

Abstract
Membrane-activity of the glycoprotein 41 membrane-proximal external region (MPER) is required for HIV-1 membrane fusion. Consequently, its inhibition results in viral neutralization by the antibody 4E10. Previous studies suggested that MPER might act during fusion by locally perturbing the viral membrane, i.e., following a mechanism similar to that proposed for certain antimicrobial peptides. Here, we explore the molecular mechanism of how MPER permeates lipid monolayers containing cholesterol, a main component of the viral envelope, using grazing incidence X-ray diffraction and X-ray reflectivity. Our studies reveal that helical MPER forms lytic pores under conditions not affecting the lateral packing order of lipids. Moreover, we observe an increment of the surface area occupied by MPER helices in cholesterol-enriched membranes, which correlates with an enhancement of the 4E10 epitope accessibility in lipid vesicles. Thus, our data support the view that curvature generation by MPER hydrophobic insertion into the viral membrane is functionally more relevant than lipid packing disruption.
Keywords: HIV gp41, MPER–lipid interaction, X-ray scattering, Lytic pore, Membrane curvature
1. Introduction
The human immunodeficiency virus type-1 (HIV-1) enters into host cells upon fusion of viral and cellular membranes induced by the envelope glycoprotein (Env) transmembrane gp41 subunit [1,2]. This fusion event depends on the concerted action of two different gp41 ectodomain structural elements: the six-helix bundle (6-HB), a helical domain that opens and closes, and two membrane-transferring hydrophobic regions, the N-terminal fusion peptide (FP) and the C-terminal membrane-proximal external region (MPER), which are postulated to insert into the cell target and viral membranes, respectively [3–5]. The most widely accepted mechanism postulates that following FP and MPER insertion, folding into the energetically stable 6-HB brings cell and viral membranes to close apposition.
Compelling mutational analysis by Salzwedel and co-workers [6] provided the first evidence to support MPER involvement in HIV-1 fusion. MPER is enriched in conserved aromatic residues that mediate its favorable partitioning from water into the membrane interface [6–9] (Fig. 1A). Peptide-based structural and functional analyses are consistent with MPER insertion into the viral membrane external monolayer as an α-helix [10–12] (Fig. 1B), which would be capable of destabilizing the lipid bilayer organization during fusion [13,14]. It has been proposed that the C-terminal “cholesterol recognition/interaction amino acid consensus” LWYIK sequence and the following transmembrane domain residues may contribute to MPER interactions with the viral membrane interface [9,15]. The functional role played in viral entry by this conserved domain is additionally supported by the fact that anti-MPER neutralizing antibodies, such as 4E10, have evolved mechanisms to recognize membrane-inserted epitopes and block MPER membrane activity [12,16–19].
Fig. 1.
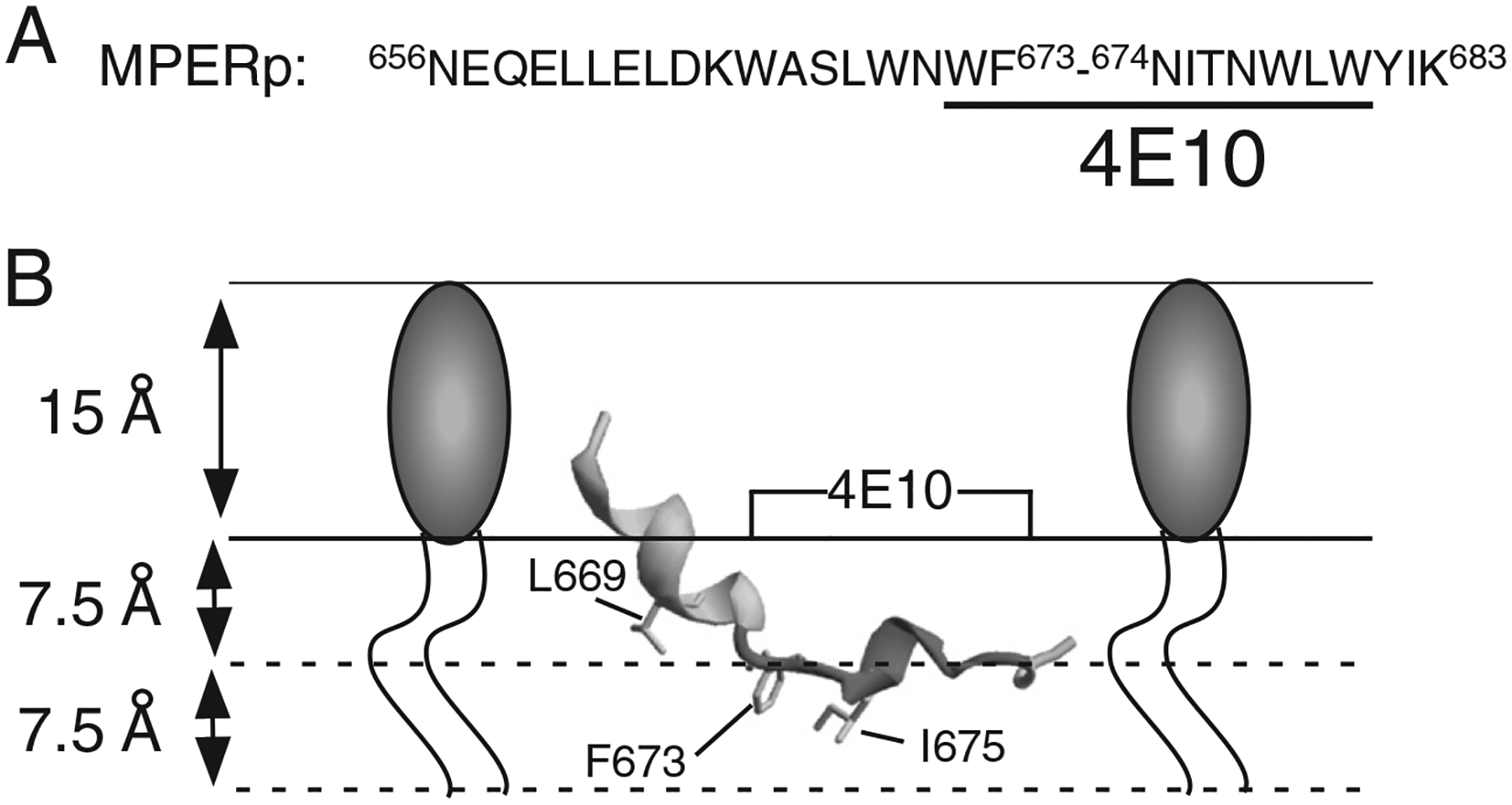
HIV MPER designation and model for its association with membranes Panel A: sequence of MPER peptide used in this study. 4E10 epitope residues underlined. Numbering is based on the prototypic HXBc2 viral isolate. Panel B: model for the cognate ELDKWASLWNWFNITNWLWYIK peptide in association with a membrane monolayer. The structure adopted in detergent micelles was obtained from the Protein Data Bank (PDB ID: 2PV6) and rendered using Swiss-PDB-viewer. The insertion depths for the depicted residues L669, F673 and I675 are based on electro paramagnetic spectroscopy determinations [12].
Mutagenesis studies corroborate that MPER insertion is not only a structure-related pattern, but also a requirement for the fulfillment of the membrane-disrupting function during fusion [20]. In those studies, MPER was replaced with sequences based on the Trp-rich, antimicrobial peptide indolicidin. Some of the gp41 mutants retained activity, thereby suggesting that MPER might disrupt lipid packing following a mechanism similar to that proposed for antimicrobial peptides [21–24]. Alternatively, it has been postulated that the shallow insertion of MPER into the envelope external leaflet before, during, or even after 6-HB formation, might poise the viral membrane for fusion [10,25]. Specifically, MPER asymmetric insertion into one leaflet might generate the bulging out of the viral membrane, while the curved end-caps of such bulges would be highly fusogenic [26].
