designed a “Schlenk-in-flow” technique for the safe handling of oxygen- and moisture-sensitive reagents, where argon is used instead of solvent to drive reagents through the tubing. Doing so, distribution phenomena are suppressed, which allowed saving both reagents and solvent. The photocatalyzed chlorination of 84.1 was one of their benchmark reactions: when an acetonitrile solution of 84.1 was pumped through the photoreactor (VR = 10 mL, tR = 100 min) in the presence of TBADT (10 mol %) and 84.2 (1.5 equiv) under UV light (365 nm), 38% of the chlorinated product 84.3 was obtained on a gram scale.
Scheme 84. Chlorination of (+)-Sclareolide via a Schlenk-in-Flow Approach.

3.6. Formation of C–D Bonds
Deuterium labeling has a range of applications, including the investigation of reaction mechanisms, drugs metabolism and distribution, as well as in spectroscopy. Accordingly, it is no surprise that photocatalyzed HAT was investigated as a direct and unique platform to access the formation of the C–D (D: deuterium) bond by the so-called hydrogen isotope exchange (HIE).514−517
In this frame, two similar approaches have been published very recently. In both cases, the photogenerated radicals (TBADT as the PCHAT) were quenched by an aromatic thiol (triisopropylbenzenethiol, TIPSH) present in catalytic amounts and involved in an acid/base equilibrium with D2O. While in one instance the deuteration of the formyl C–H bond in both aromatic and aliphatic aldehydes was achieved,518 in the other one the methodology was extended to the deuteration of benzylic sites and tertiary C(sp3)–H bonds, besides aldehydes.519
In the former case, 4-bromobenzaldehyde 85.1 was deuterated in 95% yield (D incorporation: 96%) by means of the TBADT/TIPSH system (used in 4 and 40 mol %, respectively) in DCM/D2O 1:1 upon irradiation for 24 h (Scheme 85a).518 In the latter case, the deuteration could be extended to benzylic sites and tertiary C(sp3)–H bonds simply by adding a phase transfer catalyst (TBAB, tetrabutylammonium bromide). For example, 85.2 and 85.3 were functionalized in 92% (D incorporation: 91%, 24 h) and 99% (D incorporation: 81%, 48 h) yield, respectively (Scheme 85b).519
Scheme 85. Photocatalyzed Hydrogen Isotope Exchange (HIE).

4. Formation of Other Bonds
Photocatalyzed HAT has been likewise employed for the generation of heteroradicals, including P-, Si-, and S-centered radicals.
4.1. Formation of P–C Bonds
As for P–C bond formation, the enantioselective conversion of hydrophosphine oxide 86.1 to the phosphine oxide 86.3 via EY-photocatalyzed HAT has been reported (Scheme 86).390 In particular, when a solution of amide 86.2 (1 M) in MTBE/H2O ∼ 3:1 was irradiated with a blue LED in the presence of EY (1 mol %) and 86.1 as the H-donor, 86.3 was formed in 71% yield (e.e. 68%). As mentioned before (see Scheme 19d), the enantioselectivity was entrusted to a chiral Rh complex (4 mol %). Unfortunately, the reaction required a long time to go to completion (3 days).
Scheme 86. Enantioselective Formation of a P–C Bond.

Very recently, EY was used to promote the formation of a P–C bond starting from hydrophosphine oxide as H-donor and N-(hetero)arylsulfonyl propiolamides. As already mentioned above (see Scheme 45 in section 2.5), this transformation proceeded through a cascade involving 1,4-addition of the P-centered radical onto the C≡C triple bond of propiolamide, Smiles rearrangement, and 5-endo-trig cyclization.437 In a very recent report, N-aminopyridinium salts have been used as radical traps for photogenerated P-centered radicals (AQ as the photocatalyst), resulting in the pyridylation of a handful of hydrophosphine oxides.416
4.2. Formation of Si–X Bonds (X = Cl or C)
Photocatalyzed HAT has been used also for the formation of Si–X bonds (X = Cl, C). This strategy requires that a Si–H bond is photocatalytically cleaved, which is easily predicted by the enhanced electropositivity of silicon compared to carbon. This feature contributes to make the H atom to be abstracted more hydridic.248 The forging of the Si–Cl bond gives access to chlorosilanes, currently used for the protection of alcohols and acids. The reaction occurred under visible light irradiation (EY as the PCHAT) and relied upon DCM in the double role of chlorinating agent and solvent. The range of applicability of this transformation is remarkably broad, and almost all chlorosilanes were isolated in quantitative yield. Notably, flow chemistry allowed successfully performing the process up to the gram scale. For example, when hydrosilane 87.1 (0.2 M) was irradiated with EY (1 mol %) in DCM with an 18 W blue LED strip, the corresponding chlorinated product 87.2 was obtained quantitatively after 4 h. By means of the flow technique (HPFA tubing, V = 7 mL, flow rate ∼ 0.1 mL·min–1), the same yield was achieved with a residence time of 1.1 h and 1.18 g of 87.2 was produced after 4 h (Scheme 87).248
Scheme 87. Photocatalyzed Synthesis of Chlorosilanes.

As for the formation of the Si–C bond, a handful of reports describe the use of TBADT for the activation of the Si–H bond (Scheme 88).277,378,386,520 Thus, when an acetonitrile solution of methyldiphenylsilane 88.1 was irradiated with UV light (310 nm, 10 × 15 W phosphor-coated lamps) for 24 h in the presence of TBADT (2 mol %), the expected Si-centered radical was generated via HAT.520 This intermediate was readily trapped by dimethyl maleate (88.2) to afford the corresponding silylated compound in excellent isolated yield (90%), even though it should be noted that 28% yield was observed in the absence of the PCHAT. The use of transient absorption spectroscopy and steady-state EPR experiments served to prove the nature of the activation step. Interestingly, when silanes containing more labile Si–H bonds (e.g., tris(trimethylsilyl)silane) were used, the reaction proceeded even in the absence of the photocatalyst due to radical chain processes. On the other hand, when trialkylsilanes were used, the competitive activation of the α-to-Si C–H and Si–H bonds was observed, thus generating a mixture of C- and Si-functionalized products.520
Scheme 88. TBADT-Photocatalyzed Hydrosilylation of Electron-Poor Olefins.

4.3. Formation of S–C Bonds
The formation of S–C bonds can be achieved through the addition of S-centered radicals generated via HAT onto electron-rich olefins such as styrenes. In one case, a thiol–ene process has been reported by using BP (10 mol %) as the PCHAT, which allowed formation of sulfide 89.3 in 87% yield after 12 h of irradiation of a mixture of thiophenol (89.1) and styrene (89.2) with a 18 W CFL (Scheme 89).236
Scheme 89. BP-Photocatalyzed S–C Bond Formation.

Quite recently, an asymmetric manifold for the formation of the S–C bond has been proposed.390 Therein, a sulfinic acid was activated via HAT by EY to get the corresponding S-centered radical. The latter intermediate was trapped by α,β-unsaturated amides through a radical addition manifold to forge the desired S–C bond. Notably, a Ru-based chiral Lewis acid was used to coordinate the amide with a dual objective: favoring the radical addition step and introducing asymmetry.390
Finally, given the importance of the trifluoromethylthio (−SCF3) moiety in medicinal chemistry, a methodology based on HAT has been recently proposed for the trifluoromethylation of β-ketodithioesters under visible light irradiation.251 Thus, when methyl 3-oxo-3-(3,4,5-trimethoxyphenyl)propanedithioate was irradiated with blue light in the presence of EY (2 mol %) and sodium triflinate, the corresponding trifluoromethylthiolated product was obtained as a single diastereomer (Z) in 89% yield after 15 h. It is perhaps worth mentioning that the reaction worked to some extent (37% yield) also in the absence of the PCHAT.
5. Miscellanea
This section gathers examples that do not fit into any of the categories above. In particular, net-oxidative processes based on a HAT step such as dehydrogenation and dehydroformylation reactions, as well as reactions based on a C–C oxidative cleavage, are described.
Seminal examples of such processes have been reported in the mid 1980s/early 1990s and were based on the use of polyoxometalates as PCsHAT, with particular focus on the DT anion; albeit, the reactions were usually stopped at low conversion of the starting mixtures.200 Thus, the dehydrogenation of a library of organic substrates (mostly alkanes and alcohols) based on photocatalyzed HAT has been reported.321,324,329,521,522 Indeed, specific conditions have been found to deliver the expected products with a high quantum yield (approaching unity),522 as well as to favor the formation of nonthermodynamic alkenes from alkanes.329 Furthermore, it was demonstrated that photocatalyzed HAT can also be exploited to trigger the epimerization of C–H bonds (e.g., in decalone derivatives).324,521
More recently, a dual-catalytic system consisting of TBADT (0.4 mol %) and cobaloxime pyridine chloride ([Co(dmgH)2(pyr)Cl], COPC; 0.4 mol %) was used for the preparative dehydrogenation of alkanes.523 This methodology was baptized “cooperative HAT”, since it involved the combination of photocatalyzed HAT and thermal HAT.524 In detail, the PCHAT was responsible for the activation of a strong C(sp3)–H bond, for example in cyclooctane 90.1, in what has been called a “hard” HAT step (Scheme 90a). Subsequently, the Co-based complex exerted an “easy” HAT from the α-position with respect to the radical site. The two catalytic cycles met when the reduced DT and the spent cocatalyst interacted to release a molecule of hydrogen gas. Ultimately, cyclooctene (90.2) was obtained in 19% NMR yield upon irradiation for 48 h. The very same procedure was also successfully applied to secondary alcohols, delivering the corresponding ketones in good yields.523
Scheme 90. TBADT/Cobaloxime Dual-Catalytic Approaches for the (a) Dehydrogenation of Alkanes and (b) Dehydroformylation of Aldehydes.

Later on, a dehydroformylation procedure via HAT has been developed.525 In particular, the same dual-catalytic system has been proposed to realize the decomposition of an aldehyde to an alkene, hydrogen gas, and carbon monoxide. Thus, aldehydes containing a nonenolizable quaternary α-carbon, such as 2,2-dimethyl-3-phenylpropanal 90.3, were tested at first (Scheme 90b). TBADT (4 mol %) generated an acyl radical under UV light irradiation, that spontaneously underwent decarbonylation to give a tertiary radical. The latter intermediate was in turn intercepted by cobaloxime (COPC, 4 mol %), that triggered a further dehydrogenation step, with a preference toward the less substituted olefin (terminal olefin 90.4). Aldehydes with an enolizable carbon α to the carbonyl group, however, were not competent substrates in this reaction.525
Another photocatalytic approach for the deconstructive cleavage of organic compounds via HAT dealt with the aerobic photooxidative cleavage of 1,3-diketones to give carboxylic acids.526 In such a case, ClAQ (10 mol %) was used to trigger a H-abstraction in benzoylacetones, such as 1-phenyl-1,3-butanedione 91.1 (Scheme 91a). Thus, when an acetone solution (0.06 M) was irradiated with four 22 W CFLs in the presence of oxygen and a base, the corresponding benzoic acid 91.2 was obtained in 72% yield after 20 h. The proposed mechanism underlying this transformation is quite complicated and proceeds through the formation of transient hydroperoxides and α-hydroxycarbonyl and α-dicarbonyl compounds.526 In a related instance, similar conditions were applied for the aerobic oxidative cleavage of cyclic acetals (Scheme 91b).527 As an example, irradiation of 1,3-dioxolane 91.3 in ethyl acetate solution in the presence of AQ-2-COOH (10 mol %) followed by quenching with sodium thiosulfate allowed synthesizing the corresponding ester 91.4 in good yield (69%).
Scheme 91. Aerobic Photooxidative Cleavage of (a) 1,3-Diketones and (b) 1,3-Dioxolanes.

6. Conclusions
It is apparent from the examples described in this review that the photocatalyzed hydrogen atom transfer approach has had and will have in future years a primary role in synthetic planning, in both academic and industrial settings. The versatility of the method is witnessed by the different types of bonds that can be forged, even though most of them are C–C bonds (see Table 2). In the majority of the examples reviewed, the reactions are carried out under mild conditions with no need of heating or aggressive conditions. In many instances, in the reactions developed all of the atoms of the reagents are incorporated in the desired products (100% atom economy).
Table 2. Use of the Diverse Classes of PCsHAT in the Forging of New Bonds.
| Bond formed/PCsHAT | Aromatic ketones | Anthraquinones | EY | DT | Ur | Sb-Oxo | TAC•2+ |
|---|---|---|---|---|---|---|---|
| C(sp3)–C(sp3) | √ | √ | √ | √ | √ | √ | |
| C(sp3)–C(sp2) | √ | √ | √ | √ | |||
| C(sp3)–C(sp) | √ | √ | √ | √ | |||
| C(sp2)–C(sp3) | √ | √ | √ | √ | √ | ||
| C(sp2)–C(sp2) | √ | √ | |||||
| C(sp2)–C(sp) | √ | ||||||
| C–N | √ | √ | |||||
| C–O | √ | √ | √ | √ | √ | ||
| C–S | √ | √ | √ | ||||
| C–Cl | √ | √ | |||||
| C–F | √ | √ | √ | ||||
| C–D | √ | ||||||
| Other bonds P–C, Si–Cl, Si–C, S–C | √ | √ | √ |
Table 2 gives an idea of the importance of the different classes of PCsHAT used in connection with the bond they allow to build. Decatungstate salts (either NaDT or TBADT) are by far the most employed PCsHAT, and they are involved in all of the processes described. The capability of DT-based PCsHAT to cleave even very strong bonds, such as the C–H bond in methane (BDE = 105 kcal/mol), showcases their impressive potentialities. Moreover, dual-catalytic approaches with Ni, Pd, Cu, and Co have extended even further the boundaries of the chemical space that can be explored with them. This colorless anion is robust, cheap, and easy to prepare; however, its chemistry is mainly restricted by the need for highly energetic UV light (<400 nm). Accordingly, in recent years a lot of work has been devoted to the discovery of visible light absorbing PCsHAT. Albeit colored polyoxometalates that can compete with DT are so far lacking, other classes of colored PCsHAT have been tested, including oxo-porphyrins (Sb-Oxo), uranyl salts (Ur), or MOFs. Photoorganocatalysts appear as promising derivatives worthy to be further investigated. As for aromatic ketones, early examples dealt only with the use of colorless BP, but nowadays slightly colored diaryl ketones (e.g., FL, DTX, PT) or α-diketones (PQ) are preferred. Anthraquinone derivatives could have a more important role in the future; albeit, they have been used only in selected cases so far. EY is close to the ideal candidate as PCHAT due to its wide availability and the performance showed, but its competitive role as PCSET or singlet oxygen sensitizer must be carefully considered to avoid undesired side reactions. The use of alternative hydrogen abstractors, such as the excited state of the electrogenerated tris(amino)cyclopropenium radical dication (TAC•2+), is still in its infancy as it is the photoelectrochemistry approach.528
As apparent from Table 2, the forging of C(sp3)–C(sp3) bonds is widely investigated and a lot of conditions have been tested, making use of almost all of the classes of PCsHAT available in conjunction with other catalysts, in turn resulting in interesting dual-catalytic applications. The Giese radical addition is the archetypal reactivity mode in this group.
However, in C–C bond formation limited efforts have been devoted to the formation of C(sp2)–C(sp2) and even less to C(sp2)–C(sp) bonds. The same holds for the construction of C–N and C–D bonds.
One of the main drawbacks of HAT reactions is that the homolytic cleavage of C–H bonds involves “nucleophilic” rather than “electrophilic” hydrogens, and it is quite rare to find a process that points to the generation (and ensuing reactions) of an electrophilic radical.179 However, quite recently, a new methodology based on radical polar crossover was exploited to reverse the polarity of said nucleophilic radical by generating a carbocation upon chemical oxidation.463 This concept finally made photocatalyzed HAT suitable to explore new chemical spaces, namely the world of polar chemistry. The possibility to choose the nucleophile to trap the carbocation sets up a fertile ground to develop new synthetic methodologies based on HAT.
The final comment is on selectivity in the hydrogen abstraction. As mentioned in the Introduction, there are several factors that may affect and direct the cleavage of a C–H bond out of all those present in the hydrogen donor. Dated examples typically dealt with hydrogen donors where all C–H bonds (e.g., in cyclohexane) were equivalent or the hydrogen abstraction was merely governed by the BDE (e.g., in tetrahydrofuran). Conversely, in recent times, very selective HAT reactions have been reported especially in the late-stage functionalization of complex molecules. This demonstrates that mastering the knowledge of all the factors explained in the Introduction allows chemists to develop surprisingly selective processes. Accordingly, further efforts toward a better understanding of these factors is needed to increase the potentialities of HAT reactions in the future. Finally, efforts on the development of hydrogen abstractors having peculiar shapes or a marked bulkiness as well as on the study of the cooperative effects in tuning the selectivity of the reaction are then crucial in this respect.
Acknowledgments
This review is in honor of Prof. Ilhyong Ryu on the occasion of his 70th birthday. L.C. acknowledges European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 101023615 (project name: HAT-TRICK).
Glossary
Abbreviations
- ABP
aminobenzophenone
- AP
acetophenone
- AQ
9,10-anthraquinone
- AQ-2-COOH
anthraquinone-2-carboxylic acid
- AQ-2,3-diCOOH
anthraquinone-2,3-dicarboxyliic acid
- AQ-2-SO3Na
anthraquinone-2-sulfonic acid sodium salt
- tBAQ
2-tert-butylanthraquinone
- BLB
Black light bulb
- Boc
tert-butoxycarbonyl
- BOX
bisoxazoline
- BP
benzophenone
- BPO
benzoyl peroxide
- BPSS
disodium benzophenondisulfonate
- Brsm
based on remaining starting material
- CFL
compact fluorescent lamp
- ClAQ
2-chloroantraquinone
- COPC
cobaloxime pyridine chloride
- DBS
dibenzosuberenone
- DCBP
4,4′-dichlorobenzophenone
- DCM
dichloromethane
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DIAD
diisopropyl azodicarboxylate
- DIPEA
N,N-diisopropylethylamine
- DMBP
4,4′-dimethoxybenzophenone
- DT
decatungstate anion
- DT-BPY
[Cu4(BPY)6Cl2(W10O32)]·3H2O
- DTX
3,6-dimethoxy-9H-thioxanthen-9-one
- EY
Eosin Y
- FL
9-fluorenone
- FPT
freeze pump–thaw
- HAT
hydrogen atom transfer
- h.p.
high pressure
- l.p.
low pressure
- MFC
mass flow controller
- m.p.
medium pressure
- MTBE
tert-butyl methyl ether
- NaDT
sodium decatungstate
- PC
photocatalyst
- P.E.
petroleum ether
- POC
photoorganocatalyst
- PGA
phenylglyoxylic acid
- PSMA
Prostate specific membrane antigen
- PQ
9,10-phenanthrenequinone
- PT
5,7,12,14-pentacenetetrone
- PTSA
p-toluenesulfonic acid.
- PYD
1,6-pyrenedione
- Sb-Oxo
antimony-oxo tetra-(p-methoxyphenyl)porphyrin
- SOLFIN
solar synthesis of fine chemicals
- TBAB
tetrabutyl ammonium bromide
- TBADT
tetrabutylammonium decatungstate
- TBDPS
tert-butyldiphenylsilyl
- TEA
triethylamine
- TFA
trifluoroacetic acid
- TIPSH
triisopropylbenzenethiol
- Ts
tosyl
- TX
thioxanthone
- Ur
uranyl cation
- UrN
uranyl nitrate hexahydrate
- UrP
uranyl perchlorate
- XA
xanthone
Biographies
Luca Capaldo was born in Milan, Italy, in 1991. After an Erasmus Traineeship in the De Cola group at the University of Strasbourg and a visiting period as a Ph.D. student in the Yoon Group at the University of Wisconsin—Madison, he received his doctorate in Chemical and Pharmaceutical Sciences at the University of Pavia in 2019 (advisor: Prof. M. Fagnoni). After a 2-year postdoctoral fellowship in Pavia (PI: Prof. D. Ravelli), Luca joined the Flow Chemistry Group at the University of Amsterdam as a Marie Curie Fellow (advisor: Prof. T. Noël) to develop novel synthetic methodologies based on photocatalyzed HAT in flow.
Davide Ravelli is currently Assistant Professor at the Department of Chemistry of the University of Pavia, where he also obtained his Ph.D. in Chemistry in 2012 (Prof. A. Albini as the supervisor). His main research interests focus on the generation of radical intermediates through photocatalyzed hydrogen atom transfer and their application in sustainable organic synthesis. In 2017, he received the Ciamician medal and the Vincenzo Caglioti international award.
Maurizio Fagnoni is currently a Full Professor in Organic Chemistry at the PhotoGreen Lab (Department of Chemistry, University of Pavia, Italy). His academic and professional background is in organic photochemistry, and his activity has always been focused on the exploration of the use of photochemistry and photocatalysis for organic synthesis. The photochemical generation of intermediates, for example, radicals and cations and radical ions by photochemical means, is the main topic of his research. Particular attention has been given to the significance of such mild synthetic procedures in the frame of the increasing interest for sustainable/green chemistry. He was the recipient in 2019 of the “Elsevier Lectureship Award” from the Japanese Photochemical Association. He was recently coeditor of the book Photoorganocatalysis in Organic Synthesis (World Scientific, 2019).
The authors declare no competing financial interest.
The photocatalyzed cyanation of alkanes has been reported (compare section 2.4)529 The photocatalyzed acylation of crotonic acid530 and the alkylation of aldehydes with pyridinium salts531 to give ketones have been published (compare section 2.5). The decatungstate-photocatalyzed functionalization of [60]fullerene with lactones was devised (compare section 2.8).532 The photocatalyzed C(sp3)−H sulfinylation533 or C(sp3)−H/C(sp2)−H trifluoromethylthiolation534 for the forging of C−S bonds have been obtained (compare section 3.3).
This paper was originally published ASAP on August 6, 2021, with Table 1 absent from the PDF version. The revised version was posted August 16, 2021.
