

Abstract

We present here a review of the photochemical and electrochemical applications of multi-site proton-coupled electron transfer (MS-PCET) in organic synthesis. MS-PCETs are redox mechanisms in which both an electron and a proton are exchanged together, often in a concerted elementary step. As such, MS-PCET can function as a non-classical mechanism for homolytic bond activation, providing opportunities to generate synthetically useful free radical intermediates directly from a wide variety of common organic functional groups. We present an introduction to MS-PCET and a practitioner’s guide to reaction design, with an emphasis on the unique energetic and selectivity features that are characteristic of this reaction class. We then present chapters on oxidative N–H, O–H, S–H, and C–H bond homolysis methods, for the generation of the corresponding neutral radical species. Then, chapters for reductive PCET activations involving carbonyl, imine, other X=Y π-systems, and heteroarenes, where neutral ketyl, α-amino, and heteroarene-derived radicals can be generated. Finally, we present chapters on the applications of MS-PCET in asymmetric catalysis and in materials and device applications. Within each chapter, we subdivide by the functional group undergoing homolysis, and thereafter by the type of transformation being promoted. Methods published prior to the end of December 2020 are presented.
1. Introduction to Multi-Site Proton-Coupled Electron Transfer in Organic Synthesis
1.1. Background and Significance
Proton-coupled electron transfers (PCETs) are redox mechanisms in which both an electron and proton are exchanged, often in a concerted elementary step.1−5 By virtue of transferring an electron and proton together, PCET can function as a non-traditional mechanism for homolytic bond cleavage, formally adding or removing the elements of H• to or from substrates of interest.6,7 PCET mechanisms play a key role in many distinct areas of chemistry and catalysis. For instance, familiar biological redox processes such as enzymatic C–H oxidation,8,9 ribonucleotide reduction,10 photosynthesis,11,12 and small-molecule metabolism all involve key PCET steps.13 PCET is also prevalent in inorganic technologies for the interconversion of important small molecules such as O2/H2O, N2/NH3, and CO2/alkanes.4,5,7,14 Historically the use of PCET as a mechanism for bond homolysis in organic synthesis has been less extensively investigated. However, the past decade has witnessed significant growth in this area, with organic chemists more fully recognizing the ability of PCET to enable the direct generation of free radical intermediates from numerous common organic functional groups.6,15−17 This Review aims to highlight recent progress in this area and to survey the known applications of PCET in synthetic organic chemistry, with a specific focus on photochemical and electrochemical approaches. The following sections of the introduction will give a brief description of the mechanistic features of PCET reactions and the ways in which PCET has been implemented to enable new methods for strong bond activation and catalytic radical generation.
A critical aspect of free radical chemistry is the means by which radical intermediates are generated from closed-shell starting materials. The homolytic cleavage or formation of C–H and E–H (E = N, O, S) bonds is a particularly attractive approach, as reactive radicals can be accessed directly from readily available starting materials without the need for substrate pre-functionalization.18,19 Accordingly, homolytic bond activations are powerful synthetic methods, as exemplified by the extensive use of hydrogen-atom-transfer (HAT) reactions in the selective functionalization of aliphatic C–H bonds.18−21 While powerful, the scope of radical generation via HAT is typically limited thermodynamically by the range of accessible H-atom donors and acceptors.6 The position of HAT equilibria can be expressed as the difference in the bond dissociation free energies (BDFEs) of the two bonds to hydrogen that are undergoing exchange.22 To be energetically favorable, the BDFE of the bond broken in the reactants must be lower (weaker) than the BDFE of the bond formed in the products. This simple requirement presents a significant challenge to HAT activation of many common organic functional groups, which would require either the cleavage of exceptionally strong bonds, or the formation of unusually weak bonds, to hydrogen. For example, the removal of H• from the N–H bonds in N-alkyl amides (BDFE ≈ 110 kcal mol)23,24 or the O–H bonds in aliphatic alcohols (BDFE ≈ 105 kcal mol–1)25,26 necessitates the use a H-atom abstractor that forms an even stronger bond to hydrogen (Figure 1A). Accordingly, even potent H-atom abstractors, such as the iron oxo species of cytochrome P450 enzymes (O–H BDFE ≈ 95 kcal mol–1),8,9 are thermodynamically incapable of activating those functional groups. Likewise, the addition of H• to an aromatic ketone generates a neutral ketyl radical with an exceptionally weak O–H bond (BDFE ≈ 26 kcal mol–1),16 requiring a H-atom donor with a similarly low bond strength (Figure 1B). However, metal hydrides, which are often used as catalytic H• donors, generally have M–H BDFEs ≥ 50 kcal mol–1 as complexes with lower bond strengths tend to spontaneously evolve H2.22 Conceivably, this requirement can be overcome when an unfavorable HAT is then coupled with a fast and irreversible exothermic downhill reaction of the generated radical, but this is limited to specific circumstances. In light of these limitations, general methods for the direct HAT activation of many common functional groups have yet to be reported (Figure 1C).
Figure 1.
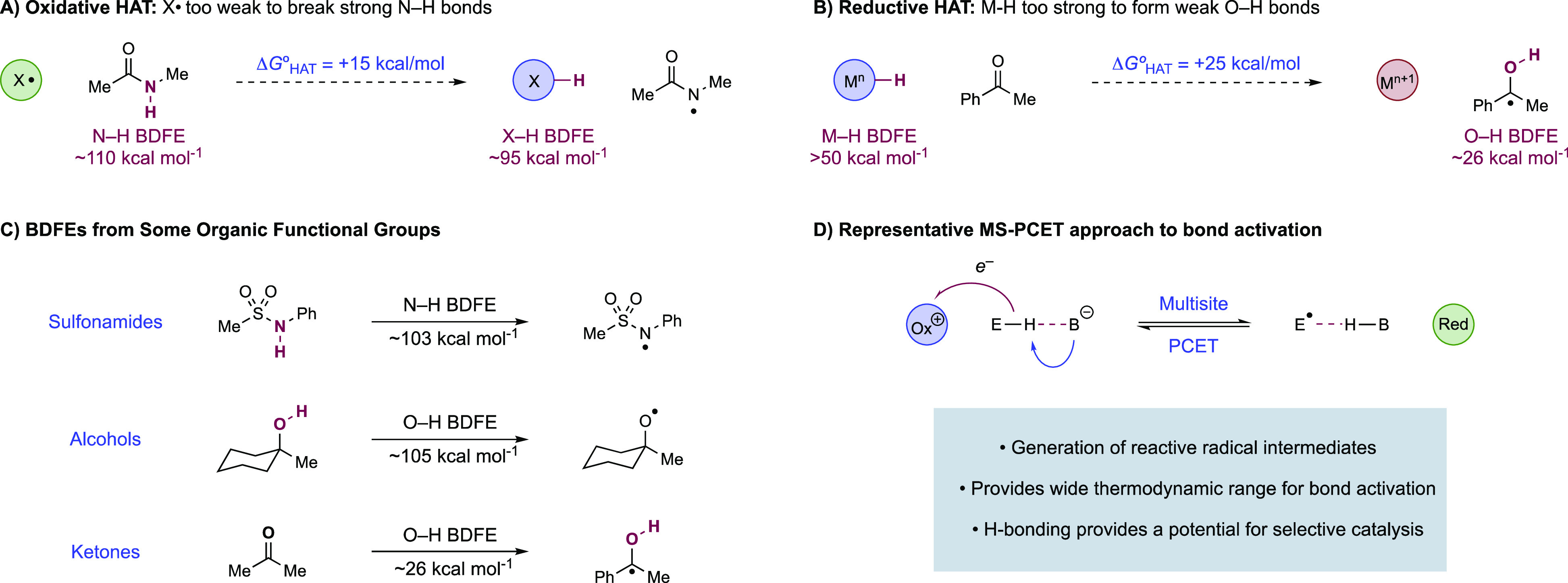
Limitations for (A) oxidative and (B) reductive HAT in synthetic chemistry. (C) BDFEs of common organic functional groups which have been difficult to activate via HAT. (D) MS-PCET approach to homolytic bond activation and formation.
Over the past decade, a subset of PCET mechanisms have emerged that address many of these thermodynamic limitations.16,27−31 Multi-site proton-coupled electron transfer (MS-PCET) involves the homolytic cleavage or formation of a C–H or E–H bond by the transfer of an electron and proton to or from completely independent reagents.6,32,33 In these MS-PCETs, the electron and proton originate from two separate donors, or travel to two distinct acceptors (Figure 1D). In practice, this typically involves the use of separate electron-transfer (ET) and proton-transfer (PT) reagents.5,16,33 For oxidative MS-PCET, the combination of a single-electron oxidant and a Brønsted base can act as a formal H• acceptor, while the combination of a single-electron reductant and Brønsted acid can act as a formal H• donor in reductive MS-PCET reactions.1,5,6 By physically separating the ET and PT reagents, MS-PCET mechanisms can span a much wider thermodynamic range than is possible using conventional platforms for HAT (vide infra), enabling much stronger formal H-atom acceptors and H-atom donors to be accessed.6,15
MS-PCET also offers orthogonal chemoselectivity from what is typically observed via HAT. HAT chemoselectivity is governed primarily by reaction driving force, polarity matching effects, and reorganization energies.1,34,35 In oxidative HAT, this typically leads to selective homolytic cleavage of the weakest or most well polarity-matched aliphatic C–H bond in a substrate.35,36 MS-PCET involving concerted transfer of an electron and proton instead requires preorganization of the PT coordinate in a hydrogen-bonding complex between the substrate and base (or acid) prior to the electron-transfer (ET) event (Figure 1D).33,37 This requirement for non-covalent pre-association allows for the selective homolysis of stronger, polar functional groups in the presence of weaker aliphatic C–H bonds, which are poor hydrogen bond donors.27,38 Similarly, through pre-association with an appropriate acid before ET, selective MS-PCET reduction of imines and carbonyl groups can be achieved in the presence of less polar olefins despite a strong thermodynamic bias for C–H bond formation.15,16
These unique features of MS-PCET (either concerted or stepwise) provide a distinct and general method for the catalytic generation of synthetically important free-radical intermediates directly from a wide range of common organic functional groups. This insight has gained traction in the synthetic community over the past decade, and forms the mechanistic basis for many of the synthetic methods highlighted in this Review. The following sections of this introduction will provide an overview on a number of topics relevant to the design and implementation of MS-PCET reactions in synthetic methods. First, we will describe the thermodynamics of MS-PCET and how to successfully design thermodynamically feasible bond activations. We will then cover topics related to chemoselectivity in MS-PCET reactions. Finally, we will briefly introduce photocatalytic and organic electrochemistry methods, as these methods have enabled the practical synthetic implementation of MS-PCET.
1.2. Thermochemistry of PCET Reactions
Knowledge of the governing thermodynamics is vital to the successful design of PCET reactions. The thermodynamics of PCET reactions are best described using the bond dissociation free energy (BDFE) of the bond involved in the PCET step.1,22 Typically, BDFEs are evaluated using a thermodynamic square scheme comprised of two readily accessible experimental parameters: pKa values and redox potentials (E°) (Figure 2A). In this scheme, the BDFE is defined as the energy required to homolytically cleave the bond of interest, yielding a radical intermediate and H•. It is often difficult to directly interrogate the energetics of the homolytic reaction along the diagonal, but the BDFE of a bond of interest can be calculated by a combination of the free energy for deprotonation (pKa) summed together with the free energies required to oxidize the resulting conjugate base (E°) to a neutral radical and to reduce proton to H•, Cg (Figure 2A, eq 1).22,39,40
Figure 2.

(A) Thermodynamic scheme describing the BDFE of an E–H bond. (B) Thermodynamic scheme describing the effective BDFE of a pair of MS-PCET reagents using the relevant pKa and E°. (C) Example of how to determine the thermodynamic driving force for a representative MS-PCET reaction.
While combinations of MS-PCET reagents do not comprise a physical bond, the relevant PCET thermochemistry can be described using the same BDFE formalism (Figure 2B).22,33,41,42 With the same combination of pKa and E°, the effective BDFE can be calculated for a pair of MS-PCET reagents (Figure 2B, eq 2). Remaining terms in these equations act as conversion factors to convert pKa values, and electrode potentials measured in volts vs Fc+/Fc, to their corresponding free energies at room temperature. For convenience, Cg = 52.6 kcal mol–1 in MeCN. For a further discussion on the derivation of this equation, see Warren, Tronic, and Mayer.22 [Recently, the literature accepted values of Cg have been revised. Many of the works discussed in this Review used the previous standard values of Cg in solving the Bordwell equation for the calculation of both E–H bond BDFEs and reagent pair effective BDFEs. This would lead to an overestimation in these values of between +1.9 and +4.8 kcal mol-1. In this Review we discuss the conclusions of the original research works without correction but make this note such that readers are aware of this recent revision. See reference (22)b for a discussion.] Oxidant and base combinations may act as formal H• acceptors, while reductant and acid combinations may act as formal H• donors. Therefore, the effective BDFE of a single-electron oxidant and base pair gives the strength of a hypothetical bond which may be cleaved in a thermoneutral reaction. Likewise, the effective BDFE of a reductant and acid combination gives the strength of a hypothetical bond which may be formed in a thermoneutral reaction. With the relevant BDFE value of the substrate bond of interest, and effective BDFE value of the MS-PCET reagent combination in hand, the thermodynamic free energy change of a proposed PCET reaction (ΔG°PCET) can then be determined by the difference in BDFE of the E–H bond to be activated and the effective BDFE of the MS-PCET reagent combination (Figure 2C, eq 3). Therefore, the judicious choice of reagents allows the researcher to design PCET reactions which are thermodynamically favorable. By convention, more thermodynamically favorable reactions are said to have greater thermodynamic driving force. This design feature has been shown to be crucial for the success of MS-PCET activations in a wide variety of synthetically useful reactions.16,27,43
The BDFE formalism described above also reveals why it is often difficult to design more reactive HAT reagents for thermodynamically difficult bond activations. To generate a more reactive molecular H• acceptor, one could make the abstracting species a stronger one-electron oxidant. Alternatively, the reduced state of the H• acceptor could be made a stronger Brønsted base. Unfortunately, within a single molecule, these two physical properties are interdependent and inversely correlated.22,44 For example, the addition of electron-withdrawing groups (EWGs) to a PCET reagent will generate a more oxidizing reagent (more positive E°) but will also lead to a compensating increase in acidity (lower pKa). Analogous difficulties arise in the design of new reductive HAT reagents, wherein any increase in the acidity of a H-atom donor will be compensated in part by a correlated decrease in the reducing power of the conjugate base.44 As a result of this thermodynamic compensation between pKa and E°, it is difficult to design more powerful HAT reagents through structural modification alone.
The physical separation of ET and PT reagents in MS-PCET overcomes this intrinsic compensation. When the pKa and E° of the PCET reagents are decoupled, each can be varied independently and cooperatively through judicious choice of reagents. For instance, a stronger Brønsted acid or a stronger one-electron reductant can be employed to decrease the effective BDFE. This type of modular construction allows for a much wider thermodynamic range of effective BDFE values for MS-PCET reagents. The expanded range of effective BDFEs for MS-PCET can also allow for the previously difficult activation of exceptionally strong E–H bonds (BDFE ≥ 100 kcal mol–1) and the formation of extremely weak E–H bonds (BDFE ≤ 30 kcal mol–1).16,29,30 The abundance of pKa, E°, and BDFE data available in the literature facilitates the rational design of new PCET reagent combinations. We would direct the reader to the useful literature on excited-state redox potentials,45,46 redox potentials in organic solvents,21,22,39,47 and pKa scales in organic solvents.22,48,49 Examples of both excited-state and ground-state MS-PCET reagent pairs and the corresponding effective BDFE values are given in Figure 3. In the main body of this Review, we give E°, pKa, and BDFE values provided by the original authors and from the available literature, as they pertain to the specific synthetic method under discussion.
Figure 3.

Representative oxidative (top) and reductive (bottom) MS-PCET reagents pairs and the corresponding effective BDFEs in kcal mol−1 using pKa and E1/2 values vs Fc+/Fc in MeCN. *Denotes photoexcited state complex as the oxidant / reductant.
An important limitation of the thermodynamic window accessible by MS-PCET reagents is the potential for incompatibilities between oxidant/base or acid/reductant pairs.50 It is important to assess the oxidative stability of a base in order to avoid unwanted ET events with the oxidant. In addition, oxidants are often good electrophiles and bases good nucleophiles which can lead to other unwanted polar side reactions. Reductants and acids can also evolve molecular hydrogen if the effective bond strength is low enough. These incompatibilities can dominate the observed reactivity of MS-PCET reagents at millimolar concentrations in organic solvents. For example, the combination of ground-state Fe(bpy)33+ and n-Bu4N+AcO– provides an effective BDFE of 103 kcal mol–1 in MeCN and could theoretically homolyze similarly strong E–H bonds.50 In practice, these two components rapidly react with one another at room temperature, precluding any use in preparative chemistry.50 As discussed further in section 1.4 of this introduction, photocatalytic and electrochemical approaches to MS-PCET provide a means to avoid many of these limitations.
1.3. Chemoselectivity of MS-PCET Reactions
The chemoselectivity of PCET is strongly influenced by the reaction mechanism.6,51,52 As noted above, a PCET reaction can proceed via either the stepwise or concerted transfer of an electron and proton (Figure 4A).5 For MS-PCET reactions, the different stepwise pathways can lead to drastically different chemoselectivities. For instance, an oxidative PT/ET mechanism can predominate if an adequately strong base is chosen such that a substantial equilibrium concentration of the substrate conjugate base exists in solution. The conjugate base form is thermodynamically much easier to oxidize than the conjugate acid, and a PT/ET mechanism can predominate.7 Such a mechanism would show selectivity for more acidic bonds. Likewise, a suitably strong oxidant which can directly oxidize the substrate could favor an ET/PT type mechanism, favoring activation of more electron-rich substrates. A potential drawback of stepwise PCET approaches in synthetic chemistry is the generation of high-energy intermediates formed by initial ET or PT activation of a substrate.5,6 Depending on the substrate, the reagents needed to generate these species are sometimes harsh and are often prone to incompatibilities described in the previous section.50 Concerted PCET avoids the generation of these high energy intermediates by delivering the electron and proton equivalent in a single kinetic step.3,5,7 When both stepwise intermediates are significantly high in energy relative to the reactants, a concerted pathway will generally dominate, providing a mechanism for cooperative substrate activation by the oxidant/base or reductant/acid pair under conditions where neither partner is able to activate the substrate alone (Figure 4B).1,52,53 In the main body of this Review, synthetic methods which involved a concerted PCET step were explicitly discussed to showcase this cooperative activation by the separated reagents.
Figure 4.

(A) Square scheme representation of PCET mechanisms available for the cleavage of an E–H bond. The stepwise transfer of electrons and protons are shown on the edges of the square. The concerted transfer of the electron and proton via concerted PCET is shown along the diagonal. (B) Reaction coordinate of stepwise vs concerted PCET mechanisms. Stepwise PCET generates high-energy charged intermediates which are avoided in a concerted PCET pathway.
When a concerted PCET activation mechanism is favored, the chemoselectivity of the PCET step is determined primarily by the PCET thermochemistry and the involvement of pre-equilibrium hydrogen bonding.6,52 The thermodynamic driving force of a PCET reaction strongly influences its kinetics.7,54 Generally, PCET reactions proceed at faster rates as they become more exergonic.5 HAT and MS-PCET reactions have both been shown to follow this relationship, and often the most exergonic PCET reaction will proceed at the fastest rate. Because of this, MS-PCET chemoselectivity is strongly influenced by the relative thermodynamic driving force of the competing bond activation or bond-forming steps.52 This is especially the case for HAT, where chemoselectivity is dominated by the driving force, self-exchange rates, and polar effects.35,36,55 Despite the sharp distinctions made between stepwise and concerted mechanisms for PCET, it is important to note that these reaction pathways are often in kinetic competition and can lead to important nuances in the observed kinetic response to thermodynamic changes in the PCET reaction.42,53
Pre-equilibrium hydrogen bonding involved in a concerted, MS-PCET reaction allows for unique chemoselectivity which departs significantly from the reactivity trends observed for HAT.6,15,51 Termolecular reactions of the substrate, base, and oxidant involved in a MS-PCET are inherently disfavored on entropic grounds. Pre-equilibrium hydrogen bonding brings the substrate and base together, and the resulting H-bonded complex can then react with the oxidant. This effectively reduces the molecularity of MS-PCET reactions and the kinetic barrier height for the concerted PCET reaction (Figure 5). The constraint of H-bonding pre-organization provides a pathway to selectively homolyze only the stronger bonds to hydrogen found in polar protic functional groups, such as alcohols, in the presence of weaker aliphatic C–H bonds that typically cannot form the requisite H-bonded complexes.27,38
Figure 5.

Pre-equilibrium hydrogen bonding can provide kinetic selectivity for homolysis of strong O–H bonds in the presence of much weaker C–H bonds.
In an analogous way, H-bond acceptor π-systems, such as ketones and imines, can be targeted selectively in the presence of less polar olefins despite a large thermochemical bias for C–H bond formation (Figure 6).16,52 Pre-PCET H-bonding can be important in other aspects as well. For example, formation of the pre-PCET H-bonded precursor (and post-PCET H-bonded successor) complexes can be a significant component of the thermochemistry for intermolecular proton–electron transfer, particularly in the low driving-force regime, and should not be neglected.52,56 Pre-PCET H-bonding is also one of the key factors that determine the chemoselectivity between different substrates eligible for MS-PCET activation, for instance N–H vs S–H activation.51 Similarly, post-PCET H-bonding successor complexes (•K–H···B, Figure 6) present intriguing opportunities to maintain non-covalent associations with open-shell intermediates.30,31 This in turn has created opportunities for MS-PCET to enable enantioselective reactions of free radicals when chiral acids and bases are employed as catalysts (see section 8).
Figure 6.

Top: Reaction scheme for MS-PCET reduction of an aryl ketone to the corresponding ketyl radical. Bottom: Reaction coordinate diagram highlighting the kinetic and thermodynamic importance of pre-equilibrium hydrogen bonding in MS-PCET.
1.4. Photocatalytic and Electrochemical Approaches to PCET
Over the past decade, the emergence of photocatalytic and electrochemical technologies in organic chemistry has greatly facilitated the implementation of MS-PCET methods by expanding the toolkit of compatible MS-PCET reagents.18,19,45,57,58 The utilization of excited-state photocatalysts in MS-PCET methods provides low and transient concentrations of potent one-electron redox equivalents while avoiding potential incompatibilities between oxidant/base or reductant/acid combinations.6,18−20,45 Similarly, the robustness of electrode materials and modularity of electrochemical methods has provided a number of new MS-PCET methods employing soluble PT reagents interfaced with heterogeneous electrode materials.59,60
Photocatalytic approaches to MS-PCET typically involve the use of visible-light absorbing molecule which can perform ET from a photoexcited state, combined with an appropriate base or acid in solution.6,15,45,61,62 Upon irradiation of the photocatalyst with visible light, an electron is promoted to a high-energy orbital, forming a singlet excited-state complex.19,45 In many cases, this initially formed singlet excited-state complex undergoes rapid intersystem crossing to form a long-lived triplet excited-state complex which can participate in ET reactions on the time scale of diffusion. The resulting excited-state complex has dual ET reactivity characteristics, wherein it typically behaves as both a potent one-electron oxidant and simultaneously a potent one-electron reductant. The high-energy electron can transfer to a substrate, oxidizing the photocatalyst in an oxidative quenching reaction. On the other hand, the empty, low-lying orbital previously occupied by the promoted electron can accept an electron from a substrate, reducing the photocatalyst in a reductive quenching reaction (Figure 7). Quenching of the excited state via either of these ET reactions forms a ground-state redox product which is also a potent ET reagent. Oxidative quenching produces a highly oxidizing ground-state species, whereas reductive quenching produces a highly reducing ground-state species. These photoproducts can participate in further ground-state redox chemistry to regenerate the original, ground-state photocatalyst.
Figure 7.

General schemes for substrate activation through (A) a photocatalytic oxidative quenching mechanism and (B) a photocatalytic reductive quenching mechanism, for overall redox-neutral transformations.
While the use of sacrificial oxidants or sacrificial reductants has been employed in a number of photocatalytic cycles of this nature, photocatalytic PCET methods are often redox-neutral, where the bond transformation is initiated by excited-state ET to/from a substrate and terminated by the follow-up ground-state ET to/from a downstream reaction intermediate. In this fashion, the photochemical approach to MS-PCET provides a wide range of one-electron redox reagents which can participate in catalytic, redox-neutral homolytic activation and functionalization of organic functional groups.19,45 The use of excited-state photooxidants and photoreductants also ensures low concentrations of the reactive redox reagent, mitigating decomposition related to nucleophilic/electrophilic character of the separated reagents. If unproductive ET between a base and an excited-state photooxidant does occur, back electron transfer (BET) can return the two components to their equilibrium positions. In addition, the short lifetime of these photosensitizers allows any unreacted excited states to relax back to the unreactive ground state without decomposition.5,50 Finally, photocatalyst structures can be modified to tune both the excited-state and ground-state redox properties in order to enable the desired redox reactions.46,63 Due to these benefits, many of the reported MS-PCET methods in this Review rely on a photochemical or photocatalytic approach to MS-PCET. We provide the structures of the photocatalysts discussed in this Review in Chart 1, and their relevant excited-state and ground-state redox potentials are given in Table 1.
Chart 1. Structures of Photocatalysts Discussed in This Reviewa.
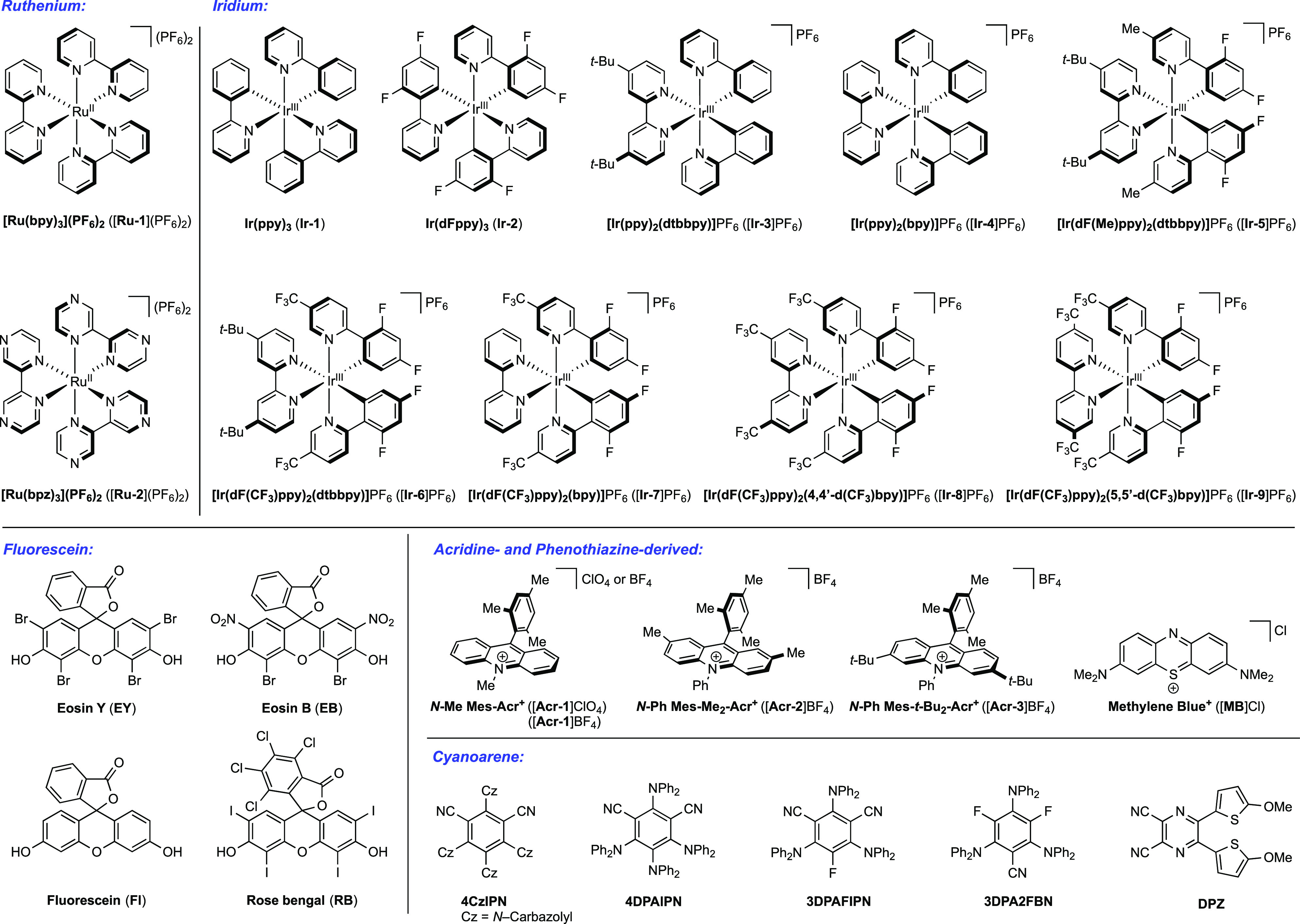
a Photocatalysts are separated into subclasses of ruthenium, iridium, fluorescein, acridine, and cyanoarene-based structures.
Table 1. Ground-State and Excited-State Redox Potentials for Photocatalysts Discussed in This Review.
| ground-state
redox potentials |
excited-state
redox potentials |
|||
|---|---|---|---|---|
| photocatalyst | E°ox(PC+/PC)a | E°red(PC/PC–)a | E°ox(PC+/*PC)a | E°red(*PC/PC–)a |
| [Ru(bpy)3](PF6)2 ([Ru-1](PF6)2) | +0.88b,64 | –1.71b,64 | –1.19b,64 | +0.39b,64 |
| [Ru(bpz)3](PF6)2 ([Ru-2](PF6)2) | +1.48b,65 | –1.18b,65 | –0.64b,65 | +1.07b,65 |
| Ir(ppy)3 (Ir-1) | +0.39b,66 | –2.57b,66 | –2.11b,66 | –0.07b,66 |
| Ir(dFppy)3 (Ir-2) | +0.6967 | –2.5167 | –1.8267 | 0.0067 |
| [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) | +0.83b,68 | –1.89b,68 | –1.34b,68 | +0.28b,68 |
| [Ir(ppy)2(bpy)]PF6 ([Ir-4]PF6) | +0.87b,46 | –2.43b,46 | –1.79b,46 | +0.23b,46 |
| [Ir(dF(Me)ppy)2(dtbbpy)]PF6 ([Ir-5]PF6) | +1.11b,68 | –1.81b,69 | –1.30b,68 | +0.59b,68 |
| [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) | +1.31b,68 | –1.75b,68 | –1.59b,68 | +0.51b,68 |
| [Ir(dF(CF3)ppy)2(bpy)]PF6 ([Ir-7]PF6) | +1.3870 | –1.6470 | –1.3070 | +1.0470 |
| [Ir(dF(CF3)ppy)2(4,4′-d(CF3)bpy)]PF6 ([Ir-8]PF6) | +1.5529 | –1.1829 | –0.8929 | +1.2729 |
| [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 ([Ir-9]PF6) | +1.5629 | –1.0729 | –0.8129 | +1.3029 |
| Eosin Y (EY) | – | –1.44b,c,71 | –1.49b,c,72 | +0.45b,c,71 |
| fluorescein (Fl) | – | –1.60b,c,71 | –1.64b,c,72 | +0.36b,c,72 |
| rose bengal (RB) | – | –1.34b,c,72 | –1.40b,c,72 | +0.47b,c,72 |
| N-Me Mes-Acr+ClO4– and N-Me Mes-Acr+BF4– | – | –0.93b,73 | – | +1.70d/+1.50e,b,73,74 |
| ([Acr-1]ClO4 and ([Acr-1]BF4) | ||||
| N-Ph Mes-Me2-Acr+ ([Acr-2]BF4) | – | –0.96b,75 | – | +1.71b,75 |
| N-Ph Mes-t-Bu2-Acr+ ([Acr-3]BF4) | – | –0.97b,76 | – | +1.70b,76 |
| methylene blue+Cl– ([MB]Cl) | +0.75b,f,45 | –0.68b,f,45 | –1.11d/–1.06e,b,f,45 | +1.18d/+1.22e,b,f,45 |
| 4CzIPN | +1.11b,77 | –1.59b,78 | –1.56b,77 | +0.97b,78 |
| 4DPAIPN | +0.65b,79 | –2.03b,79 | –1.90b,79 | +0.52b,79 |
| 3DPAFIPN | +0.92b,77 | –1.97b,78 | –1.76b,77 | +0.72b,78 |
| 3DPA2FBN | +0.86b,g,77 | –2.30b,g,77 | –1.98b,g,77 | +0.54b,g,77 |
| DPZ | +0.99b,g,80 | –1.83b,g,81 | –1.55b,82 | +0.53b,g,81 |
Potentials measured in V vs Fc+/Fc and measured in MeCN unless indicated.
Converted to Fc+/Fc from the reference electrode used in the original report according to Addison and co-workers.83
Measured in 1:1 MeCN:H2O.
From the singlet excited state.
From the triplet excited state.
Measured in H2O.
Measured in CH2Cl2.
For similar reasons, electrochemical approaches to MS-PCET have also seen noteworthy success.59,60,84 The robustness and modularity of electrode materials impart a number of advantages with respect to oxidant/base incompatibility. Depending on the potential/current applied across the electrochemical cell, the working electrode can directly act as the oxidant or reductant in a MS-PCET step (Figure 8, top). By tuning the applied potential, practitioners can exert exquisite control over the species that are oxidized/reduced in solution. Whereas species whose Eox < Eapplied (i.e., less positive) will be oxidized in an anodic reaction, species whose Ered > Eapplied (i.e., less negative) will be reduced in a cathodic reaction. For reversible redox processes, the applied potential also controls the concentration of the reduced vs oxidized form of a species. The concentration of redox intermediates can also be controlled through constant current electrolysis conditions; as the applied current becomes more positive, the concentration of oxidized species also increases. When too much or too little current is applied, however, undesired and/or no reactivity is typically seen because the concentration of reactive intermediates is too high or low, respectively, to facilitate the desired reactivity.60 An alternative electrochemical approach to substrate activation involves the use of soluble redox mediators, (Figure 8, bottom) which can be used to shuttle electrons and holes from the electrodes to substrates in solution when the direct electrolysis kinetics are inefficient. An important limitation to electrochemical separated PCET processes is the evolution of molecular hydrogen via cathode-mediated electrocatalytic proton reduction.85 As the overpotential of proton reduction is low relative to other cathodic processes, electrochemical methods for reductive MS-PCET reactions are quite rare; many electrochemical MS-PCET processes feature oxidative transformations. In these oxidative transformations, however, electrocatalytic proton reduction is typically the compensatory electrochemical reaction at the cathode.
Figure 8.

General schemes for substrate activation through direct and mediated electrolysis.
1.5. Scope of This Review
This Review aims to provide a thorough accounting of MS-PCET reactions implemented in organic synthesis through to the end of calendar year 2020. In selecting appropriate literature for this Review, we did not discriminate between stepwise or concerted MS-PCET mechanisms and deferred to the mechanistic conclusions made by the original authors of each report. Synthetic methods involving conventional HAT which do not also involve a MS-PCET step were excluded, as HAT chemistry has been reviewed extensively elsewhere.20,35,86−88 The main body of the review is organized by chapters on the bond undergoing oxidative homolytic activation (N–H, O–H, S–H, C–H), or π-system undergoing reductive homolytic activation (C=O, C=N, N=N, and other X=Y or heteroarene). Within each chapter, sections are then defined by the specific functional group containing that E–H bond undergoing homolysis. Subsections are then organized to the nature of the transformation occurring from the functional group under consideration, with material/methods being presented chronologically within a specific transformation. We have also included sections covering asymmetric catalysis and materials functionalization via MS-PCET. Where necessary, subsections begin with a brief overview of the scope to define exceptions to our overall focus on MS-PCET. As discussed above, due to the recent revitalization of photocatalysis and electrochemistry in organic synthesis, the vast majority of the content of this Review involves photocatalytic or electrochemical methods reported in the past decade. We hope this Review will exhibit the breadth and power of emerging MS-PCET methods in synthetic chemistry and prompt additional future work in this area.
2. N-Centered Radical Generation from N–H Bonds through Photochemical and Electrochemical PCET Processes
This section describes synthetic methods arising through the formal homolysis of N–H bonds in a broad number of functional groups mediated via PCET. We direct the reader to the following additional reviews, where further methods for N-centered radical generation—including those arising from prefunctionalized starting materials—are also discussed.89−9999
2.1. Transformations of Amides, Sulfonamides, Carbamates, Ureas, Imides, and Sulfonimides
2.1.1. Intramolecular C–N Bond Formation through Addition to Alkenes and Alkynes
2.1.1.1. Photochemical Reactivity
In the first proposed example of oxidative amide N–H bond activation via a photocatalytic concerted PCET manifold, Knowles and co-workers in 2015 demonstrated a catalytic intramolecular carboamidation reaction of alkene-tethered N-aryl amides, carbamates, and ureas with electron-deficient olefins (Scheme 1).100 The process was catalyzed by a combination of [Ir(dF(CF3)ppy)2(bpy)]PF6 photocatalyst ([Ir-7]PF6) and n-Bu4N+(n-BuO)2P(O)O– Brønsted base co-catalyst under visible-light irradiation in CH2Cl2 solution. A scope of 24 examples was presented with yields ranging from 50% to 95%, including preparations of lactam (1.1), oxazolidinone, cyclic urea (1.2), and thiourea heterocycles. Several examples of fused bicycles (1.3) and spirocycles (1.4) were demonstrated. The method was also extended to the functionalization of a d-glucal derivative to prepare the 3-amino-C-glycoside 1.5.
Scheme 1. Photocatalytic Alkene Carboamidation of N-Aryl Amides through Concerted PCET (Knowles, 2015).

The authors propose that this transformation proceeds via photoexcitation of the Ir(III) photocatalyst to a long-lived triplet excited state. The joint action of this photo-oxidant (E1/2 *Ir(III)/Ir(II) = +1.32 V vs SCE in MeCN)101 and Brønsted phosphate base (pKa = 13 in MeCN)100 then mediates the homolysis of the substrate N–H bond via concerted PCET. This photo-oxidant/base pair provides an effective BDFE of 97 kcal mol–1, approximately meeting the requirement to homolyze the strong N–H bond of the N-aryl amide substrates (e.g., for acetanilide, N–H BDE = 98.9 kcal mol–1 in DMSO).102 This neutral amidyl radical then undergoes 5-exo-trig cyclization onto the tethered substrate olefin, before trapping with the electron-deficient olefin partner in an intermolecular Giese-type reaction.103 Finally, reduction of the α-acyl radical with the reduced Ir(II) complex (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN)101 generates an enolate, which is protonated by phosphoric acid, yielding a closed-shell product and regenerating both catalysts. The authors conducted luminescence quenching experiments which showed that the photocatalyst excited state was only quenched by amide in the presence of phosphate base. Additionally, a first-order dependence on base and amide substrate concentration was observed in these studies. Parallel quenching experiments with N–H and N–D isotopologues of the same substrate amide revealed that kH/kD = 1.15, suggesting that homolysis of this N–H bond via PCET played a role in this quenching process. This low kinetic isotope effect (KIE) was not unexpected as MS-PCET processes have been shown to involve small isotope effects.12,32,104−107 Further mechanistic support for a PCET process came from CV studies, which revealed that direct substrate oxidation (e.g., for the amide substrate leading to 1.1, E1/2ox = +1.23 V vs Fc+/Fc in MeCN)100 in the absence of exogenous base occurs at a potential ca. 200 mV more positive than that of the Ir(III) excited state. Furthermore, the large pKa difference between substrate amide (pKa ≈ 32 in MeCN)100 and phosphate base (ΔpKaH ≈ 20) renders a discrete protonation step prior to ET unlikely. Together, these data provide evidence for a concerted PCET mechanism, as each individual PT or ET step is prohibitively endergonic.
The Knowles group also reported a photocatalytic intramolecular hydroamidation reaction of alkene-tethered N-aryl amides in 2015, proceeding through concerted PCET (Scheme 2).43 This was achieved through the inclusion of a thiophenol co-catalyst to mediate HAT to the C-centered radical generated from cyclization of the amidyl radical onto the pendant olefin, yielding the closed-shell product. Thiols have been used extensively as HAT reagents in synthetic photoredox chemistry by Nicewicz and co-workers73,108−113 due to their weak S–H bonds (e.g., for thiophenol, S–H BDE ≈ 79 kcal mol–1)114 enabling fast and favorable HAT with C-centered radicals.
Scheme 2. Photocatalytic Alkene Hydroamidation of N-Aryl Amides through Concerted PCET (Knowles, 2015).

Aside from the inclusion of thiophenol, the same combination of [Ir(dF(CF3)ppy)2(bpy)]PF6 photocatalyst ([Ir-7]PF6) and n-Bu4N+(n-BuO)2P(O)O– Brønsted base co-catalyst under visible-light irradiation was used in this report. A scope of 40 examples of catalytic intramolecular hydroamidation of N-aryl amides was demonstrated with yields ranging from 68% to 94%. The reaction tolerance for the N-aryl group was expanded beyond that of the initial report of carboamination reactivity, including across a range of arenes and heteroarenes where change had little impact on observed reactivity. The intramolecular hydroamidation of several complex substrates was demonstrated, including those derived from cis-chrysanthemic acid (2.4), progesterone, and gibberellic acid (2.6).
An analogous mechanism of reaction initiation is presented. The joint action of the Ir(III) photo-oxidant and phosphate base catalyst mediates oxidative concerted PCET activation of the amide N–H bond and formation of a neutral N-centered radical. Thereafter, 5-exo-trig cyclization generates a C-centered radical which undergoes HAT with the thiophenol co-catalyst to yield the closed-shell product. The resultant thiyl radical generated through this HAT step undergoes single-electron reduction (E1/2red = −0.22 V vs Fc+/Fc in MeCN)115 mediated by the Ir(II) state of the photocatalyst (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN)101 to give thiolate and the ground-state Ir(III) complex. Finally, PT between thiolate (e.g., for thiophenol, pKaH = 10.3 in DMSO)116 and the conjugate acid of the Brønsted base catalyst (e.g., for dibutylphosphate, pKa = 1.7 in H2O)117 regenerates all three catalytic additives.
This successful display of hydroamidation reactivity in this work is somewhat surprising, in that thiols are also established substrates for MS-PCET, and that the S–H bond is ca. 20 kcal mol–1 weaker than that of the N–H bond of the amide substrate.102,114 This observation is an exception to earlier kinetic studies of MS-PCET processes, which suggest that abstraction selectivity should track with bond strengths and react with weaker bonds preferentially.33,37,51,118−121 One would expect, based on this bond strength differential, that thiol would rapidly quench the Ir(III) photo-oxidant and inhibit the desired N–H bond activation. To better understand this unexpected selectivity, an empirical rate law for quenching of the Ir(III) photoexcited state was established through competitive luminescence quenching studies for a model hydroamidation reaction. The rate law derived from these experiments displays a first-order dependence in amide 3.1 and n-Bu4N+(n-BuO)2P(O)O– Brønsted base concentration, but is zero-order in PhSH (3.2) concentration (Scheme 3). Experiments in the absence of Brønsted base failed to quench the Ir(III) luminescence.
Scheme 3. Observed Rate Law for Ir(III) Luminescence Quenching in a Model Hydroamidation System (Knowles, 2019).

Seeking to further comprehend the observed chemoselectivity, the Knowles group in 2019 carried out a detailed kinetic analysis of competitive amide and thiol activation via PCET in the context of this hydroamidation reaction.51 The study determined hydrogen-bonding equilibrium constants between substrates and n-Bu4N+(n-BuO)2P(O)O– Brønsted base as well as rate constants for PCET substrate activation. Two different Ir(III) photo-oxidants were used in this study—[Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6, E1/2 *Ir(III)/Ir(II) = +0.89 V vs SCE in MeCN),51 and [Ir(dF(CF3)ppy)2(bpy)]PF6 ([Ir-7]PF6, E1/2 *Ir(III)/Ir(II) = +1.32 V vs SCE in MeCN).51 These catalysts provide two different effective BDFE values when in combination with the same Brønsted base—92 and 97 kcal mol–1 respectively—in order to vary the driving force for PCET bond activation. Four different N-aryl amides and four aromatic thiols were studied, which varied in their E–H bond BDFEs. This study showed that although the rate constant for oxidation via PCET of both amide (e.g., for acetanilide, kPCET = 8.4 × 109 M–1 s–1) and thiol (e.g., for thiophenol, kPCET = 9.5 × 109 M–1 s–1) substrates in some cases approach the diffusion limit in this solvent (k ≈ 1 × 1010 M–1 s–1 in 1,2-DCE),73 the amide (e.g., for acetanilide, KA = 1050 M–1) forms a more favorable pre-equilibrium hydrogen-bonded complex with the base compared to the thiol (e.g., for thiophenol, KA = 200 M–1). Because pre-association of base and substrate is required to facilitate ET, the observed chemoselectivity in this hydroamidation reaction is a result of the relative concentration of the reactive H-bonded complexes in solution. Therefore, at the concentrations of amide and thiol relevant to the synthetic hydroamidation reaction, the rate of amide activation outcompetes thiol activation by ca. 50:1, accounting for the observed empirical rate law. This work also identified a predictive model for reaction outcome of PCET-driven bond activations in the presence of thiol co-catalysts as determined by the interplay of bond strength differential and substrate-base hydrogen-bonding equilibrium constants with PCET rate constants.
The Nocera group in 2018 undertook a detailed mechanistic study of this hydroamidation reaction with the aim to reveal key insight to improve the quantum efficiency of the process (Scheme 4).122 This study focused on the hydroamination reaction of two model N-aryl amide substrates. Quantum yield measurements of the intramolecular hydroamidation of these substrates were measured using ferrioxalate actinometry. A 4-bromobenzamide substrate exhibited a quantum yield of 4.7% in this hydroamidation reaction to yield 4.1. Under the same conditions, a more electron-deficient 4-cyanobenzamide substrate displayed a marginally improved quantum yield of 5.8% in the reaction to yield 4.2, reflecting that the destabilizing effect of the electron-withdrawing nitrile on the transient amidyl radical leads to a faster rate of cyclization. This work revealed that for 4-bromobenzamide substrate leading to 4.1, the rate of PCET activation of the N–H bond was rapid (kPCET = 5.2 × 108 M–1 s–1), the low quantum efficiency of the process was a result of rapid BET (kBET = 7.4 × 109 M–1 s–1) outcompeting the relatively slow forward cyclization of the amidyl radical (kcyc = 2.4 × 104 M–1 s–1), instead returning the reactive intermediate to the closed-shell starting material.
Scheme 4. Investigation into and Improvement of the Quantum Efficiency of Photocatalytic Intramolecular Olefin Hydroamidation (Nocera, 2018).

Given this finding, the group implemented a strategy to improve the performance of these reactions through the inclusion of a disulfide additive. It was postulated that this could react with the transient N-centered amidyl radical to form an N-thioamide, which would diffuse away from the reduced Ir(II) complex, thereby impeding BET. The thioamide could reversibly reform the amidyl radical through facile N–S bond homolysis (for the N-thioamide formed in the preparation of 4.1, ΔGcalc = 1.2 kcal mol–1; for the N-thioamide formed in the preparation of 4.2, ΔGcalc = 2.6 kcal mol–1) to permit more efficient forward cyclization as the reactive amidyl radical is funneled to an intermediate where BET is no longer viable. This strategy was successful, and the quantum efficiency of the process in the presence of the disulfide additive was increased to 11.9% and 20.0% for products 4.1 and 4.2, respectively.
In a 2018 report, the Rueping group adapted these methods to now enable intramolecular amido-alkynylation, amido-alkenylation, and amido-allylation of olefins (Scheme 5).123 This transformation was achieved through the blue-light irradiation of N-aryl amide-tethered olefins in the presence of [Ir(dF(CF3)ppy)2(bpy)]PF6 ([Ir-7]PF6) photocatalyst, a stoichiometric quantity of n-Bu4N+(MeO)2P(O)O– Brønsted base, and inclusion of an alkynyl sulfone, alkenyl sulfone, or allylsulfone reagent, respectively. In this work from Rueping, 19 examples of olefin amido-alkynylation were reported in yields of 45–91% (5.1–5.3), alongside one example of amido-alkenylation (5.4) in 46% yield, and one example of amido-allylation (5.5) in 53% yield. The reaction was tolerant of variation in electron demand of the N-aryl group and permitted cyclization to form lactam, oxazolidinone, and cyclic urea products. The scope also permitted the introduction of several different arylacetylenes.
Scheme 5. Olefin Amido-alkynylation, Amido-alkenylation, and Amido-allylation of N-Aryl Amides with Sulfone Reagents (Rueping, 2018).

This method is proposed to initiate analogously to that of Knowles and co-workers, through oxidative N–H bond homolysis via PCET and subsequent 5-exo-trig cyclization onto the tethered olefin.43,100 Intermolecular radical addition into the sulfone reagent then results in distal C–C bond formation before elimination and extrusion of phenylsulfonyl radical. This radical can be readily reduced (e.g., for sodium benzenesulfinate, Ep/2ox = −0.37 V vs SCE in MeCN)124 by the Ir(II) state of the photocatalyst (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN).101 The addition–elimination reactivity of these reagents with carbon-centered radicals was originally reported by Nozaki.125 More recently, these reagents have been widely utilized for alkynylation and alkenylation in the context of photoredox catalysis since simultaneous reports from MacMillan and Inoue in 2014.126,127 In this example, stoichiometric quantities of Brønsted base are required to drive an efficient reaction, presumably due to the unfavorable equilibrium between the phosphoric acid (e.g., for dibutyl phosphate, pKa = 1.7 in H2O)117 and sulfinate (e.g., for benzene sulfinate anion, pKaH = 3.5 in H2O)116 waste product of the sulfone reagent.
In a 2019 report, Molander and co-workers reported a procedure for the amido arylation of N-aryl amide-tethered alkenes and aryl bromides, jointly mediated by a combination of [Ir(dF(CF3)ppy)2(bpy)]PF6 photocatalyst ([Ir-7]PF6), n-Bu4N+(n-BuO)2P(O)O– Brønsted base additive, and a Ni(II) complex under blue-light irradiation (Scheme 6).128 Under the optimized reaction conditions, a wide range of substituted alkenes underwent amidoarylation, including 1,1- and 1,2-disubstituted olefins 6.1 and 6.5, respectively. While trisubstituted alkenes were effective substrates in the carbo- and hydroamidation protocols of Knowles,43,100 there are no such examples reported here. In addition to N-phenyl amides, N-heteroaryl amides could be activated with this combination of photo-oxidant and Brønsted base. While most of the scope employed electron-deficient aryl and heteroaryl bromides (6.1, 6.2), the authors demonstrated that electron-rich aryl and heteroaryl bromides were also competent coupling partners (6.3, 6.4). Finally, it was shown that alkenes bearing pendant urea and carbamate functionalities were competent reaction partners, generating synthetically useful products, including an anomeric C-aryl glycoside (6.5, 6.6).
Scheme 6. Merger of Homolytic N–H Bond Activation through PCET and Ni-Catalysis Enables the Amido-arylation of Alkenes (Molander, 2019).

This reaction is proposed to proceed via the oxidative PCET homolysis of the N–H bond of the N-aryl amide substrate to generate a neutral N-centered radical. In support of this proposal, the authors note that neither the photocatalyst (for [Ir-7]PF6, *Ir(III)/Ir(II) = +1.32 V vs SCE in MeCN)101 nor the Brønsted base co-catalyst (for dibutylphosphate, pKa = 13 in MeCN),100 are each capable of driving direct ET or PT of the substrate amide respectively (e.g., for acetanilide, E1/2ox = +1.78 V vs SCE in MeCN; pKa ≈ 32 in MeCN).100,128 This electrophilic N-centered radical undergoes 5-exo-trig cyclization to a tethered olefin, generating a distal C-centered radical. This is then captured by a Ni(0) complex, formed via in situ reduction of the Ni(II) pre-catalyst, generating an alkyl-Ni(I) complex. This further reacts with the aryl bromide coupling partner in an oxidative addition step, to yield a Ni(III) intermediate, before reductive elimination to furnish the amidoarylation product and a Ni(I)-halide.129 Reduction of the resultant Ni(I) halide complex (e.g., for Ni(dtbbpy)Cl, E1/2 Ni(I)/Ni(0) = −1.17 V vs SCE in THF)130 by reduced-state Ir(II) complex (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN)101 regenerates both transition metal complexes in their active oxidation states required for catalytic turnover.
The Molander group has also recently reported a second example of combined homolytic bond activation via concerted PCET and subsequent Ni catalysis for the amido-acylation of N-aryl amide-tethered olefins (Scheme 7).131 This process occurs through intramolecular cyclization of a neutral amidyl radical onto a tethered alkene and subsequent acylation of the distal C-centered radical, through the inclusion of an acid chloride or acid anhydride coupling partner. In this work, 42 examples of olefin amido-acylation were reported in yields of 28–88%. When the substrate olefin was 1,2-disubstituted (e.g., 7.1–7.3), the reactions were typically highly selective for the formation of the kinetic, anti-isomer (e.g., for 7.1, ΔG = +1.3 kcal mol–1 relative to the syn-isomer), with d.r. typically >20:1. The reaction tolerated variation of N-aryl group electronics as well as a number of functional groups appended to the substrate olefin and allowed for a diverse number of both aryl and alkyl acid chloride coupling partners. Two examples of sugar-derived carbamate substrates were reported (e.g., 7.6), leading to interesting 3-amino-C-acyl glycosides.
Scheme 7. Catalytic Olefin Amido-acylation of N-Aryl Amides and Acyl Electrophiles through Dual PCET/Ni Catalysis (Molander, 2020).

The authors propose a similar mechanism for PCET activation of the substrate and cyclization to their previous amido arylation work.128 The same combination of [Ir(dF(CF3)ppy)2(bpy)]PF6([Ir-7]PF6) and n-Bu4N+(n-BuO)2P(O)O– as previously reported mediates PCET activation of the strong amide N–H bond, before 5-exo-trig cyclization onto the tethered olefin to form a C-centered radical. However, the Ni catalytic cycle differs in the order of oxidative addition and radical trapping at Ni. In situ reduction of a [Ni((4,4′-MeO)2bpy)]Cl2 precatalyst gives an active ligated (bpy)Ni(0) species which undergoes oxidative addition with either the acid chloride partner or an in situ generated acyl phosphate which was observed experimentally to form under the reaction conditions. The resulting Ni(II) intermediate intercepts the C-centered radical resulting from 5-exo-trig cyclization to form a Ni(III) intermediate, which undergoes a favorable, exothermic reductive elimination step to extrude the amido-acylated product and form a Ni(I) complex. This species (e.g., for Ni(dtbbpy)Cl, E1/2 Ni(I)/Ni(0) = −1.17 V vs SCE in THF)130 in turn reacts with the reduced-state Ir(II) complex (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN)101 to regenerate catalytically active Ir(III) and Ni(0). Authors note that there remains ambiguity in the order of events of oxidative addition and radical capture to yield the transient Ni(III) complex prior to reductive elimination as either a Ni(0)/Ni(II)/Ni(III) or a Ni(0)/Ni(I)/Ni(III) sequence is viable.129 In this case, the authors favor the former proposal due to the relative concentration of acyl electrophile coupling partner relative to the nascent radical. The reaction required stoichiometric quantities of phosphate base acting in a dual role for both PCET activation of the N–H bond via hydrogen-bonding and as a stoichiometric base to sequester HCl formed as a byproduct from the acid chloride coupling partner.
The group carried out DFT analysis to support this mechanistic hypothesis and to explain the high diastereoselectivity observed in these reactions products for the kinetic isomer. The products were found to be configurationally stable to the reaction conditions, ruling out any post-cyclization epimerization. Instead, the origin of this selectivity was determined at the trapping of the C-centered radical with the Ni(II) acyl complex, forming the Ni(III) acyl-alkyl complex. The transition state for trapping with the (bpy)Ni(II) acyl complex leading to the observed anti-product isomer was calculated to be 1.0 kcal mol–1 lower in energy due to reduced steric repulsion between the N-aryl group and the β-substituent of the olefin moiety in this complex, corresponding well with the observed experimental d.r. of 9:1 with the parent bpy ligand. Increasing the electron-donating ability of the 4,4′-substituents led to an increase in this selectivity, with (4,4′-MeO)2bpy providing optimal d.r. and product yield. A Hammett analysis found a linear correlation between log(d.r.) and σ+ parameters with a negative ρ value, indicating that the more electron-donating ligands, which better stabilize the putative Ni(III) intermediate in the catalytic cycle, lead to higher observed diastereoselectivity in these reactions.
Knowles and co-workers in 2019 reported on the intramolecular hydroamidation of olefin-tethered N-alkyl amides (Scheme 8).38 Compared to N-aryl amides (N–H BDFEs ≈ 100 kcal mol–1),102N-alkyl amides represent a more challenging substrate class for homolytic bond activation via PCET (N–H BDFE ≈ 110 kcal mol–1).114 A catalytic tetrad comprised of Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6([Ir-9]PF6), n-Bu4P+(n-BuO)2(O)PO– Brønsted base, TRIP thiol as a mediator of HAT, and TRIP disulfide additive was reported to enable this transformation. In this work, 34 examples of intramolecular hydroamidation reactions of olefin-tethered N-alkyl amides were reported in yields of 55–97%, to prepare lactam (8.1, 8.2), cyclic N-acylamine (8.6, 8.7), and carbamate products. In addition, three examples of the polycyclization of diene-amides were reported, where after N–H bond activation via PCET and subsequent addition across one olefin, the transient C-centered radical cyclizes further onto the second olefin before terminating HAT (e.g., 8.8). The success of these tandem processes demonstrates that the second cyclization outcompetes bimolecular quenching by thiol via HAT (kHAT ≈ 108 M–1 s–1)132 to avoid premature termination of the radical.
Scheme 8. Catalytic Intramolecular Alkene Hydroamidations of N-Alkyl Amides (Knowles, 2019).

*25 mol% n-Bu4P+(t-BuO)2P(O)O–, 80 mol% TRIP-SH. **Without TRIP-SH, 30 mol% TRIP2S2.
The combination of this Ir(III) photo-oxidant (E1/2 *Ir(III)/Ir(II) = +1.30 V vs Fc+/Fc in MeCN)29 and dibutyl phosphate Brønsted base (pKa = 13 in MeCN),100 provides an effective BDFE of 103 kcal mol–1, allowing for a marginally endergonic N–H bond homolysis to occur leading to amidyl radical generation. The requisite disulfide co-catalyst was particularly important in achieving high reaction efficiency. In line with earlier observations from Nocera,122 it is proposed to rapidly sequester the transient amidyl radical to an off-cycle N-thioamide intermediate, which can reversibly reform the amidyl radical under reaction conditions after diffusing away from the Ir(II) complex, enabling slow forward C–N bond formation to compete with otherwise rapid BET to Ir(II). Independently prepared N-thioamide was shown to form the product in an efficient manner. The requirement for this catalytic additive was more pronounced in this substrate class where the intermediate N-alkyl amidyl radical is further destabilized relative to N-aryl amidyl radical and thus is expected to have even faster rates of BET.
The groups of Chen and Huang each reported related methods enabling the photocatalytic synthesis of 3,4-dihydroisoquinolinones through amidyl radical generation and 6-endo-dig cyclization (Scheme 9).133,134 In Chen’s method, the visible-light irradiation of methanol solutions of 2-styryl-substituted N-methoxy benzamides in the presence of Na-Eosin Y (EY) photocatalyst and NaOH yielded 4-substituted dihydroisoquinolinone products. Blue LED light sources or sunlight could affect the transformation equally well. Here, nine examples were reported in yields of 66–99% (9.1–9.4). Huang later showed that the visible-light irradiation of 2-allyl-substituted N-methoxy benzamides in MeCN/H2O (1:1) with the same Na-EY and DBU as a Brønsted base yielded instead 3-substituted dihydroisoquinolinones, with 15 examples presented in yields of 43–86% (9.5–9.8). The inclusion of TEMPO led to the corresponding olefin oxyamidation product instead.
Scheme 9. (A) Photocatalytic Synthesis of 4-Substituted 3,4-Dihydroisoquinolinones (Chen, 2017), (B) Photocatalytic Synthesis of 3-Substituted 3,4-Dihydroisoquinolinones (Huang, 2019), and (C) Proposed Reaction Mechanisms.

*With 2.0 equiv of TEMPO.
Chen proposed a mechanism wherein the hydroxide base deprotonates the N-methoxyamide substrate, yielding the corresponding anion prior to single-electron oxidation by photoexcited eosin Y (E1/2 *EY/EY•– = +0.83 V vs SCE in MeCN).71 The resulting amidyl radical undergoes 6-endo-trig cyclization onto the pendant styrene, generating a doubly benzylic product radical. This radical is reduced to the corresponding anion (e.g., for the doubly benzylic radical deriving from 1,1-diphenylethane (Ph2C·Me) Ep/2red = −1.34 V vs SCE in MeCN)135 by the reduced state of the EY photocatalyst (E1/2EY/EY•– = −1.06 V vs SCE in MeCN),71 and through PT with solvent yields closed-shell product. This quoted reduction potential of the transient radical here prior to single-electron reduction likely represents a lower bound, given the presence of the electron-withdrawing acyl substituent on the substrate arene. The same pathway for amidyl radical generation is presented by Huang, but in this case, the N-centered radical undergoes 6-exo-trig cyclization onto the pendant allyl group, generating a primary alkyl radical. The mechanism for termination of this radical remains unclear. Experiments run in MeCN/D2O gave product with no deuterium incorporation, suggesting that the same ET/PT termination pathway is not operative. This may instead suggest a HAT pathway for radical termination to yield the closed-shell product. Such a pathway has been invoked by Xiao in related photocatalytic hydroamidation work where chloroform solvent acts as H-atom donor for this process,136 yet solvent participation in this work seems unlikely.
Here, we note two related reports of the reactivity of olefin-tethered N-aryl amide substrates proceeding through ground-state PCET mechanisms. Knowles and co-workers in 2015 reported a dual catalytic Ti/TEMPO system enabling the bond-weakening homolysis of substrate N–H bonds for conjugate amidation.137 Clayden and co-workers later reported an azoamination of these substrates with aryldiazonium salts.138 Initially investigated as a photocatalytic method, control experiments revealed instead ground-state reactivity. This reaction was proposed to proceed through aryl radical initiated HAT with the N–H substrate, followed by chain propagation through a PCET mechanism. A detailed discussion of these methods is beyond the scope of this Review.
The activation of N–H bonds through PCET and subsequent N-centered addition across olefins for C–N bond formation has also been extended to sulfonamide substrates. Primary and secondary sulfonamides have N–H BDFEs of ∼105 and 97 kcal mol–1, respectively,114 and thus have comparable N–H bond strengths to those of N-alkyl and N-aryl amides as discussed above. A 2018 report from Knowles and co-workers demonstrated that a catalytic triad comprising Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6([Ir-9]PF6), n-Bu4N+(n-BuO)2(O)PO– Brønsted base co-catalyst, and TRIP thiol as a HAT donor could mediate the intramolecular hydrosulfonamidation of tethered olefins with N-alkyl sulfonamides (Scheme 10).139 This report contained 33 examples of the synthesis of N-sulfonyl pyrrolidines through 5-exo-trig cyclization of a transient N-centered radical onto a tethered olefin in 47–96% yields. The reaction was highly tolerant of changes in substitution pattern across the olefin, with mono- (10.1), di- (10.2, 10.3), and trisubstituted (10.4) olefins proving to be competent. This intramolecular reaction displayed good tolerance of the sulfonamide S-substituent, with electron-rich and electron-poor S-aryl groups included, in addition to six examples of S-alkyl sulfonamides and two examples of dimethylamino sulfamides (10.6). This method also permitted the intermolecular hydrosulfonamidation of unactivated olefins (see section 2.1.4.1).
Scheme 10. Photocatalytic Intramolecular Alkene Hydrosulfonamidation (Knowles, 2018).

Cyclic voltammetry (CV) of an isolated secondary sulfonamide substrate (separate from olefin functionality) revealed that direct oxidation by this employed photo-oxidant (E1/2 *Ir(III)/Ir(II) = +1.30 V vs Fc+/Fc in MeCN)29 is prohibitively endergonic, by ca. +500 mV (e.g., for 4-methoxy-N-propylbenzenesulfonamide, Ep/2 = +1.80 V vs Fc+/Fc in MeCN).139 Less electron-rich sulfonamides displayed higher oxidation potentials still (e.g., for 4-methyl-N-propylbenzenesulfonamide, Ep/2 = +2.20 V vs Fc+/Fc in MeCN).139 Similarly, Nicewicz has shown that the oxidation potential of model 1,1-disubstituted, 1,2-disubstituted, and trisubstituted olefins are also beyond the excited-state potential of this photocatalyst (e.g., for 2-methyl-1-pentene, Ep/2ox = +2.50 V vs SCE in MeCN; for cyclohexene, Ep/2ox = +2.37 V vs SCE in MeCN; for 2-methyl-2-butene, Ep/2ox = +1.98 V vs SCE in MeCN).21 In addition, Stern–Volmer (SV) luminescence quenching studies revealed that the sulfonamide alone does not quench the Ir(III) excited state. Only when phosphate base was introduced was quenching observed. These data are all supportive of a concerted PCET mechanism being operative for N-centered radical generation, over stepwise ET or PT pathways at either of the sulfonamide or olefin sites. Following sulfonamidyl radical generation in this manner, 5-exo-trig cyclization and terminating HAT with the thiol additive yields the closed shell product. Reduction of the resulting thiyl radical by Ir(II), and protonation of the corresponding thiolate by dibutylphosphate regenerates all three catalytic mediators.
Qin and co-workers in 2017 devised a radical cascade approach to a collection of 33 indole alkaloid natural products across four families, initiating via the stepwise generation of an N-centered sulfonamidyl radical through visible-light photoredox catalysis (Scheme 11).140 This unified approach centered on an enamide-appended sulfonanilide 11.1, which was accessible in enantioenriched form in four synthetic steps from 2-nitrocinnamaldehyde, with an organocatalytic asymmetric malonate conjugate addition reaction first described by Jørgenson141 used as the key stereo-defining step. Their synthetic design envisaged N-centered radical generation at the sulfonanilide to trigger a radical cyclization cascade. This could be directed toward a number of different output structures by variation of appended substrate functionality for intramolecular cyclization or through addition of an exogenous coupling partner to trigger a subsequent intermolecular coupling. For example, aspidosperma-type intermediates were thought to be accessible via a sequence of sulfonamidyl radical generation, 5-exo-trig cyclization onto the appended enamide, and subsequent 6-exo-trig cyclization onto a tethered olefin (Scheme 11, Path A). Instead, tetrahydrocarbolinone-type intermediates were hoped to be accessible via the same combination of a sulfonamidyl radical generation and 5-exo-trig cyclization, but then intercepting an exogenous Michael acceptor in a Giese addition (Scheme 11, Path B). Finally, the authors reasoned that the combination of these approaches would offer access to corynanthe-type intermediates via an additional 6-exo cyclization after intermolecular C–C bond formation (Scheme 11, Path C). This strategy proved successful and a set of reaction conditions consisting of visible-light irradiation of sulfonanilide substrate in the presence of Ir(III) photocatalyst [Ir(ppy)2(dtbbpy)]PF6([Ir-3]PF6), KHCO3, and Brønsted base additive in THF or MeCN was generally able to trigger these cascade cyclization reactions. This reaction is understood to proceed via discrete substrate deprotonation prior to photoinduced electron transfer (PET). Notably only N-Ts sulfonanilides were successful, with N-Boc, N-Me, and -NH2 groups failing to initiate the reaction sequence. It was reasoned that the less acidic N–H bond failed to undergo PT, precluding ET and radical generation. The observed diastereomeric outcome of the cascade cyclization is understood when considering the necessarily syn-approach of the sulfonamidyl radical to the top face of the enamide as drawn (see Scheme 13). Two possible approach geometries of the tethered olefin are possible, but only that leading to the 1,2-anti-carbosulfonamidation product was observed. The upper face of the piperidone as drawn is blocked by the tosyl-protected indoline group, thus disfavoring this approach.
Scheme 11. Design Plan for the Radical Cascade Approach to a Collection of 33 Indole Alkaloid Natural Products (Qin, 2017).

Scheme 13. Stereochemical Model for Radical Cascade Cyclization (Qin, 2017).

As a representative example of a path A assembly, sulfonanilide 12.1 was subjected to optimized photocatalytic reaction conditions, yielding tetracycle 12.2 in 81% yield in 1.5:1 d.r. on 14 g scale (Scheme 12). From this common intermediate, a number of eburnamine–vincamine family alkaloids were accessible, such as (−)-eburnamine (12.3), (−)-vincamine (12.4), and (−)-vallesamidine (12.5), in six, seven, and nine further synthetic steps, respectively. Members of the yohimbine family of alkaloids were accessible through a path C assembly (Scheme 14). Sulfonanilide 14.1 was treated under optimized photocatalytic conditions in the presence of methyl vinyl ketone and conducted on 12 g scale. Subjecting the crude product of this radical cascade reaction to p-TsOH in PhMe at 60 °C facilitated Robinson annulation to give pentacycle 14.2 in 56% yield and 3.8:1 d.r. over these two steps. From here, (−)-yohimbine (14.3) was accessible in a further 11 synthetic steps, and (+)-rauwolscine (14.4) in a further nine operations. Additional access to members of the corynanthe and heteroyohimbine families were also demonstrated through similar path C assemblies, where a propargylic amine moiety underwent radical cyclization to yield these olefin containing alkaloid natural products.
Scheme 12. Path A Type Assembly for the Synthesis of Eburnamine–Vincamine Family Alkaloids (Qin, 2017).

Scheme 14. Path C-Type Assembly for the Synthesis of Yohimbine Family Alkaloids (Qin, 2017).

In 2018, Qin and co-workers extended this strategy to the synthesis of members of the eburnane indole alkaloid natural products through a Path A-type cascade polycyclization reaction (Scheme 15).142 In this report, the sulfonamidyl radical is utilized to construct the B, C, and D rings of a common intermediate 15.2 toward the alkaloid natural products. The group reported the divergent synthesis of five eburnane natural products including (−)-eburnaminol (15.3), (+)-larutenine (15.4), (−)-terengganensine B (15.5), and (−)-strempeliopine (15.6) in 6–13 steps from common intermediate 15.2. A sulfonamidyl radical is proposed to be generated though stepwise deprotonation by the KHCO3 base and oxidation by excited state of the [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) photocatalyst. The sulfonamidyl radical then undergoes a 5-exo-trig cyclization onto the pendant enamide forming the initial C–N bond, followed by a 6-exo-trig addition to the acrylonitrile acceptor. Reduction and protonation of this species furnish intermediate 15.2 in 75% yield.
Scheme 15. Path A-Type Assembly for the Synthesis of Eburnane Natural Products (Qin, 2018).

2.1.1.2. Electrochemical Reactivity
In a series of elegant synthetic and mechanistic studies, the Moeller group described their efforts toward the intramolecular cyclization of electron-rich alkenes and tethered nitrogen nucleophiles facilitated by anodic oxidation. Initial attempts centered on the oxyamidation of electron-rich alkenes such as ketene dithioacetals, vinyl sulfides, and enol ethers, with a tethered sulfonamide and exogenous methanol (Scheme 16).143,144 Anodic oxidation of these substrates was conducted under constant current conditions in an undivided cell outfitted with a reticulated vitreous carbon (RVC) anode and a Pt wire cathode in a 30% MeOH/THF solution with Et4N+TsO– as a supporting electrolyte and 2,6-lutidine as a mild Brønsted base additive. Under these conditions, high substrate conversion but low yield of the desired product was observed, and the authors noted oxidative decomposition of the alkene. The overall reaction efficiency could be improved by instead operating under strongly basic conditions. The inclusion of LiOMe or alternatively n-BuLi to generate LiOMe in situ significantly improved the yield of the desired oxyamidation product. Under the optimized reaction conditions, pendant p-toluenesulfonamides were found to cyclize onto a variety of electron-rich alkenes in a 5-exo-trig fashion, with nine examples reported in 50–90% yields (16.1–16.6).
Scheme 16. Electrochemical Intramolecular Cyclization of Sulfonamides and Electron-Rich Olefins (Moeller, 2008).

Under strongly basic conditions which promote discrete PT from the sulfonamide substrate, the authors postulated two alternative mechanisms leading to the product: (i) anodic oxidation of the deprotonated sulfonamide, generating a neutral sulfonamidyl radical, or (ii) anodic oxidation of the alkene moiety to the corresponding radical zwitterion intermediate. In scenario (i), the sulfonamidyl radical undergoes cyclization by addition onto the alkene. In scenario (ii), the deprotonated sulfonamide undergoes nucleophilic addition to the alkene radical cation. Both of these pathways generate a neutral C-centered radical which is further oxidized to the corresponding carbocation and subsequently intercepts alcohol solvent to furnish the overall oxyamidation product.
To distinguish between alkene radical cation and neutral N-centered radical pathways, the Moeller group designed an intramolecular competition experiment wherein a sulfonamide and alcohol were each appended to an enol ether, vinyl sulfide, or ketene dithioacetal (Scheme 17).145 Weaker bases such as 2,6-lutidine exclusively generated tetrahydrofuran (THF) products indicating alcohol trapping, consistent with a mechanism proceeding through an alkene radical cation intermediate. However, stronger bases such as LiOMe that deprotonate the sulfonamide favored pyrrolidine formation resulting from neutral sulfonamidyl radical cyclization. The authors found that the ratio of THF to pyrrolidine products of these two pathways decreased with increasing solvent polarity for the enol ether and ketene dithioacetal substrates. This was interpreted as evidence for the capture of an alkene radical cation by a sulfonamide anion. For the vinyl sulfide however, none of the THF product was observed regardless of the solvent polarity, leading them to suggest that the cyclization is better described as the addition of an N-centered radical to a neutral alkene.
Scheme 17. Intramolecular Competition Experiments to Study Sulfonamidyl Radical, and Olefin Radical Cation Pathways for Cyclization (Moeller, 2010).

In a joint experimental and computational study conducted in 2012, the Moeller group gained further insights into the competition between the sulfonamidyl radical and alkene radical cation pathways for cyclization.146 Based on the results of this study, it was proposed that the mechanism leading to sulfonamide cyclization products is best described as a stepwise sequence of PT and ET to form a neutral sulfonamidyl radical, which then adds to the electron-rich alkene. The alcohol trapping products, on the other hand, were hypothesized to arise from an intramolecular ET from the sulfonamidyl radical to furnish an alkene radical cation capable of capturing the alcohol. In this work, the authors also observed a temperature dependence to the ratio of alcohol vs sulfonamide trapping products under strongly basic conditions. Low reaction temperatures favored the alcohol trapping products, while higher temperatures led to a majority of sulfonamide cyclization products. This temperature dependence can be rationalized by two possible explanations: first, the entropic energy barrier (ΔS⧧) may be smaller for the sulfonamide cyclization pathway than the alcohol trapping pathway, kinetically favoring the sulfonamide products at high temperatures and alcohol products at low temperatures. Alternatively, the higher temperature reactions may allow for equilibration to the thermodynamic product, requiring reversible trapping of the olefin radical cation. To probe whether the cyclization step was reversible, the authors conducted a series of current density experiments. The degree of reversibility for the cyclization of a tethered nucleophile (sulfonamide or alcohol) onto the olefin radical cation is dictated by the competition between the forward rate of oxidation of the radical intermediate following cyclization to the cation, and the reversion of the cyclization. By varying the current density of the reaction, the rate of oxidation of the cyclized radical can be varied, and the reaction can be toggled between the kinetic and thermodynamic pathways when rate of forward oxidation outcompetes that of reversion. This was borne out experimentally, with low current densities favoring the thermodynamic sulfonamide cyclization products and high current densities favoring the kinetic alcohol trapping products.
Capitalizing on their understanding of the reaction mechanism of electrochemical olefin oxyamidation, the Moeller group expanded the substrate scope of this transformation to include N-aryl carboxamide and O-benzyl hydroxamate substrates (Scheme 18).147 The anodic oxidation of these substrates was conducted in an undivided cell outfitted with a RVC anode and a Pt wire cathode. The reaction was carried out in a methanol solution with Et4N+TsO− as the electrolyte and LiOMe as base under constant current conditions at room temperature. Under these conditions, nine different δ- and γ-lactams were obtained in 41–88% yield. When the N-phenyl amide or O-benzyl hydroxamate tethered to the ketene dithioacetal was subjected to the reaction conditions, products 18.1 and 18.2 were observed in 87% and 80% yield, respectively. High reaction efficiencies were maintained in the 6-exo-trig manifold, with γ-lactam 18.3 forming in 73% yield. When an allylic methyl group was installed on the tether, high diastereoselectivity (>20:1) was observed in the product (18.6, 78% yield). Finally, vinyl sulfides and enol ethers could be utilized as the electron-rich olefin, albeit with a lower yield (e.g., 18.4, 41% yield).
Scheme 18. Electrochemical Intramolecular Cyclization of Carboxamides and Electron-Rich Olefins (Moeller, 2014).

The authors propose that the mechanism proceeds via discrete deprotonation of the N–H bond of the substrate by methoxide and subsequent oxidation of the conjugate base of the amide or hydroxamate to the N-centered radical. Following radical addition to the tethered olefin, the resulting C-centered radical is oxidized at the anode to a carbocation. Nucleophilic addition by a molecule of the solvent furnishes the final product. O-cyclization was instead observed in the case of 18.5; with a less acidic N–H bond of the N-methylamide substrate, alkene oxidation is proposed to occur followed by nucleophilic addition of the neutral amide.
Xu and co-workers in 2014 reported an electrochemical intramolecular oxyamidation reaction of unactivated alkenes which occurred through a proposed concerted PCET pathway for N–H bond homolysis (Scheme 19).148 Earlier examples of electrochemical amide and sulfonamide cyclization reactions with alkenes reported by the Moeller group and described above were limited to electron-rich enol ethers and dithioketene acetals.143−147 This reaction system involved the electrolysis of N-aryl carbamate- and amide-tethered di- and trisubstituted olefins in the presence of TEMPO and either carbonate or hydroxide bases as stoichiometric additives. These conditions gave access to oxazolidinone (19.1, 19.2) or lactam (19.3) products respectively, with vicinal addition of the tethered N-nucleophile and TEMPO across the substrate olefin in a highly trans-selective fashion. The cell assembly consisted of an RVC anode and Pt cathode operating under constant current conditions, with n-Bu4N+BF4– as a supporting electrolyte in an aq. MeCN solvent system at 60 °C. Na2CO3 proved to be the optimal base for both reactivity and Faradaic efficiency. This reaction was typically highly efficient and unperturbed by the electronic properties of the N-aryl group. A scope of 21 examples of the electrochemical oxyamidation reaction of cyclic alkenes was reported. 1,2-Difunctionalized products were obtained in yields of 51–98% and typically exclusive trans-diastereoselectivity (19.1–19.3). An additional six examples of acyclic olefin oxyamidation were reported in yields of 58–97%, but in this substrate class, diastereoselectivity was typically poor (e.g., 19.4). The reaction was limited to 5-exo-trig cyclization reactions and N-aryl amide substrates.
Scheme 19. Electrochemical Intramolecular Oxyamidation of Olefins with N-Aryl Carbamates and Amides (Xu, 2014).

The authors observed a marked base dependency in the reaction, and the requirement for an aqueous co-solvent. CV studies revealed that TEMPO is preferentially oxidized to the corresponding oxoammonium salt (E1/2 = +0.70 V vs Ag/AgCl in MeCN)148 compared to the carbamate substrate (e.g., for cyclohexenyl N-phenyl carbamate, Ep/2ox = +1.53 V vs Ag/AgCl in MeCN).148 A mechanistic proposal therefore involved initial oxidation of TEMPO to the corresponding oxoammonium salt, which then in turn mediates a concerted PCET event with the carbamate substrate, with either exogenous carbonate or electro-generated hydroxide from the aqueous co-solvent. A stepwise pathway is unlikely to be operative due to the large offset in pKa between the substrate N–H group (e.g., for methyl N-phenyl carbamate, pKa = 21.5 in DMSO)116 and hydroxide. The resultant N-centered radical undergoes rapid 5-exo-trig cyclization and C-centered radical generation before subsequent trapping with TEMPO radical regenerated through the PCET substrate activation step. Independently prepared TEMPO oxoammonium salt in the presence of Cs2CO3 was also able to facilitate this transformation. However, no yield was observed in the absence of a basic additive.
Xu and co-workers later studied the analogous oxyamidation reaction of alkyne-tethered N-(hetero)aryl amides in a 2020 report (Scheme 20).149 Now, under similar electrolytic conditions to their first report,148in situ cleavage of the initial TEMPO adduct of the oxyamidation reaction was observed, and acylated N-aryl oxazolinone products were isolated. A scope of 26 examples was reported with yields ranging from 31% to 89%. The reaction tolerated various N-aryl substituents (20.1–20.3), including 2-pyridyl-substituted 20.2. In the case of an α,α-dimethyl propargylic carbamate substrate, where no α-proton is available to yield the oxazolinone through elimination, instead a hydration product 20.3 was formed. Furthermore, the authors reported six examples of urea substrates, which efficiently formed imidazol-2-one products.
Scheme 20. Electrochemical Intramolecular Oxyamidation of Alkynes with N-Aryl Carbamates and Ureas (Xu, 2020).

*With 2.0 equiv of NaOAc instead of NaO2CCF3. **Without NaO2CCF3.
A proposed mechanism aligns with their 2014 report,148 proceeding through N-centered radical generation and now 5-exo-dig cyclization, generating a vinyl radical intermediate which is trapped with TEMPO. TEMPO serves as a redox catalyst for mediated electrochemical concerted PCET in this proposal. The initial product undergoes N–O bond scission through a proposed polar process initiated by the N-lone pair to generate an N-acyliminium intermediate, which further reacts through deprotonation or hydration to the isolable products.
Wirth and co-workers designed an electrochemical flow microreactor and demonstrated its utility in a variety of olefin 1,2-amido functionalization reactions.150 Conducting synthetic electrochemistry in flow has potential benefits over batch-mode operations, such as (i) shorter distances between electrodes leading to improved Faradaic efficiency and reduced or eliminated quantities of supporting electrolytes, (ii) a high electrode surface area to reaction volume ratio, leading to shorter reaction times, and (iii) the more straightforward scaling out of a reaction by simply running the process for longer, as opposed to scaling up the size of the reaction vessel.151 The reactor was designed to be easily machined or 3D-printed and consisted of a 25 cm2 anode and cathode separated by a fluorinated ethylene propylene (FEP) polymer film spacer with flow channels cut into the material. The spacer acts as a seal and defines the distance between electrodes (typically 100–500 μm).
Wirth and co-workers first studied the oxyamidation of an alkene-tethered N-phenyl carbamate using this flow setup, a reaction that was first reported in a batch operation by Xu and co-workers in 2014.148 Whereas batch mode electrolysis required 1.0 equiv of Na2CO3 base and 2.9 equiv of n-Bu4N+BF4– electrolyte relative to carbamate substrate operating at 60 °C to obtain oxyamidation product 19.1 in 90% yield, in this flow apparatus a sub-stoichiometric amount of BnMe3N+OH– (50 mol%) served both of these roles, forming the same product in 84% yield at room temperature. A Pt anode and graphite cathode operating under constant current conditions in a mixed solvent system of MeCN/H2O (19:1) was optimal. These optimized conditions were then employed in the synthesis of isoindolones in flow via the cyclization of 2-styryl N-aryl benzamides, with 11 examples documented in yields of 18–96% (Scheme 21). Mechanistically, these are proposed to involve discrete deprotonation of the amide N–H bond by the hydroxide base followed by anodic oxidation to form an amidyl radical. 5-Exo-trig cyclization then generates a distal C-centered radical which is trapped by TEMPO to yield the product. The reduction of water at the cathode regenerates hydroxide and leads to the evolution of molecular hydrogen.
Scheme 21. Olefin Amido-Functionalization in an Electrochemical Flow Microreactor (Wirth, 2017).

Flow chemistry also permits the telescoping of reactor output streams into subsequent operations. In this work, two such elaborations were demonstrated. Following electrolysis and oxyamidation as above, the output stream was passed through a reactor coil held at 85 °C, leading to the elimination of the TEMPO group mediated by the same base that was required in the electrolysis reaction. This enabled the synthesis of formal vinyl C(sp2)–H amidation products, with six examples demonstrated in yields of 62–75% over two steps (21.2). Finally, the reduction of the N–O bond of the product TEMPO adducts was demonstrated by introducing an aqueous acetic acid reagent stream after electrolysis and passing this combined output through a packed Zn cartridge held at 40 °C. Here, three examples of hydroxyamidation products were given in yields of 54–80% over these two steps (21.3).
Similarly, in 2018 Ahmed and Khatoon reported the electrochemical intramolecular oxyamidation of N-allyl-N-aryl ureas (Scheme 22).152 Constant current electrolysis of these substrates was performed in an undivided cell equipped with an RVC anode and Pt cathode with the addition of TEMPO and n-Bu4N+BF4– supporting electrolyte in 5% aq. MeCN at 60 °C. Seven examples of cyclic urea products were reported in yields of 77–85%. A stepwise mechanism for N-centered radical generation was proposed involving substrate deprotonation by cathodically generated hydroxide followed by anodic oxidation of the resultant anion (22.1–22.3). Thereafter, cyclization and trapping with TEMPO in an analogous fashion to the work of Xu148 yields the closed-shell product. The group later extended this reaction to an electrochemical flow cell.153
Scheme 22. Electrochemical Synthesis of Cyclic Ureas (Ahmed, 2018).

In 2016, the Xu group disclosed a method to enable intramolecular olefin hydroamidation via formal N–H homolysis under electrocatalytic conditions, notably using ferrocene (Fc) as a redox catalyst for the generation of an N-centered radical via a stepwise PT/ET strategy (Scheme 23).154 To affect this transformation, substrate electrolysis was performed in an undivided electrochemical cell employing a glassy carbon anode and Pt cathode operating under constant current conditions in the presence of n-Bu4N+BF4– electrolyte, in a mixture of THF and MeOH (5:1) at reflux. 1,4-Cyclohexadiene was included acting as terminal reductant for HAT. In total, 23 examples were reported with yields ranging from 64%−96%. The method was shown to be effective on olefin-tethered N-aryl carbamates, amides (23.1), and ureas (23.2). The reaction tolerated primary alcohols (23.3) and sulfonamides (23.4), while also displaying a broad scope of N-aryl groups, including methoxy- (23.1, 23.2) and cyano-substituted arenes (23.5).
Scheme 23. Electrocatalytic Intramolecular Olefin Hydroamidation (Xu, 2016).

The reaction is proposed to initiate through anodic oxidation of Fc to ferrocenium and corresponding cathodic reduction of methanol to methoxide and H2. Methoxide deprotonates the substrate N–H bond, which is then oxidized by ferrocenium to the N-centered radical. This step is supported by a complete lack of reactivity observed in the absence of the Fc redox mediator. Subsequent 5-exo-trig cyclization onto a pendant olefin forms the C–N bond, affording a distal C-centered radical. This abstracts the weak C–H bond of sacrificial 1,4-cyclohexadiene (C–H BDFE = 67.8 kcal mol–1)22 to give the product. The use of THF as the primary solvent was considered to be key in the reaction by modulating the redox potentials of the catalyst and substrate. CV studies indicated that the oxidation potential of the substrate was lowered by switching to a 5:1 mixture of THF/MeOH from neat MeOH (e.g., for carbamate leading to 23.5, Ep/2 = +0.83 V vs SCE in MeOH compared to Ep/2 = +0.61 V vs SCE in THF/MeOH (5:1)), while the oxidation potential of Fc was raised (E1/2 Fc+/Fc = +0.37 V vs SCE in MeOH, compared to E1/2 Fc+/Fc = +0.55 V vs SCE in THF/MeOH (5:1)), thereby enabling mediated ET between these species. The use of methoxide generated in situ as Brønsted base was also shown to be important, as no current was observed in its absence or with Na2CO3 alone, supporting their proposal of a stepwise PT/ET mechanism.
In an extension of this work, Xu and co-workers applied the same electrocatalytic reaction conditions to amidyl radical generation for the dicyclization of acyclic dienyl carbamates in the synthesis of trans-oxazolidinone-fused cycloheptanes (Scheme 24).155 In total, 14 examples of dicyclization in this manner were given, in 40–75% yields. Only N-aryl carbamates (24.1, 24.3) and ureas (24.2) were compatible under these conditions, in line with their previous report.154 Upon formation of an amidyl radical intermediate through mediated electrolysis, consecutive diastereoselective 5-exo-trig cyclization, followed by an unusual 7-endo-trig radical cyclization onto two tethered alkenes form the fused-bicyclic skeleton, and a C-centered radical on the cycloheptane ring. This radical species then abstracts a H-atom from 1,4-cyclohexadiene to furnish the closed-shell product. DFT calculations indicated that the transition state for the 7-endo-trig cyclization favors the cis-product by 1.6 kcal mol–1. This transition state is hypothesized to avoid a repulsive interaction between the arene appended to the oxazolidinone and the substituent bonded to the carbon bearing the SOMO. It was additionally shown that the 7-endo-trig cyclization exclusively occurs with terminal olefins. Reaction of a 1,2-disubstituted alkene substrate afforded only the 5-exo-trig product 24.4 resulting from a single cyclization.
Scheme 24. Electrocatalytic Intramolecular Carbamate and Urea Dicyclization (Xu, 2018).

Later, the Xu group reported electrothermal reaction conditions to achieve the aza-Wacker-type156 intramolecular amidation of N-aryl amide- and carbamate-tethered olefins with simultaneous desaturation of the products, reforming a transposed olefin regioselectively without further reaction at this newly installed functional group (Scheme 25).157 To enable this transformation, the group used an electrochemical cell assembly consisting of RVC anode and Pt plate cathode operating under constant current conditions, with Et4N+PF6– as a supporting electrolyte in a mixed solvent system of DMA/AcOH (40:1) and at elevated temperatures of 110 °C. The authors observed severely diminished reactivity in the absence of the acid co-solvent or at ambient temperatures (41% and 45% respectively, compared to 83% under optimal conditions for 25.1). A scope of 38 examples of olefin amidation with subsequent product desaturation was reported in yields of 48–97%. Despite the high temperatures, the process possessed excellent functional group compatibility. Several heterocyclic substituents were tolerated (e.g., 25.3) in addition to alkyne conjugated olefins undergoing cyclization to yield versatile enyne products (e.g., 25.2). Typically, good to excellent levels of diastereoselectivity for trans-configured product isomers were observed. Enantiopure substrates underwent cyclization without racemization. N-aryl carbamates and amides were both competent substrate classes, though typically electron-rich PMP groups were employed for optimal reactivity. Tri- and tetra-substituted olefins were shown to undergo this transformation, but with requirement for the distal position supporting the C-centered radical post-cyclization to be tertiary. In all cases, exclusive formation of the less thermodynamically stable olefin isomer resulting from desaturation away from the newly formed C–N bond was observed.
Scheme 25. Electrothermal Intramolecular Aza-Wacker-Type Amidation of Olefins with N-PMP Carbamates and Amides (Xu, 2017).

*Reaction at 130 °C.
This process is proposed to occur through N–H bond homolysis, olefin cyclization with distal C-centered radical generation, and further anodic oxidation to the tertiary carbocation before desaturation via PT. Though the reaction bears similarity to a system described in earlier work wherein PCET is invoked in N-centered radical generation,148 details of elementary steps involved in homolytic bond activation (whether stepwise or concerted PT and ET) for amidyl radical formation and desaturation of the subsequent carbocation following further anodic oxidation remain unclear. Concomitant cathodic reduction of protons results in the liberation of dihydrogen gas as a byproduct.
Subsequently, Hu and co-workers in 2019 optimized an analogous electrocatalytic approach to this aza-Wacker-type olefin amidation (Scheme 26).158 The group investigated the inclusion of Cu(II) additives to this electrochemical transformation with the specific aim of catalyzing the olefination step, lowering the required reaction temperature and significantly expanding the scope of the transformation. Early work from Kochi and co-workers recognized the ability of Cu(II) carboxylate salts to promote the oxidative elimination of transient alkyl radicals for the preparation of olefins,159 and many groups have recently employed copper co-catalysts for the functionalization of C-centered radical intermediates generated through photocatalytic substrate activation, including olefination.160,161
Scheme 26. Electrocatalytic Aza-Wacker-Type Amidation of Olefins with N-Aryl Carbamates and Amides (Hu, 2019).

*MeOH/PhCl (1:1) as solvent, 0.1 M n-Bu4N+TsO– as electrolyte, 4.0 equiv of NaOAc as base, 65 °C. **0.1 M n-Bu4N+TsO– as electrolyte.
A divided electrochemical cell assembly was employed consisting of a carbon fiber anode and Pt cathode operating under constant current conditions, with LiClO4 as a supporting electrolyte and a mixed solvent system of MeOH/CH2Cl2 (1:1) operating at room temperature. The inclusion of a stoichiometric quantity of NaOPiv was key to achieving the desired transformation, as was a sub-stoichiometric loading of Cu(OAc)2. Under these conditions, 26 examples of the aza-Wacker-type oxidative amidation of N-aryl amide- and carbamate-tethered olefins were reported, in yields of 38–81%. Unlike previous reactions, an electron-rich PMP group was not necessary, permitting electron-poor (26.2) to electron-rich N-aryl groups without adversely affecting the reaction efficiency. Similarly, the requirement to form a tertiary radical intermediate upon olefin cyclization to permit uncatalyzed oxidative elimination was also removed, and now substrates proceeding through secondary C-centered radical intermediates (26.1) were competent. In many cases, mono-substituted olefin formation was demonstrated (e.g., 26.1, 26.2, 26.4), which was not possible previously.
The group carried out detailed mechanistic studies to understand the roles of pivalate and Cu additives. A concerted PCET mechanism is invoked wherein the pivalate additive forms a hydrogen-bonded complex to the substrate N–H bond, thereby lowering the required oxidation potential for homolysis; this association and potential shift were studied through the use of CV. The initial oxidation step was not found to be impacted by the addition of Cu(II). Instead, Cu(II) is proposed to intercept the C-centered radical, forming a transient alkyl Cu(III) carboxylate intermediate which then undergoes carboxylate-assisted elimination to yield the product olefin and a Cu(I) species. Further anodic oxidation regenerates the catalytically active Cu(II) oxidation state. The requirement for a non-polar co-solvent was based on the observations from Kochi that this facilitates the Cu-mediated oxidative elimination step. Concomitant cathodic reduction of protons results in the liberation of dihydrogen gas as a byproduct.
The electrochemical synthesis of a number of other heterocycles—indoles, indolines, imidazopyridines, benzimidazolones, and isoquinolin-1(2H)-ones—has been demonstrated through coupled PT and ET leading to N–H bond homolysis. In 2016, the Xu group in collaboration with the Lu group published an oxidative intramolecular alkyne [3+2] annulation reaction, applying a Fc-based electrocatalytic N-centered radical generation strategy to the synthesis of (aza)indoles (Scheme 27).162 Reaction conditions involved constant current electrolysis of substrates in an undivided cell with RVC anode, Pt cathode, n-Bu4N+BF4– supporting electrolyte, and solvent mixture of THF/MeOH (5:1). Here, 36 examples were reported with yields between 43% and 94%. An N,N′-diarylurea substrate was necessarily required, but the reaction was shown to be not particularly sensitive to electronics of either arene moiety, nor of the urea N-group (27.1, 27.2). The method also showed little sensitivity to the identity of the alkyne substituent (27.3–27.5), though only moderate yield was seen for a terminal alkyne (27.6).
Scheme 27. Electrocatalytic Intramolecular Indole Construction (Xu and Lu, 2016).

*From terminal alkyne (R4 = H). **From TMS-alkyne (R4 = SiMe3).
The authors propose that an amidyl radical intermediate arises through substrate deprotonation by cathodically generated methoxide and oxidation by anodically generated ferrocenium. Subsequent 6-exo-dig cyclization occurs onto the pendant alkyne, before the resultant vinyl radical adds into the N-tethered arene. Finally, oxidation and deprotonation lead to re-aromatization and formation of the indole product. DFT studies demonstrate that the cascade cyclization is energetically downhill and presents reasonable activation barriers—addition of the vinyl radical into the arene is proposed to be rate-limiting based on these studies.
In an extension, Xu and co-workers later reported the related oxidative [3+2] annulation of alkene-appended anilides, for the construction of indolines (Scheme 28).163 The reaction conditions were similar to those reported for their indole protocol.162 Here, 44 examples of indoline construction were given, in yields of 47–86% (28.1–28.4). The method was effective for di-, tri-, and tetra-substituted olefins, as well as for the formation of azaindolines from pyridine and pyrimidine-derived substrates. The method was performed on a 7 g scale as a key step for the preparation of an intermediate (28.4) toward the total synthesis of the alkaloid natural product hinckdentine A.
Scheme 28. Electrocatalytic Intramolecular Indoline Construction (Xu, 2018).

*With 1.0 equiv of K2CO3 instead of 0.5 equiv of NaOAc.
A similar mechanism consisting of sequential discrete PT and ET steps from methoxide and anodically generated ferrocenium respectively generates an amidyl radical intermediate. This undergoes 6-exo-trig cyclization with the appended alkene, resulting in a C-centered radical, which adds into the arene to form a delocalized cyclohexadienyl radical. This species undergoes sequential oxidation and deprotonation to restore aromaticity and furnish the indoline product.
The next year, the Xu group extended this method to the polycyclization of diynes, enabling a synthesis of nitrogen-containing polycyclic aromatic hydrocarbons (Scheme 29).164 This class of compounds is finding increasing importance in materials science applications and can be difficult to access in a modular fashion.165,166 With minor modifications to the above conditions reported for their indole cyclization method,162 namely a lower ratio of THF to MeOH, lowered electrolyte loading, and higher applied constant current, the researchers were able to affect the cascade polycyclization, with 37 examples reported in yields ranging from 32% to 82%. Urea substrates proved to be the only effective precursors in this protocol. The method was shown to be tolerant of a broad scope of (hetero)aromatic substitution patterns and substituents (29.1–29.2), though it was shown that when arene C is particularly electron-deficient, premature radical termination was observed, forming only the hydroamidation product. The scope was additionally extended to encompass terminal alkynes (29.3) and was shown to be effective on decagram scale with only a modest diminishment of yield (29.4). The reaction is proposed to proceed as described previously.
Scheme 29. Electrocatalytic Intramolecular Polycyclic N-Heteroaromatic Synthesis (Xu, 2017).

Xu and Lu reported an electrocatalytic synthesis of imidazo-fused N-heteroaromatic compounds from alkyne-tethered heteroarylamine carbamates, through a formal [3+2] annulation reaction featuring two C–N bond-forming steps (Scheme 30).167 The initial cyclic carbamate product of the electrocatalytic annulation was found to hydrolyze with release of CO2 under the conditions of reaction for form the final imidazo-fused N-heteroaromatic product. The authors noted that Fc, used previously by this group as a redox catalyst, was ineffective in this method, leading to the use of a tetraarylhydrazine as an organic redox catalyst. These are a promising class of redox catalysts due to their modular nature and tunability of redox properties through structural modification. Optimal conditions enabling this transformation involved the constant current electrolysis of carbamate substrates in an undivided cell equipped with RVC anode and Pt plate cathode in a mixed solvent system of MeCN/H2O (9:1) under reflux, in the presence of the tetraarylhydrazine 30.A redox catalyst, NaHCO3 additive, and Et4N+BF4– electrolyte.
Scheme 30. Electrocatalytic Synthesis of Imidazo-Fused N-Heteroaromatic Compounds (Xu and Lu, 2018).

*With 5 mol% 1, 1.0 equiv of NaHCO3, in THF/MeOH (3:1), reflux. **With 0.5 equiv of NaHCO3.
A broad substrate scope was presented, with 40 examples of imidazo-fused heteroaromatic products derived from carbamate substrates prepared in 42–94% yields. Tolerance of many functional groups to the reaction conditions was demonstrated, including halogen, nitrile, ester, ether, and trifluoromethyl groups at multiple points of substitution. Imidazo[1,2-a]pyridines (e.g., 30.1, 30.2) were accessible from 2-aminopyridine-derived substrates, imidazo[1,2-b]pyrimidines (30.3) from 3-aminopyridazines, both an imidazo[1,2-a]pyrimidine and an imidazo[1,2-c]pyrimidine (30.4, 30.5) from 2- and 4-aminopyrimidines, respectively, and imidazo[1,2-a]pyrazines (30.6) from aminopyrazines. In addition, imidazo[2,1-b]benzothiazoles (30.7) were accessible from 2-aminobenzothiazoles. Typically, substrates carried a bulky i-Pr or t-Bu group in the proximity of the carbamate linker to assist in cyclization, but a methyl-appended substrate was efficient in product formation. Urea-tethered substrates were also effective under slightly modified reaction conditions, with a further eight examples presented in 15–81% yields, giving methyl carbamate products through methanolysis of the initial cycloadducts.
A mechanism was proposed based on voltammetry and DFT studies (UB3LYP/6-31G*, gas phase) involving initial anodic oxidation of the tetraarylhydrazine redox catalyst (Ep/2 = +0.68 V vs SCE in MeCN/H2O (9:1)),167 yielding the corresponding radical cation. Meanwhile, hydroxide produced through cathodic reduction of the aqueous co-solvent deprotonates the carbamate, and the corresponding substrate anion is oxidized by the hydrazine radical cation to the key amidyl radical intermediate. The oxidation potential of the carbamate anion (e.g., for the N–H carbamate substrate leading to 30.1, Ep/2 = +0.66 V vs SCE in MeCN containing 1.0 equiv of n-Bu4OH)167 is significantly negatively shifted compared to the neutral substrate (Ep/2 = +1.76 V vs SCE in MeCN).167 A 5-exo-dig cyclization then forms the first C–N bond (ΔG⧧ = +6.5 kcal mol–1) with generation of a vinyl radical intermediate. Regioselective cyclization of the vinyl radical onto the pendant heterocycle through C–N bond formation (ΔG⧧ = +16.2 kcal mol–1), as opposed to C–C bond formation (ΔG⧧ = +22.6 kcal mol–1) yields a stabilized radical product. Finally, one-electron oxidation of the product and hydrolysis of the carbamate linker yields the product imidazopyridine. The use of the redox catalyst instead of direct electrolysis protects the final product (Ep/2 = +1.20 V vs SCE in MeCN)167 from oxidative decomposition.
A later study of this reaction with ortho-substituted arylacetylenes as the tethered-alkyne component revealed a diastereoselective approach to axially chiral imidazopyridines.168 Here, 20 examples of the atroposelective reaction variant were additionally reported, in 43–91% yields and d.r. of 5:1 to >20:1 under the same electrocatalytic conditions as above. Bulky tert-alkyl groups appended to the carbamate tether and to the ortho-position of the aryl group were necessary requirements for a highly diastereoselective outcome. A Curtin–Hammett scenario is invoked to account for the observed diastereoselectivity, wherein a low barrier to interconversion of two conformers of the vinyl radical intermediate permits rapid equilibration prior to final irreversible C–N bond formation with a higher activation barrier.
In 2019, Xu and co-workers developed a synthesis of benzimidazolones and benzoxazolones through a cascade cyclization of urea or carbamate-tethered enyne substrates, initiated via N–H bond homolysis (Scheme 31).169 The optimized reaction conditions for this transformation involved the constant current electrolysis of enyne substrates in an undivided cell equipped with RVC anode, Pt plate cathode, TFA as a Brønsted acid, and Et4N+PF6– as a supporting electrolyte in DMF at 110 °C. Exogenous basic additives were avoided, as the alkyne-tethered carbamate and urea substrates were found to be susceptible to base-mediated polar alkyne hydroamidation. The high reaction temperature is proposed to be necessary to overcome the energy barrier (ΔG⧧ = 19.0 kcal mol–1) for the substrate to adopt an unfavorable trans,cis-conformation required prior to 5-exo-dig cyclization.
Scheme 31. Electrochemical Synthesis of Benzimidazolones and Benzoxazolones through a Cascade Cyclization (Xu, 2019).

*Without TFA.
The researchers reported 36 examples of benzimidazolone synthesis from urea precursors in yields of 20–83%. Although electron-rich N-(4-methoxyphenyl)ureas were the most studied N-radical precursors in this work, cyclization also efficiently occurred from electronically neutral and electron-deficient N-aryl ureas. With respect to the enyne component, internal alkynes were the most effective substrates (31.1–31.4), with a terminal alkyne example proceeding poorly (31.5, 20%). A variety of 1,1-disubstituted and trisubstituted alkene partners were demonstrated, including alkyl olefins and styrenes. Notably, this method enables access to fully substituted benzimidazolone scaffolds. Under modified reaction conditions (constant current electrolysis in TFE solution containing AcOH as an additive, at 80 °C), 10 examples of benzoxazolone synthesis from carbamate precursors were also reported, in yields of 44–88%.
A mechanistic proposal involved anodic oxidation (Ep/2ox = +0.96 V vs SCE in DMF)169 of the neutral substrate followed by deprotonation by cathodically generated trifluoroacetate (or acetate) base. In situ generation of the weak base (e.g., for trifluoroacetate, pKaH = 3.5 in DMSO)116 was required to avoid the base-mediated polar alkyne hydroamidation side reaction. This sequence leads to N-centered radical generation, which undergoes 5-exo-dig cyclization with the tethered alkyne to generate vinyl radical. From here, two potential pathways for a second cyclization were proposed: (i) a direct 6-endo-trig cyclization or (ii) a three-step sequence involving 5-exo-trig cyclization, 3-exo-trig cyclization, and ring expansion through cyclopropylmethyl radical ring opening. DFT calculations suggested that the nature of the kinetically preferred pathway appeared to be substrate-dependent, with a 1,1-disubstituted olefin displaying preference for the 6-endo-trig mode, and a trisubstituted olefin displaying preference for the 5-exo-trig mode. However, these pathways lead to the same tertiary alkyl radical intermediate on a cyclohexene ring. After an initial ET/PT step, the cyclohexene intermediate formed undergoes dehydrogenation through two further iterations of anodic oxidation and deprotonation to yield the benzene core of the benzimidazolone (or benzoxazolone). The product of the reaction has a sufficiently higher oxidation potential (Ep/2ox = +1.26 V vs SCE in DMF)169 than substrate to shield it from oxidative degradation.
Wen and Li in 2020 leveraged the reactivity of N-centered radicals for the preparation of isoxazolidine-fused isoquinolin-1(2H)-ones via dicyclization of alkyne-tethered N-benzoyl-O-alkyl-hydroxylamines (Scheme 32).170 The conditions employed involved electrolysis of these substrates under constant current conditions in an undivided cell, consisting of carbon felt (CF) electrodes with n-Bu4N+PF6– as a supporting electrolyte in EtOH/H2O (19:1) at 80 °C. Here, 40 isoquinolin-1(2H)-ones were synthesized in yields of 35–97%. The reaction tolerates broad variation of substitution at the ortho-, meta-, and para-positions on the N-benzoyl group, though meta-substituted examples offered poor regioselectivity in cyclization. In addition, a number of arylacetylene (32.1), enyne (32.2), and diyne (32.3) substrates were competent in engaging with the N-centered radical intermediate. Also, three additional examples of homologated oxazinane-fused products via instead 6-exo-dig radical cyclization were reported in 82–87% yields (e.g., 32.4).
Scheme 32. Electrochemical Synthesis of Isoxazolidine- and Oxazinane-Fused Isoquinolin-1(2H)-ones (Wen and Li, 2020).

*Constant current = 0.5 mA.
Mechanistically, a stepwise PT/ET pathway is proposed. Electrogenerated hydroxide or ethoxide deprotonates the alkoxybenzamide substrate prior to anodic oxidation of the anion. The resultant amidyl radical undergoes 5-exo-dig cyclization onto the tethered alkyne. Supporting DFT studies indicate that this cyclization step has a free energy of activation of ΔG⧧ = 15.8 kcal mol–1. The vinyl radical thus formed adds to the arene in a 6-exo-trig cyclization. This step had a similarly high activation barrier, calculated at ΔG⧧ = 11.3 kcal mol–1. Further anodic oxidation and PT yields the heterocyclic product.
2.1.2. Intramolecular C–N Bond Formation through Addition to (Hetero)arenes
The Hong group in 2018 described a photocatalytic method for the synthesis of phenanthridinones and 2-quinolones through N–H bond activation via PCET and subsequent arene C(sp2)–H bond amidation in a net oxidative, aerobic process (Scheme 33).171 In this report, a combination of an Ir photocatalyst [Ir(dF(CF3)ppy)2(bpy)]PF6 ([Ir-7]PF6) and n-Bu3MeN+(n-BuO)2(O)PO– were found to jointly mediate the homolysis of the N–H bond in a series of N-aryl amides of biaryl 2-carboxylic acids and subsequent intramolecular arene C(sp2)–H amidation at temperatures of 60 °C. A scope of 18 examples of N-aryl phenanthridinones was reported in yields of 52–90%. Intramolecular C(sp2)–H amidation of electron-rich, -neutral, and -poor arenes (33.2) was feasible, including aryl bromide and iodide functionality. Electron-donating amide N-aryl groups were typically employed (33.2, 33.3). These reaction conditions were also found to promote oxidative quinolone synthesis from a series of trans-N-aryl cinnamides (33.4–33.6). In this substrate class, the photocatalyst is proposed to play a dual role, first enabling olefin isomerization through an energy-transfer mechanism,172 then mediating arene C(sp2)–H amination through N–H bond concerted PCET. In this report, 18 examples of quinolone synthesis were given in yields of 53–79%.
Scheme 33. Photocatalytic Intramolecular Arene C(sp2)–H Amidation with N-Aryl Amides (Hong, 2018).

A transient N-centered radical generated through concerted PCET is proposed to cyclize onto the pendant arene in a 6-exo-trig fashion, generating a stabilized allylic C-centered radical. Molecular dioxygen was proposed to facilitate re-aromatization of the arene through trapping of this radical as the allylic hydroperoxide followed by PT and elimination of hydroperoxide anion. Alternatively, a pathway involving oxidation of the delocalized cyclohexadienyl radical to the stabilized carbocation by either photoexcited-state Ir(III) or molecular oxygen and subsequent PT is also plausible and was considered. In this scenario, Ir(III) is regenerated in an ET step between Ir(II) and molecular oxygen.
Itoh and co-workers in 2020 disclosed an organo-photocatalytic method enabling intramolecular arene C(sp2)–H bond amidation through amidyl radical generation and subsequent cyclization (Scheme 34).173 The combination of 1-chloroanthraquinone (1-Cl-AQN) photocatalyst and K2CO3 Brønsted base, under an air atmosphere in CHCl3 solution under visible-light irradiation gave access to 17 phenanthridinone products, in yields of 69–99%. The reaction proceeded at ambient temperature and tolerated greater variation in the electronic properties of the amide N-aryl group than the method of Hong, including halogenation (34.3) and a benzothiazole (34.4). Notably, two substrates carrying N-alkyl groups failed. Mechanistic studies supported an analogous N–H PCET process to that proposed by Hong.171
Scheme 34. Catalytic Intramolecular Arene C(sp2)–H Amidation with N-Aryl Amides Employing an Organic Photocatalyst (Itoh, 2020).

Waldvogel and co-workers have reported related protocols for the electrochemical synthesis of N-aryl phenanthridinones and carbazoles from 2-biarylamides via the stepwise generation of an amidyl radical intermediate and subsequent intramolecular arene C(sp2)–H amidation (Scheme 35).174,175 These protocols utilize an undivided electrochemical cell assembly with a graphite anode and a Ni cathode, operating under constant current conditions with n-Bu4N+PF6– as a supporting electrolyte in HFIP. In total, 13 examples of N-aryl phenanthridinone synthesis were reported in yields of 40–85%. Typically, electron-withdrawing N-aryl substituents gave optimal reactivity (35.1, 35.2). The reaction was tolerant of halogen (35.3, 35.4), ester, nitro, nitrile, and ketone functional groups, and the transformation was demonstrated on gram scale. In the latter report, 16 examples of carbazole synthesis were presented in yields of 31–86%. In some cases, a 15% aqueous co-solvent was added. N-benzoyl (35.5–35.8) and N-acyl protecting groups both enable the efficient formation of products. An N-protected carprofen derivative (35.7) was also prepared in this work in 65% yield.
Scheme 35. Synthesis of N-Aryl Phenanthridinones (A) and Carbazoles (B) via Electrochemical Intramolecular Arene C(sp2)–H Amidation (Waldvogel, 2018 and 2020).

*With 15% H2O as cosolvent.
The authors’ proposed mechanism for N-aryl phenanthridinone and carbazole synthesis considers N–H bond homolysis through an initial discrete substrate anodic oxidation step, prior to deprotonation by HFIP anion (pKaH = 10.7 in DMSO)116 generated through cathodic reduction of solvent. The resulting amidyl radical cyclizes in a 6-exo-trig manner, then undergoes re-aromatization through further anodic oxidation and deprotonation. CV studies indicated that substrate oxidation is required prior to deprotonation to successfully yield product. Upon amidyl radical generation, a 5-exo-trig cyclization then occurs. Further anodic oxidation and deprotonation lead to re-aromatization as before. Concomitant cathodic reduction of protons leads to the evolution of molecular hydrogen.
Lei and co-workers in 2019 reported an electrocatalytic intramolecular C(sp2)–H sulfonamidation reaction of arylsulfonamide-appended N-aryl indoles for the synthesis of 10H-benzo[4,5]imidazo[1,2-a]indole derivatives (Scheme 36).176 Optimized reaction conditions involved substrate electrolysis in an undivided cell with graphite anode and Ni plate cathode under constant current conditions, with n-Bu4N+BF4– supporting electrolyte and Fc as a redox catalyst in MeCN/EtOH (1:1) at 70 °C. A scope of six examples was presented, in yields of 48–83% (36.1–36.4).
Scheme 36. Electrochemical Synthesis of 10H-Benzo[4,5]imidazo[1,2-a]indoles through Sulfonamidyl Radical Generation and Cyclization (Lei, 2019).

A proposed mechanism involves discrete sulfonamide deprotonation (e.g., for benzenesulfonanilide, pKa = 12.6 in DMSO)177 by cathodically generated ethoxide (pKa = 29.8 in DMSO),178 followed by oxidation mediated by anodically generated ferrocenium. Cyclization of the resulting sulfonamidyl radical onto the indole C2-position generates a stabilized benzylic radical, which is further oxidized and deprotonated to restore aromaticity and furnish the product.
Li and co-workers recently reported an electrochemical dehydrogenative synthesis of benzimidazolones from trisubstituted N,N′-diaryl ureas through an anodic oxidative C–N coupling reaction (Scheme 37).179 Optimized reaction conditions consisted of constant current electrolysis in an undivided cell equipped with an RVC anode, a Pt cathode, Me4N+BF4– as an electrolyte, and NaOMe as a base in MeOH at 60 °C. Carbonate bases also gave rise to product, but with reduced yields compared to methoxide. In this work, 20 examples of oxidative annulation were reported in yields of 65–87%. With respect to the arene undergoing the C–H amidation reaction, variation of the electronic nature of substituents (37.1, 37.2) and the introduction of polycyclic (37.3) and heterocyclic (37.4) aromatic systems had little impact on the reaction efficiency. N-alkyl ureas were not effective substrates, likely due to reduced N–H bond acidity compared to optimal anilide substrates.
Scheme 37. Electrochemical Dehydrogenative Synthesis of Benzimidazolones from Trisubstituted N,N′-Diaryl Ureas (Li, 2020).

Observations were consistent with a mechanism proceeding via the stepwise deprotonation and oxidation of the N–H bond for N-centered radical generation. The addition of TEMPO under otherwise standard conditions gave no product. CV experiments indicated that the oxidation potential of a model substrate (37.1) in the presence of sodium methoxide (Ep/2 = +0.72 V vs Ag/AgCl in MeOH),179 was significantly reduced from that of the neutral substrate or base alone. Following N-centered radical generation, 5-exo-trig cyclization onto the pendant arene generates a delocalized cyclohexadienyl radical, which undergoes further anodic oxidation and deprotonation to restore aromaticity and yield the benzimidazolone product. At the cathode surface, reduction of MeOH liberates molecular hydrogen and regenerates the base, though a stoichiometric quantity of sodium methoxide added at the start of the reaction was required for maximum efficiency.
2.1.3. Intramolecular C–N Bond Formation Proceeding through Aryl or Alkyl Migration
In addition to these above highlighted methods for C–N bond formation proceeding through N-centered radical generation and addition across a tethered olefin or to an arene, several creative methods for C–N bond formation have been devised which involve the migration of an aryl or alkyl group from a remote position of the substrate to the site of nitrogen centered radical generation.180,181 In 2017, Nevado and co-workers demonstrated that transient sulfonamidyl radicals promote aryl group migration with concomitant remote C-centered radical generation in a class of γ,γ-diaryl triflamides (Scheme 38).182 Following C–N bond formation and remote radical generation in this manner, the group demonstrated subsequent C–C bond formation by trapping with Michael acceptors and Nozaki allyl sulfone reagents, C–H bond formation via HAT from a silanethiol additive, and finally C–Br bond formation via HAT from BrCCl3. The combination of Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6([Ir-6]PF6) and a stoichiometric quantity of K2HPO4 Brønsted base in a mixture of PhCF3/PhCl/H2O (1:1:1) proved optimal for this transformation.
Scheme 38. Remote C-Centered Radical Generation via N–H Bond Homolysis and Subsequent Aryl Group Migration (Nevado, 2017).

*Using 3 equiv of Na2CO3 as base. **Using 2 equiv of Na2CO3 as base.
In total, 25 examples of remote C–C bond formation with aryl migration were reported, in yields of 47–95% (e.g., 38.1). A set of competition experiments between differently substituted γ,γ-diaryl sulfonamides revealed high selectivity for migration of the more electron-rich arene (e.g., 38.2), even when the arenes exhibit similar electronic properties. Replacement of the triflate group with other N-protecting groups was not tolerated. An additional four examples of remote C–H bond formation were demonstrated (38.4) in yields of 68–82%, and two examples of C–Br bond formation (38.5) in yields of 53% and 54% were reported with the addition of TIPS-SH and BrCCl3, respectively.
The group proposed a stepwise mechanism of discrete PT and subsequent ET for N–H bond homolysis due to the high N–H acidity of these triflimide substrates (e.g., for trifluoromethanesulfonamide, pKa = 9.7 in DMSO).116 Following sulfonamidyl radical generation, C–N bond formation occurs in a 5-exo-trig ipso-cyclization onto one of the arene substituents, forming a spiro radical intermediate, which collapses via C–C bond cleavage to yield an N-aryl triflamide and the remote C-centered radical. The observed preference in migratory aptitude of electron-rich arenes is ascribed to polarity matching to the electron-deficient nature of the N-centered radical. In the case of trapping with a Nozaki allyl sulfone reagent, a new C–C bond is formed through an addition–elimination pathway with extrusion of phenylsulfonyl radical. This radical is in turn reduced by the Ir(II) state of the photocatalyst (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN),68 regenerating the ground-state Ir(III) catalyst and phenylsulfinate anion. Other trapping reagents also generate readily reduced radical products (α-acyl, silanethiyl, and trichloromethyl radicals) which interact with the Ir(II) state in an analogous fashion to regenerate the Ir(III) complex. This report is noteworthy in that a mild set of conditions is found for unstrained C–C bond activation, with flexibility offered in the subsequent functionalization of the reactive intermediate.
The synthesis of medium-sized (loosely defined as 8–12-membered) saturated carbocyclic and heterocyclic rings via cyclization of a linear precursor is challenging due to entropic penalties for ring closure.183 A research team led by Ackermann and Ruan in 2020 devised an electrochemical strategy for the synthesis of 8–11-membered benzo-fused β-keto-lactams via N–H bond activation and subsequent C–C bond cleavage (Scheme 39).184
Scheme 39. Electrochemical Synthesis of Medium-Sized Benzo-Fused β-Keto-lactams via C–C Bond Cleavage (Ackermann and Ruan, 2020).

*50 °C.
An electrochemical cell assembly consisting of graphite anode and Pt cathode operating under constant current conditions with n-Bu4N+BF4– as a supporting electrolyte in aq. MeCN at room temperature was employed in this transformation. A series of N-aryl-1-acetamido-1-naphthols were prepared in a single step by adding the lithium dianion derived from the corresponding acetanilide to an α-tetralone. Electrolysis under these conditions led to ipso-arene amination with C–C bond cleavage. The alcohol moiety was further oxidized to the ketone providing β-keto-lactam products through this sequence of ring fusion/ring expansion. With this method, 24 examples of the preparation of nine-membered-ring benzo-fused N-aryl β-keto-lactams in this manner were reported in yields of 30–98% (39.1, 39.2). Typically, an electron-poor N-(3-(trifluoromethyl)phenyl) group was required for maximum efficiency, with electron-neutral and moderately electron-rich substituents giving reduced yields. Substitution at aryl and alkyl positions of the naphthol moiety was tolerated without impact on reaction efficiency. Examples of pyridine-containing substrates are included, and the reaction could be conducted on gram scale with extended reaction times and without major reductions in yield. In addition to these nine-membered-ring lactams derived from tetralones, the group reported two examples of indanone-derived eight-membered-ring lactams (39.3), one example of a chromanone-derived nine-membered-ring lactam product containing a cyclic ether, two examples of benzocycloheptanone-derived 10-membered-ring lactams, and one example of a benzocyclooctanone-derived 11-membered-ring lactam (39.4), all with efficiency comparable to that of the above nine-membered-ring examples.
A plausible mechanism for this transformation was proposed to involve the homolytic activation of the N–H bond. While the authors did not speculate on whether this occurs through a stepwise or concerted PCET mechanism, the reaction conditions are similar to those employed by Xu for the intramolecular amidooxygenation of olefins,148 who invoke a PCET step facilitated by electrogenerated hydroxide. The strong dependence seen here of the N-aryl group, which impacts the acidity of the amide N–H bond, could suggest that a stepwise pathway may be operative. Following bond activation, the resultant amidyl radical undergoes 5-exo-trig cyclization onto the arene at the ipso-position with respect to the naphthol moiety. Supporting DFT studies show that the ipso-5-exo-trig cyclization pathway is energetically favored over an ortho-6-exo-trig pathway by 5.2 kcal mol–1. The resultant radical then triggers C–C bond scission with ring expansion and neutral ketyl radical generation. Finally, this species is converted to the ketone product through further anodic oxidation and deprotonation. The authors note that the possibility of a cationic pathway for ring closure remains. Cathodic proton reduction leads to the liberation of dihydrogen.
In 2020, Guo and co-workers reported an electrocatalytic approach to the heteroarylation of sulfonamides via intramolecular aryl group migration (Scheme 40).185 Optimized reaction conditions for this transformation consisted of the constant current electrolysis of sulfonanilide substrates in an undivided cell equipped with carbon cloth anode and Pt plate cathode, in the presence of N-phenyl phenothiazine (PTZ) (40.A) redox catalyst and n-Bu4N+BF4– electrolyte in MeCN/TFE (6.5:3.5) at 70 °C. A significant decrease in yield was observed without the TFE cosolvent. A total of 23 examples of sulfonamide heteroarylation products were reported in this work in yields of 77–93%. The reaction tolerated broad variation of electronic nature of substituents on the (hetero)arylsulfonyl group: an electronically diverse scope of arenes, heteroarenes, as well as ethyl and cyclopropyl substituents were reported. Benzothiazoles (40.1, 40.2), thiazoles (40.3), and imidazoles (40.4) were all reported to be effective in migration. The reaction proceeded smoothly on gram scale as well.
Scheme 40. Electrocatalytic Sulfonamide Heteroarylation via Heteroaryl Group Migration (Guo, 2020).

The authors conducted CV experiments to probe the mechanism of this aryl migration reaction. For the neutral substrate, no oxidation peak was observed upon scanning up to +2.0 V vs Ag/AgCl in MeCN. Meanwhile, the N-Ph-PTZ catalyst showed a reversible oxidation wave at E1/2 = +0.84 V vs Ag/AgCl in MeCN.185 Since the N-Ph-PTZ catalyst 40.A cannot directly oxidize the substrate, the authors propose that the sulfonanilide is first deprotonated prior to mediated ET, with cathodically generated trifluoroethoxide (pKa = 23.6 in DMSO)116 acting as base for this process. This two-step process generates a neutral N-centered sulfonamidyl radical, which adds in a 5-exo-trig fashion to the tethered heteroarene. The spiro-heterocycle radical intermediate collapses with C–C bond cleavage to release a stabilized neutral ketyl radical, which undergoes either anodic or PTZ-mediated ET to yield the ketone product.
Guo and co-workers later reported an alternate, photocatalytic strategy to promote the same migratory heteroarylation of sulfonamides in this substrate class (Scheme 41).186 Authors note that these conditions avoided the use of stoichiometric tetraalkylammonium salts which were utilized as the electrolyte in the above protocol, which were difficult to separate from the products. N-Ph PTZ—utilized as a redox mediator in the above electrocatalytic method—now functioned as an efficient photoredox catalyst under blue-light irradiation. When subjecting sulfonanilide substrates to these conditions in the presence of Ag2CO3 as a stoichiometric oxidant and KH2PO4 as Brønsted base at 60 °C in dioxane solvent, 23 examples of sulfonamide migratory N-heteroarylation were reported with yields ranging from 19% to 88%. While varying substituents on the sulfonamide functional group did not result in large differences in yields compared to the electrocatalytic method (41.1–41.3), change in the heteroaryl group undergoing migration was significantly less tolerated (41.4, 41.5).
Scheme 41. Photocatalytic Sulfonamide Heteroarylation via Heteroaryl Group Migration (Guo, 2020).

The authors propose that upon visible-light irradiation, excitation of the PTZ photocatalyst followed by oxidative quenching with Ag2CO3 gives access to the PTZ radical cation. A similar stepwise pathway for N-centered radical generation is invoked, involving the additive base and this transient catalyst radical cation. Thereafter, the same sequence of ipso-substitution via C–N bond formation and C–C bond cleavage, and further oxidation of the resultant neutral ketyl radical, leads to product generation.
In 2020, Zhang and co-workers reported an electrocatalytic synthesis of triazolopyridones from 2-hydrazidopyridines via oxidation to the diazocarboxylate followed by cyclization and an unusual enantiospecific C-to-N alkyl migration (Scheme 42).187 Triazolopyridones feature in a number of medicinally relevant molecules, yet a general synthetic route to secondary and tertiary N-alkyl substitution was elusive prior to this report. This transformation was carried out in an undivided cell under constant current conditions with a graphite anode and Pt cathode in the presence of (4-BrC6H4)3N as a redox catalyst and n-Bu4N+BF4– as a supporting electrolyte in MeOH at 70 °C.
Scheme 42. Electrocatalytic Synthesis of N-Alkyl Triazolopyridines via C-to-N Alkyl Migration (Zhang, 2020).

A total of 62 examples were presented in yields of 27–99%. Isolated hydrazidopyridines or those prepared in situ could both be used effectively as substrates. A broad functional group tolerance was showcased. Hydrazides derived from primary (42.1), secondary (42.2), and tertiary alkyl carboxylic acids (42.3) effectively underwent oxidative cyclization and migration. However, no examples of aryl carboxylic acid substrates were presented. This reaction was demonstrated on numerous drug-, amino acid (AA)-, and natural-product-derived substrates, including from diclofenac (42.5) and clofibrate (42.6). When α-chiral secondary alkyl carboxylic acid-derived substrates were employed, an enantiospecific alkyl migration reaction occurred with complete retention of configuration ((R)-42.7). This reaction could be performed on a decagram scale and be applied to the preparation of intermediates en route to analogs of a stearoyl-CoA desaturase inhibitor analog and trazodone antidepressant.
A series of mechanistic experiments were performed. Crossover experiments involving the co-electrolysis of two different hydrazide substrates showed that the C-to-N alkyl migration was an intramolecular event. Two radical rearrangement substrates were prepared and subjected to the optimized electrocatalytic reaction conditions, but yielded products without indicative cyclopropane ring opening or allylbenzene cyclization, respectively, suggesting that the migration step also did not involve radical intermediates. Experiments were also performed to rule out a carbocation intermediate triggering migration. The authors observed a diazocarboxylate intermediate as a byproduct in one reaction. Subjecting this to optimized reaction conditions, or to simple reflux in MeOH, led to conversion to the triazolopyridine product, thus implicating this as an intermediate in the reaction. On the basis of these observations, their mechanistic model involves anodic oxidation of the triarylamine to the corresponding aminium radical cation (E1/2ox = +1.20 V vs Ag/AgCl in MeOH)187 which mediates single-electron transfer (SET) with the hydrazide substrate (Ep/2ox = +0.84 V vs Ag/AgCl in MeOH)187 to yield the hydrazidium radical cation. Two-steps of deprotonation mediated by cathodically generated methoxide yield a distonic radical anion intermediate which undergoes a further step of mediated ET to generate the diazocarboxylate intermediate. A low barrier cis/trans-isomerization and nucleophilic attack of the pyridine nitrogen onto the carbonyl then triggers the enantiospecific alkyl group migration with retention of configuration. Details on this final step remain somewhat unclear.
2.1.4. Intermolecular C–N Bond Formation through Addition to Alkenes, Alkanes, and (Hetero)arenes
2.1.4.1. With Amides and Sulfonamides through Reactions with Olefins
The following examples of intermolecular reactivity of (sulfon)amides and olefins have been reported, for olefin hydro(sulfon)amidation and the net oxidative [3+2] annulation of sulfonanilides and styrenes. In 2018, Knowles and co-workers reported photocatalytic conditions enabling the intermolecular hydrosulfonamidation of unactivated olefins with arylsulfonamides (Scheme 43).139 Reaction conditions involved Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6([Ir-9]PF6), n-Bu4N+(n-BuO)2(O)PO– Brønsted base co-catalyst, and TRIP thiol as a HAT donor under blue-light irradiation of substrate solutions in PhCF3. These reaction conditions were able to mediate the efficient intermolecular hydrosulfonamidation of a diverse series of functionalized olefins with exclusive anti-Markovnikov selectivity, with 30 examples presented in yields of 15–89%. The reaction typically required electron-rich 4-methoxybenzenesulfonamide to work with high efficiency, but primary and secondary (43.5) sulfonamides carrying this arene were tolerated. Terminal (43.7), 1,1-disubstituted (43.1, 43.3), 1,2-disubstituted (43.2, 43.6), trisubstituted, tetrasubstituted, cyclic, acyclic olefins, and silyl enol ethers (43.4) were demonstrated to serve as competent coupling partners. Silyl enol ether substrates gave facile access to valuable protected vicinal amino alcohol products. The reaction also tolerated ketone, ester, primary alkyl chloride, Boc-amine (43.3), and AA functionality on the olefin partner, and several examples of natural product-derived coupling partners were included (43.7). These reaction conditions also permitted intramolecular reactions through a 5-exo-trig mode of cyclization (see section 2.1.1.1 for details and discussion of the mechanism of operation).
Scheme 43. Intermolecular Hydrosulfonamidation of Unactivated Olefins Proceeding through Concerted PCET (Knowles, 2018).

*With 2 equiv of olefin. **From silyl enol ether.
In 2019, Knowles and co-workers reported a single example of the intermolecular hydroamidation reaction between 4-methoxybenzamide and 2,2,4-trimethyl-1-pentene, leading exclusively to the anti-Markovnikov monoalkylation product 44.1 in 67% yield (Scheme 44).38 This proceeded under the blue-light irradiation of an amide and olefin (4.0 equiv) solution in CHCl3, in the presence of Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6([Ir-9]PF6), n-Bu4N+(n-BuO)2(O)PO– Brønsted base, TRIP thiol as an HAT mediator, and TRIP disulfide additive. Similar reaction conditions also promoted the intramolecular hydroamidation of olefin-tethered N-alkyl amides (see section 2.1.1.1 for details, and discussion of the mechanism of operation). While the authors note that the reaction was limited to this electron-rich benzamide and a simple olefin in excess, this is to our knowledge the only reported example of an intermolecular anti-Markovnikov hydroamidation reaction of an unactivated olefin proceeding under photocatalytic activation.
Scheme 44. Intermolecular Hydroamidation between 4-Methoxybenzamide and 2,2,4-Trimethyl-1-pentene (Knowles, 2019).

In 2020, Zhang, Lei, and co-workers reported an electrocatalytic strategy for the [3+2] annulation of N-protected anilines with styrenes to form indoline derivatives (Scheme 45).188 The reaction was generally carried out on small scale in an undivided cell under constant current conditions with carbon rod anode, Pt cathode, in the presence of DDQ redox catalyst, AcOH Brønsted acid additive, n-Bu4N+BF4– electrolyte, in MeCN/1,2-DCE (2:1) solvent mixtures. The reaction was also shown to be scalable in a flow reactor, showing efficiency with a more economic Ni cathode and lowered loadings of DDQ and AcOH under electrolyte-free conditions. A variety of electron-rich anilides and sulfonanilides were demonstrated to be viable substrates—halogen substitution at the meta-position (45.1) and methyl substitution at the ortho-position (45.2) were both tolerated. A number of N-protecting groups were also reported, including sulfonamides (45.1, 45.2) as well as carbamates (45.3). α-Substitution of the styrene generally improved reaction yields, and the olefin scope was extended to include trans-stilbene (45.4) and 2-isopropenylthiophene (45.5). Addition of catalytic TEMPO at the end of this [3+2] annulation process and further electrolysis in a one-pot, two-step fashion allowed for the direct isolation of the corresponding indole product.
Scheme 45. Electrocatalytic Synthesis of Indolines via Intermolecular [3+2] Annulation (Zhang and Lei, 2020).

*With 1.0 equiv of Na2CO3. **Constant current = 8 mA.
Oxidation of the anilide substrate at the anode (e.g., for N-(4-methoxyphenyl)-4-toluenesulfonamide, E1/2 = +1.49 V vs Ag/AgCl in MeCN/1,2-DCE (2:1))188 followed by deprotonation results in formation of a delocalized aniline radical in a stepwise sequence of ET and PT. The aniline radical then reacts through C2 in an intermolecular C–C bond-forming step with the styrene component, generating a benzylic radical which is oxidized to the corresponding carbocation by either DDQ or at the anode. Intramolecular cyclization and subsequent deprotonation afford the indoline product. Reduced DDQH2 is re-oxidized to DDQ at the anode, while reduction of AcOH at the cathode serves to generate acetate base. Kinetic studies suggest that anilide oxidation is rate-determining. CV experiments provide evidence for the initial anilide oxidation, while radical trapping and electron paramagnetic resonance (EPR) experiments support the existence of an anilide radical intermediate where the radical character is delocalized over the arene. We note the disclosure of a related transformation proceeding through ground-state iron catalysis in the presence of stoichiometric DDQ oxidant, published in 2018 by Zhong and co-workers.189
2.1.4.2. With Amides and Sulfonamides through Alkyl C(sp3)–H and (Hetero)aryl C(sp2)–H (Sulfon)amidation
Pandey and Laha in 2015 reported a photocatalytic method enabling the oxidative inter- and intramolecular amidation of benzylic C(sp3)–H bonds through the intermediacy of amidyl radicals (Scheme 46).190 Employing the organic photocatalyst 9,10-dicyanoanthracene (DCA), irradiation at 410 nm of MeCN solutions of alkylbenzene and N-methoxyamide substrates open to air yielded the desired amidation products, with 30 examples documented in yields of 50–75%. In addition to these C–N coupling products, a minor amount of aryl ketone side product was typically observed alongside. A range of molecules containing primary (46.1), secondary (46.2), and tertiary benzylic C(sp3)–H bonds were competent substrates, with reactivity increasing with increasing substitution as expected. A series of para-substituted methylbenzenes revealed that the electronic nature of the arene has little effect on the reaction efficiency. In the case of xylenes and mesitylene substrates, exclusive mono-amidation was observed. A secondary benzylic C(sp3)–H bond underwent preferential reaction in the presence of a primary benzylic C(sp3)–H bond in 4-ethyltoluene (46.3), and a secondary benzylic C(sp3)–H bond reacted exclusively in the presence of a distal, non-benzylic tertiary alkyl C(sp3)–H bond in isopentylbenzene (46.4). Amides or amines not carrying the methoxy moiety were unreactive.
Scheme 46. Oxidative Benzylic C(sp3)–H Bond Amidation (Pandey, 2015).

A mechanistic model for this transformation involved photoexcitation of the DCA catalyst prior to reductive quenching with the methoxyamide coupling partner, yielding the amide radical cation and DCA•–. The amide radical cation is then proposed to undergo discrete PT mediated by an in situ generated base (proposed by the authors to be hydroxide arising from reaction of superoxide and water) to yield a key amidyl radical intermediate. This stepwise proposal of sequential ET and PT is feasible based on the expected oxidation potential of an amide (e.g., for N-methylacetamide, Ep/2ox = +1.81 V vs SCE in MeCN)191 and the excited-state oxidation potential of this dye (E1/2 *DCA/DCA•– = +2.17 V vs SCE in MeCN).192 However, PCET involvement cannot be ruled out, especially as the reaction progresses and the concentration of in situ-generated basic species increases, which would increase the driving force for ET. Catalyst turnover is achieved through ET between DCA•– and molecular oxygen, forming superoxide as a byproduct. Superoxide, or hydroperoxide anion, may be the base responsible for the PT step. The intermediate amidyl radical is then proposed to abstract a benzylic C–H bond in a HAT event to yield the benzylic radical and reform the closed-shell amide. Through a proposed further turn of the photocatalytic cycle, thus requiring absorption of a second photon, the benzylic radical is oxidized to the carbocation before trapping with the nucleophilic amide and forging the new C–N bond.
Support for this HAT pathway was provided by the addition of TEMPO under optimal conditions, affording the trapping product of the benzylic radical intermediate. Additionally, the appearance of ketone side products implies a minor pathway of trapping this radical with dioxygen and further oxidation. However, C–N bond formation is proposed to proceed through nucleophilic trapping of a carbocation—as opposed to a mechanism of radical–radical coupling. The addition of AcOH co-solvent as an alternative nucleophile afforded a C–H acetoxylation product alongside the C–H amidation product providing support for the nucleophilic trapping of a carbocation intermediate.
In 2018, Zeng and co-workers reported an electrocatalytic intermolecular C(sp3)–H amination of xanthenes with N-alkoxyamides (Scheme 47).193 The authors observed efficient reaction upon constant current electrolysis in an undivided cell furnished with a carbon electrodes with substrate admixtures in the presence of Fc as a redox catalyst, Na2CO3 additive, and LiClO4 serving as the supporting electrolyte in MeCN/CH2Cl2 (1:1). In total, 18 examples of C(sp3)–N coupled products were reported in 40–77% yields. A variety of N-alkoxy amides were tolerated, including aryl (47.1) and alkyl (47.2) amides. The scope of the coupling partner was limited to substituted xanthenes (47.3), as other substrates with benzylic or α-heteroatom C–H bonds afforded no product.
Scheme 47. Electrochemical Intermolecular C(sp3)–H Amidation of Xanthenes (Zeng, 2018).

The authors noted that the neutral substrate could not be oxidized by the Fc redox mediator, but a catalytic current was observed upon stoichiometric deprotonation of substrate with t-BuOK. Therefore, in the authors’ proposed mechanism of this transformation, the Fc redox mediator is first oxidized at the anode to generate ferrocenium in situ. This species undergoes SET with the amide anion, generated via discrete deprotonation of the neutral precursor by carbonate base, to yield an amidyl radical intermediate. However, there is a substantial offset between the pKa of the carbonate base (pKaH = 10.3 in DMSO) and that of the methoxy amide substrate (e.g., for N-methoxyacetamide, pKa = 17.1 in DMSO; for N-benzyloxybenzamide, pKa = 14.4 in DMSO)23 suggesting full PT is unlikely, and a concerted PCET mechanism for N-centered radical generation may be operative. The generated amidyl radical then undergoes intermolecular HAT with the xanthone substrate, generating a highly stabilized doubly benzylic radical. Further oxidation of this species affords a carbocation intermediate which undergoes nucleophilic trapping by the N-alkoxy amide. The unique reactivity of N-alkoxy amides is proposed to be due to captodative stabilization of the intermediate N-centered radical.190 Reduction of protons at the cathode generates molecular hydrogen as a byproduct.
In 2016, Yu and co-workers disclosed a photocatalytic method to achieve the net oxidative intermolecular C(sp2)–H sulfonamidation of heteroarenes via stepwise N–H bond homolysis and sulfonamidyl radical generation (Scheme 48A).194 This reaction involved blue-light irradiation of a dioxane solution of heteroarene substrate and N-alkyl or N-aryl p-toluenesulfonamide with [Ir(ppy)2(dtbbpy)]PF6([Ir-3]PF6) acting as a photocatalyst and aq. NaOCl solution as a stoichiometric terminal oxidant. In total, 48 examples of the C2-selective C(sp2)–H sulfonamidation of indoles (48.1–48.4), azaindoles, pyrroles, and benzofurans (48.5) were reported in yields of 41–92%. The reaction was broadly tolerant of many heteroarene ring- and N-substituents, and many potentially reactive functional groups were handled without incident, including alkenes, alkynes, esters, and halogens. Impressively, the electron-rich heteroarenes in this work did not suffer electrophilic chlorination or oxidation under these photocatalytic reaction conditions. These conditions also permitted intramolecular C(sp2)–H sulfonamidation when these functional groups were tethered to the heteroarene. Later, in 2017, Sun extended the scope of this transformation, under the same conditions, to include imidazo[1,2-a]pyridines, giving exclusively C3 sulfonamidation products.195 A scope of 28 examples was reported in yields of 36–92% (Scheme 48B, 48.6–48.10).
Scheme 48. (A) Photocatalytic C2-Selective C(sp2)–H Sulfonamidation of Indoles, Pyrroles, and Benzofurans (Yu, 2016) and (B) Photocatalytic C3-Selective C–H Sulfonamidation of Imidazo[1,2-a]pyridines (Sun, 2017).

*With 4.0 equiv of sulfonamide.
Interestingly, the mechanism of the reaction was shown to not involve the formation of an N-chlorosulfonamide, despite previous reports suggesting NaOCl is a competent chlorinating agent for forming these species.196−198 Subjecting an indole substrate to an independently prepared N-chlorosulfonamide under these reaction conditions gave none of the desired arene sulfonamidation product, but instead heteroarene aminochlorination with low efficiency, presumably via an electrophilic chlorination pathway. Instead, through steady-state SV studies, the authors show that NaOCl quenched the emission of the Ir(III) photocatalyst (E1/2 Ir(IV)/*Ir(III) = −0.96 V vs SCE in MeCN),68 forming an Ir(IV) species. The authors propose that joint action of this Ir(IV) oxidant (E1/2 Ir(IV)/Ir(III) = +1.21 V vs SCE in MeCN)68 and hydroxide base present in the commercial sodium hypochlorite solution effect N–H bond homolysis via PCET, generating a reactive sulfonamidyl radical intermediate. This radical then engages with the heteroarene substrate, forming a new C–N bond. Further oxidation of the resultant C-centered radical, either photocatalyst mediated or directly by sodium hypochlorite and subsequent deprotonation restores aromaticity to yield the product.
Recently, Zhang, Ackermann and co-workers disclosed a protocol for the electrochemical C(sp2)–H sulfonamidation of heteroarenes via N–H bond concerted PCET activation in an N-alkyl sulfonamide coupling partner (Scheme 49).199 Optimal conditions consisted of the constant current electrolysis of heteroarene and sulfonamide coupling partners in 1,4-dioxane/H2O (1:1) solution in an undivided electrochemical cell equipped with graphite felt anode and Pt cathode at 80 °C. K3PO4 served as both the electrolyte and the Brønsted base additive. The researchers reported 40 examples of heteroarene sulfonamidation in 15–80% yields. A variety of indoles (49.1–49.3), pyrroles, benzofurans (49.4), and benzothiophenes were demonstrated. With respect to substitution on the heterocycle, the reaction tolerated both electron-rich (49.2) and electron-deficient (49.3) substituents at various positions. Variation of the N-alkyl N-aryl sulfonamide constituent revealed that the reaction tolerated alcohols (49.5).
Scheme 49. Electrochemical C(sp2)–H Sulfonamidation of Heteroarenes (Zhang and Ackermann, 2020).

Radical trapping experiments with butylated hydroxytoluene (BHT) under the electrolytic conditions afforded a sulfonamide-BHT adduct, suggesting the involvement of a neutral N-centered sulfonamidyl radical. Furthermore, CV experiments revealed that the addition of aq. K3PO4 significantly negatively shifted the oxidation potential of the sulfonamide (e.g., for N-methyl p-toluenesulfonamide, E1/2ox = +2.50 V vs Ag/AgCl in MeCN, compared to E1/2ox = +1.00 V vs Ag/AgCl in MeCN/0.2 M aq. K3PO4 solution),199 suggesting that K3PO4 serves as both the electrolyte and base to facilitate concerted PCET activation of the sulfonamide substrate. Based on these studies, the authors propose a mechanism wherein the sulfonamide substrate undergoes concerted PCET at the anode to generate an intermediate N-sulfonamidyl radical. This radical undergoes intermolecular addition to the indole, generating a C–N bond and a stabilized C-centered radical. Further anodic oxidation to the corresponding carbocation and deprotonation re-establishes aromaticity to afford the product. The protons lost during this process are reduced at the cathode, generating molecular hydrogen as a byproduct.
Weng, Chiang, and Lei recently reported an electrochemical, oxidative formal [4+2] annulation between indole- and pyrrole-1H-carboxamides, and indoles leading to tetrahydropyrimido[5,4-b]indolinones with exquisite regioselectivity (Scheme 50).200 Such structures have several reported biological and pharmaceutical applications, including anti-HIV-1 inhibitors and α1-adrenoceptor antagonist.201−203 Prior [4+2] annulation reactions of 2,3-indole double bonds through Diels–Alder reactions have been reported, either as the 2π or part of the 4π component, but with strict limitations on substituents due to the inherent electron demand requirement for efficient cycloaddition.204−206 This work was an extension to a previously published method from this group for the oxidative annulation of phenols and indoles via O–H bond homolysis (for a discussion, see section 3.3.2.2).207 Electrolysis was carried out in an undivided cell operating under constant-current conditions with Pt plate electrodes, Et4N+BF4– as a supporting electrolyte, and NaOBz as a Brønsted base additive in a mixture of CH2Cl2/HFIP (2:1). Here, 32 examples of the oxidative annulation were reported, in yields of 50–89%. The reaction proved highly functional group tolerant with respect to indole N- and ring-substituents. Aryl and alkyl halogen, alkyl azide (50.2), unprotected primary alcohol, nitrile, alkene, and alkyne functionality (50.3) were demonstrated, whereas only N-alkoxy carboxamides were suitable substrates for N-centered radical generation. A number of complex and densely functionalized indoles were competent partners, demonstrating the ability of this method to carry out late-stage derivatization.
Scheme 50. Electrochemical Oxidative [4+2] Annulation of Indole-1H-carboxamides and N-Alkylindoles (Lei, 2020).

The authors invoke a PCET mechanism for N–H bond homolysis based on CV experiments and EPR studies. They observed that the onset of oxidation of an indole-1H-carboxamide coupling partner was lowered by ca. 500 mV upon the introduction of exogenous NaOBz (for the indole-1H-carboxamide leading to 50.1 and 50.2 in the absence of base Eonsetox = +1.10 V vs Ag/AgCl in CH2Cl2/HFIP (2:1), compared to Eonsetox = +0.60 V vs Ag/AgCl in CH2Cl2/HFIP (2:1) in the presence of NaOBz).200 The PCET activation of the carboxamide substrate lowers the oxidation potential requirement to approximately equal to that of the N-alkylindole substrate (Eonsetox = +0.60 V vs Ag/AgCl in CH2Cl2/HFIP (2:1)),200 meaning both indole radical cation and amidyl radical are formed simultaneously through anodic oxidation. In keeping with their earlier proposal,207 the authors proposed a radical–radical coupling elementary step to account for the first C–N bond-forming step of the annulation at the C3 position between a persistent indole radical cation and transient amidyl radical. Following this initial bond formation, a Friedel–Crafts-type SEAr followed by proton loss completes the formal [4+2] annulation reaction. Concomitant cathodic reduction of HFIP liberates dihydrogen, and the resultant alkoxide can either act as the base (pKa = 10.7 in DMSO)116 for PCET directly, or regenerate the additive benzoate base (pKa = 11.1 in DMSO).116
2.1.4.3. With Imides and Sulfonimides through (Hetero)aryl C(sp2)–H (Sulfon)imidation
In 2017, the Itoh group reported a photoredox strategy for a net oxidative C(sp2)–H imidation protocol of indoles and related electron-rich heteroarenes under oxidative aerobic conditions (Scheme 51).208 The method utilizes phthalimide (PhthH) or related imides as the imidation reagent, and is mediated by 2-tert-butylanthraquinone (2-t-Bu-AQN) photocatalyst and K2CO3 Brønsted base co-catalyst in DMF solution under air with visible-light irradiation. In total, 30 examples of heteroarene C(sp2)–H imidation were reported in 13–93% yields. In exploring the scope of the reaction, substitution at both indole nitrogen and C2 of the indole were shown to be essential. Substrates with electron-rich groups at C2 (51.1) generally performed the best, and the scope was extended to include pyrroles (51.2) as well as 2-phenylbenzothiophene (51.3), though with low to moderate yield. When the indole carried a C3-phenyl substituent, C2 functionalization was achieved in 54% yield (51.4). Use of nitro- and amino-substituted phthalimides as substrates resulted in little to no product, though 1,8-naphthalimide was demonstrated to generate product in moderate yield (51.5).
Scheme 51. Intermolecular Oxidative Heteroarene C(sp2)–H Imidation via Phthalimidyl Radicals (Itoh, 2017).

Formal N–H homolysis of the imide (e.g., for PhthH, pKa = 8.3 in H2O)209 is proposed to occur via stepwise deprotonation by K2CO3 and single-electron oxidation mediated by the excited state of the photocatalyst, generating the corresponding N-centered radical. This imidyl radical adds to the C3-position of the indole substrate, resulting in a C-centered radical localized at the C2-position. Meanwhile, molecular oxygen is proposed to undergo single-electron reduction by the reduced state of the photocatalyst to form superoxide. Oxidation and deprotonation or HAT of the indole radical by superoxide or hydroperoxide radical generates the re-aromatized indole product. The authors note that it was not possible to rule out an alternative mechanism where the indole is oxidized to the corresponding radical cation by the photocatalyst, followed by nucleophilic addition of PhthH, but they disfavored this pathway due to the poor nucleophilicity of the imide.
Also in 2017, Itami and co-workers reported a photocatalytic oxidative (hetero)arene C(sp2)–H sulfonimidation reaction proceeding through an N-centered radical intermediate formed through stepwise N–H bond homolysis (Scheme 52).210 Blue-light irradiation of typically equimolar mixtures of electron-rich (hetero)arene and various sulfonimides in the presence of Ru photocatalyst [Ru(bpy)3]Cl2·6H2O ([Ru-1]Cl2·6H2O) and the hypervalent I(III) reagent IBB (52.1) as a stoichiometric oxidant in 1,2-DCE solution gave access to 29 examples of C–H sulfonimidation products in yields of 35–87%. Control experiments revealed the critical requirement for both photocatalyst and visible-light irradiation. A variety of arene coupling partners underwent efficient reaction, including naphthalene (52.2, 52.3) and higher polyaromatic hydrocarbons. Electron-rich heteroarenes also underwent efficient amination, including thiophene (52.4), bithiophene (52.5), a benzofuran (52.6), and an oxazole (52.7). Naphthalene underwent regioselective mono-sulfonimidation at C1. Symmetrical and unsymmetrical sulfonimides, including those holding both aryl and alkyl substituents, were reactive under these conditions. A selective mono-desulfonylation reaction of an imide product was demonstrated using ethylenediamine to yield the corresponding sulfonamide in 98% yield, thus serving as an entry point to other aromatic amine derivatives.
Scheme 52. Photocatalytic Arene and Heteroarene C(sp2)–H Sulfonimidation (Itami, 2017).

A mechanism was proposed based on CV measurements. Photoexcitation of the Ru(II) photocatalyst produces a long-lived photoexcited state capable of engaging in productive ET (E1/2 Ru(III)/*Ru(II) = −0.81 V vs SCE in MeCN)64 with the hypervalent iodine reagent through oxidative quenching (Ep/2red IBB = −0.60 V vs Fc+/Fc in 1,2-DCE).210 This leads to the formation of an λ2-iodanyl radical intermediate and butoxide following mesolytic cleavage of the resultant radical anion and the generation of an oxidized Ru(III) complex. The Ru(III) complex was suggested to be sufficiently oxidizing (E1/2 Ru(III)/Ru(II) = +0.95 V vs Fc+/Fc in DCE)210 to engage in productive ET with the neutral sulfonimide substrate (Ep/2ox = +0.25 V vs Fc+/Fc in 1,2-DCE).210 However, the in situ generation of butoxide and benzoate bases suggests that a discrete sulfonimide PT prior to ET or a PCET mechanism is operative to accelerate this process. Following N-centered radical generation in this manner, addition to the arene component leads to a delocalized cyclohexadienyl radical intermediate. Finally, ET between the iodanyl radical and subsequent PT restores aromaticity to the arene. An intramolecular KIE experiment between deuterated and non-deuterated positions on a phenanthrene substrate was taken as evidence that C(sp2)–H bond cleavage is not rate-determining (kH/kD = 1.06). This mechanism is distinct to related work from Nicewicz, Lei, Yoshida, and Hu, demonstrating arene C(sp2)–H amination with azoles and alkylamines, where the arene component is first oxidized to its corresponding radical cation followed by addition of the closed-shell nucleophile.75,211−215
Itami and co-workers also reported a protocol for the direct C(sp2)–H sulfonimidation of naphthalene (53.1, 53.2), an oxazole (53.3), and flavone (53.4) with diarylsulfonimides upon blue-light irradiation in the presence of DDQ as a stoichiometric oxidant (Scheme 53).216 Out of a selection of eight common quinone oxidants, only DDQ gave any product formation, which was also shown through control experiments to require irradiation. While narrow in scope with respect to the (hetero)arene undergoing C(sp2)–H sulfonimidation, several symmetrical and unsymmetrical sulfonimides bearing both alkyl and aryl groups were competent coupling partners. Phthalimide and diphenylamine were unreactive coupling partners.
Scheme 53. Direct (Hetero)arene C(sp2)–H Sulfonimidation Mediated by Visible-Light Irradiation of DDQ (Itami, 2017).

A mechanistic proposal was made based on experimental observations and literature precedent of photochlorination from Fukuzumi.217 Irradiation of DDQ produces a strong photoexcited-state oxidant (E1/2 *DDQ/DDQ•– = +3.18 V vs SCE in MeCN)218 capable of engaging in ET with the sulfonimide reagent. PT, either concurrently or in a discrete second step generates two neutral radical intermediates—an N-centered sulfonimidyl radical and a semiquinone (SQ) radical. The former species adds to the arene substrate and then DDQ semiquinone radical mediates re-aromatization to yield the closed-shell product, through either HAT or ET and PT steps. Reduced DDQH2 is produced as a stoichiometric byproduct of this process. The narrow scope with respect to arene is proposed to be due to competing non-productive charge transfer (CT) to the arene rather than the sulfonimide in unsuccessful cases.
In 2019, the Lei group reported a synthetic procedure for the electrochemical intermolecular C(sp2)–H sulfonimidation of electron-rich arenes (Scheme 54).219 The reaction setup consisted of a carbon rod anode and Pt plate cathode in an undivided cell. The reaction was run under constant current conditions with a CH2Cl2/MeCN/HFIP (20:4:1) solvent mixture and n-Bu4N+AcO– as the supporting electrolyte. Under these reaction conditions, 56 examples of arene sulfonimidation were reported, ranging from 28% to 97% yields. Electron-rich heterocycles and arenes such as thiophenes (54.1) and naphthalenes (54.4) were the most effective substrates; however, 2,6-diphenylpyridine (54.2) also underwent efficient imidation. The Lei group also demonstrated that this procedure is amenable to the manipulation of natural product and drug-like molecules, such as caffeine (54.3). While benzenesulfonimide was the primary electrophile utilized in the scope studies, unsymmetrical sulfonimides (54.5) and N-acyl sulfonamides (54.6) could also be employed in the reaction.
Scheme 54. Electrochemical C(sp2)–H Sulfonimidation of (Hetero)arenes (Lei, 2019).

In addition to arene C(sp2)–H sulfonimidation, the authors demonstrated that alkene C(sp2)–H bonds were competent coupling partners (Scheme 55). Now, nine different alkenes were demonstrated in the reaction, bearing either 1,1-disubstitution (55.1) or trisubstitution (55.2). A single example of the sulfonimidation of a non-activated alkene (from methylene cyclohexane, 55.3) was included as part of the reaction scope. Although the yield in this case was modest (30%), it is noteworthy that exclusively the endocyclic trisubstituted alkene product was obtained.
Scheme 55. Electrochemical C(sp2)–H Sulfonimidation of Olefins (Lei, 2019).

The authors performed CV studies and determined that the current response for the oxidation of benzenesulfonimide increased with increasing concentrations of n-Bu4N+AcO–; based on these data, the authors proposed a plausible mechanism that begins with a concerted PCET process to generate a neutral N-centered radical intermediate. The electrophilic sulfonimidyl radical is then proposed to undergo bimolecular C–N bond formation with the arene (or alkene) to generate a stabilized cyclohexadienyl, or benzylic/tertiary alkyl radical respectively. Anodic SET followed by PT generates the product. Concomitant cathodic reduction of the protons generated in this process generates an equivalent of H2 and obviates the need for any additional external chemical oxidant.
2.1.5. Remote Bond Formation through 1,5- and 1,6-HAT Processes
In addition to the above studied methods enabling C–N bond formation to occur at the nitrogen-containing functional group, photocatalytic and electrochemical methods have been developed to instead selectively homolyze a remote C(sp3)–H bond for C-centered radical generation. From this intermediate, a variety of C–C and C–N bond-forming reactions have then been demonstrated. These pathways proceed through N–H bond homolysis for neutral N-centered radical generation, and thereafter a subsequent 1,x-HAT step leads to remote C–H bond scission. We highlight these methods in the following discussion.
2.1.5.1. C–C Bond Formation through 1,5- and 1,6-HAT under Photocatalytic Activation
In 2016, the Knowles and Rovis groups independently published concurrent reports of the remote, δ-selective, C(sp3)–H alkylation of amides with electron-deficient olefins.29,220 In these publications, an amidyl radical, generated through formal homolysis of the amide N–H bond, is leveraged to undergo selective 1,5-HAT to abstract from a distal C(sp3)–H bond—analogous to the classical Hofmann–Löffler–Freytag (HLF) reaction.221−224 The resulting C-centered radical is then trapped by a Michael acceptor in a Giese-type reaction, forming a new C–C bond remote to the site of N–H activation. While both groups utilize the innate reactivity of the amidyl radical to achieve site selectivity in C–H bond activation, the two groups applied distinct, yet complementary approaches to the generation of the amidyl radical. While the Knowles lab described a concerted PCET process to generate the amidyl radical from N-benzoyl amides, the Rovis group leveraged the increased acidity of the N–H bond in trifluoroacetamide substrates to affect formal homolysis through a stepwise deprotonation/oxidation process.
The report from Knowles and co-workers utilized a joint catalytic system comprising Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 ([Ir-9]PF6) and n-Bu4N+(n-BuO)2P(O)O– Brønsted base co-catalyst under blue-light irradiation to effect the formal homolysis of an amide N–H bond, prior to alkylation with electron-deficient olefins (Scheme 56). In this work, 27 examples of remote alkylation with yields ranging from 29% to 87% were reported. These included alkylated products 56.1 and 56.2, formed by generation of a tertiary carbon-centered radical after 1,5-HAT followed by addition into enone and methylene malonate Michael acceptors, respectively. Product 56.3 demonstrated amidyl radical abstraction to form a sterically encumbered 2° C-centered radical. Additionally, electron-deficient amide and sulfonamide substrates generate alkylated products 56.4 and 56.5 respectively. A substrate with a quaternary center blocking abstraction at the δ-position afforded instead the 1,6-HAT product in 57% yield (56.6). This group later demonstrated that 4-methoxybenzenesulfonamides also permit amidyl radical generation and 1,5-HAT using this same catalyst system.139 This combination of this Ir(III) photo-oxidant (E1/2 *Ir(III)/Ir(II) = +1.30 V vs Fc+/Fc in MeCN)29 and Brønsted base (pKa = 13 in MeCN)29 provides an effective BDFE for N–H bond abstraction of 103 kcal mol–1. While marginally endergonic, this system was sufficient to activate the N–H bond of the N-alkyl amide substrates (ca. 107 kcal mol–1)225,226 through a concerted PCET process. SV quenching experiments and the thermodynamic constraints imposed by the substrate and catalyst oxidation potentials and pKa’s support a concerted mechanism for the generation of the amidyl radical.
Scheme 56. Catalytic Remote Alkylation of C(sp3)–H Bonds in N-Acylamines (Knowles, 2016).

In contrast, the Rovis group employed a stepwise sequence of discrete PT and subsequent photocatalytic ET, enabled by the highly acidified N–H bond of a trifluoroacetamide substrate (pKa = 17.2 in DMSO)116 (Scheme 57A). Under the biphasic conditions reported, K3PO4 is sufficiently basic to deprotonate the trifluoroacetamide substrate (pKa = 12.7 in H2O).227 The resulting conjugate base is readily oxidized (e.g., for N-propyl trifluoroacetamide potassium salt, Ep = +0.77 V vs SCE in MeCN)220 by the employed Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) (E1/2 *Ir(III)/Ir(II) = +0.89 V vs SCE in MeCN),68 generating an amidyl radical which can then facilitate 1,5-HAT. The authors report 25 examples of remote alkylation in this manner with yields of 39–83%. In addition to enone-derived acceptor product 57.1, radical addition into acrylate and acrylamide Michael acceptors (57.2–57.6) was demonstrated. This was due to the increased ability of the Ir(II) state of [Ir-6]PF6 (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN)68 to reduce the resulting α-carbonyl radical and close a redox-neutral photocatalytic cycle, when compared to the more-oxidizing photocatalyst required in the method from the Knowles group (e.g., for [Ir-9]PF6, E1/2 Ir(III)/Ir(II) = −1.07 V vs Fc+/Fc in MeCN).29 Additionally, the authors presented a number of amide substrates to showcase the reactivity and selectivity of the amidyl radical for 1,5-HAT. Product 57.3 demonstrates abstraction from a methylene carbon, generating a less-stabilized secondary radical. Product 57.4 shows selectivity for 1,5-HAT over 1,6-HAT in the presence of another activated C–H at the ε carbon. Products 57.5 and 57.6 demonstrate the selectivity of this method for the 1,5-functionalized product in the presence of other tertiary C–H bonds, demonstrating complementary selectivity to other Rh- or Ir- catalyzed C–H functionalization reactions.228 Under these conditions, the authors propose a stepwise mechanism for amidyl radical generation. The similarity of the substrate and base pKa’s indicate that the amide is likely deprotonated. The observation of a minor amide conjugate addition adduct provided additional support for this anionic amide conjugate base intermediate. Furthermore, SV and CV studies demonstrated that the trifluoroacetamide conjugate base can quench the photocatalyst. These data support a primarily stepwise PT/ET mechanism under optimized conditions, although observation of product formation albeit with lower efficiency when the weaker base Cs2CO3 (pKa = 10.0 in H2O)229 was used suggests that concerted and stepwise pathways could be simultaneously operative.
Scheme 57. (A) Catalytic Remote Alkylation of C(sp3)–H Bonds in N-Trifluoroacetamides (Rovis, 2016) and (B) γ-Selective C(sp3)–H Alkylation of Imides (Rovis, 2017).

