

Abstract
Background & Aims:
Gastric chief cells, a mature cell type that secretes digestive enzymes, have been proposed to be the origin of metaplasia and cancer, through dedifferentiation or trans-differentiation. However, studies supporting this claim have had technical limitations, including issues with the specificity of chief cell markers and the toxicity of drugs used. We therefore sought to identify genes expressed specifically in chief cells and establish a model to trace these cells.
Methods:
We performed transcriptome analysis of Mist1-CreERT-traced cells, with or without chief cell depletion. Gpr30-rtTA mice were generated, crossed to TetO-Cre mice, and lineage tracing was performed after crosses to R26-TdTomato mice. Additional lineage tracing experiments were performed using Mist1-CreERT, Kitl-CreERT, Tff1-Cre, and Tff2-Cre mice crossed to reporter mice. Mice were given high-dose tamoxifen or DMP-77, or infected with Helicobacter pylori, to induce gastric metaplasia. We studied mice that expressed mutant forms of Ras in gastric cells, using TetO-KrasG12D, LSL-KrasG12D, and LSL-HrasG12V mice. We analyzed stomach tissues from GPR30-knockout mice. Mice were given dichloroacetate to inhibit pyruvate dehydrogenase kinase (PDK)-dependent cell competition.
Results:
We identified GPR30, the G-protein coupled form of the estrogen receptor, as a cell-specific marker of chief cells in gastric epithelium of mice. Gpr30-rtTA mice crossed to TetO-Cre; R26-TdTomato mice had specific expression of GPR30 in chief cells, with no expression noted in isthmus stem cells or lineage tracing of glands. Expression of mutant Kras in GPR30+ chief cells did not lead to the development of metaplasia or dysplasia, but instead led to a reduction in labeled numbers of chief cells and a compensatory expansion of neck lineage, which was derived from upper Kitl+ clones. Administration of high-dose tamoxifen, DMP-777, or H pylori decreased the number of labeled chief cells. Chief cells were eliminated from epithelia via GPR30- and PDK-dependent cell competition after metaplastic stimuli, whereas loss of GRP30 or inhibition of PDK activity preserved chief cell numbers and attenuated neck lineage cell expansion.
Conclusions:
In tracing studies of mice, we found that most chief cells are lost during metaplasia and therefore are unlikely to contribute to gastric carcinogenesis. Expansion of cells that co-express neck and chief lineage markers, known as spasmolytic polypeptide-expressing metaplasia, does not occur via dedifferentiation from chief cells, but rather through a compensatory response from neck cells to replace the eliminated chief cells.
Keywords: SPEM, gastric carcinogenesis, stomach, cell of origin
Introduction
Metaplasia in the stomach has long been considered as both a precursor of gastric cancer and a predictor of cancer risk. The most common form of metaplasia in the human stomach is intestinal metaplasia (IM), in which the glands lose stomach-specific cell types and differentiate into intestinal-like lineages. By contrast, mice with chronic gastric inflammation rarely develop IM, but instead develop spasmolytic polypeptide-expressing metaplasia (SPEM), which robustly expresses neck cell markers such as TFF2/GSII. Both IM and SPEM are associated with gastric atrophy, characterized by the loss of differentiated lineages including parietal cells and chief cells. Nevertheless, the precise relevance of SPEM to human gastric diseases remains unclear.1, 2
SPEM was first identified in mice chronically infected with Helicobacter species, with later identification in other chronic gastritis models3–6. In these models, TFF2-expressing SPEM was characterized by abundant mucins in cells that were found just below the isthmus region, often expanding to the gland base. A SPEM-like lesion also developed in mice immediately after acute epithelial injury, such as in response to drugs including DMP-777, L-635, and high-dose tamoxifen (HDT)7, 8. In these acute injury models, SPEM arose near the base of glands, without mucinous changes, and was characterized by expression of neck markers such as GSII along with chief cell markers such as gastric intrinsic factor (GIF)9. While such double positive (GSII+GIF+) cells were known to be present in normal stomach as progenitors of chief cells10, 11, their expansion after injury was believed to represent an acute form of SPEM. In addition, induction of Kras mutations in the corpus glands led to expansion of mucous-producing cells within the upper four fifths of the glands, with occasional expression of MUC412–15. In addition, a small population of cells that co-expressed chief and neck cell markers was found near the base of the mucinous glands. Together, these observations suggested the presence of three distinct histological forms of metaplasia in mouse models: acute SPEM at the gland base (aSPEM), chronic SPEM in inflammatory models (cSPEM), and SPEM induced by aberrant Ras signaling (rSPEM), with all forms demonstrating nearly complete loss of mature parietal and chief cells, which defines gastric atrophy (Fig.S1).
Considerable debate exists regarding the origin of SPEM16–19. In models of chronic inflammation, localization of SPEM near the isthmus combined with its persistent nature has long suggested a likely stem cell origin. However, studies of acute injury and Kras models have raised the possibility that SPEM is instead derived from gastric chief cells, due to its basal location and co-expression of chief cell markers7, 12, 20. Several groups have investigated this concept using CreERT-based lineage tracing experiments, and have reported that SPEM can be tracked using Mist1-CreERT and eR1-CreERT constructs that can genetically mark the chief cell population7, 12, 21. Other groups have suggested that chief cells can dedifferentiate into stem-like cells and act as a source of Kras-induced metaplasia, particularly following epithelial injury, based on experiments using Troy-CreERT and Lgr5-2A-CreERT mice23, 24.
Nevertheless, there are several limitations to the conclusions reached in these studies. First, the CreERT constructs that have been used are not specific for chief cells. While Mist1-CreERT and Troy-CreERT transgenes are robustly expressed in chief cells, isthmus stem cells also express these genes.13 eR1-CreERT predominantly marks broad isthmus progenitors, while only a subset of chief cells are also labeled21. Of the constructs that have been used, Lgr5–2A-CreERT appeared to be most specific for chief cells22. However, given the recent data from our group that Lgr5 mRNA is also expressed in the isthmus after epithelial injury, leading to gene recombination in the isthmus of Lgr5-EGFP-IRES-CreERT mice10, whether only chief cells are labeled by Lgr5-targeting mice during epithelial injury remains to be more carefully examined. Second, tamoxifen, a reagent that is used for inducible gene recombination, produces significant epithelial injury and SPEM induction in the gastric corpus, likely modifying gene expression and cell behavior.8
To address these concerns, we have now developed a new cell fate mapping system that specifically targets chief cells in a tamoxifen-independent manner. Data using this system suggest that GSII+GIF+ SPEM may not be a sign of chief cell dedifferentiation, but instead represent a regenerative expansion of neck cells in response to chief cell depletion.
Methods
Mice
Mist1-CreERT13, Lgr5-DTR-GFP23, Lox-STOP-Lox(LSL)-KrasG12D13, LSL-HrasG12V-IRES-GFP24, and LSL-GFP25 mice were previously described. R26 transgenic mice, Kitl-GFP mice, Gpr30 knockout mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Mouse and in vitro culture experiments were repeated at least twice, with at least two biological replicates per group. All animal studies and procedures were approved by the ethics committees of University of Tokyo, Gifu University, Columbia University, the Institutional Safety Committee on Recombinant DNA Experiments (approval ID 04220) and Animal Experimental Committees of the Institute for Protein Research (approval ID 29–01-2) at Osaka University and were performed in compliance with institutional guidelines. Mice were housed in a specific-pathogen free, temperature-controlled room at 22°C with a 12h ligh t/dark cycle.
Statistical Analyses
The differences between means were compared using either the Student’s t-test or the Wilcoxon test, and p < 0.05 was considered statistically significant.
See other details in Supplementary Methods section.
Results
Gpr30 is specifically expressed in gastric chief cells
To identify chief cell-specific genes, we performed transcriptome analyses using FACS-sorted cells isolated from Mist1-CreERT;R26-TdTomato mice with or without depletion of Lgr5+ chief cells (see Supplementary Methods). TdTomato+ Mist1-expressing cells were sorted from mice 2 days after tamoxifen induction and designated the chief cell-enriched group, while TdTomato+ cells sorted from induced Mist1-CreERT;Lgr5-DTR-EGFP;R26-TdTomato mice treated with DT was designated the isthmus stem cell-enriched, Lgr5-depleted Mist1+ cell group. RNA isolated from parietal cell populations was used as a third group.
Gene expression in the three groups was compared using Affymetrix GeneChip. As expected, known chief cell markers including Gif and Lgr5 were upregulated in the chief cell group, compared to the other groups. An additional chief cell-specific cell surface antigen, Cd9, was also identified by clustering and immunohistochemistry (Fig.1A, S3A). By contrast, proliferation markers including Ccnb1, Myc, and Pcna were upregulated in the isthmus stem cell-enriched, Lgr5-depleted Mist1+ cell group. We also identified several stem cell-related markers expressed in the isthmus region, including Kitl and Cd44, whose expression was confirmed by in situ hybridization (ISH) (Fig.S3B). Importantly, Gpr30, also known as Gper1, was robustly expressed in the chief cell group compared to the other two populations (Fig.1A). To independently confirm these findings, we analyzed public datasets available in the GEO database (GSE86603), in which the authors performed RNA-seq analyses of sorted Lgr5+ cells and Lgr5- cells isolated from the corpus of Lgr5-DTR-EGFP mice22. We found again that Gpr30 was robustly expressed in both Lgr5-high and Lgr5-low chief cell populations (Fig.S4A–B) in the GSE86603 dataset.