References
- Braun M.Modern Enolate Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015. [Google Scholar]
- Stolz D.; Kazmaier U.. Metal Enolates as Synthons in Organic Chemistry. In PATAI’S Chemistry of Functional Groups; John Wiley & Sons, Ltd: Chichester, UK, 2010. [Google Scholar]
- Goldman A. S.; Goldberg K. I.. Organometallic C–H Bond Activation: An Introduction. In Activation and Functionalization of C–H Bonds; ACS Symposium Series 2004; Vol. 885, pp 1–43. [Google Scholar]
- Hartwig J. F.; Larsen M. A. Undirected, Homogeneous C–H Bond Functionalization: Challenges and Opportunities. ACS Cent. Sci. 2016, 2, 281–292. 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambiagio C.; Schönbauer D.; Blieck R.; Dao-Huy T.; Pototschnig G.; Schaaf P.; Wiesinger T.; Zia M. F.; Wencel-Delord J.; Besset T.; Maes B. U. W.; Schnürch M. A Comprehensive Overview of Directing Groups Applied in Metal-Catalysed C–H Functionalisation Chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. 10.1039/C8CS00201K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasheed O. K.; Sun B. Advances in Development of C–H Activation/Functionalization Using a Catalytic Directing Group. ChemistrySelect 2018, 3, 5689–5708. 10.1002/slct.201801097. [DOI] [Google Scholar]
- Crabtree R. H. Introduction to Selective Functionalization of C–H Bonds. Chem. Rev. 2010, 110, 575–575. 10.1021/cr900388d. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Du Bois J.; Yu J.-Q. C–H Functionalization in Organic Synthesis. Chem. Soc. Rev. 2011, 40, 1855–1856. 10.1039/c1cs90010b. [DOI] [PubMed] [Google Scholar]
- Doyle M. P.; Goldberg K. I. C–H Functionalization (special issue). Acc. Chem. Res. 2012, 45, 777–777. 10.1021/ar300096z. [DOI] [PubMed] [Google Scholar]
- Rouquet G.; Chatani N. Catalytic Functionalization of C(sp2)–H and C(sp3)–H Bonds by Using Bidentate Directing Groups. Angew. Chem., Int. Ed. 2013, 52, 11726–11743. 10.1002/anie.201301451. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F. Evolution of C–H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc. 2016, 138, 2–24. 10.1021/jacs.5b08707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kärkäs M. D. Electrochemical Strategies for C–H Functionalization and C–N Bond Formation. Chem. Soc. Rev. 2018, 47, 5786–5865. 10.1039/C7CS00619E. [DOI] [PubMed] [Google Scholar]
- Wencel-Delord J.; Glorius F. C–H Bond Activation Enables the Rapid Construction and Late-Stage Diversification of Functional Molecules. Nat. Chem. 2013, 5, 369–375. 10.1038/nchem.1607. [DOI] [PubMed] [Google Scholar]
- Cernak T.; Dykstra K. D.; Tyagarajan S.; Vachal P.; Krska S. W. The Medicinal Chemist’s Toolbox for Late Stage Functionalization of Drug-like Molecules. Chem. Soc. Rev. 2016, 45, 546–576. 10.1039/C5CS00628G. [DOI] [PubMed] [Google Scholar]
- Moir M.; Danon J. J.; Reekie T. A.; Kassiou M. An Overview of Late-Stage Functionalization in Today’s Drug Discovery. Expert Opin. Drug Discovery 2019, 14, 1137–1149. 10.1080/17460441.2019.1653850. [DOI] [PubMed] [Google Scholar]
- Cannalire R.; Pelliccia S.; Sancineto L.; Novellino E.; Tron G. C.; Giustiniano M. Visible Light Photocatalysis in the Late-Stage Functionalization of Pharmaceutically Relevant Compounds. Chem. Soc. Rev. 2021, 50, 766–897. 10.1039/D0CS00493F. [DOI] [PubMed] [Google Scholar]
- Dénès F. Intermolecular Radical C–H Bond Activation: A Powerful Tool for Late Stage Functionalization. Chimia 2020, 74, 23–32. 10.2533/chimia.2020.23. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Verma V. A.; Paxton T. J.; Pillow T. H. Function-Oriented Synthesis, Step Economy, and Drug Design. Acc. Chem. Res. 2008, 41, 40–49. 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- McMurray L.; O’Hara F.; Gaunt M. J. Recent Developments in Natural Product Synthesis Using Metal-Catalysed C–H Bond Functionalisation. Chem. Soc. Rev. 2011, 40, 1885. 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]
- Qiu Y.; Gao S. Trends in Applying C–H Oxidation to the Total Synthesis of Natural Products. Nat. Prod. Rep. 2016, 33, 562–581. 10.1039/C5NP00122F. [DOI] [PubMed] [Google Scholar]
- Crabtree R. H. Alkane C–H Activation and Functionalization with Homogeneous Transition Metal Catalysts: A Century of Progress - a New Millennium in Prospect. J. Chem. Soc. Dalt. Trans. 2001, 2437–2450. 10.1039/b103147n. [DOI] [Google Scholar]
- C-H Activation and Functionalization of Alkanes and Arenes. In Organometallic Chemistry and Catalysis; Springer Berlin Heidelberg: Berlin, Heidelberg, 2007; pp 409–429. [Google Scholar]
- Caplin M. J.; Foley D. J. Emergent Synthetic Methods for the Modular Advancement of sp3 -Rich Fragments. Chem. Sci. 2021, 12, 4646–4660. 10.1039/D1SC00161B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Berendt B. G.; Latham D. E.; Dambatta M. B.; Morrill L. C. Borrowing Hydrogen for Organic Synthesis. ACS Cent. Sci. 2021, 7, 570–585. 10.1021/acscentsci.1c00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. S.; White M. C. A Predictably Selective Aliphatic C–H Oxidation Reaction for Complex Molecule Synthesis. Science 2007, 318, 783–787. 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- Cowley R. E.; Eckert N. A.; Vaddadi S.; Figg T. M.; Cundari T. R.; Holland P. L. Selectivity and Mechanism of Hydrogen Atom Transfer by an Isolable Imidoiron(III) Complex. J. Am. Chem. Soc. 2011, 133, 9796–9811. 10.1021/ja2005303. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Gao W.; Wang Y.; Wang X.; Cao F.; Shi T.; Wang Z. Recent Advances in Copper Promoted Inert C(sp3)–H Functionalization. ACS Catal. 2021, 11, 967–984. 10.1021/acscatal.0c04618. [DOI] [Google Scholar]
- Shen D.; Miao C.; Wang S.; Xia C.; Sun W. Efficient Benzylic and Aliphatic C–H Oxidation with Selectivity for Methylenic Sites Catalyzed by a Bioinspired Manganese Complex. Org. Lett. 2014, 16, 1108–1111. 10.1021/ol4037083. [DOI] [PubMed] [Google Scholar]
- Liu W.; Groves J. T. Manganese Catalyzed C–H Halogenation. Acc. Chem. Res. 2015, 48, 1727–1735. 10.1021/acs.accounts.5b00062. [DOI] [PubMed] [Google Scholar]
- Li G.; Dilger A. K.; Cheng P. T.; Ewing W. R.; Groves J. T. Selective C–H Halogenation with a Highly Fluorinated Manganese Porphyrin. Angew. Chem., Int. Ed. 2018, 57, 1251–1255. 10.1002/anie.201710676. [DOI] [PubMed] [Google Scholar]
- Baccalini A.; Vergura S.; Dolui P.; Zanoni G.; Maiti D. Recent Advances in Cobalt-Catalysed C–H Functionalizations. Org. Biomol. Chem. 2019, 17, 10119–10141. 10.1039/C9OB01994D. [DOI] [PubMed] [Google Scholar]
- Liu Y.; You T.; Wang H.-X.; Tang Z.; Zhou C.-Y.; Che C.-M. Iron- and Cobalt-Catalyzed C(sp3)–H Bond Functionalization Reactions and Their Application in Organic Synthesis. Chem. Soc. Rev. 2020, 49, 5310–5358. 10.1039/D0CS00340A. [DOI] [PubMed] [Google Scholar]
- Rej S.; Chatani N. Rhodium-Catalyzed C(sp2)– or C(sp3)–H Bond Functionalization Assisted by Removable Directing Groups. Angew. Chem., Int. Ed. 2019, 58, 8304–8329. 10.1002/anie.201808159. [DOI] [PubMed] [Google Scholar]
- Choi J.; Goldman A. S.. Ir-Catalyzed Functionalization of C–H Bonds; Andersson P. G., Ed.; Topics in Organometallic Chemistry; Springer Berlin Heidelberg: Berlin, Heidelberg, 2011; Vol. 34, pp 139–167. [Google Scholar]
- Li X.; Ouyang W.; Nie J.; Ji S.; Chen Q.; Huo Y. Recent Development on Cp*Ir(III)-Catalyzed C–H Bond Functionalization. ChemCatChem 2020, 12, 2358–2384. 10.1002/cctc.201902150. [DOI] [Google Scholar]
- Thirunavukkarasu V. S.; Kozhushkov S. I.; Ackermann L. C–H Nitrogenation and Oxygenation by Ruthenium Catalysis. Chem. Commun. 2014, 50, 29–39. 10.1039/C3CC47028H. [DOI] [PubMed] [Google Scholar]
- Dana S.; Yadav M. R.; Sahoo A. K.. Ruthenium-Catalyzed C–N and C–O Bond-Forming Processes from C–H Bond Functionalization; Topics in Organometallic Chemistry; Springer, Cham, 2015; Vol. 55, pp 189–215. [Google Scholar]
- Bryant J. R.; Mayer J. M. Oxidation of C–H Bonds by [(Bpy)2(Py)RuIVO]2+ Occurs by Hydrogen Atom Abstraction. J. Am. Chem. Soc. 2003, 125, 10351–10361. 10.1021/ja035276w. [DOI] [PubMed] [Google Scholar]
- Lyons T. W.; Sanford M. S. Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem. Rev. 2010, 110, 1147–1169. 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeldt S. R.; Sanford M. S. Controlling Site Selectivity in Palladium-Catalyzed C–H Bond Functionalization. Acc. Chem. Res. 2012, 45, 936–946. 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G.; Wang B.; Nack W. A.; Chen G. Syntheses and Transformations of α-Amino Acids via Palladium-Catalyzed Auxiliary-Directed sp3 C–H Functionalization. Acc. Chem. Res. 2016, 49, 635–645. 10.1021/acs.accounts.6b00022. [DOI] [PubMed] [Google Scholar]
- He C.; Whitehurst W. G.; Gaunt M. J. Palladium-Catalyzed C(sp3)–H Bond Functionalization of Aliphatic Amines. Chem. 2019, 5, 1031–1058. 10.1016/j.chempr.2018.12.017. [DOI] [Google Scholar]
- Das J.; Guin S.; Maiti D. Diverse Strategies for Transition Metal Catalyzed Distal C(sp3)–H Functionalizations. Chem. Sci. 2020, 11, 10887–10909. 10.1039/D0SC04676K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hydrogen-Transfer Reactions; Hynes J. T., Klinman J. P., Limbach H., Schowen R. L., Eds.; Wiley-VCH: Weinheim, 2007. [Google Scholar]
- Lai W.; Li C.; Chen H.; Shaik S. Hydrogen-Abstraction Reactivity Patterns from A to Y: The Valence Bond Way. Angew. Chem., Int. Ed. 2012, 51, 5556–5578. 10.1002/anie.201108398. [DOI] [PubMed] [Google Scholar]
- Mayer J. M. Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res. 2011, 44, 36–46. 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedder J. M. The Importance of Polarity, Bond Strength and Steric Effects in Determining the Site of Attack and the Rate of Free Radical Substitution in Aliphatic Compounds. Tetrahedron 1982, 38, 313–329. 10.1016/0040-4020(82)80168-3. [DOI] [Google Scholar]
- Newhouse T.; Baran P. S. If C–H Bonds Could Talk: Selective C–H Bond Oxidation. Angew. Chem., Int. Ed. 2011, 50, 3362–3374. 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone M.; Bietti M. Tuning Reactivity and Selectivity in Hydrogen Atom Transfer from Aliphatic C–H Bonds to Alkoxyl Radicals: Role of Structural and Medium Effects. Acc. Chem. Res. 2015, 48, 2895–2903. 10.1021/acs.accounts.5b00348. [DOI] [PubMed] [Google Scholar]
- Costas M.; Bietti M. Uncovering the Complexity of the Simplest Atom Transfer Reaction. Acc. Chem. Res. 2018, 51, 2601–2602. 10.1021/acs.accounts.8b00525. [DOI] [PubMed] [Google Scholar]
- Bietti M. Activation and Deactivation Strategies Promoted by Medium Effects for Selective Aliphatic C–H Bond Functionalization. Angew. Chem., Int. Ed. 2018, 57, 16618–16637. 10.1002/anie.201804929. [DOI] [PubMed] [Google Scholar]
- Milan M.; Salamone M.; Costas M.; Bietti M. The Quest for Selectivity in Hydrogen Atom Transfer Based Aliphatic C–H Bond Oxygenation. Acc. Chem. Res. 2018, 51, 1984–1995. 10.1021/acs.accounts.8b00231. [DOI] [PubMed] [Google Scholar]
- Luo Y.-R.Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, 2007. [Google Scholar]
- Ghosh M.; Singh K. K.; Panda C.; Weitz A.; Hendrich M. P.; Collins T. J.; Dhar B. B.; Sen Gupta S. Formation of a Room Temperature Stable FeV(O) Complex: Reactivity Toward Unactivated C–H Bonds. J. Am. Chem. Soc. 2014, 136, 9524–9527. 10.1021/ja412537m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariofillis S. K.; Shields B. J.; Tekle-Smith M. A.; Zacuto M. J.; Doyle A. G. Nickel/Photoredox-Catalyzed Methylation of (Hetero)Aryl Chlorides Using Trimethyl Orthoformate as a Methyl Radical Source. J. Am. Chem. Soc. 2020, 142, 7683–7689. 10.1021/jacs.0c02805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. S.; White M. C. Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]
- Proksch E.; de Meijere A. Oxidation of Cyclopropyl Hydrocarbons with Ozone. Angew. Chem., Int. Ed. Engl. 1976, 15, 761–762. 10.1002/anie.197607611. [DOI] [Google Scholar]
- Volker Dehmlow E.; Heiligenstädt N. Dimethyldioxirane Oxidations of Some Cyclopropanes. Tetrahedron Lett. 1996, 37, 5363–5364. 10.1016/0040-4039(96)01107-0. [DOI] [Google Scholar]
- D’Accolti L.; Dinoi A.; Fusco C.; Russo A.; Curci R. Oxyfunctionalization of Non-Natural Targets by Dioxiranes. 5. Selective Oxidation of Hydrocarbons Bearing Cyclopropyl Moieties 1. J. Org. Chem. 2003, 68, 7806–7810. 10.1021/jo034768o. [DOI] [PubMed] [Google Scholar]
- Roberts B. P. Polarity-Reversal Catalysis of Hydrogen-Atom Abstraction Reactions: Concepts and Applications in Organic Chemistry. Chem. Soc. Rev. 1999, 28, 25–35. 10.1039/a804291h. [DOI] [Google Scholar]
- Salamone M.; Basili F.; Bietti M. Reactivity and Selectivity Patterns in Hydrogen Atom Transfer from Amino Acid C–H Bonds to the Cumyloxyl Radical: Polar Effects as a Rationale for the Preferential Reaction at Proline Residues. J. Org. Chem. 2015, 80, 3643–3650. 10.1021/acs.joc.5b00549. [DOI] [PubMed] [Google Scholar]
- DiLabio G. A.; Franchi P.; Lanzalunga O.; Lapi A.; Lucarini F.; Lucarini M.; Mazzonna M.; Prasad V. K.; Ticconi B. Hydrogen Atom Transfer (HAT) Processes Promoted by the Quinolinimide- N -Oxyl Radical. A Kinetic and Theoretical Study. J. Org. Chem. 2017, 82, 6133–6141. 10.1021/acs.joc.7b00687. [DOI] [PubMed] [Google Scholar]
- Salamone M.; Ortega V. B.; Martin T.; Bietti M. Hydrogen Atom Transfer from Alkanols and Alkanediols to the Cumyloxyl Radical: Kinetic Evaluation of the Contribution of α-C–H Activation and β-C–H Deactivation. J. Org. Chem. 2018, 83, 5539–5545. 10.1021/acs.joc.8b00562. [DOI] [PubMed] [Google Scholar]
- Ravelli D.; Fagnoni M.; Fukuyama T.; Nishikawa T.; Ryu I. Site-Selective C–H Functionalization by Decatungstate Anion Photocatalysis: Synergistic Control by Polar and Steric Effects Expands the Reaction Scope. ACS Catal. 2018, 8, 701–713. 10.1021/acscatal.7b03354. [DOI] [Google Scholar]
- Bietti M.; Cucinotta E.; DiLabio G. A.; Lanzalunga O.; Lapi A.; Mazzonna M.; Romero-Montalvo E.; Salamone M. Evaluation of Polar Effects in Hydrogen Atom Transfer Reactions from Activated Phenols. J. Org. Chem. 2019, 84, 1778–1786. 10.1021/acs.joc.8b02571. [DOI] [PubMed] [Google Scholar]
- Ticconi B.; Mazzonna M.; Lanzalunga O.; Lapi A. Oxidation of α-Amino Acids Promoted by the Phthalimide N-Oxyl Radical: A Kinetic and Product Study. Tetrahedron 2019, 75, 3579–3585. 10.1016/j.tet.2019.05.026. [DOI] [Google Scholar]
- Chen K.; Baran P. S. Total Synthesis of Eudesmane Terpenes by Site-Selective C–H Oxidations. Nature 2009, 459, 824–828. 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- Gómez L.; Garcia-Bosch I.; Company A.; Benet-Buchholz J.; Polo A.; Sala X.; Ribas X.; Costas M. Stereospecific C–H Oxidation with H2O2 Catalyzed by a Chemically Robust Site-Isolated Iron Catalyst. Angew. Chem., Int. Ed. 2009, 48, 5720–5723. 10.1002/anie.200901865. [DOI] [PubMed] [Google Scholar]
- Prat I.; Gómez L.; Canta M.; Ribas X.; Costas M. An Iron Catalyst for Oxidation of Alkyl C–H Bonds Showing Enhanced Selectivity for Methylenic Sites. Chem. - Eur. J. 2013, 19, 1908–1913. 10.1002/chem.201203281. [DOI] [PubMed] [Google Scholar]
- Zou L.; Paton R. S.; Eschenmoser A.; Newhouse T. R.; Baran P. S.; Houk K. N. Enhanced Reactivity in Dioxirane C–H Oxidations via Strain Release: A Computational and Experimental Study. J. Org. Chem. 2013, 78, 4037–4048. 10.1021/jo400350v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moteki S. A.; Usui A.; Zhang T.; Solorio Alvarado C. R.; Maruoka K. Site-Selective Oxidation of Unactivated C(sp3)–H Bonds with Hypervalent Iodine(III) Reagents. Angew. Chem., Int. Ed. 2013, 52, 8657–8660. 10.1002/anie.201304359. [DOI] [PubMed] [Google Scholar]
- Schmidt V. A.; Quinn R. K.; Brusoe A. T.; Alexanian E. J. Site-Selective Aliphatic C–H Bromination Using N -Bromoamides and Visible Light. J. Am. Chem. Soc. 2014, 136, 14389–14392. 10.1021/ja508469u. [DOI] [PubMed] [Google Scholar]
- Salamone M.; Ortega V. B.; Bietti M. Enhanced Reactivity in Hydrogen Atom Transfer from Tertiary Sites of Cyclohexanes and Decalins via Strain Release: Equatorial C–H Activation vs Axial C–H Deactivation. J. Org. Chem. 2015, 80, 4710–4715. 10.1021/acs.joc.5b00636. [DOI] [PubMed] [Google Scholar]
- Milan M.; Bietti M.; Costas M. Highly Enantioselective Oxidation of Nonactivated Aliphatic C–H Bonds with Hydrogen Peroxide Catalyzed by Manganese Complexes. ACS Cent. Sci. 2017, 3, 196–204. 10.1021/acscentsci.6b00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone M.; Martin T.; Milan M.; Costas M.; Bietti M. Electronic and Torsional Effects on Hydrogen Atom Transfer from Aliphatic C–H Bonds: A Kinetic Evaluation via Reaction with the Cumyloxyl Radical. J. Org. Chem. 2017, 82, 13542–13549. 10.1021/acs.joc.7b02654. [DOI] [PubMed] [Google Scholar]
- Chen K.; Eschenmoser A.; Baran P. S. Strain Release in C–H Bond Activation?. Angew. Chem., Int. Ed. 2009, 48, 9705–9708. 10.1002/anie.200904474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondi D.; Ravelli D.; Fagnoni M.; Mella M.; Molinari A.; Maldotti A.; Albini A. Regio- and Stereoselectivity in the Decatungstate Photocatalyzed Alkylation of Alkenes by Alkylcyclohexanes. Chem. - Eur. J. 2009, 15, 7949–7957. 10.1002/chem.200900810. [DOI] [PubMed] [Google Scholar]
- De Vleeschouwer F.; Van Speybroeck V.; Waroquier M.; Geerlings P.; De Proft F. Electrophilicity and Nucleophilicity Index for Radicals. Org. Lett. 2007, 9, 2721–2724. 10.1021/ol071038k. [DOI] [PubMed] [Google Scholar]
- Li C.; Danovich D.; Shaik S. Blended Hydrogen Atom Abstraction and Proton-Coupled Electron Transfer Mechanisms of Closed-Shell Molecules. Chem. Sci. 2012, 3, 1903–1918. 10.1039/c2sc20115a. [DOI] [Google Scholar]
- Tanielian C.; Cougnon F.; Seghrouchni R. Acetone, a Substrate and a New Solvent in Decatungstate Photocatalysis. J. Mol. Catal. A: Chem. 2007, 262, 164–169. 10.1016/j.molcata.2006.08.063. [DOI] [Google Scholar]
- Dondi D.; Fagnoni M.; Albini A. Tetrabutylammonium Decatungstate-Photosensitized Alkylation of Electrophilic Alkenes: Convenient Functionalization of Aliphatic C–H Bonds. Chem. - Eur. J. 2006, 12, 4153–4163. 10.1002/chem.200501216. [DOI] [PubMed] [Google Scholar]
- Naguib Y. M. A.; Steel C.; Cohen S. G.; Young M. A. Photoreduction of Benzophenone by Acetonitrile: Correlation of Rates of Hydrogen Abstraction from RH with the Ionization Potentials of the Radicals R•. J. Phys. Chem. 1987, 91, 3033–3036. 10.1021/j100295a078. [DOI] [Google Scholar]
