

Abstract
Tissue resident memory (TRM) cells are thought to play a role in lung mucosal immunity to pathogens, but strategies to elicit TRM by mucosal vaccines have not yet been fully realized. Here, we formulated a vaccine composed of outer membrane protein (Omp) X from K. pneumoniae and LTA1 adjuvant that was administered by the intrapulmonary route. This vaccine elicited both Th1 and Th17 cells that shared transcriptional features with cells elicited by heat-killed K. pneumoniae. Antibody responses were required to prevent bacterial dissemination but dispensable for lung-specific immunity. In contrast, lung immunity required CD4+ T cells, STAT3 expression, and IL-17R signaling in fibroblasts. Lung-specific CD4+ T cells from OmpX+LTA1 immunized mice were observed homing to the lung and could mediate protection against infection in an adoptive transfer model. Vaccine-elicited Th17 cells showed reduced plasticity and were resistant to the immunosuppressant FK506 compared with Th1 cells, and Th17 cells conferred protection under conditions of transplant immunosuppression. These data demonstrate a novel vaccine strategy that elicits lung TRM cells and promotes serotype-independent immunity to K. pneumoniae.
One Sentence Summary:
A subunit mucosal vaccine conferred protection against heterologous Klebsiella pneumoniae strains, via IL-17R signaling in lung fibroblasts.
INTRODUCTION
Bacterial pneumonia remains one of the leading causes of death in children under five, and the critical protective role of CD4+ T cells in bacterial pneumonia has become especially apparent in the context of HIV infection or primary immunodeficiencies (1), IL-17 producing cells are also involved in host defense as our previous observations suggested that “relative IL-17 deficiency might partially explain the pulmonary host defense defect associated with either HIV infection or congenital immunodeficiency of CD4+ lymphocytes” (2). Analysis of human lungs has shown that lung tissue resident memory (TRM) cells accumulate with age (3), and their protective functions have been postulated to be based on their memory to prior antigen exposures (4). Several papers have shown that lung TRM cells generated from type IL-17 expressing helper CD4+ T cells (Th17 cells) can provide prompt protection to extracellular pathogens (5, 6). Thus, elicitation of lung TRMs may be instrumental in protective immunity against bacterial pneumonia and may inform strategies towards the development of vaccines or therapeutics.
Current pneumonia vaccines target polysaccharide antigens that elicit antibody responses that can be CD4+ T cell independent or dependent (7, 8). These vaccines are effective against specific vaccine serotypes, but with continued use, epidemiologic studies suggest the emergence of non-vaccine serotypes (9). Lung CD4+ T cells can recognize conserved protein antigens and thus provide broad immunity that is independent of bacterial serotype (5, 10, 11). However, much of this work has been performed with whole pathogens, heat-killed organisms, or adjuvants such as cholera toxin (CT) that have limited prospects for vaccine development due to safety concerns (12). We previously observed that intrapulmonary immunization with heat-killed K. pneumoniae induced a subset of lung CD4+ T cells that could recognize outer membrane proteins (Omps) (11). We hypothesized that recombinant bacterial Omps formulated with a mucosal Th17 adjuvant may elicit antigen-specific lung TRM cells, which could provide serotype-independent immunity in the lung. Based on prior work with influenza (13), we focused on the LTA1 adjuvant, which is derived from the A1 domain of the heat-labile toxin from E. coli. This adjuvant has recently been shown not to perturb olfaction in contrast to CT (13). LTA1 admixed with OmpX from K. pneumoniae elicited a robust population of antigen-specific CD4+ TRM cells that provided serotype-independent immunity to hypervirulent K. pneumoniae, and these CD4+ T cells required STAT3 but not IL-21R expression for their protective functions. IL-17RA/RC signaling in Dermo1+ Col1a2+ lung fibroblasts was also essential for vaccine efficacy. A large percentage of Th17 cells, as opposed to Th1 cells, were resistant to calcineurin inhibition, and these cells were associated with vaccine-induced immunity even in the presence of calcineurin immunosuppression to prevent allograft rejection. This works lays the foundation for respiratory-targeted vaccines that induce broadly protective lung TRM cells against phylogenetically related Gram-negative bacteria, including multidrug-resistant species.
RESULTS
The OmpX+LTA1 subunit vaccine generates bacterial specific Th1 and Th17 TRM cells
Prior work has shown that heat-killed K. pneumoniae elicits lung TRM cells (5, 11), and a portion of these cells recognize bacterial-encoded outer membrane proteins (11), so we sought to determine if this approach could be used to develop a subunit vaccine targeting K. pneumoniae infection. We cloned OmpX from the KP-43816 (K2) strain, due to homology with other Klebsiella spp and efficient production in E. coli. Immunized mice were challenged with a mucoid hypervirulent KP-396 (K1 strain) of K. pneumoniae to test for serotype independent immunity (11). We initially tested the mucosal adjuvant LTA1 because of its ability to elicit lung immunity and Th17 responses (13, 14). Mice were immunized twice by intratracheal oropharyngeal aspiration at three week intervals to model an inhaled vaccine schedule. One week following second immunization, mice were challenged with 104 colony forming unit (CFU) of KP-396 (Fig. 1A). Immunized mice showed a drastic reduction in lung CFU after intratracheal challenge with KP-396 that was accompanied by substantially reduced bacterial dissemination to the spleen (Fig. 1B) as well as improved survival (Fig. 1C) and oxygen saturations (Fig. S1A). Mucosal immunization with unadjuvanted OmpX or LTA1 alone limited vaccine efficacy, (Fig. S1B), and mucosal immunization was more effective than subcutaneous immunization (Fig. S1C). H&E histology analysis of immunized mice showed perivascular and peribronchial lymphocyte accumulation consistent with the architectural features of inducible bronchus-associated lymphoid tissue (iBALT) (Fig. 1D) (15). Further evaluation showed iBALT contained B cell follicles, CD3+ T cells, IgG producing plasma cells, follicular dendritic cells, CXCL13+ stromal cells, peripheral node addressin (PNAd)+ high endothelial venules, and lymphatic vessel endothelial receptor 1 (Lyve-1)+ lymphatic vessels (Fig. 1D).
Fig. 1. Intratracheal delivery of OmpX adjuvanted with LTA1 elicits tissue resident lung CD4+ Th1 and Th17 cells.
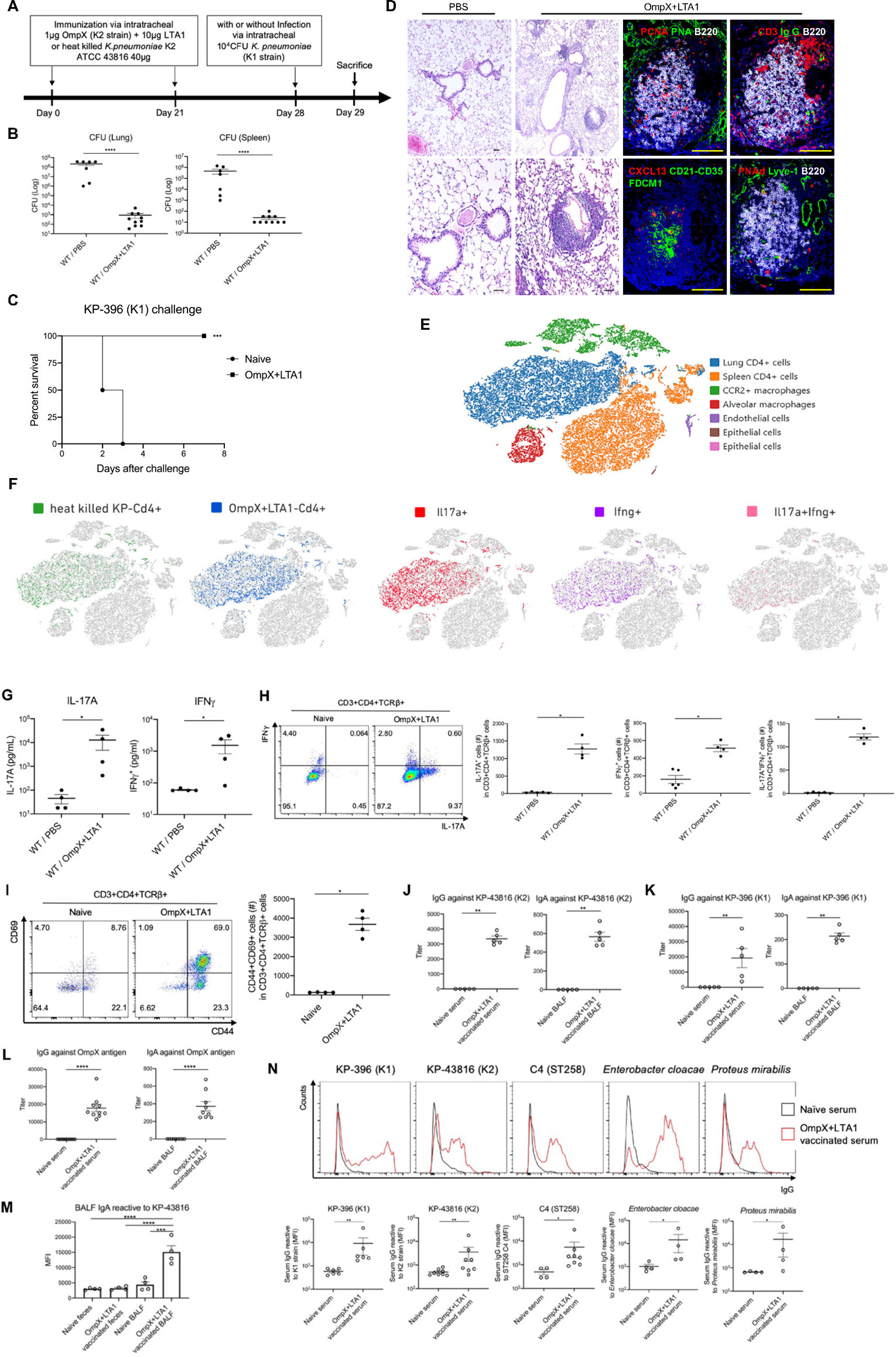
(A) Cartoon depicting immunization schedule with OmpX+LTA1 or heat killed Klebsiella pneumoniae (ATCC 43816) followed by challenge with a KP-396 (K1 strain) of K. pneumoniae. (B) C57BL/6 mice immunized and challenged as in (A) were sacrificed and bacterial burden (CFU) in lungs and spleen were determined at 24 h post infection (n = 7–10, **** denotes P < 0.0001 by Mann-Whitney U test). (C) Survival post KP-396 challenge (data were pooled from 2 independent experiment (n = 6 per group). *** denotes P < 0.01 by Log-rank test. (D) Cellular and structural features of iBALT in the lungs of immunized C57BL/6 mice, compared to naïve mice, were visualized with H&E stain (left) and multicolor immunofluorescence (right) (both panels scale bar = 100 μm). Panoramic view of iBALT structures around blood vessels and lung parenchyma with lymphocytes mainly located around blood vessels in the lungs of vaccinated mice. The representative picture shows a compact and well-organized B cell follicle with PCNA+B220+ proliferating B cells, surrounded by CD3+ T cells and IgG+ plasma cells. In the center of B cell follicle, there is a complex network of CD21+FDC-M1+ follicular dendritic cells and CXCL13+ stromal cells (Bottom middle panel). iBALT is equipped with a specialized vasculature, composed of high endothelial venules decorated with peripheral node addressing (PNAd) and lymphatics expressing lymphatic vessel endothelial receptor 1(Lyve-1, Bottom right panel). (E) t-SNE plot of scRNAseq comparing naïve splenic CD4+ T cells to lung CD4+ T cells from mice immunized with OmpX+LTA1 or heat killed K. pneumoniae (n = 2 per group) without infectious challenge (as outlined in cartoon A). (F) Data from (E) were analyzed by group and cytokine expression to detect Il17a+, Ifnγ+, and Il17a+Ifnγ+ CD4+ T cells. (G) Antigen recall responses of CD4+ T cells in lungs of naïve and OmpX+LTA1 immunized mice were analyzed after in vitro stimulation with 2 μg/mL of OmpX for three days. IL-17A and IFNγ in the supernatant were measured by Luminex. (H) IL-17A and IFNγ production by lung CD4+ T cells was estimated by intracellular cytokine analysis. (I) Co-expression of CD44 and CD69 on non-stimulated lung CD4+ T cells (gated in CD3+CD4+TCRβ+ cells). (J) Serum IgG and BALF IgA against whole KP-43816 antigen (K2 strain) and (K) whole KP-396 antigen (K1 strain) were measured by ELISA. (L) Serum IgG and BALF IgA against the OmpX antigen (M) BALF or fecal lysates to measure IgA specific to whole KP-43816 antigen (K2 strain) from naïve or OmpX+LTA1 vaccinated mice assayed by FACS of fluorescently stained (Syto+) bacteria to detect IgA binding. (N) Serum IgG binding against related Enterobacteriaceae family members by flow cytometry. Data are presented as mean ± SEM (n = 4–8). Significant differences were calculated with Mann-Whitney U test (G-L, N) and one-way ANOVA followed by Tukey’s multiple comparisons test (M). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