*DMF solvent.
Following the concurrent 2016 reports from the Rovis and Knowles groups on the remote alkylation of amine derivatives, the Rovis group disclosed a similar remote alkylation strategy in 2017 that instead proceeded through activation of carboxylic acid-derived imide functional groups (Scheme 57B).230 Under similar reaction conditions, these authors reported 35 examples of γ-alkylated carbonyl compounds, ranging in yield from 14% to 78%. A variety of olefin acceptors were employed as alkylating agents, including vinyl phosphonates, sulfonates, and amides (57.7–57.9). The authors also demonstrated a variety of substitution patterns on the distal carbon undergoing C–H abstraction; however, the majority of the examples either were tertiary C–H bonds (57.7–57.10) or secondary C–H bonds adjacent to a heteroatom (57.11). In a mechanistically related fashion, once again a stoichiometric phosphate base (pKa = 12.7 in H2O)227 deprotonates the acidic N–H bond of the imide substrate (e.g., for succinimide, pKa = 14.8 in DMSO).231 Oxidation of this conjugate base by [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) (E1/2 *Ir(III)/Ir(II) = +0.89 V vs SCE in MeCN)68 affords the N-centered radical that then carries out an intramolecular 1,5-HAT to generate the requisite γ-C-centered radical for distal alkylation.
The Rovis group later extended these methods in 2019 to include alkyl bromides as the alkylating reagent, intercepting the distal radical following 1,5-HAT with an alkyl Ni(II) complex to furnish the C(sp3)–C(sp3) cross-coupled product (Scheme 58).232 Following optimization studies, the authors found that the combination of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) as photocatalyst, Ni(glyme)Cl2 as Ni source, and (4,4′-dMe)bpy as ligand were the most effective catalysts for this remote alkylation.
Scheme 58. γ-Selective C(sp3)–H Alkylation of Trifluoroacetamides with Alkyl Bromides (Rovis, 2019).

A variety of structural variations to the trifluoroacetamide were well tolerated (15 examples, 35–82% yield, 58.1–58.6), though the authors found that increasing the degree of substitution adjacent to the nitrogen promoted a more efficient reaction, presumably through a Thorpe–Ingold effect. With N-(2-cyclohexylethyl)trifluoroacetamide as the model substrate, a variety of alkyl halides were employed in the reaction. In all cases, the observed diastereomeric ratio was >20:1 for the trans:cis isomers. Diverse functionality was tolerated in the reaction, including polar groups (58.4) and nitrogen heterocycles (58.5). It was also noteworthy that the authors observed alkylation of a substrate containing an aryl halide, highlighting the selectivity of this method for oxidative addition in alkyl halides (58.6).
In their mechanistic model, a remote C-centered radical is generated from the trifluoroacetamide substrate through discrete steps of PT, PET, and 1,5-HAT as exemplified previously. Concurrently, oxidative addition of the alkyl bromide partner occurs at a Ni(I) center, generating an alkyl Ni(III) complex. This is reduced to the corresponding alkyl Ni(II) intermediate by the Ir(II) state of the photocatalyst (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN).68 The substrate-derived C-centered radical intercepts this alkyl Ni(II) complex, resulting in a dialkyl Ni(III) intermediate, which facilitates reductive elimination to yield the product and restore Ni(I). Interestingly, Martin, Montgomery, and co-workers later found conditions for the Ni-catalyzed α-alkylation and α-arylation of similar benzamide substrates, rather than the δ-alkylation demonstrated here.233
Expanding on the scope of distal C–C bond formation through a 1,5-HAT manifold following photocatalytic N–H bond homolysis, in 2019 Tambar and co-workers reported the distal allylation of unactivated C(sp3)–H bonds (Scheme 59).234 The authors initially observed that blue-light irradiation of a model trifluoroacetamide substrate and an allyl chloride reagent with photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6([Ir-6]PF6) and K3PO4 in MeCN afforded product 59.1 in 48% yield. However, with the addition of Ni(COD)2 co-catalyst and of BiOX ligand 59.A, the yield increased to 72%. The addition of Ni(II) precursors instead of Ni(0) proved detrimental to the yield. The researchers report 25 examples with yields ranging from 38% to 78%. In addition to variation in the amide component, the authors demonstrated successful coupling with a variety of allyl chlorides, including aromatic (59.2, 59.3) substituents at the C2-position of the allyl chloride, trimethylsilane (59.4), and terminal alkene (59.5) substituted products were all incorporated efficiently. Allyl chlorides with electron-deficient substituents at the C2-position afforded the C-allylated product in lower yields, as significant amounts of N-allylation were also observed.
Scheme 59. γ-Selective C(sp3)–H Allylation of Trifluoroacetamides with Allyl Chlorides (Tambar, 2019).

The authors proposed a catalytic cycle wherein an amidyl radical intermediate is generated through a stepwise sequence of PT/ET involving K3PO4 and photoexcited-state Ir(III) (E1/2 *Ir(III)/Ir(II) = +0.89 V vs SCE in MeCN),68 respectively. This aligns with the proposal of Rovis.220 The amidyl radical then undergoes 1,5-HAT to generate the distal C-centered radical. This radical is proposed to undergo addition to the Ni(0) ligated allyl chloride reagent, resulting in a C-centered radical. This intermediate undergoes β-scission to furnish the product and chlorine radical (E1/2 Cl•/Cl– = +0.52 V vs Fc in MeCN),235 the latter of which is subsequently reduced by Ir(II) (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN)68 to regenerate the Ir(III) photocatalyst ground-state complex and chloride anion. While the exact role of the nickel catalyst was ill-defined, the authors proposed that the improvement in yield observed is due to electrophilic activation of the allyl chloride through π-coordination, rather than facilitating a Ni-mediated redox cycle involving oxidative addition, radical capture, and reductive elimination steps such as that invoked by Rovis in their related Ni co-catalyzed remote alkylation work.232
Whereas Knowles and Rovis demonstrated that N-acylamines29,220,232 and carboximides230 undergo 1,5-HAT upon concerted or stepwise N–H bond homolysis, simultaneous reports from Duan, Roizen, and Shu in 2019 showed that sulfamoyl radicals generated in this fashion instead facilitate 1,6-HAT for remote C-centered radical generation and subsequent C(sp3)–C bond formation (Scheme 60).236−238 Intramolecular 1,n-HAT processes, where n ≠ 5, are rare due to the disordered geometry of transition states for abstraction.239,240 Roizen argues that the unusual selectivity observed in these processes is a result of elongated S–O and S–N bonds (ca. 1.58 Å) and a compressed O–S–N bond angle (ca. 103 °C) which favors a seven-membered-ring transition state over the usual six-membered-ring.241,242
Scheme 60. Remote Alkylation of C(sp3)–H Bonds of Sulfamate Esters through N–H Bond Homolysis and 1,6-HAT (Duan, Roizen, and Shu, 2019).

In the first report from Duan, the optimized reaction conditions comprised of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) and K3PO4·3H2O in DMF at ambient temperature, enabling the γ-alkylation of N-tert-butyl sulfamate esters of alcohol substrates with electron-deficient olefins and styrenes (Scheme 60A). In total, 47 examples of remote alkylation were presented, in yields of 25–88% (60.1–60.3). A wide range of electron-deficient olefins were tolerated, including unsaturated esters and lactones, fumarates, ketones, amides, nitriles, sulfones and phosphates. The reaction was restricted to the use of N-tert-butyl sulfamate esters, and typically required a tertiary or otherwise stabilized C-centered radical at the distal position to work with high reaction efficiency.
The subsequent Roizen optimized conditions are similar, employing [Ir-6]PF6 as photocatalyst, K2CO3 as Brønsted base, in MeCN solution (Scheme 60B). A total of 35 examples were reported in yields of 11–98%. In this work, radical generation at secondary carbon atoms was demonstrated, but these substrates typically exhibit poor control in mono- vs di-alkylation, as the tertiary C–H bond in the initial mono-alkylation product is weaker than the secondary C–H bond of the starting substrate. Two complex natural product-derived sulfamate esters were competent substrates for this transformation (60.4, 60.5). The use of an enantiopure methylene oxazolidinone as radical acceptor, first described by Beckwith,243 enabled a highly diastereoselective Giese-type addition, yielding a protected α-AA product (60.6).
The Shu report utilized the alternative photocatalyst [Ir(dF(Me)ppy)2(dtbbpy)]PF6([Ir-5]PF6)with Na2HPO4 in a biphasic solvent system of 4-chlorotoluene/H2O (2:1) to facilitate the remote functionalization of N-trifluoroethyl sulfamate esters (Scheme 60C). A range of electron-deficient olefins and styrenes were shown to be competent partners (60.7–60.9). Here, 49 examples were presented in yields of 41–89%. Again, mostly tertiary or otherwise stabilized C-centered radicals were required for favorable 1,6-HAT to occur. Electron-deficient olefins and styrenes were successful coupling partners. In addition to the title transformation, these authors demonstrated that the sulfamate ester products of this reaction can be deprotected under mild conditions to liberate the γ-functionalized alcohol, or can act as a leaving group via SN2 displacement with iodide, azide, acetate, and thioacetate nucleophiles, increasing the synthetic utility of this method.
Mechanistically, a stepwise pathway for N–H bond homolysis is likely operative. The acidity of the N–H bond of the sulfamate ester (calculated aqueous pKa of an N-tert-butyl sulfamate = 12.7, calculated aqueous pKa of a trifluoroethyl sulfamate ester = 9.2)241 means that the three bases employed (K3PO4 pKa = 12.3, K2CO3 pKa = 10.2, Na2HPO4 pKa = 6.8) can generate an equilibrium concentration of the sulfamate anion to enable more facile oxidation by the photoexcited-state dyes (e.g., for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6), E1/2 *Ir(III)/Ir(II) = +0.89 V vs SCE in MeCN, for [Ir(dF(Me)ppy)2(dtbbpy)]PF6 ([Ir-5]PF6), E1/2 *Ir(III)/Ir(II) = +0.59 V vs Fc+/Fc).68,244 Roizen also established through SV quenching studies that the independently prepared n-Bu4N salt of N-tert-butyl sulfamate propyl ester quenches the photoexcited state of the [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) efficiently (kq = 2.7 × 109 M–1 s–1).237 Similarly, CV of this isolated salt (Ep/2ox = +0.78 V vs SCE in MeCN)237 showed thermodynamically favorable ET is possible with photoexcited [Ir-6]PF6.
Following sulfamoyl radical generation through either a stepwise or concerted manner, the authors propose distal C–H abstraction yields the C-centered radical which then adds across the electron-deficient olefin or styrene. Huang measured a KIE through parallel reactions in this 1,6-HAT reaction of kH/kD = 4.69, indicating that C(sp3)–H bond cleavage is rate-limiting in these processes. The Ir(II) state of the photocatalyst (e.g., for [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6), E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN, for [Ir(dF(Me)ppy)2(dtbbpy)]PF6 ([Ir-5]PF6), E1/2 Ir(III)/Ir(II) = −1.43 V vs SCE in MeCN)68,69 reduced the α-acyl or benzylic (e.g., for the benzylic radical deriving from ethylbenzene (PhCH·Me), E1/2red = −1.60 V vs SCE in MeCN),135 radical to the corresponding anion, which through PT steps regenerated the base co-catalyst. Relative to the Knowles report of amidyl radical generation and 1,5-HAT,29 these processes with sulfamate substrates have a greater scope with respect to the electron-deficient olefin component of the reaction and proceed through stepwise PT and ET. The use of the highly oxidizing [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 photocatalyst ([Ir-9]PF6) (E1/2 *Ir(III)/Ir(II) = +1.30 V vs Fc+/Fc in MeCN)29 to facilitate N-centered radical generation in the earlier Knowles report, meant that conversely the Ir(II) state of this photocatalyst was relatively poorly reducing (E1/2 Ir(III)/Ir(II) = −1.07 V vs Fc+/Fc in MeCN),29 and unable to mediate the reduction of ester-derived α-acyl radicals or benzylic radicals, for example, to turn over a catalytic cycle in reactions with acrylate esters or styrene coupling partners. The less photooxidizing photocatalysts employed in these processes then give a more-reducing Ir(II) ground-state complex able to engage in SET with a broader range of olefin coupling partners.
2.1.5.2. C–N Bond Formation through 1,5-HAT under Electrochemical Activation
Simultaneous reports from the groups of Muñiz and Lei in 2018 disclosed related methods to facilitate the dehydrogenative intramolecular C(sp3)–H amination of N-alkyl amides and sulfonamides via anodic oxidation (Scheme 61 and Scheme 62, respectively).245,246 Similar electrolytic cell assemblies were reported, both undivided cells equipped with carbon anode and Pt cathode, running under constant current conditions in HFIP or 1,2-DCE/HFIP solvent systems at ambient temperatures. The major difference between the two setups, however, was the electrolyte employed—n-Bu4N+BF4– in the former case, and n-Bu4N+OAc– in the latter—which impacted upon the scope of the transformation in the two cases as well as their proposed mechanism of operation. While Muñiz and co-workers proposed a mechanism of initial arene oxidation, the use of n-Bu4N+OAc– in Lei’s report suggests the substrate is activated by an N–H PCET mechanism.
Scheme 61. Electrochemical Remote C(sp3)–H Sulfonamidation through an ET/PT Manifold (Muñiz, 2018).

*Constant current = 1 mA.
Scheme 62. Electrochemical Remote C(sp3)–H Sulfonamidation through a Concerted PCET Manifold (Lei, 2018).

The Muñiz method provided access to five- and six-membered-ring N-sulfonyl and N-acylamines carrying an α-aryl substituent, with 24 examples of pyrrolidine synthesis in yields of 30–89% (61.1–61.3), and 17 examples of piperidine synthesis in yields of 24–81% (61.4, 61.5) (Scheme 61). The reaction was largely insensitive to the nature of the aryl substituent at the δ- or ε-position, with electron-rich, electron-neutral, electron-poor, and heterocyclic substituents all tolerated well. The Lei method was shown to have a broader scope with respect to substitution at the δ-position of the N-alkyl sulfonamide substrates, with benzylic (62.1–62.3), tertiary (62.4, 62.5), α-heteroatom (62.6), and a limited number of secondary centers capable of participating in cyclization, but proved to be limited to pyrrolidine products (Scheme 62). In this work, 30 examples of N-sulfonyl pyrrolidine synthesis were reported in yields of 35–98%, and through changing the solvent to MeCN/HFIP, five additional examples of N-acyl pyrrolidine synthesis in yields of 72–92% were possible. The group observed a Thorpe–Ingold effect in this reaction, with substrates carrying gem-disubstitution in the alkyl backbone performing more efficiently compared to linear, unsubstituted examples.
These differences in scope between the two reports can be understood when considering the mechanisms of operation and the effect of the supporting electrolyte. Muñiz and co-workers, working with the non-basic electrolyte n-Bu4N+BF4–, show through CV studies of model substrates featuring either only the arene moiety (e.g., for butylbenzene, Ep/2ox = +1.77 V vs Ag/AgNO3),245 or only the sulfonamide moiety (e.g., for N-(4-cyclohexylbutyl)toluenesulfonamide, Ep/2ox = +2.02 V vs Ag/AgNO3),245 that the site of initial substrate oxidation is the arene ring, forming the corresponding arene radical cation through anodic oxidation. Then, through proposed rapid PT from the now-acidified benzylic position (e.g., the pKa of toluene radical cation is ca. –9 to –13 in MeCN)247 a benzylic radical forms. Through further anodic oxidation of this radical at a lower applied potential than the initial arene oxidation (e.g., for the benzylic radical resulting from ethylbenzene (PhCH·Me), Ep/2ox = +0.37 V vs SCE in MeCN),135 a benzylic carbocation is generated. Finally, cyclization of the tethered N-nucleophile onto the carbocation and subsequent PT gives the product. Cyclization is possible in both the five- and six-membered-ring product classes. Through cathodic reduction, the liberated protons go on to evolve molecular hydrogen.
In contrast, Lei and co-workers invoke a PCET mechanism for substrate oxidation via N–H bond homolysis at the sulfonamide functional group, where the acetate additive plays a dual role of supporting electrolyte and Brønsted base.2461H NMR studies indicated hydrogen bonding between the N–H group and the acetate anion and a decrease in the oxidation potential requirement for these substrates is observed in the presence of acetate anion via CV. Control experiments support this involvement; replacing the supporting electrolyte with n-Bu4N+BF4– gave complete loss of reactivity, which could be recovered through the addition of exogenous NaOAc. Following N–H bond activation through this PCET mechanism, the resulting sulfonamidyl radical is proposed to undergo 1,5-HAT, generating a distal carbon-centered radical, provided there is some ability to stabilize the reactive intermediate at this δ-position. Relative rate data for intramolecular HAT at different sites imply that this transformation is kinetically limited to the formation of five-membered-ring products. Further anodic oxidation of this radical gives the corresponding carbocation prior to N-nucleophile cyclization and PT to yield the product. Cathodic proton reduction liberates molecular hydrogen. Comparison of these two protocols serves to highlight the effect of a relatively small change in reaction conditions to turn on a different pathway for substrate activation. This change in mechanism can in turn lead to complementary scopes, leveraging the inherent differences in reactivity and selectivity based on the nuances of each mechanism of activation.
Rueping and co-workers shortly thereafter presented a similar electrochemical method for the remote intramolecular C(sp3)–H sulfonamidation of activated benzylic and tertiary alkyl C–H bonds through a 1,5-HAT pathway (Scheme 63).248 Optimized conditions involved the constant current electrolysis of sulfonamide substrates in MeOH using an undivided cell equipped with graphite anode and stainless-steel cathode. NaOMe was employed as both the supporting electrolyte and Brønsted base additive. A total of 18 examples of pyrrolidine synthesis through C(sp3)–H sulfonamidation were reported in yields of 30–82% (63.1–63.4). The use of methoxide as base led to the proposal of discrete PT from the sulfonamide substrate prior to anodic oxidation for formation of a neutral N-centered radical intermediate. This N-centered radical promotes a 1,5-HAT process to generate a distal C-centered radical, which is further oxidized at the anode to the corresponding carbocation before intramolecular cyclization of the pendant amine group. Reduction of MeOH at the cathode surface continuously regenerates the Brønsted base and liberates molecular hydrogen. In this work, the authors also showed that the addition of KBr to the reaction system expanded the scope of this transformation to include the sulfonamidation of primary and secondary alkyl C(sp3)–H bonds through in situ N–Br formation prior to N-centered radical generation. C–N bond formation then occurred after proposed 1,5-HAT, trapping with bromine radical, and SN2 reaction in an electrochemical variant of the HLF reaction. These three comparative studies illustrate how impactful a PCET mechanism can be in lowering the energy requirement to homolyze a strong N–H bond, as compared to stepwise pathways of ET/PT or PT/ET. The choice of mechanism promoted ultimately determined the scope and mildness of these transformations.
Scheme 63. Electrochemical Remote C(sp3)–H Sulfonamidation through a PT/ET Manifold (Rueping, 2019).

2.1.6. Intra- and Intermolecular N–N Bond Formation
In 2016, Waldvogel and co-workers reported the electrochemical synthesis of pyrazolidine-3,5-diones from di-anilide precursors via intramolecular amidyl diradical coupling (Scheme 64).249 Optimized conditions consisted of an undivided cell with graphite anode and Pt cathode under constant current electrolysis. n-Bu4N+PF6– served as the supporting electrolyte in HFIP solution. Under these conditions, the authors observed N–N bond formation for a variety of symmetric di-anilide precursors, including di-anilides with electron-deficient (64.1) and electron-rich (64.2) aromatic substituents. Additionally, two examples of non-symmetric substrates were presented, including 64.3, containing aryl chloride and bromide substituents. Amidyl radical generation is proposed to occur through coupled steps of electron and proton transfers,, though the order of these events was unclear. Cathodically generated HFIP anion (pKaH = 10.7 in DMSO)116 is proposed to mediate PT steps.
Scheme 64. Electrochemical Synthesis of Pyrazolidin-3,5-diones through Intramolecular N–N Bond Formation (Waldvogel, 2016).

In a follow-up mechanistic study, the authors then dissect two possible mechanisms for N–N bond formation in this system (Scheme 65A).250 First, (i) a cationic pathway—where an amidyl radical intermediate undergoes a second anodic oxidation to generate an anilidium cation. N–N bond formation then occurs through nucleophilic trapping by the other pendant anilide. Second, (ii) a diradical pathway—involving additional steps of ET and PT at the second amide functional group to yield an N,N′-diradical intermediate, where N–N bond formation then occurs via intramolecular radical coupling. To assess these possibilities, the authors synthesized a number of unsymmetrical di-anilides and subjected them to bulk electrolysis and CV studies. In the bulk electrolysis studies, they observed two classes of products: 65.1—an adduct incorporating HFIP at the C2-position of the arene, and 65.2—the N–N coupled product (Scheme 65B). In cases where the HFIP adduct was observed, the second oxidation potential of the amidyl radical of one moiety (Ep/2ox,2) is notably lower than the first oxidation potential at the other neutral closed-shell anilide (Ep/2ox,1). This leads to two coupled steps of single-electron oxidation at the more readily oxidized aniline to the anilidium cation followed by nucleophilic attack at the aromatic ring by HFIP, rather than cyclization of the tethered amide. In the cases of the N–N bond formation, the oxidation of the second anilide occurs at a lower potential than the oxidation of the amidyl radical, resulting in the generation of a diradical and radical–radical coupling to form 65.2. As their synthetic results are consistent with these CV studies, the authors propose that a N,N-diradical coupling is a reasonable mechanism for N–N bond formation.
Scheme 65. Investigation of the Mechanism of Intramolecular N–N Bond Formation in Dianilide Substrates (Waldvogel, 2017).

Redox potentials vs Fc+/Fc in EtOH.
In 2018, Waldvogel and co-workers reported the synthesis of N,N′-diaryl phthalic hydrazides from the corresponding diamide, through dehydrogenative anodic N–N bond formation (Scheme 66).251 This was enabled via constant current electrolysis in an undivided cell, with graphite anode, Pt cathode, n-Bu4N+PF6– as a supporting electrolyte, in HFIP solution. A scope of 15 examples of this transformation was presented with yields of 34–89% yields. Variation in the N-aryl groups included chloride-substituted 66.1, and unsymmetrical N,N-diarylhydrazines 66.2 and 66.3. Additionally, variation on the phthalic acid backbone was tolerated including pyridine derivative 66.4. The authors showed that upon treatment of these reaction products with an excess of hydrazine in MeOH, and further aerobic oxidation, azobenzene products resulted. A similar diradical pathway to that discussed above was assumed to be operative.
Scheme 66. Electrochemical Synthesis of Phthalic Hydrazides through N–N Bond Formation (Waldvogel, 2018).

In 2020, Waldvogel and co-workers expanded these protocols to now enable the intermolecular N–N homo-coupling of N-aryl amides for the synthesis of N,N′-diacylhydrazines (Scheme 67).252 This reaction was developed in an undivided cell equipped with graphite electrodes under constant current electrolysis in AcOH/MeCN (5% v/v). A KOAc supporting electrolyte proved crucial for desired product formation. The authors provided 16 examples, including CF3-substituted anilide 67.1, which was isolated in 66% yield on 8.0 mmol scale. While a variety of groups on the aniline portion were tolerated, including bromo- (67.2) and cyano-substituted (67.3) anilides, other acyl protecting groups beyond acetyl (Ac), such as benzoyl (67.4), resulted in diminished yields. Additionally, the authors demonstrated removal of the Ac groups with methanolic NaOH, providing access to symmetric N,N′-diarylhydrazines.
Scheme 67. Electrochemical Intermolecular Homo-Coupling of Anilides via N–N Bond Formation (Waldvogel, 2020).

2.1.7. Other Processes of Amides, Sulfonamides, Carbamates, and Ureas
In 2017, Waldvogel and co-workers described a novel electrochemical route to benzoxazoles from readily available anilide starting materials via amidyl radical generation and C(sp2)–O bond formation (Scheme 68).253 This transformation was achieved via constant current electrolysis of HFIP solutions of anilide substrate in an undivided cell with RVC or graphite anode, Pt wire cathode, and n-Bu4N+PF6– as a supporting electrolyte. HFIP was essential to the above method, with other solvents leading to substrate degradation and low product yields. A total of nine examples were presented, in yields of 56–86% (68.1–68.4). Electron-donating substituents on either of the substrate aryl groups had a beneficial effect on the yield. Although aryl halide and triflate functionalities were tolerated, amides derived from primary or secondary alkyl carboxylic acids were not competent substrates as they led to unselective degradation.
Scheme 68. Electrochemical Synthesis of Benzoxazoles (Waldvogel, 2017).

Regarding the mechanism of reaction, the authors consider that deprotonation of the anilide substrate (e.g., for benzanilide, pKa = 18.8 in DMSO)254 by cathodically generated HFIP anion (pKaH = 10.7 in DMSO)116 may precede anodic oxidation. However, analysis of the offset here in relative pKa values between the substrate and base leads us to suggest that there may be a degree of concerted PCET involvement in this substrate activation. The resulting amidyl radical can cyclize through its C-centered resonance form, forging a C–O bond. Subsequent oxidation and deprotonation restore aromaticity, yielding the benzoxazole product. The radical character on the aryl ring is supported by the observation of undesired intermolecular para-coupling products in substrates lacking a substituent at that position. The authors also note that polar mechanism involving oxidation of the aryl radical to the corresponding carbocation, followed by an oxo-Nazarov-type electrocyclization is also a plausible mechanistic pathway.
Methods for the synthesis of 2,2′-diaminobiaryls are valuable due to the prevalence of this scaffold in ligands for transition-metal catalysts. In a 2017 report, Waldvogel and co-workers developed a method to selectively cross-couple anilide derivatives for the preparation of nonsymmetric 2,2′-diaminobiaryls (Scheme 69).255 The anodic oxidation was conducted in an undivided cell outfitted with glassy carbon electrodes in a 18% HFIP/MeOH solution with n-Bu3NMe+O3SOMe– as the electrolyte under constant current conditions at 50 °C. Unprotected anilines were incompatible under these conditions since their electron-rich nature and low oxidation potentials tended to lead to overoxidation and oligomerization. This challenge was addressed by using a number of acyl-protected anilines with higher oxidation potentials. In order to statistically favor the cross-coupling product over the homo-coupling product, the group utilized two anilides with sufficiently differentiated oxidation potentials so that one would undergo SET preferentially. The anilide bearing the lower oxidation potential was held as the limiting reagent with a 2-fold excess of the anilide with a higher oxidation potential, leading to higher yields of the cross-coupled product. Under the optimized reaction conditions, 10 different nonsymmetric 2,2′-diaminobiaryls were synthesized in moderate to good yields (38–74%). The method’s scope included a range of substituted (69.2, 69.3) and π-extended anilides (69.1) as successful heterocoupling substrates. One of the notable functional group compatibilities of this method is its tolerance of unprotected phenols on highly substituted anilides. A chloro-substituted acetanilide with a free phenol was coupled to an Ac- and a pivalyl-protected anilide (69.2, 69.3) in 56% and 51% yield, respectively. A variety of other electron-rich anilides bearing various substitution and differentiated protecting groups could be coupled with moderate efficiency (69.4–69.9, 38–74% yield). A 2018 report extended this method to the synthesis of cross-coupled formanilides under similar electrochemical conditions, with 13 examples reported in 16–56% yields.256 Coupled steps of electron and proton transfers of the lower oxidation potential substrate lead to N-centered radical generation; however, the forward pathway to C–C bond formation was unclear.
Scheme 69. Electrochemical Anodic Cross-Coupling of Anilides (Waldvogel, 2017).

*In HFIP solvent.
Xue, Chen, and Xiao reported examples of styrene 1,2-allylsulfonylation in a class of ortho-vinyl sulfonanilides. These reactions proceeded through stepwise N–H bond homolysis, but led to products where the sulfonamide functional group had not undergone C–N bond formation (Scheme 70).257 Optimized conditions consisted of blue-light irradiation of a DMF solution containing sulfonanilide substrate, Nozaki allylsulfone reagents, [Ir(ppy)2(bpy)]PF6 ([Ir-4]PF6) photocatalyst, and K2CO3 Brønsted base at room temperature. While many photocatalytic methods utilize these reagents for radical allylation, methods resulting in net incorporation of both allyl and sulfonyl components are rare. In this work, 33 examples of styrene difunctionalization were reported, in yields of 41–92%. S-Aryl- and S-alkyl-substituted sulfonanilides groups were successful substrates (70.1, 70.2). A broad number of anilide functional groups such as ether (70.3), halogen (70.4, 70.5), ester, aldehyde (70.6), and nitrile were tolerated. Although different allyl sulfone reagents were incorporated, electron-deficient olefins were necessarily required (70.8). In contrast with the ortho-vinyl substrates, meta- and para-vinyl sulfonanilide substrates underwent N-allylation in addition to styrene 1,2-difunctionalization. Notably, N-methyl sulfonanilide was unsuccessful, showcasing the necessity of an N–H bond.
Scheme 70. 1,2-Allylsulfonylation of ortho-Vinyl Sulfonanilides (Xue, Chen, and Xiao, 2019).

*In MeCN solvent.
Luminescence quenching studies revealed that sulfonanilide substrate quenches the photocatalyst only in the presence of K2CO3, which was subsequently shown through 1H NMR experiments to fully deprotonate the substrate. Further radical trapping experiments carried out with TEMPO and diphenyl diselenide implied the intermediacy of an N-centered radical in the mechanism of the reaction. These results led the researchers to propose a stepwise process of discrete PT and subsequent ET from photoexcited-state Ir(III) catalyst (E1/2 *Ir(III)/Ir(II) = +0.61 V vs SCE in MeCN)46 to generate a neutral sulfonamidyl radical intermediate. DFT experiments were used to rule out styrene oxidation and generation of the corresponding olefin radical cation. After generation of the N-centered radical, intermolecular addition to the allylsulfone reagent through an addition–elimination mechanism leads to N-allylation and extrusion of arylsulfonyl radical. Subjecting an independently prepared N-allyl sulfonanilide to optimized reaction conditions in a crossover experiment demonstrated that this was a viable intermediate in the reaction.
In other photocatalytic reactions using these allylsulfone reagents, the extruded arylsulfonyl radical is typically reduced to the corresponding sulfinate by a ground-state Ir(II) complex (E1/2 Ir(III)/Ir(II) = −2.05 V vs SCE in MeCN)46 to close a redox-neutral catalytic cycle and yield an allylated product. In this work, however, the arylsulfonyl radical adds to styrene in an anti-Markovnikov fashion, generating a benzylic radical. The researchers used DFT to study subsequent steps in the reaction pathway. Two possible pathways were identified, which varied in the order of ET and C–C bond formation. The first involved 7-endo-trig cyclization of the benzylic radical onto the sulfonamide-tethered Michael acceptor (ΔG⧧ = 12.3 kcal mol–1), then Ir(II)-mediated reduction of the resultant α-acyl radical to the enolate anion. The second pathway involved Ir(II)-mediated reduction of the benzylic radical followed by a polar Michael addition in a 7-endo-trig fashion to generate the same enolate intermediate (ΔG⧧ = 11.3 kcal mol–1). From this common enolate intermediate, a retro-Michael reaction occurs (ΔG⧧ = 5.8 kcal mol–1) to promote overall N-to-C allyl group migration and furnish the allylsulfonylated product. This research group later established that a sulfonanilide catalyst could promote styrene 1,2-allylsulfonylation in an intermolecular reaction where substrates did not necessarily incorporate a nitrogen functional group (see section 2.9.2).
2.2. Transformations of Thioamides and Thioureas
In this collection of works, the transformations of thioamides and thioureas initiated via oxidative N–H bond PCET are discussed. Whereas amides and sulfonamides subject to this activation mode then react through a N-centered radical (see section 2.1), in thioamide and thiourea substrates, reaction occurs instead through sulfur, leading to C–S, N–S, and S–S bond formation.
2.2.1. Intramolecular C–S Bond Formation through Addition to (Hetero)arenes and Alkenes
In 1979, Tabaković and co-workers reported the first examples of the electrochemical synthesis of benzothiazoles and related heterocycles through the anodic oxidation of thiocarboxamides (Scheme 71).258 Reactions were carried out using a divided cell under constant potential electrolysis with Pt gauze electrodes and Et4N+ClO4– as supporting electrolyte in MeCN. In total, nine heterocyclic products were prepared in this manner, in yields of 60–95%. The cyclization of the thiocarboxamide onto electron-deficient arenes (e.g., 71.1) in particular was demonstrated. An example of S–N bond formation in the cyclization of a thiocarboxamide of 2-aminopyridine was included (71.3). A coumarin substrate also underwent efficient C–S bond formation (71.4). The operating potentials of these reactions were typically held above the measured substrate oxidation potentials (e.g., for thiocarboxamide substrate leading to 71.1, E1/2ox = +1.55 V vs SCE in MeCN, with Ecell = +1.80 V),258 suggesting that the mechanism of this reaction involves initial ET preceding PT. Then C–S bond formation occurs, followed by re-aromatization through anodic oxidation.
Scheme 71. An Early Example of Electrochemical Benzothiazole Formation through C–S Bond Formation (Tabaković, 1979).

*Using C anode.
An early example of a photocatalytic method arising from the homolytic activation of an N–H thioamide bond came from the Li research group in 2012, which reported the oxidative synthesis of benzothiazoles from thiobenzanilides with O2 as a terminal oxidant (Scheme 72).259 Irradiation of thiobenzanilide substrates in the presence of [Ru(bpy)3](PF6)2 ([Ru-1](PF6)2) photocatalyst and DBU as a stoichiometric Brønsted base additive under a 5% O2 in N2 atmosphere in DMF resulted in efficient C–S bond formation. No reaction was observed in the absence of base. The reaction efficiency displayed a strong dependency upon the concentration of oxygen, with low concentration optimal. Using an air atmosphere (∼21% O2) resulted in partially diminished yield, and under 100% O2 no desired cyclization was observed. This was due to the competing conversion of the substrate thioanilide (C=S) to the corresponding anilide (C=O), which was impeded at lower O2 concentration. The reaction could be driven directly by sunlight as opposed to artificial light sources without great loss in efficiency (e.g., for 72.1, 84% with CFL lamps compared to 65% under sunlight irradiation). A substrate scope of 23 examples was demonstrated in yields of 10–91%. Halogenated substrates (Br and I) underwent competing protodehalogenation under these reaction conditions. Mildly electron-withdrawing and -donating groups were well tolerated on the N-aryl (e.g., 72.2 and 72.3, respectively) and 2-aryl groups (e.g., 72.4 and 72.5, respectively). A thioether functional group was tolerated without oxidation to the sulfoxide, which had previously been reported under similar photocatalytic aerobic conditions by Zen.260
Scheme 72. Aerobic Photocatalytic Synthesis of Benzothiazoles from Thiobenzanilides (Li, 2012).

A mechanism was proposed to involve stepwise PT and ET leading to S-centered radical generation and cyclization. DBU (pKaH = 13.9 in DMSO)48 promoted substrate deprotonation (pKa of thiobenzoyl anilide is approximately 10.0 in DMSO),261 prior to ET mediated by the oxidized Ru(III) complex (E1/2 Ru(III)/Ru(II) = +1.26 V vs SCE in MeCN),64 generated from oxidative quenching of the photoexcited-state Ru(II) complex (E1/2 Ru(III)/*Ru(II) = −0.81 V vs SCE in MeCN)64 with molecular oxygen (E1/2red = O2/O2•– = −0.87 V vs SCE in MeCN).262 This step was proposed based on the observation of green coloration upon irradiation of the solution under O2, which was taken as qualitative evidence for the persistence of a Ru(III) catalytic resting state.
Following S-centered radical generation, 5-exo-trig cyclization yields a delocalized cyclohexadienyl radical. Superoxide generated through Ru(III) formation mediates a HAT step to liberate aromatized product and hydroperoxyl radical as a byproduct. Hydroperoxyl radical has also been invoked in Ru(III) formation and HAT steps in related aerobic photocatalytic methods.75,213,263
Lei and co-workers in 2014 reported the oxidative synthesis of benzothiazoles from thiobenzanilides, jointly mediated by the action of a Ru(II) photosensitizer, a Co(III) proton-reduction co-catalyst, and a Brønsted base additive under visible-light irradiation (Scheme 73).264 The reaction likely proceeds via stepwise PT and ET events at the acidic N–H group, resulting in S-centered radical generation and cyclization. Hydrogen gas generation was quantified in addition to isolated benzothiazole product yield. The optimal reaction conditions for the conversion of 2-arylthiobenzanilides consisted of visible-light irradiation of substrates in the presence of [Ru(bpy)3](PF6)2 ([Ru-1](PF6)2) as a photosensitizer, [Co(dmgH)2(DMAP)Cl] as a proton-reduction co-catalyst, and sodium glycinate as a Brønsted base in MeCN solution. The pKa of the basic additive was a key parameter in the optimization, requiring one sufficiently basic to (partially) deprotonate the substrate, with its conjugate acid sufficiently acidic to act as a proton donor for the hydrogen liberation step. Thus, bases with pKa values in the range of 9–12 were optimal. While carbonate bases were effective in promoting high yields of benzothiazole formation, their use resulted in lower yield than theoretical yields of dihydrogen. The authors propose that in these cases, liberated CO2 is hydrogenated with in situ generated hydrogen gas, which has been reported to occur under ambient temperatures and pressures with [Co(dmpe)2H] as a catalyst by Linehan and co-workers.265 For 2-alkylthiobenzanilides substrates, the stronger Brønsted base co-catalyst n-Bu4N+OH– was required, due to the less acidic N–H bond in these substrates. When the stoichiometric chemical oxidants (e.g., O2, K2S2O8, or H2O2) were used in place of the Co(III) co-catalyst, significantly lower yields of the desired product were obtained, while the corresponding benzanilide was observed as the major product in these cases. Notably, this protocol required no external oxidant, with molecular hydrogen gas liberated as the sole byproduct.
Scheme 73. Dehydrogenative Synthesis of Benzothiazoles from Thiobenzanilides via N–H Bond Homolysis (Lei, 2015).

*Using 10% n-Bu4N+OH– in place of 1 equiv of Na+H2NCH2CO2–.
In this work, 20 examples of the synthesis of 2-arylbenzothiazoles in yields of 45–99%, and eight examples of 2-alkylbenzothiazole synthesis in yields of 59–99% were reported. The electronics of the N-aryl group affected the reaction efficiency, with electron-donating OMe (e.g., 73.2) and electron-withdrawing CF3 substituents leading to diminished yields, presumably due to the impact on substrate pKa. However, moderately electron-withdrawing halogen functionality was well tolerated. Substituent effects of the 2-aryl group were less impactful on product yields. tert-Butyl (e.g., 73.3) and cyclohexyl groups were competent substituents for the synthesis of 2-alkylbenzothiazoles. The reaction was demonstrated on gram scale, and the synthesis of a benzothiazole displaying potent anti-tumor activity (73.4) was reported in 78% yield.266
A series of SV quenching experiments indicated that the substrate did not quench the photoexcited state of the Ru(II) complex, but that the pre-prepared sodium salt did. The Co(III) catalyst also resulted in emission quenching, but to a lesser extent. This result was supported by CV experiments, indicating that the neutral substrate (e.g., for thiobenzanilide, Epox = +0.94 V vs SCE in MeCN)264 did not undergo thermodynamically favorable ET with the Ru dye (E1/2 *Ru(II)/Ru(I) = +0.77 V vs SCE in MeCN),64 but SET becomes feasible upon substrate ionization (e.g., for the sodium salt of thiobenzanilide, Epox = +0.53 V vs SCE in MeCN).264 Running the reaction with deuterated anilide substrate resulted in formation of mixtures of H2, HD, and D2, whereas running the reaction with non-deuterated substrate but in MeCN-d3 produced H2 exclusively. This showed that hydrogen evolution occurred ultimately from substrate, as opposed to a mechanism involving solvent as a proton-shuttle. An intramolecular KIE experiment also revealed that C(sp2)–H bond cleavage was not the rate-limiting step (kH/kD = 1.06). In order to probe reaction kinetics, in situ IR monitoring was used. Initial rate experiments showed that the reaction was zero-order in substrate and photocatalyst, but first-order in Co(III) co-catalyst. These data indicate that the interaction between the proton-reducing Co(III) co-catalyst and a proton source was likely rate-limiting.
Based on these collected observations, the following mechanism was proposed. The interaction of the basic additive and substrate generates an equilibrium concentration of the ionized form. Photoexcitation of the Ru(II) dye produces a long-lived photoexcited state capable of engaging in favorable SET with the ionized substrate, generating an S-centered radical and the reduced-state Ru(I) complex. The Ru(I) complex can regenerate the ground-state Ru(II) complex (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN)64 through reduction of the Co(III) co-catalyst (E1/2 Co(III)/Co(II) = −0.83 V vs SCE in MeCN).267 The S-centered radical undergoes cyclization onto the pendant arene, yielding a delocalized cyclohexadienyl radical. This is proposed to react through SET (e.g., for the parent cyclohexadienyl radical, Ep/2ox = −0.34 V vs SCE in MeCN)268 with the transient Co(II) complex (E1/2 Co(II)/Co(I) = −1.08 V vs SCE in MeCN),267 yielding a carbocation and a Co(I) species. Through PT, the aromatized product is furnished. The reduced-state Co(I) complex reacts with the conjugate acid of the Brønsted base additive, yielding a Co(III) hydride, which is further protonated to liberate molecular hydrogen and regenerate the ground-state Co(III) complex.
Lei and co-workers in 2017 presented an electrochemical synthesis of 2-aminobenzothiazoles from aryl isothiocyanates and secondary alkylamines (Scheme 74).269 Electrolysis of admixtures of these substrates was performed in an undivided cell equipped with a graphite rod anode and a Pt plate cathode in 10% aq. MeCN with n-Bu4N+BF4– as a supporting electrolyte at 70 °C. Here, 22 examples of 2-aminobenzothiazole synthesis were reported in yields of 70–99%. The reaction was tolerant of a broad range of functional groups including halogen (74.2), trifluoromethyl (74.3), acetal, sulfide, and ketone. Cyclic (74.1–74.3) and acyclic (74.4) secondary amines were permitted. In this work, in addition to 2-aminobenzothiazoles, under similar electrolysis conditions, 2-alkyl- and 2-arylbenzothiazole formation was demonstrated from pre-formed thioamide substrates, with 12 examples in yields of 20–98%.
Scheme 74. Synthesis of 2-Aminobenzothiazoles from Aryl Isothiocyanates and Secondary Alkylamines (Lei, 2017).

The combination of aryl isothiocyanate and amine substrates first generates an N-arylthiourea in situ through nucleophilic addition. Water is a necessary co-solvent, allowing generation of hydroxide base through cathodic reduction to enable discrete substrate deprotonation. Anhydrous MeCN provided only poor yields of the benzothiazole product. Thereafter, single-electron oxidation of the resultant anion yields an S-centered radical which cyclizes onto the pendant arene. Further anodic oxidation and deprotonation restores aromaticity to yield the product 2-aminobenzothiazole. This stepwise proposal of PT and ET is supported by CV studies. Phenyl isothiocyanate showed no oxidation onset below +2.0 V vs Ag/AgCl in MeCN/H2O (9:1).269 An authentic sample of the neutral thiourea formed between phenyl isothiocyanate and morpholine showed a peak potential Epox = +0.98 V vs Ag/AgCl in MeCN/H2O.269 Addition of hydroxide led to a negative shift in the oxidation peak potential to Epox = +0.43 V vs Ag/AgCl in MeCN/H2O,269 demonstrating initial discrete PT facilitating subsequent ET.
Wirth and Xu collaborated in the development of a catalyst- and supporting-electrolyte-free continuous-flow electrochemical synthesis of benzothiazoles and thiazolopyridines from thioamide starting materials via C(sp2)–S bond formation (Scheme 75).270 The Wirth group had used this same electrochemical flow microreactor in earlier work involving intramolecular olefin amidofunctionalization.150 Optimized electrolytic conditions involved flowing a reagent solution in MeCN/MeOH (1:1) through this cell assembly equipped with graphite anode and Pt cathode. Importantly, no supporting electrolyte or catalyst was required. This compares favorably to an earlier report from Xu, where batch mode electrolysis on the same substrate class required 20 mol% n-Bu4N+BF4– and 5 mol% TEMPO to fulfill these roles.271 A scope of 28 examples was presented in yields of 33–97%. Both aryl (75.1) and alkyl (75.2, 75.3) thioamides reacted smoothly, and many functional groups were tolerated, including unprotected alcohol, esters, carbamates, sulfonamides, and phosphine oxides. Cyclization onto both aryl and heteroaryl moieties proceeded well (75.4, 75.5), enabling access to both benzothiazoles and thiazolopyridines, and a thiazolo[4,5-c]quinoline, 75.6, which is an intermediate in the synthesis of CL075, a TLR8 receptor agonist.272
Scheme 75. Catalyst- and Electrolyte-Free Flow Electrochemical Synthesis of Benzothiazoles and Thiazolopyridines (Wirth and Xu, 2018).

*With 3% v/v H2O added as cosolvent.
The authors propose deprotonation of the thioamide substrate by electrogenerated methoxide base and oxidation of the resultant anion at the anode, yielding an S-centered thioamidyl radical. Cyclization onto the pendant arene followed by further anodic oxidation and proton loss furnishes the product. A different mechanism is proposed by Xu when a TEMPO redox catalyst is added, wherein anodic oxidation of TEMPO forms the corresponding oxoammonium ion. This then reacts with the thioamide substrate in a polar fashion to form the S-bound TEMPO adduct.271 The weak S–O bond then undergoes homolysis to generate the S-centered radical, before cyclization and aromatization in the same manner.
Gustafson and co-workers in 2019 reported a photocatalytic synthesis of benzothiazoles from the corresponding thiobenzamides (Scheme 76).273 An optimized set of reaction conditions consisted of blue-light irradiation of substrate solutions in MeCN/H2O (1:1) in the presence of [Ru(bpy)3]Cl2·6H2O ([Ru-1]Cl2·6H2O) photocatalyst, pyridine Brønsted base, and Na2S2O8 oxidant. A markedly detrimental effect on reaction performance was observed in the absence of pyridine (e.g., for example 76.1, a 57% yield was realized in the absence of pyridine, compared to 79% in its presence). A total of 15 examples of this transformation were reported, in yields of 30–79% (76.1–76.4). Primary, secondary, and tertiary alkyl and aryl substituents at the thioamide were amenable under these conditions.
Scheme 76. Photocatalytic Synthesis of Benzothiazoles (Gustafson, 2019).

Authors invoke a concerted PCET mechanism for thioamidyl radical generation, by noting that the oxidation potential of a thioamide substrate (e.g., for N-phenyl tert-butylthioamide, Ep/2 = +1.50 V vs SCE in MeCN)273 is negatively shifted in the presence of the pyridine additive (e.g., for N-phenyl tert-butylthioamide in the presence of 1 equiv of pyridine, Ep/2 = +1.20 V vs SCE in MeCN).273 Therefore, this mechanistic proposal involves initial oxidative quenching between the photoexcited Ru(II) complex (E1/2 Ru(III)/*Ru(II) = −0.96 V vs SCE in MeCN)64 and persulfate oxidant (Ep/2ox = +2.01 V vs SCE in MeCN)274 to generate an Ru(III) complex and sulfate radical anion. The joint action of this Ru(III) oxidant (E1/2 Ru(III)/Ru(II) = +1.21 V vs SCE in MeCN)64 and pyridine Brønsted base (pKaH = 12.5 in MeCN)48 mediates N–H bond homolysis via concerted PCET for thioamidyl radical generation. Thereafter, cyclization via an S-centered radical onto the pendant arene, and further steps of ET mediated by sulfate radical anion, then PT, yields the aromatized product.
Ahmed and Wirth reported the electrosynthesis of thiazolidin-2-imines from N-allylthioureas through N–H bond activation and subsequent C–S bond formation (Scheme 77).275 An electrochemical flow microreactor was used in this work, which was developed and previously demonstrated by the Wirth group.150,270 Optimized electrolytic conditions involved flowing a solution of thiourea substrate, TEMPO radical trap, and BnMe3N+ HO–, as both Brønsted base and electrolyte through this electrochemical cell equipped with a graphite anode and a Pt cathode in MeCN/H2O (19:1) at room temperature. When N-allyl-N-arylthioureas were used in this protocol, the desired thiazolidin-2-imines were formed exclusively through C–S bond formation, with 16 examples showcased in yields of 10–93% (77.1–77.4). When N-allyl-N-alkylthioureas were instead employed as substrates, mixtures of thiazolidin-2-imines and cyclic thioureas typically resulted. In three cases, comparisons to batch electrolysis in both commercial and custom electrochemical cells demonstrated that flow electrolysis provided superior yields.
Scheme 77. Electrosynthesis of Thiazolidin-2-imines from N-Allylthioureas (Ahmed and Wirth, 2019).

A plausible mechanism involves discrete deprotonation of the thiourea substrate by hydroxide base prior to mediated ET from the oxoammonium ion, produced via anodic oxidation of TEMPO (E1/2ox = +0.29 V vs Ag+/Ag in MeCN).276,277 The resulting radical can undergo a 5-exo-trig cyclization onto the pendant olefin through either sulfur or nitrogen depending on the thiourea N-substituent. Termination through trapping with TEMPO yields the major oxysulfurization or minor oxythioamidation products, respectively. Cathodic reduction of H2O regenerates the requisite hydroxide base and liberates molecular hydrogen.
2.2.2. Intramolecular S–S and N–S Bond Formation
Waldvogel and co-workers reported an electrochemical method enabling dehydrogenative S–S bond formation in an intramolecular cyclization reaction of dithioanilides, giving 3,5-diimido-1,2-dithiolanes.278 Optimized reaction conditions involved constant current electrolysis of substrates in an undivided cell equipped with graphite anode, Pt cathode, n-Bu4N+PF6– supporting electrolyte in MeOH solution (Scheme 78). A scope of 12 examples of dehydrogenative S–S bond formation were reported, including both symmetrical (78.1) and unsymmetrical (78.2) dithioanilides (33–87% yields). Substrates carried a gem-dimethyl group between the two thioamide groups to facilitate cyclization through the Thorpe–Ingold effect. A variety of substituted arenes were tolerated, including halogenated and multiply substituted. The authors do not propose a mechanism for the transformation, though PT from the N–H substrate and subsequent ET could lead to an S-centered radical. This could then add across the second, neutral thioamide, before further anodic oxidation and proton loss would lead to the product. Cathodic reduction of MeOH solvent could generate methoxide and liberate H2.
Scheme 78. Electrochemical Synthesis of 3,5-Diimido-1,2-dithiolanes (Waldvogel, 2018).

*With MeCN as solvent.
Liu, Yang, Zhou, and co-workers reported an electrochemical synthesis of 3-substituted 5-amino-1,2,4-thiadiazole heterocycles through N–S bond formation from imidoylthiourea precursors which were readily prepared through the union of an amidine and isothiocyanate (Scheme 79).279 Substrates were electrolyzed in an undivided cell with carbon rod anode and Pt plate cathode, operating under constant current conditions with n-Bu4N+BF4– supporting electrolyte in MeCN solution. A scope of 26 examples was presented, with yields of 74–92%. Imidoylthiourea substrates derived from (hetero)aryl (79.1–79.3) and alkyl amidines (79.4) were successful in this reaction. Additionally, substrates derived from both aryl and alkyl (79.5) isothiocyanates were competent. The reaction was further demonstrated on substrates containing pyridine and pyrazole heterocycles.
Scheme 79. Electrochemical Synthesis of 3-Substituted 5-Amino-1,2,4-thiadiazoles (Liu, Yang, and Zhou, 2020).

A proposed mechanism involved substrate anodic oxidation (e.g., for the substrate leading to thiadiazole 79.1, Ep/2ox = +1.10 V vs SCE in MeCN),279 and subsequent proton loss at the thiourea N–H bond to yield an S-centered radical intermediate. S–N bond formation via cyclization onto the amidine follows, before further anodic oxidation and deprotonation yield the product. Cathodic reduction of liberated protons leads to hydrogen evolution.
2.3. Transformations of Alkylamines, Anilines, and Azoles
2.3.1. Inter- and Intramolecular C–N Bond Formation through Alkyl C(sp3)–H and (Hetero)aryl C(sp2)–H Amination
2.3.1.1. With Alkylamines
Here we present two methods for heteroarene C(sp2)–H amination with alkylamines proceeding via coupled steps of ET and PT for neutral aminyl radical generation. These are nucleophilic radicals in nature, and add readily to electron-deficient π-systems.280−282 This reactivity contrasts that of aminium radical cations, which are electrophilic species and add readily to electron-rich olefins and arenes for C–N bond formation.283 In 2018, Ackermann and co-workers reported a protocol for an electrochemical dehydrogenative heteroarene C(sp2)–H amination with aliphatic amines under catalyst- and reagent-free conditions (Scheme 80).284 This amination reaction proceeded efficiently in an undivided cell equipped with a RVC anode and Pt cathode under constant current electrolysis. The authors noted that the addition of AcOH was sufficient to observe high yields of the product without additional electrolyte. Using this procedure, 40 examples of heteroarene C(sp2)–H amination products were reported, with yields of 33–95%. Primarily benzoxazoles were studied in this method (80.1–80.3), with a variety of alkyl, aryl, and halogen groups appended. Benzothiazole (requiring addition of 20 mol% Zn(OAc)2 catalyst) and several oxadiazole heterocycles were also included in the scope (80.5). A necessary requirement for successful reactivity was the use of a secondary alkylamine coupling partner, but cyclic (80.1–80.7) and acyclic amines (80.8) were permitted. Appended to this component included examples of olefin, alkyl chloride, and carbamate functionality. Additionally, anilines also provided the desired product, albeit at elevated temperatures (80 °C). An indole failed to undergo C(sp2)–H amination under these conditions.
Scheme 80. Electrochemical C(sp2)–H Amination of Heteroarenes with Alkylamines (Ackermann, 2018).

*80 °C.
The authors then studied the mechanism of the reaction through a variety of experimental and computational means. Competition experiments probing the reactivity of oxazole partners indicated preference for reaction of the substrate with electron-donating substituents. CV experiments indicate that oxidation of the amine (e.g., for morpholine, Ep/2ox = +1.19 V vs SCE in MeCN),284 occurs ca. 800 mV below that of the oxazole (e.g., for 5-methylbenzoxazole, Ep/2ox = +2.02 V vs SCE in MeCN).284 Furthermore, addition of BHT as a radical trap, suppressed C(sp2)–N product formation and afforded the BHT-amine adduct instead. In kinetic experiments, the authors observed that the rate was zero-order for both the amine and oxazole substrates, and was only dependent on the concentration of AcOH and applied operating current. The former dependency is consistent with results of DFT calculations indicating that protonation of the oxazole component facilitates addition of the aminyl radical. With these results, the authors propose a mechanism wherein the amine undergoes stepwise anodic oxidation, followed by deprotonation to generate a neutral aminyl radical. This nucleophilic aminyl radical then engages the protonated oxazole for C–N bond formation, generating a de-aromatized radical cation intermediate. Stepwise ET and PT forms the product, and protons lost during this process are reduced at the cathode, generating molecular hydrogen as a byproduct.
In 2018, Wei, Zhou, and co-workers reported a protocol for the photocatalytic C3-selective C(sp2)–H amination of quinoxalinones with primary and secondary aliphatic amines proceeding through a proposed neutral aminyl radical intermediate (Scheme 81).285 These optimal conditions consisted of visible-light irradiation of a solution containing quinoxalinone and amine substrates, and EY photocatalyst in THF under air. A total of 30 examples of 3-aminoquinoxalin-2(1H)-ones were reported with yields ranging from 45% to 84%. Variation of the N-substituent on the quinoxalinone was well tolerated, with N-propargyl (81.1) and N-aryl (81.2) substituted products produced in good yields. Additionally, an unprotected N–H quinoxalinone underwent efficient C–H amination in 81% yield (81.3). N-Acyl quinoxalinone presented a limitation in the scope, providing no amination product. A wide variety of aliphatic amines were also tolerated, including pyrrolidine (81.4) and acyclic secondary amines (81.5). Additionally, sec-butylamine reacted efficiently, affording C–N coupling product in 58% yield (81.6).
Scheme 81. Photocatalytic C(sp2)–H Amination of Quinoxalinones with Alkylamines (Wei and Zhou, 2018).

To probe the mechanism of this transformation, the authors conducted radical trapping experiments with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO). Under light irradiation, they observed a spin-trapped DMPO-morpholine adduct, indicating that the reaction proceeds through the intermediacy of a nitrogen-centered radical, as opposed to oxidation of the quinoxalinone to the corresponding radical cation and addition of the closed-shell nucleophilic amine. Furthermore, the authors determined the necessity of oxygen in the transformation, observing only trace amounts of the product when the reaction was performed under a nitrogen atmosphere. With this information, the authors proposed a mechanism where the amine substrate is oxidized (e.g., for pyrrolidine, Ep/2ox = +0.89 V vs SCE in MeCN)21 by the excited state of EY (E1/2 *EY/EY•– = +0.83 V vs SCE in MeCN),71 generating the corresponding aminium radical cation and EY•–. The reaction of the reduced photocatalyst with O2 regenerates ground-state EY and O2•–. Deprotonation of the aminium radical cation intermediate provides the neutral N-centered radical, which adds into the quinoxalinone. The authors then propose re-aromatization of this radical species by further oxidation and proton loss to generate the product.
2.3.1.2. With Arylamines
In 2017, Xia and co-workers reported a photocatalytic dehydrogenative cross-coupling reaction between diarylamines and phenols proceeding through a radical–radical coupling pathway (Scheme 82).286 Optimal conditions consisted of blue-light irradiation of acyclic diarylamine and phenol substrates in the presence of organic photocatalyst 2,4,6-triphenylpyrylium BF4 ([TPP]BF4) and (NH4)2S2O8 oxidant in CH2Cl2 solution at room temperature. Under these conditions, 33 examples of C–N bond formation were reported with yields of 23–94%. A number of electron-rich diarylamines were successful partners, but diphenylamine was not, providing the corresponding coupling product in less than 5% yield. A variety of substituted phenols were competent substrates, including those containing alkyne (82.3) and halogen (82.4) substituents. However, the phenol scope was limited to para-methoxy-substituted phenols as other substituents at this position afforded no product.
Scheme 82. Photocatalytic C(sp2)–H Amination of Phenols with Diarylamines (Xia, 2017).

The authors then turned to investigate the mechanism of this cross-coupling reaction, proposing that C–N bond formation occurs through radical–radical coupling of a neutral phenoxyl and neutral aminyl radical. Control experiments revealed that both photocatalyst and (NH4)2S2O8 were required to observe product formation. Additionally, the authors observed an EPR signal consistent with neutral aminyl radical formation based on literature precedent, which was consistent with the researchers’ observation of efficient quenching of the photocatalyst excited state by amine substrate in luminescence quenching experiments. The authors also measured a quantum yield for the reaction (Φ = 19), indicating that a chain process is operative. Based on these data, the authors propose a mechanism wherein the diarylamine is activated via single-electron oxidation from the excited state of the photocatalyst (E1/2 *TPP+/TPP• = +2.02 V vs SCE in MeCN),45 to form the corresponding aminium radical cation. Simultaneously, the phenol component is oxidized by SO4•–, which is generated via decomposition of persulfate upon light irradiation. The aminium radical cation intermediate is also proposed to act as a redox mediator of phenol oxidation. Deprotonation of the phenoxyl and aminium radical cation intermediates results in neutral phenoxyl and diaryl aminyl radicals, which then undergo radical–radical coupling to form a C–N bond. Tautomerization of the C–N coupled intermediate provides the product. The ground state of the photocatalyst is regenerated by SET (E1/2TPP+/TPP• = −0.32 V vs SCE in MeCN)45 to SO4•–.
In a 2018 report, Lei and co-workers demonstrated an electrochemical C2-selective intermolecular C(sp2)–H amination reaction of phenols with phenothiazines and phenoxazines (Scheme 83).287 Optimized reaction conditions consisted of the constant current electrolysis of substrate admixtures in an undivided cell equipped with graphite rod anode and Ni plate cathode. n-Bu4N+BF4– served as supporting electrolyte in a solvent mixture of MeCN/MeOH (4:1). A total of 20 examples of phenol C2-amination were given with yields of 21–93%. For phenols with C2-substitution, C4-amination was observed, and a further two examples given. Unsubstituted phenol reacted to form an unselective mixture mono-C2- and di-C2,C4-amination products. The reaction was highly tolerant of substitution on the phenol, with both EWGs and EDGs demonstrated. Halogen, ester, acetamide, and primary alcohol functional groups were compatible (83.1–83.4). In addition, substitution on the PTZ component was permitted. Phenoxazine was a capable amine coupling partner (83.5), but other diarylamines proceeded poorly (e.g., di-p-tolylamine gave 20% yield). A decagram-scale synthesis was performed, and the reaction could be run open to air without detriment.
Scheme 83. Electrochemical C(sp2)–H Amination of Phenols with Phenothiazines (Lei, 2018).

CV indicated that phenoxazine component (e.g., for phenoxazine, Ep/2ox = +0.70 V vs Ag/AgCl in MeCN/MeOH)287 was preferentially oxidized to the corresponding radical cation compared to the phenol substrate (e.g., for 4-methoxyphenol, Ep/2 = +0.95 V vs Ag/AgCl in MeCN/MeOH, for 4-chlorophenol, Ep/2 = +1.35 V vs Ag/AgCl in MeCN/MeOH).287 When this reaction was performed under a constant applied potential of +0.70 V, the products were still observed in good yield. Based on these observations, the authors propose that PTZ is first oxidized to its corresponding radical cation at the anode, which then reacts with the closed-shell phenol through either the aminium radical cation or neutral aminyl radical after deprotonation. Thereafter, further anodic oxidation with proton loss restores aromaticity and yields the amination product. Cathodic reduction of methanol cosolvent leads to the in situ generation of methoxide base to mediate PT steps, and the liberation of molecular hydrogen.
The next year, Lei in collaboration with Weng and Chiang, adapted the above method to now enable site-selective bioconjugation of tyrosine residues in polypeptides and proteins with PTZ and its derivatives (Scheme 84).288 Tyrosine is considered an attractive target for the labeling of biomolecules due to its low natural abundance, yet methods for the selective bioconjugation of C(sp2)–H bonds in aromatic AA side chains are rare.289 At the outset, CV studies were carried out in order to study the compatibility of an electrochemical method for protein bioconjugation of PTZ derivatives. These revealed that PTZ is oxidized (Ep/2ox = +0.80 V vs Ag/AgCl in MeCN/H2O)288 at significantly lower applied potentials than the aromatic amino acids Tyr, Trp, Phe, and His, and thus was expected to permit the selective generation of the reactive electrophilic PTZ radical. Second, the more electron-rich nature of the phenol functional group of Tyr compared to other aromatic amino acids favored this C2-position as the preferential site of bioconjugation. The reaction was proposed to occur through the same mechanism as above in small molecule phenol C2-amination.
Scheme 84. Electrochemical C2-Selective Bioconjugation of Tyrosine Residues with Phenothiazines in (A) Isolated Amino Acids, (B) Tyrosine-Containing Dipeptides, and (C) Complex Polypeptides (Lei, Weng, and Chiang, 2019).

Initial experiments devised conditions to permit the C2-selective amination of tyrosine with several functionalized PTZs on the model isolated amino acid Ac-Tyr-OMe (Scheme 84A). Constant current electrolysis in an undivided cell equipped with graphite rod anode and Ni plate cathode of AA and PTZ substrates, in the presence of Na2SO4 electrolyte in MeCN/H2O at room temperature gave access to the desired products. A series of seven substituted PTZs carrying a variety of functional groups including aryl chloride, thioether, nitrile and an azide tag were demonstrated in the C2-amination of Ac-Tyr-OMe, in yields of 33–85%. To study the selectivity for bioconjugation at tyrosine over other AA residues, a series of tyrosine-containing dipeptide models were subject to electrolysis now in mixtures of MeCN/PBS (pH 7.3), which now required no exogenous electrolyte (Scheme 84B). This demonstrated the compatibility of this method with Gly, Leu, Met, Phe, Trp, His, Lys, Ser, and Asp AA residues. However, an experiment with a Cys-containing dipeptide was unsuccessful due to incompatibility of the thiol functional group under the electrolytic conditions. Next, a series of tyrosine-containing oligopeptides from 5-mer to 29-mer were subject to this electrolytic PTZ bioconjugation method, including N-Ac-RGD peptide, kisspeptin, angiotensin Y, tyrosine kinase, and glucagon (Scheme 84C). These peptides carried tyrosine either at the N-terminus, C-terminus, or within the chain. Glucagon, containing two tyrosine residues at the i and i+3 positions, reacted with selective monoamination. These all gave >99% conversion of substrate polypeptide in 30 min at room temperature. A PTZ moiety modified with the drug probenecid, and another with a biotin tag were shown to selectively react at the tyrosine residue of the pentapeptide YAGFL. The PTZ incorporation was also shown to be fluorescent under UV irradiation, raising the prospects of use as a biological fluorophore. Finally, the electrochemical bioconjugation of insulin (5808 Da) and myoglobin (16.7 kDa) with excess PTZ was studied. Insulin displayed tagging at four distinct tyrosine sites, as revealed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) MS analysis, and myoglobin selectively incorporated only one PTZ tag, shown through liquid chromatography/mass spectrometry (LCMS) analysis. CD spectroscopy revealed that the electrochemical modification had negligible effect on the structure of either protein.
In 2019, Lei and co-workers reported a para-selective electrooxidative cross-coupling reaction between electron-rich arenes and N,N-diarylamines, for the preparation of triarylamine products via a proposed radical–radical coupling mechanism (Scheme 85).290 The optimal reaction conditions consisted of constant current electrolysis of the arene and diarylamine components in MeCN/MeOH (7:3) solvent with n-Bu4N+BF4– supporting electrolyte in an undivided cell equipped with a graphite rod anode and a Pt plate cathode. The authors provided 40 examples with 21–98% yields. The scope of diarylamine included PTZ (85.1) and phenoxazine derivatives, as well as acyclic N,N-diarylamines including estrone-derived 85.4. A variety of electron-rich arenes and heteroarenes such as N,N-dimethylaniline (85.1), indole (85.3), pyrrole, and imidazopyridine served as competent cross-coupling partners. When both PTZ and acyclic N,N-diarylamines were used as coupling partners, selective C–N bond formation of the PTZ was observed (85.2).
Scheme 85. Electrochemical C(sp2)–H Amination of Anilines and Electron-Rich (Hetero)arenes with Diarylamines (Lei, 2019).

*MeCN/AcOH (9:1) as solvent.
CV and EPR experiments indicated that under the reaction conditions both electron-rich arene (e.g., for N,N-dimethylaniline, Eonsetox = +0.65 V vs Ag/AgCl in MeCN/MeOH (7:3))290 and diarylamine (e.g., for PTZ, Eonsetox = +0.50 V vs Ag/AgCl in MeCN/MeOH (7:3))290 could be oxidized to their corresponding radical cations. Further evidence for this hypothesis is provided by the observation of homo-coupling side products of both reaction components. Based on these collected data, the authors propose a mechanism wherein both species are oxidized to their respective radical cations at the anode surface. Subsequent deprotonation of the diarylamine generates a neutral aminyl radical species, which the authors were able to observe by EPR. Radical–radical coupling between this aminyl radical and the aniline radical cation forges the C–N bond through the para-position. Proton loss generates the product, and proton reduction at the cathode generates molecular hydrogen as a byproduct.
In a similar report, Li, Song, and co-workers reported an electrooxidative para-selective C(sp2)–H amination reaction of anilines with PTZs and phenoxazines to synthesize triarylamine products (Scheme 86).291 Reported conditions consisted of constant current electrolysis of the reaction components in a solution of MeCN with n-Bu4N+PF6– supporting electrolyte in an undivided cell equipped with a carbon rod anode and Pt cathode. The authors reported 32 examples with yields ranging from 30% to 94%. Variation of the aniline was tolerated including N,N-dialkyl (86.1, 86.2) and N-naphthylen-2-yl substituents on the aniline nitrogen. Additionally, use of 3,5-dimethoxyaniline afforded product 86.3 in good yield, demonstrating tolerance to primary aniline functionality. Variation of the secondary amine was limited to variously substituted PTZ derivatives including nitrile, ketone, and alkene (86.4) functionality in addition to a single example of a phenoxazine cross-coupling partner (86.5). The authors propose an analogous mechanism to that put forth by Lei and co-workers.290
Scheme 86. Electrochemical C(sp2)–H Amination of Anilines with Diarylamines (Li and Song, 2019).

Chen and co-workers recently reported an electrochemical synthesis of medicinally important pyrido[1,2-a]benzimidazoles through the oxidative cyclization of 2-(arylamino)pyridines via neutral aminyl radical generation and C–N bond formation (Scheme 87).292 Electrolysis was performed in aqueous solutions of 2-(arylamino)pyridine substrates in an undivided cell equipped with graphite anode and Pt cathode under constant current conditions, with KPF6 as a supporting electrolyte and NaOAc as a buffer at 100 °C. A total of 32 examples of pyrido[1,2-a]benzimidazole products were reported, in yields of 30–96%. The reaction was highly functional group tolerant, including those which were sensitive to oxidation (such as an aryl thioether 87.4 and a tertiary aniline 87.5) or reduction (such as an aryl aldehyde 87.6). Substitution was permitted on either of the pyridyl or aryl rings, at multiple positions. In some cases, addition of methyl thioglycolate as a HAT catalyst was required. The antimicrobial and cytotoxic behavior of these products was screened against several cell lines, and several potent inhibitors were identified.
Scheme 87. Electrochemical Synthesis of Pyrido[1,2-a]benzimidazoles (Chen, 2020).

*With 10 mol% HSCH2CO2Me.
The authors’ proposed mechanism involves discrete substrate deprotonation by cathodically generated hydroxide base to yield the corresponding nitrogen anion. Thereafter anodic single-electron oxidation yields a stabilized nitrogen radical, which can delocalize across both arylamine and pyridine N-sites. In CV experiments, addition of n-Bu4N+OH– to a solution containing substrate resulted in a substantially negatively shifted oxidation potential (Ep/2ox = +0.44 V vs SCE in MeCN),292 compared to the neutral substrate (Ep/2ox = +1.22 V vs SCE in MeCN),292 thus demonstrating this involvement of base in initial deprotonation. After oxidation, cyclization via the pyridine nitrogen onto the tethered arene, followed by further anodic oxidation and deprotonation yields the product. An inverse secondary KIE (kH/kD = 0.89) was observed in a competition experiment, which is consistent with a change in the C-center from sp2 to sp3 hybridized, and demonstrates that C–H bond cleavage is not rate-determining. This is mechanistically distinct from a Cu-catalyzed protocol for the related synthesis of pyrido[1,2-a]benzimidazoles from the same starting materials.293 When reaction conditions were modified to use a Pt anode, n-Bu4N+I– additive, and MeCN/MeOH solvent system, intermolecular N–N bond formation from these substrates could instead be selectively promoted. Dimerization was proposed to occur through the in situ generation of an N–I intermediate, then either bond homolysis for N-radical generation and subsequent dimerization, or N–N bond formation through a two-electron pathway.
2.3.1.3. With Azoles
We present here a number of methods enabling C–N bond formation between azole nucleophiles and alkyl C(sp3)–H or aryl C(sp2)–H bonds, proceeding through the proposed intermediary of neutral azoyl radicals generated through coupled steps of PT and ET. In 2017, Lei and co-workers reported a mild, photocatalytic oxidative C2 C(sp3)–H amination of solvent quantities of THF or 2-MeTHF with a variety of pyrazoles and 1,2,3-triazoles using N-Me Mes-Acr+ClO4– ([Acr-1]ClO4) photocatalyst with air as the terminal oxidant (Scheme 88).294 In total, 17 examples of azole N-alkylation in this manner were reported in yields of 24–85%. 2-MeTHF underwent preferential alkylation at the more-hindered tertiary C–H position (88.5). A variety of substitution patterns were permitted on the azole component, and chloro- (88.4), bromo- (88.2), and iodo-functional groups (88.3) were all tolerated. The authors report that in many cases, yields were conversion limited, but that use of a pure O2 atmosphere instead led to poor results. Other ethereal solvents, such as 1,4-dioxane, tetrahydropyran, and diethyl ether, gave poor results.
Scheme 88. Photocatalytic Aerobic C(sp3)–H Amination of THF and 2-MeTHF with Azoles (Lei, 2017).

*2-MeTHF as solvent.
In order to probe the mechanism of this transformation, a series of EPR experiments were performed with the spin-trapping reagent DMPO. The EPR spectrum for the spin-adduct resulting under optimized conditions was in agreement with established literature for an N-centered radical intermediate. Also, no EPR signal was observed in the absence of the azole coupling partner. Steady-state SV emission quenching experiments indicated that the azole was able to quench the photocatalyst excited state. An observed competition KIE experiment conducted in mixtures of THF and THF-d8 (kH/kD = 5.0) suggested that C(sp3)–H bond cleavage in the THF component was rate-determining. On the basis of these data, a mechanism was proposed involving reductive quenching of the photocatalyst excited state (E1/2 *1Acr+/Acr• = +2.08 V vs SCE in MeCN; E1/2 *3Acr+/Acr• = +1.88 V vs SCE in PhCN)73,74 with the azole substrate (e.g., for 1H-pyrazole, Ep/2ox = +2.12 V vs SCE in MeCN).21 This generates the reduced acridine radical and an azole substrate radical cation, the latter of which undergoes deprotonation to yield the N-centered azole radical. This PT step can plausibly occur with superoxide or hydroperoxide generated in situ through catalyst turnover and HAT steps. ET between the reduced-state photocatalyst (E1/2 Acr+/Acr• = −0.55 V vs SCE in MeCN)73 and molecular oxygen returns this to its ground state and generates superoxide as a byproduct. Superoxide is proposed to facilitate THF radical generation through HAT with the most hydridic C(sp3)–H bond, generating hydroperoxide anion as a byproduct. Through proposed radical–radical coupling between the N-centered azole radical and THF radical, the desired product results. Alternatively, the authors consider a secondary coupling pathway involving further oxidation of the THF radical to the corresponding oxocarbenium ion and subsequent nucleophilic coupling with the closed-shell azole substrate.
Lei and co-workers later in 2017 disclosed an electrochemical strategy enabling intermolecular C(sp3)–H amination with a number of azole substrates (Scheme 89).295 Optimized conditions for this transformation involved constant current electrolysis of MeCN solutions of azole and alkane substrates in an undivided cell equipped with Pt electrodes with n-Bu4N+BF4– supporting electrolyte at 80 °C. In the model system studying the C2 C(sp3)–H amination of THF with 1H-benzotriazole (89.1), complete selectivity for N1-alkylation was observed; in prior work involving oxidative amination with stoichiometric chemical oxidants, a mixture of N1-, and N2-alkylation was observed.296−298 A scope of 24 examples of azole alkylation in this manner were reported in yields of 30–93%. In addition to a number of benzotriazole substrates, other heterocycles including tetrazole (89.2), 1,2,3-triazole (89.3), indazole, and pyrazole (89.4) substrates underwent the desired reaction. A variety of alkyl substrates carrying α-heteroatom (89.5), allylic, and benzylic C–H bonds were demonstrated, using either 20% v/v co-solvent quantity or 10 equiv of with respect to the azole component.
Scheme 89. Electrochemical Oxidative C(sp3)–H Amination with Azoles (Lei, 2017).

While the authors performed mechanistic experiments indicating the formation of an N-centered radical, the precise mechanism of C–H activation and C–N bond formation remains unclear. CV experiments established that only the azole component was capable of thermodynamically favorable ET (e.g., for 1H-benzotriazole, Ep/2ox = +2.00 V vs Ag/AgCl in MeCN),295 in preference to the ether substrate (e.g., for THF, Ep/2ox = +2.54 V vs Ag/AgCl in MeCN)295 in the operating potential range of Ecell = +1.90–2.23 V. The authors propose that following discrete anodic oxidation, the resulting azole radical cation undergoes PT to yield the corresponding neutral N-centered radical intermediate. What remains to be established is the mechanism governing the activation of the C–H bond in the alkane component, and how C–N bond formation proceeds. With respect to the C–H activation step, the authors observed a competition KIE of kH/kD = 1.22 in the reaction between 1H-benzotriazole and mixtures of THF and THF-d8. This is a distinct observation from this research group’s related photocatalytic method of C(sp3)–H bond azolation (discussed directly above), where a competition KIE of kH/kD = 5.0 was reported.294 Concomitant cathodic reduction of protons led to the generation of molecular hydrogen in a coupled process.
In 2019, Yang, Song, and Li reported an electrochemical intermolecular C(sp3)–H amination of xanthenes with heteroaromatic azole nucleophiles (Scheme 90).299 This work was enabled by use of an undivided cell operating under constant current conditions, with carbon rod anode, Pt plate cathode, n-Bu4N+BF4– supporting electrolyte, and a catalytic amount of methanesulfonic acid in MeCN. A scope of 29 examples of C–N cross-coupling products were demonstrated, in yields of 40–98%. A number of functionalized xanthenes were viable coupling partners, as were a thioxanthene (90.2) and an N-Me dihydroacridine substrate (90.3). With respect to the azole component, 1H-indazoles (90.1), 1H-pyrazoles (90.4), benzimidazoles (90.5), triazoles, benzotriazoles (90.7), a purine (90.6), and a tetrazole (90.8) underwent this transformation successfully. This method represents an advance from the earlier work of Zeng (see section 2.1.4.2) achieving intermolecular C(sp3)–H amidation of xanthenes with N-methoxyamides,193 with respect to the breadth of scope of both reaction components.
Scheme 90. Intermolecular Electrochemical C–H Amination of Xanthenes with Azole Nucleophiles (Yang, Song, and Li, 2019).

To determine the mechanism of this transformation, the authors conducted CV experiments. These studies indicated that xanthene (Epox = +1.80 V vs Ag/AgCl in MeCN)299 and azole (e.g., for 1H-indazole, Epox = +1.82 V vs Ag/AgCl in MeCN)299 are oxidized at similar potentials, suggesting that both reaction components are oxidized to their respective radical cations at the anode. Subsequent deprotonation would yield the neutral C-centered and N-centered radicals, which could undergo radical–radical cross-coupling to furnish the observed product. Concomitant cathodic reduction of the liberated protons yields molecular hydrogen. Although the role of methanesulfonic acid in this work remains unclear, the researchers note diminished yields upon excluding it from the reaction (51% compared to 81% yield of 90.1). This radical coupling mechanism is distinct from the polar reaction pathway suggested by Zeng, in which a neutral amidyl radical mediates HAT from the xanthene substrate to yield a C-centered radical which is oxidized to form a carbocation that is subsequently trapped by a closed-shell N-nucleophile.193
In 2017, Adimurthy and co-workers reported protocols for the photocatalytic C5-selective C(sp2)–H azolation of 8-aminoquinoline amides (Scheme 91A), and the C3-selective C(sp2)–H azolation of imidazo[1,2-a]pyridines with a variety of azole partners (Scheme 91B).300 Optimal reaction conditions consisted of blue-light irradiation of quinoline or imidazo[1,2-a]pyridine substrate and azole coupling partner in the presence of N-Me Mes-Acr+ClO4– photocatalyst ([Acr-1]ClO4), and K2S2O8 terminal oxidant. Under these conditions, 35 examples of quinoline azolation and 11 examples of imidazopyridine azolation were reported in yields of 27–84% and 50–82%, respectively. The scope of 8-aminoquinoline cross-coupling included a variety of azole coupling partners, including benzotriazole (91.1), imidazole (91.2), and indazole (91.3) derivatives. Additionally, alkyl (91.1–9.3) and aryl (91.4) amides of the substrate quinoline were demonstrated. For imidazo[1,2-a]pyridine amination, a similar scope with respect to azole partner was reported, including benzotriazole (91.5), pyrazole (91.6), and triazole (91.7, 91.8) derivatives. Variation of the imidazopyridine was limited to arene substituents at the C2-position.
Scheme 91. Photocatalytic C(sp2)–H Azolation of (A) 8-Aminoquinoline Amides and (B) Imidazo[1,2-a]pyridines (Adimurthy, 2017).

The authors propose a radical–radical coupling mechanism for C–N bond formation similar to that proposed by Xia and co-workers in a related photocatalytic oxidative cross-coupling reaction of phenols and diarylamines.286 The azole coupling partner is proposed to undergo oxidation (e.g., for 1H-benzotriazole, Ep/2ox = +2.00 V vs Ag/AgCl in MeCN)295 via reductive quenching of the photoexcited state of the acridinium (Acr) catalyst (E1/2 *1Acr+/Acr• = +2.08 V vs SCE in MeCN; E1/2 *3Acr+/Acr• = +1.88 V vs SCE in PhCN),73,74 followed by proton loss to generate a neutral N-centered azolyl radical. The quinoline or imidazopyridine component is simultaneously oxidized to the corresponding radical cation by persulfate radical anion generated through homolysis under blue-light irradiation. Deprotonation of this radical cation species generates a neutral radical intermediate. Radical–radical cross-coupling of these components forges the C–N bond followed by proton loss to generate the product. C5-selectivity in 8-aminoquinoline amide C(sp2)–H azolation is understood in considering stabilization of the neutral radical through an imine resonance form in the para-position.
In 2019, Feng and Chen reported an electrochemical method for the intermolecular C2-selective C(sp2)–H azolation of phenols and anilides with aromatic N-heterocycles (Scheme 92).301 Optimized reaction conditions involved the constant potential electrolysis of admixtures of these substrates in an undivided cell equipped with Pt electrodes, with n-Bu4N+PF6– supporting electrolyte, in a CH2Cl2/HFIP (3:7) solvent system. A total of 39 examples of phenol C(sp2)–H C2-azolation were reported, in yields of 38–98%. Typically, electron-rich alkoxy-substituted phenols were optimal substrates. A variety of azole partners were capable of undergoing this reaction, including pyrazoles (92.1–92.3), 1,2,4-triazoles (92.4), 1,2,3-triazoles, tetrazoles (92.5), indazoles, and benzotriazoles. Functional groups demonstrated on either component included halogen, nitro, terminal olefin, ester, carboxylic acid, thioether, and sulfonamide. A 3-hydroxypyridine substrate underwent C2-selective azolation with 4-bromopyrazole in 96% yield. In addition, under the same reaction conditions 10 examples of the C2 azolation of 4-methoxyanilide substrates were reported, in yields of 48–91% (92.6, 92.7). Sulfonamide and acetamide N-protecting groups were tolerated on the anilide, but a methyl carbamate was ineffective.
Scheme 92. Electrochemical C2 C(sp2)–H Azolation of Phenols and Anilides (Feng and Chen, 2019).

Initial mechanistic studies involved ruling out a Michael addition pathway through in situ formation of a quinone or iminoquinone intermediate from the phenol or anilide reaction partner. However, no product formation was observed with these proposed intermediates in the presence of azole, ruling out a polar mechanism. The appearance of 2,2′-biphenol and N–N linked dimerization side products indicated the generation of open-shell intermediates of both reaction partners. This was further supported by CV experiments wherein indicated that both azole and phenol components were oxidized at under the potentials applied in the reaction conditions (e.g., for pyrazole, Ep/2ox = +1.20 V vs SCE in HFIP/CH2Cl2 (7:3), and for 4-methoxyphenol, Ep/2ox = +1.00 V vs SCE in HFIP/CH2Cl2 (7:3)).301 When the applied cell potential was fixed at +1.00 V to drive oxidation of only the phenol component, no reaction product was observed, ruling out a pathway for the addition of the closed-shell azole component to the phenol radical cation. Thus, a proposed pathway involves oxidation of both components to their respective radical cation intermediates before PT steps yield the corresponding neutral radicals. Thereafter, radical–radical coupling yields the C2 C(sp2)–H amination product. Concomitant reduction of liberated protons at the cathode surface leads to the extrusion of molecular hydrogen.
Lei and co-workers in 2019 reported an electrochemical dehydrogenative intermolecular C–N coupling between 2-aryl imidazo[1,2-a]pyridines or benzo[d]imidazo[2,1-b]thiazoles and five-membered-ring azole heterocycles (Scheme 93).176 Optimized reaction conditions involved electrolysis of substrate admixtures in an undivided cell with graphite anode and Ni plate cathode under constant current conditions, with NaNO3 supporting electrolyte in EtOH/H2O (1.2:1) at 50 °C. A scope of 17 examples of C(sp2)–H amination products was presented, in yields of 40–85%. With respect to the 2-aryl imidazo[1,2-a]pyridine substrate, substitution was permitted on the 2-aryl group (93.1–93.4) and the pyridine scaffold. Similarly, a number of benzo[d]imidazo[2,1-b]thiazole substrates were shown, including a thiophene-substituted example. Several azole nucleophiles were competent partners, including imidazoles, benzimidazoles (93.6), and a pyrazole. Morpholine too gave C–N coupling product 93.7 in good yield, but n-butylamine was unreactive.
Scheme 93. Dehydrogenative C–N Cross-Coupling of Imidazo[1,2-a]pyridines or Benzo[d]imidazo[2,1-b]thiazoles with Azoles (Lei, 2019).

*With 3 equiv of azole.
A proposed mechanism involves discrete deprotonation of the azole partner by cathodically generated ethoxide or hydroxide base to yield the corresponding azole anion (e.g., for imidazole, pKa = 18.6 in DMSO).116 Thereafter, anodic oxidation generates the neutral N-centered azole radical. Simultaneously, anodic oxidation of the closed-shell electron-rich heterocycle partner yields the corresponding radical cation, which is deprotonated to yield the heteroaryl radical. Subsequent radical–radical cross-coupling yields the desired C–N coupled product. This radical–radical coupling mechanism, as opposed to azole radical addition to the heteroarene radical cation followed by deprotonation, was argued based on the observation of imidazo[1,2-a]pyridine dimerization upon electrolysis in the absence of the azole component. Although the onset potentials for azole oxidation (e.g., for imidazole, Eonsetox = +0.93 V vs Ag/AgCl in EtOH/H2O (1.2:1))176 and imidazo[1,2-a]pyridine oxidation (e.g., for 2-phenylimidazo[1,2-a]pyridine, Eonsetox = +0.94 V vs Ag/AgCl in EtOH/H2O (1.2:1))176 are similar, oxidation of the deprotonated azole anion is expected to be more facile. The ability to generate both reactive radicals simultaneously under the constant current conditions is proposed to be a prerequisite for radical–radical cross-coupling.
2.3.2. Intermolecular N–N Bond Formation
Two methods have been reported enabling the formation of N–N bonds through the homo-coupling of arylamines, and through alkylamine and azole N-nitrosation or N-nitration respectively, which proceed through the intermediacy of neutral N-centered radicals generated through coupled PT and ET. In 2019, Xu and co-workers reported a protocol for electrochemical N–N bond formation for the synthesis of symmetrical tetrasubstituted hydrazines (Scheme 94).302 The reaction proceeded in an undivided cell under constant current conditions with the addition Et4N+BF4– supporting electrolyte and pyridine as a Brønsted base, in a mixture of THF/MeOH (3:1). The authors reported 19 examples of this transformation in yields of 36–95%, including iodide-substituted 94.1, brominated carbazole dimer 94.2, as well as dimers of secondary N-aryl-N-alkylamines 94.3. The authors propose that initial oxidation of the secondary arylamine substrate at the anode, followed by deprotonation of the nascent radical cation by pyridine base, generating the N-centered aminyl radical. This intermediate is then proposed to undergo dimerization via radical–radical coupling to afford the tetrasubstituted hydrazine product. The pyridine additive can be regenerated through deprotonation with cathodically generated methoxide. As the hydrazine product of the reaction has a lower oxidation potential (e.g., for tetra(4-t-Bu-phenyl)hydrazine, Ep/2ox = +0.68 V vs SCE in MeCN)302 than the corresponding starting material (e.g., for di(4-t-Bu-phenyl)amine, Ep/2ox = +0.80 V vs SCE in MeCN),302 the authors propose that the product can then serve as a redox catalyst to facilitate the formation of the aminium radical cation in subsequent turnovers.
Scheme 94. Electrochemical Synthesis of Tetrasubstituted Hydrazines (Xu, 2019).

*With 1 equiv of KOAc instead of pyridine, at reflux.
Lu and co–workers recently reported electrochemical methods for the N-nitration of azole heterocycles and the N-nitrosation of secondary alkylamines through intermolecular N–N bond formation (Scheme 95).303 Cross-selectivity for N–N bond formation has generally been difficult to achieve, making this method noteworthy. Substrate electrolysis was performed in an undivided cell under constant current conditions, in the presence of Fe(NO3)3 nonahydrate as the nitration/nitrosation reagent and n-Bu4N+BF4– as supporting electrolyte in MeCN at 70 °C. When azole nucleophiles were subject to this method, N-nitration (N–NO2) products were obtained with 21 examples documented in yields of 15–91%. Pyrazole (95.1–95.4), imidazole, indazole (95.5), triazole, and benzotriazole heterocycles underwent this reaction successfully. When secondary alkylamines rather than heterocycles were subjected to the above conditions, N-nitrosation (N–NO) products were formed, with 27 examples reported in yields of 56–92%. Piperidines, morpholines (95.6), thiomorpholines (95.7), mono-protected piperazines (95.8), pyrrolidines, and tetrahydroisoquinolines all reacted efficiently. A number of 4-substituted piperidines were used to showcase functional group tolerance, which included primary alcohols, alkyl bromides, and alkyl iodides. Notably, diarylamines were unreactive.
Scheme 95. Electrochemical N-Nitration of Azole Heterocycles and N-Nitrosation of Secondary Alkylamines (Lu, 2020).

*60 mol% n-Bu4N+ClO4– as electrolyte.
The researchers posited that the reaction involved generation of a neutral azole N-centered radical through stepwise oxidation and deprotonation and/or the generation of a secondary alkyl aminium radical cation intermediate through ET, without coupled PT. Upon heating, Fe(NO3)3 is proposed to decompose to both nitro and nitroso radicals, which can intercept the substrate derived N-centered radicals through a radical–radical coupling mechanism. The authors hypothesize that selectivity for azole nitration instead of nitrosation is due to the reversibility of the nitrosation pathway and irreversibility of the observed nitration pathway, implying that nitration for this class of substrates is under thermodynamic rather than kinetic control. The opposite selectivity for alkylamine nitrosation instead of nitration is attributed to a higher energy transition state barrier for N–N bond formation in the nitration pathway, suggesting that nitrosation for this class of substrates is under kinetic rather than thermodynamic control.
2.4. Transformations of Imines, Amidines, and Sulfoximines
Xu and co-workers developed a synthesis of fused (aza)benzimidazoles from N-acyl amidines through oxidative cyclization reactions of electrogenerated amidinyl radicals resulting from N–H bond homolysis (Scheme 96).304 Existing photocatalytic and electrochemical methods for the generation of iminyl-type radicals prior to this work typically involved single-electron reduction of pre-functionalized N–X bond reagents (X = O, S, N, etc.), such as O-acyl or O-aryl oximes.305 Thus, the ability to generate the reactive radical directly from the N–H bond represents a more atom- and step-economical process.
Scheme 96. Synthesis of (Aza)benzimidazoles via Amidinyl Radical Generation and Cyclization (Xu, 2017).

*5:1 THF:MeOH.
Reaction conditions involved the use of an undivided electrolytic cell assembly with an RVC anode and Pt cathode, operating under constant current conditions, with Et4N+PF6– as a supporting electrolyte in MeOH at 65 °C. Under these conditions, 46 examples of (aza)benzimidazole synthesis were reported in yields of 38–91% (96.1–96.5). The reaction was highly tolerant of varied electronics of the arene group undergoing C(sp2)–H bond amination, with many types of substituents and functional groups, including 3- and 4-pyridyl rings, proving to be efficacious substrates (96.3, 96.4). Reactions involving meta-substituted arenes underwent unselective amination at two sites to yield two regioisomeric products in an equal amount. Cyclization reactions of an ortho-chloro-substituted arene also proceeded efficiently through an SRAr pathway, as opposed to a radical addition/re-aromatization pathway. Notably, reactions could also be conducted on gram scale without any deterioration in the product yield. In addition to N-acyl amidines featured predominantly in this work, N-alkyl and N-iminyl amidines were competent substrates with variation of the reaction solvent to THF/MeOH mixtures (96.5).
The group proposes the following mechanistic rationale for the observed reactivity. A stepwise generation of the amidinyl radical intermediate through anodic substrate oxidation (Ep/2ox = +1.60 V vs SCE in MeOH)304 and subsequent deprotonation mediated by electrogenerated methoxide is invoked. Following this, 5-exo-trig cyclization onto the proximal arene yields a stabilized radical intermediate. Further anodic oxidation and PT yields the re-aromatized product. Cathodic reduction of solvent liberated molecular hydrogen and methoxide.
In addition to intramolecular cyclization of iminyl radicals generated through N–H bond PCET, examples have been reported for intermolecular addition reactions with alkynes and arenes. In 2018, Li and co-workers devised a synthesis of 2-substituted isoquinolines via the dehydrogenative annulative coupling of diaryl NH-ketimines and diaryl alkynes, through a dual photoredox/Co(III) catalytic reaction manifold (Scheme 97).306 A novel cobaloxime complex bearing a 4-N,N-dimethylamidopyridine (4-Me2NCOPy) axial ligand [Co(dmgH)2(4-Me2NCOPy)Cl] was identified as being highly efficient for hydrogen evolution in this system. Blue-light irradiation of CH2Cl2 solutions of aryl ketimine and aryl alkyne substrates in the presence of the organic photocatalyst N-Me Mes-Acr+ClO4– ([Acr-1]ClO4) and this Co(III) complex under a nitrogen atmosphere proved optimal for this transformation. A scope of 41 examples of this dehydrogenative annulation was explored, with yields of 23–92% reported. Symmetrical biaryl NH-ketimine substrates were studied in an annulation reaction with diphenylacetylene (97.1–97.3). Those carrying electronically neutral and electron-rich substituents in para-positions were optimal, with slightly reduced efficiency observed in those with electron-withdrawing substituents. Ketimine substrates carrying meta-substituents reacted to yield a mixture of product regioisomers with low selectivity (97.4). Unsymmetrical aryl alkyl NH-ketimines reacted to yield 1-alkylisoquinolines in moderate yields (97.5). With respect to the alkyne partner, symmetrical diarylacetylenes were optimal and showed preference for electron-rich functionality. Additionally, a trimethylsilyl-arylacetylene was a competent alkyne substrate (97.6). The use of naphthalene and phenanthryl-substituted ketimines allowed the preparation of π-extended isoquinoline products. The reaction could be conducted efficiently on gram scale without deterioration in yield.
Scheme 97. Dual Photoredox/Co(III)-Catalyzed Dehydrogenative Annulation of Aryl NH-Ketimines and Arylacetylenes (Li, 2018).

*Minor regioisomer.
Emission quenching experiments performed showed that in addition to the diaryl NH-ketimine and diarylacetylene substrates, the Co(III) proton reduction catalyst could also quench the excited state of the Acr photocatalyst. A proposed mechanism involved reductive quenching of the photocatalyst excited state (E1/2 *PC+/PC• = +1.88 V vs SCE in PhCN)74 with the ketimine substrate (E1/2ox = +1.24 V vs Fc+/Fc in MeCN),306 generating the substrate radical cation which through proton loss yielded a neutral iminyl radical intermediate. The resulting reduced photocatalyst (E1/2 PC+/PC• = −0.49 V vs SCE in PhCN)74 then reacts with the Co(III) co-catalyst (E1/2 Co(III)/Co(II) = −1.17 V vs Fc+/Fc in MeCN),306 giving a Co(II) complex. The neutral iminyl radical adds across the acetylene partner, followed by further cyclization of the generated vinyl radical onto the aryl group of the ketimine. The resulting cyclohexadienyl radical intermediate is oxidized by Co(II) catalyst (E1/2 Co(II)/Co(I) = −1.44 V vs Fc+/Fc in MeCN),306 generating a Co(I) complex. PT to this Co(I) complex yields the closed-shell product and generates a Co(III) hydride, which reacts with another proton to evolve hydrogen and return Co(III) ready for catalytic turnover. The authors note, however, that hydrogen evolution could occur from a Co(II) hydride after protonation and that homolytic cleavage of two Co(II) hydride complexes to evolve H2 is possible.72,307 The quantum yield of the reaction was recorded via ferrioxalate actinometry (Φ = 0.19) and is supportive of a closed photoredox cycle over a chain mechanism.
Sulfoximines, the mono-aza analogues of sulfones, are emerging as important structures in pharmaceutical discovery, displaying desirable metabolic stability, physicochemical properties, and a combination of hydrogen-bond acceptor/donor functionalities.308−310 Bull and Luisi have recently reported a simple, direct method for the synthesis of NH-sulfoximines from sulfides, providing an accessible entry-point for further N-functionalization.311 Wimmer and König in 2018 reported the photocatalytic dehydrogenative N-arylation of NH-sulfoximines with electron-rich arenes (Scheme 98).312 Optimized reaction conditions showed that the joint action of N-Me Mes-Acr+ClO4– photocatalyst ([Acr-1]ClO4) and the Co(III) proton-reduction catalyst [Co(dmgH)2PyCl] in MeCN under a nitrogen atmosphere best catalyzed this transformation. Oxygen or other chemical oxidants instead of the Co co-catalyst gave significantly reduced product yields. This method expanded on work from Nicewicz, Lei, and Hu demonstrating the photocatalytic N-arylation of N-heterocycles with electron-rich arenes through a related mechanistic pathway.75,213,215 Reactions were typically run with a 10-fold excess of arene component for best results. A scope of 28 examples in yields ranging from 5% to 95% was demonstrated. Electron-rich arenes were best suited to this method and a variety of multiply alkylated arenes (e.g., 98.1, 98.2) were reported. Bromide functionality was well tolerated (98.3), enabling potential arene C(sp2)–H sulfoximidation and subsequent transition-metal catalyzed cross-coupling for further elaboration. The sulfoximine component tolerated variation of aryl and alkyl (98.4) substituents as well as cyclic sulfoximines (98.5). The reaction was demonstrated on preparative scale without a decrease in reaction efficiency or prolonged reaction times.
Scheme 98. Photocatalytic N-Arylation of NH-Sulfoximines with Electron-Rich Arenes (König, 2018).

The group conducted SV and CV experiments to determine a mechanism of this transformation. Both a model arene and sulfoximine substrates were found to quench the photoexcited state of the employed Acr photocatalyst. This Acr photocatalyst has a sufficiently oxidizing photoexcited state (E1/2 *PC+/PC• = +1.88 V vs SCE in PhCN)74 to allow for thermodynamically favorable ET with both reaction components (e.g., for diphenyl NH-sulfoximine, Ep/2ox = +2.00 V vs SCE in MeCN; for p-xylene, Ep/2ox = +2.01 V vs SCE in MeCN).312 Based on these observations and literature precedent for the operation of joint photoredox/cobaloxime hydrogen-evolution catalysis, the following mechanistic model was proposed involving two separate turns of a photoredox catalytic cycle and a two-photon requirement. Upon photoexcitation of the Acr photocatalyst, the resulting photoexcited state undergoes ET with either the sulfoximine or arene substrate to yield the respective radical cation intermediate. The resulting stabilized acridine radical (E1/2 Acr+/Acr• = −0.49 V vs SCE in PhCN)74 can regenerate the ground-state Acr catalyst through reaction with the Co(III) co-catalyst to generate a Co(II) complex (E1/2 Co(III)/Co(II) = −0.68 V vs SCE in MeCN).267 Concurrently, the photoredox catalyst also oxidizes the other reaction component to its radical cation in a second photoredox cycle. This is turned over by the Co(II) intermediate, generating the catalytically active Co(I) (Co(II)/Co(I) = −1.1 V vs SCE in MeCN)267 complex for proton reduction. The sulfoximine radical cation resulting from SET is deprotonated to yield the N-centered iminyl radical. Through radical–radical coupling between the arene radical cation and the N-centered radical, a new C–N bond forms, yielding a cyclohexadienyl cation, which is deprotonated to restore aromaticity and yield the product. Reaction of the Co(I) species with a proton gives a Co(III) hydride, which reacts with a second proton to evolve dihydrogen and regenerate the Co(III) pre-catalyst.
In a benzothiazoyl sulfoximine substrate which gave no N-arylation product under these optimized reaction conditions, the oxidation potential was recorded as E1/2ox = +2.45 V vs SCE in MeCN, and no quenching was observed in SV experiments. This is out of range of the Acr photocatalyst. The observed lack of reactivity is despite the arene component still being capable of undergoing thermodynamically favorable PET to form the radical cation. This indicates a distinct mechanism of operation as presented above to that of Nicewicz and Lei,75,213,215 which proposed that a redox-inactive N-heterocycle nucleophile can engage with the transient electrophilic arene radical cation. Observations here indicate that radical–radical cation coupling is required to yield N-arylated sulfoximine product.
Concurrently with this report, the Meier group disclosed a related method for photocatalytic N-arylation of dialkyl NH-sulfoximines with electron-rich arenes.313 A related Acr photocatalyst, N-Ph-Mes-t-Bu2-Acr+BF4– ([Acr-3]BF4) was found to be optimal in the presence of (NH4)2S2O8 as a stoichiometric oxidant. The group proposed a distinct mechanism from König, suggesting that the non-oxidizable dialkyl sulfoximine reacts as a polar nucleophile with the intermediate electrophilic arene radical cation generated through PET, akin to that proposed by Nicewicz.75 This is interesting, considering that König observed a lack of reactivity with non-oxidizable sulfoximines, in part leading to their proposal of radical–radical cation coupling being a requirement for C–N bond formation. In this work from Meier, the arene was employed as the limiting reagent, which compares favorably to the method of König.
2.5. Transformations of Sulfonylhydrazides
A variety of C–S and C–C bond-forming reactions have been developed from sulfonylhydrazide reagents. These act as a source of sulfonyl radicals under net oxidative conditions through sequential steps of ET and PT, leading ultimately to the extrusion of molecular nitrogen. In one reported example, further extrusion of sulfur dioxide from the intermediate sulfonyl radical is also observed, leading to aryl radical generation and functionalization. Reactions have been promoted through both photocatalytic and electrochemical reaction manifolds.
2.5.1. Intermolecular C–S Bond Formation through Addition to Alkenes and Alkynes
Cai and co-workers in 2016 developed a photocatalytic synthesis of β-keto sulfones from alkynes and sulfonylhydrazide reagents (Scheme 99).314 The optimal reaction conditions for this transformation involved the visible-light irradiation of a DMF/H2O (5:1) solution of an arylacetylene substrate and aryl or alkyl sulfonylhydrazide reagent in the presence of [Ru(bpy)3]Cl2 ([Ru-1]Cl2) as a photocatalyst, NaOAc as a Brønsted base, and KI as an additive at room temperature under an O2 atmosphere. The organic photocatalyst EY gave similar reactivity, though no reactivity was observed in the absence of base or iodide. A scope of 30 examples was presented with yields of 32–93%. The reaction proceeded most efficiently for arylacetylenes bearing electronically neutral or electron-donating groups (EDGs) on the arene (99.1, 99.2). With respect to the sulfonyl component, aryl, heteroaryl (99.4), and alkyl (99.3) sulfonyl hydrazides were all tolerated with similar efficiency.
Scheme 99. Photocatalytic Syntheses of β-Keto Sulfones from Alkynes and Sulfonylhydrazides (Cai, 2016).

The proposed mechanism involves the visible-light excitation of the Ru(II) complex to generate the long-lived excited state. Reductive quenching of this excited-state complex (E1/2 *Ru(II)/Ru(I) = +0.77 V vs SCE in MeCN)64 with iodide (E1/2ox I•/I– = +0.46 V vs SCE in DMF),315 generates iodine radical and a reduced-state Ru(I) complex. The iodine radical then acts as a redox mediator to generate the sulfonylhydrazide radical cation through SET. However, we note that later SV studies from this group (vide infra) demonstrates that the excited state of this photocatalyst can be directly quenched by the hydrazide substrate in the absence of iodide,316 therefore leaving the exact role of iodide unclear in the initiation of this reaction. The sulfonylhydrazide radical cation is deprotonated by the Brønsted basic acetate (pKaH = 12.3 in DMSO)116 to form the neutral sulfonylhydrazidyl radical before the second iteration of sequential ET and PT steps forms a diazene intermediate. A third and final iteration of ET and PT then leads to sulfinyl radical generation with extrusion of dinitrogen. Dioxygen is proposed to facilitate these second and third ET steps, forming superoxide in the process. Following this, anti-Markovnikov addition of the generated sulfonyl radical across the alkyne reaction partner yields a vinyl radical intermediate. This radical is trapped with dioxygen to yield a vinylhydroperoxide radical intermediate that reacts with the reduced state Ru(I) complex (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN)64 to close the photocatalytic cycle and form a closed-shell hydroperoxide anion after consecutive PT. Finally, iodide reduces the peroxide, to give the enol, which rapidly tautomerizes to the ketone product. In some cases, a vinyl iodide side product was observed, suggesting trapping of this vinyl radical intermediate with transient iodine radical. The possibility that the observed vinyl iodide was an intermediate en route to the ketone product through hydrolysis or acetolysis was investigated, but this species did not convert to product under the standard reaction conditions.
Soon after, the Cai group used similar reaction conditions to promote the photocatalytic decarboxylative sulfonylation of cinnamic acids with aryl sulfonylhydrazide reagents for the synthesis of vinylsulfones (Scheme 100).316 Optimal conditions involved visible-light irradiation of DMF/H2O (15:1) solutions of organic photocatalyst EY, cinnamic acid substrate, sulfonylhydrazide reagent, Cs2CO3 base, and KI additive under an O2 atmosphere. In total, 31 examples of vinylsulfone products were isolated, in yields of 34–92%. The method favored electron-deficient cinnamic acids (100.1–100.3), while the electronic properties of the aryl sulfonyl hydrazide component could be modified without much impact on observed product yields, including a thiophene-bearing sulfonyl hydrazide which gave vinylsulfone product in 86% yield (100.5). The reaction was tolerant of diverse functionality on the arene group of both reaction components, including fluoro, chloro, bromo, trifluoromethyl, trifluoromethoxy, nitro, acetamide (100.4), and cyano groups. No examples of α- or β-substituted cinnamic acids were reported.
Scheme 100. Decarboxylative Sulfonylation of Cinnamic Acids with Arylsulfonyl Hydrazides for the Synthesis of Vinylsulfones (Cai, 2016).

Control experiments revealed the need for light, photocatalyst, oxygen, and base for any reactivity. The addition of TEMPO to a reaction under optimal conditions led to total inhibition of reactivity. SV experiments demonstrated that both hydrazide and iodide quench the excited state of the photocatalyst employed, but that the hydrazide does so at a greater rate. Their mechanistic proposal, therefore, involved PET between photoexcited-state EY (E1/2 *EY/EY•– = +0.83 V vs SCE in MeCN)71 and hydrazide substrate leading to the generation of the aminium radical cation intermediate directly, without iodide acting as a redox mediator. Thereafter, consecutive rounds of PT and ET in a similar fashion to that described above lead to the generation of a key sulfonyl radical intermediate with extrusion of dinitrogen. The sulfonyl radical adds in an anti-Michael fashion to the deprotonated cinnamate substrate,317−320 to form a stabilized benzylic radical. Then, further oxidation and trapping of the benzylic carbocation with iodide yields a β-iodo carboxylate that undergoes a polar β-elimination process with decarboxylation to yield the (E)-vinylsulfone product. Styrene subjected to these reaction conditions gave no product, indicating that decarboxylation occurs after sulfonyl radical addition in this fashion. The photocatalyst ground state is regenerated through reaction of EY•–(E1/2EY/EY•– = −1.06 V vs SCE in MeCN)71 with molecular oxygen to complete the catalytic cycle.
In 2017, Huang and co-workers reported the electrochemical decarboxylative sulfonylation of cinnamic acids with arylsulfonyl hydrazides (Scheme 101).321 This method involved the constant current electrolysis of DMSO solutions of these substrates in an undivided cell equipped with Pt electrodes, LiOtBu Brønsted base additive, and n-Bu4N+BF4– electrolyte. A scope of 29 examples of vinylsulfonyl products in yields of 30–84% was presented (101.1–101.4). A variety of aryl-substituted cinnamic acids were studied, in addition to furan, thiophene, and pyridine (101.3) containing congeners. The reaction tolerated halogen, trifluoromethyl, ether, and acetamide functional groups on either of the cinnamic acid or arylsulfonyl hydrazide components. In all cases, exclusive E-selectivity was observed in the vinylsulfone products. Aliphatic sulfonyl hydrazide reagents were not compatible with these reaction conditions.
Scheme 101. Electrochemical Decarboxylative Sulfonylation of Cinnamic Acids (Huang, 2017).

On the basis of CV experiments, it appeared that the reaction proceeded through a stepwise mechanism for the formal homolysis of the hydrazide N–H bond and that in the presence of LiOtBu base, hydrazide oxidation occurred preferentially over the cinnamate substrate. In the absence of the Brønsted base additive, the peak potential of p-toluenesulfonylhydrazide was recorded as Ep/2 = +1.24 V vs Ag/AgCl in DMSO.321 This was negatively shifted by 710 mV on the addition of the butoxide base, indicating discrete substrate ionization (e.g., pKa of benzenesulfonyl hydrazide = 17.1 in DMSO, pKaH of LiOtBu = 29.4 in DMSO)116,178 prior to ET at the anode surface. Two further cycles of ET and PT lead to sulfonyl radical generation with extrusion of molecular nitrogen. Anti-Michael addition to the ionized cinnamate substrate yields a stabilized benzylic radical, which is further oxidized to the carbocation. Finally, decarboxylation yields the vinyl sulfone product. Cathodic reduction of tert-butanol led to the generation of molecular hydrogen. Styrene and p-tolyldisulfide, observed as side products under optimized conditions, were also subjected to the above conditions but did not yield product vinylsulfone, demonstrating that these are not viable intermediates through an alternative mechanism.
Lei and co-workers in 2018 reported the electrochemical oxidative 1,2-alkoxysulfonylation of styrenes with arylsulfonyl hydrazide reagents and alcohol co-solvents (Scheme 102).322 Optimized conditions for this transformation involved the constant current electrolysis of MeCN/alcohol (9:1) solutions of styrene and arylsulfonyl hydrazide in an undivided cell equipped with a carbon rod anode, Ni plate cathode and n-Bu4N+BF4– as supporting electrolyte. A scope of 30 examples of this three-component coupling reaction was reported in yields of 33–97%. A study of substituent effects in the reactions of arylsulfonyl hydrazide reagents with 4-methylstyrene with MeOH showed that highest yields were realized with electronically neutral substituents (102.1). Electron-withdrawing substituents gave products in good yields, but electron-donating substituents (102.2) and heteroaromatic sulfonyl hydrazides were less tolerated. Substitution of the styrene aryl group, or use of α-methylstyrenes (102.3), was permitted, but cyclohexene and allylbenzene were unreactive to these conditions. A variety of primary, secondary, and tertiary alkyl alcohols allowed for their incorporation, but all were required in 10% v/v co-solvent quantities (102.4).
Scheme 102. Electrochemical Alkoxysulfonylation of Styrenes with Arylsulfonyl Hydrazides and Alcohols (Lei, 2018).

CV experiments established that the sulfonyl hydrazide (e.g., for p-tolylsulfonyl hydrazide, Ep/2ox = +1.91 V vs Ag/AgCl in MeCN/MeOH (9:1))322 is oxidized preferentially to the styrene component (e.g., for 4-methylstyrene, Ep/2ox = +2.19 V vs Ag/AgCl in MeCN/MeOH (9:1)).322 Their mechanistic model involved three sequential cycles of anodic ET and PT of the hydrazide component, leading to arylsulfonyl radical generation with extrusion of N2. Electrogenerated alkoxide base at the cathode is responsible for deprotonation of the radical cation intermediates en route to the arylsulfonyl radical. The intermediate sulfonyl radical adds in an anti-Markovnikov fashion to the styrene to generate a stabilized benzylic radical. Further anodic oxidation yields the benzylic carbocation, which traps the alcohol co-solvent and yields the product after PT. Molecular nitrogen and hydrogen gases are the sole waste products in this process.
Zhu and co-workers in 2019 reported a photocatalytic aerobic 1,2-oxysulfonylation of styrenes for the synthesis of β-keto- and β-hydroxysulfones (Scheme 103).323 Optimal reaction conditions consisted of blue LED irradiation of EtOH solutions of styrene substrate and sulfonyl hydrazide reagent in the presence of methylene blue ([MB]Cl) photocatalyst and DABCO as a Brønsted base additive under air. When mono-substituted styrenes were employed as alkene substrates, β-ketosulfones resulted, with 15 examples reported in yields of 64–89%. Variation was tolerated on the styrene aryl group including the addition of halogen substituents (103.2), and the exclusive mono-oxysulfonylation of 1,3- and 1,4-divinylbenzene (103.3) were also realized in good yields. A variety of arylsulfonyl hydrazide reagents were also tolerated in this protocol. When 1,1-disubstituted styrenes were employed as the alkene reaction partners, β-hydroxysulfone products were obtained, with seven examples of these products reported in yields of 63–81%, including a pyridyl substrate (103.4).
Scheme 103. Photocatalytic Oxysulfonylation of Styrenes (Zhu, 2019).

A mechanism involving sequential steps of oxidative photocatalyst-mediated ET (E1/2 *MB+/MB• = +1.14 V vs SCE in MeOH)45 and subsequent PT with the hydrazide substrate was proposed, leading to extrusion of molecular nitrogen and generation of an arylsulfonyl radical intermediate. This aligns with the reaction initiation presented by Cai and co-workers.316 The authors also suggest that DABCO may serve to facilitate discrete prior PT, thereby facilitating ET in the same mode of initiation as presented by Huang.321 However, this appears unfavorable based on analysis of the relative substrate and base pKa values (e.g., pKa of benzenesulfonyl hydrazide = 17.1 in DMSO, pKaH of DABCO = 9.1 in DMSO).48,116 Methylene blue (MB) ground state is then regenerated through the reaction of the reduced form (E1/2MB+/MB• = −0.30 V vs SCE in MeCN)45 with molecular oxygen. Thereafter, anti-Markovnikov addition of the arylsulfonyl radical to the styrene substrate yields a stabilized benzylic radical, which is trapped by molecular oxygen or hydroperoxyl radical. Decomposition of the initial peroxy products yields the oxygenated products.
He, Guan, and co-workers recently reported a photocatalytic decarboxylative oxidative sulfonylation of atropic acids with sulfonyl hydrazide reagents for the synthesis of β-keto sulfones (Scheme 104).324 Optimized reaction conditions for this transformation involved visible-light irradiation of substrate admixtures in MeCN/H2O (5.5:1, v/v) solution in the presence of fluorescein photocatalyst (Fl), NaHCO3 as a Brønsted base, and KI as an additive under an oxygen atmosphere. Only trace yields were realized in the absence of base, iodide, or irradiation. A total of 20 examples of β-keto sulfone synthesis were reported, in 15–91% yields. A variety of aryl substituents on the atropic acid component were tolerated, as were a variety of aryl (104.1–104.3), alkyl (104.4), and heteroaryl (104.5) sulfonyl hydrazide reagents. A bulky mesityl sulfonyl hydrazide reacted with poor efficiency (15% yield).
Scheme 104. Photocatalytic Decarboxylative Oxidative Sulfonylation of Atropic Acids with Sulfonyl Hydrazide Reagents (He and Guan, 2020).

The proposed mechanism involved single-electron oxidation of the hydrazide component (e.g., for benzenesulfonyl hydrazide, E1/2 = −0.33 V vs Ag/AgCl in MeCN/H2O (5.5:1))324 mediated by photoexcited-state fluorescein (E1/2 *Fl/Fl•– = +1.29 V vs Ag/AgCl in MeOH).325 The reduced-state photocatalyst (E1/2Fl/Fl•– = −1.13 V vs Ag/AgCl in MeOH)325 can be regenerated through reaction with molecular oxygen. Sequential PT and ET events then lead to the extrusion of molecular nitrogen, generating a sulfonyl radical. This sulfonyl radical adds to the atropic acid in a Giese fashion and the resulting α-acyl radical product is trapped with dioxygen. Iodide is proposed to reduce the resultant hydroperoxyl radical to the corresponding anion, which fragments through loss of CO2 to yield the ketosulfone product. Iodine radical generated in the reduction of the peroxyl radical then initiates a chain reaction mechanism through HAT with another molecule of sulfonyl hydrazide. Isotopic labeling experiments with 16O2 dioxygen atmosphere with 18O2 containing aqueous co-solvent demonstrate that the ketone oxygen is derived from molecular oxygen rather than water, supporting the proposed radical trapping pathway as opposed to one involving carbocation formation and hydrolysis.
2.5.2. Intermolecular C–S and C–C Bond Formation through Addition to (Hetero)arenes
Lei and Huang reported an electrochemical dehydrogenative C(sp2)–H sulfonylation reaction between electron-rich (hetero)arenes and arylsulfonyl hydrazides (Scheme 105).326 Optimized conditions for this transformation involved the constant current electrolysis of MeCN/H2O (19:1) solutions of arene substrate and arylsulfonyl hydrazide reagent in an undivided cell equipped with a carbon rod anode and a Ni plate cathode with K2CO3 as a Brønsted base additive and n-Bu4N+BF4– as supporting electrolyte. Thiosulfonate and disulfide byproducts were observed when arylsulfonyl hydrazide was used in excess. A total of 35 examples of diaryl sulfone products were presented in yields of 24–95%. A range of arylsulfonyl hydrazide reagents was explored with 3-phenylbenzofuran as an arene coupling partner, which underwent regioselective C2-sulfonylation (105.1). Electron-deficient and electron-neutral arylsulfonyl hydrazides were best suited to this transformation, with more electron-rich examples providing moderate yields. Halogen functional groups in particular were well tolerated under this method. In addition to 3-arylbenzofurans giving C2-sulfonylation products, a 2-arylbenzofuran gave the regioisomeric C3-sulfonylation product, albeit in moderate yield. Benzothiophene (105.2), thiophene (105.4), pyrrole (105.5), and imidazo[1,2-a]pyridine (105.6) heterocycles too were competent substrates. However, indoles were not tolerated.
Scheme 105. Electrochemical C–H Sulfonylation of Electron-Rich (Hetero)arenes with Arylsulfonyl Hydrazides (Lei and Huang, 2018).

*3.0 equiv of ArSO2NHNH2.
CV experiments revealed that the carbonate base played a key role in promoting selective N-centered radical generation of the hydrazide component through a concerted PCET mechanism. In the absence of base, the oxidation of the arene component occurred preferentially (e.g., for 3-phenylbenzofuran, Ep/2ox = +1.99 V vs Ag/AgCl in MeCN/H2O (19:1), compared to 4-chlorophenylsulfonyl hydrazide, Ep/2ox = +2.21 V vs Ag/AgCl in MeCN/H2O (19:1)).326 However, the onset potential of the hydrazide oxidation was negatively shifted by ca. 400 mV upon addition of the base. Therefore, the proposed mechanism involves first hydrazidyl radical generation through a concerted PCET manifold before two further cycles of ET and PT lead to sulfonyl radical generation with loss of molecular nitrogen. The electrophilic sulfonyl radical was proposed to add to the neutral, closed-shell nucleophilic arene before further steps of oxidation and proton loss lead to re-aromatization and generation of the product. Cathodic reduction of liberated protons leads to the co-generation of molecular hydrogen.
More recently, De Sarkar and co-workers have extended the scope of this electrochemical heteroarene C(sp2)–H sulfonylation reaction to include 2-aryl-2H-indazoles (Scheme 106).327 Under similar electrolytic conditions, 29 examples of the C3-sulfonylation of 2H-indazoles were reported in yields of 60–92% (e.g., 106.1–106.3). In this work, methylsulfonyl hydrazide was also able to generate the intermediate sulfonyl radical for C(sp2)–H methylsulfonylation, yielding 106.1 in 60% yield. The same concerted PCET mechanism was operative in the initial reagent oxidation step, where the oxidation potential of the hydrazide component is negatively shifted by ca. 260 mV when 1.0 equiv of K2CO3 was included.327 Their mechanistic proposal aligns with that of Lei and Huang.326
Scheme 106. Electrochemical C–H Sulfonylation of 2-Aryl-2H-indazoles with Arylsulfonyl Hydrazides (De Sarkar, 2020).

In 2019, Lei and co-workers reported a protocol for the electrothermal deoxygenative C2-selective arylation of quinoline N-oxides with arylsulfonyl hydrazides (Scheme 107).328 Previous reports of electrochemical or photocatalytic reactivity of sulfonyl hydrazides have demonstrated their utility as a sulfonyl radical precursor through sequential steps of ET and PT with subsequent extrusion of N2 (vide supra). In this work, the unprecedented extrusion of SO2 unexpectedly also occurred, making these versatile reagents for radical arylation in addition to arylsulfonylation. This reactivity under electrochemical activation stands in contrast to an earlier reported radical C2-selective sulfonylation of quinoline N-oxides with arylsulfonylhydrazide reagents, which employed a Cu(II) catalyst in the presence of a persulfate oxidant.329
Scheme 107. Deoxygenative C2-Arylation of Quinoline N-Oxides with Arylsulfonyl Hydrazide Reagents (Lei, 2019).

Optimized conditions for this transformation involved the constant current electrolysis of MeCN/HFIP (9:1) solutions of quinoline N-oxide substrate and arylsulfonyl hydrazide reagents in an undivided cell equipped with carbon anode and Pt cathode with n-Bu4N+BF4– as supporting electrolyte at 70 °C. Here, 22 examples of C2-arylated quinolines were prepared in yields of 28–72%. With respect to the hydrazide component, a variety of electronically neutral or weakly electron-donating or -withdrawing aryl substituents were effective reagents (107.1). However, more strongly electron-donating or electron-withdrawing groups, such as methoxy and trifluoromethyl (107.2), respectively, gave poor yields. 2-Thienyl and 3-pyridylsulfonyl hydrazides (107.3) reacted with quinoline N-oxide to furnish the C2-heteroarylated products in 33% and 62% yield, respectively. An aliphatic sulfonyl hydrazide was ineffective. A variety of substituents were tolerated on the quinoline N-oxide partner, including halogen, without significant impacts on observed yields. Interestingly, an isoquinoline N-oxide was ineffective in an attempted C1-arylation.
CV experiments established that the arylsulfonyl hydrazide (Ep/2ox = +1.82 V vs Ag/AgCl in MeCN/HFIP (9:1))328 reaction component is most likely being oxidized in preference to the quinoline N-oxide (Ep/2ox = +2.24 V vs Ag/AgCl in MeCN/HFIP (9:1)).328 A proposed mechanism involved three cycles of stepwise anodic oxidation and subsequent deprotonation of the arylsulfonyl hydrazide, with the PT steps mediated by electrogenerated alkoxide base. An aryl radical then results from extrusion of N2 and SO2. C2-selective addition of the aryl radical into the quinoline N-oxide substrate creates the new C–C bond. An attempted reaction with quinoline under optimized conditions gave no product, showing radical addition occurs prior to deoxygenation. Then, cathodic reduction and PT of the resulting N-oxyl radical generates an N-hydroxydihydroquinoline, which undergoes dehydration to yield the deoxygenation product. Cathodic reduction of HFIP solvent also occurs to liberate H2. No clear rationale is provided for the selectivity of arylation over the usually observed sulfonylation, but we note this process takes place at 70 °C, whereas many of the recently reported radical sulfonylation protocols occur at lower temperatures. Indeed, the Lei group also reported the electrochemical C2-arylsulfonylation of quinoline N-oxides, using instead sodium sulfinate reagents at room temperature.330
2.5.3. Intermolecular Sulfonylation of Tertiary Amines Proceeding through C–C and C–H Bond Cleavage
Sheykhan, Abbasnia, and co-workers in 2017 reported an anodic oxidative coupling of arylsulfonyl hydrazide reagents and tertiary alkylamines, proceeding via C–N bond cleavage in the amine component, leading to aryl sulfonamide products (Scheme 108).331 Optimized reaction conditions for this transformation consisted of constant current electrolysis in an undivided cell with graphite electrodes and Na2SO4 supporting electrolyte in MeCN/H2O (1:1). A scope of 17 examples of this oxidative coupling reaction was reported, in yields of 60–78%. Tertiary alkylamines were competent reaction partners, but tertiary anilines provided only trace yields. Both symmetrical and unsymmetrical tertiary amines could be utilized with predictable selectivity for the C–N bond cleavage—for example, reactions with symmetric Et3N yielded N,N-diethylsulfonamide products (108.1) and those with unsymmetric BnNMe2 yielded exclusively N,N-dimethylsulfonamide products resulting from benzylic C–N bond cleavage (108.2–108.4). Generally, cleavage preference follows the trend of benzylic over methyl or alkyl substituents and primary or methyl cleavage over branched alkyl or aryl substituents. A variety of arenesulfonyl hydrazide functional groups were tolerated, including chloro and bromo.
Scheme 108. Oxidative Coupling of Arylsulfonyl Hydrazides and Tertiary Amines Proceeding with C–N Bond Cleavage (Sheykhan and Abbasnia, 2017).

A proposed mechanism involved anodic oxidation of the tertiary amine to the corresponding aminium radical cation (e.g., for Et3N, E1/2ox = +0.86 V vs Ag/AgCl in MeCN/H2O (1:1)),331 PT to afford the corresponding α-amino radical, and then further oxidation to the corresponding iminium ion intermediate (e.g., for the α-amino radical derived from Et3N, E1/2ox = +0.99 V vs SCE in MeCN)332 (see Scheme 110A,B). The iminium ion undergoes hydrolysis in the aqueous reaction solvent to formally cleave this C–N bond and yield a secondary amine and an aldehyde byproduct (e.g., for Et2NH, E1/2 = +1.38 V vs NHE in MeCN).333 Benzaldehyde was observed as a byproduct in the reactions of BnNMe2, supporting this route for C–N bond cleavage. Selectivity for C–N bond cleavage in unsymmetrical tertiary amines such as this appears to arise from the relative acidity of the α-protons of the aminium radical cation intermediate. The large offset in oxidation potential (∼500 mV) between the tertiary amine and hydrazide components suggests that a large proportion of the tertiary amine substrate is converted through dealkylation to the corresponding secondary amine prior to further processing of the hydrazide at higher applied (variable) potential. In addition, the sulfonyl hydrazide component also undergoes sequential steps of anodic oxidation (e.g., for benzenesulfonyl hydrazide, Ep/2ox = +1.30 V vs Ag/AgCl in MeCN/H2O (1:1))331 and PT to yield sulfonyl radical through extrusion of molecular nitrogen. Thereafter, further oxidation of the sulfonyl radical intermediate yields an electrophilic sulfonium cation, which can react with the nucleophilic secondary amine in a polar reactivity manifold.
Scheme 110. (A) Electrolytic Co-generation of Iminium Ion and Sulfonyl Radical Reactive Intermediates, (B) Mechanism of Formation of Sulfonamide Products from These Reactive Intermediates (Sheykhan and Abbasnia, 2017), and (C) Mechanism of Formation of β-Aminovinyl Sulfone Products from These Reactive Intermediates (Kim and Lee, 2019).

Kim and Lee extended this methodology in 2019 with a report on the electrochemical oxidative coupling of arylsulfonyl hydrazides and tertiary amines for the synthesis of β-aminovinyl sulfones (Scheme 109).334 This reaction outcome is distinct from the work of Sheykhan and Abbasnia,331 who observed C–N bond cleavage and arylsulfonamide products arising from these starting materials under similar electrochemical activation. Optimized conditions here consisted of the constant current electrolysis of substrate admixtures in anhydrous DMSO in an undivided cell equipped with graphite anode and Pt cathode containing AcOH Brønsted acid catalyst and n-Bu4N+BF4– supporting electrolyte open to air. A scope of 11 examples of β-aminovinyl sulfone products was reported in yields of 70–86%. iPr2NEt (109.1–109.5) and cHexNEt (109.6) were both demonstrated as efficient coupling partners, with exclusive reaction observed at the ethyl group. A variety of para-substituted arylsulfonyl hydrazide reagents were demonstrated, including those with both electron-rich and electron poor substituents.
Scheme 109. Electrochemical Synthesis of β-Aminovinyl Sulfones (Kim and Lee, 2019).

A mechanistic proposal involves anodic oxidation of both tertiary amine and arylsulfonyl hydrazide components to the corresponding iminium ion and sulfonyl radical intermediates, respectively (Scheme 110A,C). While iminium hydrolysis was observed by Sheykhan and Abbasnia,331 here the authors propose deprotonation of the iminium intermediate by cathodically-generated acetate to afford an enamine intermediate due to the anhydrous reaction conditions. The sulfonyl radical then undergoes anti-Markovnikov addition across the enamine. Thereafter, further oxidation of the resulting α-amino radical proposed to be mediated by air leads to desaturation, yielding the β-aminovinyl sulfone product. Additionally, the presence of O2 is shown to play an important role in favoring the β-aminovinyl sulfone product over the earlier observed sulfonamide product, with mixtures of these two compounds resulting from reactions carried out under a N2 atmosphere.
2.5.4. Intermolecular S–S Bond Formation in the Synthesis of Unsymmetrical Thiosulfonates
Tang, Pan, and Chen in 2018 reported the electrochemical sulfonylation of thiols for the synthesis of thiosulfonates (Scheme 111).335 This cross-coupling protocol between arylsulfonyl hydrazide reagents and aryl or alkyl thiol substrates allows for the facile preparation of unsymmetrical thiosulfonates, which are difficult to access selectively via other methods. An optimized set of reaction conditions involved constant current electrolysis of substrate admixtures in MeCN solution in an undivided cell with RVC anode and Pt cathode, with NH4I as a supporting electrolyte, which was also proposed to serve a role of HAT mediator in the reaction mechanism. Under these conditions, 26 examples of S–S bond formation for thiosulfonate synthesis were reported, in yields of 65–85%. The reaction necessarily required an arylsulfonyl hydrazide reagent to progress, with an alkyl variant providing no yield of product. However, with respect to the thiol component, both aryl (111.1–111.6) and alkyl (111.7) examples were competent. Examples of halogen (111.2), nitro, alkyl, ether, and heteroaromatic (111.8) functional groups on either reaction component were given. The thiosulfonate product formed between benzenesulfonyl hydrazide and 4-methoxythiophenol was shown to inhibit cell migration ability and tubulin polymerization in a human bladder cancer cell line (T24), with an IC50 of 8.0 ± 0.5 μM.
Scheme 111. Electrochemical Synthesis of Unsymmetrical Thiosulfonates in the Cross-Coupling of Arylsulfonyl Hydrazide Reagents and Thiols (Tang, Pan, and Chen, 2018).

TEMPO trapping experiments suggested the intermediacy of both arylsulfonyl and thiyl radicals from the two reaction components. Exposing substrate admixtures to I2 without electrolysis led to no product formation, arguing against this being the reactive oxidant generated under electrolytic conditions. Since the electrolysis of thiols typically produces disulfide products (see section 4.1), these researchers subjected an authentic model diaryl disulfide to the optimized reaction conditions in the presence of the arylsulfonyl hydrazide reagent but interestingly observed no product formation, arguing against its intermediacy here. CV experiments revealed that NH4I is oxidized at the anode (Ep/2 I–/I3– = +0.44 V vs Ag/AgCl in MeCN; Ep/2 I2/I3– = +0.69 V vs Ag/AgCl in MeCN) preferentially to either of the arylsulfonyl hydrazide (e.g., for benzenesulfonyl hydrazide, Ep/2 = +1.30 V vs Ag/AgCl in MeCN) or the thiophenol components (e.g., for 4-methoxybenzenethiol, Ep/2 = +1.31 V vs Ag/AgCl in MeCN). Based on these data, authors hypothesize that iodide is oxidized to molecular iodine at the anode surface, which exists in equilibrium with iodine radical. Iodine radical (BDE = 71.3 kcal mol–1)25 is proposed to facilitate S–H bond homolysis in the thiol component through HAT (e.g., for thiophenol, S–H BDFE = 79.1 kcal mol–1)336 to generate a thiyl radical intermediate and hydrogen iodide. Simultaneously, at the anode, the hydrazide component undergoes discrete ET prior to PT to yield a neutral sulfonamidyl radical. Iodine radical-mediated HAT for the initial activation of this reaction component is not invoked due to the offset in N–H and H–I bond strengths not permitting favorable HAT. No change in the CV behavior of the hydrazide component was observed when studied in the presence of iodide, supporting this proposal. Thereafter, steps of iodine radical or thiyl radical mediated HAT generate an azosulfonyl radical, which extrudes molecular nitrogen to form a sulfonyl radical intermediate. Thereafter, radical–radical cross-coupling yields the unsymmetrical thiosulfonate product. Concomitant reduction of liberated protons at the cathode surface releases H2.
2.6. Transformations of Hydrazines
Zhu and Zheng in 2011 reported a photocatalytic aerobic oxidative cleavage reaction of aromatic hydrazines and hydrazides (Scheme 112).337 Substrates were typically prepared through the addition of organometallic reagents to aryl azo compounds, making this an attractive method for the synthesis of secondary amines. Previous protocols for the reduction of such hydrazines to secondary amines required stoichiometric reductants such as zinc.338
Scheme 112. Photocatalytic Oxidative Cleavage of N–N Bonds in Aromatic Hydrazines and Hydrazides (Zheng, 2011).

Irradiation of MeCN/MeOH (2:1 v/v) solutions of substrate in the presence of [Ru(bpz)3](PF6)2 ([Ru-2](PF6)2) under an air atmosphere led to N–N bond cleavage and isolation of secondary amine, amide, and carbamate products depending on the starting material substrate class. The authors note that substituting the ambient air atmosphere for pure oxygen gave lower product yields. A scope of 17 examples of N–N bond cleavage in this manner was reported in yields of 21–82%. The reaction was tolerant of styrenyl (112.2), aryl chloride, bromide (112.3), and heteroaromatic functional groups, but an aromatic iodide was poorly tolerated. The method permitted the synthesis of both diaryl and aryl alkyl secondary amines.
A mechanism was proposed to involve visible-light excitation of the Ru(II) dye to a long-lived excited state. Thereafter, reductive quenching of the excited-state complex (E1/2 *Ru(II)/Ru(I) = +1.45 V vs SCE in MeCN)65 with the hydrazine or hydrazide substrate led to the generation of the corresponding substrate radical cation and a reduced state Ru(I) complex. This proposal was supported by steady-state SV luminescence quenching experiments. Deprotonation of the radical cation intermediate then yields a transient neutral N-centered hydrazinyl radical. The authors propose N–N bond cleavage occurs through trapping with molecular oxygen, rearrangement through 1,3-HAT and O–O bond cleavage, and elimination of nitrous acid to give a neutral N-centered radical. Finally, reduction of this aminyl/amidyl radical by Ru(I) (E1/2 Ru(II)/Ru(I) = −0.86 V vs SCE in MeCN)339 followed by PT from solvent or nitrous acid byproduct forms the product, regenerating ground-state Ru(II) in the process.
Pyrazoles are important heterocycles in pharmaceutical and agrochemical development and are found in the commercial drugs celecoxib, rimonabant, and sildenafil, and the insecticide fipronil.340 Methods for their synthesis often involve the cyclocondensation of hydrazines with 1,3-dicarbonyl compounds, or 1,4-addition with α,β-unsaturated carbonyl compounds followed by oxidation.341 Zhu in 2016 envisioned an alternative access through the in situ photocatalytic aerobic oxidation of N-alkyl hydrazine substrates to a diazene intermediate followed by conjugate addition to a Michael acceptor and condensation in a one-step process (Scheme 113).342 Compared to typical cyclocondensation methods which initiate through hydrazone formation prior to cyclization, this approach instead provided access to regioisomeric pyrazole products.
Scheme 113. Photocatalytic Synthesis of Pyrazoles from Hydrazines and Electron-Deficient Olefins (Zhu, 2016).

Visible-light irradiation of alkyl hydrazine and Michael acceptors in the presence of [Ru(bpy)3]Cl2 ([Ru-1]Cl2) in MeCN solution under air provided rapid access to pyrazoles in high yields at room temperature. A scope of 22 examples of pyrazole construction was reported in yields of 50–90%. Various electron-deficient olefins were competent partners, including arylidene malononitriles giving access to 3-aryl-5-aminopyrazoles (113.1), chalcones giving 3,5-diarylpyrazoles (113.2), benzylidene malonate giving a 3-aryl-5-hydroxypyrazole (113.3), and a ketoester giving a 3-styrylpyrazole (113.4). Halogen functionality was well tolerated, and reaction efficiency was only negatively impacted when an electron-donating methoxy group was appended to the arene of the benzylidene malononitrile acceptor.
A viable mechanistic proposal consists of PET between the excited-state *Ru(II) complex (E1/2 *Ru(II)/Ru(I) = +0.77 V vs SCE in MeCN)64 and the hydrazine substrate, forming an initial hydrazinium radical cation intermediate. SV quenching studies support this proposed step (KSV = 1726 M–1).342 The resulting reduced-state Ru(I) complex (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN)64 can be re-oxidized to the Ru(II) ground state upon ET with molecular oxygen, generating superoxide as a byproduct. Proton loss from the hydrazinium radical cation forms a neutral N-centered hydrazinyl radical, which undergoes a further cycle of ET and PT mediated by photoexcited-state Ru(II), or HAT mediated by superoxide to generate a reactive diazine intermediate. This adds in a polar 1,4-addition via the terminal nitrogen atom to the olefin acceptor, yielding an alkyl diazene, which rapidly tautomerizes to the hydrazone. This intermediate could be isolated from the reaction mixture in some cases, adding evidence for this route. Finally, condensation of the hydrazone nitrogen onto the pendant nitrile/carbonyl group gave the product.
Zhu and co-workers in 2019 demonstrated a dual photoredox/Cu catalysis approach to the aerobic synthesis of pyrazoles from a hydrazine and terminal alkyne via tandem photochemical Glaser reaction and intermolecular annulation (Scheme 114).343 Optimal conditions consisted of blue-light irradiation of a solution of alkyne, [Ru(bpy)3]Cl2 photocatalyst ([Ru-1]Cl2), and CuI co-catalyst in DMSO under air atmosphere facilitating alkyne dimerization. Addition of the hydrazine partway through the reaction then leads to pyrazole formation. In total, 14 examples of pyrazole synthesis in this manner were reported with yields between 34–84%. The alkyne scope was limited to terminal arylacetylenes, though substitution at the ortho-, meta-, and para-positions were all tolerated, as were electron-donating and -withdrawing substituents. The reaction was generally more effective using unsubstituted hydrazine (114.2, 114.3), though several alkyl- (114.1) and aryl-substituted hydrazines (114.4) were tolerated.
Scheme 114. Tandem Photocatalytic Aerobic Glaser Coupling/Hydrazine Annulation Cascade for the Synthesis of Pyrazoles (Zhu, 2019).

The reaction is proposed to begin with a Glaser-type coupling of 2 equiv of the arylacetylene to generate the corresponding diyne.344 Through control experiments, these authors showed that the Ru(II) photocatalyst was not necessary for this step, though visible-light irradiation was required. UV–vis spectroscopy indicated that the Cu(I) or Cu(II) acetylide intermediate absorbs blue light, suggesting that the redox processes within this homo-coupling cycle are in fact light-promoted.345 Once the alkyne homodimerization was complete, the hydrazine was added. The hydrazine is oxidized by the excited state of the Ru photocatalyst (E1/2 *Ru(II)/Ru(I) = +0.77 V vs SCE in MeCN)64 to the corresponding radical cation, then deprotonated to form the hydrazinyl radical in an overall sequential ET/PT process. This neutral N-centered radical adds to the diyne to afford a vinyl radical, which undergoes 1,5-HAT to abstract a H-atom from the hydrazine functional group and generate a second N-centered radical. This intermediate then undergoes protonation and isomerization to an allene, whereafter the N-centered radical adds to the central carbon of the allene to form a stabilized benzylic radical, completing the assembly of the pyrazole. A final reduction of this species via steps of ET and PT by either of molecular oxygen, superoxide, or the reduced state of the photocatalyst (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN)64 affords the closed-shell pyrazole product.
Zhu and co-workers in 2017 developed a photocatalytic method for the conversion of styrenes to aryl hydrazones in the presence of a hydrazine partner, proceeding through C=C bond cleavage (Scheme 115).346 Reaction conditions involved the blue-light irradiation of substrate solutions in MeCN, in the presence of [MB]Cl photocatalyst and 2,6-lutidine Brønsted base additive, under an air atmosphere. In total, 24 examples of this oxidative C=C bond cleavage reaction were reported in 52–78% yields. The reaction permitted broad structural variation in both reaction partners. In addition to hydrazine substrates (115.1, 115.2), a number of (sulfonyl)hydrazides (115.3) and carbazates (115.4) were effective also.
Scheme 115. Photocatalytic Oxidative Cleavage of C=C Bonds for the Synthesis of Hydrazones from Styrenes (Zhu, 2017).

The authors propose a mechanism initiating through photocatalytic oxidation (E1/2 *MB+/MB• = +1.60 V vs Ag/AgCl in MeOH)325 of the aryl hydrazine substrate to the corresponding diazene compound via two sequential series of ET/PT events. Initial oxidation of the hydrazine, as opposed to the styrene component, was supported by SV studies. This diazene intermediate is then proposed to undergo a photomediated [2+2] annulation with the styrene partner, generating a diazetidine intermediate. In one case, this intermediate was isolated and was converted to product when re-subjected to the optimized reaction conditions, adding support for this pathway. Thereafter a further sequence of discrete ET and PT events generates a neutral aminyl radical intermediate, triggering a ring-opening β-scission elementary step. Finally, further oxidation and proton loss leads to an iminium intermediate, which is hydrolyzed to yield the hydrazone product. Ground-state MB photocatalyst can be regenerated through reaction with molecular oxygen (E1/2MB+/MB• = −0.26 V vs Ag/AgCl in MeOH).325
These authors also developed a method for the oxyarylation of styrenes with aryl hydrazine reagents via loss of N2 under similar reaction conditions (Scheme 116).347 Blue-light irradiation of substrate solutions in MeCN in the presence of [MB]Cl photocatalyst and DABCO as a Brønsted base additive under an air atmosphere enabled this transformation. It appears the choice of Brønsted base additive in this and the previous report governs selectivity for either reaction through the neutral diazene intermediate in a [2+2] photocycloaddition or further oxidation and N2 extrusion for aryl radical generation.
Scheme 116. Photocatalytic Oxyarylation of Styrenes with Hydrazines (Zhu, 2017).

A scope of 22 examples was reported in 65–87% yields, with this method permitting variation of both styrene and hydrazine components and tolerance of several functional groups including halogen and additional olefin groups (116.1–116.4). A similar mode of reaction initiation as in the previous report is proposed, with consecutive cycles of PET and subsequent PT, generating a diazene intermediate. Then, further oxidation and deprotonation leads to extrusion of N2 and generation of an aryl radical intermediate. This adds across the styrene component in an anti-Markovnikov fashion. The C-centered radical generated is trapped with oxygen, and further oxidation leads to the ketone product.
In 2018, Hajra and co-workers presented a photocatalytic, aerobic oxidative method for unsymmetrical di(hetero)aryl and (hetero)aryl alkyl sulfide synthesis through the coupling of thiols and aryl hydrazines with loss of molecular nitrogen (Scheme 117).348 The blue-light irradiation of these substrates with rose bengal (RB) photocatalyst and Na2CO3 Brønsted base in aqueous solution under air proved optimal. A total of 33 examples of C–S coupling were presented in yields of 78–90%. Aryl hydrazines bearing electron-donating or electron-withdrawing functionality performed similarly well in this transformation (117.1, 117.2). Halogen, nitro, nitrile, and pyridyl functional groups were all tolerated. A broad range of thiols were competent coupling partners, with aryl, heteroaryl (117.6), benzylic (117.5), and primary alkyl thiols successfully demonstrated. An unsymmetrical diaryl selenide was also prepared via this method (117.4).
Scheme 117. Photocatalytic Synthesis of Sulfides and Selenides from Aryl Hydrazines and Thiols/Selenols (Hajra, 2018).

Considering the mechanism of this transformation, the authors conducted several control experiments. Subjecting thiol alone to the optimized conditions resulted in high yields of corresponding disulfide formation. Subjecting independently prepared disulfide and hydrazine substrates together to the optimized reaction conditions led to efficient product formation, suggesting the intermediacy of the disulfide in the reaction. The addition of TEMPO or BHT radical scavengers resulted in no reactivity. With the addition of DABCO as a singlet oxygen quencher, the reaction proceeded equally well. Therefore, their mechanistic proposal involves visible-light excitation of RB to its excited state (E1/2 *RB2–/RB3–• = +0.99 V vs SCE in H2O),349 which oxidizes the thiol component to the corresponding thiol radical cation (e.g., for thiophenol, E1/2ox = +0.95 V vs Fc+/Fc in MeCN),336 and forms RB3–•. The photocatalyst radical anion (E1/2RB2–/RB3–• = −0.78 V vs SCE in H2O)349 reduces molecular oxygen to regenerate the ground-state catalyst and generate superoxide. Superoxide then deprotonates the thiol radical cation to yield the thiyl radical, which dimerizes to give the closed-shell disulfide. Alternatively, the thiyl radical can form directly from thiol via HAT mediated by superoxide. Simultaneously, the aryl hydrazine substrate undergoes photocatalyst-mediated oxidation to the corresponding radical cation. Proton loss via superoxide or hydroperoxide anion leads to neutral hydrazinyl radical formation. A further two iterations of oxidation and deprotonation in the same manner leads to aryl radical generation with loss of molecular nitrogen. The resulting aryl radical engages with disulfide to yield the closed-shell sulfide product with concomitant elimination of thiyl radical.
Balaraman and co-workers in 2018 reported a dual Ru(II) visible-light photoredox/Co(III) proton reduction catalytic system for the acceptorless dehydrogenation of N,N′-diaryl hydrazines for the synthesis of aryl-azo compounds proceeding under mild conditions (Scheme 118).350 That these aryl-azo products also undergo facile hydrogenation under heterogeneous Pd/C catalysis, raises the prospects of these materials for reversible hydrogen storage. Optimized conditions for this transformation involved the blue-light irradiation of substrate solutions in ethanol in the presence of [Ru(bpy)3]Cl2 photocatalyst ([Ru-1]Cl2) and Co(dmgH)2(py)Cl proton reduction catalyst at ambient temperature. Under these conditions, 17 examples of symmetric and unsymmetric aryl-azo compound synthesis were reported, in generally excellent yields (86–97%). Functional groups demonstrated to be compatible under reaction conditions included halogen, ether, and trifluoromethyl (118.1–118.7). The synthesis of a bis(isoxazolyl)-azo product from the corresponding heteroaromatic hydrazine was included (118.8).
Scheme 118. Dual Ru(II)/Co(III) Catalytic System Enabling the Acceptorless Dehydrogenation of N,N′-Diaryl Hydrazines (Balaraman, 2018).

Based on literature precedent and CV studies, authors present a catalytic mechanism involving initial oxidative quenching of the Ru(II) complex photoexcited state (E1/2 Ru(III)/*Ru(II)= −0.81 V vs SCE in MeCN)64 with the Co(III) co-catalyst (E1/2 Co(III)/Co(II)= −0.67 V vs SCE in MeCN),350 to generate an oxidized Ru(III) complex and a reduced Co(II) complex. This Ru(III) oxidant (E1/2 Ru(III)/Ru(II)= +1.26 V vs SCE in MeCN)64 then undergoes ET with the diarylhydrazine substrate (e.g., for N,N-diphenylhydrazine, E1/2 = +0.58 V vs SCE in MeCN),350 yielding the corresponding hydrazinium radical cation and ground-state Ru(II). Through steps of ET and PT, or directly HAT, between the Co(II) species and the hydrazinium radical cation, the aryl-diazo product results with the generation of a Co(III) hydride. PT with the nascent cobalt hydride leads to hydrogen evolution and reformation of Co(III) ready for catalyst turnover. We note that plausibly reductive quenching between the Ru(II) photo-oxidant (E1/2 *Ru(II)/Ru(I)= +0.77 V vs SCE in MeCN)64 and the diarylhydrazine substrate (e.g., for N,N-diphenylhydrazine, E1/2 = +0.58 V vs SCE in MeCN)350 could also be operative for reaction initiation. Such a mode of reaction initiation in dual Ru(II) photoredox/Co(III) hydrogen evolution catalysis is presented by Lei, Li, and others in similar transformations.264,306
Yang and co-workers recently reported a photocatalytic C3-arylation and alkylation of quinoxalin-2(1H)-ones with hydrazines serving as carbon-centered radical precursors, using a hydrazone-based two-dimensional covalent organic framework (2D-COF) as an efficient and recyclable heterogeneous photocatalyst (Scheme 119).351 COFs are emerging as attractive alternatives to Ir- and Ru-based homogeneous photocatalysts, for their ease of tuning photophysical properties such as light absorption area, charge separation ability and photoexcited-state redox potentials.352
Scheme 119. Oxidative Arylation and Alkylation of Quinoxalin-2(1H)-ones with Hydrazine Reagents, Catalyzed by a 2D-COF Catalyst (Yang, 2020).

The visible-light irradiation of DMSO solutions of quinoxalinone substrate and hydrazine reagent, in the presence of 2D-COF catalyst, K2CO3 Brønsted base additive, under air was optimal for this transformation. Interestingly, in the absence of the 2D-COF photocatalyst, a significant uncatalyzed pathway to product formation was observed (e.g., for 119.1, 31% yield compared to 85% yield under optimal conditions). With aryl hydrazine reagents, 23 examples of C3-arylation were reported in yields of 41–85% (119.1, 119.2, 119.5). The reaction tolerated substitution at the ortho-, meta-, and para-positions of the arylhydrazine, and several functional groups were compatible including ester, acetamide, fluoro, and chloro. With respect to the quinoxaline component, N-H and various N-alkyl substituents were compatible, in addition to arene substituents. Primary (119.3), secondary (119.4), and tertiary alkyl hydrazines (119.6) were also viable radical precursors, giving 20 examples of the corresponding C3-alkylation products in 57–89% yields. This protocol is notable in allowing access to primary alkyl radicals from this class of precursor. The recyclability of the 2D-COF photocatalyst was demonstrated, with little variation observed in yields across six runs with the same catalyst sample.
A proposed mechanism involves oxidative quenching of the photoexcited-state catalyst with molecular oxygen, yielding the photocatalyst radical cation and a superoxide radical anion. Then, through either concerted or stepwise PCET, superoxide acts as an oxidant in a reaction with the hydrazine reagent, yielding a hydrazinium radical cation which undergoes PT to yield the neutral hydrazine radical. Two further cycles of SET and proton loss, with extrusion of molecular nitrogen leads to aryl or alkyl radical generation. Radical addition to the quinoxaline substrate, and a [1,2]-H shift gives a stabilized tertiary C-centered radical, which is oxidized to the corresponding carbocation by the oxidized-state photocatalyst. Finally, proton loss yields the closed-shell product. Quantum yield determination experiments reveal a closed photoredox cycle over a chain process is likely (Φ = 0.08–0.11).
2.7. Transformations of Sulfonyl- and Acylhydrazones
2.7.1. Intramolecular C–N Bond Formation through Addition to Alkenes
In 2014, Xiao and co-workers disclosed an intramolecular hydrosulfonamidation reaction of allylic sulfonylhydrazones to furnish 4,5-dihydropyrazole heterocycle products (Scheme 120).136 In this report, the authors sought to improve the efficiency and functional group tolerance of the process that generates the N-centered radical. Existing methods to generate reactive N-centered radical intermediates from these types of precursors required pre-functionalization of the substrate in an additional synthetic step.89 Thus, the direct conversion of the native substrate N–H bond to the corresponding hydrazonyl radical would provide a synthetic advance. Visible-light irradiation of CHCl3 solutions of allylic hydrazone substrate, [Ru(bpy)3]Cl2 photocatalyst ([Ru-1]Cl2), and NaOH Brønsted base additive gave access to 18 examples of 4,5-dihydropyrazole products with yields ranging from 38% to 88%. This transformation was amenable to arylhydrazone substrates bearing electron-donating and electron-withdrawing substituents, with little impact on reaction efficiency upon changing the electronic nature (120.1–120.4). The reaction worked equally well on alkyl hydrazone substrates, including those carrying primary, secondary, and tertiary alkyl substituents (120.5). Inclusion of TEMPO or Ph2Te2 to optimal reaction conditions led instead to olefin oxy-sulfonamidation (120.6) and telluro-sulfonamidation products of the reaction, with six examples of the former class reported in yields ranging from 56% to 95%, and one example of the latter in 56% yield.
Scheme 120. Photocatalytic Intramolecular Hydrosulfonamidation of Allylic Sulfonylhydrazones (Xiao and Chen, 2014).

*With 2 equiv of TEMPO.
Regarding the mechanism of this transformation, the authors propose that the desired neutral hydrazonyl radical was generated through formal homolysis of the N–H bond via sequential Brønsted base (pKa = 15.74 in H2O)353 mediated deprotonation of the allylic hydrazone (e.g., for acetophenone hydrazone, pKa = 4.7 in MeOH)354 and single-electron oxidation of the resulting anion by the excited state of the Ru(II) photocatalyst (E1/2 *Ru(II)/Ru(I) = +0.77 V vs SCE in MeCN).64 The resulting neutral, N-centered hydrazonyl radical undergoes a 5-exo-trig cyclization onto a pendant alkene. The nascent C-centered radical is then quenched through HAT with the chloroform solvent, affording the product and trichloromethyl radical. This radical (E1/2red = −0.3 V vs SCE, dropping Hg electrode in H2O)355 can be reduced by Ru(I) (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN),64 thus regenerating ground-state Ru(II) for catalytic turnover. Luminescence quenching experiments supported this proposal of prior substrate deprotonation being necessary to facilitate ET, with no quenching of the Ru(II) photoexcited state observed in the presence of the neutral substrate alone. Furthermore, use of CDCl3 as a solvent resulted in 50% deuterium incorporation in the product, supporting the role of chloroform in the terminating HAT step.
In 2016, Xiao, Chen, and co-workers published a report expanding on the scope of intramolecular addition of hydrazonyl radicals to tethered olefins to the synthesis of six-membered-ring dihydropyridazine products through a cyclization/allylic migration pathway (Scheme 121).356 In this work, the authors demonstrated that the regioselectivity of the hydrazonyl radical cyclization could be biased for 6-endo cyclization over the previously reported 5-exo mode by altering the substitution of the alkene. Addition of an aryl substituent at the β-position of the allylic tether favors the formation of the six-membered ring through generation of the more stable tertiary benzylic radical after cyclization of the hydrazonyl radical. Through this, 26 examples of this transformation were reported from both (hetero)aryl and alkyl hydrazone precursors in 51–96% yields (121.1–121.4). Additionally, the authors demonstrated that these products could be efficiently converted to pyridazines through base-mediated elimination.
Scheme 121. Photocatalytic Intramolecular Olefin Sulfonamidation with Allylic Migration (Xiao and Chen, 2016).

An analogous mechanism of N-centered hydrazonyl radical generation through stepwise substrate deprotonation and subsequent photocatalytic oxidation is proposed. Cyclization via a 6-endo-trig then occurs as discussed and TEMPO radical then mediates a HAT elementary step at the site adjacent to this benzylic radical to yield the dihydropyridazine product and TEMPO-H byproduct. An alternate mechanistic pathway for allylic migration was considered involving radical trapping by TEMPO followed by base-mediated elimination to form the product. Though DFT calculations disfavor this pathway, it cannot be entirely ruled out. Authors suggest that ground-state Ru(II) is regenerated through reduction of the neutral solvent CHCl3 (E1/2red = −1.09 V vs SCE in MeCN)356 by the transient Ru(I) complex (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN).64 This would form a chloroform radical anion, which readily extrudes chloride anion and dichloromethyl radical in a mesolytic cleavage step.357−359 This step was supported by the observation of a 4,5-dihydropyrazole–dichloromethyl adduct as a side product.
Later in 2016, Xiao, Chen, and co-workers reported a protocol for the intramolecular hydroxysulfonamidation of allylic hydrazones via formal homolytic activation of the precursor hydrazone N–H bond using an organophotocatalyst (Scheme 122).360 The optimal reaction conditions consisted of irradiation of a MeCN solution of hydrazone substrate in the presence of N-Me Mes-Acr+ClO4– ([Acr-1]ClO4) photocatalyst, TEMPO co-catalyst, and K2CO3 Brønsted base under an oxygen atmosphere. After irradiation, addition of PPh3 afforded a final alcohol product through reduction of an initially formed hydroperoxide. The authors reported 29 examples of hydroxysulfonamidation in this manner with yields of 4,5-dihydropyrazole products ranging from 51% to 91%. Tosyl- (122.1) and mesyl- (122.2) hydrazones were both tolerated with a variety of alkene substituents affording the 5-exo- cyclization product (21 examples). Introduction of an aryl substituent on the allylic tether instead promoted a 6-endo cyclization pathway, affording the six-membered-ring tetrahydropyridazine product (122.4) (eight examples).
Scheme 122. Photocatalytic Intramolecular Hydroxysulfonamidation of Allylic Sulfonylhydrazones (Xiao and Chen, 2016).

Mechanistic experiments suggest that TEMPO acted as a mediator of ET between the photocatalyst and the substrate conjugate base. This is in contrast to the mechanism of direct PET in the absence of TEMPO additive, proposed previously in their earlier hydrazone olefin hydrosulfonamidation method.136 The authors suggested that TEMPO is oxidized (E1/2ox = +0.62 V vs SCE in MeCN)361 to the corresponding oxoammonium ion by photoexcited Acr dye (E1/2 *Acr+/Acr• = +1.88 V vs SCE in PhCN).74 The oxoammonium species engages the hydrazone anion in a single-electron oxidation generating the N-centered radical. This is followed by cyclization of the N-centered radical onto the pendant alkene before trapping of the resulting carbon-centered radical with O2. Reduction of the resulting hydroperoxy radical, followed by protonation forms the closed-shell hydroperoxide. The mode of this reductive step is unclear. Triphenylphosphine is added after the reaction to reduce the hydroperoxide and afford the final hydroxysulfonamidation product. In support of this hypothesis, regarding the role of TEMPO as redox mediator, luminescence quenching experiments indicated that TEMPO quenches the excited state of the photocatalyst more efficiently than both the neutral and deprotonated substrate. Addition of catalytic TEMPO+BF4– under otherwise standard reaction conditions resulted in similar yields of product, indicating that this species could be catalytically competent. Furthermore, the addition of a stoichiometric quantity of TEMPO+BF4– in the absence of photocatalyst and light afforded 79% combined yield of cyclized hydroxylated and TEMPO trapped products.
Xiao and Chen in 2016 also reported the synthesis of dihydropyrazole-fused benzosultams through hydrazonyl radical initiated dicyclization (Scheme 123).362 In this report, the absence of a HAT mediator or oxidant enables a methylene radical, generated after cyclization of the N-centered hydrazonyl radical onto the pendant allyl group, to undergo further cyclization onto the electron-withdrawn sulfonyl arene. Without any additive, the authors observed significant yields of the de-aromatized 1,4-cyclohexadiene-fused sultam; however, the addition of a Co(III) co-catalyst affected re-aromatization through hydrogen evolution to furnish the benzosultam product. Notably, Stephenson later reported a related method for de-aromative 1,4-cyclohexadiene synthesis through a related dicyclization reaction of sulfonamidyl radicals.363
Scheme 123. Photocatalytic Synthesis of Benzosultams from Sulfonylhydrazones (Xiao and Chen, 2016).

Blue-light irradiation of an MeCN solution containing substrate, Ru(bpy)3Cl2·6H2O ([Ru-1]Cl2), [Co(dmgH)2(py)Cl] co-catalyst and K2CO3 afforded 22 examples of benzosultam synthesis with yields of 34–77%. Substitution of the hydrazone aryl group with electron-donating and -withdrawing substituents was well-tolerated (123.1–123.4). A variety of substituted sulfonyl arenes were competent substrates, including those carrying both electron-rich (123.5 and 123.6) and electron-deficient (123.7 and 123.8) functional groups. Of note, all substrates in the scope contained a gem-dimethyl group, presumably required to facilitate the initial hydrazonyl radical cyclization through the Thorpe-Ingold effect.
Luminescence quenching and radical trapping experiments were consistent with the authors’ previous studies, suggesting that the reaction proceeds through the stepwise generation of a hydrazonyl radical through sequential steps of discrete proton and photoexcited-state Ru(II)-mediated ETs (E1/2 *Ru(II)/Ru(I) = +0.77 V vs SCE in MeCN),64 followed by 5-exo-trig cyclization onto the pendant olefin, generating a C-centered radical. This distal radical then adds into the sulfonylarene, forming a delocalized cyclohexadienyl radical intermediate. Meanwhile, Ru(II) ground state is restored through reduction of the Co(III) co-catalyst with Ru(I) (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN);64 (E1/2red Co(III)/Co(II) = −0.47 V vs SCE in MeCN).267 The resultant Co(II) complex is proposed to mediate SET from the cyclohexadienyl radical intermediate, yielding the corresponding carbocation and a Co(I) complex (E1/2red Co(II)/Co(I) = −1.1 V vs SCE in MeCN).267 PT leads to the formation of the closed-shell reaction product and a Co(III) hydride intermediate. Finally, hydrogen evolution occurs through protonation of this hydride intermediate, with the restoration of a Co(III) complex for catalytic turnover.
In addition to this suite of olefin hydrosulfonamidation and oxysulfonamidation methods presented above, Xiao and Chen in 2017 also developed a method for the carbosulfonamidation of allylic hydrazones through radical cyclization and further reaction with Nozaki allylic sulfone reagents or Morita–Baylis–Hillman allylic acetate reagents (Scheme 124).364 This was achieved through the blue-light irradiation of hydrazone substrate and olefin reagent in the presence of [Ir(ppy)2(bpy)]PF6 ([Ir-4]PF6) photocatalyst and K2CO3 Brønsted base additive in MeCN. Through this method, 24 examples of olefin 1,2-carbosulfonamidation were reported, with yields ranging from 26% to 85% (124.1–124.3). The reaction tolerated a variety of arene groups at the hydrazone site. Cyclization onto a pendant olefin through both 5-exo- or 6-endo-trig modes was also demonstrated, with the pathway determined through substrate control. Using a Morita–Baylis–Hillman acetate as a radical allylation reagent afforded products (E)-124.4–124.6.
Scheme 124. Photocatalytic Carbosulfonamidation of Allylic Sulfonylhydrazones with (A) Nozaki Allylsulfone Reagents and (B) Morita–Baylis–Hillman Allylic Acetate Reagents (Xiao and Chen, 2017).