- Okada M.; Fukuyama T.; Yamada K.; Ryu I.; Ravelli D.; Fagnoni M. Sunlight Photocatalyzed Regioselective β-Alkylation and Acylation of Cyclopentanones. Chem. Sci. 2014, 5, 2893–2898. 10.1039/C4SC01072H. [DOI] [Google Scholar]
- Ueda M.; Kitano A.; Matsubara H. A computational study of site-selective hydrogen abstraction by sulfate radical anion. Org. Biomol. Chem. 2021, 19, 4775–4782. 10.1039/D1OB00587A. [DOI] [PubMed] [Google Scholar]
- Milan M.; Carboni G.; Salamone M.; Costas M.; Bietti M. Tuning Selectivity in Aliphatic C–H Bond Oxidation of N -Alkylamides and Phthalimides Catalyzed by Manganese Complexes. ACS Catal. 2017, 7, 5903–5911. 10.1021/acscatal.7b02151. [DOI] [Google Scholar]
- Tanko J. M.; Suleman N. K.. Solvent Effects in the Reactions of Neutral Free Radicals. In Energetics of Organic Free Radicals; Springer Netherlands: Dordrecht, 1996; pp 224–293. [Google Scholar]
- Bietti M.; Salamone M. Kinetic Solvent Effects on Hydrogen Abstraction Reactions from Carbon by the Cumyloxyl Radical. The Role of Hydrogen Bonding. Org. Lett. 2010, 12, 3654–3657. 10.1021/ol101448e. [DOI] [PubMed] [Google Scholar]
- Bietti M.; Martella R.; Salamone M. Understanding Kinetic Solvent Effects on Hydrogen Abstraction Reactions from Carbon by the Cumyloxyl Radical. Org. Lett. 2011, 13, 6110–6113. 10.1021/ol202561z. [DOI] [PubMed] [Google Scholar]
- Salamone M.; Giammarioli I.; Bietti M. Kinetic Solvent Effects on Hydrogen Abstraction Reactions from Carbon by the Cumyloxyl Radical. The Importance of Solvent Hydrogen-Bond Interactions with the Substrate and the Abstracting Radical. J. Org. Chem. 2011, 76, 4645–4651. 10.1021/jo200660d. [DOI] [PubMed] [Google Scholar]
- Salamone M.; Mangiacapra L.; Bietti M. Kinetic Solvent Effects on the Reactions of the Cumyloxyl Radical with Tertiary Amides. Control over the Hydrogen Atom Transfer Reactivity and Selectivity through Solvent Polarity and Hydrogen Bonding. J. Org. Chem. 2015, 80, 1149–1154. 10.1021/jo5026767. [DOI] [PubMed] [Google Scholar]
- Jeffrey J. L.; Terrett J. A.; MacMillan D. W. C. O-H Hydrogen Bonding Promotes H-Atom Transfer from C–H Bonds for C-Alkylation of Alcohols. Science 2015, 349, 1532–1536. 10.1126/science.aac8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewanji A.; Mück-Lichtenfeld C.; Studer A. Radical Hydrodeiodination of Aryl, Alkenyl, Alkynyl, and Alkyl Iodides with an Alcoholate as Organic Chain Reductant through Electron Catalysis. Angew. Chem., Int. Ed. 2016, 55, 6749–6752. 10.1002/anie.201601930. [DOI] [PubMed] [Google Scholar]
- Salamone M.; Giammarioli I.; Bietti M. Tuning Hydrogen Atom Abstraction from the Aliphatic C–H Bonds of Basic Substrates by Protonation. Control over Selectivity by C–H Deactivation. Chem. Sci. 2013, 4, 3255–3262. 10.1039/c3sc51058a. [DOI] [Google Scholar]
- Milan M.; Salamone M.; Bietti M. Hydrogen Atom Transfer from 1,n -Alkanediamines to the Cumyloxyl Radical. Modulating C–H Deactivation Through Acid–Base Interactions and Solvent Effects. J. Org. Chem. 2014, 79, 5710–5716. 10.1021/jo5008493. [DOI] [PubMed] [Google Scholar]
- Howell J. M.; Feng K.; Clark J. R.; Trzepkowski L. J.; White M. C. Remote Oxidation of Aliphatic C–H Bonds in Nitrogen-Containing Molecules. J. Am. Chem. Soc. 2015, 137, 14590–14593. 10.1021/jacs.5b10299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Sanford M. S. Platinum-Catalyzed, Terminal-Selective C(sp3)–H Oxidation of Aliphatic Amines. J. Am. Chem. Soc. 2015, 137, 12796–12799. 10.1021/jacs.5b09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bietti M.; Forcina V.; Lanzalunga O.; Lapi A.; Martin T.; Mazzonna M.; Salamone M. Kinetic Study of the Reaction of the Phthalimide- N -Oxyl Radical with Amides: Structural and Medium Effects on the Hydrogen Atom Transfer Reactivity and Selectivity. J. Org. Chem. 2016, 81, 11924–11931. 10.1021/acs.joc.6b02482. [DOI] [PubMed] [Google Scholar]
- Mbofana C. T.; Chong E.; Lawniczak J.; Sanford M. S. Iron-Catalyzed Oxyfunctionalization of Aliphatic Amines at Remote Benzylic C–H Sites. Org. Lett. 2016, 18, 4258–4261. 10.1021/acs.orglett.6b02003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamone M.; Carboni G.; Bietti M. Fine Control over Site and Substrate Selectivity in Hydrogen Atom Transfer-Based Functionalization of Aliphatic C–H Bonds. J. Org. Chem. 2016, 81, 9269–9278. 10.1021/acs.joc.6b01842. [DOI] [PubMed] [Google Scholar]
- Lee M.; Sanford M. S. Remote C(sp3)–H Oxygenation of Protonated Aliphatic Amines with Potassium Persulfate. Org. Lett. 2017, 19, 572–575. 10.1021/acs.orglett.6b03731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack J. B. C.; Gipson J. D.; Du Bois J.; Sigman M. S. Ruthenium-Catalyzed C–H Hydroxylation in Aqueous Acid Enables Selective Functionalization of Amine Derivatives. J. Am. Chem. Soc. 2017, 139, 9503–9506. 10.1021/jacs.7b05469. [DOI] [PubMed] [Google Scholar]
- Dantignana V.; Milan M.; Cussó O.; Company A.; Bietti M.; Costas M. Chemoselective Aliphatic C–H Bond Oxidation Enabled by Polarity Reversal. ACS Cent. Sci. 2017, 3, 1350–1358. 10.1021/acscentsci.7b00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaster E.; Kozuch S.; Pappo D. Selective Aerobic Oxidation of Methylarenes to Benzaldehydes Catalyzed by N -Hydroxyphthalimide and Cobalt(II) Acetate in Hexafluoropropan-2-ol. Angew. Chem., Int. Ed. 2017, 56, 5912–5915. 10.1002/anie.201702511. [DOI] [PubMed] [Google Scholar]
- Bietti M.; Lanzalunga O.; Lapi A.; Martin T.; Mazzonna M.; Polin M.; Salamone M. Aerobic Oxidation of 4-Alkyl-N,N-Dimethylbenzylamines Catalyzed by N-Hydroxyphthalimide: Protonation-Driven Control over Regioselectivity. J. Org. Chem. 2017, 82, 5761–5768. 10.1021/acs.joc.7b00563. [DOI] [PubMed] [Google Scholar]
- Pipitone L. M.; Carboni G.; Sorrentino D.; Galeotti M.; Salamone M.; Bietti M. Enhancing Reactivity and Site-Selectivity in Hydrogen Atom Transfer from Amino Acid C–H Bonds via Deprotonation. Org. Lett. 2018, 20, 808–811. 10.1021/acs.orglett.7b03948. [DOI] [PubMed] [Google Scholar]
- Cianfanelli M.; Olivo G.; Milan M.; Klein Gebbink R. J. M.; Ribas X.; Bietti M.; Costas M. Enantioselective C–H Lactonization of Unactivated Methylenes Directed by Carboxylic Acids. J. Am. Chem. Soc. 2020, 142, 1584–1593. 10.1021/jacs.9b12239. [DOI] [PubMed] [Google Scholar]
- Borrell M.; Gil-Caballero S.; Bietti M.; Costas M. Site-Selective and Product Chemoselective Aliphatic C–H Bond Hydroxylation of Polyhydroxylated Substrates. ACS Catal. 2020, 10, 4702–4709. 10.1021/acscatal.9b05423. [DOI] [Google Scholar]
- Salamone M.; Mangiacapra L.; DiLabio G. A.; Bietti M. Effect of Metal Ions on the Reactions of the Cumyloxyl Radical with Hydrogen Atom Donors. Fine Control on Hydrogen Abstraction Reactivity Determined by Lewis Acid–Base Interactions. J. Am. Chem. Soc. 2013, 135, 415–423. 10.1021/ja309579t. [DOI] [PubMed] [Google Scholar]
- Salamone M.; Carboni G.; Mangiacapra L.; Bietti M. Binding to Redox-Inactive Alkali and Alkaline Earth Metal Ions Strongly Deactivates the C–H Bonds of Tertiary Amides toward Hydrogen Atom Transfer to Reactive Oxygen Centered Radicals. J. Org. Chem. 2015, 80, 9214–9223. 10.1021/acs.joc.5b01661. [DOI] [PubMed] [Google Scholar]
- Salamone M.; Mangiacapra L.; Carboni G.; Bietti M. Hydrogen Atom Transfer from Tertiary Alkanamides to the Cumyloxyl Radical. The Role of Substrate Structure on Alkali and Alkaline Earth Metal Ion Induced C–H Bond Deactivation. Tetrahedron 2016, 72, 7757–7763. 10.1016/j.tet.2016.05.047. [DOI] [Google Scholar]
- Martin T.; Salamone M.; Bietti M. Hydrogen Atom Transfer from 1,2- and 1,3-Diols to the Cumyloxyl Radical. The Role of Structural Effects on Metal-Ion Induced C–H Bond Deactivation. Chem. Commun. 2019, 55, 5227–5230. 10.1039/C9CC01879D. [DOI] [PubMed] [Google Scholar]
- Schultz D. M.; Lévesque F.; DiRocco D. A.; Reibarkh M.; Ji Y.; Joyce L. A.; Dropinski J. F.; Sheng H.; Sherry B. D.; Davies I. W. Oxyfunctionalization of the Remote C–H Bonds of Aliphatic Amines by Decatungstate Photocatalysis. Angew. Chem., Int. Ed. 2017, 56, 15274–15278. 10.1002/anie.201707537. [DOI] [PubMed] [Google Scholar]
- Olivo G.; Farinelli G.; Barbieri A.; Lanzalunga O.; Di Stefano S.; Costas M. Supramolecular Recognition Allows Remote, Site-Selective C–H Oxidation of Methylenic Sites in Linear Amines. Angew. Chem., Int. Ed. 2017, 56, 16347–16351. 10.1002/anie.201709280. [DOI] [PubMed] [Google Scholar]
- Olivo G.; Capocasa G.; Lanzalunga O.; Di Stefano S.; Costas M. Enzyme-like Substrate-Selectivity in C–H Oxidation Enabled by Recognition. Chem. Commun. 2019, 55, 917–920. 10.1039/C8CC09328H. [DOI] [PubMed] [Google Scholar]
- Olivo G.; Capocasa G.; Ticconi B.; Lanzalunga O.; Di Stefano S.; Costas M. Predictable Selectivity in Remote C–H Oxidation of Steroids: Analysis of Substrate Binding Mode. Angew. Chem., Int. Ed. 2020, 59, 12703–12708. 10.1002/anie.202003078. [DOI] [PubMed] [Google Scholar]
- Stateman L.; Nakafuku K.; Nagib D. Remote C–H Functionalization via Selective Hydrogen Atom Transfer. Synthesis 2018, 50, 1569–1586. 10.1055/s-0036-1591930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S.; Cheung K. P. S.; Gevorgyan V. C–H Functionalization Reactions Enabled by Hydrogen Atom Transfer to Carbon-Centered Radicals. Chem. Sci. 2020, 11, 12974–12993. 10.1039/D0SC04881J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar G.; Pradhan S.; Chatterjee I. N-Centered Radical Directed Remote C–H Bond Functionalization via Hydrogen Atom Transfer. Chem. - Asian J. 2020, 15, 651–672. 10.1002/asia.201901744. [DOI] [PubMed] [Google Scholar]
- Costas M.Remote Oxidation of Aliphatic C–H Bonds with Biologically Inspired Catalysts. In Remote C–H Bond Functionalizations; Maiti D., Guin S., Eds.; Wiley-VCH: Weinheim, 2021; pp 383–421. [Google Scholar]
- Sambiagio C.; Maes B. U. W.. Non-Directed Functionalization of Distal C(sp3)–H Bonds. In Remote C–H Bond Functionalizations; Maiti D., Guin S., Eds.; Wiley-VCH: Weinheim, 2021; pp 343–382. [Google Scholar]
- Li W.; Zhu C.. Radically Initiated Distal C(sp3)–H Functionalization. In Remote C–H Bond Functionalizations; Maiti D., Guin S., Eds.; Wiley-VCH: Weinheim, 2021; pp 315–341. [Google Scholar]
- Li Y.; Zhang Q.; Shi B.. Directing Group Assisted Distal C(sp3)–H Functionalization of Aliphatic Substrates. In Remote C–H Bond Functionalizations; Maiti D., Guin S., Eds.; Wiley-VCH: Weinheim, 2021; pp 279–314. [Google Scholar]
- Čeković Ž. Reactions of δ-Carbon Radicals Generated by 1,5-Hydrogen Transfer to Alkoxyl Radicals. Tetrahedron 2003, 59, 8073–8090. 10.1016/S0040-4020(03)01202-X. [DOI] [Google Scholar]
- Nechab M.; Mondal S.; Bertrand M. P. 1,n -Hydrogen-Atom Transfer (HAT) Reactions in Which n ≠5: An Updated Inventory. Chem. - Eur. J. 2014, 20, 16034–16059. 10.1002/chem.201403951. [DOI] [PubMed] [Google Scholar]
- Fokin A. A.; Schreiner P. R. Selective Alkane Transformations via Radicals and Radical Cations: Insights into the Activation Step from Experiment and Theory. Chem. Rev. 2002, 102, 1551–1594. 10.1021/cr000453m. [DOI] [PubMed] [Google Scholar]
- Tsui E.; Wang H.; Knowles R. R. Catalytic Generation of Alkoxy Radicals from Unfunctionalized Alcohols. Chem. Sci. 2020, 11, 11124–11141. 10.1039/D0SC04542J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Zhu C. Recent Advances in Alkoxy Radical-Promoted C–C and C–H Bond Functionalization Starting from Free Alcohols. Chem. Commun. 2019, 55, 9747–9756. 10.1039/C9CC04785A. [DOI] [PubMed] [Google Scholar]
- Guo J.-J.; Hu A.; Zuo Z. Photocatalytic Alkoxy Radical-Mediated Transformations. Tetrahedron Lett. 2018, 59, 2103–2111. 10.1016/j.tetlet.2018.04.060. [DOI] [Google Scholar]
- Melone L.; Punta C.. N-Hydroxyphthalimide (NHPI)-Organocatalyzed Aerobic Oxidations: Advantages, Limits, and Industrial Perspectives. In Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; pp 253–265. [Google Scholar]
- Quinn R. K.; Könst Z. A.; Michalak S. E.; Schmidt Y.; Szklarski A. R.; Flores A. R.; Nam S.; Horne D. A.; Vanderwal C. D.; Alexanian E. J. Site-Selective Aliphatic C–H Chlorination Using N-Chloroamides Enables a Synthesis of Chlorolissoclimide. J. Am. Chem. Soc. 2016, 138, 696–702. 10.1021/jacs.5b12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaplyski W. L.; Na C. G.; Alexanian E. J. C–H Xanthylation: A Synthetic Platform for Alkane Functionalization. J. Am. Chem. Soc. 2016, 138, 13854–13857. 10.1021/jacs.6b09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H.; Sakai K.; Kawamura A.; Oisaki K.; Kanai M. Sulfonamides as New Hydrogen Atom Transfer (HAT) Catalysts for Photoredox Allylic and Benzylic C–H Arylations. Chem. Commun. 2018, 54, 3215–3218. 10.1039/C7CC09457D. [DOI] [PubMed] [Google Scholar]
- Ryder A. S. H.; Cunningham W. B.; Ballantyne G.; Mules T.; Kinsella A. G.; Turner-Dore J.; Alder C. M.; Edwards L. J.; McKay B. S. J.; Grayson M. N.; Cresswell A. J. Photocatalytic α-Tertiary Amine Synthesis via C–H Alkylation of Unmasked Primary Amines. Angew. Chem., Int. Ed. 2020, 59, 14986–14991. 10.1002/anie.202005294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M.; Zhang T.; Tan D.; Chen C.; Zhang Y.; Huang M.; Cai S. Visible-Light-Promoted Oxidative Alkylarylation of N-Aryl/Benzoyl Acrylamides Through Direct C–H Bond Functionalization. Adv. Synth. Catal. 2019, 361, 4237–4242. 10.1002/adsc.201900712. [DOI] [Google Scholar]
- Li W.; Duan Y.; Zhang M.; Cheng J.; Zhu C. A Photoredox Catalyzed Radical–Radical Coupling Reaction: Facile Access to Multi-Substituted Nitrogen Heterocycles. Chem. Commun. 2016, 52, 7596–7599. 10.1039/C6CC02027E. [DOI] [PubMed] [Google Scholar]
- Jin J.; MacMillan D. W. C. Alcohols as Alkylating Agents in Heteroarene C–H Functionalization. Nature 2015, 525, 87–90. 10.1038/nature14885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager D.; MacMillan D. W. C. Activation of C–H Bonds via the Merger of Photoredox and Organocatalysis: A Coupling of Benzylic Ethers with Schiff Bases. J. Am. Chem. Soc. 2014, 136, 16986–16989. 10.1021/ja5102695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbertson J. D.; MacMillan D. W. C. The Direct Arylation of Allylic sp3 C–H Bonds via Organic and Photoredox Catalysis. Nature 2015, 519, 74–77. 10.1038/nature14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qvortrup K.; Rankic D. A.; MacMillan D. W. C. A General Strategy for Organocatalytic Activation of C–H Bonds via Photoredox Catalysis: Direct Arylation of Benzylic Ethers. J. Am. Chem. Soc. 2014, 136, 626–629. 10.1021/ja411596q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P.; Hou T.; Gu X.; Li P. Visible-Light-Promoted Conversion of Alkyl Benzyl Ether to Alkyl Ester or Alcohol via O-α-sp3 C–H Cleavage. Org. Lett. 2015, 17, 1954–1957. 10.1021/acs.orglett.5b00663. [DOI] [PubMed] [Google Scholar]
- Hou T.; Lu P.; Li P. Visible-Light-Mediated Benzylic sp3 C–H Bond Functionalization to C–Br or C–N Bond. Tetrahedron Lett. 2016, 57, 2273–2276. 10.1016/j.tetlet.2016.04.036. [DOI] [Google Scholar]
- Fadeyi O. O.; Mousseau J. J.; Feng Y.; Allais C.; Nuhant P.; Chen M. Z.; Pierce B.; Robinson R. Visible-Light-Driven Photocatalytic Initiation of Radical Thiol–Ene Reactions Using Bismuth Oxide. Org. Lett. 2015, 17, 5756–5759. 10.1021/acs.orglett.5b03184. [DOI] [PubMed] [Google Scholar]
- Keylor M. H.; Park J. E.; Wallentin C.-J.; Stephenson C. R. J. Photocatalytic Initiation of Thiol–Ene Reactions: Synthesis of Thiomorpholin-3-Ones. Tetrahedron 2014, 70, 4264–4269. 10.1016/j.tet.2014.03.041. [DOI] [Google Scholar]
- Kariofillis S. K.; Doyle A. G. Synthetic and Mechanistic Implications of Chlorine Photoelimination in Nickel/Photoredox C(sp3)–H Cross-Coupling. Acc. Chem. Res. 2021, 54, 988–1000. 10.1021/acs.accounts.0c00694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P.; Chen P.; Xu H. Scalable Photoelectrochemical Dehydrogenative Cross-Coupling of Heteroarenes with Aliphatic C–H Bonds. Angew. Chem., Int. Ed. 2020, 59, 14275–14280. 10.1002/anie.202005724. [DOI] [PubMed] [Google Scholar]
- McMillan A. J.; Sieńkowska M.; Di Lorenzo P.; Gransbury G. K.; Chilton N. F.; Salamone M.; Ruffoni A.; Bietti M.; Leonori D. Practical and Selective sp3 C–H Bond Chlorination via Aminium Radicals. Angew. Chem., Int. Ed. 2021, 60, 7132–7139. 10.1002/anie.202100030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M. H.; Shurtleff V. W.; Terrett J. A.; Cuthbertson J. D.; MacMillan D. W. C. Native Functionality in Triple Catalytic Cross-Coupling: sp3 C–H Bonds as Latent Nucleophiles. Science 2016, 352, 1304–1308. 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W.; Wang X.; Liu R.; Wu J. Quinuclidine and Its Derivatives as Hydrogen-Atom-Transfer Catalysts in Photoinduced Reactions. Chin. Chem. Lett. 2021, 32, 1847–1856. 10.1016/j.cclet.2021.02.009. [DOI] [Google Scholar]
- Saito M.; Kawamata Y.; Meanwell M.; Navratil R.; Chiodi D.; Carlson E.; Hu P.; Chen L.; Udyavara S.; Kingston C.; Tanwar M.; Tyagi S.; McKillican B. P.; Gichinga M. G.; Schmidt M. A.; Eastgate M. D.; Lamberto M.; He C.; Tang T.; Malapit C. A.; Sigman M. S.; Minteer S. D.; Neurock M.; Baran P. S. N-Ammonium Ylide Mediators for Electrochemical C–H Oxidation. J. Am. Chem. Soc. 2021, 143, 7859–7867. 10.1021/jacs.1c03780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Núñez M. E.; Castellano G.; Andreu C.; Royo J.; Báguena M.; Mello R.; Asensio G. Influence of Remote Substituents on the Equatorial/Axial Selectivity in the Monooxygenation of Methylene C–H Bonds of Substituted Cyclohexanes. J. Am. Chem. Soc. 2001, 123, 7487–7491. 10.1021/ja003667u. [DOI] [PubMed] [Google Scholar]
- Mello R.; Fiorentino M.; Fusco C.; Curci R. Oxidations by Methyl(Trifluoromethyl)Dioxirane. 2. Oxyfunctionalization of Saturated Hydrocarbons. J. Am. Chem. Soc. 1989, 111, 6749–6757. 10.1021/ja00199a039. [DOI] [Google Scholar]
- Nam W. High-Valent Iron(IV)–Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions. Acc. Chem. Res. 2007, 40, 522–531. 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]
- Stephenson C.; Yoon T.; MacMillan D. W. C.. Visible Light Photocatalysis in Organic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018. [Google Scholar]