To compare the capacity of different vaccination approaches to elicit lung TRM cells (5, 11), we immunized mice with heat-killed K. pneumoniae or with the subunit vaccine (Fig. 1A) and analyzed the lung CD4+ T cells by single cell RNA sequencing (scRNAseq). Hierarchical clustering followed by T-distributed Stochastic Neighbor Embedding (t-SNE) plots of the scRNAseq data revealed that CD4+ T cells from OmpX immunized lungs clustered with the CD4+ T cells elicited by heat killed K. pneumoniae and that these cells were distinct from naïve splenic CD4+ T cells (Fig. 1E). t-SNE plots were analyzed for cytokine gene expression and showed that immunized lung CD4+ T cells mostly consisted of Il17a+, Ifnγ+, and Il17a+Ifnγ+ cells (Fig. 1F). Consistent with the scRNAseq data, CD4+ T cells secreted both IL-17A and IFNγ ex vivo in antigen specific recall responses (Fig. 1G) and displayed distinct Th17 and Th1 responses (Fig. 1H, Fig. S1P). In addition, immunized lung CD4+ T cells expressed CD44 and CD69, which are considered markers for lung TRM cells (Fig. 1I, Fig. S1P). We performed intravascular staining with anti-CD45 to further validate that subunit immunization elicited TRM cells,(16). Approximately 90% of the lung CD4+ T cells were excluded from intravascular labeling consistent with their TRM phenotype (Fig. S1D, S1E, S1Q). Moreover, analysis of the scRNAseq data also showed significant downregulation of Klf2 (Fig. S1F) and S1pr1 (Fig. S1G), and upregulation of Cd69 (Fig. S1H) and Cxcr6 (Fig. S1I), molecules expressed by TRM cells. A small portion of the cells also expressed Itgae (Fig. S1J), consistent with a TRM cell phenotype (5, 17, 18). Of note, the elicited lung CD4+ TRM cells were also resistant to depletion by intraperitoneal injection of rat-anti-CD4 antibody GK1.5 (Fig. S1K), in spite of evidence of being coated with rat IgG (Fig. S1L). Intraperitoneal treatment with FTY720 blocks egress from secondary lymphoid structures (19), and was administered one week after the second and resulted in a modest reduction in lung CD4+ T cells and B cells (Fig. S1M). FTY720-treated mice maintained protection against the KP-396 challenge, consistent with findings that TRM cells were sufficient to control pulmonary bacterial infection (Fig. S1M).
OmpX immunization resulted in KP-43816 whole antigen-specific IgG in the serum and IgA in the bronchoalveolar lavage fluid (BALF) (Fig. 1J) as well as KP-396 whole antigen specific IgG in the serum and IgA in the BALF (Fig. 1K). We also confirmed the presence of recombinant OmpX-specific serum IgG and BALF IgA (Fig. 1L). Klebsiella species are commensal bacteria, so we investigated whether intrapulmonary vaccination induced KP-43816-specific IgA in feces, but we observed no staining of KP-43816 by fecal IgA, which differs from observations in BALF (Fig. 1M, Fig. S1R). IgG reacted to whole KP-396 and KP-43816 antigen preparations, and we observed surface binding to KP-396 (K1 strain), KP-43816 (K2 strain), C4 (ST258) (20) as well as the related Enterobacteriaceae family members E. cloacae and P. mirabilis (Fig. 1N). Histologically, unvaccinated mice showed perivascular edema as early as 2 h post inoculation, with the development of severe pneumonia at 24 h, compared with minimal symptoms detected in OmpX+LTA1 immunized mice (Fig. S1N).
Vaccine-induced lung CD4+ Th17 cells are critical for bacterial control
We treated mice with anti-CD4 Ab before and during immunization to determine if CD4+ T cells were required for subunit vaccine efficacy in the lung. Vaccine induced control of bacterial growth after KP-396 challenge was substantially abrogated in both the lung and spleen of CD4 depleted mice (Fig. 2A). We observed abrogation of vaccine efficacy in Rag2/OTII mice, supporting the requirement of antigen specifc lymphocytes for vaccine efficacy (Fig. S1O). We administered anti-CD4 antibodies intraperitoneally and intratracheally following the second immunization to deplete lung TRM cells and evaluate if CD4+ T cells were required after immunization (21). This experimental approach completely abrogated vaccine protection in the lung and substantially abrogated bacterial control in the spleen after KP-396 challenge (Fig. 2B). STAT3 is required for Th17 commitment, and we immunized Stat3fl/flCd4Cre conditional knock out mice to assess the participation of Th17 cells in lung protective immunity (22). Bacterial control was substantially reduced in immunized Stat3fl/flCd4Cre+ mice (Fig. 2C), which was associated with reduced lung CD4+ T cells and B cells compared with immunized Stat3fl/flCd4Cre− mice (Fig. 2D, 2E). Moreover, antigen-specific IL-17A recall responses were abrogated in STAT3 deficient CD4+ T cells (Fig. 2F). IL-21R signaling is one mechanism of STAT3 activation in CD4+ T cells, and we observed a slight reduction of lung CD4+ T cell and B cell accumulation in vaccinated Il21r−/− mice, but this was not statistically significant (Fig. S2A). When these mice underwent bacterial challenge with KP-396, we observed a modest increase in splenic CFU but not lung CFU in Il21r−/− mice (Fig. S2B). We detected comparable IL-17A recall responses in Il21r−/− and WT mice (Fig. S2C), but antigen-specific IgA in BALF and IgG in serum were dramatically decreased in Il21r−/− mice compared to WT mice (Fig. S2D, S2E). The attenuated antibody response might be due to to reduced germinal center formation due to the loss of IL-21R signaling (23). These data support a role for B cells and antibodies against bacterial dissemination from the lung. Moreover, we tested whether vaccine efficacy required Cxcr5 which plays a key role in iBALT formation, which can be regulated by IL-17 induction of the CXCL13-CXCR5 axis (24). To this end, we found that vaccine induced bacterial control was abrogated in Cxcr5−/− mice (Fig. S2F).
Fig. 2. OmpX+LTA1 vaccine efficacy depends on CD4 T-cells and STAT3 expression by CD4 T cells.
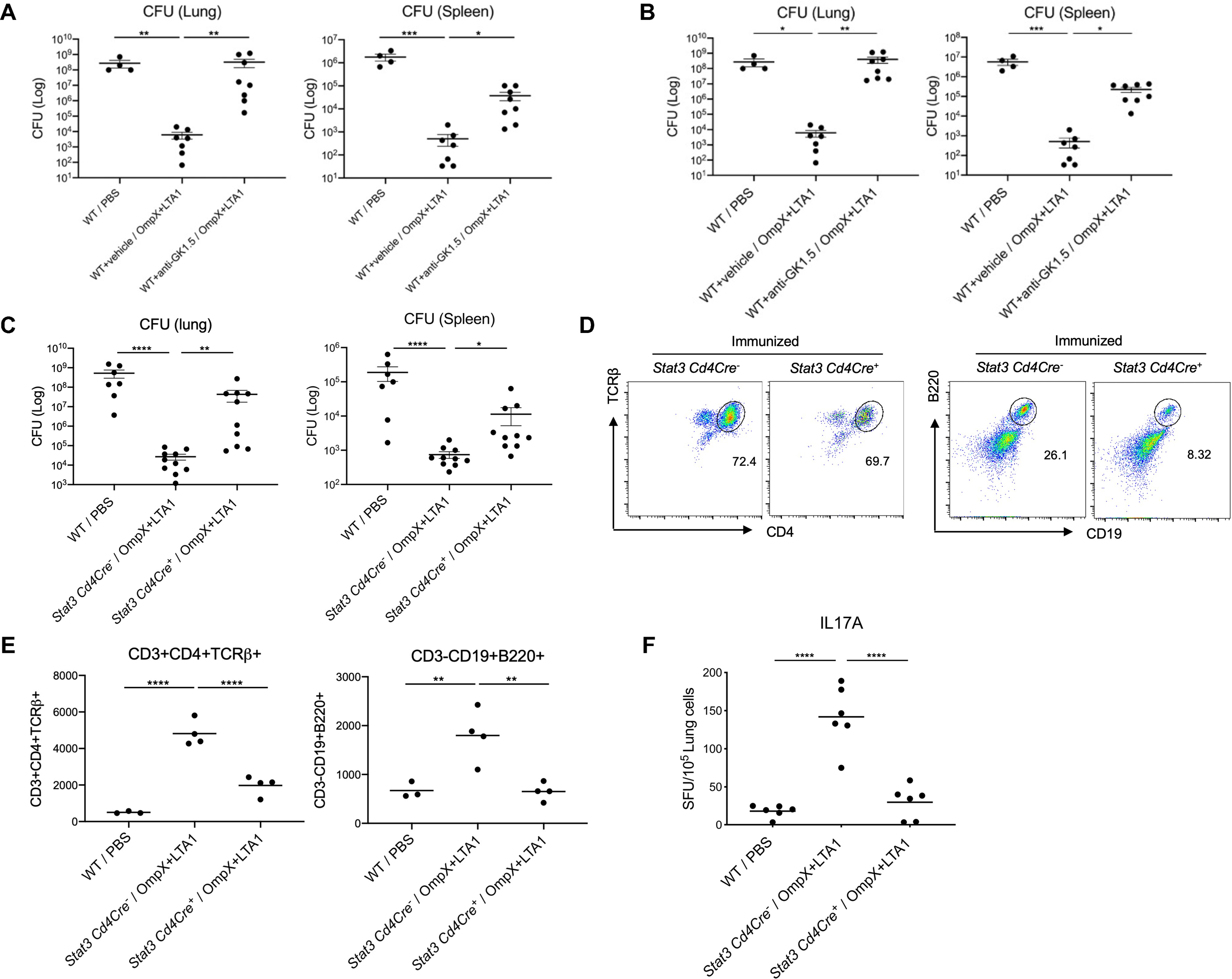
(A) Vaccinated 6–10 week old male WT mice were intraperitoneally treated with 300 μg of isotype control or anti-GK1.5 Ab (to deplete CD4+ T cells) 2 days before immunization, which was administered weekly throughout experimental period to maintain CD4+ T cell depletion. Mice were challenged with 104 CFU of the KP-396 strain, and lung and spleen CFU were assessed 24 h post challenge (n = 4–8, cumulative data of two separate experiments are shown). (B) To deplete lung resident CD4+ T cells, 500 μg of the GK1.5 Ab was intraperitoneally administered in combination with 100 μg delivered intratracheally at day 28 and 30 post-vaccination, followed by KP-396 challenge 24 h later. Lung and spleen CFU are shown (n = 7–9, cumulative data of two separate experiments are shown). (C) Lung and spleen CFU 24 h post infection in naïve WT C57BL/6 mice and OmpX+LTA1 immunized Stat3fl/fl Cd4Cre− mice and Stat3fl/fl Cd4Cre+ mice (n = 7–9, cumulative data of two separate experiments are shown). (D) Representative dot plots of lung CD3+CD4+ T cells (left) or CD19+B220+ B cells (right), in OmpX+LTA1 immunized Stat3fl/fl Cd4Cre− mice and Stat3fl/fl Cd4Cre+ mice. (E) Total lung CD4+ T cells and B cells in naïve WT C57BL/6 mice and OmpX+LTA1 vaccinated Stat3fl/fl Cd4Cre− mice and Stat3fl/fl Cd4Cre+ mice (n = 4, representative from two independent experiments). (F) Frequency of OmpX-specific IL-17A-producing CD4+ T cells in naïve or immunized lungs from Stat3fl/fl Cd4Cre− mice and Stat3fl/fl Cd4Cre+ mice was measured by ELISpot (n = 6, representative from 2 independent experiments). Data are presented as mean ± SEM. Significant differences are designated by using one-way ANOVA followed by Tukey’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Bacterial control was maintained in immunized Ifnγ−/− mice, suggesting that interferon-γ was dispensable for protective bacterial immunity (Fig. S3A). These mice also recruited similar numbers of CD4+ T cells and B cells (Fig. S3B) as well as Th17 cells (Fig. S3C) to the lung. The absence of γδ T cells did not affect vaccine efficacy, likely because other cells maintained protection in Tcrd−/− mice (Fig. S3D). Time course studies in wild-type B6 mice showed that vaccine efficacy was maintained for 1 month. Although bacterial control in the KP-396 challenge model was less efficient, it was still significant at 2, 4, or 6 months (p < 0.05) (fig S3E). Impaired efficacy correlated statistically with the contraction of lung Th17 cells over the study period (Fig. S3F).