Here, we combine structural analyses of the MPER peptide in lipid vesicles and monolayers to investigate the molecular mechanism of MPER-induced membrane perturbation and its physiological relevance. Our results reveal that MPER does not affect the lateral packing order of lipids, but changes its membrane insertion depth and topology in cholesterol-enriched membranes. This phenomenon correlates with an increment of the surface area occupied by MPER helices, and the optimal exposure of the 4E10 epitope. We conclude that insertion of helical MPER into the Chol-enriched viral envelope may modulate bilayer curvature, rather than cause bilayer rupture.
2. Materials and methods
2.1. Materials
The MPER-derived NEQELLELDKWASLWNWFNITNWLWYIK (MPERp) peptide (Fig. 1A) was produced by solid-phase synthesis using Fmoc chemistry as C-terminal carboxamides and purified by HPLC. 1-palmitoyl-2-oleoyl-sn-glycero-3-phophocholine (POPC), 1,2-dipalmitoyl-sn-glycero-3-phophocholine (DPPC) and Cholesterol (Chol) were purchased from Avanti Polar Lipids (Birmingham, AL, USA). The N-(5-dimethylaminonaphtalene-1-sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (d-DHPE), 8-aminonaphtalene-1, 3,6-trisulfonic acid sodium salt (ANTS) and p-xylenebis(pyridinium) bromide (DPX) were obtained from Molecular Probes (Junction City, OR, USA). Monoclonal 4E10 antibody (MAb4E10) was kindly donated by D. Katinger (Polynum Inc., Vienna, Austria).
2.2. Circular dichroism
Circular dichroism (CD) measurements were obtained from a thermally-controlled Jasco J-810 circular dichroism spectropolarimeter calibrated routinely with (1S)-(+)-10-camphorsulfonic acid, ammonium salt. Samples consisted of co-lyophilized peptide and lipid dissolved and sonicated in 2 mM Hepes (pH, 7.4) buffer. Spectra of the reconstituted peptide-containing vesicles were measured in a 1 mm path-length quartz cell initially equilibrated at 25 °C. Data were taken with a 1 nm band-width at 100 nm/min speed, and the results of 20 scans were averaged.
2.3. Lipid vesicle assays
Large unilamellar vesicles (LUV) were prepared according to the extrusion method in 5 mM Hepes, 100 mM NaCl (pH 7.4) using membranes with a nominal pore-size of 0.1 μm. Distribution of sizes, estimated by quasielastic light scattering using a Malvern Zeta-Sizer Nano ZS instrument (Malvern Instruments, Malvern, UK), revealed mean diameters ranging between 110 and 120 nm for the vesicles used in the experiments. The vesicle size distribution did not significantly change upon the addition of peptide at the highest tested doses (i.e., 1:100 peptide-to-lipid ratio). Chol content in vesicles was determined after extrusion by the cholesterol oxidase/peroxidase method (BioSystems, Barcelona, Spain) and found to be within the experimental error.
Vesicle permeabilization was monitored following the release to the medium of encapsulated fluorescent ANTS (ANTS/DPX assay [27]). LUV containing 12.5 mM ANTS, 45 mM DPX, 20 mM NaCl and 5 mM Hepes were obtained by separating the unencapsulated material by gel-filtration in a Sephadex G-75 column that was eluted with 5 mM Hepes and 100 mM NaCl (pH 7.4). Fluorescence measurements were performed by setting the ANTS emission at 520 nm and the excitation at 355 nm. A cutoff filter (470 nm) was placed between the sample and the emission monochromator. The baseline leakage (0%) corresponded to the fluorescence of the vesicles at time 0, while 100% leakage was the fluorescence value obtained after addition of Triton X-100 (0.5% v/v).
Partitioning into the membrane interface was measured as a function of time by energy transfer from the Trp peptide to the surface d-DHPE fluorescent probe as in reference [16]. In brief, 6 mol% of the d-DHPE probe was included in the target vesicle composition and its fluorescence was measured at an emission wavelength of 510 nm, while the excitation wavelength was that of the Trp residue (280 nm).
2.4. Grazing incidence X-ray diffraction (GIXD) and X-ray reflectivity (XR)
Both GIXD and XR are well-established techniques for studying Langmuir monolayers at the air–liquid interface. Liquid surface X-ray scattering experiments were performed at the 9-ID beam line at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL). The liquid surface spectrometer (LSS) and Langmuir trough chamber have previously been described [28]. The resolution for the in-plane scattering angle θXY was set to 1.4 mrad (9.56×10–3 Å−1) by a Soller collimator. This corresponds to an area uncertainty of 0.027 Å2. Langmuir monolayers composed of DPPC/cholesterol with 13 and 46 mol% of cholesterol were used to mimic the virion membrane with depleted and regular content of cholesterol, respectively. Lipid monolayers were formed by depositing droplets of respective solution at the air–liquid interface and after equilibrating for 15 min compressed to the surface pressure of 20 mN/m. The solution of a peptide was then evenly injected underneath the monolayers using a micro-syringe with an L-shaped needle to make up the final concentration of 0.57 μM, while the surface pressure was kept constant via proportional-integral-derivative feedback control. Injected peptides interact with the lipid monolayer and result in an increase in the surface pressure when incorporated into the film. To keep the surface pressure constant, the surface area would have to increase. The resulting relative change in mean molecular area, ΔA/A, was monitored for up to 180 min after insertion. The experiments were carried out on Dulbecco’s phosphate buffered saline without calcium and magnesium (D-PBS) (Invitrogen) at a room temperature of 22±1 °C.
The R&K trough (Riegler & Kirstein GmbH) sealed in an air-tight canister was flushed with humidified helium for X-ray measurements to reduce X-ray absorption and sample damage. The wavelength of the beam of λ=0.92017 Å was set by a cryogenically cooled Si (111) double-crystal monochromator (Kohzu Seiki Co. Ltd.). A split ion chamber monitor and feedback control of the second crystal of the Kohzu monochromator maintained the position of the beam at the monitor (9 m before the sample). Ge (111) steering crystal was used to define the striking angle of the beam onto liquid surface. Incident slits were 2 mm wide and 50 μm high; detector slits were 2×2 mm2.
In reflectivity experiments, scattering intensity is collected as a function of the out-of-plane scattering vector qZ=(2π/λ)sinαf, αf being the angle of the diffraction beam with the horizontal plane, using a single-channel scintillation detector. XR data were analyzed using both model-dependent (MD) “slab” model refinement [22,23,29] and model-independent (MI) stochastic fitting routines employing RFIT2000 (Oleg Konovalov, ESRF) and StochFit software, respectively.
To improve sensitivity of X-ray diffraction to the interface, the depth of beam penetration is limited by adjusting the incident angle αi to 0.85αc, with αc being the critical angle for total reflection. A scattering intensity was collected using linear position-sensitive Mythen detector. A scan over a range of qxy integrated over qz yields Bragg peaks of a 2D periodic structure. A position of the Bragg peak maximum defines a repeat distance dhk of the 2D lattice structure. The coherence length of the crystallinities can be estimated from the full-width at half-maximum (FWHM) of the Bragg peaks using the Scherrer formula L=0.9×2π/FWHM.
3. Results
3.1. MPERp structure and activity in Chol-containing vesicles
The synthetic sequence used in this study, MPERp can be considered as the canonical MPER peptide (Fig. 1A). The model for the structure of a cognate peptide in membranes (Fig. 1B) displays a kinked structure with the N-terminus protruding from the bilayer surface and pre-dominantly exposed to the aqueous phase, while the C-terminal region containing the 4E10 epitope remains essentially buried in the membrane [12]. Such model was rendered combining the nuclear magnetic resonance structure of the peptide dissolved in detergent, with electro paramagnetic spectroscopy determinations of insertion depths in bilayers, which included anionic phospholipids [12]. However, lipidomic analyses reveal that Chol concentrates at the HIV-1 envelope (ca. 45 mol%, [30]). These high concentrations seem to reflect a functional requirement for cell entry, since interference with [31,32], or depletion of this compound [33,34], abolishes HIV infectivity. Hence, it is inferred that MPER membrane activity, and its inhibition by the 4E10 antibody, evolve in the context of this Chol-enriched membrane. Moreover, Chol modifies the membrane-interacting properties of peptides [35], and, therefore, is predicted to exert a physiologically relevant modulation of MPER secondary structure, orientation and/or insertion level, as well as of its purported membrane-perturbing activity.