- Fagnoni M.; Protti S.; Ravelli D.. Photoorganocatalysis in Organic Synthesis; Catalytic Science Series; World Scientific (Europe), 2019; Vol. 18. [Google Scholar]
- Albini A., Fagnoni M.. Photochemically-Generated Intermediates in Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, 2013. [Google Scholar]
- Chemical Photocatalysis; König B., Ed.; De Gruyter: Berlin, Boston, 2013. [Google Scholar]
- Palmisano G.; Augugliaro V.; Pagliaro M.; Palmisano L. Photocatalysis: A Promising Route for 21st Century Organic Chemistry. Chem. Commun. 2007, 3425–3437. 10.1039/b700395c. [DOI] [PubMed] [Google Scholar]
- Fagnoni M.; Dondi D.; Ravelli D.; Albini A. Photocatalysis for the Formation of the C–C Bond. Chem. Rev. 2007, 107, 2725–2756. 10.1021/cr068352x. [DOI] [PubMed] [Google Scholar]
- Ravelli D.; Dondi D.; Fagnoni M.; Albini A. Photocatalysis. A Multi-Faceted Concept for Green Chemistry. Chem. Soc. Rev. 2009, 38, 1999–2011. 10.1039/b714786b. [DOI] [PubMed] [Google Scholar]
- Ravelli D.; Fagnoni M.; Albini A. Photoorganocatalysis. What For?. Chem. Soc. Rev. 2013, 42, 97–113. 10.1039/C2CS35250H. [DOI] [PubMed] [Google Scholar]
- Staveness D.; Bosque I.; Stephenson C. R. J. Free Radical Chemistry Enabled by Visible Light-Induced Electron Transfer. Acc. Chem. Res. 2016, 49, 2295–2306. 10.1021/acs.accounts.6b00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas J. J.; Sevrin M. J.; Stephenson C. R. J. Visible Light Photocatalysis: Applications and New Disconnections in the Synthesis of Pharmaceutical Agents. Org. Process Res. Dev. 2016, 20, 1134–1147. 10.1021/acs.oprd.6b00125. [DOI] [Google Scholar]
- Gentry E. C.; Knowles R. R. Synthetic Applications of Proton-Coupled Electron Transfer. Acc. Chem. Res. 2016, 49, 1546–1556. 10.1021/acs.accounts.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser O. Shining Light on Copper: Unique Opportunities for Visible-Light-Catalyzed Atom Transfer Radical Addition Reactions and Related Processes. Acc. Chem. Res. 2016, 49, 1990–1996. 10.1021/acs.accounts.6b00296. [DOI] [PubMed] [Google Scholar]
- Goddard J.-P.; Ollivier C.; Fensterbank L. Photoredox Catalysis for the Generation of Carbon Centered Radicals. Acc. Chem. Res. 2016, 49, 1924–1936. 10.1021/acs.accounts.6b00288. [DOI] [PubMed] [Google Scholar]
- Skubi K. L.; Blum T. R.; Yoon T. P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M. D.; Kim S.; Toste F. D. Photoredox Catalysis Unlocks Single-Electron Elementary Steps in Transition Metal Catalyzed Cross-Coupling. ACS Cent. Sci. 2016, 2, 293–301. 10.1021/acscentsci.6b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Jin H.; Hashmi A. S. K. The Recent Achievements of Redox-Neutral Radical C–C Cross-Coupling Enabled by Visible-Light. Chem. Soc. Rev. 2017, 46, 5193–5203. 10.1039/C7CS00339K. [DOI] [PubMed] [Google Scholar]
- Lee K. N.; Ngai M.-Y. Recent Developments in Transition-Metal Photoredox-Catalysed Reactions of Carbonyl Derivatives. Chem. Commun. 2017, 53, 13093–13112. 10.1039/C7CC06287G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui J. K.; Lang S. B.; Heitz D. R.; Molander G. A. Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development. ACS Catal. 2017, 7, 2563–2575. 10.1021/acscatal.7b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twilton J.; Le C.; Zhang P.; Shaw M. H.; Evans R. W.; MacMillan D. W. C. The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem. 2017, 1, 0052. 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- Zhang L.; Meggers E. Steering Asymmetric Lewis Acid Catalysis Exclusively with Octahedral Metal-Centered Chirality. Acc. Chem. Res. 2017, 50, 320–330. 10.1021/acs.accounts.6b00586. [DOI] [PubMed] [Google Scholar]
- Savateev A.; Antonietti M. Heterogeneous Organocatalysis for Photoredox Chemistry. ACS Catal. 2018, 8, 9790–9808. 10.1021/acscatal.8b02595. [DOI] [Google Scholar]
- Revathi L.; Ravindar L.; Fang W.-Y.; Rakesh K. P.; Qin H.-L. Visible Light-Induced C–H Bond Functionalization: A Critical Review. Adv. Synth. Catal. 2018, 360, 4652–4698. 10.1002/adsc.201800736. [DOI] [Google Scholar]
- Silvi M.; Melchiorre P. Enhancing the Potential of Enantioselective Organocatalysis with Light. Nature 2018, 554, 41–49. 10.1038/nature25175. [DOI] [PubMed] [Google Scholar]
- Sambiagio C.; Noël T. Flow Photochemistry: Shine Some Light on Those Tubes!. Trends Chem. 2020, 2, 92–106. 10.1016/j.trechm.2019.09.003. [DOI] [Google Scholar]
- Guillemard L.; Wencel-Delord J. When Metal-Catalyzed C–H Functionalization Meets Visible-Light Photocatalysis. Beilstein J. Org. Chem. 2020, 16, 1754–1804. 10.3762/bjoc.16.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi S.; Fagnoni M. Generation of Alkyl Radicals: From the Tyranny of Tin to the Photon Democracy. Chem. Rev. 2020, 120, 9790–9833. 10.1021/acs.chemrev.0c00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice C.; Morrisson J.; Smith A. D.; Zysman-Colman E. Recent Developments in Enantioselective Photocatalysis. Beilstein J. Org. Chem. 2020, 16, 2363–2441. 10.3762/bjoc.16.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenardon G. V. A.; Nicchio L.; Fagnoni M. Photogenerated Electrophilic Radicals for the Umpolung of Enolate Chemistry. J. Photochem. Photobiol., C 2021, 46, 100387. 10.1016/j.jphotochemrev.2020.100387. [DOI] [Google Scholar]
- Sideri I. K.; Voutyritsa E.; Kokotos C. G. Photoorganocatalysis, Small Organic Molecules and Light in the Service of Organic Synthesis: The Awakening of a Sleeping Giant. Org. Biomol. Chem. 2018, 16, 4596–4614. 10.1039/C8OB00725J. [DOI] [PubMed] [Google Scholar]
- Protti S.; Fagnoni M.; Ravelli D. Photocatalytic C–H Activation by Hydrogen-Atom Transfer in Synthesis. ChemCatChem 2015, 7, 1516–1523. 10.1002/cctc.201500125. [DOI] [Google Scholar]
- Capaldo L.; Ravelli D. Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem. 2017, 2017, 2056–2071. 10.1002/ejoc.201601485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capaldo L.; Quadri L. L.; Ravelli D. Photocatalytic Hydrogen Atom Transfer: The Philosopher’s Stone for Late-Stage Functionalization?. Green Chem. 2020, 22, 3376–3396. 10.1039/D0GC01035A. [DOI] [Google Scholar]
- Cao H.; Tang X.; Tang H.; Yuan Y.; Wu J. Photoinduced Intermolecular Hydrogen Atom Transfer Reactions in Organic Synthesis. Chem. Catal 2021, 10.1016/j.checat.2021.04.008. [DOI] [Google Scholar]
- Hoffmann N. Electron and Hydrogen Transfer in Organic Photochemical Reactions. J. Phys. Org. Chem. 2015, 28, 121–136. 10.1002/poc.3370. [DOI] [Google Scholar]
- IUPAC Compendium of Chemical Terminology; Nič M., Jirát J., Košata B., Jenkins A., McNaught A., Eds.; IUPAC: Research Triangle Park, NC, 2009. [Google Scholar]
- Capaldo L.; Ravelli D. The Dark Side of Photocatalysis: One Thousand Ways to Close the Cycle. Eur. J. Org. Chem. 2020, 2020, 2783–2806. 10.1002/ejoc.202000144. [DOI] [Google Scholar]
- Dantas J. A.; Correia J. T. M.; Paixão M. W.; Corrêa A. G. Photochemistry of Carbonyl Compounds: Application in Metal-Free Reactions. ChemPhotoChem. 2019, 3, 506–520. 10.1002/cptc.201900044. [DOI] [Google Scholar]
- Zhu D.-L.; Young D. J.; Li H.-X. Carbonyl-Photoredox/Metal Dual Catalysis: Applications in Organic Synthesis. Synthesis 2020, 52, 3493–3510. 10.1055/s-0040-1707183. [DOI] [Google Scholar]
- Perez-Prieto J.; Galian R.; Miranda M. Diaryl Ketones as Photoactivators. Mini-Rev. Org. Chem. 2006, 3, 117–135. 10.2174/157019306776819226. [DOI] [Google Scholar]
- Kamijo S.Ketones and Aldehydes. In Photoorganocatalysis in Organic Synthesis; Fagnoni M., Protti S., Ravelli D., Eds.; World Scientific (Europe), 2019; pp 1–37. [Google Scholar]
- Kamijo S.C–H Activation via Radical Processes Using Photo-Excited Ketones. Topics in Organometallic Chemistry; Springer, Cham, 2018; Vol. 54, pp 71–92. [Google Scholar]
- Das A.; Thomas K. R. J.. Facile Thiol–Ene Click Protocol Using Benzil as Sensitizer and White LED as Light Source. Eur. J. Org. Chem. 2020, 2020, 7214. 10.1002/ejoc.202001275 [DOI] [Google Scholar]
- Kuhn H. J.; Goerner H. Triplet State and Photodecarboxylation of Phenylglyoxylic Acid in the Presence of Water. J. Phys. Chem. 1988, 92, 6208–6219. 10.1021/j100333a010. [DOI] [Google Scholar]
- Itoh A.Quinones. In Photoorganocatalysis in Organic Synthesis; Fagnoni M., Protti S., Ravelli D., Eds.; World Scientific (Europe), 2019; pp 39–70. [Google Scholar]
- Cervantes-González J.; Vosburg D. A.; Mora-Rodriguez S. E.; Vázquez M. A.; Zepeda L. G.; Villegas Gómez C.; Lagunas-Rivera S. Anthraquinones: Versatile Organic Photocatalysts. ChemCatChem 2020, 12, 3811–3827. 10.1002/cctc.202000376. [DOI] [Google Scholar]
- de Lucas N. C.; Elias M. M.; Firme C. L.; Corrêa R. J.; Garden S. J.; Netto-Ferreira J. C.; Nicodem D. E. A Combined Laser Flash Photolysis, Density Functional Theory and Atoms in Molecules Study of the Photochemical Hydrogen Abstraction by Pyrene-4,5-Dione. J. Photochem. Photobiol., A 2009, 201, 1–7. 10.1016/j.jphotochem.2008.08.014. [DOI] [Google Scholar]
- Xiao W.-J.; Hu X.-Q.; Chen J.-R.. Oxygen Heterocycles: Eosin Derivatives. In Photoorganocatalysis in Organic Synthesis; Fagnoni M., Protti S., Ravelli D., Eds.; World Scientific (Europe), 2019; pp 243–286. [Google Scholar]
- Yan D.-M.; Chen J.-R.; Xiao W.-J. New Roles for Photoexcited Eosin Y in Photochemical Reactions. Angew. Chem., Int. Ed. 2019, 58, 378–380. 10.1002/anie.201811102. [DOI] [PubMed] [Google Scholar]
- Hill C. L. Introduction of Functionality into Unactivated Carbon-Hydrogen Bonds. Catalytic Generation and Nonconventional Utilization of Organic Radicals. Synlett 1995, 1995, 127–132. 10.1055/s-1995-4883. [DOI] [Google Scholar]
- Tzirakis M. D.; Lykakis I. N.; Orfanopoulos M. Decatungstate as an Efficient Photocatalyst in Organic Chemistry. Chem. Soc. Rev. 2009, 38, 2609. 10.1039/b812100c. [DOI] [PubMed] [Google Scholar]
- N. Lykakis I.; Evgenidou E.; Orfanopoulos M. Photo-Catalysis and Polyoxo-Anion Decatungstate in Organic Chemistry: A Manifold Concept for Green Chemistry. Curr. Org. Chem. 2012, 16, 2400–2414. 10.2174/138527212803520092. [DOI] [Google Scholar]
- Ravelli D.; Protti S.; Fagnoni M. Decatungstate Anion for Photocatalyzed “Window Ledge” Reactions. Acc. Chem. Res. 2016, 49, 2232–2242. 10.1021/acs.accounts.6b00339. [DOI] [PubMed] [Google Scholar]
- Lazzaroni S.; Ravelli D.; Protti S.; Fagnoni M.; Albini A. Photochemical Synthesis: Using Light to Build C–C Bonds under Mild Conditions. C. R. Chim. 2017, 20, 261–271. 10.1016/j.crci.2015.11.024. [DOI] [Google Scholar]
- Yuan X.; Yang G.; Yu B. Photoinduced Decatungstate-Catalyzed C–H Functionalization. Youji Huaxue 2020, 40, 3620–3632. 10.6023/cjoc202006068. [DOI] [Google Scholar]
- Cowie B. E.; Purkis J. M.; Austin J.; Love J. B.; Arnold P. L. Thermal and Photochemical Reduction and Functionalization Chemistry of the Uranyl Dication, [UVIO2]2+. Chem. Rev. 2019, 119, 10595–10637. 10.1021/acs.chemrev.9b00048. [DOI] [PubMed] [Google Scholar]
- Li Y.; Su J.; Mitchell E.; Zhang G.; Li J. Photocatalysis with Visible-Light-Active Uranyl Complexes. Sci. China: Chem. 2013, 56, 1671–1681. 10.1007/s11426-013-4965-y. [DOI] [Google Scholar]
- Shiragami T.; Matsumoto J.; Inoue H.; Yasuda M. Antimony Porphyrin Complexes as Visible-Light Driven Photocatalyst. J. Photochem. Photobiol., C 2005, 6, 227–248. 10.1016/j.jphotochemrev.2005.12.001. [DOI] [Google Scholar]
- Huang H.; Strater Z. M.; Lambert T. H. Electrophotocatalytic C–H Functionalization of Ethers with High Regioselectivity. J. Am. Chem. Soc. 2020, 142, 1698–1703. 10.1021/jacs.9b11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam W.; Arnold M. A.; Nau W. M.; Pischel U.; Saha-Möller C. R. A Comparative Photomechanistic Study (Spin Trapping, EPR Spectroscopy, Transient Kinetics, Photoproducts) of Nucleoside Oxidation (DG and 8-OxodG) by Triplet-Excited Acetophenones and by the Radicals Generated from α-Oxy-Substituted Derivatives through No. J. Am. Chem. Soc. 2002, 124, 3893–3904. 10.1021/ja017600y. [DOI] [PubMed] [Google Scholar]
- Ghoneim N.; Monbelli A.; Pilloud D.; Suppan P. Photochemical Reactivity of Para-Aminobenzophenone in Polar and Non-Polar Solvents. J. Photochem. Photobiol., A 1996, 94, 145–148. 10.1016/1010-6030(95)04220-2. [DOI] [Google Scholar]
- Serpa C.; Arnaut L. G. Does Molecular Size Matter in Photoinduced Electron Transfer Reactions?. J. Phys. Chem. A 2000, 104, 11075–11086. 10.1021/jp001489l. [DOI] [Google Scholar]
- Timpe H.-J.; Kronfeld K.-P. Light-Induced Polymer and Polymerization Reactions XXXIII: Direct Photoinitiation of Methyl Methacrylate Polymerization by Excited States of Ketones. J. Photochem. Photobiol., A 1989, 46, 253–267. 10.1016/1010-6030(89)80012-7. [DOI] [Google Scholar]
- Favaro G.; Romani A. Acid–Base Properties of Disodium 3,3′-Disulfonatobenzophenone (DSB) in the Ground and Excited States. J. Chem. Soc., Faraday Trans. 1993, 89, 699–702. 10.1039/FT9938900699. [DOI] [Google Scholar]
- Hamanoue K.; Nakayama T.; Kajiwara Y.; Yamaguchi T.; Teranishi H. The Lowest Triplet States of Anthraquinone and Chloroanthraquinones: The 1-chloro, 2-chloro, 1,5-dichloro, and 1,8-dichloro Compounds. J. Chem. Phys. 1987, 86, 6654–6659. 10.1063/1.452412. [DOI] [Google Scholar]
- Bergamini G.; Ceroni P.; Maestri M.; Balzani V.; Lee S.-K.; Vögtle F. Forward (Singlet–Singlet) and Backward (Triplet–Triplet) Energy Transfer in a Dendrimer with Peripheral Naphthalene Units and a Benzophenone Core. Photochem. Photobiol. Sci. 2004, 3, 898–905. 10.1039/B408659G. [DOI] [PubMed] [Google Scholar]
- Kavlakoğlu E.; Yaman A.; Bayrakçeken F. Photophysical Properties of 5H-Dibenzo(a,d)Cyclohepten-5-One in Different Solutions. Spectrochim. Acta, Part A 2000, 56, 1117–1121. 10.1016/S1386-1425(99)00209-7. [DOI] [PubMed] [Google Scholar]
- Fukuzumi S.; Itoh S.; Komori T.; Suenobu T.; Ishida A.; Fujitsuka M.; Ito O. Photochemical Reactions of Coenzyme PQQ (Pyrroloquinolinequinone) and Analogues with Benzyl Alcohol Derivatives via Photoinduced Electron Transfer. J. Am. Chem. Soc. 2000, 122, 8435–8443. 10.1021/ja001351g. [DOI] [Google Scholar]
- Elliott L. D.; Kayal S.; George M. W.; Booker-Milburn K. Rational Design of Triplet Sensitizers for the Transfer of Excited State Photochemistry from UV to Visible. J. Am. Chem. Soc. 2020, 142, 14947–14956. 10.1021/jacs.0c05069. [DOI] [PubMed] [Google Scholar]
- Fan X.; Rong J.; Wu H.; Zhou Q.; Deng H.; Tan J. Da; Xue C.; Wu L.; Tao H.; Wu J. Eosin Y as a Direct Hydrogen-Atom Transfer Photocatalyst for the Functionalization of C–H Bonds. Angew. Chem., Int. Ed. 2018, 57, 8514–8518. 10.1002/anie.201803220. [DOI] [PubMed] [Google Scholar]
- Huix-Rotllant M.; Burghardt I.; Ferré N. Population of Triplet States in Acetophenone: A Quantum Dynamics Perspective. C. R. Chim. 2016, 19, 50–56. 10.1016/j.crci.2015.10.002. [DOI] [Google Scholar]
- Bohning J. J.; Weiss K. A Kinetic Study of the Photochemical Reaction of Phenanthrenequinone with Olefins. J. Am. Chem. Soc. 1966, 88, 2893–2898. 10.1021/ja00965a002. [DOI] [Google Scholar]
- Shen T.; Zhao Z.-G.; Yu Q.; Xu H.-J. Photosensitized Reduction of Benzil by Heteroatom-Containing Anthracene Dyes. J. Photochem. Photobiol., A 1989, 47, 203–212. 10.1016/1010-6030(89)87066-2. [DOI] [Google Scholar]
- Xia J.-B.; Zhu C.; Chen C. Visible Light-Promoted Metal-Free sp3-C–H Fluorination. Chem. Commun. 2014, 50, 11701–11704. 10.1039/C4CC05650G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen A.; Renner M.; Griesbeck A. G.; Büsgen T. From 3D to 4D Printing: A Reactor for Photochemical Experiments Using Hybrid Polyurethane Acrylates for Vat-Based Polymerization and Surface Functionalization. Chem. Commun. 2020, 56, 15161–15164. 10.1039/D0CC06512A. [DOI] [PubMed] [Google Scholar]
- Manfrotto C.; Mella M.; Freccero M.; Fagnoni M.; Albini A. Photochemical Synthesis of 4-Oxobutanal Acetals and of 2-Hydroxycyclobutanone Ketals. J. Org. Chem. 1999, 64, 5024–5028. 10.1021/jo982053t. [DOI] [PubMed] [Google Scholar]
- Wang L.; Wang T.; Cheng G.-J.; Li X.; Wei J.-J.; Guo B.; Zheng C.; Chen G.; Ran C.; Zheng C. Direct C–H Arylation of Aldehydes by Merging Photocatalyzed Hydrogen Atom Transfer with Palladium Catalysis. ACS Catal. 2020, 10, 7543–7551. 10.1021/acscatal.0c02105. [DOI] [Google Scholar]
- Doohan R. A.; Hannan J. J.; Geraghty N. W. A. The Photomediated Reaction of Alkynes with Cycloalkanes. Org. Biomol. Chem. 2006, 4, 942–952. 10.1039/b517631j. [DOI] [PubMed] [Google Scholar]
- Hoshikawa T.; Inoue M. Photoinduced Direct 4-Pyridination of C(sp3)–H Bonds. Chem. Sci. 2013, 4, 3118–3123. 10.1039/c3sc51080h. [DOI] [Google Scholar]
- Hoshikawa T.; Yoshioka S.; Kamijo S.; Inoue M. Photoinduced Direct Cyanation of C(sp3)–H Bonds. Synthesis 2013, 45, 874–887. 10.1055/s-0032-1318325. [DOI] [Google Scholar]
- Kraus G. A.; Liu P. Benzophenone-Mediated Conjugate Additions of Aromatic Aldehydes to Quinones. Tetrahedron Lett. 1994, 35, 7723–7726. 10.1016/0040-4039(94)80102-9. [DOI] [Google Scholar]
- Oelgemöller M.; Schiel C.; Fröhlich R.; Mattay J. The “Photo-Friedel–Crafts Acylation” of 1,4-Naphthoquinones. Eur. J. Org. Chem. 2002, 2002, 2465–2474. 10.1002/1099-0690(200208)2002:15<2465::AID-EJOC2465>3.0.CO;2-O. [DOI] [Google Scholar]
- Fraser-Reid B.; Anderson R. C.; Hicks D. R.; Walker D. L. Synthetic Applications of the Photochemically Induced Addition of Oxycarbinyl Species to α-Enones. Part II. The Addition of Ketals, Aldehydes, and Polyfunctional Species. Can. J. Chem. 1977, 55, 3986–3995. 10.1139/v77-566. [DOI] [Google Scholar]
- Cerfontain H.; Van Noort P. C. M. Facile Photochemical Synthesis of Some 4-Oxoalkanoic Acids and Esters. Synthesis 1980, 1980, 490–492. 10.1055/s-1980-29069. [DOI] [Google Scholar]
- Masuda Y.; Tsuda H.; Murakami M. C1 Oxidation/C2 Reduction Isomerization of Unprotected Aldoses Induced by Light/Ketone. Angew. Chem., Int. Ed. 2020, 59, 2755–2759. 10.1002/anie.201914242. [DOI] [PubMed] [Google Scholar]
- Singh M.; Yadav A. K.; Yadav L. D. S.; Singh R. K. P. Visible Light Photocatalysis with Benzophenone for Radical Thiol-Ene Reactions. Tetrahedron Lett. 2017, 58, 2206–2208. 10.1016/j.tetlet.2017.04.060. [DOI] [Google Scholar]
- Matsumoto K.; Nakajima M.; Nemoto T. Visible Light-Induced Direct S0→Tn Transition of Benzophenone Promotes C(sp3)–H Alkynylation of Ethers and Amides. J. Org. Chem. 2020, 85, 11802–11811. 10.1021/acs.joc.0c01573. [DOI] [PubMed] [Google Scholar]