The mechanism of this transformation was proposed to proceed through the same sequence of prior PT and photocatalytic SET to generate a neutral N-centered hydrazonyl radical which cyclizes onto the tethered olefin. The resulting C-centered radical now intercepts either the allylsulfone or allylic acetate reagents in an additional step, forming a new C–C bond and an α-acyl radical. In the former case, the extrusion of phenylsulfonyl radical yields the closed-shell product. Reduction of phenylsulfonyl radical to the corresponding sulfinate (e.g., for sodium benzenesulfinate, Ep/2ox = +0.56 V vs Fc+/Fc in MeCN)124 by Ir(II) (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN)68 returns catalytically active ground-state Ir(III). Alternatively, in the latter case, acetoxy radical extrusion is disfavored; thus, Ir(II)-mediated single-electron reduction of the α-acyl radical is likely to occur prior to the elimination of acetate anion.
Chen and Xiao in 2018, and Nagasawa in 2019, disclosed related methods for the highly diastereoselective synthesis of 3,5-disubstituted isoxazolidines through the redox-neutral intramolecular hydrosulfonamidation of β,γ-olefin-tethered N-sulfonyl or N-acyl alkoxyamines, respectively (Scheme 125A and B, respectively).365,366 In the former work, standard reaction conditions involved the blue-light irradiation of N-sulfonyl substrates in the presence of [Ru(bpy)3]Cl2·6H2O ([Ru-1]Cl2·6H2O) photocatalyst and K2CO3 Brønsted base in CHCl3 solution. Here, 21 examples of intramolecular alkene hydrosulfonamidation were reported in yields of 53–77% (125.1–125.4). In all cases, the 3,5-disubstituted isoxazolidine products were isolated as single diastereoisomers and assigned as the 3,5-cis configuration based on 1-H NMR analysis. The reaction proceeded well on both α-aryl and α-alkyl alkoxy sulfonamides and also tolerated variation of sulfonyl substituent. The reaction proceeded equally as effectively on gram scale. An enantiopure alkoxy sulfonamide substrate gave the product without erosion of stereochemical purity. The group demonstrated that isoxazolidine 125.1, on treatment with zinc in refluxing aqueous ammonium chloride solution, was transformed into the corresponding syn-1,3-amino alcohol in 95% yield via reductive N–O bond cleavage. Finally, through inclusion of TEMPO to the standard reaction conditions, the corresponding olefin oxy-sulfonamidation product 125.5 resulted, albeit with a lower efficiency of 35%.
Scheme 125. Synthesis of 3,5-cis-Isoxazolidines through N-Centered Radical Generation and Intramolecular Hydro(sulfon)amidation (A, Chen and Xiao, 2018; B, Nagasawa, 2019).

*With 2 equiv of TEMPO, 60% RSM. **With 10 equiv of KOtBu.
In Nagasawa’s work, the N-acyl substrates required the stronger base KOH to mediate the reaction under otherwise identical conditions. Here, 15 examples of intramolecular hydroamidation were reported in yields of 16–78% with d.r. between 8:1 and >99:1 (125.6–125.12). In all cases, 3,5-cis products resulted as the major diastereoisomer. A 4,5-disubstituted isoxazolidine gave the trans-product as a single diastereomer (125.10). Again, substrates bearing α-aryl and α-alkyl substituents were competent. Variation of the amide group revealed that reaction was most efficient with acetamide or trifluoroacetamide substrates, but proceeded poorly when a benzamide was used. A Boc carbamate substrate was unreactive in the presence of KOH but underwent the desired cyclization in the presence of an excess of the stronger base KOtBu with limited efficiency (125.9, 24%). An example of a transannular hydroamidation to prepare a 12-membered isoxazolidine-fused macrocycle was included.
Chen and Xiao proposed a mechanism based on experimental observations and their previous work with hydrazone substrates.136 SV experiments indicated that the N-sulfonyl alkoxyamine substrate only quenched the photocatalyst fluorescence in the presence of the basic additive. This was taken to indicate that substrate deprotonation occurs prior to PET from photoexcited-state Ru(II) (E1/2 *Ru(II)/Ru(I)= +0.77 V vs SCE in MeCN)64 to yield a key sulfonamidyl radical intermediate and reduced-state Ru(I) complex. Then, a 5-exo-trig cyclization occurs, generating a distal C-centered radical. This abstracts a H-atom from solvent to generate the closed-shell isoxazolidine product alongside trichloromethyl radical. Finally, trichloromethyl radical (E1/2red = −0.3 V vs SCE, dropping Hg electrode in H2O)355 is reduced to the corresponding anion through ET with the Ru(I) complex (E1/2 Ru(II)/Ru(I)= −1.33 V vs SCE in MeCN)64 to regenerate the ground-state photocatalyst. The use of a 1:1 mixture CHCl3/CDCl3 resulted in a 6:1 mixture of proteo/deutero products at the distal position, indicating solvent-mediated termination in this manner. The high diastereoselectivity observed in these reactions is proposed to arise through minimization of the steric interaction between the sulfonyl group and α-substituent by placing the α-substituent into an equatorial conformation in a chairlike transition state. An analogous mechanism is invoked to explain reactivity and diastereoselectivity with the N-acyl substrates.
2.7.2. Intramolecular C–N Bond Formation through Addition to Alkynes and Subsequent Smiles Rearrangement
In 2016, Brachet, Belmont, and co-workers published a synthesis of benzyl-appended phthalazine derivatives from acetylenic sulfonyl hydrazones, through a sequence of intramolecular alkyne sulfonamidation and subsequent radical Smiles rearrangement (Scheme 126).367 This transformation proceeded through blue-light irradiation of substrate solutions in the presence of [Ru(bpy)3]Cl2 ([Ru-1]Cl2) photocatalyst and NaOH Brønsted base additive in EtOH. A scope of 23 examples was presented, in yields of 30–81%. This protocol tolerated a variety of aryl, vinyl, and alkyl substitution at the terminus of the alkyne undergoing radical addition, as well as a variety of arenes and heteroarenes undergoing migration from the sulfonyl hydrazide. This reaction was also successful in the generation of other heterocyclic backbones such as pyridazine 126.4 and pyridopyridazine 126.5.
Scheme 126. Phthalazine Synthesis through Intramolecular Alkyne Sulfonamination and Smiles Rearrangement (Brachet and Belmont, 2016).

Authors propose this transformation occurs through a mechanism consisting of the discrete deprotonation of the hydrazone N–H bond prior to SET mediated by the Ru(II) photo-oxidant (E1/2 *Ru(II)/Ru(I)= +0.77 V vs SCE in MeCN).64 The resulting neutral N-centered radical then undergoes a 6-exo-dig cyclization onto the pendant alkyne. The vinyl radical thus generated is positioned to undergo a Smiles rearrangement by adding into the ipso-carbon of the arylsulfonamide. Aryl migration occurs with fragmentation and extrusion of SO2, affording a doubly stabilized benzylic radical. Finally, this intermediate is reduced to the corresponding anion (e.g., for diphenylmethyl radical, E1/2red = −1.14 V vs SCE in MeCN)135 by the Ru(I) state of the catalyst (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN),64 regenerating Ru(II) for catalyst turnover. PT from solvent yields the neutral, closed-shell product.
In 2020, Brachet, Belmont, and co-workers introduced a phosphoramidate-appended hydrazone as a precursor to a neutral N-centered radical through stepwise PT and ET (Scheme 127).368 This functional group was designed to facilitate both N–H bond homolysis and then in situ cleavage of this moiety for traceless activation. The utility of this group was demonstrated in the synthesis of 1-substituted phthalazines. This transformation occurred in the presence of [Ru(bpy)3](PF6)2([Ru-1](PF6)2) photocatalyst, NaOt-Bu Brønsted base additive in MeOH under blue-light irradiation. Altogether, 22 examples of phthalazine synthesis were presented. This protocol accommodated a variety of substituents on the alkyne including aryl-substituted (127.1) and terminal (127.2) alkynes. Variation of the aromatic portion of the substrate was also tolerated, affording indolopyridazine 127.3 in 92% yield. Ketone-derived hydrazones were demonstrated to be compatible with the protocol, although in diminished yields compared to aldehyde-derived hydrazones, as in the case of 127.4. Notably, this protocol represents an advance on their earlier method, in permitting a broader array of phthalazines product substitution at C1.367
Scheme 127. Phthalazine Synthesis from Phosphoramidate-Appended Hydrazones (Brachet and Belmont, 2020).

The authors propose that the N-centered radical is generated via stepwise substrate deprotonation by alkoxide base, followed by single-electron oxidation mediated by photoexcited-state Ru(II) (E1/2 *Ru(II)/Ru(I) = +0.77 V vs SCE in MeCN).64 This proposal is supported by CV experiments which show no oxidation of the neutral substrate up to +1.0 V vs SCE in MeCN (for precursor to 127.1), while the corresponding anion is oxidized at milder potentials (for precursor to 127.1, E1/2 = +0.71 V vs SCE in MeCN). The hydrazonyl radical then undergoes a 6-endo-dig cyclization onto a pendant alkyne to generate a vinyl radical. Subsequent reduction to the vinyl anion by Ru(I) state of the photocatalyst (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN),64 followed by protonation by MeOH provides a phosphoramidate intermediate. When the reaction was run in deuterated methanol, the authors observed 86% deuterium incorporation at this benzylic position, supporting this proposed termination sequence of ET and PT. NaOMe generated in the previous step then cleaves the phosphoramidate to release diethyl methyl phosphate, which was observed and characterized as a byproduct of the reaction. The resulting intermediate then undergoes tautomerization to generate the final phthalazine product.
2.7.3. Intramolecular C–N and N–N Bond Formation through Addition to Heteroarenes
Triazolopyridines and related heterocycles are privileged structures in pharmaceutical molecules, with wide-ranging biological activities including antimicrobial, anti-inflammatory, antiviral, and antidepressant.369 Zhang and co-workers reported electrothermal reaction conditions for the synthesis of 1,2,4-triazole-fused heterocycles from hydrazines and aldehydes, through N–H bond homolytic activation (Scheme 128).370 Hydrazine and aldehyde substrates were premixed in MeCN solution, initially forming a hydrazone. Without isolation or purification, the mixture was then subjected to electrolysis, performed under constant current conditions in an undivided cell equipped with a graphite rod anode and Pt plate cathode, with n-Bu4N+BF4– serving as a supporting electrolyte, in a mixture of MeCN/H2O (4:1) at 70 °C. Through this one-pot sequence, 43 examples of 1,2,4-triazole synthesis were reported, with yields of 41–94%. A number of different heterocyclic hydrazine starting materials were viable, including pyridine (128.1), pyrimidine (128.2), pyrazine (128.3), pyridazine (128.4) and quinoline (128.5). A synthesis of the anxiolytic drug alprazolam (128.6) was accomplished using this method in a more efficient and direct manner than previously reported.
Scheme 128. Electrochemical Synthesis of 1,2,4-Triazole-Fused Heterocycles (Zhang, 2018).

Following hydrazone condensation, the authors propose a stepwise mechanism for N–H bond homolysis, through deprotonation with hydroxide generated through cathodic reduction of water. This was followed by anodic oxidation of the hydrazone anion to afford the N-centered hydrazonyl radical. Formation of the N-centered radical triggers a 5-exo-trig cyclization onto the tethered imine, and further anodic oxidation and PT lead to the triazole product. A series of trapping experiments with TEMPO and BHT were supportive of this proposed radical mechanism.
The Xu Group in 2019 showed that hydrazones derived from 2-acetylpyridines and 2-acetylthiazoles can undergo electrochemical oxidative cyclization through N–N bond formation to yield the corresponding 1,2,3-triazole (Scheme 129).371 This transformation is facilitated by constant-current electrolysis of substrate solutions in MeOH/H2O (4:1), with Et4N+PF6– as a supporting electrolyte and K2CO3 as a Brønsted base additive. A total of nine examples of 1,2,3-triazole preparation were reported with yields from 33–89%. Methyl and bromo substitution (129.2) of the pyridine was tolerated, and substrates derived from quinoline (129.3), thiazole, and 4,5-dihydrothiazole (129.4) underwent successful cyclization. Ketone-derived hydrazones were optimal. When instead a picolinaldehyde-derived hydrazone was subjected to these reaction conditions, triazole product 129.5 was realized in only 33% yield.
Scheme 129. Electrochemical Synthesis of 1,2,3-Triazoles (Xu, 2019).

The reaction is proposed to proceed via two separate PT and ET events at the anode, thereby oxidizing the hydrazone to the corresponding diazo compound, which undergoes spontaneous intramolecular [3+2] cyclization to afford the triazole. These oxidation events are coupled to reduction of H2O to hydroxide and molecular hydrogen at the cathode. The authors noted that the oxidation potential of a model substrate was negatively shifted by approximately 300 mV in the presence of K2CO3, indicating some degree of coupling between the PT and ET events in the substrate activation.
2.8. Transformations of Other Functional Groups—Dihydropyrimidines, Dihydropyridines, Sulfonamides, and Carbazates
Here, we collect reports of PCET activation of N–H bonds for the formal dehydrogenation of dihydropyrimidine substrates, and radical generation from dihydropyridine (DHP), sulfinamide, and carbazate precursors. Wu and Liu in 2014 devised an efficient method for the synthesis of 2-substituted pyrimidines through the photocatalytic aerobic dehydrogenation of 1,4-dihydropyrimidines via N–H bond activation mediated by the joint action of an organic photoredox catalyst and Brønsted base (Scheme 130).372 This is potentially an attractive route to access pyrimidines since the corresponding dihydropyrimidine precursors are easily accessible through the Biginelli three-component reaction.373 However, compared to Hantzsch-type DHPs, which are readily oxidized at low potentials (e.g., for dimethyl Hantzsch DHP, Ep/2ox = +0.51 V vs Fc+/Fc in MeCN),374 dihydropyrimidines are more demanding in their oxidation (E1/2ox ≈ +0.80 V vs Fc+/Fc in MeCN)375 and thus typically have required strong chemical oxidants or elevated temperatures under conventional activation to yield the heteroaromatic product. Optimized reaction conditions for this dehydrogenation reaction consisted of visible-light irradiation of a 10% aq. MeOH solution of dihydropyrimidine substrate in the presence of n-Bu4N[EY] photocatalyst and K2CO3 at room temperature under an air atmosphere. The addition of Brønsted base was critical to achieving high yields of product and reducing the incidence of oxidative side products. A scope of 24 examples of dehydrogenation was reported in yields of 70–98%. 2-Methylthio- and 2-methoxydihydropyrimidines were competent substrates (130.1, 130.2), and a variety of alkyl (130.5), aryl, and heteroaryl (130.6) substituents at the C4-, C5-, and C6-positions were well tolerated. An intermediate (130.3) to the synthesis of a rosuvastatin derivative S-4522, an HMG-CoA reductase inhibitor,376−378 was efficiently prepared using this method. Gram-scale synthesis and the ability to drive this oxidation reaction directly with sunlight were demonstrated.
Scheme 130. Photocatalytic Aerobic Oxidation of Dihydropyrimidines (Wu and Liu, 2014).

The authors conducted electron spin resonance (ESR) and transient absorption (TA) spectroscopy experiments to probe the role of K2CO3 in the dehydrogenation reaction, concluding that the inclusion of this base was critical for ET. ESR experiments were conducted by irradiation of dihydropyrimidine substrate with n-Bu4N[EY] and tetramethylpiperidine—included as a probe for 1O2 generation—in air-saturated 10% aq. MeOH. Under these conditions, the authors observed a diagnostic signal for TEMPO radical, indicating that indeed 1O2 was being produced under these conditions. However, the addition of K2CO3 completely suppressed the generation of TEMPO. TA spectroscopy experiments were then conducted in the absence and presence of K2CO3. In the absence of K2CO3, no absorption signal was observed corresponding to EY•–. However, irradiation of substrate in the presence of n-Bu4N[EY] and K2CO3 in air-saturated 10% aq. MeOH at 532 nm resulted in the appearance of absorption at 406 nm assigned to EY•–, in agreement with literature.379 Together with the results from ESR spectroscopy experiments, the authors concluded that the addition of K2CO3 is consistent with a complete switch in the role of photoexcited EY. In the absence of this additive, EY acts as a photosensitizer for singlet oxygen generation, however, the presence of K2CO3 switches its role to a single-electron photo-oxidant for the substrate.
To understand the role of K2CO3 in favoring substrate oxidation by EY over oxygen sensitization, the authors turned to CV. Voltammetry experiments revealed that the substrate oxidation potential (E1/2 = +1.10 V vs NHE in 10% aq. MeOH)372 was drastically lowered upon the introduction of base (E1/2 = +0.58 V vs NHE in 10% aq. MeOH),372 which is ascribed to the ionization of the substrate N–H bond and subsequent favorable ET from the corresponding anion. EY is a weak photo-oxidant (E1/2 *EY/EY•– = +0.83 V vs SCE in MeCN),71 and thus cannot facilitate ET from the neutral substrate. Thus, a probable mechanism of dehydrogenation involves initial PT between substrate and carbonate base to generate an equilibrium concentration of the anion before subsequent PET with EY. Then, EY•– (E1/2EY/EY•– = −1.06 V vs SCE in MeCN)71 reacts with molecular oxygen to generate superoxide and regenerate the ground-state dye. The resulting substrate N-centered radical is further oxidized through either HAT with superoxide anion to yield the aromatized product and hydroperoxyl radical, or alternatively through stepwise ET and PT steps.
In addition to their versatile reactivity as precursors for pyridines and pyrimidines as detailed above, DHPs can also serve as versatile, readily accessible sources of C-centered radicals. Saturated nitrogen heterocycles such as (thio)morpholines and piperazines feature broadly in pharmaceutical compounds.380 In 2019, Romanov-Michailidis and co-workers reported a concerted PCET-driven method for radical generation from readily accessible 4-alkyl-DHP reagents (Scheme 131).381 The group prepared a series of redox-active imino-tethered DHP reagents through a one-pot condensation reaction of an amine substrate with an aldehyde, which upon visible-light irradiation in the presence of Ir photocatalyst [Ir(dF(CF3)ppy)2(bpy)]PF6 ([Ir-7]PF6) and Li+(n-BuO)2P(O)O– Brønsted base co-catalyst, cyclized to give the saturated heterocycle products. This mode of heterocycle assembly via radical cyclization is conceptually related to the organotin (SnAP)382,383 and organosilicon (SLAP)384 reagents introduced by the Bode group, but avoids problems associated with the handling of organotin compounds in the former, and a requirement for a neighboring sulfide moiety in substrates in the latter. Additionally, the authors noted that reactivity is independent of the nature of the heteroatom proximal to the DHP moiety, and therefore this report offers a single, unified method for the synthesis of many structured heterocycles. In comparison with the aforementioned Bode group reports, the redox potential of the organotin and organosilicon reagents shifts greatly as a function of the heteroatom.
Scheme 131. Modular Assembly of Saturated Nitrogen Heterocycles through the PCET Activation of 4-Alkyldihydropyridine Precursors (Romanov-Michailidis, 2019).

Potentials measured in V vs SCE in MeCN.
A scope of 59 examples of saturated nitrogen heterocycles was reported in this work, in yields of 52–86%, which typically exhibited good levels of diastereoselectivity for equatorially disposed product isomers. Morpholine (131.1), thiomorpholine (131.2), and piperazine (131.3) products were readily formed under these conditions. A rare example of a radical-mediated seven-membered-ring cyclization to form a 1,4-oxazepane (131.4) was also included. In addition, the method exhibited a great tolerance for a broad range of ring substituents and functional groups, including many heteroarenes, and groups readily oxidized at low potentials (e.g., indole, furan, and benzothiophene).
Concerted oxidative PCET was used as a key parameter in their experimental design to enable heterocyclization with such broad functional group tolerance, by lowering of the oxidation potential required to form an alkyl radical from the DHP precursor. The hydrogen-bond donor ability of DHPs is well-recognized, and in this work the authors observed a pH dependence of the oxidation potential in a model N–H DHP. This potential was lowered significantly in basic media, compared to the pH-independent oxidation potential of the corresponding N–Et DHP (Scheme 131). Further evidence for the involvement of N–H PCET in the activation of the DHP substrate comes from the loss of reactivity in the absence of base despite the use of photocatalysts with excited-state potentials beyond that of the DHP substrate. Additionally, oxidant/base pair combinations with effective BDFE values significantly below the requirement to homolyze the N–H bond of the 1,4-DHP (ca. 90 kcal mol–1)385 were ineffective catalysts.
Mechanistically, therefore, their proposal involves concerted PCET activation of the N–H bond of the DHP moiety of the substrate through the joint action of this Ir photocatalyst (E1/2 *Ir(III)/Ir(II) = +1.32 V vs SCE in MeCN)101 and hydrogen bonding with the dibutyl phosphate base (pKaH = 13 in MeCN).100 Notably, this catalyst is unable to drive oxidation of the neutral substrate alone, nor is phosphate sufficiently basic to fully deprotonate the substrate. Thereafter, C–C bond cleavage occurs, driven by aromatization and elimination of a Hantzsch pyridine and formation of a stabilized α-heteroatom C-centered radical. This C-centered radical then undergoes cyclization onto a tethered imine. The countercation to the anionic phosphate base was observed to be important for the efficiency of this step, with Li providing the highest yields. The Li cation is postulated to improve efficiency through chelation and Lewis acid (LA) activation of the imine moiety for 6-endo-trig radical cyclization, and also stabilization of the resultant N-centered aminyl radical post-cyclization. Finally, SET reduction (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN)101 of this aminyl radical and PT yields the closed-shell product while regenerating Ir(III) and base catalysts for turnover.
These observations from Romanov-Michailidis and co-workers of PCET activation in the generation of alkyl radicals from DHP precursors raise the question of whether this mechanism is generalizable to earlier synthetic reports of radical generation from these precursors under basic conditions.386 To our knowledge, this is the first report where the PCET pathway is rigorously studied and invoked as a mechanism for substrate activation, but we note other observations from earlier reports that involve similar substrate activation. For example, Molander in 2016 reported the cross-coupling of (hetero)aryl halides with alkyl radicals generated from DHP precursors in a dual Ni/Ir catalyzed process.387 This report notes that no reactivity was observed with an N-methylated DHP precursor, and that reactivity is optimal in hydrogen-bond accepting solvents. In related processes, Nishibayashi observed a significant decrease in reaction efficiency of photocatalytic benzylic radical generation and cross-coupling in the absence of exogenous acetate base.388,389 Notably, generation of alkyl radicals from 4-alkyl-DHP precursors and subsequent intermolecular addition to imines had been reported prior to this work by Yu and co-workers, but a PCET mechanism was not considered.390 These new findings reported by Romanov-Michailidis and co-workers should now influence future reaction design to improve efficiency with these alkyl radical precursors.
In addition to DHP precursors, alkyl radical generation from sulfinamides and carbazates has been demonstrated via N–H PCET activation followed by fragmentation and concomitant extrusion of a radical intermediate. Qin and co-workers demonstrated this strategy in a 2018 report on the generation of alkyl radicals from N-acyl alkylsulfinamides, and their subsequent intermolecular reaction with electron-deficient olefins (Scheme 132).391 Optimized reaction conditions consisting of [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) photocatalyst and K2HPO4 as a Brønsted base under blue-light irradiation in acetone solution were able to affect the fragmentation of the N-acyl sulfinamide precursors for alkyl radical generation and subsequent Giese-type conjugate addition reactions. These sulfinamide reagents could be prepared from thiols in three steps. Tertiary (132.1), secondary (132.2), and primary alkyl radical generation (132.3) and conjugate addition were demonstrated, with 67 alkylation products presented in yields of 27–93%. This is notable, as unstabilized secondary and primary alkyl radical generation from more commonly employed carboxylate precursors can be challenging. Alkyl radicals generated from these sulfinamide reagents were shown to be compatible with unsaturated esters, carboxylic acids, amides, nitriles, cyclic and acyclic ketones, and sulfones as coupling partners.
Scheme 132. N-Acyl Sulfinamides as Alkyl Radical Precursors via N–H Bond Homolysis and C–S Bond Fragmentation (Qin, 2018).

The authors propose a stepwise mechanism to facilitate N–H bond homolysis via deprotonation of the acidic N-acyl sulfinamide (e.g., for an N-benzoyl S-phenyl sulfinamide, pKa = 6.5 in aq. EtOH)392 to yield the conjugate base, and subsequent facile single-electron oxidation (e.g., for neutral t-butyl N-benzoyl sulfinamide, Ep/2ox = +0.43 V vs SCE in MeCN)391 mediated by the photoexcited-state Ir(III) complex (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN).68 Upon N-centered radical generation, a C–S bond fragmentation reaction occurs, extruding an alkyl radical and a sulfinylbenzamide byproduct. Following radical Giese 1,4-addition,103 the α-acyl radical undergoes single-electron reduction mediated by the Ir(II) state of the photocatalyst (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN)68 and the resultant enolate is protonated, forming the product. This mechanism is supported by control experiments, the observation of the decomposition products of the sulfinylbenzamide, and CV experiments.
This research group later demonstrated the application of these N-acyl sulfinamide reagents for the reductive C2-alkylation of benzothiophenes (Scheme 133).393 In this report, 17 examples of the reductive alkylation of benzothiophenes and heteroarene-fused thiophenes under similar reaction conditions were reported, giving benzo-fused and heteroarene-fused dihydrothiophenes, respectively, in yields of 10–95% (133.1–133.5). In this class of radical acceptors, reactions with secondary and primary alkyl radicals generated in this manner were less efficient than with the electron-deficient olefins studied earlier. An analogous mechanism for C-centered radical generation is proposed. This radical intermediate then adds to the heteroarene, forming a benzylic radical which undergoes steps of ET and PT to furnish the product. The reaction demonstrated exclusive C2-selectivity in all cases, but no mechanistic rationale is given for this observed preference.
Scheme 133. N-Acyl Sulfinamides as Alkyl Radical Precursors for the Reductive Alkylation of Benzothiophenes (Qin, 2018).

Wang and co-workers have recently disclosed an electrochemical method for the oxidative radical alkylation of heteroarenes using O-alkyl carbazate reagents as precursors to alkyl radicals via three iterative cycles of stepwise N–H bond homolysis, with the liberation of N2, H2, and CO2 as the sole waste products (Scheme 134).394 These reagents were conveniently prepared in two-steps from primary, secondary, and tertiary alcohols through reaction with phenyl chloroformate followed by substitution with hydrazine. This is notably the first report of deoxygenative alkyl radical generation via electrochemical substrate activation, as the analogous photocatalytic methods from oxalate precursors as pioneered by Overman and MacMillan,395,396 were reportedly incompatible with electrochemical cell conditions. Prior work from Taniguchi used methyl carbazate as a precursor to methoxycarbonyl radical through an Fe(II) phthalocyanine catalyzed aerobic oxidation with liberation of molecular nitrogen, but in this case the acyloxy radical intermediate did not further extrude CO2 (presumably due to the highly destabilized methyl radical that would result).397 This acyloxy radical was efficient in additions across styrenes and electron-deficient olefins, in a net oxidative hydroxyacylation process.
Scheme 134. Alkyl Carbazates as Precursors to Alkyl Radicals via Electrochemical Activation (Wang, 2020).

*MeCN solvent, n-Bu4N+ClO4– electrolyte, constant current = 3 mA.
Reaction conditions reported by Wang consisted of constant current electrolysis of a mixture of heteroarene substrate and alkyl carbazate reagent in an undivided cell consisting of graphite anode and platinum cathode with n-Bu4N+PF6– as a supporting electrolyte in a DMSO/MeCN mixture. Tertiary alkyl radical generation could be efficiently promoted at 50 °C, whereas secondary alkyl radical generation required higher temperature of 80 °C. Only a limited collection of benzylic primary alcohol-derived alkyl carbazates were effective precursors for radical generation at 85 °C under this mode of activation. A broad scope with respect to the alkyl radical and heteroarene is described, with 46 examples given in yields of 28–91%. Functional group tolerance beyond heteroarene halogen functionality was not well defined in this initial communication. Quinoxalinones proved to be the most efficient heteroarene substrates, but additional examples of quinoxalinone (134.1), pyrazinone (134.2), quinazolinone (134.3), purine (134.4), isoquinoline (134.5), and quinazoline (134.6) alkylation are given. Benzofuran and benzothiophene were unreactive in this method. Using methyl carbazate in this work gave methoxycarbonylation, rather than alkylation, of a quinoxalinone substrate, akin to the reactivity described by Taniguchi with styrenes.397
CV established the carbazate reagent undergoes anodic oxidation (e.g., for t-Bu carbazate, Eox1/2 = +1.44 V vs AgNO3 in MeCN),394 as opposed to the heteroarene. The low potential required for radical generation from these reagents enables subsequent functionalization of this reactive intermediate. Many electrochemical methods that generate a C-centered radical intermediate do so en route to a carbocation, as they operate at potentials higher than those required for the single-electron oxidation of the alkyl radical. The authors propose that following initial anodic oxidation, the resultant carbazate radical cation loses a proton to form a hydrazinecarboxylate radical. A second and third stepwise cycle of anodic oxidation and deprotonation forms a diazenecarboxylate, and then a third cycle of oxidation and deprotonation yields an transient acyloxy radical after loss of N2. The acyloxy radical then extrudes carbon dioxide, with expected rate dependency on the stabilization of the forming alkyl radical, thus reflecting the temperature dependence of the reaction on the degree of substitution of the precursor.398 Following Minisci-type radical alkylation of the heteroarene, further anodic oxidation and deprotonation yields the product.
2.9. Catalytic Applications of N-Centered Radicals Formed through Coupled Proton- and Electron-Transfer Processes
As we have seen, the majority of methods involving N-centered radical intermediates generated via PCET unsurprisingly concern the functionalization of nitrogen-containing substrates, typically through intra- and intermolecular C–N bond formation or remote functionalization through intramolecular 1,x-HAT processes. Several groups however have recently published methods concerning catalytic applications of transient neutral N-centered radicals generated through PCET processes, which affect chemical transformations on substrates not containing the nitrogen functional group undergoing activation and not incorporating the nitrogen functional group into the product. Catalytic HAT methods enabled by thiyl radical399 and aminium radical cation intermediates processes are well established,20,58,86 but methodology leveraging the reactivity of neutral N-centered radicals for catalytic applications has been lacking. This is a promising new avenue of research and has the potential to significantly broaden the types of transformations which can be driven by photocatalytic proton-coupled X–H bond homolysis protocols.
2.9.1. Mediating Substrate HAT
In 2018, Kanai and Oisaki developed a sulfonamide catalyst to mediate intermolecular HAT for allylic and benzylic C(sp3)–H arylation in a variety of organic substrates (Scheme 135).400 These authors focused development on this class of compounds for the tunability of steric environment and N–H BDFE through facile structural modification. The group identified an optimal diaryl sulfonamide consisting of a sterically bulky and electron-donating triisopropylbenzenesulfonyl group attached to an electron-deficient 3,5-bis-trifluoromethylaniline (135.A). In the presence of weakly oxidizing Ir photosensitizer Ir(dFppy)3 (Ir-2) and Brønsted base co-catalyst K2CO3 under blue-light irradiation, this sulfonamide was found to effectively mediate the arylation of a variety of allylic and benzylic C(sp3)–H bonds with 1,4-dicyanobenzene (1,4-DCB). A scope of 20 examples in yields of 35–95% was reported. Cycloalkenes were effective substrates (e.g., 135.1), including α-pinene which underwent the allylic arylation smoothly without apparent intermediate cyclobutylmethyl radical ring opening (135.2). The highly regioselective allylic arylation of the steroid prasterone was demonstrated (135.3). Benzylic C(sp3)–H bonds carrying an additional radical-stabilizing group such as an α-heteroatom also underwent successful reaction (e.g., 135.4, 135.5).
Scheme 135. Allylic and Benzylic C–H Bond Arylation Mediated by a Diarylsulfonamide HAT Catalyst (Kanai and Oisaki, 2018).

*Mixture of uncharacterized isomers.
A proposed mechanism of this reaction involved initial deprotonation of the acidic sulfonamide N–H bond mediated by K2CO3. The photoexcited-state Ir(III) complex (E1/2 *Ir(III)/Ir(II) = 0.00 V vs Fc+/Fc in MeCN)67 then oxidizes the resultant anion (Ep/2 = +0.47 V vs SCE in MeCN)400 to yield the sulfonamidyl radical intermediate, which engages in intermolecular HAT with the substrate (e.g., for sulfonamide 135.A, calculated N–H BDFE = 95 kcal mol–1).400 The reduced Ir(II) complex (E1/2 Ir(III)/Ir(II) = −2.51 V vs Fc+/Fc in MeCN)67 engages in ET with 1,4-DCB (E1/2 = −2.02 V vs Fc+/Fc in MeCN)401 to yield the corresponding arene radical anion and reform the ground-state Ir(III) complex. Through a proposed radical–radical coupling between the persistent arene radical anion and the transient alkyl radical resulting from HAT, a new C–C bond forms. Finally, elimination of cyanide restores aromaticity to the arene and yielding the product. Cyanide anion is proposed to deprotonate the regenerated neutral sulfonamide catalyst, thus only a catalytic amount of exogenous base as an initiator is required.
Ooi and co-workers have recently reported a zwitterionic triazolium amidate catalyst to mediate photoinduced HAT in combination with an Ir photosensitizer in C(sp3)–H bond alkylation reactions (Scheme 136).402 Compared to other photocatalytic HAT mediators such as thiols, decatungstate, quinuclidine, and phosphates, the authors reason that using an amide for this purpose may permit the tuning of reactivity through more facile structural modification than in these other systems. The authors proposed that close placement of a positive charge proximal to the amide group would stabilize its conjugate base. To do this, the authors investigated a number of easily prepared, bench-stable zwitterionic triazolium amidates as catalysts. The combination of N-benzoyl-1,2,3-triazolium amidate 136.A and [Ir(dF(CF3)ppy)2(4,4′-d(CF3)bpy)]PF6 ([Ir-8]PF6) photosensitizer under visible-light irradiation proved optimal for the α-alkylation of C–H bonds in organic substrates with electron-deficient olefins in 1,2-DCE solution. An N-pivaloyl-1,2,3-triazolium amidate displayed generally poor catalytic performance, which was ascribed to the steric blocking of the reactive N-centered radical form, retarding the rate of intermolecular HAT. Compared to the sulfonamide pre-catalysts for catalytically active N-centered radicals as developed by Kanai,400 this zwitterionic amidate functioned without exogenous base.
Scheme 136. A Zwitterionic Amidate Catalyst for Photoinduced HAT Reactions (Ooi, 2020).

*With 10 equiv of C–H nucleophile.
A scope of 20 examples was reported in yields of 42–99%. Hydridic C–H bonds in carbamate (136.1), urea (136.2), ether (136.3), and alcohol substrates (136.4) were selectively functionalized. In addition, benzylic (136.5) and formyl C–H bonds (136.6) also underwent successful derivatization. A range of electron-deficient olefins were effective coupling partners. Observed diastereoselectivity in alkylation reactions preparing two contiguous stereocenters was generally poor. However, the authors demonstrated an example of excellent diastereoselectivity in the preparation of a product featuring a 1,3-relationship between two stereocenters in 136.7 (99% yield, d.r. 13:1), through the coupling of N-Boc pyrrolidine and ethyl 2-phenylacrylate. This result compares favorably to that observed when other classes of HAT catalysts and reagents were employed (d.r. between 1.3:1 and 4:1).
A proposed mechanism was made based on TA, SV, voltammetry, and quantum yield determination experiments. Single-electron oxidation of the zwitterionic amidate catalyst (for an N-benzoyl 1,2,3-triazolium amidate 136.A, E1/2 = +1.28 V vs SCE in MeCN)402 by the photoexcited Ir(III) complex (E1/2 *Ir(III)/Ir(II) = +1.27 V vs Fc+/Fc in MeCN)29 initiates the catalytic cycle, forming an amidyl radical cation intermediate. This proposal was supported by SV luminescence quenching experiments, where quenching rate constant was measured as Kq = 6.23 × 109 M–1 s–1.402 The reactive distonic amidyl radical cation participates in intermolecular HAT with the weaker substrate C–H bond to form a carbon-centered radical and a cationic amide. The BDFE of this N–H bond was estimated to be 100.2 kcal mol–1 (G3(MP2)-RAD) by computational methods.402,403 Then Giese addition with the olefin acceptor forms a new C–C bond before reduction of the resulting radical to the enolate by Ir(II) (E1/2 Ir(III)/Ir(II) = −1.07 V vs Fc+/Fc in MeCN).29 Finally, enolate protonation occurs between the cationic amide to yield product and regenerate the zwitterionic amidate catalyst. This protonation step is responsible for the diastereoselectivity observed in product 136.7, demonstrating the impact of catalyst structure on dictation of the relative stereochemistry in this step. Additionally, results from quantum yield experiments (Φ = 0.13) implied a closed photoredox cycle over a potential chain process.
In 2020, the Cresswell group reported a method for the synthesis of α-tertiary amines via the C(sp3)–H alkylation of primary amines (Scheme 137).404 Through visible-light irradiation of MeCN solutions of primary amine substrate, 4CzIPN photoredox catalyst, n-Bu4N+N3– as an electrophilic HAT co-catalyst, and electron-deficient alkenes as the alkylating reagents, a total of 37 examples of this alkylation reaction were demonstrated with yields ranging from 20% to 84%. Because of the propensity of the amino-esters to undergo thermal lactamization, the authors elected to intentionally cyclize the initial products in situ to the γ-lactams. A variety of α-secondary aliphatic amines were alkylated with methyl acrylate and isolated as the γ-lactam following cyclization in moderate to excellent yields (137.1–137.4). The reaction proved amenable to substrates containing a range of functional groups, including carbamates, esters, thioethers, sulfones, and acetals, in addition to demonstrations of efficacy on scaffolds such as oxetanes and azetidines. Sterically encumbered substrates (137.3) still provided product, albeit in slightly attenuated yields. Benzylic amines were found to be unreactive. Primary amines bearing two α-C–H bonds, including those bearing functional groups such as imidazole and sulfonamides, were also amenable substrates in the chemistry and underwent regioselective double alkylation with 3.0 equiv of the Michael acceptor, albeit with diminished efficiency (137.5, 137.6). Finally, nine different electron-deficient alkenes were used to alkylate cyclohexylamine, including a range of α- and β-substituted unsaturated ester derivatives and vinylpyridines (137.7, 137.8).
Scheme 137. Photocatalytic C(sp3)–H Alkylation of Primary Amines (Cresswell, 2020).

*Using 1 mol% [Ir-6]PF6 photocatalyst.
The authors proposed a catalytic cycle that commences with oxidation of the azide anion (Ep/2ox = +0.49 V vs Fc+/Fc in MeCN)404 to the corresponding radical by the excited state of the 4CzIPN photocatalyst (E1/2 *PC/PC•– = +1.43 V vs SCE in MeCN).78 The feasibility of this ET event was demonstrated by SV quenching studies: the azide ion (KSV = 2610 M–1) quenches the luminescence of the photocatalyst nearly 3 orders of magnitude more than cyclohexylamine (KSV = 12.5 M–1). Azide radical is then hypothesized to undergo HAT with the primary amine to furnish a nucleophilic α-amino radical and hydrazoic acid. The authors conducted a competition experiment between cyclohexylamine and cyclohexanol to determine the selectivity for abstraction by a •N3 radical. A >20:1 selectivity favoring alkylation of the cyclohexylamine was observed, consistent with their calculations that indicated the barrier for C(sp3)–H abstraction from cyclohexanol was ∼+5.3 kcal mol–1 higher than cyclohexylamine. Following conjugate addition of the α-amino radical to the alkene and reduction of the resultant C-centered radical by the reduced photocatalyst (E1/2 PC/PC•– = −1.24 V vs SCE in MeCN),78 PT between the enolate and hydrazoic acid furnishes the alkylated product and regenerates the requisite HAT catalyst. A quantum yield determination experiment (Φ = 0.04) supports the proposal of a closed photoredox cycle over an efficient chain mechanism.
2.9.2. Mediating Olefin 1,2-Difunctionalization
Xiao and Chen reported the 1,2-carbosulfonylation of alkenes, catalyzed by the combination of Ir(III) photocatalyst [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6), Brønsted base co-catalyst K2CO3, and a simple sulfonanilide (143.A) as an N-centered radical pre-catalyst (Scheme 138).405 Prior research from this group had identified reversibility in the addition of N-centered hydrazonyl radical across olefins, which raised the prospect of catalytic activity of these reactive intermediates.136,406 Visible-light irradiation of DMSO solutions of alkene substrate and allylsulfone reagent under these conditions led to an efficient regioselective insertion reaction to afford the 1,2-difunctionalized product. In all cases, the sulfone functional group is added at the terminus of the alkene component. Authors present 34 examples of alkene 1,2-carbosulfonylation in yields of 20–79%. Styrenes (138.1, 138.2) and electron-deficient olefins (138.3, 138.4) were most effective, while terminal alkyl olefins (138.5) exhibited lower efficiency.
Scheme 138. Photocatalytic Generation of an N-Centered Radical for Catalytic Alkene 1,2-Carbosulfonylation (Xiao and Chen, 2019).

A mechanistic scenario is proposed to involve initial deprotonation of the acidic sulfonanilide pre-catalyst with carbonate base. The anion then undergoes single-electron oxidation driven by the photoexcited-state Ir(III) catalyst (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 to yield the catalytically active sulfonamidyl radical. The reaction of this radical with the allylsulfone reagent through an addition–elimination mechanism forges a new C–N bond and gives an allyl sulfonamide product. This closed-shell species could be observed in the reactions, and subjecting the independently prepared putative intermediate to the same reaction conditions allowed for product formation. In this C–N bond-forming process, an arylsulfonyl radical is ejected, which can either be reduced by the Ir(II) complex (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN)68 to the corresponding sulfinate anion to regenerate the ground-state photocatalyst, or add to the styrene component in an anti-Markovnikov fashion to yield a benzylic C-centered radical. The sulfinate anion can reform the sulfinyl radical through an ET event mediated by photoexcited Ir(III). This radical is then proposed to undergo reaction with the in situ generated allyl sulfonanilide intermediate in another addition–elimination process, regenerating the N-centered radical catalyst to allow a chain-type process to occur, and yielding the alkene 1,2-carbosulfonylation product. The N-centered radical can reform the N–H pre-catalyst through ET and PT steps. The ability in this transformation to allow both N- and S-centered radicals to form stable, off-cycle intermediates which can reengage through photocatalyst-mediated activation is an impressive feature that may overcome the kinetic limitations of traditional chain-type processes.407 The authors observed dimerization of the sulfonanilide through N–N bond formation resulting from radical–radical coupling, which was catalytically active through presumed N–N bond homolysis, thus serving as a further reservoir of active N-centered radical.
A later application of the catalytic reactivity of N-centered radicals generated in a similar manner reported from Xiao and Chen involved the [3+2] cycloadditions between vinylcyclopropanes or 2-vinylaziridines and electron-rich olefins for the synthesis of cyclopentanes and pyrrolidines, respectively (Scheme 139).408 The groups of Oshima,409,410 Feldman,411−413 and Maruoka414 had previously established the catalytic activity of thiyl, stannyl, and germyl radicals for this transformation, but these protocols required heating, UV light, and/or radical initiators for radical generation. Maruoka notably demonstrated that a complex chiral thiophenol pre-catalyst rendered this process enantioselective in an early example of absolute stereocontrol through a radical manifold.415 More recently, Miyake has developed a related protocol through bromine radical catalysis generated via photosensitized energy transfer.416
Scheme 139. Photocatalytic Generation of an N-Centered Radical for Catalytic [3+2] Cycloaddition (Xiao and Chen, 2020).

In this work, sulfonyl hydrazone pre-catalyst 139.A proved optimal for the desired transformation, alongside the combination of Ir(III) photocatalyst [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) and Brønsted base additive K2CO3 under blue-light irradiation at room temperature. Here, 12 examples of the [3+2] annulation between vinylcyclopropanes and electron-rich olefins were reported in yields of 21–92%. Cyclopropanes carrying geminal electron-withdrawing functionality were uniquely effective, but several classes of electron-rich olefin were compatible, including enol ether (139.1, 139.2), enol acetate, and ene-carbamate (139.3). In addition, 19 examples of this [3+2] annulation acting upon vinyl N-tosylaziridines were reported in yields of 53–86% (139.4, 139.5). In most cases in these two substrate classes, diastereoselectivity in annulation was poor.
A mechanistic proposal involving the stepwise generation of a hydrazonyl radical through PT and ET steps is put forward, consistent with their earlier works involving hydrazonyl radical cyclization onto olefins (see section 2.7.1). This N-centered radical can then add to the vinylcyclopropane functional group, leading to C–C bond scission and formation of a stabilized tertiary C-centered radical. This electrophilic radical is polarity matched for addition across the electron-rich olefin, to afford now an α-heteroatom stabilized radical. Finally, a 5-exo-trig cyclization initiates an allylic substitution reaction in which the active hydrazonyl radical catalyst is regenerated, ready for turnover in a chain-fashion. Through reduction (BET) mediated by Ir(II) (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN),68 the N-centered radical can reform the corresponding hydrazone anion, thus serving as an off-cycle reservoir and again avoiding kinetic limitations of some traditional chain processes. This proposal was supported by the observation of the intermediate hydrazone-bound coupled products prior to cyclization in some cases where a premature termination step occurred.
2.9.3. Mediating Borylation of (Hetero)arenes
In a distinct example of the synthetic utility of N–H bond activation through PCET, Larionov and co-workers in 2020 showed how a Brønsted base can modulate the excited-state reduction potential of an N–H PTZ photocatalyst to mediate reductive SET to a number of energy-demanding arene substrates, far beyond the reducing capability of the photocatalyst in the absence of base, or that of analogous photocatalysts with N-substitution.417 During studies aiming to develop a method for the borylation of aryl phosphate esters through the reductive generation of an aryl radical intermediate and subsequent trapping with B2Pin2 (Pin = pinacolato), the group observed that an N–H PTZ photocatalyst (140.1) was uniquely effective for this transformation (64% NMR yield) when in the presence of carbonate base, compared to either N-Me (140.2) (12% NMR yield), or N-Ph (140.3) (10% NMR yield) catalysts, or without exogenous base (Scheme 140). The reduction potential of the aryl phosphate substrate (e.g., for diethyl p-tolyl phosphate, Ep/2red = −2.97 V vs SCE in MeCN)417 greatly exceeds the photoexcited-state reduction potential of this N–H PTZ catalyst in the absence of base (E1/2 *PC/PC•+ = −2.60 V vs SCE in MeCN)417 or that of the corresponding N–Me (E1/2 *PC/PC•+ = −2.58 V vs SCE in MeCN)417 or N–Ph analogues (E1/2 *PC/PC•+ = −2.51 V vs SCE in MeCN).417
Scheme 140. Distinct Reactivity of an N–H Phenothiazine Photocatalyst Attributed to N–H MS-PCET (Larionov, 2020).

Potentials measured in V vs SCE in MeCN.
This reaction was very broadly applicable across a number of different energy-demanding substrate classes, with a large scope presented (Scheme 141). Under fully optimized reaction conditions, 34 examples of the borylation of phenol-derived aryl phosphate esters were reported in yields of 42–85% (141.2–141.6). The reaction is remarkably little impacted by the arene substituents, with electron-rich and electron-poor aryl phosphates both well tolerated. In addition to aryl phosphate esters, 20 examples of the borylation of quaternary aryl trialkylammonium salts (e.g., for p-tolyl trimethylammonium iodide, Ep/2red = −2.50 V vs SCE in MeCN)417 were demonstrated in yields of 60–90% (e.g., 141.7, 141.8), 60 examples of aryl halide borylation in yields of 50–95% (e.g., 141.9–141.13) (e.g., for chlorobenzene, Ep/2red = −2.83 V vs SCE in MeCN),417 26 examples of heteroaryl halide borylation in yields of 44–99%, and finally 11 examples of the borylation of complex natural product and API-derived halides, including strychnine (141.11), indomethacin (141.12), and loratidine (141.13). Aryl triflates were not effective substrates in this protocol, instead predominantly undergoing S–O bond cleavage to regenerate the corresponding phenol. A number of different arylboronate esters could be prepared in this manner from the corresponding diboron reagents.
Scheme 141. Borylation of Energy-Demanding Arenes through MS-PCET-Induced Aryl Radical Generation (Larionov, 2020).
*With 1 equiv of H2O; ketone protected as acetal, which was removed in situ with conversion to BF3K salt.
To understand the origin of the unexpected reactivity, the group carried out mechanistic studies that implicated a base-dependency with regard to the ability of this N–H PTZ catalyst to mediate reductive aryl radical generation. NMR experiments revealed that Cs2CO3 forms a H-bonded complex to the PTZ N–H bond in the presence of B2Pin2, which acts in a dual role as a phase-transfer agent in addition to a borylation reagent. The formation of the PTZ anion under these reaction conditions is ruled out, indicating observed reactivity is not a result of PET with this anion acting as an electron donor. SV experiments revealed that quenching of the photocatalyst emission by the aryl phosphate substrate is only observed in the presence of base. These data are consistent with a concerted PCET mechanism for ET between the photoexcited-state catalyst–base hydrogen-bonded complex and substrate, which substantially expands the redox window of these series of photocatalysts to engage in productive ET with energy-demanding substrates (Scheme 142).
Scheme 142. Proposed Mechanism of Aryl Phosphate Borylation via MS-PCET As Mediated by a Phenothiazine-Carbonate Hydrogen-Bonded Complex (Larionov, 2020).

Following ET from the photoexcited PTZ-carbonate hydrogen-bonded complex to the aryl phosphate substrate, the resultant arene radical anion undergoes mesolytic bond cleavage to extrude aryl radical and phosphate anion. The aryl radical intercepts B2Pin2 to forge a new C–B bond and a boryl radical, which through ET and PT steps regenerates the catalytically active PTZ-carbonate complex. A radical rearrangement substrate indicated that the bimolecular rate constant of the reaction of the aryl radical generated in this manner from the phosphate substrate with B2Pin2 is k = 1.1 × 108 M–1 s–1, which corresponds well to a value reported previous by Studer through reductive aryl radical generation from an aryl iodide and reaction with B2Cat2 (Cat = catecholato) (k = 7.4 × 107 M–1 s–1).418 DFT calculations support this mechanistic proposal, suggesting that SET between the aryl phosphate and PTZ-carbonate complex has a low barrier to arene radical anion formation (ΔG⧧ = +2.1 kcal mol–1), and which is substantially exergonic (ΔG = −30.0 kcal mol–1).
3. O-Centered Radical Generation from O–H Bonds through Photochemical and Electrochemical PCET Processes
In this chapter, we discuss synthetic photochemical and electrochemical transformations involving MS-PCET acting upon the O–H bonds of alkyl alcohol, enol, phenol, oxime, and hydroxamic acid substrates. In an exception to our general scope, we do not explicitly discuss decarboxylative radical generation from carboxylic acid precursors, which commonly do proceed through PT/ET mechanisms. This body of literature is extensive and has been recently reviewed thoroughly by others.419−426 We also do not discuss transformations involving TEMPO, PINO, and related nitroxide radicals. Stepwise and concerted PCET pathways have been invoked as mechanisms for the regeneration of the nitroxide radical from the corresponding hydroxylamine when these compounds are applied in a catalytic context. This body of work is also large and has been thoroughly reviewed by Stahl and co-workers.277
3.1. Transformations of Alkyl Alcohols
The homolytic activation of alkyl alcohol O–H bonds for alkoxy radical generation represents a challenging elementary step. Their high bond strengths (e.g., in methanol, O–H BDE = 105 kcal mol–1)25 and the presence of weaker C–H bonds in close proximity (e.g., in methanol, C–H BDE = 96 kcal mol–1)25 often render single-site HAT approaches incompatible. Over the past decade, PCET has emerged as an applicable catalytic manifold for O–H bond activation in a variety of reaction classes. In the following section of this Review, we present a number of transformations that leverage concerted PCET activation of alkyl alcohol O–H bonds to achieve C–C bond β-scission, 1,5-HAT, and C–O bond formation. We also highlight several transformations involving cyclopropanol and cyclobutanol substrates, where an accessible substrate oxidation potential allows for the stepwise generation of an alkoxy radical intermediate through discrete ET and PT events.
3.1.1. C–C Bond β-Scission Reactions Following Alkoxy Radical Generation
As early as 2009, seminal work by Que proposed that the O–H bonds of methanol and tert-butanol can be activated via a PCET mechanism by using a diiron(IV) complex as a synthetic analogue of methane monooxygenase.427,428 However, the development of a general catalytic protocol for the homolysis of alcohol O–H bonds remained elusive. In 2016, Knowles and co-workers reported the first example of the homolytic activation of aliphatic alcohol O–H bonds enabled by oxidative MS-PCET for the redox-neutral ring-opening isomerization of unstrained cyclic alcohols to linear ketones (Scheme 143).27 The reaction system comprised an Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 ([Ir-9]PF6), collidine Brønsted base, and thiophenol HAT co-catalyst under blue-light irradiation, and the scope consisted of 28 examples of ring opening isomerization in 41–98% yields (143.1–143.4). Ring size or ring strain of the cyclic alcohol substrate was not a determining factor in successful reactivity, with 4- to 12-membered cyclic alcohols demonstrated. Although 4-methoxyphenyl (PMP) tertiary carbinols were primarily showcased, other appended electron-rich arenes were successful in promoting the desired reactivity, including benzofuran, benzothiophene, naphthalene, and phenanthrene. Natural product-derived substrates were also included, such as hecogenin derivative 143.3, which underwent ring opening with complete regioselectivity for bond scission toward the adjacent tertiary site. Moreover, when the thiophenol co-catalyst was replaced with stoichiometric quantities of either SelectFluor, CCl4, or BrCCl3, the protocol enabled distal halogenation of the ring-opened products (143.4).
Scheme 143. Photocatalytic Ring-Opening Reactions of Unstrained Cyclic Alcohols via Alkoxy Radical-Mediated C–C Bond β-Scission (Knowles, 2016).

*2 mol% [Ir-9]PF6, 1.0 equiv of collidine, 4.0 equiv of SelectFluor, 1:1 CD3CN:D2O, blue LEDs, rt.
The authors hypothesize that upon visible-light irradiation, the Ir(III) photocatalyst undergoes excitation and subsequently oxidizes (E1/2 *Ir(III)/Ir(II) = +1.30 V vs Fc+/Fc in MeCN)29 the electron-rich arene of the alcohol substrate (e.g., for 4-methoxyphenylcyclohexanecarbinol, Ep/2ox = +1.60 V vs SCE in MeCN)27 to furnish the corresponding arene radical cation. Intramolecular ET to the proximal radical cation occurs in concert with PT from the hydroxyl group to a hydrogen-bonded collidine, which serves as a weak Brønsted base in this MS-PCET event. Upon generation of the alkoxy radical in this manner, the adjacent C–C bond is sufficiently weakened to enable favorable ring-opening to generate a ketone and distal C-centered alkyl radical. DFT analysis predicted that the BDE of the C–C bond adjacent to the alkoxy radical would be reduced to ca. 0 kcal mol–1.27 Additionally, prior studies from Baciocchi and co-workers demonstrated the feasibility of this C–C bond cleavage step in aryl alkanol radical cations generated through single-electron oxidation using stoichiometric quantities of the Co(III) complex K5[CoO4W12O36].429−434 The distal C-centered radical formed upon C–C bond cleavage undergoes HAT with the thiophenol co-catalyst to furnish the ketone product. Finally, ground-state Ir(III), collidine, and thiophenol are all regenerated through successive ET and PT events.
Indeed, SV quenching studies revealed that admixtures of the alcohol substrate and collidine base efficiently quenched the excited state of the iridium photocatalyst, with a first-order dependence on alcohol concentration, and zero-order dependence on collidine concentration, suggesting that direct arene oxidation rather than concerted O–H MS–PCET, is the operative pathway for excited-state CT. The nature of the subsequent charge-transfer event between the oxidized arene and the alcohol O–H bond (through either stepwise PT/ET or concerted PCET) was studied in a series of acyclic alkanol substrates by increasing the number of methylene groups between the arene and alcohol. The pKa of the alcohol O–H bond should rapidly increase as a function of increasing distance from the arene radical cation site and ultimately approach the pKa of an isolated tertiary alcohol (e.g., for t-BuOH, pKa = 40 in MeCN),50 whose discrete deprotonation by collidine (pKa = 15.0 in MeCN)49 is prohibitively endergonic. In contrast, the thermochemistry of concerted PCET was expected to remain relatively thermoneutral irrespective of increasing distance. Indeed, this hypothesis was borne out experimentally when increasing methylene spacer units showed minimal change in observed reactivity, which strongly supports a concerted PCET mechanism for alkoxy radical generation. Finally, effective BDFE considerations were shown to accurately predict the feasibility of alkoxy radical generation across 32 unique combinations of oxidant/base pairs, whose effective BDFEs ranged from 77 to 105 kcal mol–1 (Table 2). Once the effective BDFE of the oxidant/base pair approached or exceeded that of the substrate O–H bond (e.g., in 4-methoxyphenylcyclopentanecarbinol, O–H BDFEcalc = 103 kcal mol–1),27 successful β-scission reactivity was observed, illustrating that simple thermodynamic factors govern PCET activation of O–H bonds in these substrates.
Table 2. Correlation between Effective BDFE of Oxidant/Base Pair and Observed Reactivity in 1-Arylcyclopentanol Ring Opening via β-Scission (O–H BDFEcalc ≈ 103 kcal mol–1)a.
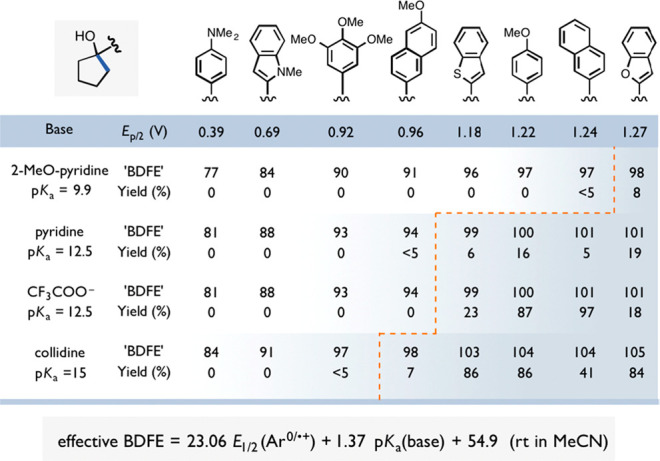
Reproduced with permission from ref (27). Copyright 2016 ACS Publications.
Seeking to broaden the scope of C–C bond β-scission reactions to include simple aliphatic alcohols, Knowles and co-workers in 2019 disclosed a protocol for the PCET activation of O–H bonds that circumvents the requirement for an oxidizable arene in the substrate alcohol (Scheme 144).28 This approach enabled the direct PCET activation of the substrate hydroxyl group through the concerted action of the photoexcited state of Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 ([Ir-9]PF6) and a Brønsted base, bypassing the formation of a radical cation intermediate as observed in the previously reported redox-relay strategy (Scheme 143).27 TRIP-SH was included as a HAT co-catalyst to trap the C-centered radical formed upon β-scission. The key change made during the development of this method was to replace the neutral collidine base with the anionic phosphate n-Bu4P+(MeO)2P(O)O–. The superiority of an anionic phosphate base in activating the O–H bonds of aliphatic alcohols is proposed to result from the propensity of such a base to form a more favorable pre-PCET hydrogen bond complex with the substrate, a mechanistic prerequisite for PCET activation.
Scheme 144. Photocatalytic C–C Bond Cleavage via Alkoxy Radical β-Scission in Aliphatic Alcohols (Knowles, 2019).

*With n-Bu4P+(PhO)2P(O)O–. **With n-Bu4P+(MeO)2P(O)O– in PhCF3.
Notably, this method now allowed for the redox-neutral isomerization of 1°, 2°, and 3° alcohols and the cleavage of carbon-substituents from both cyclic and acyclic alcohols (144.1–144.4). Lactol substrates, including protected hexose and pentose derivatives, were well tolerated under reaction conditions, yielding linear formate ester products (e.g., 144.1). Interestingly, the carbonyl-containing products showcased in this work were often found to be thermodynamically less-stable than the corresponding alcohol starting materials (e.g., for 144.2, ΔGcalc = +10.5 kcal mol–1), demonstrating the potential for excited-state PCET events to drive out-of-equilibrium transformations.
Later in 2019, Knowles and co-workers described a strategy for the catalytic conversion of cyclic n-membered-ring allylic alcohols to ring-expanded ketones by relying on the ability of alkoxy radicals to undergo C–C bond cleavage (Scheme 145).319 Much like prior β-C–C bond scission work from the Knowles group, this method employed the Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 ([Ir-9]PF6) and either n-Bu4P+CF3CO2– or collidine as a Brønsted base to jointly mediate the homolysis of the alcohol O–H bond of the cycloalkanol substrate via a concerted PCET mechanism, thereby generating an alkoxy radical intermediate primed for β-fragmentation. In this class of allylic alcohols, C–C bond scission results in ring opening and formation of an α,β-unsaturated ketone that is appended to an alkyl radical, both of which can recombine to form a new C–C bond as part of the ring expanded ketone product. A notable aspect of this strategy is its modular approach—the regioselectivity of the enone addition step can be varied to allow access to ring adducts incorporating two of the olefinic carbons (n+2) or just one of them (n+1). The selectivity of this C–C bond-forming step is controlled by the nature of the substituent on the exocyclic olefin acceptor. Alkyl olefins were shown to favor endo-trig cyclization to generate an α-acyl radical that is then reduced by the Ir(II) state of the photocatalyst to furnish the (n+2) ring-expanded product (Scheme 145A). Although this strategy could tolerate a variety of alkene substitution patterns—vinyl, mono- or di-substituted, and allenyl—this method for (n+2) expansion was limited to aza-heterocyclic substrates, possibly due to the enhanced nucleophilicity of the α-amino radical in the 1,4-conjugate addition step (145.1–145.3). In contrast, styrenyl substrates underwent exo-trig cyclization to furnish a stabilized benzylic radical via an anti-Michael addition (Scheme 145B).317 The authors noted that in the latter case, the reduction potential of the Ir(II) complex (E1/2 Ir(III)/Ir(II) = −1.07 V vs Fc+/Fc in MeCN)29 is insufficient to reduce the benzylic radical (e.g., for the benzylic radical derived from ethylbenzene, PhCH·CH3, E1/2 = −1.60 V vs SCE in MeCN),135 Consequently, TRIP-SH was employed as a co-catalyst to mediate the reduction of the benzylic radical. A variety of azacyclic, cyclic ether, and carbocyclic (n+1) products were obtained under these reaction conditions (145.4–145.6).
Scheme 145. Photocatalytic (A) (n+2) and (B) (n+1) Ring Expansion of Cyclic Allylic Alcohols via C–C Bond β-Scission (Knowles, 2019).

*With n-Bu4P+(PhO)2P(O)O–.
In 2018, Zhu and co-workers demonstrated the ring-opening bromination of unstrained cycloalkanols via alkoxy radical-induced C–C bond β-scission (Scheme 146).435 This transformation was accomplished through the use of [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) photocatalyst, N-bromosuccinimide (NBS) as a brominating reagent, and PhI(OAc)2 (PIDA) under blue-light irradiation. In this work, 45 examples were reported with yields of 24–83%. This protocol enabled the construction of distally brominated ketones from 5- to 15-membered cycloalkanols (e.g., 146.1, 146.2). Both aryl- and alkyl-bearing tertiary alcohols were effective substrates, and variations in the electronic character of aryl substrates were well tolerated. Additionally, substitution of NBS with either p-toluenesulfonyl cyanide (TsCN) or an alkynylsulfone reagent allowed for distal cyanation (146.6) and alkynylation (146.3), respectively, through an analogous ring-opening mechanism.
Scheme 146. Ring-Opening Functionalization of Cycloalkanols via C–C Bond Fragmentation (Zhu, 2018).

The authors propose two pathways for alkoxy radical generation under these reaction conditions. First, oxidative quenching of the photoexcited Ir(III) complex (E1/2 Ir(IV)/*Ir(III) = −0.96 V vs SCE in MeCN)68 with NBS forms Ir(IV), which can then engage in PCET activation of the substrate O–H bond (E1/2 Ir(IV)/Ir(III) = +1.21 V vs SCE in MeCN)68 with a weak base, such as the succinimide anion (pKaH = 14.6 in DMSO).116 The feasibility of oxidative quenching in this manner was confirmed through SV studies. The oxidation potential of the Ir(IV) complex alone was insufficient to directly oxidize the cycloalkanol substrate (e.g., for 1-phenylcyclopentanol, Ep/2ox > +2.0 V vs SCE in MeCN).435 Second, the formation of a trivalent alkoxyiodobenzene complex from PIDA and visible-light-induced O–I bond homolysis can also lead to generation of an alkoxy radical.436−439 Substantial reactivity was observed if either the Ir(III) catalyst or PIDA was removed (e.g., in a model substrate, 76% yield under optimal conditions, 53% in the absence of PIDA, 45% in the absence of [Ir-3]PF6), suggesting that both reaction pathways may be operative. Thereafter, C–C bond β-scission extrudes a distal C-centered radical, which is intercepted by NBS to yield the brominated product. The authors noted that CBr4 was also a competent brominating reagent under these conditions.
In 2018, Xia and co-workers detailed a method for the regiospecific ring-opening allylation and formylation of cycloalkanols (Scheme 147).440 The reaction system comprised similar conditions as those reported by Knowles, including the use of [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 ([Ir-9]PF6) photocatalyst and collidine Brønsted base under visible-light irradiation.27 By employing Nozaki allyl sulfone reagents as electron-deficient radical acceptors, the authors demonstrated that a variety of arylcyclobutanols (147.1–147.4) and medium- to large-sized ring systems are amenable to ring-opening allylation, with 28 examples reported in yields of 31–90%. The mechanism was proposed to involve trapping of the allyl sulfone reagent by the C-centered radical generated after β-scission, followed by elimination of the phenylsulfonyl radical. In lieu of an exogenous olefin partner, air was used as an oxidant in trifluoroethanol (TFE) to afford a range of distally formylated products in yields of 43–68% (147.5–147.7). Under this aerobic regime, it was postulated that superoxide serves as the trapping agent that intercepts the C-centered radical to deliver the aldehyde products.
Scheme 147. Photocatalytic Ring-Opening (A) Allylation and (B) Formylation of Arylcycloalkanols via C–C Bond β-Scission (Xia, 2018).

In an effort to further diversify the repertoire of C–C bond-breaking transformations, Rueping and co-workers in 2020 integrated PCET-promoted β-scission with a Ni catalytic cycle to achieve the remote, site-specific (hetero)arylation of ketones (Scheme 148).441 The authors devised a strategy wherein the highly oxidizing photocatalyst N-Me Mes-Acr+ClO4– ([Acr-1]ClO4) (E1/2 *3Acr+/Acr• = +1.88 V vs SCE in PhCN)74 and collidine base are used to promote the PCET activation of the O–H bond in an arene-containing tertiary alcohol substrate through a radical-cation redox relay event in the manner reported by Knowles.27 Rueping demonstrated 58 examples of this transformation in 38–98% yields. A range of aryl and heteroaryl halides (148.1–148.4), including pharmaceutically relevant derivatives such as a celecoxib analogue (148.5), were competent electrophilic coupling partners. Tertiary alcohols bearing a variety of oxidizable arenes (Ep/2ox > +1.38 V vs SCE in MeCN) were also suitable radical precursors in this cross-coupling system (148.2). Notably, the site of arylation could be modulated by using carbocycles of different ring sizes (e.g., 148.4), from 3- to 15-membered-ring derivatives, to afford arylated ketones of varying lengths.
Scheme 148. Dual Photoredox/Ni-Catalyzed Ring-Opening Arylation Enabled by PCET-Promoted C–C Bond Cleavage (Rueping, 2020).

The transformation was proposed to proceed through an intramolecular PCET event that generates an alkoxy radical intermediate primed for β-scission. Subsequent C–C bond cleavage provides a distal C-centered radical that is intercepted by a Ni(0) complex to give an alkyl Ni(I) intermediate. Oxidative addition with an aryl halide affords a Ni(III) species that then undergoes reductive elimination to furnish the cross-coupled product, creating a new C(sp3)–C(sp2) bond in the process. Ni(I) is then reduced to Ni(0) by the reduced-state of the photocatalyst, thereby returning both of these components to their active forms for another catalytic turnover.
Shortly after this report, Rueping and co-workers disclosed a related PCET system that achieves the ring-opening trifluoromethyl thiolation of ketones via alkoxy radical-promoted β-fragmentation (Scheme 149).442 The same combination of N-Me Mes-Acr+ClO4– photocatalyst ([Acr-1]ClO4) and collidine Brønsted base, with inclusion of N-(trifluoromethyl)thiophthalimide (Phth-SCF3), resulted in 24 examples of trifluoromethyl thiolated ketones in 34–83% yields (149.1–149.4). Following O–H bond homolysis and C–C bond cleavage as detailed above, the nascent C-centered radical intercepts Phth–SCF3 to furnish the functionalized ketone. To turn over the catalytic cycle, the resulting PhthH radical is reduced by acridine radical to the corresponding anion and protonated by collidinium, regenerating the photocatalyst and base for another PCET event.
Scheme 149. Synthesis of SCF3-Containing Ketones via Alkoxy Radical Generation and C–C Bond β-Scission (Rueping, 2020).

In addition to these photocatalytic methods for C–C bond β-scission through concerted PCET alcohol activation, several photocatalytic and electrochemical methods have targeted the ring-opening functionalization of cyclopropanols and cyclobutanols. For this substrate class, direct substrate oxidation may be feasible prior to discrete PT, and thus, stepwise and concerted homolysis pathways may operate simultaneously in O–H bond activation. These strategies are detailed in the following discussion.
Disclosing an early example of photocatalytic alcohol O–H activation, Lectka and co-workers described in 2015 a method for the regiospecific synthesis of β-fluoroketones from cyclopropanols via C–C bond β-scission (Scheme 150).443 This approach involved UV irradiation (λ = 300 nm) of substrate solutions in MeCN in the presence of 1,2,4,5-tetracyanobenzene (TCB) photocatalyst and SelectFluor as a fluorinating reagent and oxidant. Various 1,2-dialkyl- and 1-alkyl-2-arylcyclopropanols were amenable (150.1–150.5), including a cholic acid derivative. Notably, regioselective fluorination occurs even in the presence of activated benzylic C(sp3)–H bonds, with only trace amounts of benzylic fluorination products observed. This oxidative ring-opening and fluorination strategy was also employed to achieve tandem ring expansion and fluorination, yielding β-fluorocycloheptanone (150.6) in the process.
Scheme 150. Photocatalyzed Oxidative Ring-Opening and β-Fluorination of Cyclopropanols (Lectka, 2015).

UV photoexcitation of TCB results in a powerful oxidant (E1/2 *1TCB/TCB•– = +3.44 V vs SCE in MeCN)444 capable of oxidizing the cyclopropanol substrate to the corresponding radical cation, prompting elongation of the adjacent C–C bond to a calculated 2.02 Å. Accompanying proton loss and ring opening affords a β-carbonyl radical that intercepts Selectfluor to furnish the fluorinated product. The resultant tertiary aminium radical dication TEDA2+• reacts with TCB•– to regenerate the catalytically active form of the photocatalyst.445
The Zhu group developed a photocatalytic approach to the ring-opening and functionalization of 1-arylcyclopropanols in the synthesis of unsymmetric 1,8-diketone products (Scheme 151).446 This protocol relies on the coupling of ring-opened cyclopropanols with cyanohydrins in the presence of Ir(III) photocatalyst [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6), K2S2O8 oxidant, and BF3·OEt2 additive under blue-light irradiation. Successful coupling across 32 examples was demonstrated, with electron-rich and electron-deficient 1-arylcyclopropanols and aryl and alkyl olefin-tethered cyanohydrins serving as competent substrates (151.1, 151.3). Notably, this method was successfully applied to other tertiary alkenol substrates that resulted instead in the migration of benzothiazolyl (151.2) and formyl groups (151.4), albeit only in modest yields.
Scheme 151. Synthesis of Unsymmetric 1,8-Diketones Enabled by C–C Bond Fragmentation (Zhu, 2019).

*With 2-(benzothiazoyl)carbinol coupling partner. **With formyl alcohol coupling partner.
The proposed mechanism for the reaction involves oxidative quenching of photoexcited Ir(III) (E1/2 Ir(IV)/*Ir(III) = −0.96 V vs SCE in MeCN)68 by K2S2O8 and subsequent direct single-electron oxidation (E1/2 Ir(IV)/Ir(III) = +1.21 V vs SCE in MeCN) of the arylcyclopropanol substrate (e.g., for 1-(4-methoxyphenyl)-1-cyclopropanol, E1/2ox = +1.08 V vs SCE in MeCN).447 The authors noted, however, that direct oxidation of the substrate by K2S2O8 (E1/2ox = +2.08 V vs NHE in H2O)448 is thermodynamically feasible and cannot be ruled out. Indeed, significant reactivity was observed in the absence of a photocatalyst in the model studied (e.g., for a model substrate, 43% in the absence of photocatalyst compared to 72% under optimal conditions). Following oxidation, proton loss and ring-opening result in a β-keto radical. This intermediate adds into the distal olefin of the cyanohydrin coupling partner, generating another C-centered radical in a presumably thermoneutral and reversible elementary step. The authors hypothesized that the efficiency of this addition step is facilitated by a subsequent intramolecular cyano-group migration through an addition–elimination sequence, rendering this coupling irreversible. Finally, oxidation of the ketyl radical and subsequent deprotonation furnishes the final 1,8-diketone product.
Recently, von Wangelin, Majek, and co-workers reported a photocatalytic [3+2] cycloaddition of arenediazonium salts and 1-arylcyclopropanols, providing a useful platform for the synthesis of 1,5-diarylpyrazoles with complete regioselectivity (Scheme 152).447 Ru(bpy)3Cl2·6H2O ([Ru-1]Cl2) proved to be the most effective photocatalyst for the transformation, upon blue-light irradiation of MeCN solutions of these substrates. A total of 30 examples of 1,5-diarylpyrazoles were reported in yields of 8–90%. Various arenediazonium salts bearing electron-withdrawing and electron-donating substituents at any position on the aryl ring were all suitable reaction partners. Although cycloaddition of naphthalenediazonium and 4-(methylthio)benzenediazonium tetrafluoroborates was feasible, <25% of the desired product was observed in a complex product mixture. Steric hindrance on the cyclopropane moiety of 1-arylcyclopropanols did not impact the reaction efficiency (152.2), whereas electron density on the aryl moiety showed a significant effect. For example, while substrates bearing electron-donating substituents reacted smoothly to provide diarylpyrazoles in high yields (152.1), cyclopropanols with electron-withdrawing substituents exhibited much poorer reactivities under the reaction conditions (152.5). Whereas alkyl- and benzyl-substituted cyclopropanols gave low product yields (e.g., 152.6), 1-phenyl-1-cyclobutanol was unreactive in this protocol. A similar Cu-mediated oxidative cyclization reaction of these substrates to access these structures was concurrently described by Liu and co-workers.449
Scheme 152. Synthesis of 1,5-Diarylpyrazoles via Photocatalytic Cycloaddition of Arenediazonium Salts with Arylcyclopropanols (von Wangelin, 2020).

The proposed mechanism involves initiation via oxidative quenching of the Ru(II) photocatalyst (E1/2 Ru(III)/*Ru(II)= −0.81 V vs SCE in MeCN)64 with a sacrificial quantity of the arenediazonium salt (Ep/2red ≈ 0.0 V vs SCE in MeCN),447 which was evidenced by increasing SV quenching constants with decreasing electron density of the arene diazoniums. The resultant Ru(III) complex (E1/2 Ru(III)/Ru(II)= +1.26 V vs SCE in MeCN)64 is sufficiently oxidizing to convert an electron-rich 1-arylcyclopropanol to the corresponding radical cation (e.g., for 1-(4-methoxyphenyl)-1-cyclopropanol, E1/2ox = +1.08 V vs SCE in MeCN).447 However, single-electron oxidation of electron-poor 1-arylcyclopropanols is more thermodynamically challenging (e.g., for 1-(4-trifluoromethylphenyl)-1-cyclopropanol E1/2ox = +1.40 V vs SCE in MeCN),447 which is consistent with the observed poor reactivity of these substrates. The authors note that this reactivity could not be extended to 1-arylcyclobutanols because of their prohibitively endergonic oxidation potentials (e.g., for 1-phenyl-1-cyclobutanol, E1/2ox = +1.72 V vs SCE in MeCN).447 The resulting arylcyclopropanol radical cation undergoes rapid PT and ring-opening to generate a β-keto radical, which is subsequently trapped with the arenediazonium salt through C–N bond formation to afford a radical cation adduct. This intermediate can either enter a radical chain process by oxidizing another molecule of cyclopropanol or quench the photoexcited state of the Ru(II) catalyst to close the catalytic cycle and generate a diazene adduct, which subsequently undergoes tautomerization, cyclization, and dehydration to afford the desired pyrazole product. The radical chain mechanism was indeed supported through a quantum yield determination experiment (Φ = 4.2).
A regioselective electrochemical synthesis of 1-tetralones via ring-expansion reactions of functionalized 1-arylcyclobutanols was recently achieved by Parsons and co-workers (Scheme 153).450 Optimal reaction conditions required constant current electrolysis of substrate solutions in MeCN under air atmosphere at 0–25 °C in an undivided electrochemical cell fitted with carbon electrodes and LiClO4 as the supporting electrolyte. A total of 15 examples of the ring expansion of cyclobutanols to 1-tetralones were reported in yields of 13–68% (153.1–153.6). Various cyclobutanols with electron-donating, electron-withdrawing (153.3, 153.4), and electron-neutral substituents on the aromatic ring were all amenable to this protocol. Interestingly, substrates with certain electron-withdrawing substituents (e.g., o-fluoro and p-trifluoromethoxy) and more challenging heteroaromatic substrates showed no reactivity under these direct anodic oxidation conditions. Upon the addition of 10 mol% of Mn(OTf)2 catalyst, these challenging cyclobutanol substrates successfully underwent reaction to provide the corresponding tetralone products in good yields (153.4–153.6). It was hypothesized that a manganese-alkoxide complex with a lower oxidation potential is formed in these cases and that this complex can undergo a more facile inner-sphere ET to form the key alkoxy radical cation intermediate. Similar Mn-catalyzed electrochemical conditions for the deconstructive chlorination reaction of cycloalkanols were previously reported by the groups of Browne and Morrill.451
Scheme 153. Synthesis of 1-Tetralones via Regioselective Electrochemical Ring Expansion of 1-Arylcyclobutanols (Parsons, 2020).

*With 10 mol% Mn(OTf)2.
This transformation relies on a key alkoxy radical intermediate, which was postulated to form via single-electron oxidation of the arylcyclobutanol substrate (e.g., for 1-phenyl-1-cyclobutanol, Epox = +1.50 V vs Fc+/Fc in MeCN),450 followed by proton loss. Subsequent β-scission of the vicinal C–C bond leads to ring opening and generation of a C-centered radical, which undergoes intramolecular addition onto the aromatic ring to form a delocalized cyclohexadienyl radical. This intermediate, through further anodic oxidation and deprotonation, is then re-aromatized to furnish the desired tetralone product. Evolution of molecular hydrogen occurs concomitantly as a result of cathodic proton reduction.
3.1.2. Remote C–C Bond Formation through 1,5-HAT Processes
In addition to ring-opening reactions, recent progress has been made in the application of O–H bond PCET for remote C(sp3)–H bond functionalization via 1,5-HAT processes. In a report published in 2018, Zhu and co-workers disclosed a strategy for the heteroarylation of remote C(sp3)–H bonds in tertiary alcohol substrates, achieved through the intermediacy of transient alkoxy radicals (Scheme 154).452 In the presence of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 photocatalyst ([Ir-6]PF6), n-Bu4NCl additive, and stoichiometric K2S2O8 oxidant under blue-light irradiation, a variety of tertiary heteroaryl alcohols were transformed into the corresponding ketones bearing γ-C(sp3)–H bonds functionalized via migration of the heteroarene moiety. In this work, 31 examples of remote heteroarylation in yields of 31–90% were reported. With respect to the heteroarene component, benzothiazole (154.1), thiazole (154.2), and 2-pyridyl groups (154.3) proved to be amenable. Notably, electron-deficient aryl alcohols work well under the reaction conditions (154.4, 154.5), a striking departure from protocols that relied on an easily oxidizable aryl moiety to aid in the PCET activation step.27
Scheme 154. Photocatalytic Heteroarylation of Remote C(sp3)–H Bonds via Alkoxy Radical 1,5-HAT and Heteroaryl Migration (Zhu, 2018).

To elucidate the nature of alkoxy radical formation, luminescence quenching studies were carried out, and excited-state Ir(III)was shown to be oxidatively quenched (E1/2 Ir(IV)/*Ir(III) = −1.21 V vs SCE in MeCN)68 by K2S2O8, suggesting that Ir(IV) may play a role in the catalytic cycle. Moreover, the inability of the resulting Ir(IV) complex (E1/2 Ir(IV)/Ir(III) = +1.69 V vs SCE in MeCN)68 to engage in direct ET with the alcohol substrate (e.g., for alcohol substrate leading to product 154.1, Ep/2 = +2.06 V vs SCE in MeCN)452 led the authors to postulate that a ground-state PCET process involving Ir(IV) as the oxidant is operative. The identity of the base assisting this process was not determined. Following alkoxy radical generation, the electrophilic O-centered radical engages in 1,5-HAT with a remote C(sp3)–H bond to form a C-centered radical, and subsequent heteroaryl migration to the nascent alkyl radical furnishes the ketone product.
The Zhu group later leveraged a similar strategy for the activation and cyanation of remote C(sp3)–H bonds of cyanohydrins (Scheme 155).274 Under the same photocatalytic reaction conditions reported above, a number of aryl and aliphatic cyanohydrin substrates were successfully converted to δ-ketonitriles via alkoxy radical generation, 1,5-HAT, and intramolecular cyano group migration (155.1–155.4). The reaction tolerated a range of appended functionality and permitted the synthesis of α-cyanoethers (155.3). Two PCET mechanisms were invoked to explain alkoxy radical generation, one process comprising Ir(IV) as the oxidant and sulfate anion as the base and another involving a persulfate-mediated chain reaction. Based on the observed quantum yield of the reaction (Φ = 4.7) and SV analysis indicating that excited-state Ir(III) is readily oxidized by K2S2O8, the authors conclude that both mechanisms may be at play. In addition, direct abstraction of the alcoholic H-atom by a sulfate radical anion cannot be ruled out as a viable pathway.
Scheme 155. Photocatalytic Cyanation of Remote C(sp3)–H Bonds via Alkoxy Radical 1,5-HAT and Nitrile Migration (Zhu, 2019).

3.1.3. Intramolecular C–O Bond Formation via Olefin Hydroetherification
Advances in O–H bond PCET have enabled myriad alkoxy radical-mediated transformations, including, most recently, intramolecular C–O bond-forming reactions. Following the development and application of O–H PCET in the context of β-fragmentation and 1,5-HAT reactions, the Knowles group investigated the use of a dual Ir(III)/Brønsted base catalytic system for the intramolecular hydroetherification of unactivated alkenols (Scheme 156).453 In a 2020 report, the authors disclose a transformation in which activation of aliphatic alcohol O–H bonds is achieved through concerted PCET using the oxidizing Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 ([Ir-9]PF6) and n-Bu4P+(PhO)2P(O)O– base, resulting in an alkoxy radical intermediate that is leveraged for productive C–O bond formation. After the electrophilic alkoxy radical generated from PCET adds across a pendant olefin, the nascent C-centered radical is then quenched with an HAT co-catalyst to form closed-shell THF products.
Scheme 156. Catalytic Hydroetherification and Carboetherification of Unactivated Alkenes Enabled by PCET Activation of Alcohols (Knowles, 2020).

*With 2-methyl-2-oxazoline base catalyst and 4-(trifluoromethyl)thiophenol HAT co-catalyst.
This protocol allowed for the hydroetherification of substitutionally diverse olefins, including trisubstituted, disubstituted, styrenyl, and monosubstituted alkenes, and is compatible with functional groups such as alkyl halides, heterocycles (156.2), sulfonamides (156.5), and thioethers. Using this method, bridged (156.1), fused (156.3, 156.4), and spirocyclic (156.2) THF scaffolds are accessible from readily available alkenol starting materials. Remarkably, in addition to 5-exo-trig cyclizations, this strategy can achieve 6-endo-trig (156.3) and 6-exo-trig (156.6) ring closures with trisubstituted alkenes to afford tetrahydropyran products in high yields, with the latter outcompeting 1,5-HAT from comparatively weak allylic C–H bonds (e.g., for the allylic C(sp3)–H bond in propene, BDE = 89 kcal mol–1).25 Beyond hydroetherification, Knowles and co-workers extended the scope of this alkoxylation method to include olefin carboetherification reactions, wherein the C-centered radical generated following C–O bond formation is intercepted by an electron-deficient alkene, rather than by a H-atom donor, to furnish alkylated cyclic ethers, such as 156.7 in a reaction with dimethyl fumarate.
We note here a recent report from Jiao and co-workers describing a Ag-catalyzed method for the arylation of alcohol substrates proceeding through alkoxy radical generation and subsequent aryl group migration.454 A ground-state PCET mechanism is considered for O-centered radical generation, but full discussion however is outside of the scope of this Review.
3.2. Transformations of Readily Enolizable 1,3-Dicarbonyl Compounds Proceeding through the Enol/ate
Enolates generated through quantitative deprotonation of carbonyl substrates can react with stoichiometric amounts of single-electron oxidants to generate α-carbonyl radicals.455 With the development of photoredox catalysis, a number of methods have been reported for the catalytic generation of α-carbonyl radicals from readily enolizable 1,3-dicarbonyl compounds via coupled PT and ET. The reactivity of these radicals has been utilized in subsequent functionalization reactions, including α-oxyamination through C–O bond formation, alkylation through C–C bond formation for the preparation of linear adducts, and annulation through C–C bond formation. These transformations are presented in the following section.
3.2.1. C–O Bond Formation through α-Aminooxygenation
Early examples of photocatalytic activation of 1,3-dicarbonyl substrates entailed C–O bond formation in α-oxyamination processes with TEMPO radical. The first example of a photocatalytic method acting on enolizable 1,3-dicarbonyl compounds and proceeding through coupled PT and ET was reported by Tan and co-workers in 2010 (Scheme 157).456 These authors reported the photocatalytic α-oxidation of β-ketoesters upon visible-light irradiation of MeCN solutions of substrate containing RB as a photocatalyst and TEMPO as an oxidant. Under these conditions, 11 examples of the α-oxidation of β-ketoesters were reported, in yields of 64–97% (157.1–157.4). All substrates carried aryl ketones, and a preference for electron-donating substituents on this arene was observed in the reactions. A number of 2-fluoro-1,3-keto ester substrates underwent efficient oxidation in the same manner at faster rates (157.3, 157.4). In addition, a 1,3-diketone and a β-nitroketone were also compatible substrates in this work. The reaction works equally well when H2O is used as a solvent or when sunlight is used in place of fluorescent lamps. Notably, aliphatic β-keto esters were not capable substrates. Although mechanistic details were unclear, the authors reasoned that an enol radical is formed through deprotonation and subsequent photocatalyst-mediated oxidation of the resulting enolate. The more stabilized C-centered radical resonance form of the enol radical is then trapped by TEMPO radical. Although catalyst turnover was unclear, the authors noted that the reaction is insensitive to air.
Scheme 157. Photocatalytic α-Oxyamination of 1,3-Dicarbonyl Compounds Proceeding through Proposed TEMPO Radical Trapping (Tan, 2010).

In 2012, Koike, Yasu, and Akita also investigated the photocatalytic α-oxidation of 1,3-dicarbonyl compounds with TEMPO as the oxidant (Scheme 158).457 Optimal conditions consisted of visible-light irradiation of 1,3-dicarbonyl substrates in the presence of [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) photocatalyst and TEMPO in CH2Cl2 solution, with four examples demonstrated in 71–99% yields. In this method, dialkyl- (158.1) and diaryl-1,3-diketones (158.2), and alkyl (158.3) and aryl β-ketoesters (158.4) proved to be compatible substrates. However, a 1,3-diester substrate gave only trace amounts of the product. In contrast to the mechanistic proposal of Tan and co-workers, these authors reasoned that the reaction proceeds through photocatalytic disproportionation of TEMPO radical to the corresponding anion and oxoammonium cation. Whereas TEMPO anion is posited to act as a base for enolate generation, TEMPO oxoammonium cation is posited to mediate the two-electron α-oxidation of the resulting enolate. The trapping of enolates with TEMPO oxoammonium cation had been previously described by Schäfer.458 This proposal is supported by luminescence quenching experiments, which showed that TEMPO rather than the neutral dicarbonyl substrate quenched the Ir(III) photoexcited state. This reaction likely proceeds through reductive quenching of the photoexcited state of the Ir(III) complex (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 with TEMPO (E1/2ox = +0.84 V vs SCE in MeCN)277 to generate Ir(II) and TEMPO oxoammonium cation, since the alternative oxidative quenching mode (E1/2 Ir(IV)/*Ir(III) = −0.96 V vs SCE in MeCN)68 is not thermodynamically favorable (e.g., for TEMPO, E1/2red = −1.95 V vs Fc+/Fc in MeCN).22 The resulting Ir(II) complex (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN)68 reduces a second equivalent of TEMPO to the corresponding anion, which acts as a base for substrate deprotonation prior to polar α-oxyamination.
Scheme 158. Photocatalytic α-Oxyamination of 1,3-Dicarbonyl Compounds Proceeding through Proposed Catalytic Disproportionation of TEMPO (Koike, Yasu, and Akita, 2012).

In 2019, Huang, Wu, and co-workers reported the α-oxyamination of 1,3-dicarbonyl compounds with TEMPO using a heterogeneous TiO2 semiconducting photocatalyst (Scheme 159).459 The potential benefits of using a heterogeneous photocatalyst include the faster separation of charges upon visible-light irradiation to improve overall efficiency, and the recyclability of the dye when compared to homogeneous examples.460−462 This catalyst was prepared as a thin film surface on a conducting transparent support of fluorine-doped tin oxide (FTO) glass and investigated as a photocatalyst alone, or as the photoanode in a cell assembled in tandem with a Pt cathode. These researchers also studied the application of dye-sensitization of the TiO2 surface by the visible-light-absorbing dye Ru(4,4′-d(CO2H)bpy)2(NCS)2, in an effort to improve the overall efficiency. However, superior yields and improved recyclability of the material was realized in the absence of this Ru complex (e.g., for 159.1, 97% yield in the absence of Ru(II) compared to 63% yield in the presence of Ru(II)). The TiO2/FTO heterogeneous photocatalyst could be recycled for eight runs before significant deterioration in reaction performance was observed. Visible-light irradiation of MeCN solutions of 1,3-dicarbonyl and TEMPO admixtures using this photocatalyst proved optimal. The authors report 28 examples of this transformation in 32–97% yields (159.1–159.4). The reaction scope consisted predominantly of aryl β-ketoesters with varying arene substituents. The researchers noted that substituents had little impact on the reaction outcome, except in the case of ortho-substituted aryl ketones, which displayed poor performance. This observation was likely due to a lowering of the acidity of the methylene position through twisting of the arene out of conjugation with the ketone. In addition, 1,3-diketones and β-keto amide substrates were effective. The authors also provided heteroarene examples and explored α-oxyamination using a number of 4-substituted TEMPO derivatives.
Scheme 159. Heterogeneous Photocatalysis of the α-Oxyamination of 1,3-Dicarbonyls (Huang and Wu, 2019).

A series of experiments to probe the mechanism of this transformation were performed. A competition kinetic isotope experiment (KIE) was performed on ethyl benzyloxyacetate and its di-deutero isotopologue, and the results were taken as evidence that cleavage of a methylene C(sp3)–H bond was likely rate-determining (kH/kD = 8.7). Upon addition of the radical scavenger BHT, the researchers observed the adduct of ethyl benzyloxyacetate and BHT by high-resolution mass spectrometry (HRMS). This result suggested the presence of an α-carbonyl radical. However, product was still observed, albeit in reduced yield, suggesting that reaction is also permitted through an ionic pathway either exclusively or in addition to the radical pathway. UV/vis spectroscopy was performed to understand why the reaction proceeded under visible-light irradiation without dye sensitization. TiO2 nanoparticles (NPs) were found to absorb at 380 nm. However, the TiO2/FTO composite material displayed a large bathochromic shift in its absorption at features up to 550 nm. A further bathochromic shift was also observed on introduction of ethyl benzyloxyacetate or TEMPO to the solution. These features thus permit visible-light activation in the absence of the Ru(II) dye. Based on these results, it was proposed that the reaction commences with visible-light irradiation of the TiO2/FTO photocatalyst and excitation of an electron from the material valence band (VB) to the conduction band (CB). The electron in the CB is proposed to reduce TEMPO to its corresponding anion (Epred = −1.95 V vs Fc+/Fc in MeCN).22 This anion is then proposed to act as a base for enolate generation. The hole resting on the VB is proposed to oxidize both TEMPO to the corresponding oxoammonium cation (Epox = +0.68 V vs Ag/AgCl in MeCN),459 and the enolate to the corresponding radical. Both radical recombination of the α-acyl radical with TEMPO and an ionic pathway between TEMPO oxoammonium cation and the anionic enolate are proposed to generate the oxyamination product.
3.2.2. C–C Bond Formation in the Synthesis of Linear Adducts
In addition to promoting C–O bond formation through this reaction manifold, a number of photocatalytic and electrochemical methods for C2-C–C bond formation in 1,3-dicarbonyl and related substrates have been presented. We study these reactions in this section, beginning with those leading to the linear assembly of coupling products before moving on to methods for annulation.
In 2014, Fensterbank, Goddard, and Ollivier reported a number of C2-C–C bond-forming reactions of enolizable 1,3-dicarbonyl compounds proceeding through the intermediacy of the corresponding C2-radical generated from discrete PT and subsequent ET steps (Scheme 160).463 Three classes of transformation were defined in this work: (i) the dimerization of β-ketoesters at the C2 position, (ii) the intermolecular C2-allylation of β-ketoesters with allyl stannane and allyl sulfone reagents, and (iii) the annulation of olefin-tethered 1,3-diketones. The reported reaction conditions consisted of visible-light irradiation of DMF substrate solutions open to air in the presence of [Ru(bpy)3]Cl2 ([Ru-1]Cl2) photocatalyst, K2CO3 Brønsted base, and Ph3C+BF4– as a stoichiometric oxidant. Here, six examples of dimerization of β-keto esters were reported with 43–99% yields, including variously substituted electron-rich (160.1, 160.2) and electron-deficient (160.3) aryl substituents. With introduction of allyl stannane or allyl sulfone reagents (10.0 equiv,), six C2-allylation products (160.4–160.8) were reported with yields ranging from 28% to 72%. When an olefin-tethered 1,3-diketone substrate was studied, a sequence of radical generation, 6-exo-trig cyclization, and further arene annulation was observed (160.9, 160.10, 31% and 29% yields, respectively).
Scheme 160. Photocatalytic C–C Bond Formation from 1,3-Dicarbonyl Compounds (Ollivier, 2014).

The authors propose that visible-light irradiation of the photocatalyst and subsequent oxidative quenching (E1/2 Ru(III)/*Ru(II) = −0.81 V vs SCE in MeCN)64 with either molecular oxygen or Ph3C+BF4– (Ep/2ox = +0.54 vs NHE in sulfolane),464 leads to the generation of an oxidizing Ru(III) complex (E1/2 Ru(III)/Ru(II) = +1.26 V vs SCE in MeCN),64 which facilitates oxidation of the enolate formed through carbonate-mediated deprotonation of the 1,3-dicarbonyl substrate. Radical dimerization or addition to olefin results in C–C bond formation.
In 2020, Xia and co-workers reported a three-component coupling reaction of 1,3-dicarbonyl compounds, enol ethers, and isoquinolines to afford overall olefin 1,2-alkyl heteroarylation products (Scheme 161).465 Optimal reaction conditions involved blue-light irradiation of the aforementioned reaction components in i-PrOH solution in the presence of [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) photocatalyst and MsOH Brønsted acid. Under these conditions, 43 examples of this olefin difunctionalization were reported with 36–86% yields. Variation of the 1,3-dicarbonyl (161.1, 161.2), enol ether (161.3), and isoquinolone components was tolerated, including cyclic ketone, phenanthridine, and 2,3-dihydrofuran-derived, respectively. Additionally, vinyl amides also served as competent radical acceptors providing 161.4 in 77% yield.
Scheme 161. Three-Component Coupling of 1,3-Dicarbonyl Compounds, Electron-Rich Olefins, and Heteroarenes (Xia, 2020).

The authors propose that an Ir(IV) species is generated via initial oxidative quenching of the photoexcited Ir(III) catalyst (E1/2 Ir(IV)/*Ir(III) = −0.96 V vs SCE in MeCN)68 with molecular oxygen. The Ir(IV) state of the photocatalyst (E1/2 Ir(IV)/Ir(III) = +1.21 V vs SCE in MeCN)68 then oxidizes the neutral electron-rich enol form of the 1,3-dicarbonyl compound (e.g., for ethyl benzoylacetate, Ep/2ox = +1.15 V vs SCE in MeCN).465 This oxidation occurs preferentially over the oxidation of the electron-rich enol ether reaction component (e.g., for 2,3-dihydropyran, Ep/2ox = +1.51 V vs SCE in MeCN).21 This proposal of oxidative photocatalyst quenching for reaction initiation is supported by the fact that the photoexcited Ir(III) complex is not sufficiently oxidizing to drive this ET through an alternative reductive quenching pathway (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN).68 Subsequent deprotonation of the resulting enol radical cation leads to an electrophilic C-centered 1,3-dicarbonyl radical, which engages the enol ether or enamide in a C–C bond-forming step to generate a nucleophilic α-heteroatom radical. Addition of this nucleophilic radical to a protonated, electron-deficient heteroarene in Minisci fashion is followed by deprotonation and subsequent re-aromatization under aerobic conditions to generate the three-component coupled product. The selectivity in three-component coupling over direct addition of the enol-derived radical to the protonated heteroarene is rationalized through polarity matching of the reactive intermediates.
In 2020, Yamashita, Kobayashi, and Baś reported a C(sp3)–H alkylation reaction between malonate diesters and styrenes via generation and addition of a 1,3-dicarbonyl radical across the alkene acceptor (Scheme 162).466 Optimal conditions consisted of photocatalyst 4CzIPN, or the related 3DPA2FBN, and KOt-Bu Brønsted base co-catalyst in MeCN solvent under blue-light irradiation. The authors reported 33 examples of this transformation with yields ranging from 17% to 99%. 4CzIPN was a competent photocatalyst for the coupling of malonates with 1,1-diarylethylene derivatives (162.1, 162.2) and for styrenes bearing EWGs. However, 3DPA2FBN proved suitable for malonate diester coupling with more challenging styrene partners (162.3, 162.4). Finally, investigation of the malonate scope revealed that substitution on the esters was well tolerated, but alkyl substitution at the C2-position resulted in complete loss of reactivity.
Scheme 162. Photocatalytic Alkylation of Malonate Diesters with Styrenes (Kobayashi and Yamashita, 2020).

In the proposed catalytic cycle, malonate is deprotonated by the alkoxide catalyst, providing the corresponding enolate. The enolate (e.g., for diethyl malonate, Ep/2ox = +0.08 V vs Fc+/Fc in MeCN)467 is then oxidized by the photoexcited state of the catalyst (e.g., for 4CzIPN, E1/2 *PC/PC•– = +1.43 V vs SCE in MeCN; for 3DPA2FBN, E1/2 *PC/PC•– = +0.92 V vs SCE in MeCN)77,78 to the corresponding C-centered radical. Oxidation of this reaction component occurs in preference to the styrene component (e.g., for 1,1-diphenylethylene, Ep/2ox = +1.82 V vs SCE in MeCN).468 This radical intermediate adds across the styrene partner, generating a stabilized benzylic radical which undergoes single-electron reduction mediated by the reduced state of the photocatalyst (e.g., for 4CzIPN, E1/2 PC/PC•– = −1.24 V vs SCE in MeCN; for 3DPA2FBN, E1/2 *PC/PC•– = −1.92 V vs SCE in MeCN).77,78 The more reducing nature of the radical anion of 3DPA2FBN compared to 4CzIPN accounts for the observed photocatalyst preference for electron-rich styrene partners, where carbanion formation would be more thermodynamically challenging. The authors suggest this ET step for photocatalyst regeneration is rate-determining. The resulting benzylic carbanion deprotonates another 1,3-dicarbonyl substrate, thereby generating another enolate that can engage in the catalytic cycle. For this reason, only a catalytic amount of alkoxide base additive was required.
3.2.3. C–C Bond Formation in Annulation Reactions
In 2017, Xu and co-workers developed electrocatalytic annulation methods to produce oxindoles from 1,3-amido esters via intramolecular arene C(sp2)–H alkylation. The researchers first reported the dehydrogenative cyclization of 2-fluoro-1,3-amido esters upon constant current electrolysis of substrate solutions in an undivided electrochemical cell equipped with an RVC anode and Pt cathode in THF/MeOH (2:1) (Scheme 163).469 These reactions proceeded at 0 or −30 °C and contained Fc as a redox mediator, LiCp or LiOMe as Brønsted base, and n-Bu4N+PF6– as supporting electrolyte. Under these conditions, C3-fluorinated oxindoles bearing diverse functional groups (e.g., halogens, alcohols, alkynes, amino ester, silyl ethers, esters, pyrrole, carbazole, N-phenyl carbamate) could be generated in moderate to excellent yields (163.1–163.5) (33 examples, 38–91%). Interestingly, the reaction tolerated amide N-allyl (163.6) and N-propargyl substitution (163.7) as radical cyclization onto these moieties was not observed. Cyclization was demonstrated onto the C2-position of tethered pyridines for the synthesis of aza-oxindoles (163.8). These conditions were notably unsuccessful in promoting the cyclization of non-fluorinated substrates, which are significantly less acidic.
Scheme 163. Electrochemical C(sp2)–H Annulation of 2-Fluoro-1,3-amido Esters for the Synthesis of Oxindoles (Xu, 2017).

A proposed mechanism involves deprotonation of the fluoromalonate substrate by the additive or electrogenerated base prior to SET mediated by anodically-generated ferrocenium. CV studies supported this stepwise proposal, showing that the neutral fluoromalonate substrate (e.g., for the substrate leading to 163.1, Ep/2ox = +1.93 V vs SCE in THF/MeOH (2:1))469 was unable to undergo favorable ET with ferrocenium (E1/2ox = +0.52 V vs SCE in THF/MeOH (2:1)).469 CV titration studies with LiCp or LiOMe suggested that deprotonation is rate-limiting and followed by rapid ET. Following C-centered radical generation in this manner, cyclization onto the N-aryl group and further oxidation with proton loss leads to formation of the closed-shell product.
Seeking to address the prior limitation of this method and extend it to non-fluorinated substrates, these researchers in 2018 showed that running this electrolysis reaction at reflux temperature and adding Yb(OTf)3 as a LA additive enabled the preparation of both oxindoles and quinoxalinones derived from non-fluorinated 1,3-dicarbonyl compounds through five- and six-membered-ring cyclization (Scheme 164, 164.1–164.7).470 A single example of this cyclization onto a pyrrole was also included, providing a product which is an intermediate (164.8) to the anti-inflammatory drug ketorolac. This work included 35 examples of product formation in yields of 35–96%. Higher reaction temperatures and LA activation of the substrate dicarbonyl were required to facilitate the initial deprotonation step in the dehydrogenative cyclization. Otherwise, mechanistic considerations overlay with the author’s earlier proposal.
Scheme 164. Electrochemical C(sp2)–H Annulation for the Synthesis of Oxindoles, Quinoxalinones, and a Bicyclo-Fused Pyrrole (Xu, 2018).

Xu and co-workers subsequently developed electrocatalytic methods for dehydrogenative [4+1] and [4+2] annulation reactions of N-allyl amides with 1,3-dicarbonyl compounds (Scheme 165).471 A variety of N-heterocycles were produced (46 examples, 15–89%) upon constant current electrolysis of substrate solutions (t-BuOMe/MeCN/H2O (20:3:1)) in an undivided electrochemical cell with a RVC anode, Pt cathode, sodium formate Brønsted base, and an N-aryl-PTZ redox catalyst (165.A). The type of 1,3-dicarbonyl coupling partner employed determined the type of N-heterocycle that was produced; whereas the use of malonate diesters resulted in the formation of pyrrolidines through a [4+1] annulation reaction (165.1–165.6), the use of β-ketoesters resulted in the selective formation of tetrahydropyridines through a [4+2] pathway instead (165.7–165.13). The reaction proved tolerant to a variety of N-allyl amide substrates; electron-rich and electron-poor phenyl groups, as well as heteroaromatic groups (e.g., thienyl, furanyl), were well tolerated as alkene substituents. Diene and enyne substrates also underwent successful reaction. Similarly, a number of N-aryl groups of the amide moiety were demonstrated. Replacing the aryl group with an alkyl group at both positions, however, led to a drastic decrease in yield.
Scheme 165. Electrochemical [4+1] and [4+2] Annulation of N-Allyl Amides with 1,3-Dicarbonyl Compounds to Access (A) Pyrrolidine and (B) Tetrahydropyridine Derivatives (Xu, 2018).

Notably, N-aryl PTZ 165.A played a crucial role in this electrochemical transformation, as no product is formed in its absence (Scheme 166). The researchers posited that PTZ is first oxidized to the corresponding radical cation (Ep/2ox = +0.52 V vs SCE in t-BuOMe/MeCN/H2O (25:5:1))471 at the anode as water is reduced to molecular hydrogen and hydroxide at the cathode. Hydroxide-mediated deprotonation of the 1,3-dicarbonyl compound yields an enolate anion that is rapidly oxidized by the PTZ radical cation. Notably, the oxidation potential of the enol is significantly beyond that of this redox mediator (e.g., for dimethyl malonate, Ep/2ox > +2.0 V vs SCE in t-BuOMe/MeCN/H2O (25:5:1));471 however, on ionization, the oxidation potential is negatively shifted so that ET is favorable (e.g., for dimethyl malonate in the presence of 1.0 equiv of n-Bu4N+OH–, Ep/2 = +0.31 V vs SCE in t-BuOMe/MeCN/H2O (25:5:1)). Oxidation of the malonate anion generates a C-centered radical, which adds to the alkenyl moiety of the N-allyl amide in an anti-Markovnikov fashion to generate a tertiary C-centered radical. This intermediate is subsequently oxidized by 165.A+• to the corresponding carbocation, which is intramolecularly trapped by the carbonyl moiety of the N-acyl group to form an oxocarbenium ion intermediate. This oxocarbenium ion is hydrolyzed to promote an overall N- to C-acetate migration. In the absence of the aqueous reaction co-solvent, this linear adduct was isolated and characterized, supporting this pathway. Now in the case of 1,3-diester substrates leading to pyrrolidine formation, C–N bond formation occurs through additional proton- and mediated ET steps for overall [4+1] annulation. In the case of β-keto ester substrates, enamine formation occurs to yield the tetrahydropyridine product through [4+2] annulation. Concomitant reduction of liberated protons leads to the co-generation of molecular hydrogen.
Scheme 166. Proposed Mechanism of Electrochemical [4+1] and [4+2] Annulation (Xu, 2018).

Lei and co-workers were able to generate substituted dihydrofurans through an electrooxidative [3+2] annulation reaction between 1,3-dicarbonyl compounds and a variety of alkene partners (Scheme 167).472 Dihydrofurans were formed in moderate to excellent yields (26 examples, 32–95%) upon constant current electrolysis of an MeCN/HFIP (3:1) solution containing NaOAc Brønsted base catalyst and using a graphite anode. Both 1,3-diketones (167.1–167.5) and β-ketoesters (167.6) were showcased. Reaction yields tended to decrease as the steric bulk of the dicarbonyl increased (e.g., methyl (167.1) vs isopropyl (167.3) ketones). Styrenes bearing electron-donating or electron-withdrawing ortho- or para-substituents were well tolerated, as were cyclic styrenes, α- and β-methylsytrene, and vinylnaphthalene. Conjugated dienes (167.2) and vinyl ethers (167.5) also gave good yields.
Scheme 167. Electrochemical [3+2] Annulation of 1,3-Dicarbonyl Compounds and Alkenes to Yield 2,3-Dihydrofurans (Lei, 2019).

*Using 2.0 equiv of NaOt-Bu instead of NaOAc as base. **Constant current of 5 mA. ***Using 1.0 equiv of K2CO3 instead of NaOAc as base.
Interestingly, no dihydrofuran product was formed in the absence of NaOAc and lower yields were reported when it was replaced with a different base. The researchers posited that either NaOAc (pKaH = 12.3 in DMSO)39 or HFIP (pKaH = 10.7 in DMSO)116 anion generated at the cathode deprotonates the 1,3-dicarbonyl compound (e.g., for acetyl acetone, pKa = 13.3 in DMSO)473 to form the corresponding enolate. CV studies suggest that the enolate is subsequently oxidized to the radical as the oxidation potential is significantly lower than that of the neutral molecule (e.g., for sodium acetyl acetonate, Ep/2ox = +1.14 V vs Ag/AgCl in MeCN; for acetyl acetone +2.61 V vs Ag/AgCl in MeCN).472 Intermolecular radical addition to the olefin partner and subsequent anodic oxidation yields a carbocation. The authors suggest that HFIP reacts with this carbocation to yield the corresponding adduct, which then undergoes ring closure in a subsequent operation through substitution. No product was observed when HFIP was replaced with MeOH or H2O. Notably, however, product formation was observed when DMSO rather than HFIP was used as a co-solvent, suggesting this too has specific stabilizing ability through reversible trapping of the cationic intermediate. Cathodic reduction of AcOH or HFIP with liberation of molecular hydrogen regenerates the requisite Brønsted base.
Park and co-workers were recently able to generate highly substituted dihydropyrano[4,3-b]indoles from indoles and 1,3-dicarbonyl compounds (Scheme 168A, 168.1–168.4), as well as 3-amino-2,3-dihydrofurans from β-amino acrylates and 1,3-dicarbonyl compounds (Scheme 168B, 168.5–168.8), through a tandem process wherein an electrochemically mediated radical–radical cross-coupling reaction preceded a 6π-electrocyclization reaction.474 Dihydropyrano[4,3-b]indoles were synthesized in good yields (33 examples, 40–77%, yields, e.g., 168.1–168.4) upon the constant potential electrolysis of a MeCN solution of indole and 1,3-dicarbonyl compound in the presence of NaI additive using a graphite anode and RVC cathode exposed to air. A broad range of 1,3-dicarbonyl compounds and related coupling partners were suitable—1,3-diketones, and β-keto esters, amides, nitriles, sulfones, phosphonates, etc. Interestingly, unsymmetrical 1,3-diketones reacted with complete regioselectivity. The researchers noted that unsubstituted and N-substituted indoles with a variety of functional groups on the aryl ring (e.g., halides, ether, Boc-protected amines, nitro, cyano) were also well tolerated. Conditions promoting the synthesis of 3-amino-2,3-dihydrofurans involved constant current rather than constant potential electrolysis of substrate solutions and yielded products as single diastereomers. Although a variety of 1,3-dicarbonyl compounds were well tolerated, the cross-coupling of electron-rich β-amino-acrylates resulted in low yields. Here, 10 examples were reported in 29–84% yields (e.g., 168.5–168.8).
Scheme 168. Electrochemical Synthesis of Dihydropyrano[4,3-b]indoles and 2,3-Dihydrofurans (Park, 2020).

*Applied potential = 3.5 V. **2.0–3.0 equiv of active methylene compound used.
The researchers noted that neither the dihydropyrano[4,3-b]indole or 2,3-dihydrofuran product was observed in the absence of NaI, suggesting that iodine serves as a redox mediator. Further mechanistic studies suggested that iodine radical-mediated HAT is likely involved in radical generation in the indole or β-amino acrylate reaction partner. NaI was observed to undergo oxidation (E1/2ox = +0.68 V vs Ag/AgCl in MeCN)474 preferentially over the indole substrate (e.g., for dimethyl 2-(N-methylindolyl)malonate leading to product 168.3 and 168.4, Ep/2ox = +1.17 V vs Ag/AgCl in MeCN)474 in CV experiments, supporting this proposal. This observation is distinct from those of Lei and co-workers, who in related work proposed that indole substrates undergo anodic oxidation in the absence of an iodide redox mediator to yield the corresponding radical cation and subsequent C–O, C–N, or C–C bond formation occurs through radical–radical cation coupling. Additionally, electrolysis performed under inert atmosphere rather than air only yielded an indole or enamine dimer, suggesting that the oxygen in air plays a critical role in activating the dicarbonyl component. The researchers posited that superoxide formed through cathodic reduction of oxygen serves as a base to deprotonate this reaction partner and form the corresponding enolate, which subsequently undergoes anodic oxidation to form a C-centered radical. Radical–radical C2-cross-coupling between the indole/β-amino acrylate radical with the radical species derived from the dicarbonyl compound, followed by either 6π-electrocyclization furnishes the dihydropyrano[4,3-b] indole product, or alternatively oxa-Michael addition leads to the 3-amino-2,3-dihydrofuran product.
In 2020, Wu and co-workers reported a dehydrogenative benzannulation reaction of ortho-biaryl-appended 1,3-dicarbonyl compounds facilitated by a dual organophotocatalytic/Co(III) catalytic system for the synthesis of 10-phenanthrenols (Scheme 169).475 This net-oxidative reaction proceeded with N-Me Mes-Acr+ClO4– ([Acr-1]ClO4) photocatalyst and Co(dmgH)(dmgH2)Cl2 hydrogen evolution catalyst in MeCN under blue-light irradiation. In total, 33 examples of this benzannulation reaction were reported in 31–94% yields. A variety of substituents on the 2-biphenyl were tolerated, including alkyl and fluorinated (169.2, 169.4) derivatives. Electron-donating substituents such as −OMe were also found to undergo benzannulation, albeit with lower efficiencies (169.3, 169.5). Additionally, the synthesis of larger aromatic systems was demonstrated to proceed efficiently under this protocol.
Scheme 169. Dual Photoredox/Co(III) Catalysis for the Synthesis of 10-Phenanthenols via C(sp3)–H Alkylation (Wu, 2020).

In the proposed catalytic cycle, the Acr photocatalyst is excited by irradiation with visible light. The excited-state photocatalyst (E1/2 *1Acr/Acr• = +2.08 V vs SCE in MeCN; E1/2 *3Acr/Acr• = +1.88 V vs SCE in PhCN)73,74 is reductively quenched by the enol tautomer of the substrate, affording the corresponding enol radical cation and neutral acridine radical. In 1H NMR studies, authors show that the substrate adopts a low equilibrium concentration (keto/enol = 91:9) of this tautomer in solution to facilitate neutral substrate oxidation. In CV experiments with the substrate leading to 169.1, two oxidation events are observed and assigned to single-electron oxidation of the enol tautomer (Epox = +1.79 V vs SCE in MeCN)475 occurring at a lower applied potential prior to oxidation of the keto form (Epox = +2.18 V vs SCE in MeCN).475 The identity of the latter oxidation event was confirmed in an experiment where an α,α-dimethylmalonate ester substrate, which is unable to adopt the enol form, was seen to now exhibit only one oxidation overlaying with the assigned keto form of the lead substrate. Proton loss then leads to generation of an electrophilic C-centered radical which cyclizes onto the biaryl. The Co(III) catalyst undergoes ET with the acridine radical state of the photocatalyst to yield a Co(II) complex and return the ground-state Acr dye for further turnover. Product re-aromatization is facilitated by ET with subsequent PT to Co(II), forming a Co(I) complex. Finally, protonation of the Co(I) complex leads to a Co(III) hydride, which subsequently evolves molecular hydrogen and restores the Co(III) complex for further reaction. The authors noted product inhibition of further catalytic turnover through competitive quenching of the photoexcited-state Acr species (e.g., for 10-phenanthrenol 169.1, Ep/2ox = +1.43 V vs SCE in MeCN), which were observed in reaction time-course studies.
In 2020, Mei and co-workers reported the electrochemical dehydrogenative synthesis of 10-phenanthrenols from ortho-biaryl-appended 1,3-dicarbonyl compounds (Scheme 170).476 Electrolysis of these substrates in MeOH under constant current conditions in an undivided cell equipped with an RVC anode, Pt cathode, K2CO3 Brønsted base, and n-Bu4N+PF6– supporting electrolyte afforded 19 examples of these 10-phenanthrenol products in yields ranging from 41% to 90%. A variety of substitution patterns on the biaryl were tolerated, affording extended arene product 170.1 and thioether containing 170.3. Although β-ketoester (170.1–170.5) and β-ketonitriles (170.6) were competent substrates, a substrate bearing a C2-fluorine substituent afforded no product. This observation contrasts with the earlier work of Xu who demonstrated efficient radical generation and arene cyclization with similar fluoromalonate substrates for oxindole synthesis.469 Typically, transesterification of ester substrates was observed.
Scheme 170. Electrochemical Synthesis of 10-Phenanthrenols via C(sp2)–H Alkylation (Mei, 2020).

CV experiments indicated that the onset potential for oxidation of a model substrate is negatively shifted by −0.77 V in the presence of K2CO3 (e.g., for the β-ketoester substrate leading to 170.2, Eonsetox = +1.73 V vs SCE in MeOH; Eonsetox = +0.41 V vs SCE in MeOH in the presence of K2CO3).476 Experiments with the introduction of TEMPO or BHT radical scavengers led to significantly lower product yields. A modest KIE (kH/kD = 1.5) was observed in parallel experiments investigating the role of arene C(sp2)–H bond cleavage in the reaction mechanism. The authors proposed a mechanism wherein the substrate is deprotonated by K2CO3 or electrogenerated methoxide base to form the enolate, which then undergoes single-electron oxidation at the anode. The resulting C-centered radical cyclizes onto the pendant arene forming a new C–C bond. Subsequent anodic oxidation, deprotonation, and tautomerization affords the product phenanthrenol.
In 2020, Wu and co-workers described a catalytic system containing an aryl β-keto ester substrate and Ir(ppy)3 (Ir-1) in DMF under blue-light irradiation that enables the co-generation of both neutral ketyl and C2 C-centered radicals from the same substrate through reductive and oxidative PCET processes, respectively (Scheme 171A).477 Supporting the proposed generation of both of these intermediates, irradiation of β-keto ester substrate, Ir(III) photocatalyst, and 1,1-diphenylethylene in DMF afforded product 171.1 in 43% yield. In the formation of this product, the transient C-centered 1,3-dicarbonyl radical undergoes addition across the alkene component to generate a stabilized doubly benzylic C-centered radical. This intermediate then undergoes radical–radical coupling with a persistent ketyl radical to form a C–C bond. Instead, when the reaction was conducted in the presence of an alkyne partner, reaction of the β-keto ester under similar conditions afforded a mixture of naphthol 171.2 and pinacol coupling product 171.3 in a 1:1 ratio (Scheme 171B). The authors then explored the scope of this reaction with a variety of aromatic 1,3-dicarbonyl substrates and alkynes, demonstrating 51 examples of 1-naphthol synthesis in this manner in 11–88% yields (Scheme 171C). The reaction tolerated a variety of electron-donating (171.4) and -withdrawing substituents (171.5) on the aryl alkyne reaction partner. However, chloride (171.6) and bromide were not tolerated due to dehalogenation under these conditions. Other variation in the alkyne was tolerated including alkyl-substituted terminal alkynes (171.7) and an example of an internal alkyne in 32% yield (171.8). In addition to ester substrates, other 1,3-dicarbonyl substrates reacted efficiently, including those bearing amide (171.9), methyl (171.10), and phenyl ketone (171.11) functionality.
Scheme 171. Photocatalytic Generation of Both Neutral Ketyl Radicals and C2-C-Centered Radicals from Aryl β-Keto Esters (Wu, 2020).

The authors conducted a number of experiments to probe the mechanism of this reaction. Control experiments revealed that a trace amount of dimethylamine contaminant in DMF promoted this reaction, acting as a proton shuttle to render SET steps for radical generation thermodynamically feasible. In the absence of dimethylamine, the excited-state Ir(III) photocatalyst cannot facilitate either oxidative (E1/2 Ir(IV)/*Ir(III) = −1.73 V vs SCE in MeCN)66 or reductive (E1/2 *Ir(III)/Ir(II) = +0.31 V vs SCE in MeCN)66 quenching with the neutral β-keto ester substrate (Ep/2red < −1.90 V vs SCE in DMF; Ep/2ox > +1.90 V vs SCE in DMF).477 However, inclusion of 5 mol% of dimethylamine in CV experiments revealed a significant shift in the reduction and oxidation potentials of the substrate (Ep/2red = −1.05 V vs SCE in MeCN; Ep/2ox = +0.32 V vs SCE in MeCN),477 thereby favoring ET. The authors propose this shift is due to full or partial PT from the enol tautomer to the amine to generate a more easily oxidizable species, generating a 1,3-dicarbonyl radical after SET. The protonated form of dimethylamine then acts as an acid to activate the keto tautomer of another molecule of substrate for reduction. The neutral ketyl radical formed upon reduction can engage with C2-carbon-centered radicals or olefin or alkyne substrates. In the latter case, annulation occurs through cyclization of the nascent radical onto the arene ring, forming the naphthol products.
3.3. Transformations of Phenols
3.3.1. Electrochemical Phenol–Phenol Homo- and Cross-Coupling
To the best of our knowledge, the first recognition of the electrochemical generation of a phenoxy radical via the one-electron anodic oxidation of the parent phenol was made by Hedenburg and Freiser in 1953 in work aiming to develop a quantitative method for phenol analysis.478 In voltammetric studies, these researchers observed a strong pH dependence on the half-wave oxidation potential of the phenol substrate, with this value decreasing rapidly as pH increased between 5 and 9 (e.g., for phenol at pH = 5, E1/2ox = +0.76 V vs SCE in H2O; at pH = 7, E1/2ox = +0.60 V vs SCE in H2O; at pH = 9, E1/2ox = +0.52 V vs SCE in H2O),478 above which point the observed oxidation potential was independent of solution pH. At this juncture, the reversible ET was shown to be a one-electron process. The authors inferred that this observation suggested that a stepwise mechanism for substrate oxidation was operative at high pH that involved initial deprotonation of the substrate to yield the corresponding phenoxide anion, which then undergoes single-electron oxidation. During concurrent studies, Gaylor and Elving suggested that a different mechanism for ET in neutral phenol substrates was operative in a low pH regime.479 They showed that at pH = 1.2, a four-electron process occurred and that this transitioned to a two-electron process at pH = 5.2. These processes were recognized as the stepwise oxidation of phenol to hydroquinone (HQ) and then to quinone. However, it was unclear at this stage the nature of the reactive intermediate formed upon two-electron anodic oxidation.
A later systematic study from Vermillion Jr. and Pearl sought to reconcile these potentially conflicting results and showed that they were equally correct within their limiting pH regimes (Scheme 172).480 This seminal publication was the first to recognize and catalogue the duality of mechanistic possibilities resulting from the anodic oxidation of phenols with or without prior substrate ionization. This work also demonstrated the first signs of the synthetic relevance of these electrogenerated intermediates. Voltammetric studies of 2,6-di-tert-butyl-p-cresol in MeCN were performed to probe the nature of the mechanism occurring without prior substrate ionization to study the proposal of Gaylor and Elving. These researchers observed the same irreversible two-electron anodic oxidation of this neutral substrate (Ep/2ox = +1.21 V vs SCE in MeCN)480 and the generation of a phenonium ion intermediate through formal heterolysis of the O–H bond. A variety of nucleophilic trapping products of this reactive intermediate were observed. For example, on introduction of methanol to this electrolysis, 2,6-di-tert-butyl-4-methyl-4-methoxy-cyclohexanedione (172.1) was the major product observed in 65% yield. This work also recognized the further formation of a p-quinone methide resulting from benzylic deprotonation of the phenonium intermediate. Conversely, oxidation of phenoxide anion (e.g., for vanillin, E1/2ox = +1.43 V vs SCE in MeCN; for vanillin in the presence of 1.0 equiv of Et4N+OH–, E1/2ox = +0.22 V vs SCE in MeCN; vanillin pKa = 7.4 in H2O)480,481 showed the same reversible one-electron behavior as that observed by Hedenburg and Freiser and agreed with their proposal for phenoxy radical generation via stepwise formal O–H bond homolysis. The authors note that the anion of 2,6-di-tert-butyl-p-cresol decomposed in MeCN solution, necessitating the change to a different substrate. These researchers recorded the formation of 2,2′-divanillin (172.2) as the major product of the electrolysis of vanillin in 62% yield, formed through a dehydrogenative ortho,ortho-homo-coupling.
Scheme 172. Systematic Study of the Nature of the Mechanism of Phenol Oxidation under Neutral and Basic Conditions (Vermillion Jr. and Pearl, 1964).

These important early disclosures served to define the types of mechanisms that could be promoted electrochemically as well as the ability to generate either polar or radical intermediates in different pH regimes, leading to either monomeric nucleophilic trapping products or to dimeric C–C coupling products, respectively. These primary modes of electrochemical activation still define the types of synthetic electrochemical transformations of phenols to this day, with single-electron reactivity typically occurring from ionized phenols. In this section, we discuss examples of electrochemical phenoxy radical generation through coupled PT and ET and the subsequent reactivity of these intermediates. We do not include those electrochemical and photocatalytic examples for the generation of phenonium ion intermediates.
3.3.1.1. Ortho,ortho-Homo- and Cross-Coupling for 2,2′-Biphenol Synthesis
Soon after these foundational reports, a series of publications from Bobbitt and co-workers established the synthetic utility of these methods in the synthesis of isoquinoline alkaloids.482−486 A 1969 communication reported their studies on the electrolytic oxidation of the simple phenol-containing tetrahydroisoquinoline alkaloid corypalline (173.1) (Scheme 173). Electrolysis of corypalline in aqueous solution was carried out in a divided cell operating under constant potential with Pt electrodes and Na2B4O7 acting as a supporting electrolyte. Under these reaction conditions, the group isolated the symmetrical 2,2′-biphenol resulting from C–C bond formation as the major product in 55% yield (173.1a) and a C–O coupled dimer as a minor component in up to 5% yield (173.1b). Notably, the tertiary amine moiety did not interfere with the electrolysis. Later studies from Miller and Stewart showed that in this pH regime, the tertiary amine of these types of alkaloids is protonated and thus not susceptible to anodic oxidation, permitting a selective and stepwise mechanism of PT and subsequent anodic oxidation of the phenol moiety leading to phenoxy radical generation.487 Additionally, these reaction conditions did not enable the dimerization of electron-rich non-phenolic arenes, revealing the importance of substrate deprotonation prior to oxidation. Details regarding the nature of the C–C bond-forming step following radical generation were unclear at the time.
Scheme 173. An Early Example of the Homo-Coupling of a Phenol via Electrochemical Phenoxyl Radical Generation.

Regioselectivity of bond formation is dictated by steric control in the nature of the C1-substituent (Bobbitt, 1969 and 1970).
Bobbitt and co-workers then investigated the effect of C1-substituents of isoquinoline on the selectivity of the electrolytic reaction for C–C vs C–O coupling. This group had previously demonstrated that the chemical oxidant K3Fe(CN)6 mediated the oxidative dimerization and trimerization of the alkaloid lophocerine—a 1-sec-butyl tetrahydroisoquinoline.488 This reactivity occurred via C–O bond coupling, which was the minor isomer in the electrolysis of corypalline (173.1b). The trimer arising from this process is the cactus alkaloid pilocereine.489 A variety of 1-alkyltetrahydroisoquinolines were subjected to electrolytic oxidation under similar conditions to those above, and the authors observed complete steric control in reaction outcome, with total reversal in C–C vs C–O coupling selectivity on introduction of this substituent (173.2b, 173.3b).
A general method for 2,2′-biphenol synthesis through phenol ortho,ortho-homo-coupling remained elusive due to the proclivity of simple phenol substrates to yield mixtures of regioisomeric products upon anodic oxidation, with the ortho,para-isomer (the Pummerer ketone) often predominating (see section 3.3.1.2.). A 2006 study from Waldvogel revealed an unusually high chemoselectivity for the ortho,ortho-homo-coupling of 2,4-dimethylphenol using boron-doped diamond (BDD) electrodes (Scheme 174).490 In prior reports from this group the maximum yield of the 2,2′-biphenol obtained in the anodic oxidation of 2,4-dimethylphenol employing Pt electrodes was limited to 26%.491 Optimal conditions in this 2006 work involved the electrolysis of 2,4-dimethylphenol in an undivided cell operating under constant current conditions equipped with silica-supported BDD electrodes, with addition of 11 wt% water and 6 wt% Et3MeN+MeOSO3– as a supporting electrolyte. A maximum yield of 56% yield of the 2,2′-biphenol was realized, and a 17:1 selectivity for ortho,ortho-homo-coupling over ortho,para-homo-coupling and Pummerer ketone formation. The authors considered several mechanistic pathways including (i) direct substrate oxidation and proton loss, or (ii) the intermediacy of electrogenerated hydroxyl radicals and subsequent intermolecular HAT for phenoxy radical generation. The reactivity did not translate beyond this single substrate, but this report proved critical to establishing BDD electrodes as offering superior reactivity and selectivity in C–C bond coupling reactions via electrogenerated phenoxy radicals.
Scheme 174. Boron-Doped Diamond Electrodes Offered a Highly Chemoselective Ortho,ortho-Homo-Coupling of 2,4-Dimethylphenol (Waldvogel, 2006).

Waldvogel and co-workers in 2009 then showed that a combination of BDD electrode anode and HFIP solvent was uniquely effective in promoting selective and high yielding homo-coupling for the synthesis of 2,2′-biphenols across a number of phenol substrates (Scheme 175).492 Under constant current electrolysis with a BDD anode, Ni cathode, and MeEt3N+MeOSO3– as supporting electrolyte, selective formation of the ortho,ortho-homo-coupled products was observed for a variety of electron-rich and halogenated phenols (175.1–175.4) (eight examples, 13–74% yields). Notably, no such coupling was observed in the absence of HFIP.
Scheme 175. Symmetric Phenol Homo-Coupling Mediated by Boron-Doped Diamond Electrode and Hexafluoroisopropanol (Waldvogel, 2009).

Waldvogel and co-workers sought to adapt this methodology enabling selective phenol ortho,ortho-homo-coupling to more readily available electrode materials and fluorinated media in a 2011 report (Scheme 176).493 These researchers found that 2,2-biphenol products could be selectively synthesized using a graphite anode in place of a BDD anode when a fluorinated additive was present at 45 wt%. In addition to HFIP, 1,1,1-trifluoroethanol (TFE) and trifluoroacetic acid (TFA) were found to be effective fluorinated additives, facilitating the ortho,ortho-coupling in high selectivity (176.1–176.4) (five examples, 21–76% yields). In the course of this investigation, Waldvogel and co-workers noted that the anodic oxidation of 4-methylguaiacol led to the selective formation of the ortho,meta-homo-coupled product over the usual ortho,ortho-product (for details of a follow up study on unusual regioselectivity in phenol–phenol homo-coupling, see section 3.3.1.3).
Scheme 176. Electrosynthesis of Biphenols Using Graphite Electrodes and Fluorinated Media (Waldvogel, 2011).

As highlighted in previous examples, many anodic coupling reactions are limited to phenols bearing EDGs to facilitate phenoxy radical generation at lower oxidation potentials and to increase the proclivity for C–C bond formation through nucleophilic addition of the phenol to the phenoxy radical. Employing electron-poor substrates as coupling partners typically results in little to no desired coupling product. In 1999, Nishiyama and co-workers first noted the formation of halo-2,2′-biphenols upon anodic oxidation of 2,6-dihalophenols in MeOH using a glassy carbon anode and Pt cathode.494 Depending upon the nature of the halogen substituent, C2 C–C or C–O bonded coupled products were obtained through displacement of the halogen substituent. In subsequent work, Waldvogel and co-workers were able to electrochemically synthesize 3,3′-dibromo-2,2′-biphenol and 3,3′-dichloro-2,2′-biphenol in 76% and 47% yield, respectively.
Building upon this work, the researchers sought to expand the electrochemical homo-coupling of phenols to enable synthesis of symmetric 2,2′-biphenols bearing different EWGs in a 2020 report (Scheme 177A).495 Constant current electrolysis of phenol substrates in an undivided electrochemical cell using BDD or graphite electrodes in HFIP at room temperature with diisopropylethylamine (DIPEA) catalyst furnished the corresponding symmetric 2,2′-biphenols (nine examples, 14–64% yields). The researchers noted that DIPEA was a crucial component of the reaction; other bases, such as pyridine, resulted in a dramatic drop in yield and the formation of C2 C–O rather than the C–C homo-coupled product. Notably, this methodology proved robust to a variety of electron-withdrawing functional groups, including ketones (177.1), esters (177.2), sulfones (177.3), nitriles (177.4), oximes, and aldehydes. However, neither nitro nor phosphonate groups were tolerated in the reaction.
Scheme 177. (A) Electrochemical Anodic C(sp2)–H/C(sp2)–H Homo-Coupling of Phenols Bearing Electron-Withdrawing Groups and (B) Cross-Coupling in the Presence of Naphthalene (Waldvogel, 2020).

Upon electrolyzing 2-hydroxyl-5-methylacetophenone in the presence of naphthalene, the researchers observed the formation of a polycyclic phenol–naphthalene coupled product in 33% yield (Scheme 177B, 177.5). On treatment with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), this product could be further oxidized to the corresponding dibenzofuran in 83% yield. Alternatively, treatment with 1 M HCl resulted in ring opening of the polycyclic product to form the arylated phenol. Overall, this electrochemical methodology affords phenol products bearing electron-withdrawing substituents, which can serve as precursors to polydentate ligands that are ubiquitous within transition metal catalysis.496−498
As highlighted in these anodic C–C coupling methodologies, the phenol compound with greater electron density is preferentially oxidized to form the electrophilic phenoxy radical. Because electron density and nucleophilicity are intrinsically linked, the same phenol is usually then poised for nucleophilic attack to form the homo-coupled product rather than any cross-coupling product when different phenol substrates are present. In this way, the selective formation of the cross-coupled product had remained elusive as the homo-coupled product is usually formed preferentially. Inspired by their previous work, Waldvogel and co-workers showed in a 2014 study that they were able to selectively form cross-coupled 2,2′-biphenols upon constant current electrolysis of substrate admixtures in a cell equipped with BDD anode, Ni cathode, Et3MeN+MeOSO3– supporting electrolyte, and HFIP/MeOH solvent mixture at 50 °C (15 examples, 21–63%) (Scheme 178).499 Although most cross-coupled 2,2′-biphenols were produced in modest yields (ca. 30%), inclusion of sterically demanding C2- and C4-substituents resulted in an increase in yield of up to 63% (e.g., 178.4). Notably, no phenol homo-coupling products were observed. Replacement of HFIP/MeOH with 9 vol% MeOH in HCO2H also enabled selective formation of the cross-coupled product, albeit in diminished yields (e.g., for the preparation of 178.4, 34% vs 63%).
Scheme 178. Selective Anodic 2,2′-Cross-Coupling of Phenols (Waldvogel, 2014).

*In neat HFIP.
3.3.1.2. Ortho,para-Homo-Coupling for the Synthesis of Pummerer Ketones and Higher Aggregates
The oxidative dimerization of 4-alkylphenols leading to the formation of ortho,para-coupled dihydrodibenzofuranone products has long been recognized. These products are named Pummerer ketones, for the group who first systematically studied their formation from p-cresol using ferricyanide as a stoichiometric chemical oxidant.500,501 The structure assigned to the Pummerer ketone was later revised by Arkley and co-workers,502 aligning it with the structure of the lichen natural product usnic acid, which was later proposed to arise biosynthetically through a similar process.503 This mode of phenol homo-coupling has been invoked in the biosynthesis of numerous other natural products.504 The electrochemical synthesis of this enone product proceeds via oxidative phenoxy radical generation followed by ortho,para-radical–radical coupling through resonance forms of the phenoxy radical and subsequent polar conjugate addition (Scheme 179).491,505
Scheme 179. Mechanistic Proposal for the Oxidative Coupling of 4-Alkylphenols for the Synthesis of Pummerer Ketones.

The first anodic process for the synthesis of Pummerer ketones with reasonable synthetic efficiency was reported by Miller in 1978 (Scheme 180).487 These authors note that existing electrochemical protocols involving p-cresols led to no more than 10% yields of the Pummerer product. This transformation was carried out under conditions devised by Bobbit for a related set of electron-rich arene substrates.486 Notably, constant potential electrolysis of substrates in a divided electrochemical cell equipped with graphite anode, Pt cathode, Et4N+ClO4– supporting electrolyte, and NaOMe as a stoichiometric additive in MeCN/H2O (19:1) led to the synthesis of Pummerer ketones from p-cresol (180.1), 4-ethylphenol (180.2), and 3,4- and 2,4-dimethylphenol (180.3, 180.4) (four examples, 20–37% yields).
Scheme 180. An Early Example of Electrochemical Anodic Ortho,para-Homo-Coupling of 4-Alkylphenols for the Synthesis of Pummerer Ketones (Miller, 1978).

With respect to mechanism, the authors posit stepwise formation of the phenoxide anion and subsequent anodic oxidation to the phenoxy radical. Notably, the low applied potential avoids further oxidation to the phenonium cation and thus favors selective radical–radical coupling and Pummerer product formation. As this transformation takes place in a divided cell, the acidity of the anolyte increases over the course of the reaction and thus, re-protonation of phenoxide becomes favorable and limits further conversion of the phenol substrate. The authors noted high recovery of starting materials in some cases as a result of this phenomenon. This selectivity for phenoxy radical generation is notable as other reports detailing the oxidation of p-cresols with electrochemical and chemical oxidants produced monomeric products associated with phenonium cation formation and subsequent nucleophilic trapping with solvent.506−508 The positional selectivity of the isomeric forms of the products is proposed to arise through interaction with the anode surface. Though of limited efficiency, this is a seminal example of a preparative anodic oxidation of phenols that demonstrates the reactivity and selectivity of phenol–phenol homo-coupling through radical intermediates.
In 2006, Waldvogel and co-workers re-investigated the anodic oxidation of 2,4-dimethylphenol and subsequent ortho,ortho-coupling of phenoxy intermediates for the synthesis of 2,2′-biphenols. Although the desired 2,2′-biphenols were not formed, the researchers serendipitously discovered an electrochemical way to access a variety of complex, polycyclic natural product-like structures through phenol ortho,para-coupling and further aggregation (Scheme 181).491 Electrolysis was performed in an undivided cell using Pt electrodes and a variety of electrolytes and solvents. Under all screened conditions, however, the desired 2,2′-biphenol synthesis was inefficient, being present in only trace amounts. In most cases, the Pummerer ketone product was the major component of a complex mixture. Despite this setback, the authors noted the unexpected formation of an interesting higher-aggregate spirocyclic lactone, apparently formed through trimerization of the phenol substrate and further rearrangement. This compound exhibits a similar core structure to several sesquiterpene natural products from the pinguisane family.509 Synthesis of this spirocyclic lactone was most efficient when Ba(OH)2 was employed as the electrolyte in MeOH solvent. Fortuitously, the product precipitated from solution, allowing for facile isolation through filtration and subsequent purification through sublimation to yield up to 18% of the spirolactone product (181.1).
Scheme 181. Unexpected Formation of a Trimeric Spirocyclic Lactone Product during the Electrolysis of 2,4-Dimethylphenol (Waldvogel, 2006).

The spirolactone product 181.1 is understood to form through a total of four consecutive rounds of oxidation and deprotonation that are first initiated by phenoxy radical generation and subsequent addition to the enolate derived from the in situ formed Pummerer ketone. The resultant species undergoes further oxidation and deprotonation to reform the enolate before the phenoxy radical cyclizes via oxygen onto the proximal enolate. A final oxidative step forms a carbocation and triggers a Meerwein-type rearrangement to yield the spirolactone. The atroposelectivity observed in product formation is proposed to be a result of chelation of the intermediate 2,2′-biphenolate by the barium cation en route to the spirocycle, leading to the observed diastereoisomer where the sterically demanding body of the Pummerer ketone is facing away from the projecting arene methyl group.
Given the similarity of this spirolactone product to a natural product family, the group further studied its electrochemical synthesis and published an improved preparation in 2008 (Scheme 182).509 These authors showed that the material isolated after precipitation and filtration was, in fact, a tetrameric adduct formed through nucleophilic trapping of the carbocationic intermediate prior to Meerwein rearrangement with a further equivalent of the phenol (182.1). The sublimation method for product purification in their prior work was shown to facilitate the subsequent rearrangement to the spirolactone. Pure hemiacetal could be isolated via filtration in 52% yield and up to 23 g scale per batch. By subjecting the hemiacetal to a solution of sulfuric acid in toluene at room temperature, the cationic rearrangement could be promoted to give spirolactone 183.1 in 75% yield.
Scheme 182. Improved Preparation of a Tetrameric Adduct via Anodic Oxidation of 2,4-Dimethylphenol (Waldvogel, 2008).

Notably, this hemiketal proved to be a robust precursor to a host of other complex natural product-like dibenzofuran compounds that could be accessed through a variety of one- or two-step transformations, of which a representative set is discussed here (Scheme 183).510 The authors discovered that through initial silylation of the hemiketal and subsequent treatment with BF3·OEt2 as a LA promotor, the stereochemical course of the Meerwein rearrangement could be almost completely reversed, allowing access to both diastereomers of spirolactone product (183.2). Treatment with thionyl chloride led to elimination, demethylation, and aromatization to yield a pentacyclic bis-dibenzofuran structure (183.3). Treatment with the potent nucleophile cyanide led to a skeletal rearrangement, involving ring-opening of the hemiketal, elimination to the α,β-unsaturated system, and subsequent ring closure that led to formation of 183.4. Treatment with NH4F or amines in the presence of a CsF promotor yielded amino-functionalized Pummerer ketones (e.g., 183.5 and 183.6).511 Treatment with anisole in the presence of BF3·OEt2 gave Friedel–Crafts product 183.7, a scaffold which is remarkably similar to naturally occurring rocaglamide—an antileukemic flavagline.512 Finally, treatment with ketones or isocyanates in the presence of BF3·OEt2 led to [3+2] cycloaddition products (e.g., 183.8). Use of a chiral ketone permitted chromatographic separation of the resultant diastereoisomers, which could then be transformed into enantiomerically enriched spirolactones through the same Meerwein rearrangement as discussed above.513 These reports from Waldvogel demonstrate that anodic oxidation of a simple phenol can serve as a powerful entry point to a large series of complex polycyclic compounds.
Scheme 183. Diversity-Oriented Synthesis of a Series of Complex Polycyclic Structures from Tetrameric Adduct 182.1 Resulting from Anodic Oxidation of 2,4-Dimethylphenol (Waldvogel, 2006–2016).

3.3.1.3. Other Modes of Phenol–Phenol Homo- and Cross-Coupling
As detailed above, the anodic oxidation of phenols normally leads to ortho,ortho- or ortho,para-homo- or cross-coupled products (section 3.3.1.1 and 3.3.1.2, respectively). Several reports describe the selective preparation of other regioisomeric coupling products of particular subsets of phenol and naphthol substrates. These methods are discussed herein.
In the course of studying the selective ortho,ortho-homo-coupling of phenols, Waldvogel and co-workers noted that constant current electrolysis of 4-methylguaiacol in TFA solution in an electrochemical cell equipped with graphite electrodes resulted in the formation of the ortho,meta-homo-coupling product (184.1) rather than the ortho,ortho-homo-coupling product (184.2) in 57% yield.493 Encouraged by this reactivity, these researchers further explored the regioselectivity of homo-coupling reactions of these types of methoxy-substituted (i.e., guaiacol) substrates (Scheme 184).514 The researchers noted that the formation of the symmetrical (ortho,ortho) or non-symmetrical (ortho,meta) biphenol products was influenced by the steric bulk of the substituent at the 4-position, as increasing steric demand at this position lowered the selectivity for ortho,meta-coupling and increased selectivity of ortho,ortho-coupling (Scheme 184A). For example, under these electrolysis conditions, 4-methylguaiacol underwent exclusive ortho,meta-coupling in 33% yield. When 4-isopropylguaiacol was used in place of 4-methylguaiacol, a 1:1 ratio of the ortho,meta-coupling product and ortho,ortho-coupling product was observed in a cumulative 10% yield. Furthermore, 4-tert-butylguaiacol led to the selective formation of the ortho,ortho-coupling product in 18% yield.
Scheme 184. Electrochemical Anodic Oxidative Coupling of Guaiacol Derivatives (Waldvogel, 2011).

The researchers observed other modes of regioselectivity depending on the guaiacol substrate used in the homo-coupling reaction. For example, anodic oxidation of 4-methoxyguaiacol led to the selective formation of the meta,meta-homo-coupling product 184.3 in 44% yield (Scheme 184B), whereas anodic oxidation of 6-methoxyguaiacol led to exclusive formation of the meta,para-biphenol in 44% yield. Furthermore, oxidation of 2,5-dimethoxyphenol led to the selective formation of the para,para-coupled biphenol 184.4 in 83% yield (Scheme 184C). The researchers posited that oxidation and subsequent deprotonation of the phenol substrate leads to the formation of a phenoxy radical, which is resonance stabilized. The resulting electrophilic carbon-centered radical can be trapped by the un-oxidized nucleophilic phenol, furnishing the biphenol product upon subsequent oxidation and re-aromatization. In this way, the steric bulk and nucleophilicity of the phenol influence the regioselectivity of the C–C bond formation step.
In a 2017 study, Waldvogel and co-workers sought to benchmark their previously reported electrochemical phenol–phenol homo- and cross-coupling499,515 methodologies against chemical dehydrogenative coupling methods and extend these strategies to phenol–naphthol cross-coupling (Scheme 185).516 Constant current electrolysis of substrate admixtures with BDD electrodes, n-Bu3MeN+MeOSO3– supporting electrolyte, and 18 vol% MeOH in HFIP at 30–50 °C resulted in the selective formation of the cross-coupled products (eight examples, 35–86% yields). Regioselectivity for phenol–naphthol cross-coupling could be biased between ortho,ortho- and ortho,para-coupling through substrate selection, with 4-methylguiacaol tending to the former (185.2) and 2,6-dimethoxyphenol tending toward the latter (185.1). Encouragingly, the researchers found that the yields offered by this electrochemical method are in most cases comparable to the yields obtained through the use of an Fe(III) catalyst in combination with di-tert-butyl peroxide (DTBP), as reported by Pappo and co-workers.517 Notably, the anodic cross-coupling methodology underperformed with respect to the chemical approach when the naphthol coupling partner contained an aryl bromide (185.4) (35% vs 96% yield).
Scheme 185. Electrochemical Anodic C(sp2)–H/C(sp2)–H Cross-Coupling of Phenol and Naphthol Derivatives, and Comparison to a Fe(III)/DTBP-Mediated Oxidative Coupling Reaction (Waldvogel, 2017).

*At 30 °C. **HFIP + 18 vol% MeOH solvent.
The selective dehydrogenative cross-coupling of two different phenol substrates for the synthesis of para,para-coupled 4,4′-biphenols is a challenging goal. A successful method must overcome potential problems of cross- vs homo-coupling, of undesired 2,2′- and 2,4′-coupling, and overoxidation of the desired 4,4′-biphenol to the corresponding para-diphenoquinones. While a general solution is yet to be found, Waldvogel and co-workers recently reported a method enabling the dehydrogenative homo- and cross-coupling of 2,5- and 2,6-disubstituted phenols for 4,4′-biphenol synthesis (Scheme 186).518 These substitution patterns successfully block undesired C2-coupling and force the product biaryl to adopt a twisted geometry about the newly formed C–C bond, thereby preventing diphenoquinone formation.
Scheme 186. Selective Cross- and Homo-4,4′-Coupling of 2,5- and 2,6-Disubstituted Phenols (Waldvogel, 2019).

*6 vol% aqueous co-solvent.
Constant current electrolysis was performed in an undivided cell equipped with BDD electrodes in aq. HFIP solvent mixtures with n-Bu3MeN+MeOSO3– as a supporting electrolyte. Here, seven examples of the 4,4′-cross-coupling of phenols were presented, in yields of 10–77% (186.1–186.3), in addition to five examples of 4,4′-homo-coupling, in 14–60% yields. When less sterically demanding C5-substituents (e.g., fluoro-, 186.3) were present, lower yields were realized as these substituents enforce less of a torsional angle across the C–C bond, and therefore overoxidation is a feasible deleterious process. The anodically generated phenoxy radical intermediate is proposed to couple with a neutral, closed-shell phenol at the 4-position, and the coupled product is subsequently oxidized and deprotonated to furnish the aromatized product. Reduction of liberated protons at the cathode releases molecular hydrogen. Selectivity for cross- over homo-coupling arises from a sufficient offset in oxidation potential between the two reaction partners (e.g., for 2,6-dimethoxyphenol, Epox = +1.19 V vs Fc+/Fc in HFIP, compared to 2,5-dimethylphenol, Epox = +1.38 V vs Fc+/Fc in HFIP).518 In some cases, product overoxidation or other side reactions could be successfully avoided through selective precipitation of the 4,4′-biphenol products directly from the reaction mixture. This preparation was used in the synthesis of all-para teraryl-based α-helix mimics; anodic coupling yielded 4,4′-biphenols which were converted to the bis-nonaflate and were used in subsequent Pd-catalyzed Suzuki cross-coupling to access the teraryl target molecules of interest.519
3.3.2. Electrochemical Phenol–Arene and Phenol–Heteroarene Cross-Coupling
3.3.2.1. Phenol–Arene Cross-Coupling
In a 2010 report, Waldvogel and co-workers sought to expand upon their prior electrochemical methodology describing phenol ortho,ortho- and ortho,para-homo-coupling (see sections 3.3.1.1 and 3.3.1.2) to enable the selective dehydrogenative C(sp2)–H/C(sp2)–H cross–coupling of phenols with electron-rich arenes 187.1–187.4 (Scheme 187).520 Under constant current electrolysis with a BDD anode, Ni cathode, and HFIP solvent, eight examples of the phenol–arene cross-coupled product were produced in modest yields (11–47%) and good cross-selectivity (>2.5:1 ratio for the phenol–arene cross-coupling product compared to the arene–arene homo-coupling byproduct). However, a large excess (10.0 equiv) of arene coupling partner was required to achieve modest yields of the cross-coupled phenol–arene products.
Scheme 187. Electrochemical C(sp2)–H/C(sp2)–H Cross-Coupling of Phenols with Electron-Rich Arenes (Waldvogel, 2010).

The researchers conducted CV studies to interrogate the mechanism of this transformation. Although the phenol substrate and arene substrate are oxidized at similar potentials, the researchers posited that the phenol substrate is preferentially oxidized and deprotonated at the BDD anode. The resulting electrophilic phenoxyl radical is proposed to preferentially attack the nucleophilic, electron-rich arene rather than the closed-shell phenol, as the arene substrate is present in large excess. The radical resulting from phenol–arene coupling is subsequently oxidized and deprotonated to furnish the aromatized cross-coupling product.
In a follow-up study published in 2012, Waldvogel and co-workers investigated the effect that polar additives such as MeOH and H2O had on the selectivity and efficiency of this anodic phenol–arene cross-coupling reaction (Scheme 188).521 Constant current electrolysis of an HFIP/MeOH (4.5:1) solution of phenol and arene substrates in an undivided electrochemical cell fitted with BDD anode and Ni cathode led to the phenol–arene cross-coupled product (188.1–188.3, 18 examples, 6–69% yield). Compared to their previously reported methodology that required 10 equiv of electron-rich arene relative to phenol, this improved methodology only required 3 equiv of arene substrate. In the course of reaction screening, the researchers noted that the desired cross-coupled phenol–arene product was formed in relatively low yield when conducted in pure HFIP (e.g., for 188.1, 21% yield). Notably, the yield of the desired phenol–arene cross coupled product could be drastically increased by performing the reaction with 9 vol% H2O (e.g., for 188.1, 67% yield) or 18 vol% MeOH (e.g., for 188.1, 69% yield) in HFIP. These polar additives also facilitated selective formation of the cross-coupled product in other fluorinated solvents (e.g., trifluoroethanol, octafluoropentanol), albeit with diminished yields, suggesting that the identity of fluorinated solvent and quantity of polar additive are equally important in facilitating the selectivity and efficiency of this transformation. During this investigation, the authors also noted that this electrochemical transformation did not depend on the use of BDD anode; although BDD anode facilitated superior yields, other anodes (e.g., graphite, glassy carbon, Pt) produced the desired cross-coupled product with excellent selectivity. Notably, this methodology was tolerant of alkyl groups, halogens, and polycyclic aromatic moieties (e.g., naphthalene, anthracene), which are often prone to overoxidation.
Scheme 188. A Study on the Effect of MeOH on Electrochemical C(sp2)–H/C(sp2)–H Cross-Coupling of Phenols with Electron-Rich Arenes (Waldvogel, 2012).

The researchers revised their previous mechanistic hypothesis to account for the improved efficiency and selectivity in formation of the phenol–arene cross-coupling product in the presence of such polar additives. Specifically, the researchers posited that direct oxidation of the polar additive (MeOH or H2O) on the surface of the BDD anode generates a highly reactive methoxy (O–H BDE = 105 kcal mol–1)25 or hydroxy radical (O–H BDE = 119 kcal mol–1)25 which is stabilized via interaction with HFIP solvent. The researchers hypothesize that this stabilized radical chemoselectively abstracts the phenolic H-atom (e.g., for phenol, O–H BDE = 90 kcal mol–1).25 The resulting phenoxyl radical then electrophilically attacks an electron-rich arene to furnish the cross-coupled product upon subsequent re-aromatization, which is proposed to proceed via subsequent HAT.
In this same year, Nishiyama and co-workers carried out ESR spin trapping studies to determine if alkoxy radical intermediates are generated in electrochemical reactions employing MeOH and BDD, Pt, and glassy carbon electrodes.522 The researchers found that methoxy radicals are formed in electrochemical oxidation reactions of MeOH using all three anodes. Notably, however, the efficiency with which methoxy radical generation occurs varies depending upon the anode with BDD > Pt > glassy carbon. These results from Nishiyama and co-workers support the hypothesis of Waldvogel and co-workers regarding the involvement of methoxy radicals in the selective formation of phenol–arene cross-coupled products when MeOH is used as a polar additive in electrochemical cells fitted with BDD anodes.521
To gain further insight into the mechanistic origins of the selectivity of this phenol–arene cross-coupling reaction, Waldvogel and co-workers systematically examined the electrochemical properties (e.g., Eox) of phenol and arene substrates in HFIP as a function of MeOH concentration in a follow-up study published in 2015.523 The researchers noted that the addition of MeOH decreases the oxidation potential of a model phenol substrate (e.g., for guaiacol, Eox ≈ +0.84 V vs Ag/AgCl in pure HFIP, to Eox ≈ +0.76 V vs Ag/AgCl in 18 vol% MeOH/HFIP)523 and increases the oxidation potential of a model electron-rich arene substrate (e.g., for 1,2,4-trimethoxybenzene, Eox ≈ +0.73 V vs Ag/AgCl in pure HFIP to Eox ≈ +0.83 V vs Ag/AgCl in 18 vol% MeOH/HFIP).523 The researchers note that 1,2,4-trimethoxybenzene is preferentially oxidized in pure HFIP, thereby facilitating the formation of its homo-coupled biaryl product. Upon the addition of MeOH, however, guaiacol is preferentially oxidized. Notably, the preferential oxidation of guaiacol does not lead to the formation of its homo-coupled biphenol product. Instead, preferential oxidation of guaiacol in MeOH/HFIP leads to the formation of the cross-coupled phenol–arene product, as previously described in the authors’ 2010 and 2012 reports.520,521 The formation of the cross-coupled product instead of the homo-coupled biphenol product is surprising since nucleophilicity is typically linked to oxidation potential, i.e., the substrate that is preferentially oxidized is also the most nucleophilic. Given that the addition of MeOH leads to the formation of the cross-coupled product rather than the homo-coupled biphenol product, the researchers posited that the addition of MeOH changes the substrates’ solvation environments so that their nucleophilicity is not as strongly coupled to their oxidation potential. As a result, the phenol substrate guaiacol is most easily oxidized whereas the arene substrate 1,2,4-trimethoxybenzene is the most nucleophilic in MeOH/HFIP. In contrast, the arene substrate 1,2,4-trimethoxybenzene is the most easily oxidized and the most nucleophilic in pure HFIP, hence the formation of the homo-coupled biaryl product under these conditions. Based upon these results, the researchers concluded that changes to both the oxidation potential and the nucleophilicity of the coupling components upon addition of polar additives, such as MeOH facilitate selective cross-coupling over homo-coupling. Although this conclusion seems to contradict the researchers’ previous mechanistic hypothesis regarding the involvement of electrogenerated methoxy radicals in facilitating selective phenol–arene cross-coupling reactions,521 it suggests that many different mechanisms may be operating concurrently and collectively contributing to the selectivity of these anodic phenol–arene cross-coupling reactions.
The plurality of mechanistic proposals related to the source of selectivity in these anodic phenol–arene cross-coupling reactions have not stopped researchers from utilizing and expanding the scope of these transformations. In 2016, Waldvogel and co-workers reported the electrochemical C(sp2)–H/C(sp2)–H cross-coupling of phenols with aryl triisopropylsilyl (TIPS) ethers (Scheme 189). Constant current electrolysis of an HFIP solution of phenol and aryl TIPS ether at 50 °C in an undivided electrochemical cell fitted with BDD anode and Ni cathode resulted in the desired cross-coupled product in generally excellent yield (189.1–189.4) (12 examples, 70–92%). The researchers noted that this bulky silyl group confers a twist across the biaryl product of the electrochemical reaction and that this group also increases the oxidation potential of the heterocoupled product relative to the unprotected 2,2′-biphenol, thereby protecting the product from overoxidation. Consequently, TIPS protection enabled phenols that typically undergo rapid homo-coupling (e.g., 3,4-dimethoxyphenol, 5-hydroxybenzo-1,2-dioxole) to be selectively cross-coupled with electron-rich arenes. TIPS protection proved superior to other silyl protecting groups such as trimethylsilyl (TMS) and tert-butyldimethylsilyl (TDBMS), which were generally unstable to electrolysis conditions. The Waldvogel group later extended this methodology employing a similar electrochemical cell assembly to the synthesis of both symmetric (ABA-type) and non-symmetric (ABC-type) meta-524 and para-terphenyl-2,2′-diols,525 through two-fold cross-coupling of phenols and electron-rich arenes. ABA-type products could be prepared in a one-pot fashion, and ABC-type products were accessible via protection of the intermediate biaryl product, prior to a further iterative electrochemical cross-coupling.
Scheme 189. Synthesis of Mono-TIPS-Protected 2,2′-Biphenols via Electrochemical Anodic Phenol–Arene Cross-Coupling (Waldvogel, 2016).

*With Ni cathode.
3.3.2.2. Phenol–Heteroarene Cross-Coupling
In 2017, Lei and co-workers developed an electro-oxidative [3+2] intermolecular annulation between phenols and N-Ac-indoles for the synthesis of benzofuroindolines (Scheme 190).207 Substrate control in these reactions provides access to both isomeric benzofuro[2,3-b]indoline and benzofuro[3,2-b]indoline products. This heterocyclic structure exists in several bioactive natural products such as the diazonamides, azonazines, and phalarine. Typical existing oxidative approaches to these core structures required stoichiometric quantities of chemical oxidants, such as iodobenzene diacetate, Fe(III), and DDQ.526 In addition, these stoichiometric methods usually provide benzofuro[2,3-b]indolines; methods accessing the isomeric benzofuro[3,2-b]indolines are less common.527,528
Scheme 190. Electrochemical Oxidative [3+2] Annulation of Phenols and N-Acetyl Indoles (Lei, 2017).

Electrolysis was carried out in an undivided cell consisting of a graphite rod anode and platinum plate cathode operating under constant current conditions with n-Bu4N+BF4– as a supporting electrolyte in a mixture of HFIP/CH2Cl2 (3:2). Electron-rich phenols were competent coupling partners in this transformation. When a 2-substituted N-Ac-indole substrate was used, a benzofuro[3,2-b]indoline product was obtained (e.g., 190.1–190.3, 16 examples, 35–99%). When 3-substituted or 2,3-disubstituted N-Ac-indoles were used, the isomeric benzofuro[2,3-b]indolines were formed instead (e.g., 190.4, 190.5, seven examples, 71–98%). The regiosisomeric preference observed in 2,3-disubstituted indoles could be altered by adding ZnCl2, which provided the benzofuro[3,2-b]indoline as a minor product. The reaction tolerated phenol and indole ring substitution, as well as alkyl ester, nitrile, alkene, and primary bromide functionality on the indole.
The researchers posited that anodic oxidation of both the phenol (e.g., for 4-methoxyphenol, E1/2 = +1.16 V vs Ag/AgCl in HFIP/CH2Cl2 (3:2))207 and the N-Ac-indole component (e.g., for 3-methyl-N-Ac-indole, E1/2 = +1.10 V vs Ag/AgCl in HFIP/CH2Cl2 (3:2))207 occurs at similar potentials, thus forming phenol and indole radical cations in solution. The phenol radical cation is then deprotonated by electrogenerated hexafluoroisopropoxide to yield the phenoxy radical. HFIP has previously been shown to stabilize aromatic radical cations,529−531 and thus a radical–radical coupling mechanism between a persistent indolyl radical cation, and a transient phenoxy radical is invoked here to describe the first C–C bond-forming step.532 A trapping experiment with triethylphosphite also implicated an indoyl radical cation intermediate. Annulation subsequently occurs via polar cyclization onto the indole carbocation. Deprotonation of the resulting cationic species restores aromaticity and yields the product. Cathodic reduction of the HFIP solvent liberates molecular hydrogen.
Waldvogel and co-workers have reported related methods for electrochemical dehydrogenative phenol–heteroarene cross-coupling. These share common mechanistic features, namely (i) the generation of a reactive phenoxy radical from the parent phenol through anodic oxidation and proton loss, (ii) the addition of the transient phenoxy radical to a neutral, closed-shell heteroarene coupling partner, and (iii) selectivity for cross-coupling over homo-coupling determined by the relative ease of phenol oxidation compared to the oxidation of other reaction components.
A 2017 report disclosed a method for the C2-selective dehydrogenative cross-coupling of phenols with thiophenes (Scheme 191).533 Being electron-rich heterocycles prone to oxidation themselves, the ability to selectively cross-couple these two systems is noteworthy. The optimal undivided electrochemical cell assembly consisted of BDD electrodes operating under constant current conditions with n-Bu3MeN+MeOSO3– as a supporting electrolyte in HFIP. Here, seven examples of thiophene C2-mono-arylation were reported, in yields of 8–60%. Typically, electron-rich 4-methoxyphenols were optimal and required ortho-alkyl substituents for an efficient reaction. A variety of thiophene C3- and C5- substituents were demonstrated (191.1). By variation of the reaction stoichiometry, thiophene C2,5-diarylation successfully yielded ABA-type teraryl systems (e.g., 191.3), and five examples were presented in yields of 33–53%.
Scheme 191. Dehydrogenative Cross-Coupling of Phenols and Thiophenes or Benzothiophenes (Waldvogel, 2017 and 2018).