The circular dichroism (CD) analysis displayed in Fig. 2A indicate that the main secondary structure adopted in contact with membranes by MPERp was similar upon inclusion of high quantities of Chol. The CD spectra showed major band components at 208 and 222 nm for MPERp in POPC, POPC:Chol (2:1, molar ratio) and POPC: Chol (1:1, molar ratio) vesicles, therefore indicating that an overall helical conformation was preserved in all cases. In addition, the lower absorption suggests that non-helical conformations (strand/coil) may contribute more to the peptide structure in the latter samples. The leakage data revealed the existence of different membrane activities for these helical structures (Fig. 2B). The leakage process exhibited a transition from slow to fast kinetics upon inclusion of Chol in the lipid composition (solid lines). As judged from the energy transfer from Trp-s to membrane-interface residing dansyl moieties (dotted lines), the slower leakage observed for vesicles devoid of Chol was not a consequence of a reduction in the rate of peptide incorporation into vesicles.
Fig. 2.
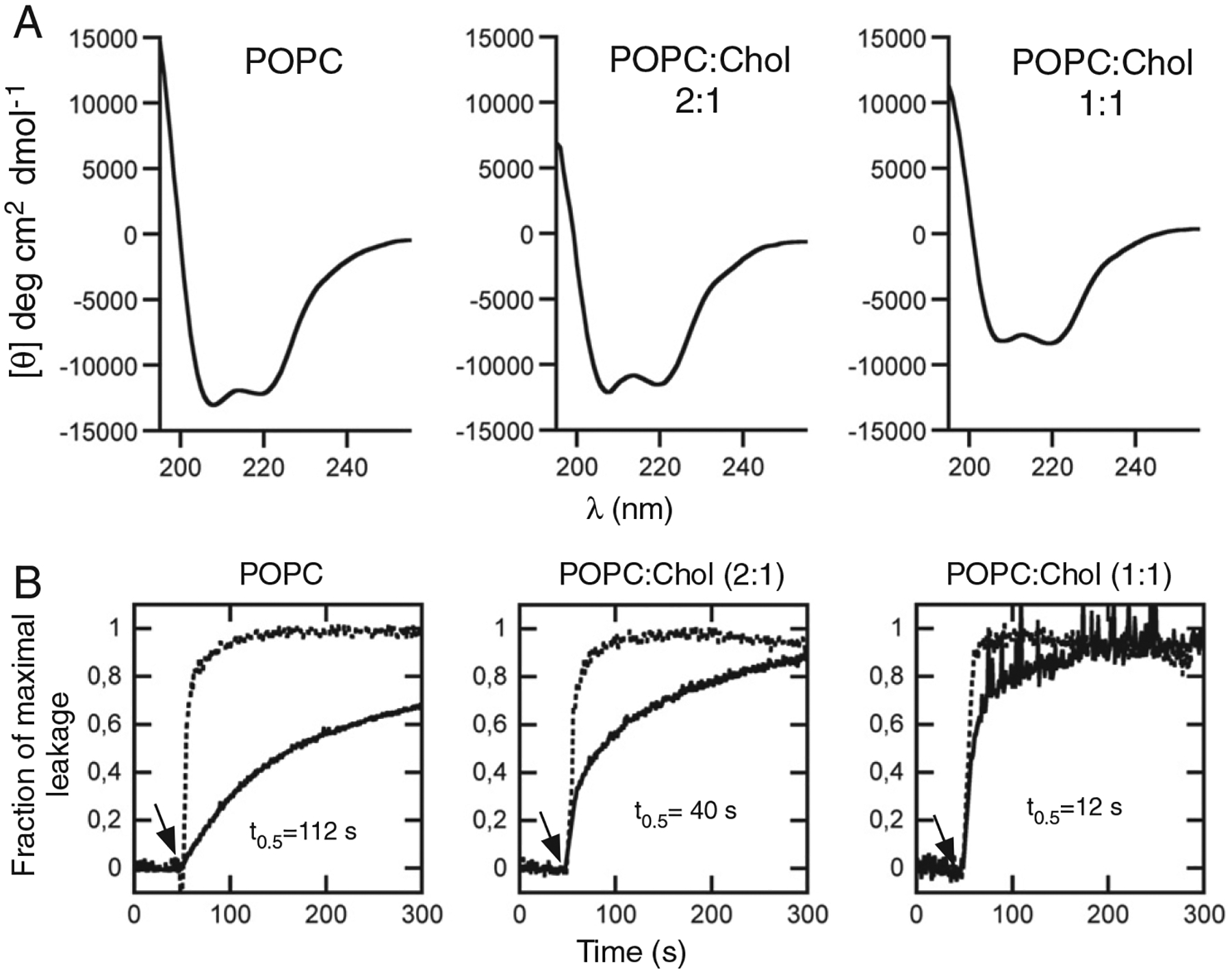
Secondary structure and MPERp activity as a function of the Chol content in the bilayer. A) CD spectra of MPERp in association with PC vesicles containing different Chol mole ratios as indicated in the panels. The lipid and peptide concentrations were 1 mM and 30 μM, respectively. B) Effect of Chol on MPERp-induced ANTS leakage kinetics. The peptide was added to a vesicle suspension (100 μM lipid) at the time indicated by the arrow (t=50 s). The peptide-to-lipid ratio was 1:150. Chol mole fractions are indicated for each curve. The dotted traces follow peptide incorporation into the vesicles monitored through energy transfer from tryptophans to membrane-residing d-DHPE.
3.2. Capacity for perturbing lipid packing in phospholipid monolayers containing Chol
The use of Langmuir monolayers allows us to imitate the in vivo situation when MPER comes in contact with the outer leaflet of the virion membrane. The GIXD technique provides direct structural information on a nanoscale lateral molecular order in Langmuir monolayers and has been earlier applied to the analysis of membrane permeabilization mechanisms by antimicrobial peptides [22,23,29,36,37]. Particularly, GIXD measurements of peptide-containing phospholipid monolayers disclosed the capacity of LL-37 or protegrin for selectively disrupting the structure of monolayers made of anionic phospholipid [22,23]. Hence, to gain insights into the mechanism of MPER-induced membrane perturbation and its modulation by Chol, we performed GIXD analyses of phospholipid monolayers with low and high levels of Chol, in the presence and absence of MPERp (Fig. 3 and Table 1).
Fig. 3.