- Dondi D.; Caprioli I.; Fagnoni M.; Mella M.; Albini A. A Convenient Route to 1,4-Monoprotected Dialdehydes, 1,4-Ketoaldehydes, γ-Lactols and γ-Lactones through Radical Alkylation of α,β-Unsaturated Aldehydes in Organic and Organic-Aqueous Media. Tetrahedron 2003, 59, 947–957. 10.1016/S0040-4020(02)01659-9. [DOI] [Google Scholar]
- Dondi D.; Protti S.; Albini A.; Carpio S. M.; Fagnoni M. Synthesis of γ-Lactols, γ-Lactones and 1,4-Monoprotected Succinaldehydes under Moderately Concentrated Sunlight. Green Chem. 2009, 11, 1653–1659. 10.1039/b904427b. [DOI] [Google Scholar]
- Kamijo S.; Takao G.; Kamijo K.; Tsuno T.; Ishiguro K.; Murafuji T. Alkylation of Nonacidic C(sp3)–H Bonds by Photoinduced Catalytic Michael-Type Radical Addition. Org. Lett. 2016, 18, 4912–4915. 10.1021/acs.orglett.6b02391. [DOI] [PubMed] [Google Scholar]
- Tada N.; Ikebata Y.; Nobuta T.; Hirashima S.; Miura T.; Itoh A. Direct Aerobic Photo-Oxidative Syntheses of Aromatic Methyl Esters from Methyl Aromatics Using Anthraquinone-2,3-Dicarboxylic Acid as Organophotocatalyst. Photochem. Photobiol. Sci. 2012, 11, 616–619. 10.1039/c2pp05387j. [DOI] [PubMed] [Google Scholar]
- Yavorskyy A.; Shvydkiv O.; Hoffmann N.; Nolan K.; Oelgemöller M. Parallel Microflow Photochemistry: Process Optimization, Scale-up, and Library Synthesis. Org. Lett. 2012, 14, 4342–4345. 10.1021/ol301773r. [DOI] [PubMed] [Google Scholar]
- Bume D. D.; Pitts C. R.; Jokhai R. T.; Lectka T. Direct, Visible Light-Sensitized Benzylic C H Fluorination of Peptides Using Dibenzosuberenone: Selectivity for Phenylalanine-like Residues. Tetrahedron 2016, 72, 6031–6036. 10.1016/j.tet.2016.08.018. [DOI] [Google Scholar]
- Zhang Y.; Ji P.; Hu W.; Wei Y.; Huang H.; Wang W. Organocatalytic Transformation of Aldehydes to Thioesters with Visible Light. Chem. - Eur. J. 2019, 25, 8225–8228. 10.1002/chem.201900932. [DOI] [PubMed] [Google Scholar]
- Papadopoulos G. N.; Kokotos C. G. Photoorganocatalytic One-Pot Synthesis of Hydroxamic Acids from Aldehydes. Chem. - Eur. J. 2016, 22, 6964–6967. 10.1002/chem.201600333. [DOI] [PubMed] [Google Scholar]
- Tripathi C. B.; Ohtani T.; Corbett M. T.; Ooi T. Photoredox Ketone Catalysis for the Direct C–H Imidation and Acyloxylation of Arenes. Chem. Sci. 2017, 8, 5622–5627. 10.1039/C7SC01700F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K.; Ohtani T.; Tripathi C. B.; Uraguchi D.; Ooi T. Formal Hydroformylation of α,β-Unsaturated Carboxylic Acids under Photoexcited Ketone Catalysis. Chem. Lett. 2019, 48, 715–717. 10.1246/cl.190197. [DOI] [Google Scholar]
- Fan X.; Xiao P.; Jiao Z.; Yang T.; Dai X.; Xu W.; Tan J. Da; Cui G.; Su H.; Fang W.; Wu J. Neutral-Eosin-Y-Photocatalyzed Silane Chlorination Using Dichloromethane. Angew. Chem., Int. Ed. 2019, 58, 12580–12584. 10.1002/anie.201908556. [DOI] [PubMed] [Google Scholar]
- Wang H.; Li W.; Zeng K.; Wu Y.; Zhang Y.; Xu T.; Chen Y. Photocatalysis Enables Visible-Light Uncaging of Bioactive Molecules in Live Cells. Angew. Chem., Int. Ed. 2019, 58, 561–565. 10.1002/anie.201811261. [DOI] [PubMed] [Google Scholar]
- Inoa J.; Patel M.; Dominici G.; Eldabagh R.; Patel A.; Lee J.; Xing Y. Benzylic Hydroperoxidation via Visible-Light-Induced C(sp3)–H Activation. J. Org. Chem. 2020, 85, 6181–6187. 10.1021/acs.joc.0c00385. [DOI] [PubMed] [Google Scholar]
- Soni S.; Pali P.; Ansari M. A.; Singh M. S. Visible-Light Photocatalysis of Eosin Y: HAT and Complementing MS-CPET Strategy to Trifluoromethylation of β-Ketodithioesters with Langlois’ Reagent. J. Org. Chem. 2020, 85, 10098–10109. 10.1021/acs.joc.0c01355. [DOI] [PubMed] [Google Scholar]
- Xia Q.; Shi Z.; Yuan J.; Bian Q.; Xu Y.; Liu B.; Huang Y.; Yang X.; Xu H. Visible-Light-Enabled Selective Oxidation of Primary Alcohols through Hydrogen-Atom Transfer and Its Application in the Synthesis of Quinazolinones. Asian J. Org. Chem. 2019, 8, 1933–1941. 10.1002/ajoc.201900491. [DOI] [Google Scholar]
- König B.; Ramm S.; Bubenitschek P.; Jones P. G.; Hopf H.; Knieriem B.; Meijere A. De. [2.2](4,7)Isobenzofuranophanes - Synthesis, Characterisation, and Reactivity. Chem. Ber. 1994, 127, 2263–2266. 10.1002/cber.1491271126. [DOI] [Google Scholar]
- Zhang X.; Cunningham M. M.; Walker R. A. Solvent Polarity at Polar Solid Surfaces: The Role of Solvent Structure. J. Phys. Chem. B 2003, 107, 3183–3195. 10.1021/jp021071i. [DOI] [Google Scholar]
- Ohta A.; Hattori K.; Kusumoto Y.; Kawase T.; Kobayashi T.; Naito H.; Kitamura C. Effects of Alkoxy Substitution on the Optical Properties of 9,10-Anthraquinone and Anthracene: 2,3,6,7-Tetrapropoxy-Substituted vs. 2,6-Dipropoxy-Substituted Derivatives. Chem. Lett. 2012, 41, 674–676. 10.1246/cl.2012.674. [DOI] [Google Scholar]
- Chen S.-C.; Fang T.-S. Observation of a Novel Emission from an Exciplex of Triplet 4-Phenylbenzophenone with Triethylamine. Chem. Phys. Lett. 2007, 450, 65–70. 10.1016/j.cplett.2007.11.011. [DOI] [Google Scholar]
- Peters R. H.; Sumner H. H. 430. Spectra of Anthraquinone Derivatives. J. Chem. Soc. 1953, 2101–2110. 10.1039/jr9530002101. [DOI] [Google Scholar]
- Kuzmanich G.; Simoncelli S.; Gard M. N.; Spänig F.; Henderson B. L.; Guldi D. M.; Garcia-Garibay M. A. Excited State Kinetics in Crystalline Solids: Self-Quenching in Nanocrystals of 4,4′-Disubstituted Benzophenone Triplets Occurs by a Reductive Quenching Mechanism. J. Am. Chem. Soc. 2011, 133, 17296–17306. 10.1021/ja204927s. [DOI] [PubMed] [Google Scholar]
- Rajendiran N.; Sankaranarayanan R. K.; Venkatesh G. Excimer Emission in Inclusion Complexes of Dibenzofuran and 5-Dibenzosuberenone with α- and β-Cyclodextrins. Bull. Chem. Soc. Jpn. 2014, 87, 797–808. 10.1246/bcsj.20140057. [DOI] [Google Scholar]
- Gautrot J. E.; Hodge P.; Helliwell M.; Raftery J.; Cupertino D. Synthesis of Electron-Accepting Polymers Containing Phenanthra-9,10-Quinone Units. J. Mater. Chem. 2009, 19, 4148–4156. 10.1039/b901853k. [DOI] [Google Scholar]
- Togashi D. M.; Nicodem D. E. Photophysical Studies of 9,10-Phenanthrenequinones. Spectrochim. Acta, Part A 2004, 60, 3205–3212. 10.1016/j.saa.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Fischer G.; Oehme G.; Schellenberger A. Zur Theorie Der α-Ketosäuren. Tetrahedron 1971, 27, 5683–5696. 10.1016/S0040-4020(01)91733-8. [DOI] [Google Scholar]
- Pirenne V.; Kurtay G.; Voci S.; Bouffier L.; Sojic N.; Robert F.; Bassani D. M.; Landais Y. Eosin-Mediated Alkylsulfonyl Cyanation of Olefins. Org. Lett. 2018, 20, 4521–4525. 10.1021/acs.orglett.8b01828. [DOI] [PubMed] [Google Scholar]
- Waele V. De; Poizat O.; Fagnoni M.; Bagno A.; Ravelli D. Unraveling the Key Features of the Reactive State of Decatungstate Anion in Hydrogen Atom Transfer (HAT) Photocatalysis. ACS Catal. 2016, 6, 7174–7182. 10.1021/acscatal.6b01984. [DOI] [Google Scholar]
- Capaldo L.; Ertl M.; Fagnoni M.; Knör G.; Ravelli D. Antimony–Oxo Porphyrins as Photocatalysts for Redox-Neutral C–H to C–C Bond Conversion. ACS Catal. 2020, 10, 9057–9064. 10.1021/acscatal.0c02250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira G. C.; Schmitt C. C.; Neumann M. G. Dependence of the Thioxanthone Triplet-Triplet Absorption Spectrum with Solvent Polarity and Aromatic Ring Substitution. J. Braz. Chem. Soc. 2006, 17, 905–909. 10.1590/S0103-50532006000500013. [DOI] [Google Scholar]
- Iyer A.; Clay A.; Jockusch S.; Sivaguru J. Evaluating Brominated Thioxanthones as Organo-Photocatalysts. J. Phys. Org. Chem. 2017, 30, e3738 10.1002/poc.3738. [DOI] [Google Scholar]
- Pant D. D.; Tripathi H. B. Spectra, Lifetimes and Energy Transfer for Uranyl Species in Solution. J. Lumin. 1974, 8, 492–501. 10.1016/0022-2313(74)90015-5. [DOI] [Google Scholar]
- Scaiano J. C. Solvent Effects in the Photochemistry of Xanthone. J. Am. Chem. Soc. 1980, 102, 7747–7753. 10.1021/ja00546a018. [DOI] [Google Scholar]
- Ghosh R.; Mondal J. A.; Ghosh H. N.; Palit D. K. Ultrafast Dynamics of the Excited States of the Uranyl Ion in Solutions. J. Phys. Chem. A 2010, 114, 5263–5270. 10.1021/jp912039r. [DOI] [PubMed] [Google Scholar]
- Raviola C.; Ravelli D. Efficiency and Selectivity Aspects in the C–H Functionalization of Aliphatic Oxygen Heterocycles by Photocatalytic Hydrogen Atom Transfer. Synlett 2019, 30, 803–808. 10.1055/s-0037-1612079. [DOI] [Google Scholar]
- Xia J.-B.; Zhu C.; Chen C. Visible Light-Promoted Metal-Free C–H Activation: Diarylketone-Catalyzed Selective Benzylic Mono- and Difluorination. J. Am. Chem. Soc. 2013, 135, 17494–17500. 10.1021/ja410815u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodwell M. B.; Yang H.; Čolović M.; Yuan Z.; Merkens H.; Martin R. E.; Bénard F.; Schaffer P.; Britton R. 18 F-Fluorination of Unactivated C–H Bonds in Branched Aliphatic Amino Acids: Direct Synthesis of Oncological Positron Emission Tomography Imaging Agents. J. Am. Chem. Soc. 2017, 139, 3595–3598. 10.1021/jacs.6b11533. [DOI] [PubMed] [Google Scholar]
- Dondi D.; Fagnoni M.; Molinari A.; Maldotti A.; Albini A. Polyoxotungstate Photoinduced Alkylation of Electrophilic Alkenes by Cycloalkanes. Chem. - Eur. J. 2004, 10, 142–148. 10.1002/chem.200305285. [DOI] [PubMed] [Google Scholar]
- Laudadio G.; Govaerts S.; Wang Y.; Ravelli D.; Koolman H. F.; Fagnoni M.; Djuric S. W.; Noël T. Selective C(sp3)–H Aerobic Oxidation Enabled by Decatungstate Photocatalysis in Flow. Angew. Chem., Int. Ed. 2018, 57, 4078–4082. 10.1002/anie.201800818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protti S.; Ravelli D.; Fagnoni M.; Albini A. Solar Light-Driven Photocatalyzed Alkylations. Chemistry on the Window Ledge. Chem. Commun. 2009, 47, 7351–7353. 10.1039/b917732a. [DOI] [PubMed] [Google Scholar]
- Capaldo L.; Ravelli D. Decatungstate as Direct Hydrogen Atom Transfer Photocatalyst for SOMOphilic Alkynylation. Org. Lett. 2021, 23, 2243–2247. 10.1021/acs.orglett.1c00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter I.; Cameron D. W.; Titman R. B. Formation of Some Linear Polycyclic Diquinones via Novel Dimerization. J. Chem. Soc. D 1969, 563–564. 10.1039/c29690000563. [DOI] [Google Scholar]
- Weigel W. K.; Dang H. T.; Yang H.-B.; Martin D. B. C. Synthesis of Amino-Diamondoid Pharmacophores via Photocatalytic C–H Aminoalkylation. Chem. Commun. 2020, 56, 9699–9702. 10.1039/D0CC02804E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.-B.; Feceu A.; Martin D. B. C. Catalyst-Controlled C–H Functionalization of Adamantanes Using Selective H-Atom Transfer. ACS Catal. 2019, 9, 5708–5715. 10.1021/acscatal.9b01394. [DOI] [Google Scholar]
- Li Y.; Lei M.; Gong L. Photocatalytic Regio- and Stereoselective C(sp3)–H Functionalization of Benzylic and Allylic Hydrocarbons as Well as Unactivated Alkanes. Nat. Catal. 2019, 2, 1016–1026. 10.1038/s41929-019-0357-9. [DOI] [Google Scholar]
- Zhang Y.; Yang X.; Wu J.; Huang D. Aerobic C-H Functionalization Using Pyrenedione as the Photocatalyst. Synthesis 2020, 52, 2512–2520. 10.1055/s-0040-1707135. [DOI] [Google Scholar]
- Capaldo L.; Merli D.; Fagnoni M.; Ravelli D. Visible Light Uranyl Photocatalysis: Direct C–H to C–C Bond Conversion. ACS Catal. 2019, 9, 3054–3058. 10.1021/acscatal.9b00287. [DOI] [Google Scholar]
- West J. G.; Bedell T. A.; Sorensen E. J. The Uranyl Cation as a Visible-Light Photocatalyst for C(sp3)–H Fluorination. Angew. Chem., Int. Ed. 2016, 55, 8923–8927. 10.1002/anie.201603149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida N.; Masuda Y.; Uemoto S.; Murakami M. A Light/Ketone/Copper System for Carboxylation of Allylic C–H Bonds of Alkenes with CO2. Chem. - Eur. J. 2016, 22, 6524–6527. 10.1002/chem.201600682. [DOI] [PubMed] [Google Scholar]
- Cantillo D.; de Frutos O.; Rincón J. A.; Mateos C.; Kappe C. O. A Continuous-Flow Protocol for Light-Induced Benzylic Fluorinations. J. Org. Chem. 2014, 79, 8486–8490. 10.1021/jo5016757. [DOI] [PubMed] [Google Scholar]
- Yatsuhashi T.; Nakajima Y.; Shimada T.; Inoue H. Photophysical Properties of Intramolecular Charge-Transfer Excited Singlet State of Aminofluorenone Derivatives. J. Phys. Chem. A 1998, 102, 3018–3024. 10.1021/jp980158u. [DOI] [Google Scholar]
- Tzirakis M.; Lykakis I.; Panagiotou G.; Bourikas K.; Lycourghiotis A.; Kordulis C.; Orfanopoulos M. Decatungstate Catalyst Supported on Silica and γ-Alumina: Efficient Photocatalytic Oxidation of Benzyl Alcohols. J. Catal. 2007, 252, 178–189. 10.1016/j.jcat.2007.09.023. [DOI] [Google Scholar]
- Kamijo S.; Kamijo K.; Maruoka K.; Murafuji T. Aryl Ketone Catalyzed Radical Allylation of C(sp3)–H Bonds under Photoirradiation. Org. Lett. 2016, 18, 6516–6519. 10.1021/acs.orglett.6b03586. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Yang X.; Tang H.; Liang D.; Wu J.; Huang D. Pyrenediones as Versatile Photocatalysts for Oxygenation Reactions with in Situ Generation of Hydrogen Peroxide under Visible Light. Green Chem. 2020, 22, 22–27. 10.1039/C9GC03152A. [DOI] [Google Scholar]
- Moriconi E. J.; Rakoczy B.; O’Connor W. F. Oxidation-Reduction Potentials and Absorption Spectra of Polycyclic Aromatic Quinones 1. J. Org. Chem. 1962, 27, 2772–2776. 10.1021/jo01055a016. [DOI] [Google Scholar]
- Chaumont A.; Klimchuk O.; Gaillard C.; Billard I.; Ouadi A.; Hennig C.; Wipff G. Perrhenate Complexation by Uranyl in Traditional Solvents and in Ionic Liquids: A Joint Molecular Dynamics/Spectroscopic Study. J. Phys. Chem. B 2012, 116, 3205–3219. 10.1021/jp209476h. [DOI] [PubMed] [Google Scholar]
- Meinrath G.; Kato Y.; Kimura T.; Yoshida Z. Stokes Relationship in Absorption and Fluorescence Spectra of U(VI) Species. Radiochim. Acta 1998, 82, 115–120. 10.1524/ract.1998.82.special-issue.115. [DOI] [Google Scholar]
- Karbowiak M.; Fourest B.; Hubert S.; Moulin C. Complexation of U(VI) with Iodate Ions: Determination of Stability Constants by Using Spectroscopic Methods and Capillary Zone Electrophoresis. Radiochim. Acta 2003, 91, 505–512. 10.1524/ract.91.9.505.20002. [DOI] [Google Scholar]
- Williams J. D.; Nakano M.; Gérardy R.; Rincón J. A.; de Frutos Ó.; Mateos C.; Monbaliu J.-C. M.; Kappe C. O. Finding the Perfect Match: A Combined Computational and Experimental Study toward Efficient and Scalable Photosensitized [2 + 2] Cycloadditions in Flow. Org. Process Res. Dev. 2019, 23, 78–87. 10.1021/acs.oprd.8b00375. [DOI] [Google Scholar]
- Wagner P. J.; Hammond G. S.. Properties and Reactions of Organic Molecules in Their Triplet States. In Adv. Photochem.; 1968; pp 21–156. [Google Scholar]
- Formosinho S. J. Photochemical Hydrogen Abstractions as Radiationless Transitions. Part 2. - Thioketones, Quinones, Aza-Aromatics, Olefins and Azobenzenes. J. Chem. Soc., Faraday Trans. 2 1976, 72, 1332–1339. 10.1039/F29767201332. [DOI] [Google Scholar]
- Formosinho S. J. Photochemical Hydrogen Abstractions as Radiationless Transitions. Part 1. - Ketones, Aldehydes and Acids. J. Chem. Soc., Faraday Trans. 2 1976, 72, 1313–1331. 10.1039/F29767201313. [DOI] [Google Scholar]
- Anpo M.; Kubokawa Y. Reactivity of Excited Triplet Alkyl Ketones in Solution. I. Quenching and Hydrogen Abstraction of Triplet Acetone. Bull. Chem. Soc. Jpn. 1977, 50, 1913–1916. 10.1246/bcsj.50.1913. [DOI] [Google Scholar]
- Walling C.; Gibian M. J. Hydrogen Abstraction Reactions by the Triplet States of Ketones 1. J. Am. Chem. Soc. 1965, 87, 3361–3364. 10.1021/ja01093a014. [DOI] [Google Scholar]
- Wagner P.; Park B.-S.. Organic Photochemistry; Padwa A., Ed.; CRC Press: Routledge, 2017. [Google Scholar]
- Wu Y.; Kim D.; Teets T. S. Photophysical Properties and Redox Potentials of Photosensitizers for Organic Photoredox Transformations. Synlett 2021, 10.1055/a-1390-9065. [DOI] [Google Scholar]
- Wagner P. J.; Kemppainen A. E.; Schott H. N. Photoreactivity of p-Methoxyphenyl Ketones. Evidence for Hydrogen Abstraction from Equilibrium Concentrations of Upper n,π* Triplets. J. Am. Chem. Soc. 1970, 92, 5280–5281. 10.1021/ja00720a081. [DOI] [Google Scholar]
- Wagner P. J.; Truman R. J.; Scaiano J. C. Substituent Effects on Hydrogen Abstraction by Phenyl Ketone Triplets. J. Am. Chem. Soc. 1985, 107, 7093–7097. 10.1021/ja00310a056. [DOI] [Google Scholar]
- Wagner P. J.; Truman R. J.; Puchalski A. E.; Wake R. Extent of Charge Transfer in the Photoreduction of Phenyl Ketones by Alkylbenzenes. J. Am. Chem. Soc. 1986, 108, 7727–7738. 10.1021/ja00284a041. [DOI] [PubMed] [Google Scholar]
- Wagner P. J.; Thomas M. J.; Puchalski A. E. Photoreactivity of α-Fluorinated Phenyl Alkyl Ketones. J. Am. Chem. Soc. 1986, 108, 7739–7744. 10.1021/ja00284a042. [DOI] [PubMed] [Google Scholar]
- Ortica F.; Romani A.; Favaro G. Light-Induced Hydrogen Abstraction from Isobutanol by Thienyl Phenyl, Dithienyl, and Thienyl Pyridyl Ketones. J. Phys. Chem. A 1999, 103, 1335–1341. 10.1021/jp983839y. [DOI] [Google Scholar]
- Chandra A. K. The Perturbational Treatment of the Process of Hydrogen Abstraction by Ketones. J. Photochem. 1979, 11, 347–360. 10.1016/0047-2670(79)85023-6. [DOI] [Google Scholar]
- Fréneau M.; Lefebvre C.; Gómez Fernández M. A.; Richard C.; Hoffmann N. Photochemical Reactivity of Phenyl (Methyl-Tetrazolyl) Ketone - Hydrogen Atom Transfer vs. Electron Transfer. New J. Chem. 2019, 43, 17151–17158. 10.1039/C9NJ03061A. [DOI] [Google Scholar]
- Péter Á.; Agasti S.; Knowles O.; Pye E.; Procter D. J. Recent Advances in the Chemistry of Ketyl Radicals. Chem. Soc. Rev. 2021, 50, 5349–5365. 10.1039/D0CS00358A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yayla H.; Knowles R. Proton-Coupled Electron Transfer in Organic Synthesis: Novel Homolytic Bond Activations and Catalytic Asymmetric Reactions with Free Radicals. Synlett 2014, 25, 2819–2826. 10.1055/s-0034-1379304. [DOI] [Google Scholar]
- Cardarelli A. M.; Fagnoni M.; Mella M.; Albini A. Hydrocarbon Activation. Synthesis of β-Cycloalkyl (Di)Nitriles through Photosensitized Conjugate Radical Addition. J. Org. Chem. 2001, 66, 7320–7327. 10.1021/jo010400k. [DOI] [PubMed] [Google Scholar]
- Papaconstantinou E.; Dimotikali D.; Politou A. Photochemistry of Heteropoly Electrolytes. The 18-Molybdodiphosphate. Inorg. Chim. Acta 1980, 43, 155–158. 10.1016/S0020-1693(00)90521-8. [DOI] [Google Scholar]
- Papaconstantinou E. Photocatalytic Oxidation of Organic Compounds Using Heteropoly Electrolytes of Molybdenum and Tungsten. J. Chem. Soc., Chem. Commun. 1982, 12–13. 10.1039/c39820000012. [DOI] [Google Scholar]