*With 1.0 equiv of phenol and 3.0 equiv of thiophene; **With 1.0 equiv of phenol and 1.5 equiv of benzothiophene; ***With 3.0 equiv of phenol and 1.0 equiv of thiophene.
Regarding the mechanism of the transformation, the authors propose that the phenol substrate is oxidized preferentially to the thiophene component to generate the corresponding phenol radical cation, then subsequent proton loss yields the neutral phenoxy radical (e.g., for 4-methoxy-2-methylphenol, Epox = +1.22 V vs Fc+/Fc in HFIP, compared to 3-hexylthiophene, Epox = +1.59 V vs Fc+/Fc in HFIP).533 The selectivity in cross- vs homo-coupling is understood to arise from this observed offset in oxidation potentials of the two components. Addition of the phenoxy radical to the thiophene coupling partner forges a new C–C bond before further oxidation and deprotonation restores aromaticity to yield the product. Concomitant reduction of the liberated protons at the cathode surface generates molecular hydrogen.
In a follow-up study published in 2018, the Waldvogel group presented a method for the cross-coupling of phenols with benzothiophenes (Scheme 191).534 A variety of pharmaceutical agents carry this heterobiaryl motif, including the osteoporosis medication raloxifene. The same electrochemical cell assembly utilized in their protocol for thiophene arylation was employed.533 The choice of HFIP solvent was key to achieving selective cross-coupling and avoiding oligomeric or polymeric side products. This method permitted regioselective mono-arylation at both C2- or C3-positions of benzothiophenes through either ortho- or para-positions of the phenol moiety, which was achieved by blocking the alternative positions. A total of 18 examples of phenol–benzothiophene cross-coupling were presented in yields of 38–88%. A range of alkyl (191.2, 191.4), alkoxy and halogen (191.2) substituents were permitted on either coupling partner. In all cases, no homo-coupling side products were observed. The protocol could easily be scaled to multigram quantities without re-optimization of the electrochemical parameters. The researchers propose a similar mechanism entailing the same proposal of stepwise phenoxy radical generation, bimolecular addition to the benzothiophene partner, and further oxidation and PT to restore aromaticity to the product. Again, phenol oxidation occurs preferentially over benzothiophene oxidation (e.g., for 4-methoxy-2-methylphenol, Epox = +1.22 V vs Fc+/Fc in HFIP, compared to 2-methylbenzothiophene, Epox = +1.38 V vs Fc+/Fc in HFIP).534
When benzofurans were investigated for their reactivity in electrochemical dehydrogenative cross-coupling with phenols, a unique furan metathesis reaction was identified (Scheme 192).535 This procedure was carried out using the same electrochemical cell assembly and reaction conditions as employed for the phenol–benzothiophene cross-couplings. A total of 12 examples of benzofuran–phenol cross-coupling with furan metathesis were reported in yields of 24–61% (192.1–192.3). Electron-rich phenols were optimal substrates in this work and tolerated alkyl and alkoxy substituents. Various alkyl, alkoxy, and halo-substituted benzofuran partners were tolerated.
Scheme 192. Convergent Synthesis of C3-(2-Hydroxyphenyl)benzofurans from Both C2- and C3-Substituted Benzofurans, with Furan Metathesis (Waldvogel, 2018).

Curiously, both C2- and C3-substituted benzofurans undergo this coupling and furan metathesis sequence to exclusively yield C2-substituted benzofuran products. Consequently, two different mechanistic pathways are proposed for these two substrate classes. The first involves anodic phenoxy radical generation through discrete ET and PT steps followed by phenoxy radical addition to the C3-position of a C2-substituted benzofuran, forming a stabilized α-oxy radical (Scheme 193, A). Further anodic oxidation yields an oxocarbenium ion intermediate, which can be nucleophilically attacked by the tethered phenol to form a dihydrobenzofuro[2,3-b]benzofuran intermediate. This acetal can reopen and does so with cleavage of the original C–O bond of the benzofuran substrate to form the alternative oxocarbenium ion intermediate. Finally, deprotonation leads to re-aromatization and product formation. The second possibility involves the same phenoxy radical generation and addition to the C2-position of a C3-substituted benzofuran (Scheme 193, B). Anodic oxidation now affords a benzylic carbocation, which cyclizes onto the pendant phenol to yield a dihydrobenzofuro[3,2-b]benzofuran. Although ring opening also occurs, a subsequent phenonium ion shift occurs to furnish the observed product. Thus, apparent migration of the C3-substituent to the C2-position is observed. The driving force for the metathesis reaction in both of these modes is the greater stability of the resultant oxocarbenium ion intermediate following metathesis and the higher nucleophilicity of the incoming phenol (both achieved through judicious choice of electron-rich phenol substrates). Concomitant reduction of protons at the cathode surface liberates molecular hydrogen. Again, the source of selectivity in cross-coupling arises from the observed offset in oxidation potentials between the two components (e.g., for 4-methoxy-2-methylphenol, Epox = +1.22 V vs Fc+/Fc in HFIP, compared to 3-methylbenzofuran, Epox = +1.56 V vs Fc+/Fc in HFIP).535
Scheme 193. Furan metathesis in the oxidative coupling of phenols and benzofurans (Waldvogel, 2018).
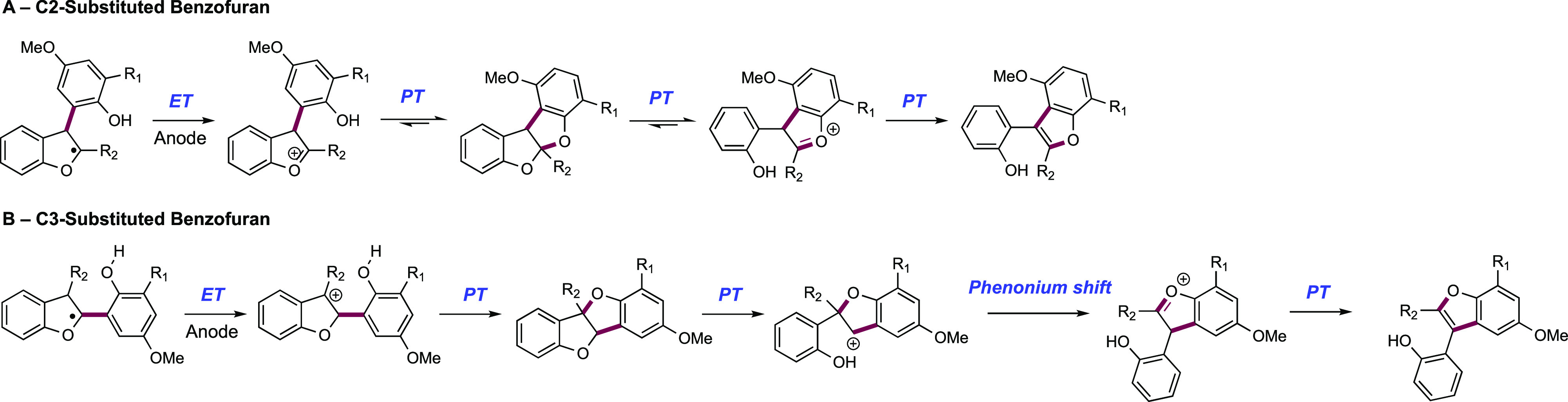
A 2019 report disclosed a method for the synthesis of functionalized N,N-diarylamides through the cross-coupling of phenols and benzoxazoles (Scheme 194).536 The initial cycloadduct formed through the electrochemical reaction was found to hydrolyze under acidic conditions to ring-open the benzoxazole component. A similar cell assembly was used, with n-Bu4N+PF6– as supporting electrolyte. Here, eight examples of electrolysis and subsequent hydrolysis were presented in 26–57% yields (194.1, 194.2). The protocol could easily be scaled to multigram without re-optimization of the electrochemical parameters. The researchers proposed that electrogenerated phenoxy radical couples with the benzoxazole component through the nucleophilic nitrogen atom, forming a C–N bond as opposed to a C–C bond observed in other classes of heteroarenes. Further anodic oxidation of the resultant radical forms an N-arylbenzoxazolium cation, which cyclizes via the tethered phenol to yield the initial coupling product. Concomitant reduction of protons at the cathode surface generates molecular hydrogen.
Scheme 194. Synthesis of N,N-Diarylamides through Anodic Coupling of Phenols and Benzoxazoles (Waldvogel, 2019).

3.3.3. Other Electrochemical Processes of Phenol Substrates
In this section, we present a small collection of methods arising from the electrochemical activation of phenol substrates and subsequent reaction with olefin and sulfinate cross-coupling partners for C–C and C–S bond formation, respectively. In 2018, Einaga and Waldvogel developed an electrochemical method for the stereoselective synthesis of the polyphenol plant lignan α-diisoeugenol (195.2) via the oxidative dimerization of isoeugenol (195.1) (Scheme 195).537 This natural product is of interest for its diverse biological activities which, along with derivatives, have been widely studied.538 An undivided electrochemical cell assembly consisting of BDD electrodes with n-Bu3MeN+MeOSO3– as supporting electrolyte in HFIP operating under constant current conditions at low current densities (j = 0.7 mA/cm2) was employed in this work. Higher charge density led to lower yields through over oxidation of the product. Under the optimized reaction conditions, a 57% yield of diisoeugenol was obtained. Notably, only a single diastereomer of product was produced.
Scheme 195. Electrochemical Synthesis of Diisoeugenol (195.2) (Einaga and Waldvogel, 2018).

The reaction was proposed to proceed via stepwise ET/PT to generate a phenoxy radical intermediate, which is in resonance with a p-quinone methide radical. Through this resonance structure, radical addition occurs across the styrene functional group of another molecule of isoeugenol. The C-centered radical formed in this step initiates a radical chain process with another molecule of isoeugenol via HAT with the phenol moiety. The authors observed that a sub-stoichiometric amount of charge enabled full conversion due to the chain process. Finally, intramolecular Friedel–Crafts-type alkylation of the electron-rich guaiacol onto the tethered quinone methide gives the dimeric product. Stereoselectivity is proposed to arise from a solvent–cage interaction.
Zhang and Wang have recently published an electrochemical method to enable the intermolecular oxidative [3+2] annulation between phenols and cinnamate-type electron-deficient olefins, leading to anti-2-aryl-2,3-dihydrobenzofurans with exclusive α-addition (anti-Michael addition) observed in the initial bond-forming step (Scheme 196).539 This protocol for electron-deficient olefins contrasts with existing photocatalytic and electrochemical methods for [3+2] annulations, which are typically restricted to electron-rich olefins.540−543 Optimized reaction conditions consisted of constant current electrolysis of a mixture of phenol and cinnamate ester substrates in an undivided electrochemical cell consisting of graphite rod electrodes and n-Bu4N+BF4– supporting electrolyte in a mixed solvent system of HFIP/CH2Cl2 (3:2). In total, 30 examples of dihydrobenzofuran synthesis were reported in yields of 30–95%. Electron-rich phenols and cinnamate esters bearing electron-rich aryl groups proved to be the most competent substrates. Notably, exclusive anti-diastereoselectivity was observed in all cases where 2,3-disubstituted products were formed (196.1). A 2,2,3-trisubstituted example, however, gave a mixture of diastereoisomers with little control (196.3). In addition to cinnamate esters, an example of a β-aryl acrylonitrile (196.2) and a β-trifluoromethylstyrene (196.4) also reacted with anti-Michael selectivity. The synthetic utility of this method was established in a short total synthesis of the bioactive natural product 3′,4-di-O-methylcedrusin, which entailed electrooxidative [3+2] annulation and subsequent global reduction with LiAlH4.
Scheme 196. Synthesis of Benzodihydrofurans via Electrochemical, Intermolecular [3+2] Annulation between Phenols and Electron-Deficient Olefins (Zhang and Wang, 2020).

The researchers proposed stepwise oxidation and deprotonation of the phenol to form a reactive phenoxy radical intermediate, which was supported by the observation that electron-rich phenols are typically required. This intermediate reacts with the cinnamate partner in an anti-Michael fashion, leading to a stabilized benzylic radical. Further anodic oxidation forms a benzylic carbocation, which is trapped through polar cyclization of the hydroxyl group. Cathodic reduction of solvent liberates molecular hydrogen and produces the requisite base for radical generation. DFT studies show that the energy of activation (ΔG⧧) for β-addition of the phenoxy radical (Michael-type) is destabilized by ca. 6.4 kcal mol–1 relative to the observed α-addition. Also, the benzylic radical intermediate resulting from α-addition (ΔGC–C) is stabilized by ca. 4.4 kcal mol–1 relative to the isomeric α-acyl radical, thereby accounting for the observed radical addition selectivity. Such anti-Michael radical addition reactions with cinnamate esters have been noted by other groups using photoredox and stoichiometric methods for radical generation.317−320
Waldvogel reported the electrosynthesis of aryl sulfones through the sulfonylation of phenols with sodium sulfinates (Scheme 197).544 Aryl-alkyl-, and diarylsulfones were produced (10 examples, 11–55%) upon constant current electrolysis of HFIP/H2O (15 vol%) solution of phenol substrate and sodium sulfinate reagent in an undivided electrochemical cell using BDD electrodes. The researchers noted that the amount of water co-solvent had a pronounced effect on the yield of the reaction. Notably, BDD electrodes could be substituted with graphite or glassy carbon electrodes with only a small decrease in yield (≤10%). The reaction was tolerant of halogenated aryl sulfinates (197.1, 197.3) and aliphatic sulfinates (197.4). When the phenol coupling partner was unsubstituted at the 4- and 6-positions, a mixture of regioisomeric sulfone substitution products were typically observed. Notably, scale up of the reaction did not substantially diminish yield.
Scheme 197. Electrochemical Sulfonylation of Phenols Using Sulfinates (Waldvogel, 2019).

3.3.4. Photocatalytic Transformations of Phenols
Compared to electrochemical methods, there are relatively few photocatalytic methods that enable the homolytic activation of phenol O–H bonds. The few methods that exist are described herein: phenol–arene and phenol–phenol cross- and homo-coupling through C–C and C–O bond formation, Heck-type olefin arylation, and orcinaldehyde de-aromatization.
3.3.4.1. Phenol–Arene Cross-Coupling
In 2017, König and co-workers developed an efficient photocatalytic method enabling C–C and C–O cross-coupling of electron-rich phenols with arenes (Scheme 198).545 The researchers found that phenol–arene cross-coupling could be achieved upon blue-light irradiation of an MeCN solution containing phenol and arene substrates, Ru(bpz)3(PF6) ([Ru-2](PF6)2) photocatalyst, and (NH4)2S2O8 oxidant. The use of MeCN is notable as most dehydrogenative cross-coupling reactions involving phenol and arene substrates proceeding through electrochemical activation have necessitated the use of highly fluorinated solvents such as HFIP to achieve good yields and high selectivities (see section 3.3.1). The desired heterocoupled biaryls were generated with high functional group tolerance, for example, halides and alkenes are conserved (14 examples, 7–79%).
Scheme 198. Photocatalytic Oxidative Phenol–Arene Cross-Coupling (König, 2017).
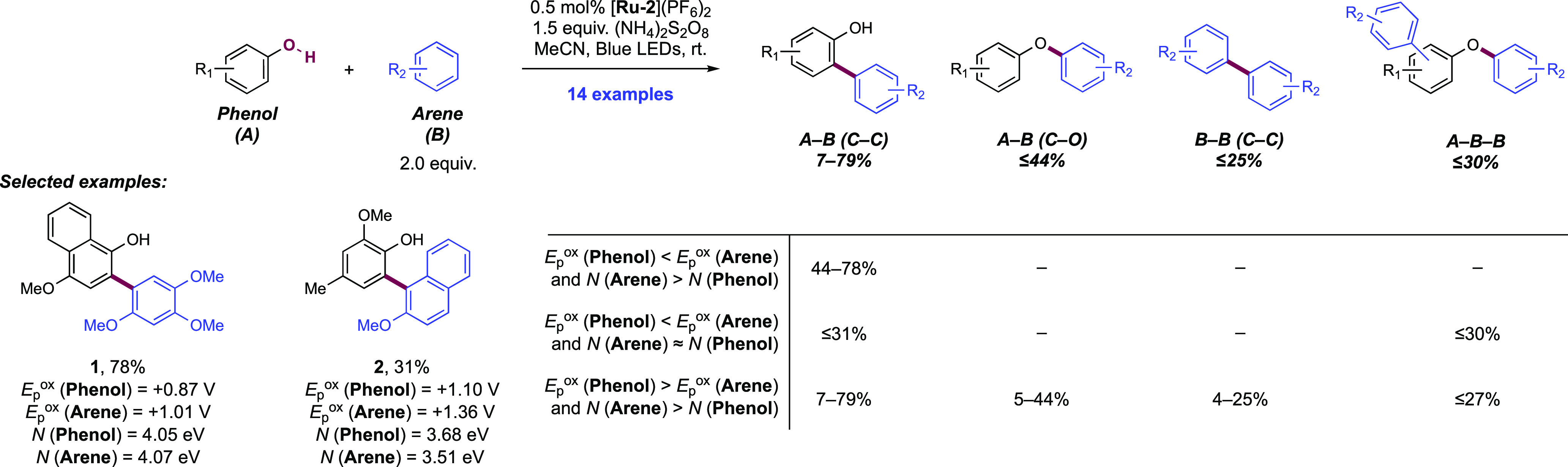
Epox/V vs SCE in MeCN.
The authors propose that the reaction commences with persulfate-mediated oxidative quenching of the photoexcited state of the Ru(II) photocatalyst, thereby generating the strongly oxidizing Ru(III) ground state (E1/2 Ru(II)/Ru(III) = +1.86 V vs SCE in MeCN) and sulfate radical anion. Notably, the sulfate radical anion is oxidizing enough (Ep/2 (SO4•–/SO42–) = +2.18 V vs SCE in MeCN) to oxidize the ground state of the Ru(II) photocatalyst to its Ru(III) state. Either the sulfate radical anion or the Ru(III) species can oxidize either the phenol or arene substrate and thus initiate phenol–arene cross-coupling. The researchers noted that the outcome of C–C vs C–O cross-coupling was dependent upon the oxidation potential (Epox/V) and calculated global nucleophilicity value (N/eV) of both phenol and arene coupling partners. If Epox of the phenol was less than that of the arene, phenol oxidation is thermodynamically favored. When the nucleophilicity of the arene is greater than the phenol, the resulting phenoxyl radical is preferentially attacked by the arene, leading to selective formation of the C–C heterocoupled biaryl in excellent yield (e.g., 198.1, three examples, 44–78%). When the nucleophilicity of the arene and phenol are similar, the C–C heterocoupled biaryl is formed in modest yields, e.g., 198.2 (four examples, up to 31%), with most of the phenol substrate undergoing oligomerization. Although arene oxidation is thermodynamically favored when Epox of the phenol is greater than that of the arene, the C–C heterocoupled biaryl was still formed in modest to excellent yields (seven examples, 7–79%). Notably, the amount of C–C heterocoupled biaryl decreased as the oxidation potential of the phenol increased. As the difference in the coupling partners’ oxidation potentials (i.e., ΔEox = Epox(phenol) – Epox(arene)) increased past 0.1 and 0.2 V, the C–O heterocoupled biaryl product and the homo-coupled biaryl product, respectively, were formed in non-negligible yields. Whereas phenols with high nucleophilicity values favored the formation of the C–O heterocoupled biaryl product, use of less nucleophilic phenols resulted in the formation of both the C–O heterocoupled biaryl product and the homo-coupled biaryl product.
In an effort to establish a non-enzymatic protocol for the challenging ortho-ortho coupling of tyrosine, Kozlowski and co-workers developed a method for homo- and cross-coupling of phenols by pairing a photocatalyst with a terminal oxidant (Scheme 199).546N-Me Mes-Acr+BF4– ([Acr-1]BF4) as the photocatalyst, O2 in an air atmosphere as the oxidant, HFIP as the solvent, and 4,4′-di-tert-butylbiphenyl as an additive proved to be crucial to obtaining high yields of the desired dimer product. Interestingly, the role of biphenyl is two-fold—it can act as a photocatalyst itself, but it can also serve as a radical mediator or co-sensitizer of [Acr-1]BF4. Fluorescence quenching experiments suggested that biphenyl quenches [Acr-1]BF4 to form a biphenyl radical cation, which then oxidizes the phenol monomer more effectively than the photocatalyst does, presumably due to the longer lifetime of the oxidized biphenyl intermediate. The authors proposed that the homo-coupling of phenols begins with phenol oxidation to the corresponding radical cation by either excited-state Acr photocatalyst or the biphenyl radical cation. Molecular oxygen reoxidizes the reduced state of the photocatalyst, forming superoxide anion in the process. The phenol radical cation is then deprotonated by superoxide to afford a phenoxy radical that combines with a molecule of the neutral phenol. An in situ formed peroxyl radical subsequently oxidizes and deprotonates the biphenol intermediate to furnish the dimer product and hydrogen peroxide as a byproduct.
Scheme 199. Photocatalytic Homo- and Cross-Coupling of Phenols (Kozlowski, 2020).

*With 2.0 mol% [Ru-2](PF6)2 photocatalyst.
The authors reported 22 examples of phenol homo-coupling and demonstrated that phenols with 2,4-, 2,4,5-, and 3,4,5-substitution patterns (199.1–199.3) performed well under optimized reaction conditions, providing yields of up to 76%. With adjustments to the photocatalyst, the protocol was extended to enable the catalytic coupling of tyrosine (199.5). Given the characteristics of radical-nucleophile coupling, it was hypothesized that cross-coupling can be achieved if a more readily oxidizable phenol is paired with a phenol containing a more nucleophilic reactive site. By leveraging the more nucleophilic para-position of one phenol partner and employing another substrate with a blocked para site, ortho–para cross-coupling was realized with high yields and regioselectivities (199.6–199.8), with 27 examples of cross-coupled product in yields of 25–97%. Notably, monosubstituted phenols (199.8) were selectively coupled at the para-position despite having multiple reactive sites available.
3.3.4.2. Phenol–Alkene Reactivity
In 2020, the Xia group relied on the visible-light activation of vinylphenols to achieve regiospecific and stereoselective Heck-type arylation with aryl and heteroaryl halide coupling partners (Scheme 200).547 Using a system comprising just Cs2CO3 as a stoichiometric Brønsted base, the arylation of vinylphenols was accomplished with aryl iodides, bromides, and chlorides, with iodides exhibiting higher reactivity than the other halides. In total, 72 examples were presented in yields of 39–92%. Notably, this photochemical method was compatible with a range of functional groups, including carboxylic acids (200.1) and nitro groups (200.2) as well as heteroaryl iodides and bromides (200.3). A variety of vinylphenols could be employed in the transformation without any prior protecting group manipulation, enabling the synthesis of arylated products such as congested tetrasubstituted olefins (200.4) and carbohydrate derivatives like esculin (200.5).
Scheme 200. Photochemical Heck-Type Arylation of Vinylphenols via an Excited-State Phenolate Intermediate (Xia, 2020).

Melchiorre and co-workers had previously shown that excited-state phenolate anions can serve as photoreductants in the activation of perfluoroalkyl iodides.548 Inspired by this precedent, the Xia group investigated whether excited-state phenolate anions (e.g., for the photoexcited state of methyl 4-hydroxycinnamate cesium salt, estimated reduction potential E1/2red = −2.48 V vs SCE in DMSO)547 can similarly reduce aryl halides (e.g., for 4-bromoacetophenone, E1/2 = −1.83 V vs SCE in DMSO)547 to generate aryl radical intermediates. A series of mechanistic experiments revealed that upon addition of base, in situ formed phenolate anion absorbs in the visible spectral region, and the excited state of this anion is effectively quenched with the addition of aryl halide. Ruling out formation of a ground-state EDA complex between the phenolate anion and aryl halide, the authors proposed a mechanism wherein vinylphenol is deprotonated by Cs2CO3 to form the colored phenolate anion, which is then excited under blue-light irradiation. The excited state of the anion initiates one-electron reduction of the aryl halide, forming a phenoxyl radical and a corresponding aryl radical. The latter adds to another molecule of the phenolate anion to afford a radical anion intermediate, which can proceed via HAT or ET/PT to furnish the final arylated product.
3.3.4.3. Aerobic Phenol De-aromatization
Narayan and Baker Dockrey recently presented an elegant, bioinspired solution to the oxidative de-aromatization of orcinaldehyde derivatives, developing a method with air as the terminal oxidant, a natural flavin photocatalyst FMN, and water as solvent (Scheme 201).549 This method avoids the use of synthetic noble metal photocatalysts and the generation of stoichiometric quantities of waste products, which occurs when traditional chemical oxidants are employed for phenol de-aromatization (Pb(OAc)4 and hypervalent iodine reagents, for example).550 The optimized conditions here consisted of blue-light irradiation of orcinaldehyde substrates in the presence of flavin mononucleotide (FMN) photocatalyst in tris pH = 8 aqueous buffer solution under an O2 atmosphere. The use of a mildly alkaline buffered aqueous solution was essential to achieving the desired reactivity, with only a trace amount of conversion of the substrate observed in organic solvents that contained hydride and alkoxide bases or in neutral aqueous conditions. FMN was favored over riboflavin because of solubility.
Scheme 201. Photocatalytic Oxidative De-aromatization of Orcinaldehyde Derivatives (Narayan, 2020).

IBX conditions: 1.1 equiv of IBX, 0.1 equiv of n-Bu4N+I–, 5.0 equiv of TFA, 1,2-DCE, rt.
A scope of seven successful examples of orcinaldehyde de-aromatization was presented in yields of 42–91%. Comparison was made to an IBX-mediated de-aromatization protocol,551 and in all cases this photocatalytic method was superior in reaction yields and ease of product isolation. A C1-EWG was a necessary requirement for reactivity, though aldehyde (201.1–201.3), ketone (201.4), and ester functionality (201.5) were permitted. A phenol (single O–H group) scaffold, as opposed to the resorcinol (two O–H groups) scaffold, was unreactive. An example of a tandem de-aromatization/cyclization of a keto-aldehyde substrate was demonstrated providing azaphilone bicycle 201.6 in an 85% yield. Though currently limited in scope, these highly substituted o-quinols have potential as intermediates toward numerous natural product families.
The observed dependence of reactivity on the use of an alkaline buffer (pH = 8) led the authors to propose that substrate ionization (e.g., for β-orcinaldehyde, pKa = 7.2 in H2O)552 precedes PET. SV fluorescence emission studies indicate that the resulting phenoxide quenches the photocatalyst approximately 200 times faster than the tris buffer (KSV phenoxide = 206/M, compared to KSV tris = 1.13/M, under air).549 Therefore, the photoexcited-state FMN most likely undergoes direct ET with the phenoxide to deliver the phenoxyl radical and FMNSQ semiquinone radical anion. The phenoxy radical reacts with triplet oxygen to yield a peroxy radical, which is reduced to the peroxide anion by the SQ form of the photocatalyst. Notably, this peroxide intermediate was observed by MS. Reduction of the peroxide, which could be mediated by tris buffer,553 subsequently yields the o-quinol product. An alternative mechanism involving a second oxidation of the phenoxy radical to form a phenonium ion intermediate543 and subsequent trapping with water was discounted in experiments using H218O additive as no 18O was incorporated into the product. A flavin photocatalyst had previously been reported to mediate arene-de-aromatizing epoxidation through singlet oxygen generation and nucleophilic attack of the electron-rich arene on singlet oxygen.554 This possibility was also shown to be unlikely as the addition of sodium azide—a quencher of singlet oxygen555—still allowed for the efficient formation of the hydroxylation product.
3.4. Transformations of Oxime and Hydroxamic Acids
In this section of the review, we consider photocatalytic and electrochemical methods involving oximes and hydroxamic acids for the synthesis of nitriles via dehydration of the former class and in C–O and C–N bond formation reactions. An additional review of methods concerning the electrochemistry of these compounds has been presented by Onomura.556 We note that we do not discuss transformations involving TEMPO, PINO, and related nitroxide radicals. Stepwise and concerted PCET pathways have been invoked as mechanisms for the regeneration of the nitroxide radical from the corresponding hydroxylamine when these compounds are applied in a catalytic context. This body of work is extensive and has been thoroughly reviewed by Stahl and co-workers.277
In 2015, Hartmer and Waldvogel were able to generate aryl nitriles in generally good yields (eight examples, 40–81%) through constant current electrolysis of an MeCN solution of aryl aldoxime substrate in an electrochemical cell equipped with graphite anode and Pb cathode (Scheme 202).557 High yields were achieved with substrates bearing electron-donating (e.g., methyl-, methoxy-) ortho-substituents (202.1 and 202.2, respectively); lower yields were observed using 2,6-dichlorobenzaldoxime (202.4) as a substrate and no product was observed using 2,6-dibromobenzaldoxime as a substrate, presumably due to reductive dehalogenation.
Scheme 202. Electrochemical Synthesis of Nitriles via Dehydration of Oximes (Waldvogel, 2015).

Anodic oxidation of the oxime generates an iminoxyl radical that can be further oxidized to produce the nitrile oxide or that can dimerize to produce an aldazine bis-N-oxide, which can subsequently liberate dinitrogen to form benzaldehyde. Across all substrates, this benzaldehyde byproduct was observed. Notably, however, benzaldehyde formation was significantly higher for substrates that lacked ortho-substituents. The researchers posited that the dimerization pathway was favored due to the lack of steric hindrance. In contrast, steric hindrance in ortho-substituted substrates disfavors this dimerization pathway and prompts further oxidation to form nitrile oxides, which are subsequently deoxygenated at the cathode to produce the nitrile product. The choice of cathode material significantly influences deoxygenation of the nitrile oxide intermediate; whereas Pb furnished the desired nitrile product in good yields, other cathode materials such as stainless steel, Ni, Pt, and BDD performed poorly.
To address the scalability of this domino oxidation–reduction reaction, Waldvogel and co-workers adapted this batch reaction to a flow electrochemical cell (Scheme 203).558 The researchers sought to demonstrate this flow process using dichlorobenzaldehyde oxime, which was a challenging substrate in the batch reaction due to over-reduction to the monochloro byproduct. Upon optimization, the yield of the dichlorobenzonitrile increased from 41% in batch (202.4) to 63% in an undivided electrochemical flow-cell (203.1). The use of the flow cell enabled removal of supporting electrolyte from the process. By simply adding a Nafion membrane, the researchers were able to switch the electrochemical cell from operating in an undivided mode to a divided mode, which enabled them to intercept the nitrile-N-oxide intermediate generated upon oxidation. Rather than being reduced at the cathode to form the corresponding nitrile, the researchers used the nitrile-N-oxide in a 1,3-dipolar cycloaddition with methyl acetoacetate to generate a functionalized isoxazole (203.2) in 60% yield. This isoxazole is used in the preparation of the antibiotic dicloxacillin.
Scheme 203. Electrochemical Dehydration, and 1,3-Dipolar Cycloaddition of 2,6-Dichlorobenzaldoxime in a Flow Electrochemical Cell (Waldvogel, 2017).

In 2016, Chen and co-workers reported a protocol for the 1,2-dioxygenation of alkenes via the generation of an electrophilic O-centered radical from the formal homolysis of an oxime O–H bond (Scheme 204).360 The optimized conditions consisted of photocatalyst N-Me Mes-Acr+ ClO4–, ([Acr-1]ClO4), TEMPO co-catalyst, and K2CO3 Brønsted base in MeCN solution under an atmosphere of oxygen and blue-light irradiation. Addition of PPh3 after the photocatalytic reaction is complete afforded the final product via reduction of a peroxide intermediate. The authors reported 10 examples of this transformation with 65–97% yields. The reaction performed well with a variety of substituted alkenes including terminal (204.1, 204.2), 1,1-disubstituted (204.3) and trisubstituted (204.4) alkenes.
Scheme 204. Photocatalytic 1,2-Dioxygenation of Alkenes: The Cyclization of Oximes (Chen, 2016).

The authors proposed that TEMPO acts as a redox mediator between the photocatalyst and oxime substrate. They posited that the excited state of the photocatalyst (E1/2 *1Acr/Acr• = +2.08 V vs SCE in MeCN; *3Acr/Acr• = +1.88 V vs SCE in PhCN)73,74 is quenched by ET from TEMPO (E1/2 = +0.62 V vs Ag/AgCl in MeCN),559 thereby generating the corresponding TEMPO-oxoammonium cation. This species then engages a deprotonated oxime in a single-electron oxidation, generating an O-centered radical. Subsequent cyclization onto the pendant alkene generates a C–O bond and C-centered radical, which is trapped with oxygen to form the hydroperoxy radical. Further ET and PT yields the hydroperoxide product. This work also includes alkene amino-oxygenation from similar hydrazone substrates (for a further mechanistic discussion and details thereof, see section 2.7.1).
In 2018, Xu and co-workers reported the synthesis of polycyclic nitrogen-containing aromatic heterocycles via the generation of oxime radicals and subsequent cyclization onto a pendant aryl ring (Scheme 205).560 The authors observed that the choice of cathode material determined whether they observed the N-heteroaromatic N-oxide product or the corresponding deoxygenated product. Constant current electrolysis of the oxime substrate, TEMPO catalyst, and KOH co-catalyst in MeCN/H2O (1:1) in an undivided electrochemical cell equipped with a RVC anode and Pt cathode at room temperature or 80 °C afforded the phenanthridine N-oxide product. Use of a Pb cathode and increased charge consumption under otherwise similar conditions resulted in deoxygenation. A similar observation was made by Waldvogel in earlier work on the unique ability of Pb cathode material to allow for the reductive deoxygenation of nitrile oxides.557 In this work, Xu proposed that deoxygenation is likely due to the higher overpotential for proton reduction on the Pb cathode, which makes reductive cleavage of the N–O bond more favorable. Under the Pt cathode conditions, the substrate scope included 38 examples of N-heteroaromatic N-oxide products in 21–98% yields and demonstrated functional group tolerance for a variety of halogen (205.2a), alcohol, and heterocyclic substituents. Arene scope is exemplified by the high regioselectivities (>20:1) observed with 2-naphthalene (205.1a) and 3-thiophene (205.3a) aromatic rings and the ability to cyclize substrates derived from variously substituted phenylpyridines. Upon switching to the Pb cathode, a similar scope for the deoxygenated product was demonstrated, albeit with significant amounts of dehalogenation observed in bromide-containing substrates and loss of compatibility with a thiophene-containing scaffold (205.3b).
Scheme 205. Electrochemical Synthesis of N-Heterocycles via Oxime Cyclization (Xu, 2018).

The authors proposed a mechanism wherein TEMPO radical is oxidized at the anode to generate the corresponding oxoammonium salt; this species then undergoes ET from the deprotonated oxime substrate (e.g., for acetone oxime, pKa = 26.0 in DMSO; for the corresponding anion, E1/2ox = −0.57 V vs Fc+/Fc in DMSO)561 to regenerate TEMPO• in addition to the neutral oxime radical. The nascent oxime radical cyclizes onto the pendant arene to generate a C–N bond. Subsequent HAT with TEMPO• restores aromaticity. In an intramolecular competition experiment between cyclization onto a pendant arene or a pendant alkene, cyclization via a 5-exo-trig process occurs exclusively in a manner similar to the photocatalytic reactivity reported by Chen.360 If Pt was used as the cathode, water was reduced to generate hydroxide and molecular hydrogen. If instead Pb was used as the cathode, the phenanthridine N-oxide product underwent coupled reductive deoxygenation.
The benzo[1,2]oxazine motif is a critical component of a number of natural products and pharmaceuticals, including the antiviral raistrickindole A and the antibiotic paeciloxazine. This moiety also provides rapid access to δ-aminoalcohols via facile reductive cleavage of the N–O bond. Traditional syntheses of these cores often rely upon [4+2] cycloaddition strategies, especially Diels–Alder and Povarov-type reactions.562−565 These methods are limited by the use of a preformed diene as the 4π partner and a nitroso compound as the 2π partner. In 2020, Han and co-workers hypothesized that a radical approach might provide access to benzo[1,2]oxazines by using an N-aryl hydroxamic acid as the 4π partner in reactions with olefin partners (Scheme 206).566 These researchers disclosed an electrochemical method for this benzoxazine synthesis. Optimal conditions were reported to consist of a solution of the hydroxamic acid and alkene substrates in a CH2Cl2/HFIP/MeCN (3.5:0.4:0.1) mixture with n-Bu4N+BF4– electrolyte. The reaction was run in an undivided electrochemical cell with an RVC anode and Pt cathode under constant current conditions. A total of 47 examples were reported with yields ranging from 33% to 94%. A wide range of alkenes, composed mostly of terminal and 1,2-disubstituted styrenes (206.1), were shown to be effective coupling partners, as were indoles with substituents at both the C2- and C3-positions (206.2). Dienes and norbornene (206.3) derivatives were also shown to be competent substrates. A number of N-aryl N-hydroxyurethanes were shown to perform well in the reaction. Ortho-, meta-, and para-substitution patterns on the arene were all tolerated, as were electron-donating and -withdrawing moieties.
Scheme 206. Electrochemical [4+2] Annulation and Dioxygenation of Olefins with Hydroxamic Acids (Han, 2020).

The authors proposed sequential oxidation and deprotonation of the hydroxamic acid (e.g., for neutral N-phenyl-N-hydroxy ethyl carbamate, E1/2ox = +1.32 V vs Ag/Ag+ in CH2Cl2/HFIP/MeCN (3.5:0.4:0.1))566 to generate an amidoxyl radical. Anti-Markovnikov addition of this intermediate to the olefin partner affords a C-centered radical species, which undergoes a second oxidation to the corresponding carbocation. In cases where the olefin is oxidized at low potentials (e.g., indole partners), the authors propose the olefin is oxidized to the corresponding radical cation and undergoes subsequent radical–radical coupling with the amidoxyl species to reach the same carbocation species. Finally, an intramolecular electrophilic aromatic substitution completes the annulation to afford the product. This mechanism is supported by EPR data which showed a triplet signal corresponding to an amidoxyl radical upon electrolysis of a solution of the hydroxamic acid.567 Although no change was observed upon the addition of radical trap 2-methyl-2-nitrosopropane (MNP), the addition of both alkene and MNP resulted in the appearance of a sextuplet that corresponded to MNP trapping of the C-centered radical species formed upon addition of the aminoxyl radical to the alkene.
In this same report,566 Han and co-workers were able to extend this platform to a related 1,2-dioxygenation of alkenes. By simply changing the identity of the anode from RVC to C felt under otherwise identical conditions, the authors showed a complete switch in reaction outcome to olefin 1,2-dioxygenation instead of annulation (206.5–206.7) (10 examples, 35–81% yields). Styrenes and unactivated olefins were shown to be effective, though indoles were not. Interestingly, 1,4-dioxygenation (206.8) was achieved when a 1,3-diene was used as the substrate. The reaction is proposed to go through a similar mechanism. After C–O bond formation, however, the resultant C-centered radical is not able to undergo anodic oxidation with the C felt anode and instead undergoes radical–radical coupling with a second equivalent of the hydroxamic acid-derived amidoxyl radical to afford the dioxygenated product.
We note here that a large body of work has been reported by Schmidt, Alexanian, and co-workers demonstrating the synthetic utility of O-centered radicals of hydroxamic acids for 1,2-olefin functionalization, including dioxygenation,568−570 aminooxygenation,571 carboetherification,572 and hydroetherification.573 These proceed through aerobic activation of the weak substrate O–H bond (e.g., for N-tert-butyl N-hydroxybenzamide, O–H BDE = 80 kcal mol–1)26 and, thus, are not explicitly discussed in this Review.
3.5. Catalytic Applications of O-Centered Radicals Formed through Coupled Proton- and Electron-Transfer Processes for Substrate HAT
Instead of relying upon PCET-based strategies to directly activate substrate bound O–H bonds and thereafter bond-forming or -breaking steps, a complementary strategy utilizes a PCET elementary step to activate the O–H bond of a reagent or catalyst. O-centered radicals generated in this manner have found utility as catalytic mediators of substrate HAT, leading to C-centered radical generation and further functionalization. These methods are the subject of this section.
In 2016, Glorius and co-workers demonstrated a catalytic application of the formal homolysis of an O–H bond through coupled PT and ET in devising a strategy for the trifluoromethyl thiolation of unactivated C(sp3)–H bonds (Scheme 207).574 Using a dual catalytic system of Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) and NaOBz in MeCN solution with blue-light irradiation, the authors demonstrated that methine C–H bonds could be selectively activated via intermolecular HAT in the presence of many other methylene and methyl C–H bonds. The C-centered radical generated in this manner then reacted with N-trifluoromethylthiophthalimide (Phth−SCF3) to afford the C(sp3)–H trifluoromethyl thiolation product. In this work, 26 examples of this transformation were reported with yields ranging from 35% to 96% (207.1–207.4). The method was reported to be selective for hydridic C–H bonds (e.g., methine or methylene C–H bonds adjacent to heteroatoms) and generally favored the most sterically accessible site. Unactivated methylene C–H bonds were shown to react only slowly in the absence of other hydridic C–H bonds. Esters, amides, ketones, ethers, and heteroarenes were all well tolerated. However, the major highlight of this work is in its selectivity: for all but four of 24 relevant examples, selectivity proved to be ≥19:1.
Scheme 207. Photocatalytic Trifluoromethyl Thiolation of C(sp3)–H Bonds (Glorius, 2016).

The authors propose a mechanism beginning with single-electron oxidation of the benzoate anion (e.g., for n-Bu4N+BzO–, E1/2ox = +1.40 V vs SCE in MeCN)21 by the photoexcited state of the Ir(III) photocatalyst (E1/2 *Ir(III)/Ir(II) = +0.89 V vs SCE in MeCN),68 thereby generating the potent HAT mediator benzoyloxy radical (O–H BDE = 111 kcal mol–1).574 Despite the apparent thermodynamic unfavorability of this step, SV luminescence quenching studies revealed that n-Bu4N+BzO– does efficiently quench the photoexcited state (KSV = 128 M–1). No photocatalyst quenching was observed from alkane substrate or Phth–SCF3. Benzoyloxy radical subsequently undergoes fast and selective HAT with the substrate’s most hydridic C(sp3)–H bond, which results in the formation of benzoic acid and a C-centered radical species. The authors noted that the rate of HAT (e.g., the rate of C(sp3)–H abstraction from cyclohexane by 4-ClBzO•, k = 1.2 × 107 M–1 s–1)575,576 outcompetes unimolecular decarboxylation of the benzoyloxy radical (e.g., for 4-ClBzO•, k = 1.4 × 106s−1).575 The C-centered radical reacts with Phth–SCF3 to yield product and Phth•, the latter of which undergoes ET with the Ir(II) state of the photocatalyst (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN)68 to regenerate ground-state Ir(III) photocatalyst and phthalimide anion. Finally, phthalimide anion (pKaH = 8.3 in H2O)574 deprotonates benzoic acid (pKa = 4.2 in H2O)574 to restore the benzoate pre-catalyst and produce PhthH as a byproduct. The authors noted that phthalimidyl radical may participate in a radical chain process and could serve to abstract a H-atom from the substrate. Previous literature has suggested that phthalimidyl radical is unselective for methine over methylene C(sp3)–H bond abstraction (3°/2° ≈ 4).577 The researchers observed a quantum yield of Φ = 1.76, suggesting an average chain length of roughly 2.0. These data indicate that the chain mechanism is likely in effect, although not to a significant degree.
The next year, Glorius and co-workers extended this method of benzoyloxy radical-mediated HAT to the alkynylation of formyl C(sp2)–H bonds (Scheme 208).578 Optimal reaction conditions consisted of the blue-light irradiation of formyl substrate and an ethynylbenziodoxolone (EBX) reagent in the presence of [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 ([Ir-9]PF6) photocatalyst, sodium 2-iodobenzoate co-catalyst 208.A, and benziodoxolone acetate (BIOAc) additive in 1,2-DCE solution. A total of 36 examples of formyl C(sp2)–H alkynylation were reported in 46–90% yields. The scope consisted primarily of substrate aldehydes (208.1, 208.2), but formate esters (208.4) and formamides (208.3) also proved to be suitable substrates. Both alkyl or aryl aldehydes performed well in the reaction, and ethers, esters, sulfonamides, indoles, and (benzo)thiophenes were all well tolerated. Aryl- and silyl-substituted EBX reagents were demonstrated to be successful alkynylation reagents.
Scheme 208. Photocatalytic Alkynylation of Formyl C(sp2)–H Bonds (Glorius, 2017).

The proposed mechanism of the transformation relies upon the generation of a benzoyloxy radical from the corresponding benzoate through a similar activation mechanism as described in the authors’ previous report on C(sp3)–H trifluoromethyl thiolation reactions.574 This radical species then abstracts the formyl H-atom to generate an acyl radical and regenerate benzoic acid. The former species then adds to the EBX reagent to give alkynylated product after elimination of benziodoxolonyl radical, which exists in equilibrium with 2-iodobenzoyloxy radical. This species can undergo ET with the Ir(II) complex to generate additional 2-iodobenzoate anion. Alternatively, the additive BIOAc can serve as a sacrificial oxidant to regenerate ground-state photocatalyst and an equivalent of acetate anion. SV quenching studies confirm the role of 2-iodobenzoate sodium salt; no other reaction component was reported to quench the excited state of the photocatalyst. Generation of the acyl radical is supported by isolation of the corresponding TEMPO adduct in radical trapping studies.
In recent years, the phosphoryl radical, generated from one-electron oxidation of a phosphate anion, has been implicated in several transformations involving HAT from and functionalization of aliphatic C(sp3)–H bonds. The Nicewicz and Alexanian groups proposed that the use of highly oxidizing Acr photocatalyst N-Ph Mes-t-Bu2-Acr+BF4– ([Acr-3]BF4) enables the generation of an oxygen-centered phosphoryl radical, which then serves as a HAT mediator for the abstraction of unactivated C(sp3)–H bonds (Scheme 209).579 The use of stoichiometric K3PO4 or pH 8 aqueous phosphate buffer proved to be key to the catalytic system; luminescence quenching experiments demonstrated that an analogous dibasic phenyl phosphate, employed as a substitute for K3PO4 due to the latter’s insolubility in a range of solvents, quenched the excited state of Acr while the conjugate acid did not. In total, 31 examples demonstrating this catalytic system were reported in 30–91% yield. The authors first investigated the compatibility of this protocol with C(sp3)–H azidation, employing a sulfonyl azide as the radical trap, and found that azidation of cycloalkanes (209.1), adamantane (209.3, 209.4), derivatives of dihydrocitronellol (209.2), and drug analogs (209.3, 209.5) proceeded smoothly. Site-selectivity generally aligned with the activation of the most electron-rich C(sp3)–H bond in the substrate. Further C–H functionalization of cyclooctane with a variety of radical trapping reagents—halogenation, trifluoromethyl thiolation, and alkylation—was also demonstrated (209.6–209.8). Additionally, dibasic phenyl phosphate was examined as a sub-stoichiometric base for the C–H azidation of cyclooctane and was shown to produce up to 48% yield of product, consistent with catalytic activity of the base; however, optimal yields generally required stoichiometric quantities of K3PO4 or pH 8 phosphate buffer.
Scheme 209. Photocatalytic Aliphatic C(sp3)–H Functionalization via Phosphoryl Radical-Mediated HAT (Alexanian, 2018).

The transformation is hypothesized to proceed through the photoexcitation of the Acr photocatalyst (E1/2 *Acr+/Acr• = +2.08 V vs SCE in MeCN)76 and oxidation of the phosphate salt to generate a phosphoryl radical intermediate. This species then abstracts a C(sp3)–H bond from the substrate, generating a C-centered radical that engages with arylsulfonyl azide to furnish the desired product and a sulfonyl radical. The authors propose that the ejected sulfonyl radical oxidizes the reduced state of the photocatalyst to regenerate the active Acr catalyst, thus turning over the catalytic cycle.
Interestingly, concurrent with this report, Kanai and co-workers disclosed a similar C(sp3)–H cyanation strategy with a few notable differences (Scheme 210).580 Rather than using a stoichiometric amount of pre-formed phosphate base, the authors employ a catalytic amount of a BINOL-derived hydrogen phosphate ((±)-210.A) in the presence of [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 ([Ir-9]PF6) photocatalyst and K2CO3 Brønsted base. In the transformation, it is proposed that excited-state [Ir-9]PF6 (E1/2 *Ir(III)/Ir(II) = +1.30 V vs Fc+/Fc in MeCN)29 oxidizes the in situ formed phosphate anion (e.g., for BINOL potassium phosphate, E1/2ox = +1.12 V vs Fc+/Fc in MeCN)580 to the phosphoryl radical as it is unable to oxidize the corresponding phosphoric acid (e.g., for BINOL hydrogen phosphate, E1/2ox = +1.34 V vs Fc+/Fc in MeCN)580 in the absence of a base. The phosphoryl radical (O–H BDE = 102.4 kcal mol–1)580 abstracts the C(sp3)–H bond from the α-position of a heteroatom-containing substrate, and the resulting C-centered radical traps TsCN to afford the cyanation product. The extruded p-toluenesulfonyl radical is reduced by the Ir(II) species, thereby regenerating the photocatalyst. Although this protocol was only applied to the activation of α-heteroatom C–H bonds, it was able to achieve good catalytic efficiency in a range of heterocycles and acyclic N-Boc amines (210.1–210.4) as well as a methionine-containing dipeptide (210.5, 210.5′). In the latter example, while cyanation of the thioether group was generally unselective between the two α-positions, all other C–H bonds in the molecule, including weak benzylic sites, were unaffected.
Scheme 210. Photocatalytic Aliphatic C(sp3)–H Cyanation via Phosphoryl Radical-Mediated HAT (Kanai, 2018).

*With 3.0 equiv of TsCN.
4. S-Centered Radical Generation from S–H Bonds through Photochemical and Electrochemical PCET Processes
4.1. Synthesis of S(II) Functional Groups
The generation of thiyl radical through the homolysis of aryl and alkyl S–H bonds is a well-studied transformation in radical chemistry, often accessed through use of small molecule radical initiators, UV photolysis, or thermolysis. In the following section of this Review, we introduce a number of synthetic transformations that utilize concerted or stepwise MS-PCET homolysis of S–H bonds to accomplish C–S, S–S, S–N, and S–P bond formation toward products in the S(II) oxidation state.
4.1.1. C–S Bond Formation through Alkene and Alkyne Hydrothiolation, Oxythiolation, and Aminothiolation
Beginning with a survey of recent archetypal examples of thiol–ene and thiol–yne reactions using PCET-generated thiyl radicals, we progress then to alkene and alkyne oxy- and aminothiolation.
4.1.1.1. Photocatalytic Alkene and Alkyne Hydrothiolation (Thiol–Ene)
In an early example of the photocatalytic activation of thiol S–H bonds via stepwise PCET, Yoon and co-workers in 2013 disclosed a system enabling the intermolecular thiol–ene coupling reaction of thiols and olefins to generate sulfide and thioester products (Scheme 211).581 The blue-light irradiation of a MeCN solution of these components in the presence of [Ru(bpz)3](PF6)2 ([Ru-2](PF6)2) photocatalyst proved optimal. Notably, earlier work from Matsuda established that the fluorescence of the more-common dye [Ru(bpy)3]2+ ([Ru-1]2+) is not quenched by neutral thiols, demonstrating the necessity of the more oxidizing Ru photocatalyst used in this work.582,583 Using this procedure, a broad array of thiol and olefin partners were accommodated in the transformation. Primary, secondary, and tertiary alkyl thiols, including benzyl thiol (211.1, 211.2) and Boc-Cys-OMe (211.3) all proved to be competent substrates for the reaction. Initial optimization demonstrated excellent performance with styrene derivatives (211.3–211.5). In addition, aliphatic olefins were also amenable to functionalization (211.1, 211.2). Notably, the thiol–ene reaction was tolerant of unprotected hydroxyl groups (211.2) and hydrolytically sensitive functionality such as vinyl esters and allylic halides. Despite the efficacy of the reaction, one of the primary limitations is the requirement of excess of thiol (4.0 equiv) with respect to the olefin starting material.
Scheme 211. Visible-Light Photocatalytic Thiol–Ene Reaction (Yoon, 2013).

The reaction is proposed to proceed via direct reductive quenching of the photoexcited-state Ru(II)complex (E1/2 *Ru(II)/Ru(I) = +1.45 V vs SCE in MeCN)65 by the thiol substrate (e.g., for benzyl mercaptan, E1/2ox = +0.45 V vs Fc+/Fc in MeCN)336 to produce a reduced Ru(I) complex and the corresponding thiol radical cation. The latter readily loses a proton (e.g., for the radical cation deriving from benzyl mercaptan, pKa = 2.4 in DMSO)336 to form a thiyl radical, which then adds across the olefin partner with anti-Markovnikov regioselectivity. The resultant carbon-centered radical abstracts a H-atom from another equivalent of thiol yielding thiol–ene product while concomitantly regenerating thiyl radical, thus propagating a chain reaction.584 The reduced Ru(I) complex (E1/2 Ru(II)/Ru(I) = −0.86 V vs SCE in MeCN)339 is oxidized to Ru(II) by reduction of O2, regenerating the ground-state photocatalyst and superoxide. Superoxide generated in this manner may act as a base to enable deprotonation of the thiol radical cation or as a HAT reagent to further initiate thiyl radical.
In an effort to develop a more efficient photocatalytic thiol–ene reaction, Yoon and co-workers later examined conditions with thiol as limiting reagent instead of olefin, as was the case in their earlier method, and found drastically reduced reactivity (Scheme 212).585 Despite the significant thermodynamic driving force for ET from the thiol to the excited-state Ru-photocatalyst (e.g., for benzyl mercaptan, E1/2ox = +0.45 V vs Fc+/Fc in MeCN);336E1/2 *Ru(II)/Ru(I) = +1.45 V vs SCE in MeCN),65 Yoon and co-workers observed that thiols did not significantly quench the luminescence of [Ru(bpz)3](PF6)2,581 suggesting a significant kinetic barrier for oxidation. To address this challenge, the authors began investigating additives which could better mediate this ET event. Initial optimization examined inorganic base additives to drive formation of more easily oxidized thiolate anions, but was met with no success. When p-toluidine, a potential redox mediator, was instead used as an additive, the reaction rapidly proceeded to complete conversion in only 30 min. To unearth the mechanistic rationale for this dramatic rate enhancement, BDE, basicity, and oxidation potential were systematically varied across a series of aniline additives to determine any correlation with reaction performance. It was found that neither N–H BDE nor pKa correlated significantly with yield, arguing against this additive acting as either a HAT mediator or Brønsted base additive, respectively. However, the oxidation potential of the aniline additive showed good correlation, suggesting its role as a redox mediator.
Scheme 212. Dual Photocatalytic/Aniline-Catalyzed Thiol–Ene Reaction (Yoon, 2014).

*Reaction conducted in water.
In this proposed mechanism, the excited-state Ru(II) photocatalyst instead oxidizes p-toluidine to the corresponding aminium radical cation which then undergoes either sequential single-site ET and PT or concerted single-site PCET to generate the key thiyl radical intermediate. Under these conditions, reactivity for substrates which previously exhibited low yields with limiting thiol was drastically improved by addition of p-toluidine. Notably, the reaction is also compatible with unprotected amine and carboxylic acid functionalities and allows for efficient thiol–ene conjugation using glutathione and peptide-derived olefins (212.3) in water. Several other research groups have since utilized these mediated photooxidative reaction conditions for thiol–ene processes in a variety of contexts.586−589 However, due to the single-site nature of the oxidant and base in this method, we consider these works to be outside of the scope of this Review, and hence an in-depth discussion is not offered.
In 2015, Greaney and co-workers reported a protocol for a photocatalytic thiol–ene reaction utilizing TiO2 NPs as a heterogeneous photocatalyst to promote PET (Scheme 213).590 The authors reported the use of TiO2 (P25) to achieve this transformation, and demonstrated 19 examples of alkene hydrothiolation in 30–98% yields. The optimized conditions consisted of visible-light irradiation of alkene and thiol (4 equiv) in the presence of TiO2 in MeCN under an air atmosphere. Under these conditions, primary (213.1, 213.2, 213.4) and secondary thiols were tolerated, along with an example using thiophenol (213.3). However, reaction of t-butyl mercaptan afforded no product. Additionally, the authors observed varying degrees of background reactivity in the absence of TiO2 photocatalyst. The degree of background reactivity depended on the thiol identity, ranging from 0% (213.1) to 93%. Control reactions revealed this reactivity was due to ambient light, as reactions with titania in the dark afforded no conversion to product.
Scheme 213. Photocatalytic Titanium Dioxide-Promoted Thiol–Ene (Greaney, 2015).

*With 10 mol% TiO2. **In the absence of TiO2. ***With 100 mol% TiO2.
The authors propose that the reaction is initiated by light absorption by TiO2, promoting an electron from the VB into the CB of the material. The hole thus formed in the VB is quenched by oxidation of thiol to the corresponding thiyl radical cation. The electron promoted to the CB reduces oxygen to regenerate the ground state of the titania photocatalyst. Deprotonation of the thiyl radical cation generates a neutral thiyl radical that adds into alkene, forming a C–S bond and a C-centered radical. This C-centered radical undergoes HAT with another molecule of thiol, affording product and generating another thiyl radical, which can then propagate a chain mechanism.
In 2016, Ananikov and co-workers disclosed a photoredox-mediated thiol-yne reaction for the preparation of sulfenylated dienes (Scheme 214).591 In an effort to develop a metal-free variant of this transformation, green light irradiation of EY was employed with alkyne and thiophenol substrates and pyridine Brønsted base co-catalyst, in hexane solution. Hexanes were found to be the optimal solvent due to the metered dissolution of the sparingly soluble EY, counteracting the loss of reactivity due to gradual photocatalyst decomposition.
Scheme 214. Organophotocatalytic Thiol–Yne Reaction (Ananikov, 2016).

The reaction was demonstrated for 16 examples of vinyl arylsulfide product formation in yields of 55–91%, with very high anti-Markovnikov regioselectivity and E-olefin stereoselectivity observed. Both electron-rich (214.1) and electron-deficient aromatic thiols (214.2) were well tolerated. Sterically hindered alkynes also performed well (214.3, 214.4). However, use of aliphatic thiols resulted in a significant decrease in stereoselectivity (<10:1 E/Z). The reaction is proposed to be initiated via reductive quenching of the excited state of EY (E1/2 *EY/EY•– = +0.83 V vs SCE in MeCN)71 with the thiol substrate (e.g., for thiophenol, E1/2ox = +0.95 V vs Fc+/Fc in MeCN),336 generating a thiol radical cation. Facile deprotonation of this radical cation (e.g., for the radical cation of thiophenol, pKa = −11.7 in DMSO)336 by the basic pyridine additive (pKaH = 12.5 in MeCN)48 affords a thiyl radical that undergoes anti-Markovnikov addition to the alkyne substrate. HAT between the resulting vinyl radical and another equivalent of thiol yields the closed-shell product, generating an equivalent of thiyl radical in what is likely a radical chain mechanism. Turnover of the photocatalyst is proposed to result from reduction of a trace amount of molecular oxygen in solution by the reduced photocatalyst (E1/2EY/EY•– = −1.06 V vs SCE in MeCN).71 This same catalytic system was utilized in 2020 by Weaver and co-workers for the prenylation of thiophenol using a sulfone reagent, affording the corresponding sulfide in 91% yield.592 After thiyl radical generation and addition across the olefin moiety as in the above manner, elimination of phenylsulfonyl radical affords the product. Reduction of this radical by EY•– returns the ground-state photocatalyst with phenylsulfinate anion as a byproduct.
In 2017, Wang and co-workers reported a protocol for a visible-light-mediated organophotocatalytic thiol–ene reaction for the coupling of mercaptan and alkene derivatives (Scheme 215).593 Optimal conditions consisted of irradiation of thiol and alkene substrates with N-Me Mes-Acr+BF4–([Acr-1]BF4) photocatalyst in MeCN at room temperature. In total, 28 examples of thiol–ene products were given with yields ranging from 73% to 90%. A variety of alkenes were tolerated, including disubstituted (215.1), terminal (215.2) and styrenyl (215.3) olefins. In addition to benzyl mercaptan (215.1–215.3), secondary (215.4) and tertiary (215.6) aliphatic thiols performed well. An example using thiophenol was also demonstrated, forming thiol–ene product in 88% yield (215.5). The authors also demonstrated the utility of this thiol–ene protocol as a “click” reaction, conjugating a variety of biologically relevant molecules including S-linked nucleoside 215.7 and glycoconjugate 215.8.
Scheme 215. Light-Mediated Thiol–Ene Reaction through Organic Photoredox Catalysis (Wang, 2017).

The authors propose the reaction is initiated via a stepwise sequence of ET and PT. Initially, the excited-state photocatalyst (E1/2 PC•/*PC+ = +1.88 V vs SCE in PhCN)74 oxidizes the neutral thiol (e.g., for benzyl mercaptan, Ep/2ox = +0.45 V vs Fc+/Fc in MeCN)336 to generate the corresponding thiyl radical cation. Deprotonation forms the neutral thiyl radical (e.g., for the radical cation derived from benzyl mercaptan, pKa = 2.3 in DMSO),336 which undergoes anti-Markovnikov addition across an alkene, forging a C–S bond. The resulting C-centered radical engages another molecule of thiol in HAT, forming the product and another molecule of thiyl radical. This thiyl radical can then form additional product, thereby facilitating a chain propagation mechanism. The reduced state of the photocatalyst subsequently reduces molecular oxygen to regenerate the photocatalyst ground state for another initiation process. Superoxide formed in the process likely acts as a base to mediate PT steps.
In 2019, the authors expanded the scope of this transformation to obtain thiol–yne and 1,2-bis-hydrothiolation products from the coupling of thiols with alkynes (Scheme 216).594 Irradiation of thiol (4.0 equiv) and alkyne with N-Me Mes-Acr+BF4–([Acr-1]BF4) photocatalyst in MeCN afforded eight examples of thiol–yne reactivity with yields ranging from 70% to 95%. Hindered thiols such as tertiary thiols (216.1) afforded vinyl sulfide products through the single addition of the thiol to the alkyne, whereas secondary and primary mercaptans afforded double addition products. For example, tert-butyl mercaptan afforded 83% of the vinyl sulfide product (216.1, E/Z = 1:3.5); however, cyclohexyl- and benzyl-substituted mercaptans underwent a second addition to afford the 1,2-bis-hydrothiolation products 216.2 and 216.3, respectively. For reactions of unhindered thiols, mono-addition was difficult to achieve, even with sub-stoichiometric quantities of mercaptan. The authors further extended this protocol to form glycoconjugation products. Irradiation of thiosugar and alkyne substrates in MeCN containing N-Me Mes-Acr+BF4– ([Acr-1]BF4) photocatalyst afforded the mono-thiolated vinyl sulfide products, providing seven examples with yields ranging from 61% to 68%. This protocol was demonstrated for conjugation reactions of glucosyl thiol with a variety of alkyne-tagged amino acids and peptides, including Boc-protected glycine (216.4) and Fmoc-protected serine (216.5) derivatives.
Scheme 216. Photocatalytic Thiol–Yne Reactions (Wang, 2019).

*With 1 mol% [Acr-1]BF4, 1.0 equiv of thiol, 1.7 equiv of alkyne.
In 2019, Dilman and co-workers reported a light-mediated thiol–ene reaction that was proposed to proceed through a concerted PCET mechanism for thiol S–H bond activation (Scheme 217).595 The authors employed two different organic photocatalysts with moderate Brønsted basicity, a benzimidazole-containing compound 217.A and acridine orange 217.B, such that the single catalyst could serve as both the photooxidant and Brønsted base to mediate concerted PCET for S–H bond activation. Catalyst 217.A was found to be optimal for relatively acidic thiols, whereas the more-basic 217.B was best for less acidic thiols, notably aliphatic thiols. A low dielectric solvent, cyclohexane, was employed to favor concerted PCET by mitigating the formation of charged intermediates resulting from stepwise pathways for CT and PT. In this work, 32 examples of olefin hydrothiolation were reported in yields of 58–99%. A diverse array of thiols was amenable to this transformation, including aromatic (217.1) and aliphatic (217.2) thiols. Notably, the presence of unprotected alcohols and amines on the thiol coupling partner did not hinder reactivity. Thioacetic acid also proved to be a competent substrate, providing access to a variety of thioacetyl-substituted products (217.3-217.5) using 217.A as a catalyst. The alkene scope was similarly broad, including examples of terminal (217.4) and internal (217.3, 217.5) olefins as well as ones containing heterocycles and sensitive functionality, such as alkyl halides.
Scheme 217. Light-Mediated Thiol–Ene Reaction through Concerted PCET (Dilman, 2019).

The authors propose that the key thiyl radical intermediate is generated through a concerted PCET event between the photoexcited catalyst and the thiol substrate, which is consistent with results from UV–vis absorption and SV quenching experiments. Due to the combined Brønsted basicity and oxidizing capability of the photocatalyst excited state, the proton and electron from the thiol substrate are transferred to different sites within the same catalyst. The concerted pathway was suggested over stepwise ET/PT or PT/ET pathways through the observation that the UV–vis absorption profile of 217.B in CH2Cl2 was not altered through the addition of thioacetic acid, implying the absence of an initial PT step. However, addition of thioacetic acid in MeCN resulted in a new absorption feature at 490 nm, which was assigned to be the conjugate acid of 217.B. After PCET-mediated S–H bond homolysis, the resulting thiyl radical adds across the olefin to give the regioisomer consistent with formation of the most stabilized carbon-centered radical. To form the closed-shell product, this intermediate radical then engages with either the open-shell catalyst species, turning over the catalytic cycle, or with another molecule of thiol, propagating a radical chain. Indeed, the quantum yield (Φ = 6) indicated significant participation of a radical chain process.
Expanding on the utility of the photocatalytic thiol–yne reaction, Volla and co-workers in 2019 disclosed a protocol for a thiol–yne annulation cascade for the synthesis of sulfenylated dihydrochromenones (Scheme 218).596 Green light irradiation of DMF solutions containing EY photocatalyst, alkyne-tethered cyclohexadienone substrate, and thiol coupling partner, afforded 35 examples of the cascade annulated products in excellent yield (80–96%). With regard to the thiol component, thiophenols containing EDGs and EWGs were well tolerated (218.1–218.3). The lone exceptions to this were pentafluorothiophenol and thioacetic acid, both of which afforded only a trace amount of the C–S coupled product. Additionally, aliphatic thiols were also competent reaction partners, with examples including chiral pool-derived thiols such as N-Ac cysteine (218.4) and glucose-derived thiols. The requisite cyclohexadienone partner also showed tolerance to a range of substitution, including substitution to the aryl alkyne (218.5–218.7). Aromatic groups tolerated included those substituted with esters, ethers, ketones, and halides, in addition to nitroarenes and thiophenes. However, terminal alkynes only afforded traces of the desired product. A variety of alkyl (primary and secondary) and aryl substituents were also accommodated at the R1 position on the dieneone substrate.
Scheme 218. Thiol–Yne Annulation Cascade for the Synthesis of Sulfenylated Dihydrochromenones (Volla, 2019).

The authors conducted a number of experiments to probe the mechanism of this transformation. Luminescence quenching experiments indicated that the emission intensity of the excited-state photocatalyst was reduced with increasing concentrations of thiol but was unaffected by increasing alkyne. Furthermore, addition of radical quenchers such as TEMPO, BHT, and CuCl2 inhibited the reaction, which the authors attributed to the generation of radical species in the reaction. Additionally, no product was observed upon addition of disulfide to the reaction instead of thiol, suggesting that disulfide is not an on-cycle intermediate in this reaction. Based on these experiments, the authors proposed a mechanism wherein the excited state of the photocatalyst (E1/2 *EY/EY•– = +0.83 V vs SCE in MeCN)71 oxidizes the thiol substrate to generate the corresponding thiol radical cation. The neutral thiyl radical is then formed through deprotonation by O2•–, which is generated by reaction of O2 with EY•–. This thiyl radical undergoes addition to the alkyne in a thiol–yne reaction, forming a C–S bond and a C-centered vinyl radical. The vinyl radical is then positioned to undergo addition to the pendant cyclohexadienone, forming the bicyclic product. The authors then propose that the resulting α-acyl radical can then undergo HAT with another equivalent of thiol to propagate the reaction in a chain process.
In 2020, Du and co-workers disclosed a system for the conversion of enones to α-sulfenylated aldehydes following an unexpected C–C bond cleavage (Scheme 219).597 Blue-light irradiation of a MeCN solution of enone and thiol, in the presence of [Ir(ppy)3] (Ir-1) as photocatalyst and under an O2 atmosphere, afforded 26 examples of functionalized β-sulfenylated alcohols (13–86% yields), after reductive workup with NaBH4. Reaction scope investigation was primarily conducted using 4-chlorothiophenol, but several other aryl thiols substituted with halogens, alkyl groups, or esters also proved amenable to the transformation. In contrast, pyridine-derived thiols afforded no product and alkyl thiols afforded exclusively products of conjugate addition without C–C bond cleavage. Enone components were typified by a range of substituted chalcones (219.1, 219.2), tolerating aryl substitution with nitriles and halogens, but a single example of a cyclohexyl-substituted enone also proceeded in good yield (219.3). Substitution with heteroarenes abrogated reactivity. Generally, the use of substrates wherein electron-rich 4-methoxyphenyl ketones resulted in more efficient reactivity than the corresponding phenyl compound. Product diversification was then demonstrated through functionalization of the resultant α-sulfenylated aldehyde toward hydrazone formation, reduction, and Grignard addition. The authors also included a three-step synthesis of a PPAR modulator analogue through enone formation, the titular decarbonylative sulfenylation, and a Suzuki–Miyaura cross-coupling in 61% overall yield.
Scheme 219. Preparation of β-Sulfenylated Alcohols through Photoredox Catalysis (Du, 2020).

Mechanistically, the authors propose an initial oxidation of thiol (e.g., for 4-chlorothiophenol, Ep/2ox = +1.33 V vs Fc+/Fc in MeCN)336 by photoexcited-state Ir(III) (E1/2 *Ir(III)/Ir(II) = +0.31 V vs SCE in MeCN)66 or autoxidation, followed by proton loss to generate thiyl radical. Oxidation of the thiol by photocatalyst is unlikely given the large potential difference. Following thiyl radical generation, this reactive intermediate adds to the enone, affording an α-carbonyl radical, which is subsequently trapped by 3O2 to afford an alkyl peroxide radical intermediate. Trace amounts of conjugate addition product observed are rationalized as resulting from polarity mismatched HAT to the α-carbonyl radical instead of from thiol. The alkyl peroxide radical is proposed to then fragment to yield α-sulfenylated aldehyde product through multiple possible mechanisms. Observed byproducts of this cleavage included benzaldehyde and benzoic acid. Photocatalyst turnover is posited to occur through reduction of disulfide (formed through thiyl radical dimerization) or of alkyl peroxide radical to the corresponding anion.
Huang and co-workers reported in 2019 a formal oxidative [3+2] cycloaddition between 2-mecaptobenzimidazoles and ketones via a photocatalytic α-thiolation/annulation sequence. Thiyl radical addition across an intermediate aluminum enolate provides access to thiazolidine-containing products (Scheme 220).598 This reaction utilized molecular oxygen as a terminal oxidant as well as a stoichiometric LA additive to activate the carbonyl substrate to enolate formation. Optimal conditions used RB as a photocatalyst, AlCl3 as the LA, and MeCN as the solvent under an atmosphere of O2 with blue-light irradiation. A scope of 23 examples of α-thiolation/annulation was disclosed with yields of 29–70%. Both cyclic (220.1, 220.2) and acyclic ketones (220.3) were tolerated. All reactions proceeded with high selectivity for C–S bond formation over the alternative α-amination. Acyclic aliphatic ketones also proved effective, though in lower yields than cyclic ketones. The scope further included aldehydes, such as 5-chloropentanal (220.4) and 2-ethylhexanal, which performed comparably to the cyclic ketone substrates. Tetralone and cyclooctanone were shown to undergo successful α-thiolation, though subsequent annulation was not observed.
Scheme 220. Photocatalytic Tandem α-Thiolation/Annulation of Carbonyls with 2-Mercaptobenzimidazoles (Huang, 2019).

The inclusion of radical inhibitors such as TEMPO and BHT resulted in precipitous drops in yield, suggesting the involvement of open-shell intermediates. While SV quenching experiments established that both 2-mercaptobenzimidazole and ketone substrates quench the RB photoexcited state, the 2-mercaptobenzimidazole substrate was much more efficient quencher. Therefore, the authors proposed that excited-state RB (E1/2 *RB2–/RB3–• = +0.99 V vs SCE in H2O)349 undergoes SET with the 2-mercaptobenzimidazole substrate (Ep/2ox = +0.81 V vs SCE in MeCN),21 forming the corresponding substrate radical cation and RB3–•. Subsequent deprotonation of the radical cation affords the thiyl radical that can engage the in situ formed aluminum enolate in a radical addition step. HAT with hydroperoxyl radical and subsequent tautomerization yields an oxocarbenium ion that can be trapped by the tethered imidazole moiety to afford the desired annulated product after dissociation of the LA and PT. The ground-state RB photocatalyst is regenerated through reaction with molecular oxygen (E1/2RB2–/RB3–• = −0.78 V vs SCE in H2O).349
4.1.1.2. Photocatalytic and Electrochemical Alkene and Alkyne Oxy- and Aminothiolation
In 2018, Kshirsagar and co-workers disclosed a protocol for the 1,2-aminothiolation of alkynes via a radical cascade annulation reaction (Scheme 221).599 This reaction begins with the addition of thiyl radical across the pendant alkyne of a 1-azido-2-(phenylethynyl)benzene substrate, followed by subsequent C–N bond formation with loss of molecular nitrogen to afford an indole product. Reaction of the alkyne and thiophenol substrates with EY photocatalyst in a solution of MeCN under air atmosphere afforded 21 examples of 2-substituted 3-sulfenylindole products in 58–86% yields. The reaction tolerated electron-deficient substituents on the aryl azide (221.2), but substrates with EDGs were not reported. Aryl (221.1, 221.2) and aliphatic substituents (221.3) on the alkyne were also tolerated. In addition to thiophenols, heteroaromatic thiols (221.4) and aliphatic thiols (221.5) also performed well, affording product in 70% and 72% yields, respectively.
Scheme 221. Photocatalytic 1,2-Thioamination of Alkynes for the Synthesis of 3-Sulfenylindoles (Kshirsagar, 2018).

Control reactions revealed only trace or no formation of product in the absence of light, and only traces of the product in the absence of photocatalyst and oxygen. Additionally, the authors noted suppression of the reaction in the presence of added TEMPO. Based on these control experiments, the authors propose a mechanism for this transformation, starting with initial oxidation of thiol (e.g., for thiophenol, E1/2ox = +0.95 V vs Fc+/Fc in MeCN)336 by the photoexcited-state EY photocatalyst (E1/2 *EY/EY•– = +0.83 V vs SCE in MeCN).71 This is followed by deprotonation of the resulting radical cation by O2•–, generated via reduction of O2 by reduced state EY (E1/2EY/EY•– = −1.06 V vs SCE in MeCN),71 affording the neutral thiyl radical intermediate. This radical then adds across the alkyne substrate yielding a vinyl radical, which cyclizes onto the pendant azide to form a C–N bond and release molecular nitrogen. The resulting N-centered radical undergoes HAT to form the closed-shell product.
In contrast to extensive research on catalytic alkene dihydroxylation and aminohydroxylation,600,601 direct three-component oxysulfenylation and aminosulfenylation of alkenes using thiols and O- or N-nucleophiles respectively has remained relatively underexplored due to the ease with which thiophenols and thiols can be overoxidized to sulfoxides and sulfones.602−604 Lei and co-workers posited that the overoxidation of thiophenols and thiols could be avoided by replacing a chemical oxidant with an electrochemical cell, whose current and potential can be precisely set (Scheme 222).605 Constant-current electrolysis of an MeCN solution of a thiol, alkene, and alcohol substrates in an undivided electrochemical cell using a graphite anode and a Pt cathode at 40 °C furnished the desired oxysulfenylated product (38 examples, 20–95%). With respect to the alkene substrate, a variety of styrenes bearing electron-donating (222.1, 222.2) or halogen (222.3) substituents in the ortho, meta, or para positions were well tolerated. Notably, α-methylstyrene also proved amenable to oxysulfenylation. In addition to aryl-substituted alkenes, aliphatic alkenes such as cyclohexene also proved to be competent substrates (222.4). With respect to the thiol, a variety of thiophenols bearing electron-neutral or halogen (222.1–222.6) substituents were effective, as were aliphatic thiols such as benzyl mercaptan and cyclohexyl mercaptan (222.7). Though initially optimized with a variety of aliphatic alcohols, the reaction also proved to be compatible with other O-nucleophiles (e.g., AcOH, H2O). Having established this electrochemical oxysulfenylation methodology, the authors extended the scope to include aminosulfenylation by replacing the O-nucleophile with an amine under the same reaction conditions. A variety of primary aliphatic (222.8) as well as primary and secondary (hetero)aromatic amines (222.9, 222.11, 222.12) furnished the aminosulfenylated product (12 examples, 30–74%).
Scheme 222. Electrochemical Oxysulfenylation and Aminosulfenylation of Alkenes (Lei, 2018).

*With 10.0 equiv of amine.
Control electrolysis experiments in the presence of TEMPO furnished a TEMPO-thiol adduct and those in the absence of alkene substrate afforded the disulfide, suggesting the formation of thiyl radicals under the electrochemical reaction conditions. The authors posited that oxidation and subsequent deprotonation of thiophenol or thiol at the anode generates these thiyl radicals, which subsequently add to the alkene in an anti-Markovnikov fashion. The resulting carbon-centered radical is then oxidized at the anode to form a carbocation intermediate and subsequently trapped by the O/N-nucleophile to generate the oxysulfenylated or aminosulfenylated product. Concomitant reduction of protons at the cathode generates molecular hydrogen.
Mei, Du, Han, and co-workers contemporaneously developed an electrochemical methodology for the three-component hydroxy- and alkoxysulfenylation of alkenes (Scheme 223).606 Constant potential electrolysis of an MeCN solution of thiol, alkene, and water or alcohol, in an undivided electrochemical cell using Pt electrodes at room temperature furnished the oxysulfenylated product (43 examples, 38–85%). A variety of thiophenols bearing electron-rich, electron-neutral, or halogen substituents (223.1–223.3). With respect to the alkene substrate, a variety of α-substituted styrenes bearing electron-donating or halogen substituents in the para- and meta-position were well tolerated. Ortho-substituents on the styrene were also tolerated albeit in diminished yields relative to the para- and meta- derivatives. Though initially optimized with water, the reaction also proved to be compatible with other O-nucleophiles such as primary alcohols. Notably, the authors were able to synthesize thio-substituted γ-lactones by tethering a carboxylic acid to the alkene substrates and utilizing modified conditions with added TsOH hydrate as Brønsted acid catalyst (223.9–223.11). The authors posited that oxidation and subsequent deprotonation of thiophenol–thiol at the anode generates thiyl radicals, which subsequently add to the alkene in an anti-Markovnikov fashion. The resulting carbon radical is then oxidized at the anode to form a carbocation, which is trapped by the O-nucleophile to generate the oxysulfenylated product. Concomitant reduction of protons at the cathode generates molecular hydrogen. This proposal aligns with that of Lei in their related work.
Scheme 223. Electrochemical Hydroxy- and Alkoxysulfenylation of Alkenes (Mei, Du, and Han, 2018).

*With 3.0 equiv of thiophenol, 1.0 equiv of p-TsOH·H2O, in MeCN.
4.1.2. C–S Bond Formation through (Hetero)arene C(sp2)–X Thiolation
We present here a variety of methods for the thiolation of (hetero)aryl halides through photochemical and electrochemical activation. Many of these are complementary in nature to existing transition-metal-mediated C(sp2)–S cross-coupling methodologies.607 In 2016, Oderinde, Johannes, and co-workers reported a dual photoredox/Ni-catalyzed cross-coupling of thiols and (hetero)aryl iodides via the intermediacy of a thiyl radical generated through a proposed stepwise combination of ET and PT (Scheme 224).608 The combination of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) photocatalyst, NiCl2·glyme co-catalyst, dtbbpy ligand, and pyridine base, in MeCN under blue LED irradiation was optimal for the desired C(sp2)–S bond-forming reaction. Reactivity was insensitive to oxygen concentration. Control experiments revealed a total lack of reactivity in the absence of the pyridine base. Regarding the scope of the transformation, a total of 30 examples were given in 30–97% yields. Iodoarenes were a necessary requirement, with only trace amounts of product formation observed when the corresponding bromoarenes were employed. However, with this requirement met, a broad functional group tolerance was demonstrated—for example, primary aniline (224.1), carboxylic acid (224.2), aldehyde (224.3), and BPin (224.4) substituents examples given. Heteroaromatic iodides were showcased, including pyrazole (224.5), pyridine, pyrimidine (224.6), indole, and thiophene examples given. With respect to the thiol component, aryl, benzyl, and alkyl thiols were competent coupling partners. Only when 1-adamantyl thiol was used was a significant decrease in reaction yield observed (224.7). Boc-protected cysteine (Cys) underwent the desired arene thiolation reaction without competitive decarboxylation (224.8).
Scheme 224. Dual Photoredox/Ni Catalysis for the Cross-Coupling of (Hetero)aryl Iodides and Thiols (Oderinde and Johannes, 2016).

In a departure to typical observations of dual photoredox/Ni-catalysis for cross-coupling with C-centered radical intermediates,62 authors here noted that Ni(0) precatalysts were ineffective at promoting the desired transformation, in the absence or presence of molecular oxygen. Authors demonstrated through time-resolved SV luminescence studies that the thiol component of the reaction efficiently quenches the excited state of the employed Ir(III) photocatalyst (kSV = 7.6 × 105 M–1 s–1). On the basis of these data and of CV experiments, the following mechanistic proposal was made. Visible-light excitation of the Ir(III) chromophore to its excited state and ISC yields a long-lived triplet excited-state complex. This engages in single-electron oxidation (E1/2ox *Ir(III)/Ir(II) = +1.21 V vs SCE in MeCN)68 with the thiol reaction component (e.g., for benzyl thiol, Ep/2ox = +0.83 V vs SCE in MeCN)336 to generate the corresponding thiol radical cation and a ground-state Ir(II) complex. Deprotonation of the acidified thiol radical cation (e.g., for benzyl thiol radical cation, pKa = 2.4 in DMSO)336 by pyridine base (pKa = 3.4 in DMSO)48 yields the neutral thiyl radical. Authors also note that a concerted PCET mechanism may be operative in this transformation, but that prior discrete ionization of the thiol to generate thiolate prior to ET is unlikely (e.g., for neutral benzyl thiol, pKa = 15.4 in DMSO).336 The thiyl radical intercepts an in situ generated Ni(I) complex, formed through an off-cycle single-electron reduction event presumably mediated by the Ir(II) state of the photocatalyst. The resulting Ni(II) thiolate intermediate is proposed to be reduced to the corresponding Ni(I) complex in reaction with the Ir(II) ground-state complex (E1/2ox Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN).68 Oxidative addition with the aryl iodide partner generates a Ni(III) aryl thiolate intermediate, which undergoes reductive elimination via C(sp2)–S bond formation to generate the product and catalytically active Ni(I). Authors also postulate that pyridine is effective in stabilizing coordinatively unsaturated Ni(I) halide and thiolate intermediates.
In 2017, Fu and co-workers disclosed a photocatalytic system for the arylation of thiols using aryl halides (Scheme 225).609 Using homoleptic photocatalyst Ir(ppy)3 (Ir-1) in conjunction with Cs2CO3 as a base in DMF under visible-light irradiation at ambient temperature, Fu and co-workers demonstrated 42 examples of aryl sulfide formation with yields ranging between 40% and 94%. During initial investigations of scope of thiol arylation with aryl bromides and iodides, the system was found to be similarly efficacious with both electron-rich and -poor aryl and heteroaryl halides (225.1, 225.2). Driven by greater availability of aryl chlorides compared to bromides and iodides, Fu and co-workers then subjected a range of aryl and heteroaryl chlorides to the arylation conditions. Intriguingly, despite the sluggish cross-coupling reactivity of aryl chlorides under more traditional transition metal cross-coupling conditions, aryl chlorides (225.3) and even fluorides proved to be a competent substrate class for this photocatalytic arylation, albeit with somewhat diminished yields compared to the corresponding aryl bromides and iodides. Generally, electron-poor aryl chlorides performed on par with analogous bromides and iodides, while electron-rich chloroarenes were less effective. The substrate scope focuses on aryl thiols with a single example of an alkyl thiol, n-BuSH, which performed significantly less efficiently than thiophenols (225.4).
Scheme 225. Photocatalytic Cross-Coupling of Aryl Halides and Thiols (Fu, 2017).

The reaction is proposed to proceed through initial thiophenol deprotonation (e.g., for 3-methylbenzenethiol, pKa = 10.6 in DMSO)336 by Cs2CO3 (pKa = 10 in H2O) and subsequent oxidation of thiolate anion (e.g., for 3-methylbenzenethiolate, Ep/2ox = −0.39 V vs Fc+/Fc in DMSO)336 by the photoexcited Ir(III) catalyst (E1/2 *Ir(III)/Ir(II) =+0.35 V vs SCE in CH2Cl2)610 to generate thiyl radical and a reduced-state Ir(II) complex. The strongly reducing Ir(II) complex (E1/2 Ir(III)/Ir(II) = −2.19 V vs SCE in MeCN)66 then undergoes SET with the aryl halide substrate to form the corresponding arene radical anion (e.g., for chlorobenzene, Ep/2red = −1.60 V vs Ag+/Ag in DMF),609 which then undergoes nucleophilic displacement with another equivalent of thiolate to yield the diarylsulfide radical anion and liberated halide anion. Finally, ET to thiyl radical forms the product diarylsulfide while generating another equivalent of thiolate. As is typical for radical processes, TEMPO inhibits the reaction and TEMPO adducts of the thiyl radical were observed by HRMS. The formation of multiple sulfur-centered radical species (thiyl radical and sulfide radical anion) is supported by the observation of a second sulfur-centered spin in ESR trapping assays of reaction mixtures with spin trap 5,5-dimethylpyrroline N-oxide. Additionally, no evidence of carbon-centered radical species was observed in the ESR, ruling out discrete aryl radical formation through the mesolytic cleavage of the intermediate radical anion.611
In a similar vein, Glorius and co-workers disclosed a system for the selective arylation of thiols with heteroaryl chlorides and bromides (Scheme 226).612 Using the combination of thiol, and heteroaryl chloride or bromide substrates and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) as a photocatalyst, 39 examples of thiol heteroarylation were achieved with yields between 40% and 99%. Initial exposition of heteroaryl halide scope was performed with methyl 3-mercaptopropionate as a thiol and showed efficacy with a range of five-membered-ring heterocycles including thiazoles (226.1), imidazoles (226.2–226.6) pyrroles, thiophenes, and furans, while concomitantly demonstrating tolerance of ester, amide, and polyhalogenated arenes without significant amounts of products arising from multiple C–S couplings. Interestingly, the reaction shows complete chemoselectivity for the arylation of a dichloroimidazole in the presence of a chlorinated pyridine amide. Thiol scope exploration using N-methyl-2,3-dichloroimidazole as haloarene partner revealed broad compatibility with a range of electronically diverse alkyl and aryl thiols, including thiosaccharides (226.4) and thiophenol derivatives (226.5), along with peptide, steroid, and biotin-derived alkyl thiols.
Scheme 226. Photocatalytic Cross-Coupling of Heteroaryl Chlorides and Thiols (Glorius, 2020).

*Indicates minor site of thiolation. **With 1.0 equiv of thiol, 1.5 equiv of aryl halide, 1.2 equiv of K2HPO4. ***With 3.0 equiv of thiol.
Mechanistically, this reaction is proposed to take a different course than that put forth by Fu and co-workers.609 After demonstrating the intermediacy of thiyl radical through competition experiments with thiol–ene, and demonstration of arylation reactivity in the presence of radical initiators, Glorius and co-workers put forth a preliminary mechanistic proposal. A thiyl radical is initially formed through oxidation of the neutral thiol substrate (e.g., for benzyl mercaptan, Ep/2ox = +0.45 V vs Fc+/Fc in MeCN)336 by Ir(IV) (E1/2 Ir(IV)/Ir(III) = +1.69 V vs SCE in MeCN)68 and subsequent loss of a proton (e.g., for the radical cation deriving from benzyl mercaptan, pKa = 2.4 in DMSO).336 Neither aryl halide nor thiol substrates were found to quench the luminescence of the photoexcited-state Ir(III) complex as demonstrated in SV quenching experiments, precluding generation of Ir(IV) through on-cycle substrate-mediated oxidative quenching. Also, addition of an external oxidant (K2S2O8) in equal stoichiometry to photocatalyst removed an induction period otherwise observed, lending credence to the proposed role of Ir(IV). The thiyl radical derived from thiol oxidation and proton loss then adds to the halogenated heteroarene to generate a delocalized radical, which ejects halide anion to yield the corresponding radical cation of the sulfenylated heteroarene. From here, the authors propose two possible pathways for further reactivity: (i) propagation of a radical chain by PCET between the aforementioned arene radical cation and another equivalent of thiol to generate closed-shell products and thiyl radical or (ii) ET from photoexcited-state Ir(III) (E1/2 (Ir(IV)/*Ir(III) = −1.21 V vs SCE in MeCN)68 to regenerate Ir(IV). The radical chain mechanism is supported by the high reaction quantum yield (Φ > 8) and the previous observation of productive C(sp2)–S bond formation with initiation by AIBN.
In 2019, Wang and co-workers disclosed a system for the thiolation of aryl iodides using a electrochemically promoted Ni catalysis (Scheme 227).613 Under constant voltage conditions of thiol and aryl iodide substrates in DMA solution in an undivided cell (graphite felt anode, foamed Ni cathode, LiBr electrolyte) with NiBr2(glyme), di-tert-butylbipyridine (dtbbpy) as ligand, and pyridine as Brønsted base additive, 42 examples of C(sp2)–S bond formation were demonstrated in 38–99% yields. Thiol scope as canvassed using 4-cyano- and 4-trifluoromethoxy iodobenzene revealed the conditions to be amenable for use with electron-rich and -neutral aryl thiols (227.1, 227.2), including those bearing unprotected anilines and phenols, along with alkyl thiols (227.3). Electron-poor aryl thiols are absent from the disclosed scope. The authors then demonstrate the aryl iodide scope to include both aryl and heteroaryl iodides as competent substrates. Additionally, a range of functionality is tolerated on the aryl iodide partner without incident, including ketones, esters, aldehydes, aryl bromides and pinacol boronates.
Scheme 227. Electrocatalytic Cross-Coupling of (Hetero)aryl Iodides and Thiols (Wang, 2019).

The proposed mechanism initiates with cathodic reduction of the Ni(II) precatalyst to reactive Ni(0), which then undergoes oxidative addition with the aryl iodide substrate to form a Ni(II) aryl complex. Thiol substrate is oxidized at the anode to the corresponding radical cation, which, following deprotonation by pyridine, affords thiyl radical. CV experiments with 4-methoxythiophenol showed multiple oxidation events between +0.5 and 2.0 V vs SCE in MeCN, but addition of pyridine coalesced these features to a single oxidation event centered at Ep/2 = +1.04 V vs SCE in MeCN.613 The authors do not invoke concerted PCET generation of thiyl radical, rather favoring a discrete oxidation–deprotonation mechanism. The Ni(II) aryl complex is then intercepted by the thiyl radical to form a Ni(III) aryl thiolate complex, which subsequently undergoes reductive elimination to liberate product sulfides and Ni(I). The requisite Ni(0) active catalyst can then be regenerated via cathodic reduction. The authors ruled out the intermediacy of bromine radicals (generated by either Ni–Br homolysis,614,615 or anodic oxidation of bromide—Epox (Br•/Br–) = +0.69 V vs SCE in MeCN)616—through an extensive series of trapping experiments with TEMPO, 1,1-diphenylethylene, and diallyl diethylmalonate.
Contemporaneously, Mei and co-workers disclosed a similar system for (hetero)arene thiolation using aryl bromides and chlorides instead of aryl iodides as coupling partners (Scheme 228).617 Under constant current electrolysis of aryl halide and thiol substrates in DMF solution in an undivided cell (sacrificial Mg anode, Ni foam cathode, LiBr electrolyte), Mei and co-workers described 47 examples of C(sp2)–S bond formation in 50–95% yields. The method proved amenable to use of a wide range of heteroaryl (228.1, 228.2) and aryl bromides (228.3, 228.4, 228.5), while tolerating ethers, esters (228.3, 228.5), nitro groups, aldehydes, ketones, phenols and nitriles. Aryl chloride scope was less extensively explored, but the system showed comparable efficiency to disclosed aryl and heteroaryl bromides. Aryl halide substrate scope was canvassed using 4-methylthiophenol, but a number of other electron-rich, electron-poor, and heteroaryl thiols also proved compatible.
Scheme 228. Electrochemical Cross-Coupling of (Hetero)aryl Halides and Thiols (Mei, 2019).

Contrary to the mechanism proposed by Wang and co-workers,613 Mei and co-workers instead propose a Ni(II)/Ni(0) mechanism. The proposed mechanism again begins with cathodic reduction of Ni(II) to Ni(0) followed by oxidative addition to the aryl halide substrate to form Ni(II) aryl complex. At this point, thiolate, generated by cathodic reduction of thiol substrate (e.g., for 4-methylbenzenethiol, Ep/2red = +0.07 V vs SCE in MeCN)115 with H2 evolution, displaces halide to form an aryl Ni(II) thiolate, which subsequently undergoes reductive elimination to form product sulfide and reform Ni(0). The authors mention that a thiyl radical-centered mechanism through Ni(I)/Ni(III) aligning to the above proposal of Wang613 could not be ruled out. Disulfide was consistently observed in addition to the desired C–S coupling product. As this could potentially arise from thiyl radical dimerization, Mei and co-workers probed for thiyl radical generation through CV and TEMPO trapping experiments. Using the same cell assembly used in the preparative reaction, CV of 4-methylthiophenol was attempted to determine oxidation potential of the thiolate. Instead of thiolate oxidation, they observed oxidation of the Mg anode (Eonsetox = −0.7 V vs Ag/Ag+ in MeCN),617 but upon changing anode material to Al or glassy carbon, oxidation of the thiolate was observed at Eonsetox = −1.2 and −0.8 V vs Ag/Ag+, respectively. From these data, Mei and co-workers conclude that thiyl generation from anodic thiolate oxidation is feasible under the reaction conditions.
4.1.3. C–S Bond Formation through (Hetero)arene and Alkene C(sp2)–H Thiolation
Lei and co-workers reported in 2017 an electrochemical method for the cross-coupling of electron-rich arenes and (hetero)aryl thiols (Scheme 229).618 Using Pt electrodes in an undivided cell operating under constant-current conditions with LiClO4 as an electrolyte, this method provides access to a variety of diaryl sulfide products, with examples demonstrated on gram scale. The metal-free nature of this protocol avoids complications from catalyst poisoning by thiol or thiolate ligation.619 In this work, 37 examples of C(sp2)–H thiolation were presented with yields of 24–99%. With N-methylindole as the C–H coupling partner, thiophenols bearing electron-donating (229.2) as well as electron-withdrawing (229.3, 229.5) substituents were well tolerated. Heteroaryl thiols also underwent cross-coupling, yielding products containing a variety of nitrogen-, oxygen-, and sulfur-containing heterocycles (229.4). Unprotected indoles proved to be competent coupling partners as well as other electron-rich arenes such as alkoxy-and amine-substituted benzenes (229.5, 229.6). Diminished yields were observed with less electron-rich arenes, such as pentamethylbenzene, which afforded a 24% yield of product with 4-chlorothiophenol.
Scheme 229. Electrochemical C(sp2)–H Thiolation of Indoles and Electron-Rich Arenes (Lei, 2017).

Constant current = 6 mA.
Support for the involvement of open-shell intermediates came from the observation that TEMPO and BHT shut down reactivity. Additionally, a radical trapping experiment with triethyl phosphite implicated an indole radical cation intermediate in the mechanism through isolation of the product of indole 3-phosphorylation. Based on these observations, the authors propose that this coupling reaction proceeds through oxidation of both the thiol (e.g., for thiophenol, Ep/2ox = +1.51 V vs SCE in MeCN)21 and electron-rich arene substrates (e.g., for indole, Ep/2ox = +1.16 V vs SCE in MeCN)21 at the anode, producing the corresponding radical cations of each species. Deprotonation of the thiol radical cation (e.g., for the radical cation deriving from thiophenol, pKa = −11.7 in DMSO)336 generates a thiyl radical that then engages with the arene radical cation in radical–radical C–S bond formation. Deprotonation then yields the product. Reduction of thiol and liberation of hydrogen gas is proposed to complete the circuit in this cell. We note that an aerobic photocatalytic variant of such an indole C(sp3)–H sulfenylation reaction was published by Fan, Chen, and Guo in 2017, proceeding through proposed singlet oxygen generation and S–H bond HAT for thiyl radical generation.620
In 2018, Barman and co-workers disclosed a photocatalytic method for the C(sp2)–H sulfenylation of imidazopyridines with thiols (Scheme 230).621 Blue-light (450 nm) irradiation of a mixture of imidazopyridine, thiol, and RB as photocatalyst in DMSO under an ambient air atmosphere afforded 28 examples of sulfenylated imidazopyridines with good to excellent yields (69–96%) (230.1–230.4). With regards to imidazopyridine scope, substitution was tolerated at several positions, with functional groups including halogens, nitriles, ethers and arenes. Additionally, four examples of indole C(sp2)–H sulfenylation are also demonstrated, affording C3-selective C–S bond formation. Compatible thiols included several substituted electron-rich and electron-poor thiophenols, naphthalene thiols, and two primary alkyl thiols with no major diminution in yield noted for specific substitution patterns.
Scheme 230. Photocatalytic C(sp2)–H Sulfenylation of Imidazopyridines (Barman, 2018).

Mechanistically, the authors propose initial ET from the thiol substrate to the excited state of the RB photocatalyst to generate thiol radical cation and the reduced photocatalyst. ET from the reduced state of the photocatalyst to molecular oxygen then affords superoxide and regenerates ground-state RB. Thiol radical cation is then proposed to be deprotonated by superoxide to yield thiyl radical and hydroperoxide radical. Following this, thiyl radical adds to imidazopyridine forming a carbon centered radical which then undergoes ET to hydroperoxide radical to afford a carbocation. Hydroperoxide deprotonates the carbocation intermediate to re-aromatize and yield the sulfenylated imidazopyridine product.
In 2019, Lei, Tang, and co-workers disclosed a method for the C3-selective C(sp2)–H sulfenylation of imidazo[1,2-a]pyridines under electrochemical conditions (Scheme 231).622 Under constant current conditions in an undivided cell (carbon rod anode, Ni plate cathode, n-Bu4N+PF6– supporting electrolyte) in 5% MeOH in MeCN at 40 °C under N2, 28 examples of this oxidative cross-coupling between thiols and imidazopyridines were disclosed with yields of 16–90%. Thiol scope, assessed with 2-phenylimidazopyridine as substrate, consisted of substituted select examples of alkyl thiols (231.1) and a broader range of aryl thiols (231.2–231.5). Arene substitution and electronics had minimal impact on reaction performance, except in the case of 4-methoxybenzenethiol, which was less efficient (231.2, 231.3, 231.4). Aryl, alkyl, and halogen substitution at the 5-, 6-, 7-, and 8-positions are tolerated on the imidazopyridine coupling partner (231.5, 231.6). Fully unsubstituted imidazopyridine performed poorly, yielding only 16% of the desired sulfenylated product. The authors also demonstrated the method on gram scale with only minor diminution in yield. The mechanistic proposal aligns with their 2017 work on the C3-selective C(sp2)–H thiolation of indoles.618
Scheme 231. Electrochemical C3 C(sp2)–H Sulfenylation of Imidazopyridines (Lei and Tang, 2019).

*Constant current = 6 mA, 55 °C.
Li and co-workers disclosed a similar system in 2020 for the C(sp2)–H sulfenylation of quinoxaline-2(1H)-ones with thiols under electrochemical conditions (Scheme 232).623 With constant current electrolysis of substrate solutions in DMF in an undivided cell (Pt electrodes, n-Bu4N+BF4– supporting electrolyte), in the presence of AcOH Brønsted acid activator at ambient temperature, 31 examples of oxidative cross-coupling between thiols and quinoxalinones were disclosed in yields of 15–81%. Li and co-workers observe that alkyl thiols are more efficacious coupling partners than aryl thiols in this method. Primary, secondary, and tertiary alkyl thiols (232.1, 232.2, 232.3), including those bearing arene, ester, and olefin substituents, were demonstrated to be successful substrates. Lower yields were observed with furfuryl thiol and allyl thiol (232.3) than with the other alkyl thiols examined. Aryl thiols performed poorly, with sulfenylation yields not exceeding 20%. The quinoxaline scope demonstrated tolerance of a range of core substitutions (including halogens, nitriles, alkyl groups, esters) (232.4, 232.5), albeit all with lower yields (23–73%) than the parent unsubstituted quinoxaline. N-alkylated and arylated (232.6) quinoxalines were also compatible with the transformation. Finally, the transformation is demonstrated on multigram scale with only slightly decreased performance.
Scheme 232. Electrochemical C(sp2)–H Sulfenylation of Quinoxalin-2-ones (Li, 2020).

The mechanism was proposed to begin with anodic oxidation and PT of the thiol substrate to generate thiyl radical. Thiyl radical then adds to the protonated quinoxaline to yield a radical cation, which can be anodically oxidized further with loss of a proton to form the sulfenylated product. Generated protons are finally reduced to molecular hydrogen at the cathode surface. The authors determined that the corresponding disulfides are not competent thiolating reagents and yield no C–S coupling product when used under standard reaction conditions.
In 2020, Lenardão, Silviera and co-workers disclosed a photocatalytic method for the C(sp2)–H sulfenylation of indolizines (Scheme 233).624 Using EY as a photocatalyst, blue-light irradiation of DMSO solutions of indolizine and thiol substrates under an air atmosphere, 30 examples of C3-selective C(sp2)–H mono-sulfenylation were described with yields of 24–88%. With respect to the thiol component, six thiophenols (233.1, 233.2) and one aliphatic thiol were demonstrated to be efficacious reaction partners, with lower yields observed for 2-chlorobenzenethiol and n-propanethiol. For the 2-arylindolidizine component, different para-substituents (H, Me, OMe, Cl) (233.3, 233.4) on the peripheral arene are included as substrates, though diminished yields are observed with 2-(4-methoxyphenyl)indolizine. A similar mechanistic proposal to that of the 2018 work of Barman on the C–H sulfenylation of imidazopyridines was made.621
Scheme 233. Photocatalytic aerobic C(sp2)–H sulfenylation of indolizines (Lenardão and Silviera, 2020).

In 2020, Wu and co-workers expanded on this suite of arene C(sp2)–H thiolation methods with the publication of a cross-dehydrogenative electrochemical coupling of thiols and arenes rather than heteroarenes (Scheme 234).625 Reaction conditions consisted of constant current electrolysis of thiol and arene substrates in undivided cell with Pt electrodes, n-Bu4N+PF6– in DCE/HFIP (3:1) at ambient temperature. A total of 40 examples of arene C(sp2)–H sulfenylation were disclosed with 42–99% yields. Though predominantly electron-rich arene substrates are a prerequisite, a number of substitution patterns and functional groups are tolerated under this mode of activation, including aryl halides (F, Cl, Br, I) (234.2), carbamates (234.3), unprotected secondary and tertiary amines (234.4), alcohols, indoles (234.5), and amides. Efficient C–S bond formation was observed even with hindered 2,6-disubstituted thiophenols, with major diminution in yields only seen with ortho- and meta-electron-withdrawing substituents. The scope of the methodology is underscored by demonstration of arene sulfenylation on a number of active pharmaceutical ingredients (APIs) including atomoxetine, metaxalone (234.3), ranolazine (234.4), and tadalafil (234.5).
Scheme 234. Electrochemical C(sp2)–H Thiolation of Arenes (Wu, 2020).

*With n-Bu4N+BF4– electrolyte in HFIP solvent. **Indicates minor site of arylation.
Mechanistically, the transformation is proposed to proceed through a different route than prior examples of heteroarene sulfenylation. Initially, thiyl radical is generated through sequential anodic oxidation (e.g., for 4-methoxybenzenethiol, Ep/2ox = +0.67 V vs Fc+/Fc in MeCN)336 and proton loss (e.g., for the radical cation deriving from 4-methoxybenzenethiol, pKa = −8.6 in DMSO).336 The nascent thiyl radical then dimerizes to form the corresponding disulfide. The disulfide is then further oxidized at the anode to the corresponding radical cation (e.g., for bis(4-methoxybenzene) disulfide, Ep/2ox = +1.28 V vs SCE in MeCN).21 Following this, arene substrate engages the disulfide radical cation in an SEAr step. The finding that fluoroalcohol solvents such as HFIP or TFE were critical to the success of the reaction was rationalized through stabilization of the radical cation intermediates. Finally, the C–S cross-coupled product is formed following loss of a proton and thiyl radical. Concomitant reduction of protons at the cathode liberated molecular hydrogen. Wu and co-workers include multiple pieces of evidence to support the proposed electrophilic aromatic substitution mechanism. By monitoring concentrations of thiol substrate, disulfide and C–S coupled product, it was demonstrated that the reaction has an induction period for product formation which coincides with nearly full conversion of thiol to disulfide. When disulfide was used as a substrate instead of thiol, no induction period for product formation was seen. Additionally, regioselectivity for C(sp2)–H thiolation are consistent with other Pummerer-type electrophilic aromatic substitution reactions.626 However, the authors concede that they cannot rule out the further oxidation of the disulfide radical cation to trisulfur cationic species which would also be a competent electrophile for the observed sulfenylation.
Lei and co-workers were able to generate highly functionalized heterocycle 2,3-fused indolines through an electrooxidative [4+2] de-aromative annulation (Scheme 235).627 Although the reaction was initially optimized using diols (i.e., ethylene glycol), it also proved to be highly efficient with mercaptoalcohols (e.g., mercaptoethanol, 3-mercaptopropan-1-ol) and a dithiol (i.e., ethane-1,2-dithiol). Constant current electrolysis of an MeCN/CH2Cl2 (5:4) solution containing the indole substrate, mercapto alcohol or dithiol, and n-Bu4N+BF4– as supporting electrolyte using carbon cloth electrodes afforded the S,O- or S,S-substituted indoline products in generally good yields (nine examples, 52–76%). The reaction tolerated a bromide substituent on the indole substrate (235.3) and proved effective on C3-substituted and 2,3-disubstituted indoles (235.1–235.4). CV studies revealed that the indole substrate (e.g., N-Ac-3-methylindole), mercaptoethanol, and ethane-1,2-dithiol begin to be oxidized around Eonset = +1.20 V vs Ag/AgCl in MeCN,627 suggesting that all of these components are oxidized under electrolysis conditions. EPR experiments with the radical trapping agent DMPO under standard electrolysis conditions provided evidence for thiol oxidation as an alkyl thiol radical adduct was observed. Subsequent electrolysis experiments in the presence of triethyl phosphite resulted in indole phosphorylation, suggesting the formation of an indole radical cation under electrolysis conditions. Based on these mechanistic studies, the authors posited that the [4+2] de-aromative annulation proceeded through coupling of the thiyl radical with the indole radical cation. The authors speculated that C–S bond formation exclusively occurred at the C3 position due to the stabilization of the indole radical cation. Subsequent intramolecular nucleophilic attack by −OH (in the case of mercaptoethanol) or −SH (in the case of dithiol) to the iminium cation affords the corresponding product. The authors noted that the reaction was first-order in indole and zero-order in mercaptoethanol. This result suggests that anodic oxidation of the indole substrate to the radical cation is the rate-limiting step within this transformation and that thiol oxidation to thiyl radical, radical–radical coupling, and cascade nucleophilic attack are relatively fast elementary steps.
Scheme 235. Electrochemical [4+2] De-aromative Annulation of Indoles (Lei, 2020).

In 2019, Lei and co-workers utilized a similar scheme to achieve the sulfenylation of β-aminoacrylates, a class of enamines (Scheme 236).628 Under constant current electrolysis of enamine and thiol substrates in MeCN in an undivided cell with carbon anode, Fe cathode, and n-Bu4N+BF4– supporting electrolyte at ambient temperature, 28 examples of sulfenylation were disclosed in yields of 53–92%. Demonstrated thiol scope, with (Z)-ethyl 3-(allylamino)-3-phenylacrylate as a model enamine, included both aryl (236.1, 236.3), and heteroaryl (236.2) thiols with performance largely unperturbed by structural variation, with the notable exception of p-(trifluoromethyl)thiophenol (236.1). Enamine scope tolerates alkyl and aryl substitution on both nitrogen and β-carbon sites, again with only minor diminution in performance compared to the model substrate.
Scheme 236. Electrochemical C(sp2)–H Sulfenylation of β-Aminoacrylates (Lei, 2019).

Lei and co-workers demonstrate the generation of both thiyl and carbon-centered radicals under the reaction conditions through trapping with DMPO and fitting of observed EPR spectra to calculated spectra of the anticipated trapping products. As such, the authors propose a mechanism in which thiyl radical (formed through successive anodic oxidation and proton loss) undergoes radical–radical coupling with enamine radical cation (formed through anodic oxidation) to forge a new C–S bond. The resulting α-sulfenylated iminium loses a proton to afford the sulfenylated enamine product. The authors also posit that thiyl radical could also be generated by reduction of disulfide to the radical anion followed by mesolytic cleavage, as evidenced by the competence of disulfide in place of thiol under the electrolytic conditions, while showing no reactivity without electricity.
Following this, in 2020, Lei, Lu, Gao, and co-workers demonstrated the α-sulfenylation of N,N-dimethylenaminone derivatives under similar electrochemical conditions (Scheme 237).629 Under constant current electrolysis of enamine and thiol substrates in an undivided cell (C rod anode, Pt plate cathode, n-Bu4N+BF4– supporting electrolyte in MeCN) at ambient temperature, 28 examples of this C(sp2)–H oxidative cross-coupling were disclosed in yields of 24–87%. The scope includes a range of aryl thiols and aryl enaminones. Notably, alkyl and heteroaryl thiols were incompatible with the reaction and gave no C–S bond formation. To demonstrate the method’s selectivity for enaminone sulfenylation, thiol scope was assessed using an enaminone bearing a distal olefin (237.1, 237.2), and in all cases, the distal olefin remained untouched. Use of disulfide instead of thiol also yielded productive C–S bond formation, albeit in reduced yield. The reaction could also be performed on gram-scale with slightly reduced yield. Finally, enaminone products could be readily converted to 3-sulfenylisoxazoles upon treatment with methanolic hydroxylamine hydrochloride in excellent yield. A mechanistic proposal similar to their 2019 work was made.628
Scheme 237. Electrochemical C(sp2)–H Sulfenylation of N,N-Dimethylenaminones (Lei, Lu, and Gao, 2020).

4.1.4. C–S Bond Formation Involving Nitrile, Isonitrile, and Hydrazone Thiolation
In 2018, Natarajan and co-workers disclosed a photocatalytic system for the modular synthesis of 2-substituted benzothiazoles from nitriles and thiophenols (Scheme 238).630 Contrary to many literature methods for the preparation of these scaffolds, which rely upon intramolecular SNAr or cyclization of 2-aminothiophenols onto carboxylic acid derivatives,631 Natarajan’s method obviates the need for pre-installed reactive functionality. Using a combination of thiophenol, nitrile, and EY as a photocatalyst in DMSO under air with green-light irradiation, the authors demonstrated 21 examples of benzothiazole synthesis, with good to excellent yields (68–92%). The reaction proved tolerant of electron-releasing and electron-withdrawing substituents on both the nitrile and thiophenol components (238.1, 238.2) without substantial depression of yield. Alkyl nitriles also engage fruitfully in the reaction (238.4), affording benzothiazoles in comparable yields to the aryl nitrile examples. The method was very scalable and proceeded equally efficiently at 25 mmol scale with respect to nitrile.
Scheme 238. Benzothiazole Synthesis through Photocatalytic Aerobic Oxidative Thiyl Radical Generation (Natarajan, 2018).

Mechanistically, the reaction is proposed to begin with ET from thiophenol substrate (e.g., for thiophenol, E1/2ox = +0.95 V vs Fc+/Fc in MeCN),336 to the photoexcited state of the EY dye (E1/2EY*/EY•– = +0.83 V vs SCE in MeCN)71 to generate both the thiol radical cation and EY•–. Molecular oxygen is reduced by EY•– (E1/2EY/EY•– = −1.06 V vs SCE in MeCN)71 to form superoxide and regenerate the ground-state dye. Superoxide deprotonates the thiol radical cation to form neutral thiyl radical and hydroperoxide radical, the former of which adds into the aryl nitrile substrate. The resultant nitrogen-centered iminyl radical further adds into the 2-position of the thiophenol and following ET and PT to hydroperoxide radical yields the aromatized benzothiazole product.
In 2018, Wei and co-workers reported a method to prepare thiocarbamates from thiophenols, isonitriles, and water through visible-light photoredox catalysis (Scheme 239).632 The authors employed RB as photocatalyst in a mixture of EtOAc/H2O (1:1) as solvent under air and blue-light irradiation, proposing that molecular oxygen is required to achieve efficient turnover of the photocatalyst. Thiocarbamates are found in numerous molecules relevant to the pharmaceutical or agrochemical industries, including an array of herbicides, pesticides, and antifungal agents. This work involved the preparation of 22 thiocarbamate products, which were reported in yields of 60–88%. Both electron-rich and electron-poor thiophenols participated in this transformation, affording aromatic thiocarbamates bearing methoxy (239.1), trifluoromethyl (239.3), and halogen (239.2) substituents. The demonstration of isonitriles included mostly aliphatic examples, such as ethyl isocyanoacetate and tert-butyl isocyanide (239.3), as well as one example of an aromatic isonitrile (239.4).
Scheme 239. Photocatalytic Aerobic Synthesis of Thiocarbamates through the Reaction of Thiols and Isonitriles (Wei, 2018).

The authors probed the mechanism of this transformation through SV studies in EtOAc solution, which suggested that a representative thiophenol substrate quenched the excited-state photocatalyst (E1/2 *RB2–/RB3–• = +0.99 V vs SCE in H2O)349 through single-electron oxidation (e.g., for 4-methoxybenzenethiol, Ep/2ox = +1.15 V vs SCE in MeCN),21 and that the isocyanide component did not. The resulting radical cation is then proposed to undergo deprotonation by superoxide generated from reduction of molecular oxygen during turnover of the photocatalyst (E1/2RB2–/RB3–• = −0.78 V vs SCE in H2O).349 Addition of the thiyl radical to the isonitrile substrate is proposed to follow, producing a C-centered radical that is then oxidized by the excited-state photocatalyst and attacked by water to generate the thiocarbamate product. Using H218O resulted in full 18O incorporation at the carbonyl group oxygen atom, supporting this proposal. Support for the key role of oxygen and water in this reaction came from the observation that only traces of the product formed under a nitrogen atmosphere or in anhydrous EtOAc.
In 2019, Singh and co-workers reported a protocol for the synthesis of allylic and benzylic thioethers through the denitrogenative coupling of thiophenols and hydrazone precursors (Scheme 240).633 Here, 25 examples of thioether synthesis were demonstrated through green light irradiation of a solution of DMF containing EY photocatalyst, hydrazone, thiophenol derivative, and NaOH as Brønsted base (66–90% yields). Acetophenone hydrazone and derivatives thereof containing EWGs and EDGs were tolerated (240.1–240.3). Additionally, heteroaromatic hydrazones (240.4) and a cinnamaldehyde-derived hydrazone (240.6) also proved to be competent substrates for C–S coupling. The authors noted that only thiophenol derivatives were competent substrates, with trace amounts of the product observed when aliphatic thiols were subjected to the reaction conditions.
Scheme 240. Photocatalytic Synthesis of Thioethers from Thiols and Hydrazones (Singh, 2019).

The proposed mechanism begins with deprotonation of the hydrazone substrate by hydroxide, affording a diazo intermediate. The authors then propose two potential mechanisms for C–S bond formation. In the first pathway, thiyl radical, generated by joint action of the photoexcited state of the photocatalyst and hydroxide base, adds into the diazo functional group at carbon. Liberation of molecular nitrogen generates an α-sulfur radical, which can be quenched by subsequent HAT. In the second proposed pathway, the diazo decomposes to the carbene followed by C–S bond formation generating the same α-sulfur radical as in the first pathway, which can then undergo HAT to form the product.
In 2019, Huang and co-workers reported an electrochemical method for the formation of sulfide-containing enaminonitriles through the coupling of thiols and 2 equiv of MeCN (Scheme 241A).315 Optimization studies revealed the use of an undivided cell with Pt electrodes operating under constant current electrolysis in the presence of n-Bu4N+ClO4– as an electrolyte with substoichiometric loadings of KI, citric acid, and 1,2-bis(diphenylphosphino)ethane (DPPE) as additives. Exclusion of citric acid resulted in a diminished yield of product, whereas the reaction failed completely in the absence of KI. The DPPE additive resulted in an increase in the stereoselectivity of the transformation in favor of the Z-olefin product through proposed in situ formation of the corresponding bisphosphine oxide. Under these conditions, 28 examples of enaminonitrile formation were reported in yields of 24–96%. Thiophenols bearing both electron-withdrawing and electron-donating substituents performed well regardless of their position on the ring relative to the thiol. Notably, substituents that could be oxidatively sensitive, such as amines, were also tolerated, though in diminished yield (241.3). Primary aliphatic thiols were competent substrates in this transformation despite exhibiting lower reactivity than aromatic thiols, with 2-phenylethanethiol affording the corresponding product in 53% yield, albeit with low stereoselectivity. Secondary aliphatic thiols were much less successful, producing only trace amounts of the desired product. Diselenides could serve as coupling partners to yield olefinic products bearing selenium in high yield and selectivity (241.5).
Scheme 241. Electrochemical Synthesis of Sulfur-Containing Enaminonitriles through the Coupling of Thiols and Acetonitrile. (Huang, 2019; Lei, 2019).

*60 mol% KI, **from dimethyl diselenide, 60 mol% KI
Soon after the publication of this transformation by Huang and co-workers, Lei and co-workers independently reported slightly modified conditions that allowed the preparation of the same class of products also from MeCN and thiols (Scheme 241B).634 Optimization studies revealed the use of an undivided cell containing a carbon rod anode and a Pt plate cathode operating under constant current conditions in the presence of excess KI and substoichiometric cyclohexanecarboxylic acid as additives. As is the case in the earlier work, the yield drops precipitously without the acid additive, and no product was formed when KI was substituted with n-Bu4N+I– or n-Bu4N+BF4–. In their method, 17 examples of enaminonitrile formation were reported in yields of 25–95%. Thiophenols bearing electron-neutral (241.6) and electron-withdrawing (241.7) substituents performed well in this transformation. However, electron-rich thiophenols (241.8) exhibited a much poorer reactivity. Beyond thiophenols, 2-naphthalenethiol and 2-thiophenethiol proved to be competent substrates, providing olefin products (241.9, 241.10) in moderate to good yields. No examples of aliphatic thiols were included, though the reaction was noted to be successful using aryl disulfides in place of aryl thiols.
Lei and co-workers developed an electrochemical methodology that enabled the gem-difunctionalization of isocyanides with thiols and alcohols (Scheme 242).635 Constant current electrolysis of a MeCN solution of a thiol, isocyanide, and alcohol substrates in an undivided electrochemical cell using graphite anode and Pt cathode furnished imino sulfide ethers (85 examples, 30–87%). The reaction accommodated a variety of thiol substrates, including electron-rich (242.1) and electron-poor thiophenols (242.2), primary and secondary alkyl thiols (242.3), while also demonstrating a variety of primary (242.1–242.3), secondary (242.4), and allylic alcohols (242.5). The corresponding disulfide could be used in place of the thiol component to similar effect. Similarly, a variety of aryl and sterically hindered alkyl isocyanides were competent substrates within the reaction. Notably, the reaction displayed remarkable functional group tolerance, leaving halides, alkenes, alkynes, nitriles, esters, and silyl groups untouched.
Scheme 242. Electrochemical Oxidative gem-Difunctionalization of Isocyanides (Lei, 2020).

Control electrolysis experiments in the presence of TEMPO/BHT or in the absence of isocyanide substrate suggested that thiyl radicals were formed under the electrochemical reaction conditions. Additionally, an S-centered radical was observed in EPR experiments. The authors posited that oxidation and subsequent deprotonation of thiophenol–thiol at the anode generates these electrophilic thiyl radicals, which subsequently add to the nucleophilic carbon of the isocyanide. The resulting carbon-centered radical is then oxidized at the anode to form a nitrilium ion, which is trapped by the O/N-nucleophile to generate the imino-sulfide ether product. Concomitant reduction of protons at the cathode generates molecular hydrogen.
4.1.5. S–S Bond Formation in the Synthesis of Symmetrical and Unsymmetrical Disulfides and Thiosulfonates
Finally, we present a number of examples of sulfur(II)–heteroatom bond formation enabled by thiyl radical resulting from PCET homolysis. In 2014, Wu and co-workers reported the near quantitative homo-coupling reaction of thiols to disulfides and molecular hydrogen upon blue-light irradiation of an aqueous solution containing CdSe quantum dots (QDs) and NiCl2 (12 examples, 83–99% yield) (Scheme 243).636 The reaction tolerated a variety of aliphatic thiols and thiophenols including those bearing carboxyl, amino, methoxy, and halide substituents. In the case of aliphatic thiols, the length of the alkyl chain had no effect on disulfide formation (243.1). In the case of thiophenols, both EDGs and EWGs in the para-position were well tolerated (243.2–243.7). Although there was not an electronic effect with respect to the coupling of thiophenols, there was a clear steric effect when the substituent was moved from the para to the ortho or meta position, the yield of disulfide decreased, and in some cases (i.e., 2-chlorothiophenol) virtually no disulfide was observed.
Scheme 243. Photocatalytic Conversion of Thiols into Disulfides and H2 Using CdSe Quantum Dots (Wu, 2014).

To probe the mechanism of the reaction, the authors studied the interaction of the thiol with the CdSe QDs. They found that thiolate was bound to the CdSe QD in solutions whose pH was near the pKa of the thiol (e.g., 4–7 for aromatic thiols, 9–11 for aliphatic thiols). EPR experiments in the presence of the radical trap DMPO suggested that thiyl radicals are formed within the reaction. The authors next performed a crossover experiment in which they mixed CdSe QDs formulated with 3-mercaptopropionic acid (MPA) with CdSe QDs formulated with p-toluenethiol. Notably, the authors only observed the MPA disulfide and the p-toluenethiol; no heterocoupled disulfide was observed. The authors posit that thiyl radicals are generated at the CdSe surface, wherein they couple to one another before diffusing away as the homo-coupled disulfide. Isotope labeling experiments with D2O instead of H2O generated D2 rather than H2, suggesting that the source of H-atoms in the H2 product stems from water rather than thiol. Based upon these observations, the authors proposed the following mechanism. Visible-light excitation of the CdSe QDs promotes an electron from the VB to the CB. The hole that is subsequently within the VB is quenched through oxidation of the thiolate that is bound to the QD surface. The resulting bound thiyl radicals that couple to one another to form the homo-coupled disulfide, which is not as strongly bound to the QD surface and diffuses into solution. Meanwhile, the excited-state CB is quenched through reduction of protons to molecular hydrogen. The Ni(II) salt further facilitates proton reduction to molecular hydrogen.
In 2018, Dethe and co-workers reported the photocatalytic formation of symmetrical and unsymmetrical disulfides using thiol substrates (Scheme 244).637 Blue-light irradiation of an acetone solution containing a single thiol and [Ir(ppy)3] (Ir-1) photocatalyst led to the formation of the symmetrical disulfides (30 examples, 20–94%). Although the reaction tolerated a variety of aliphatic and aromatic thiols, thiophenols bearing electron-withdrawing substituents and heterocyclic thiols reacted at a slower rate. When a second thiol was then added to the reaction mixture, the cross-coupled unsymmetrical disulfide was generated in good yields (14 examples, 66–83%). Reactions between aromatic and aliphatic thiols predominately furnished the asymmetric disulfides and only trace amounts of the symmetrical disulfides (244.1–244.3), as did reactions between aromatic thiols when one of the coupling partners contained an electron-withdrawing substituent (244.4). Although the unsymmetrical disulfide was formed when neither aromatic thiol contained an EWG, a moderate amount of symmetrical disulfide was also formed.
Scheme 244. Photocatalytic Synthesis of Symmetric and Unsymmetric Disulfides (Dethe, 2018).

*≤6% symmetrical disulfide observed. **≤15% symmetrical disulfide observed.
In probing the mechanism of the reaction, the authors noted that these reaction conditions facilitated disulfide–thiol exchange. Under the optimal conditions, the symmetrical bis(2-methoxyphenyl)disulfide was converted to an unsymmetric disulfide when an aliphatic thiol (e.g., with cyclopentanethiol, 86% yield) or aromatic thiol (e.g., with 4-chlorothiophenol, 63% yield) was present. The authors posit that a thiol substrate is oxidized by the excited state of the Ir-1 photocatalyst (E1/2 *Ir(III)/Ir(II) = +0.31 V vs SCE in MeCN)66 and subsequently deprotonated to generate thiyl radical. Radical–radical coupling generates the disulfide, which can be further substituted by other thiyl radicals. Reduction of protons by the reduced state of the photocatalyst closes the catalytic cycles, generating molecular hydrogen as a byproduct. The co-generation of molecular hydrogen was supported by GC detection.
Building on their work in electrochemical C(sp3)–H/S–H cross-coupling, Lei and co-workers reported in 2018 an electrochemical method of direct S–H/S–H cross-coupling for the selective formation of unsymmetrical disulfides from thiols (Scheme 245).638 Using an undivided cell with Pt electrodes operating under constant current, with n-Bu4N+PF6– as an electrolyte and DMF as solvent, this method provided access to 24 aryl-alkyl disulfides in yields of 43–87%. No chemical oxidant was needed for efficient reactivity under these conditions and dihydrogen gas generated from cathodic reduction of protons represented the only side product. The difference in oxidation potentials between the aryl and alkyl thiol substrates was postulated to be the source of high cross-selectivity. The scope of the aryl thiol component included many examples of mercapto-heterocycles, such as 2-mercaptobenzoxazole (245.1) and 2-mercaptopyridine (245.2). Thiophenols also proved to be effective coupling partners, providing access to disulfides containing phenyl groups of various substitution patterns (245.3, 245.4). Changing the solvent from DMF to 3:1 MeCN:CH2Cl2 and increasing the current density allowed alkyl thiols to participate in this transformation (245.4, 245.5). The coupling still proceeded efficiently in the presence of an unprotected primary alcohol (245.4).
Scheme 245. Electrochemical Synthesis of Unsymmetrical Aryl-alkyl Disulfides through S–S Bond Formation (Lei, 2018).

*MeCN/CH2Cl2 (3:1) solvent. Constant current = 16 mA.
The mechanism of this reaction was investigated through control experiments in which symmetrical disulfides of each starting thiol were independently used as substrates. Only a trace amount of unsymmetrical disulfide product was formed with di-tert-butyl disulfide and 2-mercaptobenzothioazole, whereas efficient reaction was observed between the symmetrical 2-mercaptobenzothiazole disulfide and tert-butyl thiol. EPR measurement of mixtures of 2-mercaptobenzothiazole and t-BuSH electrolyzed in the presence of radical trap DMPO showed only evidence of 2-mercaptobenzothiazolyl radical trapping. Electrolysis of DMPO and t-BuSH showed a distinct EPR signal to that observed when both thiols were present. Additionally, while diaryl disulfides proved competent for unsymmetric aryl-alkyl disulfide formation, use of dialkyl disulfides in place of alkyl thiols resulted in only a trace amount of the product. Together, these data suggested that diaryl disulfide could serve as a precursor for aryl thiyl radical under the reaction conditions, while dialkyl disulfides could not, providing rationale for cross-selectivity. CV established the oxidation potentials of the 2-mercaptobenzothiazole and tert-butyl thiol substrates to be Ep/2ox = +1.15 V and +1.44 V vs Ag/AgCl in DMF,638 respectively, suggesting that both substrates can undergo oxidation at the anode. Based on these data, the authors propose that the mechanism initiates via anodic ET followed by PT to generate the thiyl radicals of both coupling partners. The desired unsymmetrical product may form at this stage from radical–radical cross-coupling, or instead homodimerization may lead to the formation of an inactive dialkyl disulfide and a viable diaryl disulfide. Cathodic reduction of this diaryl disulfide (e.g., for 4-chlorophenyl disulfide, E1/2red = −1.43 V vs SCE in DMF)639 facilitates cleavage into the corresponding thiyl radical and thiolate anion via mesolytic cleavage,639 the former of which can engage in product formation while the latter can undergo protonation to reform starting aryl thiol. Redox balance is achieved through the reduction of protons to hydrogen gas at the cathode.
In 2019, Volla and co-workers reported a protocol for the synthesis of thiosulfonates, via the three-component coupling of aryl diazonium salts, SO2, and thiols, mediated by visible-light photocatalysis (Scheme 246).640 Green-light irradiation of EY in the presence of thiol, aryl diazonium salt, and DABSO—the charge-transfer complex formed between DABCO and sulfur dioxide641—afforded 34 examples of thiosulfonate products in 54–89% yield. The authors demonstrated a wide scope of thiols, including thiophenols containing EWGs (246.1) and EWGs (246.2). Additionally, alkyl thiols were competent reaction partners, with thiosugar 246.3 obtained in 82% yield. Several diazonium salts were also tolerated, including those with electron-donating (246.1–246.4), nitro (246.5), and ortho- (246.6) substituents.
Scheme 246. Sulfonylation of thiols enabled by visible-light generation of thiyl radical (Volla, 2019).

The authors propose that a DABSO-aryl diazonium salt complex forms, which then undergoes ET reaction, resulting in the generation of aryl radical via extrusion of molecular nitrogen, SO2, and O2S–DABCO+•. An aryl sulfonyl radical intermediate is then formed through the reaction of the generated aryl radical with SO2. The authors propose that S–S bond formation results from radical–radical coupling between this sulfonyl radical intermediate and thiyl radical, which is generated through oxidation by the excited-state EY photocatalyst, followed by deprotonation to generate the neutral thiyl radical. This coupling reaction forms the closed-shell product, and the EY photocatalyst is regenerated through single-electron reduction of O2S–DABCO•+.
4.1.6. S–N Bond and S–P Bond Formation
An early example of processes mediated by thiol PCET comes from Torii and co-workers for the direct synthesis of benzothiazolyl sulfenamides from thiols and alkylamines (Scheme 247).642 The transformation was accomplished through constant potential electrolysis of a DMF mixture of either 2-mercaptobenzothiazole and amine, or 2-benzothiazolyl disulfide and amine in an undivided cell using Pt or stainless-steel electrodes with Et4N+ClO4– as a supporting electrolyte. The authors demonstrated 10 examples of alkylamine-derived 2-benzothiazolyl sulfenamide synthesis (25–98%). An array of simple primary (247.1) and secondary alkylamines (247.2) proved to be competent substrates; only sterically encumbered secondary amine substrates such as diisopropyl- and dicyclohexylamine gave diminished yields (247.3).
Scheme 247. Electrochemical Synthesis of 2-Benzothiazolyl Sulfenamides (Torii, 1978).

*With stainless steel electrodes.
Mechanistically, the transformation begins with sequential oxidation and deprotonation of 2-mercaptobenzothiazole (e.g., for 2-mercaptobenzothiazole, Ep/2ox = +1.03 V vs SCE in MeCN)21 to generate the corresponding thiyl radical. The nascent thiyl radical dimerizes to form 2-benzothiazolyl disulfide, which is subsequently attacked by neutral amine substrate to form the sulfenamide product along with an equivalent of alkylammonium thiolate. The thiolate can be anodically oxidized to regenerate thiyl radical and concomitant proton reduction of the ammonium salt at the cathode liberates molecular hydrogen and regenerates neutral amine substrate. Evaporative workup of non-electrolyzed solutions containing the two substrates exclusively yields the disulfide, suggesting that sulfenamide formation from disulfide and alkylamine is reversible. This early work from Torii portends the prominence of electrochemical thiyl radical generation and subsequent dimerization to the disulfide in more recent examples of sulfenamide formation and other thiol functionalizations.
Li, Yuan, and co-workers have developed an electrochemical method for synthesis of sulfenamides from heteroaromatic thiols and amines (Scheme 248).643 Constant current electrolysis of a MeCN solution of thiol and amine substrates in an undivided electrochemical cell equipped with Ni electrodes at ambient temperature furnished the desired sulfenamides, with 20 examples reported in 10–95% yields. Primary (248.1, 248.2) and secondary aliphatic amines (248.3–248.5) were competent substrates within the reaction, as were benzylic amines. Yields tended to diminish with increasing steric bulk (e.g., diisopropylamine only gave 20% yield in a reaction with 2-mercaptobenzothiazole), and arylamines did not furnish the desired sulfenamide product. The scope of the transformation was limited to heteroaryl thiols such as 2-mercaptobenzothiazole (248.1–248.3), 2-mercaptobenzoxazole (248.4), and 2-mercaptopyridine (248.5). Although the reaction was insensitive to air and moisture, sufficient yields were only achieved with 10:1 amine:thiol. Notably the electrolysis could be performed on 50 g scale, furnishing the desired sulfenamide product in up to 95% yield.
Scheme 248. Electrochemical Dehydrogenative Coupling of S–H/N–H Bonds (Li and Yuan, 2019).

The authors posited that anodic oxidation of the thiol substrate and subsequent deprotonation generates a thiyl radical, which can dimerize to form the disulfide. The intermediacy of this disulfide was supported by the observation of sulfenamide when the thiol substrate was replaced with the corresponding disulfide. Similarly, it was proposed that a neutral aminyl radical is generated through oxidation and subsequent deprotonation. This is supported by CV data and control electrolysis experiments conducted in the presence of a spin-trapping reagent (e.g., DMPO). Subsequent coupling of the aminyl radical intermediate with the disulfide or the corresponding thiyl radical generates the sulfenamide. Concomitant reduction of protons at the cathode generates molecular hydrogen.
Zheng and co-workers in 2019 developed an electrochemical dehydrogenative cross-coupling reaction of diary phosphine oxide with aryl or alkyl thiols (Scheme 249).644 Constant current electrolysis of a MeCN solution of substrates in an undivided electrochemical cell using Pt electrodes at room temperature furnished the desired product, with 21 examples documented in 46–90% yields. Thiophenols bearing halides, electron-donating (e.g., methoxy, 249.4), or electron-neutral (e.g., methyl, 249.1) substituents were well tolerated under the reaction conditions. Although naphthalene thiol was also tolerated, the desired product was produced in diminished yield (46%). Primary and secondary aliphatic thiols were competent substrates (249.7), as were benzylmercaptan (249.8) and methyl thioglycolate (249.6). With respect to the diphenylphosphine oxide substrate, both methyl and fluoride substituents were well tolerated.
Scheme 249. Electrochemical Dehydrogenative Cross-Coupling of Diaryl Phosphine Oxides and Thiols (Zheng, 2019).

*DMF used as solvent.
The authors posited that oxidation and subsequent deprotonation of the thiol generates a thiyl radical (e.g., for thiophenol, S–H BDFE = 79.1 kcal mol–1),336 which can abstract a H-atom from the diaryl phosphine oxide (e.g., for Ph2P(O)H P–H BDFE = 79 kcal mol–1)645 or reversibly dimerize to form the disulfide. Based on control electrolysis experiments conducted in the presence of a radical scavenger (i.e., TEMPO), the authors hypothesized that HAT between the thiyl radical and the diarylphosphine oxide occurs to regenerate thiol and produce a phosphorus radical intermediate. EPR experiments confirmed the formation of this phosphorus radical intermediate. Subsequent coupling of the phosphorus radical intermediate with either of the in situ-generated disulfide or thiyl radical intermediates furnished the product. Concomitant reduction of protons at the cathode generates molecular hydrogen. The authors also in this work expanded the scope of the reaction to allow for the cross-coupling of diarylphosphine oxides with alcohols through modified reaction conditions involving iodide additives, which likely mediated a polar pathway for P–O bond formation.
Xia and Wu reported in 2020 the photocatalytic dehydrogenative cross-coupling of phosphines and thiophenols to generate mono- and bis-thiophosphanes (Scheme 250).646 This method expands on the presently limited repertoire of photoredox-mediated bond formations between two heteroatoms and offers a room temperature alternative to existing methods for the coupling of phosphines and thiophenols.647,648 Optimization studies revealed the requirement for a hydrogen acceptor in order to obtain acceptable yields of the formal dehydrogenation product, and that benzaldehyde was optimal in this role. With Ir(ppy)3 (Ir-1) as photocatalyst and benzaldehyde as oxidant, in MeCN solution under irradiation by white LEDs, 37 examples of phosphine-thiophenol cross-coupling to yield P(III) thiophosphanes were reported in yields of 50–93%. In reactions with diphenylphosphine, thiophenols bearing methoxy (250.1), alkyl, and halide (250.2) substituents are all well tolerated. When primary aryl phosphines are employed as substrates with thiophenols, two sequential S–P bond-forming events occur to afford bis-thiophosphanes as products. A similar scope of thiophenols was reported for the bis-thiophosphane formation (250.3). Interestingly, the application of di- and trialkyl phosphines under these reaction conditions leads to the dealkylative coupling with thiophenols, producing alkyl bis-thiophosphanes in the case of di-tert-butylphosphine, from one dehydrogenative and one dealkylative coupling event, and dialkylthiophosphanes (250.4) in the case of tri-tert-butylphosphine, resulting from one dealkylative coupling. The authors do not comment on the mechanism of dealkylation or the byproduct deriving from the liberated alkyl group. We note however that several groups have reported alkyl radical generation from thiol substrates in the presence of phosphines under oxidative photocatalysis, via an α-scission pathway.649
Scheme 250. Photocatalytic Cross-Coupling between Phosphines and Thiophenols (Wu and Xia, 2020).

*3.0 equiv of thiol, 2.0 equiv of benzaldehyde.
Control experiments established the necessity of the photocatalyst, hydrogen acceptor, and light, with only a trace amount of the product obtained in the absence of any of these components. Significantly attenuated yields were observed for reactions conducted with excess TEMPO, implying the formation of radical intermediates. Exclusion of the phosphine coupling partner in otherwise standard conditions led to the formation of the disulfide derived from the thiophenol component in good yield, which could be isolated and converted to product through separate reaction with phosphine under the optimized conditions. Based on these mechanistic observations, the authors propose that excited-state Ir(ppy)3 (E1/2 (Ir(IV)/*Ir(III) = −2.19 V vs SCE in MeCN),66 reduces benzaldehyde (e.g., for benzaldehyde, Ep/2red = −1.93 V vs SCE in MeCN)21 to the corresponding ketyl radical anion, which is then protonated to form the neutral ketyl radical. HAT between this radical and the thiophenol coupling partner generates thiyl radical. Alternatively, the authors further propose that the Ir(IV) state of the photocatalyst (E1/2 (Ir(IV)/Ir(III) = +0.77 V vs SCE in MeCN)66 could mediate oxidation of thiophenol (e.g., for thiophenol. Ep/2ox = +1.51 V vs SCE in MeCN)21 to form thiyl radical after deprotonation of the radical cation (e.g., for thiophenol, pKa = −11.7 in DMSO).336 The significantly endergonic nature of this process implies a proton-coupled element is operative, whereby thiolate generated through discrete PT with ketyl radical anion, or through a concerted mechanism, may assist in this step. Thiyl radical dimerization yields the characterized disulfide intermediate which can then be attacked by the phosphine, affording the final thiophosphane product and thiophenol.
4.2. Synthesis of S(IV) and S(VI) Functional Groups through C–S, S–O, and S–N Bond Formation
Having demonstrated the wealth of bond-forming reactivity of thiyl radical toward S(II)-containing products, in this section we detail systems which instead access S(IV) and S(VI) products through series of C–S, S–O, S–N, and S–F bond formations. Oxidative transformations analogous to the thiol–ene and thiol–yne reactivity detailed in section 4.1.1 open this section, though formation of sulfinate esters, sulfonate esters, sulfonyl fluorides, and sulfonamides is also described.
4.2.1. Photocatalytic Sulfoxide Synthesis through Alkene and Alkyne Hydrothiolation and Aerobic Oxidation
In 2017, Alemán, Fraile, and co-workers described a modular system for the visible-light-driven one-pot preparation of sulfoxides from thiols and olefins through sequential steps of thiol–ene and sulfide oxidation (Scheme 251A).650 Green light irradiation of a CH2Cl2 solution containing thiol, olefin, and EY as photocatalyst under an air atmosphere afforded 21 examples of sulfoxide products in 24–93% yields. The method accommodates several alkyl (primary and tertiary) and electron-rich and -poor aryl and benzyl thiols (251.1–251.4). Both mono- and disubstituted (both 1,1- and 1,2-) styrenes were compatible substrates, although 1,2-disubstituted styrenes showed diminished yields. A range of arene substituents, including carbamates (251.2), ethers, aryl halides (251.1, 251.2), nitro and trifluoromethyl (251.4) groups, were demonstrated. Products could be derivatized through Pummerer reaction with trifluoroacetic anhydride to afford nucleophilic substitution α- to sulfur.
Scheme 251. Tandem Photocatalytic Thiol–Ene/S-Oxidation for the Synthesis of Sulfoxides (Alemán and Fraile, 2017; Wang and Wei, 2017).

These authors propose a mechanism initiating via ET from thiol substrate (e.g., for BnSH, E1/2ox = +0.45 V vs SCE in MeCN)336 to the excited state of EY (E1/2 *EY/EY•– = +0.83 V vs SCE in MeCN)71 to yield thiyl radical after deprotonation, and the reduced state of EY. Thiyl radical addition across olefin substrate proceeds in an anti-Markovnikov fashion to furnish a benzylic radical. Given the high quantum yield observed for the process (Φ = 5.8), a chain mechanism is operative in this thiol–ene reaction. This phenomenon manifests itself as HAT from another equivalent of thiol to the benzylic radical to regenerate thiyl radical in addition to a sulfide intermediate. The authors suggest, however, that EY•– (E1/2EY/EY•– = −1.06 V vs SCE in MeCN)71 could potentially reduce the benzylic radical resulting from this olefin addition (e.g., for the benzylic radical derived from ethylbenzene, PhCH·CH3, E1/2red = −1.45 V vs SCE in MeCN).135 Control experiments under argon atmosphere yielded sulfide, rather than sulfoxide product, and subsequently subjecting isolated sulfide to these reaction conditions under O2 atmosphere allowed for efficient conversion to the sulfoxide product. Singlet oxygen (1O2) is likely responsible as oxidant, as revealed in control experiments performed with 1O2 scavenger DABCO, or the superoxide scavenger benzoquinone led to no sulfide oxidation.651 These authors propose that singlet oxygen is generated via energy transfer from photoexcited EY (ET = 1.89 eV)652 to 3O2 (ET = 0.95 eV).653
Contemporaneously, Wang, Wei and co-workers reported a similar system for sulfoxide synthesis (Scheme 251B).654 White light irradiation of an EtOH/H2O (1:1) solution of olefin, thiol, and RB as photocatalyst under an air atmosphere afforded 26 sulfoxide products in 16–90% yields. Though olefin scope shares similarities with the work of Alemán (styrenes with electron-rich and electron-poor ring substitution), some of the key differentiating features between these works include the use of heteroarene-derived olefins (251.5) and olefins bearing reactive electrophiles, such as a benzylic chloride, and limited performance with aliphatic olefins (251.6). Alkyl (primary, benzylic) and aryl thiols (251.7, 251.8) both proved amenable to the reaction. Aryl thiols were demonstrated with a range of electron-rich and electron-poor substituents with slight difference noted between ortho, meta, and para-tolyl thiols’ reaction with styrene.
Mechanistically, the reaction is proposed to proceed in a similar fashion to the above process from Alemán,650 commencing with initial ET from thiol substrate (e.g., for BnSH, E1/2 = +0.45 V vs SCE in MeCN)336 to excited-state RB (E1/2 *RB2–/RB3–• = +0.99 V vs SCE in H2O)349 to yield thiyl radical after deprotonation and RB3–•. ET from RB3–• to molecular oxygen regenerates ground-state RB and concomitantly produces superoxide radical anion. After the thiyl radical adds across the olefin, the resultant alkyl radical is proposed to chain propagate through HAT from another equivalent of thiol substrate to form sulfide intermediate and additional thiyl radicals. In this work, sulfide is proposed to undergo photocatalyst-mediated oxidation (e.g., for thioanisole, Ep/2ox = +1.44 V vs SCE in MeCN)21 to the corresponding radical cation and subsequent trapping with superoxide radical anion to generate a peroxysulfide species, which can further yield the sulfoxide. Given the endergonic nature of this proposed ET, authors also concede that singlet-oxygen-mediated oxidation akin to that described by Alemán and co-workers may also be operative.
The work of Singh and co-workers further explored this transformation, using N-hydroxyphthalimide (NHPI) as an oxidizing photocatalyst (Scheme 252).655 Irradiation of an MeCN solution of olefin, thiol, and NHPI under an air atmosphere using white lights afforded 18 examples of sulfoxide products (28–96% yields). Olefin scope is typified by substituted styrene derivatives, displaying tolerance for pyridyl (252.1), aryl halide, nitro and aryl ether derivatives. Notably, this system showed improved performance with non-styrenyl olefins, with 1-hexene derivative isolated in 28% yield. Aryl (252.2) and alkyl thiols (252.3) both proved compatible in this transformation, with yields of sulfoxide largely insensitive to thiol structure given the same styrene substrate. Substitution with halides, aryl ethers, and alkyl groups were tolerated with aryl thiols, including 2-naphthyl thiol 252.4.
Scheme 252. Tandem Photocatalytic Thiol–Ene/S-Oxidation for the Synthesis of Sulfoxides (Singh, 2018).

In contrast to the works of Alemán and Wang, the reaction developed by Singh and co-workers is proposed to proceed through initial photolysis of NHPI to generate PINO radical. Thiol substrate then undergoes ET to the PINO radical (E1/2red = +0.69 V vs SCE in MeCN),656 yielding PINO anion and thiyl radical after deprotonation. S–H bond HAT with PINO radical (for the corresponding hydroxylamine, O–H BDE = 86 kcal mol–1)657 is thermodynamically feasible, but argued to be polarity mismatched, thus the ET pathway is preferred.658 Thiyl radical proceeds to sulfide intermediate via olefin addition and chain propagation through HAT from another equivalent of thiol. Oxidation of sulfide by the PINO radical and trapping of the resulting sulfur radical cation by superoxide generates a peroxysulfide intermediate. Although this oxidation would be highly endergonic (e.g., for thioanisole, Ep/2ox = +1.44 V vs SCE in MeCN),21 subjecting isolated sulfide to standard reaction conditions furnished sulfoxide product.
In 2020, Shah and co-workers reported the photocatalytic synthesis of functionalized sulfoxides from terminal alkynes and thiols (Scheme 253).659 Blue-light irradiation of a MeOH solution containing [Ru(bpy)3]Cl2 ([Ru-1]Cl2) photocatalyst and DIPEA as a Brønsted base under an air atmosphere led to the formation of α-alkoxy-β-ketosulfoxides when aromatic alkynes were used as substrates (e.g., 253.1–253.3, 25 examples, 44–70% yield) and α,β-dimethoxysulfoxides when aliphatic alkynes were used as substrates (e.g., 253.8–253.10, nine examples, 52–68% yield). Notably, the anti-diastereomers were formed with good diastereoselectivity (>20:1 d.r.). Substituted phenylacetylenes bearing halide, alkyl, or EDGs were well tolerated, as were substituted thiophenols and aliphatic thiols. A variety of alcohols (e.g., i-PrOH, d3-MeOH, PhOH) proved to be competent nucleophiles when they were used in place of MeOH as solvent (253.4–253.6). The authors found that use of non-alcohol solvents impacted the functionalized sulfoxide product that was formed; β-ketosulfoxides were formed when the reaction was run in MeCN (e.g., 253.11–253.18, 26 examples, 49–72% yield) and vinyl sulfoxides were formed when the reaction was run in water (e.g., 253.19, eight examples, 43–55% yield). Similar to their substrate scope studies carried out in MeOH, the authors observed excellent functional group tolerance with respect to the terminal alkyne substrate, with both aliphatic alkynes and substituted phenylacetylenes affording the β-ketosulfoxide or vinyl sulfoxide in good yields depending on solvent conditions. Additionally, both reactions tolerated aliphatic thiols and a variety of substituted thiophenols.
Scheme 253. Photoredox-Mediated Synthesis of Functionalized Sulfoxides from Terminal Alkynes (Shah, 2020).

*H2O solvent.
The authors conducted several experiments to probe the mechanism of these transformations. Upon adding TEMPO to their standard reaction conditions, no product formation was observed. Both the addition of benzoquinone as a superoxide radical scavenger and the replacement of an air atmosphere with argon shut down the reaction. Subsequent isotope labeling experiments with H218O did not afford isotopically enriched product, implying that molecular oxygen rather than an O-nucleophile (e.g., water, alcohol) was responsible for oxygen incorporation at sulfur. SV quenching experiments in MeOH with equimolar quantities of thiophenol, 1-octanethiol, DIPEA, and phenylacetylene suggested that thiophenol effectively quenched the excited state of the photocatalyst.
Based upon these observations, the authors posited that thiophenol is oxidized by the photoexcited state of the Ru(II) complex and subsequently deprotonated to generate the thiyl radical. Reduction of molecular oxygen by the resulting Ru(I) complex is responsible for photocatalyst turnover. Thiyl radical addition to the alkyne substrates generates a vinyl radical, which can engage in two different reaction pathways. In the first pathway, the vinyl radical is trapped by hydroperoxide to yield a peroxy intermediate. Subsequent peroxide cleavage yields a β-ketosulfide that can be oxidized by the photoexcited state of the Ru(II) photocatalyst. In the absence of nucleophiles, the resulting radical cation undergoes radical coupling with superoxide to yield the β-ketosulfoxide product. However, the β-ketosulfide radical cation can also undergo additional proton- and ET steps to generate a thiocarbenium intermediate, which can be trapped by a nucleophile to form α-alkoxy-β-ketosulfide intermediate. Subsequent oxidation by the photoexcited state of the Ru(II) photocatalyst and trapping with hydroperoxide yields another peroxide intermediate that upon peroxide cleavage furnishes the α-alkoxy-β-ketosulfoxide product. The authors attributed the anti diastereoselectivity of the reaction to gauche effects within the α-alkoxy-β-ketosulfide radical cation intermediate.
Conversely, the vinyl radical formed upon thiyl radical addition to the alkyne undergoes HAT with thiol to generate a vinyl sulfide. Further photoinduced oxidation of the vinyl sulfide to the radical cation and subsequent radical coupling with superoxide yield the vinyl sulfoxide product. In the presence of a nucleophile, the vinyl sulfoxide undergoes a Pummerer reaction at the α- and β-positions to furnish a dimethoxy sulfide intermediate. Subsequent oxidation by the photoexcited state of the Ru(II) photocatalyst and trapping with superoxide furnishes the α,β-dialkoxysulfoxide product.
4.2.2. Electrochemical Synthesis of Sulfinate Esters from Thiols
Zhong and co-workers have developed an electrochemical methodology that enables the synthesis of sulfinic acid esters through the oxidative coupling of thiols and alcohols (Scheme 254).660 Constant current electrolysis of a CH2Cl2 solution of thiol, alcohol (20% by volume), and n-Bu4N+BF4– as a supporting electrolyte in an undivided electrochemical cell using Pt electrodes at room temperature under N2 furnished the desired sulfinic acid esters (27 examples, 58–90%). A variety of thiophenols bearing electron-donating, alkyl, or halide substituents were well tolerated (254.1–254.4). The reaction also proved to be amenable to 2-mercaptopyridine (254.5), 1-naphthalenethiol, and cyclohexanethiol (254.6) as substrates. With respect to alcohol, both primary and secondary aliphatic alcohols were competent substrates, as was benzyl alcohol (254.7, 254.8).
Scheme 254. Electrochemical Synthesis of Sulfinic Acid Esters through the Oxidative Coupling of Thiols and Alcohols (Zhong, 2019).

*MeCN as solvent.
The authors posited that anodic oxidation (e.g., for thiophenol, E1/2ox = +0.95 V vs Fc+/Fc in MeCN),336 and subsequent deprotonation (e.g., for the radical cation of thiophenol, pKa = −11.7 in DMSO)336 of the thiol generates a thiyl radical, which dimerizes to form the corresponding disulfide. The authors noted that substituting the thiol with the corresponding disulfide still allowed for product formation. The authors also observed the formation of thiosulfinate as a minor side product under standard electrolytic conditions. Electrolysis of this thiosulfinate side product under standard electrolytic conditions resulted in the formation of the sulfinic acid ester, suggesting that it is a viable intermediate within the reaction pathway. Notably, sulfinic acid ester was formed in good yields when the electrolysis was performed with anhydrous methanol under an insert atmosphere. Further experiments in the presence of 18O-labeled water led to the formation of 18O-enriched sulfinic ester, suggesting that O-nucleophiles may serve as oxidants in addition to reactants in this transformation. Based on these results, the authors posit that anodic oxidation of the disulfide (e.g., for diphenyl disulfide, Ep = +1.90 V vs Ag/AgCl in CH2Cl2)661 leads to formation of the thiosulfinate ester, with which alcohol partner reacts as a nucleophile to generate the sulfinic ester product. Concomitant reduction of protons at the cathode generates molecular hydrogen.
Kaboudin and co-workers have developed an electrocatalytic methodology that enabled the synthesis of sulfinate esters through the oxidative coupling of thiols and alcohols (Scheme 255).662 Constant potential electrolysis of a MeCN solution of thiol and alcohol substrates, Ni(ClO4)2 transition metal catalyst, bipyridine as a ligand, and Li+ClO4– as a supporting electrolyte in an undivided electrochemical cell using a modified graphite anode and Ni cathode at room temperature under air furnished the desired sulfinate ester (255.1–255.5,11 examples, 35–92%). Notably, only thiophenol derivatives were tolerated in the reaction; aliphatic and benzylic thiols did not yield the desired sulfinate ester product. The reaction did not appear robust to substitution on the thiophenol, as amine and chloride substituents were not tolerated. With respect to alcohol, both primary and secondary aliphatic alcohols were competent substrates. However, benzyl alcohol and furfuryl alcohol did not furnish the desired sulfinate ester product. The authors reasoned that oxidation and subsequent deprotonation of the thiol generates a thiyl radical, which dimerizes to form the disulfide. The authors noted that substituting the thiol with the corresponding disulfide also enabled product formation. CV data indicated that the Ni(II) pre-catalyst is likely reduced to Ni(0) (Ni(II)/Ni(0) Ep = −1.26 V vs Ag/AgCl in MeCN) at the cathode. The mechanism for sulfinate ester thereafter remains unclear but authors considered a Ni(0)/Ni(I)/Ni(III) mechanism involving thiyl radical trapping, oxidative addition, and reductive elimination leading to S–O bond formation.
Scheme 255. Electrochemical Synthesis of Sulfinate Esters via Ni(II)-Catalyzed Oxidative Esterification of Thiols with Alcohols (Kaboudin, 2019).

4.2.3. Photochemical and Electrochemical Synthesis of Sulfonate Esters, Sulfonamides, and Sulfonyl Fluorides from Thiols
Lei and co-workers reported in 2017 a photochemical method for the preparation of benzenesulfonate esters from aryl thiols and aliphatic alcohols (Scheme 256).663 The transformation is mediated by the organic photocatalyst N-Me-Mes-Acr+ClO4– ([Acr-1]ClO4), under blue-light irradiation of thiol substrate solution in an alcohol solvent under an O2 atmosphere. Reactions conducted under air or N2 atmosphere resulted in no product formation, indicating the crucial role of high concentrations of molecular oxygen in this transformation. Under these conditions, 12 examples of benzenesulfonate ester synthesis were reported in yields of 45–70%. Both primary and secondary aliphatic alcohols reacted with comparable efficiency (256.1–256.4), although tertiary alcohols were unsuccessful in this coupling reaction. TFE reacted with 4-methoxythiophenol to afford the corresponding sulfonate product (256.3) in good yield. On the thiophenol coupling partner, substitution at the ortho-position (256.5) was tolerated as was the presence of an electron-withdrawing p-fluoro substituent (256.6), though with diminished yields relative to the more electron-rich thiophenols.
Scheme 256. Light-Induced Oxidative Cross-Coupling of Thiophenols and Alcohols (Lei, 2017).

Through probing a model reaction between 4-methoxythiophenol and methanol at early time points, the authors were able to isolate and establish the validity of 4-methoxybenzene disulfide and the S(IV) intermediate methyl 4-methoxybenzenesulfinate ester as intermediates in this transformation. Both species afforded the expected sulfonate ester product in good yield when exposed to the standard reaction conditions. The addition of TEMPO fully inhibited product formation, implying the involvement of open-shell intermediates. Based on these observations, the authors propose that reaction initiates with oxidation of the thiophenol substrate (e.g., for 4-methoxythiophenol, E1/2ox = +1.15 V vs SCE in MeCN),21 by the photoexcited-state Mes-Acr+ photocatalyst (E1/2 *Acr+/Acr• = +1.88 V vs SCE in PhCN),74 which generates the corresponding thiyl radical after PT. Oxidation of the resulting acridine radical (E1/2 PC+/PC• = −0.49 V vs SCE in PhCN)74 by molecular oxygen turns over the photocatalyst and generates an equivalent of superoxide radical anion. Dimerization of the thiyl radical leads to the isolable disulfide intermediate (e.g., for 4-methoxybenzene disulfide E1/2ox = +1.28 V vs SCE in MeCN),21 which is then proposed to undergo a further oxidation mediated by photoexcited N-Me-Mes-Acr+ to generate the corresponding disulfide radical cation, before reaction with superoxide radical anion. The resulting thiopersulfinate intermediate reacts with the alcohol coupling partner and undergoes an oxidative cleavage of the S–S bond to give the isolable sulfinate ester intermediate and a sulfonic acid fragment that was observable by HRMS. Lastly, photocatalyst-mediated oxidation of this sulfinate, as shown in model studies, affords the final sulfonate product.
Noël and co-workers have developed an electrochemical methodology that enables the oxidative coupling of thiols and amines (Scheme 257).664 Constant current electrolysis of MeCN/0.3 M aq. HCl solution (3:1 v/v) of thiol and amine substrates in a microfluidic electrochemical flow reactor with graphite anode and stainless-steel cathode furnished the targeted sulfonamides (57 examples, 19–97%). This electrochemical reaction could be completed in 5 min and accommodated a variety of structurally and electronically distinct amines and thiols. Primary and secondary amines were competent substrates within the reaction (257.1–257.6), as were amino acids (257.8). Notably, racemization of the chiral center in these AA-derived substrates was not observed under the electrochemical reaction conditions. Although heteroarylamines gave only a trace amount of the product under above conditions, the addition of 1 equiv of pyridine to the electrochemical reaction enabled the targeted sulfonamide to be synthesized in modest yields (257.7). With respect to the thiol component, thiophenols bearing electron-neutral, -donating, or -withdrawing substituents in the ortho-, meta-, and para-position were well tolerated, as were heterocyclic thiols and aliphatic thiols (257.9).
Scheme 257. Electrochemical Synthesis of Sulfonamides from Thiols and Amines (Noël, 2020).

*Batch reactor, 1 equiv of thiol, 1.5 equiv of amine, 1 equiv of Me4N+BF4–. **1 equiv of pyridine added.
Kinetic experiments suggested that the thiol was consumed within the first 30 s of the reaction. The authors posited that oxidation (e.g., for 4-chlorothiophenol, Eonsetox = +1.33 V vs SCE in MeCN)336 and subsequent deprotonation (e.g., for the radical cation of 4-chlorothiophenol, pKa = −18.0 in DMSO)336 of the thiol generates a thiyl radical that dimerizes to form a disulfide. Notably, disulfides proved to be competent coupling partners under identical reaction conditions. Based on CV data and control electrolysis experiments conducted in the presence of a radical scavenger (e.g., TEMPO), the authors posited the formation of a neutral aminyl radical intermediate through amine oxidation (e.g., for piperidine, Ep/2ox = +0.94 V vs SCE in MeCN; for isopropylamine, Ep/2ox = +1.54 V vs SCE in MeCN)21,665 and subsequent deprotonation. Reaction of the aminyl radical intermediate with the disulfide generates a sulfenamide, which is subsequently oxidized first to a sulfinamide intermediate, then onward to the sulfonamide product. Presumably these oxygenation steps entail anodic oxidation and subsequent trapping with water. Concomitant reduction of protons at the cathode generates molecular hydrogen. Exposing independently prepared sulfenamide to these conditions also allowed for product formation. This electrochemical methodology enabled a library of sulfonamides to be rapidly synthesized from readily available starting materials.
Noël and co-workers have developed an electrochemical synthesis of sulfonyl fluorides via the oxidative coupling of thiols and potassium fluoride (Scheme 258).666 Constant current electrolysis of an MeCN/1.0 M aq. HCl solution (1:1 v/v) containing thiol, KF, and pyridine in an undivided electrochemical cell using a graphite anode and a stainless-steel cathode furnished the targeted sulfonyl fluoride (27 examples, 19–94%). Although other alkali fluorides such as NaF and electrophilic fluorine sources such as Selectfluor were effective, KF proved to be the cheapest and most effective fluoride source. The authors posit that KF serves as both a nucleophilic fluoride source and an electrolyte, as lower concentrations of KF proved equally effective if a different supporting electrolyte was added. This electrochemical reaction accommodated a variety of structurally and electronically distinct thiols. Thiophenols bearing electron-neutral, -donating, or -withdrawing substituents were well tolerated (258.1–258.6), as were heterocyclic thiols (258.7) and a variety of primary and secondary aliphatic thiols (258.8, 258.9).
Scheme 258. Electrochemical Synthesis of Sulfonyl Fluorides through the Oxidative Coupling of Thiols and Potassium Fluorides (Noël, 2019).

Kinetic experiments suggested that the thiol substrate was rapidly consumed within the reaction. The authors posited that oxidation and subsequent deprotonation of the thiol generates a thiyl radical that dimerizes to form a disulfide. Notably, disulfides proved to be competent substrates under the reaction conditions. Previous studies suggested that the disulfide can be oxidized to the corresponding radical cation and react with the nucleophilic fluoride to yield the corresponding sulfenyl fluoride, which furnishes the sulfonyl fluoride upon further oxidation.667 Concomitant reduction of protons at the cathode generates molecular hydrogen. The authors suggested that pyridine functions as a phase-transfer reagent, enabling fluoride to pass from the aqueous phase into the organic phase as the pyridinium fluoride salt.
To enable the rapid synthesis of a library of sulfonyl fluorides, the authors adapted this methodology to a flow electrochemical cell.668 Using a home-built electrochemical flow reactor under the same electrochemical conditions as the batch reaction, the authors were able to reduce reaction time from 24–36 h to ca. 5 min while producing the targeted sulfonyl fluoride in similar yield (seven examples, 37–85%). The authors attributed the reduced reaction time to an increase in reactor surface-to-volume ratio, a high interfacial area between the organic and aqueous phase, and an increase in mass transport. Because many sulfonyl fluorides are volatile and difficult to isolate, the authors paired this flow electrochemical technique with a sulfur(VI) fluoride exchange reaction (SuFEx)669,670 to produce a phenyl sulfonate derivative. This tandem flow process enables a library of sulfonic ester-containing compounds to be rapidly and efficiently synthesized.
4.3. Acylation and Heteroarylation of Amines from S–H Precursors under Visible-Light Activation
In addition to allowing a wide range of C–S and heteroatom-sulfur bond formations, PCET generation of thiyl radicals can also enable desulfurative functionalization of thioacids, thiolated arenes and heteroarenes through in situ transformation to more nucleofugal groups.
4.3.1. Synthesis of Amides from Amines and Thiocarboxylate Precursors
In 2016, Tan, Qiu, and Chen disclosed a photocatalytic method for the acylation of amines with potassium thiocarboxylate reagents (Scheme 259).671 Upon visible-light irradiation of MeCN substrate solutions with [Ru(bpy)3]Cl2 ([Ru-1]Cl2) photocatalyst under air, the authors disclosed 30 examples of amide bond formation in 66–94% yields. A wide assortment of potassium thiocarboxylate salts—aliphatic (259.2), aromatic, AA-derived (259.1)—and amines—primary (259.1) and secondary aliphatic (259.4), anilines—proved amenable to use in the transformation. Functionality tolerated included unprotected aliphatic alcohols (259.4), phenols (259.3) and carboxylic acids with selective acylation of only the amine functionality. These authors later applied a similar set of reaction conditions (2 mol% Ru(bpy)3Cl2 photocatalyst, thioacid reagent, K2CO3, and catalyst, under air) to achieve the acylation of phenols in a two-pot process by first generating diacyl disulfide using visible-light photocatalysis (seven examples) and then utilizing these as reagents for phenol acylation in a separate operation under non-photochemical conditions.672
Scheme 259. Photocatalytic Aerobic Oxidative Acylation of Amines with Thiocarboxylate Salts (Tan, 2016).