Figure 1.
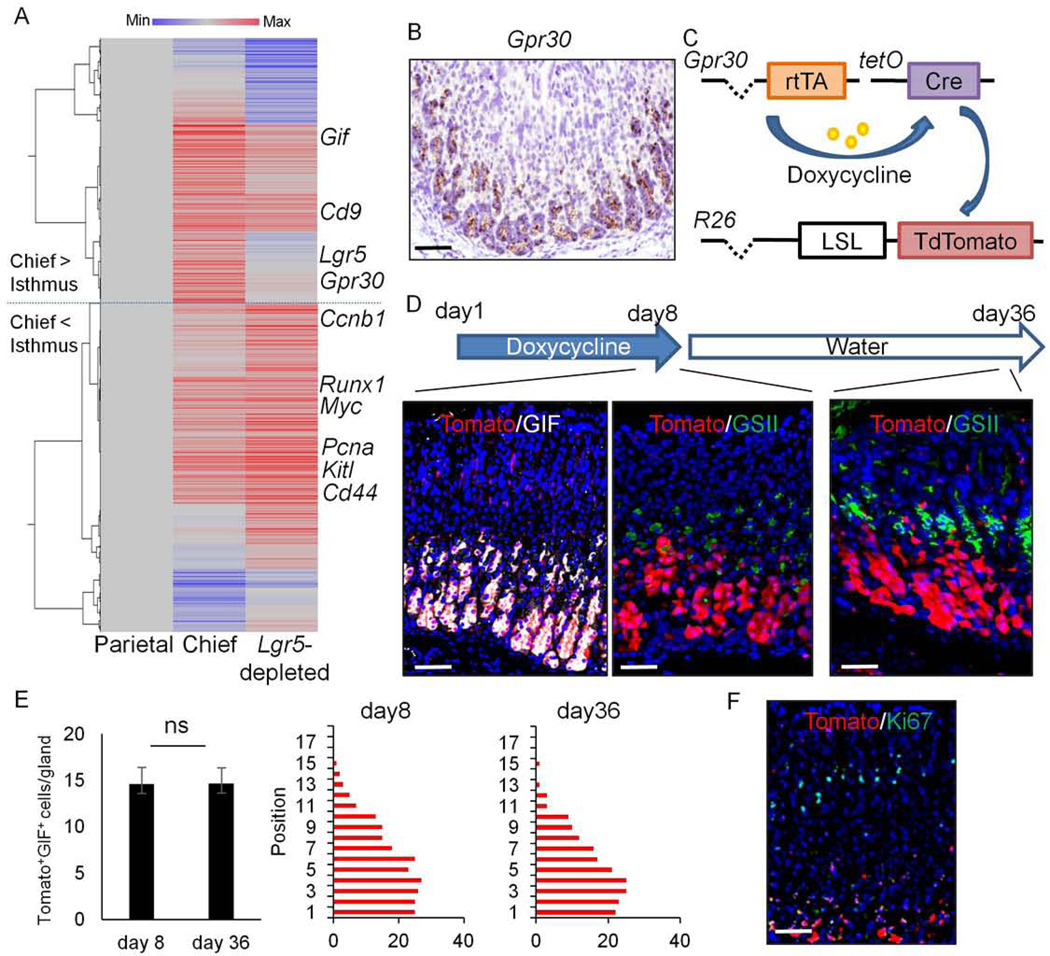
Gpr30 is specifically expressed in gastric chief lineage.
(A)Hierarchical clustering of average gene expression in Mist1+ chief cell-enriched group (Chief)and Lgr5-depleted, Mist1+ isthmus cell-enriched group (Lgr5-depleted) compared to parietal cell group (n=3/group). (B) ISH of Gpr30(brown) in the mouse corpus. (C-D)Schema (C) and protocol (D, top) of Gpr30-rtTA;TetO-Cre;R26-TdTomato mice treatment. Immunostainings at the indicated time points are shown (Tomato(red) and GIF(gray) at the left, Tomato(red) and GSII(green) at the middle and right). (E)Numbers of Tomato+GIF+ cells (left) and cell position (right) at day 8 and 36. Total 30 glands were quantified in each panel. Mean±SEM. (F)Immunostaining of Ki67(green) and Tomato(red) on Gpr30-rtTA;TetO-Cre;R26-TdTomato mice at day 8. Bars=50μm.
These observations were further confirmed by ISH, revealing strong lineage-specific expression of Gpr30 in chief cells (Fig.1B). We compared the expression pattern of Gpr30 and Lgr5 in the corpus by double ISH technique, and confirmed that Gpr30 was expressed in a much broader chief cell population than Lgr5 under normal conditions (Fig.S4C). Co-staining with GIF demonstrated that Gpr30 transcripts were found in nearly all (98.6%) GIF+ cells, while Lgr5 was expressed in only a subset of GIF+ cells (Fig.S4D). Instead, Lgr5 transcripts were found in a subset of H/K-ATPase+ parietal cells, particularly near the base, although the Lgr5-GFP protein in Lgr5-DTR-EGFP mice was not expressed in parietal cells (Fig.S4D–E). No Gpr30 expression was observed in parietal cells (Fig.S4D). After ablation of Lgr5+ cells following DT treatment of Lgr5-DTR-EGFP mice, the number of Gpr30+ cells was decreased but a subset of Gpr30+ cells still persisted in the upper chief cell region, indicating that Gpr30 is expressed in both Lgr5+ and Lgr5- chief cell populations (Fig.S4F). Thus, GPR30 is a highly representative marker of chief cells and marks much broader but more specific chief cell population than Lgr5.
Following epithelial injury by HDT, both Lgr5 and Gpr30 expression expands to the upper mucosal region (Fig.S4C), suggesting the inherent difficulty of selective chief cell labeling when employing the tamoxifen-dependent Cre/LoxP system. In fact, up to 12.0% of cells with Lgr5 transcripts were found in the upper or middle part of glands shortly after HDT. Thus, we generated a tamoxifen-independent GPR30+ chief cell fate mapping system using the doxycycline-dependent inducible Tet-ON constructs (Fig.1C). The targeting vector was generated by inserting an rtTA sequence into the translational start site of a Gpr30 BAC clone to produce Gpr30-rtTA transgenic mice. We crossed the Gpr30-rtTA mice to TetO-Cre mice, then crossed the Gpr30-rtTA;TetO-Cre mice to R26-TdTomato reporter mice, and induced the mice with doxycycline. After 7 days of doxycycline treatment, TdTomato expression was found in the gastric corpus glands at position +15 and below, with TdTomato expressed in the vast majority (85.8%) of the GIF+ chief cell lineage (Fig.1D–E, S5A). While a very small subset of GSII+ neck lineage cells and stromal αSMA+ myofibroblasts at upper mucosal region were also found to express TdTomato, no TdTomato expression was found in H/K-ATPase+ parietal, chromogranin A+ enterochromaffin-like, TFF1+ surface pit, Dclk1+ tuft, or Ki67+ proliferating cells (Fig.1F, S5B–E). There was no TdTomato expression in epithelial stem or progenitor cells, such that there was no full glandular lineage tracing observed during the 4-week observation period (Fig.1D–E). To compare the labeling pattern of Gpr30+ and Lgr5+ cells in this system, we generated Gpr30-rtTA;TetO-Cre;R26-TdTomato;Lgr5-DTR-EGFP mice. After 1 week administration of doxycycline, all Lgr5-GFP+ basal chief cells start to express TdTomato, suggesting that Lgr5+ chief cells belong to a part of Gpr30-lineage (Fig.S5F). We found that a few Lgr5-GFP+ cells (<5%) expressed Dclk1, suggesting that a subset of tuft cells may express Lgr5 in the normal state (Fig.S5E). There are no histological damage and no changes in the numbers of proliferating cells and chief cells following doxycycline treatment (Fig.3B,S5G). Together, these data indicate that the epithelial Gpr30+ cell population consists primarily of the chief cell lineage.
Figure 3.