- Darwent J. R. Photocatalytic Hydrogen Evolution from Alcohols Using Dodecawolframosilicic Acid Colloidal Platinum. J. Chem. Soc., Chem. Commun. 1982, 798–799. 10.1039/c39820000798. [DOI] [Google Scholar]
- Dimotikali D.; Papaconstantinou E. Photochemistry of Heteropoly Electrolytes: The 1:12 Tungstates. Inorg. Chim. Acta 1984, 87, 177–180. 10.1016/S0020-1693(00)81833-2. [DOI] [Google Scholar]
- Ward M. D.; Brazdil J. F.; Grasselli R. K. Photocatalytic Alcohol Dehydrogenation Using Ammonium Heptamolybdate. J. Phys. Chem. 1984, 88, 4210–4213. 10.1021/j150663a004. [DOI] [Google Scholar]
- Hill C. L.; Kozik M.; Winkler J.; Hou Y.; Prosser-McCartha C. M.. Polyoxometalates in Catalytic Photochemical Hydrocarbon Functionalization and Photomicrolithography. In Photosensitive Metal—Organic Systems; Kutal C., Serpone N., Eds.; American Chemical Society, 1993; pp 243–259. [Google Scholar]
- Yamase T.; Takabayashi N.; Kaji M. Solution Photochemistry of Tetrakis(Tetrabutylammonium) Decatungstate(VI) and Catalytic Hydrogen Evolution from Alcohols. J. Chem. Soc., Dalton Trans. 1984, 5, 793–799. 10.1039/dt9840000793. [DOI] [Google Scholar]
- Yamase T.; Usami T. Photocatalytic Dimerization of Olefins by Decatungstate(VI), [W10O32]4–, in Acetonitrile and Magnetic Resonance Studies of Photoreduced Species. J. Chem. Soc., Dalton Trans. 1988, 183–190. 10.1039/DT9880000183. [DOI] [Google Scholar]
- Hill C. L.; Bouchard D. A. Catalytic Photochemical Dehydrogenation of Organic Substrates by Polyoxometalates. J. Am. Chem. Soc. 1985, 107, 5148–5157. 10.1021/ja00304a019. [DOI] [Google Scholar]
- Hill C. L.; Bouchard D. A.; Kadkhodayan M.; Williamson M. M.; Schmidt J. A.; Hilinski E. F. Catalytic Photochemical Oxidation of Organic Substrates by Polyoxometalates. Picosecond Spectroscopy, Photochemistry, and Structural Properties of Charge-Transfer Complexes between Heteropolytungstic Acids and Dipolar Organic Compounds. J. Am. Chem. Soc. 1988, 110, 5471–5479. 10.1021/ja00224a035. [DOI] [Google Scholar]
- Chambers R. C.; Hill C. L. Excited States of Polyoxometalates as Oxidatively Resistant Initiators of Hydrocarbon Autoxidation. Selective Production of Hydroperoxides. Inorg. Chem. 1989, 28, 2509–2511. 10.1021/ic00312a002. [DOI] [Google Scholar]
- Combs-Walker L. A.; Hill C. L. Use of Excited-State and Ground-State Redox Properties of Polyoxometalates for Selective Transformation of Unactivated Carbon-Hydrogen Centers Remote from the Functional Group in Ketones. J. Am. Chem. Soc. 1992, 114, 938–946. 10.1021/ja00029a022. [DOI] [Google Scholar]
- Jaynes B. S.; Hill C. L. Selective Ethylation and Vinylation of Alkanes via Polyoxotungstate Photocatalyzed Radical Addition Reactions. J. Am. Chem. Soc. 1993, 115, 12212–12213. 10.1021/ja00078a089. [DOI] [Google Scholar]
- Jaynes B. S.; Hill C. L. Radical Carbonylation of Alkanes via Polyoxotungstate Photocatalysis. J. Am. Chem. Soc. 1995, 117, 4704–4705. 10.1021/ja00121a028. [DOI] [Google Scholar]
- Zheng Z.; Hill C. L. Alkanes to Nitriles and α-Iminoesters. Polyoxotungstate Photocatalytic Radical Chain Initiation. Chem. Commun. 1998, 2467–2468. 10.1039/a805036h. [DOI] [Google Scholar]
- Prosser-McCartha C. M.; Hill C. L. Direct Selective Acylation of an Unactivated Carbon-Hydrogen Bond in a Caged Hydrocarbon. Approach to Systems for Carbon-Hydrogen Bond Functionalization That Proceed Catalytically and Selectively at High Substrate Conversion. J. Am. Chem. Soc. 1990, 112, 3671–3673. 10.1021/ja00165a069. [DOI] [Google Scholar]
- Renneke R. F.; Pasquali M.; Hill C. L. Polyoxometalate Systems for the Catalytic Selective Production of Nonthermodynamic Alkenes from Alkanes. Nature of Excited-State Deactivation Processes and Control of Subsequent Thermal Processes in Polyoxometalate Photoredox Chemistry. J. Am. Chem. Soc. 1990, 112, 6585–6594. 10.1021/ja00174a020. [DOI] [Google Scholar]
- Chambers R. C.; Hill C. L. Redox Catalysis Involving Substrate Photooxidation with Catalyst Regeneration by Substrate Reduction. Simultaneous Oxidative C–H Bond Cleavage and Reductive C–S Bond Cleavage in Thioethers Catalyzed by Polyoxotungstate, W10O324–. J. Am. Chem. Soc. 1990, 112, 8427–8433. 10.1021/ja00179a029. [DOI] [Google Scholar]
- Chemseddine A.; Sanchez C.; Livage J.; Launay J. P.; Fournier M. Electrochemical and Photochemical Reduction of Decatungstate: A Reinvestigation. Inorg. Chem. 1984, 23, 2609–2613. 10.1021/ic00185a014. [DOI] [Google Scholar]
- Kothe T.; Martschke R.; Fischer H. Photoreactions of the Decatungstate Anion W10O324– with Organic Substrates in Solution Studied by EPR and Kinetic Absorption Spectroscopy: An Example for the Persistent Radical Effect. J. Chem. Soc., Perkin Trans. 2 1998, 503–508. 10.1039/a708191j. [DOI] [Google Scholar]
- Tanielian C. Decatungstate Photocatalysis. Coord. Chem. Rev. 1998, 178–180, 1165–1181. 10.1016/S0010-8545(98)00160-X. [DOI] [Google Scholar]
- Ermolenko L. P.; Delaire J. A.; Giannotti C. Laser Flash Photolysis Study of the Mechanism of Photooxidation of Alkanes Catalysed by Decatungstate Anion. J. Chem. Soc., Perkin Trans. 2 1997, 25–30. 10.1039/a604691f. [DOI] [Google Scholar]
- Tanielian C.; Duffy K.; Jones A. Kinetic and Mechanistic Aspects of Photocatalysis by Polyoxotungstates: A Laser Flash Photolysis, Pulse Radiolysis, and Continuous Photolysis Study. J. Phys. Chem. B 1997, 101, 4276–4282. 10.1021/jp970475l. [DOI] [Google Scholar]
- Duncan D. C.; Fox M. A. Early Events in Decatungstate Photocatalyzed Oxidations: A Nanosecond Laser Transient Absorbance Reinvestigation. J. Phys. Chem. A 1998, 102, 4559–4567. 10.1021/jp980723t. [DOI] [Google Scholar]
- Tanielian C.; Schweitzer C.; Seghrouchni R.; Esch M.; Mechin R. Polyoxometalate Sensitization in Mechanistic Studies of Photochemical Reactions: The Decatungstate Anion as a Reference Sensitizer for Photoinduced Free Radical Oxygenations of Organic Compounds. Photochem. Photobiol. Sci. 2003, 2, 297–305. 10.1039/b210786b. [DOI] [PubMed] [Google Scholar]
- Tanielian C.; Lykakis I. N.; Seghrouchni R.; Cougnon F.; Orfanopoulos M. Mechanism of Decatungstate Photocatalyzed Oxygenation of Aromatic AlcoholsPart I. Continuous Photolysis and Laser Flash Photolysis Studies. J. Mol. Catal. A: Chem. 2007, 262, 170–175. 10.1016/j.molcata.2006.08.092. [DOI] [Google Scholar]
- Texier I.; Delaire J. A.; Giannotti C. Reactivity of the Charge Transfer Excited State of Sodium Decatungstate at the Nanosecond Time Scale. Phys. Chem. Chem. Phys. 2000, 2, 1205–1212. 10.1039/a908588b. [DOI] [Google Scholar]
- Yamase T.; Ohtaka K. Photochemistry of Polyoxovanadates. Part 1. Formation of the Anion-Encapsulated Polyoxovanadate [V15O36(CO3)]7– and Electron-Spin Polarization of α-Hydroxyalkyl Radicals in the Presence of Alcohols. J. Chem. Soc., Dalton Trans. 1994, 53, 2599–2608. 10.1039/DT9940002599. [DOI] [Google Scholar]
- Duncan D. C.; Netzel T. L.; Hill C. L. Early-Time Dynamics and Reactivity of Polyoxometalate Excited States. Identification of a Short-Lived LMCT Excited State and a Reactive Long-Lived Charge-Transfer Intermediate Following Picosecond Flash Excitation of [W10O32]4– in Acetonitrile. Inorg. Chem. 1995, 34, 4640–4646. 10.1021/ic00122a021. [DOI] [Google Scholar]
- Texier I.; Delouis J. F.; Delaire J. A.; Giannotti C.; Plaza P.; Martin M. M. Dynamics of the First Excited State of the Decatungstate Anion Studied by Subpicosecond Laser Spectroscopy. Chem. Phys. Lett. 1999, 311, 139–145. 10.1016/S0009-2614(99)00803-9. [DOI] [Google Scholar]
- Jen S. F.; Anderson A. B.; Hill C. L. Alkane Reactions with Photoactivated Decatungstate in Neutral and Acid Solution: Molecular Orbital Theory. J. Phys. Chem. 1992, 96, 5658–5662. 10.1021/j100192a083. [DOI] [Google Scholar]
- Ravelli D.; Dondi D.; Fagnoni M.; Albini A.; Bagno A. Electronic and EPR Spectra of the Species Involved in [W10O32]4– Photocatalysis. A Relativistic DFT Investigation. Phys. Chem. Chem. Phys. 2013, 15, 2890–2896. 10.1039/c2cp43950f. [DOI] [PubMed] [Google Scholar]
- McGlynn S. P.; Smith J. K. The Electronic Structure, Spectra, and Magnetic Properties of Actinyl Ions. J. Mol. Spectrosc. 1961, 6, 164–187. 10.1016/0022-2852(61)90237-5. [DOI] [Google Scholar]
- Rabinowitch E.; Belford R. L.. Spectroscopy and Photochemistry of Uranyl Compounds; Pergamon, The MacMillan Company: New York, 1964. [Google Scholar]
- Burrows H. D.; Kemp T. J. The Photochemistry of the Uranyl Ion. Chem. Soc. Rev. 1974, 3, 139–165. 10.1039/cs9740300139. [DOI] [Google Scholar]
- Hill R. J.; Kemp T. J.; Allen D. M.; Cox A. Absorption Spectrum, Lifetime and Photoreactivity towards Alcohols of the Excited State of the Aqueous Uranyl Ion (UO2+2). J. Chem. Soc., Faraday Trans. 1 1974, 70, 847–857. 10.1039/f19747000847. [DOI] [Google Scholar]
- Jørgensen C. K.; Reisfeld R.. Uranyl Photophysics. In Topics in Inorganic and Physical Chemistry. Structure and Bonding; 1982; Vol. 50, pp 121–171. [Google Scholar]
- Knör G. Bionic Catalyst Design: A Photochemical Approach to Artificial Enzyme Function. ChemBioChem 2001, 2, 593–596. 10.1002/1439-7633(20010803)2:7/8<593::AID-CBIC593>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Hoffmann N. Photochemical Electron and Hydrogen Transfer in Organic Synthesis: The Control of Selectivity. Synthesis 2016, 48, 1782–1802. 10.1055/s-0035-1561425. [DOI] [Google Scholar]
- Morton C. M.; Zhu Q.; Ripberger H.; Troian-Gautier L.; Toa Z. S. D.; Knowles R. R.; Alexanian E. J. C–H Alkylation via Multisite-Proton-Coupled Electron Transfer of an Aliphatic C–H Bond. J. Am. Chem. Soc. 2019, 141, 13253–13260. 10.1021/jacs.9b06834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L.; Tada N.; Okubo H.; Miura T.; Itoh A. Efficient Synthesis of Gem-Dihydroperoxides with Molecular Oxygen and Anthraquinone under Visible Light Irradiation with Fluorescent Lamp. Green Chem. 2011, 13, 2347–2350. 10.1039/c1gc15437k. [DOI] [Google Scholar]
- Tada N.; Cui L.; Okubo H.; Miura T.; Itoh A. An Efficient Synthesis of Gem-Dihydroperoxides with Molecular Oxygen and Anthracene under Light Irradiation. Adv. Synth. Catal. 2010, 352, 2383–2386. 10.1002/adsc.201000357. [DOI] [Google Scholar]
- Cui L.; Furuhashi S.; Tachikawa Y.; Tada N.; Miura T.; Itoh A. Efficient Generation of Hydrogen Peroxide by Aerobic Photooxidation of 2-Propanol Using Anthraquinone-2-Carboxylic Acid and One-Pot Epoxidation of α,β-Unsaturated Ketones. Tetrahedron Lett. 2013, 54, 162–165. 10.1016/j.tetlet.2012.10.119. [DOI] [Google Scholar]
- Voutyritsa E.; Garreau M.; Kokotou M. G.; Triandafillidi I.; Waser J.; Kokotos C. G. Photochemical Functionalization of Heterocycles with EBX Reagents: C–H Alkynylation versus Deconstructive Ring Cleavage. Chem. - Eur. J. 2020, 26, 14453–14460. 10.1002/chem.202002868. [DOI] [PubMed] [Google Scholar]
- Papadopoulos G. N.; Kokotou M. G.; Spiliopoulou N.; Nikitas N. F.; Voutyritsa E.; Tzaras D. I.; Kaplaneris N.; Kokotos C. G. Phenylglyoxylic Acid: An Efficient Initiator for the Photochemical Hydrogen Atom Transfer C–H Functionalization of Heterocycles. ChemSusChem 2020, 13, 5934–5944. 10.1002/cssc.202001892. [DOI] [PubMed] [Google Scholar]
- Voutyritsa E.; Kokotos C. G. Green Metal-Free Photochemical Hydroacylation of Unactivated Olefins. Angew. Chem., Int. Ed. 2020, 59, 1735–1741. 10.1002/anie.201912214. [DOI] [PubMed] [Google Scholar]
- Yagci Y.; Jockusch S.; Turro N. J. Photoinitiated Polymerization: Advances, Challenges, and Opportunities. Macromolecules 2010, 43, 6245–6260. 10.1021/ma1007545. [DOI] [Google Scholar]
- Christmann J.; Allonas X.; Ley C.; Croutxé-Barghorn C. The Role of Ketyl Radicals in Free Radical Photopolymerization: New Experimental and Theoretical Insights. Polym. Chem. 2019, 10, 1099–1109. 10.1039/C8PY01185K. [DOI] [Google Scholar]
- Sanai Y.; Kagami S.; Kubota K. Cross-Linking Photopolymerization of Monoacrylate Initiated by Benzophenone. J. Polym. Sci., Part A: Polym. Chem. 2018, 56, 1545–1553. 10.1002/pola.29038. [DOI] [Google Scholar]
- Tretinnikov O. N.; Pilipenko V. V.; Prikhodchenko L. K. Benzophenone-Initiated Grafting Photopolymerization of Acrylic Acid on the Surface of Polyethylene from the Monomer Aqueous Solution without Its Deaeration. Polym. Sci., Ser. B 2012, 54, 427–433. 10.1134/S1560090412090060. [DOI] [Google Scholar]
- Stache E. E.; Kottisch V.; Fors B. P. Photocontrolled Radical Polymerization from Hydridic C–H Bonds. J. Am. Chem. Soc. 2020, 142, 4581–4585. 10.1021/jacs.0c00287. [DOI] [PubMed] [Google Scholar]
- Lalevée J.; Blanchard N.; Tehfe M.-A.; Fouassier J. P. Decatungstate (W10O324–)/Silane: A New and Promising Radical Source Under Soft Light Irradiation. Macromol. Rapid Commun. 2011, 32, 838–843. 10.1002/marc.201100099. [DOI] [PubMed] [Google Scholar]
- Bolland J. L.; Cooper H. R. The Photo-Sensitized Oxidation of Ethanol. Proc. R. Soc. London. Ser. A. Math. Phys. Sci. 1954, 225, 405–426. [Google Scholar]
- Schenck G. O.; Koltzenburg G.; Grossmann H. Durch Benzophenon Photosensibilisierte Synthese Der Terebinsäure. Angew. Chem. 1957, 69, 177–178. 10.1002/ange.19570690506. [DOI] [Google Scholar]
- Vaßen R.; Runsink J.; Scharf H.-D. Chirale Induktion Bei Photochemischen Reaktionen, VI. Asymmetrische Steuerung Der Addition von Semipinakol-Radikalen an Chirale Malein- Und Fumarsäureester. Chem. Ber. 1986, 119, 3492–3497. 10.1002/cber.19861191125. [DOI] [Google Scholar]
- Fraser-Reid B.; Holder N. L.; Yunker M. B. Ground and Excited State 1,4-Addition Reactions of Some Carbohydrate Enones. J. Chem. Soc., Chem. Commun. 1972, 1286–1287. 10.1039/c39720001286. [DOI] [Google Scholar]
- Fraser-Reid B.; Holder N. L.; Hicks D. R.; Walker D. L. Synthetic Applications of the Photochemically Induced Addition of Oxycarbinyl Species to α-Enones. Part I. The Addition of Simple Alcohols. Can. J. Chem. 1977, 55, 3978–3985. 10.1139/v77-565. [DOI] [Google Scholar]
- Benko Z.; Fraser-Reid B.; Mariano P. S.; Beckwith A. L. J. Conjugate Addition of Methanol to α-Enones: Photochemistry and Stereochemical Details. J. Org. Chem. 1988, 53, 2066–2072. 10.1021/jo00244a039. [DOI] [Google Scholar]
- Graalfs H.; Fröhlich R.; Wolff C.; Mattay J. Diastereoselective Addition of Radicals to Chiral 1,3-Dioxin-4-Ones. Eur. J. Org. Chem. 1999, 1999, 1057–1073. 10.1002/(SICI)1099-0690(199905)1999:5<1057::AID-EJOC1057>3.0.CO;2-A. [DOI] [Google Scholar]
- Fukuyama T.; Yamada K.; Nishikawa T.; Ravelli D.; Fagnoni M.; Ryu I. Site-Selectivity in TBADT-Photocatalyzed C(sp3)–H Functionalization of Saturated Alcohols and Alkanes. Chem. Lett. 2018, 47, 207–209. 10.1246/cl.171068. [DOI] [Google Scholar]
- Oelgemöller M.; Hoffmann N. Photochemically Induced Radical Reactions with Furanones. Pure Appl. Chem. 2015, 87, 569–582. 10.1515/pac-2014-0902. [DOI] [Google Scholar]
- Shvydkiv O.; Yavorskyy A.; Nolan K.; Youssef A.; Riguet E.; Hoffmann N.; Oelgemöller M. Photosensitized Addition of Isopropanol to Furanones in a 365 Nm UV-LED Microchip. Photochem. Photobiol. Sci. 2010, 9, 1601–1603. 10.1039/c0pp00223b. [DOI] [PubMed] [Google Scholar]
- Yavorskyy A.; Shvydkiv O.; Nolan K.; Hoffmann N.; Oelgemöller M. Photosensitized Addition of Isopropanol to Furanones in a Continuous-Flow Dual Capillary Microreactor. Tetrahedron Lett. 2011, 52, 278–280. 10.1016/j.tetlet.2010.11.018. [DOI] [Google Scholar]
- Ghosh A. K.; Leshchenko S.; Noetzel M. Stereoselective Photochemical 1,3-Dioxolane Addition to 5- Alkoxymethyl-2(5 H)-Furanone: Synthesis of Bis-Tetrahydrofuranyl Ligand for HIV Protease Inhibitor UIC-94017 (TMC-114). J. Org. Chem. 2004, 69, 7822–7829. 10.1021/jo049156y. [DOI] [PubMed] [Google Scholar]
- Ravelli D.; Albini A.; Fagnoni M. Smooth Photocatalytic Preparation of 2-Substituted 1,3-Benzodioxoles. Chem. - Eur. J. 2011, 17, 572–579. 10.1002/chem.201002546. [DOI] [PubMed] [Google Scholar]
- Prieto A.; Taillefer M. Visible-Light Decatungstate/Disulfide Dual Catalysis for the Hydro-Functionalization of Styrenes. Org. Lett. 2021, 23, 1484–1488. 10.1021/acs.orglett.1c00189. [DOI] [PubMed] [Google Scholar]
- Murphy J. J.; Bastida D.; Paria S.; Fagnoni M.; Melchiorre P. Asymmetric Catalytic Formation of Quaternary Carbons by Iminium Ion Trapping of Radicals. Nature 2016, 532, 218–222. 10.1038/nature17438. [DOI] [PubMed] [Google Scholar]
- Ravelli D.; Zoccolillo M.; Mella M.; Fagnoni M. Photocatalytic Synthesis of Oxetane Derivatives by Selective C–H Activation. Adv. Synth. Catal. 2014, 356, 2781–2786. 10.1002/adsc.201400027. [DOI] [Google Scholar]
- Bonassi F.; Ravelli D.; Protti S.; Fagnoni M. Decatungstate Photocatalyzed Acylations and Alkylations in Flow via Hydrogen Atom Transfer. Adv. Synth. Catal. 2015, 357, 3687–3695. 10.1002/adsc.201500483. [DOI] [Google Scholar]
- Ravelli D.; Montanaro S.; Zema M.; Fagnoni M.; Albini A. A Tin-Free, Radical Photocatalyzed Addition to Vinyl Sulfones. Adv. Synth. Catal. 2011, 353, 3295–3300. 10.1002/adsc.201100591. [DOI] [Google Scholar]
- Yu J.; Zhao C.; Zhou R.; Gao W.; Wang S.; Liu K.; Chen S.; Hu K.; Mei L.; Yuan L.; Chai Z.; Hu H.; Shi W. Visible-Light-Enabled C–H Functionalization by a Direct Hydrogen Atom Transfer Uranyl Photocatalyst. Chem. - Eur. J. 2020, 26, 16521–16529. 10.1002/chem.202003431. [DOI] [PubMed] [Google Scholar]
- Yamada K.; Fukuyama T.; Fujii S.; Ravelli D.; Fagnoni M.; Ryu I. Cooperative Polar/Steric Strategy in Achieving Site-Selective Photocatalyzed C(sp3)–H Functionalization. Chem. - Eur. J. 2017, 23, 8615–8618. 10.1002/chem.201701865. [DOI] [PubMed] [Google Scholar]
- Angioni S.; Ravelli D.; Emma D.; Dondi D.; Fagnoni M.; Albini A. Tetrabutylammonium Decatungstate (Chemo)Selective Photocatalyzed, Radical C–H Functionalization in Amides. Adv. Synth. Catal. 2008, 350, 2209–2214. 10.1002/adsc.200800378. [DOI] [Google Scholar]
- Capaldo L.; Fagnoni M.; Ravelli D. Vinylpyridines as Building Blocks for the Photocatalyzed Synthesis of Alkylpyridines. Chem. - Eur. J. 2017, 23, 6527–6530. 10.1002/chem.201701346. [DOI] [PubMed] [Google Scholar]
- Abadie B.; Jardel D.; Pozzi G.; Toullec P.; Vincent J. Dual Benzophenone/Copper-Photocatalyzed Giese-Type Alkylation of C(sp3)–H Bonds. Chem. - Eur. J. 2019, 25, 16120–16127. 10.1002/chem.201904111. [DOI] [PubMed] [Google Scholar]
- Paul S.; Guin J. Radical C(sp3)–H Alkenylation, Alkynylation and Allylation of Ethers and Amides Enabled by Photocatalysis. Green Chem. 2017, 19, 2530–2534. 10.1039/C7GC00840F. [DOI] [Google Scholar]