Mechanistically, the reaction is proposed to initiate via oxidation of the thiocarboxylate anion (e.g., for thiobenzoic acid potassium salt, Epox = +0.65 V vs SCE in MeCN),673 by the photoexcited Ru(II) photocatalyst (E1/2 *Ru(II)/Ru(I) = +0.77 V vs SCE in MeCN)64 to generate a thiyl radical intermediate, which then dimerizes to form a diacyl disulfide. The amine reacts as a polar nucleophile with the diacyl disulfide, displacing an equivalent of a thiopercarboxylate, which itself undergoes acylation with another equivalent of the amine partner. Catalyst turnover is achieved via oxidation of the transient Ru(I) complex (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN)64 with atmospheric oxygen (Epred = −0.92 V vs SCE in MeCN).674
In related work Biswas and co-workers demonstrated similar reactivity could be achieved with a semiconducting heterogeneous photocatalyst in place of molecular homogeneous transition-metal photocatalysts (Scheme 260).675 Using CdS NPs as heterogeneous photocatalyst, with amine and thioacid substrates in water under visible-light irradiation with a CFL bulb, 14 examples of amide bond formation were disclosed in 73–96% yields, including an example conducted on 40 mmol scale. Thioacid scope was demonstrated with thioacetic and thiobenzoic acids. The scope of nitrogen nucleophile was comparatively broad (ammonia, aryl (260.1, 260.2) and heteroarylamines (260.3), aliphatic amines (260.4), hydrazines (260.5), and tolerated a range of functionality (esters, ketones, aryl halides, phenols). The CdS NP photocatalyst could be recycled up to seven cycles before significant diminution in performance was noted. The mechanism proposed mirrors that of the small molecule system used by Tan and co-workers above, with the semiconductor acting as photooxidant. Now, the neutral alkyl or aryl thioacid (e.g., for thiobenzoic acid, Ep/2ox = +1.36 V vs NHE in H2O)676 is oxidized by the VB of the irradiated CdS NPs (Ep/2VB = +1.59 V vs NHE in H2O),675 yielding thiyl radical after proton loss which dimerizes to form the reactive diacyl disulfide reagent for amine acylation. Biswas and co-workers demonstrate that oxidation by holes on the CdS NPs were pivotal to reaction success by demonstrating that the reaction was poisoned by addition of noted hole scavenger ammonium oxalate.
Scheme 260. Photocatalytic Acylation of Amines with Thiocarboxylic Acids Using a Heterogeneous Catalyst (Biswas, 2018).

In 2020, Song and co-workers also reported amide bond-forming reactions between thioacids and amines using an organic photocatalyst (Scheme 261).677 Using a combination of thioacid, and aliphatic or aromatic amine in MeCN with N-Me Mes-Acr+BF4– ([Acr-1]BF4) as a photocatalyst under open air with blue-light irradiation, the authors disclosed 35 examples of amide bond formation with 71–95% yields. A range of aliphatic and aromatic thioacids proved amenable to the transformation, including thioacids derived from Nα protected amino acids (261.4), with minimal variance in performance with structure. Regarding amine scope, aliphatic, aromatic, and heteroaromatic amines (261.5) all coupled efficiently under the reaction conditions with selectivity observed for N-acylation over phenol, aliphatic alcohol, sulfonamide, or indole acylation. Embedded within reaction scope are numerous examples of API preparation including moclobemide, melatonin, and acetazolamide.
Scheme 261. Photocatalytic Acylation of Amines with Thiocarboxylic Acids Using an Organic Photocatalyst (Song, 2020).

The mechanism is proposed to begin with deprotonation of thioacid by the amine substrate to generate the corresponding thiolate, which is subsequently oxidized by the excited-state N-Me Mes-Acr+BF4– ([Acr-1]+BF4–) photocatalyst (E1/2 *Acr+/Acr• = +1.88 V vs SCE in PhCN)74 to the corresponding thiyl radical. Following this, the thiyl radical dimerizes to form diacyl disulfide, which can acylate the amine substrate with concomitant loss of thioperacid, itself an acylating agent for another equivalent of amine. Photocatalyst turnover is achieved by reduction of oxygen to superoxide, which can act as a proton scavenger. Mechanistic experiments demonstrated inhibition of acylation by addition of TEMPO or stoichiometric reducing agent dithiothreitol (DTT), and that diacyl disulfide is generated in the absence of amine substrate when irradiated with [Acr-1]+BF4–. Finally, the authors show that by running acylation with thioacetic acid in the presence of dibenzoyl disulfide both acetylated and benzoylated products were observed, supporting activity of the disulfide as a potential acylating agent.
4.3.2. Synthesis of Amino-Heterocycles from Amines and Heteroaryl Thiols
In 2017, Wacharasindhu and co-workers reported a protocol for the synthesis of 2-aminobenzoxazoles and 4-aminoquinazolines through a photoactivated SNAr reaction of heterocyclic thiols (Scheme 262).678 This protocol bypasses the necessity of converting the 2-mercaptobenzoxazole into a heteroaryl halide, sulfide, or sulfone prior to amination since it makes use of the in situ oxidation of the thiol to an oxidized sulfur intermediate, which can act as a leaving group for SNAr after nucleophilic attack by the amine partner. Scope studies were conducted by irradiation of an MeCN solution of RB photocatalyst, DBU base, and 2-mercaptobenzoxazole, under white LED irradiation in the presence of amine partner (5.0 equiv). Under these conditions, the authors reported 11 examples of benzoxazole amination with yields ranging from 61% to 88%. A number of primary (262.1) and secondary (262.2) amines were competent coupling partners, including those containing free hydroxyl groups (262.3) and acid labile functionality such as acetal 262.4. Notably, reaction with a less nucleophilic aniline afforded no product. Additionally, the authors reported three examples of 4-mercaptoquinazoline amination (262.5, 262.6) under similar conditions in DMSO solvent.
Scheme 262. Organophotocatalytic Amination of Heteroaromatic Thiols (Wacharasindhu, 2017).

*DMSO solvent.
The authors propose a mechanism wherein thiyl radical generation is accomplished via stepwise deprotonation of the mercaptan by DBU (pKa = 24.3 in MeCN, 13.9 in DMSO)48 and subsequent oxidation by the excited-state rose bengal photocatalyst (E1/2 *RB2–/RB3–• = +0.99 V vs SCE in H2O).349 Reaction of the thiyl radical and O2•–, generated by reduction of O2 by the reduced-state rose bengal photocatalyst (E1/2RB2–/RB3–• = −0.78 V vs SCE in H2O),349 affords the proposed peroxysulfur intermediate, which can act as a leaving group after nucleophilic addition of the amine to the heteroarene, in analogy to the use of methyl sulfone as a leaving group for nucleophilic aromatic substitution.679 The authors also considered an alternative mechanism proceeding through a disulfide intermediate as this compound was observed in their optimization studies. However, subjecting the mercaptobenzoxazole disulfide to the reaction conditions only afforded a trace amount of the product and a sulfenamide side product resulting from S–N bond formation (for similar reactivity, see section 4.1.6). Based on this result, the authors proposed that the formation of the disulfide species is not the major pathway for product formation.
4.4. Catalytic Applications of Thiol PCET in Mediating Substrate HAT
In this final section describing S–H PCET, we describe examples in which thiyl radical is not involved in formation of new bonds to sulfur, but rather is used as a mediator of substrate HAT for formation of new C–C, C–Si, and C–B bonds. The included examples demonstrate C(sp3)–H functionalization from readily available precursors.
4.4.1. Thiyl Radical Mediating Substrate HAT for C–C Bond Formation
In an early example of synthetic application of S–H bond activation via photocatalytic oxidative PCET, MacMillan and co-workers in 2014 disclosed a thiyl radical-mediated C(sp3)–H arylation of benzylic alcohols and ethers with electron-poor (hetero)aryl nitriles (Scheme 263).680 Irradiation of a DMA solution of 3.0 equiv of benzylic alcohol or ether substrate, cyanoarene, methylthioglycolate thiol (263.A) co-catalyst, Brønsted base K2HPO4, and octanal with Ir(ppy)3(Ir-1) as photocatalyst afforded the desired benzylic C(sp3)–H arylation products. In this work, 28 examples of C(sp3)–H arylation with yields ranging from 51% to 86% were reported, including preparations of arylated benzylic ethers (263.1), alcohols (263.2) and silyl ethers (263.3, 263.4). The protocol was also tolerant of unprotected primary alcohols, tertiary amines, and alkyl halides with minimal diminution in yields. Heterobenzylic C(sp3)–H bonds were also selectively functionalized in this work. Both parent and substituted 1,4-DCBs, and 4-cyanopyridine coupling partners were documented.
Scheme 263. Photocatalytic C(sp3)–H Arylation of Benzylic Alcohols and Ethers (MacMillan, 2014).

*DMA/DMSO (1:1) solvent.
This transformation proceeds via a proposed initial oxidative quenching of the photoexcited state of the Ir(III) catalyst (E1/2 Ir(IV)/*Ir(III) = −1.73 V vs SCE in MeCN)66 by the aryl nitrile (e.g., for 1,4-DCB, E1/2red = −2.02 V vs Fc+/Fc in MeCN)401 to generate an aryl nitrile radical anion, and an Ir(IV) complex. Methyl thioglycolate undergoes S–H bond homolysis through a concerted PCET event, jointly mediated by the action of this Ir(IV) oxidant (E1/2 Ir(IV)/Ir(III) = +0.77 V vs SCE in MeCN)66 and K2HPO4 Brønsted base additive (pKa = 12.67 in H2O),227 resulting in thiyl radical generation. Notably, the photooxidant is incapable of driving an efficient direct ET for a typical alkyl thiol (e.g., for Cys, E1/2ox = +0.85 V vs SCE in H2O),681 nor is the Brønsted base co-catalyst sufficiently basic to generate significant concentration of thiolate anion (e.g., for methyl thioglycolate, pKa = 7.9 in H2O)682 in a discrete PT step, thus demonstrating the concerted PCET homolysis pathway to be operative.
The nascent thiyl radical (e.g., for methyl thioglycolate, S–H BDFEcalc = 87.2 kcal mol–1)683 then abstracts a benzylic C(sp3)–H from the ether substrate (e.g., for the benzylic C–H bond of benzyl methyl ether, C–H BDFE = 85.8 kcal mol–1)684 in a HAT elementary steps to generate a benzylic radical. Finally, the benzylic radical undergoes radical–radical cross-coupling with the persistent aryl nitrile radical anion, expelling cyanide anion to re-aromatize and provide the arylated products. Octanal is added to these reactions as an in situ trapping agent for the cyanide generated during re-aromatization. This additive is also proposed to serve as an in situ masking group for benzylic alcohol substrates via hemiacetalization. Additionally, neither of the photoexcited-state Ir(III) complex (E1/2 *Ir(III)/Ir(II) = +0.35 V vs SCE in MeCN)66 or Ir(IV) complex generated in situ are capable of direct oxidation of the benzylic ether substrate (e.g., for benzyl methyl ether, Epox = +2.23 V vs SCE in MeCN),685 thus both the thiol and base are key for efficient relay of the potential of the catalyst into a HAT mediator. The PCET system allows the use of catalytic, rather than stoichiometric HAT reagents.
The use of a catalytic PCET-generated thiyl radical as an entry point to mediate carbon-centered radical formation via HAT was expanded to the preparation of vicinal amino alcohol motifs from benzylic ethers and imines, in subsequent work from MacMillan and co-workers in 2014 (Scheme 264).686 Irradiation of a DMA solution of 5.0 equiv of benzylic alcohol or ether substrate, imine, methylthioglycolate (264.A) co-catalyst, Brønsted base LiOAc with Ir(ppy)3(Ir-1) as photocatalyst afforded the desired benzylic C(sp3)–H cross-coupled vicinal amino alcohol products. Scope with respect to the benzylic ether component is reminiscent of that described in the prior report, with alkyl (264.1, 264.2, 264.4) and silyl ethers (264.3) derived from benzylic (264.1, 264.2, 264.4) and heterobenzylic (264.3) alcohols well-represented. Arene moieties embedded in the benzylic ether partner could tolerate alkyl, alkoxy, and trifluoromethyl substitution at the ortho-, meta-, and para-positions without notable diminution in yield. Imine partners were C,N-diaryl aldimines, with heteroarene substitution amenable at both sites. Arene and heteroarene moieties in the imine partners could also be substituted with halogens, esters, and alkyl ethers. In contrast with their initial disclosure,680 in this work photoexcited-state Ir(III) complex (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 and base (AcOH pKa = 4.76 in H2O; 13.63 in DMF)687,688 synergistically activate the S–H bond of methyl thioglycolate via concerted PCET to generate thiyl radical and the reduced Ir(II) complex. Next, SET between the imine substrate and the Ir(II) complex (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN)68 forms an α-amino radical anion, and regenerates the ground-state Ir(III) photocatalyst. The thiyl radical then abstracts a benzylic C(sp3)–H from the ether substrate (here used in 5 or 10-fold excess) to generate a benzylic radical, which subsequently undergoes radical–radical cross-coupling to yield, after protonation, the amino alcohol product.
Scheme 264. Photocatalytic C(sp3)–H Aminoalkylation of Benzylic Ethers (MacMillan, 2014).

*10 equiv of silyl ether.
MacMillan and co-workers later extended their dual photoredox/thiol catalytic system toward the activation and subsequent arylation of allylic C(sp3)–H bonds with cyano (hetero)arenes (Scheme 265).689 Irradiation of an acetone solution of 1.0–5.0 equiv of olefin substrate, cyanoarene, triisopropylsilanethiol co-catalyst, Brønsted base K2CO3 with Ir(ppy)3(Ir-1) as photocatalyst afforded the desired allylic C(sp3)–H arylated products. A variety of arylated products could be produced via this method, including those derived from cyclic (265.1–265.4), acyclic (265.5), and heteroatom-substituted alkenes. In the case of non-symmetrical olefin substrates, a mixture of product regioisomers was typically obtained. Electron-deficient pyridine heteroaryl groups could be introduced (265.3) in addition to arenes bearing other electron-withdrawing substituents (e.g., halides, phenyl sulfones). Through the application of allylic alcohols, in which the hydridic α-oxy H-atom is abstracted, β-aryl ketones could be afforded (265.4) via arylation at the distal allylic position.
Scheme 265. Dual Photoredox/Thiol-Catalyzed Allylic C(sp3)–H Arylation (MacMillan, 2015).

*Indicates minor regioisomer. **25% K2CO3 base. ***From 2-cyclohexene-1-ol. ****25 mol% A.
A postulated mechanism involves oxidative quenching of the Ir(III) photoexcited-state complex (E1/2 Ir(IV)/*Ir(III) = −1.73 V vs SCE in MeCN)66 with cyanoarene partner (e.g., for 1,4-DCB, E1/2red = −2.02 V vs Fc+/Fc in MeCN),401 yielding an Ir(IV) complex and arene radical anion. This Ir(IV) complex (E1/2 Ir(IV)/Ir(III) = +0.77 V vs SCE in MeCN)66 oxidizes the silanethiol co-catalyst (e.g., for TIPS-SH, E1/2ox = +0.28 V vs SCE in MeCN)690 to the corresponding radical cation, which then undergoes deprotonation by the carbonate base to generate the key thiyl radical intermediate. For reactions utilizing cyanopyridine coupling partners, significant reactivity was observed in the absence of the exogenous carbonate base, the basic site on the heteroarene can fulfill this role. The electrophilic thiyl radical then selectively abstracts the allylic H-atom from the alkene substrate, producing a transient allylic radical that engages in radical–radical coupling with the persistent arene radical anion to yield the final, C(sp3)–H functionalized product after elimination of cyanide anion. Competition experiments employing ethylbenzene (benzylic C–H BDE. = 85.4 kcal mol–1)691 and cyclohexene (allylic C–H BDE = 83.2 kcal mol–1)692 as substrates, afforded arylated cyclohexene as the sole product. The allylic–benzylic C(sp3)–H bond of the product, despite being weaker than the starting material C–H bond, is less hydridic and thus deactivated toward HAT with the electrophilic thiyl radical. This experiment established the selectivity of the thiyl radical for the weakest, most hydridic H-atom and polarity-based selectivity additionally explains the absence of doubly arylated products.
Building upon their suite of transformations utilizing dual photoredox/thiol organocatalysis, in 2015 MacMillan and co-workers disclosed a system enabling the C(sp2)–H alkylation of heteroarenes, utilizing alcohol feedstocks as the alkylating regent with loss of water (Scheme 266).693 Using [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) photocatalyst, ethyl 2-mercaptopropionate co-catalyst (266.A), and p-TsOH Brønsted acid activator in DMSO, a range of nitrogen-containing heterocycles were efficiently alkylated, with 29 examples presented in 43–98% yields. With respect to the heteroaromatic substrate, pyridines (266.1, 266.2), quinolines (266.3), and isoquinolines (266.4, 266.5), cinnolines, and phenanthridines (266.6) were all demonstrated to be competent reaction partners. In addition to methanol and ethanol, the alcohol scope included a small array of other primary aliphatic alcohols. Notably, secondary alcohols and polyols were tolerated and no evidence of regioisomeric C–H alkylation products were noted. Cyclic ethers could also be used in select examples to afford linear C–H alkylated products.
Scheme 266. C(sp2)–H Alkylation of N-Heteroaromatics with Alcohols via Dual Photoredox/Thiol Catalysis (MacMillan, 2015).

*From 1,3-butanediol.
The authors proposed that the reaction initiates through single-electron reduction of a sacrificial amount of protonated heteroarene (e.g., for pyridinium perchlorate, Ep/2 = −1.0 V vs Ag+/Ag in H2O)694 by the excited-state Ir(III) photocatalyst (E1/2 Ir(IV)/*Ir(III) = −0.96 V vs SCE in MeCN)68 to generate an Ir(IV) intermediate. This Ir(IV) complex (E1/2 Ir(IV)/Ir(III) = +1.21 V vs SCE in MeCN)68 then oxidizes the thiol co-catalyst to the corresponding radical cation which, following loss of a proton, forms the key thiyl radical. The thiyl radical then abstracts the hydridic α-C–H of the primary alcohol (e.g., for H–CH2OH, BDE = 97.2 kcal mol–1)695 or ether substrate to form a C-centered radical. The selectivity for the α-C–H over the O–H can be rationalized by polarity matching between the electrophilic thiyl radical and hydridic C–H, compared to the protic O–H. The nascent C-centered radical then adds to the protonated heteroarene substrate in a Minisci fashion,696 to generate an aminium radical cation which loses a proton to form an α-amino radical intermediate. This undergoes a spin-centered shift (SCS) elementary step,697 to yield a heterobenzylic radical with loss of H2O, which is reduced to the corresponding anion by the photoexcited-state Ir(III) catalyst, and PT produces the alkylated heteroarene.
Since these initial reports from MacMillan, other groups have subsequently expanded the scope and utility of this thiol PCET-based carbon-radical generation methodology to other substrate classes. For example, Wang and co-workers in 2016 reported the C(sp3)–H arylation of N-protected tetrahydroisoquinolines and tetrahydro-β-carbolines with (hetero)aryl nitriles (Scheme 267).698 Under visible-light irradiation of DMA solutions of N-Boc tetrahydroisoquinoline (2.0 equiv) and cyanoarene substrates with Ir(ppy)3 (Ir-1) as a photocatalyst, triphenylsilanethiol as HAT co-catalyst (267.A), and K2HPO4 as base, the authors disclosed 20 examples of arylation (38–96% yields). Though the authors found that triphenylsilanethiol (267.A) was the optimal HAT co-catalyst and K2HPO4 the optimal base, control studies revealed that the reaction is successful in the absence of an exogenous base, albeit with diminished yields (for 267.1, 78%). Given the requisite reduction of the aryl coupling partner by the excited-state photocatalyst, only electron-deficient aryl nitriles gave efficient reactivity. Nonetheless, various electron-withdrawing substituents were tolerated on the aryl nitrile coupling partner, including sulfonyl groups (267.2), methyl esters (267.3), and halides (267.4). Derivatives of 4-cyanopyridine also proved to be competent coupling partners (267.4). Tetrahydroisoquinolines bearing electron-withdrawing as well as electron-donating substituents were tolerated, providing access to arylated substrates containing bromine and methoxy groups. Furthermore, benzylic α-amino positions on alternative partially saturated heterocycles could be arylated using this method to afford arylated tetrahydro-β-carboline (267.5) and dihydroisoindole derivatives. A mechanistic proposal analogous to that of Qvortrup, Rankic, and MacMillan was proposed.680
Scheme 267. C(sp3)–H Arylation of N-Protected Tetrahydroisoquinoline and Tetrahydro-β-carbolines (Wang, 2016).

Liu and co-workers in 2019 disclosed a system for the synthesis of tertiary alcohols from ketones and allylic or benzylic C(sp3)–H bonds via radical–radical coupling (Scheme 268).699 This transformation involved the blue-light irradiation of substrate solutions in acetone in the presence of Ir(ppy)3 (Ir-1) photocatalyst, and KSAc or TIPS-SH with K2CO3 as HAT co-catalyst. A scope of 51 examples in 22–85% yields was demonstrated. A range of aryl (268.1, 268.2), heteroaryl (268.3), and alkyl-aryl (268.4) ketones proved amenable to the transformation, allowing efficient formation of hindered tertiary alcohols, including some precursors of APIs Tamoxifen and Fenofenadine. While chiefly symmetric, cyclic olefins were utilized, the reaction also tolerated aliphatic olefins, alkyl benzene substrates (268.5), and a single example with an aldehyde to generate an α-hydroxyketone product (268.6). With unsymmetric olefin substrates, regioisomeric mixtures were often obtained, except in select case—notably allylic ethers and alcohols.
Scheme 268. Dual Photoredox/Thiyl Catalysis for the Synthesis of Tertiary Alcohol via Allylic and Benzylic C(sp3)–H Cleavage (Liu, 2019).

*Indicates minor regioisomer.
Aligning with the mechanism previously proposed by MacMillan and co-workers,686 this reaction initiates with photoexcitation of the Ir(III) photocatalyst (E1/2 Ir(IV)/*Ir(III) = −1.73 V vs SCE in MeCN),66 which then engages in single-electron reduction with the aryl ketone substrate (e.g., for acetophenone, Ep/2red = −2.14 V vs SCE in MeCN; for benzophenone, Ep/2red = −1.83 V vs SCE in MeCN)700 to generate the corresponding ketyl radical anion and an Ir(IV) complex. Thiloate—either present as KSAc or through in situ deprotonation of TIPS-SH by K2CO3—is then oxidized by the transient Ir(IV) complex (E1/2 Ir(IV)/Ir(III) = +0.77 V vs SCE in MeCN)66 to generate a thiyl radical intermediate while regenerating the ground-state Ir(III). The incipient thiyl radical (for TIPS-SH, S–H BDFE = 88.2 kcal mol–1)690 abstracts an allylic (e.g., for cyclohexene, allylic C–H BDFEcalc = 83.2 kcal mol–1)692 or benzylic (e.g., for toluene, benzylic C–H BDFEcalc = 89.9 kcal mol–1)701 C–H bond from the olefin or alkyl benzene substrate to form a new C-centered radical, which then undergoes radical–radical coupling with the ketyl radical anion to yield the tertiary alcohol product after protonation.
A further exploration of the use of PCET-generated thiyl radicals to effect carbon-centered radical formation is seen in subsequent work by the groups of Rueping and Huang, focusing on the formation of homoallylic amines by redox-neutral coupling of imines and olefins via allylic C(sp3)–H abstraction (Scheme 269).702 The combination of Ir(III) photocatalyst [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6), Li2CO3 base, and TIPS-SH (269.A) HAT co-catalyst was found optimal for the allylic coupling of imines and olefins, which allowed for the efficient preparation of a range of homoallylic amine derivatives (32 examples, 46–91% yields). Imine scope chiefly includes N-aryl aromatic aldimines (269.1–269.3), with select examples of N-alkyl aldimines (269.4), and N-aryl ketimines (269.3). Heterocyclic substrates such as pyridine (269.2) and thiophene derivatives were well-tolerated, as were a variety of functionalized aromatic imines bearing pinacol boronate, ester, sulfide, ether, and halide substituents. Prototypical allylic substrates a variety of cyclic (269.1–269.4) and acyclic (269.5, 269.6) olefins, including monoterpene and steroid derivatives also proved competent substrates under the reaction conditions. Tri- and tetrasubstituted aliphatic olefin substrates gave products reflective of allylic radical rearrangement (269.5) with steric effects favoring coupling at the less hindered allylic radical.
Scheme 269. Allylic C(sp3)–H Alkylation with N-Aryl Aldimines and Ketimines (Huang and Rueping, 2020).

*Indicates minor regioisomer.
The reaction proceeds via proposed initial photoexcitation of [Ir-3]PF6 (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 which then oxidizes TIPS-SH (E1/2ox = +0.28 V vs SCE in MeCN)690 via concerted PCET in the presence of Li2CO3 to generate a thiyl radical intermediate along with an Ir(II) complex. Following this, thiyl radical (e.g., for TIPS-SH, S–H BDFE = 88.2 kcal mol–1)690 abstracts an allylic C(sp3)–H bond to form a C-centered allylic radical. In contrast to the earlier proposal of MacMillan for benzylic radical coupling with imines,686 the authors here favor a route in which the allylic C-centered radical adds to the neutral closed-shell imine substrate to generate an N-centered radical, which is subsequently reduced by the Ir(II) photocatalyst (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN)68 to form the product homoallylic amine following protonation. The rationale for this proposal rests upon three observations: (1) the lack of informative side products that would be suggestive of single-electron reduction of the imine substrate (e.g., vicinal diamines or amines), (2) lack of yield dependence on electronic character of the imine substrate, and (3) CV studies indicating that reduction potentials of the imine substrates (e.g., for N-benzylidene aniline, Ep/2red = −1.91 V vs SCE in MeCN)21 are typically 400–500 mV more negative than that of the Ir(III)/Ir(II) couple of [Ir-3]PF6. We note here a report from Romanov-Michailidis and co-workers, who proposed LA activation of an imine by Li cation for subsequent radical addition, but in an intramolecular fashion (see section 2.8).381 In this work from Rueping and Huang, however, significant reactivity is observed in a model substrate in the absence of Li2CO3 (for 269.1, 55% compared to 81% under optimal conditions) suggesting LA activation plays less of a role here.
In 2017, Kanai and co-workers reported a ternary catalytic system capable of room temperature, acceptorless dehydrogenation of tetrahydronaphthalenes to afford the corresponding naphthalenes and 2 equiv of hydrogen gas (Scheme 270).703 Along with 430 nm light irradiation, the authors employed the organic photocatalyst N-Me Mes-Acr+ClO4([Acr-1]ClO4), a BINOL-derived thiophosphoric imide HAT co-catalyst ((±)-270.A), and a Pd(II) co-catalyst to drive this transformation. Optimization studies revealed that the BINOL scaffold as well as the highly electrophilic nature of the corresponding thiyl radical of the phosphoric imide HAT co-catalyst were crucial aspects for effective reactivity. No reactivity was observed with more conventional thiol HAT co-catalysts, such as thiophenol or methyl thioglycolate. Earlier methods for hydrocarbon desaturation often required forcing conditions for efficient reactivity.704 Recent related work from Sorensen and co-workers established key precedent in mild acceptorless, photocatalytic alkane desaturation employing n-Bu4N decatungstate and a Co(III) co-catalyst to cooperatively abstract two adjacent H-atoms from simple alkane and alcohol substrates to yield the corresponding alkene or carbonyl-containing products, respectively.705 Lei and co-workers have recently reported mild acceptorless electrochemical conditions enabling this transformation also through the use of TEMPO as a HAT reagent.706 More broadly, the development of mild, catalytic acceptorless desaturation methods could allow the use of saturated hydrocarbons as transportable hydrogen precursors for alternative fuel applications.707 In this work, Kanai and co-workers reported 11 examples of tetrahydronaphthalene desaturation in yields of 46–82%. This desaturation protocol tolerated both electron-withdrawing and electron-donating substituents on the aromatic ring of the starting material, including aryl groups (270.1), halogens (270.2), and amides (270.3). Substitution at the site of desaturation was also tolerated (270.4). Substrates other than tetrahydronaphthalenes also participated in this transformation, such as 3-methylcyclohexene which afforded toluene (270.5) in 17% GC yield after 47 h of irradiation.
Scheme 270. Triple Photoredox/Thiyl/Pd(II) Catalytic Dehydrogenation of Tetrahydronaphthalenes (Kanai, 2017).

In the proposed mechanism, the photoexcited state of the Acr photocatalyst (E1/2ox *Acr+/Acr• = +1.88 V vs SCE in PhCN)73 undergoes steps of ET and PT with the thiophosphoric imide co-catalyst to generate a thiyl radical that then abstracts a H-atom from a benzylic site on the substrate, similar to reactivity previously observed by MacMillan and co-workers.680,686,689,693 The benzylic radical then intercepts the Pd(II) co-catalyst to give a putative alkyl-Pd(III) species that is reduced to the corresponding alkyl-Pd(II) complex by the neutral acridine radical form of the photocatalyst (E1/2 = −0.49 vs SCE, MeCN). Subsequent rapid β-hydride elimination from the resulting Pd(II) intermediate liberates the desaturated product.708 Protonation of the resulting Pd(II)–H intermediate, presumably ultimately from the thiol radical cation, generates molecular hydrogen and closes the transition-metal catalytic cycle. Further iteration of this scheme yields the naphthalene product. The generation of 2 equiv of hydrogen gas in the overall reaction with tetrahydronaphthalene was confirmed through a two-pot transfer hydrogenation reaction wherein the atmosphere of the desaturation reaction was used to carry out the catalytic hydrogenation of 2 equiv of cyclododecene, which afforded cyclododecane in 82% NMR yield.
Kanai and Mitsunuma in 2020 then disclosed a catalytic tetrad for the diastereoselective allylation of aldehydes with unactivated C(sp3)–H olefin precursors (Scheme 271).709 Reaction conditions comprised of Acr photocatalyst N-Ph-Mes-Me2-Acr+-BF4 ([Acr-2]BF4), a BINOL-derived thiophosphoric imide HAT co-catalyst ((±)-271.A), K2HPO4 Brønsted base co-catalyst, and CrCl2 transition metal catalyst, under blue-light irradiation. While previous disclosures from the Glorius710 and Kanai711 groups have utilized Cr(II) complexes as co-catalysts for photocatalytic radical polar crossover allylation, in these prior works generation of the requisite allylic radical was accomplished via α-deprotonation of olefin radical cations. As such, the scope of these transformations was chiefly restricted to comparatively electron-rich olefin partners. Kanai and Mitsunuma have overcome this by instead generating the key allylic radical through HAT mediated by the thiophosphoric imide additive ((±)-271.A). Kainai and Mistunuma reported 38 examples of aldehyde allylation to yield homoallylic alcohol products in yields of 55–98% and diastereoselectivities ranging from 1.2:1 to >20:1. Both aliphatic (271.1, 271.2) and aromatic (271.4) aldehydes were efficient partners in the allylation reaction, with tolerance of boronate esters, aryl halides, nitriles, thioethers, tertiary ammonium salts (271.3), dipeptides (271.4), a protected nucleoside, and allylic and benzylic C–H bonds. Though much of the scope exposition is demonstrated with 2-butene, a range of olefins proved efficacious substrates, including disubstituted styrenes, allylic ethers, cyclic and acyclic aliphatic olefins. Isolated products were reflective of C–C bond formation at the more substituted position of the nascent allyl radical. Through the inclusion of a chiral bisoxazoline ligand, this allylation reaction could also be rendered enantioselective (59–88% e.e.).
Scheme 271. Photocatalytic Aldehyde Allylation via Ternary Catalysis (Kanai and Mitsunuma, 2020).

As in their previous report,703 this transformation initiates through generation of a thiophosphoric imide radical intermediate through steps of PET (E1/2 *PC+/PC• = +2.09 V vs SCE in MeCN)75 and subsequent PT. This thiophosphoric imide radical then abstracts an allylic C–H bond to yield an allylic radical. The nascent allylic radical combines with Cr(II) to form an allyl Cr(III) intermediate which adds to the aldehyde substrate through a six-member-ring chair transition state as in the classic Nozaki–Hiyama–Kishi reaction.712 The resultant Cr(III) alkoxide undergoes protonolysis to liberate the product. The reduced state of the photocatalyst (E1/2 = −0.58 V vs SCE in MeCN)112 facilitates ET to Cr(III) (E1/2 Cr(III)/Cr(II) = −0.51 V vs SCE in DMF),713 returning both of these components to their respective active oxidation states ready for catalytic turnover. Mechanistic studies ruled out involvement of a ketyl radical intermediate through observation of no ring cleavage when cyclopropyl carbaldehyde was used as a substrate. Radical involvement was substantiated by complete inhibition of product formation upon addition of TEMPO. Finally, an intermolecular KIE analysis using 3,3-d2-decene revealed a kH/kD = 3.7, consistent with C(sp3)–H abstraction being rate-limiting.
In 2020, Kanai and co-workers also reported a redox-neutral hydroxyalkylation of heteroarenes, utilizing a thiophosphoric acid based co-catalyst ((±)-272.A) to generate an acyl radical intermediate from an aldehyde via formyl C(sp2)–H bond HAT (Scheme 272).714 Visible-light irradiation of admixtures of heteroarene and aldehyde substrates in the presence of organic photocatalyst N-Me Mes-Acr+ClO4 ([Acr-1]ClO4), BINOL-derived thiophosphoric acid HAT co-catalyst ((±)-272.A), and TFA additive in CH2Cl2 solution afforded 37 examples of the corresponding hydroxyalkylation product in 42–94% yields. Both aryl (272.1) and alkyl (272.2–272.7) aldehydes underwent this transformation. A number of pyridine (272.5), quinoline (272.3, 272.4, 272.6), and isoquinolines (272.1, 272.2, 272.7) underwent hydroxyalkylation in this manner. Additionally, both alkyl and aryl halides were well tolerated. The authors also demonstrated this protocol for a number of late-stage modifications of more complex substrates, including camptothecin (272.6) and fasudil (272.7).
Scheme 272. Dual Photoredox/Thiyl Catalytic Hydroxyalkylation of Heteroarenes (Kanai, 2020).

The authors propose a mechanism wherein the thiophosphoric acid is oxidized by the photoexcited-state Acr catalyst (E1/2 *PC+/PC• = +1.88 V vs SCE in PhCN)74 and deprotonated to generate a neutral thiyl radical, which then engages an aldehyde in formyl C(sp2)–H HAT (e.g., in for the formyl C–H bond in acetaldehyde, C–H BDFE = 89.4 kcal mol–1)25 to form an acyl radical. This radical undergoes addition to a protonated isoquinoline in a Minisci-like fashion.696 Loss of a proton is followed by a SCS process,697 which re-aromatizes the isoquinoline and forms a neutral ketyl radical. Subsequent ET (E1/2 PC+/PC• = −0.49 V vs SCE in PhCN)74 and PT form the product and regenerate the ground-state photocatalyst. Deuterium labeling experiments were conducted to obtain preliminary information on the mechanism of this transformation. When the authors subjected deuterium labeled aldehyde to the reaction conditions, the product only retained 15% deuterium at the α-hydroxy position. However, when the reaction was conducted with F3CCO2D and a CH2Cl2/D2O (20:1) co-solvent mixture, the authors observed 95% deuterium incorporation at this position. This, the authors suggest, indicates that protonation is responsible for forming this C–H bond in the product.
König and co-workers recently reported the photocatalytic generation of benzylic carbanions through dual photoredox and thiyl radical HAT catalysis, providing an entry point to high-energy carbanions from C(sp3)–H bonds without the requirement for substrate pre-functionalization, as is the case with traditional Grignard and Barbier-type methodologies (Scheme 273).715,716 The carbanions produced by this catalytic system then reacted with CO2 to yield C(sp3)–H carboxylation products. Optimized reaction conditions consisted of irradiation of a DMF solution of substrate with 4CzIPN photocatalyst, TIPS-SH as a thiol co-catalyst (273.A) under CO2 (4 atm.). This method allowed the preparation of 2-arylpropanoic acids with a variety of substitution patterns on the aromatic ring, including 4-fluoro (273.1) and 4-chloro substituents (273.2), in moderate to good yields (27 examples, 36–89% yields). However, more sensitive functionality, such as Br- and I-substituents as well as ketones and aldehydes, were not well tolerated. Photocatalysts more reducing in their radical anion form than 4CzIPN (E1/2 PC/PC•– = −1.24 V vs SCE in MeCN)78 were required for efficient reaction with less readily reduced substrates such as 4-ethylanisole (273.3), which for example required 3DPA2FBN (E1/2 PC/PC•– = −1.92 V vs SCE in CH2Cl2).77 A variety of heteroaromatic substrates could be carboxylated in moderate to good yields (273.4, 273.5). An alternative photocatalyst, 3DPAFIPN (E1/2 PC/PC•– = −1.59 V vs SCE in MeCN)77 was required to achieve good yields in the case of 2-ethylthiophene (273.5).
Scheme 273. Photocatalytic Carboxylation of Benzylic C(sp3)–H Bonds (König, 2019).

*3DPA2FBN photocatalyst. **3DPAFIPN photocatalyst.
The authors demonstrated that the 4CzIPN photocatalyst first undergoes in situ alkylation to form the active 4CzPEBN, derived from 4CzIPN through substitution of one cyano group for a 1-phenylethyl group, in the case with ethylbenzene as substrate. The photoexcited state of this 1-phenylethyl-substituted photocatalyst (E1/2 *PC/PC•– = +1.19 V vs SCE in DMF)715 undergoes reductive quenching with TIPS-SH (E1/2ox = +0.28 V vs SCE in MeCN)690 to produce the corresponding thiol radical cation which is subsequently deprotonated to form a thiyl radical. A benzylic C–H bond (e.g., for the benzylic C–H bond in ethylbenzene, C–H BDFE = 85.4 kcal mol–1)717 is then abstracted by this thiyl radical (e.g., for TIPS-SH, S–H BDFE = 88.2 kcal mol–1)690 and the resulting benzylic radical (e.g., for the benzylic radical deriving from ethylbenzene (PhCH·CH3) E1/2red = −1.60 V vs SCE in MeCN)135 is reduced by the radical anion of the 4CzPEBN photocatalyst (E1/2 PC/PC•– = −1.69 V vs SCE in DMF).715 Finally, the resultant benzylic carbanion reacts with dissolved CO2 to form a carboxylate intermediate that is eventually protonated to form a carboxylic acid product. The inhibition of product formation by TEMPO and BHT suggested the intermediacy of radical species. When a substrate with a doubly deuterated benzylic position was subjected to the reaction, no proton exchange was observed at the benzylic position, suggesting an irreversible HAT of the substrate.
The König group shortly thereafter reported the extension of this photocatalytic carbanion generation to the nucleophilic addition to ketones and aldehydes for the synthesis of secondary and tertiary homobenzylic alcohols (Scheme 274).718 Here, the authors found the more reducing 3DPA2FBN photocatalyst to be universally more effective than 4CzIPN. An exogenous base, K2CO3, was found to greatly improve yields and the use of a large excess of the electrophile (10 equiv to solvent quantities) was found to be beneficial in most cases in terms of yield. However, yields suffered due to inhibition from the product alcohols, presumably due to competition for quenching of the carbanion intermediate by the protic hydroxyl groups. Nonetheless, this method provided access to a broad range of homobenzylic alcohols in low to good yields (38 examples, 11–87% yields). Electron-rich arenes (274.1) proved to be more efficient for carbanion generation than electron-deficient arenes (274.2), likely due to slower H-atom abstraction or attenuated carbanion reactivity in the case of electron-deficient arenes. More sterically encumbered benzylic carbanions (274.3) were found to exhibit decreased yields. Similarly, sterically accessible ketones generally outperformed longer-chain and more bulky ketones (274.4). A few successful examples using aldehydes as electrophiles were reported (274.5), though in these reactions, a 3-fold excess of the ethylbenzene substrate was required to limit competitive H-atom abstraction of the formyl C–H bond. Similar trends were observed with aldehydes as with ketones, where steric encumbrance on the aldehyde resulted in lowered yields.
Scheme 274. Photocatalytic Addition of Benzylic Carbanions to Ketones and Aldehydes (König, 2019).

*5 mol% 3DPA2FBN, 20 mol% 274.A, 50 mol% K2CO3, 3 equiv of aldehyde in MeCN.
This homobenzylic alcohol formation follows a similar set of mechanistic steps as their previously described benzylic carboxylation.715 However, in the case of the homobenzylic alcohol formation, the thiyl radical is proposed to originate from the thiol co-catalyst via PT followed by ET, unlike the alternative ET/PT sequence invoked in the benzylic carboxylation mechanism. Potentially, this mechanistic alteration arises because the less oxidizing photoexcited state of the 3DPA2FBN photocatalyst (E1/2 *PC/PC•– = +0.92 V vs SCE in CH2Cl2)77 likely cannot as readily oxidize the thiol to the radical cation (E1/2ox = +0.28 V vs SCE in MeCN)690 as the 4CzIPN (E1/2 *PC/PC•– = +1.43 V vs SCE in MeCN)78 photocatalyst used in the former report.
More recently, the König group published an extension of this photocatalytic carbanion generation methodology to include the formation of 1,3-dithiane-substituted carbanions enabling a photocatalytic variant of the Corey–Seebach reaction (Scheme 275).719 Through the reaction of the dithiane-stabilized carbanions with ketones and aldehydes, this method provided access to a variety of dithiane products that can be converted to valuable α-hydroxy ketones or aldehydes. Investigation of the reaction conditions revealed that the use of 3DPA2FBN as photocatalyst, TIPS-SH (275.A) as HAT co-catalyst, and bis(neopentylglycolato)diboron (B2Neop2) as a LA co-catalyst in dry DMF under blue-light irradiation was optimal. Generally, a large excess of the ketone or aldehyde substrate was required for adequately efficient conversion. Thirty-seven examples of dithiane alkylation were demonstrated in yields of 9–65%. Dithianes containing alkyl substitution at the site of carbanion generation were well tolerated (275.1). The reaction also demonstrated good functional group tolerance as dithianes bearing more sensitive functionality, such as cyano and ester (275.2) groups, also proved competent in this reaction, indicating high chemoselectivity for reaction of the dithiane carbanion. With respect to the electrophile, a variety of functional groups on the ketone proved amenable to this transformation, including tertiary amines (275.3) and alkenes (275.4), despite the potential for thiol–ene side products with the latter. Aldehydes also were effective electrophiles (275.5).
Scheme 275. Photocatalytic Corey–Seebach Reaction (König, 2020).

*3 equiv of dithiane, 1 equiv of aldehyde.
The authors propose a mechanism analogous to previous work is also operable in the case of dithiane substrates. SV studies indicated minimal quenching of the photocatalyst excited state by the dithianes or electrophiles, whereas the thiol HAT co-catalyst was observed to efficiently quench. Thus, oxidation and deprotonation of the thiol co-catalyst affords thiyl radical, which then abstracts the hydridic C–H bond of dithiane substrate to afford a carbon-centered radical. The dithiane radical intermediate (e.g., for 2-methyl-1,3-dithiane E1/2red(calc) = −1.87 V vs SCE)719 generated through HAT with the open-shell thiyl can be reduced by the radical anion of the photocatalyst (E1/2 PC/PC•– = −1.92 V vs SCE in CH2Cl2),77 giving rise to the key dithiane carbanion intermediate. The relevance of this carbanionic intermediate was established through deuterium labeling experiments with t-BuOD in the absence of electrophiles, which showed 70% deuterium incorporation into the recovered dithiane substrate even though the dithiane radical (e.g., for 2-methyl-1,3-dithiane, C2 C–H BDEcalc = 88.2 kcal mol–1)719 should not be able to abstract deuterium from t-BuOD (O–H BDE = 106.3 kcal mol–1).720
Though used less frequently than alkyl and aryl thiols, thioacids have also seen use as reagents mediating substrate HAT through S–H bond homolysis via PCET. Thioacids are both more acidic (e.g., for thioacetic acid, pKa = 3.2 in H2O)682 and have stronger S–H bonds (e.g., for thioacetic acid, S–H BDE = 88 kcal mol–1; for thiobenzoic acid, S–H BDE of thiobenzoic acid = 87.4 kcal mol–1)399 than analogous alkyl and aryl thiols (e.g., for thiophenol, pKa = 10.3 in DMSO, S–H BDE = 79.1 kcal mol–1).336 In 2018, Hamashima and co-workers disclosed a photocatalytic system for the regioselective α-arylation of benzylamines with cyanoarenes jointly catalyzed by thiobenzoic acid (276.A), K2HPO4 as a stoichiometric base and Ir(ppy)3 photocatalyst (Ir-1) (Scheme 276).673 Using this system, these authors described 34 examples of α-arylation with 27–98% yields. The method proved amenable to the functionalization of a variety of benzylic and heterobenzylic (pyridine, indole) primary, secondary, and tertiary amine derivatives. To underscore the selectivity of the thioacid HAT catalyst, the authors also demonstrated regioselective α-arylation of a benzylic amine over a benzylic alcohol (276.4) as well as a benzylic aniline over a benzylic amine (276.5). Though initial scope was explored with terephthalonitrile as the arylating reagent, the authors also showed cyanopyridines and cyanoquinolines were competent partners.
Scheme 276. Photoredox/Thiobenzoic Acid Dual Catalysis for the α-Arylation of Benzylamines (Hamashima, 2018).

Akin to earlier systems, the mechanism of this transformation is proposed to proceed through initial ET from excited-state Ir(III) photocatalyst (E1/2 Ir(IV)/*Ir(III) = −1.73 V vs SCE in MeCN)66 to the cyanoarene substrate (e.g., for 1,4-DCB, E1/2red = −1.64 V vs SCE in MeCN)401 to generate the corresponding persistent arene radical anion and the Ir(IV) state of the photocatalyst. Subsequently, thiobenzoate, formed from the deprotonation of thiobenzoic acid by KH2PO4, is oxidized (e.g., for potassium thiobenzoate, E1/2ox = +0.80 V vs AgCl/Ag in DMA)673 to the corresponding thiyl radical which then abstracts the benzylic α-amino C–H (e.g., for N,N-dimethylbenzylamine, BDE = 84.9 kcal mol–1)87 to form an α-amino radical. The oxidation of the thiobenzoate component was found to be more favorable than that of the amine component (e.g., for N-benzyl tetrahydroquinoline, Epox = +0.97 V vs AgCl/Ag in DMA).673 The nascent α-amino radical undergoes radical–radical coupling with the cyanoarene radical anion. Finally, loss of cyanide forms the α-arylated product.
Hamashima and co-workers in 2020 described a method for the homo-coupling of benzylic amines and alcohols under visible-light irradiation to form vicinal diamines and diols (Scheme 277).721 Through blue-light irradiation of a DMA solution of benzylic alcohol or amine substrate, thiobenzoic acid HAT co-catalyst (277.A), pivaladehyde, and MgSO4 as desiccant with [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) as photocatalyst, the authors disclosed 12 examples of amine and alcohol homo-coupling with 23–80% yields (277.1–277.4). The scope included a small number of tertiary benzylic amines as well as primary and secondary benzylic alcohols differentiated primarily by arene substitution.
Scheme 277. Photoredox/Thiobenzoic Acid Co-catalyzed Reductive Homo-Coupling of Benzylic Amines and Alcohols (Hamashima, 2020).

The authors propose this reaction proceeds through protonation of pivalaldehyde by thiobenzoic acid to generate the thiolate and an oxocarbenium. While the authors propose a stepwise PT/ET mechanism to generate this thiyl radical, we reason that this process may be better described as a concerted PCET process given the significant offset between the relative acidities of the carbonyl substrate and thiocarboxylic acid catalyst (e.g., for 9-formylfluorene, pKa = −4.5 in H2O; for thioacetic acid, pKa = 3.2 in H2O).682,722 Thiolate is then oxidized by the excited-state Ir(III) photocatalyst (E1/2*Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 to form thiyl radical and the Ir(II) state of the photocatalyst. The nascent thiyl radical then abstracts an α-heteroatom benzylic C–H bond to form the corresponding α-amino or α-oxy carbon-centered radical, which dimerizes with another equivalent of radical to form the meso diamine or diol products. Finally, the ground-state Ir(III) photocatalyst is regenerated through a concerted reductive PCET event from the Ir(II) species to the aldehyde to form neutral ketyl radical. As no pivalaldehyde derived byproducts were detectable, the authors ran an experiment with 2-methyl-2-phenylpropionaldehyde to determine the fate of the ketyl radical generated in the reaction from the sacrificial oxidant. In these experiments, the authors observed byproducts consistent with decarbonylation and radical–radical coupling with the alcohol substrate, suggesting similar pathways may be operative in conversion of pivalaldehyde to volatile products.
4.4.2. Thiyl Radical Mediating Substrate HAT for Si–C and B–C Bond Formation
In addition to these above highlighted for C–H bond activation through thiyl radical-mediated HAT, we now present two methods for Si–H and B–H bond activation and subsequent functionalization. In 2017, Wu and co-workers disclosed conditions for the anti-Markovnikov hydrosilylation of both electron-rich and electron-poor olefins enabled by dual photoredox and thiol HAT co-catalysis (Scheme 278).690 Blue-light irradiation of a mixture of electron-rich olefin and silane in the presence of 4CzIPN as photocatalyst, TIPS-SH HAT co-catalyst (278.A), and DIPEA Brønsted base co-catalyst, in dioxane or THF afforded 21 examples of olefin hydrosilylation in 35–95% yields. A range of secondary and tertiary alkyl, mixed aryl-alkyl (278.1, 278.2, 278.4), and aryl silanes proved amenable as silane reagents, with notable diminution in yield seen only for sterically encumbered (i-Pr)3Si-H. Electron-rich olefin scope was restricted to terminal olefins, but within this, the reaction tolerated several functional groups including esters, ketones, amides, epoxides, silanes and alkyl chlorides without competing reduction or dehalogenation noted. Electron poor olefins required the use of a quinuclidine HAT co-catalyst in place of TIPS-SH (278.A).
Scheme 278. Dual Photoredox/Thiol Co-catalytic Hydrosilylation of Olefins (Wu, 2017).

The authors propose initial oxidation of 278.A (E1/2ox = +0.28 V vs SCE in MeCN)690 by the excited state of the photocatalyst (E1/2 *PC/PC•– = +1.35 V vs SCE in MeCN),78 followed by deprotonation to form the corresponding thiyl radical. This thiyl radical abstracts the hydridic Si–H bond of the silane substrate to generate silyl radical and regenerate TIPS-SH. The silyl radical adds to the less-sterically encumbered site of the electron-rich olefin substrate to afford a nucleophilic, secondary C-centered radical. Product is then formed via a mildly endergonic HAT process between the TIPS-SH (S–H BDEcalc = 88.2 kcal mol–1)690 and this C-centered radical (e.g., for the β-Si C–H bond of n-octyl dimethylphenylsilane, C–H BDEcalc = 93.2 kcal mol–1).690 The resulting thiyl radical from this process can then close the photocatalytic cycle by undergoing ET with the reduced photocatalyst (E1/2 PC/PC•– = −1.24 V vs SCE in MeCN),78 followed by PT to regenerate the ground-state 4CzIPN and TIPS-SH. The authors concede that they are unable to rule out the possibility of a chain process with termination achieved by reaction of thiyl radical with another equivalent of silane substrate, though a low quantum yield (Φ = 0.02) does not support the dominance of a chain process.
In 2018, Xie and Zhu reported a dual photoredox/thiol co-catalytic system to achieve the regioselective hydroboration of imines using N-heterocyclic carbene (NHC)-boranes (Scheme 279).723 Curran and co-workers first reported on the discovery of boryl radical generation from this class of NHC-ligated boranes in 2009.724 In contrast to polar imine hydroboration methods that preferentially form nitrogen-bound boryl intermediates en route to amines,725 this dual photocatalytic method selectively yields the inverse, carbon-bound boryl regioisomer. A key challenge in the development of this chemistry was in overcoming the uncatalyzed polar background reaction affording the undesired regioisomer. To achieve this, the thiol co-catalyst must be able to quickly abstract a H-atom from the NHC-borane and that proper polarity matching between the thiyl radical and the NHC-borane (e.g., for N,N′-dimethylimidazolium borane, B–H BDE = 74.5 kcal mol–1)723 would be crucial. Optimization of the thiol co-catalyst revealed benzyl mercaptan was most effective, able, almost exclusively, to favor the single-electron hydroboration pathway. Under the optimized reaction conditions, consisting of visible-light irradiation of Ir(ppy)2(dtbbpy)PF6([Ir-3]PF6) with thiol co-catalyst 279.A and catalytic NaH in MeCN, this method allowed for the preparation of a range of α-amino NHC borane products, including those with electron-rich (279.1–279.2), electron-neutral (279.3), and electron-deficient arenes (279.4). Different substituents on the borane-NHC (279.5) were also well tolerated. The authors demonstrated that the products of this reaction can be readily converted to N-aryl α-amino boronates through hydrolysis, highlighting the potential to access N-aryl α-AA mimics using this strategy. Many α-amino boronates have shown valuable biological activity and a few examples are even FDA-approved pharmaceuticals, such as bortezomib and varborbactam.726
Scheme 279. Joint Photoredox/Thiol-Catalyzed Inverse Hydroboration of Imines (Xie and Zhu, 2018).

Mechanistically, the authors propose that the excited state of the [Ir-3]PF6 photocatalyst (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 oxidizes the thiolate anion (e.g., for benzyl thiolate, E1/2ox = −0.32 V vs Fc+/Fc in MeCN)336—generated in situ through the reaction of NaH and the neutral thiol co-catalyst 279.A—to a thiyl radical (e.g., for benzyl mercaptan, S–H BDFE = 86.9 kcal mol–1)336 which then undergoes B–H HAT with the NHC borane to generate an NHC-boryl radical intermediate. Evidence for the intermediacy of the NHC boryl radical was provided by the observation of a TEMPO-boryl adduct in trapping experiments. Subsequent B–C formation is proposed to occur by two potential pathways. In the first (Path A) the reduced Ir(II) state of the photocatalyst (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN)68 facilitates single-electron reduction to the imine substrate (e.g., for N-benzylidene aniline, E1/2red = −1.91 V vs SCE in MeCN)21 to generate an α-amino radical anion which subsequently deprotonates the thiol co-catalyst, giving rise to a neutral α-amino radical. This intermediate undergoes radical–radical coupling with the boryl radical to generate the product. Alternatively, in Path B the boryl radical undergoes direct radical addition into the closed-shell imine. This is followed by a series of PT and ET to yield the final hydroboration product. DFT studies indicated a similar overall ΔG° for both C–B bond-forming pathways, and further studies are required to decipher which mechanistic pathway predominates.
5. C-Centered Radical Generation from C–H Bonds through Concerted PCET
Previous sections highlighted the multitude of ways that N–H, O–H, and S–H bonds can be homolyzed through the stepwise or concerted transfer of electrons and protons. However, the activation of C–H bonds via concerted proton-coupled electron-transfer mechanisms is much rarer. C–H bonds are typically poor H-bond donors, which renders them inert to homolysis under MS-PCET conditions utilizing base and oxidant.13 Instead, these bonds are cleaved through a variety of other distinct mechanisms, including HAT,9,727−734 and photochemical735−740 and electrochemical98,741−744 stepwise ET/PT. Many of these methods have been reviewed recently; therefore, for this section, we chose to focus on strategies that cleave C–H bonds in a concerted fashion only. Concerted C–H bond homolysis via PCET requires the pre-association of at least one of the three reactions components (oxidant, base, and substrate) to reduce the activation barrier of the termolecular, bond-breaking elementary step. The following examples highlight the various ways that this can be accomplished, including surface adsorption, intramolecular deprotonation, or ion-pairing. Furthermore, we include examples of C–H PCET utilizing thermal redox mediators due to the rarity of this elementary step in the literature.
5.1. Concerted PCET Involvement in Alkyl Radical Generation through Heterogeneous Photoredox Catalysis
While the use of alkylamines, alcohols, and cyclic ethers as sacrificial electron donors in zinc sulfide-based hydrogen evolution had been shown previously,745,746 Kisch and co-workers first demonstrated in 1985 that zinc sulfide NPs can be used to form C–C bonds on a preparative scale with concomitant evolution of hydrogen gas (Scheme 280).747 Upon UV irradiation of ZnS in a mixture of 2,5-dihydrofuran and water, dehydrodimerization products were formed in a 1:2:1 ratio of the 2,2′-, 2,3′-, and 3,3′-dimers (280.1). Then, in follow-up studies,748−750 these researchers extended this C–C coupling methodology across a series of cyclic ethers and alkenes, including the efficient dimerization of 4-methyl-3,6-dihydro-2H-pyran (280.2) and cyclohexene (280.3).
Scheme 280. Dehydrodimerization of Cyclic Ethers and Olefins Catalyzed by ZnS (Kisch, 1985–1999).

Initial experiments to probe the mechanism of this transformation demonstrated that water was needed to observe the C–C coupled product, and substrate was needed to observe efficient hydrogen evolution. Additionally, irradiation of ZnS and alkane in the presence of D2O resulted in the initial formation of D2 gas, with HD and H2 increasing in concentration with increasing turnovers. This suggested that while water initially serves as the source of hydrogen, as the reaction progresses, the substrate increasingly becomes the hydrogen donor through the reduction of proton to hydrogen gas. Furthermore, these observations suggested that the reduction of water to hydroxide and hydrogen gas occurs at the CB of the semiconductor, while oxidation of the substrate occurs at the VB, which is followed by deprotonation to generate the alkyl radical.
In a follow-up mechanistic study, the researchers provided evidence that oxidation/deprotonation of the alkane by the semiconductor is a concerted PCET process.750 The authors first postulated that since oxidation of the alkane by the VB is endergonic by several hundred millivolts, a stepwise oxidation–deprotonation mechanism was unlikely. Further evidence supporting this hypothesis was found through measuring the quantum yield of dimerization (Φapp) and plotting it against either the oxidation potential of the alkane (Eox) (Figure 9A) or the C–H BDFE of the alkane (Figure 9B). While there was no relationship between Eox and Φapp, the decreasing Φapp with increasing C–H BDFE strongly suggested that the alkyl radical generation occurs via concerted PCET. Taken together, these experiments allowed the authors to propose the following mechanism. UV irradiation of the ZnS NP promotes an electron (e–) into the CB, leaving an oxidizing electron hole (h+) in the VB. Water is reduced at the CB to produce hydroxide and a H-atom, while adsorbed 2,5-dihydrofuran is oxidized at the VB via concerted PCET to yield an adsorbed alkyl radical. This radical then dimerizes with a second equivalent of alkyl radical on the NP surface to produce the dehydrodimerized product.
Figure 9.

Dependence of apparent quantum yield (Φapp) on (A) substrate oxidation potential (Eox) or (B) calculated C–H bond dissociation energies implies a PCET mechanism for H-atom abstraction. Reproduced with permission from ref (750). Copyright 1999 John Wiley and Sons.
Kisch and co-workers have also demonstrated the allylation of 1,2-diazenes with enol ethers and olefins using a cadmium sulfide heterogeneous photocatalyst under UV irradiation.751,752 Here, oxidative C–H PCET and reductive N=N PCET operate in tandem to forge a new C–N bond via radical–radical coupling (for a full discussion of this process, see section 6.4.1).
Kisch and co-workers further applied heterogeneous semiconductor photocatalysis to the sulfoxidation of alkanes under visible-light irradiation (Scheme 281).753,754 The sulfoxidation of alkanes was originally demonstrated with the direct excitation of sulfur dioxide by UV light.755,756 The authors here demonstrated that this transformation was promoted by visible light with a TiO2 heterogeneous catalyst. Under these conditions, the sulfoxidation of adamantane proceeds to greater than 40% yield after 10 h of irradiation (281.1).753N-heptane and cyclohexane were also shown to be competent alkanes for this oxidation protocol.
Scheme 281. Sulfoxidation of Adamantane via a Titania–SO2 Charge-Transfer Complex (Kisch, 2008 and 2012).

In proposing a mechanism for the formation of the alkyl radical, the authors first noted that adsorption of sulfur dioxide to the semiconductor resulted in a color change, with a distinct charge-transfer feature between 400 and 420 nm appearing in the UV/vis absorption spectrum.754 Upon irradiation, CT occurs from the adsorbed sulfur dioxide to the semiconductor, generating reduced TiO2 and a highly oxidizing sulfur radical cation. This radical cation then oxidizes the alkane to an alkyl radical and a proton via concerted PCET, while the VB electron reduces oxygen to superoxide to turn over the TiO2 catalyst. The observation of only a trace amount of alcohol and ketone side products highlighted the distinct selectivity for this alkane oxidation protocol for C–S bond formation. An additional mechanism for alkyl radical formation that was not discounted involves HAT by a hydroperoxyl radical generated from superoxide. While the inclusion of HQ as a radical scavenger inhibited product formation, turning off irradiation had no effect on the production of the alkylsulfonic acid. This indicates that initial radical generation from the charge-transfer state propagates an efficient chain through either a concerted PCET or HAT pathway. A detailed and more extensive account of these works has recently been presented by the author.757
5.2. Concerted PCET Involvement in Alkyl Radical Generation through Homogeneous Ground-State Redox Catalysis
In 2017, Ooi and co-workers reported a strategy to homolytically cleave C(sp3)–H bonds through concerted PCET using a redox-active catalyst with an appended basic site (Scheme 282).758 Through the action of a zwitterionic Acr phenoxide catalyst under air, this strategy was applied in the dimerization of 3-aryl oxindoles, with 15 examples proceeding in good to excellent yields. The Acr-betaine catalyst was designed to accept both electron and proton from the oxindole substrate α-carbonyl C–H bond. Electron-rich and -poor arenes at the α-carbon were well tolerated under the reaction conditions (282.1 and 282.2), as was electron-withdrawing substitution at the 7-position of the oxindole (282.3).
Scheme 282. Dimerization of Oxindoles with an Acridinium Betaine Catalyst (Ooi, 2017).

The authors proposed a mechanism beginning with a concerted PCET step to oxidize the oxindole α-carbonyl C–H bond. The catalyst provides an effective BDFE of 65 kcal mol–1 (E1/2 Acr+/Acr• = −0.98 V vs Fc+/Fc in MeCN; phenoxide pKa = 23.7 in MeCN),758 enabling thermoneutral activation of the α-carbonyl C–H bond (BDFEcalc = 65 kcal mol–1).758 The oxidation step produces an enol radical that dimerizes to yield product, while the catalyst is turned over via stepwise ET/PT reduction of oxygen to hydrogen peroxide. The authors also considered a stepwise mechanism that entailed deprotonation of the substrate C–H bond (e.g., for N-(4-methoxyphenyl) 3-phenyloxindole, pKa = 23.5 in MeCN)758 followed by single-electron oxidation. However, enolate oxidation is endergonic with the Acr (e.g., for the enolate derived from for N-(4-methoxyphenyl)-3-phenyloxindole, Ep/2ox = +0.18 V vs Fc+/Fc in MeCN),758 and inclusion of a catalytic amount of KOt-Bu to generate enolate in situ did not improve the reaction rate. Therefore, the authors ruled out this possibility as the dominant reaction pathway. Additionally, using separated Acr redox catalyst N-Ph Mes-Acr+BF4– and base catalyst significantly diminished the reaction rate and yield, demonstrating the efficiency gained from lowering the PCET molecularity with Acr-betaine.
In 2018, Mayer and co-workers demonstrated that MS-PCET was a viable strategy to homolyze C–H bonds with the judicious placement of a base near the C(sp3)–H bond of interest (Scheme 283).37 In this system, the benzylic C–H bond of 2-(9-fluorenyl)benzoate is cleaved in a concerted fashion via PT to the neighboring benzoate base and ET to an exogenous oxidant. The authors showed that oxidants with reduction potentials spanning 800 mV of driving force were competent in promoting this reactivity, ranging from tris(4-bromophenyl)aminium radical cation (E1/2ox = +0.67 V vs Fc+/Fc in MeCN)37 to the mildly oxidizing decamethylferrocenium cation (FeCp*2+, Eox = −0.48 V vs Fc+/Fc in MeCN).37 By monitoring the disappearance of the absorbance of the colored oxidant using stopped-flow techniques, the researchers determined second-order PCET rate constants, which spanned over 4 orders of magnitude and were dependent upon the strength of the oxidant. The thermochemistry for stepwise ET/PT and PT/ET mechanisms were calculated to be unfavorable (ΔG°ET = +38 kcal mol–1, for FeCp2+, and ΔG°PT = +19 kcal mol–1). Therefore, taken together with the linear relationship between kPCET and driving force with a Brønsted α slope of 0.21 ± 0.01, the authors proposed a concerted mechanism of C–H bond homolysis.
Scheme 283. Demonstration of Benzylic C(sp3)–H Bond Homolysis via Concerted MS-PCET (Mayer, 2018).

In the same report, Mayer and co-workers showed that C–H bonds could be formed through reductive PCET in an analogous fashion to the above oxidative examples. Treating 2-(1-phenylvinyl)benzoic acid with 2 equiv of decamethylcobaltocene (CoCp*2) resulted in the formation of the hydrogenated product, 2-(1-phenylethyl)benzoic acid, in 30% yield. Thermochemical analysis revealed that rate-limiting ET to the alkene is prohibitively endergonic (for 1,1-diphenylethylene, Ered = −3.08 V vs Fc+/Fc in THF;47,759 and E1/2 Co(III)/Co(II) = −1.95 V vs Fc+/Fc in THF, – 1.91 V vs Fc+/Fc in MeCN)47,760 while the calculated effective BDFE of the combination of CoCp*2 and benzoic acid is 40 kcal mol–1, rendering the concerted pathway 6 kcal mol–1 exergonic.37 Similar results were seen in the reduction of rhodamine B with 2 equiv of CoCp*2, providing reduced leuco-rhodamine B in quantitative yield.
After the initial report of C–H bond cleavage in a fluorenyl benzoate substrate via concerted PCET, subsequent kinetic and theoretical studies of this system revealed the intricacies of this particular mode of activation. In 2018, Hammes-Schiffer and co-workers performed theoretical calculations on this PCET reaction to explain the anomalously small, experimentally measured α value of 0.2, based on vibronically nonadiabatic PCET theory.761 In these calculations, inner sphere and outer sphere reorganization allows for simultaneous tunneling of the proton and the electron, with significant contributions of excited vibronic states. By calculating the rate of PCET at different driving forces, the authors computed an α value of 0.37, larger than the experimental value (α = 0.2), but still significantly smaller than the typical value of 0.5 for HAT reactions of C–H bonds762 and concerted PCET reactions of O–H and N–H bonds.33,51,120 They then proposed that this small α value could be explained by the negative reaction free energies associated with the major pairs of vibronic states and larger overlap integrals in excited vibronic states between reactant and product.
To further explore the rate versus driving force relationship in this system, Mayer and co-workers synthesized a series of fluorenyl benzoates differing by the para-substitution of the benzoate in order to vary the pKa of the base (Scheme 284).763 By determining the second-order rate constants of C–H oxidation for each of these substrates in the presence of various aminium and metallocene oxidants, a series of rate versus driving force plots were produced. To fully analyze the results, two sets of linear free energy relationships were utilized: one to describe sensitivity to base pKa and one to describe sensitivity to reduction potential of the oxidants. For each of the substrates, while an α value of approximately 0.2 was found across the series of oxidants, in agreement with the previous disclosure from Mayer (Figure 10A),37 a much larger α value was observed across the series of base pKa’s. This shallow slope for the oxidant series was interpreted to mean C–H bond cleavage is relatively insensitive to the driving force for ET. However, when the PCET rate constants for each substrate were plotted versus driving force for a single oxidant, a much steeper slope was obtained with an α value of 0.58, implying that the C–H bond cleavage is particularly sensitive to base pKa (Figure 10B).
Scheme 284. Second-Order PCET Rate Constants for the Reaction of Fluorenyl Benzoates with Various pKa’s and Oxidants of Various Potentials (Mayer, 2019).

Figure 10.
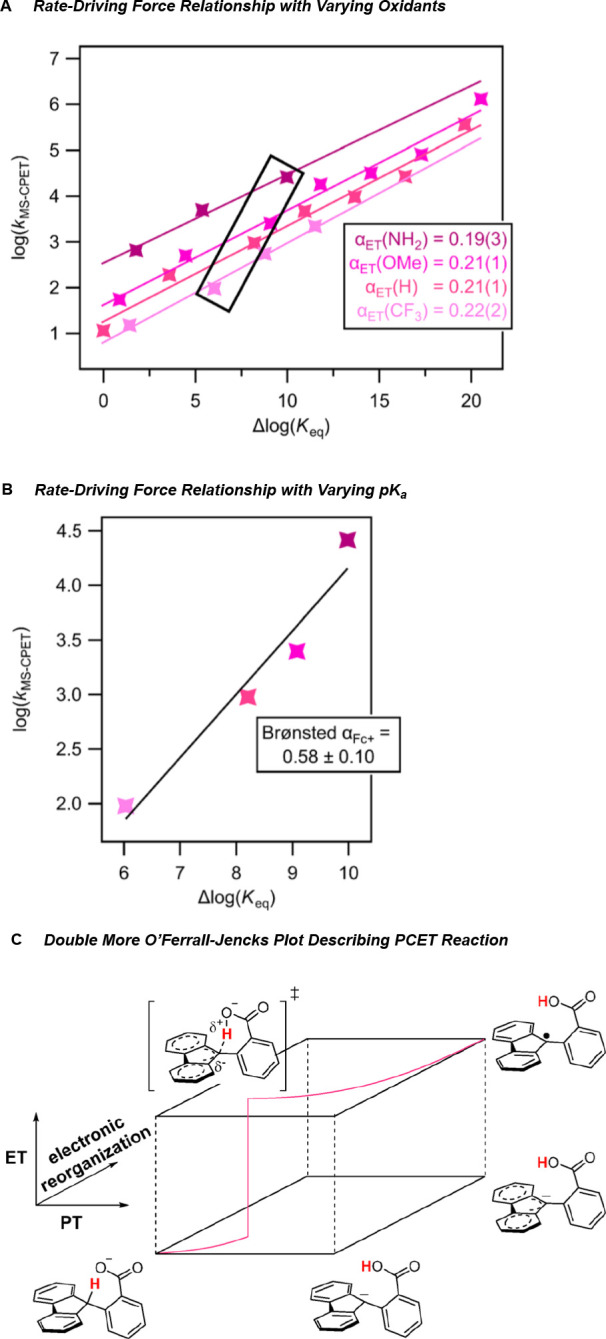
Rate–driving force relationships in the concerted PCET activation of fluorenyl benzoates. (A) Plot of PCET rate constant versus driving force for PCET across a series of fluorenyl benzoates and oxidants. From top line to bottom line: R = NH2, OMe, H, CF3. The slope of each plot is the α value for ET for that substrate. (B) Plot of PCET rate constant versus driving force for PCET across a series of fluorenyl benzoates with a single oxidant (FeCp2+). The slope of the plot is the α value for PT. (C) Double More O’Ferrall–Jencks plot for the PCET activation of a fluorenyl benzoate. The horizontal planes represent progress in the PT coordinate and electronic reorganization coordinate, while the jump between the two planes represents the instantaneous ET that occurs between the fluorenyl benzoate and the oxidant. Reproduced with permission from ref (763). Copyright 2019 ACS Publishing.
The authors explained this result by proposing that the PCET transition state is imbalanced, with PT being more advanced than ET. DFT calculated potential energy surfaces for the concerted PCET event supported this hypothesis, with the transition state found further along the PT internal reaction coordinate than ET. This means that the α value can be used as a qualitative readout on the position of the transition state in a Hammond postulate analysis, with the higher α value for PT being consistent with a later transition state. The authors finally illustrate this conclusion by generating a double More O’Ferrall–Jenks plot with two stacked planes, with the bottom plane representing the reactant internal reaction coordinate and the top plane representing the product internal reaction coordinate (Figure 10C). The two axes in the plane describe PT and electronic reorganization energy, and the vertical axis between the planes describes ET from the reactant. In line with the Franck–Condon principle, ET moves instantaneously from the reactant to the product at the transition state, represented in the plot by the vertical move from the bottom to the top plane without any change in PT or reorganization energy.
In a follow-up to Mayer and co-workers’ kinetic study on the linear free energy relationships involved in the C–H bond activation of fluorenyl benzoates via PCET, Hammes-Schiffer and co-workers in 2019 looked for a more complete description of the higher α value for PT by using the same vibrationally nonadiabatic PCET theory as in their 2018 study.764 By computing the rate of PCET for each of the substrates with a single oxidant as disclosed by Mayer,763 the authors were able to reproduce the experimental α value within error (αtheory = 0.63). Importantly, they note that the calculation of the α value is determined by two terms: one that is a function of ET and PT driving forces, and one that is a function of the energy needed to access shorter proton donor–acceptor distances, or the distance between the benzoate oxygen and the C–H bond, which affects only PT. Changing the para-substitution of the benzoate from EWGs to EDGs increases its basicity. This not only makes PT more thermodynamically favorable but also decreases the equilibrium bond distance for benzoate and the activated C–H bond, making it more energetically favorable to access the shorter proton donor–acceptor distances required for abstraction. The authors postulated that it was the sum of these effects that makes the PCET activation of fluorenyl benzoate C–H bonds more sensitive to PT than ET.
However, a 2020 report from Constentin and Savéant discussing the mechanism of activation in these fluorenyl benzoates casts doubt on the need to use an imbalanced transition state hypothesis to explain the observed low α value for the system (Figure 11).765 Instead, the authors proposed that a change in mechanism from concerted PCET to stepwise PT/ET can also explain the shallow slope in the rate-drive force relationship. Illustrating this point, the authors found that it was difficult to reasonably fit all of the experimental data presented by Mayer763 when a standard Marcus model was used to predict the rate constants of concerted PCET. However, when the rate constants of pre-equilibrium PT followed by ET were considered, it was found that this expression could fit the experimental data at low driving force well, with a change in mechanism to the concerted pathway occurring at higher driving forces. By considering different mechanistic regimes at different driving forces, the authors were then able to fit the kinetic data for each substrate in the Mayer report.763
Figure 11.
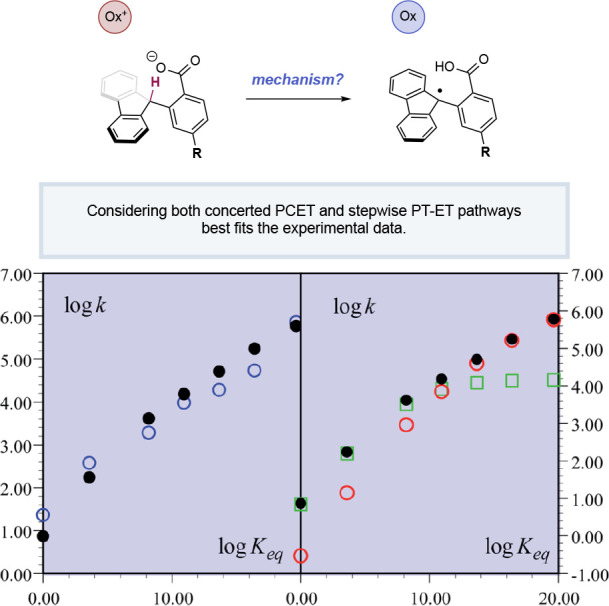
Rate constants of the reaction of the parent fluorenyl benzoate. Left: open blue circles, experimental data; black circles, predicted rate constant of the global reaction. Right: open red circles, predicted concerted PCET rate constant; open green circles, predicted stepwise PT-ET rate constant; black circles, predicted rate constant of the global reactions. Reproduced with permission from ref (765). Copyright 2020 RCS Publishing.
5.3. Concerted PCET Involvement in Alkyl Radical Generation through Homogeneous Photoredox Catalysis
Recently, Mayer and co-workers applied their 2018 disclosure of C–H PCET involving fluorenyl benzoates with ground-state redox catalysts37 to the photocatalytic deuteration and oxidative lactonization of benzylic C(sp3)–H bonds (Scheme 285).70 In this system (Scheme 285A), the joint action of Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) or [Ir(dF(CF3)ppy)2(bpy)]PF6([Ir-7]PF6) excited state and a tethered benzoate, acetate, or sulfonamide Brønsted base homolyzes the C(sp3)–H bond, resulting in the efficient deuteration of a variety of tertiary benzylic positions, including fluorenyl (285.1) and trityl C–H bonds (285.2). For these types of substrates, the inclusion of a stoichiometric amount of Co(acac)3 resulted in the formation of lactone (Scheme 285B, observed by NMR). Luminescence quenching experiments demonstrated that the Co(III) oxidant rapidly quenches the Ir(III) photoexcited state (kq ≅ 108 M–1 s–1), generating an Ir(IV) complex that is capable of oxidizing the tertiary benzylic C–H bond with concomitant deprotonation by the tethered Brønsted base. Further oxidation of the generated alkyl radical results in the lactone product.
Scheme 285. Photocatalytic Deuteration and Oxidative Lactonization of Benzylic C(sp3)–H Bonds via Concerted PCET (Mayer, 2020).

Substrates with stronger C(sp3)–H bonds were also deuterated efficiently, including the allylic position of 3-methylbutenoic acid (285.3). Primary benzylic C(sp3)–H bonds were also competent, but with deuteration proceeding to lower conversion. Interestingly, replacing the benzoic acid moiety for a primary sulfonamide also resulted in deuteration (285.4); however, in this case, a small amount of a doubly deuterated product was formed as well. The lack of deuterium incorporation in substrates such as 4-benzylbenzoic acid (285.5) implies that the positioning of the base proximal to the desired site of C–H activation is vital for reactivity. Further mechanistic studies were consistent with the authors’ previous work on this system,37,763 where two regimes of bond homolysis are plausible depending on the strength of the activated C–H bond. For weaker C–H bonds, a concerted MS-PCET was proposed, while for stronger C–H bonds, a stepwise ET/HAT mechanism is likely based on luminescence quenching experiments demonstrating that unsubstituted benzoate base quenches photoexcited Ir(III) (kq = 4 × 105 M–1 s–1).70,574 Therefore, at slower rates of MS-PCET, this stepwise pathway becomes competitive.
Building on the initial disclosures of C–H PCET reactivity from Mayer37 and Ooi,758 Alexanian, Knowles, and co-workers provided evidence of concerted PCET of a C–H bond in a photocatalytic context in 2019 through the development of an intermolecular C(sp3)–H alkylation (Scheme 286).766 This transformation was jointly catalyzed by Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 ([Ir-9]PF6) and n-Bu4N+(n-BuO)2P(O)O– Brønsted base co-catalyst, where the cooperative action of the photocatalyst excited-state and base affects the homolysis of the C–H bond. Twenty-four examples of this transformation, with yields ranging from 40% to 92%, were reported. The conditions were amenable to efficient functionalization of a broad scope of aliphatic substrates, including α-heteroatom (286.1), allylic (286.2), benzylic (286.3), and unactivated (286.4) C(sp3)–H bonds. The reaction site-selectivity was illustrated in the alkylation of 1-methyl-4-isopropylbenzene (286.3) and 2,3-dihydrobenzofuran (286.5), where radical generation favored the weakest C–H bond. Subjecting ibuprofen methyl ester (286.6) to the reaction conditions further demonstrated the high regioselectivity of the transformation, forming a single regioisomer resulting from alkylation at the secondary benzylic position (benzylic C–H bond BDFE ≈ 85 kcal mol–1)717 over the adjacent tertiary position (tertiary C–H bond BDFE ≈ 93 kcal mol–1). In the same substrate, abstraction from this secondary benzylic position is likely favored over the α-carbonyl, tertiary benzylic position due to a high barrier for abstraction resulting from polarity mismatching in the transition state.
Scheme 286. PCET Enables Catalytic C(sp3)–H Alkylation (Alexanian and Knowles, 2019).

Mechanistic studies of the reaction highlighted the unique nature of this MS-PCET process, where molecularity is lowered via ion-pairing of the photocatalyst and base, rather than through hydrogen-bonding between substrate and base, as is typically the case with polar N–H, O–H, and S–H bonds. 1H NMR spectroscopy studies and SV quenching experiments demonstrated the strong association of photocatalyst and base in solution (KA ≈ 103 – 104 M–1 s–1). Titration of n-Bu4N+(n-BuO)2P(O)O– into solutions of [Ir-9]PF6 showed a pronounced downfield shift of the 3,3′-bipyridyl proton resonances in the 1H NMR spectrum, demonstrating that the base was associating to these photocatalyst ligand protons. This affinity was confirmed in the solid state, where the single-crystal X-ray structure of the photocatalyst–phosphate complex featured close association of the phosphate oxygen atom to the 3,3′-bipyridyl protons. Kinetic analysis of the reaction demonstrated that fluoride anion had an inhibitory effect on the reaction rate by acting as a competitive binder for the 3,3′-bipyridyl position of the photocatalyst, highlighting the requirement of photocatalyst–phosphate association for the reaction to proceed.
Further time-resolved spectroscopic experiments showed that not only does alkane quench the photocatalyst excited state in the presence of base (for ethylbenzene, kq = 2.3 × 106 M–1 s–1), but that this quenching has an isotopic dependency (kH/kD = 2.0 ± 0.2), providing strong evidence that C–H bond cleavage is involved in excited-state quenching. Taking into consideration these mechanistic experiments, the proposed catalytic cycle for this transformation begins with photoexcitation of the photocatalyst-base complex, which then engages substrate in a concerted PCET abstraction of the C–H bond, with the electron reducing the photocatalyst and the proton transferring to the phosphate. The alkyl radical generated in this concerted PCET event then reacts with an electron-deficient olefin in solution for C–C bond formation. Product is formed via reduction of the stabilized α-acyl radical to the corresponding anion followed by protonation, regenerating both oxidant and base catalysts in the process. Contemporaneous reports of photocatalytic C(sp3)–H activation for substrate azidation and cyanation using a similar combination of either Acr or Ir(III) photocatalyst and phosphate Brønsted base catalyst were disclosed by Alexanian and Nicewicz,579 as well as Kanai.580 The proposed mechanisms in these transformations involve a stepwise ET/HAT mechanism, where the phosphate base is first oxidized to an O-centered phosphoryl radical, before HAT with substrate to generate an alkyl radical (for an expanded discussion, see section 3.5).
In 2019, Glorius and co-workers reported a photochemical alkylation of indoles and other electron-rich heterocycles with Katritzky pyridinium salts767−769 (Scheme 287).770 The reaction proceeded through visible-light excitation of an electron donor–acceptor (EDA) complex that is formed between the heteroarene donor and the pyridinium salt acceptor. Various AA derivatives served as competent electron acceptors in this chemistry, including the pyridinium salt of N-Cbz-protected lysine, affording the alkylated-indole product in good yield (287.1). Besides indoles, other electron-rich arenes, including 2-phenylbenzofuran (287.2) and benzothiophene-3-boronic acid pinacol ester (287.3), were found to be efficient electron donors in this reaction. Furthermore, the authors reported that cyclohexanone could be alkylated through the in situ formation of an enamine with an additional equivalent of morpholine (287.4). Similarly, aldehydes also served as acceptable donors with the inclusion of a catalytic amount of pyrrolidine and triethylamine as enamine precursor and base, respectively (287.5).
Scheme 287. Alkylation of Indoles, Benzofurans, Benzothiophenes, and Enamines Enabled by Excitation of an EDA Complex (Glorius, 2019).

The authors proposed that this reaction proceeds through a radical chain mechanism that is initiated by the formation of an EDA complex between the electron-rich arene and the pyridinium salt, evidenced by the brightly colored solutions of the arene and pyridinium salt, as well as a large red-shift in the absorption spectrum of the reaction mixture. Visible-light excitation of this EDA complex results in ET from the arene to the pyridinium, which rapidly fragments to release pyridine and an α-carbonyl radical. This radical intermediate reversibly adds into the arene, with ET from the benzylic radical to an additional equivalent of the pyridinium salt, releasing product from the catalytic cycle and propagating the chain. The authors postulated that the unfavorable ET thermodynamics (ΔEET = 0.50 V) could be modulated by simultaneous PT. This PCET-based propagation step could be further assisted by pre-coordination of the morpholine base to the pyridinium acceptor, which would decrease the molecularity of the collision. Clayden and co-workers reported a related proposal of chain-propagation via concerted PCET in the homolysis of an amide N–H bond for olefin azocycloamidation.138
6. Reductive Transformations of Carbonyls, Imines, and Other X=Y Functional Groups through Photochemical and Electrochemical PCET Processes
6.1. Reductive Transformations of Carbonyls and Imines (C=O and C=N)
In seminal work, several examples of the reductive activation of carbonyl compounds under photochemical conditions were detailed by Pac and co-workers. In a 1983 study, Pac and co-workers described a system utilizing the NADH model compound 1-benzyl-1,4-dihydronicotinamide (BNAH), in combination with [Ru(bpy)3]Cl2 ([Ru-1]Cl2) as a photocatalyst to generate a number of adducts resulting from the single-electron reduction of this functional group—including alcohols via further reduction, 1,2-diols via pinacol coupling, and C–C coupling between the carbonyl substrate and the NADH derivative (Scheme 288).771 Pac and co-workers note that the primary products observed with a range of ester, ketone, and aldehyde substrates were adducts of BNAH and the carbonyl compound, identified as the C–C coupling product 288.1. It was hypothesized that this adduct arose from radical–radical coupling. Initially, reductive quenching via ET from BNAH to the photoexcited-state Ru(II) catalyst formed BNAH•+ and Ru(I), which forms BNA• after proton loss. ET from Ru(I) to the carbonyl substrate would then afford ketyl radical anion, which yielded neutral ketyl radical after PT. Radical–radical coupling between the neutral ketyl radical and BNA• forms the C–C coupled product (288.1). With select ketone substrates bearing electron-withdrawing substituents, such as 2-pyridyl ketone, reduction to the alcohol instead proved dominant, proceeding through further ET between BNA• and the neutral ketyl, coupled with subsequent PT.
Scheme 288. Photocatalytic Carbonyl Reduction with an NADH Model (Pac, 1983 and 1987).

In subsequent work, Pac and co-workers demonstrated that for aldehyde substrates bearing EWGs, the observed C–C coupled products undergo bond cleavage to afford overall carbonyl reduction products when irradiated in the presence of [Ru(bpy)3]Cl2 ([Ru-1]Cl2) and BNAH.772 Comparison of carbonyl substrate polarographic half-wave potentials in MeOH and MeCN solutions revealed a substantial positive shift of reduction potential, along with the presence of a second reduction feature ascribed to speciation of the ketyl radical anion, on moving from the aprotic to the protic solvent (e.g., for di(2-pyridyl)ketone, E1/2red,1 = −1.8 V vs Ag+/Ag in MeCN, E1/2red,2 = −1.9 V vs Ag+/Ag in MeCN; E1/2red = −1.46 V vs Ag+/Ag in MeOH).772 In MeCN solution, two reduction events with similar diffusion current constants are observed for carbonyl substrates, indicative of two sequential, reversible electron transfers. By contrast, in MeOH solution, these reduction events are proposed to be coupled with PT, in the parlance of contemporary work referred to as ECE (electron transfer–chemical reaction–electron transfer),773,774 and as such show a positive shift in polarographic potential and/or a coalescence of the two reduction event peaks.
In a 1987 kinetic study from Fukuzuki, these researchers showed that the ET rate of reaction between both ground-state Co(III) and excited-state Ru(II) single-electron reductants, with quinone and ketone substrate was strongly correlated with the solution pH. This too is evidence of protonation or H-bonding between the acid additive and the carbonyl substrate facilitating ET.775
In later work, Pac and co-workers described another system capable of affording carbonyl reduction using alkylamines as sacrificial reductants (Scheme 289).776 Using poly(p-phenylene) (PPP) particles as a photocatalyst in MeOH solution, with 1.0 M Et3N and 1 mM of carbonyl substrate, irradiation with >400 nm light afforded carbonyl reduction for a range of substrates including aryl aldehydes, aryl α-ketoesters, and diaryl ketones (e.g., 289.1, 289.2). However, more difficult-to-reduce alkyl aryl ketones showed limited efficacy. This system makes use of a catalytic scheme present in many more recent reductive proton-coupled ET systems insofar as a potent photoreductant is generated through use of a sacrificial stoichiometric reductant. In this system, photoexcitation generates a hole in the VB of PPP and promotes an electron to the CB. ET from the tertiary amine reductant to this hole regenerates the filled VB state in addition to the aminium radical cation. ET from the CB to the carbonyl substrate affords the ketyl radical anion and then neutral ketyl radical following PT. Subsequent ET from the VB and CB respectively, paired with PT, affords the alcohol product and iminium, which hydrolyzes to acetaldehyde and diethylamine in situ. Electron-poor olefins could be reduced by the system as well. Interestingly, although pinacol coupling product was observed for some carbonyl substrates, this could be largely suppressed by impregnation of the PPP with Ru.
Scheme 289. Photoreduction of Carbonyl Compounds Using a Semiconducting Organic Polymer Photocatalyst (Pac, 1990).

These early reports laid the foundations for much of the later photocatalytic reductive PCET methods, which are the subject of this section. We begin here with a discussion of those protocol involving addition of reductively generated neutral ketyl radicals, and α-amino radicals across olefins, before moving to (aza)pinacol homo-coupling, and radical–radical cross-coupling methods.
6.1.1. Intra- and Intermolecular C–C Bond Formation through Reactions with Alkenes and Alkynes
An early example of the photocatalytic reductive activation of an aryl ketone came from Ollivier and Fensterbank, who in 2011 developed a method for the reduction and reductive allylation of the epoxide or aziridine moiety of α-epoxy- and α-aziridino-ketones, respectively (Scheme 290).777 The ability to functionalize these two substrate classes is notable, as existing Ti-mediated reductive ring opening methods had not previously been possible with aziridines.778 The direct single-electron reduction of these functional groups is challenging (e.g., for stilbene oxide, E1/2red = −2.35 V vs Ag+/Ag in DMF),779 leading the authors to instead study this substrate class where single-electron reduction can be focused on the ketone functionality, likely facilitated by PCET, to enable epoxide ring opening after a 1,2-SCS process. This follows notable earlier reports of α-epoxy-ketone fragmentation upon UV irradiation in the presence of tertiary amine donors.780−782
Scheme 290. Epoxide and Aziridine Reduction and Reductive Allylation through Aryl Ketone PCET Activation (Ollivier and Fensterbank, 2011).

Optimized reaction conditions for reductive ring-opening consisted of visible-light irradiation of DMSO solutions of the epoxy- or aziridino-chalcone substrates in the presence of [Ru(bpy)3]Cl2 ([Ru-1]Cl2) as a photocatalyst and Hantzsch ester (HE) as a stoichiometric terminal reductant. Under these conditions, 14 examples of β-hydroxy and β-amino ketone synthesis were reported in yields of 42–94% (e.g., 290.1–290.4). Exclusive reduction at the α-position was observed in all cases. In the case of α-substituted α-epoxy-ketones, the more-reducing photocatalyst [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) was required and reduction products were obtained with poor diastereoselectivity. In addition, reductive allylation was achieved through inclusion of an electron-deficient allylsulfone reagent (290.A). In this transformation, use of 4-Me-HE was important to achieving selectivity for reductive allylation over direct reduction of the radical intermediate. With these conditions, eight examples of the reductive allylation of these substrates were demonstrated in yields of 43–73% (e.g., 290.5–290.8). In all cases, a very high syn-diastereoselectivity was observed, which is consistent with Guindon’s model for radical allylation of β-alkoxy esters operating under non-chelate control.783,784
The authors present a mechanism whereby the photoexcited Ru(II) state catalyst is reductively quenched by HE, forming corresponding HE•+ and Ru(I). The reducing Ru(I) complex then engages with the ketone in an ET step, forming a ketone radical anion intermediate. However, we note that since this early report on photocatalytic reductive transformations of ketones in the presence of HE, PCET has been invoked as an enabling tool to facilitate an otherwise significantly endergonic ET step, and thus we reason that it may be operative here also; the direct reduction of an acetophenone (e.g., for acetophenone E1/2red = −2.11 V vs SCE in MeCN)21 is ca. 780 mV more demanding than this Ru(I) complex is capable of delivering (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN).64 This process is made significantly more favorable through PCET activation via hydrogen bonding between the ketone functional group and acidic additives, such as the intermediate HE radical cation or the pyridinium byproduct of its oxidation, as proposed by others.785−788 In this work, the authors note that reaction efficiency is reduced in the presence of basic additives. Following reductive radical generation, a SCS reaction occurs with opening of the epoxide/aziridine ring, yielding an α-acyl radical. In the reduction reaction manifold, HAT with the HE radical cation yields the closed-shell product and Hantzsch pyridine as a byproduct. Alternatively, the radical intermediate arising from SCS reacts in an addition–elimination reaction with the allylsulfone reagent, eliminating an arylsulfonyl radical which can be reduced to the corresponding sulfinate through an ET step mediated by Ir(II). The Seeberger group later translated this method to a continuous flow reactor.789 In this work, a combination of DIPEA as a terminal reductant and HCO2H as a Brønsted acid additive was employed, akin to work from Yoon promoting reductive cyclization reactions of bis-enones.790 Under these conditions, β-hydroxy ketone 290.1 was prepared in 84% yield in a residence time of 10 min and with a reduced loading of [Ru(bpy)3]Cl2 ([Ru-1]Cl2) photocatalyst to 0.5 mol%.
In 2011, Yoon and co-workers reported the Brønsted acid-mediated reductive cyclization of enones to form cyclic diketones (Scheme 291).790 In contrast to the group’s previous work on Lewis-acid-catalyzed enone cyclization to form [2+2] cycloadducts,791,792 the use of a Brønsted acid for substrate activation in this work led to selective 5-exo-trig cyclization. This divergent reactivity was attributed to the formation of distinct intermediates under the two modes of substrate activation, with enone radical anions forming under LA catalysis and neutral ketyl radicals forming under Brønsted acid catalysis. In the case of aryl enone substrates, the optimal conditions involved the visible-light irradiation of the enone substrate in the presence of [Ru(bpy)3]Cl2 photocatalyst ([Ru-1]Cl2), formic acid Brønsted acid additive, and DIPEA as a terminal reductant in MeCN solution. With aliphatic enone substrates, which generally exhibit more negative reduction potentials, the more reducing [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) proved to be a more effective photocatalyst.
Scheme 291. Dual Photoredox/Brønsted Acid-Catalyzed Reductive Cyclization of Enones (Yoon, 2011).

The authors reported 24 examples of enone cyclization in yields of 60–95%. Symmetric aryl bis(enone)s with electron-rich (291.1) and electron-poor (291.2) arene moieties underwent efficient cyclization with high trans-diastereoselectivity. α-Substitution is tolerated, albeit without control over product diastereoselectivity. A variety of radical acceptors in addition to enones participated in this transformation, including styrenes (291.3), alkynoates (291.4), and vinyl nitriles (291.5). This scope is expanded compared to the group’s Lewis-acid-catalyzed work, showcasing the greatly expanded scope achieved under the Brønsted-acid-catalyzed conditions.
The mechanism of this enone cyclization was interrogated through a set of control experiments. To exclude the formation of a [2+2] cycloadduct as an intermediate en route to the observed product, the authors subjected authentic [2+2] product to the reaction conditions and observed a 32% yield of the cis diastereomer of the 5-exo-trig product, which is inconsistent with the high trans diastereoselectivity observed starting from the bis(enone) substrate. Given the reductive nature of the transformation, the identity of the terminal reductant was also investigated. Using formic acid deuterated at carbon, the authors noticed no deuterium incorporation in the final product, suggesting that the DIPEA additive instead serves as the terminal reductant while formic acid functions solely as a Brønsted acid. Based on these observations, the authors posit that the excited-state photocatalyst (for [Ru-1]Cl2, E1/2 *Ru(II)/Ru(I) = +0.77 V vs SCE in MeCN; for [Ir-3]PF6, E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)64,68 undergoes reductive quenching with DIPEA to afford the reduced ground-state photocatalyst (for [Ru-1]Cl2, E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN; for [Ir-3]PF6, E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN) and DIPEA•+. Substrate protonation then facilitates single-electron reduced by Ru(I) or Ir(II) to form a vinylogous ketyl radical intermediate that engages with the olefin in a 5-exo-trig cyclization. The resulting C-centered radical finally abstracts a H-atom from the DIPEA•+ to afford the closed-shell product.
Shortly after the publication of this enone cyclization method from Yoon, Xia, and co-workers reported an intermolecular chalcone coupling to form similar five-membered-ring products through the dimerization of the chalcone radical anions generated via photoreduction by [Ru-1](PF6)2 in the presence of Sm(OTf)3 LA additive.793 An intramolecular aldol addition then afforded the final product. Zeitler and co-workers later demonstrated that hydrogen-bonding thioureas could be used to activate the enone substrate toward reduction in place of the Brønsted acid employed by Yoon.794 Here, the authors employed EY as a photocatalyst and HE as a terminal reductant.
In 2013, Knowles and co-workers published a report describing the reductive cyclization reaction between ketones and olefins (Scheme 292).16 In this method, the ketone was activated as a neutral ketyl radical through reductive PCET wherein the excited state of [Ru(bpy)3](BArF)2 ([Ru-1](BArF)2), and diphenyl phosphoric acid donate an electron and a proton, respectively. Though the individual ET and PT steps are both considerably endergonic, the concerted nature of MS-PCET provides a lower energy pathway for the formation of ketyl radical due to the avoidance of high-energy intermediates. In effect, the excited-state photoreductant and Brønsted acid function jointly as a bimolecular H-atom donor capable of donating a H-atom to the ketone starting material. Because both the photocatalyst excited-state oxidation potential and the pKa of the Brønsted acid can be independently varied, a broad range of thermodynamic driving forces are accessible through light-driven PCET. This report by Knowles and co-workers demonstrated one of the first applications of concerted PCET-based substrate activation in a synthetic context.
Scheme 292. Photocatalytic Intramolecular Cyclization of Ketones with Olefins Enabled by Reductive PCET (Knowles, 2013).

Optimized conditions involved irradiation of a THF solution containing ketone substrate in the presence of [Ru-1](BArF)2 as photocatalyst, diphenylphosphoric acid, and 2-phenyldihydrobenzothiazoline (BT) as the stoichiometric reductant. The authors reported 10 examples of ketyl-olefin cyclizations in yields of 64–96%. Heteroatoms were tolerated as substituents on the α-position of the ketone, providing access to substituted THF (292.1) and pyrrolidine (292.2) products. Branching at the α-position was similarly well tolerated (292.3). Benzylic ketones containing more electron-rich arenes, which make the ketone moiety more challenging to reduce, underwent efficient cyclization in high diastereoselectivity (292.4), demonstrating the thermodynamic advantages of concerted PCET over stepwise alternatives. In addition to acrylate acceptors, styrenyl acceptors proved to be viable reaction partners, affording dehydrated product (292.5) in good yield.
The mechanism of this transformation was investigated via SV fluorescence quenching experiments using Ir(ppy)3 (Ir-1) as a substitute for Ru(II) due to its larger excited-state oxidation potential (E1/2 Ir(IV)/*Ir(III)= −1.73 V vs SCE in MeCN, compared to E1/2 Ru(III)/*Ru(II) = −0.81 V SCE in MeCN).64,66 These experiments revealed that the photocatalyst excited state is not quenched by acetophenone (E1/2red = −2.11 V vs SCE in MeCN)21 or the phosphoric acid alone, though admixtures of these two compounds resulted in quenching. A first-order kinetic dependence on each component was observed for this quenching. Together with a small isotope effect (1.22 ± 0.02) in the presence of deuterated phosphoric acid, these results excluded the possibility of direct electron transfer to substrate. Direct PT from the Brønsted acid to the substrate was also ruled out on the basis of the large pKa difference between the acetophenone (pKaH = −0.1 in MeCN)795 and the phosphoric acid (pKa ≈ 13 in MeCN).796 Therefore, the authors proposed that the ketone is activated through concerted, reductive PCET to form a ketyl radical intermediate. The electron donor in this PCET step was proposed to be the reduced ground-state Ru(I) complex (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN)64 generated via reductive quenching of the Ru(II) photoexcited state by BT. The phosphoric acid was posited to be the proton donor. The nucleophilic ketyl radical undergoes 5-exo-trig cyclization with the electrophilic acceptor. The resulting α-carbonyl radical then abstracts a H-atom from the BT additive, generating the closed-shell product. SET from the BT radical to the Ru(II) photocatalyst followed by PT from the 2-phenylbenzothiazolium turns over all the catalysts.
This reductive ketyl-olefin cyclization reaction has also been investigated using photocatalysts based on metals more earth-abundant than Ru. The design and application of photocatalysts prepared from earth-abundant metals, such as Cu,797,798 Zr,799 Cr,800 and Fe801 is an emerging area of research.802 Collins and co-workers in 2018 reported a combinatorial approach to the preparation of a library of 50 cationic heteroleptic Cu(I)(diamine)(bisphosphine) complexes and evaluated their performance in three mechanistically distinct classes of photoredox reactions: reductive SET, ET, and reductive PCET.803 Such heteroleptic Cu(I) dyes have only recently been reported and remain underexplored in synthetic photoredox reactions compared to homoleptic Cu-polypyridyl complexes and other dyes.
These authors studied the application of this library in catalyzing the intramolecular reductive-coupling reaction between an aryl ketone and olefin, a transformation first reported by Knowles and co-workers in 2013 (Scheme 293).16 In the initial report from Knowles, [Ru(bpy)3](BArF)2 ([Ru-1](BArF)2) photocatalyst was used, and under optimal conditions gave a 78% yield of cyclization product 293.1. Through screening of this library of heteroleptic Cu(I) dyes, these researchers identified two Cu(I) photocatalysts with comparable efficiency to the Ru(II) dye: [Cu(dq)(BINAP)]BF4, yielding 71% of the product cyclopentane, and [Cu(quintri)(Xantphos)]BF4, proceeding in 79% yield. The facile synthesis of these cationic Cu(I) complexes—simply involving consecutive treatment of THF solutions of [Cu(MeCN)4]BF4 with bisphosphine, then diamine ligands—and comparable reaction efficiency make them attractive alternatives to Ru and Ir photocatalysts for promoting such PCET transformations.
Scheme 293. Photocatalytic Intramolecular Ketyl-Olefin Cyclization, Catalyzed by Cationic Heteroleptic Cu(I)(diamine)(bisphosphine) Complexes (Collins, 2018).

In a 2016 report, Rueping and co-workers disclosed a photocatalytic method for the synthesis of chromanols via ketyl-olefin cyclization of O-cinnamyl salicylaldehydes (Scheme 294).804 This is notable, in that cyclization proceeds in a 6-exo-trig fashion, distinct from the 5-exo-trig mode reported in 2013 by the Knowles group in other ketyl-olefin cyclization reactions.16 After optimization, [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) was identified to be the most effective photocatalyst for this cyclization reaction along with DIPEA as a reductant in MeCN solution under blue-light irradiation. In this work, 16 examples of chromanol synthesis were provided in yields of 43–92%. The reaction could tolerate electron-rich and electron-poor aryl aldehyde substrates (294.1 and 294.2, respectively) as well as sterically encumbered arenes (294.3). Alkynes also served as efficient radical acceptors through 6-exo-dig cyclization, providing benzylidene-substituted chromanols in good yields (294.4).
Scheme 294. Intramolecular ketyl-olefin coupling for the synthesis of chromanols via reductive PCET (Rueping, 2016).

In line with a previous report from this group on the reductive activation of benzaldehyde substrates for pinacol coupling (see section 6.1.2),805 the authors propose reductive quenching of the photoexcited-state Ir(III) catalyst (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 by DIPEA (E1/2 = +0.31 V vs Fc+/Fc in MeCN)806 to generate a reducing Ir(II) complex and DIPEA•+. This aminium radical cation, or closed-shell DIPEA-H+ generated through bimolecular PT between DIPEA and DIPEA•+, serves the role of Brønsted acid to facilitate an otherwise endergonic substrate activation through PCET, affording a neutral ketyl radical. Olefin cyclization ensues, generating a benzylic C-centered radical, which yields the closed-shell chromanol product through HAT.
The ketyl and α-amino radicals resulting from reductive PCET activation of carbonyls and imines, respectively, exhibit nucleophilic polarity due to resonance donation from the heteroatom lone pairs adjacent to the site of spin density. As a result, reductive PCET can be used as a method to achieve umpolung reactivity of typically electrophilic ketones, aldehydes, and imines. This polarity reversal was demonstrated by Chen and co-workers in 2016 in the context of a carbonyl and imine allylation reaction wherein an allyl sulfone reagent serves as the electrophile (Scheme 295).788 [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) was found to be the optimal photocatalyst along with HE and DIPEA as reductants under blue-light irradiation in CH2Cl2. The efficiency of the reaction was found to drop dramatically when only DIPEA was used as a reductant. Notably, the reaction was also highly tolerant of air and water, occurring with only a minor decrease in product yield relative to anhydrous conditions under inert atmosphere.
Scheme 295. Umpolung Allylation of Carbonyls and Imines via Reductive PCET (Chen, 2016).

Across a range of aldehyde, ketone, and imine substrates, 28 examples of polarity-reversed allylation were reported in yields of 44–95%. Aryl and heteroaryl aldehydes efficiently underwent this transformation with an allyl sulfone, including those bearing potentially sensitive functionality such as halogens (295.1). A thiophene substituent was also well tolerated (295.2). Ketones containing esters and amide functional groups exclusively underwent allylation at the ketone moiety (e.g., 295.3), with no competitive conversion of the more difficult-to-reduce carbonyl. An aryl imine bearing a propargyl ether substituent was able to participate in this reaction without interference from the alkyne (295.4). Products derived from less-stable alkyl imines could also be prepared through a one-pot, three-component coupling variation whereby the imine intermediate is formed in situ from the corresponding aldehyde and amine precursors. In this protocol, an imine derived from cyclopropanecarboxaldehyde was tolerated and afforded the desired allylation product (295.5) in 83% yield without evidence of ring opening.
SV quenching experiments revealed that both DIPEA and the HE quench the luminescence of the photocatalyst, while neither the substrate nor the olefin acceptor are effective quenchers. Given the demanding reduction potentials of carbonyl and imine substrates (e.g., for p-tolualdehyde, E1/2red = −2.28 V vs SCE in MeCN;807 for the imine derived from isobutyraldehyde and aniline, Epred = −2.90 V vs Ag+/Ag in MeCN),808 concerted PCET was invoked as the mode of reduction for these substrates with potentially the HE radical cation or the corresponding pyridinium cation acting as acid activators during this process, though the authors note that there remains some ambiguity about which species is involved in substrate activation and how. The addition of a Brønsted base (Na2CO3) resulted in a decrease in the overall rate of reaction, while the addition of AcOH served to increase the reaction rate. With these observations in mind, the authors propose that photoexcited Ir(III) catalyst (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 is reduced by the HE (Ep/2ox = +0.51 V vs Fc+/Fc in MeCN)374 to form the Ir(II) state of the photocatalyst, which can mediate reductive PCET of the substrate activated as previously described. The resulting neutral ketyl or α-amino radical intermediate then adds into the olefin acceptor through a radical addition–elimination mechanism to form the desired product.
The Dixon group has developed several synthetic methods for α-amine functionalization initiating via the reductive PCET generation of α-amino radicals from in situ formed imines.809 Whereas α-amino radical generation via amine single-electron oxidation and subsequent α-deprotonation is widely established, these methods can be prone to overoxidation to the iminium ion.810 Generating this same intermediate via reductive initiation overcomes this challenge. This mode of activation allows for either net-reductive reactions of imines through inclusion of a stoichiometric reducing agent, or redox-neutral reactions when coupled to an oxidative process later in the reaction mechanism. This group has explored both of these types of transformation.
In a 2016 publication, Dixon and co-workers demonstrated the photocatalytic reductive three-component coupling of aryl aldehydes, anilines, and Nozaki allyl sulfone reagents for the synthesis of homoallylic N-arylamines (Scheme 296).785 Optimized conditions for this transformation consisted of green light irradiation of DMF solutions of aldehyde and aniline substrates with allylsulfone reagents in the presence of EY photocatalyst and HE terminal reductant. A scope of 27 examples of reductive coupling were reported in yields of 47–99%. Electron-rich aromatic aldehydes were optimal substrates in this work (296.2), whereas electron-deficient examples required prolonged reaction times and proceeded with more limited yields (296.3). Heteroaromatic aldehydes were well explored (296.4). The reaction was not affected by steric bulk in the ortho-position of either the aldehyde or the aniline component. Similarly, electron-rich anilines were the most competent coupling partners. In addition to ester-substituted allyl sulfone reagents, those with nitrile and phosphonate groups were also effective (296.6 and 296.7, respectively). In some cases, when desired reductive coupling was slow, competing reductive amination (without allylation) was observed as a byproduct.
Scheme 296. Three-Component Coupling of Aryl Aldehydes, Anilines, and Allyl Sulfones for the Synthesis of Homoallylic N-Arylamines (Dixon, 2016).

A mechanistic proposal was made involving reductive quenching of the excited-state photocatalyst (E1/2 *EY/EY•– = +0.83 V vs SCE in MeCN)71 with HE (Ep/2ox = +0.51 V vs Fc+/Fc in MeCN),374 to generate EY•– and HE radical cation. Reductive quenching in this manner was supported by SV experiments. HE was proposed to fulfill two roles in this reaction, acting as both a stoichiometric reductant in this quenching process and as an activator of the imine toward PCET through hydrogen bonding by the resultant acidic aminium radical cation. Thus, coordination of the radical cation to the in situ generated imine (through condensation of aldehyde and aniline) leads to thermodynamically favorable ET mediated by EY•–. The resulting α-amino radical engages with the allylsulfone reagent in a radical addition–elimination reaction to yield the product and phenylsulfonyl radical. This radical can be further reduced to the corresponding sulfinate by HE or EY•–. Another potential mechanism was considered on the basis of an observed reductive amination side product. This alternative involves imine formation and reductive amination mediated by HE811 before subsequent oxidative generation of the α-amino radical via ET/PT or HAT steps. However, subjecting this independently prepared possible intermediate to optimized reaction conditions with EY and allylsulfone reagent yielded no allylated product, thus favoring the reductive PCET mechanism.
Soon after, the Dixon group published an extension to this methodology in the preparation of α,γ-diamino acid esters, in a three-component reductive coupling of (hetero)aryl and alkyl aldehydes, (hetero)arylamines, and dehydroalanine electrophiles (Scheme 297).786 Optimal conditions for this work involved blue-light irradiation of mixed MeOH/DMSO (3:2) solutions of aldehyde and aniline substrates, bis-N-Boc-dehydroalanine methyl ester, and 4-Ph-HE reducing agent in the presence of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) photocatalyst at room temperature. In all cases, the anti-1,3-diamine resulted as the major isomer in low to moderate levels of d.r. The mixed solvent system and use of the substituted HE proved important in improving the diastereoselectivity of the reductive coupling reaction. A total of 35 examples of 1,3-diamine synthesis in this fashion were recorded, in yields of 42–99%. With respect to the aldehyde component, broad variation was tolerated, allowing electron-rich, -neutral (297.5), and -poor (297.1, 297.4, 297.6) aryl groups to participate as well as many heterocycles including pyridine, furan, thiophene, pyrrole, and imidazole (297.2). Alkyl aldehydes were competent substrates, whether α-primary, -secondary or -tertiary (e.g., 297.3). With respect to the aniline component, variation of electronic properties and position of substituents, or the inclusion of heteroaromatic amines (297.4) did not significantly impact the efficiency of the reaction. When the aniline component was excluded from the reaction under the same conditions, an anti-1,3-amino alcohol (297.6) was obtained in 85% yield and a d.r. of 1.5:1 via proton-coupled single-electron reduction of the aldehyde. Finally, a two-step synthesis of the core γ-lactam structure of a CB-1 receptor inverse agonist was demonstrated through this reductive coupling, yielding a diamine product prior to refluxing in a methanolic HCl solution to simultaneously remove the Boc protecting groups and cyclize to form the corresponding amide.
Scheme 297. Three-Component Coupling of Aldehydes, Anilines, and a Dehydroalanine Derivative for the Synthesis of anti-1,3-Diamines (Dixon, 2018).

The authors’ mechanistic proposal aligns with their previous investigations,785 involving reductive quenching of the Ir(III) photoexcited state with HE prior to a PCET reduction of the imine mediated by the acidic HE radical cation and reduced-state Ir(II) complex. Radical Giese-type addition to the dehydroalanine, followed by HAT with HE radical yields the closed-shell product.
In 2018, the Dixon group further extended the utility of α-amino radicals generated through this reductive PCET reaction manifold to enable the synthesis of 2-aryl-4-sulfonyltetrahydroquinolines (e.g., 298.1) through a type of reverse-polarity Povarov annulation reaction (Scheme 298).812 Subjecting DMSO solutions of pre-formed or in situ generated N-aryl aldimine substrates and vinylsulfone reagents to visible-light irradiation in the presence of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 photocatalyst ([Ir-6]PF6) and a sub-stoichiometric quantity of 4-cyclohexyl-HE at room temperature gave the desired products, with 24 examples documented in yields of 40–95%. Despite literature precedent that 4-alkyl-HEs act as alkyl radical reservoirs in group-transfer reactions, these types of products were not observed in this work.387,813 The reaction was tolerant to variation of the electronic properties of the N-aryl moiety, but typically required an electronically neutral or electron-rich aryl group on the aldimine to proceed efficiently. An aldimine derived from a 3-formylpyridine gave an excellent yield (298.2). In addition to arylvinylsulfone electrophiles, three examples of maleimide coupling partners were included (298.3). In all cases, good to excellent levels of diastereoselectivity were observed, forming the trans-isomer preferentially. This is opposite the stereochemical preference in typical polar Brønsted or LA catalyzed Povarov reactions. An interesting observation was made in that the observed d.r. of the product was a function of the reaction time and conversion, where increasing d.r. was observed only at high conversion. For example, in the case of tetrahydroquinoline 298.1, 19F NMR monitoring indicated that the product was forming with an intrinsic and stable d.r. of 10:1 during the course of reaction. At the point of approximately 90% conversion of the imine substrate at 18 h, through epimerization the d.r. increased gradually up to a final ratio of 20:1 after 48 h. This epimerization was dependent upon the presence of the iridium photocatalyst under visible-light irradiation.
Scheme 298. Synthesis of Tetrahydroisoquinolines in a Photocatalytic Reverse Povarov Reaction (Dixon, 2018).

The requirement for only a sub-stoichiometric quantity of reducing agent and an overall redox-neutral transformation implied a mechanism different from the group’s earlier reports was operative.785,786 The authors propose that initial reductive quenching of the Ir(III) excited state by the HE yields the corresponding acidic radical cation and a reduced-state Ir(II) complex. Through a PCET process, these two species jointly mediate the reduction of the imine substrate, yielding a neutral α-amino radical, HE radical, and ground-state Ir(III) complex. Addition of this nucleophilic α-amino radical to the vinylsulfone electrophile in a Giese fashion then occurs before subsequent cyclization onto the N-aryl group. A KIE study revealed that C(sp2)–H bond cleavage is not rate-determining (kH/kD = 1.1). Thereafter, deprotonation of the resultant delocalized cyclohexadienyl radical yields the product radical anion intermediate in an overall base-assisted homolytic aromatic substitution mechanism.814,815 This process generates an arene radical anion which is postulated to propagate a radical chain process where single-electron reduction of the imine yields the closed-shell product and a neutral α-amino radical for chain propagation. It is unclear whether the reduction event in this propagation step occurs through concerted or stepwise ET and PT. Therefore, the photoredox cycle acts as an initiator of the radical chain process. A quantum yield determination experiment supports their hypothesis of a chain process (Φ = 18). The observed epimerization is proposed to result from PET between photoexcited Ir(III) and the product secondary amine, yielding the corresponding aminium radical cation. Then, facile α-deprotonation yields the planar product α-amino radical, which can return to the closed-shell species through reprotonation, a [1,2]-H shift, and SET reduction mediated by Ir(II).
Dixon and co-workers utilized a biomimetic approach to enable the general α-C(sp3)–H functionalization of α,α-disubstituted primary amines for the synthesis of α-tertiary primary amines (Scheme 299).816 Included among several methods was a protocol for the α-allylation of these primary amines through photocatalytic reductive PCET activation of an in situ generated ketimine intermediate. In designing this transformation, the authors took inspiration from copper amine oxidase enzymes (CuAOs), a family of metalloenzymes which mediate the oxidation of primary amines to aldehydes using molecular oxygen, through the combination of a quinone cofactor and a Cu(II) species.817 The mechanism of this transformation involves condensation of the amine with the quinone cofactor and a subsequent formal [1,5]-H shift to generate an imine prior to hydrolysis and liberation of the aldehyde. These researchers reasoned that a small-molecule mimic of this quinone cofactor would condense with an α,α-disubstituted primary amine and undergo the same [1,5]-H shift, revealing a reactive ketimine intermediate which could be intercepted by both nucleophilic and electrophilic reagents through normal and umpolung modes of reactivity, respectively. A three-step sequence of (i) amine condensation with quinone, (ii) functionalization of the resulting ketimine, and (iii) oxidative removal of the aminophenol group deriving from the quinone auxiliary could be carried out in a telescoped fashion without isolation of intermediates or solvent exchange.
Scheme 299. Biomimetic Strategy for the α-C(sp3)–H Allylation of α,α-Disubstituted Primary Amines (Dixon, 2019).

A photocatalytic protocol for the synthesis of α-allylic- α-tertiary primary amines through an umpolung mode of reactivity involved initial amine condensation with quinone 299.A then exposure of the resulting ketimine to blue-light irradiation in the presence of Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6([Ir-6]PF6), HE, and allylsulfone reagents. This work represents an extension to their 2016 method of homoallylic amine synthesis in this manner, which gave N-arylated products.785 Now, six examples of primary amine products obtained after the oxidative removal of the quinone auxiliary were reported in combined yields of 30–43% over three sequential steps. Substrates deriving from α-methylbenzylamines (299.1, 299.2) and 1-aminoindane (299.3) were included. In situ lactamization of the product amino esters could be triggered through addition of NaOH (299.4). An analogous mechanism to their 2016 work is proposed to account for this transformation, involving reductive PCET to the imine substrate through the joint action of the Ir(II) reductant and HE radical cation.785 Allylation then occurs through a radical addition–elimination mechanism with the sulfone reagent. Other modes of primary amine α-C(sp3)–H functionalization in this work with nucleophilic reagents included alkylation and arylation through addition of organolithium and Grignard reagents, and cyanation through addition of TMSCN. We note a contemporaneous report from the Cresswell group achieving the α-alkylation of primary amines with electron-deficient olefins through the joint action of an organic photocatalyst and n-Bu4N+N3– co-catalyst to mediate a HAT event for substrate radical generation (see section 2.9.1).404
In 2017, Ngai and co-workers reported the β-selective reductive coupling of aldehydes or imines with alkenylpyridines proceeding through joint Brønsted acid/Lewis acid activation of the two coupling partners, respectively (Scheme 300).787 The β-selectivity offered in this photocatalytic approach is complementary to existing transition-metal methods from Krische and Lam where α-selectivity results.818−820 The optimized reaction conditions for this transformation consisted of visible-light irradiation of MeCN solutions of aryl aldehyde or N-phenyl aryl aldimines and vinylpyridine substrates in the presence of [Ru(bpy)3](PF6)2 photocatalyst ([Ru-1](PF6)2), La(OTf)3 LA co-catalyst, 2,2′-bipyridine (bpy) ligand, and HE terminal reductant at room temperature. The inclusion of the LA additive was essential to achieving the desired reductive cross-coupling, with pinacol homo-coupling product observed in its absence.805 In the absence of photocatalyst, a low yield of the desired reductive coupling product was still observed (16% in a model system), which was proposed to arise through direct PET between HE and the aldehyde substrate.821
Scheme 300. β-Selective Reductive Coupling of Aldehydes or Imines with Alkenylpyridines (Ngai, 2017).

A scope of 36 examples of reductive coupling was presented in yields of 37–99%. Aryl aldehydes carrying diverse substituents including fluoro (300.1), trifluoromethyl, trifluoromethoxy, nitrile, and ether functionality (300.2) were competent substrates. Notably, bromo- and iodobenzaldehydes reacted without dehalogenation. A number of heteroaromatic aldehydes including thiophene, benzofuran, furan (300.4), pyridine, and pyrimidine were demonstrated. Aryl aldehydes carrying diacetone-d-glucose, estrone, and dipeptide functionality all reacted efficiently, providing alcohol products as single diastereoisomers. Reactions of N-phenyl aryl aldimines under the same conditions also proceeded smoothly (e.g., 300.3). With respect to the vinylpyridine reaction component, 2- (300.2) and 4-vinylpyridines (300.1, 300.3) were effective coupling partners. α-Alkyl, α-aryl, and α-heteroaryl alkenylpyridines too were reactive, albeit poorly diastereoselective (e.g., 300.2). No examples of β-functionalized alkenylpyridine substrates were included.
Interestingly, the proposed mechanism of reaction is different from many net-reductive photocatalytic transformations of carbonyl and imine compounds with tertiary amine or HE reductants. These are commonly proposed to proceed through a reductive quenching manifold between photoexcited-state photocatalyst and amine. However, in this work, HE (Ep/2ox = +0.51 V vs Fc+/Fc in MeCN)374 was found not to quench the photoexcited state of [Ru(bpy)3](PF6)2 (E1/2 *Ru(II)/Ru(I) = +0.77 V vs SCE in MeCN)64 in SV experiments, which is in agreement with the endergonic nature of this ET. Aldehyde, aldimine, or vinylpyridine components similarly did not quench this photoexcited-state Ru(II) complex in the presence or absence of La(OTf)3. Instead, the authors propose that the reaction initiates via direct PET and concomitant PT between photoexcited-state HE (Ep/2 *HE/HE•+ = −2.28 V vs SCE in MeCN, λmax = 380 nm)821 and aldehyde substrate to yield a neutral ketyl radical and HE radical. This is proposed based on the appearance of low product yields in the absence of photocatalyst. Then, the intermediate HE radical (Ep/2 = −1.14 V vs Fc+/Fc in MeCN)374 undergoes facile reductive quenching with the photoexcited Ru(II) complex, generating a reduced-state Ru(I) complex and Hantzsch pyridinium byproduct. The joint action of this Ru(I) complex (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN)64 and the in situ generated pyridinium Brønsted acid mediate reductive PCET with the aldehyde substrate through hydrogen bond activation, yielding a neutral ketyl radical intermediate and returning ground-state Ru(II). Addition of this radical intermediate to the LA-activated vinylpyridine substrate forges a new C–C bond and produces a stabilized benzylic radical. This benzylic radical is then proposed to abstract a H-atom from additional HE in a HAT step, yielding the closed-shell product and further HE radical for catalyst turnover.
In a related report published soon after, Liu and co-workers demonstrated the intermolecular reductive coupling of 2,2,2-tri- and 2,2-difluoroacetophenones with 2-vinylpyridine (Scheme 301).822 In the previously discussed Ngai report,787 the authors noted low background reactivity in the absence of a photocatalyst, which was proposed to arise through direct PET between photoexcited HE and the aldehyde substrate.787 This step was required to initiate the photoredox catalytic cycle. This work from Liu makes use of this noted observation to develop a catalyst-free reductive coupling, which proceeds to high levels of efficiency for this class of fluorinated ketone substrates.
Scheme 301. Photocatalyst-Free Intermolecular Reductive Coupling of 2,2,2-Tri- and 2,2-Difluoroacetophenones with 2-Vinylpyridine (Liu, 2018).

Optimized reaction conditions consisted of irradiation at 395 nm of a CH2Cl2 solution of fluorinated acetophenone and 2-vinylpyridine substrates in the presence of (PhO)2P(O)OH catalyst and HE reductant. Interestingly, no exogenous LA was required in this work, which was found to be essential in Ngai’s report and was proposed to activate the alkenylpyridine component through chelation at the pyridine nitrogen.787 This wavelength of light was chosen as it gave optimal spectral overlap with the absorption maximum of HE (λmax = 380 nm).821 No reactivity was observed in the absence of the acid catalyst or light. In total, 26 examples of this photocatalyst-free reductive cross-coupling were reported in yields of 33–99%. A range of diversely substituted 2,2,2-trifluoro- and 2,2-difluoroacetophenones were viable in this transformation (301.1–301.3), but the acceptor olefin was limited to 2-vinylpyridine. When a reaction with 4-vinylpyridine was attempted, exclusive reductive pinacol homo-coupling product was obtained.
The proposed mechanism consists of oxidative quenching of the photoexcited state of HE by the fluorinated ketone substrate facilitated by hydrogen-bonding with the Brønsted acid additive, similar to the initiation step in the Ngai proposal above.787 This mechanism was supported by SV quenching studies, which also showed that vinylpyridine does not quench the excited state. UV–vis spectroscopy experiments indicated that no discrete EDA complex is generated prior to ET.823 This direct photoinduced CT is proposed to involve a PCET elementary step given that no reactivity was observed in the absence of acid additives. However, we note that the direct reduction of the trifluoroacetophenone (e.g., for α,α,α-trifluoroacetophenone, Ep/2red = −1.42 V vs SCE in MeCN)824 by the photoexcited-state HE (Ep/2 *HE/HE•+ = −2.28 V vs SCE in MeCN)821 is thermodynamically viable. In this reaction, the more strongly acidic phosphoric acid potentially facilitates radical addition to the vinylpyridine component through N-protonation, similar to Minisci-type reactivity.696 In any case, following neutral ketyl radical generation, radical addition to the olefin component and HAT with another equivalent of HE generates the product.
Many current methods using commonly employed reducing Ir and Ru photocatalysts (e.g., for Ir(ppy)3, E1/2 Ir(III)/Ir(II) = −2.19 V vs SCE in MeCN)66 enabling the reductive generation of neutral ketyl radicals through PCET are limited to aryl ketone and aldehyde substrates. With aliphatic ketones, these methods often fail due to the more-demanding reduction potentials of these substrates (e.g., for cyclohexanone Ep/2red = −2.33 V vs SCE in MeCN; for isobutyraldehyde, Ep/2red = −2.24 V vs SCE in MeCN).21 Addressing these limitations requires catalytic systems that exhibit stronger reduction potentials. One such system developed by Seo and Jamison utilizing a strongly reducing p-terphenyl photocatalyst in the presence of a stoichiometric amine electron donor under UV irradiation was successful for neutral ketyl radical generation from aliphatic ketones and aldehydes (Scheme 302).825 This system was applied to achieve net-reductive intermolecular hydroalkoxyalkylation of aliphatic ketones or aldehydes with styrenes via neutral ketyl radical generation. This catalyst system had been previously demonstrated by the Jamison group to mediate the demanding reduction of CO2 (E1/2red = −2.21 V vs SCE in DMF)826 for tertiary amine α-carboxylation,827 and styrene hydrocarboxylation.828
Scheme 302. Reductive Coupling of Aliphatic Carbonyl Compounds and Styrenes (Jamison, 2019).

*With 4.0 equiv of acetaldehyde.
In this work, the optimal conditions for reductive ketyl-olefin coupling consisted of UV irradiation (wavelength >280 nm) of DMA solutions of ketone and styrene substrates with p-terphenyl photocatalyst, 4-hydroxypentamethylpiperidine (4-hydroxy-PMP) as terminal reductant, and 4 equiv of H2O as a proton source. Reactions could be carried out equally well in flow (residence time = 35 min) or batch (reaction time = 2 h) operations. A scope of 20 ketone (38–99% yields) and 7 aldehyde (38–68% yields) substrates was included with primarily electron-rich styrene coupling partners (302.1–302.4). In some cases, Norrish type I and II fragmentation byproducts were observed. An example of a tandem styrene addition/cyclization of a tethered alkyl chloride for synthesis of tetrahydrofuran 302.5 was included.
Control experiments in the absence of p-terphenyl yielded no product, ruling out a possible mechanism involving direct CT between the ketone and amine under the UV irradiation.829 On thermodynamic grounds, both the styrene (e.g., for styrene E1/2red = −2.58 V vs SCE in DMF)830 and carbonyl (for example, cyclohexanone E1/2red = −2.33 V vs SCE in MeCN)21 reaction components can undergo ET from the reduced-state photocatalyst (E1/2 PC/PC•– = −2.63 V vs SCE in DMF)776 which is generated after initial reductive quenching of the photoexcited state with tertiary amine. To test whether the ketone or styrene partners are initially reduced, a cyclopropyl methyl ketone (E1/2red = −3.38 V vs SCE in DMF)831 radical rearrangement experiment was conducted. Ring-opened reductive coupling products were observed (34%), consistent with the intermediacy of a ketyl radical.831−833 Furthermore, when more easily reduced styrene partners were employed, such as 4-vinylbiphenyl (E1/2red(calc) = −2.26 V vs SCE in DMF),825 reduction to 4-ethylbiphenyl was observed in low yield (15%) with none of the ketone coupling product. This implied a change in mechanism to one involving preferential reduction of the styrene to the corresponding radical anion and further reduction to the saturated product. With electron-rich styrenes (preferred in this reductive coupling reaction) which are more difficult to reduce, ketyl radical instead appears to form preferentially. The authors invoked a PCET mechanism for this observed chemoselective (and in this case significantly endergonic) ketyl radical generation in the presence of styrenes via coordination of the acidic aminium radical cation intermediate,805 or water additive,834 with the hydrogen bond acceptor carbonyl group. Therefore, a mechanistic model consisted of UV irradiation of p-terphenyl photocatalyst, leading to reductive quenching with the tertiary amine donor, to generate the strongly reducing terphenyl radical anion and an aminium radical cation. The joint action of this reduced-state photocatalyst and proton source enables PCET with the aliphatic carbonyl substrate, yielding the neutral ketyl radical. Addition across the styrene leads to a stabilized benzylic radical, which is reduced to the carbanion through a further turn of this photoredox cycle, with a net two-photon requirement. Finally, PT yields the reductive coupling product. The α-amino radical generated through photocatalyst reductive quenching and PCET is ultimately quenched through dimerization, and this byproduct was observed.
Gaunt and co-workers reported the total syntheses of the polycyclic alkaloid natural products (−)-FR901483 (303.1) and (+)-TAN1251C (303.2) utilizing a photocatalytic reductive olefin hydroaminoalkylation reaction as a method of synthesizing an early common intermediate to both target molecules (Scheme 303).835 (−)-FR901483 was isolated from the fermentation broth of Cladobotryum sp. No. 11231 by Fujisawa Pharmaceutical Co. in 1996, and displayed potent immunosuppressant activity.836 (+)-TAN1251C was first isolated by Takeda in 1991 and displayed activity as muscarinic antagonists. Hydroaminoalkylation was affected through blue-light irradiation of a CH2Cl2 solution of l-tyrosine methyl ester, 1,4-cyclohexanedione monoethylene acetal, and the Karady–Beckwith alkene,243 in the presence of fac-Ir(ppy)3 (Ir-1) photocatalyst, TFA as a Brønsted acid co-catalyst, HE terminal reductant, and molecular sieves, yielding 303.3. Then, without isolation, the reaction mixture was treated with aqueous hydrochloric acid in THF at 50 °C to effect lactamization and ketal deprotection, leading to gram-quantities of spirocyclic lactam 303.4 in 73% overall yield. From this intermediate, (−)-FR901483 (303.1) is accessed in a further seven synthetic operations and (+)-TAN1251C (303.2) in just three further steps.
Scheme 303. Olefin Hydroaminoalkyation Used to Access a Common Intermediate Toward the Total Synthesis of Alkaloids (−)-FR901483 (303.1) and (+)-TAN1251C (303.2) (Gaunt, 2020).
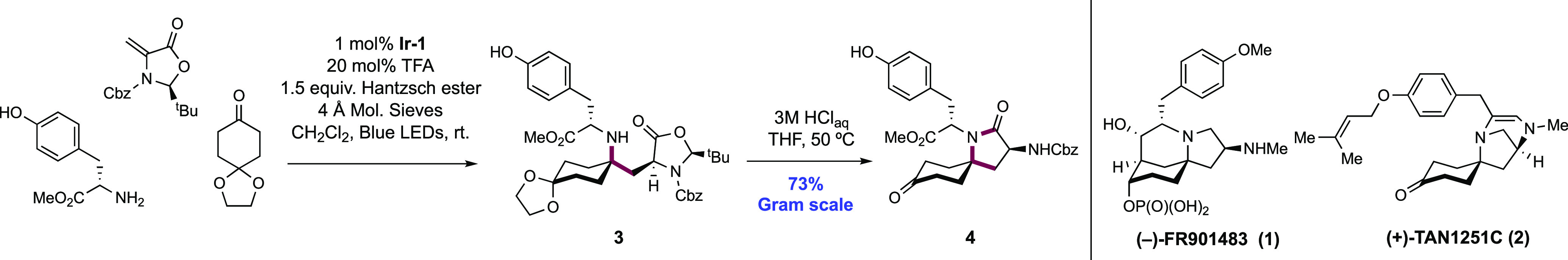
A likely mechanism of hydroaminoalkyation involves initial imine condensation between l-tyrosine methyl ester and the cyclohexanone (Scheme 304). Then, protonation of the newly formed imine by the TFA acid catalyst yields the corresponding iminium ion. Reductive quenching of the photoexcited-state Ir(III) complex (E1/2 *Ir(III)/Ir(II) = +0.31 V vs SCE in MeCN)66 with HE (Ep/2ox = +0.51 V vs Fc+/Fc in MeCN)374 generates a strongly reducing Ir(II) complex (E1/2 Ir(III)/Ir(II) = −2.19 V vs SCE in MeCN)66 which is proposed to engage in SET with the protonated imine. In the absence of TFA additive, only trace yields of product were realized, suggesting the necessary protonation prior to reduction. This generates a nucleophilic α-amino radical intermediate which undergoes Giese-type radical addition to the Karady–Beckwith alkene, resulting in a stabilized α-carbonyl radical species. Finally, diastereoselective HAT to this intermediate from HE radical cation yields the closed-shell amine product and Hantzsch pyridinium as a byproduct.
Scheme 304. Proposed Hydroaminoalkylation Mechanism through Stepwise PT/ET (Gaunt, 2020).

An intermolecular reductive coupling of imines and electron-deficient olefins was reported by Rueping and co-workers in 2020 (Scheme 305).837 Using [Ir(4F(CF3)ppy)2(bpy)]PF6 as photocatalyst, n-Bu3N as the sole additive, and EtOH as solvent under visible-light irradiation, 26 examples of imine–olefin reductive coupling were reported in yields of 35–89%. A range of electron densities on the aldehyde-derived aryl component of the imine substrate were tolerated (305.1, 305.2) as was a similar range on the aniline-derived N-aryl component (305.2, 305.3). In addition to acrylates, acrylonitrile, acrylamide, and phenyl vinyl sulfone (305.4) could function as radical acceptors. The authors also were able to adapt this reaction to flow conditions, which provided a significant reduction in reaction time and allowed for a drop in photocatalyst loading.
Scheme 305. Photocatalytic Intermolecular Imine–Olefin Reductive Coupling (Rueping, 2020).

Here, photoexcitation of the Ir(III) catalyst produces a long-lived photoexcited state, capable of reductive quenching with n-Bu3N additive (Epox = +0.62 V vs Ag+/Ag in MeCN)838 to generate a reducing Ir(II) complex and n-Bu3N•+. Through a proposed formal [1,2]-H shift, this adopts a distonic aminium radical cation structure, which then acts as the Brønsted acid activator of the imine component for PCET, mediated by Ir(II). In a 2016 proposal, these authors also reasoned that under similar reaction conditions, R3NH+ is generated in situ through bimolecular PT between R3N•+ and R3N and this closed-shell Brønsted acid may also serve to activate the substrate.804 The substrate α-amino radical thus generated adds across the electron-deficient olefin prior to termination of the resulting radical through a HAT event with the α-amino radical derived from n-Bu3N. Thus, n-Bu3N serves three roles in this mechanistic proposal—stoichiometric reductant, in situ generated Brønsted acid activator, and HAT mediator. Support for the activating role of n-Bu3N•+ or n-Bu3NH+ was provided by the observation that catalytic loadings of Brønsted bases could entirely inhibit the reaction.
Ye and co-workers in 2020 developed a visible-light-mediated radical ynamide Smiles rearrangement for the synthesis of 2-benzhydrylindoles and 3-benzhydrylisoquinolines initiated via reductive PCET of acetophenone starting materials (Scheme 306A,B).839 Optimized reaction conditions for the synthesis of indoles consisted of visible-light irradiation of DMF solutions of an acetophenone substrate carrying an aniline-derived N-arylsulfonyl ynamide in the ortho-position in the presence of [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) as a photocatalyst and HE as a stoichiometric terminal reductant. The organic photocatalyst 4CzIPN was almost equally as effective. A scope of 30 examples of benzhydrylindole synthesis was reported in yields of 38–88% (306.1–306.3). 3-Benzhydrylisoquinoline products could also be achieved for substrates bearing a homologated benzylamine-derived N-arylsulfonyl ynamide employed under modified reaction conditions consisting with 1,2-DCE solvent and an air atmosphere (306.4–306.6). Now, 20 examples of this class were reported in yields of 23–83% yields.
Scheme 306. Photocatalytic Synthesis of Indoles and Isoquinolines through Reductive PCET-Mediated Neutral Ketyl Radical Generation and Subsequent Smiles Rearrangement (Ye, 2020).

The authors propose a two-step sequence for the formation of the product. First, the formation of a 3-hydroxyindolenine intermediate occurs via cyclization of a PCET-generated neutral ketyl radical and Smiles rearrangement (Scheme 307). This intermediate then undergoes a second reduction by the photocatalyst followed by a SCS and re-aromatization to afford the final indole product. The authors favor a mechanism for neutral ketyl radical generation similar to that proposed by Chen,788 involving PCET activation of the acetophenone substrate mediated by Ir(II) (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN)68 through hydrogen bonding with the transient acidic aminium radical cation formed on reductive quenching of the photoexcited-state Ir(III) catalyst (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 with HE (E1/2 = +0.51 V vs Fc+/Fc in MeCN).374 SV quenching studies show that ET occurs between the Ir(III) excited state and HE, but not directly with the ynamide substrate. The resulting ketyl radical then undergoes 5-exo-dig cyclization onto the tethered ynamide. The nascent vinyl radical undergoes a second 5-exo-trig spirocyclization onto the sulfonylarene, initiating a radical Smiles rearrangement. This spiro-intermediate then collapses to achieve net aryl group migration and aminosulfonyl radical formation, which rapidly extrudes SO2 leading to a stabilized aza-allyl radical. Either a stepwise single-electron reduction/protonation sequence or a direct HAT step yields a neutral, closed-shell 3-hydroxyindolenine as the initial product of the reaction. DFT calculations support the stepwise ET/PT pathway for radical termination. This intermediate was observed and isolated in one example.
Scheme 307. Mechanism of Photocatalytic Indole Formation via Neutral Ketyl Radical Initiated Smiles Rearrangement (Ye, 2020).
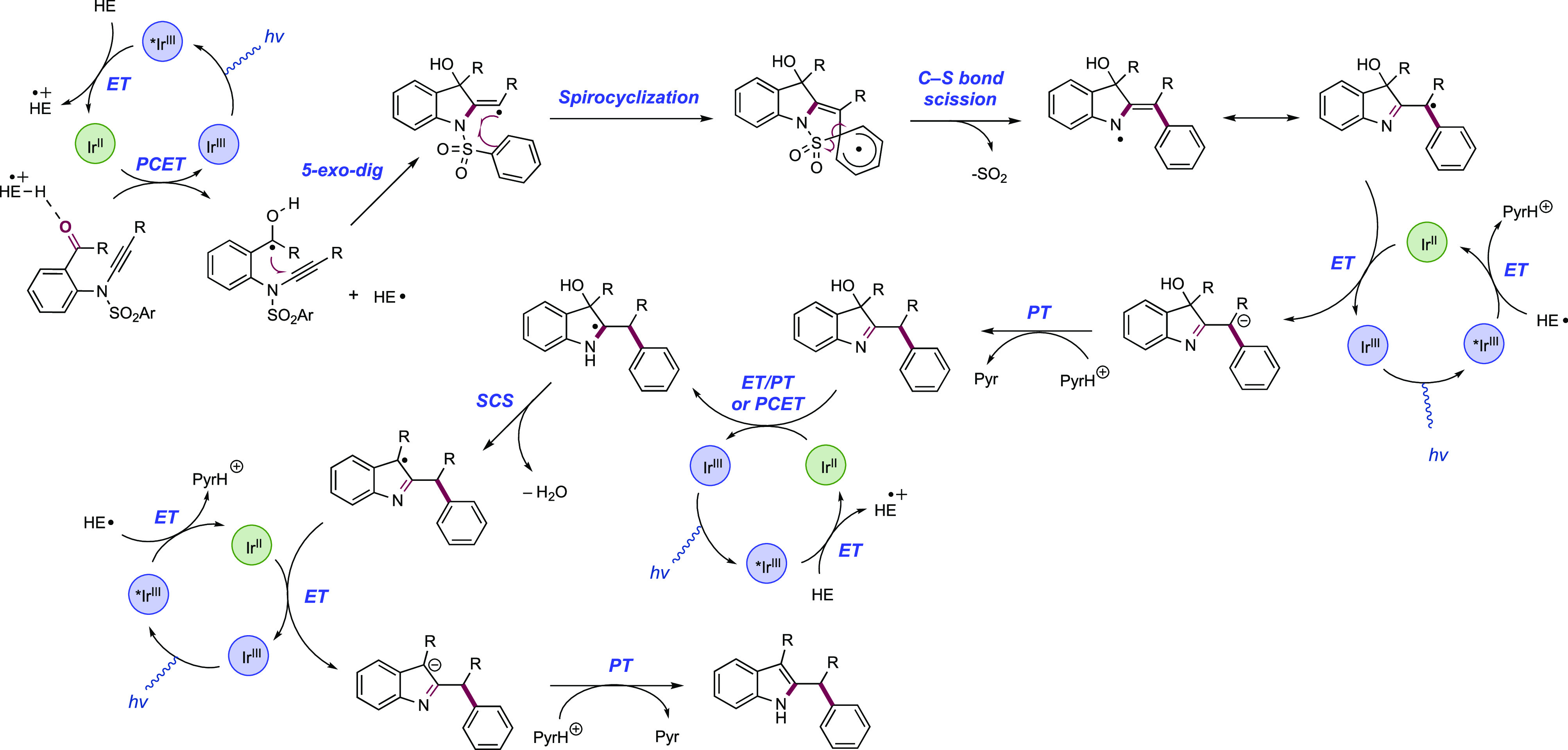
Then, through a second reductive radical generation step, the imine is transformed to the corresponding α-amino radical. It is unclear whether this intermediate is directly accessible through single-electron reduction mediated by Ir(II) or if it requires PCET involvement as has been invoked in other work describing reductive radical generation in imines (for example by Dixon and others as discussed in this section).785 This radical undergoes a SCS reaction, generating a stabilized benzylic aza-allyl radical with elimination of water. Finally, a stepwise ET/PT sequence is invoked to yield the indole product through these two sequential reductive transformations.
In the case of isoquinoline synthesis, an analogous pathway is followed for a homologated benzylamine-derived N-arylsulfonyl ynamide substrate. The reaction sequence is initiated through 6-exo-dig cyclization, leading to a dihydroisoquinoline product initially. Under the aerobic conditions of the reaction, a photocatalytic aerobic oxidation is proposed to yield the final product, for which there is literature precedent under similar conditions.840−842
Soon after, the authors extended this method to the synthesis of 2-hydroxymethylindoles using a class of N-methanesulfonyl ynamides which are unable to undergo Smiles rearrangement (Scheme 306C).843 With this substrate class, exposure to similar photocatalytic conditions initially yielded an eneindolin-3-ol product, which could be converted in a one-pot fashion to a 3-hydroxyindole by promoting an allylic 1,3-transposition reaction at elevated temperature after aerobic oxidation of the excess HE. A scope of 15 examples of 2-hydroxymethylindole synthesis were reported in yields of 23–86% (306.7–306.9).
An analogous mechanism was presented involving reductive-PCET-mediated neutral ketyl radical generation through the action of Ir(II) and HE radical cation followed by 5-exo-dig cyclization. In this class, the vinyl radical was unable to promote an aryl group migration, so radical termination occurs at this stage via HAT with HE radical. A polar pathway for allylic 1,3-transposition is invoked, wherein in situ-generated methanesulfonic acid promotes ionization at elevated temperatures.844 This pathway is supported by isotopic labeling studies, where 18O incorporation is seen in the product with addition of H218O.
6.1.2. Intermolecular C–C Bond Formation through Pinacol and Aza-pinacol Processes
The often significantly endergonic single-electron reduction of carbonyl and imine substrates (e.g., for p-tolualdehyde, E1/2red = −2.28 V vs SCE in MeCN; for N-isobutylidene aniline, Epred = −2.90 V vs Ag/Ag+ in MeCN)807,808 can place a strict limitation on the chemoselectivity of traditional approaches to ketyl and α-amino radicals from these substrates, respectively. These reduction protocols have previously called for the use of reducing metal salts derived from metals such as samarium, titanium, and zirconium.845−848 However, the activation of mildly Lewis-basic carbonyl and imine substrates toward single-electron reduction with Brønsted acidic and/or hydrogen-bond-donating species can offer an alternative to these traditionally demanding procedures. By coordinating to the substrate during reduction, these activators can stabilize the development of negative charge as well as provide access to lower-barrier PCET processes that avoid the need for extremely reducing species accessing radical anion intermediates. In 2015, Rueping and co-workers demonstrated that tertiary amines act in the role of this activator, in addition to the role of stoichiometric reductant for the reductive pinacol coupling of aldehydes, ketones, and imines in the presence of a reducing photocatalyst (Scheme 308).805 In the presence of Ir(III) photocatalyst [Ir(4F(CF3)ppy)2(bpy)]PF6 and a stoichiometric loading of n-Bu3N in DMF, blue-light irradiation gave access to 23 examples of reductive pinacol couplings of carbonyl derivatives in yields of 41–91%. Diols derived from the reductive dimerization of electron-rich as well as electron-poor benzaldehydes were obtained in good yield (308.1 and 308.2, respectively). Acetophenone- and benzophenone-derived ketones also proved to be viable substrates (308.3 and 308.4, respectively). With a lowered loading of n-Bu3N and in MeCN, 13 examples of the reductive coupling of benzyl-protected imine substrates (308.5) were also achieved in good to excellent yields (42–91%).
Scheme 308. Photocatalytic Reductive Pinacol-Type Coupling of Aldehydes, Ketones, and Imines (Rueping, 2015).

The authors propose that this reductive coupling reaction was initiated through reductive quenching of the Ir(III) photoexcited state with the tertiary amine additive (e.g., for n-Bu3N, Epox = +0.62 V vs Ag+/Ag in MeCN)838 to yield an Ir(II) complex. Given the modest reduction potential of this Ir(II) form of the photocatalyst, (E1/2 Ir(III)/Ir(II) = −1.69 V vs Fc+/Fc),805 which alone provides insufficient driving force for substrate activation (e.g., for benzaldehyde E1/2red = −1.93 V vs SCE in MeCN; for acetophenone, E1/2red = −2.14 V vs SCE in MeCN; for benzophenone, E1/2red = −1.83 V vs SCE in MeCN),21,700 the authors here assumed that the oxidation product of n-Bu3N facilitates ET. Two modes of substrate binding of this activator were proposed, (i) the formation of a 2-center-3-electron bond between the localized aminium radical cation n-Bu3N•+ and the carbonyl oxygen, or (ii) initially a formal [1,2]-hydride shift occurring to generate the corresponding distonic aminium radical cation which then serves as a Brønsted acid activator through hydrogen bonding with the carbonyl substrate. In a 2016 report, these authors also proposed that the closed-shell ammonium species n-Bu3NH+, generated through bimolecular PT between n-Bu3N and n-Bu3N•+, may act as a Brønsted acid for the in situ activation of similar benzaldehyde substrates.804 This proposal may be more reasonable when considering lifetime of these open-shell intermediates. This hypothesis was supported by the detrimental effect of Brønsted bases on the reaction performance, providing no product when K3PO4 was added. However, the yield of the desired product was improved over the previously optimized conditions by the inclusion of catalytic amounts of Brønsted acids. With the aid of this acid activator, a reductive PCET event occurs to generate a neutral ketyl radical or α-amino radical intermediate which goes on to yield C–C coupling product.
In addition to the use of Ir and Ru photocatalysts in reductive homo-coupling reactions of aldehydes and imines, organic photocatalysts have also found success under similar conditions. For example, Sudo and co-workers reported in 2016 the use of catalytic quantities of perylene as a photocatalyst in the reductive dimerization of aldehydes and imines (Scheme 309).849 Other organic molecules possessing highly extended π-systems have also been reported to catalyze reductive photochemical transformations. Of note, Zhang and co-workers disclosed the use of p-phenylene, a polymer of para-linked phenyl monomers, in the reductive coupling of benzaldehyde under visible-light irradiation.850 More recently, Miyake and co-workers were able to achieve the initiation of the radical polymerization of functionalized vinyl monomers through the oxidative quenching of the perylene photoexcited state with α-bromo ester initiators.851 Using 12 mol% perylene in the presence of 8 equiv of DIPEA in MeCN solution under white light irradiation, seven examples of reductive homo-coupling of aldehydes and ketones were provided in yields of 23–67% yields. Benzaldehydes of varying electronic character were amenable to this transformation (309.1, 309.2), though more difficult-to-reduce benzophenone and acetophenone afforded the desired coupling products (309.3 and 309.4, respectively) in low to moderate yields. In the case of benzophenone, a 28% yield of the reduction byproduct 1,1-diphenylmethanol formed alongside the desired homo-coupling product, although no such byproduct formed with acetophenone as substrate. Under otherwise identical conditions with 1 mol% perylene, Sudo and co-workers were later able to extend the scope of this methodology to include a broader range of imines (309.5–309.7), with 13 examples of symmetrical 1,2-diamine synthesis in yields of 41–80%.852
Scheme 309. Reductive Coupling of Aldehydes, Ketones, and Imines via Reductive PCET Using Perylene as a Photocatalyst (Sudo, 2016 and 2017).

This reaction is proposed to initiate with the absorption of visible light by the perylene π-system (λmax = 430 nm),853 producing an oxidizing photoexcited state which undergoes reductive quenching with the DIPEA additive (Epox = +0.68 V vs SCE in MeCN)854 to generate DIPEA•+ and form perylene radical anion (PC•–). This radical anion (E1/2 PC/PC•– = −1.66 V vs SCE in MeCN)855 is proposed to reduce the substrate to the corresponding ketyl or α-amino radical anion that then dimerizes and undergoes PT (or vice versa) to afford the diol or diamine product. However, the endergonic nature of the direct ET (e.g., for 4-chlorobenzaldehyde, Ep/2red = −1.85 V vs SCE in MeCN)21 suggests a PCET involvement, where amine reductants such as DIPEA generate acidic aminium radical cation intermediates and ammonium salts in situ that allow for concerted PT also.805 In the context of this reductive pinacol-type coupling from Sudo and co-workers, perylenes modified with electron-donating and electron-withdrawing substituents were investigated in the reductive pinacol coupling, but none offered an advantage over unsubstituted perylene. Control experiments revealed the necessity of all the reaction components.
In 2017, the groups of Wang and Yao reported a protocol for the reductive pinacol homo-coupling reaction of α-ketoesters (Scheme 310).856 Optimal reaction conditions enabling this transformation consisted of white light irradiation of ketoester substrate in the presence of Na2-EY photocatalyst, HE reductant in a 95:5 (v/v) mixture of THF/H2O. The researchers reported 21 examples of the synthesis of 2,3-dialkylated tartaric acid esters with yields of 40–92%. A broad range of functional groups were well tolerated in this protocol, including 4-methoxybenzyl ether, alkene (310.2), alkyne, aldehyde acetal, silyl ether, secondary amine-derived carbamate, cyclic ether/thioether (310.3), bromoalkane, and N-alkoxyphthalimide (310.4). For ketoesters bearing bulky substituents (e.g., adamantyl) or two EWGs (such as ketomalonic acid dimethyl ester), no reductive coupling was observed and only carbonyl reduction occurred. Bulkier alkoxy groups on the ester moiety did not have any significant effect on the reaction yield or the diastereomeric ratio (310.5). The researchers were able to achieve comparably good yield on a 60 mmol scale, highlighting the scalability of this protocol.
Scheme 310. Visible-Light Photocatalytic Pinacol Coupling of α-Ketoesters (Wang and Yao, 2017).

This reductive coupling transformation was proposed to initiate through visible-light irradiation of the reaction mixture to produce the long-lived photoexcited state (E1/2 *EY/EY•– = +0.83 V vs SCE in MeCN),71 which oxidizes HE (Ep/2ox = +0.51 V vs Fc+/Fc in MeCN)374 to generate the photocatalyst radical anion EY•– and HE radical cation. Through HAT or deprotonation and further oxidation of HE radical cation, a pyridinium species is proposed to form. The joint action of the EY•– reductant (E1/2 PC/PC•– = −1.06 V vs SCE in MeCN)71 and this pyridinium Brønsted acid mediates reductive PCET to the α-ketoester substrate to produce a neutral ketyl radical, which rapidly undergoes self-coupling to furnish the dimerized product. The authors demonstrated that the reduction potential of the ketoester substrate in the absence of acid additives (e.g., for methyl 3-methyl-2-oxobutanoate, E1/2red = −1.75 V vs SCE in DMF)856 was appreciably endergonic compared to that of EY•–, thus supporting the involvement of PCET.
As an alternative to Ru- and Ir-based photocatalysts, Cozzi and co-workers reported in 2018 the use of substituted coumarin dyes in the reductive pinacol coupling of aldehydes and ketones (Scheme 311).857 The authors investigated several coumarin derivatives and determined that sulfonated thiophene-substituted catalyst 311.A, which could be prepared in two steps from commercially available starting materials, was optimal. This catalyst featured a λmax = 413 nm, an excited-state lifetime of 2.9 ns, and an excited-state reduction potential of E1/2 PC•+/*PC = −1.89 V vs SCE in DMF.857 To demonstrate the utility of this photocatalyst, the authors reported 17 examples of pinacol-type couplings of aldehydes, ketones, and imines in yields of 31–95%. The optimized conditions involved blue-light irradiation of the substrate in the presence of 5 mol% 311.A and Et3N in DMF solution. Electron-rich and electron-poor aldehydes efficiently underwent coupling (311.1, 311.2) as did a fluorinated benzophenone derivative (311.3). Imines could also be reductively coupled in moderate to good yields (311.4). Similar to the reports by Rueping and co-workers,804,805 hydrogen bonding between the substrate and the Et3N•+ was proposed to be crucial in activating the substrates toward reduction. The authors propose oxidative catalyst quenching to facilitate substrate reduction.
Scheme 311. Pinacol Coupling of Aldehydes, Ketones, and Imines Using a Coumarin Photocatalyst (Cozzi, 2018).

Primary amines constitute an essential class of molecules in natural and synthetic compounds, especially in pharmaceuticals, agrochemicals, and materials with industrially useful properties. In 2018, Gilmore and co-workers reported a photoredox-mediated protocol for the synthesis of a variety of unprotected primary amines from NH3 and carbonyl starting materials via the intermediacy of an α-amino carbon radical (Scheme 312).858 Irradiation of carbonyl substrates in methanolic NH3 solution in the presence of [Ru(bpy)3]Cl2 photocatalyst ([Ru-1]Cl2), TFA or Sc(OTf)3 Brønsted or Lewis acid co-catalyst, and HE terminal reductant, generated an unprotected α-amino radical intermediate through acid-promoted condensation of ammonia and an aldehyde/ketone substrate followed by chemoselective single-electron reduction mediated by the excited-state photocatalyst. Single-electron reduction was proposed to be facilitated by either Brønsted or Lewis acid activation of the in situ generated imine. This resulting radical intermediate could then undergo a variety of subsequent transformations depending on the specific reaction conditions. These aldehyde and ketone starting materials provided 1,2-diamine products (e.g., 312.1, 312.2) via dimerization of the α-amino radical intermediates (17 examples with 54–82% yields and 14 examples with 41–99% yields, respectively). The addition of thiophenol HAT co-catalyst successfully mediated overall carbonyl reductive amination for primary amine synthesis (e.g., 312.3) via chemoselective polarity-matched HAT (three examples with 71–80% yields). The authors also demonstrated that the addition of 4-cyanopyridine allows for the synthesis of α-arylated unprotected primary amine products (e.g., 312.4) via radical–radical cross-coupling (three examples with 72–99% yields).
Scheme 312. Photocatalytic Synthesis of Primary Amines from NH3 and Aldehyde or Ketone Starting Materials (Gilmore, 2018).

In 2019, Collins and co-workers reported a bifunctional heteroleptic Cu(I)(diamine)(bisphosphine) complex capable of fulfilling the dual roles of photoreductant and Brønsted acid required to mediate the reductive PCET activation of aromatic ketones and aldehydes for neutral ketyl radical generation for pinacol homo-coupling reactions (Scheme 313).859 By appending the acidic activating group to the photoreductant, the acidity required to effectively drive reductive PCET can be significantly less than when separate reaction components are employed. Initially, the catalyst [Cu(dq)(BINAP)]BF4 (where dq = 2,2′-biquinoline (313.L1), BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl) was shown to effectively act as a photoreductant when combined with (PhO)2P(O)OH as a Brønsted acid co-catalyst (pKa = 3.7 in DMSO),860 delivering pinacol coupling product 313.1 in 90% yield when irradiated with acetophenone solutions in THF in the presence of HE reductant. However, no product formation resulted when the acid additive was omitted. Replacing the dq ligand with a 2-(3-pyrazoyl)pyridine ligand (pypz, 313.L2) (pKa for pyrazole N–H bond = 14.2)861 and irradiating under the same reaction conditions in the absence of any exogenous acid led to product 313.1 formation in 25% yield. Through further catalyst optimization, appending a sulfonamide group (e.g., for PhSO2NH, pKa = 16.1 in DMSO)23 carrying a 4-fluorophenyl group to the pyrazole ligand (313.L3) arrived at lead catalyst [Cu(pypzs)(BINAP)]BF4, which afforded product 313.1 in 76% yield in the absence of any exogenous acid. Under optimized conditions, 24 examples of aromatic ketone and aldehyde pinacol homo-coupling were reported in yields of 46–99%. Substrates bearing halogen (313.2), boronate (313.5), thioether (313.4), thiophene, pyridine (313.6), and quinoline groups were all tolerated. Notably, this bifunctional Cu(I) catalyst was successful in promoting product formation where an existing Ir(III) protocol was poorly efficient ([Ir-7]PF6, NBu3, DMF, Blue LED, 15% of 313.4; < 10% of 313.5).805
Scheme 313. A Bifunctional Cu(I) Complex Mediates Photocatalytic Reductive PCET for Pinacol Coupling (Collins, 2019).

Luminescence quenching studies indicated that photoexcited-state Cu(I) (E1/2 *Cu(I)/Cu(0) = +1.86 V vs SCE in MeCN)859 first underwent reductive quenching with HE (Ep/2ox = +0.51 V vs Fc+/Fc in MeCN)374 to deliver a weakly reducing Cu(0) complex (E1/2 Cu(I)/Cu(0) = −0.04 V vs SCE in MeCN).859 Thereafter, coordination of the carbonyl substrate through hydrogen bonding occurs. 1H NMR titration experiments carried out with increasing equivalents of acetophenone indicated that the substrate adopts a two-point binding association with both the sulfonamide N–H and pyrazole N–H groups. Reductive PCET results in the generation of a ketyl radical, which undergoes homo-coupling and termination via HAT with the HE radical cation. Finally, PT between the Hanztsch pyridinium byproduct and the deprotonated photocatalyst returns the latter to its active form. It is notable that this acidic sulfonamide-ligated Cu(0) complex can drive reductive PCET to carbonyl substrates when more strongly reducing Ru(I) or Ir(II) complexes are typically used in tandem with exogenous acid co-catalysts for neutral ketyl radical generation (e.g., for [Ru(bpy)3](PF6)2 ([Ru-1](PF6)2), E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN, and for [Ir(dF(CF3)ppy)2(bpy)]PF6 ([Ir-7]PF6), E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN),64,101 and reflects the benefits of proximity between the substrate and the photoreductant as well as the two-point binding mode of substrate chelation.
Ternary semiconducting compounds of the type A+xB+y2C–z4 have garnered considerable interest in the field of optoelectronics.862 In particular, ZnIn2S4 has also received attention in photocatalysis due to the fact that it absorbs visible light (λmax ≈ 360 nm with a low-energy absorption edge of 490 nm), offers a band gap of 2.5 eV with a VB edge of 1.3 eV vs NHE, and displays good chemical stability.863 This ternary semiconductor has been previously applied synthetically toward the photocatalytic oxidative dimerization of amines to imines,863 as well as the oxidation of benzyl alcohol to benzaldehyde.864 In 2020, Sun and co-workers reported the use of ZnIn2S4 for the reductive pinacol homo-coupling of benzaldehydes to produce a variety of hydrobenzoin derivatives (Scheme 314).865 The optimized reaction conditions consisted of blue-light irradiation of the benzaldehyde substrate in the presence of the ZnIn2S4 photocatalyst (100 mg/mmol substrate) and approximately 70 equiv of Et3N as a sacrificial reductant in H2O/MeCN (7:3 v/v) at room temperature. The inclusion of water was crucial for obtaining high conversion and selectivity for the desired hydrobenzoin products over reduced benzyl alcohol byproducts. Additionally, the heterogeneous photocatalyst could be recycled in subsequent reactions without significant loss of activity (>90% yield of hydrobenzoin after a third cycle).
Scheme 314. Reductive Pinacol Coupling of Aryl Aldehydes Using a ZnIn2S4 Semiconductor as Photocatalyst (Sun, 2020).