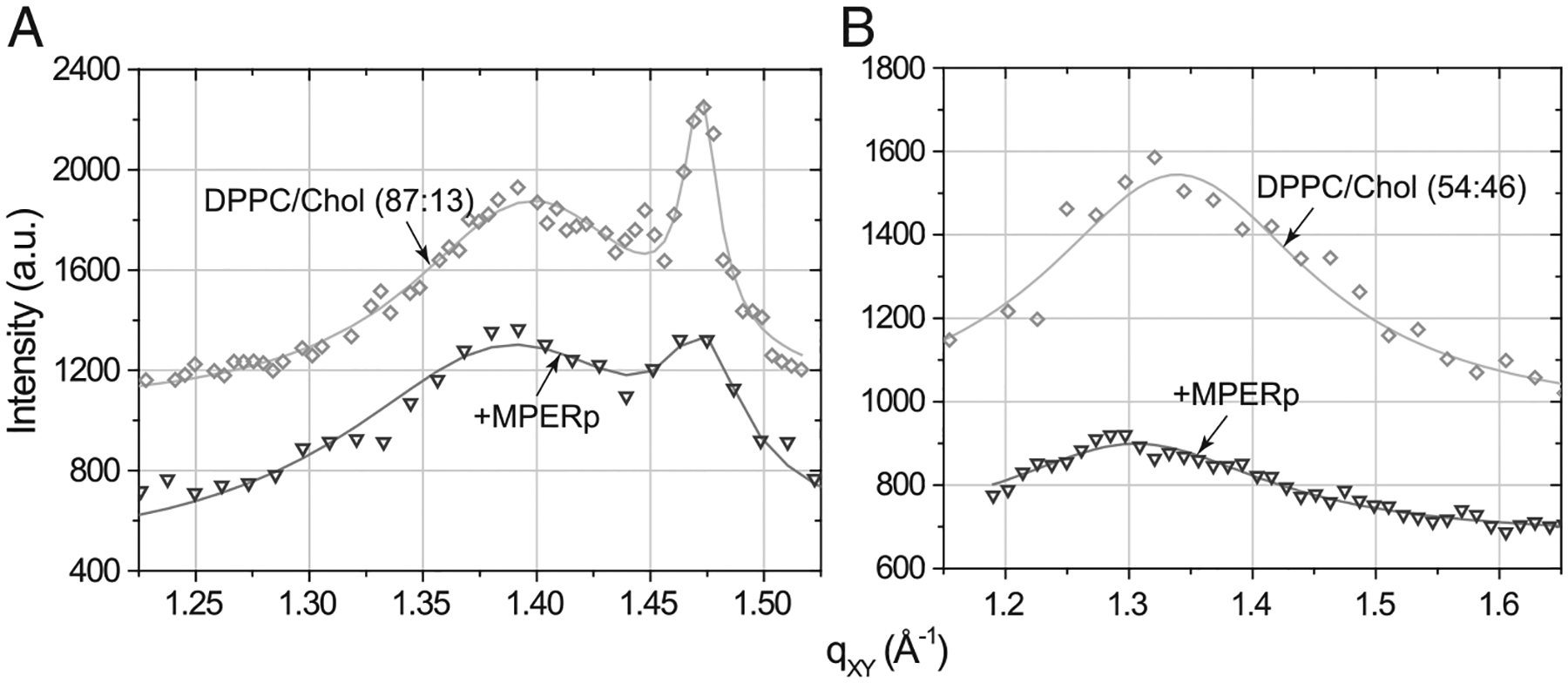
Grazing incidence X-ray diffraction data (symbols) and corresponding fits (lines): scattering intensity, integrated over qZ range, against scattering vector qXY of (A) DPPC/Chol (87:13, molar ratio) monolayer before (rhombs) and after MPERp (inverted triangles) injection; (B) DPPC/Chol (54:46, molar ratio) monolayer before (rhombs) and after MPERp (inverted triangles) injection.
Table 1.
Grazing incidence X-ray diffraction data.
| Experiment | d-Spacing, Å | Unit cell parameters a, b (Å), γ (°), and area A (Å2) | Coherence length (Å) |
|---|---|---|---|
| 13:87 |
d(1,1),(1,−1)=4.55 d(0,2)=4.30 |
a=5.37, b=8.6, γ=90, A=46.2 |
L(1,1),(1,−1)=52 L(0,2)=301 |
| + MPERp |
d(1,1),(1,−1)=4.53 d(0,2)=4.27 |
a=5.34, b=8.54, γ=90, A= 45.6 |
L(1,1),(1,−1)=36 L(0,2)=174 |
| 46:54 | 4.69 |
a=b=5.41, γ=120, A=25.3 |
58 |
| + MPERp | 4.81 |
a=b=5.55, γ=120, A=26.7 |
23 |
Using constant-pressure insertion assays we first tested the propensity of the peptide to incorporate into DPPC/cholesterol (87:13, molar ratio) and (54:46, molar ratio) mixed monolayers at the constant surface pressure of 20 mN/m. The final relative increase in mean molecular area, ΔA/A, upon introduction of MPERp comprised 22% in DPPC/cholesterol (87:13) monolayer and 71% in DPPC/cholesterol (54:46) film. GIXD analyses of these films revealed that inserted MPERp was not capable of disrupting the in-plane order of the mixed monolayers completely, as follows from the preserved diffraction peaks (Fig. 4). This is in contrast to what was found for LL-37 [22], which induced disappearing of Bragg peaks, consistent with lipid packing disruption by the peptide under conditions of effective membrane permeabilization.
Fig. 4.
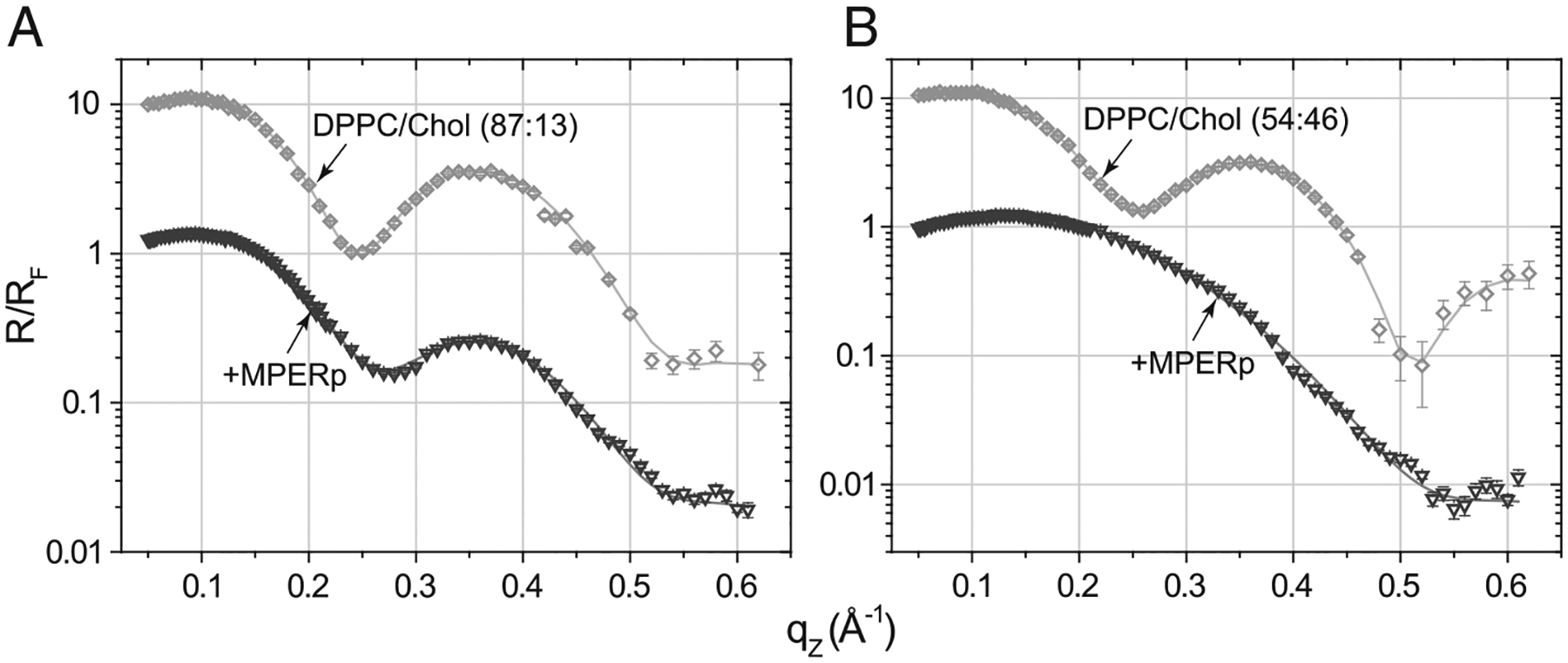
X-ray reflectivity data (symbols) and corresponding fits (lines) normalized by Fresnel reflectivity plotted against scattering vector qZ of (A) DPPC/Chol (87:13, molar ratio) monolayer before (rhombs) and after MPERp (inverted triangles) injection; (B) DPPC/Chol (54:46, molar ratio) monolayer before (rhombs) and after MPERp (inverted triangles) injection.