We hypothesized that cross-reactive T cell responses may be present given the cross reactivity of OmpX-elicited IgG,. Analysis of OmpX sequences or orthologues showed close alignment of OmpX in members of the Enterobacteriaceae family (Fig. 3A) with dramatic divergence between P. aeruginosa and S. pneumoniae. This correlated with Th17 ELISpot responses in CD4+ T cells from OmpX+LTA1 vaccinated lungs (Fig. 3B), in which spot frequency was highest using whole bacterial antigens from Enterobacteriaceae. OmpX+LTA1 immunized mice were not protected against a challenge with serotype 3 S. pneumoniae (OmpX ortholog has < 32% amino acid indentity) but were protected against a challenge with KP-43816 (serotype K2) (Fig. 3C), which is consistent with the requirement for Ag-specific T cells. Moreover, we observed vaccine mediated protection following challenge with 2 x 106 Serratia marcescens (OmpX homolog has > 80% amino acid identity) (Fig. S4).
Fig. 3. Specificity and oligloclonality of lung CD4+ T-cells in LTA1/OmpX and OmpC vaccinated mice.
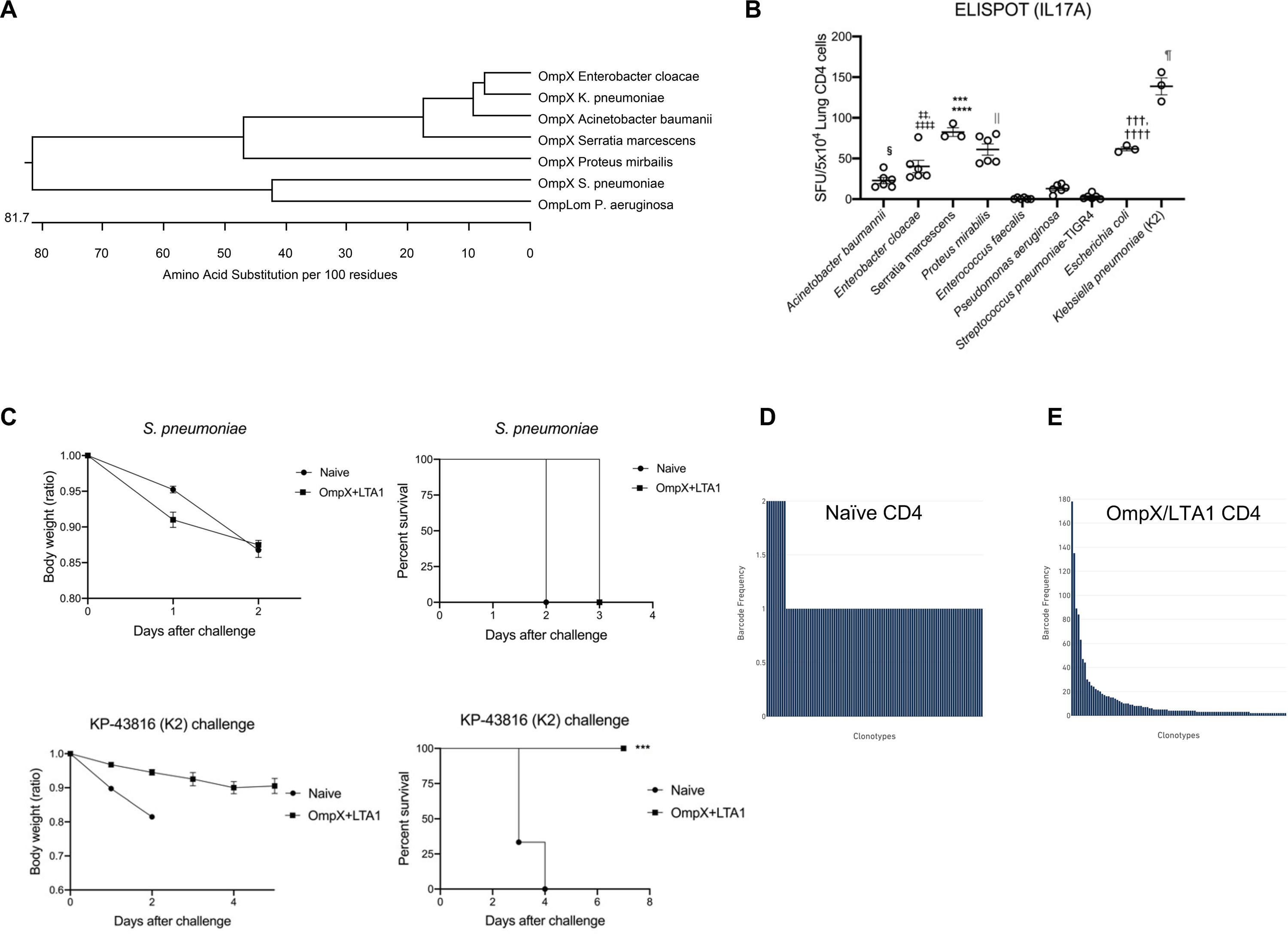
(A) Dendrogram analysis of OmpX protein sequences across members of the Enterobacteriaceae family, as well as the OmpLom orthologues in P. aeruginosa and S. pneumoniae. (B) Lung CD4+ T cells from OmpX+LTA1 immunized mice were isolated with MACS kit and stimulated with extracts of sonicated whole bacteria to enumerate IL-17A-producing cells by ELISpot. Significant differences were calculated by ANOVA followed by Tukey’s multiple comparisons test (n = 3–6, representative from 2 independent experiments). ****, P < 0.0001 (vs Acinetobacter, Enterococcus, Pseudomonas and Pneumococcus), ***, P < 0.001 (vs Enterobacter), ‡‡‡‡ P < 0.0001 (vs Enterococcus, Pseudomonas and Pneumococcus), ‡‡P < 0.01 (vs Acinetobacter), ††††P < 0.0001 (vs Enterococcus, Pseudomonas and Pneumococcus), †††P < 0.01 (vs Acinetobacter), §P < 0.05 (vs Enterococcus, Pneumococcus), || P < 0.0001 (vs Acinetobacter, Enterococcus, Pseudomonas and Pneumococcus), and ¶P < 0.0001 (vs all other bacteria). (C) Weight loss (as a ratio of starting weight) and survival in the naïve and OmpX+LTA1 vaccinated mice after challenge with 106 CFU of type 3 Streptococcus pneumoniae or KP-43816 (K2 strain) (n = 6, cumulative data of two separate experiments are shown). T cell clonotypes and single cell TCR seq for (D) naïve CD4+ T cells from spleen or (E) OmpX+LTA1 immunized lung CD4+ T cells were performed. Significant differences were estimated by one-way ANOVA followed by Tukey’s multiple comparisons test ***, P < 0.001.
Consistent with prior studies (25, 26), in vitro-differentiated Th17 cells showed plasticity towards Th1 cells after incubation with IL-12p70, but in vivo immunized Th17 cells did not differentiate in this manner (Fig. S5A). Consistent with this observation, STAT4 phosphorylation was detected in in vitro differentiated Th17 cells after incubation with IL-12p70 but not seen in Th17 cells from immunized mice (Fig. S5B). To determine oligoclonality, we investigated the TCRβ repertoire of these cells by flow cytometry. Compared with naïve lung CD4+ T cells, lung CD4+ T cells from subunit immunized mice showed a substantial increase in the TCRVβ14+ cell population (Fig. S5C, S5D). This was also supported by single cell TCR analysis that showed a dramatic left-shift of TCR clonotypes in OmpX immunized mice compared with control naïve spleen (Fig. 3D and E).
Adjuvants play a critical role in vaccines by enhancing development of protective immune responses in both healthy and immunocompromised hosts. To this end, we investigated if OmpX immunization with TLR-based adjuvants could elicit lung TRM cells similar to LTA1. We compared the TLR2/6/9-agonist Pam2-ODN (27), and the TLR1/2-agonist diprovocim (28) against LTA1. The LTA1-based adjuvant induced more lung TRM compared with TLR-based adjuvants (Fig. S5E, S5F) and was more protective against KP-396 bacterial challenge (Fig. S5G).
Cooperative roles of B cells and neutrophils in vaccine efficacy
We immunized μMT mice, which lack mature B cells, to elucidate the role of B cells in protective responses induced by subunit vaccination.. OmpX+LTA1 immunization elicited lung CD4+ T cells (Fig. S6A, S6C) in spite of the absence of B cells (Fig. S6B, S6C), and this was associated with bacterial control in immunized μMT mice (Fig. S6D). Lung CD4+ T cells in immunized μMT mice showed comparable levels of IL-17A secretion relative to cells from immunized WT mice upon antigen stimulation (Fig. S6E).
We observed less protection against bacterial dissemination and increased splenic CFU in vaccinated μMT mice (Fig. S6D), which suggested a critical role for B cells and antibodies to prevent bacterial dissemination from the lungs. To determine the protective role of neutrophils and B cells in OmpX elicited immunization, we immunized WT C57BL/6 and μMT mice and depleted neutrophils 24 h before challene using the anti-Ly6G mAb. Neutrophil depletion exacerbated lung CFU in immunized μMT but not in wild type mice, suggesting a cooperative role of neutrophils and B cells in lung and systemic immunity (Fig. 4A), and depletion of recruited Gr-1+CD11b+ neutrophils were confirmed by flow cytometry (Fig. 4B). We analyzed the kinetics of neutrophil recruitment into the BAL fluid of naïve and subunit-immunized mice upon K. pneumoniae infection since neutrophils have been linked to Th17 mediated lung protection against bacterial infection (29). Similar to a prior report with heat-killed K. pneumoniae exposed mice (5), we observed aaccelerated neutrophil recruitment into the lung in subunit immunized mice that contracted by 24 h post-infection (Fig. 4C), suggesting the accelerated neutrophil accumulation into lungs might be a key mechanism to protection against infection. Representative gating strategies are shown in Figure S7A.
Fig. 4. Cooperation between neutrophils and B cells is required for vaccine efficacy.