The authors report eight examples of aryl aldehyde pinacol coupling in high-performance liquid chromatography (HPLC) yields of 16–99%. Electron-withdrawing substituents greatly enhanced the efficiency of the reaction (314.1, 314.2), whereas EDGs diminished reactivity (314.3), trending with the ease of substrate reduction. 2-Furfural was also a competent substrate, providing hydrofuroin (314.5) in greater than 90% yield by HPLC. When run in the absence of a sacrificial reductant, benzyl alcohol was able to undergo dimerization and further oxidation to a mixture of benzoin (58% yield) and deoxybenzoin (40% yield). A similar mixture of products was observed when an equimolar mixture of benzaldehyde and benzyl alcohol was subjected to the optimized reaction conditions, demonstrating the photocatalyst’s ability to mediate both benzaldehyde reduction and benzyl alcohol oxidation.
In analogy to mechanisms described previously for semiconductor-catalyzed photoredox reactions, the authors propose that visible-light absorption by the ZnIn2S4 photocatalyst promotes a VB electron to the CB. The resulting VB hole oxidizes Et3N to the corresponding aminium radical cation. The benzaldehyde substrate (e.g., for benzaldehyde, E1/2red = −1.93 V vs SCE in MeCN)21 is then postulated to undergo reductive PCET with the excited CB electron and a proton liberated from Et3N•+, affording a neutral ketyl radical intermediate. Inclusion of 1,1-diphenylethylene as a radical trap produced the expected adduct with the ketyl radical intermediate as determined by mass spectrometry (MS). Coupling between two ketyl radicals generates the desired hydrobenzoin product. The reactivity and selectivity enhancement observed upon increasing volume fractions of water in the solvent mixture is proposed to maximize the concentration of lipophilic ketyl radicals at the surface of the semiconductor photocatalyst via the hydrophobic effect, thereby increasing the likelihood of dimerization over further reduction. The use of a mixture of MeCN/D2O led to deuterium incorporation only in the hydroxyl groups. The lack of deuterium incorporation at other positions within the hydrobenzoin product or the benzyl alcohol byproduct suggests that Et3N•+ acts as the proton source in the reduction mechanism.
When benzyl alcohol was employed as the substrate, the key α-hydroxy radical intermediate was postulated to arise from oxidation of benzyl alcohol (Ep/2ox = +1.90 V vs Ag/AgNO3 in MeCN)866 by a VB hole followed by benzylic deprotonation. Due to the lack of a sacrificial reductant with benzyl alcohol as substrate, the hydrobenzoin that is formed from this dimerization can undergo further oxidation to yield benzoin or dehydration to afford deoxybenzoin. Minor quantities of benzaldehyde were observed under these conditions. As a mechanistic probe of the role of surface-bound protons in the PCET reduction of benzaldehyde produced during the benzyl alcohol coupling reaction, ZnIn2S4 doped with 2 wt% Ni as a hydrogen evolution catalyst was investigated and found to yield benzaldehyde nearly quantitatively through the removal of protons from the system as dihydrogen. This observation further supports the key role of protons in activating benzaldehyde toward reduction by the CB electron.
A complicating side-reaction that can occur alongside electrochemical reductive PCET processes is the unproductive cathodic reduction of protons to molecular hydrogen, which is often thermodynamically and kinetically favored over formation of a weak neutral ketyl radical O–H bond-forming through PCET. In order to achieve productive reductive PCET, this side reaction must be suppressed as loss of protons in this manner slows down the reaction through the removal of a necessary component of the desired process. A specially designed catalytic mediator of the electrochemical reduction could potentially bias the rate of substrate reduction through PCET relative to proton reduction via its ability to co-localize a proton and an electron, thereby facilitating their transfer to substrate in a single elementary step.
In 2020, Peters and co-workers reported the successful design and application of an anilinium-substituted cobaltocenium derivative, [(Cp)Co(CpN)]+, as one such mediator of electrocatalytic PCET (Scheme 315).867 This mediator was able to accelerate the reductive PCET of acetophenone to the corresponding neutral ketyl radical while suppressing hydrogen evolution. In an undivided cell under constant potential conditions (Ecell = −1.30 V vs Fc+/Fc) with a BDD cathode and a glassy carbon anode, 83% yield of the acetophenone pinacol coupling product 315.1 was obtained in the presence of this [(Cp)Co(CpN)]+ catalyst, p-TsOH additive, and n-Bu4N+PF6– electrolyte. DFT calculations revealed the concerted PCET activation of acetophenone under these conditions to be thermodynamically favored (ΔGrel = 0 kcal mol–1) over activation by initial discrete ET (ΔGrel = +26 kcal mol–1) or PT (ΔGrel = +19 kcal mol–1). Mechanistically, the authors propose that the mediator is protonated by p-TsOH at the aniline nitrogen to form [(Cp)Co(CpNH)]2+ (pKa = 8.6 in MeCN)867 which can then undergo reduction at the cathode (E°red = −1.21 V vs Fc+/Fc in MeCN)867 to generate the active PCET catalyst, [(Cp)Co(CpNH)]+. Reduction of the protonated mediator significantly lowers the BDFE of the anilinium N–H from 79 kcal mol–1 in the Co(III) complex to 39 kcal mol–1 in the Co(II) complex in accordance with the Bordwell equation. This marked labilization of the N–H bond facilitates the reductive PCET of acetophenone wherein an electron is donated from the Co(II) center as the anilinium moiety donates a proton. Finally, radical recombination of the acetophenone neutral ketyl radicals leads to product formation. A KIE of 4.9 ± 0.7 was observed when 4-NCC6H4ND3+ was used as the stoichiometric acid additive, suggesting that PCET reduction is rate-determining. Additionally, Hammett analysis indicated the accumulation of a slight positive charge on the acetophenone substrate during PCET (ρ = −0.50), implying some asynchronicity with PT moderately more advanced at the transition state than ET. The rate of reaction is decreased 10-fold with 2-pentanone as substrate, though the success of the reaction with this more challenging substrate demonstrates the ability of this system to effect challenging PCET reactions.
Scheme 315. Electrochemical Reductive PCET of Acetophenone with a Cobaltocenium-Anilinium PCET Mediator (Peters, 2020).

6.1.3. Intermolecular C–C Bond Formation through Reactions with (Hetero)arenes
As detailed above, photocatalytic methods enabling the reductive PCET activation of carbonyls and imines have primarily involved addition reactions with olefins and alkynes (see section 6.1.1) as well as (aza)pinacol homo-coupling (see section 6.1.2). In 2017, Xia and co-workers expanded on the types of transformations amenable to this activation mode, and reported the first photocatalytic direct (hetero)arylation of aldehydes, ketones, and imines with 1,4-DCB and 4-cyanopyridine (Scheme 316).868 This transformation offers a catalytic alternative to stoichiometric organometallic methods for the synthesis of secondary and tertiary alcohols. An optimized set of reaction conditions consisted of the blue-light irradiation of MeOH solutions containing substrate, [Ir(ppy)2(dtbbpy)]PF6([Ir-3]PF6) photocatalyst, and Et3N reductant. In total, 16 examples of aromatic aldehyde arylation for the synthesis of secondary alcohols, 24 examples of aromatic ketone arylation for the synthesis of tertiary alcohols, and six examples of aromatic imine arylation giving benzhydrylamine products were reported in yields of 23–96%. In general, electron-rich aryl aldehydes were preferable substrates in reactions with 1,4-DCB, with lower efficiency observed for arenes carrying EWGs (316.2 and 316.3, respectively). In contrast, for the reductive arylations of acetophenones, electronic effects on the reaction efficiency were somewhat reversed, with electron-poor acetophenones demonstrating higher efficiency (316.5 and 316.6, respectively). Other cyclic and acyclic aryl alkyl ketones were also compatible substrates, and examples of indanone (316.7), tetralone, and chromanone (316.8) arylation were reported. N-aryl and N-alkyl aldimines and ketimines were also demonstrated (316.9, 316.10).
Scheme 316. Photocatalytic Reductive Arylation of Aldehydes, Ketones, and Imines (Xia, 2017).

A mechanistic proposal involved two reductive quenching cycles of the Ir(III) photocatalyst and therefore a two-photon requirement. In the first cycle, reductive quenching of the Ir(III) photoexcited state (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 with Et3N (Ep/2ox = +0.83 V vs SCE in MeCN)21 yields an Ir(II) complex (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN)68 and Et3N•+ (pKa = 14.7 in MeCN).735 The joint action of these two species mediates reductive PCET activation of the carbonyl/imine substrate to produce a neutral ketyl or α-amino radical respectively and regenerate the ground-state Ir(III) complex. Notably, the reducing Ir(II) complex alone is incapable of thermodynamically favorable ET with aldehyde (e.g., for benzaldehyde, E1/2red = −1.93 V vs SCE in MeCN),21 ketone (e.g., for acetophenone, E1/2red = −2.11 V vs SCE in MeCN),21 or imine (e.g., for N-phenyl benzaldimine, E1/2red = −1.91 V vs SCE in MeCN)21 functional groups, highlighting the concerted PCET involvement. In the second catalytic cycle, an additional reductive quenching by Et3N occurs to generate Ir(II), which mediates SET to the 1,4-DCB (E1/2red = −1.64 V vs SCE in MeCN)21 or 4-cyanopyridine (E1/2red = −1.64 V vs SCE in MeCN)869 component to form the corresponding radical anion. These two reactive species then undergo a radical–radical cross-coupling event, followed by the extrusion of cyanide anion to yield the desired product. The observation of pinacol and triethylamine α-arylation side products supported this proposal.
In 2019, Wang and co-workers reported a photocatalytic heteroarene alkylation reaction in which aldehydes and ketones served as the alkylating agents, affording a variety of alkylated heteroarenes with the net loss of an equivalent of water (Scheme 317).870 The optimal conditions involved the blue-light irradiation of the heteroarene substrate in MeCN solution with 30 equiv of ketone or aldehyde, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) photocatalyst, (TMS)3SiH as a terminal reductant, and TFA as a Brønsted acid additive. Reactions in which acetone served as the alkylating agent were run in acetone solvent. The authors invoked a SCS step to account for the loss of the carbonyl oxygen as water. Though recognized as a key step in the mechanism of class I ribonucleotide reductase enzymes,697 SCS has been more rarely applied in synthesis.610,693,714,777 The authors reported 49 examples of heteroarene alkylation in yields of 33–98%. With 4-hydroxyquinazoline as a model substrate, both cyclic and acyclic ketones proved to be competent substrates, with yield decreasing as chain length increases (317.1, 317.2). A variety of aldehydes also afforded alkylated product in good to excellent yields that were less sensitive to the sterics of aldehyde (317.3). In addition to quinazolines bearing EDGs and EWGs, the heteroarene scope included benzothiazoles, imidazo[1,2-b]pyridazines (317.4), and substituted pyridines, which were all alkylated at the most electrophilic position. Quinolines other than 4-methoxyquinoline were unsuccessful in this alkylation reaction. Upon reaction with 4-hydroxyquinazoline, pivaldehyde gave a mixture of products resulting from the direct addition of the ketyl radical as well as the addition of tert-butyl radical, presumably formed through acyl radical decarbonylation.871
Scheme 317. Heteroarene Alkylation with Ketones and Aldehydes via Reductive PCET and Spin-Centered Shift (Wang, 2019).

To probe the mechanism of this heteroarene alkylation reaction, the authors conducted SV fluorescence quenching experiments and observed that the (TMS)3SiH reductant (Ep/2ox = +0.71 V vs SCE in MeCN)870 is an effective quencher of the photocatalyst excited state (E1/2 *Ir(III)/Ir(II) = +0.89 V vs SCE in MeCN).68 Reduction of the carbonyl component to the corresponding alcohol under the reaction conditions was ruled out by the failure of cyclohexanol to afford the desired product. The authors propose that the excited-state Ir(III) photocatalyst undergoes single-electron reduction by the silane to afford the corresponding Ir(II) complex (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN).68 A ketyl radical is then generated via reductive PCET between the TFA-coordinated carbonyl substrate and the reduced photocatalyst. Addition of the resulting ketyl radical to the protonated heteroarene followed by deprotonation affords an α-amino radical that can expel a molecule of water through SCS, forming a heterobenzylic radical. Subjecting independently prepared closed-shell hydroxyl compound to optimized reaction conditions allowed for efficient product formation, supportive of this path. Hydrogen atom abstraction then yields the closed-shell alkylated heteroarene product. This post-SCS radical was proposed to abstract a H-atom from the solvent given that with deuterated acetone, a near quantitative yield of the product containing deuterium at the benzylic position was produced.
More recently, Dixon, Duarte, and co-workers disclosed a method to prepare bridged 1,3-diazepanes, a motif that appears in the core of various natural products and pharmaceuticals,872 via the coupling of N-aryl imines and 4-substituted quinolines (Scheme 318).873 An initially formed de-aromatized adduct between the imine and quinoline underwent a cyclization through a two-electron manifold to form the bridged bicyclic framework of the observed products. As in previous reports from the group,785,786,812 the imine was activated through reductive PCET to afford a key α-amino radical intermediate. However, with this report, the authors demonstrate that the scope of electrophiles capable of reacting with the α-amino radical intermediate extends beyond the more traditional Michael acceptor-type electrophiles. The optimized conditions involved blue-light irradiation of the N-aryl imine and quinoline substrates in the presence of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) as photocatalyst, 4-aryl-HE 318.A as a terminal reductant, and 30 mol% Zn(NTf2)2 as a catalytic LA additive in DMSO solution. The authors reported 31 examples of 1,3-diazepane synthesis in yields of 40–95% and diastereoselectivity up to 8.0:1. Quinolines bearing branched substituents at the 4-position reacted efficiently (318.1) as did quinolines substituted with potentially sensitive functionality at the 7- and 8-positions, including bromide (318.2) and BP in groups (318.3). Branching at the 4-position led to improvements in the endo-diastereoselectivity of the transformation. The aniline component of the N-aryl imine substrate could accommodate both electron-donating and electron-withdrawing substituents (318.3 and 318.4, respectively). A similarly broad range of electronic profiles was also demonstrated on the aldehyde-derived component of the N-aryl imine (e.g., 318.5). N-Arylquinolinium substrates were tolerated as a quinoline alternative, though with reduced diastereoselectivity (1:1). Across all substrates investigated, near exclusive regioselectivity in favor of radical addition at the 4-position of the quinoline was observed.
Scheme 318. Synthesis of 1,3-Diazepanes from N-Aryl Imines and 4-Substituted Quinolines via Reductive PCET (Dixon and Duarte, 2020).

The authors interrogated the mechanism of this transformation through both experimental and computational approaches. In line with their previous studies, the authors invoked reductive PCET activation of the N-aryl imine substrate as the mechanism for the formation of the α-amino radical. To rule out reductive PCET occurring at the protonated quinoline substrate, a 4-cyclopropyl-substituted quinoline was subjected to the reaction conditions and was observed to afford 1,3-diazepane product without concomitant ring opening, suggesting that a quinoline-based radical is not formed. The reduction potentials of both the quinoline (e.g., for 4-methylquinoline, Ep/2red = −2.14 V vs SCE in DMSO)873 and imine (e.g., for the imine formed from 4-methoxyaniline and 4-fluorobenzaldehyde, Ep/2red = −1.95 V vs SCE in DMSO)873 substrates were found to lay outside of the reach of the reduced-state Ir(II) complex (E1/2 *Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN).68 However, upon protonation, the reduction potentials of these quinoline and imine substrates were calculated to positively shift to Ep/2red = −1.27 V vs SCE in DMSO and Ep/2red = −0.97 V vs SCE in DMSO, respectively, implicating protonation or hydrogen bonding as a key component of substrate activation. Under the reaction conditions, the authors posit that either the Zn(NTf2)2 additive or the HE radical cation can serve in this substrate activation role. The Ir(II) state of the photocatalyst, generated through reductive quenching of the Ir(III) photoexcited-state complex (E1/2 *Ir(III)/Ir(II) = +0.89 V vs SCE in MeCN)68 by the 4-aryl-HE (e.g., for 318.A, E1/2ox(calc) = +0.61 V vs SCE),873 serves as the single-electron reductant during the PCET step. Given the more accessible reduction potential of the protonated N-aryl imine substrate relative to the quinolinium, the authors propose that the reaction involves reductive PCET to the iminium, producing an α-amino radical that then adds to the 4-position of the quinoline. The resulting aromatic radical intermediate (E1/2red(calc) = −0.84 V vs SCE in DMSO)873 is postulated to undergo a second reductive PCET event with the HE radical to afford the closed-shell product. This de-aromatized addition product then undergoes a slightly endergonic (ΔG° = +1.7 kcal mol–1) tautomerization followed by exothermic cyclization (ΔG° = −11.2 kcal mol–1) with the pendant amine to yield the 1,3-diazepane product. The high regioselectivity for addition at C4 was rationalized through an oxidative fragmentation reaction available to the C2-addition product that returns the α-amino radical and quinoline.
The Scheidt group in 2019 presented a method for the β-heteroarylation of arylidene malonates with cyanopyridines and other heterocycles, which proceeds through a reductive PCET mechanism for vinylogous radical generation (Scheme 319).874 This research group had previously published methods for the reductive alkylation875 and reductive annulation,876 of arylidene malonates through a mechanistically distinct pathway under dual photoredox/LA catalysis. A high-throughput experimentation (HTE) approach was used here to discover and optimize reaction conditions for this reductive coupling. Optimized conditions consisted of visible-light irradiation of MeCN solutions of arylidene malonate substrate and cyanoheteroarene coupling partner in the presence of the organic photocatalyst 4DPAIPN(78,877) with Et3N as a terminal reductant. In contrast with their previous work, LA additives, which promoted annulation and alkylation, were found to be deleterious to the desired reactivity in this system. The method presented compares favorably to existing conjugate arylations proceeding through organometallic intermediates in scope and mildness of conditions. A scope of 24 examples with yields of 62–91% was reported. A range of electron-donating, electron-withdrawing, and heteroaromatic (e.g., 319.4) arene substituents were well tolerated on the arylidene malonate substrate. Pyridine (319.1 and 319.2), pyrimidine, pyrrole, and indole nitrile coupling partners (319.3) were competent, but notably 1,4-DCB, often used in similar arylation protocols, was ineffective. An acyclic diester substrate was necessary, as diketone and Meldrum acid derivatives were poorly reactive, and an alkylidene malonate substrate was unreactive due to the more demanding single-electron reduction. The telescoping of prior Knoevenagel condensation and subsequent Krapcho decarboxylation into a single-flask process was performed to demonstrate this method’s synthetic utility.
Scheme 319. Reductive Vinylogous Arylation of Arylidene Malonates with Cyanoheteroarenes (Scheidt, 2019).

The authors propose the following mechanism based on combined spectroscopic and electrochemical observations. Photoexcitation of the 4DPAIPN dye produces an excited state which reacts through a reductive quenching pathway (E1/2 *PC/PC•– = +0.90 V vs SCE in MeCN)79 with Et3N (Ep/2ox = +0.83 V vs SCE in MeCN),21 generating a strongly reducing photocatalyst radical anion (E1/2 PC/PC•– = −1.65 V vs SCE in MeCN)79 and Et3N•+. The aminium radical cation undergoes deprotonation with another equivalent of tertiary amine to give triethylammonium cation and the neutral α-amino radical. This proposal is supported by SV quenching studies, demonstrating that neither the arylidene malonate (Ep/2red = −1.57 V vs SCE in MeCN)874 nor the 4-cyanopyridine (E1/2red = −1.87 V vs SCE in MeCN)878 quench the catalyst fluorescence. Hydrogen bonding of the in situ generated ammonium salt (pKa = 18.5 in MeCN)879 to the malonate substrate facilitates PCET mediated by reduction from the photocatalyst radical anion, leading to regeneration of the photocatalyst ground state and generation of a stabilized vinylogous ketyl radical after PT from the ammonium salt. Alternatively, the authors postulate that the acidic tertiary aminium radical cation also could serve as the acid mediator of this transformation.805 The former proposal is preferred based on the transient nature of the radical cation and the relative concentration of the Et3N leading to rapid deprotonation of this intermediate and generation of the stable salt.
The acidic ammonium salt is also proposed to activate the cyanopyridine component through discrete protonation prior to single-electron reduction in a second separate photoredox cycle. The reduction potential of 4-cyanopyridine was positively shifted in the presence of a stoichiometric amount of Et3NH+Cl– in voltammetry studies (E1/2 = −1.51 V vs SCE in MeCN with addition of 1.0 equiv of Et3NHCl),874 and a distinct change to the absorption profile of cyanopyridine in the presence of Et3NH+Cl– was observed in UV–vis studies. The requisite protonation required to allow favorable ET accounts for the observed lack of reactivity with non-basic 1,4-DCB (E1/2 = −1.67 V vs SCE in MeCN).874 The same voltammetry experiment with addition of Et3NH+Cl– to 1,4-DCB showed only modest change in the reduction potential (E1/2 = −1.60 V vs SCE in MeCN with addition of 1.0 equiv of Et3NH+Cl–).874 The resulting neutral hydrocyanopyridine radical can be considered persistent880 and thus allows for selective radical–radical cross-coupling with the transient β-malonate radical, forming a new C–C bond. Elimination of HCN restores aromaticity and yields the product. The fate of the α-amino radical generated through amine oxidation and deprotonation is unclear. This proposal requires absorption of two photons and is supported by a quantum yield determination experiment (Φ = 0.28), where now the theoretical maximum quantum efficiency is 0.5.
6.1.4. Intra- and Intermolecular C(sp3)–C Bond Formation through Radical–Radical Coupling
Early photocatalytic proton-coupled methods for neutral ketyl radical generation from the groups of Ollivier,777 Yoon,790 Knowles,16 and Rueping,805 required stoichiometric quantities of exogenous amine electron-donors to facilitate overall net-reductive transformations. Later, several research groups demonstrated how reductive neutral ketyl radical generation can be coupled with oxidative radical generation in other substrates (for example, enamines, amines, etc.) to then permit C–C bond formation through radical–radical cross-coupling pathways. In this way, an overall redox-neutral transformation could be envisaged. We now highlight these C–C bond-forming methods. MacMillan and co-workers in 2015 reported a formal β-Mannich reaction of cyclic ketones with imines via the synergistic combination of photoredox catalysis and organocatalysis (Scheme 320).881 Methods for the direct β-functionalization of saturated ketones remain rare,882 in contrast to many enabling ipso- or α-functionalization strategies. Optimized conditions consisted of irradiation of DMPU solutions of ketone and imine substrates in the presence of [Ir(ppy)2(dtbbpy)]PF6 photocatalyst ([Ir-3]PF6), secondary amine organocatalyst (320.A), TFA Brønsted acid catalyst, and DABCO additive as a redox-mediator at room temperature.
Scheme 320. Photocatalytic β-Mannich Reaction of Cyclic Ketones with Preformed Imines (MacMillan, 2015).

*40 mol% Morpholine used in place of azepane. **20 mol% pyrrolidine used in place of azepane.
In total, 18 examples of γ-aminoketone synthesis were demonstrated in yields of 16–90%. Various imines derived from both ketones (320.1) and aldehydes (320.2) furnished the desired γ-aminoketone products with high levels of efficiency. N-aryl (320.2) and N-alkylimines (320.3) were both competent partners. Various cyclic ketones were demonstrated substrates, including a cyclopentanone (320.4) and substituted cyclohexanones (320.5). Substitution at the ketone α-position led to poor reactivity, likely due to slower rates of enamine formation. In all cases, low diastereoselectivity was observed, typically between 1:1 and 1.5:1. A three-component reaction where the imine was formed in situ, subjecting cyclohexanone, p-anisidine and benzaldehyde to the optimized reaction conditions, gave the β-Mannich product (320.2) in a 70% yield, comparable to a reaction with preformed imine (74%). For all substrates, complete selectivity was observed for the β-Mannich product over the α-Mannich product, highlighting the capability of photoredox catalysis to selectively partition transformations to non-classical pathways.
A proposed mechanism involved enamine formation through condensation of the amine organocatalyst and cyclic ketone substrate followed by relay single-electron oxidation and deprotonation to afford a transient C-centered radical at the β-position. The iminium component is reduced to a persistent α-amino radical and the two open-shell species undergo selective radical–radical coupling to form the product. The lack of observed reaction diastereoselectivity indicated this mode of bond formation. The use of azepane (320.A) gave more readily oxidized enamines than those derived from piperidine, another common organocatalyst for enamine formation. Visible-light irradiation of the Ir(III) photocatalyst gave an excited state (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 which underwent reductive quenching with the DABCO additive (Ep/2ox = +0.69 V vs SCE in MeCN)881 to yield a reduced-state Ir(II) complex and DABCO radical cation. DABCO radical cation oxidizes the enamine (e.g., for the enamine formed between azepane and cyclohexanone, Epox = +0.39 V vs SCE in MeCN)883 to its corresponding radical cation, which acidifies the β-protons toward subsequent deprotonation by DABCO. While DABCO serves a catalytic role in these elementary steps, improved reaction rates were offered through use of a stoichiometric quantity. SV quenching studies support this proposal, which showed that both DABCO and preformed enamine quench the excited state of this photocatalyst, but preformed imine did not. Reduced yields were observed in the absence of DABCO, highlighting its important role as a redox mediator. The Ir(II) complex generated through reductive catalyst quenching (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN)68 alone is not sufficiently reducing to interact with the neutral imine coupling partner. However, TFA acts to protonate the imine prior to ET, which significantly improves the thermodynamic driving force in this step, to give a neutral α-amino radical and return the ground-state Ir(III) complex. TFA is also proposed to assist in enamine condensation.
The MacMillan group has also demonstrated related synergistic photoredox catalysis and organocatalysis for the β-arylation,884 β-alkylation,885 and β-crossed aldol886 reactions of ketones and aldehydes as well as for the dynamic kinetic resolution of β-functionalized ketones in combination with enzymatic ketone reduction, the latter study in collaboration with the Hyster group.887 In their crossed-aldol report, the possibility of PCET activation of the ketone coupling partner was considered but was shown not to be involved in the mechanism of reaction.
Zhu and co-workers later hypothesized that incorporating a ketone (acting as an electron-acceptor) and an amine (acting as an electron-donor) into the same molecule could potentially generate a biradical intermediate and effect a cyclization via radical–radical coupling to yield hydroxylated cyclic amines (Scheme 321).888 Optimized reaction conditions were developed consisting of visible-light irradiation of a DMF solution of N-benzyl-aminoketone substrates in the presence of [Ir(ppy)2(dtbbpy)]PF6 photocatalyst ([Ir-3]PF6), methylthioglycolate as a HAT co-catalyst, and K2HPO4 as a Brønsted acid co-catalyst. Under these conditions, a total of 47 examples of cyclic amine synthesis were demonstrated in yields of 51–95%. Regioselective cyclization occurred exclusively between the ketone functional group and the benzylic position of the amine substituent, yielding 2-aryl-3-hydroxylamines. Diastereoselectivity in cyclization was typically modest to good for formation of the trans-1,2-diaryl products. Impressively, the method provided access to piperidine (321.5), pyrrolidine (321.1–321.4), and azetidine rings (321.6) through the reaction of γ-, β-, and α-aminoketones, respectively. Also, in a class of 2-aminoacetophenone-derived substrates, indole formation was possible through redox-neutral cyclization and in situ dehydration under the reaction conditions.
Scheme 321. Redox-Neutral Cyclization of Amino-ketones for the Synthesis of 3-Hydroxy Azetidines, Pyrrolidines, and Piperidines (Zhu, 2016).

In the authors’ mechanistic proposal, the photoexcited-state Ir(III) complex acts as a photooxidant in a PCET activation of the methyl thioglycolate co-catalyst, generating thiyl radical and reduced state Ir(II). An oxidative-PCET mechanism, rather than stepwise ET/PT or PT/ET mechanisms for thiyl radical generation is invoked in earlier work by MacMillan,680,686 noting that the potential of the excited-state photooxidant (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 is insufficient to oxidize the thiol directly (e.g., for Cys, E1/2ox = +0.85 V vs SCE in H2O),681 and that the weakly acidic thiol (e.g., for methyl thioglycolate, pKa = 7.9 in H2O)682 would not generate a substantial concentration of thiolate in the presence of this base. We also note that this Ir dye is insufficiently oxidizing in its photoexcited state to drive efficient ET to the arylamine group (e.g., for dimethylaniline, Ep/2ox = +0.74 V vs SCE in MeCN),21 thus oxidation likely occurs through PCET at the thiol. The resulting thiyl radical (e.g., for methyl thioglycolate, S–H BDFEcalc = 87.2 kcal mol–1)683 engages in a HAT step at the benzylic position of the benzylamine moiety, yielding a stabilized α-amino benzylic radical intermediate and regenerating the thiol co-catalyst (for more examples of S–H bond PCET mediating substrate HAT elementary steps, see section 4.4.1). This strategy is similar to that used in earlier studies from MacMillan and co-workers for radical generation in benzylic ethers.680,686 Concurrently, reductive PCET activation of the ketone functional group generates a neutral ketyl radical intermediate. The Ir(II) complex here is not sufficiently reducing (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN)68 to drive reduction of the aryl ketone function group (e.g., for acetophenone E1/2red = −2.11 V vs SCE in MeCN).21 Following proposed biradical formation, a radical–radical coupling ensues to yield the cyclic product. The observed diastereoselectivity is due to steric repulsion between the two aryl groups across the forming C–C bond placing them into a staggered conformation.
However, we note that two inconsistencies arise in this mechanistic proposal. First, it is unclear what governs biradical formation on the same molecule. An HAT step mediating benzylic radical generation and a reductive PCET step mediating neutral ketyl radical generation are decoupled and thus could presumably occur on two different substrate molecules. Second, in four examples, the redox-neutral coupling of aliphatic ketones is achieved in excellent yields (e.g., 321.4). This is inconsistent with a reductive PCET mechanism for neutral ketyl radical generation, as methodology employing this iridium dye is limited to aryl ketone substrates with intrinsically less-negative reduction potentials. Aliphatic ketones are very demanding in their single-electron reduction (e.g., for cyclohexanone, E1/2red = −2.33 V vs SCE in MeCN).21
In 2018, Wang and co-workers reported a photocatalytic, redox-neutral cross-coupling reaction between N-arylamines and aldehydes, ketones, or imines, demonstrating a valuable strategy for the synthesis of 1,2-amino alcohols and vicinal diamines respectively (Scheme 322).889 The optimal reaction conditions involved white light irradiation of substrate solutions in the presence of [Ru(bpy)3]Cl2·6H2O ([Ru-1]) photocatalyst, oxalic acid co-catalyst, DABCO, and LiBF4. The authors reported 40 examples of this cross-coupling reaction with yields of 27–83%. With respect to N-arylamine scope, a wide range of N-aryl (e.g., 322.3 and 322.4) and N-heteroaryl (322.1) tetrahydroisoquinolines bearing substituents with varying electronic properties performed well in this protocol. Moreover, secondary amines and arylglycine ester substrates also underwent efficient coupling (322.2). With respect to aldehyde scope, benzaldehydes bearing various EWGs afforded the desired products in high yields. Ketones (322.3) and imines (322.4) were also competent substrates in this transformation. This protocol was successful in the late-stage functionalization of a derivative of the antidepressant fluoxetine (322.5).
Scheme 322. Photocatalytic Intermolecular Cross-Coupling Reaction between N-Arylamines and Aldehydes, Ketones, and Imines for the Synthesis of 1,2-Amino Alcohols and Vicinal Diamines (Wang, 2018).

The authors proposed that the reaction mechanism involves the formation of a neutral ketyl or α-amino radical intermediate from the carbonyl/imine functional groups through concerted reductive PCET mediated by the joint action of the Ru(II) photoreductant (E1/2 Ru(III)/*Ru(II)= −0.81 V vs SCE in MeCN)64 and the in situ generated DABCO oxalate salt. This proposal was evidenced by SV luminescence quenching experiments, wherein quenching was observed only in the presence of all three of the substrate benzaldehyde, DABCO, and oxalic acid; omitting one of these components resulted in no luminescence quenching. Following reductive PCET, the oxidized Ru(III) complex (E1/2 Ru(III)/Ru(II)= +1.26 V vs SCE in MeCN)64 then reacts with the N-arylamine substrate (e.g., for N-Ph tetrahydroisoquinoline, Ep/2ox = +0.43 V vs Fc+/Fc in MeCN)890 to the corresponding aminium radical cation and regenerates ground-state Ru(II). The aminium radical cation is deprotonated by DABCO, resulting in an α-amino radical, which rapidly combines with the neutral radical generated through reductive PCET in a radical–radical cross-coupling step to furnish the desired product. The authors note that on thermodynamic grounds, the DABCO radical cation (E1/2ox = +0.69 V vs SCE in MeCN),881 which could form through reductive photocatalyst quenching or through reaction with transient Ru(III) with this Ru(II) dye, cannot oxidize the N-arylamine partner, therefore excluding the possibility of this species mediating ET as was proposed in work from MacMillan and co-workers as discussed above.881
Cho and co-workers recently reported a reductive coupling between N-benzylidene-[1,1′-biphenyl]-2-amines and N,N-dicyclohexylmethylamine (Cy2NMe) (Scheme 323).891 The authors noted the importance of the phenyl substituent in the aniline moiety on the imine to promote this transformation, as simple N-benzylideneaniline did not furnish the desired product under several conditions. Cho suggested that the presence of this additional phenyl group stabilizes the α-amino radical intermediate and modulates its reactivity. Optimization of this coupling protocol revealed that [Ir(ppy)2dtbbpy)]PF6([Ir-3]PF6) was the most efficient photocatalyst. Both a major cross-coupling product and a minor imine-derived homo-coupling product were observed. Optimal selectivity for the cross-coupling product was achieved when 2 equiv of Cy2NMe (one equivalent acting as coupling partner and the other as a sacrificial reductant) were used. The product selectivity is also sensitive to the choice of solvent, as DMF was found to favor cross-coupling while MeOH favored homo-coupling. This work included 22 examples of the synthesis of 1,2-diamines in yields of 36–96%. All examples show high diastereoselectivity, which was attributed to the bulky substitution pattern of the substrates. For the cross-coupling reaction between the aryl aldimine substrate and Cy2NMe, substrates with both electron-donating and electron-withdrawing substituents at any position on the aromatic ring were well-tolerated (323.1–323.4). Electron density on the N-aryl moiety of the imine also did not have a large impact on the reaction efficiency. Other aliphatic amines such as TMEDA, Et3N, and DIPEA were also assessed; however, a complex mixture of products resulted. Several examples for the homo-coupling reactions of these imine substrates were also reported; these reactions proceeded smoothly regardless of electron density or position of the substituents on the benzylidene moiety.
Scheme 323. Synthesis of Vicinal Diamines via Photocatalytic Reductive Coupling of N-Aryl Aldimines and N,N-Dicyclohexlmethylamine (Cho, 2020).

The proposed mechanism involves the reductive quenching of the excited-state Ir(III) photocatalyst by Cy2NMe to generate the Ir(II) complex and Cy2NMe•+. This is supported by SV experiments, in which the emission intensity of the excited-state photocatalyst significantly decreased with increasing concentrations of Cy2NMe. The luminescence quenching was much less affected by the concentration of the imine substrate. Reductive PCET activation of imine via the joint action of the resulting Ir(II) complex and Cy2MeN•+ produces two α-amino radical intermediates, which rapidly undergo radical–radical coupling to furnish the desired unsymmetric vicinal diamine product. When MeOH was used as the solvent, it serves as the proton donor in the reductive PCET step instead of Cy2MeN•+, preventing the formation of α-amino radical from Cy2NMe and instead promoting the homo-coupling of the imine-derived α-amino radical to selectively afford the symmetric diamine product.
In 2017, Yu and co-worker disclosed a photocatalytic protocol for the radical alkylation of both aldimines and ketimines with 4-alkyl-1,4-dihydropyridines (4-alkyl-DHPs) as precursors to alkyl radicals (Scheme 324).390 Optimal conditions consisted of irradiation of the imine substrate and 4-alkyl-DHP reagent in the presence of [Ir(ppy)2(dtbbpy)]PF6([Ir-3]PF6) photocatalyst and PhCO2H co-catalyst in MeCN under white light irradiation at room temperature. A total of 57 examples of amine products were reported in yields ranging from 50% to 98%. N-aryl aldimines with electron-neutral and electron-donating substituents on the aniline moiety were successfully alkylated in excellent yields, while electron-deficient N-aryl aldimines were less reactive (324.1–324.3). N-aryl aldimines bearing ortho-substituents on the aniline ring also underwent alkylation, albeit with lower efficiency compared with meta- and para-substituted substrates (324.4). In addition, aldimines derived from differently functionalized benzaldehydes bearing hydroxyl, ester, amide, cyanide, and heterocycles were also successfully coupled with aniline (70–98%) (324.6–324.8). The authors also demonstrated that various ketimines derived from aryl ketones with aniline were also amenable substrates in this transformation, affording structurally diverse and sterically hindered amines in good to excellent yields (64–97%) (324.10–324.12). It is worth noting that an alternative set of conditions, which consisted of [Ru(bpy)3]Cl2 ([Ru-1]Cl2) and TFA in MeOH solution, was shown to be optimal for a number of ketimine substrates. The scope further included amino-acid-derived (324.13) and isatin-derived ketimines (324.14). Gram-scale synthesis was demonstrated on the latter substrate with only slight diminution in yield. Finally, the authors showed that a number of 4-alkyl-1,4-DHPs bearing isopropyl, benzhydryl, and benzyl groups with electron-donating and electron-withdrawing substituents at different positions of the ring also showed good reactivity (324.5, 324.9, 324.15).
Scheme 324. Photocatalytic Radical Alkylation of Aldimines/Ketimines with 4-Alkyl-1,4-dihydropyridines (Yu, 2017).

Several control reactions and spectroscopic studies were conducted to gain insight into the reaction mechanism. Trace amount of the desired amine product was observed when TEMPO was introduced to the reaction and the TEMPO-adduct of the DHP component was also detected by HRMS, suggesting the involvement of radical processes. SV quenching experiments revealed that 4-benzyl-DHP was able to quench the excited-state of the Ir(III)photocatalyst, while neutral imine showed much less pronounced quenching. With these observations, the authors proposed the mechanism of this transformation to begin with a reductive quenching of the excited-state photocatalyst by 4-alkyl-DHP to generate a reduced transition-metal complex (Ir(II) or Ru(I)) and the corresponding alkyl radical. From here, two pathways for C–C bond formation are considered: (i) addition of the alkyl radical to the neutral, closed-shell imine to afford an N-centered radical, which then undergoes ET and PT with the reduced state photocatalyst and acid, respectively, to afford the desired amine product; and (ii) reduction of the imine via a concerted PCET mechanism through the combination of the reducing state of the photocatalyst and acid co-catalyst, generating the corresponding α-amino alkyl radical. This proposal was supported by the observation of the aza-pinacol dimer of this α-aminoalkyl radical in the reaction mixture. The radical–radical coupling between the α-aminoalkyl radical and the alkyl radical generated from 4-alkyl-DHP then provided the desired amine product. These dual mechanistic proposals for imine alkylation were also noted by Molander and co-workers, where alkyl silicates were used as alkyl radical precursors through single-electron oxidation.892 This reaction is carried out in the absence of any acidic or hydrogen-bonding additive. In this work, generally non-basic N-sulfonyl imine substrates were used, which have accessible reduction potentials not requiring coupling with PT to facilitate ET, yielding instead the corresponding radical anion (e.g., for N-methanesulfonyl p-tolualdehyde imine, Ep/2red = −1.39 V vs SCE in MeCN).892
In 2019, the Yu group also reported a catalyst-free visible-light-induced radical acylation of aldimines with α-ketoacids enabled by the generation of an EDA complex (Scheme 325).893 This catalyst-free protocol was carried out with 1 equiv of imine and 1.5 equiv of α-ketoacid in CH2Cl2 solvent under irradiation with blue light. Under these reaction conditions, 30 examples of the desired α-amino ketone were synthesized, with yields ranging from 50% to 90%. A variety of aldimines were tolerated, including those derived from both electron-rich and electron-deficient benzaldehydes (325.1–325.5). With regard to the α-ketoacid scope, both aromatic (325.6) and aliphatic substrates (325.7) were tolerated. Notably, α-amino amides could also be synthesized using this decarboxylative acylation procedure (325.8). These reactions could be scaled up to 5 mmol without significant diminution of yield.
Scheme 325. Photoinduced Acylation of Imines with α-Ketoacids (Yu, 2019).

To further understand the elementary steps involved in this transformation, the authors conducted a series of mechanistic experiments. Conducting the reaction in the presence of TEMPO led to the formation of TEMPO-trapping adducts, providing support for a radical mechanism. No reaction was observed in the absence of irradiation, even at elevated temperatures (100 °C). Mixing the imine and α-ketoacid led to a bathochromic shift and appearance of a charge-transfer band in the UV–vis spectrum. The stoichiometry of the complex was confirmed to be 1:1 using Job’s method.823 Based on these experiments, the authors propose that the reaction proceeds via formation of an EDA complex. Irradiation of this association complex leads to proton-coupled SET and generation of a radical pair. Decarboxylation furnished an acyl radical which undergoes radical–radical coupling with the α-amino radical to furnish the desired α-amino ketone product. The quantum yield of the reaction was found to be Φ = 0.08, suggesting that the reaction proceeds through a radical–radical coupling rather than a radical chain mechanism.
In 2019, Lu, Gong, and co-workers reported a photocatalytic, redox-neutral net formylation reaction of fluoroalkyl imines, with 1,3-dioxolane serving as a formyl equivalent (Scheme 326).894 The optimal reaction conditions require the use of 2,3-butanedione and N-hydroxysuccinimide (NHS), which were found to be the most efficient organic photocatalyst and HAT reagent for this transformation, respectively. In total, 29 examples were reported, affording various α-amino 1,3-dioxolanes in yields of 51–96%. A series of primary, secondary, and tertiary N-alkyl and N-aryl groups on the imine component were well tolerated (326.1–326.4), although imines with more hindered substituents on the nitrogen underwent slower reactions than those with the linear substituents. A number of fluoroalkyl groups too were demonstrated, including CF3, CF2H, and C2F5. Ketimines prepared from ethyl 3,3,3-trifluoropyruvate were also highly efficient reaction partners, providing access to fluoroalkyl AA derivatives with a quaternary carbon center (326.5). Finally, 2-ethyl-1,3-dioxolane and THF were also shown to engage in productive HAT and imine addition (326.6, 326.7).
Scheme 326. Photocatalytic Synthesis of Masked Fluoroalkyl Amino Aldehydes from 1,3-Dioxoline and Fluoroalkyl Aldimines/Ketimines (Lu and Gong, 2019).

The proposed mechanism involves initial complexation through hydrogen bonding between 2,3-butanedione and NHS. Excitation with blue light generates a triplet ketyl diradical of the diketone, which undergoes either a HAT or PCET event to generate an N-oxy radical from NHS. This electrophilic alkoxy radical then abstracts a H-atom from 1,3-dioxolane, affording a nucleophilic 1,3-dioxolan-2-yl radical that undergoes addition into the imine to afford an N-centered radical adduct. To furnish the desired product, the resulting N-centered radical could feasibly undergo either a PCET step with the reduced form of the photocatalyst, regenerating butanedione, or abstract a H-atom from another NHS molecule, producing an N-oxy radical intermediate which could establish a chain process. The mechanism for this C–C bond-forming step is proposed to involve imine addition rather than radical–radical coupling as invoked in previous work.686,892,895 This proposal is supported by the absence of a reductive aza-pinacol-type homo-coupling product during the course of the reaction. Furthermore, the authors suggested that the rate-determining step changes over the course of the reaction. At the beginning of the reaction when imine concentration is high, H-atom abstraction from 1,3-dioxolane is rate-limiting. However, at the later stages of the reaction when imine concentration is low, the radical addition step becomes rate-limiting.
Zhong and Zeng recently presented a synthesis of vicinal amino alcohols via the decarboxylative coupling of N-arylglycines with aldehydes or ketones (Scheme 327).896 The optimized reaction conditions consisted of blue LED irradiation of aqueous solutions of these substrates in the presence of [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) photocatalyst at room temperature. In this work, 40 examples of this decarboxylative coupling were reported in yields of 45–98%. Aryl and heteroaryl aldehyde coupling partners were the most efficient substrate class in this work (327.1–327.6), and variation of the electronic and steric properties of the arene through substitution at the ortho-, meta-, and para-positions had minimal impact on the reaction yields. Chloride, bromide (327.3), and unprotected phenol functionality were tolerated. Furan (327.6) and thiophene aldehydes were also efficiently transformed to the corresponding amino alcohol products. Additionally, four examples of alkyl-aldehyde coupling were included, proceeding in moderate yields (e.g., 327.7). Aryl ketone substrates underwent reaction with moderate yields (e.g., 327.8). Finally, N-phenylalanine, N-phenylvaline, and N-phenylproline were demonstrated in this cross-coupling reaction with benzaldehyde to give α-substituted vicinal amino alcohols (327.9), but with poor diastereocontrol.
Scheme 327. Photocatalytic Decarboxylative Synthesis of Vicinal Amino Alcohols (Zeng and Zhong, 2020).

A mechanism was proposed involving visible-light excitation of the Ir photocatalyst (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 and subsequent reductive quenching with the glycine carboxylate (Eonsetox = +0.90 V vs SCE in aq. NaOH (pH 13)),897 initially forming the acyloxy radical which rapidly extrudes CO2 and generates a stabilized α-amino radical. This process generates a reduced-state Ir(II) complex (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN),68 which alone is not sufficiently reducing to permit SET with carbonyl substrates (e.g., for 4-benzaldehyde, Ep/2red = −1.93 V vs SCE in MeCN).21 However, through hydrogen bonding with either water or the acidic glycine substrate, a PCET mechanism is invoked to account for the otherwise significantly endergonic ET and generate a neutral ketyl radical intermediate. If the glycine component was omitted and Et3N added instead, pinacol reductive homo-coupling products were observed, which is indicative of ketyl radical generation in this manner.805 Various exogenous acidic additives were screened but none was found to be beneficial. Finally, radical–radical coupling between the α-amino radical and the neutral ketyl radical yields the product.
It is noteworthy that alkyl aldehydes were competent substrates in this work. These are very demanding in their direct reduction (e.g., for 3-methylbutyraldehyde, Ep/2red = −2.24 V vs SCE in MeCN).21 Even with PCET to facilitate this step, there are only a few examples of successful reactivity with this substrate class. For example, Jamison has recently reported the reductive cross-coupling of alkyl aldehydes and ketones with styrenes involving reductive PCET activation of the carbonyl component, but employing an extremely reducing p-terphenyl photocatalyst (E1/2 PC/PC•– = −2.63 V vs SCE in DMF).825 It is unclear if the Ir(III) dye employed in this work has sufficient driving force in its Ir(II) state to permit this demanding reductive radical generation and therefore other pathways may be operative in this subset of substrates.
Recently, Sun and co-workers have disclosed a photocatalytic Barbier-type preparation of propargylic alcohols through the reductive coupling of acetylenic ketones and potassium benzyltrifluoroborate proceeding through a proposed PCET mechanism for neutral ketyl radical generation (Scheme 328).898 The use of potassium trifluoroborate salts for oxidative alkyl radical generation in photoredox reactions has extensive precedent.899−902 To effect this transformation, acetone solutions of ynone substrate and benzyl trifluoroborate reagent in the presence of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]) photocatalyst and NH4+AcO– Brønsted acid additive were irradiated with visible light. Significantly reduced reactivity was observed in the absence of the acidic additive. A scope of 25 examples of propargylic alcohol synthesis was defined with yields of 24–73%. Those ynone substrates carrying aryl (e.g., 328.1) or heteroaryl (e.g., 328.2) moieties at the carbonyl group were most reactive in this work, though alkyl substituents could be tolerated with lower product yields (e.g., 328.3). The electronic nature of the aryl group in this position impacted reactivity, with electron-rich arenes most well suited. Similarly, reactivity was optimal when the alkyne was conjugated to an aryl group but was attenuated when an alkyl ynone was used (e.g., 328.5, 328.6). In this position, the electronic properties of the arene group exerted little impact on the observed yields. Notably, exclusive 1,2-addition is observed in these α,β-unsaturated ketones, which is contrary to the usually observed 1,4-addition products when in the presence of organic radicals.103 This overriding selectivity preference can be understood when considering the mechanism of the transformation.
Scheme 328. Intermolecular Reductive Coupling of Acetylenic Ketones and Potassium Benzyltrifluoroborate (Sun, 2020).

The authors propose that an initial reductive quenching of the excited-state photocatalyst (E1/2 *Ir(III)/Ir(II) = +0.89 V vs SCE in MeCN)68 occurs with the potassium trifluoroborate reagent (e.g., for potassium benzyltrifluoroborate, Ep/2 = +0.67 V vs Fc+/Fc in MeCN),899 to extrude BF3 and generate the benzylic radical. This proposal was based on steady-state SV experiments, which indicate that the potassium trifluoroborate quenches the excited state of the photocatalyst in preference to the ketone in the presence or absence of NH4+AcO–. Then, the reduced state Ir(II) complex (E1/2 Ir(III)/Ir(II) = −1.37 V vs SCE in MeCN)68 and the NH4+AcO– additive mediate reductive PCET of the ynone substrate through hydrogen-bond chelation to generate a neutral ketyl radical intermediate. The reduction potential of the ketone substrate in the absence of the acidic additive was too negative (e.g., for ynone substrate leading to product 328.1, Ep/2red = −1.70 V vs SCE in MeCN)898 to enable a discrete ET step by this Ir dye. This PCET event regenerates the ground-state Ir(III) complex for catalyst turnover. The appearance of trace amounts of pinacol and 1,2-diphenylethane side products in reaction mixtures was highlighted as evidence for the discrete formation of both benzyl and ketyl radical intermediates. Finally, a radical–radical cross-coupling step yields the 1,2-addition product. The coupling of the oxidative generation of the benzylic radical intermediate to the reductive generation of the neutral ketyl radical intermediate is what apparently governs the 1,2-selectivity observed through this radical–radical coupling step over an alternative mechanism of 1,4-addition through radical addition to a closed-shell α,β-unsaturated ketone.
6.1.5. C–H Bond Formation through Reduction to Alcohols and Amines, and Reductive Amination of Carbonyl Compounds
We describe here a number of processes for the reduction of carbonyl compounds and imines to alcohols and amines, respectively, proceeding under photocatalytic or electrochemical activation. Notably, PCET has also been invoked as a key mechanism for the reduction of carbonyl compounds by SmI2–H2O complexes, as studied by Flowers and co-workers.903 The hydrogenation of imines provides access to amines that have value as pharmaceuticals, agrochemicals, and natural products. However, many traditional hydrogenation methods exhibit limited chemoselectivity for imines in the presence of other functional groups such as olefins and carbonyls. In 2018, Polyzos and co-workers reported a photoredox-transfer hydrogenation method that takes advantage of the facile single-electron reduction of diaryl ketimines to achieve chemoselectivity for imines (Scheme 329).838 The optimized conditions included irradiation of the imine substrate, [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) photocatalyst, and 5.0 equiv of Et3N as a terminal reductant and H-atom donor in MeCN at room temperature. A notable drop in reaction efficiency was observed upon reducing the loading of Et3N. Under these conditions, 31 examples of imine-transfer hydrogenation were reported in yields of 71–99%. Reaction efficiency was largely agnostic to the electron character of aryl group of N-aryl imines, with electron-rich and electron-poor substrates giving similarly excellent yields (329.1, 329.3). Substrates bearing ketone (329.2) and nitrile (329.4) functionality selectively afforded products resulting from imine hydrogenation, demonstrating the chemoselectivity of this protocol. Pyridine-containing imines also underwent efficient hydrogenation (329.3). In addition to N-aryl imines, N-alkyl imines such as N-benzyl (329.6) and N-hexyl were also tolerated in this protocol. The authors note that this reaction could be efficiently conducted in a flow reactor, resulting in a significant reduction in reaction time (e.g., from 3 h to 12 min).
Scheme 329. Photocatalytic Transfer Hydrogenation of Diarylimines (Polyzos, 2018).

The authors conducted steady-state SV, TA, and isotopic labeling experiments to probe the mechanism of this transformation. SV luminescence quenching studies demonstrated that both Et3N and the diarylimine substrate quench the excited-state photocatalyst; however, the latter is much less competitive as a quencher. TA experiments of the photocatalyst in the presence of Et3N resulted in a new persistent absorption feature at 380 nm, which was assigned to the reduced Ir(II) photocatalyst. This suggests that Et3N reduces the excited-state photocatalyst. As a further probe the role of Et3N in the reaction, use of perdeuterated Et3N-d15 led to 75% deuterium incorporation at the benzhydryl position of the amine, while use of MeCN-d3 led to no deuterium incorporation, establishing Et3N as the ultimate source of an H-atom. These deuterium labeling experiments thus supported a HAT termination pathway as opposed to an alternative ET/PT pathway involving solvent. Based on these mechanistic experiments, the authors propose a reaction mechanism involving photoexcited Ir(III)-mediated single-electron oxidation (E1/2 *Ir(III)/Ir(II) = +0.66 V vs SCE in MeCN)68 of Et3N (Ep/2 = +0.83 V vs SCE in MeCN)21 to generate Et3N•+ and an Ir(II) complex. This reduced state photocatalyst (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN)68 then donates an electron to the diarylimine substrate (e.g., for N-phenyl benzophenone imine, Epred = −1.88 V vs SCE in MeCN)808 to generate the corresponding iminyl radical anion which after protonation gives an α-amino radical. This observed offset in reduction potential between Ir(II) and substrate may imply some degree of PCET assistance in this ET step. Tertiary amine radical cations have been invoked in this role in several other works (see section 6.1.2).804,805 Finally, HAT from Et3N•+ (α-C–H BDFE = 42 kcal mol–1)735 affords the amine product.
A curious observation made by these researchers in this work led to the discovery and further development of several other synthetic methods extending more generally beyond transfer hydrogenation. The successful reactivity of N-alkyl imines (e.g., 329.6) is somewhat surprising given that they are significantly more challenging to reduce (e.g., N-benzyl benzophenone imine (Epred = −2.18 V vs SCE in MeCN) than N-aryl imines.904 This is some 670 mV beyond the capability of the Ir(II) complex generated through reductive quenching of the photoexcited state of [Ir-3]PF6 (E1/2 Ir(III)/Ir(II) = −1.51 V vs SCE in MeCN).68 While concerted PCET is a mechanism by which otherwise endergonic ETs can proceed in hydrogen bond acceptor substrates (such as imines), these authors note of other supposedly endergonic ETs being successfully driven by this dye, for which there can be no hydrogen-bonding contribution. For example, in a 2017 report from Martin, Juliá-Hernández, and co-workers demonstrated the visible-light promoted atom-transfer radical cyclization reactions of unactivated alkyl iodide-tethered alkynes proceeding in the presence of [Ir-3]PF6.905 The alkyl iodide substrates featured in this work are extremely difficult to reduce (Ep/2red ≤ – 2.5 V vs SCE in MeCN)905 and have no ability to hydrogen bond to any donor to assist in the SET. These observations led Connell, Polyzos, and Francis to further investigate the combination of [Ir(ppy)2(dtbbpy)]PF6 photocatalyst ([Ir-3]PF6) and tertiary amine reductants to better understand how this reagent combination can facilitate such endergonic ETs.904
This work identified that on visible-light irradiation of this Ir(III) complex in the presence of Et3N reductant, a ligand modification ensues, resulting in partial reduction of the dtbbpy ligand (L2-type) to a mono-dihydropyridine ligand (LX-type) (Scheme 330). The resulting Ir(ppy)2(H3dtbbpy) complex also absorbs visible light and produces a reducing photoexcited state (E1/2 Ir(IV)/*Ir(III) = −1.70 V vs SCE in MeCN),904 which is a stronger photoreductant than the parent Ir(III) photocatalyst ([Ir-3]PF6) from which it is derived (E1/2 Ir(IV)/*Ir(III) = −0.96 V vs SCE in MeCN).68 The photoexcited state of Ir(ppy)2(H3dtbbpy) is the proposed in situ generated photoreductant responsible for the observed anomalous reactivity with energy-demanding substrates such as N-alkyl imines838 and unactivated alkyl iodides.905 This ligand-reduced photocatalyst is proposed to react through oxidative quenching with the substrate to generate an Ir(IV) complex (E1/2 Ir(IV)/Ir(III) = +0.88 V vs SCE in MeCN).904 The Ir(IV) state of the Ir(ppy)2(H3dtbbpy)complex and the Ir(II) state of the parent [Ir(ppy)2(dtbbpy)]PF6 complex (generated through reductive quenching with Et3N) undergo a comproportionation reaction to return the Ir(III) ground state of both complexes. Researchers also suggest that the H3dtbbpy can reform the dtbbpy ligand through a HAT event with substrate, allowing the complex to reversibly partition between the two forms. This permits a tandem photoredox mechanism to develop where in the presence of Et3N reductant, a [Ir(ppy)2(dtbbpy)]PF6 reductive quenching cycle and a Ir(ppy)2(H3dtbbpy) oxidative quenching cycle can mutually operate to drive demanding single-electron reductions through a two-photon process (Scheme 329). These researchers showed that this tandem photoredox catalysis can successfully drive the demanding single-electron reduction of aryl halides (e.g., methyl 4-chlorobenzoate, Epred = −1.98 V vs SCE in MeCN)904 and alkyl iodides (e.g., cyclohexyl iodide, Epred = −2.15 V vs SCE in MeCN)904 for substrate hydrodehalogenation. This tandem photoredox catalysis also allowed for the development of a carbonylative amidation of aryl and alkyl halides, and of a reduction/reductive functionalization of olefins, in more recent reports from this group.906,907
Scheme 330. A Tandem Photoredox Mechanism Operates upon Irradiation of [Ir-3]PF6 in the Presence of Et3N Reductant, Permitting the In Situ Generation of the Potent Photoreductant [Ir(ppy)2(H3dtbbpy)]PF6 for the Activation of Energy-Demanding Substrates (Connell, Polyzos, and Francis, 2019).

In 2018, Guo and Wenger reported the visible-light-driven photocatalytic reductive amination of carbonyl compounds (Scheme 331).908 Visible-light irradiation of a MeOH solution containing carbonyl and amine substrates, [Ru(bpy)3]Cl2 ([Ru-1]Cl2) photocatalyst, and ascorbic acid (AscH2) and MPA as HAT reagents led to formation of the desired reductive amination products, with 25 examples documented in 32–97% yields. With regard to the amine scope, electron-rich (331.2), electron-poor (331.3), and halogenated (331.4) anilines were well tolerated. Additionally, both primary (331.5, 331.6) and secondary (331.7) aliphatic amines were competent substrates under the reaction conditions. With regard to the carbonyl scope, aliphatic (331.1–331.7) and aromatic aldehydes (331.9) as well as ketones (331.8) reacted to furnish the desired reductive amination products. Notably, the reaction afforded complete retention of stereochemistry as demonstrated by the reaction of pivaldehyde with L-tryptophan methyl ester (331.6).
Scheme 331. Reductive Amination Enabled by Photoredox Catalysis and Polarity-Matched Hydrogen Atom Transfer (Guo and Wenger, 2018).

Under these reaction conditions, the carbonyl and amine substrates are in equilibrium with the imine and protonated iminium cation. The researchers postulated that AscH2-promoted reductive quenching of the excited state of the Ru(II) photocatalyst generates the reducing Ru(I) state, which can facilitate ET to the iminium cation to generate an α-aminoalkyl radical. Hydrogen atom transfer to this radical then furnishes the reductive amination product. During their optimization studies with isobutyraldehyde and aniline, the researchers noted that no product was observed when only the thiol component (MPA) was used as the HAT reagent. When AscH2 was the only HAT reagent present, the desired amination product was only produced in 34% yield. In contrast, the desired amination product was produced in 74% yield when 3 equiv of MPA and 20 mol% AscH2 were present, highlighting the synergistic effect of these two HAT reagents.
To examine the specific role of these reagents, the researchers performed radical rearrangement experiments with methyl cyclopropyl ketone as the carbonyl substrate. In the presence of both MPA and AscH2, the major product was the ring-retention reductive amination product. In contrast, the major product in the absence of MPA was 2-pentanone. Notably, the ring-retention reductive amination product was not observed in the absence of MPA. Based upon these observations, the researchers posited that HAT from MPA to the α-aminoalkyl radical is faster than intramolecular ring opening. The researchers were able to estimate that the rate of HAT from MPA is about 100 times faster than the rate of HAT from AscH2/AscH– (ca. 107 M–1 s–1 vs 105 M–1 s–1, respectively). These results suggest that MPA is the primary H-atom donor that enables formation of the reductive amination product from the photogenerated α-aminoalkyl radical. In addition to acting as a reductive quencher of the Ru(II) photoexcited state, the researchers posited that AscH2/AscH– intercepts the thiyl radicals formed upon HAT to regenerate MPA and produce dehydroascorbic acid, which can be reduced back to AscH2/AscH– through two-electron/two-proton oxidation of MPA to the corresponding disulfide. This proposed series of HAT events explains why excess MPA and sub-stoichiometric AscH2 are used in the reaction. The researchers posited that the proposed mechanism relies on polarity-matched HAT. Although HAT to the α-aminoalkyl radical from MPA is not as thermodynamically favorable as HAT from AscH2, it is kinetically favored due to the nucleophilicity of the radical substrate and electrophilicity of the thiyl radical product. Subsequent HAT between the electrophilic thiyl radical and the nucleophilic AscH2 is also a polarity-matched process.
In 2018, Pu, Shen, and co-workers reported the light-driven reduction of imines to amines using CdSe/CdS core/shell QDs (Scheme 332).909 Green light irradiation of a PhMe solution containing imine substrate, CdSe/CdS QDs, and a super stoichiometric quantity of 4-fluorothiophenol led to formation of the amine reduction product (33 examples, 40–94%). A variety of different aldimines were competent substrates; electron-donating (332.1), electron-neutral, and electron-withdrawing substituents (332.2) were well tolerated at all positions on the N-aryl and C-aryl groups. N-alkyl aldimines as well as diaryl and aryl alkyl ketimines (332.3) were also competent substrates within the reaction. Notably, the reaction worked when the imine was replaced with the corresponding aldehyde and amine precursors for in situ substrate preparation, and when green LEDs were replaced with sunlight.
Scheme 332. Photocatalytic Reduction of Imines Using Semiconducting Quantum Dots (Pu and Shen, 2018).

Fluorescence quenching experiments showed that 4-fluorothiophenol strongly quenches the excited state of the CdSe/CdS QDs. The researchers noted the oleylamine ligands decorating the QDs are able to deprotonate the thiol (e.g., for 4-fluorothiophenol, pKa(calc) = 8.0 in H2O)910 to form the thiolate anion and that this thiolate anion likely plays an important role in quenching the hole within the VB. Additional experiments using stoichiometric amounts of TEMPO yielded the TEMPO-thiophenol adduct, suggesting that thiyl radicals are generated over the course of the reaction. Based upon these observations, the researchers posit that visible-light irradiation of the QDs leads to the formation of an electron–hole pair. Either thiol or thiolate anion can quench the hole in the VB to directly or indirectly generate thiyl radical (Pathway I and Pathway II, respectively). Stepwise or concerted reductive PCET of the imine substrate is proposed to yield the α-aminoalkyl radical, with oxidized thiophenol as the acid and the CB electron as the reductant. Subsequently, this intermediate undergoes HAT with an additional equivalent of thiol. Thiyl dimerization leads to the formation of the disulfide as a byproduct. This report indicates that further band structure and wave function engineering will enable QDs to exhibit versatile photocatalytic activity.
In 2020, Lercher, Gutiérrez, Karkamkar, and co-workers demonstrated that a CF-based cathode surface-functionalized with acidic groups and Pd NPs could catalyze the reduction of benzaldehyde to benzyl alcohol through a mechanism involving reductive concerted PCET (Scheme 333).911 Beyond the introduction of Pd NPs, the electrodes used in this study were derivatized through the introduction of nitrogen- and oxygen-containing functionality as well as through inclusion of colloidal silica to modify total surface area. The incorporation of acidic groups (i.e., carboxylic acids) on the cathode surface was found to increase rates of benzaldehyde hydrogenation under electrochemical conditions, whereas when H2 was used as the reductant, no rate effect was observed upon surface modification of the electrode. This observation led the authors to hypothesize that a PCET mechanism may be operative in which the acidic site on the electrode surface transfers a proton to the benzaldehyde carbonyl in concert with ET from the adsorbed Pd NP. Given that the reaction exhibited zero-order rate dependence on benzaldehyde concentration, the authors investigated whether binding site saturation by benzaldehyde limited H2 adsorption, a complication not relevant to the electrochemical mode of reduction. Relative to the rate of H2 adsorption in the absence of benzaldehyde, H2 adsorption was reduced by only 30% with benzaldehyde, indicating that binding sites remain available for H2 whether substrate is present or not. Thus, the authors proposed a Langmuir–Hinshelwood-type mechanism in which proximity of the substrate, Pd NP, and acidic group on the cathode surface facilitates reductive PCET. The requirement for these three reactants to exist in proximity of one another explains the direct rate increase observed upon increasing the concentration of surface Brønsted acidic sites. Electrodes with higher concentration of Brønsted acidic sites also showed higher turnover frequencies than electrodes with fewer surface acidic groups, especially at high pH (i.e., pH = 10.5) where turnover frequency becomes more dependent on the rate of proton exchange with hydronium in the buffer medium.
Scheme 333. Electrochemical, Pd-Catalyzed Reduction of Benzaldehyde through Concerted PCET (Lercher, 2020).

6.2. Reductive Transformations of NHPI Esters (C=O)
Photocatalytic decarboxylative alkyl radical generation has become a central tool for the modern synthetic chemist and is used in a very wide variety of chemical transformations.419−426,912 Two modes of initiation can be promoted: (i) decarboxylative alkyl radical generation via the single-electron oxidation of a carboxylate anion, or (ii) decarboxylative alkyl radical generation via the single-electron reduction of a suitably pre-functionalized carboxylate derivative. N-(Acyloxy)phthalimide esters (NHPI esters) are commonly employed functionalized precursors in this respect.913 Okada and Oda reported the first examples of photosensitized decarboxylative radical generation from these compounds using 1,6-bis(dimethylamino)pyrene (BDMAP) (in 1988)914 and later Ru(bpy)3Cl2 ([Ru-1]) (in 1992) as photosensitizers.915 Following alkyl radical generation in this manner, a thiol additive mediates overall reduction to the alkane through HAT. Later work from these authors demonstrated a variety of further functionalization reactions of the alkyl radical generated in this manner, including chlorination with inclusion of CCl4 as co-solvent,916 Michael addition with electron-deficient olefins,917 and selenation with diphenyldiselenide.918
Despite their widespread use, these precursors are demanding in the reduction potential requirement to drive single-electron reduction (e.g., for a primary carboxylic acid-derived NHPI ester, Epred = −1.37 V vs SCE in MeCN).914 This has typically necessitated photocatalysts which are strongly reducing in their photoexcited state, such as Ir(ppy)3 (Ir-1) (E1/2red Ir(IV)/*Ir(III) = −1.73 V vs SCE in MeCN).19 This limits the range of compatible functional groups to those not also susceptible to reduction and limits the scope of oxidative processes which can be coupled to this radical generation (e.g., for Ir-1, E1/2 Ir(IV)/Ir(III) = +0.77 V vs SCE).919 Alternatively, one may use stoichiometric quantities of terminal reductants, such as HEs or tertiary amines, to promote the initial formation of a more-reducing Ir(II) center through oxidative quenching, thus catalyzing overall net-reductive transformations.
Reductive decarboxylative radical generation from these precursors which couple with a broad range of oxidative elementary steps for an overall redox-neutral transformation remained largely unexplored until 2017, when Glorius and co-workers showed that the simple addition of water as a co-solvent to these transformations could modulate the reduction potential of these N-(acyloxy)phthalimides through hydrogen bonding.834 Interestingly, by looking back to the initial Okada and Oda report of 1988,914 we see possible evidence of the same modulation of the reduction potential of these NHPI substrates through hydrogen bonding. The authors note that “while the sensitized reaction proceeded efficiently in anhydrous iPrOH–t-BuSH, a more sluggish reaction was observed in dry THF–t-BuSH or in dry acetonitrile–t-BuSH”, suggesting the same ability of water to facilitate ET.914 This mode of hydrogen bond activation of NHPI ester substrates then facilitating ET for decarboxylative radical generation has since been extended to a variety of redox-neutral and net-reductive transformations proceeding through photocatalytic activation, and these are the subject of this section.
6.2.1. Intermolecular C–C Bond Formation through Reactions with Alkenes and Alkynes
In a demonstration of the attenuation of NHPI ester reduction potential through hydrogen bonding, Glorius and co-workers developed a protocol for the redox-neutral decarboxylative oxyalkylation of styrenes with water or alcohols using NHPI esters (Scheme 334).834 This transformation was catalyzed by [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6), a dye that is not sufficiently reducing in its Ir(III) excited state to drive this process in the absence of hydrogen bond activation and that does not require a terminal reductant to access an Ir(II) state. Notably, Ir(ppy)3 (Ir-1) was poorly effective in this process despite being able to drive efficient substrate reduction, possibly reflecting the poor ability of the resultant Ir(IV) complex to drive the coupled oxidative elementary step. Visible-light irradiation of styrene and NHPI ester substrates in MeCN solution containing 50 equiv of H2O at room temperature led to efficient hydroxyalkylation, with exclusive selectivity for hydroxylation at the benzylic site. In total, 30 examples of this transformation were reported in yields of 20–85%. Primary (334.3), secondary (334.1, 334.2), and tertiary (334.4) alkyl radicals could be generated from their corresponding NHPI esters under this mode of activation. Alternatively, an alcohol co-solvent, though a weaker hydrogen bond donor than water (e.g., based on the Taft–Kamlet parameter, water has a hydrogen bond donor ability of 1.17, greater than that of methanol at 0.98, ethanol at 0.86, or isopropanol at 0.76),920 still gave alkoxyalkylation products (334.5), with four examples documented in yields of 44–68%.
Scheme 334. Photocatalytic Oxyalkylation of Styrenes with NHPI Esters (Glorius, 2017).

A mechanism was proposed involving formation of a hydrogen-bonded complex between water and a carbonyl group of the phthalimide moiety which lowers the substrate reduction potential, leading to single-electron reduction mediated by *Ir(III). It is unclear whether a PT occurs either following or concurrent with ET to generate a neutral ketyl-type radical or if a hydrogen bonded ketyl radical-anion results. Okada and Oda favor a mechanism involving stepwise ET then PT in protic media, forming a neutral ketyl radical prior to N–O bond scission.914 Glorius favors a mechanism involving single-electron reduction with neutral water hydrogen-bonding to the resultant radical anion.834 Evidence for the involvement of hydrogen bonding in activating this substrate to accept an electron came from SV measurements, where emission quenching by the NHPI substrate is absent in anhydrous MeCN, but then is “switched on” by addition of H2O, with a first-order dependence on water concentration and NHPI ester concentration observed. Following ET, the N–O bond of the substrate cleaves to yield a carboxyl radical and PhthH, the former of which extrudes CO2 to yield the key alkyl radical intermediate. The alkyl radical adds to the styrene in an anti-Markovnikov fashion, forming a benzylic radical (e.g., for the benzylic radical derived from ethylbenzene (PhCH·CH3), E1/2ox = +0.37 V vs SCE in MeCN)135 that can be oxidized by the Ir(IV) state of the photocatalyst (E1/2ox Ir(III)/Ir(IV) = +1.21 V vs SCE in MeCN) to yield the corresponding carbocation, which is trapped by the protic co-solvent to deliver the product.
Since Glorius identified the advantageous effect of hydrogen-bonding additives in reductive radical generation from NHPI esters, other groups have used these conditions to promote a range of transformations. Later in 2017, Pan and co-workers demonstrated an alkylation/lactonization sequence of carboxylic-acid-tethered styrenes with NHPI esters (Scheme 335).921 The same combination of [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) and aq. MeCN under visible-light irradiation proved optimal for this transformation, with addition of p-TsOH co-catalyst to promote lactonization. As above, Ir(ppy)3 (Ir-1) and [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) in the absence of an aqueous co-solvent were ineffective catalysts. Here, 24 examples of 1,2-alkylation/lactonization of carboxylic-acid-tethered styrenes were reported in yields of 33–84%. The scope with respect to NHPI ester is expanded in this work, with methyl (335.1), ethyl, n-propyl, and cyclobutyl (335.2) radical generation and subsequent alkylation shown to be feasible. Five- (335.1, 335.2) and six-membered-ring (335.3) lactonization following styrene alkylation was demonstrated. An analogous mechanism for alkyl radical generation through hydrogen-bonding activation of the NHPI ester substrate and subsequent alkylation/carbocation formation was proposed. Isotopic labeling studies with H218O showed that trapping of the carbocation occurs through two pathways: (i) direct lactonization of the tethered carboxylic acid onto the transient carbocation, or (ii) hydroxylation of the carbocation with H2O followed by acid-catalyzed lactonization in a subsequent step. These pathways occur in a ratio of 1.8:1.
Scheme 335. Photocatalytic 1,2-Alkylation/Lactonization of Carboxylic Acid Tethered Styrenes (Pan, 2017).

Shortly thereafter, Song and co-workers applied these reaction conditions to the alkylation of acetophenone-derived silyl enol ethers with NHPI esters, yielding aryl alkyl ketone products (Scheme 336).922 This disconnection achieves the same overall transformation as ketone enolate α-alkylation with alkyl electrophiles but avoids problems of mono-alkylation selectivity and cryogenic reaction temperatures. The combination of [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) in aq. MeCN under visible-light irradiation proved optimal for this transformation. Here, 34 examples of alkylated ketone products were reported in yields of 32–90%. A range of substituted acetophenone-derived silyl enol ether substrates were competent, but pyridine- and thiophene-containing silyl enol ethers, in addition to an alkyl ketone-derived silyl enol ether, were unreactive. In addition to simple, aliphatic acid-derived NHPI esters, an indomethacin-derived NHPI ester was also a successful substrate (336.3). This method achieved efficient tert-alkylation of ketones (e.g., 336.2), which is not possible through a typical polar enolate SN2 manifold.
Scheme 336. Decarboxylative Alkylation of Acetophenone-Derived Silyl-Enol Ethers for the Synthesis of Alkylated Ketones (Song, 2018).

The same proposed mechanism of activation of the PhthH carbonyl group through hydrogen bonding with water was invoked to account for alkyl radical generation through single-electron reduction given that diminished reactivity was observed in anhydrous solvents. The transient alkyl radical then adds across the electron-rich silyl enol ether in an anti-Markovnikov fashion before oxidation mediated by Ir(IV) and desilylation yield the ketone product.
Xiao and co-workers in 2018 developed a domino reaction sequence for the synthesis of benzazepine derivatives from acrylamide-tethered styrenes initiated via hydrogen-bond activation of NHPI esters for reductive decarboxylative alkyl radical generation (Scheme 337).923 These heterocycles are core structures to many pharmaceutical compounds, including benzazepril (ACE inhibitor),924 ivabradine (HCN channel blocker),925 and varenicline (nACh-R agonist).926 The optimized reaction conditions consisted of visible-light irradiation of substrate solutions in aq. MeCN in the presence of [Ir(ppy)2(dtbbpy)]PF6([Ir-3]PF6)photocatalyst and pyridine as a stoichiometric Brønsted base. Here, 23 examples of benzazepine formation were reported in yields of 28–83%. The reaction required an N-alkyl acrylamide substrate, with an analogous N-H substrate showing no reactivity. A variety of primary (337.1), secondary (337.2, 337.3), and tertiary (337.4) alkyl radical precursors were effective in initiating this cascade cyclization. Furthermore, an N-allyl acrylamide was tolerated, forming benzazepine 337.3 in 64% yield. In addition, one example was reported of the cascade cyclization of an acrylate-tethered styrene yielding benzoxepine 337.5 in 31% yield.
Scheme 337. Cascade Cyclization for the Synthesis of Benzazepines Initiated via Reductive Radical Generation from NHPI Esters (Xiao, 2018).

A mechanistic scenario involving single-electron reduction of the hydrogen-bonded complex between water and the NHPI ester substrate mediated by photoexcited-state Ir(III) was invoked. In the absence of water, the reaction proceeded with poor efficiency, highlighting the importance of hydrogen bonding in facilitating radical generation. After single-electron reduction, extrusion of PhthH and CO2 reveals the reactive alkyl radical, which adds chemoselectively to the acrylamide Michael acceptor in a Giese-fashion. The resultant α-acyl radical then adds via a 7-endo-trig cyclization to the styrene moiety before Ir(IV)-mediated oxidation of the product benzylic radical yields a carbocation. Interestingly, under these reaction conditions, the carbocation is not trapped by water to yield hydroxylated products, as is the case in other transformations under this mode of activation, but instead undergoes a base-mediated elimination reaction to reform the alkene.
Paixão and co-workers extended this concept in 2019 when they reported the [2+2+1] polycyclization of 1,7-enynes initiated by reductive decarboxylative alkyl radical generation upon hydrogen-bonding activation of NHPI esters, yielding complex dihydroquinolinone derivatives (Scheme 338).927 Similarly mild reaction conditions were used, consisting of visible-light irradiation of the enyne substrates in the presence of [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) as a photocatalyst and 2,6-lutidine as a Brønsted base additive in aq. MeCN. In this work, 29 examples of these [2+2+1] polycyclization products were obtained in yields of 22–82% (e.g., 338.1–338.4). Predominantly secondary alkyl radical precursors were required for efficient reaction, with primary radical precursors giving low yields.
Scheme 338. Synthesis of Complex Dihydroquinolinone Derivatives via the [2+2+1] Polycyclization of 1,7-Enynes (Paixão, 2019).

Similar to the above Xiao report,923 an initiation sequence of reductive alkyl radical generation and chemoselective Giese addition to the acrylamide moiety is proposed. Control experiments demonstrate the requirement for H2O in facilitating substrate activation, with substantially lower yields in anhydrous solvents. After alkyl radical addition, a 6-exo-dig cyclization occurs, yielding a vinyl radical intermediate which triggers a 1,5-HAT event at the C–H bond of the original site of radical generation. Presumably, the low efficiency observed in primary alkyl radical precursors is due to an unfavorable driving force for this HAT step. In this way, the reactive radical undergoes translocation to the initial site of generation, allowing two C–C bonds to form at one carbon center.928 The resulting alkyl radical undergoes a rebound-type mechanism whereby it then adds back across the styrene component in a 5-endo-trig manner, finally yielding a stabilized benzylic, tertiary radical. Oxidation of this radical is mediated by Ir(IV) to afford a carbocation, which undergoes elimination to reform an alkene in the presence of the basic additive. Again, the absence of hydroxylation products of the incipient carbocation is notable.
Shortly thereafter, Xu and Hu reported the related [2+2+1] polycyclization of 1,6-enynes for complex indene synthesis (Scheme 339).929 Reaction conditions consisted of visible-light irradiation of admixtures of 1,6-enyne substrates and NHPI ester reagent in the presence of Ir(ppy)3 photocatalyst (Ir-1) and TFA as a Brønsted acid co-catalyst in DMF solution. The absence of TFA or replacement of TFA with H2O resulted in lower yields in a model system. A scope of 27 examples of decarboxylative radical generation and polycyclization were reported in yields of 31–93%. Cyclic and acyclic secondary NHPI ester reagents were effective in promoting this transformation (339.1–339.4). Acrylate esters and acrylonitrile (e.g., 339.5) acceptor-olefins on the 1,6-enyne partner were both competent.
Scheme 339. [2+2+1] Polycyclization of 1,6-Enynes via Decarboxylative Radical Generation (Xu and Hu, 2019).

The authors propose that pre-equilibrium hydrogen bonding between the TFA co-catalyst and the NHPI ester substrate facilitates PCET activation of this reagent with photoexcited Ir(III) for decarboxylative radical generation. Thereafter, an analogous mechanistic sequence to that described by Paixão is invoked—intermolecular Giese-type radical addition, 5-exo-dig cyclization, 1,5-HAT, 5-endo-trig cyclization, then termination via oxidation of the resultant radical by Ir(IV) and subsequent PT. However, quantum yield determination experiments support a radical chain mechanism (Φ = 12.7), where the radical intermediate following 5-endo-trig cyclization can reduce further NHPI ester. This mode of reaction is distinct from the observations reported by Paixão.
π-Conjugated phospholes are emerging as important components of organic electronic materials and devices with uniquely favorable properties.930 Zhou and Dong developed a synthesis of benzo[b]phosphole oxides through the alkylation and subsequent arene C(sp2)–H cyclization of alkynylphosphine oxides (Scheme 340).931 This method compares favorably to existing preparations of phospholes in the ease of access to the synthetic precursor and in avoiding the use of transition metals, which are problematic for this class of ligating products.
Scheme 340. Synthesis of Benzo[b]phosphole Oxides from Alkynylphosphine Oxides and NHPI Esters (Zhou and Dong, 2019).

Optimized conditions consisted of the visible-light irradiation of DMSO solutions of alkynylphosphine oxide substrate and NHPI ester reagent in the presence of eosin B (EB) photocatalyst, DIPEA as a redox mediator, and H2O as a hydrogen-bond donor under a nitrogen atmosphere at room temperature. A scope of 40 examples was presented in yields of 8–76%. Primary (340.1), secondary (340.2), and tertiary NHPI esters (340.3) were well tolerated, though poor reaction efficiency was realized in the case of tertiary NHPI esters. Cyclic four- to seven-membered-ring and acyclic secondary reagents were demonstrated. Variation of para-substituents of the aryl group of the tethered arylacetylene showed the reaction was invariant to this change. The heteroarenes pyridine and thiophene (340.5) were tolerated in this position. In addition to alkynylphosphine oxides, a single example of an alkynylphosphinite ester was included, proceeding in poor yield (340.4). The authors report that in the cases of poor yielding reactions, low conversion of the substrate alkynylphosphine oxide was the cause.
The proposed mechanism consisted of PET between the EB excited state and tertiary amine additive in a reductive quenching elementary step to generate EB•– and DIPEA•+. This mode of initiation is supported by steady-state SV quenching experiments. Hydrogen-bonding between the water additive and the NHPI ester at the phthalimide carbonyl group facilitates SET from the reduced-state photocatalyst before extrusion of PhthH and CO2 to form a nascent alkyl radical. Compared to other reports which invoke hydrogen-bond activation of NHPI esters toward SET (other works under consideration in this section), where little reactivity is observed in the absence of water or other hydrogen-bonding additives, in this work significant reactivity is observed under anhydrous conditions, with only a modest boost in reactivity observed in the presence of water (83% compared to 88% yield by 31P NMR). Following reductive radical generation, regioselective addition to the electron-deficient alkynylphosphine oxide yields a vinyl radical intermediate. A di-p-tolyl alkynylphosphine oxide yielded two regioisomeric products, with the methyl group in the para- and meta-positions with respect to the phosphine oxide substituent in an approximate 5:1 ratio. This observation implied two modes of cyclization of the intermediate vinyl radical onto the arene. The major pathway involves 5-exo-trig cyclization followed by oxidation of the resultant delocalized cyclohexadienyl radical mediated by DIPEA•+ and final deprotonation to re-aromatize the arene. Thus, DIPEA is proposed to serve a catalytic role as a redox mediator in this transformation as opposed to a terminal reductant as is often the case. The minor pathway is proposed to involve instead 4-exo-trig cyclization to yield a spiro-phosphetane intermediate before ring opening, P-centered radical generation, and recyclization through a 5-exo-trig mode prior to stepwise ET/PT to yield the aromatized migration product. A quantum yield determination experiment through actinometry (Φ = 1.43) revealed that a radical chain mechanism may also be operative.
6.2.2. Intra- and Intermolecular C–C Bond Formation through Reactions with (Hetero)arenes
In 2019, Sherwood and Xiao, in collaboration with the Knowles group devised a decarboxylative intramolecular arene alkylation reaction for the synthesis of benzannulated carbocycles via the reductive PCET activation of an NHPI ester substrate (Scheme 341).932 Whereas previous methods for reductive decarboxylative radical generation from Glorius and others (see section 6.2.1) have used hydrogen bond donor co-solvents such as water and alcohols to facilitate ET to NHPI ester substrates, in this work TFA is used as a stoichiometric additive to fill this role. While the aforementioned methods typically give hydroxylation and alkoxylation products through trapping of a carbocationic intermediate with the protic co-solvent, the use of TFA allowed for an alternative reaction outcome.
Scheme 341. Intramolecular Alkylation of Arenes via Reductive Decarboxylative Radical Generation from NHPI Esters (Sherwood and Xiao, 2019).

A HTE approach to reaction optimization arrived at a set of conditions consisting of purple-light irradiation of an NHPI ester-tethered arene substrate in the presence of 4CzIPN photocatalyst and 10 equiv of TFA promoter in DMSO at room temperature. Control experiments carried out in the absence of TFA revealed a significant reduction in substrate conversion and product formation in addition to a significant amount of a linear decarboxylation product resulting from termination of the alkyl radical prior to cyclization. In this work, 25 examples of intramolecular alkylation were reported in yields of 11–81%. Aryl and heteroaryl ketone-derived substrates were most optimal in this work (341.1, 341.2), though several examples of carbamate (341.3) or sulfone (341.4) containing linkers were reported. The reaction scope with respect to the arene component is complementary to Friedel–Craft methods for arene alkylation, which favor electron-rich arene substrates. Formation of six-membered-ring carbocycles was most effective, with five- and seven-membered rings operating with low efficiency. NHPI esters yielding primary, secondary, and tertiary alkyl radicals were all competent. The ability of a flow chemistry platform to scale this process and the in situ generation of the NHPI ester from a carboxylic acid precursor prior to telescoping into the photocatalytic reaction were also demonstrated.
SV quenching studies revealed NHPI substrate did not quench the excited state of this photocatalyst in the presence or absence of TFA. This implies that the photoexcited state of this photocatalyst does not engage with the substrate in productive forward ET. This is despite CV studies showing that an NHPI-tethered aryl ketone substrate has a reduction potential of Ep/2red = −1.06 V vs SCE in MeCN,932 which would suggest the possibility of thermoneutral ET with the excited state of this dye (E1/2 PC•+/*PC = −1.18 V vs SCE in MeCN).78 Upon addition of the TFA additive, the reduction potential of the substrate was positively shifted by +40 mV, showing only modest PCET activation toward reduction. To determine the feasibility of PCET reduction mediated by the reduced-state of the photocatalyst instead (E1/2 PC/PC•– = −1.24 V vs SCE in MeCN),78 further voltammetry was performed on solutions of photocatalyst, substrate, and TFA combined. While voltammograms of photocatalyst alone were reversible, on combining all three, a strong catalytic current was then observed characterized by an earlier onset potential, increased current response, and lack of reversibility for the PC/PC•– couple. These combined data suggest a PCET activation of the substrate mediated by the reduced-state of the photocatalyst PC•–.
The mechanistic proposal therefore begins with reductive activation of the NHPI ester substrate by PC•– in the presence of TFA through PCET, then decarboxylation and extrusion of PhthH to generate the alkyl radical. This radical undergoes a cyclization onto the arene to generate a delocalized cyclohexadienyl radical. Since TFA was shown to retard premature radical termination prior to cyclization in a model substrate, it appears to play a role in facilitating this cyclization in addition to radical initiation. Then, facile oxidation of the resultant delocalized cyclohexadienyl radical (e.g., for the parent cyclohexadienyl radical, E1/2ox = −0.34 V vs SCE in MeCN)268 by the photoexcited-state dye (E1/2 *PC/PC•– = +1.43 V vs SCE in MeCN)78 furnishes a carbocation through which deprotonation (pKa = −2.6 in DMSO)39 yields the re-aromatized arene. This stepwise ET/PT pathway for termination, as opposed to a radical chain mechanism, is supported by a quantum yield determination experiment (Φ = 0.022). For this mechanism to be operative, an initial off-cycle reductive initiation step is required to generate the active photocatalyst radical anion from its ground state to facilitate decarboxylative radical generation. The exact nature of this initiation remains unclear. We note here a related transformation reported in 2018 from Reiser and co-workers demonstrating similar reductive radical generation and subsequent arene cyclization from NHPI ester precursors. Authors propose a distinct energy-transfer mechanism leading to decarboxylative radical generation.933,934
The involvement of PCET in the reductive decarboxylative generation of an alkyl radical from NHPI esters by acid additives raises questions for other processes involving these same components. Several groups have reported methods enabling the redox-neutral alkylation of heteroarenes with NHPI esters in the presence of either stoichiometric or catalytic acid additives. In these Minisci reactions, these acid additives are understood to facilitate radical addition to the heteroarene through protonation and electrophilic activation. Fu and Shang reported the photocatalytic decarboxylative alkylation of heteroarenes with α-amino-acid-derived935 or alkyl carboxylic acid-derived NHPI esters936 using an Ir photosensitizer in the presence of a phosphoric acid catalyst. Phipps and co-workers later adapted these conditions to enable the enantioselective alkylation of heteroarenes with α-amino-acid-derived NHPI esters using a chiral phosphoric acid (CPA) catalyst.937 Sherwood and co-workers also demonstrated the in situ activation of carboxylic acids as their NHPI esters for decarboxylative alkylation of heteroarenes using the organic photocatalyst 4CzIPN and a TFA additive.938,939
Mechanistically, these processes are all understood to proceed via an Ir(III)/Ir(II) cycle (or corresponding 4CzIPN/4CzIPN•– cycle), similar to that invoked for the intramolecular alkylation of arenes by Sherwood and co-workers above,932 whereby an off-cycle catalyst activation via reductive quenching generates the reduced-state photocatalyst capable of engaging in ET with the PhthH moiety. Given the observations in this work from Sherwood and co-workers that acid additives accelerate this single-electron reduction via PCET, it is reasonable to consider that acids may play a beneficial role in this elementary step of these other processes too.
Contemporaneous reports from the groups of Jin, Sun, and Li also invoked a concerted reductive PCET mechanism for photocatalytic decarboxylative radical generation from NHPI esters for the intermolecular C3-selective C(sp2)–H alkylation of coumarins and quinoxalin-2(1H)-ones. For the alkylation of coumarins, the reaction conditions consisted of visible-light irradiation of DMSO solutions of these substrates and NHPI ester radical precursor in the presence of Ir(ppy)3 as photocatalyst (Ir-1) and TFA as a Brønsted acid co-catalyst (Scheme 342).940 Significantly diminished reactivity was observed in the absence of TFA (e.g., 29% compared to 81% yields for 342.3). A scope of 27 examples of alkylated coumarin products was presented with yields ranging from 30% to 92%. The process tolerated a variety of coumarin substituents, including fluoro, mono- and dichloro, mono- and dibromo (e.g., 342.4); and dimethylamino (e.g., 342.5). Primary, secondary, and tertiary alkyl NHPI ester reagents were competent alkyl radical precursors. Additionally, an acetate-NHPI ester provided access to a methylated coumarin product (342.1). Primary alkyl bromide, terminal olefin, and Boc carbamate (342.6) functional groups appended to the NHPI ester reagent were tolerated by these conditions.
Scheme 342. Photocatalytic C3-Alkylation of Coumarins with NHPI Ester Reagents (Jin and Sun, 2019).

Jin, Sun, and co-workers also reported the C3-selective C(sp2)–H alkylation of quinoxalin-2(1H)-ones (Scheme 343A).941 The reaction conditions consisted of visible-light irradiation of DMSO solutions of these substrates and NHPI ester reagents in the presence of Na2-EY as photocatalyst and TFA as a Brønsted acid co-catalyst. Again, significantly attenuated reactivity was seen without the acid co-catalyst (e.g., 11% compared to 97% for 343.3). A scope of 31 examples was reported in yields of 49–99%. Similar functional group tolerance to their earlier coumarin alkylation method was observed.940 Both N-H and N-alkyl quinoxalin-2(1H)-ones were viable substrates. Additionally, the synthesis of an intermediate toward an aldose reductase inhibitor was demonstrated using this protocol (343.6). A 10 mmol scale experiment was performed without deterioration in yield.
Scheme 343. Photocatalytic C3-Alkylation of Quinoxalin-2(1H)-ones with NHPI Ester Reagents (Jin and Sun, 2019; Li, 2019).

A similar transformation was reported by Li and co-workers and consisted of the visible-light irradiation of DMSO solutions of these substrates and NHPI ester reagents in the presence of Ir(ppy)3 as photocatalyst (Ir-1) and TFA as a Brønsted acid co-catalyst (Scheme 343B).942 A total of 27 examples of quinoxalin-2(1H)-one C3-alkylation were reported in yields of 27–95% (343.7–343.12). Primary, secondary, and tertiary alkyl NHPI ester reagents were capable reagents, and a variety of quinoxalin-2(1H)-one N- (343.10) and ring-substitution (343.11) were tolerated. An amino-acid-derived NHPI ester was also demonstrated (343.12).
As previously reported, hydrogen bonding between the TFA additive and NHPI ester reagent activates this moiety toward single-electron reduction through PCET mediated by photoexcited Ir(III) or EY. Thereafter, fragmentation of the reagent reveals the reactive alkyl radical which adds to the coumarin or quinoxalin-2(1H)-one. Li and co-workers also postulated that hydrogen bonding with TFA may activate the quinoxalin-2(1H)-one substrate toward radical addition. Oxidation of the resulting benzylic radical to the corresponding carbocation by Ir(IV) and PT mediated by trifluoroacetate yields the product. Jin, Sun, and co-workers showed that this transformation likely proceeds through a closed photoredox cycle as opposed to an alternative chain mechanism based on a quantum yield determination experiment with a quinoxalin-2(1H)-one substrate (Φ = 0.23).
6.2.3. Other Processes for C–C, C–N, and C–S Bond Formation
Ren and Cong in 2018 developed a decarboxylative benzylic C(sp3)–H alkylation reaction of N-aryl tetrahydroisoquinolines with NHPI esters using a dye-sensitized semiconductor as an efficient visible-light photocatalyst (Scheme 344).943 The standard reaction conditions consisted of blue LED irradiation of TFE solutions of these substrates in the presence of TiO2 (P25) semiconductor catalyst and erythrosine B sensitizer at 40 °C. Protic solvents were essential and TFE was found to be optimal. The combination of this semiconductor and sensitizer pair was necessary to achieve high yields, with significantly diminished reactivity observed if one was omitted or other materials were used. Using these conditions, 19 examples of tetrahydroisoquinoline C–H alkylation were reported in yields of 44–90% (344.1–344.4). The reaction permitted variation of the tetrahydroisoquinoline N-aryl groups and use of primary, secondary, and tertiary NHPI ester C-centered radical precursors.
Scheme 344. Decarboxylative Benzylic C(sp3)–H Alkylation of Tetrahydroisoquinolines with NHPI Esters (Ren and Cong, 2018).

A plausible mechanistic scenario was proposed, involving visible-light excitation of erythrosine B and charge injection into the titania CB. This in turn facilitates reductive PCET activation of the substrate assisted by hydrogen bonding to TFE. Boyington and Jui made a similar observation of solvent-dependent reactivity in the reductive PCET activation of halopyridines in TFE solution (see section 7.2).944 Extrusion of CO2 and PhthH yields the C-centered radical intermediate. The photocatalyst radical cation resulting from charge injection oxidizes the amine substrate to the aminium radical cation followed by a subsequent PT which yields the α-amino radical. Finally, radical–radical coupling yields the alkylated product. We note here a related visible-light-mediated radical alkylation of tetrahydroisoquinolines using alkyl halides in concert with a Pd catalyst published at this time from Yu and co-workers.945
In 2019, Yu and co-workers developed a method enabling the decarboxylative N-alkylation of α-diazoacetates with alkyl NHPI esters, leading to functionalized N-alkyl hydrazones (Scheme 345).946 Optimized reaction conditions consisted of yellow light irradiation of CH2Cl2 solutions of diazoacetate substrate and NHPI ester reagent in the presence of RB as a photocatalyst, HE as a terminal reductant, and DBU as a Brønsted base additive at room temperature. The authors noted that the UV–vis absorption profile of the photocatalyst exhibited a DBU concentration dependence, featuring a prominent absorption at 565 nm in the presence of excess DBU. This absorption maximum necessitated the use of a yellow light source, an unusual wavelength for photoredox chemistry.
Scheme 345. Photocatalytic Decarboxylative N-Alkylation of α-Diazoacetates with Alkyl NHPI Esters (Yu, 2019).

A broad substrate scope was presented, with 61 examples of N-alkyl hydrazone synthesis demonstrated in yields of 33–99%. With respect to the diazo component, α-aryl (345.1) and α-heteroaryl (345.4) diazoacetates were ideal substrates, providing good to excellent yields in cross-coupling with a cyclopentanecarboxylate-derived NHPI ester. A variety of arene substituents were screened and the reaction showed only minor changes in efficiency with variation of electronic and steric nature. Several alkyl and allylic diazoesters were tolerated. An α-phenyl trifluorodiazoethane substrate underwent N-alkylation in 42% yield (345.5). α-Alkyldiazoacetates were less tolerated in this reaction, yielding the desired products in 33–49% NMR yields (e.g., 345.6). With respect to the NHPI ester component, a variety of primary, secondary, and tertiary alkyl radical precursors were compatible (345.1–345.3). Reactants derived from abietic acid and gemfibrozil enabled the corresponding N-alkylation reaction. A citronellic acid-derived NHPI ester underwent an initial 5-exo-trig radical cyclization onto the pendant prenyl group prior to diazoester N-alkylation with the resultant tertiary C-centered radical. When α-amino acid-derived NHPI esters were employed, the initial α-amino hydrazone product underwent spontaneous cyclodehydration under the conditions of reaction to yield the corresponding 1,2,4-triazole (e.g., 345.7).
Reductive quenching of the RB excited state (E1/2 *RB2–/RB3–• = +0.99 V vs SCE in H2O)349 with HE (Ep/2ox = +0.51 V vs Fc+/Fc in MeCN)374 was proposed to initiate this transformation, leading to generation of RB3–• and HE•+. This is supported by steady-state SV luminescence quenching experiments, where no degree of quenching was observed by either NHPI ester substrate or DBU. RB3–• (E1/2RB2–/RB3–• = −0.78 V vs SCE in H2O)349 alone is not sufficiently reducing to engage in SET with the NHPI ester substrate (e.g., for cyclopentane carboxylic acid-derived NHPI ester, Ep/2red = −1.32 V vs SCE in MeCN),946 so the authors propose that protonated DBU acts as a Brønsted acid mediator of this process through hydrogen-bonding and subsequent PCET. Upon reductive activation, the NHPI ester extrudes PhthH and CO2 to liberate an alkyl radical intermediate, which then adds across the diazo substrate. Termination occurs through HAT to the resultant radical from the HE•+ to yield the closed-shell product and HE pyridinium salt, which is in turn deprotonated by DBU.
In 2020, the groups of Li and Liu reported a dual photoredox/Cu-catalyzed protocol enabling the decarboxylative radical S-alkylation of organic sulfinate salts with NHPI esters leading to valuable sulfone products (Scheme 346).947 A standard set of reaction conditions involved the blue-light irradiation of a DME/MeCN (1:1) solution of sodium sulfinate salt and NHPI ester reagent in the presence of 4CzIPN as photocatalyst, a Cu(OTf)2 co-catalyst, and a stoichiometric amount of (n-BuO)2P(O)OH additive at room temperature. The phosphoric acid additive proved key to achieving the desired reactivity and suppressing unproductive decarboxylative desaturation of the NHPI ester. TFA could be used in place of the phosphoric acid with similarly high efficiency, but weaker acids or the absence of an acid altogether led to lower yields (e.g., 65% yield compared to 95% yield in the presence of acid for 346.1).
Scheme 346. Dual Photoredox/Cu-Catalyzed Alkyl Radical Sulfonylation (Li and Liu, 2020).

*TFA used in place of (n-BuO)2PO2H.
In total, 44 examples of decarboxylative sulfonylation in this manner were reported in yields of 23–98%. A variety of functionalized primary (e.g., 346.1) and secondary (e.g., 346.2) NHPI ester radical precursors were competent reagents, delivering products in good to excellent yields. A single example of the sulfonylation of a tertiary radical generated from a bicyclopentane-derived NHPI ester was included (346.3), albeit in reduced efficiency (47%). A broad range of functional groups were tolerated, including terminal alkenes and alkynes, aryl and alkyl bromides, ketones, aldehydes, esters, amides, sulfonamides, α-amino acids, indoles (346.4), and pyrroles. Both aryl and alkyl sulfinate salts were viable substrates, all with similar reaction efficiency. Pyridine 3-sulfinate gave 23% yield of the sulfone product (346.5) due to competing Minisci reactivity. The late-stage sulfonylation of a variety of natural products and pharmaceutical molecules, including gibberellic acid diacetate (346.6), indomethacin, and dehydrocholic acid, was also demonstrated. Finally, a synthesis of the anti-prostate cancer drug bicalutamide was performed using this methodology to introduce the sulfone functional group, providing synthetic intermediate 346.7 in 60% yield from a malic acid-derived NHPI ester.
The proposed mechanism involves initial oxidative quenching between photoexcited PC and the NHPI ester substrate, leading to PC•+ and alkyl radical generation upon extrusion of PhthH and CO2. Analysis of the reduction potential of this photocatalyst (E1/2 PC•+/*PC = −1.18 V vs SCE in MeCN)948 compared to an NHPI ester substrate (e.g., for the NHPI ester substrate leading to product 346.1, Ep/2red = −1.32 V vs SCE in MeCN)914 suggests this direct ET is unfavorable. The acid additive is suggested to facilitate this step through hydrogen-bonding with the NHPI substrate, priming it for reductive SET. The acid was also hypothesized to effectively buffer the reaction to slow the rate of alkyl radical desaturation through deprotonation of the α-C–H bonds. The Cu(II) co-catalyst undergoes ligand exchange with the substrate sulfinate to yield a Cu(II) sulfinate complex, which is then proposed to enable RSO2 group transfer with the alkyl radical from the NHPI ester followed by elimination of Cu(I). The successful incorporation of SO2 into the products of reactions with alkylsulfinates here suggests that a sulfonyl radical does not form in the mechanism, as these readily extrude SO2 to yield the corresponding alkyl radical.949−951 However, formation of a Cu(III) alkyl sulfinate intermediate prior to reductive elimination is also a possibility.160 Finally, SET between PC•+ and Cu(I) readies the system for turnover.
6.3. Reductive Transformations of Peroxyacetates (C=O)
PCET as a mechanism for otherwise endergonic ET has also been utilized in the reductive activation of peroxyacetate reagents toward alkyl radical generation following O–O bond homolysis and β-scission or decarboxylation. We study several methods involving these compounds from DiRocco and Wang in this subsection.
Introduction of small alkyl and fluoroalkyl groups—in particular methyl—can have drastic effects on adsorption, distribution, metabolism, and excretion (ADME) properties as well as the selectivity and potency of biologically active molecules.952,953 However, alkylation is particularly challenging to accomplish in complex molecules at a late stage without substrate pre-functionalization.954 The Minisci reaction955—the addition of a C-centered radical to heteroarenes assisted by Brønsted or Lewis acid activation—is a common method used to this effect, but it traditionally has suffered limitations such as requiring high temperatures, UV irradiation, or strong oxidants to generate the reactive alkyl radical.696 In seeking to address these issues, DiRocco and co-workers used a HTE approach956−958 to develop a protocol for the late-stage methylation, ethylation, and cyclopropanation of heterocyclic pharmaceuticals wherein PCET was used to facilitate reductive alkyl radical generation from peroxyacetates under mild conditions (Scheme 347).959
Scheme 347. Photocatalytic Late-Stage C(sp2)–H Methylation, Ethylation, and Cyclopropanation of Pharmaceuticals Mediated by PCET Activation of Peroxyacetates (DiRocco, 2014).

*Indicated minor site of alkylation.
A combination of an Ir(III) photocatalyst [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) or [Ir(dF(CF3)ppy)2(dtbbpy)]PF6([Ir-6]PF6), stoichiometric quantity of TFA or AcOH as an acid additive or cosolvent, and tert-butyl peracetate (TBPA) as methyl radical source was effective for the room-temperature methylation of a diverse collection of complex heterocyclic pharmaceutical and agrochemical molecules, including camptothecin (347.1), fasudil (347.2), voriconazole (347.3), diflufenican (347.4), and eszopiclone (347.5). Often, slow addition of reagents for radical generation was required in traditional Minisci protocols. This was not the case in this work, as kinetic studies demonstrate that the degree of alkylation and mono- vs dialkylation selectivity was controlled solely as a factor of light intensity, with more intense light inducing a higher amount of dialkylation. In addition to methylation, heteroarene ethylation was described using tert-amyl peracetate (TAPA) and cyclopropanation using bis(cyclopropanecarbonyl)peroxide, all taking place at room temperature.
The direct single-electron reduction of peresters is thermodynamically challenging (e.g., for tert-butyl perbenzoate (TBPB), E1/2red = −1.95 V vs SCE in MeCN)960 and is inaccessible to the Ir(III) photooxidants employed here (for [Ir-3]PF6, E1/2 Ir(IV)/*Ir(III)= −0.96 V vs SCE in MeCN; for [Ir-6]PF6, E1/2 Ir(IV)/*Ir(III)= −1.21 V vs SCE in MeCN).68 However, a PCET mechanism was invoked to rationalize the observed reactivity. The acid additive serves a dual role, both protonating the heteroarene to promote Minisci radical addition and also facilitating the single-electron reduction of the peracetate by the photoexcited Ir(III) complex through hydrogen bonding. The resultant unstable α-peroxy radical undergoes facile O–O bond scission to generate AcOH and tert-butoxy radical. tert-Butoxy radical then undergoes a further C–C bond β-scission step to liberate methyl radical and acetone,961 the former of which engages with the heteroarene. Oxidation of the resulting radical intermediate mediated by Ir(IV) and subsequent PT restores aromaticity and yields the product. Exclusive ethylation occurs from TAPA as the more stable ethyl radical is ejected. Cyclopropyl radical generation occurs via a similar process of PCET-driven reduction and subsequent decarboxylation.
This team later extended this method to achieve mild late-stage hydroxymethylation of heteroaromatic pharmaceuticals through sequential reductive PCET and HAT elementary steps (Scheme 348).962 Under modified reaction conditions, the combination of Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6), benzoyl peroxide (BPO) as a stoichiometric oxidant and HAT mediator, TFA as an acid additive, and MeOH as both solvent and hydroxymethylating reagent enabled the hydroxymethylation of 19 heteroarenes at room temperature, including those containing pyridine (348.3), pyrazine (348.1, 348.4), pyrimidine, quinoline, isoquinoline (348.2), benzimidazole, and thiazole moieties. Several pharmaceutical compounds were competent substrates. These conditions were shown to be superior to existing thermal conditions reported for Minisci hydroxymethylation. In a limited number of substrates, arene or benzylic methoxylation was observed in preference to hydroxymethylation arising from a proposed arene radical cation intermediate or a benzylic HAT/ET mechanism, respectively. The use of BPO, as opposed to TBPA, was key to achieving selective hydroxymethylation while avoiding methylation.
Scheme 348. Photocatalytic, Late-Stage Hydroxymethylation of Heteroarene Pharmaceuticals (DiRocco, 2016).

The authors propose a mechanism involving reductive activation of benzoyl peroxide (Epred = −1.01 V vs SCE in DMF),963 forming an α-peroxy radical, which fragments to yield benzoyloxy radical and benzoic acid as a byproduct. In related work, Knowles, DiRocco, and co-workers established PCET as a key driver for the photoexcited-state Ir(III)-mediated reductive activation of peroxybenzoates in the presence of TFA.964 Decarboxylation of benzoyl radical yields benzene radical (C–H BDE = 112.9 kcal mol–1)25 which acts as a HAT mediator to abstract a weak C–H bond (for MeOH, C–H BDE = 96.1 kcal mol–1)25 from MeOH solvent and generate nucleophilic hydroxymethyl radical. This radical then adds to the protonated heteroarene in a Minisci fashion followed by re-aromatization mediated by Ir(IV).
Wang and co-workers later used a similar combination of reductive PCET and HAT steps for alkyl radical generation and heteroarene alkylation beyond the above methylation, ethylation, cyclopropanation, and hydroxymethylation protocols (Scheme 349).965 Reaction conditions were similar to those first established by DiRocco and co-workers959 involving the blue-light irradiation of heteroarene and coupling-partner in acetone in the presence of Ir(III) photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6), TBPA as an oxidant and HAT mediator, and TFA as an additive. Reductive PCET activation of TBPA and generation of tert-butoxy radical through the same reaction manifold was found to promote intermolecular HAT and alkyl radical formation in a number of carbamate (349.1), ether (349.2), formamide (349.3), and alkane (349.4) cross-coupling partners before Minisci addition to heteroarenes. A scope of 47 examples of alkylation in yields of 28–95% was reported. For α-amino radical generation, only 2 equiv of amine coupling partner were typically required; however, for other examples of α-ether and secondary alkyl radical generation, co-solvent quantities were necessary. In the case of the bifunctional substrate N-Boc morpholine, exclusive heteroarylation at the α-amino position was observed. Benzothiazole (349.2–349.4), quinoline, isoquinoline, quinazoline, benzimidazole, pyridazine (349.1), and pyrimidine functionalization was described. We note that an analogous method for HAT-driven alkyl radical generation and further Minisci addition using persulfate as a stoichiometric oxidant was developed at this time by Jin and MacMillan.966
Scheme 349. Heteroarene Minisci Alkylation through Dual Reductive PCET and HAT (Wang, 2018).

Wang and co-workers in 2019 also reported a protocol enabling a photocatalytic Minisci C(sp2)–H alkylation reaction of N-heteroarenes with alkyl iodides (Scheme 350).967 PCET was leveraged as a mechanism for the reductive activation of TBPA; subsequent O–O bond homolysis and β-scission generates methyl radical in a similar fashion to that reported by DiRocco and co-workers.959 In this report, Wang demonstrated that methyl radical can facilitate halogen atom abstraction in the alkyl iodide partner to generate a more stable alkyl radical.968 The specific reaction conditions consisted of irradiating MeCN solutions of heteroarene, alkyl iodide, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) photocatalyst, TBPA, and TFA. In this work, 27 examples of Minisci alkylation were reported in yields of 24–73%. Alkyl radical generation from primary, secondary, and tertiary alkyl iodides was reported. Typically, 4-substituted quinolines (350.1) were preferred substrates, but isoquinolines (350.2), a quinazoline (350.3), and a phenanthridine also underwent regioselective C(sp2)–H alkylation. The late-stage alkylation of quinoxyfen (350.4) was demonstrated.
Scheme 350. Photocatalytic Minisci C(sp2)–H Alkylation Reaction of N-Heteroarenes with Alkyl Iodides, Proceeding through Halogen Atom Transfer (Wang, 2019).

6.4. Reductive Transformations of Diazenes, Diazoacetates, Nitro Compounds, and N-Oxides (N=N and N=O)
6.4.1. Heterogeneous Photocatalysis for C–C and C–N Bond Formation between Diazenes and C(sp3)–H Bonds
In addition to small molecule photocatalysis, semiconductors have also been well documented to catalyze light-driven reductions involving PCET. In semiconductor photocatalysts, as in the case of their small molecular analogs, absorption of a photon results in the promotion of an electron from the VB of the semiconductor to the higher-energy CB, leaving an electron hole (h+) in the VB. The photoexcited CB electron and the VB hole then migrate to the surface of the semiconductor crystal where these separated charges may each mediate redox chemistry, with the electron hole acting as an oxidant and the photoexcited electron acting as a reductant.757 Commonly employed semiconductor photocatalysts include CdS, ZnS, WO3, and TiO2, which are often employed as NPs.
In 1992, Kisch and co-workers reported an example of a reductive PCET process mediated by a semiconducting photocatalyst, involving the allylic C(sp3)–H cross-coupling of cyclic enol ethers and 1,2-diazenes to form trisubstituted hydrazines (Scheme 351).751 The authors found that tungsten lamp (λ ≥ 350 nm) irradiation of a CdS photocatalyst suspended in a MeOH solution of the enol ether and 1,2-diazene substrates was optimal. ZnS also proved to be an effective photocatalyst. In addition to the desired reaction, the reduction of the 1,2-diazene to the corresponding disubstituted hydrazine and the dehydrodimerization of the enol ether at the allylic position were noted byproducts. In the absence of photocatalyst, only the cis/trans isomerization of the 1,2-diazene was observed. Here, four examples of enol ether allylic coupling with 1,2-diazenes were reported in assay yields of 44–75%. Both 3,4-dihydropyran (351.1) and 2,3-dihydrofuran (351.2) afforded the desired trisubstituted hydrazine. Later, the authors expanded this scope to include cyclic olefins, such as cyclopentene and cyclohexene, under identical conditions (351.4).752 1,2-Diazenes bearing phenyl, p-tolyl (351.5), and t-butyl (351.3) substituents were all tolerated.
Scheme 351. Addition of Olefins and Enol Ethers to 1,2-Diazenes through Semiconductor Photoredox Catalysis (Kisch, 1992 and 1995).

The mechanism of this transformation was borne out over multiple reports. Prior to undergoing any redox events, the substrates are proposed to adsorb onto the surface of the CdS photocatalyst. In the case of the enol ether and olefin substrates interacting with silica-supported CdS, substrate binding likely occurs through hydrogen bonding between the olefin and surface −OH and −SH groups, whereas the 1,2-diazenes bind at Brønsted acidic sites on the semiconductor. Absorption of a photon by the CdS photocatalyst excites an electron from the VB to the CB, creating a charge-separated electron/hole pair. The 1,2-diazene substrate undergoes reductive PCET with the CB electron as the reductant and MeOH acting as the proton donor, forming an N-centered hydrazyl radical. Protic solvents are reported to significantly modify the reduction potential of 1,2-diphenyldiazene, which is reported to be Ep/2red = −1.10 V vs NHE in anhydrous DMF and −0.75 V vs NHE in aqueous dioxane.969 At neutral pH, the reduction potential of CB electrons in CdS is Ered = −0.9 V vs NHE,970 suggesting that the ET from photoexcited CdS to 1,2-diphenyldiazene is only favorable in the presence of hydrogen bond donors. Indeed, the rate of the desired addition reaction was decreased in aprotic media such as THF and petroleum ether. A second reductive PCET event to the hydrazyl radical or HAT results in formation of the undesired hydrazine byproduct. The use of Pt as a conducting support for CdS was found to promote hydrazine formation via overall substrate reduction. Given that the mechanism of hydrazine formation requires two electron/hole pairs whereas the desired cross-coupling reaction requires only one, the enhanced rate of charge separation and ET induced by the conducting support favors 1,2-diazene reduction.752
Simultaneously, the olefin substrate is proposed to undergo oxidative C(sp3)–H PCET activation with the VB hole acting as the oxidant, yielding a surface adsorbed allylic radical. Evidence for olefin oxidation via concerted PCET over stepwise ET/PT came from the lack of observed alcohol trapping products, which would have likely formed from a radical cation intermediate (for a more detailed discussion of concerted PCET involvement in C–H bond homolysis in similar heterogeneous photocatalytic systems, see section 5.1). Still bound to the surface of the semiconductor, the allylic and hydrazyl radicals diffuse through the surface-solvent interface and eventually couple through radical recombination, forming the desired C–N coupling product. Experiments run under high pressure revealed the activation volume for this reaction to be ΔV⧧ = +17.4 ± 3.4 mL mol−1, suggesting that the diffusion of the surface-bound radicals is the rate-determining step, given that other steps in the proposed mechanism are expected to involve negative activation volumes.971 Radical recombination between two allylic radicals within the surface–solvent interface leads to the formation of the observed dehydrodimerization byproduct.
Subsequent studies from the Kisch group further broadened the scope of this photocatalytic cross-coupling to include aryl imines for the synthesis of homoallylic amines through coupling with olefins and enol ethers (Scheme 352).972 Across 19 examples of this olefin–imine cross-coupling, yields of 40–80% were reported. Using CdS as photocatalyst under conditions otherwise identical to this group’s prior work involving diazene cross-coupling, electron-poor (352.1) and electron-rich imines (352.2) could efficiently undergo alkylation. The desired addition product could also be formed using the trisubstituted N-phenylbenzophenone imine (352.3).973,974 With a silica-supported CdS photocatalyst shown to accelerate the reaction rate by 3-fold, imines bearing α-ester substituents could undergo addition with various hydrocarbon olefins (352.4, 352.5) in yields of 11–49% along with significant quantities of imine reduction and reductive dimerization byproducts.972 This silica-supported CdS photocatalyst was later found to be ineffective for imines bearing N-benzoyl and α-cyano groups, but switching to a ZnS-supported CdS photocatalyst allowed the isolation of homoallylic N-benzoyl-α-amine nitriles in yields of 65–84% through the addition of cyclopentene or cyclohexene.975,976 The use of a semiconducting support such as ZnS for CdS likely increases the lifetime of the charge-separated state through interparticle electron transfer (IPET) and enhanced spatial separation between excited electrons and holes. With alumina-grafted CdS, which provided a longer excited-state lifetime (1.2 μs vs 0.75 μs) and a slightly increased bandgap relative to unsupported CdS, sterically hindered N-adamantyl homoallylamines could be prepared through the addition of olefin-derived allylic radicals to N-adamantyl imines.977 This later work from the Kisch group on the cross-coupling of imines with olefins is proposed to follow a similar mechanism to that described in their earlier reports of 1,2-diazene reduction, involving a reductive PCET process of the imine component.751,752,971
Scheme 352. Addition of Olefins and Enol Ethers to Imines through Semiconductor Photoredox Catalysis (Kisch, 1996, 1997, and 2002).

6.4.2. Traceless Olefin Hydroalkylation Enabled by Reductive PCET with α-Diazoacetates
As we have seen previously in this section, the majority of transformations reported for aldehydes, ketones, imines, and diazenes proceeding through reductive PCET processes—leading to neutral α-heteroatom (O or N, respectively) radical intermediates—eventually yield products incorporating alcohol, amine, or hydrazine functionality. Doyle and co-workers have recently demonstrated access to primary and secondary C-centered radicals without neighboring heteroatoms via the reductive PCET activation of α-diazoacetates and select diazoalkanes (Scheme 353).978 Two modes of reactivity were developed through complementary photocatalytic and iron-catalyzed modes of substrate activation. These substrates extrude dinitrogen upon ET, allowing for traceless incorporation of the attached group. C-Centered radicals generated in this manner were found to efficiently add across olefins, allowing for the development of anti-Markovnikov hydroalkylation and azidoalkylation processes, respectively. This too marks a distinct departure from the standard reactivity of diazo compounds and olefins, where cyclopropanation and X–H bond insertion pathways, via the intermediary of metal carbene complexes or [3+2] cycloaddition, are dominant.979,980
Scheme 353. Photocatalytic Traceless Olefin Hydroalkylation with α-Diazoacetates (Doyle, 2020).

Reaction conditions enabling the photocatalytic anti-Markovnikov olefin hydroalkylation with α-diazoacetates involved blue-light irradiation of CH2Cl2 solutions of these substrates in the presence of [Ru(bpy)3]Cl2 ([Ru-1]Cl2) photocatalyst, p-tolyl thiol as a HAT co-catalyst, and a stoichiometric quantity of HE as a terminal reductant. Control experiments revealed the need for photocatalyst, HAT co-catalyst, reductant, and blue-light irradiation. This reaction proved highly tolerant of olefin substitution patterns and additional substrate functional groups, all without sign of cyclopropanation. A total of 60 examples of olefin hydroalkylation in this manner were presented in yields of 34–99%. Styrenes (353.1–353.5), vinylheteroarenes (353.6), terminal alkyl (353.8), 1,1- and 1,2-disubstituted (353.7), and trisubstituted alkenes were all competent substrates. Tetrasubstituted olefins however proved unreactive. Acyclic 1,4-dienes reacted with excellent 1,4-addition selectivity, but the cyclic 1,4-diene cyclohexadiene gave a mixture of 1,2- and 1,4-addition. The presence of protic functional groups such as primary alkyl alcohol (353.8), aromatic carbocyclic acid (353.3), primary sulfonamide (353.4), and even primary aniline functionality (353.5) was tolerated without apparent X–H carbene insertion byproducts. Sulfide functionality was tolerated without ylide formation. An aromatic aldehyde, proven substrates for photocatalytic reductive PCET activation, also survived the hydroalkylation reaction (353.2). With respect to the diazo component, acceptor diazo compounds showed higher reactivity than did acceptor/acceptor or donor/acceptor diazo compounds. A diverse collection of acceptor diazo compounds, including esters, amide, cyano (353.10), sulfonyl, and trifluoromethyl (353.11), gave hydroalkylation products in good yields.
The authors attempted to modify these conditions to enable a photocatalytic azidoalkylation protocol through replacement of the thiol HAT co-catalyst with azidation reagents but were unsuccessful. However, they were instead able to promote the desired transformation through an alternative Fe-catalyzed reaction manifold. The combination of Fe(OAc)2 acting as both a redox catalyst and azide-transfer catalyst, tert-butylhydroperoxide as a H-atom donor and oxidant, and Et3N as a Brønsted base in IPA at 50 °C promoted the azidoalkylation of alkenes with acceptor diazo compounds and TMSN3, with a similar substrate scope to the above photocatalytic hydroalkylation reaction.
A proposed mechanism of the photocatalytic hydroalkylation reaction involved reductive quenching of the Ru(II) photoexcited-state complex (E1/2 *Ru(II)/Ru(I) = +0.77 V vs SCE in MeCN)64 with the sacrificial HE reductant (Ep/2ox = +0.51 V vs Fc+/Fc in MeCN)374 to yield a reducing Ru(I) species and HE radical cation. The combination of this Ru(I) complex and a proton source ultimately proposed to arise from the HE pyridinium byproduct mediates the reductive PCET activation of the diazoacetate reagent followed by extrusion of molecular nitrogen to yield the α-acyl radical intermediate. Addition of this electrophilic radical intermediate occurs across the nucleophilic olefin acceptor before terminal HAT with the thiol co-catalyst. The resulting thiyl radical reacts with the transient HE radical cation to reform the closed-shell thiol catalyst and a HE pyridinium salt.
6.4.3. Reduction of Nitro Compounds and N-Oxides to Amines
The reduction of a nitroarene to an aniline is a commonplace transformation for pharmaceutical and agrochemical synthesis.981 However, often this process requires stoichiometric quantities of reducing agents such as zinc or iron, or as an alternative, heterogeneous hydrogenation over precious metal catalysts. Fukuzumi and co-workers first reported an efficient photoreduction of nitrobenzene and derivatives by a dihydroacridinium reagent under UV irradiation.982 Notably, the reaction was accelerated in the presence of perchloric acid, which the authors suggested facilitated ET.983,984 Helaja and co-workers in 2019 reported a sustainable method for the photocatalytic reduction of nitroquinolines to the corresponding aminoquinolines instead using AscH2 as the reductant (Scheme 354).985 Aminoquinolines are important core heterocycles for antimalarial drugs such as chloroquine, primaquine, and tafenoquine.986 Notably, a photocatalytic method for nitroarene reduction developed by Tung and co-workers employing EY photocatalyst and triethanolamine (TEOA) as a reductant failed in this substrate class.987 The optimized reaction conditions for nitro reduction here involved blue-light irradiation of nitroquinoline substrates in the presence of [Ru(bpy)3]Cl2 ([Ru-1]Cl2) photocatalyst and AscH2 reductant in MeOH/H2O (4:1). Notable observations from the optimization trials indicated that aprotic solvents were poorly suited and that use of sodium ascorbate in place of AscH2 gave no reactivity. A scope of 21 examples of nitroheteroarene reduction to the corresponding heteroarylamine was reported in yields of 16–99%. Primarily nitroquinolines were studied, with examples of 4-, 5-, 6-, 7-, and 8-nitroquinoline regioisomers all undergoing efficient reduction (354.1–354.6). Reductions of nitro-isoquinoline and nitro-benzothiazole also proved successful. Three examples of the reduction of both nitro and N-oxide functional groups of nitroquinoline N-oxides to the corresponding aminoquinolines were also demonstrated, which is notable in that N-oxidation can be used to bias the regioselectivity of the heteroarene electrophilic nitration reaction and is often removed in an operation separate from nitro group reduction. A nitroindazole reacted to yield the corresponding hydroxylamine without further reduction under these reaction conditions. A number of other functional groups were tolerated in this process, including alkenes, alkynes, aryl triflates, aryl acetates, and aryl bromides (354.3), highlighting the chemoselectivity offered via this method.
Scheme 354. Photocatalytic Reduction of Nitroquinolines to the Corresponding Aminoquinolines (Helaja, 2019).

Through reaction monitoring by 1H NMR, the authors determined that a hydroxylamine is formed as an intermediate on the path from the nitro compound to the corresponding amine and is slowly consumed relative to the rate of consumption of the initial nitro compound. However, a putative nitroso intermediate was not observed, suggesting it is rapidly consumed if formed. No intermediates indicating bimolecular condensation pathways for nitro group reduction were observed. SV studies indicated that a combination of nitroquinoline substrate and AscH2 reductant quenched the photoexcited state of Ru(II) more efficiently than either component alone. Also, the direct reductive quenching of photoexcited Ru(II) (E1/2 *Ru(II)/Ru(I) = +0.77 V vs SCE in MeCN)64 by neutral AscH2 (E1/2ox(calc) = +1.42 V vs SCE in MeOH)985 was calculated to be appreciably endergonic, while oxidative quenching of *Ru(II) (E1/2 Ru(III)/*Ru(II) = −0.81 V vs SCE in MeCN)64 by the neutral substrate quinoline too was endergonic (e.g., for 4-methoxy-8-nitroquinoline, E1/2red (calc) = −0.95 V vs SCE in MeOH).985 These combined data suggest that the initiation of the reaction mechanism involves reductive quenching of photoexcited-state Ru(II) complex through an MS-PCET process, jointly mediated by AscH2 as reductant and quinoline substrate as Brønsted base. The resulting Ru(I) complex then mediates reduction of the protonated quinoline substrate. This protonation step was shown to assist single-electron reduction of the nitro group by Ru(I) compared to the neutral substrate, thereby highlighting the observed reactivity preference for the AscH2 reductant compared to the corresponding sodium salt. This sequence leads initially to a nitroso intermediate, which through direct reaction with AscH2, yields the corresponding hydroxylamine. A similar sequence of elementary steps as described above then leads to the aniline product. Deoxygenation is achieved through dehydration upon PT.
Recently, von Wangelin, Konev, and co-workers reported conditions enabling the catalyst-free N-deoxygenation of heteroaryl N-oxides upon blue-light irradiation of substrate solutions in the presence of HE (Scheme 355).988 In addition to acting as a terminal reductant in many reported photocatalytic processes, direct visible-light irradiation of HE results in the generation of a potent single-electron reductant (E1/2red = −2.28 V vs SCE in DMF)821 capable of mediating direct substrate reduction in the absence of any photocatalyst. The optimized reaction conditions consisted of irradiation of substrate and HE in CH2Cl2 with 410 nm lamps. This wavelength offered optimal spectral overlap with the HE absorption. This deoxygenative transformation was demonstrated on 21 examples of heteroarene N-oxide substrates in yields of 20–89%. A number of 3- and/or 4-substituted pyridines (355.1–355.6) were showcased in addition to quinoline (355.7), isoquinoline (355.8), 7-azaindole (355.9), and benzo[c]cinnoline (355.10) substrates. Also, the 2-fold deoxygenation of 2,2′-bipyridine bis-N-oxide and quinoxaline bis-N-oxide proceeded smoothly (355.11, 355.12). In addition, nitroarene reduction proceeded under these reaction conditions to afford the corresponding hydroxylamine (e.g., 355.13), with five examples in yields of 38–86%. No further reduction to the amine was observed even in the presence of excess HE or elevated temperatures.
Scheme 355. Photocatalyst-Free Direct Deoxygenation of Pyridine N-Oxides and Nitroarenes with Hantzsch Ester (von Wangelin and Konev, 2020).

*With 2.4 equiv of Hantzsch ester. **With 2.2 equiv of Hantzsch ester.
In order to understand the mechanism of this transformation, the authors carried out UV–vis spectroscopy experiments, which indicated that a bathochromic shift occurred in the absorption of HE in the presence of the pyridine N-oxide substrate, suggesting the formation of a ground-state EDA complex which facilitates visible-light absorption. Similarly, 1H NMR experiments supported this proposed EDA complex. A Job plot was constructed, indicating a 1:1 ratio of donor to acceptor in the EDA complex. The mode of association was unclear but postulated to involve either π-stacking or a hydrogen bond complex between the HE N–H bond and the pyridine N-oxide.989,990 Thereafter, visible-light irradiation of the EDA complex leads to photoinduced PCET and the formation of a neutral N-hydroxy-1,4-DHP and neutral HE radical. These then react further to form deoxygenated products, but the nature of these subsequent elementary steps was unclear.
7. Reductive Transformations of Heteroarenes through Photochemical and Electrochemical PCET Processes
7.1. Reductive Transformations of Heteroaryl Cyanides
Early work on the photochemistry of cyanopyridines centered on radical substitution reactions in which the cyanide group was substituted for an alkyl group derived from a ketone (e.g., benzophenone),991−993 alcohol,994,995 or olefin996 coupling partner. These methods employed UV light to photoexcite either the cyanopyridine (λmax = 281 nm for 2,4-dicyanopyridine)997 or the ketone component (in the case of benzophenone). The photoexcited pyridine was proposed to engage with the coupling partner through either HAT with an α-oxy or allylic C–H bond, or ET pathways with alcohol substrates, where the photoexcited pyridine could effect the single-electron oxidation of the alcohol. Alternatively, with a UV-absorbing ketone such as benzophenone, the excited state of benzophenone was proposed to reduce the pyridine substrate to a pyridyl radical. Radical–radical coupling between the pyridyl radical and substrate-derived radicals followed by elimination of cyanide gave rise to a variety of C–C coupled products. Many of these transformations were facilitated in the presence of an acid additive, which enhanced the reactivity of the cyanopyridine substrates via protonation.
These early reports largely focused on mechanistic details and did not explore the synthetic utility of this light-induced radical substitution reactivity of cyanopyridines. However, a more recent publication from MacMillan and co-workers outlined the use of heteroaryl cyanides as coupling partners in an α-amino C–H arylation reaction in which the heteroarene was activated for radical–radical coupling with an α-amino radical through single-electron reduction by an excited-state photocatalyst.998 The activation of cyanopyridines and other electron-deficient nitrile-bearing arenes via photoinduced single-electron reduction has since been developed as a useful approach to arylation reactions.680,689,698,884,999 In this section, we focus our attention on several recent photocatalytic and electrochemical methods specifically involving the proton-coupled reductive reactivity of these heteroaryl cyanides, where ET to the heteroarene is facilitated by a preceding or concurrent PT from an acid additive.
Hoshikawa and Inoue developed conditions enabling the 4-pyridination of benzylic C(sp3)–H bonds with 4-cyanopyridine, employing benzophenone as a photocatalyst in aq. MeCN (2:1) under UV light irradiation (Scheme 356).1000 In this work, 31 examples of benzylic C(sp3)–H bond pyridination in yields of 47–90% were recorded. To study the functional group tolerance of this reaction, a series of substituted butylbenzenes was subjected to the optimized reaction conditions. Aromatic ether, ester (356.1), acetamide, chloride, and bromide functional groups were compatible, as were primary aliphatic esters and a primary aliphatic bromide (356.2). Benzylic primary (356.3), secondary, and tertiary C(sp3)–H bonds were all competent coupling partners in this work. The latter transformation provides access to sterically cumbersome quaternary centers adjacent to the pyridine ring, which are difficult to access through alternative C–H activation technologies. Benzylic C–H bonds adjacent to heteroatom functionality were arylated preferentially to those with no additional neighboring group (356.4, 356.5). C–H bonds adjacent to the heteroarenes thiophene and furan also underwent successful pyridination, as did the secondary allylic C–H bond in cyclohexene (356.6). The benzylic C–H pyridination of derivatives of the natural products podocarpic acid (356.7) and totarol (356.8) were demonstrated. Interestingly, a secondary benzylic C–H bond undergoes selective functionalization in the presence of a weaker tertiary benzylic C–H bond in the latter example. It was argued that the neighboring pivaloyl group sterically shielded the isopropyl position. Finally, the application of this methodology to the functionalization of aliphatic secondary and tertiary C–H bonds was demonstrated, albeit with reduced efficiency relative to benzylic positions.
Scheme 356. Photoinduced 4-Pyridination of C(sp3)–H Bonds (Inoue, 2013).

The authors’ mechanistic model involves photoexcitation of the benzophenone catalyst to its triplet diradical excited state. The electrophilic alkoxy radical engages in HAT with the substrate C–H bond to yield a stabilized benzylic radical and a neutral ketyl radical. Coupling of these two radical intermediates was observed as only a minor pathway (approximately 5%). The resulting neutral ketyl radical then facilitates reduction of the cyanopyridine coupling partner through a PCET mechanism, where hydrogen bonding with the aqueous co-solvent activates the cyanopyridine component for ET. This elementary step regenerates the benzophenone sensitizer and generates a neutral 4-cyanopyridyl radical intermediate. Through radical–radical coupling of this persistent pyridyl radical and the transient benzylic radical, a new C–C bond is formed. Elimination of HCN then restores aromaticity to the heteroarene, yielding the product. Significantly attenuated yields were observed under anhydrous conditions (55% compared to 91% yields in a model substrate). Chemoselectivity in C–H bond functionalization was determined by relative C–H bond strengths, with weaker C–H bonds undergoing reaction preferentially.
In addition to C–H bond pyridination as described here, the Inoue group has developed related methodology with benzophenone sensitizers to mediate substrate HAT in the context of C–H bond cyanation,1001,1002 alkenylation,126 and alkynylation.1003 The latter method was employed in their total synthesis of the natural product (+)–lactacystin.1004
Access to hindered primary amines carrying fully substituted α-carbon centers is an underdeveloped area. Rovis and co-workers, in collaboration with Lehnherr, developed an electrochemical synthesis of these valuable targets through the reductive coupling of bench-stable, pre-formed ketiminium hydrochloride salts and cyanoheteroarenes (Scheme 357).869 Optimization of electrode materials, solvent, and electrolyte arrived at reaction conditions consisting of the constant current electrolysis of DMSO solutions of iminium and cyanoheteroarene substrates in an undivided cell equipped with a graphite anode and a Zn cathode with Et4N+Cl– as a supporting electrolyte. A mixture of an iminium hydrochloride salt and a neutral cyanopyridine performed better than a mixture of hydrochloride salts of both iminium and cyanopyridinium components. A total of 51 examples of ketimine reductive heteroarylation were presented in yields of 6–99%. A diverse collection of substituted 4-cyanopyridines were effective coupling partners with a phenyl butyl ketone-derived iminium salt. Substituents tolerated in this transformation included other aryl and heteroaryl ring (357.1) as well as functional groups such as alkyne (357.2), trifluoromethyl (357.3), ester (357.4), amide, and unprotected primary alcohol. 2-Cyanopyridines were also effective, as were a cyanopyrazine (357.5), cyanopyrimidines (357.6), and a cyanopurine. DCB and cyanonaphthalene gave no product under optimized conditions. A range of substituted acetophenone-derived iminium substrates took part in this transformation, but dialkyliminium salts were ineffective. A clear electronic effect in the aryliminium para-substituent was observed, with electron-withdrawing substituents giving lower product yields than electron-donating substituents.
Scheme 357. Electrochemical Synthesis of Hindered Primary Amines through Iminium Heteroarylation with Cyanoheteroarene Reagents (Rovis and Lehnherr, 2020).

The group proposed a plausible mechanism based on experimental observations and DFT calculations. The observation of 1,2-diamines as a major byproduct resulting from reductive homo-coupling of the iminium component suggested the intermediacy of an α-amino radical. 1H NMR analysis of mixtures of iminium salt and neutral cyanopyridine was used to determine the protonation state of the substrates in the reaction, revealing that the iminium component is more basic than the cyanopyridine, with the equilibrium position resting >98% in favor of imine protonation. CV revealed that the reduction of isolated 4-cyanopyridine (Ep/2red = −1.8 V vs Ag/AgNO3 in DMSO)869 is significantly more demanding than that of the isolated iminium hydrochloride salt (Ep/2red = −1.1 V vs Ag/AgNO3 in DMSO).869 However, the admixture displayed an onset potential for reduction more accessible than that of either isolated component (Eonsetred = −1.0 V vs Ag/AgNO3 in DMSO).869 These observations suggested a PCET mechanism for cyanopyridine single-electron reduction at the cathode surface to yield a persistent neutral 4-cyanodihydropyridine radical intermediate. This is further supported by the lack of reactivity with non-basic 1,4-DCB. A similar observation was made by Scheidt and co-workers in the conjugate heteroarylation of arylidene malonates (for a complete discussion, see section 6.1.3).874 This PCET mechanism is favored over a Curtin–Hammett-type situation involving ET to the protonated pyridinium reagent due to its low equilibrium population. Simultaneously at the cathode surface, iminium reduction occurs to generate a neutral α-amino radical intermediate prior to radical–radical coupling and elimination of HCN to give the product. The authors observed that reaction yields were strongly dependent on the cathode material’s overpotential required to drive H+ reduction to H2. High overpotential materials such as Zn (η = 1.09 V) outperformed low overpotential materials such as Pt (η = 0.38 V) (85% compared to 10% ultraperformance liquid chromatography (UPLC) assay yield for a model substrate), indicating a high proton concentration was important for substrate activation. The observed trend between reaction yields and the electronic nature of the aryliminium para-substituent could be understood in terms of matching of the reduction potential of the iminium salt to the onset potential for PCET reduction of the cyanopyridine to better favor radical–radical cross-coupling over homo-coupling processes. The coupled anodic process was unclear but was postulated to involve oxidation of chloride or the DMSO solvent.
Rovis, Lehnherr, DiRocco, and co-workers also reported a photocatalytic synthesis of hindered primary amines achieved through the coupling of either pre-formed iminium salts or O-benzoyl oximes and cyanoheteroarenes (Scheme 358).1005 High-throughput screening revealed the optimal conditions for the reaction with iminium salts to include [Ir(dF(Me)ppy)2(dtbbpy)]PF6 ([Ir-5]PF6) as photocatalyst and DIPEA as the terminal reductant in DMSO under 455 nm light irradiation. Additionally, the cyano heteroarene was added in excess relative to the iminium salt to limit unproductive α-amino radical dimerization. Optimal conditions for O-benzoyl oxime substrates were otherwise identical except for the inclusion of 1 equiv of benzoic acid to overcome an induction phase in the activation of the cyanopyridine substrates. Of the photocatalysts investigated during optimization, oxidizing Ir photocatalysts appeared to be uniquely successful, with Ru and organic photocatalysts of similar redox properties providing only trace yields of product.
Scheme 358. Synthesis of α,α-Disubstituted Primary Amines through the Cross-Coupling of Imines or Oximes with Cyanoheteroarenes (Rovis, Lehnherr, and DiRocco, 2020).

Across the scope of iminium chloride salt and O-benzoyl oxime substrates, 48 examples of α,α-disubstituted primary amines were reported in assay yields of 9–98%. The efficiency of the coupling was unaffected by varied electronic properties of the O-benzoyl oxime substrates (358.1, 358.2), although the product yield was negatively affected by electron-withdrawing substituents on the iminium chloride coupling partner. The presence of an ortho-methyl group on the oxime substrate led to diminished product yield, whereas the same substitution pattern was well tolerated on the iminium chloride partner (358.3). An oxime bearing a cyclopropyl group afforded desired product without competitive ring opening (358.4). Investigation of the scope of cyanoarenes demonstrated that a variety of heterocyclic cyanoarenes were tolerated under both sets of conditions (358.5, 358.6), though non-heterocyclic cyanoarenes failed to provide product. The reaction could be scaled up 12-fold without significant decreases in yield.
The mechanism of this transformation is postulated to initiate via reductive quenching of the excited-state Ir(III) photocatalyst with DIPEA. Although iminoyl radical generation via reductive activation of O-benzoyl oximes is known,1006 the authors in this work propose that Dexter energy transfer leads to triplet sensitization of the O-benzoyl oxime substrates (e.g., for the O-benzoyl oxime of acetophenone, ET(calc) = 54.2 kcal mol–1)1005 by the excited-state Ir(III) photosensitizer ([Ir-5]PF6, ET = 55.8 kcal mol–1)1005 provides access to iminyl radical intermediate through N–O bond homolysis from the triplet sensitized oxime. Poor reaction performance with strongly reducing photocatalyst Ir(ppy)3 (Ir-1) (E1/2 Ir(IV)/*Ir(III)= −1.73 V vs SCE in MeCN; E1/2 Ir(III)/Ir(II)= −2.19 V vs SCE in MeCN)66 as well as the endergonic nature of the single-electron reduction of oxime (E1/2red (calc) = −2.03 V vs SCE)1005 by the excited state of the optimal Ir(III) photocatalyst [Ir-5]PF6 (E1/2 Ir(III)/Ir(II)= −1.43 V vs SCE in MeCN)69 argue against reductive oxime activation. The iminyl radical intermediate is proposed to undergo HAT with i-Pr2NH and protonation by benzoic acid generating an iminium salt in situ. Alternatively, pre-formed iminium hydrochloride salts could be utilized directly. Single-electron reduction of this iminium ion mediated by the α-amino radical-derived from DIPEA (e.g., for acetophenone iminium chloride, ΔE(calc) = −14.4 kcal mol–1)1005 gives rise to the key substrate-derived α-amino radical. The authors also considered Ir(II)-mediated reduction for the generation of this intermediate. The cyano heteroarene coupling partner, likely activated by benzoic acid or iminium cation, is proposed to undergo reductive PCET with the reduced-state Ir(II) complex to generate a persistent neutral 4-cyanodihydropyridine radical. Radical recombination between this persistent radical and the transient α-amino radical leads to a de-aromatized intermediate that undergoes elimination of HCN to afford the desired primary amine product.
7.2. Reductive Transformations of Heteroaryl Halides and Ethers
Early voltammetry studies from Volke, Saveant, and Daasbjerg established that, upon single-electron reduction, halopyridines form neutral pyridyl radicals through mesolytic cleavage of the initially formed radical anion.611,1007−1009 Modern photocatalytic methods for aryl radical generation1010 from arenediazonium salts1011−1013 and arene boronates899,1014−1017 typically fail in the case of pyridines and other heteroarenes due to the instability of the requisite precursors.1018,1019 Tin-mediated halogen atom abstraction for heteroaryl radical generation has been established but is limited to hydrodehalogenation due to rapid HAT with further tin hydride reagent.1020 In this section, we discuss recent photocatalytic methods for heteroaryl radical generation from halide and ether substrates for olefin hydro-heteroarylation, and pyridone synthesis respectively, where reductive PCET is invoked or considered as a mechanism facilitating single-electron reduction.
In 2015, Weaver and co-workers reported the alkylation of five-membered-ring heterocycles, including thiazoles, oxazoles, and imidazoles, with alkenes via the photoinduced single-electron reduction of the corresponding heteroaryl bromides (Scheme 359).1021 This method provides a modular approach to azole functionalization that could streamline SAR studies on bioactive, azole-containing compounds. The optimal conditions were revealed to include blue-light irradiation of the bromoazole and alkene substrates with Ir(ppy)3 (Ir-1) as photocatalyst and 3 equiv each of n-Bu3N and HCO2H in MeCN. The protodebrominated azole was found to be a significant byproduct of the reaction under all conditions. This work expanded upon the group’s 2013 report describing the α-aminoalkylation of 2-chloroazole heterocycles using trialkylamines, which used imidazole as a sacrificial base and a reduced photocatalyst loading under otherwise identical conditions.1022 Here, the researchers reported 31 examples of azole alkylation in yields of 43–93%. With dihydropyran as the acceptor, the reaction tolerated 2-bromothiazoles, benzothiazoles, imidazoles, and benzimidazoles with varying substituents, including esters (359.1) and other halogens (359.2). Alkylation occurred only at the activated C2-bromide and did not affect halogens at other positions on the azole. 2-Bromobenzoxazole failed to give any of the desired alkylation product under these conditions. The alkene scope was very broad and included examples of terminal, 1,1-disubstituted (359.3), trisubstituted (359.4), and styrenyl alkenes. Substrates containing a vinylcyclobutane moiety—for example α-pinene—afforded alkylated thiazole products resulting from ring opening (359.5).
Scheme 359. Photocatalytic Hydro-heteroarylation of Alkenes with 2-Bromoazoles (Weaver, 2015).

*Indicates minor site of heteroarylation
The authors speculate on two mechanistic possibilities: (i) oxidative quenching of the photoexcited-state Ir(III) complex (E1/2 Ir(IV)/*Ir(III)= −1.73 V vs SCE in MeCN)66 with the 2-bromoazole substrate (E1/2red = −1.97 V vs SCE in MeCN)1023 to directly form the corresponding heteroaryl radical anion; or (ii) reductive quenching of the photoexcited-state Ir(III) complex(E1/2 *Ir(III)/Ir(II)= +0.31 V vs SCE in MeCN)66 with the tertiary amine additive (e.g., for n-Bu3N, Epox = +0.62 V vs Ag+/Ag in MeCN)838 to form the corresponding radical cation. The Ir(II) state resulting from this latter pathway (E1/2 Ir(III)/Ir(II)= −2.19 V vs SCE in MeCN)66 is then proposed to reduce the 2-bromoazole substrate. The substrate radical anion, formed through either quenching pathway, then eliminates bromide through mesolytic cleavage to give a 2-azoyl radical and bromide anion. This heteroaryl radical adds across the alkene coupling partner and subsequent HAT to the resulting C-centered radical with either n-Bu3N•+ or n-Bu3N furnishes the final product. Monitoring the ratio of the desired alkylated azole product relative to undesired protodebrominated azole revealed that the product mixture becomes enriched in the desired product as the reaction progresses. The authors rationalize this observation by postulating that acidic species generated through the course of the reaction, such as aminium radical cation,805 may accelerate the formation of alkylated azole product via reductive PCET to the azole heterocycle (e.g., for thiazole, pKaH = 29.4 in DMSO).116 In addition, formic acid (pKa = 20.9 in MeCN)1024 may activate the azole substrate toward reductive PCET at lower conversion. SV emission quenching experiments were attempted to further study this proposal, but results proved inconclusive due to the weak quenching observed by the azole substrate in the presence or absence of amine or acid additives. Weaver and co-workers later expanded this methodology to include azole C2-arylation in the presence of electron-rich arenes,1025 and azole C2-prenylation in the presence of a prenyl sulfone reagent.592
The Jui group has published a collection of elegant photocatalytic olefin hydro-heteroarylation protocols proceeding through heteroarene radical generation via the reductive PCET activation of heteroaryl halides.1026 In a 2017 report, the group demonstrated a reductive Meerwein-type heteroarylation of electron-deficient alkenes with heteroaryl halides via radical generation and conjugate addition (Scheme 360).1027 The combination of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) photocatalyst and HE as a stoichiometric reductant in 25% aq. DMSO proved optimal in this reaction. Authors report 39 examples of alkylated pyridine products in yields of 39–96%. Heteroaryl iodides and bromides were competent precursors in this transformation, and pyridine (360.1–360.4), pyrimidine (360.5), pyrrolopyrimidine (360.6), pyrazine (360.7), and azaindole (360.8) substrates were demonstrated. Notably, 3-halopyridines (e.g., 360.4) are competent precursors to radical generation, enabling access to the 3-alkylpyridine regioisomer which is inaccessible through Minisci radical-addition protocols. A range of unsaturated ketones (360.2), esters, nitriles, and amides (360.3) were effective olefin partners.
Scheme 360. Photocatalytic Meerwein-Type Hydro-heteroarylation of Electron-Deficient Olefins (Jui, 2017).

Selectivity for heteroaryl radical alkylation vs protodehalogenation was shown to be a function of the solvent water content. In the presence of water as co-solvent, the protodehalogenation pathway was suppressed by reducing the effective concentration of the HE reductant due to its limited water solubility. Under the optimal conditions, a 19:1 preference for alkylation over protodehalogenation was achieved. At this stage, mechanistic details regarding generation of the pyridyl radical and termination of the reaction were unclear.
The Jui group then extended this method to achieve a reductive heteroarylation of dehydroalanine derivatives for the synthesis of β-heteroaryl α-amino acids (Scheme 361).1028 Heteroarene incorporation into amino acids and peptides has dramatic effects on their biological activities. For example, azatyrosine displays potent antibiotic and antitumor properties1029,1030 and the (3-pyridyl)alanine residue incorporated into the gonadotropin-releasing hormone antagonist cetrorelix improved aqueous solubility and receptor affinity.1031 Optimized reaction conditions consisted of [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) photocatalyst and HE as a stoichiometric reductant in 17% aq. DMSO. Reactions could be run open to air without consequence and the reaction was demonstrated on a multigram scale with reduced photocatalyst loadings. Here, 34 examples of β-heteroaryl α-AA synthesis were reported in yields of 34–97%. The authors demonstrated 2-, 3-, and 4-pyridyl radical generation from pyridyl iodide and bromide starting materials. Electron-donating (361.1, 361.2) and electron-withdrawing (361.3), hydroxy, and chloride substituents were supported on the pyridine substrates without significant change in reaction efficiency. Pyridine, pyrimidine (361.2), isoquinoline (361.4), and pyrrolopyrimidine heterocycles were competent substrates. A variety of dehydroalanine derivatives were supported, including those carrying a β-substituent (361.5) as well as other electron-deficient carbamate (361.6) and aniline-substituted alkenes. Reaction with the Kardy–Beckwith alkene,243 enabled a highly diastereoselective synthesis of oxazolidinone tethered heteroarenes (361.6), which could be hydrolyzed to the AA, enabling an asymmetric synthesis of heteroarene amino acids.
Scheme 361. Photocatalytic Synthesis of Heteroaryl Amino Acids (Jui, 2017).

Deuterium labeling studies supported a mechanism for radical termination involving single-electron reduction and subsequent protonation of the α-acyl-α-amino radical resulting from conjugate addition of pyridyl radical with dehydroalanine. Complete deuterium incorporation in this position was observed with D2O as co-solvent. In the case of the reaction with the Kardy–Beckwith alkene, the observed diastereoselectivity therefore arises from facial selectivity in a protonation step.
These articles revealed the nucleophilic nature of the neutral pyridyl radical generated under the above reaction conditions, in their addition reactions across electron-deficient olefins. The Jui group discovered that the reactivity of this radical could be inverted by carrying out the reaction under similar conditions to the above but in HFIP solution; this enabled the hydro-heteroarylation of simple, nucleophilic alkenes (Scheme 362).944 This observation implied electrophilic character to the intermediate pyridyl radical in this mildly acidic solvent. Addition of acidic additives AcOH or NH4Cl further boosted the reaction efficiency for the desired transformation, whereas basic additives in this solvent retarded product formation almost entirely. In this work, 31 examples of unbiased olefin hydro-heteroarylation were reported, in yields of 40–91%. With respect to the scope of the halo-heteroarene component, 2-, 3-, and 4-halopyridines were all reactive (362.1–362.3), again highlighting the departure from traditional Minisci-type heteroarylation protocols. Pyridones (362.3), as well as pyridines bearing trifluoromethyl and Boc carbamate (362.2) functional groups remained intact under reaction conditions. In the case of dihalopyridine substrates, mono-alkylation proceeded in moderate yields of 41–57% (e.g., 362.4). Regarding olefin scope, a broad range of terminal olefins were demonstrated carrying pendant carbamate (362.5), primary alkyl alcohol (362.6), primary alkyl chloride (362.7), and phosphonate groups. Both 1,1- and 1,2-disubstituted olefins were also amenable to this transformation (362.8, 362.9). Exclusive anti-Markovnikov heteroarylation of alkenes was observed—this is in contrast to the recent methods for olefin hydro-heteroarylation from Herzon1032 and Shenvi,1033 which initiate instead via HAT to an olefin substrate and subsequent trapping with a heteroarene to yield branched products. When allylbenzene was examined as an olefin substrate, the desired anti-Markovnikov hydro-heteroarylation product was obtained in 76% yield; however, a minor arene heteroarylation product was also obtained in 12% yield. Under modified conditions with excess arene partner (5.0 equiv), these conditions allowed for direct arene C(sp2)–H pyridination, with 3 examples reported in 67–91% yields (e.g., 362.10).
Scheme 362. Photocatalytic Anti-Markovnikov Hydro-heteroarylation of Unbiased Olefins and Arenes (Jui, 2017).

*Indicates minor site of arylation.
Authors conducted competition experiments, where combinations of 2-bromo-6-methylpyridine, ethyl crotanoate, and 1-octene were subjected to their previously reported conditions for electron-deficient olefin hydro-heteroarylation (25% H2O/DMSO) and obtained exclusive crotanoate arylation. Simply reverting to HFIP solution with the addition of NH4Cl completely reversed the selectivity, now enabling octene heteroarylation in good yield, showing how strongly solvent effects dictate the observed reactivity of these pyridyl radicals generated under photoredox catalysis.
When the Jui group later in 2018 sought to expand the scope of their previously published hydro-heteroarylation protocol,944 now to nucleophilic olefins (e.g., in the synthesis of 363.1) they isolated the desired product in low yield (Scheme 363).1034 The authors however noted the formation of oligomeric side products resulting from multiple alkene additions before terminating HAT. In order to accelerate the HAT process, they investigated the use of a polarity-reversal catalyst and identified cyclohexyl thiol as the optimal species to perform the HAT. This protocol was demonstrated on 39 examples with yields ranging from 26% to 83%. The finalized conditions consisted of the halopyridine, functionalized olefin, HE reductant, [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) photocatalyst, and cyclohexyl thiol HAT co-catalyst in TFE solvent under irradiation with blue light. Several functionalized alkenes were amenable coupling partners in the reaction, including halogenated alkenes (363.2), vinyl boronates, silanes (363.3), phosphonates, and sulfides, enol ethers, and even alkynes (363.4).
Scheme 363. Photocatalytic Hydro-heteroarylation of Functionalized Olefins through Reductive PCET (Jui, 2018).

The authors proposed that this transformation proceeds through the following series of elementary steps: first, reductive quenching by a sacrificial amount of HE generates reduced Ir(II) from photo-excited Ir(III).30 Next, reductive PCET involving TFE solvent and the reduced Ir(II) regenerates the Ir(III) photocatalyst and an electrophilic pyridyl radical. Anti-Markovnikov addition of this species to the nucleophilic olefin partner furnishes an electron-rich radical poised to undergo polarity matched HAT with cyclohexyl thiol. Subsequent HAT from HE regenerates the thiol co-catalyst. The reduction potential of 2-bromopyridine has been challenging to measure directly but has been reported to be Ep/2red ≈ – 2.29 V vs SCE in MeCN,1009 which is well out of range of the Ir(II) complex generated through reductive quenching (E1/2 Ir(III)/(II) = −1.51 V vs SCE in MeCN).68 Based on the pKa values of TFE (pKa = 12 in DMSO)116 and 2-bromopyridinium (pKa = 0.5 in DMSO),1034 full PT between solvent and the substrate is unlikely. The authors posited that a concerted PCET mechanism for pyridine reduction followed by rapid mesolytic bond cleavage to expel bromide anion is responsible to the generation of the pyridyl radical. The lowest energy barrier to the concerted pathway was computed to be ΔG⧧(calc) = +13 kcal mol–1, a reasonable barrier for the proposed PCET event. The rapid rate of the reactions (complete in about 10 min) prompted the authors to investigate whether a radical chain mechanism may be responsible for the observed reactivity. However, the relatively low quantum yield (Φ = 0.31) of the process indicates that any radical chain processes that are present are inefficient.
In 2017, Helaja and co-workers disclosed a method for the photocatalytic deprotection of 2- and 4-benzyloxypyridines to yield the corresponding pyridone via a C–C bond cleavage reaction proceeding through a proposed stepwise mechanism of protonation and subsequent single-electron reduction (Scheme 364).1035 In the presence of either Ru(bpy)Cl2·6H2O ([Ru-1]Cl2), [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6), or [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-6]PF6) as a photoredox catalyst and AscH2 as both a proton source and sacrificial reductant in MeCN/H2O (1:1) solution, the benzyloxypyridines were converted to the corresponding pyridones in yields ranging from 36% to quantitative, with most substrates forming product in >90% yield (364.1). The authors demonstrated a scope of 21 substrates. Quinoline and quinazoline derivatives (364.2, 364.3) proved to be particularly effective. Pyridines, pyrimidines (364.4), and isoquinolines (364.5) bearing no substitution other than the ether group also proved amenable to debenzylation. Aryl halides, acetals, esters, and methyl ethers were well-tolerated under the reaction conditions. Additionally, the benzyl group on the ether could be replaced by other leaving groups such as allyl, geranyl, and methylene ester with no loss of yield.
Scheme 364. Photoreductive Deprotection of Benzyloxypyridines for the Synthesis of Pyridones (Helaja, 2017).

The authors propose a mechanism beginning with reductive quenching of the catalyst photoexcited state by AscH2 (E1/2red = −0.41 V vs SCE in MeCN),22 generating reduced photocatalyst (Ru(I) or Ir(II)). The pyridine (e.g., for 2-methoxypyridine, pKaH = 9.98 in MeCN, 3.28 in H2O)1036 is then proposed to undergo protonation by AscH2 (pKa = 18.3 in MeCN; 4.12 in H2O),1037,1038 followed by reduction of the resultant pyridinium by the reduced state of the photocatalyst. The resultant radical species then undergoes β-scission with the exocyclic ether, forming pyridone product and liberating tolyl radical. This mechanism is supported by SV quenching studies, which suggest AscH2 to be the primary quencher of the photocatalyst excited state.
8. Asymmetric Synthesis Enabled by Photochemical PCET
This section discusses several examples of the merger of photochemical substrate activation for reactive radical generation through PCET mechanisms, with both small molecule and enzymatic methods for achieving absolute stereochemical control in the promoted transformations. We also draw the reader’s attention to a report of a CPA-catalyzed asymmetric α-oxidation of cyclic ketones with 1,4-benzoquinone from List and co-workers,1039 and of a Cu-catalyzed asymmetric olefin phosphinoylcyanation reaction from Zou, Liu, and co-workers.1040 These processes proceed through ground-state PCET mechanisms for the generation of reactive intermediates, and thus are outside of the explicit scope of this Review.
8.1. Merging PCET Substrate Activation with Small-Molecular Asymmetric Catalysis
Despite the recent growth in photoredox catalysis and renewed interest in radical-based transformations, few strategies have been developed to provide high enantioselectivities in these processes. One challenge with controlling enantioselectivity is maintaining a meaningful association between a radical intermediate and a chiral catalyst during the selectivity-determining step(s). To address this challenge, Knowles and co-workers reported a highly enantioselective example of catalytic reductive coupling between ketones and hydrazones (Scheme 365).30 These reactions were proposed to proceed through a neutral ketyl radical intermediate generated via concerted PCET jointly mediated by a CPA ((R)-365.A) and [Ir(ppy)2(dtbbpy)]PF6 ([Ir-3]PF6) as the photocatalyst under visible-light irradiation. Analogous to prior work from the same group,16 thermochemical analysis is consistent with a concerted PCET mechanism for ketyl formation—both direct ET from the Ir(II) state of the photocatalyst to the ketone substrate and direct PT from phosphoric acid (R)-365.A are significantly endergonic. The key hydrogen-bonding interaction that is proposed to occur between the neutral ketyl radical and chiral conjugate base of (R)-365.A is supported by (i) the excellent enantioselectivity observed in the reaction, (ii) a linear relationship between the e.e. of the chiral catalyst (R)-365.A and the e.e. of the product, and (iii) DFT studies of the ketyl-phosphate complex. The reaction tolerated various electron-rich and electron-poor acetophenone derivatives to furnish syn-1,2-amino alcohol derivatives with moderate to excellent levels of diastereo- and enantioselectivity (365.1–365.4). Substrates containing heteroatoms in the newly formed ring also gave efficient reactivity and good enantioselectivity (365.3). This work provided the first demonstration of the feasibility and potential benefits of concerted PCET activation in asymmetric catalysis.
Scheme 365. Photocatalytic Asymmetric Aza-pinacol Cyclization Enabled by Concerted PCET (Knowles, 2013).

In 2016, Meggers and co-workers reported an enantioselective method for the synthesis of 1,2-amino alcohols from trifluoromethyl ketones and tertiary amines through stereocontrolled radical–radical recombination (Scheme 366).1041 In the presence of the chiral-at-metal iridium catalyst Δ-[Ir] and under visible-light irradiation, a range of radical cross-coupling products were obtained with good yields, enantioselectivities, and diastereoselectivities (15 examples, 57–97% yield, 91–99% e.e., and up to 10:1 d.r.). The reaction utilized trifluoromethyl ketones with a chelating functional group, such as 1-phenyl-2-imidazolyl or 2-pyridinyl moieties, as well as aniline-derived tertiary amines that can act as good electron donors (366.1–366.4).
Scheme 366. Enantioselective Synthesis of 1,2-Amino Alcohols Enabled by Visible-Light-Activated Iridium Catalysis (Meggers, 2016).

The proposed mechanism begins with the photoexcitation of the iridium-chelated ketone substrate which is then reduced via single-electron reduction from the tertiary amine substrate, generating a reduced iridium–ketyl complex and an aminium radical cation. Following PT between the aminium radical cation and ketyl anion, the nascent Ir-bound neutral ketyl and α-amino alkyl radicals subsequently recombine to form a C–C bond in which the stereochemistry is determined by the chiral environment of the iridium complex. This study represents another example in asymmetric syntheses where the key open-shell intermediates are generated through a stepwise ET/PT sequence. The ketyl radical is only generated upon coordination to the chiral-at-metal iridium catalyst, ensuring that C–C bond formation occurs in an enantioselective manner.
In 2017, Gong and Meggers reported an enantioselective conjugate amination of α,β-unsaturated carbonyl compounds employing [Ir(dF(CF3)ppy)2(5,5′-d(CF3)-bpy)]PF6 ([Ir-9]PF6), phosphate base, and a chiral-at-rhodium LA (Scheme 367).1042 This reaction is proposed to proceed via concerted PCET activation of the N-aryl carbamate substrate to generate the conjugate acid of the base, reduced Ir(II) photocatalyst, and an N-centered radical. Following substrate coordination to Δ-[Rh]PF6, the Ir(II) complex then transfers an electron, furnishing an electron-rich Rh enolate radical. Radical–radical coupling between this intermediate and the electrophilic N-centered radical in the chiral environment of the LA produces a new C–N bond in an asymmetric fashion. Protonation of the Rh enolate by the phosphoric acid and exchange of product by another equivalent of starting material regenerates all the catalytic components.
Scheme 367. Photocatalytic Enantioselective Conjugate Amination Enabled by Concerted PCET (Gong and Meggers, 2017).

Under the optimized reaction conditions, a diverse set of α,β-unsaturated 2-acylimidazoles could be employed in the transformation. Electronically differentiated para-substituted arenes in the β-position all gave excellent yields and enantioselectivities (367.1, 367.2). Meta-substituted arenes (367.3) were tolerated, but ortho-substituted arenes (367.4) provided only trace yield. Several N-aryl carbamates were utilized in the reaction, including a menthol-derived carbamate (367.5). Limitations to the amine scope include N-alkyl carbamates and N-aryl amides.
As a continuation of their protocol for enantioselective conjugate amination,1042 Gong, Meggers, and co-workers reported the enantioselective alkylation of remote, unactivated C(sp3)–H bonds with α,β-unsaturated 2-acylimidazoles (Scheme 368).1043 The reaction proceeds through initial N–H PCET, generating an N-centered amidyl radical.29,220 This amidyl radical then facilitates 1,5-HAT, affording a distal C-centered radical, which can subsequently engage in an enantioselective radical–radical coupling with a chiral Rh-stabilized homoenolate radical to afford the enantioenriched alkylated product. The authors observe a significant dependence on reaction yield depending on the electronic nature of the β-substituent of the 2-acyl imidazole partner. While electron-neutral and electron-deficient (368.1, 368.2) arenes at this position are well tolerated, no product is observed with electron-rich arenes (368.3). This was taken as evidence for the initial reduction of the α,β-unsaturated 2-acyl imidazoles to the homoenolate radical, although the authors note that direct conjugate addition to the Michael acceptor cannot be conclusively ruled out. In addition to β-substitution, the authors also examined the effect of benzamide and alkyl substitution. While PMP and 3-(MeO)C6H4(368.4) substituents are tolerated, 2-(MeO)C6H4 and phenyl substituents yield no product (368.5, 368.6). Additionally, variations on the alkyl substituents afforded products 368.7–368.9 in good yield and selectivity.
Scheme 368. Enantioselective Remote Alkylation of Benzamides with α,β-Unsaturated 2-Acylimidazoles (Gong and Meggers, 2017).

In 2018, Knowles and co-workers reported the synthesis of several pyrroloindoline natural products via indole activation through PCET (Scheme 369).1044 Inspired by PCET activation in enzymatic systems, hydrogen bonding to a tryptophan residue induces ET in an oxidative PCET step. As many of these enzymatic hydrogen-bonding partners are not basic enough to fully deprotonate the resulting indolyl radical cation, these species remain associated as an ionic H-bonded adduct. Based on these observations, the authors proposed that this post-PCET hydrogen-bonding interaction could be leveraged to induce asymmetry in subsequent bond-forming steps.
Scheme 369. Enantioselective Pyrroloindoline Synthesis via Photocatalytic Indole PCET (Knowles, 2018).