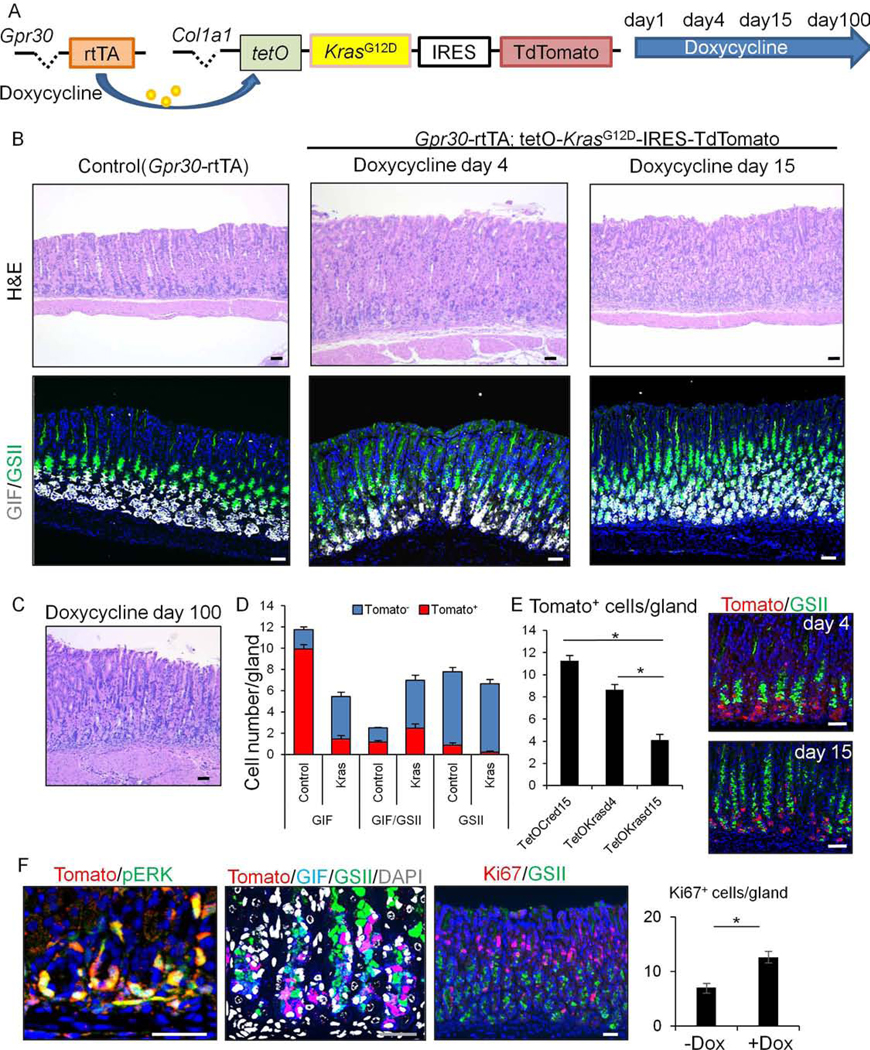
Gpr30-expressing chief cells are lost after Kras activation and do not contribute to metaplasia and dysplasia
(A) Gene constructs of Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato mice and protocol. (B)H&E and GIF(gray) and GSII(green) staining on control (Gpr30-rtTA) mice treated with doxycyline until day15 (left) and Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato mice at day 4 and 15 (middle/right). (C)H&E staining of Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato mice treated with doxycycline for 100 days. (D-E)Numbers of GIF+, GIF+GSII+, and GSII+ cells (D) and TdTomato+ cells (E) per gland in Gpr30-rtTA;TetO-Cre;R26-TdTomato (control) and Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato (Kras) mice treated with doxycycline for 4 or 15 days. Tomato(red) and GSII(green) staining on Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato mice at day 4 and 15 are shown in (E). Total 20 glands per group are quantified. (F) (Left)Tomato(red) and p-ERK(green), (Middle)Tomato(red), GIF(blue), GSII(green), and DAPI(gray), (Right)Ki67(red) and GSII(green) staining on Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato mice at day 15. Ki67+ cell numbers per gland are quantified in total 30 glands per group. Bars=50μm. Mean±SEM. *p<0.05.
Inducible Kras activation via the Tet-ON system causes gastric metaplasia and dysplasia
Next we generated a new transgenic mouse line (TetO-KrasG12D-IRES-TdTomato mice), in which the KrasG12D-IRES-TdTomato cassette was inserted into the Col1a1 locus under the control of the TetO promoter, which allowed for inducible mutant Kras expression in rtTA-expressing cells without tamoxifen treatment. We mated the TetO-KrasG12D-IRES-TdTomato mice with Tff2-Cre;R26-rtTA mice that express rtTA in the entire corpus gland26, as well as with Tff1-Cre;R26-rtTA mice that express rtTA predominantly in surface pits and upper isthmus progenitors14(Fig.2A–B).
Figure 2.
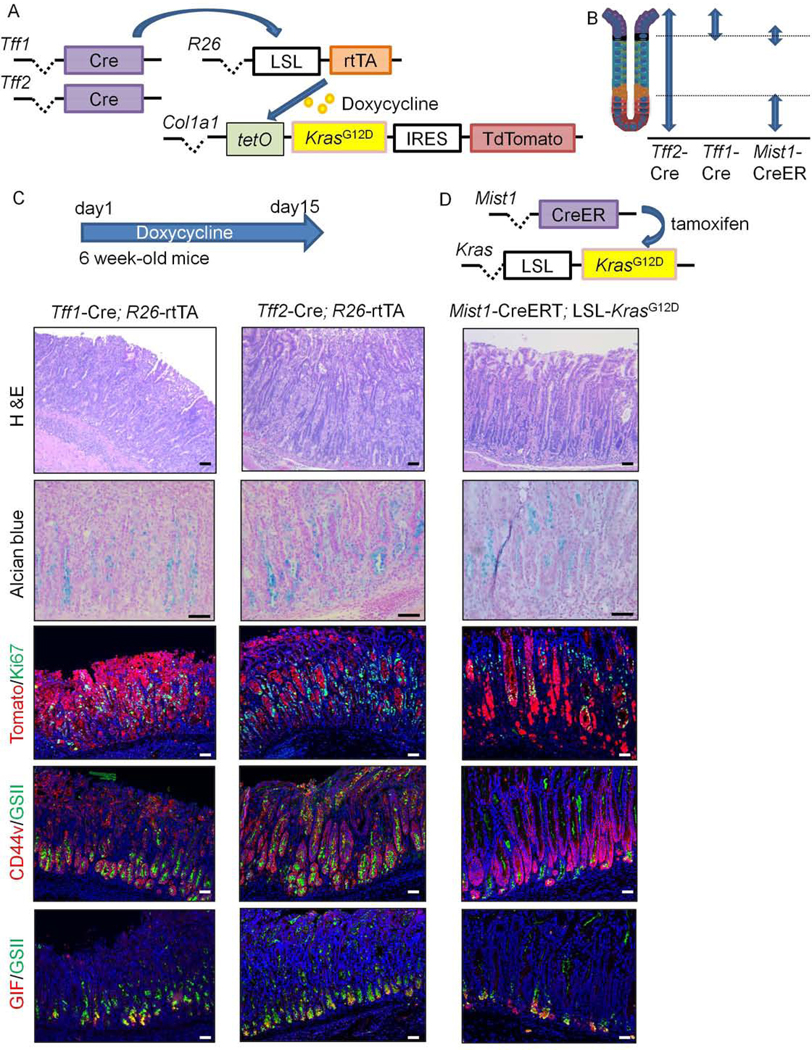
Inducible Kras activation via tetON system causes gastric metaplasia and dysplasia
(A)Schema of Tff1-Cre; or Tff2-Cre;R26-rtTA;TetO-KrasG12D-IRES-TdTomato mice. (B)Expression sites of Tff1-Cre, Tff2-Cre, and Mist1-CreERT constructs in corpus glands. (C)Corpus images of Tff1-Cre; or Tff2-Cre;R26-rtTA;TetO-KrasG12D-IRES-TdTomato mice treated with doxycycline for 14 days. First row, H&E; second row, Alcian blue; third row, Tomato(red) and Ki67(green); fourth row, CD44v9(red) and GSII(green); fifth row, GIF(red) and GSII(green) staining. (D)Gene constructs and images of Mist1-CreERT;LSL-KrasG12D;R26-TdTomato mice 42 days after tamoxifen induction shown in the same order as in (C). Bars=50μm.
After 14 days of doxycycline treatment, both Tff2-Cre;R26-rtTA;TetO-KrasG12D-IRES-TdTomato mice and Tff1-Cre;R26-rtTA;TetO-KrasG12D-IRES-TdTomato mice developed dysplastic glands with abundant mucin-rich metaplasia stained by Alcian blue in the corpus (Fig.2C). Similar to previous observations in Tff1-Cre;LSL-KrasG12D mice14, the gastric mucosa was dominated by Ki67+TdTomato+ Kras-mutated cells and developed CD44v9+ cells that are normally absent in this region. In addition, mature parietal and chief cells were lost, with only a few cells at the gland base that co-expressed GIF and GSII. These histological changes were equivalent to those seen in Mist1-CreERT crossed to LSL-KrasG12D mice (Fig.2C–D), as well as in other Kras models that used eR1-CreERT, Lgr5–2A-CreERT, or Tff1-Cre mice.12–14, 21, 22 Thus, these new rtTA-inducible models recapitulate the gastric phenotype previously seen in Kras-mutated mice. After cessation of doxycycline treatment, dysplastic corpus glands gradually recovered to a normal structure, with rapid expansion of a mucous-rich neck lineage near the base, followed by the reappearance of parietal and chief lineages (Fig.S6A–B). During the entire recovery phase, proliferation was found specifically above the neck region and not at the gland base (Fig.S6B).
Gpr30-expressing chief cells are lost after Kras activation and do not contribute to metaplasia and dysplasia
Given that Tff1-Cre and Tff2-Cre are not specific for one gastric cell type, it was not immediately possible to identify the origins of metaplasia and dysplasia in these models. Thus, to determine whether chief cells can serve as the origin of Kras-induced metaplasia and dysplasia, we mated Gpr30-rtTA to TetO-KrasG12D-IRES-TdTomato mice, followed by treatment with doxycycline (Fig.3A). In contrast to the other Kras models described above (Fig.2C–D), Gpr30-rtTA; TetO-KrasG12D-IRES-TdTomato mice did not develop either metaplasia or dysplasia, even after continuous administration of doxycycline for up to 100 days (Fig.3B–C). These findings suggest that metaplasia and dysplasia seen in previous Kras models do not originate from Gpr30-expressing chief cells. There were also no changes in parietal and surface pit lineages (Fig.S5D), further highlighting the difference from previous Kras models.