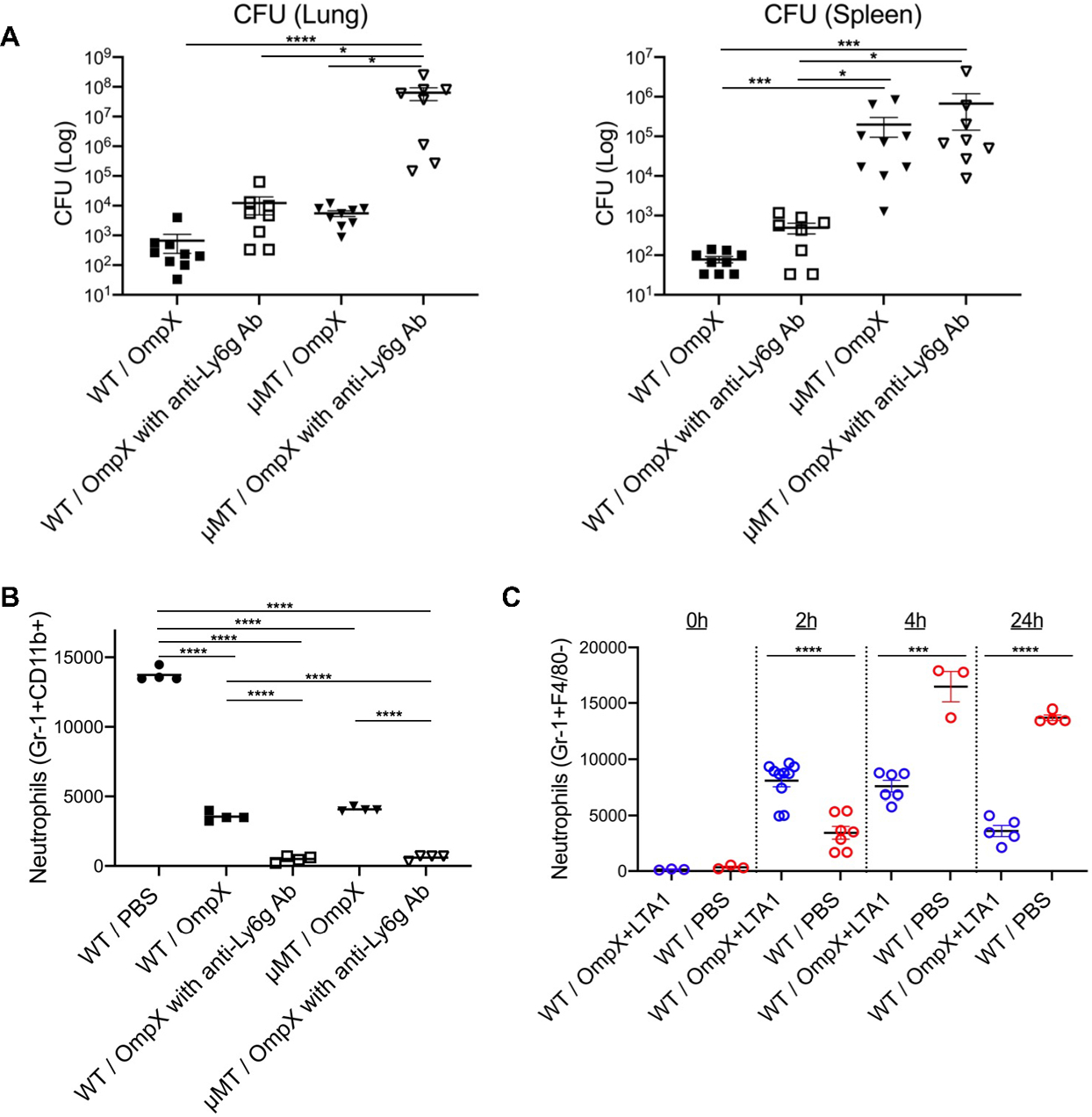
(A) CFU in the lungs and spleens of naïve C57BL/6 and vaccinated C57BL/6 and μMT mice, with or without anti-Ly6g Ab treatment, were estimated at 24 h post infection (n = 8–9). (B) Gr-1+CD11b+ cells in BALF of naïve C57BL/6 mice and vaccinated C57BL/6 and μMT mice, with or without anti-Ly6g Ab treatment, were enumerated at 24 h post challenge. (C) The kinetics of neutrophil recruitment into BALF of naïve and OmpX+LTA1 vaccinated C57BL/6 mice post-bacterial challenge are shown (n = 3–8, cumulative data of two separate experiments are shown). Significant differences were calculated with Kruskal-Wallis followed by Dunn’s multiple comparisons test (A, B) or unpaired t-test at 2, 4, and 24 h post-challenge (C). *, P < 0.05, ***, P < 0.001, ****, P < 0.0001.
Homing of TRM cells
We performed adoptive transfer of lung CD4+ T cells subunit-immunized into naïve mice prior to KP-396 challenge as further validation of the role of these cells in protection. Lung or splenic CD4+ T cells were purified from immunized CD45.1+ mice, and transferred intravenously to CD45.2+ C57BL/6 mice (30). 24 h after transfer, flow cytometry gating on CD45.1 showed that splenic CD4+ T cells from immunized mice accumulated in the spleen (yellow in Fig. 5A), and transferred immunized lung CD4+ T cells efficiently accumulated and proliferated in the recipient lung (red in Fig. 5A, 5B). Representative gating strategies are shown in Figure S7B. IL-17A ELISpot assays showed that the transferred lung CD4+ T cells that accumulated in the lung were OmpX-specific (red in Fig. 5C) and conferred protection against KP-396 challenge in the lung and spleen (Fig. 5D).
Fig. 5. Adoptively transferred lung CD4+ T cells but not splenic CD4+ T cells mediate vaccine protection.
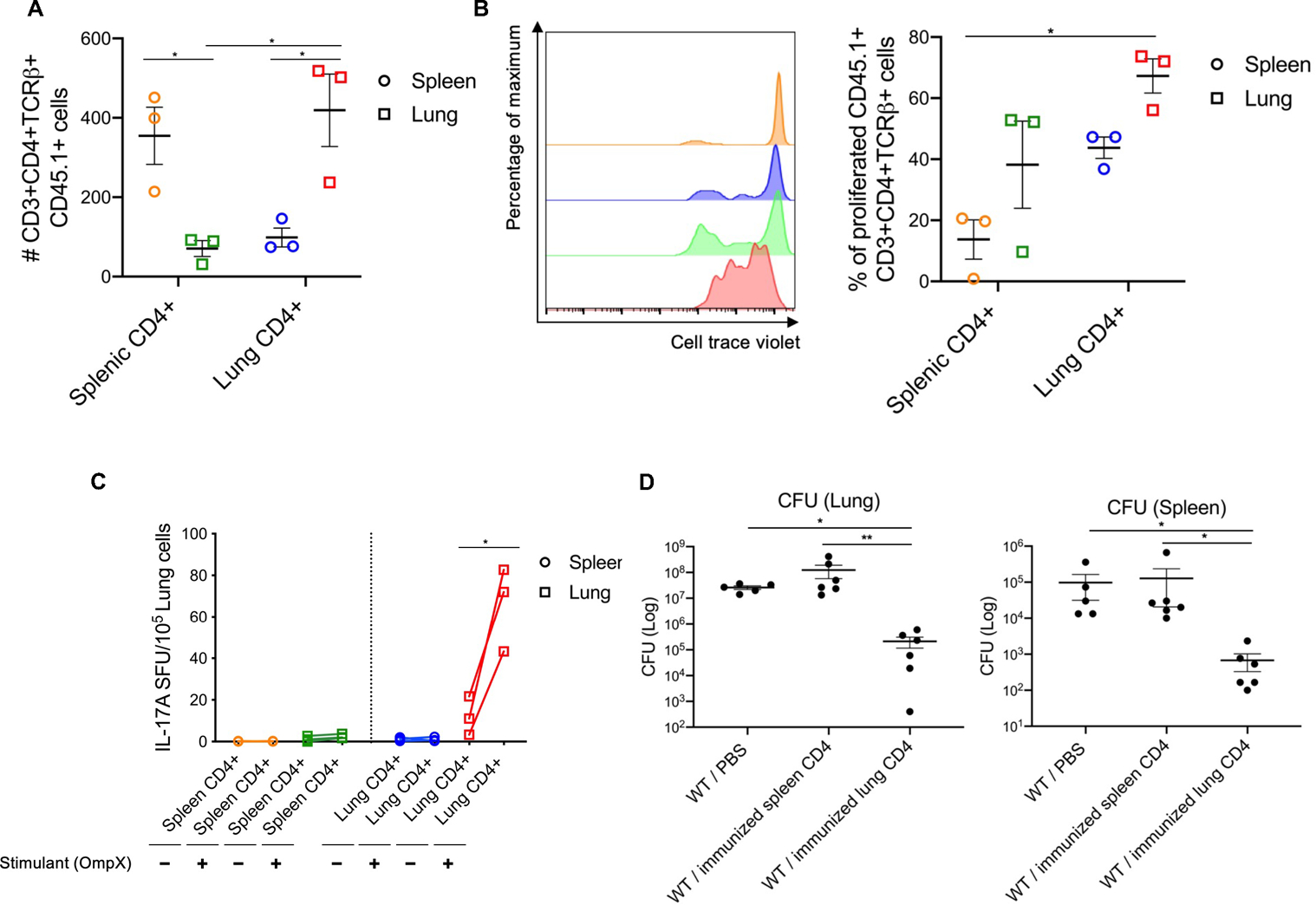
Lung and splenic CD4+ T cells from OmpX+LTA1 vaccinated CD45.1+ congenic mice were purified on day 29 post-vaccination as in Fig.1A. 5 x 105 CD4+ T cells were intravenously transferred into CD45.2+ C57BL/6 mice. Lymphocytes from lungs and spleens of recipient mice were enumerated by flow cytometry, and representative dot plots of cells 1 day post transfer are shown. (A) Absolute number and (B) proliferation of splenic or lung CD45.1+CD3+CD4+TCRβ+ cells of in CD45.2+ C57BL/6 mice post transfer. Yellow: spleen samples from CD45.2+ mice receiving splenic CD4+ T cells in vaccinated CD45.1+ congenic mice. Blue: spleen samples from CD45.2+ mice receiving lung CD4+ T cells in vaccinated CD45.1+ congenic mice. Green: lung samples from CD45.2+ mice receiving splenic CD4+ T cells in vaccinated CD45.1+ congenic mice. Red: lung samples from CD45.2+ mice receiving lung CD4+ T cells in vaccinated CD45.1+ congenic mice. Significant differences were calculated with one-way ANOVA followed by Tukey’s multiple comparisons test. *, P < 0.05. (C) IL-17A ELISpot frequency to detect OmpX-specific CD4+ T cells from the transferred CD45.2+ mice. Significant differences calculated with paired t-test. *, P < 0.05 (A, B, C; n = 3, representative from 2 independent experiments). (D) CFU in lung and spleen were estimated at 24 h post challenge with KP-396, 2 days after CD4+ T cell transfer (n = 6, representative from 2 independent experiments) . Data are presented as mean ± SEM. Significant differences were calculated with Kruskal-Wallis followed by Dunn’s multiple comparisons test. *, P < 0.05, **, P < 0.01.
Serotype independent vaccine efficacy requires IL-17R signaling in Dermo1+ fibroblasts and lung Th17 cells are resistant to the immunosuppressant FK506
To confirm the importance of IL-17R signaling in serotype independent immunity, we challenged subunit vaccine immunized Il17rafl/fl E2a-Cre+ mice. Compared with littermate controls, subunit immunized Il17rafl/fl E2a-Cre+ mice showed loss of bacterial control after KP-396 bacterial challenge (Fig. S8A), whereas bacterial control was maintained in Il22ra1-E2a-Cre+ mice (Fig. S8B). The loss of bacterial control was associated with a modest reduction in lung CD4+ T cells (Fig. S8C, S8E) and B cells (Fig. S8D, S8E). Subunit vaccine efficacy was completely abrogated in Il17rafl/fl E2a-Cre+ mice (Fig. S8A), in spite of a modest reduction in cellular responses, Deletion of Il22ra1 globally in Il22ra1-E2a-Cre+ mice did not affect vaccine-elicited changes in lung CD4+ T cells or B cells (Fig. S8F).