The authors envisioned a prospective catalytic cycle wherein oxidation of a tryptophan-phosphate H-bonded adduct by an excited-state photocatalyst would afford an ionic indolyl radical cation-phosphate adduct. The ability of the pre-PCET hydrogen bond to modulate the redox potential of the indole would ensure that oxidation occurs only in the presence of an associated chiral base. The indolyl radical cation would then be trapped at carbon by TEMPO in a radical–radical coupling step and subsequent C–N bond formation would liberate the product and phosphoric acid. PCET between the phosphoric acid and TEMPO, facilitated by the reduced state of the photocatalyst, would regenerate both catalysts and form TEMPO-H as a byproduct. In later synthetic steps, the TEMPO-substituted pyrroloindoline adducts could be derivatized into a variety of natural products through oxidative mesolytic cleavage, generating TEMPO• and a stabilized carbocation that could be trapped by a variety of nucleophiles.1045
The optimal conditions for the synthesis of these enantioenriched TEMPO-pyrroloindoline adducts consisted of blue-light irradiation of N′-Cbz-protected tryptamine in the presence of Ir(ppy)3 (Ir-1) and (S)-H8-TRIP BINOL phosphate tetrabutylammonium salt (n-Bu4N+(S)-369.A–). The addition of 2 equiv of TEMPO served to trap the indolyl radical. TIPS-EBX served as stoichiometric oxidant and base in this reaction, preventing the buildup of TEMPO–H during the reaction. Under these conditions, the authors demonstrated eight examples of enriched pyrroloindoline synthesis with yields ranging from 59% to 81% and enantioselectivities ranging from 87% to 93% e.e. Reaction with N′-Cbz tryptophan provided 369.1 in 81% yield and 93% e.e. on a 10 mmol scale synthesized in a flow reactor. Electron-rich (369.2) and electron-deficient substituents on the indole were tolerated. Additionally, ester (369.3) and BPin (369.4) substituents were amenable to the reaction, providing handles for further elaboration of the products.
The authors then sought to investigate the mechanism of the indole PCET activation. The authors noted that the addition of TIPS-EBX, an iodonium reagent, was required to observe high levels of reactivity. However, this reagent (Ep/2red = −1.12 V vs SCE in MeCN)1044 is not sufficiently oxidizing to interact directly with the indole substrate (e.g., for N-Cbz tryptamine, Ep/2ox = +1.16 V vs SCE in THF).1044 Instead, the authors proposed that TIPS-EBX quenches the excited state of the Ir(III) photocatalyst (E1/2 Ir(IV)/*Ir(III) = −1.73 V vs SCE in THF),66 generating the more oxidizing ground-state Ir(IV)species (E1/2 Ir(IV)/Ir(III) = +0.77 V vs SCE in MeCN).66 This ground-state Ir(IV)complex then serves as the oxidant, generating the phosphate-associated indolyl radical cation in a PCET step. The H-bonding interactions modulate the indole potential but the proton remains associated with the substrate following ET.
Luminescence quenching experiments of photoexcited state of [Ir(ppy)2(dtbbpy)]PF6([Ir-3]PF6) were used to approximate the ground-state PCET reaction of Ir(IV). Quenching of the excited state of this Ir(III) complex was not observed in the presence of the tryptamine substrate. However, the addition of phosphate base resulted in strong luminescence quenching, suggesting that the indole is only oxidized when it is associated with the phosphate base. CV experiments further supported this proposal. Increasing concentrations of phosphate negatively shifted the onset of the oxidation potential of the tryptophan substrate. Furthermore, voltammograms of Ir(ppy)3 revealed that the reversible Ir(IV)/Ir(III) couple was unaffected by the addition of either phosphate or indole alone. However, in the presence of phosphate and tryptophan, the reversible peak was replaced by a new catalytic current feature. These results indicate that phosphate association is required to observe oxidation of the indole to the radical cation, suggesting that this could potentially be a general strategy for substrate activation in asymmetric catalysis. We note that a similar transformation was reported by Xia and co-workers concurrent to this report, involving proposed direct visible-light excitation of the nitroxide additive.1046
In 2018, Jiang and co-workers demonstrated that 1,2-amino tertiary alcohols can be synthesized enantioselectively from N-aryl glycines and 1,2-diketones in the presence of a dicyanopyrazine-derived chromophore (DPZ) and a CPA catalyst (S)-370.A (Scheme 370).81 Under these reaction conditions, N-aryl glycines react with symmetric 1,2-diketones and isatin derivatives to provide two series of chiral 1,2-amino alcohol products in high yields and enantioselectivities. The researchers reported 30 examples of 1,2-amino alcohol synthesis in isolates yields of 61–91% and e.e. of 78–97%. 1,2-Diaryl ketone substrates bearing electron-rich and electron-poor (370.1, 370.2) arenes efficiently afforded product with good to excellent control over the newly formed stereocenters. A similar tolerance of electronic character was observed in the N-aryl group, with electron-donating (370.3, 370.4) and electron-withdrawing substituents all being well tolerated. Various isatin derivatives also underwent efficient and selective reaction. The authors suggest that a stepwise PCET mechanism is operative, wherein the excited-state DPZ photocatalyst oxidizes the N-aryl glycine substrate and subsequently undergoes a PT and decarboxylation to generate an α-amino alkyl radical intermediate. Simultaneously, the reduced state of the DPZ chromophore then reduces the 1,2-diketone, generating a ketyl radical intermediate after a PT. These key open-shell intermediates subsequently undergo radical–radical coupling to provide the desired 1,2-amino alcohol product, in which the stereoselectivity is determined by the bifunctional H-bonding catalyst 370.A–370.C.
Scheme 370. Enantioselective Synthesis of 1,2-Amino Alcohols through Catalytic Radical Coupling of Activated Ketones and N-Arylglycines (Jiang, 2018).

In 2019, Jiang and co-workers reported a photochemical method for the enantioselective preparation of stereocenters in the γ-position of alkylpyridines using reductive activation of aldehydes, ketones, and imines (Scheme 371).82 Previous light-dependent approaches to asymmetric pyridines have established the efficacy of chiral Brønsted acids in asymmetric Minisci-type reactions of prochiral radicals to set stereocenters in the α-position of the pyridine.937,1047 In these methods, asymmetric induction is achieved by coordination of the conjugate base of the chiral acid catalyst to the substrate pyridinium during prochiral radical addition to the pyridine ring. Aiming to develop a method for establishing stereocenters in the more remote γ-position of alkylpyridines, the authors investigated the use of chiral SPINOL-derived spirocyclic phosphoric acids in the addition of prochiral radicals to 2- and 4-vinylpyridines. In the case of aldehydes or ketones as radical precursors, the optimized conditions were established to include DPZ, (S)-371.A as a CPA, a HE terminal reductant, and 3 Å MS as an additive in a mixture of CHCl3 and C6F5H under blue-light irradiation at low temperature. With imine substrates, a higher loading of CPA (S)-371.A (20 mol%) was required for high stereoselectivity to be achieved in toluene as the solvent for adequate reactivity. Across all substrate classes, 49 examples of chiral γ-functionalized alkylpyridines were obtained in yields ranging from 40% to 90% and e.e. of 75–99%. With 2-vinylpyridines as coupling partners, electron-rich (371.1) and electron-poor (371.2) benzaldehydes performed well, as did heteroaryl aldehydes. In addition, α,β-unsaturated aldehydes also were viable substrates, as evidenced by the success of cinnamaldehyde (371.3) as substrate. Moderately high enantioselectivity could still be achieved under further modification of the conditions with 4-vinylpyridines (371.4), where the stereocenter is considerably removed from the site of catalyst coordination. However, alkyl aldehydes were uniformly unsuccessful. A similar scope of ketones proved successful under the modified conditions, including a ketone substrate derived from oestrone (371.6). Imines bearing 4-ethoxyphenyl as an N-substituent participated in this transformation to provide access to chiral γ-amino-substituted pyridines (371.5, 371.7) in a scope similar to those reported for aldehyde and ketone substrates. 4-Vinylpyridines were able to be functionalized with imines in overall good enantioselectivity.
Scheme 371. Enantioselective Synthesis of γ-Functionalized Alkylpyridines via Addition of Prochiral Radicals to Vinylpyridines (Jiang, 2019).

*Reaction performed in toluene.
SV quenching experiments revealed that the photoexcited state of DPZ is not quenched by the HE, benzaldehyde, or 2-vinylpyridine. Control experiments run in the absence of vinylpyridine substrate implicated photoexcited HE as a necessary intermediate, as no pinacol coupling product derived from ketyl radical dimerization was formed when a laser line filter was employed to remove all irradiation below the absorption cut off of the HE at 445 nm. These results led the authors to propose that this reaction initiates via single-electron reduction of the aldehyde substrate (e.g., for benzaldehyde, E1/2red = −1.93 V vs SCE in MeCN)21 by the excited-state HE (E1/2 *HE/HE•+ = −2.28 V vs SCE in MeCN)821 to provide HE radical and the benzaldehyde ketyl radical. Photoexcited-state DPZ can then be reduced by the HE radical (Ered = −0.76 V vs SCE in MeCN)787 to give the radical anion of the DPZ photocatalyst. With the CPA catalyst, the DPZ radical anion can facilitate reductive PCET with the benzaldehyde substrate, generating a phosphate-bound neutral ketyl radical. Conjugate addition of this radical species into the vinylpyridine substrate is proposed to pass through a ternary transition state where the ketyl radical and vinylpyridine substrate are both bound to the chiral phosphate catalyst, thus allowing this key C–C bond-forming step to occur enantioselectively. HAT with another equivalent of HE then affords the closed-shell product. Homo-coupling of the ketyl/iminyl radicals or pyridine-derived radicals was not observed in reactions containing all components, indicating chemoselectivity for conjugate addition of the reductively generated radicals over other competing pathways.
In 2019, Jiang and co-workers reported the asymmetric synthesis of azaarene-substituted secondary alcohols by photocatalytic reduction of ketones (Scheme 372).1048 In this protocol, the authors propose that the initial reduction of the ketone to the ketyl radical anion is facilitated by H-bonding between the 2-quinolyl nitrogen and a CPA ((S)-372.A). Subsequent transfer of two electrons and a proton from a sacrificial amine reductant furnishes the enantioenriched product. A variety of 2-quinolyl ketones were tolerated, including aromatic (372.1) and aliphatic (372.2) substituents. However, polyheteroaromatic substituents were crucial to observe high selectivity (e.g., 372.3), demonstrated by the lower enantioenrichment observed for pyridine derivative 372.4.
Scheme 372. Enantioselective Reduction of Azaarene Ketones by Cooperative Photoredox and Hydrogen-Bonding Catalysis (Jiang, 2019).

*Using modified chiral phosphoric acid. **Reaction run at rt.
The proposed mechanism for this transformation begins with the excitation of the DPZ photocatalyst by visible light. SV quenching experiments reveal that photocatalyst excited state then oxidizes N-phenyl piperidine to the corresponding radical cation, generating DPZ radical anion. The resulting radical anion can then engage the substrate–CPA complex in a reductive PCET step to generate a ketyl radical anion. CV studies revealed that the addition of 10% CPA positively shifted the reduction potential of the ketone substrate by ca. 0.1 V.1049 This ketyl radical anion is then protonated, which the authors proposed forms the second point of non-covalent interaction with the CPA for enantioinduction. This adduct is further reduced to form a benzylic carbanion subsequent with enantioselective protonation affording the enantioenriched product. The authors observed 91% deuterium incorporation at the heterobenzylic position with the addition of deuterium oxide (200 equiv), suggesting that protonation is a feasible mechanism for product formation.
Radical C–H functionalization to form C–C bonds is an active area of research,1050 but examples in asymmetric synthesis are rare. Melchiorre and co-workers reported a visible-light-mediated protocol for the synthesis of β-benzylated aldehydes from the corresponding enals and toluene derivatives in the presence of the chiral gem-difluorinated diarylprolinol silyl ether catalyst (S)-373.A (Scheme 373).739 The reaction is proposed to proceed through an iminium intermediate formed by condensation of the enal substrate with organocatalyst (S)-373.A, catalyzed by LA additive Zn(OTf)2. This photoactive intermediate is then excited (E1/2 ≈ +2.40 V vs Ag/Ag+ in MeCN)1051 and oxidizes the toluene derivative (e.g., for toluene, E1/2ox = +2.36 V vs SCE in MeCN),21 forming a tolyl-derived radical cation and β-enaminyl radical. The radical cation is then deprotonated by the triflate counteranion (e.g., for toluene, pKa of the benzylic C–H bond in the radical cation = −13 in MeCN)247 to generate a benzylic radical by means of a sequential MS-PCET mechanism. The nascent benzylic radical and β-enaminyl radical finally recombine to afford the enantioenriched β-alkylated aldehyde product after hydrolysis. The authors reported 31 examples of β-benzylated aldehyde synthesis in yields of 26–91% and e.e. of 53–91%. Various cinnamaldehyde derivatives with different electronic properties are tolerated as the enal coupling partner with moderate to high yields and enantioselectivities. A range of toluene and xylene derivatives with diverse alkyl substituents as well as ones bearing halogens, esters, PhthHs, and NHPI esters are also competent substrates with good yields and enantioselectivities (373.1–373.4). This study represents an interesting example of a photochemical organocatalytic protocol where stepwise PCET is utilized to activate a C–H bond and the following C–C bond formation is rendered enantioselective through the condensation of a substrate and chiral amine catalyst.
Scheme 373. Enantioselective Synthesis of β-Benzylated Aldehydes via Photocatalytic C(sp3)–H Functionalization of Toluene and Derivatives (Melchiorre, 2018).

Building upon their prior disclosures of racemic intramolecular olefin hydrosulfonamidation,139 Knowles and co-workers reported in 2020 a protocol for the enantioselective intramolecular hydrosulfonamidation of alkenes utilizing a chiral phosphate base to induce selectivity in the C–N bond-forming step to generate enriched pyrrolidine products (Scheme 374).31 Standard reaction conditions consisted of blue-light irradiation of sulfonamide substrate with [Ir(dF(CF3)ppy)2(5,5′-d(CF3)-bpy)]PF6 ([Ir-9]PF6), chiral phosphate (S)-374.A, and TRIP thiol in a solution of PhCF3 at −20 °C. The authors reported 29 pyrrolidine products with 50–98% yields and 87:13 to 98:2 e.r. A variety of sulfonamides were tolerated, including those bearing heteroarene (374.1), arene, and alkyl substituents 374.2. Additionally, sulfamides (374.3) and sulfamate esters proved to be competent substrates in the reaction. Despite reaction selectivity depending upon specific hydrogen-bonding interactions, the reaction also tolerated substituents with other hydrogen-bond-donating and -accepting sites, such as a sildenafil derivative 374.4 which was afforded in 95:5 e.r. Under modified conditions (i.e., higher temperature and substitution of TRIP disulfide for TRIP thiol), disubstituted alkenes could also be accommodated (374.5).
Scheme 374. Photocatalytic Enantioselective Hydrosulfonamidation of Alkenes (Knowles, 2020).

*Reaction at 0 °C with TRIP2S2.
The authors proposed a mechanism involving a hydrogen-bonding association between the substrate and chiral catalyst before PCET-mediated radical generation. After PCET, enantioselective bond formation was affected by the residual neutral non-covalent interactions between the resulting amidyl radical and phosphoric acid. The authors considered another mechanism wherein the trisubstituted alkene is oxidized to the radical cation. In this case, ion-pairing of the alkene radical cation and the phosphate anion would be the primary interaction for enantioinduction. However, comparison of the excited-state oxidation potential of the photocatalyst (E1/2 *Ir(III)/Ir(II) = +1.30 V vs Fc+/Fc in MeCN)29 and the disubstituted alkenes (e.g., for cyclopentene, Ep/2ox = +1.99 V vs Fc+/Fc in MeCN),31 suggests that this oxidation step is significantly endergonic. Additionally, the minimal effect of solvent dielectric on enantioselectivity disfavors an ion-pairing mechanism. The results from these experiments are consistent with a neutral catalyst–radical interaction governing selectivity in the C–N bond-forming step.
8.2. Merging PCET Substrate Activation with Enzymatic Asymmetric Catalysis
The laboratories of Ward and Wenger reported a method for the enantioselective synthesis of secondary amines by merging photoredox catalysis with enzymatic catalysis in an aqueous solution (Scheme 375).1052 The reaction proceeds through visible-light-driven reduction of an imine substrate to form the racemic amine product followed by enantioselective aerobic oxidation of the minor amine enantiomer product, which occurs concurrently in the same reaction pot. In this protocol, Na3[Ir(sppy)3] (sppy = 3-(pyridin-2-yl)benzenesulfonate) is used as a new water-soluble variant of the widely used Ir(ppy)3 (Ir-1). The excited state of this photocatalyst in a buffered aqueous solution leads to the reduction of cyclic imine substrates via a proposed PCET mechanism. The resultant α-amino alkyl radical intermediate is intercepted by HAT with AscH2 to afford the corresponding pyrrolidine derivatives in a racemic fashion. Simultaneously, monoamine oxidase (MAO-N-9) preferentially catalyzes the aerobic oxidation of the (S)-product amine into the corresponding imine starting material. Under continuous light irradiation, this cyclic reaction network provides the desired (R)-enantiomer of the amine product in excellent yield and variable enantioselectivity, 375.1–375.3 (five examples, 92–95% yield and 8–99% ee). Cyclic amines with aliphatic substituents gave excellent enantioselectivity, while those with benzylic substituents provide low enantioselectivity. Time-course studies show that the light-driven reduction of imine reaches full conversion within 2 h, whereas enantioenrichment takes up to 10 h, suggesting that enzymatic oxidation is rate-determining. This study provides a proof-of-concept that the overall transformation can be rendered enantioselective through the introduction of a separate kinetic resolution process that reverts the product to the starting material when the desired reaction pathway and the PCET event are non-selective.
Scheme 375. Enantioselective Synthesis of Pyrrolidine Derivatives through a Concurrent Photoredox and Enzymatic Catalysis (Ward and Wenger, 2018).

In 2018, Hyster and co-workers reported an enantioselective reductive deoxygenation reaction of α-acetoxyketones, combining photoredox- and biocatalysis to achieve non-natural reactivity of a nicotinamide-dependent double-bond reductase (DBR) enzyme (Scheme 376).1053 The optimized catalytic system consisted of a DBR from Nicotiana tabacum(Nt), NADP+, and RB as the photocatalyst in a solution of glucose dehydrogenase (GDH-105) and glucose in KPi buffer. This reaction tolerated a variety of alkyl-substituted tetralones (376.1) and provided moderate selectivity for a seven-membered-ring benzocycloheptanone (376.2) substrate. Acyclic m-methoxy ketones were also promising substrates, providing de-acetoxylated product 376.3 in 44% yield and 70:30 er.
Scheme 376. Enantioselective Reductive Deoxygenation of α-Acetoxy Ketones (Hyster, 2018).

*With glucose-d1
The catalytic cycle commences with excitation of the RB photocatalyst (E1/2 *RB2–/RB3–• = +0.99 V vs SCE in H2O),349 which is quenched by NADPH, generating the reduced state of the photocatalyst and NADPH•+ (E1/2ox = +0.57 V vs SCE in MeCN).1054 The reduced photocatalyst can then reduce the enzyme-bound α-acetoxy tetralone substrate. The authors calculated that the hydrogen-bonding interactions between the substrate and enzyme active site can positively shift the reduction potential of the substrate by +157 mV, effectively enabling reduction only when the substrate is enzyme-bound. The resulting ketyl radical anion then undergoes a spin-center shift process, releasing acetate and forming an α-ketone radical. This radical engages NADPH in enantioselective HAT, generating the deoxygenated product and NADP•. The role of NADPH as a HAT co-catalyst was supported by a deuterium incorporation study, where deuterated NADPD was generated in situ from d-glucose-d1. Example 376.4 was isolated with 80% deuterium incorporation, which was only observed at the α-position, thus confirming the source of the H-atom donor. Oxidation by the RB photocatalyst generates NADP+ and the catalytic cycle is completed by exchange of NADP+ and the product for another equivalent of substrate and NADPH. This report highlights the ability of hydrogen bonding to modulate the reduction potential of a substrate such that the ET only occurs in a chiral environment, thus enabling enantioselective downstream bond-forming events.
In 2019, Hyster and co-workers reported an enantioselective ketone reduction through a photoenzymatic catalytic system, inducing non-natural reactivity in a flavin-dependent ene-reductase (FDER) biocatalyst (Scheme 377).1055 In this manifold, H-bonding in the active site of a morphinone reductase from P. pudita (MorB) induces ET from a Ru(I)species, generated through reduction of the excited-state Ru(II) photocatalyst. The resulting ketyl radical anion then undergoes enantioselective HAT with the flavin HQ (FMNhq) cofactor to generate the reduced alcohol product (377.1–377.3). In this report, 10 examples were presented, ranging from 15% to 99% yields and 4–76% e.e. Of particular interest, the authors highlighted the differences in product formation with and without light. The reaction of ketone 377.4 with MorB in the absence of light afforded the alkene reduction product 377.5 in 98% yield. Upon light irradiation, reduction of both the alkene and ketone to product 377.6 was observed in 89% yield and 66% e.e. This example serves to highlight the change in product selectivity under photoenzymatic conditions.
Scheme 377. Non-natural Reactivity of Ene-Reductases Induced by Synergistic Photoenzymatic Catalysis (Hyster, 2019).

The catalytic cycle begins with the reduction of the flavin cofactor to the HQ species (FMNhq) by NADH. This is followed by activation of the ketone substrate by a double hydrogen-bonding interaction with a His/Asn pair of AA residues in the enzyme active site. This hydrogen-bonding activation serves to modulate the reduction potential of the ketone substrate (e.g., for acetophenone, E1/2red = −2.11 V vs SCE in MeCN)21 for single-electron reduction by the strongly reducing ground-state Ru(I) species (E1/2 Ru(II)/Ru(I) = −1.33 V vs SCE in MeCN).64 The resulting ketyl radical anion can then undergo HAT with the FMNhq cofactor to generate the alcohol product and FMNsq. Then, single-electron oxidation of FMNsq to FMNox by the excited-state Ru(II)complex completes the catalytic cycle. In this system, there is a discrepancy in the reduction potentials of the free acetophenone substrate and the Ru(I) reductant. This highlights the role of hydrogen-bonding interactions in the enzyme active site in promoting single-electron reduction of the ketone in a PCET step without PT to the substrate.
9. Materials and Devices Applications of PCET
9.1. Materials Applications of PCET
The utility of PCET as an efficient platform for the homolytic activation of strong bonds is not restricted to the synthesis of small molecules; it has also proved to be crucial in a wide range of material and device applications. We presented here a number of examples employing PCET for (1) the synthesis of NPs and peptide-based hydrogels, (2) depolymerization of native lignin biopolymer into useful chemical feedstocks, (3) triggering switching behaviors in stimuli-responsive materials, and (4) enhancing the performance of energy conversion and energy storage devices.
9.1.1. Synthesis of Metal Nanoparticles
In 2006, Scaiano and co-workers reported a method to synthesize Au NPs through reduction of an Au(III) precursor using ketyl radicals generated via irradiation with UV light (350 nm) (Scheme 378).1056 This method employs Irgacure-2959 (378.1), an α,α-disubstituted hydroxy ketone, as a ketyl radical precursor. UV light irradiation of 378.1 induces a Norrish type I α-cleavage reaction of the weak α-ketol C–C bond, leading to an acyl radical and a ketyl radical. The highly reducing ketyl radical (e.g., for the ketyl radical derived from acetone, E1/2red = −1.8 V vs NHE in H2O, pH = 7)1057 reduces tetrachloroaurate to Au(II) which spontaneously disproportionates to give an equivalent of Au(I). Another equivalent of ketyl radical finally reduces Au(I) to form Au(0) NPs. The authors observed that the acyl radical fragment from 378.1 is oxidized on exposure to air to produce a benzoic acid derivative that stabilizes the product NPs in solution through weak coordination with the NP surface. As a result, the Au NPs produced by this method are stable for months in aqueous suspension without the need for conventional exogenous stabilizing ligands (e.g., thiols, amines, phosphines).
Scheme 378. Photochemical Generation of Metal Nanoparticles (Scaiano, 2006).

Protic solvents with relatively high pKa values were found to be necessary for rapid reduction, leading to the proposal that the reduction of the gold ions by ketyl radical occurred through concerted PCET to produce acetone and a solvated proton as side products.1058 A pre-PCET hydrogen bond, in which the ketyl radical acts as the hydrogen bond donor and the solvent acts as the hydrogen bond acceptor, was established to be crucial.1059 The key to the success of Irgacure-2959 (378.1) in the synthesis of Au NP is the short lifetime of its triplet excited state (10 ns) due to rapid α-cleavage, thus preventing the gold ions in solution from unproductively quenching the pre-scission excited state. Consequently, reduction of Au(III) can be accomplished in minutes to yield NPs of 12 nm diameter on average with good monodispersity (increased light intensity affords larger NPs). In addition to Au NPs, this method also allows the preparation of NPs of a variety of metals, including Ag,1060 Cu,1061 Ru,1062 and Co.1063 In the case of less-readily reduced metals, Irgacure 907 (378.2), a precursor to more reducing α-amino radicals, was observed to be more efficient.1064 However, the innocuous byproducts of reduction with Irgacure-2959 (i.e., acetone) results in the production of NPs with fewer surface ligands than does Irgacure 907 (378.2), which forms an equivalent of morpholine that can bind strongly to the surface of the NPs.
9.1.2. Photo-Cross-Linking of Peptide Hydrogels
Peptide-based supramolecular hydrogels have many biomaterial applications due to their simple synthesis, stimuli-responsiveness, and high biocompatibility.1065−1067 However, because these hydrogels are self-assembled via weak non-covalent interactions, they often exhibit poor mechanical properties and are prone to breakage and solvent erosion.1067 In 2013, Wang and co-workers reported a cross-linking strategy to enhance the mechanical strength and stability of tyrosine-containing peptide hydrogels, with key steps of coupled ET and PT responsible for the generation of a reactive phenoxyl radical intermediate (Figure 12A).1068 This approach found its basis on the well-known [Ru(bpy)3]2+ catalyzed photo-cross-linking reaction of two tyrosine residues in close proximity to generate dityrosine adducts.1069 The proposed mechanism of this reaction involves the initial formation of the photoexcited state of Ru(II) upon visible-light irradiation of the Ru(II) complex (Figure 12B). Single-electron reduction of ammonium persulfate from this species results in decomposition to sulfate radical anion and a sulfate dianion. Oxidation of a tyrosine residue by the resulting Ru(III) complex followed by PT generates a tyrosinyl radical, which subsequently undergoes cross-linking with a proximal tyrosine residue to furnish the dityrosine adduct.
Figure 12.
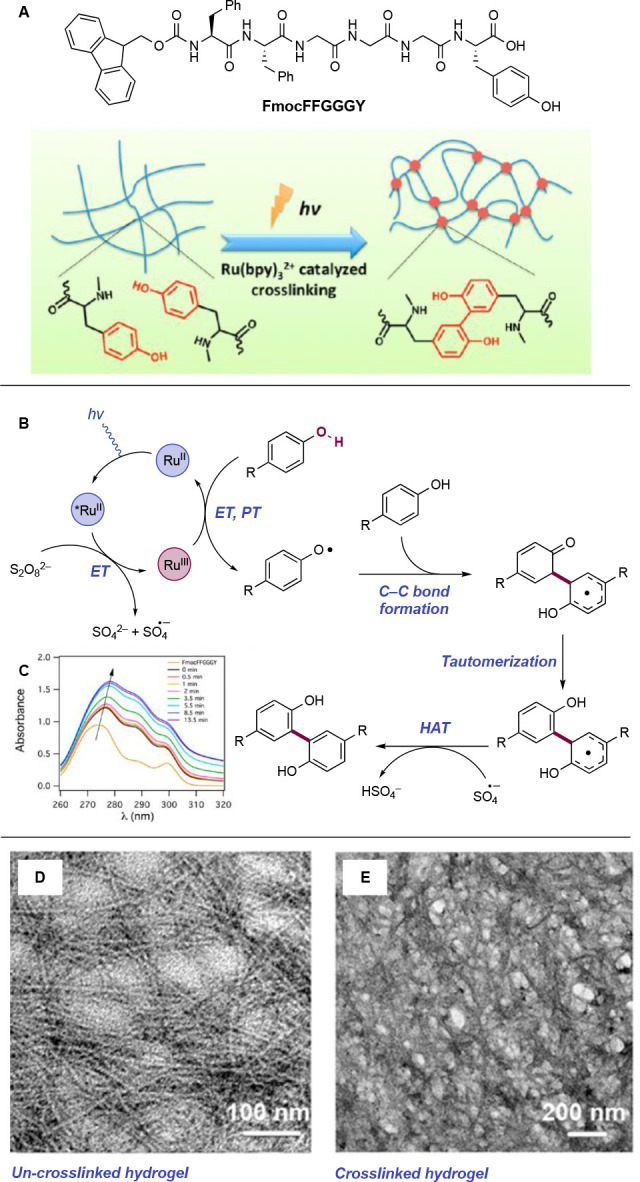
Ru-catalyzed photo-cross-linking reaction of tyrosine-containing peptide hydrogels. (A) Structure of FmocFFGGGY and schematic illustration of hydrogelation and Ru-catalyzed photo-cross-linking of hydrogel. (B) Mechanism of photomediated cross-linking reaction of tyrosine to form the cross-linked dityrosine adduct. (C) UV–vis spectra experiments showing evidence of the formation of peptide dimer through a dityrosine bond. (D, E) TEM images of uncross-linked and cross-linked hydrogels, suggesting increased entanglement of the peptide fibrils upon photo-cross-linking. Reproduced with permission from ref (1068). Copyright 2013 ACS Publications.
In this work, the authors found that Fmoc-Phe-Phe-Gly-Gly-Gly-Tyr-OH (FmocFFGGGY) is the optimal peptide sequence for the hydrogel to achieve efficient photo-cross-linking without material deformation. The triglycine (GGG) linker helped to prevent the dityrosine adducts from interfering with the assembly between FmocFF moiety and other phenyl groups, while also increasing the hydrophilicity of the hydrogelator. Visible-light irradiation of this peptide-based hydrogel in the presence of [Ru(bpy)3]Cl2 ([Ru-1]Cl2) and (NH4)2S2O8 resulted in the formation of the cross-linked hydrogel within 2 min. The cross-linked hydrogel exhibited a storage modulus of 108 kPa, nearly 104 times higher than that of the un-cross-linked hydrogel (∼8 Pa). The improvement in storage modulus was attributed to the dimerization of the peptide, which was evidenced by UV–vis spectroscopy showing that the major peak of tyrosine at 274 nm, upon light irradiation, gradually red-shifted to 278 nm corresponding to the absorbance of dityrosine (Figure 12C). The cross-linked material also showed much higher stability compared to the un-cross-linked material, remaining in the gel form without significant change in shape for at least 13 days, whereas the un-cross-linked hydrogel eroded within 3 days. Studies of the molecular arrangement of the peptide hydrogel using circular dichroism (CD) spectroscopy revealed that photo-cross-linking reaction stabilized the β-sheet structures of FmocFFGGGY assemblies. Atomic force microscopy (AFM) and transmission electron microscopy (TEM) experiments confirmed that the enhancement of the mechanical strength and stability of the hydrogel upon cross-linking was due to the formation of densely entangled fibrous networks of peptide dimers through the proposed dityrosine linkage (Figure 12D). This rapid switch in the mechanical strength and stability of peptide-based hydrogels via a simple light-mediated cross-linking reaction renders this approach amenable to a wide range of biomedical applications in which time is a factor. The employment of this photo-cross-linking reaction to improve the material properties of other tyrosine-containing hydrogels was also reported.1070−1073
9.1.3. Depolymerization of Lignin
Lignin biopolymer, accounting for ∼30% of the organic carbon on earth, is often considered as a renewable alternative resource for petroleum to produce arene feedstocks. Due to the robust and complex structure of the polymer, traditional lignin depolymerization methods require harsh conditions, consume stoichiometric reagents and generate poorly defined mixtures of inseparable products. Stephenson and co-workers employed the use of consecutive electrocatalysis and photoredox catalysis to achieve the depolymerization of native lignin in a two-step, one-pot process at ambient temperature (Scheme 379).92 The first step involved electrocatalytic benzylic oxidation to the corresponding ketone product, which is mediated by C–H HAT with the in situ generated phthalimide N-oxyl radical (PINO) from NHPI. In the second step, the resulting ketone undergoes a previously reported photocatalytic reductive cleavage of the C–O bond to cleave the β-O-4 linkages.1074 Notably, during the optimization for the first oxidation step, the authors found that the use of 2,6-lutidine was beneficial, which helped to dramatically lower the oxidation potential of the PINO/NHPI redox couple via PCET. Furthermore, PCET was also suggested to be the operative mechanism for the oxidation of the important ketyl radical intermediate to provide the desired ketone product (379.1). In addition to the oxidative cleavage of various lignin model substrates, this two-step protocol was also successfully deployed in the depolymerization of native pine lignin into two major monomeric products (379.M1 and 379.M2) in a total yield of 2.4 wt%.
Scheme 379. Two-Step, One-Pot Depolymerization of Native Lignin via Electrocatalysis and Photoredox Catalysis (Stephenson, 2017).
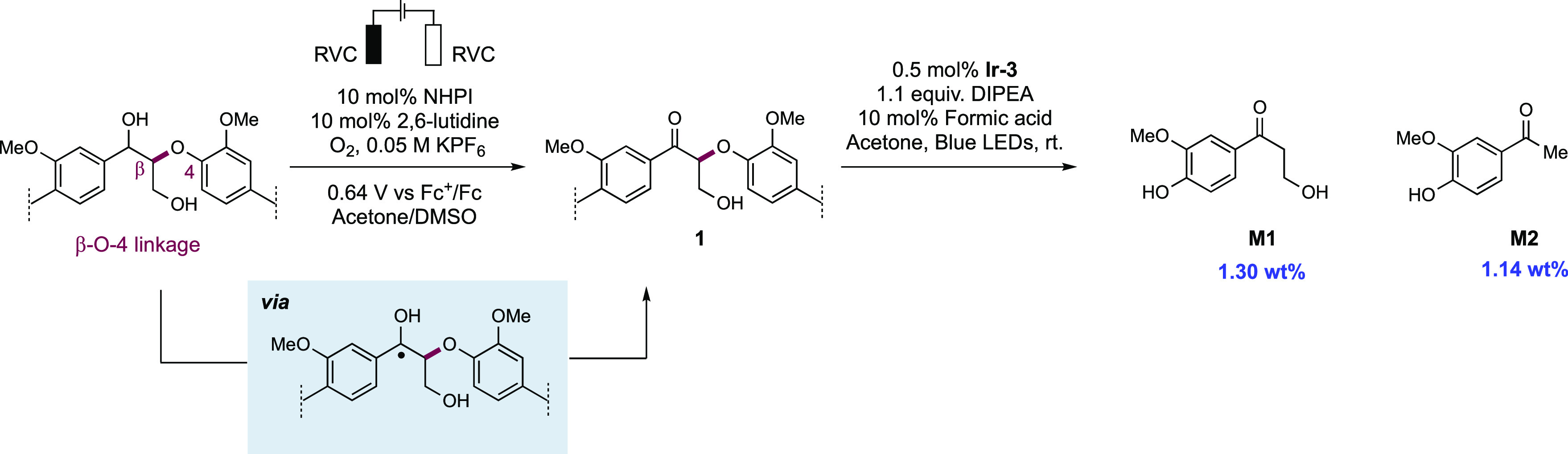
More recently, Knowles and co-workers demonstrated the application of PCET to develop an alternative approach for the photocatalytic depolymerization of unfunctionalized native lignin (Scheme 380).1075 This transformation proceeds via a PCET activation of a benzylic O–H bond to generate a key alkoxy radical intermediate, which triggers the β-scission of a vicinal C–C bond in the lignin β-O-4 linkages in a redox-neutral fashion. The protocol was examined on the fragmentation of β-O-4 and β-1 lignin model dimers and four different isolated softwood lignin materials. When lignin samples were subjected to blue-light irradiation under the optimal catalytic conditions including [Ir(dF(CF3)ppy)2(5,5′-d(CF3)bpy)]PF6 photocatalyst ([Ir-9]PF6), n-Bu4P+(t-BuO)2P(O)O– as a Brønsted base co-catalyst, and 3,4-difluorothiophenol as a HAT co-catalyst, two monomers (380.M2 and 380.M3) were obtained as the major monomeric products in the total yields of 3.4–5.1 wt%, corresponding to the cleavage of at least 22–41% of the total β-ether bonds present in the lignin sample. The feasibility of this depolymerization protocol on a preparative scale (480 mg of lignin polymer) was also demonstrated, from which vanillin (380.M2) could be isolated directly from the reaction mixture. Around the same time, Zhang and co-workers demonstrated that similar PCET conditions were also efficient in the depolymerization of pretreated O-methylated birch lignin, affording monomers with a total yield of 5.0 wt%.1076
Scheme 380. Photocatalytic Depolymerization of Native Lignin through O–H Bond PCET (Knowles, 2020).

9.1.4. Stimuli-Responsive Materials
9.1.4.1. Redox-Switching in Rosarins and Octaphyrins
In 2013, Sessler and co-workers reported the unusual redox properties of a planar 24 π-electron, antiaromatic β,β′-phenylene-bridged hexaphyrin 381.1, an example of a compound class referred to as rosarins (Scheme 381A).1077 Upon protonation by HI, the planar rosarin underwent a proton-coupled reduction to a 26 π-electron, aromatic congener through a stable 25 π-electron intermediate. The use of acids with less redox active conjugate bases, such as perchloric acid, led simply to the protonated starting material, but subsequent introduction of n-Bu4N+I– led to rapid spectroscopic changes consistent with the 25 and 26 π-electron reduced products. Similarly, in the absence of acid, no reduction occurred, even with the reducing iodide anion present. These observations led the authors to propose that protonation is a necessary elementary step in the reduction of 381.1, evidenced by the significant anodic shift of the two one-electron reduction potentials of 381.1 (E1/2red,1 = −0.62 V; E1/2red,2 = −0.92 V vs Fc+/Fc in CH2Cl2)1077 upon protonation to form the triply protonated form H3-381.1+ (E1/2red,1 = +0.42 V vs Fc+/Fc in CH2Cl2; E1/2red,2 = +0.04 V vs Fc+/Fc in CH2Cl2).1077 Interest in this system was prompted by the potential for such protonation-activated redox properties to be applied as a switching mechanism for stimuli-responsive redox activity, such as in smart molecular electronic devices.1078
Scheme 381. Proton-Coupled Redox Switching in Rosarins and Octaphyrins (Sessler, 2013 and 2018).
Later, in 2018, Sessler and co-workers expanded on this work through the study of a larger, 32 π-electron, antiaromatic octaphyrin 381.2 rigidified into a planar geometry via the incorporation of inflexible naphthobipyrrole and dithienothiophene moieties (Scheme 381B).1079 This structural rigidity is believed to play a key role in facilitating the PCET activity of these macrocycles through suppression of competitive processes, such as conformational changes that could arise in the presence of acids or reductants.1080 In fact, the authors found in their previous study that a less rigid, non-annulated rosarin derivative failed to undergo proton-coupled reduction upon treatment with HI or TFA.1077 X-ray diffraction analysis of 381.2 confirmed the desired planar geometry, with atomic deviations from the mean plane not exceeding 0.065 Å. Differential pulse voltammetry (DPV) measurements revealed that 381.2 is difficult to be reduced in the free base form, with reduction potentials at E1/2red,1 = −0.88 V vs Fc+/Fc in CH2Cl2, and E1/2red,2 = −0.99 V vs Fc+/Fc in CH2Cl2.1079 However, in analogy to the 24 π-electron rosarin mentioned previously, the protonated form shows a marked anodic shift in the first and second reduction potentials (ΔE1/2red,1 = +0.56 V and ΔE1/2red,2 = +0.25 V vs Fc+/Fc in CH2Cl2 relative to the free base form). Indeed, upon exposure of 381.2 to HCl, a new fully aromatic, 34 π-electron species H4-381.22+ was observed via UV–Vis–NIR absorption spectroscopy. Relative to the starting octaphyrin, this reduced species has gained four protons and two electrons. In addition to HCl, both HBr and HI both proved to be competent acids for the proton-coupled reduction of 381.2. Acids with less redox active conjugate bases, such as TFA, yielded an EPR-active, 33 π-electron nonaromatic radical resulting from the addition of four protons and one electron to the octaphyrin core. This intermediate radical species could yield the 34 π-electron aromatic species upon further one-electron reduction by FeCp*2. Non-redox-active acids, such as AcOH, led to no notable change in the absorption spectrum of 381.2, as did treatment of 381.2 with tetrathiafulvene (TTF), an organic reductant, in the absence of acid. Reduction readily occurred, however, when 381.2 was exposed to both TTF and AcOH, providing further support for the proton-dependent redox properties of 381.2. Mechanistically, it remains unclear whether protonation occurs prior to or concurrently with reduction, though the authors note that strong redox-active hydrohalic acids may facilitate concerted PCET to 381.2.
9.1.4.2. Electrochromic and Photochromic Behavior
In 2019, Zhang and co-workers developed bistable electrochromic devices based on concerted intramolecular PCET (Figure 13).1081 The color change induced from these devices is controlled by redox reactions of electrochromic materials consisting of copolymers of poly(methyl methacrylate) grafted with functional subunits containing a redox-active proton donor (e.g., p-phenylenediamine) tethered to a proton acceptor acting as a molecular switch (e.g., a fluoran derivative) (Figure 13A). Upon application of a positive voltage, synchronous transfer of two electrons and one proton between the p-phenylenediamine and fluoran moieties results in the color change associated with the formation of the fluoran’s ring-opened form (Figure 13B). Several functionalized copolymers bearing different substituents on the fluoran moiety were synthesized to achieve color switching between transparent and magenta, blue, yellow, and black. Multilayer electrochromic devices of these polymeric materials were also fabricated to study their potential use in display applications (Figure 13C). Overall, the devices exhibit excellent performance compared with others previously reported, including on-switching time shorter than 1.5 s, off-switching time shorter than 7.0 s, 70% transmittance change, a coloration efficiency of 1240 cm2/C, bistability lasting for more than 52 h, and reversibility for more than 12 000 cycles. In addition, a variety of solid-state electronic shelf label (ESL) prototypes with different colors and patterns were also realized (Figure 13D).
Figure 13.
Design of bistable electrochromic devices based on concerted intramolecular PCET. (A) General structure of polymeric electrochromic materials used in the study and proposed PCET mechanism responsible for color switching between a transparent state and magenta (R1 = R3 = NEt2, R2 = H). (B) Schematic of an electrochromic device. (C) Multicolor ESL prototypes (the colored outlines represent the rim of ESLs). (D) A single-color ESL prototype (dashed blue line represents the ongoing process for the ESL display, and the solid blue line represents the completed process). Reproduced with permission from ref (1081). Copyright 2019 Springer Nature.
An additional report by Hou and co-workers described a host–guest synergistic metal–organic framework (MOF) material {[ZnL(bpe)0.5]·0.5(H2bpe)·3(H2O)}n (F14.1, H4LCl = 3,5-dicarboxylic-1-(3,5-dicarboxybenzyl)pyridinium chloride, bpe = 1,2-di(pyridin-4-yl)ethylene) with photochromic properties originating from light-induced PCET (Figure 14).1082 Crystallizing in the triclinic P1̅ space group, F14.1 consists of the host framework [Zn2L2(bpe)]2– and the guest molecule H2bpe2+ which interacts with the uncoordinated carboxylate group on the skeleton via a stable hydrogen bond (Figure 14A). Irradiation of F14.1 with a 365 nm light source triggered a reversible color change from pale yellow to deep red. This photochromism is attributed to the formation of the H2bpe2+• radical in a light-induced PCET process wherein electrons on the uncoordinated carboxylate group are transferred to the guest pyridinium nitrogen through a synergy-coupling interaction of protons in the N–H···O hydrogen bonds (Figure 14B). A slight shift of the proton toward the – COO– group and the appearance of −COOH on the host framework were also indicated via IR analysis. Hence, this redox event was proposed to proceed via PCET, as it was observed that NH3-treated F14.1 exhibited a complete loss of photoresponsive behavior due to a competitive interaction of NH3 with the protons of F14.1. Overall, F14.1 is stable in air and different chemical environments, and exhibits excellent photochromic performance, including good reversibility and fatigue resistance. Different patterns were successfully recorded on the same sample repeatedly, demonstrating the potential application of F14.1 as a photoprinting and anti-counterfeiting material (Figure 14C).
Figure 14.
Development of a host–guest collaborative MOF as a photochromic material. (A) The host–guest collaborative framework structure of F14.1. (B) Proposed mechanism for the photoinduced formation of H2 bpe2+• radical, which is responsible for photochromic behavior of N. (C) Generation of different patterns on the same film of F14.1. Reproduced with permission from ref (1082). Copyright 2019. The Royal Society of Chemistry.
9.2. Applications of PCET in Devices
9.2.1. Energy Conversion and Energy Storage Devices
PCET has also proved to be an important mechanism by which the performance of supercapacitors (SCs) can be enhanced. Traditional supercapacitors are electrical double layer capacitors, in which charge storage is accomplished by ion adsorption at the electrode–electrolyte interface. To this end, high surface area materials (e.g., graphene) are typically coated with the electrode to enhance ion adsorption near the electrode surface.1083,1084 In 2011, Santamaria and co-workers successfully demonstrated that adding redox-active molecules (e.g., HQ) to the electrolyte significantly improved the overall capacitance of these carbon-based SCs.1085 It was shown that specific capacitances (Ce) of four different SCs were at least 2 times higher after the addition of HQ to the supporting electrolyte (Figure 15A,B). Notably, chemically activated carbon material AC-KOH exhibited the greatest increase in its capacitance, which tripled the original Ce value and reached 901 F g–1 at 2.65 mA cm–2. This Ce was the highest value reported for carbon-based capacitors at the time, and was also greater than the best value reported for SCs (720 F g–1) prior to this work.1086 This remarkable improvement in the capacitance was attributed to the additional pseudocapacitive contribution from the redox reaction of the HQ/BQ system, which involves a transfer of 2H+ and 2e– (Figure 15C). The kinetics of the ET process of this redox couple strongly depends on the surface characteristics of the electrode, giving different carbon electrodes distinct electrochemical responses. In the long term cyclic test of the best performing material AC-KOH in the presence of HQ, a 65% drop in Ce was observed with the main loss occurring during the first 1000 cycles. This loss of capacity is related to the fact that the redox reaction of HQ is not completed within the operating voltage window of the cell.
Figure 15.
Use of redox molecules for the performance improvement of the carbon-based SCs. (A) Specific capacitances with current density in 0.1 M H2SO4 solution of four carbon-based SCs. (B) Specific capacitances with current density of four carbon-based SCs in HQ/H2SO4 solution (prepared by dissolving 0.38 M HQ in 0.1 M H2SO4). (C) Redox reaction of HQ/BQ system. Reproduced with permission from ref (1085). Copyright 2011 John Wiley and Sons.
Considering the low long-term stability of this system, Singh and Paul developed an alternative method to improve the capacitance retention of AC-based supercapacitor.1087 The electrode material used in this study was produced by physisorbing HQ on AC via a simple shaking method. Thermogravimetric analysis (TGA) and scanning electron microscopy (SEM) measurements showed that the resulting HQ-AC material contains ∼8 wt% HQ and is more porous than the original AC. By depositing the material on a Pt electrode, an HQ-AC composite material was produced, which was studied electrochemically. Interestingly, the redox chemistry of HQ-AC was determined to be reversible at all scan rates investigated (5–100 mV/s) (Figure 16A), and the ET kinetics were also found to be very fast, as evidenced by the narrow separation of the anodic and cathodic peaks.1088 The electrochemical reversibility and the fast ET kinetics of HQ-AC were attributed to PCET processes catalyzed by the oxide sites present on the surface of this material. It was proposed that even though the carbonyl groups of AC are only weakly basic, the hydrogen bonds between these functionalities and the physisorbed HQ molecules can lower the O–H BDFE of HQ, facilitating the PT from HQ to AC during the oxidation step of the PCET event (Figure 16B). The overall reaction involves the transfer of two electrons to the electrode and two protons to the oxides of AC from HQ. Similar ET and PT events occur during the reverse process to regenerate HQ from BQ. Overall, the Ce value of HQ-AC was nearly three times higher than that of AC alone, which was attributed to pseudocapacitance (∼40%) from the HQ/BQ redox reaction (Figure 16C), as well as to the improvement of the double layer capacitance of HQ-AC due to its better morphology (e.g., larger pore size) that allows more ions to diffuse inside the pore of the material. Moreover, the performance of HQ-AC was also assessed in solutions of pH ranging from 2.2 to 7.2. While the total Ce systematically decreased with increasing pH (Figure 16C), the redox chemistry of HQ remained chemically reversible. The material also exhibited excellent capacitance retention, as at least 84% of the capacitance was retained after 1000 cycles at various pH values, suggesting long-term stability of HQ during electrochemical cycling. In short, these examples from the Santamaria and Paul groups highlight the significance of PCET in the supercapacitor applications.
Figure 16.
Study of physisorbed HQ on activated charcoal material as a supercapacitor. (A) Cyclic voltammograms at scan rates of 5 (green trace), 10 (blue), 20 (red), and 50 (black) mV/s. (B) Proposed overall mechanism of HQ redox chemistry on electrode surface when HQ is physisorbed on AC. (C) Total capacitance, double layer capacitance, and pseudocapacitances calculated from cyclic voltammograms at 5 mV/s scan rate for HQ-AC in different pH electrolyte solutions. Reproduced with permission from ref (1087). Copyright 2015 ACS Publications.
In 2014, Xie, Bakker and co-workers demonstrated a preliminary application of PCET reactivity to the conversion of light energy to electrical energy (Figure 17).1089 The photoelectric cell used in this process employed a tetrabromoquinone/-hydroquinone (F17.Q/F17.H2Q) redox couple in DMF or MeCN solvent that engages in PCET with transparent indium tin oxide electrodes. In order to create a potential difference between the two electrodes, a photochromic spiropyran F17.SP was included. Upon irradiation with UV light (365 nm ±10 nm), spiropyran F17.SP spontaneously ring-opens to the more basic merocyanine (MC) isomer F17.MC, which creates a drop in proton concentration and a resultant drop in potential on the irradiated electrode in accordance with the Nernst equation. Protonated MC F17.MCH+ then diffuses to the opposite dark electrode where it reverts to spiropyran and releases a proton, raising the electrode’s potential through an increase in local proton concentration. By mass action, the PCET reaction of the redox couple is driven to oppose this proton gradient. A steady-state short circuit current density of 9 μA/cm2 was obtained using the cell setup depicted. The authors noted that a decrease in the kinetic barrier to PCET, such as through catalyst impregnation of the electrode surface, modification of the redox pair, and altering reaction media, would result in larger theoretical photocurrent.
Figure 17.

Photoelectric conversion cell set up and reactions (Xie and Bakker 2014).
In addition to the conversion of light energy into electric potential energy, PCET has also been applied in the charging mechanism of a redox battery. For example, Haga and co-workers reported an redox battery based on the PCET reactions of benzimidazole-ligated Ru complexes (Figure 18).1090 Similar to the cell developed by Bakker and co-workers,1089 this cell is based on the creation of a proton gradient between the two electrodes. The Ru complexes studied contained derivatives of the tridentate ligand 2,6-bis(benzimidazol-2-yl)pyridine [H2-BIMP] and the more electron-donating bis(benzimidazolyl)benzene [H2-BIMB]. The imino N–H groups of these ligands are Brønsted acidic and exhibit pKa values that depend on the oxidation state of the Ru center. Complexes in the Ru(III) state showed pKa values several orders of magnitude lower than the corresponding Ru(II) complexes (e.g., pKa,1 of F18.1/Ru(II) = 6.31; pKa,1 of F18.1/Ru(III) < 2 in buffered MeCN). In addition, complexes containing the more electron-donating H2-BIMB ligand were found to be weaker Brønsted acids than the analogous H2-BIMP complexes (e.g., pKa,1 of F18.1/Ru(II) = 6.31; pKa,1 of F18.2/Ru(II) = 10.91). Complexes containing H2-BIMB were also more reducing (e.g., for F18.2, E1/2Ru(III)/Ru(II) = −0.16 V vs Fc+/Fc in buffered MeCN) than the analogous H2-BIMP complexes (e.g., for F18.1, Ru(III)/Ru(II)) = 0.47 V vs Fc+/Fc in buffered MeCN). Because the acidities and redox potentials of these Ru complexes can be tuned so substantially through the choice of ligand and oxidation state of Ru, placement of a Ru(II) H2-BIMP complex F18.1 (low pKa) at the cathode of a divided cell and a Ru(III) H2-BIMB complex F18.2 (high pKa) at the anode produces a cell capable of creating and maintaining a proton gradient as a means of energy storage. Upon constant-current charging of the cell, F18.1 undergoes oxidative PCET with the cathode, donating an electron to the electrode and concomitantly releasing a proton from the H2-BIMP ligand to solution. At the same time, F18.2 undergoes reductive PCET with the anode, accepting an electron from the electrode while concomitantly protonating one of the benzimidazole moieties of the H2-BIMB ligand. As a result of these PCET processes during charging, the pH at the cathode of the cell decreases from 7.0 to 4.2 while the pH at the anode increases from 7.0 to 8.7. The cell reaches full charge when these two half-cell pH values maximally diverge. The cell containing the F18.1/F18.2 redox pair underwent a change in open-circuit potential (OCP) from 0.1 V prior to charging to 0.62 V after a full charge. This cell was demonstrated to reliably undergo at least five completely reversible charging/discharging cycles without a decrease in OCP at full charge. The importance of the proton gradient for energy storage in the cell was underscored through the observation that no change in OCP occurred when Britton–Robinson buffer was used as the electrolyte. Under such conditions, no proton gradient could be maintained upon charging. Use of methyl-substituted ligands Me2-BIMP andMe2-BIMB resulted in no change of pH during the charging and discharging cycles, establishing the importance of PCET with the imino N–H groups of the ligands.
Figure 18.