Data in Table 1 further indicate that MPERp’s insertion into (54:46) mixture results in the increased unit cell area, whereas insertion into (87:13) mixed film leads to an opposite effect on the unit cell area. The diffraction of DPPC/cholesterol monolayers is due to the lipid hydrophobic region spanning DPPC acyl chains and cholesterol ring body. Peptide insertion into the headgroup region would create voids in the monolayer hydrophobic region resulting in a less dense packing of the acyl chains and cholesterol molecules in the ordered domains. Therefore, an increase in the unit cell area would be consistent with MPERp insertion mainly taking place at the lipid headgroups of the (54:46) mixture. On the other hand, deep peptide penetration into the film hydrophobic core may create additional stress on the lipid acyl chains and force them to pack denser. Thus, the GIXD data indirectly support deeper MPERp insertion into the (87:13) mixture.
3.3. Insertion depth and topology in phospholipid monolayers containing Chol
Changes in MPERp’s depth of penetration were directly measured by X-ray reflection measurements (XR) of the films. Analysis of XR data yields information on the electron density distribution in a monolayer in a direction perpendicular to the interface ρ(z) averaged over beam footprint and thus over ordered and disordered regions of the film. Changes in ρ(z) after introduction of MPERp can be related to a depth of membrane insertion, orientation within a film, and interfacial concentration, lipid-to-peptide ratio, of the peptide (Table 2). Fig. 4 displays the reflectivity curves from the monolayers before and after introduction of MPERp. The XR curves for the films were fitted using a previously introduced three-slab model for Chol-containing monolayers [28]. Following the notation in Table 2, the DPPCAC+CHOLAC slab corresponds to the layer closest to the air, which includes the acyl chains, the DPPCAC+CHOLRB slab denotes an intermediate region that includes the rest of DPPC acyl chains and the cholesterol rigid-ring body structure, and the DPPCHG+CHOLHG slab corresponds to the bottom layer closest to aqueous buffer comprising the headgroups. Each of these slabs is characterized by an individual electron density (ρi) and thickness (Li) (Table 2). Variation of the number of extra electrons (e−extra) among the different slabs indicates that MPERp peptide penetrated deep into the hydrophobic core of DPPC/cholesterol (87:13) film (most of the extra electrons are within the 1st and 2nd slabs). However, when cholesterol concentration was increased to 46 mol%, the number of electrons increased significantly at the headgroup region (3rd slab), consistent with a shallow insertion of MPERp into the film. In addition, the surface area occupied by each peptide (Apeptide) was comparatively higher in this system (Table 2). The lower penetration and the higher surface occupancy in the (54:46) monolayer were matched by a peptide-induced decrease of the monolayer’s thickness (LT). Such thickness reduction actually occurred within the DPPCAC+CHOLAC and, DPPCAC+CHOLRB regions (compare Li values).
Table 2.
Results of X-ray reflectivity data analysis.
| Slab | Li (Å) | ρi (e−/Å3)exp | ρi (e−/Å3)est | e−extra | e−extra total | σ (Å) | LT (Å) | A (Å2) | L:P 100 | L:P 50 | Apeptide (Å2) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DPPC/cholesterol (87:13) | |||||||||||
| DPPCAC+CHOLAC | 7.9 | 0.311 | NA | NA | NA | 3.3±0.2 | 24.3 | 47.7 | NA | NA | NA |
| DPPCAC+CHOLRB | 7.5 | 0.331 | |||||||||
| DPPCHG+CHOLHG | 8.9 | 0.424 | |||||||||
| DPPC/cholesterol (87:13)+MPERp | |||||||||||
| DPPCAC+CHOLAC+MPERp | 8.3 | 0.301 | 0.243 | 28 | 71.9 | 3.3±0.1 | 23.6 | 58.2 | 36:1 | 32:1 | 336–378 |
| DPPCAC+CHOLRB+MPERp | 7.6 | 0.359 | 0.268 | 40.3 | |||||||
| DPPCHG+CHOLHG+MPERp | 7.7 | 0.410 | 0.402 | 3.6 | |||||||
| DPPC/cholesterol (54:46) | |||||||||||
| DPPCAC+CHOLAC | 7.5 | 0.281 | NA | NA | NA | 3.1±0.2 | 24.5 | 40.9 | NA | ||
| DPPCAC+CHOLRB | 9.9 | 0.359 | |||||||||
| DPPCHG+CHOLHG | 7.1 | 0.417 | |||||||||
| DPPC/cholesterol (54:46)+MPERp | |||||||||||
| DPPCAC+CHOLAC+MPERp | 5.7 | 0.26 | 0.216 | 17.5 | 176.5 | 3.2±0.4 | 20.5 | 69.9 | 15:1 | 13:1 | 377–435 |
| DPPCAC+CHOLRB+MPERp | 6.8 | 0.463 | 0.306 | 74.6 | |||||||
| DPPCHG+CHOLHG+MPERp | 8.0 | 0.367 | 0.216 | 84.4 | |||||||
AC — acyl chains; RB — ring body of cholesterol; HG — headgroups; L — slab thickness; ρexp — experimental average electron density of a slab; ρest — estimated average electron density of a slab; e−extra — number of extra electrons in a slab; e−extra total — total number of extra electrons in a film; LT — total thickness of the film; A — area per an average lipid molecule; L:P100 and L:P50 — lipid-to-peptide ratio assuming 100% and 50% hydration of peptide’s polar groups; Apeptide — estimated area per a peptide molecule.
3.4. Chol effect on 4E10 epitope accessibility
The previous monolayer results suggest that MPERp topology was significantly altered upon inclusion of Chol. The physiological relevance of this phenomenon was further tested using the 4E10 antibody (Fig. 5).
Fig. 5.
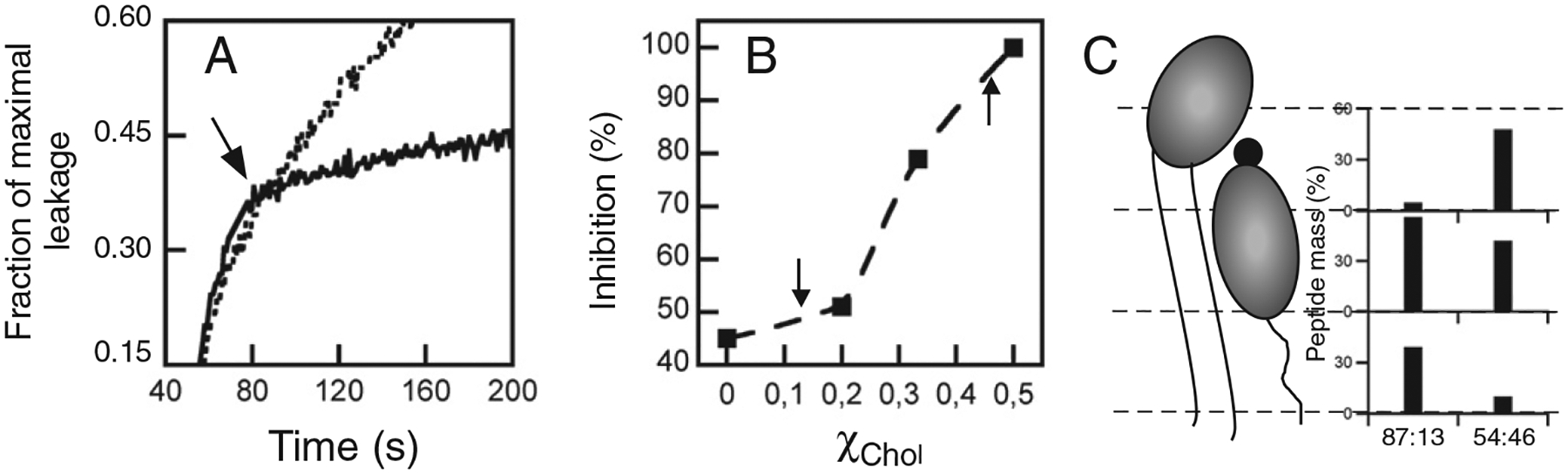
Inhibition of MPERp-induced vesicle contents leakage by 4E10. A) Effect of antibody addition to the ongoing leakage. POPC:Chol (2:1, molar ratio) vesicle samples (100 μM lipid) were treated with 1 μM peptide and, subsequently supplemented with 10 μg/ml of 4E10 (addition time indicated by the arrow). The dotted traces follow the leakage kinetics in the absence of antibody. B) 4E10-induced inhibition percentages plotted as a function of the Chol mole fraction. Rate reduction caused by antibody with respect to the leakage control without antibody was calculated by correcting 0% extent of leakage to the time point of antibody addition, and subsequently measuring increment of leakage after 20 s in both samples. C) Peptide mass percentage distribution between the monolayer slabs under the experimental conditions used for X-ray scattering assays (indicated by the arrows in the previous panel). Preferential location of the peptide into the HG slab correlates with better 4E10 epitope recognition-blocking.