Given the striking difference from the previous Mist1-dependent Kras model, we compared Gpr30 expression with Mist1, whose expression was confirmed in 87.1% of GIF+ chief cells (Fig.S7A). Co-ISH/IF experiments confirmed that almost all (99.2%) Mist1+ cells were labeled by Gpr30-rtTA constructs, and that Gpr30 expression was detected in all Mist1-CreERT+ chief cells (Fig.S7B–D). Therefore, the phenotypic differences between Mist1-targeting and GPR30-targeting models are likely due to Kras induction in Mist1+ non-chief cells (e.g. Mist1+ isthmus stem cells), as previously documented10, 13.
Interestingly, there was a remarkable decrease of basophilic chief cells at the gland base in doxycycline-treated Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato mice (Fig.3A–B). We previously reported that such a loss of basophilic chief cells occurred when Lgr5+ chief cells were selectively ablated in Lgr5-DTR-GFP mice10. While the number of chief cells that express GIF alone was decreased after ≥14 days of doxycycline treatment, there was an expansion of cells co-expressing GIF and GSII, similar to that seen after Lgr5+ chief cell ablation10 (Fig.3B–D, S7E). To determine whether the expanded GIF+GSII+ cells harbored the Kras mutation, we examined phosphorylated ERK and TdTomato expression. Interestingly, the number of TdTomato+ cells in glands was dramatically decreased following Kras activation in Gpr30+ cells, while TdTomato and phospho-ERK were expressed in the few remaining basal GIF+ chief cells, but not in the expanded GSII+ neck lineage (Fig.3D–F). Gland proliferation was increased in doxycycline-treated animals, although the vast majority of proliferating (Ki67+) cells were found in the isthmus and neck region, with few Ki67+ cells at the gland base where phospho-ERK was activated by Kras (Fig.3F). Together, these results suggest that induction of Kras mutation in Gpr30+ chief cells results in the loss of those chief cells, with a compensatory expansion of GIF+GSII+ cells likely derived from neck cells located above.
We confirmed the sustained expression of Gpr30 at the gland base in Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato mice even after doxycycline treatment (Fig.S7F). Furthermore, ISH studies confirmed the sustained expression of Lgr5 in basal cells including GIF/GSII lineage, indicating that the loss of Gpr30+ chief cells occurred independent of any effects mediated by Lgr5 signaling (Fig.S7G). Histology remained consistent over time even in mice repeatedly challenged with HDT, suggesting that HDT pulse treatment of this line does not convert Kras-mutated Gpr30+ chief cells into stem-like, cancer-initiating cells (Fig.S7H). Finally, we employed a commonly used mutant Kras transgenic mouse line, LSL-KrasG12D, in combination with Mist1-CreERT;R26-mTmG mice. As reported previously13, at 7 days after low-dose tamoxifen treatment in Mist1-CreERT;R26-mTmG mice without Kras mutations, GFP+ recombination occurs in basal Mist1+ chief cells and Mist1+ isthmal cells (Fig.S8A). However, the addition of LSL-KrasG12D to these mice resulted in rapid loss of GFP-labeled GIF+Mist1+ chief cells, while Mist1+GFP+ isthmal clones become more evident (Fig.S8A–B). Therefore, Kras-mediated loss of chief cells likely occurs independent of the specific transgene or fluorescent reporter strain used.
Next, in order to trace the cell fate of Kras-mutated Gpr30+ chief cells, we generated Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato;TetO-Cre;R26-TdTomato mice (Fig.4A). After 14 days of doxycycline treatment, the TdTomato expression in these mice remained limited to the gland base, with no signs of upward lineage tracing (Fig.4B), verifying that the expanded GIF+GSII+ cells were not derived from Gpr30+ chief cells. To confirm that such GIF+GSII+ cells are derived from the isthmus-neck region, we generated new BAC transgenic Kitl-CreERT mice. Analogous to the Kitl gene expression (Fig.S3B) and GFP expression in the Kitl-GFP mice (Fig.S8C), gene recombination occurred predominantly in the upper part of glands including the isthmus in Kitl-CreERT;R26-TdTomato mice shortly after tamoxifen induction (Fig.4C). TdTomato+ cells were also found in rare stromal cells that express Vimentin or CD31 within the mucosa (Fig.4C–D). After tamoxifen administration, the Kitl-derived TdTomato+ clones gradually expanded and migrated towards the lower part of corpus glands (Fig.4E), suggesting that Kitl-CreERT marks isthmus progenitor cells that supply differentiated cell types. We then crossed Kitl-CreERT;R26-TdTomato mice to Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato mice and treated them with tamoxifen and doxycycline (Fig.4F). As a result, we observed fully traced glands including expanded GIF+GSII+ cells from Kitl-derived TdTomato+ clones, suggesting that the GIF+GSII+ SPEM-like cells found in Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato mice arise from Kitl+ isthmus progenitors.
Figure 4.
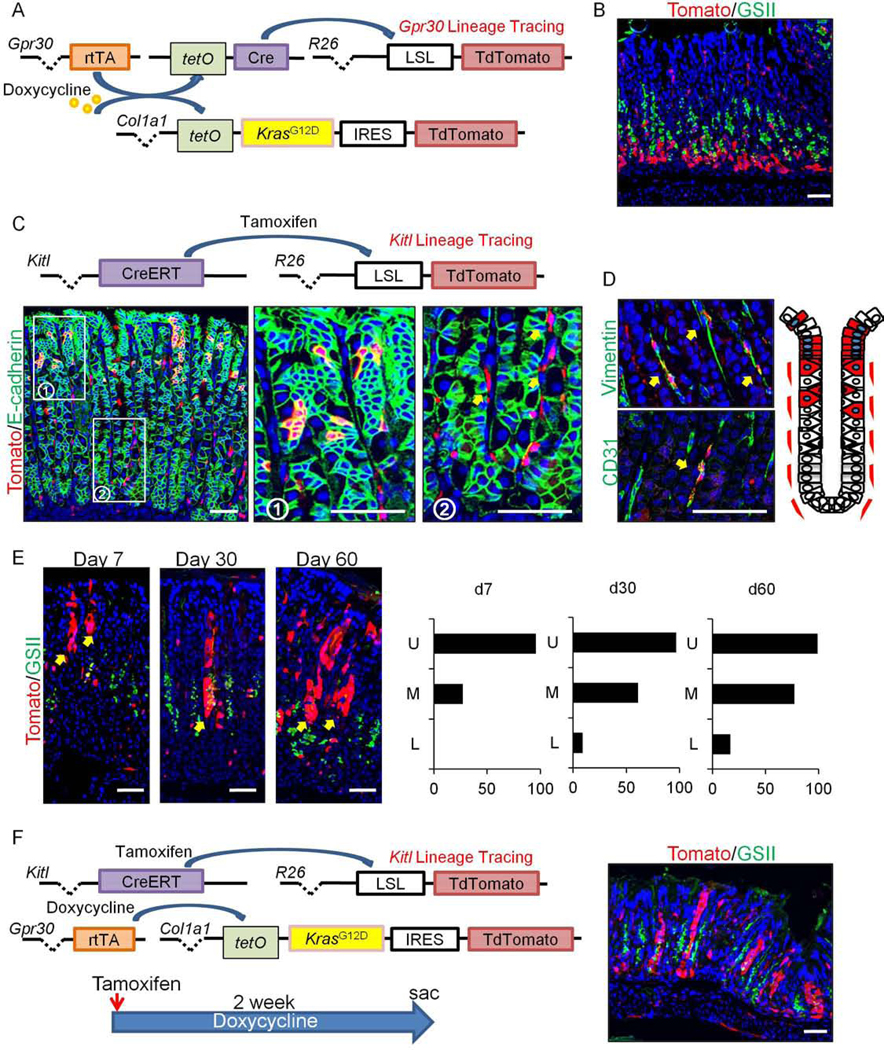
GIF+GSII+ SPEM is not derived from Gpr30-expressing chief cells, but from isthmal progenitors.
(A)Schema of gene constructs. (B)Tomato(red) and GSII(green) staining on Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato;TetO-Cre;R26-TdTomato mice after 14 days doxycycline treatment. (C-D)Gene constructs and E-cadherin(green, C), Vimentin and CD31(green, D) staining on Kitl-CreERT;R26-TdTomato(red) mice 4 days after tamoxifen. While box area is enlarged. Schematic TdTomato expression pattern in Kitl-CreERT;R26-TdTomato mouse stomach is shown. (E)Tomato(red) and GSII(green) staining on Kitl-CreERT;R26-TdTomato mice 7, 30, and 60 days after tamoxifen. Location of TdTomato+ clones (U, upper; M, middle; L, lower part of glands) is shown. Total 100 TdTomato+ glands at each point were counted. (F)(Left) Schema of gene constructs and protocol are shown. (Right) Tomato(red) and GSII(green) staining on Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato;Kitl-CreERT;R26-TdTomato mice 14 days after tamoxifen and doxycycline treatment. Bars=50μm.