- Bonciolini S.; Di Filippo M.; Baumann M. A Scalable Continuous Photochemical Process for the Generation of Aminopropylsulfones. Org. Biomol. Chem. 2020, 18, 9428–9432. 10.1039/D0OB01801E. [DOI] [PubMed] [Google Scholar]
- Kuang Y.; Wang K.; Shi X.; Huang X.; Meggers E.; Wu J. Asymmetric Synthesis of 1,4-Dicarbonyl Compounds from Aldehydes by Hydrogen Atom Transfer Photocatalysis and Chiral Lewis Acid Catalysis. Angew. Chem., Int. Ed. 2019, 58, 16859–16863. 10.1002/anie.201910414. [DOI] [PubMed] [Google Scholar]
- Srivastava V.; Singh P. K.; Singh P. P. Photocatalysed Eosin Y Mediated C(sp3)-H Alkylation of Amine Substrates via Direct HAT. Tetrahedron Lett. 2019, 60, 1333–1336. 10.1016/j.tetlet.2019.04.016. [DOI] [Google Scholar]
- Meanwell N. A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- Sarver P. J.; Bacauanu V.; Schultz D. M.; DiRocco D. A.; Lam Y.; Sherer E. C.; MacMillan D. W. C. The Merger of Decatungstate and Copper Catalysis to Enable Aliphatic C(sp3)–H Trifluoromethylation. Nat. Chem. 2020, 12, 459–467. 10.1038/s41557-020-0436-1. [DOI] [PubMed] [Google Scholar]
- Yamada K.; Okada M.; Fukuyama T.; Ravelli D.; Fagnoni M.; Ryu I. Photocatalyzed Site-Selective C–H to C–C Conversion of Aliphatic Nitriles. Org. Lett. 2015, 17, 1292–1295. 10.1021/acs.orglett.5b00282. [DOI] [PubMed] [Google Scholar]
- Shi D.; He C.; Sun W.; Ming Z.; Meng C.; Duan C. A Photosensitizing Decatungstate-Based MOF as Heterogeneous Photocatalyst for the Selective C-H Alkylation of Aliphatic Nitriles. Chem. Commun. 2016, 52, 4714–4717. 10.1039/C6CC00862C. [DOI] [PubMed] [Google Scholar]
- Fukuyama T.; Nishikawa T.; Yamada K.; Ravelli D.; Fagnoni M.; Ryu I. Photocatalyzed Site-Selective C(sp3)–H Functionalization of Alkylpyridines at Non-Benzylic Positions. Org. Lett. 2017, 19, 6436–6439. 10.1021/acs.orglett.7b03339. [DOI] [PubMed] [Google Scholar]
- Oliva M.; Coppola G. A.; Van der Eycken E. V.; Sharma U. K. Photochemical and Electrochemical Strategies towards Benzylic C–H Functionalization: A Recent Update. Adv. Synth. Catal. 2021, 363, 1810–1834. 10.1002/adsc.202001581. [DOI] [Google Scholar]
- Qrareya H.; Ravelli D.; Fagnoni M.; Albini A. Decatungstate Photocatalyzed Benzylation of Alkenes with Alkylaromatics. Adv. Synth. Catal. 2013, 355, 2891–2899. 10.1002/adsc.201300598. [DOI] [Google Scholar]
- Wen Z.; Maheshwari A.; Sambiagio C.; Deng Y.; Laudadio G.; Van Aken K.; Sun Y.; Gemoets H. P. L.; Noël T. Optimization of a Decatungstate-Catalyzed C(sp3)–H Alkylation Using a Continuous Oscillatory Millistructured Photoreactor. Org. Process Res. Dev. 2020, 24, 2356–2361. 10.1021/acs.oprd.0c00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondi D.; Cardarelli A. M.; Fagnoni M.; Albini A. Photomediated Synthesis of β-Alkylketones from Cycloalkanes. Tetrahedron 2006, 62, 5527–5535. 10.1016/j.tet.2006.03.028. [DOI] [Google Scholar]
- Dai Z.-Y.; Nong Z.-S.; Wang P.-S. Light-Mediated Asymmetric Aliphatic C–H Alkylation with Hydrogen Atom Transfer Catalyst and Chiral Phosphoric Acid. ACS Catal. 2020, 10, 4786–4790. 10.1021/acscatal.0c00610. [DOI] [Google Scholar]
- Dai Z.-Y.; Nong Z.-S.; Song S.; Wang P.-S. Asymmetric Photocatalytic C(sp3)–H Bond Addition to α-Substituted Acrylates. Org. Lett. 2021, 23, 3157–3161. 10.1021/acs.orglett.1c00801. [DOI] [PubMed] [Google Scholar]
- Laudadio G.; Deng Y.; van der Wal K.; Ravelli D.; Nunõ M.; Fagnoni M.; Guthrie D.; Sun Y.; Noël T. C(sp3)-H Functionalizations of Light Hydrocarbons Using Decatungstate Photocatalysis in Flow. Science 2020, 369, 92–96. 10.1126/science.abb4688. [DOI] [PubMed] [Google Scholar]
- Xu S.; Chen H.; Zhou Z.; Kong W. Three-Component Alkene Difunctionalization by Direct and Selective Activation of Aliphatic C–H Bonds. Angew. Chem., Int. Ed. 2021, 60, 7405–7411. 10.1002/anie.202014632. [DOI] [PubMed] [Google Scholar]
- Campbell M. W.; Yuan M.; Polites V. C.; Gutierrez O.; Molander G. A. Photochemical C–H Activation Enables Nickel-Catalyzed Olefin Dicarbofunctionalization. J. Am. Chem. Soc. 2021, 143, 3901–3910. 10.1021/jacs.0c13077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Dai Z.-Y.; Zhang C.; Wang P.-S. Palladium-Catalyzed Allylic Alkylation via Photocatalytic Nucleophile Generation. ACS Catal. 2021, 11, 6757–6762. 10.1021/acscatal.1c01500. [DOI] [Google Scholar]
- Supranovich V. I.; Levin V. V.; Dilman A. D. Radical Addition to N -Tosylimines via C–H Activation Induced by Decatungstate Photocatalyst. Org. Lett. 2019, 21, 4271–4274. 10.1021/acs.orglett.9b01450. [DOI] [PubMed] [Google Scholar]
- Qi X.-K.; Guo L.; Yao L.-J.; Gao H.; Yang C.; Xia W. Multicomponent Synthesis of α-Branched Tertiary and Secondary Amines by Photocatalytic Hydrogen Atom Transfer Strategy. Org. Lett. 2021, 23, 4473–4477. 10.1021/acs.orglett.1c01412. [DOI] [PubMed] [Google Scholar]
- Yahata K.; Sakurai S.; Hori S.; Yoshioka S.; Kaneko Y.; Hasegawa K.; Akai S. Coupling Reaction between Aldehydes and Non-Activated Hydrocarbons via the Reductive Radical-Polar Crossover Pathway. Org. Lett. 2020, 22, 1199–1203. 10.1021/acs.orglett.0c00096. [DOI] [PubMed] [Google Scholar]
- Doohan R. A.; Geraghty N. W. A. A Comparative Analysis of the Functionalisation of Unactivated Cycloalkanes Using Alkynes and Either Sunlight or a Photochemical Reactor. Green Chem. 2005, 7, 91–96. 10.1039/b411786g. [DOI] [Google Scholar]
- Geraghty N. W.; Hannan J. J. Functionalisation of Cycloalkanes: The Photomediated Reaction of Cycloalkanes with Alkynes. Tetrahedron Lett. 2001, 42, 3211–3213. 10.1016/S0040-4039(01)00390-2. [DOI] [Google Scholar]
- Geraghty N. W. A.; Hernon E. M. The Reaction of Photochemically Generated α-Hydroxyalkyl Radicals with Alkynes: A Synthetic Route to γ-Butenolides. Tetrahedron Lett. 2009, 50, 570–573. 10.1016/j.tetlet.2008.11.067. [DOI] [Google Scholar]
- Adak T.; Hoffmann M.; Witzel S.; Rudolph M.; Dreuw A.; Hashmi A. S. K. Visible Light-Enabled sp3-C–H Functionalization with Chloro- and Bromoalkynes: Chemoselective Route to Vinylchlorides or Alkynes. Chem. - Eur. J. 2020, 26, 15573–15580. 10.1002/chem.202001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H.; Kuang Y.; Shi X.; Wong K. L.; Tan B. B.; Kwan J. M. C.; Liu X.; Wu J. Photoinduced Site-Selective Alkenylation of Alkanes and Aldehydes with Aryl Alkenes. Nat. Commun. 2020, 11, 1956. 10.1038/s41467-020-15878-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo S.; Kamijo K.; Murafuji T. Synthesis of Alkylated Pyrimidines via Photoinduced Coupling Using Benzophenone as a Mediator. J. Org. Chem. 2017, 82, 2664–2671. 10.1021/acs.joc.6b03058. [DOI] [PubMed] [Google Scholar]
- Lee W.; Jung S.; Kim M.; Hong S. Site-Selective Direct C–H Pyridylation of Unactivated Alkanes by Triplet Excited Anthraquinone. J. Am. Chem. Soc. 2021, 143, 3003–3012. 10.1021/jacs.1c00549. [DOI] [PubMed] [Google Scholar]
- Perry I. B.; Brewer T. F.; Sarver P. J.; Schultz D. M.; DiRocco D. A.; MacMillan D. W. C. Direct Arylation of Strong Aliphatic C–H Bonds. Nature 2018, 560, 70–75. 10.1038/s41586-018-0366-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Gu Y.; Martin R. sp3 C–H Arylation and Alkylation Enabled by the Synergy of Triplet Excited Ketones and Nickel Catalysts. J. Am. Chem. Soc. 2018, 140, 12200–12209. 10.1021/jacs.8b07405. [DOI] [PubMed] [Google Scholar]
- Dewanji A.; Krach P. E.; Rueping M. The Dual Role of Benzophenone in Visible-Light/Nickel Photoredox-Catalyzed C–H Arylations: Hydrogen-Atom Transfer and Energy Transfer. Angew. Chem., Int. Ed. 2019, 58, 3566–3570. 10.1002/anie.201901327. [DOI] [PubMed] [Google Scholar]
- Ren C.; Wang T.; Zhang Y.; Peng D.; Liu X.; Wu Q.; Liu X.; Luo S. Photoinduced Activation of Unactivated C(sp3)-H Bonds and Acylation Reactions. ChemistrySelect 2021, 6, 2523–2528. 10.1002/slct.202100225. [DOI] [Google Scholar]
- Li C.-J. Cross-Dehydrogenative Coupling (CDC): Exploring C–C Bond Formations beyond Functional Group Transformations. Acc. Chem. Res. 2009, 42, 335–344. 10.1021/ar800164n. [DOI] [PubMed] [Google Scholar]
- Quattrini M. C.; Fujii S.; Yamada K.; Fukuyama T.; Ravelli D.; Fagnoni M.; Ryu I. Versatile Cross-Dehydrogenative Coupling of Heteroaromatics and Hydrogen Donors via Decatungstate Photocatalysis. Chem. Commun. 2017, 53, 2335–2338. 10.1039/C6CC09725A. [DOI] [PubMed] [Google Scholar]
- Capaldo L.; Quadri L. L.; Merli D.; Ravelli D. Photoelectrochemical Cross-Dehydrogenative Coupling of Benzothiazoles with Strong Aliphatic C–H Bonds. Chem. Commun. 2021, 57, 4424–4427. 10.1039/D1CC01012C. [DOI] [PubMed] [Google Scholar]
- Chatgilialoglu C.; Crich D.; Komatsu M.; Ryu I. Chemistry of Acyl Radicals. Chem. Rev. 1999, 99, 1991–2070. 10.1021/cr9601425. [DOI] [PubMed] [Google Scholar]
- Sumino S.; Fusano A.; Fukuyama T.; Ryu I. Carbonylation Reactions of Alkyl Iodides through the Interplay of Carbon Radicals and Pd Catalysts. Acc. Chem. Res. 2014, 47, 1563–1574. 10.1021/ar500035q. [DOI] [PubMed] [Google Scholar]
- Singh J.; Sharma S.; Sharma A. Photocatalytic Carbonylation Strategies: A Recent Trend in Organic Synthesis. J. Org. Chem. 2021, 86, 24–48. 10.1021/acs.joc.0c02205. [DOI] [PubMed] [Google Scholar]
- Boese W. T.; Goldman A. S. Alkane Carbonylation Photocatalyzed by Aromatic Ketones under High CO Pressure. Tetrahedron Lett. 1992, 33, 2119–2122. 10.1016/0040-4039(92)88155-X. [DOI] [Google Scholar]
- Boese W. T.; Goldman A. S. Photochemical Cyclohexane Carbonylation Cocatalyzed by d8 Transition Metal Carbonyls and Aromatic Ketones and Aldehydes. J. Am. Chem. Soc. 1992, 114, 350–351. 10.1021/ja00027a049. [DOI] [Google Scholar]
- Ryu I.; Tani A.; Fukuyama T.; Ravelli D.; Fagnoni M.; Albini A. Atom-Economical Synthesis of Unsymmetrical Ketones through Photocatalyzed C–H Activation of Alkanes and Coupling with CO and Electrophilic Alkenes. Angew. Chem., Int. Ed. 2011, 50, 1869–1872. 10.1002/anie.201004854. [DOI] [PubMed] [Google Scholar]
- Ryu I.; Tani A.; Fukuyama T.; Ravelli D.; Montanaro S.; Fagnoni M. Efficient C–H/C–N and C–H/C–CO–N Conversion via Decatungstate-Photoinduced Alkylation of Diisopropyl Azodicarboxylate. Org. Lett. 2013, 15, 2554–2557. 10.1021/ol401061v. [DOI] [PubMed] [Google Scholar]
- Azam M.; Al-Resayes S. I.; Trzesowska-Kruszynska A.; Kruszynski R.; Kumar P.; Jain S. L. Seven-Coordinated Chiral Uranyl(VI) Salen Complex as Effective Catalyst for C–H Bond Activation of Dialkylanilines under Visible Light. Polyhedron 2017, 124, 177–183. 10.1016/j.poly.2016.12.033. [DOI] [Google Scholar]
- Esposti S.; Dondi D.; Fagnoni M.; Albini A. Acylation of Electrophilic Olefins through Decatungstate-Photocatalyzed Activation of Aldehydes. Angew. Chem., Int. Ed. 2007, 46, 2531–2534. 10.1002/anie.200604820. [DOI] [PubMed] [Google Scholar]
- Ravelli D.; Zema M.; Mella M.; Fagnoni M.; Albini A. Benzoyl Radicals from (Hetero)Aromatic Aldehydes. Decatungstate Photocatalyzed Synthesis of Substituted Aromatic Ketones. Org. Biomol. Chem. 2010, 8, 4158–4164. 10.1039/c0ob00066c. [DOI] [PubMed] [Google Scholar]
- Fagnoni M.; Bonassi F.; Palmieri A.; Protti S.; Ravelli D.; Ballini R. Flow Synthesis of Substituted γ-Lactones by Consecutive Photocatalytic/Reductive Reactions. Adv. Synth. Catal. 2014, 356, 753–758. 10.1002/adsc.201300859. [DOI] [Google Scholar]
- Garbarino S.; Protti S.; Gabrielli S.; Fagnoni M.; Palmieri A.; Ravelli D. Multi-Step Continuous Flow Synthesis of β/γ-Substituted Ketones. ChemPhotoChem. 2018, 2, 847–850. 10.1002/cptc.201800107. [DOI] [Google Scholar]
- Okada M.; Yamada K.; Fukuyama T.; Ravelli D.; Fagnoni M.; Ryu I. Photocatalytic One-Pot Synthesis of Homoallyl Ketones via a Norrish Type I Reaction of Cyclopentanones. J. Org. Chem. 2015, 80, 9365–9369. 10.1021/acs.joc.5b01850. [DOI] [PubMed] [Google Scholar]
- Yan J.; Cheo H. W.; Teo W. K.; Shi X.; Wu H.; Idres S. B.; Deng L.-W.; Wu J. A Radical Smiles Rearrangement Promoted by Neutral Eosin Y as a Direct Hydrogen Atom Transfer Photocatalyst. J. Am. Chem. Soc. 2020, 142, 11357–11362. 10.1021/jacs.0c02052. [DOI] [PubMed] [Google Scholar]
- Fan P.; Zhang C.; Lan Y.; Lin Z.; Zhang L.; Wang C. Photocatalytic Hydroacylation of Trifluoromethyl Alkenes. Chem. Commun. 2019, 55, 12691–12694. 10.1039/C9CC07285C. [DOI] [PubMed] [Google Scholar]
- Liu H.; Xue F.; Wang M.; Tang X.; Wu J. Neutral-Eosin Y-Catalyzed Regioselective Hydroacylation of Aryl Alkenes under Visible-Light Irradiation. Synlett 2021, 32, 406–410. 10.1055/a-1319-6237. [DOI] [Google Scholar]
- Wang X.; Chen Y.; Song H.; Liu Y.; Wang Q. Synthesis of Unnatural α-Amino Acids via Photoinduced Decatungstate-Catalyzed Giese Reactions of Aldehydes. Org. Lett. 2021, 23, 2199–2204. 10.1021/acs.orglett.1c00345. [DOI] [PubMed] [Google Scholar]
- Fan P.; Lan Y.; Zhang C.; Wang C. Nickel/Photo-Cocatalyzed Asymmetric Acyl-Carbamoylation of Alkenes. J. Am. Chem. Soc. 2020, 142, 2180–2186. 10.1021/jacs.9b12554. [DOI] [PubMed] [Google Scholar]
- Fan P.; Zhang C.; Zhang L.; Wang C. Acylation of Aryl Halides and α-Bromo Acetates with Aldehydes Enabled by Nickel/TBADT Cocatalysis. Org. Lett. 2020, 22, 3875–3878. 10.1021/acs.orglett.0c01121. [DOI] [PubMed] [Google Scholar]
- Kawaai K.; Yamaguchi T.; Yamaguchi E.; Endo S.; Tada N.; Ikari A.; Itoh A. Photoinduced Generation of Acyl Radicals from Simple Aldehydes, Access to 3-Acyl-4-Arylcoumarin Derivatives, and Evaluation of Their Antiandrogenic Activities. J. Org. Chem. 2018, 83, 1988–1996. 10.1021/acs.joc.7b02933. [DOI] [PubMed] [Google Scholar]
- Wang H.; Li T.; Hu D.; Tong X.; Zheng L.; Xia C. Acylation of Arenes with Aldehydes through Dual C–H Activations by Merging Photocatalysis and Palladium Catalysis. Org. Lett. 2021, 23, 3772–3776. 10.1021/acs.orglett.1c01184. [DOI] [PubMed] [Google Scholar]
- Mateo-Alonso A.; Guldi D. M.; Paolucci F.; Prato M. Fullerenes: Multitask Components in Molecular Machinery. Angew. Chem., Int. Ed. 2007, 46, 8120–8126. 10.1002/anie.200702725. [DOI] [PubMed] [Google Scholar]
- Vougioukalakis G. C.; Roubelakis M. M.; Orfanopoulos M. Open-Cage Fullerenes: Towards the Construction of Nanosized Molecular Containers. Chem. Soc. Rev. 2010, 39, 817–844. 10.1039/B913766A. [DOI] [PubMed] [Google Scholar]
- Thompson B. C.; Fréchet J. M. J. Polymer–Fullerene Composite Solar Cells. Angew. Chem., Int. Ed. 2008, 47, 58–77. 10.1002/anie.200702506. [DOI] [PubMed] [Google Scholar]
- Guldi D. M.; Illescas B. M.; Atienza C. M.; Wielopolski M.; Martín N. Fullerene for Organic Electronics. Chem. Soc. Rev. 2009, 38, 1587–1597. 10.1039/b900402p. [DOI] [PubMed] [Google Scholar]
- Nakamura E.; Isobe H. Functionalized Fullerenes in Water. The First 10 Years of Their Chemistry, Biology, and Nanoscience. Acc. Chem. Res. 2003, 36, 807–815. 10.1021/ar030027y. [DOI] [PubMed] [Google Scholar]
- Krusic P. J.; Wasserman E.; Keizer P. N.; Morton J. R.; Preston K. F. Radical Reactions of C60. Science 1991, 254, 1183–1185. 10.1126/science.254.5035.1183. [DOI] [PubMed] [Google Scholar]
- Tzirakis M. D.; Orfanopoulos M. Decatungstate-Mediated Radical Reactions of C60 with Substituted Toluenes and Anisoles: A New Photochemical Functionalization Strategy for Fullerenes. Org. Lett. 2008, 10, 873–876. 10.1021/ol7030125. [DOI] [PubMed] [Google Scholar]
- Tzirakis M. D.; Orfanopoulos M. Photochemical Addition of Ethers to C60 : Synthesis of the Simplest [60]Fullerene/Crown Ether Conjugates. Angew. Chem., Int. Ed. 2010, 49, 5891–5893. 10.1002/anie.201002285. [DOI] [PubMed] [Google Scholar]
- Tzirakis M. D.; Alberti M. N.; Orfanopoulos M. Hydroxyalkylation of [60]Fullerene: Free Radical Addition of Alcohols to C60. Chem. Commun. 2009, 46, 8228–8230. 10.1039/c0cc02836c. [DOI] [PubMed] [Google Scholar]
- Tzirakis M. D.; Orfanopoulos M. Acyl Radical Reactions in Fullerene Chemistry: Direct Acylation of [60]Fullerene through an Efficient Decatungstate-Photomediated Approach. J. Am. Chem. Soc. 2009, 131, 4063–4069. 10.1021/ja808658b. [DOI] [PubMed] [Google Scholar]
- Ravelli D.; Montanaro S.; Tomasi C.; Galinetto P.; Quartarone E.; Merli D.; Mustarelli P.; Fagnoni M. One-Step Decatungstate-Photomediated PEGylation of Single-Walled Carbon Nanotubes. ChemPlusChem 2012, 77, 210–216. 10.1002/cplu.201100040. [DOI] [Google Scholar]
- Ravelli D.; Merli D.; Quartarone E.; Profumo A.; Mustarelli P.; Fagnoni M. PEGylated Carbon Nanotubes: Preparation, Properties and Applications. RSC Adv. 2013, 3, 13569–13582. 10.1039/c3ra40852c. [DOI] [Google Scholar]
- Papadopoulos G. N.; Kokotos C. G. One-Pot Amide Bond Formation from Aldehydes and Amines via a Photoorganocatalytic Activation of Aldehydes. J. Org. Chem. 2016, 81, 7023–7028. 10.1021/acs.joc.6b00488. [DOI] [PubMed] [Google Scholar]
- Zwick C. R.; Renata H. Remote C–H Hydroxylation by an α-Ketoglutarate-Dependent Dioxygenase Enables Efficient Chemoenzymatic Synthesis of Manzacidin C and Proline Analogs. J. Am. Chem. Soc. 2018, 140, 1165–1169. 10.1021/jacs.7b12918. [DOI] [PubMed] [Google Scholar]
- Zwick C. R.; Renata H. Evolution of Biocatalytic and Chemocatalytic C–H Functionalization Strategy in the Synthesis of Manzacidin C. J. Org. Chem. 2018, 83, 7407–7415. 10.1021/acs.joc.8b00248. [DOI] [PubMed] [Google Scholar]
- Niu L.; Jiang C.; Liang Y.; Liu D.; Bu F.; Shi R.; Chen H.; Chowdhury A. D.; Lei A. Manganese-Catalyzed Oxidative Azidation of C(sp3)–H Bonds under Electrophotocatalytic Conditions. J. Am. Chem. Soc. 2020, 142, 17693–17702. 10.1021/jacs.0c08437. [DOI] [PubMed] [Google Scholar]
- Pitzer L.; Schwarz J. L.; Glorius F. Reductive Radical-Polar Crossover: Traditional Electrophiles in Modern Radical Reactions. Chem. Sci. 2019, 10, 8285–8291. 10.1039/C9SC03359A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiles R. J.; Molander G. A. Photoredox-Mediated Net-Neutral Radical/Polar Crossover Reactions. Isr. J. Chem. 2020, 60, 281–293. 10.1002/ijch.201900166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan T.; Capaldo L.; Laudadio G.; Nyuchev A.; Rincon J.; Garcia-Losada P.; Mateos Gutierrez C.; Frederick M. O.; Nuno M.; Noël T. Decatungstate-mediated C(sp3)–H Heteroarylation via Radical-Polar Crossover in Batch and Flow. Angew. Chem., Int. Ed. 2021, 10.1002/anie.202104682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Wang Q.; Xiong Z. Method for preparing KA oil through cyclohexane oxidation. CN105315128A, 2016-02-10.