To determine if IL-17R signaling was required in specific cells mediating crosstalk with vaccine-elicited TRM cells, we assessed subunit vaccine immunity in which IL-17R was deleted in club cells using CcspCre (31) or in fibroblasts using Dermo1-cre (32). Vaccine-mediated protection was mildly compromised in CcspCre+ mice but was dramatically compromised in Il17rafl/fl DermoCre+ mice (Fig. 6A), suggesting IL-17R signaling in lung Dermo1+ fibroblasts is critical for vaccine efficacy. Deletion of IL-17RA in club cells did not affect vaccine-induced accumulation of lung CD4+ T cells (Fig. 6B) or Bcells (Fig. 6C), deletion of Il17ra in fibroblasts resulted in a modest reduction in lung CD4+ T cells (Fig. 6B) and Bcells (Fig. 6C). To examine the TRM niche, we performed H&E and RNAscope analysis for Il17a and observed similar positioning of Il17a+ cells in the Il17rafl/flDermoCre+ or Cre− mice, but there was a modest reduction in cell number (Fig. 6D, 6E) that was confirmed by flow cytometry analysis of whole lung tissue (Fig. 6B, 6C). Moreover, less OmpX-specific IgA was detected in the BALF of DermoCre+ mice but not in CcspCre+ mice (Fig. 6F, 6G). We also observed decreased neutrophil recruitment into the lung in the Il17rafl/flDermoCre+ mice (Fig. 6H), suggesting the neutrophil accumulation in lungs might be perturbed in Il17rafl/flDermoCre+ mice. We compared whole lung tissue from Il17rafl/fl mice and Il17rafl/flDermoCre+ mice by RNA-seq and observed downregulation of multiple immune system-related genes in Dermo1 Cre+ mice (Fig. S9). These included genes involved in macrophage and dendritic cell maturation (Csf2, Csf3), neutrophil chemotaxis and activation-related genes (S100a8, S100a9, Cxcr2, Cxcl1, Cxcl5), monocyte chemotaxis (Ccl2, Ccl7), and B cell maturation (Tnfrsf17, Cd79a, Pou2af1). Importantly, Il1r1 was significantly downregulated (p = 0.026), which may explain attenuated LTA1-mediated Th17 response (33). We observed downregulation of Cxcl13, which may alter iBALT formation in light of evidence suggesting a role of CXCL13 in the formation of ectopic lymphoid-like structures (15, 34, 35). We independently verifed that lung fibroblasts were an important target of IL-17R signaling by mining scRNAseq lung data and observing that IL-17RC (the receptor for IL-17A and IL-17F signaling) was expressed in Dermo1+ fibroblasts that were enriched for expression of Col1a2. Thus, we crossed Il17rcfl/fl mice to inducible Col1a2-cre mice, and Col1a2-cre was induced by ingestion of tamoxifen-supplemented chow. Tamoxifen-fed Cre+ and Cre− mice were immunized twice and then challenged as outlined in Fig 1A. Col1a2cre+ mice phenocopied global Il17r−/− mice, which strongly implicated fibroblasts as key mediators of vaccine efficacy in Dermo1Cre+ mice (Fig. 6I).
Fig. 6. IL-17R signaling in fibroblasts is required for vaccine efficacy.
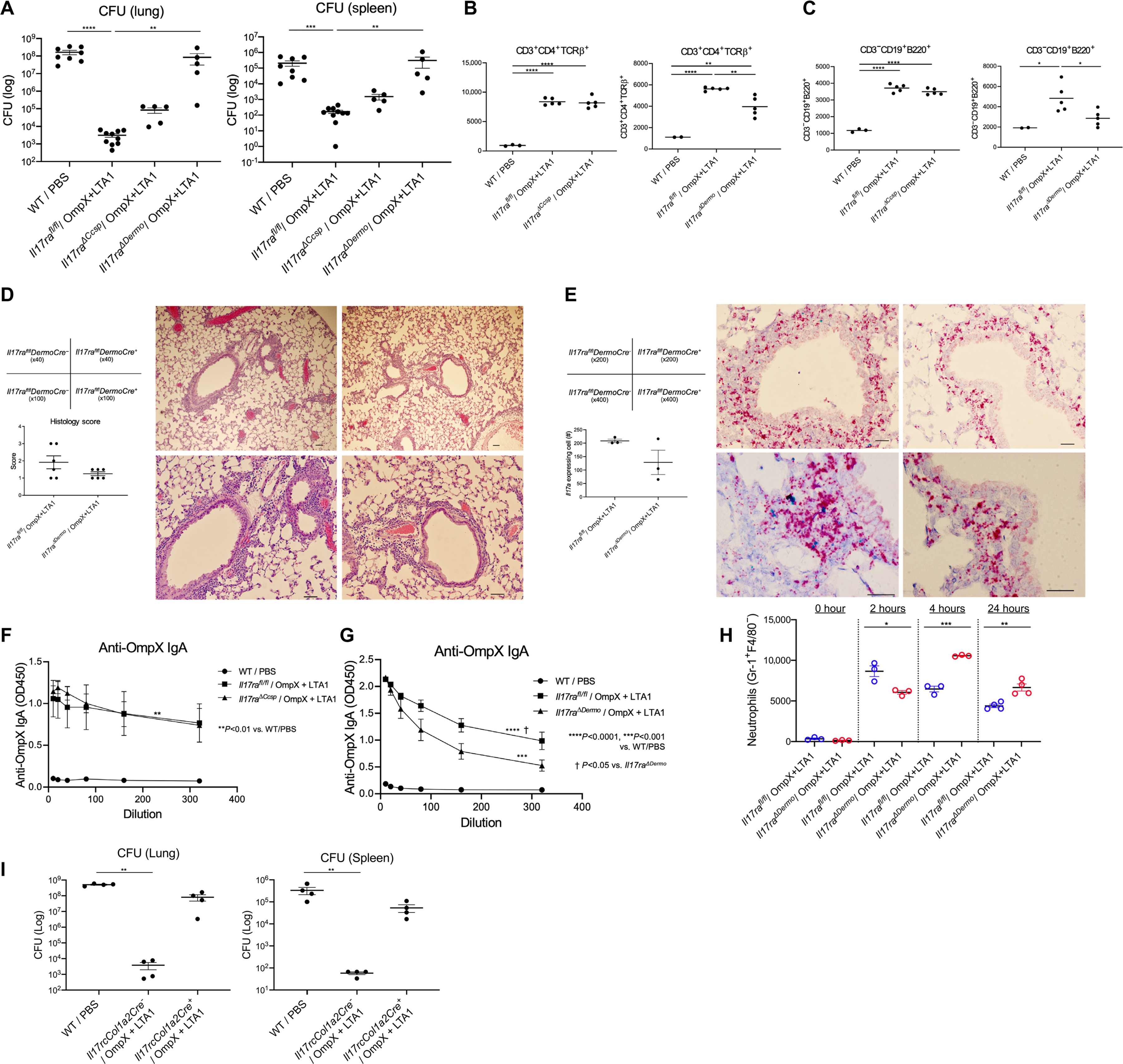
(A) Lung and spleen CFU in naïve C57BL/6, vaccinated Il17ra fl/fl mice, Il17rafl/fl CcspCre+ mice (Il17ra⊿ccsp mice), and Il17rafl/fl DermoCre+ mice (Il17ra⊿Dermo mice) were estimated at 24 h post infection (n = 5–9, representative from 2 independent experiments). (B) Lung CD3+CD4+ T cells and (C) CD19+ B cells in naïve C57BL/6, OmpX+LTA1 vaccinated Il17rafl/fl, Il17ra⊿ccsp and Il17ra⊿Dermo mice were enumerated at 29 days post first immunization, as is Fig.1A. (D) H&E stain and histology scoring of pathology (magnification x40, x100, add scale bars) in lungs of OmpX+LTA1 vaccinated Il17rafl/fl DermoCre− and Il17rafl/fl DermoCre+ mice. (E) RNAScope for Il17a in lungs of OmpX+LTA1 vaccinated Il17rafl/fl DermoCre− and Il17rafl/fl DermoCre+ mice (D and E, scale bar = 100 μm). Total number of Il17a-expressing cells was determined by morphometry (lower left). Data are presented as mean ± SEM (n = 3–6). (F) OmpX-specific IgA in BALF of naïve C57BL/6, vaccinated Il17ra fl/fl and vaccinated Il17ra⊿ccsp mice. (G) OmpX-specific IgA in BALF of naïve C57BL/6, vaccinated Il17ra fl/fl and vaccinated Il17ra⊿Dermo mice (n = 5, representative from 2 independent experiments). (H) Kinetics of neutrophil recruitment in Il17ra DermoCre− and Il17ra DermoCre+ mice (n = 3–4, cumulative data of two separate experiments is shown). (I) Lung and spleen CFU in naïve C57BL/6, vaccinated Il17rc fl/fl mice and Il17rcfl/fl Col1a2Cre+ mice at 24 h post infection (n = 4, the data is representative from two independent experiment). Data are presented as mean ± SEM. Significant differences were calculated with one-way ANOVA followed by Tukey’s multiple comparisons test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Immunosuppressive drugs are essential to prevent organ and tissue rejection in transplant recipients, but these individuals are at an increased risk of multidrug-resistant (MDR) infection, particularly in solid organ transplantation (36). We next investigated if subunit vaccine-elicited cells were sensitive to calcineurin inhibition-based immunosuppression based on recent evidence that that the calcineurin inhibitor FK506 inhibits γδ Il-17+ cells (20). To test the sensitivity of vaccine-elicited T cells, mice were immunized as outlined in Fig. 1A and then treated with FK506 or vehicle 24 h before KP-396 challenge. FK506 treatment did not alter the total number of lung CD4+ T cells in vaccinated mice (Fig. 7A). Lung Th1 cells were dramatically reduced (Fig. 7A, 7B), but vaccine-elicited Th17 cells only decreased by 35% (Fig. 7A, 7B). Preservation of FK506-resistant Th17 cells was associated with significant decrease of bacterial burden in the KP-396 challenge model (p < 0.05) (Fig. 7C), but IL-17A+ and IFNγ+ γδ cells were dramatically decreased after FK506 treatment, confirming that γδ cells are more sensitive to FK506 (Fig. 7D). Importantly, vaccine efficacy was maintained in FK506-treated mice, as measured indirectly by weight loss (Fig. 7E) and bacterial control (Fig. 7F) in a K.pneumoniae carbapenemase (KPC) - producing K. pneumoniae-challenge model using a mucoid I1 (ST258 strain) (20).
Fig. 7. Calcineurin inhibition does not affect OmpX vaccine efficacy and lung Th17 cells.