Ras-mutated chief cells are extruded from the epithelium through PDK-dependent cell competition
Based on the above results, we wondered why Gpr30+ chief cells were lost from the gastric epithelium after induction of Kras mutation. It has been previously recognized that mature villus cells in the small intestine that acquire Ras mutation are rapidly extruded by surrounding normal cells via a cell competition-dependent mechanism.24 Thus, we explored this possibility in Ras-mutated stomachs. For this experiment, we used LSL-HrasG12V-IRES-GFP and LSL-GFP mice mated with Mist1-CreERT mice to induce GFP with or without Hras expression in Mist1+ cells. Immediately after administration of tamoxifen, we observed strong GFP expression in Mist1+ chief cells near the gland base, with fewer Mist1+GFP+ cells present at the isthmus (Fig.5A), consistent with previous studies10, 13. The number of Mist1+GFP+ chief cells in the Mist1-CreERT;LSL-GFP mice did not change for up to 7 days, while a slight expansion of isthmus-derived Mist1+GFP+ clones was observed (Fig.5A–B). By contrast, Mist1-CreERT;LSL-HrasG12V-IRES-GFP mice exhibited a dramatic decrease in the number of Mist1+ chief cells, with only Mist1+GFP+ isthmus cell clusters remaining (Fig.5A–B).
Figure 5.
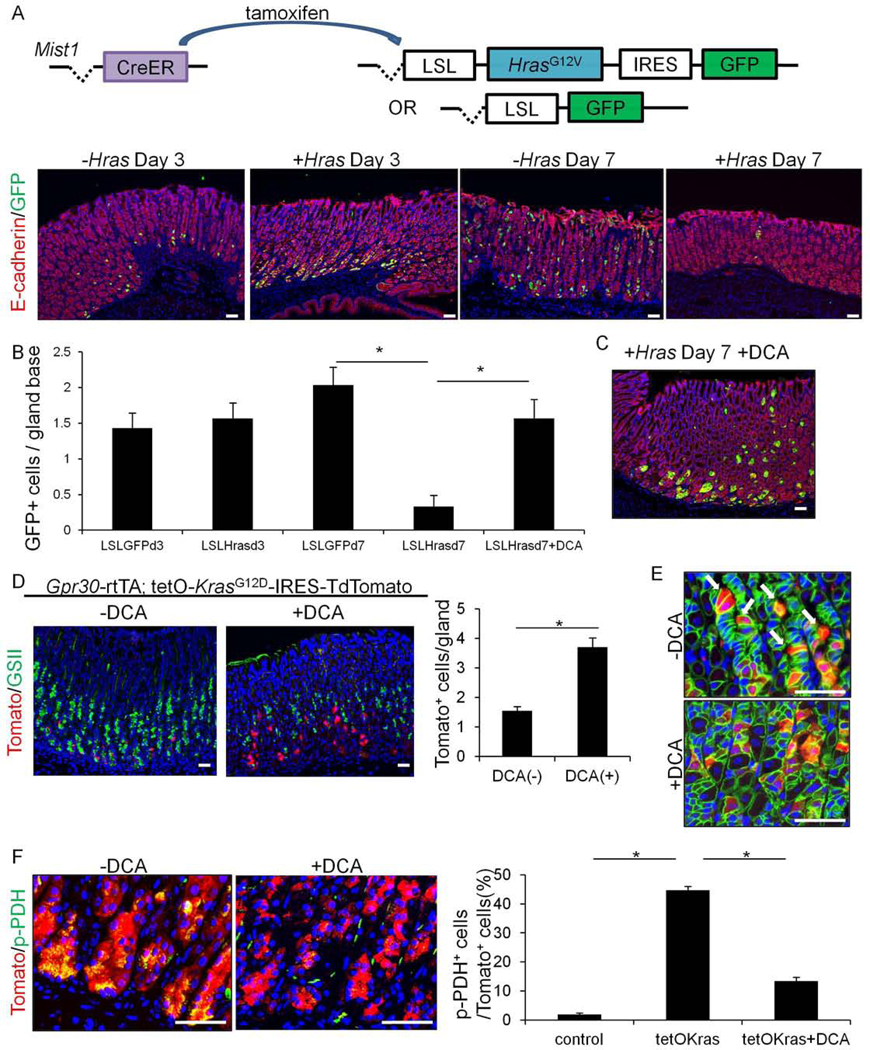
Ras-mutated chief cells are extruded from the epithelium through PDK-dependent cell competition.
(A)Schema of gene constructs and E-cadherin(red) and GFP(green) staining of Mist1-CreERT;LSL-GFP (-Hras) and Mist1-CreERT;LSL-HrasG12V-IRES-GFP (+Hras) mice 3 or 7 days after tamoxifen. (B)Numbers of GFP+ cell numbers per gland (below position +15) in Mist1-CreERT;LSL-GFP and Mist1-CreERT;LSL-HrasG12V-IRES-GFP mice 3 or 7 days after tamoxifen. (C)E-cadherin(red) and GFP(green) staining of day 7 Mist1-CreERT;LSL-HrasG12V-IRES-GFP mice treated with DCA. (D)Tomato(red) and GSII(green) staining on Gpr30-rtTA;tetO-KrasG12D-IRES-TdTomato mice after 14 days doxycycline. Mice were treated with or without DCA. Number of Tomato+ cells per gland is quantified in total 30 glands. (E-F)E-cadherin(green) and Tomato(red) staining (E) and p-PDH(green) and Tomato(red) staining (F) on Gpr30-rtTA;tetO-KrasG12D-IRES-TdTomato mice after 4 days doxycycline treatment with or without DCA. Arrows in (E) indicate extruding chief cells. Percentage of p-PDH+ cells in Tomato+ cells are shown in (F). Total 300 cells per group are quantified. Bars=50μm. Mean±SEM. *p<0.05.
Ras-dependent cell competition occurs as a result of PDK-dependent metabolic changes in extruded cells24. Therefore, we treated Hras-mutated animals with a PDK inhibitor, DCA, which inhibits Ras-dependent cell competition in the gut24. There was a significant increase in Mist1+GFP+ chief cells in DCA-treated Mist1-CreERT;LSL-HrasG12V-IRES-GFP mice compared to untreated controls (Fig.5B–C). Next, we investigated the effects by DCA in Kras mutant models by treating Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato mice and Mist1-CreERT;LSL-KrasG12D;R26-mTmG mice. As seen in the Hras-mutated mice, DCA treatment significantly increased the number of Kras-mutated Gpr30+ and Mist1+ chief cells remaining near the gland base (Fig.5D, S8B,D–E). At an earlier time point (4days), extruding Gpr30+Tomato+ chief cells were evident in Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato mice while DCA treatment inhibited this extrusion (Fig.5E). While more Kras-mutated Gpr30+ chief cells were retained at the gland base in DCA-treated Gpr30-rtTA;TetO-KrasG12D-IRES-TdTomato mice, no histological metaplasia or dysplasia were observed, and there was less prominent expansion of GIF+GSII+ cells (Fig.S8E). These data suggest that Kras mutation in Gpr30+ chief cells does not in any way contribute to metaplasia or dysplasia, even if Kras-mutant cells are allowed survive over longer periods of time. In Kras-mutated Gpr30+ chief cells, there was marked phosphorylation of PDH, a downstream of PDK, which was not observed in non-mutated controls or DCA-treated Kras mutants (Fig.5F,S8F). Thus, Ras-mediated, PDK-dependent metabolic changes in chief cells are a key initial event during the cell extrusion process and a major cause of chief cell loss.
Gpr30-expressing chief cells are lost after injury and do not contribute to regeneration
Next, we used our Gpr30 fate mapping system to examine whether Gpr30+ chief cells can acquire progenitor-like features and contribute to gland regeneration following acute and chronic injury. We labeled Gpr30+ chief cells by administering doxycycline to Gpr30-rtTA;TetO-Cre;R26-TdTomato mice for 1 week, followed by 3 weeks of washout, and then treatment with HDT or DMP-777 to induce acute injury (Fig.6A). We observed a significant increase in GIF+GSII+ cells 3 days after administration of HDT, although these increases quickly diminished, and the number of double positive cells returned to normal levels by day 7 (Fig.6B–C), consistent with previous reports8. During this acute injury and regeneration process, there was a continuous decrease in labeled chief cells, with no upward lineage tracing events to indicate progenitor conversion (Fig.6B–C). A similar decrease in labeled chief cells was observed in DMP-777-treated Gpr30-rtTA;TetO-Cre;R26-TdTomato mice (Fig.S9A), as previously seen in Mist1+ chief cells7, 13. Therefore, Gpr30+ and Mist1+ chief cells appear to be lost after acute injury, with the transient increase in GIF+GSII+ cells in these models likely comprising a compensatory expansion of neck lineage cells and not indicative of transdifferentiation or dedifferentiation.
Figure 6.