This antibody specifically recognizes the C-terminal sequence of MPER (Fig. 1), and is capable of arresting peptide-induced membrane permeabilization (Fig. 5A) [16,19,38]. As shown in Fig. 5B, the inhibitory effect of the antibody increased upon inclusion of Chol in the membrane composition. The data also show a correlation between antibody effectiveness and MPERp insertion level into the monolayers (Fig. 5B and C). Thus, results displayed in this figure demonstrate a Chol-induced enhancement of 4E10 epitope accessibility, which correlates with a preferential location of the MPERp within the headgroup region of the membrane.
4. Discussion
A widely accepted model proposes that MPER might help in remodeling of the merging membranes along the HIV-1 fusion pathway by sustaining transient disruption of lipid continuity [5,7,20,39]. Alternatively, it has been suggested that MPER hydrophobic insertion into one monolayer might alter the bilayer elastic properties, thereby priming the viral membrane for fusion [26,40]. In this work, using Langmuir lipid monolayers and X-ray scattering techniques, we have investigated the mechanism underlying the membrane activity of MPER and evaluated the effect exerted by high Chol levels. The fact that the peptide–lipid interactions that we measured were indeed modulated by Chol underscores the physiological relevance of our findings.
In a previous single-vesicle study [41], we found that MPERp (termed as NpreTM in that study) induced transient (“graded”) permeabilization of giant unilamellar vesicles (GUVs) made of pure POPC. This phenomenon was characterized by slow kinetics and resulted in most instances in partial permeabilization of the individual vesicles. Notably, inclusion of Chol in the lipid composition increased the fraction of the vesicle population permeabilized according to an “all-or-none” mechanism, a process characterized by fast kinetics and total permeabilization of vesicles. Results in Fig. 2 suggest that a similar Chol-dependent change in the permeabilization mechanism occurred within the bulk vesicle population, but that such phenomenon was not due to an alteration of the overall helical conformation adopted by the peptide. In the case of the POPC:Chol (1:1 molar ratio) vesicles, the decrease in CD absorption further suggests that an increase of MPERp flexibility might contribute to enhanced permeability rate.
Our X-ray scattering experiments provided a molecular mechanism to explain Chol effects on MPERp membrane activity. The GIXD data displayed in Fig. 3 and Table 1 reveal that, in contrast to what is observed for antimicrobial peptides [22,23], MPERp did not disrupt the integrity of the monolayers. Thus, MPER peptide did not seem to perturb the organization of lipids according to the mechanisms proposed for antimicrobial peptides [21,24,42]. Furthermore, X-ray reflectivity (XR) experiments of lipid monolayers revealed that Chol stimulated MPERp reorientation (Fig. 4 and Table 2). The peptide was located deeply into the acyl-chain region with low Chol content (>8 Å), while insertion was preferentially at the head-group region with high Chol content (≤8 Å) (Fig. 5 and Table 2). Thus, our data are consistent with transient or stable lipidic pores being assembled by MPERp preferentially inserting at the level of the acyl chain or head-group regions, respectively [41]. Chol-promoted relocation was accompanied by increases of the unit cell area, and of the area occupied by the peptide, and a reduction of the monolayer thickness (Tables 1 and 2). Moreover, the process resulted in better exposure of the 4E10 epitope at the surface of membranes (Fig. 5, and model depicted in Fig. 6).
Fig. 6.
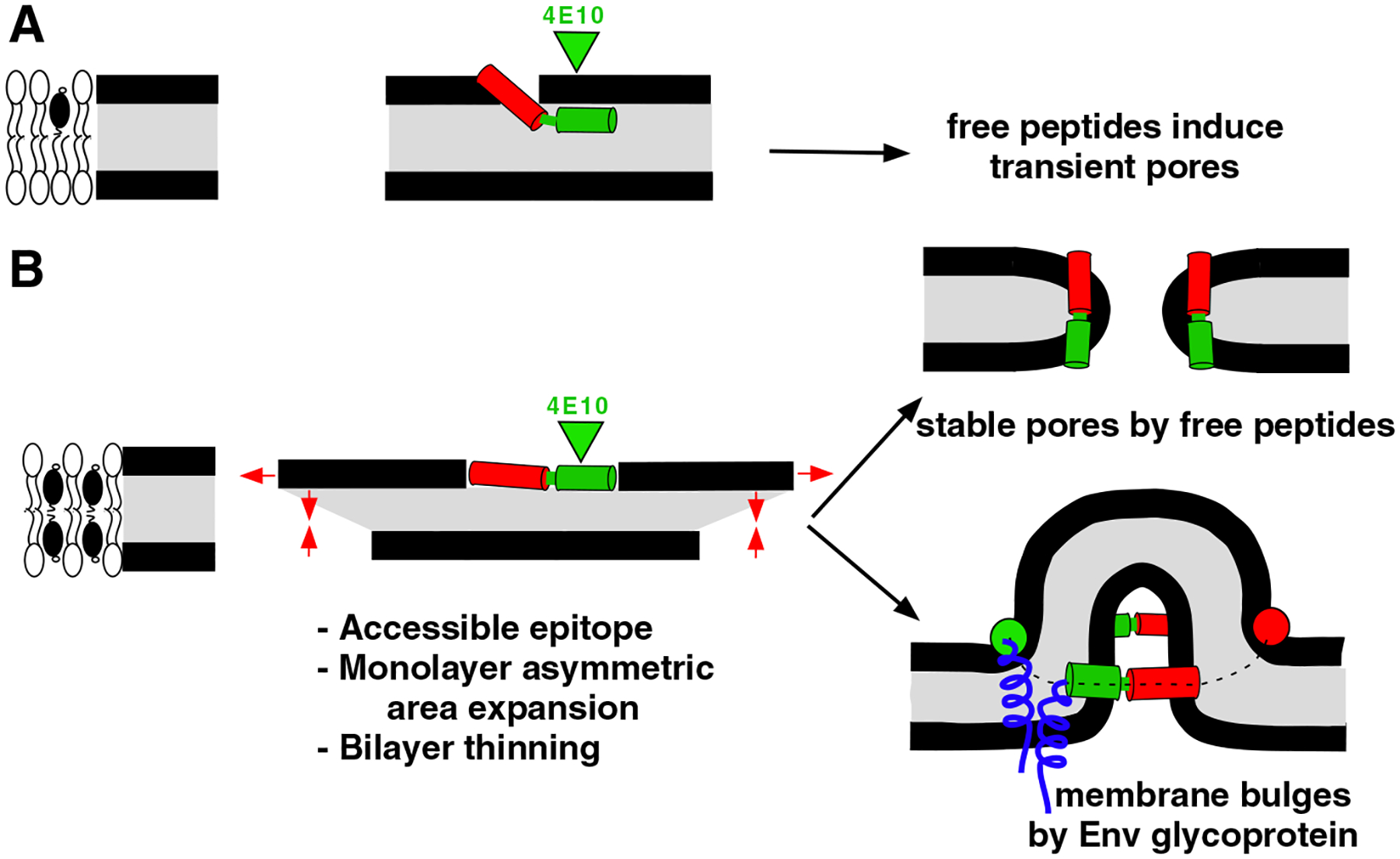
Model to explain MPER-induced bilayer perturbation and its dependence on Chol. A) When Chol levels are low the peptide penetrates deeper into the monolayer, the accessibility to 4E10 epitope is hindered and the free peptides induce transient permeabilization of the bilayer [41]. B) Chol levels as those existing at the viral envelope lead to shallower MPER insertion and increased accessibility to the 4E10 epitope. Moreover, the surface occupied by each peptide increases and the monolayer thickness decreases. We surmise that the elastic stress generated in bilayers by the expansion of one monolayer can be relaxed in two ways: 1) free peptides may generate toroidal, stable aqueous pores [41,43]; and 2) in the context of the viral gp41, transmembrane domains lock MPER sequences into a ring-like configuration at the membrane interface. Experimental evidence for involvement of 5–7 trimers at the fusion site has been obtained by electron tomography [49]. Elastic stress is released in this case through the formation of the protruding bulges (see reference [26] for a discussion on membrane fusion driven by curvature generation).