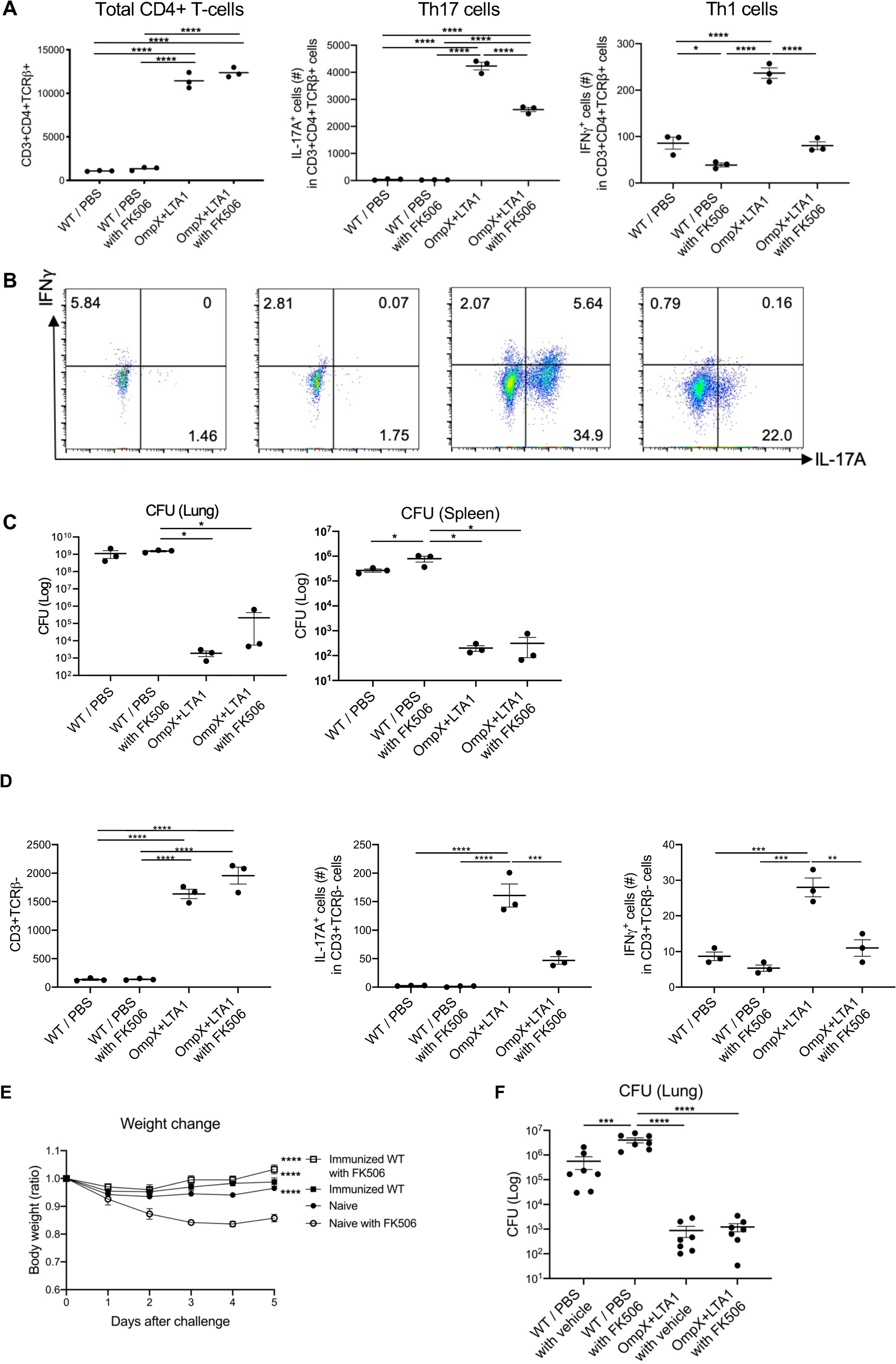
(A) Total lung CD3+CD4+ T cells, IL-17A+ cells (Th17 cells) and IFNγ+ cells (Th1 cells) in naïve C57BL/6 mice and OmpX+LTA1 vaccinated mice after treatment with FK506 or vehicle (intraperitoneal dosing from 24 h prior to challenge, followed two doses 24 h apart). (n = 3). (B) Representative dot plots of IL-17A+ and IFNγ+ lung CD3+CD4+TCRβ+ T cells in each group are shown. (C) Lung and spleen CFU in naïve and OmpX+LTA1 vaccinated C57BL/6 mice, with or without FK506 treatment, were estimated at 24 h post infection (n = 3–4). Data are representative from two independent experiments. (D) Total lung γδ cells, IFNγ+ and IL-17A+ cells in naïve and OmpX+LTA1 vaccinated after treatment with FK506 or vehicle (intraperitoneal dosing from 24 h prior to challenge, followed two doses 24 h apart). (n = 3). (E, F) Vaccine efficacy in a KPC-producing K.pneumoniae-challenge model using 107 CFU of a mucoid I1 (ST258 strain), in the presence or absence of 10 mg/kg FK506. (E) Weight change over time (D, n = 6 per group) and (F) lung CFU (E, n = 7 per group) are shown. Data are pooled from two independent experiments and presented as mean ± SEM. Significant differences were calculated with one-way ANOVA followed by Tukey’s multiple comparisons test. *, P < 0.05; ****, P < 0.0001
DISCUSSION
K. pneumoniae is a leading cause of hospital-acquired pneumonia with a high incidence of serious infections (37) and a high mortality rate in immunocompromised individuals, such as transplant recipients and patients with hematologic malignancies that acquire MDR strains, including K. pneumoniae (38). MDR organisms continue to pose a severe threat to public health (39) (40), and vaccines targeting these pathogens may represent a critical strategy to reduce the incidence of these infections. To date, few candidate vaccines have been evaluated in pre-clinical studies, including candidates comprised of bacterial outer membrane vesicles (41) and surface polysaccharides (42, 43), and there are currently no licensed K. pneumoniae-specific vaccines. Lung-specific CD4+ T cells play critical roles in pulmonary immunity (44), but there is no licensed vaccine that specifically target this tissue. TRM cells are an attractive cell type to target for MDR-specific vaccines due to their ability to generate a rapid immune response against invasive lung pathogens and their ability to be maintained in mucosal tissues for prolonged periods of time (45).
Preclinical models using whole cell immunization have demonstrated serotype-independent CD4+ T cell immunity to Gram-positive bacteria (10, 46), Gram-negative bacteria (5, 11), and fungi (47). The selection of specific antigens and adjuvant combinations is critical to advancing the development of these vaccines (48). LTA1 is from a bacterially-derived ADP-ribosylating enterotoxin but lacks the B-subunit of the native toxin, which enables safe mucosal delivery without the potential for neurologic toxicity observed in older nasal vaccines (13). Here we demonstrated that mucosal immunization with OmpX formulated with LTA1 could promote iBALT formation and conferred heterologous protection against autologous KP-43816 (K2 strain), a hypervirulent KP-396 (K1 strain), or a mucoid I1 (ST258 strain) of K. pneumoniae. The role of iBALT formation in TRM cell activation and vaccine immunity is unclear, but we did observe reduced vaccine efficacy in Cxcr5−/− mice, although Cxcr5 has other functions in germinal center reactions independent of iBALT formation.
Transcriptomic analysis of CD4+ T cells elicited by this subunit vaccine approach showed a clustering pattern similar to cells elicited by whole bacterial cell immunization ScRNA-seq data were consistent with multipotent lung CD4+ TRM cells with high expression of Cd69 and reduced expression of surface marker Klf2 and S1pr1 (3), and these cells were predominantly oligoclonal based on single-cell TCR analysis. OmpX-elicited cells recognized phylogenetically similar members of the Enterobacteriaceae consistent with the known conservation of OmpX within these species, and this vaccine protected against a challenge with phylogenetically related Serratia marcescens but not S. pneumoniae. These data suggest that this vaccine strategy may be able to provide protection beyond Klebsiella spp. to phylogenetically related Gram-negative bacteria, but future studies are needed to validate this concept in outbred MHC II mouse models.
LTA1 admixed with OmpX elicited robust Th17 responses as well as local humoral immunity with antigen specific IgA and IgG (11, 13). Compared with other adjuvants such as TLR2/6/9-agonist Pam2-ODN and TLR1/2-agonist diprovocim, LTA1 was associated with superior memory CD4+ T cell response in the lung. These findings suggest that the innate immune reponses initiated by LTA1 may be critical for protection and the generation of lung Th17 TRMs. This subunit vaccination elicited mucosal IgA in the lung but not in the gut, which suggested compartmentalization of this response. The lung Th17 cells elicited by subunit immunization did not show plasticity to Th1-like cells with IL-12 stimulation in vitro compared with in vitro differentiated Th17 cells, likely due to reduced STAT4 activation. Adoptive transfer of these cells could confer protection, which suggests that potential use of adoptive T cell therapy for MDR K. pneumoniae infection, which is a concept that we are currently investigating further.
Vaccine-elicited CD4+ lung TRM cells appear to occupy a submucosal niche that requires fibroblast IL-17RA/IL-17RC signaling for efficacy. These data contrast with data showing that a primary challenge with K. pneumoniae elicited a T cell IL-17 response that required signaling to club cells in the airway to establish a chemokine gradient for lumen neutrophil recruitment (29). RNAscope analysis showed that vaccine-elicited Il17a+ cells resided deeper in the peribronchial submucosa and in close proximity to the bronchial lymphatics, which was source of IL-7, essential for the maintenance of TRM CD4+ T cells (5). Fibroblast IL-17R signaling regulated the expression of several chemokines that are likely to contribute to vaccine efficacy, including CXCL1 and CXCL13. Additionally, IL-17R signaling in Twist2+ fibroblasts was also required for mucosal IgA production.
Of note, protection induced by subunit vaccine immunization was effective against the both hypervirulent K. pneumoniae and Carbapenem-resistant K. pneumoniae ST258 I1 strain infection in the presence of FK506 immunosuppression. This result implies the subunit vaccine immunization might be effective in patients, including transplant recipients, that being treated with calcineurin inhibitors like FK506, and this is a particularly meaningful finding given the recent study suggesting that these patients are at higher risk of MDR K. pneumoniae infection (36). Mechanisms of FK506 resistance are unclear, but preliminary transcriptomic analysis showed higher levels of the anti-apoptotic gene Bcl2 and the transcription factor Nfatc4 in TRM cells and a previous study showed that Th17 are less dependent on IL-2 and STAT5 (49), which may be related to the FK506 resistance observed in this study. The cellular and molecular mechanisms of FK506 resistance is a subject of ongoing investigation.
OmpX formulated with LTA1 enhanced cellular and humoral immunity and conferred heterologous protection against several strains of K. pneumoniae but not S. pneumoniae, and these findings suggest that similar immunization strategies may be useful for protection against other MDR Gram-negative bacteria. Our findings also elucidate cooperative roles between immune cell compartments and indicate that the TRM niche requires IL-17R signaling in Dermo+1/Col1a2-expressing lung fibroblasts. We observed a dramatic contraction of the Th17 response over time following vaccination. This response was associated with impaired vaccine efficacy and this contraction is similar to the CD8+ T cell responses following influenza infection in the lung (50, 51), but lung CD8+ T cell contraction was not observed after recombinant adenovirus vaccination for influenza (51). We hypothesize that other adjuvants or vaccine platforms may lead to more sustained CD4+ TRM responses, and additional studies on the molecular mechanisms involved in CD4+ T cell contraction may provide insights that guide the development of durable vaccine-induced T cell responses. Further development of vaccines targeting mucosal responses such as those described here will advance the field of therapeutic vaccination.
The study has a number of limitations. First, we cannot completely exclude a role for some aspect of trained immunity also playing a role in vaccine efficacy. Generating K. pneumoniae strains with deletions of OmpX may be useful in determining the role of antigen specific immunity, however, several papers have suggested that mutants of OmpX in E.coli have altered fitness and virulence (52, 53) and thus these studies may be difficult to interpret in vivo. Secondly, we used Tcrd−/− mice to determine if vaccine efficacy involves γδ T cells. However, it is well known that the constitutive Tcrd−/− mice can be functionally compensated by other lymphocytes (54). Future studies are needed that use conditional knock-in mice tohelp define the role of this cell population in vaccine efficacy.