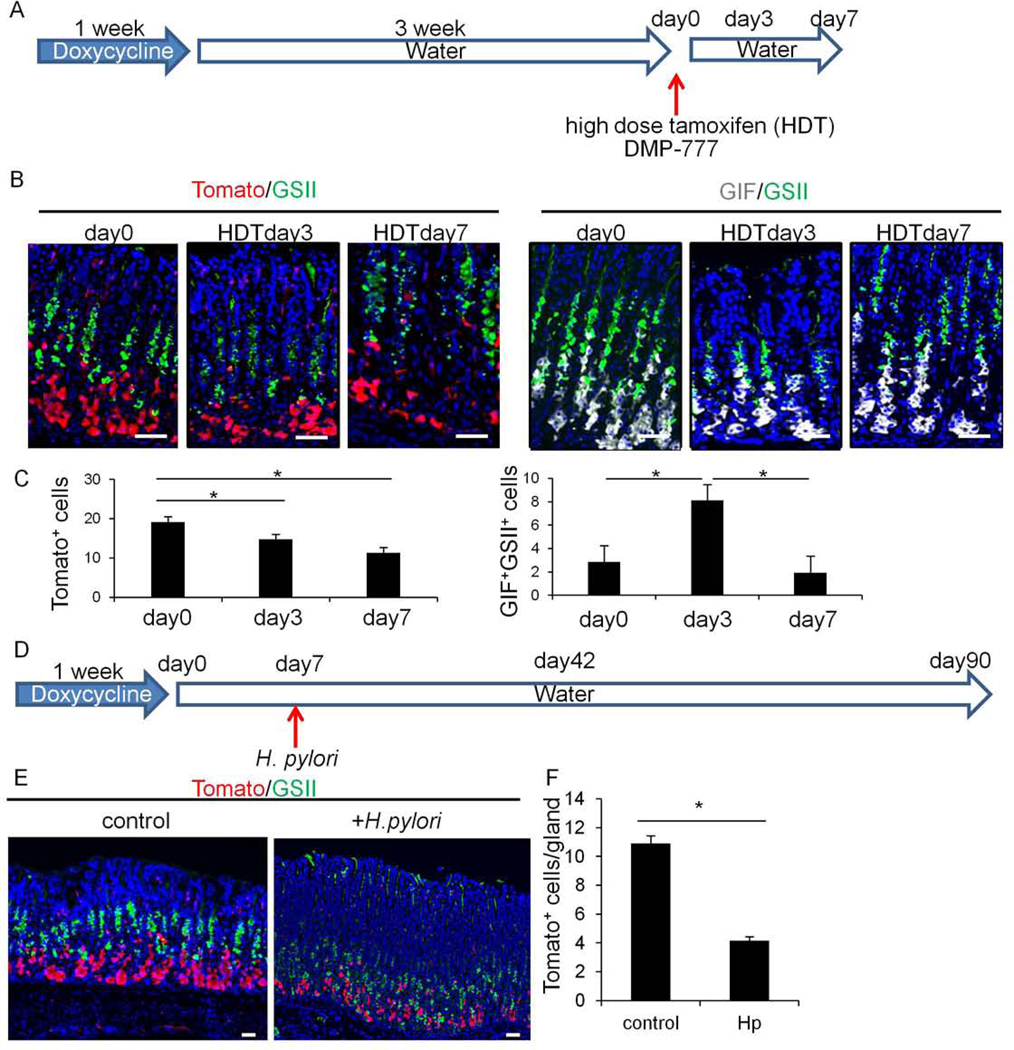
GPR30-expressing chief cells are lost after injury and do not contribute to regeneration.
(A)Protocol. (B)Staining of Tomato(red, left) and GIF(gray, right) together with GSII(green) on Gpr30-rtTA;TetO-Cre;R26-TdTomato mice at day 0, 3, and 7 after HDT treatment. (C)Numbers of Tomato+ cells and GIF+GSII+ cells per gland at each time point. Total 20 glands per group were quantified. (D)Protocol. (E-F)Tomato(red) and GSII(green) staining on Gpr30-rtTA;TetO-Cre;R26-TdTomato mice with or without H pylori infection (E) and numbers of Tomato+ cells and GIF+GSII+ cells (F). Total 30 glands per group were quantified. Mean±SEM. *p<0.05.
To further test these conclusions, we investigated a chronic (H pylori infection) injury model using the same mouse lines. Compared to uninfected controls, there was a significant (>60%) decrease in TdTomato+ chief cells without any upward lineage tracing in H pylori-infected mice at day 42 (Fig.6D–F). Even at day 90, the vast majority of GSII+ SPEM cells was not traced from the Gpr30-derived chief cell lineage (Fig.S9B). Therefore, as in the acute injury models, Gpr30+ chief cells are lost during chronic H pylori infection without converting to progenitor-like cells or giving rise to metaplasia. However, in H pylori infection models, we did not find phosphorylation of PDH in GIF+ chief cells, suggesting that the mechanisms for chief cell loss in H pylori infection model is likely distinct from Kras models.
GPR30 governs PDK-dependent chief cell extrusion following acute injury
Finally, we analyzed the functional role of GPR30 in the stomach using GPR30 knockout (Gpr30−/−) mice. The gastric histology of Gpr30−/− mice appeared normal, with no significant changes in the numbers of GIF+ and GSII+ cells prior to epithelial injury (Fig.S10A). However, upon treatment with HDT, GIF+ chief cells in Gpr30−/− mice were relatively preserved at the gland base compared to control mice, while the number of GIF+GSII+ cells was decreased (Fig.7A–B). Apoptosis in parietal cells was equally observed following HDT administration in both control and Gpr30−/− mice (Fig.S10B), suggesting that Gpr30 deficiency was specifically protective for GIF+ chief cells. Further, the findings suggest that the increases in the GSII+ lineage depend predominantly on the degree of chief cell loss. Similar to the HDT administration model, the reduction of GIF+ chief cells and expansion of neck lineage cells after DMP-777 treatment was less prominent in Gpr30−/− mice than in control mice, without any significant changes in parietal cell numbers (Fig.S11A–B). Nevertheless, in the chronic H pylori infection model, there was no significant difference in SPEM development between Gpr30−/− mice and their control littermates (Fig.S11D), suggesting that GPR30 plays a role primarily in the acute and Kras SPEM models. In contrast, in the more chronic Helicobacter models, metaplasia may be less dependent on GPR30 signaling, and driven more by inflammatory responses in other epithelial cell types.
Figure 7.
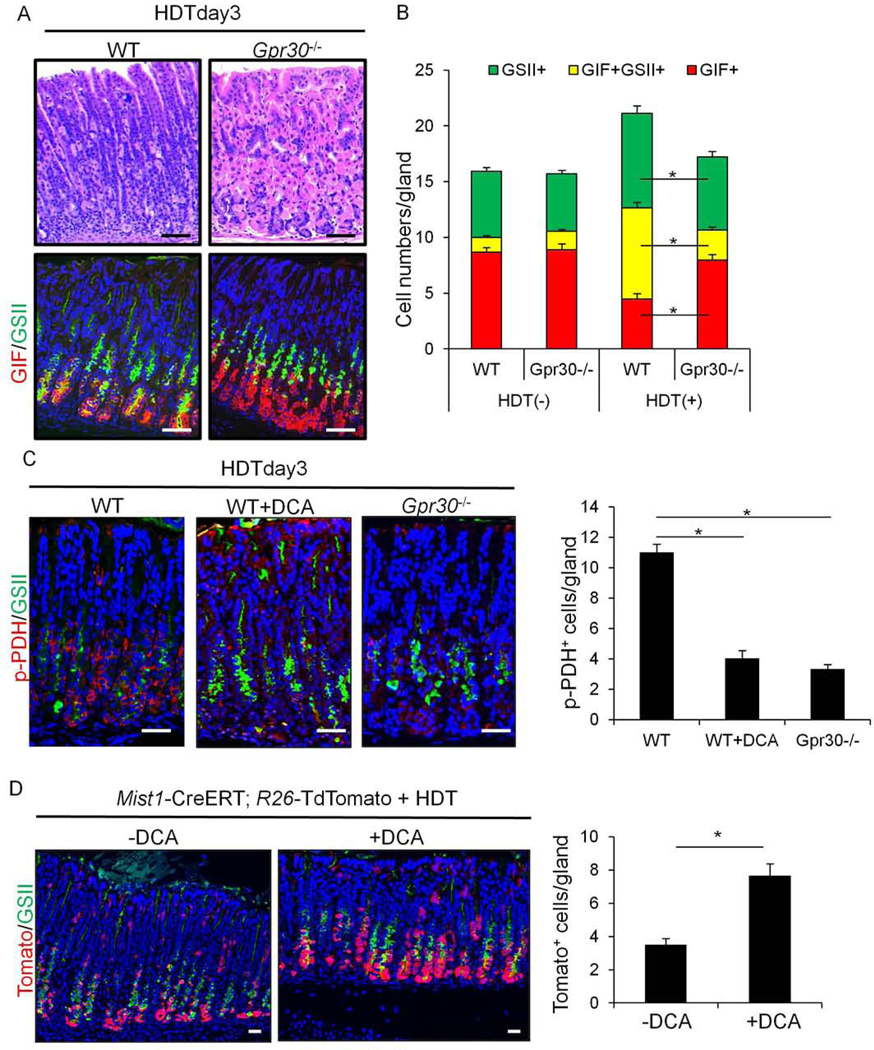
GPR30 governs PDK-dependent chief cell extrusion following acute injury.
(A)H&E and GIF(red) and GSII(green) staining on WT and Gpr30−/− mice 3 days after HDT treatment. (B)Numbers of GIF+, GIF+GSII+, and GSII+ cells per gland in WT and Gpr30−/− mice with or without tamoxifen treatment. (C)Staining of p-PDH(red) and GSII(green) and numbers of p-PDH+ cells per gland in WT and Gpr30−/− mice 3 days after HDT, with or without DCA. (D)Staining of Tomato(red) and GSII(green) and numbers of Tomato+ chief cells per gland in Mist1-CreERT;R26-TdTomato mice 3 days after HDT with or without DCA. Total 30 glands per group were quantified. Mean±SEM. *p<0.05.