- Alini S.; Babini P.. The Industrial Oxidation of KA Oil to Adipic Acid. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez D., Cavani F., Eds.; Imperial College Press, 2014; pp 320–333. [Google Scholar]
- Mao Y.; Bakac A. Photocatalytic Oxidation of Toluene to Benzaldehyde by Molecular Oxygen. J. Phys. Chem. 1996, 100, 4219–4223. 10.1021/jp9529376. [DOI] [Google Scholar]
- Ohkubo K.; Hirose K.; Fukuzumi S. Photooxygenation of Alkanes by Dioxygen with p-Benzoquinone Derivatives with High Quantum Yields. Photochem. Photobiol. Sci. 2016, 15, 731–734. 10.1039/C6PP00102E. [DOI] [PubMed] [Google Scholar]
- Wells C. F. Hydrogen Transfer to Quinones. Part 1. - Kinetics of the Deactivation of the Photo-Excited Quinone. Trans. Faraday Soc. 1961, 57, 1703–1718. 10.1039/TF9615701703. [DOI] [Google Scholar]
- Wells C. F. Hydrogen Transfer to Quinones. Part 2. - Reactivities of Alcohols, Ethers and Ketones. Trans. Faraday Soc. 1961, 57, 1719–1731. 10.1039/TF9615701719. [DOI] [Google Scholar]
- Clark K. P.; Stonehill H. I. Photochemistry and Radiation Chemistry of Anthraquinone-2-Sodium-Sulphonate in Aqueous Solution. Part 1. - Photochemical Kinetics in Aerobic Solution. J. Chem. Soc., Faraday Trans. 1 1972, 68, 577–590. 10.1039/f19726800577. [DOI] [Google Scholar]
- Clark K. P.; Stonehill H. I. Photochemistry and Radiation Chemistry of 9,10-Anthraquinone-2-Sodium Sulphonate in Aqueous Solution. Part 2. - Photochemical Products. J. Chem. Soc., Faraday Trans. 1 1972, 68, 1676–1686. 10.1039/f19726801676. [DOI] [Google Scholar]
- Lykakis I. N.; Tanielian C.; Seghrouchni R.; Orfanopoulos M. Mechanism of Decatungstate Photocatalyzed Oxygenation of Aromatic Alcohols Part II. Kinetic Isotope Effects Studies. J. Mol. Catal. A: Chem. 2007, 262, 176–184. 10.1016/j.molcata.2006.08.064. [DOI] [Google Scholar]
- Lykakis I. N.; Orfanopoulos M. Deuterium Kinetic Isotope Effects in Homogeneous Decatungstate Catalyzed Photooxygenation of 1,1-Diphenylethane and 9-Methyl-9H-Fluorene: Evidence for a Hydrogen Abstraction Mechanism. Tetrahedron Lett. 2005, 46, 7835–7839. 10.1016/j.tetlet.2005.09.017. [DOI] [Google Scholar]
- Lykakis I. N.; Vougioukalakis G. C.; Orfanopoulos M. Homogeneous Decatungstate-Catalyzed Photooxygenation of Tetrasubstituted Alkenes: A Deuterium Kinetic Isotope Effect Study. J. Org. Chem. 2006, 71, 8740–8747. 10.1021/jo061238u. [DOI] [PubMed] [Google Scholar]
- Lykakis I.; Orfanopoulos M. Decatungstate-Catalyzed Photooxygenation of S-2-Phenylbutane and Cumene via a Free Carbon-Radical Intermediate. Curr. Org. Chem. 2009, 13, 1737–1745. 10.2174/138527209789578018. [DOI] [Google Scholar]
- Nomiya K.; Sugie Y.; Miyazaki T.; Miwa M. Catalysis by Heteropolyacids - IX. Photocatalytic Oxidation of Isopropyl Alcohol to Acetone under Oxygen Using Tetrabutylammonium Decatungstate. Polyhedron 1986, 5, 1267–1271. 10.1016/S0277-5387(00)83470-1. [DOI] [Google Scholar]
- Carraro M.; Gardan M.; Scorrano G.; Drioli E.; Fontananova E.; Bonchio M. Solvent-Free, Heterogeneous Photooxygenation of Hydrocarbons by Hyflon? Membranes Embedding a Fluorous-Tagged Decatungstate. Chem. Commun. 2006, 4533–4535. 10.1039/b610551c. [DOI] [PubMed] [Google Scholar]
- Nieweg J. A.; Lemma K.; Trewyn B. G.; Lin V. S.-Y.; Bakac A. Mesoporous Silica-Supported Uranyl: Synthesis and Photoreactivity. Inorg. Chem. 2005, 44, 5641–5648. 10.1021/ic050130e. [DOI] [PubMed] [Google Scholar]
- Hermans I.; Nguyen T. L.; Jacobs P. A.; Peeters J. Autoxidation of Cyclohexane: Conventional Views Challenged by Theory and Experiment. ChemPhysChem 2005, 6, 637–645. 10.1002/cphc.200400211. [DOI] [PubMed] [Google Scholar]
- Maldotti A.; Amadelli R.; Carassiti V.; Molinari A. Catalytic Oxygenation of Cyclohexane by Photoexcited (nBu4N)4W10O32: The Role of Radicals. Inorg. Chim. Acta 1997, 256, 309–312. 10.1016/S0020-1693(96)05432-1. [DOI] [Google Scholar]
- Maldotti A.; Molinari A.; Bergamini P.; Amadelli R.; Battioni P.; Mansuy D. Photocatalytic Oxidation of Cyclohexane by (nBu4N)4W10O32Fe(III)Prophyrins Integrated Systems. J. Mol. Catal. A: Chem. 1996, 113, 147–157. 10.1016/S1381-1169(96)00107-0. [DOI] [Google Scholar]
- Wu W.; Fu Z.; Tang S.; Zou S.; Wen X.; Meng Y.; Sun S.; Deng J.; Liu Y.; Yin D. (nBu4N)4W10O32-Catalyzed Selective Oxygenation of Cyclohexane by Molecular Oxygen under Visible Light Irradiation. Appl. Catal., B 2015, 164, 113–119. 10.1016/j.apcatb.2014.08.045. [DOI] [Google Scholar]
- Giannotti C.; Richter C. Photocatalysed Oxidation of Cyclohexane by W10O32–4 Irradiation with Natural Sunlight. Int. J. Photoenergy 1999, 1, 69–73. 10.1155/S1110662X99000136. [DOI] [Google Scholar]
- Fornal E.; Giannotti C. Photocatalyzed Oxidation of Cyclohexane with Heterogenized Decatungstate. J. Photochem. Photobiol., A 2007, 188, 279–286. 10.1016/j.jphotochem.2006.12.023. [DOI] [Google Scholar]
- Molinari A.; Amadelli R.; Andreotti L.; Maldotti A. Heterogeneous Photocatalysis for Synthetic Purposes: Oxygenation of Cyclohexane with H3PW12O40 and (nBu4N)4W10O32 Supported on Silica. J. Chem. Soc., Dalton Trans. 1999, 1203–1204. 10.1039/a901051c. [DOI] [Google Scholar]
- Maldotti A.; Molinari A.; Varani G.; Lenarda M.; Storaro L.; Bigi F.; Maggi R.; Mazzacani A.; Sartori G. Immobilization of (n-Bu4N)4W10O32 on Mesoporous MCM-41 and Amorphous Silicas for Photocatalytic Oxidation of Cycloalkanes with Molecular Oxygen. J. Catal. 2002, 209, 210–216. 10.1006/jcat.2002.3618. [DOI] [Google Scholar]
- Molinari A.; Amadelli R.; Mazzacani A.; Sartori G.; Maldotti A. Tetralkylammonium and Sodium Decatungstate Heterogenized on Silica: Effects of the Nature of Cations on the Photocatalytic Oxidation of Organic Substrates. Langmuir 2002, 18, 5400–5405. 10.1021/la0110141. [DOI] [Google Scholar]
- Arnold P. L.; Purkis J. M.; Rutkauskaite R.; Kovacs D.; Love J. B.; Austin J. Controlled Photocatalytic Hydrocarbon Oxidation by Uranyl Complexes. ChemCatChem 2019, 11, 3786–3790. 10.1002/cctc.201900037. [DOI] [Google Scholar]
- Symeonidis T. S.; Tamiolakis I.; Armatas G. S.; Lykakis I. N. Green Photocatalytic Organic Transformations by Polyoxometalates vs. Mesoporous TiO2 Nanoparticles: Selective Aerobic Oxidation of Alcohols. Photochem. Photobiol. Sci. 2015, 14, 563–568. 10.1039/C4PP00268G. [DOI] [PubMed] [Google Scholar]
- Lykakis I. N.; Tanielian C.; Orfanopoulos M. Decatungstate Photocatalyzed Oxidation of Aryl Alkanols. Electron Transfer or Hydrogen Abstraction Mechanism?. Org. Lett. 2003, 5, 2875–2878. 10.1021/ol0349211. [DOI] [PubMed] [Google Scholar]
- Molinari A.; Bratovcic A.; Magnacca G.; Maldotti A. Matrix Effects on the Photocatalytic Oxidation of Alcohols by [nBu4N]4W10O32 Incorporated into Sol–Gel Silica. Dalt. Trans. 2010, 39, 7826–7833. 10.1039/c003282d. [DOI] [PubMed] [Google Scholar]
- Maldotti A.; Molinari A.; Bigi F. Selective Photooxidation of Diols with Silica Bound W10O324–. J. Catal. 2008, 253, 312–317. 10.1016/j.jcat.2007.11.008. [DOI] [Google Scholar]
- Farhadi S.; Momeni Z. Zirconia-Supported Sodium Decatungstate (Na4W10O32/ZrO2): An Efficient, Green and Recyclable Photocatalyst for Selective Oxidation of Activated Alcohols to Carbonyl Compounds with O2. J. Mol. Catal. A: Chem. 2007, 277, 47–52. 10.1016/j.molcata.2007.07.024. [DOI] [Google Scholar]
- Fujiya A.; Nobuta T.; Yamaguchi E.; Tada N.; Miura T.; Itoh A. Aerobic Photooxidative Direct Asymmetric Aldol Reactions of Benzyl Alcohols Using Water as the Solvent. RSC Adv. 2015, 5, 39539–39543. 10.1039/C5RA05155J. [DOI] [Google Scholar]
- Zhang W.; Gacs J.; Arends I. W. C. E.; Hollmann F. Selective Photooxidation Reactions Using Water-Soluble Anthraquinone Photocatalysts. ChemCatChem 2017, 9, 3821–3826. 10.1002/cctc.201700779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada N.; Hattori K.; Nobuta T.; Miura T.; Itoh A. Facile Aerobic Photooxidation of Methyl Group in the Aromatic Nucleus in the Presence of an Organocatalyst under VIS Irradiation. Green Chem. 2011, 13, 1669–1671. 10.1039/c1gc15154a. [DOI] [Google Scholar]
- Taguchi M.; Nagasawa Y.; Yamaguchi E.; Tada N.; Miura T.; Itoh A. One-Pot Epoxidation of Alkenes Using Aerobic Photoperoxidation of Toluenes. Tetrahedron Lett. 2016, 57, 230–232. 10.1016/j.tetlet.2015.12.027. [DOI] [Google Scholar]
- Dong J.; Yue F.; Wang X.; Song H.; Liu Y.; Wang Q. Light-Mediated Difluoromethylthiolation of Aldehydes with a Hydrogen Atom Transfer Photocatalyst. Org. Lett. 2020, 22, 8272–8277. 10.1021/acs.orglett.0c02902. [DOI] [PubMed] [Google Scholar]
- Wang X.; Dong J.; Liu Y.; Song H.; Wang Q. Decatungstate as a Direct Hydrogen Atom Transfer Photocatalyst for Synthesis of Trifluromethylthioesters from Aldehydes. Chin. Chem. Lett. 2021, 10.1016/j.cclet.2021.03.070. [DOI] [Google Scholar]
- Cao S.; Hong W.; Ye Z.; Gong L. Photocatalytic Three-Component Asymmetric Sulfonylation via Direct C(sp3)-H Functionalization. Nat. Commun. 2021, 12, 2377. 10.1038/s41467-021-22690-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerien D. E.; Bonesi S.; Postigo A. Fluorination Methods in Drug Discovery. Org. Biomol. Chem. 2016, 14, 8398–8427. 10.1039/C6OB00764C. [DOI] [PubMed] [Google Scholar]
- Yakubov S.; Barham J. P. Photosensitized Direct C–H Fluorination and Trifluoromethylation in Organic Synthesis. Beilstein J. Org. Chem. 2020, 16, 2151–2192. 10.3762/bjoc.16.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda-Becerril M.; Chatalova Sazepin C.; Leung J. C. T.; Okbinoglu T.; Kennepohl P.; Paquin J.-F.; Sammis G. M. Fluorine Transfer to Alkyl Radicals. J. Am. Chem. Soc. 2012, 134, 4026–4029. 10.1021/ja211679v. [DOI] [PubMed] [Google Scholar]
- Spencer A. R. A.; Grainger R.; Panigrahi A.; Lepper T. J.; Bentkowska K.; Larrosa I. Transition Metal-Free Cross-Dehydrogenative Arylation of Unactivated Benzylic C–H Bonds. Chem. Commun. 2020, 56, 14479–14482. 10.1039/D0CC06212J. [DOI] [PubMed] [Google Scholar]
- Nodwell M. B.; Bagai A.; Halperin S. D.; Martin R. E.; Knust H.; Britton R. Direct Photocatalytic Fluorination of Benzylic C–H Bonds with N-Fluorobenzenesulfonimide. Chem. Commun. 2015, 51, 11783–11786. 10.1039/C5CC04058B. [DOI] [PubMed] [Google Scholar]
- Halperin S. D.; Fan H.; Chang S.; Martin R. E.; Britton R. A Convenient Photocatalytic Fluorination of Unactivated C–H Bonds. Angew. Chem., Int. Ed. 2014, 53, 4690–4693. 10.1002/anie.201400420. [DOI] [PubMed] [Google Scholar]
- Liu W.; Huang X.; Cheng M.-J.; Nielsen R. J.; Goddard W. A.; Groves J. T. Oxidative Aliphatic C–H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin. Science 2012, 337, 1322–1325. 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]
- Halperin S. D.; Kwon D.; Holmes M.; Regalado E. L.; Campeau L.-C.; DiRocco D. A.; Britton R. Development of a Direct Photocatalytic C–H Fluorination for the Preparative Synthesis of Odanacatib. Org. Lett. 2015, 17, 5200–5203. 10.1021/acs.orglett.5b02532. [DOI] [PubMed] [Google Scholar]
- Yuan Z.; Nodwell M. B.; Yang H.; Malik N.; Merkens H.; Bénard F.; Martin R. E.; Schaffer P.; Britton R. Site-Selective, Late-Stage C–H 18F-Fluorination on Unprotected Peptides for Positron Emission Tomography Imaging. Angew. Chem., Int. Ed. 2018, 57, 12733–12736. 10.1002/anie.201806966. [DOI] [PubMed] [Google Scholar]
- Yuan Z.; Yang H.; Malik N.; Čolović M.; Weber D. S.; Wilson D.; Bénard F.; Martin R. E.; Warren J. J.; Schaffer P.; Britton R. Electrostatic Effects Accelerate Decatungstate-Catalyzed C–H Fluorination Using [18F]- and [19F]NFSI in Small Molecules and Peptide Mimics. ACS Catal. 2019, 9, 8276–8284. 10.1021/acscatal.9b02220. [DOI] [Google Scholar]
- Meanwell M.; Lehmann J.; Eichenberger M.; Martin R. E.; Britton R. Synthesis of Acyl Fluorides via Photocatalytic Fluorination of Aldehydic C–H Bonds. Chem. Commun. 2018, 54, 9985–9988. 10.1039/C8CC06375C. [DOI] [PubMed] [Google Scholar]
- Yang L.; Tan D.; Fan W.; Liu X.; Wu J.; Huang Z.; Li Q.; Wang H. Photochemical Radical C–H Halogenation of Benzyl N-Methyliminodiacetyl (MIDA) Boronates: Synthesis of Α-Functionalized Alkyl Boronates. Angew. Chem., Int. Ed. 2021, 60, 3454–3458. 10.1002/anie.202011872. [DOI] [PubMed] [Google Scholar]
- Kleoff M.; Schwan J.; Christmann M.; Heretsch P. A Modular, Argon-Driven Flow Platform for Natural Product Synthesis and Late-Stage Transformations. Org. Lett. 2021, 23, 2370–2374. 10.1021/acs.orglett.1c00661. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Ji P.; Dong Y.; Wei Y.; Wang W. Deuteration of Formyl Groups via a Catalytic Radical H/D Exchange Approach. ACS Catal. 2020, 10, 2226–2230. 10.1021/acscatal.9b05300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh Y. Y.; Nagao K.; Hoover A. J.; Hesk D.; Rivera N. R.; Colletti S. L.; Davies I. W.; MacMillan D. W. C. Photoredox-Catalyzed Deuteration and Tritiation of Pharmaceutical Compounds. Science 2017, 358, 1182–1187. 10.1126/science.aap9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R.; Li J.; Cheo H. W.; Chua R.; Zhan G.; Hou Z.; Wu J. Visible-Light-Mediated Deuteration of Silanes with Deuterium Oxide. Chem. Sci. 2019, 10, 7340–7344. 10.1039/C9SC02818H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R.; Ma L.; Yang X.; Cao J. Recent Advances in Visible-Light Photocatalytic Deuteration Reactions. Org. Chem. Front. 2021, 8, 426–444. 10.1039/D0QO01299H. [DOI] [Google Scholar]
- Dong J.; Wang X.; Wang Z.; Song H.; Liu Y.; Wang Q. Formyl-Selective Deuteration of Aldehydes with D2O via Synergistic Organic and Photoredox Catalysis. Chem. Sci. 2020, 11, 1026–1031. 10.1039/C9SC05132E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang Y.; Cao H.; Tang H.; Chew J.; Chen W.; Shi X.; Wu J. Visible Light Driven Deuteration of Formyl C–H and Hydridic C(sp3)–H Bonds in Feedstock Chemicals and Pharmaceutical Molecules. Chem. Sci. 2020, 11, 8912–8918. 10.1039/D0SC02661A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qrareya H.; Dondi D.; Ravelli D.; Fagnoni M. Decatungstate-Photocatalyzed Si–H/C–H Activation in Silyl Hydrides: Hydrosilylation of Electron-Poor Alkenes. ChemCatChem 2015, 7, 3350–3357. 10.1002/cctc.201500562. [DOI] [Google Scholar]
- Hill C. L.; Renneke R. F.; Combs L. Anaerobic Functionalization of Remote Unactivated Carbon-Hydrogen Bond by Polyoxometalates. Tetrahedron 1988, 44, 7499–7507. 10.1016/S0040-4020(01)86244-X. [DOI] [Google Scholar]
- Renneke R. F.; Hill C. L. Selective Photochemical Dehydrogenation of Saturated Hydrocarbons with Quantum Yields Approaching Unity. Angew. Chem., Int. Ed. Engl. 1988, 27, 1526–1527. 10.1002/anie.198815261. [DOI] [Google Scholar]
- West J. G.; Huang D.; Sorensen E. J. Acceptorless Dehydrogenation of Small Molecules through Cooperative Base Metal Catalysis. Nat. Commun. 2015, 6, 10093. 10.1038/ncomms10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattamuri P. V.; West J. G. Cooperative Hydrogen Atom Transfer: From Theory to Applications. Synlett 2021, 32, 1179. 10.1055/a-1463-9527. [DOI] [Google Scholar]
- Abrams D. J.; West J. G.; Sorensen E. J. Toward a Mild Dehydroformylation Using Base-Metal Catalysis. Chem. Sci. 2017, 8, 1954–1959. 10.1039/C6SC04607J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachikawa Y.; Cui L.; Matsusaki Y.; Tada N.; Miura T.; Itoh A. Aerobic Photooxidative Cleavage of 1,3-Diketones to Carboxylic Acids Using 2-Chloroanthraquinone. Tetrahedron Lett. 2013, 54, 6218–6221. 10.1016/j.tetlet.2013.09.015. [DOI] [Google Scholar]
- Yamaguchi T.; Kudo Y.; Hirashima S.; Yamaguchi E.; Tada N.; Miura T.; Itoh A. Facile and Efficient Synthesis of Hydroxyalkyl Esters from Cyclic Acetals through Aerobic Photo-Oxidation Using Anthraquinone-2-Carboxylic Acid. Tetrahedron Lett. 2015, 56, 1973–1975. 10.1016/j.tetlet.2015.02.104. [DOI] [Google Scholar]
- Hardwick T.; Ahmed N. C–H Functionalization via Electrophotocatalysis and Photoelectrochemistry: Complementary Synthetic Approach. ACS Sustainable Chem. Eng. 2021, 9, 4324–4340. 10.1021/acssuschemeng.0c08434. [DOI] [Google Scholar]
- Kim K.; Lee S.; Hong S. H. Direct C(sp3)−H Cyanation Enabled by a Highly Active Decatungstate Photocatalyst. Org. Lett. 2021, 23, 5501–5505. 10.1021/acs.orglett.1c01846. [DOI] [PubMed] [Google Scholar]
- Parodi A.; Jorea A.; Fagnoni M.; Ravelli D.; Samorì C.; Torri C; Galletti P. Bio-based crotonic acid from polyhydroxybutyrate: synthesis and photocatalyzed hydroacylation. Green Chem. 2021, 23, 3420–3427. 10.1039/D1GC00421B. [DOI] [Google Scholar]
- Murugesan V.; Ganguly A.; Karthika A.; Rasappan R. C−H Alkylation of Aldehydes by Merging TBADT Hydrogen Atom Transfer with Nickel Catalysis. Org. Lett. 2021, 23, 5389–5393. 10.1021/acs.orglett.1c01716. [DOI] [PubMed] [Google Scholar]
- Malliaros N. G.; Kellner I. D.; Drewello T.; Orfanopoulos M. Decatungstate-Photocatalyzed Addition of Lactones to [60]Fullerene. J. Org. Chem. 2021, 86, 9876–9882. 10.1021/acs.joc.1c00954. [DOI] [PubMed] [Google Scholar]
- Sarver P. J.; Bissonnette N. B.; MacMillan D. W. C. Decatungstate-Catalyzed C(sp3)−H Sulfinylation: Rapid Access to Diverse Organosulfur Functionality. J. Am. Chem. Soc. 2021, 143, 9737–9743. 10.1021/jacs.1c04722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer T. E.; Rolka A. B.; Karl T. A.; Holzhausen F.; König B. Photocatalytic C−H Trifluoromethylthiolation by the Decatungstate Anion. Org. Lett. 2021, 23, 5729–5733. 10.1021/acs.orglett.1c01870. [DOI] [PubMed] [Google Scholar]