Area expansion and concomitant monolayer thinning are in line with the idea that, in the presence of Chol, pore formation by MPERp primarily results from the asymmetric increase of the monolayer surface and further generation of elastic stresses in the bilayer [43–45] (Fig. 6). We surmise that the accumulated elastic energy, which dissipates through pore-formation in vesicles [43], can be coupled to membrane merger in the context of the Env glycoprotein complexes (see caption for Fig. 6). In that regard our results would sustain previous Kozlov’s model, which postulates that MPER insertion into the external monolayer of the viral membrane could generate curvature, even when this element is in great part recruited into the 6-HB [25]. According to these authors Trp 678, Trp 680 and Tyr 681 of the gp41, are exposed toward the membrane and well positioned to insert their side chains into the bilayer. Using a model for membrane bending by hydrophobic insertions [45], one can estimate the membrane curvature that these MPER residues generate by a shallow insertion. One gp41 chain produces local curvature of ~0.65 nm−1; thus a gp41 trimer might stabilize a membrane cylinder of about 15 nm diameter, which would facilitate fusion considerably.
Reconstructions of native Env glycoprotein particles either at the viral surface of intact virions [46], or purified and detergent-stabilized in solution [47], suggest that MPER might insert into the viral envelope also in a native pre-fusion state. In the context of the native Env “tripod” model derived from those studies, MPER would submerge into the external membrane monolayer as an independent element [46,48]. Our XR data suggest that MPER domain might potentially occupy larger membrane areas under those conditions (Fig. 4 and Table 2). Particularly, the canonical MPERp sequence might occupy an area three times larger than that calculated for the MPER region exposed in the 6-HB [25], and this area would further increase in the presence of high Chol concentrations (Table 2). Thus, our experimental results complement Kozlov’s model [25] by establishing the putative pre-fusion conditions required for optimal induction of membrane deformations by MPER inserted into the viral envelope. Firstly, they identify the complete MPER sequence as an important determinant of curvature generation. Secondly, they confirm that the high Chol content of the viral envelope ensures a more favorable level of insertion. Lower Chol concentrations would result in deeper insertion of this element, reduced interfacial occupancy and, consequently, limited thinning of the external monolayer. These effects would translate into a lower capacity for generating curvature at the viral envelope. Together, our observations would be consistent with the putative adaptation of MPER for actively deforming the Chol-enriched viral envelope as an element of a “tripod”-like structure [46,48].
Acknowledgements
We acknowledge the financial support of the Spanish “Ministerio de Economia y Competitividad”, Basque Government and the University of the Basque Country (BIO2011-29792 and GIU 06/42 grants to JLN). BA was recipient of a pre-doctoral fellowship of the Spanish MEU. D.G. was supported by the National Institutes of Health R01 AI073892 and DARPA W911NF-09-1-378 grants. The authors are indebted to Binhua Lin and Mati Meron for their help with X-ray measurements at APS. ChemMatCARS Sector 15 is principally supported by the National Science Foundation/Department of Energy under grant number NSF/CHE-0822838. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
References
- [1].Gallo SA, Finnegan CM, Viard M, Raviv Y, Dimitrov A, Rawat SS, Puri A, Durell S, Blumenthal R, The HIV Env-mediated fusion reaction, Biochim. Biophys. Acta 1614 (2003) 36–50. [DOI] [PubMed] [Google Scholar]
- [2].Melikyan GB, Common principles and intermediates of viral protein-mediated fusion: the HIV-1 paradigm, Retrovirology 5 (2008) 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Peisajovich SG, Shai Y, Viral fusion proteins: multiple regions contribute to membrane fusion, Biochim. Biophys. Acta 1614 (2003) 122–129. [DOI] [PubMed] [Google Scholar]
- [4].Nieva JL, Agirre A, Are fusion peptides a good model to study viral cell fusion? Biochim. Biophys. Acta 1614 (2003) 104–115. [DOI] [PubMed] [Google Scholar]
- [5].Lorizate M, Huarte N, Saez-Cirion A, Nieva JL, Interfacial pre-transmembrane domains in viral proteins promoting membrane fusion and fission, Biochim. Biophys. Acta 1778 (2008) 1624–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Salzwedel K, West JT, Hunter E, A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity, J. Virol 73 (1999) 2469–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Suarez T, Gallaher WR, Agirre A, Goni FM, Nieva JL, Membrane interface-interacting sequences within the ectodomain of the human immunodeficiency virus type 1 envelope glycoprotein: putative role during viral fusion, J. Virol 74 (2000) 8038–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen SS, Yang P, Ke PY, Li HF, Chan WE, Chang DK, Chuang CK, Tsai Y, Huang SC, Identification of the LWYIK motif located in the human immunodeficiency virus type 1 transmembrane gp41 protein as a distinct determinant for viral infection, J. Virol 83 (2009) 870–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Apellaniz B, Ivankin A, Nir S, Gidalevitz D, Nieva JL, Membrane-proximal external HIV-1 gp41 motif adapted for destabilizing the highly rigid viral envelope, Biophys. J 101 (2011) 2426–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Saez-Cirion A, Arrondo JL, Gomara MJ, Lorizate M, Iloro I, Melikyan G, Nieva JL, Structural and functional roles of HIV-1 gp41 pretransmembrane sequence segmentation, Biophys. J 85 (2003) 3769–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schibli DJ, Montelaro RC, Vogel HJ, The membrane-proximal tryptophan-rich region of the HIV glycoprotein, gp41, forms a well-defined helix in dodecylphosphocholine micelles, Biochemistry 40 (2001) 9570–9578. [DOI] [PubMed] [Google Scholar]
- [12].Sun ZY, Oh KJ, Kim M, Yu J, Brusic V, Song L, Qiao Z, Wang JH, Wagner G, Reinherz EL, HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane, Immunity 28 (2008) 52–63. [DOI] [PubMed] [Google Scholar]
- [13].Suarez T, Nir S, Goni FM, Saez-Cirion A, Nieva JL, The pre-transmembrane region of the human immunodeficiency virus type-1 glycoprotein: a novel fusogenic sequence, FEBS Lett 477 (2000) 145–149. [DOI] [PubMed] [Google Scholar]
- [14].Shnaper S, Sackett K, Gallo SA, Blumenthal R, Shai Y, The C- and the N-terminal regions of glycoprotein 41 ectodomain fuse membranes enriched and not enriched with cholesterol, respectively, J. Biol. Chem 279 (2004) 18526–18534. [DOI] [PubMed] [Google Scholar]
- [15].Epand RM, Thomas A, Brasseur R, Epand RF, Cholesterol interaction with proteins that partition into membrane domains: an overview, Subcell. Biochem 51 (2010) 253–278. [DOI] [PubMed] [Google Scholar]
- [16].Lorizate M, Cruz A, Huarte N, Kunert R, Perez-Gil J, Nieva JL, Recognition and blocking of HIV-1 gp41 pre-transmembrane sequence by monoclonal 4E10 antibody in a Raft-like membrane environment, J. Biol. Chem 281 (2006) 39598–39606. [DOI] [PubMed] [Google Scholar]