We hypothesized that the loss of chief cells in acute injury models is caused by PDK-dependent cell competition, consistent with that seen in Ras mutation models. Indeed, we observed robust phosphorylation of PDH in chief cells following HDT or DMP-777 treatment, which was attenuated following treatment with DCA (Fig.7C, S11C). Phosphorylation of PDH was similarly attenuated in Gpr30−/− mice treated with HDT or DMP-777. The numbers of Mist1+ chief cells labeled by TdTomato increased in DCA-treated Mist1-CreERT;R26-TdTomato mice compared to that of untreated mice after HDT treatment (Fig.7D). Knockout of GPR30 similarly preserved labeled Mist1+ chief cell number in Mist1-CreERT;Gpr30−/−;R26-TdTomato mice (Fig.S12). Taken together, these data suggest that GPR30 regulates PDK-dependent PDH phosphorylation, which triggers loss of chief cells and subsequent expansion of neck lineage.
Discussion
Chief cells have attracted considerable attention after a number of studies suggested they might be the cellular origin of cancer and precancerous metaplasia7, 12, 22. However, this proposed model of chief cell transdifferentiation/dedifferentiation rested largely on two major observations: namely, lineage tracing events in CreERT mice and co-expression of neck cell markers with chief cell markers on immunostaining. There are several critical limitations to lineage tracing experiments, including the specificity of lineage markers and injurious effects by tamoxifen to activate CreERT constructs14, 17, 25. It remains uncertain whether the co-expression of neck and chief cell markers directly reflects stem cell function or the cell-of-origin of cancer.
Recent single cell transcriptome analyses in the stomach revealed that there could be significant heterogeneity in certain cell populations, such as Lgr5+ cells 27–29. In fact, our analysis confirmed that Lgr5 is expressed not only in chief cells but also in other cell types. In turn, it can be easily assumed that there is a transcriptional heterogeneity in chief cell population, for instance, Lgr5-low and Lgr5-high subsets. While this type of transcriptional analysis can at times provide new insights in the field, the results sometimes present their own sets of challenges with respect to data interpretation, and such data do not provide functional evidence. Based on our analysis, Gpr30 expression overlaps nearly completely with GIF (98.6%) and Mist1 (99.2%) expression, and covers all Lgr5+ populations within the chief cell lineage. Therefore, the likely explanation for the phenotypic differences in the Lgr5 or Mist1-targeted Kras models compared to the Gpr30-targeted Kras model is that Lgr5 and Mist1 are expressed in non-chief cell populations particularly when tamoxifen-induced injury is given. It seems rather unlikely that the presence of a rare Gpr30-negative chief cell subset that retains this divergent progenitor function could account for these findings. However, further studies are needed to more precisely address possible cellular heterogeneity in the chief cell population.
The data presented here argue against the theory of chief cell transdifferentiation/dedifferentiation, and raise important questions as to the current definition of SPEM in mouse models. We found no evidence of lineage expansion from Gpr30+ chief cells, with these cells being lost and replaced, rather than interconverting to progenitor cells during metaplasia development. Instead, Kitl+ isthmus progenitors expand and give rise to GIF+GSII+ SPEM lineage when Ras mutation induces chief cell loss. Similarly, ablation of Lgr5+ chief cells results in expansion of neck lineage, including GIF+GSII+ pre-chief progenitors10. While detailed characterization of Kitl+ progenitors would be needed in future studies, these findings suggest that the reason that GIF+GSII+ cells expand in SPEM models would be because of the expansion of upper progenitors, rather than due to chief cell dedifferentiation. Our data using Gpr30-rtTA and Mist1-CreERT mouse models suggest that in Ras-induced rSPEM models, aSPEM can co-exist near the base soon after mutant Ras is induced in chief cells which are rapidly extruded. Given that Kras activation in Gpr30+ chief cells induces only aSPEM but does not lead to parietal cell atrophy, MUC4+ metaplasia, and dysplasia afterwards, the role of aSPEM in gastric carcinogenesis remains unclear and further studies are needed to determine whether aSPEM is directly relevant to the later cancer lineage.
We found that Ras mutation in chief cells resulted in rapid elimination from the epithelium via PDK-dependent cell competition. Recent studies have shown that newly emerging transformed cells with gene mutations in Ras, Tp53 and Erbb2, are often extruded by surrounding normal epithelial cells24, 30, 31, suggesting a form of anti-tumor activity by the normal epithelium, independent of immune cell involvement: a process termed epithelial defense against cancer. During this process, PDK-dependent Warburg-like metabolic changes in transformed cells play a central role24. PDK phosphorylates and thus inactivates PDH, which catalases conversion of pyruvate into acetyl-CoA, thereby blocking entry into the tricarboxylic acid cycle. Because PDH phosphorylation was enhanced in chief cells following injury and Ras mutation, this pathway may be relevant in chief cell loss, a fundamental histological feature of atrophic gastritis. Several proteins are potential upstream regulators of PDK, including FOXO, PPAR, glucocorticoid receptors, and estrogen-related receptors32. Among the more interesting activators of GPR30 may be the binding of estrogen receptor to tamoxifen33. One explanation would be that the binding of tamoxifen to the chief cell-specific receptor GPR30 triggers PDK-dependent metabolic changes and promotes cell competition. However, given that GPR30 signaling also plays a role in DMP-777-mediated chief cell loss, activation of GPR30 might be regulated by other factors, such as inflammatory pathways.
Previous studies have suggested the existence of cellular plasticity between stem cells and progenitors. In the intestine, lineage-committed progenitors have been shown to dedifferentiate into long-lived stem-like cells when the active stem cells are damaged, therefore acting as potential cancer-initiating cells34–38. More recent studies have suggested that a subset of mature cell types may have the capacity to dedifferentiate in response to stem cell injury suggesting a broader concept of plasticity39–42. Although such a concept requires further confirmation, a distinct feature of gastric injury models is that stem/progenitor cells in the isthmus region are largely preserved and even more active within the niche at the time that SPEM emerges13. Indeed, a recent work using multi-color labeling system concluded that isthmus-derived clones rapidly expand and contribute to mucosal regeneration following injury43. This multi-color labeling study, as well as a recently published long-term BrdU labeling study9, also proposed that chief cells may comprise a set of basal stem cells, distinct from the isthmus stem cell. While these studies have established that some basal chief cells are extremely long-lived with slow turnover, it remains uncertain whether such basal glandular cells have self-renewal or multipotential ability, the defining features of tissue stem cells44. Given that the vast majority of corpus glands are monoclonal in mice and humans45–48, further evidence will be helpful to prove that such ling-lived cells are a type of stem cells or cancer-initiating cells, rather than long-lived post-mitotic cells.
Cancer arises after accumulation of multiple gene mutations, thus long-lived stem cells are thought to be a cell-of-origin of cancers in most organs44, 49. Given that some gastric chief cells are also long-lived, the acquisition of multiple oncogenic mutations by chief cells could in theory lead to their transformation. In fact, in an experimental model, the simultaneous induction of multiple gene mutations is able to convert certain mature cell types to cancer-initiating cells by enabling cell-autonomous signaling that compensates for niche dependency outside of the stem cell niche50. However, given that most gene mutations occur gradually or in stepwise fashion, and that Ras mutations alone in mature cell types likely eliminates mutated cells via cell competition, chief cells should be regarded as an unlikely cell of origin for gastric cancer. In contrast, stem cells have the ability to escape from cell competition after acquisition of initial gene mutation, and such mechanisms should be addressed in future studies.
Supplementary Material
What you need to know:
BACKGROUND AND CONTEXT:
Gastric chief cells have been proposed to be the origin of metaplasia and cancer, through dedifferentiation. However, studies supporting this claim have had technical limitations.
NEW FINDINGS:
This study identified a new chief cell marker, GPR30. Lineage tracing experiments revealed that Ras mutation or drug-induced injury extrudes chief cells from epithelia via pyruvate dehydrogenase kinase-dependent cell competition. Blocking GPR30 or PDK preserves chief cell loss and inhibits expansion of neck lineage.
LIMITATIONS:
This study was performed in mice and focuses on the acute phase of metaplasia development.
IMPACT:
Findings from this study contradict the theory of chief cell dedifferentiation. This study found that expansion of cells that are used as a marker of spasmolytic polypeptide-expressing metaplasia are a compensatory response by the neck lineage to replace the eliminated chief cells.
Lay Summary:
We identified cells in stomach of mice that might serve as the precursors to metaplasia and gastric cancer.
Acknowledgements
Y.H. is supported by the KAKENHI Grant-in-Aid for Scientific Research,17K09347 and 17H05081, P-CREATE from AMED, the Pharmacological Research Foundation, the research grant of Bristol-Myers Squibb, the Kowa Life Science Foundation, the Senshin Medical Research Foundation, the Yokoyama Clinical Pharmacological Research Foundation, the Kanae Foundation of the Promotion of Medical Science, the Inoue Science Research Award, and the Takeda Science Foundation Visionary Research Grant, the Princess Takamatsu Cancer Research Fund, and the Advanced Research and Development Programs for Medical Innovation (PRIME). M.K. is supported by the research grant from the Institute for Adult Diseases, Asahi Life Foundation. T.C.W. received the NIH grants (R35CA210088 and 5U01DK103155-04).
Footnotes
Disclosures:
The authors disclose no conflicts.
Transcript profiling:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Author names in bold designate shared co-first authorship.