MATERIALS AND METHODS
Study design
The aim of this study was to determine if vaccines could be formulated to generate CD4+ TRM cells that may be broadly effective against hypervirulent and antibiotic-resistant K. pneumoniae. To achieve this aim, we defined the best strategies to use heat-labile toxin E.coli (LT)-based adjuvants and conducted experiments to determine immunological mechanisms responsible for adjuvant-mediated vaccine efficacy, including the use of genetically engineered mice or pharmacological intervention. To understand mucosal immunity, we performed single-cell RNA sequencing, RT-PCR, flow cytometry, ELISA, ELISpot, and histology.
Mice
Male mice (6–10 weeks old) were used for in vivo studies. Wild-type C57BL/6J mice were bred in house or purchased from The Jackson Laboratory. Stat3fl/flCd4Cre mice, μMT mice, Il17ra−/− mice, Il22ra1−/− mice, Il21r−/− mice, Cxcr5−/− mice, Tcrd−/− mice, Ifnγ−/− mice, B6.SJL-Ptprca Pepcb/BoyJ mice (CD45.1+ C57BL/6), IL-17F-Thy1.1 reporter mice, which have been previously described (55), Il17rafl/flCcspCre mice, Il17rafl/flDermoCre mice, Il17rcfl/flCol1a2Cre mice, and Rag2/OT-II mice (Taconic) were housed and bred under specific pathogen-free conditions within Tulane University. All experiments were performed using sex- and age-matched controls and approved by the Institutional Animal Care and Use Committee of Tulane University.
Antigen and adjuvants preparation for immunization
Heat-killed K. pneumoniae-43816 (ATCC) (K2 strain) was prepared by incubating suspensions for 2 h at 70°C as previously described (5). Recombinant OmpX were generated from K. pneumoniae-43816 and purified by using E. coli expression systems. The adjuvant LTA1 was also purified from E. coli expression systems as previously described (13). All immunization was performed with isoflurane-anesthetized mice using the oropharyngeal aspiration-tongue pull technique (referred as intratracheal, i.t.) and boosted with the same vaccine 3 weeks later based on prior work with influenza (13).
Experimental K. pneumoniae and S. pneumoniae infection
K. pneumoniae-396 (K1 strain), 43816 (ATCC) (K2 strain), C4 (ST258), and I1 (ST258) were grown in 30 ml tryptic soy broth (Difco) for 18 h at 37°C. Cultures was then diluted at 1:100 and grown for an additional 2.5 h to reach the early logarithmic phase. The concentration of K. pneumoniae was determined by measuring the absorbance at 600 nm. Bacteria were pelleted by centrifugation and washed twice in cold PBS, and then resuspended to achieve the desired density. Mice were infected with 1 x 104 CFU (KP-396 and KP-43816) or 1 x 107 CFU (I1 strain) live K. pneumoniae via intratracheal and sacrificed at 24 h after infection. For pneumococcal pneumoniae, mice were challenged with 1 x 106 CFU of serotype 3 pneumococcus (Strain ATCC 6303). For all of the survival studies, we observed weight change and survival for 7 days. For analyzing mouse arterial oxygen saturation (SpO2), we used MouseOX Plus® (STARR Life Sciences Corp.). Whole lungs were harvested following perfusion of 10 ml of heparinized cold PBS into the right ventricle for removing RBCs and leukocytes from the lung vasculature. Bacterial burdens in left lung and spleen was estimated by plating organ homogenates in PBS and counting CFU. Simultaneously, the right middle lobe of the spleen was resected in Trizol for RNA extraction, and the other lobes were harvested in medium for single cell digestion. To induce expression of Cre recombinase in Il17rc fl/flCol1a2Cre mice, mice were initiated on a diet containing tamoxifen (500 mg/kg; Envigo) from 2 weeks before the initiation of immunization and throughout the experiment. For anti-CD4 antibody (GK1.5) depletion in vivo, we compared 300 μg GK1.5 administered intraperitoneally 2 days before challenge, pre-treatment (300 μg anti-GK1.5 Ab intraperitoneally 2 days before immunization and administered once a week throughout the experiment), and post-treatment (500 μg via intraperitoneal and 100 μg via intratracheal at Day 28 and Day 30 followed by bacterial challenge at Day31).
Bacterial antigen preparation including K. pneumoniae for ELISA and ELISpot
K. pneumoniae-43816 (ATCC) (K2 strain), K. pneumoniae-396 (K1 strain) was cultured in 30 ml tryptic soy broth (Difco) for 18 hours at 37°C and then harvested. The pellet was re-suspended in 5 ml of lysis buffer [50 mM Tris pH 8.0, 10% glycerol, 0.1% Triton X-100, 100 μg/ml lysozyme (Thermo Scientific) with 1 mM EDTA, 1 mM PMSF and proteases inhibitor cocktail (Thermo Scientific). After incubation on ice for 30 min, the bacterial lysate was sonicated for 20 s (5% Duty cycle and 50% Power output) on and off for 8–10 times until sample was no longer viscous, and finally centrifuged at 10,000 rpm for 30 min at 4°C. The protein concentration in supernatant was measured by BCA protein assay (Pierce). A similar antigen preparation was performed for the following bacteria: Acinetobacter baumannii-NR-17783 (BEI), Enterobacter cloacae-NR-50391 (BEI), Proteus mirabillis-HM-752 (BEI), Enterococcus faecalis-HM-201 (BEI), Pseudomonas aeruginosa Migula (ATCC® 29260™), Streptococcus pneumoniae (Klein) Chester (ATCC® BAA-334™), and Serratia marcescens subsp. marcescens Bizio (ATCC® 13880™).
ELISA for K. pneumoniae antibody
ELISA plates were coated with 2 μg/ml of K. pneumoniae antigen at 4°C, overnight (The antigen preparation is mentioned in the supplemental file). Coated plates were rinsed with washing buffer (0.05% Tween 20 in PBS), incubated for 2 hours with blocking buffer (1% BSA and 0.1% Tween 20 in PBS), and washed before the addition of serially diluted serum, bronchial alveolar lavage (BAL), or fecal supernatants. After 2 hours of incubation at room temperature and plate washing, bacterial specific antibodies were detected with 1/5000 goat anti-mouse IgG or IgA conjugated with horseradish peroxidase (Cat #1030-05, #1040-05, Southern Biotech), diluted in assay diluent (0.5% BSA and 0.05% Tween 20 in PBS), and incubated for 1 hour at room temperature. After washing, TMB peroxidase substrate (Cat #0410-01, Southern Biotech) was added to each well. Absorbance was read at 450 nm on a microplate reader (BioTek).
IL-17A ELISpot assays
ELISpot assays were conducted in Millipore pore plates to evaluate the frequency of background and antigen stimulated SFU following the recommendations of a previously described protocol (Millipore, USA). 105 lung cells per well were stimulated with 2 μg/ml of different kinds of stimulants including of OmpX in duplicate or triplicate. Plates were read on a CTL ImmunoSpot S4 Core Analyzer, and analyzed with ImmunoSpot Software (Cellular Technology Ltd., Shaker Heights, OH, USA).
Intracellular cytokine staining
Single cell suspension from mouse lungs were stimulated for 6 hours with 50 ng/ml of Phorbol 12-myristate 13-acetate (Sigma) and 750 ng/ml ionomycin (Sigma). After 1 hour of incubation, 1 mg/ml GolgiStop (BD Bioscience) was added to block cytokine secretion. Cells were then fixed and stained with antibodies specific for surface markers, followed by permeabilization with Cytofix/Cytoperm (BD Bioscience) and staining with antibodies against intracellular molecules. The details of antibodies are shown in the supplemental.
Single Cell 3’ RNA sequencing and V(D)J Target Enriched and 5’ Gene Expression sequencing
1x106 cells per condition were collected as whole lung single cell populations. The remaining cells were subjected enrichment by using a CD4 positive selection kit (Cat #130-104-454, Miltenyi Biotec). Those cells were treated with 100 μL of TrypLE for 1 minute to dissociate single cells from small aggregates or clusters. Cell numbers and viability were validated by Cellometer prior to preparation of single cell RNAseq library. 10x single cell 3’ RNAseq assay. 5000 live cells per sample were targeted by using 10x single cell RNAseq technology provided by 10x Genomics (10X Genomics Inc, CA). Viable single cell suspensions were partitioned into nanoliter-scale Gel Beads-In-EMulsion (GEMs). Full-length barcoded cDNAs were then generated and amplified by PCR to obtain sufficient mass for library construction. Following enzymatic fragmentation, end-repair, A-tailing, and adaptor ligation, single cell 3’ libraries comprising standard Illumina P5 and P7 paired-end constructs were generated. For 10x single cell V(D)J seq assay, 5000 live cells per sample were targeted to generate GEMs by combining barcoded Single Cell 5’ Gel Beads. Following GEM-RT, RT cleanup, full-length cDNA amplification and QC, T-cell receptor enriched libraries and 5’ gene expression libraries containing standard Illumina P5 and P7 paired-end constructs were generated. Library quality controls were performed by using Agilent High Sensitive DNA kit with Agilent 2100 Bioanalyzer and quantified by Qubit 2.0 fluorometer. Pooled libraries at a final concentration of 1.8 pM were sequenced with paired end single index configuration by Illumina NextSeq 550. Cell Ranger version 2.1.1 (10x Genomics) was used to process raw sequencing data and Loupe Cell Browser (10x Genomics) to obtain differentially expressed genes between specified cell clusters. In addition, Seurat suite version 2.2.1 (47, 48) was used for quality control and downstream analysis. Filtering was performed to remove multiplets and broken cells. Also, uninteresting sources of variation were regressed out. Variable genes were determined by iterative selection based on the dispersion vs. average expression of the gene. For clustering, principal-component analysis was performed for dimension reduction. Top 10 principal components (PCs) were selected by using a permutation-based test implemented in Seurat and passed to t-SNE for clustering visualization. GEO accession number is GSE178385.
Immunofluorescence staining and histologic analysis
Lung tissues were fixed in 10% neutral buffered formalin for 48–96 hours and embedded in paraffin. 4 μm lung sections were then stained with hematoxylin and eosin. Serial unstained sections were incubated at 60°C to melt the paraffin. Remaining paraffin was removed by immersion in xylenes. Lung sections were gradually hydrated by sequential immersion in graded alcohols (absolute ethanol, 95% Ethanol, 70% alcohol) and finally in water. Antigens were unmasked by boiling slides in a coplin jar filled with DAKO antigen retrieval solution (S1699, DAKO). Slides were cool down for 10 minutes and washed with deionized water. Non-specific antibody binding was blocked with 5% normal and 2 μg/μl of rat anti-mouse CD16/CD32 (Fc block, clone 2.4G2, BioXCell). To detect CD3+ T cells, IgG+ plasma cells and B220+ B cells, lung sections were incubated with primary antibodies against CD3-epsilon (Clone M-20, Santa Cruz Biotechnology), FITC-donkey anti-mouse IgG (715-096-150, Jackson ImmunoResearch Laboratories) and APC-rat anti-mouse CD45R/B220 (clone RA3-6B2, BD Biosciences). Proliferating B cells were detected with a combination of FITC peanut agglutinin (L7381-5MG, SIGMA) and antibodies specific for proliferating cell nuclear antigen (PCNA, clone C-20, Santa Cruz Biotechnology) and APC-rat anti-mouse CD45R/B220. CXCL13 producing cells and CD21-CD35/FDCM1+ follicular dendritic cells were visualized with goat anti-mouse CXCL13 (AF470, R&D Systems), biotin-rat anti-mouse CD21-CD35 (clone 7E9, Biolegend) and rat anti-mouse follicular dendritic cell (clone FDCM1, BD Biosciences). Finally, we incubated lung sections with rat ant-peripheral node addressin (PNAd, MECA-79, BD Biosciences) and rabbit anti-Lymphatic Vessel Endothelial Receptor 1 (Lyve-1, DP3513, Acris), in combination with APC-rat anti-mouse CD45R/B220 to detect PNAd+ high endothelial venules and LYVE-1+ lymphatics in iBALT areas. Primary antibodies were revealed with Alexa Fluor 568 donkey anti-goat IgG (A11057, Thermo Fisher Scientific), Alexa Fluor 488 donkey anti-rat IgG (A-21208, Thermo Fisher Scientific), Cy3-donkey anti-rat IgM (112-165-020, Jackson ImmunoResearch Laboratories), and Alexa Fluor 488 streptavidin (S11223, Thermo Fisher Scientific). Slides were washed in PBS and mounted with Vectashield with DAPI (H-1200-10, Vector Laboratories). Representative pictures were taken with a Zeiss Axioplan Microscope and recorded with a Hamamatsu camera.