- [17].Montero M, van Houten NE, Wang X, Scott JK, The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: dominant site of antibody neutralization and target for vaccine design, Microbiol. Mol. Biol. Rev 72 (2008) 54–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Montero M, Gulzar N, Klaric KA, Donald JE, Lepik C, Wu S, Tsai S, Julien JP, Hessell AJ, Wang S, Lu S, Burton DR, Pai EF, Degrado WF, Scott JK, Neutralizing epitopes in the membrane-proximal external region of HIV-1 gp41 are influenced by the transmembrane domain and the plasma membrane, J. Virol 86 (2012) 2930–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nieva JL, Apellaniz B, Huarte N, Lorizate M, A new paradigm in molecular recognition? Specific antibody binding to membrane-inserted HIV-1 epitopes, J. Mol. Recognit 24 (2011) 642–646. [DOI] [PubMed] [Google Scholar]
- [20].Vishwanathan SA, Hunter E, Importance of the membrane-perturbing properties of the membrane-proximal external region of human immunodeficiency virus type 1 gp41 to viral fusion, J. Virol 82 (2008) 5118–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shai Y, Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides, Biochim. Biophys. Acta 1462 (1999) 55–70. [DOI] [PubMed] [Google Scholar]
- [22].Neville F, Cahuzac M, Konovalov O, Ishitsuka Y, Lee KY, Kuzmenko I, Kale GM, Gidalevitz D, Lipid headgroup discrimination by antimicrobial peptide LL-37: insight into mechanism of action, Biophys. J 90 (2006) 1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Neville F, Ishitsuka Y, Hodges CS, Konovalov O, Waring AJ, Lehrer R, Lee KY, Gidalevitz D, Protegrin interaction with lipid monolayers: grazing incidence X-ray diffraction and X-ray reflectivity study, Soft Matter 4 (2008) 1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Epand RM, Vogel HJ, Diversity of antimicrobial peptides and their mechanisms of action, Biochim. Biophys. Acta 1462 (1999) 11–28. [DOI] [PubMed] [Google Scholar]
- [25].Buzon V, Natrajan G, Schibli D, Campelo F, Kozlov MM, Weissenhorn W, Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions, PLoS Pathog. 6 (2010) e1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kozlov MM, McMahon HT, Chernomordik LV, Protein-driven membrane stresses in fusion and fission, Trends Biochem. Sci 35 (2010) 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ellens H, Bentz J, Szoka FC, H+- and Ca2 +-induced fusion and destabilization of liposomes, Biochemistry 24 (1985) 3099–3106. [DOI] [PubMed] [Google Scholar]
- [28].Ivankin A, Kuzmenko I, Gidalevitz D, Cholesterol–phospholipid interactions: new insights from surface x-ray scattering data, Phys. Rev. Lett 104 (2010) 108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Neville F, Hodges CS, Liu C, Konovalov O, Gidalevitz D, In situ characterization of lipid A interaction with antimicrobial peptides using surface X-ray scattering, Biochim. Biophys. Acta 1758 (2006) 232–240. [DOI] [PubMed] [Google Scholar]
- [30].Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Krausslich HG, The HIV lipidome: a raft with an unusual composition, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 2641–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sarin PS, Gallo RC, Scheer DI, Crews F, Lippa AS, Effects of a novel compound (AL 721) on HTLV-III infectivity in vitro, N. Engl. J. Med 313 (1985) 1289–1290. [DOI] [PubMed] [Google Scholar]
- [32].Schaffner CP, Plescia OJ, Pontani D, Sun D, Thornton A, Pandey RC, Sarin PS, Anti-viral activity of amphotericin B methyl ester: inhibition of HTLV-III replication in cell culture, Biochem. Pharmacol 35 (1986) 4110–4113. [DOI] [PubMed] [Google Scholar]
- [33].Graham DR, Chertova E, Hilburn JM, Arthur LO, Hildreth JE, Cholesterol depletion of human immunodeficiency virus type 1 and simian immunodeficiency virus with beta-cyclodextrin inactivates and permeabilizes the virions: evidence for virion-associated lipid rafts, J. Virol 77 (2003) 8237–8248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liao Z, Graham DR, Hildreth JE, Lipid rafts and HIV pathogenesis: virion-associated cholesterol is required for fusion and infection of susceptible cells, AIDS Res. Hum. Retroviruses 19 (2003) 675–687. [DOI] [PubMed] [Google Scholar]
- [35].McIntosh TJ, Simon SA, Roles of bilayer material properties in function and distribution of membrane proteins, Annu. Rev. Biophys. Biomol. Struct 35 (2006) 177–198. [DOI] [PubMed] [Google Scholar]
- [36].Gidalevitz D, Ishitsuka Y, Muresan AS, Konovalov O, Waring AJ, Lehrer RI, Lee KY, Interaction of antimicrobial peptide protegrin with biomembranes, Proc. Natl. Acad. Sci. U. S. A 100 (2003) 6302–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Neville F, Ivankin A, Konovalov O, Gidalevitz D, A comparative study on the interactions of SMAP-29 with lipid monolayers, Biochim. Biophys. Acta 1798 (2010) 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Huarte N, Lorizate M, Maeso R, Kunert R, Arranz R, Valpuesta JM, Nieva JL, The broadly neutralizing anti-human immunodeficiency virus type 1 4E10 monoclonal antibody is better adapted to membrane-bound epitope recognition and blocking than 2F5, J. Virol 82 (2008) 8986–8996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Saez-Cirion A, Nir S, Lorizate M, Agirre A, Cruz A, Perez-Gil J, Nieva JL, Sphingomyelin and cholesterol promote HIV-1 gp41 pretransmembrane sequence surface aggregation and membrane restructuring, J. Biol. Chem 277 (2002) 21776–21785. [DOI] [PubMed] [Google Scholar]
- [40].Chernomordik LV, Kozlov MM, Protein–lipid interplay in fusion and fission of biological membranes, Annu. Rev. Biochem 72 (2003) 175–207. [DOI] [PubMed] [Google Scholar]
- [41].Apellaniz B, Nieva JL, Schwille P, Garcia-Saez AJ, All-or-none versus graded: single-vesicle analysis reveals lipid composition effects on membrane permeabilization, Biophys. J 99 (2010) 3619–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wimley WC, Describing the mechanism of antimicrobial peptide action with the interfacial activity model, ACS Chem. Biol 5 (2010) 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Huang HW, Molecular mechanisms of peptide-induced pores in membranes, Phys. Rev. Lett 92 (2004) 198304. [DOI] [PubMed] [Google Scholar]
- [44].Zemel A, Ben-Shaul A, May S, Membrane perturbation induced by interfacially adsorbed peptides, Biophys. J 86 (2004) 3607–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Campelo F, McMahon HT, Kozlov MM, The hydrophobic insertion mechanism of membrane curvature generation by proteins, Biophys. J 95 (2008) 2325–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhu P, Liu J, Bess J Jr., Chertova E, Lifson JD, Grise H, Ofek GA, Taylor KA, Roux KH, Distribution and three-dimensional structure of AIDS virus envelope spikes, Nature 441 (2006) 847–852. [DOI] [PubMed] [Google Scholar]
- [47].Wu SR, Loving R, Lindqvist B, Hebert H, Koeck PJ, Sjoberg M, Garoff H, Single-particle cryoelectron microscopy analysis reveals the HIV-1 spike as a tripod structure, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 18844–18849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhu P, Winkler H, Chertova E, Taylor KA, Roux KH, Cryoelectron tomography of HIV-1 envelope spikes: further evidence for tripod-like legs, PLoS Pathog 4 (2008) e1000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sougrat R, Bartesaghi A, Lifson JD, Bennett AE, Bess JW, Zabransky DJ, Subramaniam S, Electron tomography of the contact between T cells and SIV/HIV-1: implications for viral entry, PLoS Pathog. 3 (2007) e63. [DOI] [PMC free article] [PubMed] [Google Scholar]