- 1.Kinoshita H, Hayakawa Y, Koike K. Metaplasia in the Stomach-Precursor of Gastric Cancer? Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayakawa Y, Fox JG, Wang TC. The Origins of Gastric Cancer From Gastric Stem Cells: Lessons From Mouse Models. Cellular and Molecular Gastroenterology and Hepatology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayakawa Y, Fox J, Gonda T, et al. Mouse Models of Gastric Cancer. Cancers 2013;5:92–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang TC, Dangler CA, Chen D, et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000;118:36–47. [DOI] [PubMed] [Google Scholar]
- 5.Wang TC, Goldenring JR, Dangler C, et al. Mice lacking secretory phospholipase A2 show altered apoptosis and differentiation with Helicobacter felis infection. Gastroenterology 1998;114:675–89. [DOI] [PubMed] [Google Scholar]
- 6.Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008;14:408–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nam KT, Lee HJ, Sousa JF, et al. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 2010;139:2028–2037.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huh WJ, Khurana SS, Geahlen JH, et al. Tamoxifen induces rapid, reversible atrophy, and metaplasia in mouse stomach. Gastroenterology 2012;142:21–24 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burclaff J, Willet S, Saenz JB, et al. Proliferation and Differentiation of Gastric Mucous Neck and Chief Cells During Homeostasis and Injury-induced Metaplasia. Gastroenterology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kinoshita H, Hayakawa Y, Niu Z, et al. Mature gastric chief cells are not required for the development of metaplasia. Am J Physiol Gastrointest Liver Physiol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramsey VG, Doherty JM, Chen CC, et al. The maturation of mucus-secreting gastric epithelial progenitors into digestive-enzyme secreting zymogenic cells requires Mist1. Development 2007;134:211–22. [DOI] [PubMed] [Google Scholar]
- 12.Choi E, Hendley AM, Bailey JM, et al. Expression of Activated Ras in Gastric Chief Cells of Mice Leads to the Full Spectrum of Metaplastic Lineage Transitions. Gastroenterology 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayakawa Y, Ariyama H, Stancikova J, et al. Mist1 Expressing Gastric Stem Cells Maintain the Normal and Neoplastic Gastric Epithelium and Are Supported by a Perivascular Stem Cell Niche. Cancer Cell 2015;28:800–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinoshita H, Hayakawa Y, Konishi M, et al. Three types of metaplasia model through Kras activation, Pten deletion, or Cdh1 deletion in the gastric epithelium. J Pathol 2019;247:35–47. [DOI] [PubMed] [Google Scholar]
- 15.Okumura T, Ericksen RE, Takaishi S, et al. K-ras mutation targeted to gastric tissue progenitor cells results in chronic inflammation, an altered microenvironment, and progression to intraepithelial neoplasia. Cancer Res 2010;70:8435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayakawa Y, Fox JG, Wang TC. Isthmus Stem Cells Are the Origins of Metaplasia in the Gastric Corpus. Cellular and Molecular Gastroenterology and Hepatology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mills JC, Goldenring JR. Metaplasia in the Stomach Arises From Gastric Chief Cells. Cell Mol Gastroenterol Hepatol 2017;4:85–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldenring JR, Mills JC. Isthmus time is here: Runx1 identifies mucosal stem cells in the gastric corpus. Gastroenterology 2016. [DOI] [PubMed] [Google Scholar]
- 19.Hayakawa Y, Wang TC. Isthmus Progenitors, Not Chief Cells, Are the Likely Origin of Metaplasia in eR1-CreERT; LSL-KrasG12D Mice. Gastroenterology 2017;152:2078–2079. [DOI] [PubMed] [Google Scholar]
- 20.Radyk MD, Burclaff J, Willet SG, et al. Metaplastic Cells in the Stomach Arise, Independently of Stem Cells, via Dedifferentiation or Transdifferentiation of Chief Cells. Gastroenterology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuo J, Kimura S, Yamamura A, et al. Identification of Stem Cells in the Epithelium of the Stomach Corpus and Antrum of Mice. Gastroenterology 2016. [DOI] [PubMed] [Google Scholar]
- 22.Leushacke M, Tan SH, Wong A, et al. Lgr5-expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat Cell Biol 2017. [DOI] [PubMed] [Google Scholar]
- 23.Tian H, Biehs B, Warming S, et al. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 2011;478:255–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kon S, Ishibashi K, Katoh H, et al. Cell competition with normal epithelial cells promotes apical extrusion of transformed cells through metabolic changes. Nat Cell Biol 2017;19:530–541. [DOI] [PubMed] [Google Scholar]
- 25.Kawamoto S, Niwa H, Tashiro F, et al. A novel reporter mouse strain that expresses enhanced green fluorescent protein upon Cre-mediated recombination. FEBS Lett 2000;470:263–8. [DOI] [PubMed] [Google Scholar]
- 26.Hayakawa Y, Sakitani K, Konishi M, et al. Nerve Growth Factor Promotes Gastric Tumorigenesis through Aberrant Cholinergic Signaling. Cancer Cell 2017;31:21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bockerstett KA, Lewis SA, Wolf KJ, et al. Single-cell transcriptional analyses of spasmolytic polypeptide-expressing metaplasia arising from acute drug injury and chronic inflammation in the stomach. Gut 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sigal M, Reines MDM, Mullerke S, et al. R-spondin-3 induces secretory, antimicrobial Lgr5(+) cells in the stomach. Nat Cell Biol 2019;21:812–823. [DOI] [PubMed] [Google Scholar]
- 29.Tabula Muris C, Overall c, Logistical c, et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018;562:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe H, Ishibashi K, Mano H, et al. Mutant p53-Expressing Cells Undergo Necroptosis via Cell Competition with the Neighboring Normal Epithelial Cells. Cell Rep 2018;23:3721–3729. [DOI] [PubMed] [Google Scholar]
- 31.Leung CT, Brugge JS. Outgrowth of single oncogene-expressing cells from suppressive epithelial environments. Nature 2012;482:410–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeong JY, Jeoung NH, Park KG, et al. Transcriptional regulation of pyruvate dehydrogenase kinase. Diabetes Metab J 2012;36:328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol 2011;7:715–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asfaha S, Hayakawa Y, Muley A, et al. Krt19(+)/Lgr5(−) Cells Are Radioresistant Cancer-Initiating Stem Cells in the Colon and Intestine. Cell Stem Cell 2015;16:627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayakawa Y, Tsuboi M, Asfaha S, et al. BHLHA15-Positive Secretory Precursor Cells Can Give Rise to Tumors in Intestine and Colon in Mice. Gastroenterology 2019;156:1066–1081 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yan KS, Gevaert O, Zheng GXY, et al. Intestinal Enteroendocrine Lineage Cells Possess Homeostatic and Injury-Inducible Stem Cell Activity. Cell Stem Cell 2017;21:78–90 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tetteh PW, Basak O, Farin HF, et al. Replacement of Lost Lgr5-Positive Stem Cells through Plasticity of Their Enterocyte-Lineage Daughters. Cell Stem Cell 2016;18:203–13. [DOI] [PubMed] [Google Scholar]
- 38.Tomic G, Morrissey E, Kozar S, et al. Phospho-regulation of ATOH1 Is Required for Plasticity of Secretory Progenitors and Tissue Regeneration. Cell Stem Cell 2018;23:436–443 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmitt M, Schewe M, Sacchetti A, et al. Paneth Cells Respond to Inflammation and Contribute to Tissue Regeneration by Acquiring Stem-like Features through SCF/c-Kit Signaling. Cell Rep 2018;24:2312–2328 e7. [DOI] [PubMed] [Google Scholar]
- 40.Yu S, Tong K, Zhao Y, et al. Paneth Cell Multipotency Induced by Notch Activation following Injury. Cell Stem Cell 2018;23:46–59 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westphalen CB, Asfaha S, Hayakawa Y, et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J Clin Invest 2014;124:1283–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harnack C, Berger H, Antanaviciute A, et al. R-spondin 3 promotes stem cell recovery and epithelial regeneration in the colon. Nat Commun 2019;10:4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han S, Fink J, Jorg DJ, et al. Defining the Identity and Dynamics of Adult Gastric Isthmus Stem Cells. Cell Stem Cell 2019;25:342–356 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010;327:542–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bjerknes M, Cheng H. Multipotential stem cells in adult mouse gastric epithelium. Am J Physiol Gastrointest Liver Physiol 2002;283:G767–77. [DOI] [PubMed] [Google Scholar]
- 46.Tatematsu M, Fukami H, Yamamoto M, et al. Clonal analysis of glandular stomach carcinogenesis in C3H/HeN<==>BALB/c chimeric mice treated with N-methyl-N-nitrosourea. Cancer Lett 1994;83:37–42. [DOI] [PubMed] [Google Scholar]
- 47.Nomura S, Esumi H, Job C, et al. Lineage and clonal development of gastric glands. Dev Biol 1998;204:124–35. [DOI] [PubMed] [Google Scholar]
- 48.McDonald SA, Greaves LC, Gutierrez-Gonzalez L, et al. Mechanisms of field cancerization in the human stomach: the expansion and spread of mutated gastric stem cells. Gastroenterology 2008;134:500–10. [DOI] [PubMed] [Google Scholar]
- 49.Hayakawa Y, Sethi N, Sepulveda AR, et al. Oesophageal adenocarcinoma and gastric cancer: should we mind the gap? Nat Rev Cancer 2016;16:305–18. [DOI] [PubMed] [Google Scholar]
- 50.Schwitalla S, Fingerle AA, Cammareri P, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013;152:25–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.