Adoptive transfer of CD4+ T cells
WT C57BL/6 mice (CD45.1) were immunized as in Figure 1A. One week post second immunization, the lung and spleen were removed and digested into the single-cell suspensions, and CD4+ T cells were enriched by using a CD4 positive selection kit (Cat #130-104-454, Miltenyi Biotec). WT C57BL/6 mice (CD45.2) were transferred with the enriched 5 x 105 immunized spleen or lung CD4+ T cells via tail vein or retro-orbital vein one day post inoculation with 1μg OmpX and 10μg LTA1 via i.t. route. Three to four mice per condition of these mice were euthanized to evaluate the immune response at one day post-transfer, and the remaining mice were challenged with 104 CFU KP-396 (K1 strain) via i.t. route two days post-transfer and euthanized to measure bacterial load (CFU) in lungs and spleen 24 h post challenge.
RNAscope
Paraffin-embedded lung sections from Il17rafl/fl and Il17rafl/fl x DermoCre+ mice underwent in situ hybridization according to the manufacturer’s instructions (ACD). Briefly, after hydrogen peroxide treatment, we performed target retrieval, created a hydrophobic barrier and applied AP conjugated-Il17a probe (RNAscope® Probe Mm-Il17a-O1, ACD) for hybridization. After hybridization, tissues were stained with 50% Hematoxylin.
Pathology Scores
Slides stained with hematoxylin and eosin were coded and blindly scored from 0 (absent) to 4 (severe). 0: no obvious pathology, 1: Inflammation around the airways, 2: Moderate airway inflammation with diffuse/patchy mild parenchymal involvement, 3: Moderate to severe airway inflammation with moderate patchy parenchymal involvement, 4: Moderate to severe airway inflammation with severe parenchymal involvement. The total histology score was expressed as the sum of the scores for each parameter.
Statistical analysis
Statistical analysis was performed with Prism (Graphpad) software. P values < 0.05 was considered statistically significant. Comparisons between two normally distributed groups were performed by simple 2-tailed unpaired student’s t-test, and other analyses for unequal variance were determined by Mann-Whitney U test. For multiple groups comparisons, we used one-way ANOVA with Tukey’s post-hoc analysis for equally distributed groups and Kruskal-Wallis followed by Dunn’s multiple comparison test for unequally distributed groups. Values are represented as means ± SEM. P values are annotated as follows (*) ≤0.05, (**) ≤0.01, (***) ≤0.001, and (****) ≤0.0001.
Other methodology is in the supplement.
Supplementary Material
Acknowledgments:
We thank Scott Evans for providing a TLR2/6/9-agonist Pam2-ODN, and thank Dale Boger, Scripps Research Institute for providing a TLR1/2-agonist diprovocim.
Funding:
This work was supported by the Louisiana Board of Regents Endowed Chairs for Eminent Scholars program, as well as by PHS grant R35HL139930 and NIAID/NIH awards R01AI114697.
Footnotes
Declaration of interests: Tulane University has filed a PCT application to the US Patent and Trademark Office on the vaccine described here.
Data and materials availability: The sequencing data for this study have been deposited in the database Gene Expression Omnibus and can be accessed using accession numbers GSE178385 (scRNA-seq) and GSE174849 (bulk RNA-seq). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Il17ra and Il17rc floxed mice are available from the Jackson Laboratory or they, and related materials can be made available from the corresponding author upon completion of a standard material transfer agreement (MTA) in accordance with Tulane technology transfer policy.
References
- 1.Hirschtick RE et al. , Bacterial pneumonia in persons infected with the human immunodeficiency virus. Pulmonary Complications of HIV Infection Study Group. N Engl J Med 333, 845–851 (1995). [DOI] [PubMed] [Google Scholar]
- 2.Ye P et al. , Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J.Exp.Med 194, 519–527 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar BV et al. , Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep 20, 2921–2934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farber DL, Form and function for T cells in health and disease. Nature reviews. Immunology 20, 83–84 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Amezcua Vesely MC et al. , Effector TH17 Cells Give Rise to Long-Lived TRM Cells that Are Essential for an Immediate Response against Bacterial Infection. Cell 178, 1176–1188 e1115 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shenoy AT et al. , Lung CD4(+) resident memory T cells remodel epithelial responses to accelerate neutrophil recruitment during pneumonia. Mucosal Immunol, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isturiz RE et al. , Pneumococcal conjugate vaccine use for the prevention of pneumococcal disease in adults <50 years of age. Expert Rev Vaccines 17, 45–55 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Avci FY, Li X, Tsuji M, Kasper DL, A mechanism for glycoconjugate vaccine activation of the adaptive immune system and its implications for vaccine design. Nature medicine 17, 1602–1609 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lo SW et al. , Pneumococcal lineages associated with serotype replacement and antibiotic resistance in childhood invasive pneumococcal disease in the post-PCV13 era: an international whole-genome sequencing study. Lancet Infect Dis 19, 759–769 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith NM et al. , Regionally compartmentalized resident memory T cells mediate naturally acquired protection against pneumococcal pneumonia. Mucosal immunology 11, 220–235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen K et al. , Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity. 35, 997–1009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malley R, Anderson PW, Serotype-independent pneumococcal experimental vaccines that induce cellular as well as humoral immunity. Proc.Natl.Acad.Sci.U.S.A 109, 3623–3627 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valli E et al. , LTA1 is a safe, intranasal enterotoxin-based adjuvant that improves vaccine protection against influenza in young, old and B-cell-depleted (muMT) mice. Sci Rep 9, 15128 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maciel M Jr. et al. , Intradermal or Sublingual Delivery and Heat-Labile Enterotoxin Proteins Shape Immunologic Responses to a CFA/I Fimbria-Derived Subunit Antigen Vaccine against Enterotoxigenic Escherichia coli. Infect Immun 87, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rangel-Moreno J et al. , The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat.Immunol 12, 639–646 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson KG et al. , Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc 9, 209–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mackay LK et al. , The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol 14, 1294–1301 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Skon CN et al. , Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol 14, 1285–1293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruane D et al. , Lung dendritic cells induce migration of protective T cells to the gastrointestinal tract. J Exp Med 210, 1871–1888 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwanaga N et al. , Host immunology and rational immunotherapy for carbapenem-resistant Klebsiella pneumoniae infection. JCI Insight, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shenoy AT et al. , Lung CD4(+) resident memory T cells remodel epithelial responses to accelerate neutrophil recruitment during pneumonia. Mucosal immunology 13, 334–343 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egwuagu CE, STAT3 in CD4+ T helper cell differentiation and inflammatory diseases. Cytokine 47, 149–156 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutloff A, T Follicular Helper-Like Cells in Inflamed Non-Lymphoid Tissues. Front Immunol 9, 1707 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rangel-Moreno J et al. , The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol 12, 639–646 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basu R, Hatton RD, Weaver CT, The Th17 family: flexibility follows function. Immunol.Rev 252, 89–103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee YK et al. , Late developmental plasticity in the T helper 17 lineage. Immunity 30, 92–107 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alfaro VY et al. , Safety, tolerability, and biomarkers of the treatment of mice with aerosolized Toll-like receptor ligands. Front Pharmacol 5, 8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su L et al. , Structural Basis of TLR2/TLR1 Activation by the Synthetic Agonist Diprovocim. J Med Chem 62, 2938–2949 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen K et al. , IL-17 Receptor Signaling in the Lung Epithelium Is Required for Mucosal Chemokine Gradients and Pulmonary Host Defense against K. pneumoniae. Cell Host Microbe 20, 596–605 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKinley L et al. , TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol 181, 4089–4097 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon DM et al. , Epithelial cell PPAR[gamma] contributes to normal lung maturation. FASEB J 20, 1507–1509 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Swonger JM, Liu JS, Ivey MJ, Tallquist MD, Genetic tools for identifying and manipulating fibroblasts in the mouse. Differentiation 92, 66–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brereton CF et al. , Escherichia coli heat-labile enterotoxin promotes protective Th17 responses against infection by driving innate IL-1 and IL-23 production. J Immunol 186, 5896–5906 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Eddens T et al. , Pneumocystis-Driven Inducible Bronchus-Associated Lymphoid Tissue Formation Requires Th2 and Th17 Immunity. Cell Rep 18, 3078–3090 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshitomi H et al. , Human Sox4 facilitates the development of CXCL13-producing helper T cells in inflammatory environments. Nat Commun 9, 3762 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satlin MJ, Jenkins SG, Walsh TJ, The global challenge of carbapenem-resistant Enterobacteriaceae in transplant recipients and patients with hematologic malignancies. Clin Infect Dis 58, 1274–1283 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin RM, Bachman MA, Colonization, Infection, and the Accessory Genome of Klebsiella pneumoniae. Front Cell Infect Microbiol 8, 4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Satlin MJ, Walsh TJ, Multidrug-resistant Enterobacteriaceae, Pseudomonas aeruginosa, and vancomycin-resistant enterococci: Three major threats to hematopoietic stem cell transplant recipients. Transpl Infect Dis, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Exner M et al. , Antibiotic resistance: What is so special about multidrug-resistant Gram-negative bacteria? GMS Hyg Infect Control 12, Doc05 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Munoz-Price LS et al. , Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis 13, 785–796 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiu CW et al. , Clinical impact of Gram-negative nonfermenters on adults with community-onset bacteremia in the emergency department. J Microbiol Immunol Infect 48, 92–100 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Adamo R, Margarit I, Fighting Antibiotic-Resistant Klebsiella pneumoniae with “Sweet” Immune Targets. mBio 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi M, Tennant SM, Simon R, Cross AS, Progress towards the development of Klebsiella vaccines. Expert Rev Vaccines 18, 681–691 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen K, Kolls JK, T cell-mediated host immune defenses in the lung. Annu.Rev.Immunol 31, 605–633 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takamura S, Niches for the Long-Term Maintenance of Tissue-Resident Memory T Cells. Front Immunol 9, 1214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Hara JM et al. , Generation of protective pneumococcal-specific nasal resident memory CD4(+) T cells via parenteral immunization. Mucosal Immunol 13, 172–182 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wuthrich M et al. , Vaccine-induced protection against 3 systemic mycoses endemic to North America requires Th17 cells in mice. J Clin.Invest 121, 554–568 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clements JD, Norton EB, The Mucosal Vaccine Adjuvant LT(R192G/L211A) or dmLT. mSphere 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laurence A et al. , Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 26, 371–381 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Uddback I, Kohlmeier JE, Thomsen AR, Christensen JP, Harnessing Cross-Reactive CD8(+) TRM Cells for Long-Standing Protection Against Influenza A Virus. Viral Immunol 33, 201–207 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uddback I et al. , Long-term maintenance of lung resident memory T cells is mediated by persistent antigen. Mucosal Immunol 14, 92–99 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirakawa H, Suzue K, Takita A, Kamitani W, Tomita H, Roles of OmpX, an Outer Membrane Protein, on Virulence and Flagellar Expression in Uropathogenic Escherichia coli. Infect Immun 89, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otto K, Hermansson M, Inactivation of ompX causes increased interactions of type 1 fimbriated Escherichia coli with abiotic surfaces. J Bacteriol 186, 226–234 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sandrock I et al. , Genetic models reveal origin, persistence and non-redundant functions of IL-17-producing gammadelta T cells. J Exp Med 215, 3006–3018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Conti HR et al. , Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206, 299–311 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.