Electrochemical cell assembly and half-cell reactions during charging. Constant current = 0.1 mA; electrolyte = 0.1 M NaCl; OCP = 0.62 V; solvent = 1:1 v/v H2O/CH3CN (Haga, 2017).
10. Summary
In this Review, we aimed to present a thorough accounting of the photochemical and electrochemical applications of multi-site proton-coupled electron transfer (MS-PCET) in organic synthesis. A central theme throughout was the notion that MS-PCET can serve as a broadly applicable mechanism for H-atom exchange and the generation of useful free radical intermediates. For oxidative MS-PCET, the combination of a single-electron oxidant and a Brønsted base can act as a formal H• acceptor, while the combination of a single-electron reductant and Brønsted acid can act as a formal H• donor in reductive MS-PCET reactions. By physically separating the ET and PT reagents, MS-PCET mechanisms can access a much wider thermodynamic range than is possible using conventional reagents for HAT, enabling the cleavage of very strong E–H bonds in an oxidative manifold, or formation of very weak E–H bonds in a reductive manifold. MS-PCET also offers orthogonal chemoselectivity from what is typically observed via HAT. A requirement for hydrogen-bonding pre-association between substrate and base or acid allows for the selective homolysis of stronger, polar functional groups in the presence of weaker aliphatic C–H bonds, which are poor hydrogen bond donors. Further, these non-covalent interactions have been utilized to enable new opportunities in catalytic, asymmetric bond formation using free radical intermediates generated through PCET events. The emergence of modern photochemical and electrochemical methods in organic chemistry has greatly facilitated the implementation of MS-PCET methods by expanding the toolkit of compatible MS-PCET reagents. The utilization of excited-state photocatalysts in MS-PCET methods provides low and transient concentrations of potent one-electron redox while avoiding potential incompatibilities between oxidant/base or reductant/acid combinations. Similarly, the robustness of electrode materials and modularity of electrochemical methods has provided a number of new MS-PCET methods employing soluble PT reagents interfaced with heterogeneous electrode materials.
We have shown how widely methods involving MS-PCET have been applied in the area of organic synthesis. Combinations of oxidants and bases have been used to affect the homolysis of numerous functional groups containing N–H, O–H, and S–H bonds, for generation of the corresponding neutral radicals (sections 2, 3, and 4 respectively). These MS-PCET methods not only function to generate substrate-bound radicals for subsequent intra- and intermolecular bond formation, but also function to generate reactive radical forms of a catalytic component of the reaction, to then mediate a subsequent substrate activation—for example in the generation of neutral N-centered (section 2.9), O-centered (section 3.5), and S-centered radicals (section 4.4) for use as H-atom abstractors in C–H functionalization reactions. As well as targeting these polar functional groups, an emerging area of C–H bond homolysis via MS-PCET is now demonstrating the utility of the method for bond activation in substrates unable to participate in pre-equilibrium hydrogen bonding (section 5). We have also seen how proper selection of pairs of reductants and acids have enabled the generation of neutral ketyl and α-amino radicals from carbonyl and imine substrates (section 6), and of neutral heteroarene radicals, from heteroaryl nitrile and heteroaryl halide starting materials (section 7). We explored the utility of MS-PCET methods in determining the absolute stereochemical course of a synthetic transformation, through the interfacing of MS-PCET processes for bond homolysis, with both small-molecule and enzymatic methods enabling enantioselective synthesis (section 8). Finally, the application of MS-PCET in materials and devices applications was explored (section 9).
In closing, given that the great majority of the work discussed in this Review was reported only in the past 7–8 years, we anticipate significant future growth in the applications of MS-PCET in organic synthesis. We hope for increased recognition and understanding of concerted PCET in the synthetic community, then becoming a considered aspect in the design of novel transformations. Further study on the unique attributes of C–H PCET and how these differ from canonical HAT would expand the availability of C–H bonds amenable to functionalization, and the types of coupled processes from the C-centered radical. One could envisage extension to other functional groups, to more complex substrates, and to other E–H bonds not yet documented. The further development of these methods for bond homolysis in tandem with molecular catalysts enabling control of the stereochemical course of a transformation is also of particular interest. We are optimistic that these mechanisms may provide novel solutions to synthetic challenges in a wide variety of contexts and are excited to see how these advances unfold in the years to come.
Acknowledgments
Financial support was provided by the NIH (R35 GM134893). P.R.D.M wishes to thank the European Commission for a Marie-Skłodowska-Curie Individual Fellowship (Grant number: 886224). J.H.C. is supported by the National Science Foundation Graduate Research Fellow Program (#DGE-2039656). N.D.C. thanks the NIH for support through a Ruth L. Kirschstein National Research Service Award (F32GM142190). S.T.N. thanks the Andlinger Center for Energy and the Environment (Princeton University) for a Maeder Graduate Fellowship. The authors would like to acknowledge Prof. Dian Wang for early contributions to this project.
Glossary
Abbreviations
- 1-Cl-AQN
1-chloroanthraquinone
- 2-t-Bu-AQN
2-tert-butylanthraquinone
- 3DPA2FBN
2,4,6-tris(diphenylamino)-3,5-difluorobenzonitrile
- 3DPAFIPN
2,4,6-tris(diphenylamino)-5-fluoroisophthalonitrile
- 4CzIPN
1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene
- 4CzPEBN
2,3,4,6-tetra(9H-carbazol-9-yl)-5-(1-phenylethyl)benzonitrile
- 4DPAIPN
2,4,5,6-tetrakis(diphenylamino)isophthalonitrile
- AA
amino acid
- AC
activated carbon
- Ac
acetyl
- ACE
angiotensin-converting enzyme
- Acr
acridinium
- ACS
American Chemical Society
- ADME
adsorption, distribution, metabolism, and excretion
- AFM
atomic force microscopy
- AIBN
2,2′-azobis(isobutyronitrile)
- AM1
Austin Model 1
- API
active pharmaceutical ingredient
- aq.
aqueous
- AQN
anthraquinone
- Ar
aryl
- AscH2
ascorbic acid
- Asn
asparagine
- Asp
aspartic acid
- atm
atmospheres
- BDD
boron-doped diamond
- BDE
bond dissociation energy
- BDFE
bond dissociation free energy
- BDMAP
1,6-bis(dimethylamino)pyrene
- BET
back electron transfer
- BHT
butylated hydroxytoluene
- BIMB
1,3-bis(benzimidazol-2-yl)benzene
- BIMP
2,6-bis(benzimidazol-2-yl)pyridine
- BINAP
2,2-bis(diphenylphosphino)-1,1-binaphthyl
- BINOL
1,1′-bi-2-naphthol
- BIOAc
benzodioxolane acetate
- Bn
benzyl
- BNAH
1-benzyl-1,4-dihydronicotinamide
- Boc
tert-butoxycarbonyl
- bpe
1,2-di(pyridin-4-yl)ethane
- BPO
benzoyl peroxide
- bpy
2,2-bipyridine
- bpz
2,2′-bipyrazine
- BQ
benzoquinone
- BT
2-phenyl-dihydrobenzothiazoline
- Bz
benzoyl
- calc
calculated
- Cat
catecholato
- CB
conduction band
- Cbz
benzyloxycarbonyl
- CD
circular dichroism
- CDC
U.S. Centers for Disease Control and Prevention
- CF
carbon felt
- CFL
compact fluorescent light
- COD
1,5-cyclooctadiene
- COF
covalent organic framework
- Cp
cyclopentadienyl
- CPA
chiral phosphoric acid
- CpN
4-N,N-dimethylanilinecyclopentadienyl
- CT
charge transfer
- CuAO
copper amine oxidase
- CV
cyclic voltammetry
- Cys
cysteine
- Cz
N-carbazolyl
- DABCO
1,4-diazabicyclo[2.2.2]octane
- DABSO
1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) adduct
- DBR
double-bond reductase
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCA
9,10-dicyanoanthracene
- DCB
1,2-dicyanobenzene
- DCE
1,2-dichloroethane
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DDQH
2,3-dichloro-5,6-dicyano-1,4-hydroquinone
- DFT
Density Functional Theory
- DHP
1,4-dihydropyridine
- DIPEA
diisopropylethylamine
- DMA
N,N-dimethylacetamide
- DMAP
4-dimethylaminopyridine
- DME
1,2-dimethoxyethane
- DMF
N,N-dimethylformamide
- dmgH
dimethylglyoxime
- DMPO
5,5-dimethyl-1-pyrroline-N-oxide
- DMPU
N,N′-dimethylpropyleneurea
- DMSO
dimethyl sulfoxide
- DOE
U.S. Department of Energy
- DOI
digital object identifier
- DPPE
1,2-bis(diphenylphosphino)ethane
- DPV
differential pulse voltammetry
- DPZ
dicyanopyrazine
- dq
2,2′-biquinoline
- d.r.
diastereoisomeric ratio
- dtbbpy
4,4′-di-tert-butyl-2,2′-bipyridine
- DTBP
di-tert-butyl peroxide
- DTT
dithiothreitol
- e.e.
enantiomeric excess
- e.r.
enantiomeric ratio
- eq
equation
- EB
Eosin B
- EBX
ethynylbenziodoxolone
- ECE
electron transfer–chemical reaction–electron transfer
- EDA
electron donor–acceptor
- EDG
electron-donating group
- EPR
electron paramagnetic resonance
- equiv
equivalents
- ERFC
Energy Frontier Research Center
- ESL
electronic shelf label
- ESR
electron spin resonance
- ET
electron transfer
- EWG
electron-withdrawing group
- EY
eosin Y
- F-TEDA
1-(chloromethyl)-4-fluorotriethylenediammonium bis(tetrafluoroborate)
- Fc
ferrocene
- FDA
U.S. Food and Drug Administration
- FDER
flavin-dependent ene-reductase
- FEP
fluorinated ethylene propylene
- Fl
fluorescein
- FMN
flavin mononucleotide
- Fmoc
fluorenylmethoxycarbonyl
- FTO
fluorine-doped tin oxide
- Gly
glycine
- GC
gas chromatography
- GDH
glucose dehydrogenase
- HAT
hydrogen-atom transfer
- HCN
hyperpolarization-activated cyclic nucleotide-gated
- HE
Hantzsch ester
- HFIP
hexafluoroisopropanol
- His
histidine
- HIV
human immunodeficiency virus
- HLF
Hofmann–Löffler–Freytag
- HMG-CoA
β-hydroxy-β-methylglutaryl coenzyme A
- HOMO
highest occupied molecular orbital
- HPLC
high-performance liquid chromatography
- HQ
hydroquinone
- HRMS
high-resolution mass spectrometry
- HTE
high-throughput experimentation
- IBB
1-butoxy-1,3-benzo[d][1,2]iodaoxol-3(1H)-one
- IBX
2-iodoxybenzoic acid
- IC50
half-maximal inhibitory concentration
- IPA
isopropanol
- IPET
interparticle electron transfer
- IR
infrared
- KIE
kinetic isotope effect
- KPi
potassium phosphate buffer
- LA
Lewis acid
- LCMS
liquid chromatography/mass spectrometry
- LED
light-emitting diode
- Leu
leucine
- Lys
lysine
- MALDI
matrix-assisted laser desorption/ionization
- MAO
monoamine oxidase
- MB
Methylene blue
- MC
merocyanine
- Mes
mesityl
- Met
methionine
- MNP
2-methyl-2-nitrosopropane
- MOF
metal–organic framework
- MorB
morphinone reductase B
- MPA
3-mercaptopropionic acid
- MS
mass spectrometry
- MS-PCET
multisite proton-coupled electron transfer
- NADH
nicotinamide adenine dinucleotide
- NADP
nicotinamide adenine dinucleotide phosphate
- NBS
N-bromosuccinimide
- NCS
N-chlorosuccinimide
- Neop
neopentylglycolato
- NHC
N-heterocyclic carbene
- NHE
normal hydrogen electrode
- NHPI
N-hydroxyphthalimide
- NHS
N-hydroxysuccinimide
- NIH
U.S. National Institutes of Health
- NIR
near-infrared
- NMR
nuclear magnetic resonance
- NP
nanoparticle
- NSC
Cancer Chemotherapy National Service Center
- OCP
open-circuit potential
- PBS
phosphate-buffered saline
- PC
photocatalyst
- PCET
proton-coupled electron transfer
- PE
polyethylene
- PET
photoinduced electron transfer
- Ph
phenyl
- Phe
phenylalanine
- phen
1,10-phenanthroline
- PhthH
phthalimide
- PIDA
(diacetoxyiodo)benzene
- Pin
pinacolato
- PINO
phthalimide N-oxyl radical
- Piv
pivalate
- PMP
4-methoxyphenyl
- PP
polypropylene
- PPAR
peroxisome proliferator–activator receptor
- PPP
poly(p-phenylene)
- ppy
2-phenylpyridine
- PT
proton transfer
- PTZ
phenothiazine
- QD
quantum dot
- quintri
1-(1-(p-tolyl)-1H-1,2,3-triazol-4-yl)isoquinoline
- RB
rose bengal
- RGD
Arg-Gly-Asp
- r.r.
regioisomeric ratio
- rt
room temperature
- RSC
Royal Society of Chemistry
- RVC
reticulated vitreous carbon
- SAR
structure–activity relationship
- SCE
saturated calomel electrode
- SCS
spin-centered shift
- SEAr
electrophilic aromatic substitution
- SEM
scanning electron microscopy
- Ser
serine
- SET
single-electron transfer
- SLAP
silicon amine protocol
- SnAP
stannylamine protocol
- SNAr
nucleophilic aromatic substitution
- SOMO
singly occupied molecular orbital
- SPINOL
1,1-spirobiindane-7,7′-diol
- SQ
semiquinone
- SRAr
homolytic aromatic substitution
- SuFEx
sulfur fluoride exchange
- SV
Stern–Volmer
- TA
transient absorption
- TADDOL
α,α,α′,α′-tetraaryl-2,2-dimethyl-1,3-dioxolane-4,5-dimethanol
- TAPA
tert-amyl peracetate
- TBPA
tert-butyl peracetate
- TBPB
tert-butyl perbenzoate
- TCB
1,2,4,5-tetracyanobenzene
- TDBMS
tert-butyldimethylsilyl
- TEM
transmission electron microscopy
- TEMPO
2,2,6,6-(tetramethylpiperidin-1-yl)oxyl
- TEOA
triethanolamine
- Tf
triflyl
- TFA
trifluoroacetic acid
- TFE
1,1,1-trifluoroethanol
- TGA
thermogravimetric analysis
- THF
tetrahydrofuran
- TIPS
triisopropylsilyl
- TLR8
human toll-like receptor 8
- TMEDA
N,N,N′,N′-tetramethylethylenediamine
- TMS
trimethylsilyl
- ToF
time-of-flight
- TPP
triphenylpyrilium
- TRIP
2,4,6-triisopropylphenyl
- Trp
tryptophan
- Ts
tosyl
- TTF
tetrathiafulvene
- Tyr
tyrosine
- UPLC
ultraperformance liquid chromatography
- UV
ultraviolet
- VB
valence band
- Vis
visible
- Xantphos
4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
Biographies
Philip R. D. Murray received his MSci. degree in chemistry from Imperial College London, UK, in 2012. Philip moved to the University of Cambridge, UK, to pursue a Ph.D. degree under the supervision of Professor Steven V. Ley CBE FRS FMedSci, where he spent time developing new methods involving flow chemistry and working on natural product total synthesis. After graduating in 2016, he moved to the University of Oxford, UK, as a postdoctoral scholar in the group of Professor Michael Willis, developing catalytic methods for sulfonylation and heteroarene cross-coupling. In 2019, Philip moved to Princeton University, NJ, USA, where he is currently a Marie Skłodowska-Curie research fellow in the group of Professor Robert Knowles. Philip’s research interests include developing novel photocatalytic transformations and achieving relative and absolute stereocontrol through non-traditional mechanisms such as catalytic de-racemization.
James H. Cox earned his B.S. degree in chemistry from the University of Minnesota, Twin Cities, in 2019 after growing up in Orono, MN. James conducted undergraduate research on the sigmatropic rearrangement of allylic azides under the supervision of Joseph Topczewski. During the summer of 2019, he joined the lab of Robert Knowles at Princeton University as a Ph.D. student. James’s current research interests include the application of proton-coupled electron transfer to depolymerization and to the development of synthetic methods.
Nicholas D. Chiappini received his B.A. degree in chemistry at Drew University in 2014. Following this, he completed a Ph.D. in 2019 under the guidance of Prof. Justin Du Bois at Stanford University. Nicholas then moved to Princeton University for postdoctoral work with Prof. Robert R. Knowles. His research interests broadly include transition metal- and non-transition metal-catalyzed group-transfer chemistry and utilization of photoredox catalysis thereto.
Casey B. Roos is a graduate student at Princeton University in the lab of Prof. Robert Knowles. Previously, Casey earned a B.S. degree in chemistry from the University of California, Berkeley. She is interested in asymmetric catalysis with radical intermediates.
Elizabeth A. McLoughlin is a postdoctoral researcher in the lab of Professor Robert Knowles in the Department of Chemistry at Princeton University. Elizabeth completed her doctoral training under the supervision of Professor Robert M. Waymouth in the Department of Chemistry at Stanford University, receiving her Ph.D. in 2019. During her graduate studies, she developed electrocatalytic hydrogen atom transfer (ecHAT) as a strategy to improve the thermodynamic efficiency of homogeneous electrocatalytic systems. Currently, Elizabeth’s research focuses on using light-mediated excited-state redox processes to spatiotemporally control polymer properties in collaboration with Professor Brett Fors in the Department of Chemistry at Cornell University.
Benjamin G. Hejna is a graduate student working in the lab of Prof. Robert Knowles in the Department of Chemistry at Princeton University. Before joining the Knowles group at Princeton, Ben graduated from Swarthmore College with a B.A. in chemistry in 2019, under the advising of Prof. Bob Paley. Ben’s current research focuses on the development and understanding of radical-based methods for asymmetric catalysis.
Suong T. Nguyen received her B.S. in chemistry in 2017 at the University of Mississippi, where she conducted undergraduate research with Prof. Davita L. Watkins. Suong is currently a Ph.D. candidate at Princeton University under the mentorship of Prof. Robert R. Knowles, and she is a member of the DOE-sponsored Bioinspired Light Escalated Chemistry (BioLEC) Energy Frontier Research Center (ERFC). Her research explores the use of photoredox catalysis for the development of olefin hydroamidation reactions, valorization of lignin biopolymers, chemical upcycling of synthetic polymers, and surface functionalization of single-crystalline diamonds. Suong is currently the Maeder Graduate Fellow and is supported by the Andlinger Center for Energy and the Environment at Princeton University.
Hunter H. Ripberger is a Ph.D. candidate at Princeton University. Hunter obtained his B.S. in chemistry from the University of North Carolina at Chapel Hill in 2016, where he conducted undergraduate research in the laboratory of Professor David A. Nicewicz. In 2016, Hunter began his graduate studies at Princeton University in the laboratory of Professor Robert R. Knowles. His research focuses on elucidating the mechanism of photoredox transformations enabled by proton-coupled electron transfer.
Jacob M. Ganley obtained his B.S. in chemistry from Northeastern University in 2017, where he completed co-ops at Cubist Pharmaceuticals and Merck Research Laboratories. Jacob began graduate studies at Princeton University later that year, where he joined the Knowles group. His graduate research has focused on the development of photocatalytic methods for hydroamination, ranging from reaction development to asymmetric catalysis.
Elaine Tsui is from Temple City, CA. She obtained a B.A. in chemistry and English in 2015 from Wesleyan University, where she conducted research under the guidance of Professor Albert J. Fry. Since 2017, Elaine has been pursuing her Ph.D. in chemistry at Princeton University under the supervision of Professor Robert R. Knowles. Her research currently focuses on the development of new reactions using proton-coupled electron transfer, with a particular emphasis on the chemistry of alkoxy radicals.
Nick Y. Shin studied chemistry and obtained his B.S. degree under the supervision of Thomas J. Maimone and Richard A. Andersen at the University of California, Berkeley. Nick is currently a graduate student working with Robert R. Knowles in the Department of Chemistry at Princeton University. Nick’s research focuses on developing photocatalytic methods for asymmetric syntheses and studying the mechanism and reactivities of transient open-shell intermediates in organic reactions. He is a recipient of the James Y. and Harriet P. Tong Chemistry Award from the University of California, Berkeley, the Centennial Fellowship from Princeton University, and the Bristol-Myers Squibb Graduate Fellowship in Organic Synthesis.
Brian Koronkiewicz is a postdoctoral researcher at Princeton University. Brian received his Ph.D. in inorganic chemistry from Yale University in 2020 with Professor James Mayer. His graduate research focused on experimental investigations of the thermodynamic and kinetic parameters controlling proton-coupled electron transfers in biology, C–H activation, and catalysis. Brian’s current research focuses on developing new methods for C–H homolysis and functionalization.
Guanqi Qiu obtained her B.A. honors degree in natural sciences at the University of Cambridge in 2015. After studying oxathiazole fragmentation for one year as a research associate with Dr. Brendan Burkett at A*STAR, Singapore, she joined the Knowles group at Princeton University in 2016 for her Ph.D. studies. In the Knowles group, Guanqi works on understanding the mechanism and chemoselectivity of PCET in organic synthesis.
Robert R. Knowles is a Professor of Chemistry at Princeton University. A native of the Shenandoah Valley of Virginia, Rob received a B.S. degree in chemistry from The College of William and Mary in 2003, working with David Kranbuehl and Robert Hinkle. He carried out his doctoral studies on natural product total synthesis at Caltech with David MacMillan, earning his Ph.D. in 2008. Following his graduate studies, Rob was a NIH postdoctoral fellow at Harvard University with Eric Jacobsen, where he studied asymmetric catalysis using chiral hydrogen bond donors. Rob joined the chemistry faculty at Princeton University in the 2011, where his research has focused primarily on the applications of proton-coupled electron transfer in organic synthesis and asymmetric catalysis.
The authors declare no competing financial interest.
References
- Mayer J. M. Proton-Coupled Electron Transfer: A Reaction Chemist’s View. Annu. Rev. Phys. Chem. 2004, 55, 363–390. 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]
- Weinberg D. R.; Gagliardi C. J.; Hull J. F.; Murphy C. F.; Kent C. A.; Westlake B. C.; Paul A.; Ess D. H.; McCafferty D. G.; Meyer T. J. Proton-Coupled Electron Transfer. Chem. Rev. 2012, 112, 4016–4093. 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]
- Savéant J. M. Concerted Proton-Electron Transfers: Fundamentals and Recent Developments. Annu. Rev. Anal. Chem. 2014, 7, 537–560. 10.1146/annurev-anchem-071213-020315. [DOI] [PubMed] [Google Scholar]
- Cukier R. I.; Nocera D. G. Proton-Coupled Electron Transfer. Annu. Rev. Phys. Chem. 1998, 49, 337–369. 10.1146/annurev.physchem.49.1.337. [DOI] [PubMed] [Google Scholar]
- Darcy J. W.; Koronkiewicz B.; Parada G. A.; Mayer J. M. A Continuum of Proton-Coupled Electron Transfer Reactivity. Acc. Chem. Res. 2018, 51, 2391–2399. 10.1021/acs.accounts.8b00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry E. C.; Knowles R. R. Synthetic Applications of Proton-Coupled Electron Transfer. Acc. Chem. Res. 2016, 49, 1546–1556. 10.1021/acs.accounts.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer J. M.; Rhile I. J. Thermodynamics and Kinetics of Proton-Coupled Electron Transfer: Stepwise vs. Concerted Pathways. Biochim. Biophys. Acta, Bioenerg. 2004, 1655, 51–58. 10.1016/j.bbabio.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Mittra K.; Green M. T. Reduction Potentials of P450 Compounds I and II: Insight into the Thermodynamics of C-H Bond Activation. J. Am. Chem. Soc. 2019, 141, 5504–5510. 10.1021/jacs.9b00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosca T. H.; Rittle J.; Krest C. M.; Onderko E. L.; Silakov A.; Calixto J. C.; Behan R. K.; Green M. T. Iron(IV)Hydroxide pKa and the Role of Thiolate Ligation in C-H Bond Activation by Cytochrome P450. Science 2013, 342, 825–829. 10.1126/science.1244373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minnihan E. C.; Nocera D. G.; Stubbe J. A. Reversible, Long-Range Radical Transfer in E. Coli Class Ia Ribonucleotide Reductase. Acc. Chem. Res. 2013, 46, 2524–2535. 10.1021/ar4000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger O. S. Proton-Coupled Electron Transfer with Photoexcited Ruthenium(II), Rhenium(I), and Iridium(III) Complexes. Coord. Chem. Rev. 2015, 282–283, 150–158. 10.1016/j.ccr.2014.03.025. [DOI] [Google Scholar]
- Megiatto Jr J. D.; Méndez-Hernández D. D.; Tejeda-Ferrari M. E.; Teillout A.-L.; Llansola-Portolés M. J.; Kodis G.; Poluektov O. G.; Rajh T.; Mujica V.; Groy T. L.; et al. A Bioinspired Redox Relay That Mimics Radical Interactions of the Tyr-His Pairs of Photosystem II. Nat. Chem. 2014, 6, 423–428. 10.1038/nchem.1862. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Mayer J. M. Moving Protons and Electrons in Biomimetic Systems. Biochemistry 2015, 54, 1863–1878. 10.1021/acs.biochem.5b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh M. H. V.; Meyer T. J. Proton-Coupled Electron Transfer. Chem. Rev. 2007, 107, 5004–5064. 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yayla H. G.; Knowles R. R. Proton-Coupled Electron Transfer in Organic Synthesis: Novel Homolytic Bond Activations and Catalytic Asymmetric Reactions with Free Radicals. Synlett 2014, 25, 2819. 10.1055/s-0034-1379304. [DOI] [Google Scholar]
- Tarantino K. T.; Liu P.; Knowles R. R. Catalytic Ketyl-Olefin Cyclizations Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2013, 135, 10022–10025. 10.1021/ja404342j. [DOI] [PubMed] [Google Scholar]
- Miller D. C.; Tarantino K. T.; Knowles R. R. Proton-Coupled Electron Transfer in Organic Synthesis: Fundamentals, Applications, and Opportunities. Top. Curr. Chem. 2016, 374, 30. 10.1007/s41061-016-0030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skubi K. L.; Blum T. R.; Yoon T. P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capaldo L.; Ravelli D. Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem. 2017, 2017, 2056–2071. 10.1002/ejoc.201601485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth H. G.; Romero N. A.; Nicewicz D. A. Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. 10.1055/s-0035-1561297. [DOI] [Google Scholar]
- a Warren J. J.; Tronic T. A.; Mayer J. M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev. 2010, 110, 6961–7001. 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Agarwal R. G.; Coste S. C.; Groff B. D.; Heuer A. M.; Noh H.; Parada G. A.; Wise C. F.; Nichols E. M.; Warren J. J.; Mayer J. M. Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications. Chem. Rev. 2021, 10.1021/acs.chemrev.1c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordwell F. G.; Fried H. E.; Hughes D. L.; Lynch T. Y.; Satish A. V.; Whang Y. E. Acidities of Carboxamides, Hydroxamic Acids, Carbohydrazides, Benzenesulfonamides, and Benzenesulfonohydrazides in DMSO Solution. J. Org. Chem. 1990, 55, 3330–3336. 10.1021/jo00297a063. [DOI] [Google Scholar]
- Zhao Y.; Bordwell F. G.; Cheng J.-P.; Wang D. Equilibrium Acidities and Homolytic Bond Dissociation Energies (BDEs) of the Acidic H-N Bonds in Hydrazines and Hydrazides. J. Am. Chem. Soc. 1997, 119, 9125–9129. 10.1021/ja971230p. [DOI] [Google Scholar]
- Blanksby S. J.; Ellison G. B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255–263. 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- Bordwell F. G.; Liu W. Z. Solvent Effects on Homolytic Bond Dissociation Energies of Hydroxylic Acids. J. Am. Chem. Soc. 1996, 118, 10819–10823. 10.1021/ja961469q. [DOI] [Google Scholar]
- Yayla H. G.; Wang H.; Tarantino K. T.; Orbe H. S.; Knowles R. R. Catalytic Ring-Opening of Cyclic Alcohols Enabled by PCET Activation of Strong O-H Bonds. J. Am. Chem. Soc. 2016, 138, 10794–10797. 10.1021/jacs.6b06517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota E.; Wang H.; Frye N. L.; Knowles R. R. A Redox Strategy for Light-Driven, Out-of-Equilibrium Isomerizations and Application to Catalytic C-C Bond Cleavage Reactions. J. Am. Chem. Soc. 2019, 141, 1457–1462. 10.1021/jacs.8b12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi G. J.; Zhu Q.; Miller D. C.; Gu C. J.; Knowles R. R. Catalytic Alkylation of Remote C-H Bonds Enabled by Proton-Coupled Electron Transfer. Nature 2016, 539, 268–271. 10.1038/nature19811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rono L. J.; Yayla H. G.; Wang D. Y.; Armstrong M. F.; Knowles R. R. Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization. J. Am. Chem. Soc. 2013, 135, 17735–17738. 10.1021/ja4100595. [DOI] [PubMed] [Google Scholar]
- Roos C. B.; Demaerel J.; Graff D. E.; Knowles R. R. Enantioselective Hydroamination of Alkenes with Sulfonamides Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2020, 142, 5974–5979. 10.1021/jacs.0c01332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrauben J. N.; Cattaneo M.; Day T. C.; Tenderholt A. L.; Mayer J. M. Multiple-Site Concerted Proton-Electron Transfer Reactions of Hydrogen-Bonded Phenols Are Nonadiabatic and Well Described by Semiclassical Marcus Theory. J. Am. Chem. Soc. 2012, 134, 16635–16645. 10.1021/ja305668h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris W. D.; Mayer J. M. Separating Proton and Electron Transfer Effects in Three-Component Concerted Proton-Coupled Electron Transfer Reactions. J. Am. Chem. Soc. 2017, 139, 10312–10319. 10.1021/jacs.7b03562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts B. P. Polarity-Reversal Catalysis of Hydrogen-Atom Abstraction Reactions: Concepts and Applications in Organic Chemistry. Chem. Soc. Rev. 1999, 28, 25–35. 10.1039/a804291h. [DOI] [Google Scholar]
- Milan M.; Salamone M.; Costas M.; Bietti M. The Quest for Selectivity in Hydrogen Atom Transfer Based Aliphatic C-H Bond Oxygenation. Acc. Chem. Res. 2018, 51, 1984–1995. 10.1021/acs.accounts.8b00231. [DOI] [PubMed] [Google Scholar]
- Salamone M.; Bietti M. Tuning Reactivity and Selectivity in Hydrogen Atom Transfer from Aliphatic C-H Bonds to Alkoxyl Radicals: Role of Structural and Medium Effects. Acc. Chem. Res. 2015, 48, 2895–2903. 10.1021/acs.accounts.5b00348. [DOI] [PubMed] [Google Scholar]
- Markle T. F.; Darcy J. W.; Mayer J. M. A New Strategy to Efficiently Cleave and Form C-H Bonds Using Proton-Coupled Electron Transfer. Sci. Adv. 2018, 4, eaat5776. 10.1126/sciadv.aat5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen S. T.; Zhu Q.; Knowles R. R. PCET-Enabled Olefin Hydroamidation Reactions with N-Alkyl Amides. ACS Catal. 2019, 9, 4502–4507. 10.1021/acscatal.9b00966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordwell F. G.; Cheng J. P.; Harrelson J. A. Homolytic Bond Dissociation Energies in Solution from Equilibrium Acidity and Electrochemical Data. J. Am. Chem. Soc. 1988, 110, 1229–1231. 10.1021/ja00212a035. [DOI] [Google Scholar]
- Wayner D. D. M.; Parker V. D. Bond Energies in Solution from Electrode Potentials and Thermochemical Cycles. A Simplified and General Approach. Acc. Chem. Res. 1993, 26, 287–294. 10.1021/ar00029a010. [DOI] [Google Scholar]
- Liu T.; Guo M.; Orthaber A.; Lomoth R.; Lundberg M.; Ott S.; Hammarström L. Accelerating Proton-Coupled Electron Transfer of Metal Hydrides in Catalyst Model Reactions. Nat. Chem. 2018, 10, 881–887. 10.1038/s41557-018-0076-x. [DOI] [PubMed] [Google Scholar]
- Bourrez M.; Steinmetz R.; Ott S.; Gloaguen F.; Hammarström L. Concerted Proton-Coupled Electron Transfer from a Metal-Hydride Complex. Nat. Chem. 2015, 7, 140–145. 10.1038/nchem.2157. [DOI] [PubMed] [Google Scholar]
- Miller D. C.; Choi G. J.; Orbe H. S.; Knowles R. R. Catalytic Olefin Hydroamidation Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2015, 137, 13492–13495. 10.1021/jacs.5b09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt D. A.; Dilabio G. A.; Mulder P.; Ingold K. U. Bond Strengths of Toluenes, Anilines, and Phenols: To Hammett or Not. Acc. Chem. Res. 2004, 37, 334–340. 10.1021/ar010010k. [DOI] [PubMed] [Google Scholar]
- Romero N. A.; Nicewicz D. A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Kim D.; Teets T. S.. Photophysical Properties and Redox Potentials of Photosensitizers for Organic Photoredox Transformations. Synlett, 2021. 10.1055/a-1390-9065 [DOI] [Google Scholar]
- Connelly N. G.; Geiger W. E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- Tshepelevitsh S.; Kütt A.; Lõkov M.; Kaljurand I.; Saame J.; Heering A.; Plieger P. G.; Vianello R.; Leito I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735–6748. 10.1002/ejoc.201900956. [DOI] [Google Scholar]
- Kaljurand I.; Kütt A.; Sooväli L.; Rodima T.; Mäemets V.; Leito I.; Koppel I. A. Extension of the Self-Consistent Spectrophotometric Basicity Scale in Acetonitrile to a Full Span of 28 pKa Units: Unification of Different Basicity Scales. J. Org. Chem. 2005, 70, 1019–1028. 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
- Waidmann C. R.; Miller A. J. M.; Ng C. W. A.; Scheuermann M. L.; Porter T. R.; Tronic T. A.; Mayer J. M. Using Combinations of Oxidants and Bases as PCET Reactants: Thermochemical and Practical Considerations. Energy Environ. Sci. 2012, 5, 7771–7780. 10.1039/c2ee03300c. [DOI] [Google Scholar]
- Qiu G.; Knowles R. R. Understanding Chemoselectivity in Proton-Coupled Electron Transfer: A Kinetic Study of Amide and Thiol Activation. J. Am. Chem. Soc. 2019, 141, 16574–16578. 10.1021/jacs.9b08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu G.; Knowles R. R. Rate-Driving Force Relationships in the Multisite Proton-Coupled Electron Transfer Activation of Ketones. J. Am. Chem. Soc. 2019, 141, 2721–2730. 10.1021/jacs.8b13451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyburski R.; Liu T.; Glover S. D.; Hammarström L. Proton-Coupled Electron Transfer Guidelines, Fair and Square. J. Am. Chem. Soc. 2021, 143, 560–576. 10.1021/jacs.0c09106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J. P.; Yoder J. C.; Won T. J.; Mayer J. M. Application of the Marcus Cross Relation to Hydrogen Atom Transfer Reactions. Science 2001, 294, 2524–2526. 10.1126/science.1066130. [DOI] [PubMed] [Google Scholar]
- Mayer J. M.; Hrovat D. A.; Thomas J. L.; Borden W. T. Proton-Coupled Electron Transfer versus Hydrogen Atom Transfer in Benzyl/Toluene, Methoxyl/Methanol, and Phenoxyl/Phenol Self-Exchange Reactions. J. Am. Chem. Soc. 2002, 124, 11142–11147. 10.1021/ja012732c. [DOI] [PubMed] [Google Scholar]
- Mader E. A.; Mayer J. M. The Importance of Precursor and Successor Complex Formation in a Bimolecular Proton-Electron Transfer Reaction. Inorg. Chem. 2010, 49, 3685–3687. 10.1021/ic100143s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo L.; Pagire S. K.; Reiser O.; König B. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis?. Angew. Chem., Int. Ed. 2018, 57, 10034–10072. 10.1002/anie.201709766. [DOI] [PubMed] [Google Scholar]
- Capaldo L.; Quadri L. L.; Ravelli D. Photocatalytic Hydrogen Atom Transfer: The Philosopher’s Stone for Late-Stage Functionalization?. Green Chem. 2020, 22, 3376–3396. 10.1039/D0GC01035A. [DOI] [Google Scholar]
- Yan M.; Kawamata Y.; Baran P. S. Synthetic Organic Electrochemical Methods since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319. 10.1021/acs.chemrev.7b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston C.; Palkowitz M. D.; Takahira Y.; Vantourout J. C.; Peters B. K.; Kawamata Y.; Baran P. S. A Survival Guide for the “Electro-Curious.. Acc. Chem. Res. 2020, 53, 72–83. 10.1021/acs.accounts.9b00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas J. J.; Sevrin M. J.; Stephenson C. R. J. Visible Light Photocatalysis: Applications and New Disconnections in the Synthesis of Pharmaceutical Agents. Org. Process Res. Dev. 2016, 20, 1134–1147. 10.1021/acs.oprd.6b00125. [DOI] [Google Scholar]
- Twilton J.; Le C. C.; Zhang P.; Shaw M. H.; Evans R. W.; MacMillan D. W. C. The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem. 2017, 1, 0052. 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- Arias-Rotondo D. M.; McCusker J. K. The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design. Chem. Soc. Rev. 2016, 45, 5803–5820. 10.1039/C6CS00526H. [DOI] [PubMed] [Google Scholar]
- Kalyanasundaram K. Photophysics, Photochemistry and Solar Energy Conversion with Tris(Bipyridyl)Ruthenium(II) and Its Analogues. Coord. Chem. Rev. 1982, 46, 159–244. 10.1016/0010-8545(82)85003-0. [DOI] [Google Scholar]
- Haga M. A.; Dodsworth E. S.; Eryavec G.; Seymour P.; Lever A. B. P. Luminescence Quenching of the Tris(2,2’-Bipyrazine)Ruthenium(II) Cation and Its Monoprotonated Complex. Inorg. Chem. 1985, 24, 1901–1906. 10.1021/ic00206a041. [DOI] [Google Scholar]
- Flamigni L.; Barbieri A.; Sabatini C.; Ventura B.; Barigelletti F.. Photochemistry and Photophysics of Coordination Compounds: Iridium. In Photochemistry and Photophysics of Coordination Compounds II; Springer, 2007; pp 143–203. [Google Scholar]
- Doeven E. H.; Zammit E. M.; Barbante G. J.; Francis P. S.; Barnett N. W.; Hogan C. F. A Potential-Controlled Switch on/off Mechanism for Selective Excitation in Mixed Electrochemiluminescent Systems. Chem. Sci. 2013, 4, 977–982. 10.1039/C2SC21707D. [DOI] [Google Scholar]
- Lowry M. S.; Goldsmith J. I.; Slinker J. D.; Rohl R.; Pascal R. A.; Malliaras G. G.; Bernhard S. Single-Layer Electroluminescent Devices and Photoinduced Hydrogen Production from an Ionic Iridium(III) Complex. Chem. Mater. 2005, 17, 5712–5719. 10.1021/cm051312+. [DOI] [Google Scholar]
- Ladouceur S.; Fortin D.; Zysman-Colman E. Enhanced Luminescent Iridium(III) Complexes Bearing Aryltriazole Cyclometallated Ligands. Inorg. Chem. 2011, 50, 11514–11526. 10.1021/ic2014013. [DOI] [PubMed] [Google Scholar]
- Ener M. E.; Darcy J. W.; Menges F. S.; Mayer J. M. Base-Directed Photoredox Activation of C-H Bonds by PCET. J. Org. Chem. 2020, 85, 7175–7180. 10.1021/acs.joc.0c00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M.; Füldner S.; König B.; Zeitler K. Metal-Free, Cooperative Asymmetric Organophotoredox Catalysis with Visible Light. Angew. Chem., Int. Ed. 2011, 50, 951–954. 10.1002/anie.201002992. [DOI] [PubMed] [Google Scholar]
- Lazarides T.; McCormick T.; Du P.; Luo G.; Lindley B.; Eisenberg R. Making Hydrogen from Water Using a Homogeneous System without Noble Metals. J. Am. Chem. Soc. 2009, 131, 9192–9194. 10.1021/ja903044n. [DOI] [PubMed] [Google Scholar]
- Romero N. A.; Nicewicz D. A. Mechanistic Insight into the Photoredox Catalysis of Anti-Markovnikov Alkene Hydrofunctionalization Reactions. J. Am. Chem. Soc. 2014, 136, 17024–17035. 10.1021/ja506228u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuzumi S.; Kotani H.; Ohkubo K.; Ogo S.; Tkachenko N. V.; Lemmetyinen H. Electron-Transfer State of 9-Mesityl-10-Methylacridinium Ion with a Much Longer Lifetime and Higher Energy Than That of the Natural Photosynthetic Reaction Center. J. Am. Chem. Soc. 2004, 126, 1600–1601. 10.1021/ja038656q. [DOI] [PubMed] [Google Scholar]
- Romero N. A.; Margrey K. A.; Tay N. E.; Nicewicz D. A. Site-Selective Arene C-H Amination via Photoredox Catalysis. Science 2015, 349, 1326–1330. 10.1126/science.aac9895. [DOI] [PubMed] [Google Scholar]
- Joshi-Pangu A.; Lévesque F.; Roth H. G.; Oliver S. F.; Campeau L. C.; Nicewicz D.; DiRocco D. A. Acridinium-Based Photocatalysts: A Sustainable Option in Photoredox Catalysis. J. Org. Chem. 2016, 81, 7244–7249. 10.1021/acs.joc.6b01240. [DOI] [PubMed] [Google Scholar]
- Speckmeier E.; Fischer T. G.; Zeitler K. A Toolbox Approach to Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor-Acceptor Cyanoarenes. J. Am. Chem. Soc. 2018, 140, 15353–15365. 10.1021/jacs.8b08933. [DOI] [PubMed] [Google Scholar]
- Luo J.; Zhang J. Donor-Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp3)-C(sp2) Cross-Coupling. ACS Catal. 2016, 6, 873–877. 10.1021/acscatal.5b02204. [DOI] [Google Scholar]
- Garreau M.; Le Vaillant F.; Waser J. C-Terminal Bioconjugation of Peptides through Photoredox Catalyzed Decarboxylative Alkynylation. Angew. Chem., Int. Ed. 2019, 58, 8182–8186. 10.1002/anie.201901922. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Zhang C.; Chin K. F.; Pytela O.; Wei G.; Liu H.; Bureš F.; Jiang Z. Dicyanopyrazine-Derived Push-Pull Chromophores for Highly Efficient Photoredox Catalysis. RSC Adv. 2014, 4, 30062–30067. 10.1039/C4RA05525J. [DOI] [Google Scholar]
- Liu Y.; Liu X.; Li J.; Zhao X.; Qiao B.; Jiang Z. Catalytic Enantioselective Radical Coupling of Activated Ketones with N-Aryl Glycines. Chem. Sci. 2018, 9, 8094–8098. 10.1039/C8SC02948B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao K.; Tan S. M.; Lee R.; Yang S.; Jia H.; Zhao X.; Qiao B.; Jiang Z. Catalytic Enantioselective Addition of Prochiral Radicals to Vinylpyridines. J. Am. Chem. Soc. 2019, 141, 5437–5443. 10.1021/jacs.9b00286. [DOI] [PubMed] [Google Scholar]
- Pavlishchuk V. V.; Addison A. W. Conversion Constants for Redox Potentials Measured versus Different Reference Electrodes in Acetonitrile Solutions at 25°C. Inorg. Chim. Acta 2000, 298, 97–102. 10.1016/S0020-1693(99)00407-7. [DOI] [Google Scholar]
- Francke R.; Little R. D. Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments. Chem. Soc. Rev. 2014, 43, 2492–2521. 10.1039/c3cs60464k. [DOI] [PubMed] [Google Scholar]
- McCarthy B. D.; Martin D. J.; Rountree E. S.; Ullman A. C.; Dempsey J. L. Electrochemical Reduction of Brønsted Acids by Glassy Carbon in Acetonitrile-Implications for Electrocatalytic Hydrogen Evolution. Inorg. Chem. 2014, 53, 8350–8361. 10.1021/ic500770k. [DOI] [PubMed] [Google Scholar]
- Protti S.; Fagnoni M.; Ravelli D. Photocatalytic C-H Activation by Hydrogen-Atom Transfer in Synthesis. ChemCatChem 2015, 7, 1516–1523. 10.1002/cctc.201500125. [DOI] [Google Scholar]
- Luo Y. R.Comprehensive Handbook of Chemical Bond Energies; CRC Press, 2007. [Google Scholar]
- Cao H.; Tang X.; Tang H.; Yuan Y.; Wu J. Photoinduced Intermolecular Hydrogen Atom Transfer Reactions in Organic Synthesis. Chem. Catal. 2021, 1, 523. 10.1016/j.checat.2021.04.008. [DOI] [Google Scholar]
- Zard S. Z. Recent Progress in the Generation and Use of Nitrogen-Centred Radicals. Chem. Soc. Rev. 2008, 37, 1603. 10.1039/b613443m. [DOI] [PubMed] [Google Scholar]
- Nguyen L. Q.; Knowles R. R. Catalytic C-N Bond-Forming Reactions Enabled by Proton-Coupled Electron Transfer Activation of Amide N-H Bonds. ACS Catal. 2016, 6, 2894–2903. 10.1021/acscatal.6b00486. [DOI] [Google Scholar]
- Xiong T.; Zhang Q. New Amination Strategies Based on Nitrogen-Centered Radical Chemistry. Chem. Soc. Rev. 2016, 45, 3069–3087. 10.1039/C5CS00852B. [DOI] [PubMed] [Google Scholar]
- Bosque I.; Magallanes G.; Rigoulet M.; Kärkäs M. D.; Stephenson C. R. J. Redox Catalysis Facilitates Lignin Depolymerization. ACS Cent. Sci. 2017, 3, 621–628. 10.1021/acscentsci.7b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T. Recent Advances in Reactions of Heteroatom-Centered Radicals. Synthesis 2017, 49, 3511–3534. 10.1055/s-0036-1588481. [DOI] [Google Scholar]
- Luo J.; Wei W. T. Recent Advances in the Construction of C-N Bonds Through Coupling Reactions between Carbon Radicals and Nitrogen Radicals. Adv. Synth. Catal. 2018, 360, 2076–2086. 10.1002/adsc.201800205. [DOI] [Google Scholar]
- Zhao Y.; Xia W. Recent Advances in Radical-Based C-N Bond Formation: Via Photo-/Electrochemistry. Chem. Soc. Rev. 2018, 47, 2591–2608. 10.1039/C7CS00572E. [DOI] [PubMed] [Google Scholar]
- Xiong P.; Xu H. C. Chemistry with Electrochemically Generated N-Centered Radicals. Acc. Chem. Res. 2019, 52, 3339–3350. 10.1021/acs.accounts.9b00472. [DOI] [PubMed] [Google Scholar]
- Chan C. M.; Chow Y. C.; Yu W. Y. Recent Advances in Photocatalytic C-N Bond Coupling Reactions. Synthesis 2020, 52, 2899–2921. 10.1055/s-0040-1707136. [DOI] [Google Scholar]
- Kärkäs M. D. Electrochemical Strategies for C-H Functionalization and C-N Bond Formation. Chem. Soc. Rev. 2018, 47, 5786–5865. 10.1039/C7CS00619E. [DOI] [PubMed] [Google Scholar]
- Chen J. R.; Hu X. Q.; Lu L. Q.; Xiao W. J. Visible Light Photoredox-Controlled Reactions of N-Radicals and Radical Ions. Chem. Soc. Rev. 2016, 45, 2044–2056. 10.1039/C5CS00655D. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Studer A. Chemistry With N-Centered Radicals Generated by Single-Electron Transfer-Oxidation Using Photoredox Catalysis. CCS Chem. 2019, 1, 38–49. 10.31635/ccschem.019.20180026. [DOI] [Google Scholar]
- Choi G. J.; Knowles R. R. Catalytic Alkene Carboaminations Enabled by Oxidative Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2015, 137, 9226–9229. 10.1021/jacs.5b05377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanss D.; Freys J. C.; Bernardinelli G.; Wenger O. S. Cyclometalated Iridium(III) Complexes as Photosensitizers for Long-Range Electron Transfer: Occurrence of a Coulomb Barrier. Eur. J. Inorg. Chem. 2009, 2009, 4850–4859. 10.1002/ejic.200900673. [DOI] [Google Scholar]
- Cheng J. P.; Zhao Y. Homolytic Bond Cleavage Energies of the Acidic NH Bonds in Dimethyl Sulfoxide Solution and Properties of the Corresponding Radicals and Radical Cations. Tetrahedron 1993, 49, 5267–5276. 10.1016/S0040-4020(01)82376-0. [DOI] [Google Scholar]
- Giese B. Formation of CC Bonds by Addition of Free Radicals to Alkenes. Angew. Chem., Int. Ed. Engl. 1983, 22, 753–764. 10.1002/anie.198307531. [DOI] [Google Scholar]
- Warren J. J.; Menzeleev A. R.; Kretchmer J. S.; Miller T. F.; Gray H. B.; Mayer J. M. Long-Range Proton-Coupled Electron-Transfer Reactions of Bis(Imidazole) Iron Tetraphenylporphyrins Linked to Benzoates. J. Phys. Chem. Lett. 2013, 4, 519–523. 10.1021/jz400029w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren J. J.; Mayer J. M. Proton-Coupled Electron Transfer Reactions at a Heme-Propionate in an Iron-Protoporphyrin-IX Model Compound. J. Am. Chem. Soc. 2011, 133, 8544–8551. 10.1021/ja201663p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S.; Soudackov A. V. Proton-Coupled Electron Transfer in Solution, Proteins, and Electrochemistry. J. Phys. Chem. B 2008, 112, 14108–14123. 10.1021/jp805876e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S. J.; Soudackov A. V.; Hammes-Schiffer S. Analysis of Kinetic Isotope Effects for Proton-Coupled Electron Transfer Reactions. J. Phys. Chem. A 2009, 113, 2117–2126. 10.1021/jp809122y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilger D. J.; Gesmundo N. J.; Nicewicz D. A. Catalytic Hydrotrifluoromethylation of Styrenes and Unactivated Aliphatic Alkenes via an Organic Photoredox System. Chem. Sci. 2013, 4, 3160–3165. 10.1039/c3sc51209f. [DOI] [Google Scholar]
- Nguyen T. M.; Nicewicz D. A. Anti-Markovnikov Hydroamination of Alkenes Catalyzed by an Organic Photoredox System. J. Am. Chem. Soc. 2013, 135, 9588–9591. 10.1021/ja4031616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkowski A. J.; Nicewicz D. A. Direct Catalytic Anti-Markovnikov Addition of Carboxylic Acids to Alkenes. J. Am. Chem. Soc. 2013, 135, 10334–10337. 10.1021/ja4057294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. M.; Manohar N.; Nicewicz D. A. Anti-Markovnikov Hydroamination of Alkenes Catalyzed by a Two-Component Organic Photoredox System: Direct Access to Phenethylamine Derivatives. Angew. Chem., Int. Ed. 2014, 53, 6198–6201. 10.1002/anie.201402443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilger D. J.; Grandjean J. M. M.; Lammert T. R.; Nicewicz D. A. The Direct Anti-Markovnikov Addition of Mineral Acids to Styrenes. Nat. Chem. 2014, 6, 720–726. 10.1038/nchem.2000. [DOI] [PubMed] [Google Scholar]
- Margrey K. A.; Nicewicz D. A. A General Approach to Catalytic Alkene Anti-Markovnikov Hydrofunctionalization Reactions via Acridinium Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1997–2006. 10.1021/acs.accounts.6b00304. [DOI] [PubMed] [Google Scholar]
- Bordwell F. G.; Ji G. Z.; Satish A. V.; Zhang X.; Cheng J. P. Bond Dissociation Energies in DMSO Related to the Gas Phase. J. Am. Chem. Soc. 1991, 113, 9790–9795. 10.1021/ja00026a012. [DOI] [Google Scholar]
- Larsen A. G.; Holm A. H.; Roberson M.; Daasbjerg K. Substituent Effects on the Oxidation and Reduction Potentials of Phenylthiyl Radicals in Acetonitrile. J. Am. Chem. Soc. 2001, 123, 1723–1729. 10.1021/ja003811b. [DOI] [PubMed] [Google Scholar]
- Bordwell F. G. Equilibrium Acidities in Dimethyl Sulfoxide Solution. Acc. Chem. Res. 1988, 21, 456–463. 10.1021/ar00156a004. [DOI] [Google Scholar]
- Kumler W. D.; Eiler J. J. The Acid Strength of Mono and Diesters of Phosphoric Acid. The N-Alkyl Esters from Methyl to Butyl, the Esters of Biological Importance, and the Natural Guanidine Phosphoric Acids. J. Am. Chem. Soc. 1943, 65, 2355–2361. 10.1021/ja01252a028. [DOI] [Google Scholar]
- Nomrowski J.; Wenger O. S. Photoinduced PCET in Ruthenium-Phenol Systems: Thermodynamic Equivalence of Uni- and Bidirectional Reactions. Inorg. Chem. 2015, 54, 3680–3687. 10.1021/acs.inorgchem.5b00318. [DOI] [PubMed] [Google Scholar]
- Rhile I. J.; Mayer J. M. One-Electron Oxidation of a Hydrogen-Bonded Phenol Occurs by Concerted Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2004, 126, 12718–12719. 10.1021/ja031583q. [DOI] [PubMed] [Google Scholar]
- Markle T. F.; Rhile I. J.; DiPasquale A. G.; Mayer J. M. Probing Concerted Proton-Electron Transfer in Phenol-Imidazoles. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 8185–8190. 10.1073/pnas.0708967105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markle T. F.; Tronic T. A.; Dipasquale A. G.; Kaminsky W.; Mayer J. M. Effect of Basic Site Substituents on Concerted Proton-Electron Transfer in Hydrogen-Bonded Pyridyl-Phenols. J. Phys. Chem. A 2012, 116, 12249–12259. 10.1021/jp311388n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruccolo S.; Qin Y.; Schnedermann C.; Nocera D. G. General Strategy for Improving the Quantum Efficiency of Photoredox Hydroamidation Catalysis. J. Am. Chem. Soc. 2018, 140, 14926–14937. 10.1021/jacs.8b09109. [DOI] [PubMed] [Google Scholar]
- Jia J.; Ho Y. A.; Bülow R. F.; Rueping M. Brønsted Base Assisted Photoredox Catalysis: Proton Coupled Electron Transfer for Remote C-C Bond Formation via Amidyl Radicals. Chem. - Eur. J. 2018, 24, 14054–14058. 10.1002/chem.201802907. [DOI] [PubMed] [Google Scholar]
- Meyer A. U.; Jäger S.; Prasad Hari D.; König B. Visible Light-Mediated Metal-Free Synthesis of Vinyl Sulfones from Aryl Sulfinates. Adv. Synth. Catal. 2015, 357, 2050–2054. 10.1002/adsc.201500142. [DOI] [Google Scholar]
- Miyamoto N.; Fukuoka D.; Utimoto K.; Nozaki H. The Reaction of Styryl Sulfoxides or Sulfones with Boranes. Bull. Chem. Soc. Jpn. 1974, 47, 503–504. 10.1246/bcsj.47.503. [DOI] [Google Scholar]
- Amaoka Y.; Nagatomo M.; Watanabe M.; Tao K.; Kamijo S.; Inoue M. Photochemically Induced Radical Alkenylation of C(sp3)-H Bonds. Chem. Sci. 2014, 5, 4339–4345. 10.1039/C4SC01631A. [DOI] [Google Scholar]
- Noble A.; MacMillan D. W. C. Photoredox α-Vinylation of α-Amino Acids and N-Aryl Amines. J. Am. Chem. Soc. 2014, 136, 11602–11605. 10.1021/ja506094d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S.; Gutiérrez-Bonet Á.; Molander G. A. Merging Photoredox PCET with Ni-Catalyzed Cross-Coupling: Cascade Amidoarylation of Unactivated Olefins. Chem. 2019, 5, 339–352. 10.1016/j.chempr.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc. 2015, 137, 4896–4899. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields B. J.; Doyle A. G. Direct C(sp3)-H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc. 2016, 138, 12719–12722. 10.1021/jacs.6b08397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S.; Zhang S. Q.; Saeednia B.; Zhou J.; Anna J. M.; Hong X.; Molander G. A. Diastereoselective Olefin Amidoacylation via photoredox PCET/Nickel-Dual Catalysis: Reaction Scope and Mechanistic Insights. Chem. Sci. 2020, 11, 4131–4137. 10.1039/D0SC01459A. [DOI] [Google Scholar]
- Horner J. H.; Musa O. M.; Bouvier A.; Newcomb M. Absolute Kinetics of Amidyl Radical Reactions. J. Am. Chem. Soc. 1998, 120, 7738–7748. 10.1021/ja981244a. [DOI] [Google Scholar]
- Yu X.; Zhou F.; Chen J.; Xiao W. Visible Light Photocatalytic N-Radical-Based Intramolecular Hydroamination of Benzamides. Acta. Chimica. Sinica 2017, 75, 86–91. 10.6023/A16070367. [DOI] [Google Scholar]
- Zou S.; Geng S.; Chen L.; Wang H.; Huang F. Visible Light Driven Metal-Free Intramolecular Cyclization: A Facile Synthesis of 3-Position Substituted 3,4-Dihydroisoquinolin-1(2H)-One. Org. Biomol. Chem. 2019, 17, 380–387. 10.1039/C8OB02560F. [DOI] [PubMed] [Google Scholar]
- Wayner D. D. M.; McPhee D. J.; Griller D. Oxidation and Reduction Potentials of Transient Free Radicals. J. Am. Chem. Soc. 1988, 110, 132–137. 10.1021/ja00209a021. [DOI] [Google Scholar]
- Hu X.-Q.; Chen J.-R.; Wei Q.; Liu F.-L.; Deng Q.-H.; Beauchemin A. M.; Xiao W.-J. Photocatalytic Generation of N-Centered Hydrazonyl Radicals: A Strategy for Hydroamination of β,γ-Unsaturated Hydrazones. Angew. Chem., Int. Ed. 2014, 53, 12163–12167. 10.1002/anie.201406491. [DOI] [PubMed] [Google Scholar]
- Tarantino K. T.; Miller D. C.; Callon T. A.; Knowles R. R. Bond-Weakening Catalysis: Conjugate Aminations Enabled by the Soft Homolysis of Strong N-H Bonds. J. Am. Chem. Soc. 2015, 137, 6440–6443. 10.1021/jacs.5b03428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrams R.; Lefebvre Q.; Clayden J. Transition Metal Free Cycloamination of Prenyl Carbamates and Ureas Promoted by Aryldiazonium Salts. Angew. Chem., Int. Ed. 2018, 57, 13587–13591. 10.1002/anie.201809323. [DOI] [PubMed] [Google Scholar]
- Zhu Q.; Graff D. E.; Knowles R. R. Intermolecular Anti-Markovnikov Hydroamination of Unactivated Alkenes with Sulfonamides Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2018, 140, 741–747. 10.1021/jacs.7b11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Xia D.; Qin W.; Zhou R.; Zhou X.; Zhou Q.; Liu W.; Dai X.; Wang H.; Wang S.; et al. A Radical Cascade Enabling Collective Syntheses of Natural Products. Chem. 2017, 2, 803–816. 10.1016/j.chempr.2017.04.007. [DOI] [Google Scholar]
- Brandau S.; Landa A.; Franzén J.; Marigo M.; Jørgensen K. A. Organocatalytic Conjugate Addition of Malonates to α,β- Unsaturated Aldehydes: Asymmetric Formal Synthesis of (−)-Paroxetine, Chiral Lactams, and Lactones. Angew. Chem., Int. Ed. 2006, 45, 4305–4309. 10.1002/anie.200601025. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Dai X.; Song H.; He H.; Wang X.; Liu X. Y.; Qin Y. Concise Syntheses of Eburnane Indole Alkaloids. Chem. Commun. 2018, 54, 9510–9512. 10.1039/C8CC05374J. [DOI] [PubMed] [Google Scholar]
- Xu H. C.; Moeller K. D. Intramolecular Anodic Olefin Coupling Reactions: The Use of a Nitrogen Trapping Group. J. Am. Chem. Soc. 2008, 130, 13542–13543. 10.1021/ja806259z. [DOI] [PubMed] [Google Scholar]
- Xu H. C.; Moeller K. D. Intramolecular Anodic Olefin Coupling Reactions and the Synthesis of Cyclic Amines. J. Am. Chem. Soc. 2010, 132, 2839–2844. 10.1021/ja910586v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H. C.; Moeller K. D. Intramolecular Anodic Olefin Coupling Reactions: Using Competition Studies to Probe the Mechanism of Oxidative Cyclization Reactions. Org. Lett. 2010, 12, 1720–1723. 10.1021/ol100317t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J. M.; Xu H. C.; Moeller K. D. Investigating the Reactivity of Radical Cations: Experimental and Computational Insights into the Reactions of Radical Cations with Alcohol and p-Toluene Sulfonamide Nucleophiles. J. Am. Chem. Soc. 2012, 134, 18338–18344. 10.1021/ja307046j. [DOI] [PubMed] [Google Scholar]
- Xu H. C.; Campbell J. M.; Moeller K. D. Cyclization Reactions of Anode-Generated Amidyl Radicals. J. Org. Chem. 2014, 79, 379–391. 10.1021/jo402623r. [DOI] [PubMed] [Google Scholar]
- Xu F.; Zhu L.; Zhu S.; Yan X.; Xu H. C. Electrochemical Intramolecular Aminooxygenation of Unactivated Alkenes. Chem. - Eur. J. 2014, 20, 12740–12744. 10.1002/chem.201404078. [DOI] [PubMed] [Google Scholar]
- Hou Z. W.; Xu H. C. Electrochemically Enabled Intramolecular Aminooxygenation of Alkynes via Amidyl Radical Cyclization. Chin. J. Chem. 2020, 38, 394–398. 10.1002/cjoc.201900500. [DOI] [Google Scholar]
- Folgueiras-Amador A. A.; Philipps K.; Guilbaud S.; Poelakker J.; Wirth T. An Easy-to-Machine Electrochemical Flow Microreactor: Efficient Synthesis of Isoindolinone and Flow Functionalization. Angew. Chem., Int. Ed. 2017, 56, 15446–15450. 10.1002/anie.201709717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts K.; Baker A.; Wirth T. Electrochemical Synthesis in Microreactors. J. Flow Chem. 2015, 4, 2–11. 10.1556/JFC-D-13-00030. [DOI] [Google Scholar]
- Ahmed N.; Khatoon S. Facile Electrochemical Intramolecular Amination of Urea-Tethered Terminal Alkenes for the Synthesis of Cyclic Ureas. ChemistryOpen 2018, 7, 576–582. 10.1002/open.201800064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N.; Vgenopoulou A. Flow Electrochemical Cyclizations via Amidyl Radicals: Easy Access to Cyclic Ureas. SynOpen 2019, 3, 46–48. 10.1055/s-0037-1611772. [DOI] [Google Scholar]
- Zhu L.; Xiong P.; Mao Z. Y.; Wang Y. H.; Yan X.; Lu X.; Xu H. C. Electrocatalytic Generation of Amidyl Radicals for Olefin Hydroamidation: Use of Solvent Effects to Enable Anilide Oxidation. Angew. Chem., Int. Ed. 2016, 55, 2226–2229. 10.1002/anie.201510418. [DOI] [PubMed] [Google Scholar]
- Long H.; Song J.; Xu H. C. Electrochemical Synthesis of 7-Membered Carbocycles through Cascade 5-exo-tri/7-endo-trig Radical Cyclization. Org. Chem. Front. 2018, 5, 3129–3132. 10.1039/C8QO00803E. [DOI] [Google Scholar]
- Kotov V.; Scarborough C. C.; Stahl S. S. Palladium-Catalyzed Aerobic Oxidative Amination of Alkenes: Development of Intra- and Intermolecular Aza-Wacker Reactions. Inorg. Chem. 2007, 46, 1910–1923. 10.1021/ic061997v. [DOI] [PubMed] [Google Scholar]
- Xiong P.; Xu H. C. H. H.; Xu H. C. H. H. Metal- and Reagent-Free Intramolecular Oxidative Amination of Tri- and Tetrasubstituted Alkenes. J. Am. Chem. Soc. 2017, 139, 2956–2959. 10.1021/jacs.7b01016. [DOI] [PubMed] [Google Scholar]
- Yi X.; Hu X. Formal Aza-Wacker Cyclization by Tandem Electrochemical Oxidation and Copper Catalysis. Angew. Chem., Int. Ed. 2019, 58, 4700–4704. 10.1002/anie.201814509. [DOI] [PubMed] [Google Scholar]
- Kochi J. K.; Bemis A.; Jenkins C. L. Mechanism of Electron Transfer Oxidation of Alkyl Radicals by Copper (II) Complexes. J. Am. Chem. Soc. 1968, 90, 4616–4625. 10.1021/ja01019a018. [DOI] [Google Scholar]
- Hossain A.; Bhattacharyya A.; Reiser O. Copper’s Rapid Ascent in Visible-Light Photoredox Catalysis. Science 2019, 364, eaav9713. 10.1126/science.aav9713. [DOI] [PubMed] [Google Scholar]
- Tlahuext-Aca A.; Candish L.; Garza-Sanchez R. A.; Glorius F. Decarboxylative Olefination of Activated Aliphatic Acids Enabled by Dual Organophotoredox/Copper Catalysis. ACS Catal. 2018, 8, 1715–1719. 10.1021/acscatal.7b04281. [DOI] [Google Scholar]
- Hou Z. W.; Mao Z. Y.; Zhao H. B.; Melcamu Y. Y.; Lu X.; Song J.; Xu H. C. Electrochemical C-H/N-H Functionalization for the Synthesis of Highly Functionalized (Aza)Indoles. Angew. Chem., Int. Ed. 2016, 55, 9168–9172. 10.1002/anie.201602616. [DOI] [PubMed] [Google Scholar]
- Hou Z. W.; Yan H.; Song J. S.; Xu H. C. Electrochemical Synthesis of (Aza)Indolines via Dehydrogenative [3+2] Annulation: Application to Total Synthesis of (±)-Hinckdentine A. Chin. J. Chem. 2018, 36, 909–915. 10.1002/cjoc.201800301. [DOI] [Google Scholar]
- Hou Z. W.; Mao Z. Y.; Song J.; Xu H. C. Electrochemical Synthesis of Polycyclic N-Heteroaromatics through Cascade Radical Cyclization of Diynes. ACS Catal. 2017, 7, 5810–5813. 10.1021/acscatal.7b02105. [DOI] [Google Scholar]
- Jiang W.; Li Y.; Wang Z. Heteroarenes as High Performance Organic Semiconductors. Chem. Soc. Rev. 2013, 42, 6113–6127. 10.1039/c3cs60108k. [DOI] [PubMed] [Google Scholar]
- Stępień M.; Gońka E.; Żyła M.; Sprutta N. Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications. Chem. Rev. 2017, 117, 3479–3716. 10.1021/acs.chemrev.6b00076. [DOI] [PubMed] [Google Scholar]
- Hou Z. W.; Mao Z. Y.; Melcamu Y. Y.; Lu X.; Xu H. C. Electrochemical Synthesis of Imidazo-Fused N-Heteroaromatic Compounds through a C-N Bond-Forming Radical Cascade. Angew. Chem., Int. Ed. 2018, 57, 1636–1639. 10.1002/anie.201711876. [DOI] [PubMed] [Google Scholar]
- Yan H.; Mao Z. Y.; Hou Z. W.; Song J.; Xu H. C. A Diastereoselective Approach to Axially Chiral Biaryls via Electrochemically Enabled Cyclization Cascade. Beilstein J. Org. Chem. 2019, 15, 795–800. 10.3762/bjoc.15.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F.; Long H.; Song J.; Xu H. C. De Novo Synthesis of Highly Functionalized Benzimidazolones and Benzoxazolones through an Electrochemical Dehydrogenative Cyclization Cascade. Angew. Chem., Int. Ed. 2019, 58, 9017–9021. 10.1002/anie.201904931. [DOI] [PubMed] [Google Scholar]
- Zhang L. B.; Geng R.-S.; Wang Z. C.; Ren G. Y.; Wen L. R.; Li M. Electrochemical Intramolecular C-H/N-H Functionalization for the Synthesis of Isoxazolidine-Fused Isoquinolin-1(2H)-Ones. Green Chem. 2020, 22, 16–21. 10.1039/C9GC03290H. [DOI] [Google Scholar]
- Moon Y.; Jang E.; Choi S.; Hong S. Visible-Light-Photocatalyzed Synthesis of Phenanthridinones and Quinolinones via Direct Oxidative C-H Amidation. Org. Lett. 2018, 20, 240–243. 10.1021/acs.orglett.7b03600. [DOI] [PubMed] [Google Scholar]
- Metternich J. B.; Gilmour R. A Bio-Inspired, Catalytic E - Z Isomerization of Activated Olefins. J. Am. Chem. Soc. 2015, 137, 11254–11257. 10.1021/jacs.5b07136. [DOI] [PubMed] [Google Scholar]
- Usami K.; Yamaguchi E.; Tada N.; Itoh A. Transition-Metal-Free Synthesis of Phenanthridinones through Visible-Light-Driven Oxidative C-H Amidation. Eur. J. Org. Chem. 2020, 2020, 1496–1504. 10.1002/ejoc.201900536. [DOI] [Google Scholar]
- Kehl A.; Breising V. M.; Schollmeyer D.; Waldvogel S. R. Electrochemical Synthesis of 5-Aryl-Phenanthridin-6-One by Dehydrogenative N,C Bond Formation. Chem. - Eur. J. 2018, 24, 17230–17233. 10.1002/chem.201804638. [DOI] [PubMed] [Google Scholar]
- Kehl A.; Schupp N.; Breising V. M.; Schollmeyer D.; Waldvogel S. R. Electrochemical Synthesis of Carbazoles by Dehydrogenative Coupling Reaction. Chem. - Eur. J. 2020, 26, 15847–15851. 10.1002/chem.202003430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Yuan Y.; Liu H.; He M.; Yang M.; Liu P.; Yu B.; Dong X.; Lei A. Electrochemical Oxidative C-H/N-H Cross-Coupling for C-N Bond Formation with Hydrogen Evolution. Chem. Commun. 2019, 55, 1809–1812. 10.1039/C8CC09899A. [DOI] [PubMed] [Google Scholar]
- Kravtsov D. N.; Peregudov A. S.; Petrov E. S.; Terekhova M. I.; Shatenshtein A. I. Equilibrium NH Acidity of Benzenesulfonamide and Its Derivatives in Dimethyl Sulfoxide. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1981, 30, 993–997. 10.1007/BF00950279. [DOI] [Google Scholar]
- Olmstead W. N.; Margolin Z.; Bordwell F. G. Acidities of Water and Simple Alcohols in Dimethyl Sulfoxide Solution. J. Org. Chem. 1980, 45, 3295–3299. 10.1021/jo01304a032. [DOI] [Google Scholar]
- Li J. S.; Yang P. P.; Xie X. Y.; Jiang S.; Tao L.; Li Z. W.; Lu C. H.; Liu W. D. Catalyst-Free Electrosynthesis of Benzimidazolones through Intramolecular Oxidative C-N Coupling. Adv. Synth. Catal. 2020, 362, 1977–1981. 10.1002/adsc.202000198. [DOI] [Google Scholar]
- Studer A.; Bossart M. Radical Aryl Migration Reactions. Tetrahedron 2001, 57, 9649–9667. 10.1016/S0040-4020(01)00990-5. [DOI] [Google Scholar]
- Chen Z. M.; Zhang X. M.; Tu Y. Q. Radical Aryl Migration Reactions and Synthetic Applications. Chem. Soc. Rev. 2015, 44, 5220–5245. 10.1039/C4CS00467A. [DOI] [PubMed] [Google Scholar]
- Shu W.; Genoux A.; Li Z.; Nevado C. γ-Functionalizations of Amines through Visible-Light-Mediated, Redox-Neutral C-C Bond Cleavage. Angew. Chem., Int. Ed. 2017, 56, 10521–10524. 10.1002/anie.201704068. [DOI] [PubMed] [Google Scholar]
- Molander G. A. Diverse Methods for Medium Ring Synthesis. Acc. Chem. Res. 1998, 31, 603–609. 10.1021/ar960101v. [DOI] [Google Scholar]
- Xu Z.; Huang Z.; Li Y.; Kuniyil R.; Zhang C.; Ackermann L.; Ruan Z. Catalyst-Free, Direct Electrochemical Synthesis of Annulated Medium-Sized Lactams through C-C Bond Cleavage. Green Chem. 2020, 22, 1099–1104. 10.1039/C9GC03901E. [DOI] [Google Scholar]
- Liu C.; Jiang Q.; Lin Y.; Fang Z.; Guo K. C- To N-Center Remote Heteroaryl Migration via Electrochemical Initiation of N-Radical by Organic Catalyst. Org. Lett. 2020, 22, 795–799. 10.1021/acs.orglett.9b04141. [DOI] [PubMed] [Google Scholar]
- Liu C.; Cai C.; Yuan C.; Jiang Q.; Fang Z.; Guo K. Visible-Light-Promoted N-Centered Radical Generation for Remote Heteroaryl Migration. Org. Biomol. Chem. 2020, 18, 7663–7670. 10.1039/D0OB01594F. [DOI] [PubMed] [Google Scholar]
- Ye Z.; Wu Y.; Chen N.; Zhang H.; Zhu K.; Ding M.; Liu M.; Li Y.; Zhang F. Enantiospecific Electrochemical Rearrangement for the Synthesis of Hindered Triazolopyridinone Derivatives. Nat. Commun. 2020, 11, 3628. 10.1038/s41467-020-17389-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Wang P.; Gao X.; Wang D.; Wang S.; Liang X.; Wang L.; Zhang H.; Lei A. Regioselective/Electro-Oxidative Intermolecular [3+2] Annulation for the Preparation of Indolines. Chem. Sci. 2020, 11, 2181–2186. 10.1039/C9SC05729C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y.; Yu Q.; Liu Q.; Zuo H.; Yu H.-B.; Wei W. J.; Liao R. Z.; Zhong F. Iron-Catalyzed Regiospecific Intermolecular Radical Cyclization of Anilines: Strategy for Assembly of 2,2-Disubstituted Indolines. Org. Lett. 2018, 20, 1404–1408. 10.1021/acs.orglett.8b00176. [DOI] [PubMed] [Google Scholar]
- Pandey G.; Laha R. Visible-Light-Catalyzed Direct Benzylic C(sp3)-H Amination Reaction by Cross-Dehydrogenative Coupling. Angew. Chem., Int. Ed. 2015, 54, 14875–14879. 10.1002/anie.201506990. [DOI] [PubMed] [Google Scholar]
- O’Donnell J. F.; Mann C. K. Controlled-Potential Oxidation of Aliphatic Amides. J. Electroanal. Chem. Interfacial Electrochem. 1967, 13, 157–162. 10.1016/0022-0728(67)80108-6. [DOI] [Google Scholar]
- Blanc S.; Pigot T.; Cugnet C.; Brown R.; Lacombe S. A New Cyanoaromatic Photosensitizer vs. 9,10-Dicyanoanthracene: Systematic Comparison of the Photophysical Properties. Phys. Chem. Chem. Phys. 2010, 12, 11280–11290. 10.1039/c002705g. [DOI] [PubMed] [Google Scholar]
- Lin M. Y.; Xu K.; Jiang Y. Y.; Liu Y. G.; Sun B. G.; Zeng C. C. Intermolecular Electrochemical C(sp3)-H/N-H Cross-Coupling of Xanthenes with N-Alkoxyamides: Radical Pathway Mediated by Ferrocene as a Redox Catalyst. Adv. Synth. Catal. 2018, 360, 1665–1672. 10.1002/adsc.201701536. [DOI] [Google Scholar]
- Tong K.; Liu X.; Zhang Y.; Yu S. Visible-Light-Induced Direct Oxidative C-H Amidation of Heteroarenes with Sulfonamides. Chem. - Eur. J. 2016, 22, 15669–15673. 10.1002/chem.201604014. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Chen S.; Lu W.; Gu W.; Liu P.; Sun P. Visible Light-Induced C3-Sulfonamidation of Imidazopyridines with Sulfamides. Org. Biomol. Chem. 2017, 15, 8102–8109. 10.1039/C7OB02029E. [DOI] [PubMed] [Google Scholar]
- Challis B. C.; Iley J. N. N-ethyl-N-methyltoluene-4-sulphonamide ad its relevance to the nucleophilic properties of neutral sulphonamides. J. Chem. Soc., Perkin Trans. 2 1985, 2, 699–703. 10.1039/p29850000699. [DOI] [Google Scholar]
- Pastoriza C.; Antelo J. M.; Crugeiras J. Use of N-Chloro-N-Methyl-p-Toluenesulfonamide in N-Chlorination Reactions. J. Phys. Org. Chem. 2013, 26, 551–559. 10.1002/poc.3127. [DOI] [Google Scholar]
- Dzandzi J. P. K.; Beckford Vera D. R.; Genady A. R.; Albu S. A.; Eltringham-Smith L. J.; Capretta A.; Sheffield W. P.; Valliant J. F. Fluorous Analogue of Chloramine-T: Preparation, X-Ray Structure Determination, and Use as an Oxidant for Radioiodination and s-Tetrazine Synthesis. J. Org. Chem. 2015, 80, 7117–7125. 10.1021/acs.joc.5b00988. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lin Z.; Ackermann L. Electrochemical C-H Amidation of Heteroarenes with N-Alkyl Sulfonamides in Aqueous Medium. Chem. - Eur. J. 2021, 27, 242–246. 10.1002/chem.202004229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C.; Liu K.; Jiang X.; Dong X.; Weng Y.; Chiang C. W.; Lei A. Electrooxidation Enables Selective Dehydrogenative [4+2] Annulation between Indole Derivatives. Angew. Chem., Int. Ed. 2020, 59, 7193–7197. 10.1002/anie.202000226. [DOI] [PubMed] [Google Scholar]
- Romeo G.; Materia L.; Manetti F.; Cagnotto A.; Mennini T.; Nicoletti F.; Botta M.; Russo F.; Minneman K. P. New Pyrimido[5,4-b]Indoles as Ligands for A1-Adrenoceptor Subtypes. J. Med. Chem. 2003, 46, 2877–2894. 10.1021/jm0307741. [DOI] [PubMed] [Google Scholar]
- Ismail M. A. H.; Aboul-Enein M. N. Y.; Abouzid K. A. M.; Serya R. A. T. Ligand Design and Synthesis of New Imidazo[5,1-b]Quinazoline Derivatives as A1-Adrenoceptor Agonists and Antagonists. Bioorg. Med. Chem. 2006, 14, 898–910. 10.1016/j.bmc.2005.07.037. [DOI] [PubMed] [Google Scholar]
- Zagni C.; Guimarães D. M.; Salerno L.; Punzo F.; Squarize C. H.; Mineo P. G.; Romeo G.; Rescifina A. An A1-Adrenergic Receptor Ligand Repurposed as a Potent Antiproliferative Agent for Head and Neck Squamous Cell Carcinoma. RSC Adv. 2015, 5, 6536–6542. 10.1039/C4RA11856A. [DOI] [Google Scholar]
- Chataigner I.; Hess E.; Toupet L.; Piettre S. R. Activation of the Dienophilicity of Indoles in Normal Electron Demand [4+2] Cycloadditions under High Pressure. Org. Lett. 2001, 3, 515–518. 10.1021/ol0067989. [DOI] [PubMed] [Google Scholar]
- Han-Ya Y.; Tokuyama H.; Fukuyama T. Total Synthesis of (−)-Conophylline and (−)-Conophyllidine. Angew. Chem., Int. Ed. 2011, 50, 4884–4887. 10.1002/anie.201100981. [DOI] [PubMed] [Google Scholar]
- Ziegler R. E.; Tan S. J.; Kam T. S.; Porco J. A. Development of an Alkaloid-Pyrone Annulation: Synthesis of Pleiomaltinine. Angew. Chem., Int. Ed. 2012, 51, 9348–9351. 10.1002/anie.201204093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Tang S.; Huang P.; Lei A. External Oxidant-Free Electrooxidative [3+2] Annulation between Phenol and Indole Derivatives. Nat. Commun. 2017, 8, 775. 10.1038/s41467-017-00873-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T.; Yamaguchi E.; Itoh A. Cross-Dehydrogenative C-H Amination of Indoles under Aerobic Photo-Oxidative Conditions. Org. Lett. 2017, 19, 1282–1285. 10.1021/acs.orglett.7b00026. [DOI] [PubMed] [Google Scholar]
- Kondo Y.; Kondo K.; Kusabayashi S. Nucleophilic Reactivity and Solvation of Succinimide and Phthalimide Anions in Acetonitrile-Methanol Mixtures. J. Chem. Soc., Perkin Trans. 2 1993, 1141–1145. 10.1039/P29930001141. [DOI] [Google Scholar]
- Ito E.; Fukushima T.; Kawakami T.; Murakami K.; Itami K. Catalytic Dehydrogenative C-H Imidation of Arenes Enabled by Photo-Generated Hole Donation to Sulfonimide. Chem. 2017, 2, 383–392. 10.1016/j.chempr.2017.02.006. [DOI] [Google Scholar]
- Margrey K. A.; McManus J. B.; Bonazzi S.; Zecri F.; Nicewicz D. A. Predictive Model for Site-Selective Aryl and Heteroaryl C-H Functionalization via Organic Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 11288–11299. 10.1021/jacs.7b06715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margrey K. A.; Levens A.; Nicewicz D. A. Direct Aryl C-H Amination with Primary Amines Using Organic Photoredox Catalysis. Angew. Chem., Int. Ed. 2017, 56, 15644–15648. 10.1002/anie.201709523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu L.; Yi H.; Wang S.; Liu T.; Liu J.; Lei A. Photo-Induced Oxidant-Free Oxidative C-H/N-H Cross-Coupling between Arenes and Azoles. Nat. Commun. 2017, 8, 14226. 10.1038/ncomms14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morofuji T.; Shimizu A.; Yoshida J. Electrochemical C-H Amination: Synthesis of Aromatic Primary. J. Am. Chem. Soc. 2013, 135, 5000–5003. 10.1021/ja402083e. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Liardet L.; Luo J.; Ren D.; Grätzel M.; Hu X. Photoelectrocatalytic Arene C-H Amination. Nat. Catal. 2019, 2, 366–373. 10.1038/s41929-019-0231-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara Y.; Ito E.; Kawakami T.; Yamada S.; Murakami K.; Itami K. Direct Coupling of Naphthalene and Sulfonimides Promoted by DDQ and Blue Light. Chem. Lett. 2017, 46, 1014–1016. 10.1246/cl.170321. [DOI] [Google Scholar]
- Ohkubo K.; Fujimoto A.; Fukuzumi S. Aromatic Monochlorination Photosensitized by DDQ with Hydrogen Chloride under Visible-Light Irradiation. Chem. - Asian J. 2016, 11, 996–999. 10.1002/asia.201600083. [DOI] [PubMed] [Google Scholar]
- Das S.; Natarajan P.; König B. Teaching Old Compounds New Tricks: DDQ-Photocatalyzed C-H Amination of Arenes with Carbamates, Urea, and N-Heterocycles. Chem. - Eur. J. 2017, 23, 18161–18165. 10.1002/chem.201705442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Zhang G.; Nie L.; Kong T.; Lei A. Electrochemical Oxidation Induced Intermolecular Aromatic C-H Imidation. Nat. Commun. 2019, 10, 5467. 10.1038/s41467-019-13524-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J. C. K.; Rovis T. Amide-Directed Photoredox-Catalysed C-C Bond Formation at Unactivated sp3 C-H Bonds. Nature 2016, 539, 272–275. 10.1038/nature19810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freytag C.; Loffler K. Über Eine Neue Bildungsweise von N-alkylierten Pyrrolidinen. Ber. Dtsch. Chem. Ges. 1909, 42, 3427–3431. 10.1002/cber.19090420377. [DOI] [Google Scholar]
- Chow Y. L.; Mojelsky T. W.; Magdzinski L. J.; Tichý M. Chemistry of Amido Radicals: Intramolecular Hydrogen Abstraction as Related to Amido Radical Configurations. Can. J. Chem. 1985, 63, 2197–2202. 10.1139/v85-361. [DOI] [Google Scholar]
- Hernández R.; Rivera A.; Salazar J. A.; Suárez E. Nitroamine Radicals as Intermediates in the Functionalization of Non-Activated Carbon Atoms. J. Chem. Soc., Chem. Commun. 1980, 958–959. 10.1039/C39800000958. [DOI] [Google Scholar]
- Jeffrey J. L.; Sarpong R. Intramolecular C(sp3)-H Amination. Chem. Sci. 2013, 4, 4092–4106. 10.1039/c3sc51420j. [DOI] [Google Scholar]
- Bordwell F. G.; Zhang S.; Zhang X. M.; Liu W. Z. Homolytic Bond Dissociation Enthalpies of the Acidic H-A Bonds Caused by Proximate Substituents in Sets of Methyl Ketones, Carboxylic Esters, and Carboxamides Related to Changes in Ground State Energies. J. Am. Chem. Soc. 1995, 117, 7092–7096. 10.1021/ja00132a008. [DOI] [Google Scholar]
- Bordwell F. G.; Singer D. L.; Satish A. V. Effects of Structural Changes on Acidities and Homolytic Bond Dissociation Energies of the N-H Bonds in Pyridones and Related Heterocycles. J. Am. Chem. Soc. 1993, 115, 3543–3547. 10.1021/ja00062a019. [DOI] [Google Scholar]
- Zhang L.; He L.; Hong C.-B.; Qin S.; Tao G. H. Brønsted Acidity of Bio-Protic Ionic Liquids: The Acidic Scale of [AA]X Amino Acid Ionic Liquids. Green Chem. 2015, 17, 5154–5163. 10.1039/C5GC01913C. [DOI] [Google Scholar]
- Archambeau A.; Rovis T. Rhodium(III)-Catalyzed Allylic C(sp3)-H Activation of Alkenyl Sulfonamides: Unexpected Formation of Azabicycles. Angew. Chem., Int. Ed. 2015, 54, 13337–13340. 10.1002/anie.201504150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad M.; Masohan A.; Sawhney S. S. Alkanolmonoamines as Activators for the Hot Potash Process for CO2 Capture. Asian J. Chem. 2014, 26, 975–980. 10.14233/ajchem.2014.15687. [DOI] [Google Scholar]
- Chen D. F.; Chu J. C. K.; Rovis T. Directed γ-C(sp3)-H Alkylation of Carboxylic Acid Derivatives through Visible Light Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 14897–14900. 10.1021/jacs.7b09306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordwell F. G.; Hughes D. L. Isomerization of 3-Butenenitrile. J. Am. Chem. Soc. 1985, 107 (8), 4737–4744. 10.1021/ja00302a023. [DOI] [Google Scholar]
- Thullen S. M.; Treacy S. M.; Rovis T. Regioselective Alkylative Cross-Coupling of Remote Unactivated C(sp3)-H Bonds. J. Am. Chem. Soc. 2019, 141, 14062–14067. 10.1021/jacs.9b07014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand A. W.; Yin H.; Xu L.; Giacoboni J.; Martin-Montero R.; Romano C.; Montgomery J.; Martin R. Dual Catalytic Platform for Enabling sp3 α C-H Arylation and Alkylation of Benzamides. ACS Catal. 2020, 10, 4671–4676. 10.1021/acscatal.0c01318. [DOI] [Google Scholar]
- Xu B.; Tambar U. K. Remote Allylation of Unactivated C(sp3)-H Bonds Triggered by Photogenerated Amidyl Radicals. ACS Catal. 2019, 9, 4627–4631. 10.1021/acscatal.9b00563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkind S. I.; Vander Griend D. A.; Swager T. M. Electroactive Anion Receptor with High Affinity for Arsenate. J. Org. Chem. 2020, 85, 10050–10061. 10.1021/acs.joc.0c01206. [DOI] [PubMed] [Google Scholar]
- Ma Z. Y.; Guo L. N.; You Y.; Yang F.; Hu M.; Duan X. H. Visible Light Driven Alkylation of C(sp3)-H Bonds Enabled by 1,6-Hydrogen Atom Transfer/Radical Relay Addition. Org. Lett. 2019, 21, 5500–5504. 10.1021/acs.orglett.9b01804. [DOI] [PubMed] [Google Scholar]
- Kanegusuku A. L. G.; Castanheiro T.; Ayer S. K.; Roizen J. L. Sulfamyl Radicals Direct Photoredox-Mediated Giese Reactions at Unactivated C(3)-H Bonds. Org. Lett. 2019, 21, 6089–6095. 10.1021/acs.orglett.9b02234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu W.; Zhang H.; Huang Y. γ-Alkylation of Alcohols Enabled by Visible-Light Induced 1,6-Hydrogen Atom Transfer. Org. Lett. 2019, 21, 6107–6111. 10.1021/acs.orglett.9b02255. [DOI] [PubMed] [Google Scholar]
- Nechab M.; Mondal S.; Bertrand M. P. 1, n-Hydrogen-Atom Transfer (HAT) Reactions in Which n≠5: An Updated Inventory. Chem. - Eur. J. 2014, 20, 16034–16059. 10.1002/chem.201403951. [DOI] [PubMed] [Google Scholar]
- Sathyamoorthi S.; Banerjee S.; Du Bois J.; Burns N. Z.; Zare R. N. Site-Selective Bromination of sp3 C-H Bonds. Chem. Sci. 2018, 9, 100–104. 10.1039/C7SC04611A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short M. A.; Blackburn J. M.; Roizen J. L. Sulfamate Esters Guide Selective Radical-Mediated Chlorination of Aliphatic C-H Bonds. Angew. Chem., Int. Ed. 2018, 57, 296–299. 10.1002/anie.201710322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayer S. K.; Roizen J. L. Sulfamate Esters Guide C(3)-Selective Xanthylation of Alkanes. J. Org. Chem. 2019, 84, 3508–3523. 10.1021/acs.joc.9b00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axon J. R.; Beckwith A. L. J. Diastereoselective Radical Addition to Methyleneoxazolidinones: An Enantioselective Route to α-Amino Acids. J. Chem. Soc., Chem. Commun. 1995, (5), 549–550. 10.1039/C39950000549. [DOI] [Google Scholar]
- Musacchio A. J.; Lainhart B. C.; Zhang X.; Naguib S. G.; Sherwood T. C.; Knowles R. R. Catalytic Intermolecular Hydroaminations of Unactivated Olefins with Secondary Alkyl Amines. Science 2017, 355, 727–730. 10.1126/science.aal3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold S.; Bafaluy D.; Muñiz K. Anodic Benzylic C(sp3)-H Amination: Unified Access to Pyrrolidines and Piperidines. Green Chem. 2018, 20, 3191–3196. 10.1039/C8GC01411F. [DOI] [Google Scholar]
- Hu X.; Zhang G.; Bu F.; Nie L.; Lei A. Electrochemical-Oxidation-Induced Site-Selective Intramolecular C(sp3)-H Amination. ACS Catal. 2018, 8, 9370–9375. 10.1021/acscatal.8b02847. [DOI] [Google Scholar]
- Nicholas A. M. de P.; Boyd R. J.; Arnold D. R. Thermochemical Parameters for Organic Radicals and Radical Ions. Part 2. The Protonation of Hydrocarbon Radicals in the Gas Phase. Can. J. Chem. 1982, 60, 3011–3018. 10.1139/v82-431. [DOI] [Google Scholar]
- Nikolaienko P.; Jentsch M.; Kale A. P.; Cai Y.; Rueping M. Electrochemical and Scalable Dehydrogenative C(sp3)-H Amination via Remote Hydrogen Atom Transfer in Batch and Continuous Flow. Chem. - Eur. J. 2019, 25, 7177–7184. 10.1002/chem.201806092. [DOI] [PubMed] [Google Scholar]
- Gieshoff T.; Schollmeyer D.; Waldvogel S. R. Access to Pyrazolidin-3,5-Diones through Anodic N-N Bond Formation. Angew. Chem., Int. Ed. 2016, 55, 9437–9440. 10.1002/anie.201603899. [DOI] [PubMed] [Google Scholar]
- Gieshoff T.; Kehl A.; Schollmeyer D.; Moeller K. D.; Waldvogel S. R. Insights into the Mechanism of Anodic N-N Bond Formation by Dehydrogenative Coupling. J. Am. Chem. Soc. 2017, 139, 12317–12324. 10.1021/jacs.7b07488. [DOI] [PubMed] [Google Scholar]
- Kehl A.; Gieshoff T.; Schollmeyer D.; Waldvogel S. R. Electrochemical Conversion of Phthaldianilides to Phthalazin-1,4-Diones by Dehydrogenative N-N Bond Formation. Chem. - Eur. J. 2018, 24, 590–593. 10.1002/chem.201705578. [DOI] [PubMed] [Google Scholar]
- Breising V. M.; Kayser J. M.; Kehl A.; Schollmeyer D.; Liermann J. C.; Waldvogel S. R. Electrochemical Formation of: N,N’-Diarylhydrazines by Dehydrogenative N-N Homocoupling Reaction. Chem. Commun. 2020, 56, 4348–4351. 10.1039/D0CC01052A. [DOI] [PubMed] [Google Scholar]
- Gieshoff T.; Kehl A.; Schollmeyer D.; Moeller K. D.; Waldvogel S. R. Electrochemical Synthesis of Benzoxazoles from Anilides-a New Approach to Employ Amidyl Radical Intermediates. Chem. Commun. 2017, 53, 2974–2977. 10.1039/C7CC00927E. [DOI] [PubMed] [Google Scholar]
- Bordwell F. G.; Algrim D. Nitrogen Acids. 1. Carboxamides and Sulfonamides. J. Org. Chem. 1976, 41, 2507–2508. 10.1021/jo00876a042. [DOI] [Google Scholar]
- Schulz L.; Enders M.; Elsler B.; Schollmeyer D.; Dyballa K. M.; Franke R.; Waldvogel S. R. Reagent- and Metal-Free Anodic C-C Cross-Coupling of Aniline Derivatives. Angew. Chem., Int. Ed. 2017, 56, 4877–4881. 10.1002/anie.201612613. [DOI] [PubMed] [Google Scholar]
- Schulz L.; Franke R.; Waldvogel S. R. Direct Anodic Dehydrogenative Cross- and Homo-Coupling of Formanilides. ChemElectroChem 2018, 5, 2069–2072. 10.1002/celc.201800422. [DOI] [Google Scholar]
- Zhao Q. Q.; Li M.; Xue X. S.; Chen J. R.; Xiao W. J. Visible-Light-Driven Neutral Nitrogen Radical Mediated Intermolecular Styrene Difunctionalization. Org. Lett. 2019, 21, 3861–3865. 10.1021/acs.orglett.9b01362. [DOI] [PubMed] [Google Scholar]
- Tabaković I.; Trkovnik M.; Batušić M.; Tabaković K. Electrochemical Synthesis of Heterocyclic Compounds: VII. Anodic Synthesis of 1,3-Thiazole Derivatives. Synthesis 1979, 1979, 590–592. 10.1055/s-1979-28768. [DOI] [Google Scholar]
- Cheng Y.; Yang J.; Qu Y.; Li P. Aerobic Visible-Light Photoredox Radical C-H Functionalization: Catalytic Synthesis of 2-Substituted Benzothiazoles. Org. Lett. 2012, 14, 98–101. 10.1021/ol2028866. [DOI] [PubMed] [Google Scholar]
- Zen J. M.; Liou S. L.; Kumar A. S.; Hsia M. S. An Efficient and Selective Photocatalytic System for the Oxidation of Sulfides to Sulfoxides. Angew. Chem., Int. Ed. 2003, 42, 577–579. 10.1002/anie.200390166. [DOI] [PubMed] [Google Scholar]
- Bordwell F. G.; Fried H. E. Heterocyclic Aromatic Anions with 4n+2 π-Electrons. J. Org. Chem. 1991, 56, 4218–4223. 10.1021/jo00013a027. [DOI] [Google Scholar]
- Sawyer D. T.; Calderwood T. S.; Yamaguchi K.; Angelis C. T. Synthesis and Characterization of Tetramethylammonium Superoxide. Inorg. Chem. 1983, 22, 2577–2583. 10.1021/ic00160a022. [DOI] [Google Scholar]
- Maity S.; Zheng N. A Visible-Light-Mediated Oxidative C-N Bond Formation/Aromatization Cascade: Photocatalytic Preparation of N-Arylindoles. Angew. Chem., Int. Ed. 2012, 51, 9562–9566. 10.1002/anie.201205137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Liu C.; Yi H.; Meng Q.; Bian C.; Chen H.; Jian J. X.; Wu L. Z.; Lei A. External Oxidant-Free Oxidative Cross-Coupling: A Photoredox Cobalt-Catalyzed Aromatic C-H Thiolation for Constructing C-S Bonds. J. Am. Chem. Soc. 2015, 137, 9273–9280. 10.1021/jacs.5b05665. [DOI] [PubMed] [Google Scholar]
- Jeletic M. S.; Mock M. T.; Appel A. M.; Linehan J. C. A Cobalt-Based Catalyst for the Hydrogenation of CO2 under Ambient Conditions. J. Am. Chem. Soc. 2013, 135, 11533–11536. 10.1021/ja406601v. [DOI] [PubMed] [Google Scholar]
- Mortimer C. G.; Wells G.; Crochard J. P.; Stone E. L.; Bradshaw T. D.; Stevens M. F. G.; Westwell A. D. Antitumor Benzothiazoles. 26.1 2-(3,4-Dimethoxyphenyl)-5- Fluorobenzothiazole (GW 610, NSC 721648), a Simple Fluorinated 2-Arylbenzothiazole, Shows Potent and Selective Inhibitory Activity against Lung, Colon, and Breast Cancer Cell Lines. J. Med. Chem. 2006, 49, 179–185. 10.1021/jm050942k. [DOI] [PubMed] [Google Scholar]
- Du P.; Schneider J.; Luo G.; Brennessel W. W.; Eisenberg R. Visible Light-Driven Hydrogen Production from Aqueous Protons Catalyzed by Molecular Cobaloxime Catalysts. Inorg. Chem. 2009, 48, 4952–4962. 10.1021/ic900389z. [DOI] [PubMed] [Google Scholar]
- Anderson R. F. Oxidation of the Cyclohexadienyl Radical by Metal Ions: A Pulse Radiolysis Study. Radiat. Phys. Chem. 1979, 13, 155–157. 10.1016/0146-5724(79)90039-6. [DOI] [Google Scholar]
- Wang P.; Tang S.; Lei A. Electrochemical Intramolecular Dehydrogenative C-S Bond Formation for the Synthesis of Benzothiazoles. Green Chem. 2017, 19, 2092–2095. 10.1039/C7GC00468K. [DOI] [Google Scholar]
- Folgueiras-Amador A. A.; Qian X. Y.; Xu H. C.; Wirth T. Catalyst- and Supporting-Electrolyte-Free Electrosynthesis of Benzothiazoles and Thiazolopyridines in Continuous Flow. Chem. - Eur. J. 2018, 24, 487–491. 10.1002/chem.201705016. [DOI] [PubMed] [Google Scholar]
- Qian X. Y.; Li S. Q.; Song J.; Xu H. C. TEMPO-Catalyzed Electrochemical C-H Thiolation: Synthesis of Benzothiazoles and Thiazolopyridines from Thioamides. ACS Catal. 2017, 7, 2730–2734. 10.1021/acscatal.7b00426. [DOI] [Google Scholar]
- Kokatla H. P.; Yoo E.; Salunke D. B.; Sil D.; Ng C. F.; Balakrishna R.; Malladi S. S.; Fox L. M.; David S. A. Toll-like Receptor-8 Agonistic Activities in C2, C4, and C8Modified Thiazolo[4,5-c]Quinolines. Org. Biomol. Chem. 2013, 11, 1179–1198. 10.1039/c2ob26705e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh A. N.; Nguyen A. D.; Aceves E. M.; Albright S. T.; Cedano M. R.; Smith D. K.; Gustafson J. L. Photocatalytic Oxidative C-H Thiolation: Synthesis of Benzothiazoles and Sulfenylated Indoles. Synlett 2019, 30, 1648–1655. 10.1055/s-0039-1690107. [DOI] [Google Scholar]
- Wang M.; Huan L.; Zhu C. Cyanohydrin-Mediated Cyanation of Remote Unactivated C(sp3)-H Bonds. Org. Lett. 2019, 21, 821–825. 10.1021/acs.orglett.8b04104. [DOI] [PubMed] [Google Scholar]
- Islam M.; Kariuki B. M.; Shafiq Z.; Wirth T.; Ahmed N. Efficient Electrosynthesis of Thiazolidin-2-Imines via Oxysulfurization of Thiourea-Tethered Terminal Alkenes Using the Flow Microreactor. Eur. J. Org. Chem. 2019, 2019, 1371–1376. 10.1002/ejoc.201801688. [DOI] [Google Scholar]
- Shibuya M.; Pichierri F.; Tomizawa M.; Nagasawa S.; Suzuki I.; Iwabuchi Y. Oxidation of Nitroxyl Radicals: Electrochemical and Computational Studies. Tetrahedron Lett. 2012, 53, 2070–2073. 10.1016/j.tetlet.2012.02.033. [DOI] [Google Scholar]
- Nutting J. E.; Rafiee M.; Stahl S. S. Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions. Chem. Rev. 2018, 118, 4834–4885. 10.1021/acs.chemrev.7b00763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breising V. M.; Gieshoff T.; Kehl A.; Kilian V.; Schollmeyer D.; Waldvogel S. R. Electrochemical Formation of 3,5-Diimido-1,2-Dithiolanes by Dehydrogenative Coupling. Org. Lett. 2018, 20, 6785–6788. 10.1021/acs.orglett.8b02904. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Zhang J.; Hu L.; Li L.; Liu K.; Yang T.; Zhou C. Electrochemical Oxidative Intramolecular N-S Bond Formation: Synthesis of 3-Substituted 5-Amino-1,2,4-Thiadiazoles. J. Org. Chem. 2020, 85, 3358–3363. 10.1021/acs.joc.9b03155. [DOI] [PubMed] [Google Scholar]
- Danen W. C.; Neugebauer F. A. Aminyl Free Radicals. Angew. Chem., Int. Ed. Engl. 1975, 14, 783–789. 10.1002/anie.197507831. [DOI] [Google Scholar]
- Michejda C. J.; Campbell D. H. Addition of Complexed Amino Radicals to Conjugated Alkenes. J. Am. Chem. Soc. 1979, 101, 7687–7693. 10.1021/ja00520a011. [DOI] [Google Scholar]
- Michejda C. J.; Campbell D. H. Amphoteric Amino Radicals. Tetrahedron Lett. 1977, 18, 577–580. 10.1016/S0040-4039(01)92698-X. [DOI] [Google Scholar]
- Ganley J. M.; Murray P. R. D.; Knowles R. R. Photocatalytic Generation of Aminium Radical Cations for C-N Bond Formation. ACS Catal. 2020, 10, 11712–11738. 10.1021/acscatal.0c03567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y.; Struwe J.; Meyer T. H.; Oliveira J. C. A.; Ackermann L. Catalyst- and Reagent-Free Electrochemical Azole C-H Amination. Chem. - Eur. J. 2018, 24, 12784–12789. 10.1002/chem.201802832. [DOI] [PubMed] [Google Scholar]
- Wei W.; Wang L.; Bao P.; Shao Y.; Yue H.; Yang D.; Yang X.; Zhao X.; Wang H. Metal-Free C(sp2)-H/N-H Cross-Dehydrogenative Coupling of Quinoxalinones with Aliphatic Amines under Visible-Light Photoredox Catalysis. Org. Lett. 2018, 20, 7125–7130. 10.1021/acs.orglett.8b03079. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Huang B.; Yang C.; Li B.; Gou B.; Xia W. Photocatalytic Cross-Dehydrogenative Amination Reactions between Phenols and Diarylamines. ACS Catal. 2017, 7, 2446–2451. 10.1021/acscatal.7b00192. [DOI] [Google Scholar]
- Tang S.; Wang S.; Liu Y.; Cong H.; Lei A. Electrochemical Oxidative C-H Amination of Phenols: Access to Triarylamine Derivatives. Angew. Chem., Int. Ed. 2018, 57, 4737–4741. 10.1002/anie.201800240. [DOI] [PubMed] [Google Scholar]
- Song C.; Liu K.; Wang Z.; Ding B.; Wang S.; Weng Y.; Chiang C. W.; Lei A. Electrochemical Oxidation Induced Selective Tyrosine Bioconjugation for the Modification of Biomolecules. Chem. Sci. 2019, 10, 7982–7987. 10.1039/C9SC02218J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGruyter J. N.; Malins L. R.; Baran P. S. Residue-Specific Peptide Modification: A Chemist’s Guide. Biochemistry 2017, 56, 3863–3873. 10.1021/acs.biochem.7b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Tang S.; Wu T.; Wang S.; Zou M.; Cong H.; Lei A. Electrooxidative Para-Selective C-H/N-H Cross-Coupling with Hydrogen Evolution to Synthesize Triarylamine Derivatives. Nat. Commun. 2019, 10, 639. 10.1038/s41467-019-08414-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. C.; Jiang S. S.; Song R. J.; Li J. H. A Metal- and Oxidizing-Reagent-Free Anodic Para-Selective Amination of Anilines with Phenothiazines. Chem. Commun. 2019, 55, 4371–4374. 10.1039/C9CC01332F. [DOI] [PubMed] [Google Scholar]
- Lv S.; Han X.; Wang J. Y.; Zhou M.; Wu Y.; Ma L.; Niu L.; Gao W.; Zhou J.; Hu W.; et al. Tunable Electrochemical C-N versus N-N Bond Formation of Nitrogen-Centered Radicals Enabled by Dehydrogenative Dearomatization: Biological Applications. Angew. Chem., Int. Ed. 2020, 59, 11583–11590. 10.1002/anie.202001510. [DOI] [PubMed] [Google Scholar]
- Wang H.; Wang Y.; Peng C.; Zhang J.; Zhu Q. A Direct Intramolecular C-H Amination Reaction Cocatalyzed by Copper(II) and Iron(III) as Part of an Efficient Route for the Synthesis of Pyrido[1,2-a]Benzimidazoles from N-Aryl-2-Aminopyridines. J. Am. Chem. Soc. 2010, 132, 13217–13219. 10.1021/ja1067993. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Yi H.; Wang J.; Lei A. Visible-Light Mediated Oxidative C-H/N-H Cross-Coupling between Tetrahydrofuran and Azoles Using Air. J. Org. Chem. 2017, 82, 10704–10709. 10.1021/acs.joc.7b01841. [DOI] [PubMed] [Google Scholar]
- Wu J.; Zhou Y.; Zhou Y.; Chiang C. W.; Lei A. Electro-Oxidative C(sp3)-H Amination of Azoles via Intermolecular Oxidative C(sp3)-H/N-H Cross-Coupling. ACS Catal. 2017, 7, 8320–8323. 10.1021/acscatal.7b03551. [DOI] [Google Scholar]
- Dian L.; Wang S.; Zhang-Negrerie D.; Du Y.; Zhao K. Organocatalytic Amination of Alkyl Ethers via n-Bu4NI/t-BuOOH-Mediated Intermolecular Oxidative C(sp3)-N Bond Formation: Novel Synthesis of Hemiaminal Ethers. Chem. Commun. 2014, 50, 11738–11741. 10.1039/C4CC05758A. [DOI] [PubMed] [Google Scholar]
- Singh M. K.; Akula H. K.; Satishkumar S.; Stahl L.; Lakshman M. K. Ruthenium-Catalyzed C-H Bond Activation Approach to Azolyl Aminals and Hemiaminal Ethers, Mechanistic Evaluations, and Isomer Interconversion. ACS Catal. 2016, 6, 1921–1928. 10.1021/acscatal.5b02603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruri H.; Singh U.; Sharma S.; Gudup S.; Bhogal M.; Kumar S.; Singh D.; Gupta V. K.; Kant R.; Vishwakarma R. A.; et al. Cross-Dehydrogenative Coupling of Azoles with α-C(sp3)-H of Ethers and Thioethers under Metal-Free Conditions: Functionalization of H-N Azoles via C-H Activation. J. Org. Chem. 2015, 80, 1929–1936. 10.1021/jo502477r. [DOI] [PubMed] [Google Scholar]
- Yang Y. Z.; Song R. J.; Li J. H. Intermolecular Anodic Oxidative Cross-Dehydrogenative C(sp3)-N Bond-Coupling Reactions of Xanthenes with Azoles. Org. Lett. 2019, 21, 3228–3231. 10.1021/acs.orglett.9b00947. [DOI] [PubMed] [Google Scholar]
- Samanta S.; Ravi C.; Rao S. N.; Joshi A.; Adimurthy S. Visible-Light-Promoted Selective C-H Amination of Heteroarenes with Heteroaromatic Amines under Metal-Free Conditions. Org. Biomol. Chem. 2017, 15, 9590–9594. 10.1039/C7OB02504A. [DOI] [PubMed] [Google Scholar]
- Feng P.; Ma G.; Chen X.; Wu X.; Lin L.; Liu P.; Chen T. Electrooxidative and Regioselective C-H Azolation of Phenol and Aniline Derivatives. Angew. Chem., Int. Ed. 2019, 58, 8400–8404. 10.1002/anie.201901762. [DOI] [PubMed] [Google Scholar]
- Feng E.; Hou Z.; Xu H. Electrochemical Synthesis of Tetrasubstituted Hydrazines by Dehydrogenative N-N Bond Formation. Youji Huaxue 2019, 39, 1424–1428. 10.6023/cjoc201812007. [DOI] [Google Scholar]
- Zhao J. P.; Ding L.-j.; Wang P. C.; Liu Y.; Huang M. J.; Zhou X. L.; Lu M. Electrochemical Nonacidic N-Nitrosation/N-Nitration of Secondary Amines through a Biradical Coupling Reaction. Adv. Synth. Catal. 2020, 362, 5036–5043. 10.1002/adsc.202000267. [DOI] [Google Scholar]
- Zhao H. B.; Hou Z. W.; Liu Z. J.; Zhou Z. F.; Song J.; Xu H. C. Amidinyl Radical Formation through Anodic N-H Bond Cleavage and Its Application in Aromatic C-H Bond Functionalization. Angew. Chem., Int. Ed. 2017, 56, 587–590. 10.1002/anie.201610715. [DOI] [PubMed] [Google Scholar]
- Yin W.; Wang X. Recent Advances in Iminyl Radical-Mediated Catalytic Cyclizations and Ring-Opening Reactions. New J. Chem. 2019, 43, 3254–3264. 10.1039/C8NJ06165C. [DOI] [Google Scholar]
- Tian W. F.; Wang D. P.; Wang S. F.; He K. H.; Cao X. P.; Li Y. Visible-Light Photoredox-Catalyzed Iminyl Radical Formation by N-H Cleavage with Hydrogen Release and Its Application in Synthesis of Isoquinolines. Org. Lett. 2018, 20, 1421–1425. 10.1021/acs.orglett.8b00193. [DOI] [PubMed] [Google Scholar]
- Dempsey J. L.; Brunschwig B. S.; Winkler J. R.; Gray H. B. Hydrogen Evolution Catalyzed by Cobaloximes. Acc. Chem. Res. 2009, 42, 1995–2004. 10.1021/ar900253e. [DOI] [PubMed] [Google Scholar]
- Lücking U. Sulfoximines: A Neglected Opportunity in Medicinal Chemistry. Angew. Chem., Int. Ed. 2013, 52, 9399–9408. 10.1002/anie.201302209. [DOI] [PubMed] [Google Scholar]
- Frings M.; Bolm C.; Blum A.; Gnamm C. Sulfoximines from a Medicinal Chemist’s Perspective: Physicochemical and in Vitro Parameters Relevant for Drug Discovery. Eur. J. Med. Chem. 2017, 126, 225–245. 10.1016/j.ejmech.2016.09.091. [DOI] [PubMed] [Google Scholar]
- Mäder P.; Kattner L. Sulfoximines as Rising Stars in Modern Drug Discovery? Current Status and Perspective on an Emerging Functional Group in Medicinal Chemistry. J. Med. Chem. 2020, 63, 14243–14275. 10.1021/acs.jmedchem.0c00960. [DOI] [PubMed] [Google Scholar]
- Tota A.; Zenzola M.; Chawner S. J.; John-Campbell S. S.; Carlucci C.; Romanazzi G.; Degennaro L.; Bull J. A.; Luisi R. Synthesis of NH-Sulfoximines from Sulfides by Chemoselective One-Pot N- and O-Transfers. Chem. Commun. 2017, 53, 348–351. 10.1039/C6CC08891K. [DOI] [PubMed] [Google Scholar]
- Wimmer A.; König B. Visible-Light-Mediated Photoredox-Catalyzed N-Arylation of NH-Sulfoximines with Electron-Rich Arenes. Adv. Synth. Catal. 2018, 360, 3277–3285. 10.1002/adsc.201800607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lämmermann H.; Sudau A.; Rackl D.; Weinmann H.; Collins K.; Wortmann L.; Candish L.; Hog D. T.; Meier R. Late-Stage Sulfoximidation of Electron-Rich Arenes by Photoredox Catalysis. Synlett 2018, 29, 2679–2684. 10.1055/s-0037-1609656. [DOI] [Google Scholar]
- Cai S.; Chen D.; Xu Y.; Weng W.; Li L.; Zhang R.; Huang M. Visible-Light-Promoted Syntheses of β-Keto Sulfones from Alkynes and Sulfonylhydrazides. Org. Biomol. Chem. 2016, 14, 4205–4209. 10.1039/C6OB00617E. [DOI] [PubMed] [Google Scholar]
- He T. J.; Ye Z.; Ke Z.; Huang J. M. Stereoselective Synthesis of Sulfur-Containing β-Enaminonitrile Derivatives through Electrochemical Csp3-H Bond Oxidative Functionalization of Acetonitrile. Nat. Commun. 2019, 10, 833. 10.1038/s41467-019-08762-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S.; Xu Y.; Chen D.; Li L.; Chen Q.; Huang M.; Weng W. Visible-Light-Enabled Decarboxylative Sulfonylation of Cinnamic Acids with Sulfonylhydrazides under Transition-Metal-Free Conditions. Org. Lett. 2016, 18, 2990–2993. 10.1021/acs.orglett.6b01353. [DOI] [PubMed] [Google Scholar]
- Lovett G. H.; Sparling B. A. Decarboxylative Anti-Michael Addition to Olefins Mediated by Photoredox Catalysis. Org. Lett. 2016, 18, 3494–3497. 10.1021/acs.orglett.6b01712. [DOI] [PubMed] [Google Scholar]
- Gu F.; Huang W.; Liu X.; Chen W.; Cheng X. Substituted Hantzsch Esters as Versatile Radical Reservoirs in Photoredox Reactions. Adv. Synth. Catal. 2018, 360, 925–931. 10.1002/adsc.201701348. [DOI] [Google Scholar]
- Zhao K.; Yamashita K.; Carpenter J. E.; Sherwood T. C.; Ewing W. R.; Cheng P. T. W. W.; Knowles R. R. Catalytic Ring Expansions of Cyclic Alcohols Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2019, 141, 8752–8757. 10.1021/jacs.9b03973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinet S.; Méou A.; Brun P. Access to Enantiopure 2,5-Diaryltetrahydrofurans - Application to the Synthesis of (−)-Virgatusin and (+)-Urinaligran. Eur. J. Org. Chem. 2009, 2009, 2306–2311. 10.1002/ejoc.200900099. [DOI] [Google Scholar]
- Zhao Y.; Lai Y. L.; Du K. S.; Lin D. Z.; Huang J. M. Electrochemical Decarboxylative Sulfonylation of Cinnamic Acids with Aromatic Sulfonylhydrazides to Vinyl Sulfones. J. Org. Chem. 2017, 82, 9655–9661. 10.1021/acs.joc.7b01741. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Cao Y.; Lin Y.; Li Y.; Huang Z.; Lei A. Electrochemical Oxidative Alkoxysulfonylation of Alkenes Using Sulfonyl Hydrazines and Alcohols with Hydrogen Evolution. ACS Catal. 2018, 8, 10871–10875. 10.1021/acscatal.8b03302. [DOI] [Google Scholar]
- Wu J.; Zhang Y.; Gong X.; Meng Y.; Zhu C. Visible-Light Promoted Aerobic Difunctionalization of Alkenes with Sulfonyl Hydrazides for the Synthesis of β-Keto/Hydroxyl Sulfones. Org. Biomol. Chem. 2019, 17, 3507–3513. 10.1039/C9OB00278B. [DOI] [PubMed] [Google Scholar]
- Chen J.; Allyson Z. G.; Xin J. R.; Guan Z.; He Y. H. Photo-Mediated Decarboxylative Ketonization of Atropic Acids with Sulfonyl Hydrazides: Direct Access to β-Ketosulfones. Adv. Synth. Catal. 2020, 362, 2045–2051. 10.1002/adsc.201901525. [DOI] [Google Scholar]
- Shen T.; Zhao Z. G.; Yu Q.; Xu H. J. Photosensitized Reduction of Benzil by Heteroatom-Containing Anthracene Dyes. J. Photochem. Photobiol., A 1989, 47, 203–212. 10.1016/1010-6030(89)87066-2. [DOI] [Google Scholar]
- Yuan Y.; Yu Y.; Qiao J.; Liu P.; Yu B.; Zhang W.; Liu H.; He M.; Huang Z.; Lei A. Exogenous-Oxidant-Free Electrochemical Oxidative C-H Sulfonylation of Arenes/Heteroarenes with Hydrogen Evolution. Chem. Commun. 2018, 54, 11471–11474. 10.1039/C8CC06451B. [DOI] [PubMed] [Google Scholar]
- Mahanty K.; Maiti D.; De Sarkar S. Regioselective C-H Sulfonylation of 2H-Indazoles by Electrosynthesis. J. Org. Chem. 2020, 85, 3699–3708. 10.1021/acs.joc.9b03330. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Jiang M.; Wang T.; Xiong Y.; Li J.; Guo H.; Lei A. Synergy of Anodic Oxidation and Cathodic Reduction Leads to Electrochemical Deoxygenative C2 Arylation of Quinoline: N-Oxides. Chem. Commun. 2019, 55, 11091–11094. 10.1039/C9CC05841A. [DOI] [PubMed] [Google Scholar]
- Du B.; Qian P.; Wang Y.; Mei H.; Han J.; Pan Y. Cu-Catalyzed Deoxygenative C2-Sulfonylation Reaction of Quinoline N-Oxides with Sodium Sulfinate. Org. Lett. 2016, 18, 4144–4147. 10.1021/acs.orglett.6b02289. [DOI] [PubMed] [Google Scholar]
- Jiang M.; Yuan Y.; Wang T.; Xiong Y.; Li J.; Guo H.; Lei A. Exogenous-Oxidant-and Catalyst-Free Electrochemical Deoxygenative C2 Sulfonylation of Quinoline: N-Oxides. Chem. Commun. 2019, 55, 13852–13855. 10.1039/C9CC07777D. [DOI] [PubMed] [Google Scholar]
- Sheykhan M.; Khani S.; Abbasnia M.; Shaabanzadeh S.; Joafshan M. An Approach to C-N Activation: Coupling of Arenesulfonyl Hydrazides and Arenesulfonyl Chlorides with: Tert -Amines via a Metal-, Oxidant- and Halogen-Free Anodic Oxidation. Green Chem. 2017, 19, 5940–5948. 10.1039/C7GC03141F. [DOI] [Google Scholar]
- Wayner D. D. M.; Dannenberg J. J.; Griller D. Oxidation Potentials of α-Aminoalkyl Radicals: Bond Dissociation Energies for Related Radical Cations. Chem. Phys. Lett. 1986, 131, 189–191. 10.1016/0009-2614(86)80542-5. [DOI] [Google Scholar]
- Jonsson M.; Wayner D. D. M.; Lusztyk J. Redox and Acidity Properties of Alkyl- and Arylamine Radical Cations and the Corresponding Aminyl Radicals. J. Phys. Chem. 1996, 100, 17539–17543. 10.1021/jp961286q. [DOI] [Google Scholar]
- Kim H. S.; Lee S. Electrochemical Coupling of Arylsulfonyl Hydrazides and Tertiary Amines for the Synthesis of β-Amidovinyl Sulfones. Eur. J. Org. Chem. 2019, 2019, 6951–6955. 10.1002/ejoc.201901277. [DOI] [Google Scholar]
- Mo Z. Y.; Swaroop T. R.; Tong W.; Zhang Y. Z.; Tang H. T.; Pan Y. M.; Sun H.-B.; Chen Z. F. Electrochemical Sulfonylation of Thiols with Sulfonyl Hydrazides: A Metal- and Oxidant-Free Protocol for the Synthesis of Thiosulfonates. Green Chem. 2018, 20, 4428–4432. 10.1039/C8GC02143K. [DOI] [Google Scholar]
- Bordwell F. G.; Zhang X. M.; Satish A. V.; Cheng J. P. Assessment of the Importance of Changes in Ground-State Energies on the Bond Dissociation Enthalpies of the O-H Bonds in Phenols and the S-H Bonds in Thiophenols. J. Am. Chem. Soc. 1994, 116, 6605–6610. 10.1021/ja00094a015. [DOI] [Google Scholar]
- Zhu M.; Zheng N. Photoinduced Cleavage of N-N Bonds of Aromatic Hydrazines and Hydrazides by Visible Light. Synthesis 2011, 14, 2223–2236. 10.1055/s-0030-1260082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapountzis I.; Knochel P. A General Animation Method Based on the Addition of Polyfunctional Arylmagnesium Reagents to Functionalized Arylazo Tosylates. Angew. Chem., Int. Ed. 2004, 43, 897–900. 10.1002/anie.200353241. [DOI] [PubMed] [Google Scholar]
- Crutchley R. J.; Lever A. B. P. Ruthenium(II) Tris(Bipyrazyl) Dication - a New Photocatalyst. J. Am. Chem. Soc. 1980, 102, 7128–7129. 10.1021/ja00543a053. [DOI] [Google Scholar]
- Ansari A.; Ali A.; Asif M.; Shamsuzzaman Review: Biologically Active Pyrazole Derivatives. New J. Chem. 2017, 41, 16–41. 10.1039/C6NJ03181A. [DOI] [Google Scholar]
- Fustero S.; Sánchez-Roselló M.; Barrio P.; Simón-Fuentes A. From 2000 to Mid-2010: A Fruitful Decade for the Synthesis of Pyrazoles. Chem. Rev. 2011, 111, 6984–7034. 10.1021/cr2000459. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Zhang T.; Chen Q. Y.; Zhu C. Visible-Light Photocatalytic Aerobic Annulation for the Green Synthesis of Pyrazoles. Org. Lett. 2016, 18, 4206–4209. 10.1021/acs.orglett.6b01867. [DOI] [PubMed] [Google Scholar]
- Meng Y.; Zhang T.; Gong X.; Zhang M.; Zhu C. Visible-Light Promoted One-Pot Synthesis of Pyrazoles from Alkynes and Hydrazines. Tetrahedron Lett. 2019, 60, 171–174. 10.1016/j.tetlet.2018.12.009. [DOI] [Google Scholar]
- Sindhu K. S.; Anilkumar G. Recent Advances and Applications of Glaser Coupling Employing Greener Protocols. RSC Adv. 2014, 4, 27867–27887. 10.1039/C4RA02416H. [DOI] [Google Scholar]
- Sagadevan A.; Lyu P. C.; Hwang K. C. Visible-Light-Activated Copper(I) Catalyzed Oxidative Csp-Csp Cross-Coupling Reaction: Efficient Synthesis of Unsymmetrical Conjugated Diynes without Ligands and Base. Green Chem. 2016, 18, 4526–4530. 10.1039/C6GC01463A. [DOI] [Google Scholar]
- Ding Y.; Li H.; Meng Y.; Zhang T.; Li J.; Chen Q. Y.; Zhu C. Direct Synthesis of Hydrazones by Visible Light Mediated Aerobic Oxidative Cleavage of the C = C Bond. Org. Chem. Front. 2017, 4, 1611–1614. 10.1039/C7QO00276A. [DOI] [Google Scholar]
- Ding Y.; Zhang W.; Li H.; Meng Y.; Zhang T.; Chen Q. Y.; Zhu C. Metal-Free Synthesis of Ketones by Visible-Light Induced Aerobic Oxidative Radical Addition of Aryl Hydrazines to Alkenes. Green Chem. 2017, 19, 2941–2944. 10.1039/C7GC01083D. [DOI] [Google Scholar]
- Kibriya G.; Mondal S.; Hajra A. Visible-Light-Mediated Synthesis of Unsymmetrical Diaryl Sulfides via Oxidative Coupling of Arylhydrazine with Thiol. Org. Lett. 2018, 20, 7740–7743. 10.1021/acs.orglett.8b03549. [DOI] [PubMed] [Google Scholar]
- Lambert C. R.; Kochevar I. E. Electron Transfer Quenching of the Rose Bengal Triplet State. Photochem. Photobiol. 1997, 66, 15–25. 10.1111/j.1751-1097.1997.tb03133.x. [DOI] [PubMed] [Google Scholar]
- Sahoo M. K.; Saravanakumar K.; Jaiswal G.; Balaraman E. Photocatalysis Enabling Acceptorless Dehydrogenation of Diaryl Hydrazines at Room Temperature. ACS Catal. 2018, 8, 7727–7733. 10.1021/acscatal.8b01579. [DOI] [Google Scholar]
- Tian M.; Liu S.; Bu X.; Yu J.; Yang X. Covalent Organic Frameworks: A Sustainable Photocatalyst toward Visible-Light-Accelerated C3 Arylation and Alkylation of Quinoxalin-2(1H)-ones. Chem. - Eur. J. 2020, 26, 369–373. 10.1002/chem.201903523. [DOI] [PubMed] [Google Scholar]
- Wang H.; Wang H.; Wang Z.; Tang L.; Zeng G.; Xu P.; Chen M.; Xiong T.; Zhou C.; Li X.; et al. Covalent Organic Framework Photocatalysts: Structures and Applications. Chem. Soc. Rev. 2020, 49, 4135–4165. 10.1039/D0CS00278J. [DOI] [PubMed] [Google Scholar]
- Um I. H.; Kim M. Y.; Cho H. J.; Dust J. M.; Buncel E. The α-Effect in the SNAr Reaction of 1-(4-Nitrophenoxy)-2,4-Dinitrobenzene with Anionic Nucleophiles: Effects of Solvation and Polarizability on the α-Effect. Can. J. Chem. 2015, 93, 1109–1114. 10.1139/cjc-2015-0073. [DOI] [Google Scholar]
- Harnsberger H. F.; Cochran E. L.; Szmant H. H. The Basicity of Hydrazones. J. Am. Chem. Soc. 1955, 77, 5048–5050. 10.1021/ja01624a032. [DOI] [Google Scholar]
- Bansal K. M.; Henglein A.; Sellers R. M. Pulse Radiolysis and Polarography: Polarograms and Structure of Short-Lived Free Radicals. Berichte der Bunsengesellschaft für Phys. Chemie 1974, 78, 569–575. 10.1002/bbpc.19740780610. [DOI] [Google Scholar]
- Hu X. Q.; Qi X.; Chen J. R.; Zhao Q. Q.; Wei Q.; Lan Y.; Xiao W. J. Catalytic N-Radical Cascade Reaction of Hydrazones by Oxidative Deprotonation Electron Transfer and TEMPO Mediation. Nat. Commun. 2016, 7, 11188. 10.1038/ncomms11188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran J.; Gallardo I.; Moreno M.; Savéant J. M. Dissociative Electron Transfer. Ab Initio Study of the Carbon-Halogen Bond Reductive Cleavage in Methyl and Perfluoromethyl Halides. Role of the Solvent. J. Am. Chem. Soc. 1992, 114, 9576–9583. 10.1021/ja00050a042. [DOI] [Google Scholar]
- Zhang W.; Yang L.; Wu L. M.; Liu Y. C.; Liu Z. L. Photoinduced Electron Transfer Retropinacol Reaction of 4-(N,N-Dimethylamino)Phenyl Pinacols in Chloroform. J. Chem. Soc., Perkin Trans. 2 1998, 1189–1193. 10.1039/a709169i. [DOI] [Google Scholar]
- Costentin C.; Robert M.; Savéant J. M. Successive Removal of Chloride Ions from Organic Polychloride Pollutants. Mechanisms of Reductive Electrochemical Elimination in Aliphatic Gem-Polychlorides, α,β-Polychloroalkenes, and α,β -Polychloroalkanes in Mildly Protic Medium. J. Am. Chem. Soc. 2003, 125, 10729–10739. 10.1021/ja036141t. [DOI] [PubMed] [Google Scholar]
- Hu X. Q.; Chen J.; Chen J. R.; Yan D. M.; Xiao W. J. Organophotocatalytic Generation of N- and O-Centred Radicals Enables Aerobic Oxyamination and Dioxygenation of Alkenes. Chem. - Eur. J. 2016, 22, 14141–14146. 10.1002/chem.201602597. [DOI] [PubMed] [Google Scholar]
- Marx L.; Schöllhorn B. Intramolecular Charge Effects in the Electrochemical Oxidation of Aminoxyl Radicals. New J. Chem. 2006, 30, 430–434. 10.1039/b515677g. [DOI] [Google Scholar]
- Zhao Q. Q.; Hu X. Q.; Yang M. N.; Chen J. R.; Xiao W. J. A Visible-Light Photocatalytic N-Radical Cascade of Hydrazones for the Synthesis of Dihydropyrazole-Fused Benzosultams. Chem. Commun. 2016, 52, 12749–12752. 10.1039/C6CC05897C. [DOI] [PubMed] [Google Scholar]
- McAtee R. C.; Noten E. A.; Stephenson C. R. J. Arene Dearomatization through a Catalytic N-Centered Radical Cascade Reaction. Nat. Commun. 2020, 11, 2528. 10.1038/s41467-020-16369-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q. Q.; Chen J.; Yan D. M.; Chen J. R.; Xiao W. J. Photocatalytic Hydrazonyl Radical-Mediated Radical Cyclization/Allylation Cascade: Synthesis of Dihydropyrazoles and Tetrahydropyridazines. Org. Lett. 2017, 19, 3620–3623. 10.1021/acs.orglett.7b01609. [DOI] [PubMed] [Google Scholar]
- Chen J. J. R.; Guo H. M.; Zhao Q. Q.; Chen J. J. R.; Xiao W. J. Visible Light-Driven Photocatalytic Generation of Sulfonamidyl Radicals for Alkene Hydroamination of Unsaturated Sulfonamides. Chem. Commun. 2018, 54, 6780–6783. 10.1039/C7CC09871E. [DOI] [PubMed] [Google Scholar]
- Ishizuka H.; Adachi K.; Odagi M.; Nagasawa K. Synthesis of Isoxazolidines by Intramolecular Hydroamination of N-Alkoxyamides in the Presence of a Visible-Light Photoredox Catalyst. Bull. Chem. Soc. Jpn. 2019, 92, 1447–1449. 10.1246/bcsj.20190127. [DOI] [Google Scholar]
- Brachet E.; Marzo L.; Selkti M.; König B.; Belmont P. Visible Light Amination/Smiles Cascade: Access to Phthalazine Derivatives. Chem. Sci. 2016, 7, 5002–5006. 10.1039/C6SC01095D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Abreu M.; Selkti M.; Belmont P.; Brachet E. Phosphoramidates as Transient Precursors of Nitrogen-Centered Radical Under Visible-Light Irradiation: Application to the Synthesis of Phthalazine Derivatives. Adv. Synth. Catal. 2020, 362, 2216–2222. 10.1002/adsc.202000018. [DOI] [Google Scholar]
- Kharb R.; Sharma P. C.; Yar M. S. Pharmacological Significance of Triazole Scaffold. J. Enzyme Inhib. Med. Chem. 2011, 26, 1–21. 10.3109/14756360903524304. [DOI] [PubMed] [Google Scholar]
- Ye Z.; Ding M.; Wu Y.; Li Y.; Hua W.; Zhang F. Electrochemical Synthesis of 1,2,4-Triazole-Fused Heterocycles. Green Chem. 2018, 20, 1732–1737. 10.1039/C7GC03739B. [DOI] [Google Scholar]
- Xu P.; Xu H. C. Electrochemical Synthesis of [1,2,3]Triazolo[1,5-a]Pyridines through Dehydrogenative Cyclization. ChemElectroChem 2019, 6, 4177–4179. 10.1002/celc.201900080. [DOI] [Google Scholar]
- Wang L.; Ma Z. G.; Wei X. J.; Meng Q. Y.; Yang D. T.; Du S. F.; Chen Z. F.; Wu L. Z.; Liu Q. Synthesis of 2-Substituted Pyrimidines and Benzoxazoles via a Visible-Light-Driven Organocatalytic Aerobic Oxidation: Enhancement of the Reaction Rate and Selectivity by a Base. Green Chem. 2014, 16, 3752–3757. 10.1039/C4GC00337C. [DOI] [Google Scholar]
- de Fátima ł.; Braga T. C.; Neto L. da S.; Terra B. S.; Oliveira B. G. F.; da Silva D. L.; Modolo L. V. A Mini-Review on Biginelli Adducts with Notable Pharmacological Properties. J. Adv. Res. 2015, 6, 363–373. 10.1016/j.jare.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X. Q.; Li H. R.; Li Q.; Ai T.; Lu J. Y.; Yang Y.; Cheng J. P. Determination of the C4-H Bond Dissociation Energies of NADH Models and Their Radical Cations in Acetonitrile. Chem. - Eur. J. 2003, 9, 871–880. 10.1002/chem.200390108. [DOI] [PubMed] [Google Scholar]
- Meng F.-k.; Zhu X. q. Elemental Steps of the Thermodynamics of Dihydropyrimidine: A New Class of Organic Hydride Donors. Org. Biomol. Chem. 2017, 15, 197–206. 10.1039/C6OB02195F. [DOI] [PubMed] [Google Scholar]
- Watanabe M.; Koike H.; Ishiba T.; Okada T.; Seo S.; Hirai K. Synthesis and Biological Activity of Methanesulfonamide Pyrimidine- and N-Methanesulfonyl Pyrrole-Substituted 3,5-Dihydroxy-6-Heptenoates, a Novel Series of HMG-CoA Reductase Inhibitors. Bioorg. Med. Chem. 1997, 5, 437–444. 10.1016/S0968-0896(96)00248-9. [DOI] [PubMed] [Google Scholar]
- Ahmad S.; Madsen C. S.; Stein P. D.; Janovitz E.; Huang C.; Ngu K.; Bisaha S.; Kennedy L. J.; Chen B. C.; Zhao R.; et al. (3R,5S,E)-7-(4-(4-Fluorophenyl)-6-Isopropyl-2-(Methyl(1-Methyl-1H-1,2, 4-Triazol-5-Yl)Amino)Pyrimidin-5-Yl)-3,5-Dihydroxyhept-6-Enoic Acid (BMS-644950): A Rationally Designed Orally Efficacious 3-Hydroxy-3- Methylglutaryl Coenzyme-A Reductase Inhibitor Wi. J. Med. Chem. 2008, 51, 2722–2733. 10.1021/jm800001n. [DOI] [PubMed] [Google Scholar]
- Hobson L. A.; Akiti O.; Deshmukh S. S.; Harper S.; Katipally K.; Lai C. J.; Livingston R. C.; Lo E.; Miller M. M.; Ramakrishnan S.; et al. Development of a Scaleable Process for the Synthesis of a Next-Generation Statin. Org. Process Res. Dev. 2010, 14, 441–458. 10.1021/op100010n. [DOI] [Google Scholar]
- Joselevich E.; Willner I. Photoinduced Electron Transfer from Eosin and Ethyl Eosin to Fe(CN)63- in AOT Reverse Micelles: Separation of Redox Products by Electron-Transfer-Induced Hydrophobicity. J. Phys. Chem. 1995, 99, 6903–6912. 10.1021/j100018a022. [DOI] [Google Scholar]
- Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- Bissonnette N. B.; Ellis J. M.; Hamann L. G.; Romanov-Michailidis F. Expedient Access to Saturated Nitrogen Heterocycles by Photoredox Cyclization of Imino-Tethered Dihydropyridines. Chem. Sci. 2019, 10, 9591–9596. 10.1039/C9SC03429C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo C. V. T.; Mikutis G.; Bode J. W. SnAP Reagents for the Transformation of Aldehydes into Substituted Thiomorpholines - An Alternative to Cross-Coupling with Saturated Heterocycles. Angew. Chem., Int. Ed. 2013, 52, 1705–1708. 10.1002/anie.201208064. [DOI] [PubMed] [Google Scholar]
- Siau W. Y.; Bode J. W. One-Step Synthesis of Saturated Spirocyclic N-Heterocycles with Stannyl Amine Protocol (SnAP) Reagents and Ketones. J. Am. Chem. Soc. 2014, 136, 17726–17729. 10.1021/ja511232b. [DOI] [PubMed] [Google Scholar]
- Hsieh S. Y.; Bode J. W. Lewis Acid Induced Toggle from Ir(II) to Ir(IV) Pathways in Photocatalytic Reactions: Synthesis of Thiomorpholines and Thiazepanes from Aldehydes and SLAP Reagents. ACS Cent. Sci. 2017, 3, 66–72. 10.1021/acscentsci.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout D. M.; Meyers A. I. Recent Advances in the Chemistry of Dihydropyridines. Chem. Rev. 1982, 82, 223–243. 10.1021/cr00048a004. [DOI] [Google Scholar]
- Wang P. Z.; Chen J. R.; Xiao W. J. Hantzsch Esters: An Emerging Versatile Class of Reagents in Photoredox Catalyzed Organic Synthesis. Org. Biomol. Chem. 2019, 17, 6936–6951. 10.1039/C9OB01289C. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Bonet Á.; Tellis J. C.; Matsui J. K.; Vara B. A.; Molander G. A. 1,4-Dihydropyridines as Alkyl Radical Precursors: Introducing the Aldehyde Feedstock to Nickel/Photoredox Dual Catalysis. ACS Catal. 2016, 6, 8004–8008. 10.1021/acscatal.6b02786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K.; Nojima S.; Nishibayashi Y. Nickel- and Photoredox-Catalyzed Cross-Coupling Reactions of Aryl Halides with 4-Alkyl-1,4-Dihydropyridines as Formal Nucleophilic Alkylation Reagents. Angew. Chem., Int. Ed. 2016, 55, 14106–14110. 10.1002/anie.201606513. [DOI] [PubMed] [Google Scholar]
- Nakajima K.; Nojima S.; Sakata K.; Nishibayashi Y. Visible-Light-Mediated Aromatic Substitution Reactions of Cyanoarenes with 4-Alkyl-1,4-Dihydropyridines through Double Carbon-Carbon Bond Cleavage. ChemCatChem 2016, 8, 1028–1032. 10.1002/cctc.201600037. [DOI] [Google Scholar]
- Zhang H. H.; Yu S. Radical Alkylation of Imines with 4-Alkyl-1,4-Dihydropyridines Enabled by Photoredox/Brønsted Acid Cocatalysis. J. Org. Chem. 2017, 82, 9995–10006. 10.1021/acs.joc.7b01425. [DOI] [PubMed] [Google Scholar]
- Xue F.; Wang F.; Liu J.; Di J.; Liao Q.; Lu H.; Zhu M.; He L.; He H.; Zhang D.; et al. A Desulfurative Strategy for the Generation of Alkyl Radicals Enabled by Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed. 2018, 57, 6667–6671. 10.1002/anie.201802710. [DOI] [PubMed] [Google Scholar]
- Yee Y. K.; Bernstein P. R.; Adams E. J.; Brown F. J.; Cronk L. A.; Hebbel K. C.; Vacek E. P.; Krell R. D.; Snyder D. W. A Novel Series of Selective Leukotriene Antagonists: Exploration and Optimization of the Acidic Region in 1,6-Disubstituted Indoles and Indazoles. J. Med. Chem. 1990, 33, 2437–2451. 10.1021/jm00171a018. [DOI] [PubMed] [Google Scholar]
- Di J.; He H.; Wang F.; Xue F.; Liu X. Y.; Qin Y. Regiospecific Alkyl Addition of (Hetero)Arene-Fused Thiophenes Enabled by a Visible-Light-Mediated Photocatalytic Desulfuration Approach. Chem. Commun. 2018, 54, 4692–4695. 10.1039/C8CC02052C. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Wu Z.; Yu L.; Wang Y.; Pan Y. Alkyl Carbazates for Electrochemical Deoxygenative Functionalization of Heteroarenes. Angew. Chem. 2020, 132, 10951–10955. 10.1002/ange.202001571. [DOI] [PubMed] [Google Scholar]
- Lackner G. L.; Quasdorf K. W.; Overman L. E. Direct Construction of Quaternary Carbons from Tertiary Alcohols via Photoredox-Catalyzed Fragmentation of Tert-Alkyl N-Phthalimidoyl Oxalates. J. Am. Chem. Soc. 2013, 135, 15342–15345. 10.1021/ja408971t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrat C. C.; Jamison C. R.; Slutskyy Y.; MacMillan D. W. C.; Overman L. E. Oxalates as Activating Groups for Alcohols in Visible Light Photoredox Catalysis: Formation of Quaternary Centers by Redox-Neutral Fragment Coupling. J. Am. Chem. Soc. 2015, 137, 11270–11273. 10.1021/jacs.5b07678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T.; Sugiura Y.; Zaimoku H.; Ishibashi H. Iron-Catalyzed Oxidative Addition of Alkoxycarbonyl Radicals to Alkenes with Carbazates and Air. Angew. Chem., Int. Ed. 2010, 49, 10154–10157. 10.1002/anie.201005574. [DOI] [PubMed] [Google Scholar]
- Hilborn J. W.; Pincock J. A. Rates of Decarboxylation of Acyloxy Radicals Formed In the Photocleavage of Substituted 1-Naphthylmethyl Alkanoates. J. Am. Chem. Soc. 1991, 113, 2683–2686. 10.1021/ja00007a049. [DOI] [Google Scholar]
- Dénès F.; Pichowicz M.; Povie G.; Renaud P. Thiyl Radicals in Organic Synthesis. Chem. Rev. 2014, 114, 2587–2693. 10.1021/cr400441m. [DOI] [PubMed] [Google Scholar]
- Tanaka H.; Sakai K.; Kawamura A.; Oisaki K.; Kanai M. Sulfonamides as New Hydrogen Atom Transfer (HAT) Catalysts for Photoredox Allylic and Benzylic C-H Arylations. Chem. Commun. 2018, 54, 3215–3218. 10.1039/C7CC09457D. [DOI] [PubMed] [Google Scholar]
- Mori Y.; Sakaguchi Y.; Hayashi H. Magnetic Field Effects on Chemical Reactions of Biradical Radical Ion Pairs in Homogeneous Fluid Solvents. J. Phys. Chem. A 2000, 104, 4896–4905. 10.1021/jp0007437. [DOI] [Google Scholar]
- Ohmatsu K.; Suzuki R.; Furukawa Y.; Sato M.; Ooi T. Zwitterionic 1,2,3-Triazolium Amidate as a Catalyst for Photoinduced Hydrogen-Atom Transfer Radical Alkylation. ACS Catal. 2020, 10, 2627–2632. 10.1021/acscatal.9b04491. [DOI] [Google Scholar]
- Šakić D.; Zipse H. Radical Stability as a Guideline in C-H Amination Reactions. Adv. Synth. Catal. 2016, 358, 3983–3991. 10.1002/adsc.201600629. [DOI] [Google Scholar]
- Ryder A. S. H.; Cunningham W. B.; Ballantyne G.; Mules T.; Kinsella A. G.; Turner-Dore J.; Alder C. M.; Edwards L. J.; McKay B. S. J.; Grayson M. N.; et al. Photocatalytic α-Tertiary Amine Synthesis via C-H Alkylation of Unmasked Primary Amines. Angew. Chem., Int. Ed. 2020, 59, 14986–14991. 10.1002/anie.202005294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q. Q.; Chen J.; Zhou X. S.; Yu X. Y.; Chen J. R.; Xiao W. J. Photogenerated Neutral Nitrogen Radical Catalyzed Bifunctionalization of Alkenes. Chem. - Eur. J. 2019, 25, 8024–8029. 10.1002/chem.201901665. [DOI] [PubMed] [Google Scholar]
- Yu X. Y.; Chen J. R.; Wang P. Z.; Yang M. N.; Liang D.; Xiao W. J. A Visible-Light-Driven Iminyl Radical-Mediated C-C Single Bond Cleavage/Radical Addition Cascade of Oxime Esters. Angew. Chem., Int. Ed. 2018, 57, 738–743. 10.1002/anie.201710618. [DOI] [PubMed] [Google Scholar]
- Studer A.; Curran D. P. Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed. 2016, 55, 58–102. 10.1002/anie.201505090. [DOI] [PubMed] [Google Scholar]
- Zhao Q.-Q.; Zhou X.-S.; Xu S.-H.; Wu Y.-L.; Xiao W.-J.; Chen J.-R. Visible-Light-Driven Nitrogen Radical-Catalyzed [3+2] Cyclization of Vinylcyclopropanes and N-Tosyl Vinylaziridines with Alkenes. Org. Lett. 2020, 22, 2470–2475. 10.1021/acs.orglett.0c00712. [DOI] [PubMed] [Google Scholar]
- Miura K.; Fugami K.; Oshima K.; Utimoto K. Triphenylstannyl Radical or Benzenethiyl Radical Promoted Transformation of 1,1-Dialk Oxycarbonyl-2-(1,3-Butadienyl)Cyclopropanes into 2-Ethenyl-3-Cyclopentenes. Tetrahedron Lett. 1988, 29, 1543–1546. 10.1016/S0040-4039(00)80347-0. [DOI] [Google Scholar]
- Miura K.; Fugami K.; Oshima K.; Utimoto K. Synthesis of Vinylcyclopentanes from Vinylcyclopropanes and Alkenes Promoted by Benzenethiyl Radical. Tetrahedron Lett. 1988, 29, 5135–5138. 10.1016/S0040-4039(00)80701-7. [DOI] [Google Scholar]
- Feldman K. S.; Romanelli A. L.; Ruckle R. E.; Miller R. F. Cyclopentane Synthesis via Free-Radical-Mediated Addition of Functionalized Alkenes to Substituted Vinylcyclopropanes. J. Am. Chem. Soc. 1988, 110, 3300–3302. 10.1021/ja00218a050. [DOI] [Google Scholar]
- Feldman K. S.; Romanelli A. L.; Ruckle R. E.; Jean G. Vinylcyclopentane Synthesis via Phenylthio Radical Catalyzed Alkenylation of Vinylcyclopropanes: Preparative and Mechanistic Studies. J. Org. Chem. 1992, 57, 100–110. 10.1021/jo00027a020. [DOI] [Google Scholar]
- Feldman K. S.; Berven H. M.; Weinreb P. H. 2,2-Dihalovinylcyclopropanes as Highly Diastereoselective Three-Atom Addends in Phenylthio Radical Mediated Vinylcyclopentane Synthesis. J. Am. Chem. Soc. 1993, 115, 11364–11369. 10.1021/ja00077a040. [DOI] [Google Scholar]
- Hashimoto T.; Takino K.; Hato K.; Maruoka K. A Bulky Thiyl-Radical Catalyst for the [3+2] Cyclization of N-Tosyl Vinylaziridines and Alkenes. Angew. Chem., Int. Ed. 2016, 55, 8081–8085. 10.1002/anie.201602723. [DOI] [PubMed] [Google Scholar]
- Hashimoto T.; Kawamata Y.; Maruoka K. An Organic Thiyl Radical Catalyst for Enantioselective Cyclization. Nat. Chem. 2014, 6, 702–705. 10.1038/nchem.1998. [DOI] [PubMed] [Google Scholar]
- Chen D. F.; Chrisman C. H.; Miyake G. M. Bromine Radical Catalysis by Energy Transfer Photosensitization. ACS Catal. 2020, 10, 2609–2614. 10.1021/acscatal.0c00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.; Dang H. T.; Haug G. C.; He R.; Nguyen V. D. V. T.; Nguyen V. D. V. T.; Arman H. D.; Schanze K. S.; Larionov O. V. Visible Light-Induced Borylation of C-O, C-N, and C-X Bonds. J. Am. Chem. Soc. 2020, 142, 1603–1613. 10.1021/jacs.9b12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Mück-Lichtenfeld C.; Studer A. Metal-Free Radical Borylation of Alkyl and Aryl Iodides. Angew. Chem., Int. Ed. 2018, 57, 16832–16836. 10.1002/anie.201810782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan J.; Zhang Z. G.; Xiao W. J. Visible-Light-Induced Decarboxylative Functionalization of Carboxylic Acids and Their Derivatives. Angew. Chem., Int. Ed. 2015, 54, 15632–15641. 10.1002/anie.201505731. [DOI] [PubMed] [Google Scholar]
- Li Y.; Ge L.; Muhammad M. T.; Bao H. Recent Progress on Radical Decarboxylative Alkylation for C(sp3)-C Bond Formation. Synthesis 2017, 49, 5263–5284. 10.1055/s-0036-1590935. [DOI] [Google Scholar]
- Jin Y.; Fu H. Visible-Light Photoredox Decarboxylative Couplings. Asian J. Org. Chem. 2017, 6, 368–385. 10.1002/ajoc.201600513. [DOI] [PubMed] [Google Scholar]
- Patra T.; Maiti D. Decarboxylation as the Key Step in C-C Bond-Forming Reactions. Chem. - Eur. J. 2017, 23, 7382–7401. 10.1002/chem.201604496. [DOI] [PubMed] [Google Scholar]
- Schwarz J.; König B. Decarboxylative Reactions with and without Light-A Comparison;. Green Chem. 2018, 20, 323–361. 10.1039/C7GC02949G. [DOI] [Google Scholar]
- Zhou Q. Q.; Wei Y.; Lu L. Q.; Xiao W. J. Decarboxylative Coupling Reactions. Sci. Synth. 2018, 6, 167–218. 10.1055/sos-SD-229-00103. [DOI] [Google Scholar]
- McMurray L.; McGuire T. M.; Howells R. L. Recent Advances in Photocatalytic Decarboxylative Coupling Reactions in Medicinal Chemistry. Synthesis 2020, 52, 1719–1737. 10.1055/s-0039-1690843. [DOI] [Google Scholar]
- Zheng Y.; Shao X.; Ramadoss V.; Tian L.; Wang Y. Recent Developments in Photochemical and Electrochemical Decarboxylative C(sp3)-N Bond Formation. Synthesis 2020, 52, 1357–1368. 10.1055/s-0039-1690839. [DOI] [Google Scholar]
- Wang D.; Farquhar E. R.; Stubna A.; Münck E.; Que L. A Diiron(IV) Complex That Cleaves Strong C-H and O-H Bonds. Nat. Chem. 2009, 1, 145–150. 10.1038/nchem.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Que L. Oxidation of Water by a Nonhaem Diiron(IV) Complex via Proton-Coupled Electron Transfer. Chem. Commun. 2013, 49, 10682–10684. 10.1039/c3cc46391e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester A. W. Oxidation of Alkyl Aromatic Hydrocarbons by Potassium 12-Tungstocobaltate(III). J. Org. Chem. 1970, 35, 1797–1800. 10.1021/jo00831a017. [DOI] [Google Scholar]
- Baciocchi E.; Bietti M.; Putignani L.; Steenken S. Side-Chain Fragmentation of Arylalkanol Radical Cations. Carbon-Carbon and Carbon-Hydrogen Bond Cleavage and the Role of α- and β-OH Groups. J. Am. Chem. Soc. 1996, 118, 5952–5960. 10.1021/ja954236s. [DOI] [Google Scholar]
- Baciocchi E.; Bietti M.; Steenken S. Base-Catalyzed C-H Deprotonation of 4-Methoxybenzyl Alcohol Radical Cations in Water: Evidence for a Carbon-to-Oxygen 1,2-H-Shift Mechanism. J. Am. Chem. Soc. 1997, 119, 4078–4079. 10.1021/ja970259q. [DOI] [Google Scholar]
- Baciocchi E.; Bietti M.; Lanzalunga O.; Steenken S. Oxygen Acidity of 1-Arylalkanol Radical Cations. 4-Methoxycumyloxyl Radical as -C(Me)2-O-to-Nucleus Electron-Transfer Intermediate in the Reaction of 4-Methoxycumyl Alcohol Radical Cation with OH–. J. Am. Chem. Soc. 1998, 120, 11516–11517. 10.1021/ja980645n. [DOI] [Google Scholar]
- Baciocchi E.; Bietti M.; Manduchi L.; Steenken S. Oxygen versus Carbon Acidity in the Side-Chain Fragmentation of 2-, 3-, and 4-Arylalkanol Radical Cations in Aqueous Solution: The Influence of the Distance between the OH Group and the Aromatic Ring. J. Am. Chem. Soc. 1999, 121, 6624–6629. 10.1021/ja990352+. [DOI] [Google Scholar]
- Baciocchi E.; Bietti M.; Lanzalunga O. Mechanistic Aspects of β-Bond-Cleavage Reactions of Aromatic Radical Cations. Acc. Chem. Res. 2000, 33, 243–251. 10.1021/ar980014y. [DOI] [PubMed] [Google Scholar]
- Wang D.; Mao J.; Zhu C. Visible Light-Promoted Ring-Opening Functionalization of Unstrained Cycloalkanols via Inert C-C Bond Scission. Chem. Sci. 2018, 9, 5805–5809. 10.1039/C8SC01763H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concepción J. I.; Francisco C. G.; Hernández R.; Salazar J. A.; Suárez E. Intramolecular Hydrogen Abstraction. Iodosobenzene Diacetate, an Efficient and Convenient Reagent for Alkoxy Radical Generation. Tetrahedron Lett. 1984, 25, 1953–1956. 10.1016/S0040-4039(01)90085-1. [DOI] [Google Scholar]
- Martín A.; Salazar J. A.; Suárez E. Synthesis of Chiral Spiroacetals from Carbohydrates. Tetrahedron Lett. 1995, 36, 4489–4492. 10.1016/0040-4039(95)00766-6. [DOI] [Google Scholar]
- Martín A.; Salazar J. A.; Suárez E. Synthesis of Chiral Spiroacetals from Carbohydrates. J. Org. Chem. 1996, 61, 3999–4006. 10.1021/jo960060g. [DOI] [PubMed] [Google Scholar]
- Martín A.; Pérez-Martín I.; Suárez E. Intramolecular Hydrogen Abstraction Promoted by Amidyl Radicals. Evidence for Electronic Factors in the Nucleophilic Cyclization of Ambident Amides to Oxocarbenium Ions. Org. Lett. 2005, 7, 2027–2030. 10.1021/ol050526u. [DOI] [PubMed] [Google Scholar]
- Wang J.; Huang B.; Shi C.; Yang C.; Xia W. Visible-Light-Mediated Ring-Opening Strategy for the Regiospecific Allylation/Formylation of Cycloalkanols. J. Org. Chem. 2018, 83, 9696–9706. 10.1021/acs.joc.8b01225. [DOI] [PubMed] [Google Scholar]
- Huang L.; Ji T.; Rueping M. Remote Nickel-Catalyzed Cross-Coupling Arylation via Proton-Coupled Electron Transfer-Enabled C-C Bond Cleavage. J. Am. Chem. Soc. 2020, 142, 3532–3539. 10.1021/jacs.9b12490. [DOI] [PubMed] [Google Scholar]
- Ji T.; Chen X. Y.; Huang L.; Rueping M. Remote Trifluoromethylthiolation Enabled by Organophotocatalytic C-C Bond Cleavage. Org. Lett. 2020, 22, 2579–2583. 10.1021/acs.orglett.0c00493. [DOI] [PubMed] [Google Scholar]
- Bloom S.; Bume D. D.; Pitts C. R.; Lectka T. Site-Selective Approach to β-Fluorination: Photocatalyzed Ring Opening of Cyclopropanols. Chem. - Eur. J. 2015, 21, 8060–8063. 10.1002/chem.201501081. [DOI] [PubMed] [Google Scholar]
- Protti S.; Fagnoni M.; Monti S.; Réhault J.; Poizat O.; Albini A. Activation of Aliphatic C-H Bonds by Tetracyanobenzene Photosensitization. A Time-Resolved and Steady-State Investigation. RSC Adv. 2012, 2, 1897–1904. 10.1039/c2ra01054b. [DOI] [Google Scholar]
- Aguilar Troyano F. J.; Merkens K.; Gómez-Suárez A. Selectfluor® Radical Dication (TEDA2+) - A Versatile Species in Modern Synthetic Organic Chemistry. Asian J. Org. Chem. 2020, 9, 992–1007. 10.1002/ajoc.202000196. [DOI] [Google Scholar]
- Ji M.; Wu Z.; Zhu C. Visible-Light-Induced Consecutive C-C Bond Fragmentation and Formation for the Synthesis of Elusive Unsymmetric 1,8-Dicarbonyl Compounds. Chem. Commun. 2019, 55, 2368–2371. 10.1039/C9CC00378A. [DOI] [PubMed] [Google Scholar]
- Cardinale L.; Neumeier M.; Majek M.; Jacobi Von Wangelin A. Aryl Pyrazoles from Photocatalytic Cycloadditions of Arenediazonium. Org. Lett. 2020, 22, 7219–7224. 10.1021/acs.orglett.0c02514. [DOI] [PubMed] [Google Scholar]
- Lee J.; Von Gunten U.; Kim J. H. Persulfate-Based Advanced Oxidation: Critical Assessment of Opportunities and Roadblocks. Environ. Sci. Technol. 2020, 54, 3064–3081. 10.1021/acs.est.9b07082. [DOI] [PubMed] [Google Scholar]
- Liu J.; Xu E.; Jiang J.; Huang Z.; Zheng L.; Liu Z. Q. Copper-Mediated Tandem Ring-Opening/Cyclization Reactions of Cyclopropanols with Aryldiazonium Salts: Synthesis of N-Arylpyrazoles. Chem. Commun. 2020, 56, 2202–2205. 10.1039/C9CC09657D. [DOI] [PubMed] [Google Scholar]
- Petti A.; Natho P.; Lam K.; Parsons P. J. Regioselective Electrochemical Cyclobutanol Ring Expansion to 1-Tetralones. Eur. J. Org. Chem. 2021, 2021, 854–858. 10.1002/ejoc.202001535. [DOI] [Google Scholar]
- Allen B. D. W.; Hareram M. D.; Seastram A. C.; McBride T.; Wirth T.; Browne D. L.; Morrill L. C. Manganese-Catalyzed Electrochemical Deconstructive Chlorination of Cycloalkanols via Alkoxy Radicals. Org. Lett. 2019, 21, 9241–9246. 10.1021/acs.orglett.9b03652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Wang M.; Huan L.; Wang D.; Wang J.; Zhu C. Tertiary-Alcohol-Directed Functionalization of Remote C(sp3)-H Bonds by Sequential Hydrogen Atom and Heteroaryl Migrations. Angew. Chem., Int. Ed. 2018, 57, 1640–1644. 10.1002/anie.201709025. [DOI] [PubMed] [Google Scholar]
- Tsui E.; Metrano A. J.; Tsuchiya Y.; Knowles R. R. Catalytic Hydroetherification of Unactivated Alkenes Enabled by Proton-Coupled Electron Transfer. Angew. Chem., Int. Ed. 2020, 59, 11845–11849. 10.1002/anie.202003959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Zhang Z.; Jin R.; Liu J.; Liu G.; Han B.; Jiao N. DMSO-Enabled Selective Radical O-H Activation of 1,3(4)-Diols. Angew. Chem., Int. Ed. 2020, 59, 19851–19856. 10.1002/anie.202007187. [DOI] [PubMed] [Google Scholar]
- Röck M.; Schmittel M. Controlled Oxidation of Enolates to α-Carbonyl Radicals and α-Carbonyl Cations. J. Chem. Soc., Chem. Commun. 1993, 1739–1741. 10.1039/C39930001739. [DOI] [Google Scholar]
- Liu H.; Feng W.; Kee C. W.; Zhao Y.; Leow D.; Pan Y.; Tan C. H. Organic Dye Photocatalyzed α-Oxyamination through Irradiation with Visible Light. Green Chem. 2010, 12, 953–995. 10.1039/b924609f. [DOI] [Google Scholar]
- Koike T.; Yasu Y.; Akita M. Visible-Light-Driven Oxidation of 1,3-Dicarbonyl Compounds via Catalytic Disproportionation of TEMPO by Photoredox Catalysis. Chem. Lett. 2012, 41, 999–1001. 10.1246/cl.2012.999. [DOI] [Google Scholar]
- Schämann M.; Schäfer H. J. Alkoxyamines by Reaction of 2,2,6,6-Tetramethylpiperidine-1-Oxoammonium Tetrafluoroborate with Enolates. Synlett 2004, 1601–1603. 10.1055/s-2004-829073. [DOI] [Google Scholar]
- Gong M.; Kim J. K.; Zhao X.; Li Y.; Zhang J.; Huang M.; Wu Y. Visible-Light-Induced α-Oxyamination of 1,3-Dicarbonyls with TEMPO via a Photo(Electro)Catalytic Process Applying a DSSC Anode or in a DSSC System. Green Chem. 2019, 21, 3615–3620. 10.1039/C9GC01154D. [DOI] [Google Scholar]
- Kisch H. Semiconductor Photocatalysis—Mechanistic and Synthetic Aspects. Angew. Chem., Int. Ed. 2013, 52, 812–847. 10.1002/anie.201201200. [DOI] [PubMed] [Google Scholar]
- Lang X.; Chen X.; Zhao J. Heterogeneous Visible Light Photocatalysis for Selective Organic Transformations. Chem. Soc. Rev. 2014, 43, 473–486. 10.1039/C3CS60188A. [DOI] [PubMed] [Google Scholar]
- Friedmann D.; Hakki A.; Kim H.; Choi W.; Bahnemann D. Heterogeneous Photocatalytic Organic Synthesis: State-of-the-Art and Future Perspectives. Green Chem. 2016, 18, 5391–5411. 10.1039/C6GC01582D. [DOI] [Google Scholar]
- Daniel M.; Fensterbank L.; Goddard J. P.; Ollivier C. Visible-Light Photocatalytic Oxidation of 1,3-Dicarbonyl Compounds and Carbon-Carbon Bond Formation. Org. Chem. Front. 2014, 1, 551–555. 10.1039/c4qo00071d. [DOI] [Google Scholar]
- Arnett E. M.; Flowers R. A.; Ludwig R. T.; Meekhof A. E.; Walek S. A. Triarylmethanes and 9-Arylxanthenes as Prototypes Amphihydric Compounds for Relating the Stabilities of Cations, Anions and Radicals by C-H Bond Cleavage and Electron Transfer. J. Phys. Org. Chem. 1997, 10, 499–513. . [DOI] [Google Scholar]
- Li T.; Liang K.; Zhang Y.; Hu D.; Ma Z.; Xia C. Three-Component Minisci Reaction with 1,3-Dicarbonyl Compounds Induced by Visible Light. Org. Lett. 2020, 22, 2386–2390. 10.1021/acs.orglett.0c00584. [DOI] [PubMed] [Google Scholar]
- Baś S.; Yamashita Y.; Kobayashi S. Development of Brønsted Base-Photocatalyst Hybrid Systems for Highly Efficient C-C Bond Formation Reactions of Malonates with Styrenes. ACS Catal. 2020, 10, 10546–10550. 10.1021/acscatal.0c02716. [DOI] [Google Scholar]
- Niyazymbetov M. E.; Rongfeng Z.; Evans D. H. Oxidation Potential as a Measure of the Reactivity of Anionic Nucleophiles. Behaviour of Different Classes of Nucleophiles. J. Chem. Soc., Perkin Trans. 2 1996, 9, 1957–1961. 10.1039/p29960001957. [DOI] [Google Scholar]
- Arnold D. R.; Du X.; Chen J. The Effect of Meta- or Para-Methoxy Substitution on the Reactivity of the Radical Cations of Arylalkenes and Alkanes. Radical Ions in Photochemistry. Part 26. Can. J. Chem. 1995, 73, 307–318. 10.1139/v95-042. [DOI] [Google Scholar]
- Wu Z. J.; Xu H. C. Synthesis of C3-Fluorinated Oxindoles through Reagent-Free Cross-Dehydrogenative Coupling. Angew. Chem., Int. Ed. 2017, 56, 4734–4738. 10.1002/anie.201701329. [DOI] [PubMed] [Google Scholar]
- Wu Z. J.; Li S. R.; Long H.; Xu H. C. Electrochemical Dehydrogenative Cyclization of 1,3-Dicarbonyl Compounds. Chem. Commun. 2018, 54, 4601–4604. 10.1039/C8CC02472C. [DOI] [PubMed] [Google Scholar]
- Wu Z. J.; Li S. R.; Xu H. C. Synthesis of N-Heterocycles by Dehydrogenative Annulation of N-Allyl Amides with 1,3-Dicarbonyl Compounds. Angew. Chem., Int. Ed. 2018, 57, 14070–14074. 10.1002/anie.201807683. [DOI] [PubMed] [Google Scholar]
- Xiong M.; Liang X.; Liang X.; Pan Y.; Lei A. Hexafluoro-2-Propanol-Promoted Electro-Oxidative [3+2] Annulation of 1,3-Dicarbonyl Compounds and Alkenes. ChemElectroChem 2019, 6, 3383–3386. 10.1002/celc.201900753. [DOI] [Google Scholar]
- Olmstead W. N.; Bordwell F. G. Ion-Pair Association Constants in Dimethyl Sulfoxide. J. Org. Chem. 1980, 45, 3299–3305. 10.1021/jo01304a033. [DOI] [Google Scholar]
- Choi S.; Park J.; Yu E.; Sim J.; Park C. M. Electrosynthesis of Dihydropyrano[4,3-b]Indoles Based on a Double Oxidative [3+3] Cycloaddition. Angew. Chem., Int. Ed. 2020, 59, 11886–11891. 10.1002/anie.202003364. [DOI] [PubMed] [Google Scholar]
- Guo J.-D.; Yang X.-L.; Chen B.; Tung C.-H.; Wu L.-Z. Photoredox/Cobalt-Catalyzed C(sp3)-H Bond Functionalization toward Phenanthrene Skeletons with Hydrogen Evolution. Org. Lett. 2020, 22, 9627–9632. 10.1021/acs.orglett.0c03665. [DOI] [PubMed] [Google Scholar]
- Mei R.; Yang C.; Xiong F.; Mao M.; Li H.; Sun J.; Zou L.; Ma W.; Ackermann L. Access to 10-Phenanthrenols via Electrochemical C-H/C-H Arylation. Adv. Synth. Catal. 2021, 363, 1120–1125. 10.1002/adsc.202001431. [DOI] [Google Scholar]
- Yang X. L.; Guo J. D.; Xiao H.; Feng K.; Chen B.; Tung C. H.; Wu L. Z. Photoredox Catalysis of Aromatic β-Ketoesters for in Situ Production of Transient and Persistent Radicals for Organic Transformation. Angew. Chem., Int. Ed. 2020, 59, 5365–5370. 10.1002/anie.201916423. [DOI] [PubMed] [Google Scholar]
- Hedenburg J. F.; Freiser H. Anodic Voltammetry of Phenols. Anal. Chem. 1953, 25, 1355–1358. 10.1021/ac60081a015. [DOI] [Google Scholar]
- Gaylor V. F.; Elving P. J.; Conrad L. Polarographic Oxidation of Phenolic Compounds. Anal. Chem. 1953, 25, 1078–1082. 10.1021/ac60079a022. [DOI] [Google Scholar]
- Vermillion F. J.; Pearl I. A. Anodic Reactions of Simple Phenolic Compounds. J. Electrochem. Soc. 1964, 111, 1392. 10.1149/1.2426012. [DOI] [Google Scholar]
- Serjeant E. P.; Dempsey B.. Ionisation Constants of Organic Acids in Aqueous Solution; Pergamon Press, 1979. [Google Scholar]
- Kirkbright G. F.; Stock J. T.; Pugliese R. D.; Bobbitt J. M. The Anodic Voltammetry and Electrolytic Oxidation of Corypalline. J. Electrochem. Soc. 1969, 116, 219. 10.1149/1.2411800. [DOI] [Google Scholar]
- Bobbitt J. M.; Weisgraber K. H.; Steinfeld A. S.; Weiss S. G. Synthesis of Isoquinoline Alkaloids. IV. Steric Effects in the Electrolytic and Catalytic Oxidative Coupling of Phenolic Tetrahydroisoquinolines. J. Org. Chem. 1970, 35, 2884–2888. 10.1021/jo00834a005. [DOI] [Google Scholar]
- Bobbitt J. M.; Yagi H.; Shibuya S.; Stock J. T. Electrochemistry of Natural Products. II. Electrolytic Oxidation of Some Simple 1,2,3,4-Tetrahydroisoquinoline Phenols. J. Org. Chem. 1971, 36, 3006–3010. 10.1021/jo00819a021. [DOI] [Google Scholar]
- Bobbitt J. M.; Noguchi I.; Yagi H.; Weisgraber K. H. Electrochemistry of Natural Products. III. A Stereoselective, Stereospecific Phenol Coupling Reaction. J. Am. Chem. Soc. 1971, 93, 3551–3552. 10.1021/ja00743a056. [DOI] [Google Scholar]
- Bobbitt J. M.; Noguchi I.; Yagi H.; Weisgraber K. H. Electrochemistry of Natural Products. IV. Electrochemical and Chemical Oxidative Dimerization of 1,2-Dimethyl-7-Hydroxy-6-Methoxy-1,2,3,4-Tetrahydroisoquinoline. J. Org. Chem. 1976, 41, 845–850. 10.1021/jo00867a021. [DOI] [Google Scholar]
- Miller L. L.; Stewart R. F.; Gillespie J. P.; Ramachandran V.; So Y. H.; Stermitz F. R. Synthesis of Morphinandienones, a Dihydrophenanthrone, and Pummerer’s Ketones by Anodic Coupling. J. Org. Chem. 1978, 43, 1580–1586. 10.1021/jo00402a024. [DOI] [Google Scholar]
- Bobbitt J. M.; Ebermann R.; Schubert M. Synthesis of Isoquinoline Alkaloids - III Pilocereine and “Isopilocereine.. Tetrahedron Lett. 1963, 4, 575–577. 10.1016/S0040-4039(01)90676-8. [DOI] [Google Scholar]
- Djerassi C.; Figdor S. K.; Bobbitt J. M.; Markley F. X. Alkaloid Studies. XVII. The Structure of the Cactus Alkaloid Pilocereine. J. Am. Chem. Soc. 1957, 79, 2203–2210. 10.1021/ja01566a047. [DOI] [Google Scholar]
- Malkowsky I. M.; Griesbach U.; Pütter H.; Waldvogel S. R. Unexpected Highly Chemoselective Anodic Ortho-Coupling Reaction of 2,4-Dimethylphenol on Boron-Doped Diamond Electrodes. Eur. J. Org. Chem. 2006, 2006, 4569–4572. 10.1002/ejoc.200600466. [DOI] [Google Scholar]
- Malkowsky I. M.; Rommel C. E.; Wedeking K.; Fröhlich R.; Bergander K.; Nieger M.; Quaiser C.; Griesbach U.; Pütter H.; Waldvogel S. R. Facile and Highly Diastereoselective Formation of a Novel Pentacyclic Scaffold by Direct Anodic Oxidation of 2,4-Dimethylphenol. Eur. J. Org. Chem. 2006, 2006, 241–245. 10.1002/ejoc.200500517. [DOI] [Google Scholar]
- Kirste A.; Nieger M.; Malkowsky I. M.; Stecker F.; Fischer A.; Waldvogel S. R. Ortho-Selective Phenol-Coupling Reaction by Anodic Treatment on Boron-Doped Diamond Electrode Using Fluorinated Alcohols. Chem. - Eur. J. 2009, 15, 2273–2277. 10.1002/chem.200802556. [DOI] [PubMed] [Google Scholar]
- Kirste A.; Hayashi S.; Schnakenburg G.; Malkowsky I. M.; Stecker F.; Fischer A.; Fuchigami T.; Waldvogel S. R. Highly Selective Electrosynthesis of Biphenols on Graphite Electrodes in Fluorinated Media. Chem. - Eur. J. 2011, 17, 14164–14169. 10.1002/chem.201102182. [DOI] [PubMed] [Google Scholar]
- Takahashi M.; Konishi H.; Iida S.; Nakamura K.; Yamamura S.; Nishiyama S. Phenolic Oxidation of O,O’-Dihalogenated Phenols. Tetrahedron 1999, 55, 5295–5302. 10.1016/S0040-4020(99)00217-3. [DOI] [Google Scholar]
- Röckl J. L.; Schollmeyer D.; Franke R.; Waldvogel S. R. Dehydrogenative Anodic C-C Coupling of Phenols Bearing Electron-Withdrawing Groups. Angew. Chem., Int. Ed. 2020, 59, 315–319. 10.1002/anie.201910077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke R.; Selent D.; Börner A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. 10.1021/cr3001803. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Ren W. M.; Liu J.; Lu X. B. Asymmetric Copolymerization of CO2 with Meso-Epoxides Mediated by Dinuclear Cobalt(III) Complexes: Unprecedented Enantioselectivity and Activity. Angew. Chem., Int. Ed. 2013, 52, 11594–11598. 10.1002/anie.201305154. [DOI] [PubMed] [Google Scholar]
- Zhang H. C.; Huang W. S.; Pu L. Biaryl-Based Macrocyclic and Polymeric Chiral (Salophen)Ni(II) Complexes: Synthesis and Spectroscopic Study. J. Org. Chem. 2001, 66, 481–487. 10.1021/jo001276s. [DOI] [PubMed] [Google Scholar]
- Elsler B.; Schollmeyer D.; Dyballa K. M.; Franke R.; Waldvogel S. R. Metal- and Reagent-Free Highly Selective Anodic Cross-Coupling Reaction of Phenols. Angew. Chem., Int. Ed. 2014, 53, 5210–5213. 10.1002/anie.201400627. [DOI] [PubMed] [Google Scholar]
- Pummerer R.; Melamed D.; Puttfarcken H. Die Dehydrierung von P-Kresol (VII. Mitteilung Über Die Oxydation Der Phenole). Ber. Dtsch. Chem. Ges. B 1922, 55, 3116–3132. 10.1002/cber.19220550921. [DOI] [Google Scholar]
- Pummerer R.; Puttfarcken H.; Schopflocher P. Die Dehydrierung von P-Kresol. (VIII. Mitteilung Über Die Oxydation Der Phenole). Ber. Dtsch. Chem. Ges. B 1925, 58, 1808–1820. 10.1002/cber.19250580876. [DOI] [Google Scholar]
- Arkley V.; Dean F. M.; Robertson A.; Sidisunthorn P. Usnic Acid. Part XII Pummerer’s Ketone. J. Chem. Soc. 1956, 2322–2328. 10.1039/jr9560002322. [DOI] [Google Scholar]
- Taguchi H.; Sankawa U.; Shibata S. Biosynthesis of Natural Products. VI. Biosynthesis of Usnic Acid in Lichens. (1). A General Scheme of Biosynthesis of Usnic Acid. Chem. Pharm. Bull. 1969, 17, 2054–2060. 10.1248/cpb.17.2054. [DOI] [PubMed] [Google Scholar]
- Taylor W. I.; Battersby A. R.. Oxidative Coupling of Phenols. Organic substances of natural origin; Marcel Dekker, 1967. [Google Scholar]
- Dhar D.; Yee G. M.; Markle T. F.; Mayer J. M.; Tolman W. B. Reactivity of the Copper(III)-Hydroxide Unit with Phenols. Chem. Sci. 2017, 8, 1075–1085. 10.1039/C6SC03039D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler E.; Junghahn L.; Lindberg U.; Berggren B.; Westin G. Periodate Oxidation of Phenols. V. 2,4-Dimethylphenol. Acta Chem. Scand. 1960, 14, 1261–1273. 10.3891/acta.chem.scand.14-1261. [DOI] [Google Scholar]
- Bruice T. C.; Gregory M. J.; Walters S. L. Reactions of Tetranitromethane. I. Kinetics and Mechanism of Nitration of Phenols by Tetranitromethane. J. Am. Chem. Soc. 1968, 90, 1612–1619. 10.1021/ja01008a035. [DOI] [Google Scholar]
- Haynes C. G.; Turner A. H.; Waters W. A. 552. The Oxidation of Monohydric Phenols by Alkaline Ferricyanide. J. Chem. Soc. 1956, 2823–2831. 10.1039/jr9560002823. [DOI] [Google Scholar]
- Barjau J.; Königs P.; Kataeva O.; Waldvogel S. R. Reinvestigation of Highly Diastereoselective Pentacyclic Spirolactone Formation by Direct Anodic Oxidation of 2,4-Dimethylphenol. Synlett 2008, 2008, 2309–2312. 10.1055/s-2008-1078276. [DOI] [Google Scholar]
- Barjau J.; Schnakenburg G.; Waldvogel S. R. Diversity-Oriented Synthesis of Polycyclic Scaffolds by Modification of an Anodic Product Derived from 2,4-Dimethylphenol. Angew. Chem., Int. Ed. 2011, 50, 1415–1419. 10.1002/anie.201006637. [DOI] [PubMed] [Google Scholar]
- Barjau J.; Fleischhauer J.; Schnakenburg G.; Waldvogel S. R. Installation of Amine Moieties into a Polycyclic Anodic Product Derived from 2,4-Dimethylphenol. Chem. - Eur. J. 2011, 17, 14785–14791. 10.1002/chem.201102722. [DOI] [PubMed] [Google Scholar]
- King M. L.; Chiang C. C.; Ling H. C.; Fujita E.; Ochiai M.; McPhail A. T. X-Ray Crystal Structure of Rocaglamide, a Novel Antileukemic 1H-Cyclopenta[b]Benzofuran from Aglaia Elliptifolia. J. Chem. Soc., Chem. Commun. 1982, 260, 1150–1151. 10.1039/c39820001150. [DOI] [Google Scholar]
- Mirion M.; Andernach L.; Stobe C.; Barjau J.; Schollmeyer D.; Opatz T.; Lützen A.; Waldvogel S. R. Synthesis and Isolation of Enantiomerically Enriched Cyclopenta[b]Benzofurans Based on Products from Anodic Oxidation of 2,4-Dimethylphenol. Eur. J. Org. Chem. 2015, 2015, 4876–4882. 10.1002/ejoc.201500600. [DOI] [Google Scholar]
- Kirste A.; Schnakenburg G.; Waldvogel S. R. Anodic Coupling of Guaiacol Derivatives on Boron-Doped Diamond Electrodes. Org. Lett. 2011, 13, 3126–3129. 10.1021/ol201030g. [DOI] [PubMed] [Google Scholar]
- Wiebe A.; Schollmeyer D.; Dyballa K. M.; Franke R.; Waldvogel S. R. Selective Synthesis of Partially Protected Nonsymmetric Biphenols by Reagent- and Metal-Free Anodic Cross-Coupling Reaction. Angew. Chem., Int. Ed. 2016, 55, 11801–11805. 10.1002/anie.201604321. [DOI] [PubMed] [Google Scholar]
- Riehl B.; Dyballa K. M.; Franke R.; Waldvogel S. R. Electroorganic Synthesis as a Sustainable Alternative for Dehydrogenative Cross-Coupling of Phenols and Naphthols. Synthesis 2016, 49, 252–259. 10.1055/s-0036-1588610. [DOI] [Google Scholar]
- Libman A.; Shalit H.; Vainer Y.; Narute S.; Kozuch S.; Pappo D. Synthetic and Predictive Approach to Unsymmetrical Biphenols by Iron-Catalyzed Chelated Radical-Anion Oxidative Coupling. J. Am. Chem. Soc. 2015, 137, 11453–11460. 10.1021/jacs.5b06494. [DOI] [PubMed] [Google Scholar]
- Dahms B.; Kohlpaintner P. J.; Wiebe A.; Breinbauer R.; Schollmeyer D.; Waldvogel S. R. Selective Formation of 4,4’-Biphenols by Anodic Dehydrogenative Cross- and Homo-Coupling Reaction. Chem. - Eur. J. 2019, 25, 2713–2716. 10.1002/chem.201805737. [DOI] [PubMed] [Google Scholar]
- Vareka M.; Dahms B.; Lang M.; Hoang M. H.; Trobe M.; Weber H.; Hielscher M. M.; Waldvogel S. R.; Breinbauer R. Synthesis of a Bcl9 Alpha-Helix Mimetic for Inhibition of Ppis by a Combination of Electrooxidative Phenol Coupling and Pd-Catalyzed Cross Coupling. Catalysts 2020, 10, 340. 10.3390/catal10030340. [DOI] [Google Scholar]
- Kirste A.; Schnakenburg G.; Stecker F.; Fischer A.; Waldvogel S. R. Anodic Phenol-Arene Cross-Coupling Reaction on Boron-Doped Diamond Electrodes. Angew. Chem., Int. Ed. 2010, 49, 971–975. 10.1002/anie.200904763. [DOI] [PubMed] [Google Scholar]
- Kirste A.; Elsler B.; Schnakenburg G.; Waldvogel S. R. Efficient Anodic and Direct Phenol-Arene C,C Cross-Coupling: The Benign Role of Water or Methanol. J. Am. Chem. Soc. 2012, 134, 3571–3576. 10.1021/ja211005g. [DOI] [PubMed] [Google Scholar]
- Sumi T.; Saitoh T.; Natsui K.; Yamamoto T.; Atobe M.; Einaga Y.; Nishiyama S. Anodic Oxidation on a Boron-Doped Diamond Electrode Mediated by Methoxy Radicals. Angew. Chem., Int. Ed. 2012, 51, 5443–5446. 10.1002/anie.201200878. [DOI] [PubMed] [Google Scholar]
- Elsler B.; Wiebe A.; Schollmeyer D.; Dyballa K. M.; Franke R.; Waldvogel S. R. Source of Selectivity in Oxidative Cross-Coupling of Aryls by Solvent Effect of 1,1,1,3,3,3-Hexafluoropropan-2-ol. Chem. - Eur. J. 2015, 21, 12321–12325. 10.1002/chem.201501604. [DOI] [PubMed] [Google Scholar]
- Lips S.; Wiebe A.; Elsler B.; Schollmeyer D.; Dyballa K. M.; Franke R.; Waldvogel S. R. Synthesis of Meta-Terphenyl-2,2’’-Diols by Anodic C-C Cross-Coupling Reactions. Angew. Chem., Int. Ed. 2016, 55, 10872–10876. 10.1002/anie.201605865. [DOI] [PubMed] [Google Scholar]
- Lips S.; Franke R.; Waldvogel S. R. Electrochemical Synthesis of 2-Hydroxy-Para-Terphenyls by Dehydrogenative Anodic C-C Cross-Coupling Reaction. Synlett 2019, 30, 1174–1177. 10.1055/s-0037-1611942. [DOI] [Google Scholar]
- Zhao J. C.; Yu S. M.; Liu Y.; Yao Z. J. Biomimetic Synthesis of ent-(−)-Azonazine and Stereochemical Reassignment of Natural Product. Org. Lett. 2013, 15, 4300–4303. 10.1021/ol4015706. [DOI] [PubMed] [Google Scholar]
- Li C.; Chan C.; Heimann A. C.; Danishefsky S. J. On the Rearrangement of an Azaspiroindolenine to a Precursor to Phalarine: Mechanistic Insights. Angew. Chem., Int. Ed. 2007, 46, 1444–1447. 10.1002/anie.200604071. [DOI] [PubMed] [Google Scholar]
- Tomakinian T.; Guillot R.; Kouklovsky C.; Vincent G. Direct Oxidative Coupling of N-Acetyl Indoles and Phenols for the Synthesis of Benzofuroindolines Related to Phalarine. Angew. Chem., Int. Ed. 2014, 53, 11881–11885. 10.1002/anie.201404055. [DOI] [PubMed] [Google Scholar]
- Eberson L.; Hartshorn M. P.; Persson O. 1,1,1,3,3,3-Hexafluoropropan-2-ol as a Solvent for the Generation of Highly Persistent Radical Cations. J. Chem. Soc., Perkin Trans. 2 1995, 1735–1744. 10.1039/p29950001735. [DOI] [Google Scholar]
- Eberson L.; Persson O.; Hartshorn M. P. Detection and Reactions of Radical Cations Generated by Photolysis of Aromatic Compounds with Tetranitromethane in 1,1,1,3,3,3-Hexafluoro-2-propanol at Room Temperature. Angew. Chem., Int. Ed. Engl. 1995, 34, 2268–2269. 10.1002/anie.199522681. [DOI] [Google Scholar]
- Eberson L.; Hartshorn M. P.; Persson O. Generation of Solutions of Highly Persistent Radical Cations by 4-Tolylthallium(III) Bis(Trifluoroacetate) in 1,1,1,3,3,3-Hexafluoropropan-2-ol. J. Chem. Soc., Chem. Commun. 1995, 1131–1132. 10.1039/c39950001131. [DOI] [Google Scholar]
- Fischer H. The Persistent Radical Effect: A Principle for Selective Radical Reactions and Living Radical Polymerizations. Chem. Rev. 2001, 101, 3581–3610. 10.1021/cr990124y. [DOI] [PubMed] [Google Scholar]
- Wiebe A.; Lips S.; Schollmeyer D.; Franke R.; Waldvogel S. R. Single and Twofold Metal- and Reagent-Free Anodic C-C Cross-Coupling of Phenols with Thiophenes. Angew. Chem., Int. Ed. 2017, 56, 14727–14731. 10.1002/anie.201708946. [DOI] [PubMed] [Google Scholar]
- Lips S.; Schollmeyer D.; Franke R.; Waldvogel S. R. Regioselective Metal- and Reagent-Free Arylation of Benzothiophenes by Dehydrogenative Electrosynthesis. Angew. Chem., Int. Ed. 2018, 57, 13325–13329. 10.1002/anie.201808555. [DOI] [PubMed] [Google Scholar]
- Lips S.; Frontana-Uribe B. A.; Dörr M.; Schollmeyer D.; Franke R.; Waldvogel S. R. Metal- and Reagent-Free Anodic C-C Cross-Coupling of Phenols with Benzofurans Leading to a Furan Metathesis. Chem. - Eur. J. 2018, 24, 6057–6061. 10.1002/chem.201800919. [DOI] [PubMed] [Google Scholar]
- Dörr M.; Lips S.; Martínez-Huitle C. A.; Schollmeyer D.; Franke R.; Waldvogel S. R. Synthesis of Highly Functionalized N,N-Diarylamides by an Anodic C,N-Coupling Reaction. Chem. - Eur. J. 2019, 25, 7835–7838. 10.1002/chem.201901442. [DOI] [PubMed] [Google Scholar]
- Yamamoto T.; Riehl B.; Naba K.; Nakahara K.; Wiebe A.; Saitoh T.; Waldvogel S. R.; Einaga Y. A Solvent-Directed Stereoselective and Electrocatalytic Synthesis of Diisoeugenol. Chem. Commun. 2018, 54, 2771–2773. 10.1039/C8CC00794B. [DOI] [PubMed] [Google Scholar]
- Kouznetsov V. V.; Merchan Arenas D. R. First Green Protocols for the Large-Scale Preparation of γ-Diisoeugenol and Related Dihydro(1H)Indenes via Formal [3+2] Cycloaddition Reactions. Tetrahedron Lett. 2009, 50, 1546–1549. 10.1016/j.tetlet.2009.01.038. [DOI] [Google Scholar]
- Zhao Q.; Jin J. K.; Wang J.; Zhang F. L.; Wang Y. F. Radical α-Addition Involved Electrooxidative [3+2] Annulation of Phenols and Electron-Deficient Alkenes. Chem. Sci. 2020, 11, 3909–3913. 10.1039/D0SC01078B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates B. D.; Dalidowicz P.; Tebben A.; Wang S.; Swenton J. S. Mechanistic Aspects and Synthetic Applications of the Electrochemical and Iodobenzene Bis(Trifluoroacetate) Oxidative 1,3-Cycloadditions of Phenols and Electron-Rich Styrene Derivatives. J. Org. Chem. 1992, 57, 2135–2143. 10.1021/jo00033a040. [DOI] [Google Scholar]
- Kerns M. L.; Conroy S. M.; Swenton J. S. Dihydrobenzofuran Derivatives via the Anodic Cycloaddition Reaction of p-Methoxyphenols and Vinyl Sulfides. Tetrahedron Lett. 1994, 35, 7529–7532. 10.1016/S0040-4039(00)78335-3. [DOI] [Google Scholar]
- Chiba K.; Fukuda M.; Kim S.; Kitano Y.; Tada M. Dihydrobenzofuran Synthesis by an Anodic [3+2] Cycloaddition of Phenols and Unactivated Alkenes. J. Org. Chem. 1999, 64, 7654–7656. 10.1021/jo9908243. [DOI] [Google Scholar]
- Blum T. R.; Zhu Y.; Nordeen S. A.; Yoon T. P. Photocatalytic Synthesis of Dihydrobenzofurans by Oxidative [3+2] Cycloaddition of Phenols. Angew. Chem., Int. Ed. 2014, 53, 11056–11059. 10.1002/anie.201406393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikl J.; Lips S.; Schollmeyer D.; Franke R.; Waldvogel S. R. Direct Metal- and Reagent-Free Sulfonylation of Phenols with Sodium Sulfinates by Electrosynthesis. Chem. - Eur. J. 2019, 25, 6891–6895. 10.1002/chem.201900850. [DOI] [PubMed] [Google Scholar]
- Eisenhofer A.; Hioe J.; Gschwind R. M.; König B. Photocatalytic Phenol-Arene C-C and C-O Cross-Dehydrogenative Coupling. Eur. J. Org. Chem. 2017, 2017, 2194–2204. 10.1002/ejoc.201700211. [DOI] [Google Scholar]
- Niederer K. A.; Gilmartin P. H.; Kozlowski M. C. Oxidative Photocatalytic Homo- and Cross-Coupling of Phenols: Nonenzymatic, Catalytic Method for Coupling Tyrosine. ACS Catal. 2020, 10, 14615–14623. 10.1021/acscatal.0c04515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K.; Li T.; Li N.; Zhang Y.; Shen L.; Ma Z.; Xia C. Redox-Neutral Photochemical Heck-Type Arylation of Vinylphenols Activated by Visible Light. Chem. Sci. 2020, 11, 2130–2135. 10.1039/C9SC06184C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini G.; Nappi M.; Melchiorre P. Photochemical Direct Perfluoroalkylation of Phenols. Tetrahedron 2015, 71, 4535–4542. 10.1016/j.tet.2015.02.034. [DOI] [Google Scholar]
- Baker Dockrey S. A.; Narayan A. R. H. Photocatalytic Oxidative Dearomatization of Orcinaldehyde Derivatives. Org. Lett. 2020, 22, 3712–3716. 10.1021/acs.orglett.0c01207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche S. P.; Porco J. A. Dearomatization Strategies in the Synthesis of Complex Natural Products. Angew. Chem., Int. Ed. 2011, 50, 4068–4093. 10.1002/anie.201006017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenderski T. A.; Hoarau C.; Mejorado L.; Pettus T. R. R. Dearomatization Applications of I(III) Reagents and Some Unusual Reactivity amongst Resorcinol Derived Cyclohexadienones. Tetrahedron 2010, 66, 5873–5883. 10.1016/j.tet.2010.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedy S. E.; Rodríguez Benítez A.; Narayan A. R. H.; Zimmerman P. M.; Brooks C. L.; Wymore T. Hydroxyl Radical-Coupled Electron-Transfer Mechanism of Flavin-Dependent Hydroxylases. J. Phys. Chem. B 2019, 123, 8065–8073. 10.1021/acs.jpcb.9b08178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach R. D.; Su M.-D.; Schlegel H. B. Oxidation of Amines and Sulfides with Hydrogen Peroxide and Alkyl Hydrogen Peroxide. The Nature of the Oxygen-Transfer Step. J. Am. Chem. Soc. 1994, 116, 5379–5391. 10.1021/ja00091a049. [DOI] [Google Scholar]
- Péault L.; Nun P.; Le Grognec E.; Coeffard V. Multicatalytic Dearomatization of Phenols into Epoxyquinols via a Photooxygenation Process. Chem. Commun. 2019, 55, 7398–7401. 10.1039/C9CC03068A. [DOI] [PubMed] [Google Scholar]
- Bancirova M. Sodium Azide as a Specific Quencher of Singlet Oxygen during Chemiluminescent Detection by Luminol and Cypridina Luciferin Analogues. Luminescence 2011, 26, 685–688. 10.1002/bio.1296. [DOI] [PubMed] [Google Scholar]
- Onomura O.Electrochemistry of Hydroxylamines, Oximes and Hydroxamic Acids; John Wiley & Sons, Ltd., 2010. [Google Scholar]
- Hartmer M. F.; Waldvogel S. R. Electroorganic Synthesis of Nitriles via a Halogen-Free Domino Oxidation-Reduction Sequence. Chem. Commun. 2015, 51, 16346–16348. 10.1039/C5CC06437F. [DOI] [PubMed] [Google Scholar]
- Gütz C.; Stenglein A.; Waldvogel S. R. Highly Modular Flow Cell for Electroorganic Synthesis. Org. Process Res. Dev. 2017, 21, 771–778. 10.1021/acs.oprd.7b00123. [DOI] [Google Scholar]
- Baur J. E.; Wang S.; Brandt M. C. Fast-Scan Voltammetry of Cyclic Nitroxide Free Radicals. Anal. Chem. 1996, 68, 3815–3821. 10.1021/ac960603m. [DOI] [PubMed] [Google Scholar]
- Zhao H. B.; Xu P.; Song J.; Xu H. C. Cathode Material Determines Product Selectivity for Electrochemical C-H Functionalization of Biaryl Ketoximes. Angew. Chem., Int. Ed. 2018, 57, 15153–15156. 10.1002/anie.201809679. [DOI] [PubMed] [Google Scholar]
- Bordwell F. G.; Zhang S. Structural Effects on Stabilities of Iminoxy Radicals. J. Am. Chem. Soc. 1995, 117, 4858–4861. 10.1021/ja00122a016. [DOI] [Google Scholar]
- Eschenbrenner-Lux V.; Kumar K.; Waldmann H. The Asymmetric Hetero-Diels-Alder Reaction in the Syntheses of Biologically Relevant Compounds. Angew. Chem., Int. Ed. 2014, 53, 11146–11157. 10.1002/anie.201404094. [DOI] [PubMed] [Google Scholar]
- Yamamoto H.; Momiyama N. Rich Chemistry of Nitroso Compounds. Chem. Commun. 2005, 3514–3525. 10.1039/b503212c. [DOI] [PubMed] [Google Scholar]
- Huple D. B.; Ghorpade S.; Liu R. S. Recent Advances in Gold-Catalyzed N- and O-Functionalizations of Alkynes with Nitrones, Nitroso, Nitro and Nitroxy Species. Adv. Synth. Catal. 2016, 358, 1348–1367. 10.1002/adsc.201600018. [DOI] [Google Scholar]
- Chen C. N.; Liu R. S. Gold-Catalyzed [4+2] Annulations of Dienes with Nitrosoarenes as 4π Donors: Nitroso-Povarov Reactions. Angew. Chem., Int. Ed. 2019, 58, 9831–9835. 10.1002/anie.201903615. [DOI] [PubMed] [Google Scholar]
- Wei B.; Xie D.; Lai S.; Jiang Y.; Fu H.; Wei D.; Han B. Electrochemically Tuned Oxidative [4+2] Annulation and Dioxygenation of Olefins with Hydroxamic Acids. Angew. Chem. 2021, 133, 3219–3225. 10.1002/ange.202012209. [DOI] [PubMed] [Google Scholar]
- Recupero F.; Punta C. Free Radical Functionalization of Organic Compounds Catalyzed by N-Hydroxyphthalimide. Chem. Rev. 2007, 107, 3800–3842. 10.1021/cr040170k. [DOI] [PubMed] [Google Scholar]
- Schmidt V. A.; Alexanian E. J. Metal-Free, Aerobic Dioxygenation of Alkenes Using Hydroxamic Acids. Angew. Chem., Int. Ed. 2010, 49, 4491–4494. 10.1002/anie.201000843. [DOI] [PubMed] [Google Scholar]
- Giglio B. C.; Schmidt V. A.; Alexanian E. J. Metal-Free, Aerobic Dioxygenation of Alkenes Using Simple Hydroxamic Acid Derivatives. J. Am. Chem. Soc. 2011, 133, 13320–13322. 10.1021/ja206306f. [DOI] [PubMed] [Google Scholar]
- Schmidt V. A.; Alexanian E. J. Metal-Free, Aerobic Ketooxygenation of Alkenes Using Hydroxamic Acids. Chem. Sci. 2012, 3, 1672–1674. 10.1039/c2sc01042a. [DOI] [Google Scholar]
- Schmidt V. A.; Alexanian E. J. Metal-Free Oxyaminations of Alkenes Using Hydroxamic Acids. J. Am. Chem. Soc. 2011, 133, 11402–11405. 10.1021/ja204255e. [DOI] [PubMed] [Google Scholar]
- Quinn R. K.; Schmidt V. A.; Alexanian E. J. Radical Carbooxygenations of Alkenes Using Hydroxamic Acids. Chem. Sci. 2013, 4, 4030–4034. 10.1039/c3sc51466h. [DOI] [Google Scholar]
- Giglio B. C.; Alexanian E. J. Alkene Hydrofunctionalization Using Hydroxamic Acids: A Radical-Mediated Approach to Alkene Hydration. Org. Lett. 2014, 16, 4304–4307. 10.1021/ol5020202. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Maji B.; Tlahuext-Aca A.; Glorius F. Visible-Light-Promoted Activation of Unactivated C(sp3)-H Bonds and Their Selective Trifluoromethylthiolation. J. Am. Chem. Soc. 2016, 138, 16200–16203. 10.1021/jacs.6b09970. [DOI] [PubMed] [Google Scholar]
- Chateauneuf J.; Lusztyk J.; Ingold K. U. Spectroscopic and Kinetic Characteristics of Aroyloxyl Radicals. 2. Benzoyloxyl and Ring-Substituted Aroyloxyl Radicals. J. Am. Chem. Soc. 1988, 110, 2886–2893. 10.1021/ja00217a032. [DOI] [Google Scholar]
- Fokin A. A.; Schreiner P. R. Selective Alkane Transformations via Radicals and Radical Cations: Insights into the Activation Step from Experiment and Theory. Chem. Rev. 2002, 102, 1551–1593. 10.1021/cr000453m. [DOI] [PubMed] [Google Scholar]
- Day J. C.; Govindaraj N.; McBain D. S.; Skell P. S.; Tanko J. M. The Chemistry of 1,8-Naphthalenedicarboximidyl and Phthalimidyl Radicals. J. Org. Chem. 1986, 51, 4959–4963. 10.1021/jo00375a038. [DOI] [Google Scholar]
- Mukherjee S.; Garza-Sanchez R. A.; Tlahuext-Aca A.; Glorius F. Alkynylation of Csp2(O)-H Bonds Enabled by Photoredox-Mediated Hydrogen-Atom Transfer. Angew. Chem., Int. Ed. 2017, 56, 14723–14726. 10.1002/anie.201708037. [DOI] [PubMed] [Google Scholar]
- Margrey K. A.; Czaplyski W. L.; Nicewicz D. A.; Alexanian E. J. A General Strategy for Aliphatic C-H Functionalization Enabled by Organic Photoredox Catalysis. J. Am. Chem. Soc. 2018, 140, 4213–4217. 10.1021/jacs.8b00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakaki T.; Sakai K.; Enomoto T.; Kondo M.; Masaoka S.; Oisaki K.; Kanai M. C(sp3)-H Cyanation Promoted by Visible-Light Photoredox/Phosphate Hybrid Catalysis. Chem. - Eur. J. 2018, 24, 8051–8055. 10.1002/chem.201801746. [DOI] [PubMed] [Google Scholar]
- Tyson E. L.; Ament M. S.; Yoon T. P. Transition Metal Photoredox Catalysis of Radical Thiol-Ene Reactions. J. Org. Chem. 2013, 78, 2046–2050. 10.1021/jo3020825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita T.; Matsuda M. Electron-Transfer Reaction from Sodium Benzenethiolate to Acceptors Assisted by Photo-Excited Tris(2,2’-Bipyridine)Ruthenium(II). Bull. Chem. Soc. Jpn. 1981, 54, 1740–1742. 10.1246/bcsj.54.1740. [DOI] [Google Scholar]
- Miyashita T.; Matsuda M. Photoinduced Electron-Transfer from Thiols to [Ru(bpy)3]2+ in Aqueous Solutions. Bull. Chem. Soc. Jpn. 1985, 58, 3031–3032. 10.1246/bcsj.58.3031. [DOI] [Google Scholar]
- Cismesia M. A.; Yoon T. P. Characterizing Chain Processes in Visible Light Photoredox Catalysis. Chem. Sci. 2015, 6, 5426–5434. 10.1039/C5SC02185E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson E. L.; Niemeyer Z. L.; Yoon T. P. Redox Mediators in Visible Light Photocatalysis: Photocatalytic Radical Thiol-Ene Additions. J. Org. Chem. 2014, 79, 1427–1436. 10.1021/jo500031g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Boyer C. Visible Light Photocatalytic Thiol-Ene Reaction: An Elegant Approach for Fast Polymer Postfunctionalization and Step-Growth Polymerization. Macromolecules 2015, 48, 520–529. 10.1021/ma502460t. [DOI] [Google Scholar]
- Liu H.; Chung H. Visible-Light Induced Thiol-Ene Reaction on Natural Lignin. ACS Sustainable Chem. Eng. 2017, 5, 9160–9168. 10.1021/acssuschemeng.7b02065. [DOI] [Google Scholar]
- Santandrea J.; Kairouz V.; Collins S. K. Continuous Flow Science in an Undergraduate Teaching Laboratory: Photocatalytic Thiol-Ene Reaction Using Visible Light. J. Chem. Educ. 2018, 95, 1073–1077. 10.1021/acs.jchemed.7b00639. [DOI] [Google Scholar]
- Zhang P.; Li Y.; Yan Z.; Gong J.; Yang Z. Asymmetric Total Synthesis of (−)-Pavidolide B via a Thiyl-Radical-Mediated [3+2] Annulation Reaction. J. Org. Chem. 2019, 84, 15958–15971. 10.1021/acs.joc.9b02230. [DOI] [PubMed] [Google Scholar]
- Bhat V. T.; Duspara P. A.; Seo S.; Abu Bakar N. S. B.; Greaney M. F. Visible Light Promoted Thiol-Ene Reactions Using Titanium Dioxide. Chem. Commun. 2015, 51, 4383–4385. 10.1039/C4CC09987G. [DOI] [PubMed] [Google Scholar]
- Zalesskiy S. S.; Shlapakov N. S.; Ananikov V. P. Visible Light Mediated Metal-Free Thiol-Yne Click Reaction. Chem. Sci. 2016, 7, 6740–6745. 10.1039/C6SC02132H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathnayake M. D.; Weaver J. D. A General Photocatalytic Route to Prenylation. Eur. J. Org. Chem. 2020, 2020, 1433–1438. 10.1002/ejoc.201900356. [DOI] [Google Scholar]
- Zhao G.; Kaur S.; Wang T. Visible-Light-Mediated Thiol-Ene Reactions through Organic Photoredox Catalysis. Org. Lett. 2017, 19, 3291–3294. 10.1021/acs.orglett.7b01441. [DOI] [PubMed] [Google Scholar]
- Kaur S.; Zhao G.; Busch E.; Wang T. Metal-Free Photocatalytic Thiol-Ene/Thiol-Yne Reactions. Org. Biomol. Chem. 2019, 17, 1955–1961. 10.1039/C8OB02313A. [DOI] [PubMed] [Google Scholar]
- Levin V. V.; Dilman A. D. Visible-Light-Mediated Organocatalyzed Thiol-Ene Reaction Initiated by a Proton-Coupled Electron Transfer. J. Org. Chem. 2019, 84, 8337–8343. 10.1021/acs.joc.9b01331. [DOI] [PubMed] [Google Scholar]
- Nair A. M.; Kumar S.; Volla C. M. R. Visible Light Mediated Sulfenylation-Annulation Cascade of Alkyne Tethered Cyclohexadienones. Adv. Synth. Catal. 2019, 361, 4983–4988. 10.1002/adsc.201900874. [DOI] [Google Scholar]
- Ma R.; Feng J.; Zhang K.; Zhang B.; Du D. Photoredox β-Thiol-α-Carbonylation of Enones Accompanied by Unexpected Csp2-C(CO) Bond Cleavage. Org. Biomol. Chem. 2020, 18, 7549–7553. 10.1039/D0OB01349H. [DOI] [PubMed] [Google Scholar]
- Ji X.; Tan M.; Fu M.; Deng G. J.; Huang H. Photocatalytic Aerobic α-Thiolation/Annulation of Carbonyls with Mercaptobenzimidazoles. Org. Biomol. Chem. 2019, 17, 4979–4983. 10.1039/C9OB00625G. [DOI] [PubMed] [Google Scholar]
- Tambe S. D.; Rohokale R. S.; Kshirsagar U. A. Visible-Light-Mediated Eosin Y Photoredox-Catalyzed Vicinal Thioamination of Alkynes: Radical Cascade Annulation Strategy for 2-Substituted-3-Sulfenylindoles. Eur. J. Org. Chem. 2018, 2018, 2117–2121. 10.1002/ejoc.201800287. [DOI] [Google Scholar]
- Kolb H. C.; VanNieuwenhze M. S..; Sharpless K. B. Catalytic Asymmetric Dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. 10.1021/cr00032a009. [DOI] [Google Scholar]
- Bodkin J. A.; McLeod M. D. The Sharpless Asymmetric Aminohydroxylation. J. Chem. Soc. Perkin Trans. 1 2002, 2733–2746. 10.1039/b111276g. [DOI] [Google Scholar]
- Trost B. M.; Shibata T. Nucleophilic Attack on Olefins Initiated by Dimethyl(Methylthio)Sulfonium Fluoroborate (DMTSF). Azasulfenylation. J. Am. Chem. Soc. 1982, 104, 3225–3228. 10.1021/ja00375a054. [DOI] [Google Scholar]
- Roth A.; Denmark S. E. Enantioselective, Lewis Base-Catalyzed, Intermolecular Sulfenoamination of Alkenes. J. Am. Chem. Soc. 2019, 141, 13767–13771. 10.1021/jacs.9b07019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matviitsuk A.; Panger J. L.; Denmark S. E. Catalytic, Enantioselective Sulfenofunctionalization of Alkenes: Development and Recent Advances. Angew. Chem., Int. Ed. 2020, 59, 19796–19819. 10.1002/anie.202005920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.; Chen Y.; Tang S.; Huang Z.; Lei A. Electrochemical Oxidative Oxysulfenylation and Aminosulfenylation of Alkenes with Hydrogen Evolution. Sci. Adv. 2018, 4, eaat5312. 10.1126/sciadv.aat5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Deng L.; Mei H.; Du B.; Han J.; Pan Y. Electrochemical Oxidative Radical Oxysulfuration of Styrene Derivatives with Thiols and Nucleophilic Oxygen Sources. Green Chem. 2018, 20, 3444–3449. 10.1039/C8GC01337C. [DOI] [Google Scholar]
- Beletskaya I. P.; Ananikov V. P. Transition-Metal-Catalyzed C-S, C-Se, and C-Te Bond Formation via Cross-Coupling and Atom-Economic Addition Reactions. Chem. Rev. 2011, 111, 1596–1636. 10.1021/cr100347k. [DOI] [PubMed] [Google Scholar]
- Oderinde M. S.; Frenette M.; Robbins D. W.; Aquila B.; Johannes J. W. Photoredox Mediated Nickel Catalyzed Cross-Coupling of Thiols With Aryl and Heteroaryl Iodides via Thiyl Radicals. J. Am. Chem. Soc. 2016, 138, 1760–1763. 10.1021/jacs.5b11244. [DOI] [PubMed] [Google Scholar]
- Jiang M.; Li H.; Yang H.; Fu H. Room-Temperature Arylation of Thiols: Breakthrough with Aryl Chlorides. Angew. Chem. Int. Ed. 2017, 56, 874–879. 10.1002/ange.201610414. [DOI] [PubMed] [Google Scholar]
- Nacsa E. D.; MacMillan D. W. C. Spin-Center Shift-Enabled Direct Enantioselective α-Benzylation of Aldehydes with Alcohols. J. Am. Chem. Soc. 2018, 140, 3322–3330. 10.1021/jacs.7b12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costentin C.; Robert M.; Savéant J. M. Fragmentation of Aryl Halide π Anion Radicals. Bending of the Cleaving Bond and Activation vs Driving Force Relationships. J. Am. Chem. Soc. 2004, 126, 16051–16057. 10.1021/ja045989u. [DOI] [PubMed] [Google Scholar]
- Sandfort F.; Knecht T.; Pinkert T.; Daniliuc C. G.; Glorius F. Site-Selective Thiolation of (Multi)Halogenated Heteroarenes. J. Am. Chem. Soc. 2020, 142, 6913–6919. 10.1021/jacs.0c01630. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Deng L.; Wang X.; Wu Z.; Wang Y.; Pan Y. Electrochemically Promoted Nickel-Catalyzed Carbon-Sulfur Bond Formation. ACS Catal. 2019, 9, 1630–1634. 10.1021/acscatal.8b04633. [DOI] [Google Scholar]
- Sun R.; Qin Y.; Ruccolo S.; Schnedermann C.; Costentin C.; Nocera D. G. Elucidation of a Redox-Mediated Reaction Cycle for Nickel-Catalyzed Cross Coupling. J. Am. Chem. Soc. 2019, 141, 89–93. 10.1021/jacs.8b11262. [DOI] [PubMed] [Google Scholar]
- Kariofillis S. K.; Doyle A. G. Synthetic and Mechanistic Implications of Chlorine Photoelimination in Nickel/Photoredox C(sp3)-H Cross-Coupling. Acc. Chem. Res. 2021, 54, 988–1000. 10.1021/acs.accounts.0c00694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariq M. Electrochemistry of Br–/Br2 Redox Couple in Acetonitrile, Methanol and Mix Media of Acetonitrile-Methanol: An Insight into Redox Behavior of Bromide on Platinum (Pt) and Gold (Au) Electrode. Z. Phys. Chem. 2020, 234, 295–312. 10.1515/zpch-2018-1321. [DOI] [Google Scholar]
- Liu D.; Ma H. X.; Fang P.; Mei T. S. Nickel-Catalyzed Thiolation of Aryl Halides and Heteroaryl Halides through Electrochemistry. Angew. Chem., Int. Ed. 2019, 58, 5033–5037. 10.1002/anie.201900956. [DOI] [PubMed] [Google Scholar]
- Wang P.; Tang S.; Huang P.; Lei A. Electrocatalytic Oxidant-Free Dehydrogenative C-H/S-H Cross-Coupling. Angew. Chem., Int. Ed. 2017, 56, 3009–3013. 10.1002/anie.201700012. [DOI] [PubMed] [Google Scholar]
- Fernández-Rodríguez M. A.; Shen Q.; Hartwig J. F. A General and Long-Lived Catalyst for the Palladium-Catalyzed Coupling of Aryl Halides with Thiols. J. Am. Chem. Soc. 2006, 128, 2180–2181. 10.1021/ja0580340. [DOI] [PubMed] [Google Scholar]
- Guo W.; Tan W.; Zhao M.; Tao K.; Zheng L. Y.; Wu Y.; Chen D.; Fan X. L. Photocatalytic Direct C-S Bond Formation: Facile Access to 3-Sulfenylindoles: Via Metal-Free C-3 Sulfenylation of Indoles with Thiophenols. RSC Adv. 2017, 7, 37739–37742. 10.1039/C7RA08086G. [DOI] [Google Scholar]
- Rahaman R.; Das S.; Barman P. Visible-Light-Induced Regioselective Sulfenylation of Imidazopyridines with Thiols under Transition Metal-Free Conditions. Green Chem. 2018, 20, 141–147. 10.1039/C7GC02906C. [DOI] [Google Scholar]
- Yuan Y.; Cao Y.; Qiao J.; Lin Y.; Jiang X.; Weng Y.; Tang S.; Lei A. Electrochemical Oxidative C-H Sulfenylation of Imidazopyridines with Hydrogen Evolution. Chin. J. Chem. 2019, 37, 49–52. 10.1002/cjoc.201800405. [DOI] [Google Scholar]
- Zhou J.; Li Z.; Sun Z.; Ren Q.; Zhang Q.; Li H.; Li J. Electrochemically C-H/S-H Oxidative Cross-Coupling between Quinoxalin-2(1H)-Ones and Thiols for the Synthesis of 3-Thioquinoxalinones. J. Org. Chem. 2020, 85, 4365–4372. 10.1021/acs.joc.0c00050. [DOI] [PubMed] [Google Scholar]
- Penteado F.; Gomes C. S.; Monzon L. I.; Perin G.; Silveira C. C.; Lenardão E. J. Photocatalytic Synthesis of 3-Sulfanyl- and 1,3-Bis(Sulfanyl)Indolizines Mediated by Visible Light. Eur. J. Org. Chem. 2020, 2020, 2110–2115. 10.1002/ejoc.202000162. [DOI] [Google Scholar]
- Wang J. H.; Lei T.; Wu H. L.; Nan X. L.; Li X. B.; Chen B.; Tung C. H.; Wu L. Z. Thiol Activation toward Selective Thiolation of Aromatic C-H Bond. Org. Lett. 2020, 22, 3804–3809. 10.1021/acs.orglett.0c01050. [DOI] [PubMed] [Google Scholar]
- Berger F.; Plutschack M. B.; Riegger J.; Yu W.; Speicher S.; Ho M.; Frank N.; Ritter T. Site-Selective and Versatile Aromatic C-H Functionalization by Thianthrenation. Nature 2019, 567, 223–228. 10.1038/s41586-019-0982-0. [DOI] [PubMed] [Google Scholar]
- Liu K.; Song W.; Deng Y.; Yang H.; Song C.; Abdelilah T.; Wang S.; Cong H.; Tang S.; Lei A. Electrooxidation Enables Highly Regioselective Dearomative Annulation of Indole and Benzofuran Derivatives. Nat. Commun. 2020, 11, 3. 10.1038/s41467-019-13829-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Li S.; Peng C.; Lu L.; Wang S.; Wang P.; Chen Y. H.; Cong H.; Lei A. Electrochemical Oxidative C-H/S-H Cross-Coupling between Enamines and Thiophenols with H2 Evolution. Chem. Sci. 2019, 10, 2791–2795. 10.1039/C8SC05143G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F.; Zhang K.; Wang X.; Yao Y.; Li L.; Hu J.; Lu L.; Gao Z.; Lei A. Electrochemical Oxidative Cross-Coupling of Enaminones and Thiophenols to Construct C-S Bonds. Chem. - Asian J. 2020, 15, 4005–4008. 10.1002/asia.202001116. [DOI] [PubMed] [Google Scholar]
- Natarajan P.; Manjeet M.; Muskan M.; Brar N. K.; Jot Kaur J. Visible Light Photoredox Catalysis: Conversion of a Mixture of Thiophenols and Nitriles into 2-Substituted Benzothiazoles via Consecutive C-S and C-N Bond Formation Reactions. Org. Chem. Front. 2018, 5, 1527–1531. 10.1039/C8QO00219C. [DOI] [Google Scholar]
- Prajapati N. P.; Vekariya R. H.; Borad M. A.; Patel H. D. Recent Advances in the Synthesis of 2-Substituted Benzothiazoles: A Review. RSC Adv. 2014, 4, 60176–60208. 10.1039/C4RA07437H. [DOI] [Google Scholar]
- Wei W.; Bao P.; Yue H.; Liu S.; Wang L.; Li Y.; Yang D. Visible-Light-Enabled Construction of Thiocarbamates from Isocyanides, Thiols, and Water at Room Temperature. Org. Lett. 2018, 20, 5291–5295. 10.1021/acs.orglett.8b02231. [DOI] [PubMed] [Google Scholar]
- Chand S.; Pandey A. K.; Singh R.; Kumar S.; Singh K. N. Eosin-Y-Catalyzed Photoredox C-S Bond Formation: Easy Access to Thioethers. Chem. - Asian J. 2019, 14, 4712–4716. 10.1002/asia.201901060. [DOI] [PubMed] [Google Scholar]
- Lu F.; Yang Z.; Wang T.; Wang T.; Zhang Y.; Yuan Y.; Lei A. Electrochemical Oxidative Csp3-H/S-H Cross-Coupling with Hydrogen Evolution for Synthesis of Tetrasubstituted Olefins. Chin. J. Chem. 2019, 37, 547–551. 10.1002/cjoc.201900113. [DOI] [Google Scholar]
- Guan Z.; Zhu S.; Wang S.; Wang H.; Wang S.; Zhong X.; Bu F.; Cong H.; Lei A. Electrochemical Oxidative Carbon-Atom Difunctionalization: Towards Multisubstituted Imino Sulfide Ethers. Angew. Chem., Int. Ed. 2021, 60, 1573–1577. 10.1002/anie.202011329. [DOI] [PubMed] [Google Scholar]
- Li X. B.; Li Z. J.; Gao Y. J.; Meng Q. Y.; Yu S.; Weiss R. G.; Tung C. H.; Wu L. Z. Mechanistic Insights into the Interface-Directed Transformation of Thiols into Disulfides and Molecular Hydrogen by Visible-Light Irradiation of Quantum Dots. Angew. Chem., Int. Ed. 2014, 53, 2085–2089. 10.1002/anie.201310249. [DOI] [PubMed] [Google Scholar]
- Dethe D. H.; Srivastava A.; Dherange B. D.; Kumar B. V. Unsymmetrical Disulfide Synthesis through Photoredox Catalysis. Adv. Synth. Catal. 2018, 360, 3020–3025. 10.1002/adsc.201800405. [DOI] [Google Scholar]
- Huang P.; Wang P.; Tang S.; Fu Z.; Lei A. Electro-Oxidative S-H/S-H Cross-Coupling with Hydrogen Evolution: Facile Access to Unsymmetrical Disulfides. Angew. Chem., Int. Ed. 2018, 57, 8115–8119. 10.1002/anie.201803464. [DOI] [PubMed] [Google Scholar]
- Antonello S.; Daasbjerg K.; Jensen H.; Taddei F.; Maran F. Formation and Cleavage of Aromatic Disulfide Radical Anions. J. Am. Chem. Soc. 2003, 125, 14905–14916. 10.1021/ja036380g. [DOI] [PubMed] [Google Scholar]
- Nair A. M.; Kumar S.; Halder I.; Volla C. M. R. Visible-Light Mediated Sulfonylation of Thiols via Insertion of Sulfur Dioxide. Org. Biomol. Chem. 2019, 17, 5897–5901. 10.1039/C9OB01040H. [DOI] [PubMed] [Google Scholar]
- Emmett E. J.; Willis M. C. The Development and Application of Sulfur Dioxide Surrogates in Synthetic Organic Chemistry. Asian J. Org. Chem. 2015, 4, 602–611. 10.1002/ajoc.201500103. [DOI] [Google Scholar]
- Torii S.; Tanaka H.; Ukida M. Electrosynthesis of Hetero-Hetero Atom Bonds. 2. An Efficient Preparation of (2-Benzothiazolyl)- and Thiocarbamoylsulfenamides by Electrolytic Cross-Coupling Reaction of 2-Mercaptobenzothiazole, Bis(2-Benzothiazolyl) Disulfide, and/or Bis(Dialkylthiocarba. J. Org. Chem. 1978, 43, 3223–3227. 10.1021/jo00410a025. [DOI] [Google Scholar]
- Tang S.; Liu Y.; Li L.; Ren X.; Li J.; Yang G.; Li H.; Yuan B. Scalable Electrochemical Oxidant-and Metal-Free Dehydrogenative Coupling of S-H/N-H. Org. Biomol. Chem. 2019, 17, 1370–1374. 10.1039/C8OB03211D. [DOI] [PubMed] [Google Scholar]
- Li Y.; Yang Q.; Yang L.; Lei N.; Zheng K. A Scalable Electrochemical Dehydrogenative Cross-Coupling of P(O)H Compounds with RSH/ROH. Chem. Commun. 2019, 55, 4981–4984. 10.1039/C9CC01378D. [DOI] [PubMed] [Google Scholar]
- Jessop C. M.; Parsons A. F.; Routledge A.; Irvine D. J. Radical Addition Reactions of Phosphorus Hydrides: Tuning the Reactivity of Phosphorus Hydrides, the Use of Microwaves and Horner-Wadsworth-Emmons-Type Reactions. Eur. J. Org. Chem. 2006, 2006, 1547–1554. 10.1002/ejoc.200500907. [DOI] [Google Scholar]
- Wang X.; Xia C.; Wu L. Visible-Light-Promoted Photoredox Dehydrogenative Coupling of Phosphines and Thiophenols. Org. Lett. 2020, 22, 7373–7377. 10.1021/acs.orglett.0c02746. [DOI] [PubMed] [Google Scholar]
- Han L. B.; Tilley T. D. Selective Homo- and Heterodehydrocouplings of Phosphines Catalyzed by Rhodium Phosphido Complexes. J. Am. Chem. Soc. 2006, 128, 13698–13699. 10.1021/ja065346+. [DOI] [PubMed] [Google Scholar]
- Wu L.; Annibale V. T.; Jiao H.; Brookfield A.; Collison D.; Manners I. Homo- and Heterodehydrocoupling of Phosphines Mediated by Alkali Metal Catalysts. Nat. Commun. 2019, 10, 2786. 10.1038/s41467-019-09832-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi-Ashton J. A.; Clarke A. K.; Unsworth W. P.; Taylor R. J. K. Phosphoranyl Radical Fragmentation Reactions Driven by Photoredox Catalysis. ACS Catal. 2020, 10, 7250–7261. 10.1021/acscatal.0c01923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Corella A.; María Martinez-Gualda A.; Ahmadi F.; Ming E.; Fraile A.; Alemán J. Thiol-Ene/Oxidation Tandem Reaction under Visible Light Photocatalysis: Synthesis of Alkyl Sulfoxides. Chem. Commun. 2017, 53, 10463–10466. 10.1039/C7CC05672A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner R.; Kümmel S.; König B. Visible Light Flavin Photo-Oxidation of Methylbenzenes, Styrenes and Phenylacetic Acids. Photochem. Photobiol. Sci. 2010, 9, 1367–1377. 10.1039/c0pp00202j. [DOI] [PubMed] [Google Scholar]
- Hari D. P.; König B. Synthetic Applications of Eosin Y in Photoredox Catalysis. Chem. Commun. 2014, 50, 6688–6699. 10.1039/C4CC00751D. [DOI] [PubMed] [Google Scholar]
- Baciocchi E.; Del Giacco T.; Elisei F.; Gerini M. F.; Guerra M.; Lapi A.; Liberali P. Electron Transfer and Singlet Oxygen Mechanisms in the Photooxygenation of Dibutyl Sulfide and Thioanisole in MeCN Sensitized by N-Methylquinolinium Tetrafluoborate and 9,10-Dicyanoanthracene. The Probable Involvement of a Thiadioxirane Intermediate in Electron Transfer Photooxygenations. J. Am. Chem. Soc. 2003, 125, 16444–16454. 10.1021/ja037591o. [DOI] [PubMed] [Google Scholar]
- Cui H.; Wei W.; Yang D.; Zhang Y.; Zhao H.; Wang L.; Wang H. Visible-Light-Induced Selective Synthesis of Sulfoxides from Alkenes and Thiols Using Air as the Oxidant. Green Chem. 2017, 19, 3520–3524. 10.1039/C7GC01416C. [DOI] [Google Scholar]
- Singh M.; Yadav A. K.; Yadav L. D. S.; Singh R. K. P. Visible-Light-Activated Selective Synthesis of Sulfoxides via Thiol-Ene/Oxidation Reaction Cascade. Tetrahedron Lett. 2018, 59, 450–453. 10.1016/j.tetlet.2017.12.066. [DOI] [Google Scholar]
- Baciocchi E.; Bietti M.; Di Fusco M.; Lanzalunga O. A Kinetic Study of the Electron-Transfer Reaction of the Phthalimide-N-Oxyl Radical (PINO) with Ferrocenes. J. Org. Chem. 2007, 72, 8748–8754. 10.1021/jo071211q. [DOI] [PubMed] [Google Scholar]
- Koshino N.; Cai Y.; Espenson J. H. Kinetic Study of the Phthalimide N-Oxyl (PINO) Radical in Acetic Acid. Hydrogen Abstraction from C-H Bonds and Evaluation of O-H Bond Dissociation Energy of N-Hydroxyphthalimide. J. Phys. Chem. A 2003, 107, 4262–4267. 10.1021/jp0276193. [DOI] [Google Scholar]
- Coseri S.; Mendenhall G. D.; Ingold K. U. Mechanisms of Reaction of Aminoxyl (Nitroxide), Iminoxyl, and Imidoxyl Radicals with Alkenes and Evidence That in the Presence of Lead Tetraacetate, N-Hydroxyphthalimide Reacts with Alkenes by Both Radical and Nonradical Mechanisms. J. Org. Chem. 2005, 70, 4629–4636. 10.1021/jo0504060. [DOI] [PubMed] [Google Scholar]
- Kumar J.; Ahmad A.; Rizvi M. A.; Ganie M. A.; Khajuria C.; Shah B. A. Photoredox-Mediated Synthesis of Functionalized Sulfoxides from Terminal Alkynes. Org. Lett. 2020, 22, 5661–5665. 10.1021/acs.orglett.0c02055. [DOI] [PubMed] [Google Scholar]
- Ai C.; Shen H.; Song D.; Li Y.; Yi X.; Wang Z.; Ling F.; Zhong W. Metal- And Oxidant-Free Electrochemical Synthesis of Sulfinic Esters from Thiols and Alcohols. Green Chem. 2019, 21, 5528–5531. 10.1039/C9GC02125F. [DOI] [Google Scholar]
- Yamamoto K.; Tsuchida E.; Nishide H.; Yoshida S.; Park Y.-S. Anodic Oxidation of Diphenyl Disulfides for Preparation of Oligo(P-phenylene Sulfide)s in Acidic Media. J. Electrochem. Soc. 1992, 139, 2401–2406. 10.1149/1.2221239. [DOI] [Google Scholar]
- Kaboudin B.; Behrouzi L.; Kazemi F.; Najafpour M. M.; Aoyama H. Electrochemical Synthesis of Sulfinate Esters: Nickel(II)-Catalyzed Oxidative Esterification of Thiols with Alcohols in an Undivided Cell. ACS Omega 2020, 5, 17947–17954. 10.1021/acsomega.0c00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. K.; Yi H.; Zhang G.; Bian C.; Pei P.; Lei A. Photoinduced Oxidative Cross-Coupling for O-S Bond Formation: A Facile Synthesis of Alkyl Benzenesulfonates. Synlett 2017, 28, 1558–1563. 10.1055/s-0036-1588728. [DOI] [Google Scholar]
- Laudadio G.; Barmpoutsis E.; Schotten C.; Struik L.; Govaerts S.; Browne D. L.; Noël T. Sulfonamide Synthesis through Electrochemical Oxidative Coupling of Amines and Thiols. J. Am. Chem. Soc. 2019, 141, 5664–5668. 10.1021/jacs.9b02266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdelande J. L.; Gallardo I.; Guirado G. Inductive vs Solvation Effects in Primary Alkyl Amines: Determination of the Standard Potentials. J. Am. Chem. Soc. 2007, 129, 2817–2821. 10.1021/ja0656245. [DOI] [PubMed] [Google Scholar]
- Laudadio G.; Bartolomeu A. D. A.; Verwijlen L. M. H. M.; Cao Y.; De Oliveira K. T.; Noël T. Sulfonyl Fluoride Synthesis through Electrochemical Oxidative Coupling of Thiols and Potassium Fluoride. J. Am. Chem. Soc. 2019, 141, 11832–11836. 10.1021/jacs.9b06126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam K.; Geiger W. E. Anodic Oxidation of Disulfides: Detection and Reactions of Disulfide Radical Cations. J. Org. Chem. 2013, 78, 8020–8027. 10.1021/jo401263z. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Adriaenssens B.; de A. Bartolomeu A.; Laudadio G.; de Oliveira K. T.; Noël T. Accelerating Sulfonyl Fluoride Synthesis through Electrochemical Oxidative Coupling of Thiols and Potassium Fluoride in Flow. J. Flow Chem. 2020, 10, 191–197. 10.1007/s41981-019-00070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Krasnova L.; Finn M. G.; Sharpless K. B. Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. 10.1002/anie.201309399. [DOI] [PubMed] [Google Scholar]
- Barrow A. S.; Smedley C. J.; Zheng Q.; Li S.; Dong J.; Moses J. E. The Growing Applications of SuFEx Click Chemistry. Chem. Soc. Rev. 2019, 48, 4731–4758. 10.1039/C8CS00960K. [DOI] [PubMed] [Google Scholar]
- Liu H.; Zhao L.; Yuan Y.; Xu Z.; Chen K.; Qiu S.; Tan H. Potassium Thioacids Mediated Selective Amide and Peptide Constructions Enabled by Visible Light Photoredox Catalysis. ACS Catal. 2016, 6, 1732–1736. 10.1021/acscatal.5b02943. [DOI] [Google Scholar]
- Shi L.; Liu H.; Huo L.; Dang Y.; Wang Y.; Yang B.; Qiu S.; Tan H. Site-Selective Phenol Acylation Mediated by Thioacids via Visible Light Photoredox Catalysis. Org. Chem. Front. 2018, 5, 1312–1319. 10.1039/C8QO00041G. [DOI] [Google Scholar]
- Ide T.; Barham J. P.; Fujita M.; Kawato Y.; Egami H.; Hamashima Y. Regio- and Chemoselective Csp3-H Arylation of Benzylamines by Single Electron Transfer/Hydrogen Atom Transfer Synergistic Catalysis. Chem. Sci. 2018, 9, 8453–8460. 10.1039/C8SC02965B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan D.; Wendt H. Electroreduction of Oxygen in Aprotic Media. J. Electroanal. Chem. 1995, 392, 69–74. 10.1016/0022-0728(95)04044-O. [DOI] [Google Scholar]
- Das S.; Ray S.; Ghosh A. B.; Samanta P. K.; Samanta S.; Adhikary B.; Biswas P. Visible Light Driven Amide Synthesis in Water at Room Temperature from Thioacid and Amine Using CdS Nanoparticles as Heterogeneous Photocatalyst. Appl. Organomet. Chem. 2018, 32, e4199. 10.1002/aoc.4199. [DOI] [Google Scholar]
- Zhao R.; Lind J.; Merényi G.; Eriksen T. E. One-Electron Redox Potential of Thiobenzoic Acid. Kinetic Characteristics of Benzoylthiyl Radical β-Fragmentation. J. Am. Chem. Soc. 1998, 120, 2811–2816. 10.1021/ja974136h. [DOI] [Google Scholar]
- Song W.; Dong K.; Li M. Visible Light-Induced Amide Bond Formation. Org. Lett. 2020, 22, 371–375. 10.1021/acs.orglett.9b03905. [DOI] [PubMed] [Google Scholar]
- Rattanangkool E.; Sukwattanasinitt M.; Wacharasindhu S. Organocatalytic Visible Light Enabled SNAr of Heterocyclic Thiols: A Metal-Free Approach to 2-Aminobenzoxazoles and 4-Aminoquinazolines. J. Org. Chem. 2017, 82, 13256–13262. 10.1021/acs.joc.7b02357. [DOI] [PubMed] [Google Scholar]
- Guan Y.; Wang C.; Wang D.; Dang G.; Chen C.; Zhou H.; Zhao X. Methylsulfone as a Leaving Group for Synthesis of Hyperbranched Poly(Arylene Pyrimidine Ether)s by Nucleophilic Aromatic Substitution. RSC Adv. 2015, 5, 12821–12823. 10.1039/C5RA00634A. [DOI] [Google Scholar]
- Qvortrup K.; Rankic D. A.; MacMillan D. W. C. A General Strategy for Organocatalytic Activation of C-H Bonds via Photoredox Catalysis: Direct Arylation of Benzylic Ethers. J. Am. Chem. Soc. 2014, 136, 626–629. 10.1021/ja411596q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaidarova L. G.; Ziganshina S. A.; Budnikov G. K. Electrocatalytic Oxidation of Cysteine and Cystine at a Carbon-Paste Electrode Modified with Ruthenium(IV) Oxide. J. Anal. Chem. 2003, 58, 577–582. 10.1023/A:1024124421268. [DOI] [Google Scholar]
- Jencks W. P.; Salvesen K. Equilibrium Deuterium Isotope Effects on the Ionization of Thiol Acids. J. Am. Chem. Soc. 1971, 93, 4433–4436. 10.1021/ja00747a016. [DOI] [Google Scholar]
- Escoubet S.; Gastaldi S.; Vanthuyne N.; Gil G.; Siri D.; Bertrand M. P. Thiyl Radical Mediated Racemization of Nonactivated Aliphatic Amines. J. Org. Chem. 2006, 71, 7288–7292. 10.1021/jo061033l. [DOI] [PubMed] [Google Scholar]
- Ochiai M.; Yamane S.; Hoque M. M.; Saito M.; Miyamoto K. Metal-Free α-CH Amination of Ethers with Hypervalent Sulfonylimino-Λ3-Bromane That Acts as an Active Nitrenoid. Chem. Commun. 2012, 48, 5280–5282. 10.1039/c2cc31523h. [DOI] [PubMed] [Google Scholar]
- Del Giacco T.; Lipparoni L.; Ranchella M.; Rol C.; Sebastiani G. V. Mechanism of the Oxidation of Benzylic Ethers Photosensitized by a 2,4,6-Triphenylpyrylium Salt. J. Chem. Soc. Perkin Trans. 2 2001, 1, 1802–1807. 10.1039/b103267b. [DOI] [Google Scholar]
- Hager D.; Macmillan D. W. C. Activation of C-H Bonds via the Merger of Photoredox and Organocatalysis: A Coupling of Benzylic Ethers with Schiff Bases. J. Am. Chem. Soc. 2014, 136, 16986–16989. 10.1021/ja5102695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Wakchaure V.; Kraft P.; List B. Primary-Amine-Catalyzed Enantioselective Intramolecular Aldolizations. Angew. Chem., Int. Ed. 2008, 47, 7656–7658. 10.1002/anie.200802497. [DOI] [PubMed] [Google Scholar]
- Bartnicka H.; Bojanowska I.; Kalinowski M. K. Solvent Effect on the Dissociation Constants of Aliphatic Carboxylic Acids. Aust. J. Chem. 1991, 44, 1077–1084. 10.1071/CH9911077. [DOI] [Google Scholar]
- Cuthbertson J. D.; MacMillan D. W. C. The Direct Arylation of Allylic sp3 C-H Bonds via Organic and Photoredox Catalysis. Nature 2015, 519, 74–77. 10.1038/nature14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R.; Goh Y. Y.; Liu H.; Tao H.; Li L.; Wu J. Visible-Light-Mediated Metal-Free Hydrosilylation of Alkenes through Selective Hydrogen Atom Transfer for Si-H Activation. Angew. Chem., Int. Ed. 2017, 56, 16621–16625. 10.1002/anie.201711250. [DOI] [PubMed] [Google Scholar]
- Xue X. S.; Ji P.; Zhou B.; Cheng J. P. The Essential Role of Bond Energetics in C-H Activation/Functionalization. Chem. Rev. 2017, 117, 8622–8648. 10.1021/acs.chemrev.6b00664. [DOI] [PubMed] [Google Scholar]
- Khursan S. L.; Mikhailov D. A.; Yanborisov V. M.; Borisov D. I. AM1 Calculations of Bond Dissociation Energies. Allylic and Benzylic C-H Bonds. React. Kinet. Catal. Lett. 1997, 61, 91–95. 10.1007/BF02477518. [DOI] [Google Scholar]
- Jin J.; MacMillan D. W. C. Alcohols as Alkylating Agents in Heteroarene C-H Functionalization. Nature 2015, 525, 87–90. 10.1038/nature14885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucio A. J.; Shaw S. K. Pyridine and Pyridinium Electrochemistry on Polycrystalline Gold Electrodes and Implications for CO2 Reduction. J. Phys. Chem. C 2015, 119, 12523–12530. 10.1021/acs.jpcc.5b03355. [DOI] [Google Scholar]
- Berkowitz J.; Ellison G. B.; Gutman D. Three Methods to Measure RH Bond Energies. J. Phys. Chem. 1994, 98, 2744–2765. 10.1021/j100062a009. [DOI] [Google Scholar]
- Proctor R. S. J.; Phipps R. J. Recent Advances in Minisci-Type Reactions. Angew. Chem., Int. Ed. 2019, 58, 13666–13699. 10.1002/anie.201900977. [DOI] [PubMed] [Google Scholar]
- Wessig P.; Muehling O. Spin-Center Shift (SCS) - A Versatile Concept in Biological and Synthetic Chemistry. Eur. J. Org. Chem. 2007, 2007, 2219–2232. 10.1002/ejoc.200600915. [DOI] [Google Scholar]
- Yan C.; Li L.; Liu Y.; Wang Q. Direct and Oxidant-Free Electron-Deficient Arylation of N-Acyl-Protected Tetrahydroisoquinolines. Org. Lett. 2016, 18, 4686–4689. 10.1021/acs.orglett.6b02326. [DOI] [PubMed] [Google Scholar]
- Vu M. D.; Das M.; Guo A.; Ang Z.-E.; Dokic M.; Soo H. S.; Liu X.-W. Visible-Light Photoredox Enables Ketone Carbonyl Alkylation for Easy Access to Tertiary Alcohols. ACS Catal. 2019, 9, 9009–9014. 10.1021/acscatal.9b02401. [DOI] [Google Scholar]
- Wagner P. J.; Truman R. J.; Puchalski A. E.; Wake R. Extent of Charge Transfer in the Photoreduction of Phenyl Ketones by Alkylbenzenes. J. Am. Chem. Soc. 1986, 108, 7727–7738. 10.1021/ja00284a041. [DOI] [PubMed] [Google Scholar]
- Wu Y. D.; Wong C. L.; Chan K. W. K.; Ji G. Z.; Jiang X. K. Substituent Effects on the C-H Bond Dissociation Energy of Toluene. A Density Functional Study. J. Org. Chem. 1996, 61, 746–750. 10.1021/jo951212v. [DOI] [PubMed] [Google Scholar]
- Jia J.; Kancherla R.; Rueping M.; Huang L. Allylic C(sp3)-H Alkylation via Synergistic Organo- And Photoredox Catalyzed Radical Addition to Imines. Chem. Sci. 2020, 11, 4954–4959. 10.1039/D0SC00819B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S.; Saga Y.; Kojima M.; Fuse H.; Matsunaga S.; Fukatsu A.; Kondo M.; Masaoka S.; Kanai M. Hybrid Catalysis Enabling Room-Temperature Hydrogen Gas Release from N-Heterocycles and Tetrahydronaphthalenes. J. Am. Chem. Soc. 2017, 139, 2204–2207. 10.1021/jacs.7b00253. [DOI] [PubMed] [Google Scholar]
- Göttker-Schnetmann I.; White P.; Brookhart M. Iridium Bis(Phosphinite) p-XPCP Pincer Complexes: Highly Active Catalysts for the Transfer Dehydrogenation of Alkanes. J. Am. Chem. Soc. 2004, 126, 1804–1811. 10.1021/ja0385235. [DOI] [PubMed] [Google Scholar]
- West J. G.; Huang D.; Sorensen E. J. Acceptorless Dehydrogenation of Small Molecules through Cooperative Base Metal Catalysis. Nat. Commun. 2015, 6, 1–7. 10.1038/ncomms10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Yi H.; Lei A. Electrochemical Acceptorless Dehydrogenation of N-Heterocycles Utilizing TEMPO as Organo-Electrocatalyst. ACS Catal. 2018, 8, 1192–1196. 10.1021/acscatal.7b04137. [DOI] [Google Scholar]
- Preuster P.; Papp C.; Wasserscheid P. Liquid Organic Hydrogen Carriers (LOHCs): Toward a Hydrogen-Free Hydrogen Economy. Acc. Chem. Res. 2017, 50, 74–85. 10.1021/acs.accounts.6b00474. [DOI] [PubMed] [Google Scholar]
- Deluca R. J.; Stokes B. J.; Sigman M. S. The Strategic Generation and Interception of Palladium-Hydrides for Use in Alkene Functionalization Reactions. Pure Appl. Chem. 2014, 86, 395–408. 10.1515/pac-2014-5041. [DOI] [Google Scholar]
- Tanabe S.; Mitsunuma H.; Kanai M. Catalytic Allylation of Aldehydes Using Unactivated Alkenes. J. Am. Chem. Soc. 2020, 142, 12374–12381. 10.1021/jacs.0c04735. [DOI] [PubMed] [Google Scholar]
- Schwarz J. L.; Schäfers F.; Tlahuext-Aca A.; Lückemeier L.; Glorius F. Diastereoselective Allylation of Aldehydes by Dual Photoredox and Chromium Catalysis. J. Am. Chem. Soc. 2018, 140, 12705–12709. 10.1021/jacs.8b08052. [DOI] [PubMed] [Google Scholar]
- Mitsunuma H.; Tanabe S.; Fuse H.; Ohkubo K.; Kanai M. Catalytic Asymmetric Allylation of Aldehydes with Alkenes through Allylic C(sp3)-H Functionalization Mediated by Organophotoredox and Chiral Chromium Hybrid Catalysis. Chem. Sci. 2019, 10, 3459–3465. 10.1039/C8SC05677C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargaden G. C.; Guiry P. J. The Development of the Asymmetric Nozaki-Hiyama-Kishi Reaction. Adv. Synth. Catal. 2007, 349, 2407–2424. 10.1002/adsc.200700324. [DOI] [Google Scholar]
- Schwarz J. L.; Huang H. M.; Paulisch T. O.; Glorius F. Dialkylation of 1,3-Dienes by Dual Photoredox and Chromium Catalysis. ACS Catal. 2020, 10, 1621–1627. 10.1021/acscatal.9b04222. [DOI] [Google Scholar]
- Fuse H.; Nakao H.; Saga Y.; Fukatsu A.; Kondo M.; Masaoka S.; Mitsunuma H.; Kanai M. Photocatalytic Redox-Neutral Hydroxyalkylation of N-Heteroaromatics with Aldehydes. Chem. Sci. 2020, 11, 12206–12211. 10.1039/D0SC04114A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q. Y.; Schirmer T. E.; Berger A. L.; Donabauer K.; König B. Photocarboxylation of Benzylic C-H Bonds. J. Am. Chem. Soc. 2019, 141, 11393–11397. 10.1021/jacs.9b05360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donabauer K.; König B. Strategies for the Photocatalytic Generation of Carbanion Equivalents for Reductant-Free C-C Bond Formations. Acc. Chem. Res. 2021, 54, 242–252. 10.1021/acs.accounts.0c00620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillen D. F.; Golden D. M. Hydrocarbon Bond Dissociation Energies. Annu. Rev. Phys. Chem. 1982, 33, 493–532. 10.1146/annurev.pc.33.100182.002425. [DOI] [Google Scholar]
- Berger A. L.; Donabauer K.; König B. Photocatalytic Carbanion Generation from C-H Bonds-Reductant Free Barbier/Grignard-Type Reactions. Chem. Sci. 2019, 10, 10991–10996. 10.1039/C9SC04987H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donabauer K.; Murugesan K.; Rozman U.; Crespi S.; König B. Photocatalytic Reductive Radical-Polar Crossover for a Base-Free Corey-Seebach Reaction. Chem. - Eur. J. 2020, 26, 12945–12950. 10.1002/chem.202003000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervin K. M.; DeTuri V. F. Anchoring the Gas-Phase Acidity Scale. J. Phys. Chem. A 2002, 106, 9947–9956. 10.1021/jp020594n. [DOI] [Google Scholar]
- Fujita M.; Kobayashi F.; Ide T.; Egami H.; Hamashima Y. Oxidative and Redox-Neutral Approaches to Symmetrical Diamines and Diols by Single Electron Transfer/Hydrogen Atom Transfer Synergistic Catalysis. Eur. J. Org. Chem. 2020, 2020, 7151–7155. 10.1002/ejoc.202001329. [DOI] [Google Scholar]
- More O’Ferrall R. A.; O’Brien D. M.; Murphy D. G. Rate and Equilibrium Constants for Formation and Hydrolysis of 9-Formylfluorene Oxime: Diffusion-Controlled Trapping of a Protonated Aldehyde by Hydroxylamine. Can. J. Chem. 2000, 78, 1594–1612. 10.1139/v00-129. [DOI] [Google Scholar]
- Zhou N.; Yuan X. A.; Zhao Y.; Xie J.; Zhu C. Synergistic Photoredox Catalysis and Organocatalysis for Inverse Hydroboration of Imines. Angew. Chem., Int. Ed. 2018, 57, 3990–3994. 10.1002/anie.201800421. [DOI] [PubMed] [Google Scholar]
- Ueng S. H.; Solovyev A.; Yuan X.; Geib S. J.; Fensterbank L.; Lacôte E.; Malacria M.; Newcomb M.; Walton J. C.; Curran D. P. N-Heterocyclic Carbene Boryl Radicals: A New Class of Boron-Centered Radical. J. Am. Chem. Soc. 2009, 131, 11256–11262. 10.1021/ja904103x. [DOI] [PubMed] [Google Scholar]
- Kobayashi S.; Ishitani H. Catalytic Enantioselective Addition to Imines. Chem. Rev. 1999, 99, 1069–1094. 10.1021/cr980414z. [DOI] [PubMed] [Google Scholar]
- Yang W.; Gao X.; Wang B. Boronic Acid Compounds as Potential Pharmaceutical Agents. Med. Res. Rev. 2003, 23, 346–368. 10.1002/med.10043. [DOI] [PubMed] [Google Scholar]
- White M. C.; Zhao J. Aliphatic C-H Oxidations for Late-Stage Functionalization. J. Am. Chem. Soc. 2018, 140, 13988–14009. 10.1021/jacs.8b05195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newhouse T.; Baran P. S. If C-H Bonds Could Talk: Selective C-H Bond Oxidation. Angew. Chem., Int. Ed. 2011, 50, 3362–3374. 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le C.; Liang Y.; Evans R. W.; Li X.; MacMillan D. W. C. Selective sp3 C-H Alkylation via Polarity-Match-Based Cross-Coupling. Nature 2017, 547, 79–83. 10.1038/nature22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzirakis M. D.; Lykakis I. N.; Orfanopoulos M. Decatungstate as an Efficient Photocatalyst in Organic Chemistry. Chem. Soc. Rev. 2009, 38, 2609–2621. 10.1039/b812100c. [DOI] [PubMed] [Google Scholar]
- Liu W.; Groves J. T. Manganese Catalyzed C-H Halogenation. Acc. Chem. Res. 2015, 48, 1727–1735. 10.1021/acs.accounts.5b00062. [DOI] [PubMed] [Google Scholar]
- Huang X.; Groves J. T. Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. 10.1021/acs.chemrev.7b00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittra K.; Green M. T. Reduction Potentials of P450 Compounds I and II: Insight into the Thermodynamics of C-H Bond Activation. J. Am. Chem. Soc. 2019, 141, 5504–5510. 10.1021/jacs.9b00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kal S.; Xu S.; Que L. Bio-inspired Nonheme Iron Oxidation Catalysis. Growing Evidence for the Involvement of Oxoiron(V) Oxidants in Cleaving Strong C-H Bonds. Angew. Chem., Int. Ed. 2020, 59 (19), 7332–7349. 10.1002/anie.201906551. [DOI] [PubMed] [Google Scholar]
- Beatty J. W.; Stephenson C. R. J. Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc. Chem. Res. 2015, 48, 1474–1484. 10.1021/acs.accounts.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz Espelt L.; Wiensch E. M.; Yoon T. P. Brønsted Acid Cocatalysts in Photocatalytic Radical Addition of α-Amino C-H Bonds across Michael Acceptors. J. Org. Chem. 2013, 78, 4107–4114. 10.1021/jo400428m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Das A.; Bui L.; Zhou H.; Curran D. P.; Rueping M. Oxygen Switch in Visible-Light Photoredox Catalysis: Radical Additions and Cyclizations and Unexpected C-C-Bond Cleavage Reactions. J. Am. Chem. Soc. 2013, 135, 1823–1829. 10.1021/ja309580a. [DOI] [PubMed] [Google Scholar]
- McManus J. B.; Onuska N. P. R.; Nicewicz D. A. Generation and Alkylation of α-Carbamyl Radicals via Organic Photoredox Catalysis. J. Am. Chem. Soc. 2018, 140, 9056–9060. 10.1021/jacs.8b04890. [DOI] [PubMed] [Google Scholar]
- Mazzarella D.; Crisenza G. E. M.; Melchiorre P. Asymmetric Photocatalytic C-H Functionalization of Toluene and Derivatives. J. Am. Chem. Soc. 2018, 140, 8439–8443. 10.1021/jacs.8b05240. [DOI] [PubMed] [Google Scholar]
- Lee B. J.; DeGlopper K. S.; Yoon T. P. Site-Selective Alkoxylation of Benzylic C-H Bonds by Photoredox Catalysis. Angew. Chem. Int. Ed. 2020, 59, 197–202. 10.1002/ange.201910602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shono T.; Hamaguchi H.; Matsumura Y. Electroorganic Chemistry. XX. Anodic Oxidation of Carbamates. J. Am. Chem. Soc. 1975, 97, 4264–4268. 10.1021/ja00848a020. [DOI] [Google Scholar]
- Shono T.; Matsumura Y.; Tsubata K. Electroorganic Chemistry. 46. A New Carbon-Carbon Bond Forming Reaction at the α-Position of Amines Utilizing Anodic Oxidation as a Key Step. J. Am. Chem. Soc. 1981, 103, 1172–1176. 10.1021/ja00395a029. [DOI] [Google Scholar]
- Suga S.; Okajima M.; Fujiwara K.; Yoshida J. Cation Flow” Method: A New Approach to Conventional and Combinatorial Organic Syntheses Using Electrochemical Microflow Systems. J. Am. Chem. Soc. 2001, 123, 7941–7942. 10.1021/ja015823i. [DOI] [PubMed] [Google Scholar]
- Yoshida J. I.; Suga S. Basic Concepts of “Cation Pool” and “Cation Flow” Methods and Their Applications in Conventional and Combinatorial Organic Synthesis. Chem. - Eur. J. 2002, 8, 2650–2658. . [DOI] [PubMed] [Google Scholar]
- Yanagida S.; Azuma T.; Kawakami H.; Kizumoto H.; Sakurai H. Photocatalytic Carbon-Carbon Bond Formation with Concurrent Hydrogen Evolution on Colloidal Zinc Sulphide. J. Chem. Soc., Chem. Commun. 1984, 21–22. 10.1039/C39840000021. [DOI] [Google Scholar]
- Bücheler J.; Zeug N.; Kisch H. Zinc Sulfide as Catalyst for the Heterogenous Photoreduction of Water. Angew. Chem., Int. Ed. Engl. 1982, 21, 783–784. 10.1002/anie.198207831. [DOI] [Google Scholar]
- Zeug N.; Bücheler J.; Kisch H. Catalytic Formation of Hydrogen and C-C Bonds on Illuminated Zinc Sulfide Generated from Zinc Dithiolenes. J. Am. Chem. Soc. 1985, 107, 1459–1465. 10.1021/ja00292a001. [DOI] [Google Scholar]
- Hetterich W.; Kisch H. Heterogene Photokatalyse, V. Cadmium-Zinksulfide Als Katalysatoren Der Photodehydrodimerisierung von 2,5-Dihydrofuran. Chem. Ber. 1988, 121, 15–20. 10.1002/cber.19881210104. [DOI] [Google Scholar]
- Künneth R.; Twardzik G.; Emig G.; Kisch H. Heterogeneous Photocatalysis XI. Zinc Sulphide Catalysed Dehydrodimerization of Dihydropyrans and Cyclohexene. J. Photochem. Photobiol., A 1993, 76, 209–215. 10.1016/1010-6030(93)80137-X. [DOI] [Google Scholar]
- Hörner G.; Johne P.; Künneth R.; Twardzik G.; Roth H.; Clark T.; Kisch H. Semiconductor Type A Photocatalysis: Role of Substrate Adsorption and the Nature of Photoreactive Surface Sites in Zinc Sulfide Catalyzed C-C Coupling Reactions. Chem. - Eur. J. 1999, 5, 208–217. . [DOI] [Google Scholar]
- Künneth R.; Feldmer C.; Kisch H. Semiconductor-Catalyzed Photoaddition of Cyclic Enol Ethers to 1, 2-Diazenes. Angew. Chem., Int. Ed. Engl. 1992, 31, 1039–1040. 10.1002/anie.199210391. [DOI] [Google Scholar]
- Künneth R.; Feldmer C.; Knoch F.; Kisch H. Semiconductor-Catalysed Photoaddition of Olefins and Enol Ethers to 1,2-Diazenes: A New Route to Allylhydrazines. Chem. - Eur. J. 1995, 1, 441–448. 10.1002/chem.19950010709. [DOI] [Google Scholar]
- Parrino F.; Ramakrishnan A.; Kisch H. Semiconductor-Photocatalyzed Sulfoxidation of Alkanes. Angew. Chem., Int. Ed. 2008, 47, 7107–7109. 10.1002/anie.200800326. [DOI] [PubMed] [Google Scholar]
- Parrino F.; Ramakrishnan A.; Damm C.; Kisch H. Visible-Light-Induced Sulfoxidation of Alkanes in the Presence of Titania. ChemPlusChem 2012, 77, 713–720. 10.1002/cplu.201200097. [DOI] [Google Scholar]
- Ramloch H.; Täuber G. Moderne Verfahren Der Großchemie: Die Sulfoxidation. Chem. Unserer Zeit 1979, 13, 157–162. 10.1002/ciuz.19790130505. [DOI] [Google Scholar]
- Graf R. Der Reaktionsmechanismus Der Sulfoxydation. Justus Liebigs Ann. Chem. 1952, 578, 50–82. 10.1002/jlac.19525780110. [DOI] [Google Scholar]
- Kisch H. Semiconductor Photocatalysis for Chemoselective Radical Coupling Reactions. Acc. Chem. Res. 2017, 50, 1002–1010. 10.1021/acs.accounts.7b00023. [DOI] [PubMed] [Google Scholar]
- Uraguchi D.; Torii M.; Ooi T. Acridinium Betaine as a Single-Electron-Transfer Catalyst: Design and Application to Dimerization of Oxindoles. ACS Catal. 2017, 7, 2765–2769. 10.1021/acscatal.7b00265. [DOI] [Google Scholar]
- Wang H. C.; Lillie E. D.; Slomkowski S.; Levin G.; Szwarc M. Flash Photolysis of Na+,C–(Ph)2CH2CH2C–(Ph)2,Na+. Redox Potential of 1,1-Diphenylethylene and Rate Constant of Dimerization of Its Radical Anion. J. Am. Chem. Soc. 1977, 99, 4612–4617. 10.1021/ja00456a015. [DOI] [Google Scholar]
- Valdez C. N.; Schimpf A. M.; Gamelin D. R.; Mayer J. M. Proton-Controlled Reduction of ZnO Nanocrystals: Effects of Molecular Reductants, Cations, and Thermodynamic Limitations. J. Am. Chem. Soc. 2016, 138, 1377–1385. 10.1021/jacs.5b12182. [DOI] [PubMed] [Google Scholar]
- Sayfutyarova E. R.; Goldsmith Z. K.; Hammes-Schiffer S. Theoretical Study of C-H Bond Cleavage via Concerted Proton-Coupled Electron Transfer in Fluorenyl-Benzoates. J. Am. Chem. Soc. 2018, 140, 15641–15645. 10.1021/jacs.8b10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer J. M. Hydrogen Atom Abstraction by Metal-Oxo Complexes: Understanding the Analogy with Organic Radical Reactions. Acc. Chem. Res. 1998, 31, 441–450. 10.1021/ar970171h. [DOI] [Google Scholar]
- Darcy J. W.; Kolmar S. S.; Mayer J. M. Transition State Asymmetry in C-H Bond Cleavage by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2019, 141, 10777–10787. 10.1021/jacs.9b04303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayfutyarova E. R.; Lam Y. C.; Hammes-Schiffer S. Strategies for Enhancing the Rate Constant of C-H Bond Cleavage by Concerted Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2019, 141, 15183–15189. 10.1021/jacs.9b06849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costentin C.; Savéant J. M. Hydrogen and Proton Exchange at Carbon. Imbalanced Transition State and Mechanism Crossover. Chem. Sci. 2020, 11, 1006–1010. 10.1039/C9SC05147C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton C. M.; Zhu Q.; Ripberger H.; Troian-Gautier L.; Toa Z. S. D.; Knowles R. R.; Alexanian E. J. C-H Alkylation via Multisite-Proton-Coupled Electron Transfer of an Aliphatic C-H Bond. J. Am. Chem. Soc. 2019, 141, 13253–13260. 10.1021/jacs.9b06834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapat J. B.; Blade R. J.; Boulton A. J.; Epsztajn J.; Katritzky A. R.; Lewis J.; Molina-Buendia P.; Nie P. L.; Ramsden C. A. Pyridines as Leaving Groups in Synthetic Transformations: Nucleophilic Displacements of Amino Groups, and Novel Preparations of Nitriles and Isocyanates. Tetrahedron Lett. 1976, 17, 2691–2694. 10.1016/S0040-4039(00)77797-5. [DOI] [Google Scholar]
- Katritzky A. R.; Marson C. M. Pyrylium Mediated Transformations of Primary Amino Groups into Other Functional Groups. New Synthetic Methods (41). Angew. Chem., Int. Ed. Engl. 1984, 23, 420–429. 10.1002/anie.198404201. [DOI] [Google Scholar]
- Klauck F. J. R.; James M. J.; Glorius F. Deaminative Strategy for the Visible-Light-Mediated Generation of Alkyl Radicals. Angew. Chem., Int. Ed. 2017, 56, 12336–12339. 10.1002/anie.201706896. [DOI] [PubMed] [Google Scholar]
- James M. J.; Strieth-Kalthoff F.; Sandfort F.; Klauck F. J. R.; Wagener F.; Glorius F. Visible-Light-Mediated Charge Transfer Enables C-C Bond Formation with Traceless Acceptor Groups. Chem. - Eur. J. 2019, 25, 8240–8244. 10.1002/chem.201901397. [DOI] [PubMed] [Google Scholar]
- Ishitani O.; Pac C.; Sakurai H. Formation of a Novel Type of Adduct between an NADH Model and Carbonyl Compounds by Photosensitization Using Ru(bpy)32+. J. Org. Chem. 1983, 48, 2941–2942. 10.1021/jo00165a037. [DOI] [Google Scholar]
- Ishitani O.; Yanagida S.; Takamuku S.; Pac C. Redox-Photosensitized Reactions. 13. Ru(bpy)32+-Photosensitized Reactions of an NADH Model, l-Benzyl-l,4-Dihydronicotinamide, with Aromatic Carbonyl Compounds and Comparison with Thermal Reactions. J. Org. Chem. 1987, 52, 2790–2796. 10.1021/jo00389a027. [DOI] [Google Scholar]
- Given P. H.; Peover M. E. 74. Polarographic Reduction of Aromatic Hydrocarbons and Carbonyl Compounds in Dimethylformamide in the Presence of Proton-Donors. J. Chem. Soc. 1960, 385–393. 10.1039/jr9600000385. [DOI] [Google Scholar]
- Michielli R. F.; Elving P. J. Electrochemical Reduction of Benzophenone in Aprotic Medium. Effect of Proton Availability. J. Am. Chem. Soc. 1968, 90, 1989–1995. 10.1021/ja01010a011. [DOI] [Google Scholar]
- Fukuzumi S.; Ishikawa K.; Hironaka K.; Tanaka T. Acid Catalysis in Thermal and Photoinduced Electron-Transfer Reactions. J. Chem. Soc., Perkin Trans. 2 1987, (6), 751–760. 10.1039/p29870000751. [DOI] [Google Scholar]
- Shibata T.; Kabumoto A.; Shiragami T.; Ishitani O.; Pac C.; Yanagida S. Novel Visible-Light-Driven Photocatalyst. Poly(p-Phenylene)-Catalyzed Photoreductions of Water, Carbonyl Compounds, and Olefins. J. Phys. Chem. 1990, 94, 2068–2076. 10.1021/j100368a063. [DOI] [Google Scholar]
- Larraufie M. H.; Pellet R.; Fensterbank L.; Goddard J. P.; Lacôte E.; Malacria M.; Ollivier C. Visible-Light-Induced Photoreductive Generation of Radicals from Epoxides and Aziridines. Angew. Chem., Int. Ed. 2011, 50, 4463–4466. 10.1002/anie.201007571. [DOI] [PubMed] [Google Scholar]
- McCallum T.; Wu X.; Lin S. Recent Advances in Titanium Radical Redox Catalysis. J. Org. Chem. 2019, 84, 14369–14380. 10.1021/acs.joc.9b02465. [DOI] [PubMed] [Google Scholar]
- Boujlel K.; Martigny P.; Simonet J. Cathodic Redox Catalysis of Aromatic Epoxides. J. Electroanal. Chem. Interfacial Electrochem. 1983, 144, 437–442. 10.1016/S0022-0728(83)80174-0. [DOI] [Google Scholar]
- Cossy J.; Aclinou P.; Bellosta V.; Furet N.; Baranne-Lafont J.; Sparfel D.; Souchaud C. Radical Anion Ring Opening Reactions via Photochemically Induced Electron Transfer. Tetrahedron Lett. 1991, 32, 1315–1316. 10.1016/S0040-4039(00)79654-7. [DOI] [Google Scholar]
- Hasegawa E.; Ishiyama K.; Horaguchi T.; Shimizu T. Exploratory Study on Photoinduced Single Electron Transfer Reactions of α,β-Epoxy Ketones with Amines. J. Org. Chem. 1991, 56, 1631–1635. 10.1021/jo00004a052. [DOI] [Google Scholar]
- Hasegawa E.; Ishiyama K.; Horaguchi T.; Shimizu T. Free Radical Trapping of α-Keto Radicals Derived from α,β-Epoxy Ketones via Photoinduced Single Electron Transfer Process. Tetrahedron Lett. 1991, 32, 2029–2032. 10.1016/S0040-4039(00)78899-X. [DOI] [Google Scholar]
- Guindon Y.; Lavallée J. F.; Boisvert L.; Chabot C.; Delorme D.; Yoakim C.; Hall D.; Lemieux R.; Simoneau B. Stereoselective Radical-Mediated Reduction and Alkylation of α-Halo Esters. Tetrahedron Lett. 1991, 32, 27–30. 10.1016/S0040-4039(00)71209-3. [DOI] [Google Scholar]
- Guindon Y.; Guérin B.; Chabot C.; MacKintosh N.; Ogilvie W. W. Stereoselective Radical Allylation of α-Iodo-β-Alkoxy Esters: Reversal of Facial Selectivity by Lewis Acid Complexation. Synlett 1995, 1995, 449–451. 10.1055/s-1995-4995. [DOI] [Google Scholar]
- Fuentes De Arriba A. L.; Urbitsch F.; Dixon D. J. Umpolung Synthesis of Branched α-Functionalized Amines from Imines via Photocatalytic Three-Component Reductive Coupling Reactions. Chem. Commun. 2016, 52, 14434–14437. 10.1039/C6CC09172E. [DOI] [PubMed] [Google Scholar]
- Rossolini T.; Leitch J. A.; Grainger R.; Dixon D. J. Photocatalytic Three-Component Umpolung Synthesis of 1,3-Diamines. Org. Lett. 2018, 20, 6794–6798. 10.1021/acs.orglett.8b02923. [DOI] [PubMed] [Google Scholar]
- Lee K. N.; Lei Z.; Ngai M. Y. β-Selective Reductive Coupling of Alkenylpyridines with Aldehydes and Imines via Synergistic Lewis Acid/Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 5003–5006. 10.1021/jacs.7b01373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L.; Chen Y. Polarity-Reversed Allylations of Aldehydes, Ketones, and Imines Enabled by Hantzsch Ester in Photoredox Catalysis. Angew. Chem., Int. Ed. 2016, 55, 13312–13315. 10.1002/anie.201607813. [DOI] [PubMed] [Google Scholar]
- Bou-Hamdan F. R.; Seeberger P. H. Visible-Light-Mediated Photochemistry: Accelerating Ru(bpy)32+-Catalyzed Reactions in Continuous Flow. Chem. Sci. 2012, 3, 1612–1616. 10.1039/c2sc01016j. [DOI] [Google Scholar]
- Du J.; Espelt L. R.; Guzei I. A.; Yoon T. P. Photocatalytic Reductive Cyclizations of Enones: Divergent Reactivity of Photogenerated Radical and Radical Anion Intermediates. Chem. Sci. 2011, 2, 2115–2119. 10.1039/c1sc00357g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischay M. A.; Anzovino M. E.; Du J.; Yoon T. P. Efficient Visible Light Photocatalysis of [2+2] Enone Cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]
- Du J.; Yoon T. P. Crossed Intermolecular [2+2] Cycloadditions of Acyclic Enones via Visible Light Photocatalysis. J. Am. Chem. Soc. 2009, 131, 14604–14605. 10.1021/ja903732v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G.; Yang C.; Guo L.; Sun H.; Lin R.; Xia W. Reactivity Insight into Reductive Coupling and Aldol Cyclization of Chalcones by Visible Light Photocatalysis. J. Org. Chem. 2012, 77, 6302–6306. 10.1021/jo300796j. [DOI] [PubMed] [Google Scholar]
- Neumann M.; Zeitler K. A Cooperative Hydrogen-Bond-Promoted Organophotoredox Catalysis Strategy for Highly Diastereoselective, Reductive Enone Cyclization. Chem. - Eur. J. 2013, 19, 6950–6955. 10.1002/chem.201204573. [DOI] [PubMed] [Google Scholar]
- Kolthoff I. M.; Chantooni M. K. Protonation Constants of Very Weak Uncharged Bases in Acetonitrile. J. Am. Chem. Soc. 1973, 95, 8539–8546. 10.1021/ja00807a007. [DOI] [Google Scholar]
- Rueping M.; Nachtsheim B. J.; Ieawsuwan W.; Atodiresei I. Modulating the Acidity: Highly Acidic Brønsted Acids in Asymmetric Catalysis. Angew. Chem., Int. Ed. 2011, 50, 6706–6720. 10.1002/anie.201100169. [DOI] [PubMed] [Google Scholar]
- Hernandez-Perez A. C.; Collins S. K. Heteroleptic Cu-Based Sensitizers in Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1557–1565. 10.1021/acs.accounts.6b00250. [DOI] [PubMed] [Google Scholar]
- Zhong M.; Pannecoucke X.; Jubault P.; Poisson T. Recent Advances in Photocatalyzed Reactions Using Well-Defined Copper(I) Complexes. Beilstein J. Org. Chem. 2020, 16, 451–481. 10.3762/bjoc.16.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Petersen J. L.; Milsmann C. A Luminescent Zirconium(IV) Complex as a Molecular Photosensitizer for Visible Light Photoredox Catalysis. J. Am. Chem. Soc. 2016, 138, 13115–13118. 10.1021/jacs.6b05934. [DOI] [PubMed] [Google Scholar]
- Büldt L. A.; Wenger O. S. Chromium Complexes for Luminescence, Solar Cells, Photoredox Catalysis, Upconversion, and Phototriggered NO Release. Chem. Sci. 2017, 8, 7359–7367. 10.1039/C7SC03372A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjær K. S.; Kaul N.; Prakash O.; Chábera P.; Rosemann N. W.; Honarfar A.; Gordivska O.; Fredin L. A.; Bergquist K. E.; Häggström L.; et al. Luminescence and Reactivity of a Charge-Transfer Excited Iron Complex with Nanosecond Lifetime. Science 2019, 363, 249–253. 10.1126/science.aau7160. [DOI] [PubMed] [Google Scholar]
- Hockin B. M.; Li C.; Robertson N.; Zysman-Colman E. Photoredox Catalysts Based on Earth-Abundant Metal Complexes. Catal. Sci. Technol. 2019, 9, 889–915. 10.1039/C8CY02336K. [DOI] [Google Scholar]
- Minozzi C.; Caron A.; Grenier-Petel J. C.; Santandrea J.; Collins S. K. Heteroleptic Copper(I)-Based Complexes for Photocatalysis: Combinatorial Assembly, Discovery, and Optimization. Angew. Chem., Int. Ed. 2018, 57, 5477–5481. 10.1002/anie.201800144. [DOI] [PubMed] [Google Scholar]
- Fava E.; Nakajima M.; Nguyen A. L. P.; Rueping M. Photoredox-Catalyzed Ketyl-Olefin Coupling for the Synthesis of Substituted Chromanols. J. Org. Chem. 2016, 81, 6959–6964. 10.1021/acs.joc.6b01006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M.; Fava E.; Loescher S.; Jiang Z.; Rueping M. Photoredox-Catalyzed Reductive Coupling of Aldehydes, Ketones, and Imines with Visible Light. Angew. Chem., Int. Ed. 2015, 54, 8828–8832. 10.1002/anie.201501556. [DOI] [PubMed] [Google Scholar]
- Alligrant T. M.; Alvarez J. C. The Role of Intermolecular Hydrogen Bonding and Proton Transfer in Proton-Coupled Electron Transfer. J. Phys. Chem. C 2011, 115, 10797–10805. 10.1021/jp108339k. [DOI] [Google Scholar]
- Amemiya F.; Fuse K.; Fuchigami T.; Atobe M. Chemoselective Reaction System Using a Two Inlet Micro-Flow Reactor: Application to Carbonyl Allylation. Chem. Commun. 2010, 46, 2730–2732. 10.1039/b926943f. [DOI] [PubMed] [Google Scholar]
- Root D. K.; Smith W. H. Electrochemical Behavior of Selected Imine Derivatives, Reductive Carboxylation, α-Amino Acid Synthesis. J. Electrochem. Soc. 1982, 129, 1231–1236. 10.1149/1.2124092. [DOI] [Google Scholar]
- Leitch J. A.; Rossolini T.; Rogova T.; Maitland J. A. P.; Dixon D. J. α-Amino Radicals via Photocatalytic Single-Electron Reduction of Imine Derivatives. ACS Catal. 2020, 10, 2009–2025. 10.1021/acscatal.9b05011. [DOI] [Google Scholar]
- Hu J.; Wang J.; Nguyen T. H.; Zheng N. The Chemistry of Amine Radical Cations Produced by Visible Light Photoredox Catalysis. Beilstein J. Org. Chem. 2013, 9, 1977–2001. 10.3762/bjoc.9.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C.; You S. L. Transfer Hydrogenation with Hantzsch Esters and Related Organic Hydride Donors. Chem. Soc. Rev. 2012, 41, 2498–2518. 10.1039/c1cs15268h. [DOI] [PubMed] [Google Scholar]
- Leitch J. A.; Fuentes De Arriba A. L.; Tan J.; Hoff O.; Martínez C. M.; Dixon D. J. Photocatalytic Reverse Polarity Povarov Reaction. Chem. Sci. 2018, 9, 6653–6658. 10.1039/C8SC01704B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez-Bonet Á.; Remeur C.; Matsui J. K.; Molander G. A. Late-Stage C-H Alkylation of Heterocycles and 1,4-Quinones via Oxidative Homolysis of 1,4-Dihydropyridines. J. Am. Chem. Soc. 2017, 139, 12251–12258. 10.1021/jacs.7b05899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz S.; Leifert D.; Studer A. Cross Dehydrogenative Coupling via Base-Promoted Homolytic Aromatic Substitution (BHAS): Synthesis of Fluorenones and Xanthones. Org. Lett. 2013, 15, 928–931. 10.1021/ol4000857. [DOI] [PubMed] [Google Scholar]
- Leifert D.; Daniliuc C. G.; Studer A. 6-Aroylated Phenanthridines via Base Promoted Homolytic Aromatic Substitution (BHAS). Org. Lett. 2013, 15, 6286–6289. 10.1021/ol403147v. [DOI] [PubMed] [Google Scholar]
- Vasu D.; Fuentes de Arriba A. L.; Leitch J. A.; De Gombert A.; Dixon D. J. Primary α-Tertiary Amine Synthesis via α-C-H Functionalization. Chem. Sci. 2019, 10, 3401–3407. 10.1039/C8SC05164J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copper Amine Oxidases. Structures, Catalytic Mechanisms and Role in Pathophysiology; International Institute of Anticancer Research, 2010; p 30. [Google Scholar]
- Komanduri V.; Grant C. D.; Krische M. J. Branch-Selective Reductive Coupling of 2-Vinyl Pyridines and Imines via Rhodium Catalyzed C-C Bond Forming Hydrogenation. J. Am. Chem. Soc. 2008, 130, 12592–12593. 10.1021/ja805056g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A.; Choi B.; Lam H. W. Enantioselective Copper-Catalyzed Reductive Coupling of Alkenylazaarenes with Ketones. J. Am. Chem. Soc. 2012, 134, 8428–8431. 10.1021/ja3036916. [DOI] [PubMed] [Google Scholar]
- Choi B.; Saxena A.; Smith J. J.; Churchill G. H.; Lam H. W. Enantioselective Copper-Catalyzed Reductive Coupling of Vinylazaarenes with n -Boc Aldimines. Synlett 2015, 26, 350–354. 10.1055/s-0034-1379548. [DOI] [Google Scholar]
- Jung J.; Kim J.; Park G.; You Y.; Cho E. J. Selective Debromination and α-Hydroxylation of α-Bromo Ketones Using Hantzsch Esters as Photoreductants. Adv. Synth. Catal. 2016, 358, 74–80. 10.1002/adsc.201500734. [DOI] [Google Scholar]
- Xu X.; Min Q. Q.; Li N.; Liu F. Visible Light-Promoted Umpolung Coupling of Aryl Tri-/Difluoroethanones with 2-Alkenylpyridines. Chem. Commun. 2018, 54, 11017–11020. 10.1039/C8CC06748A. [DOI] [PubMed] [Google Scholar]
- Crisenza G. E. M.; Mazzarella D.; Melchiorre P. Synthetic Methods Driven by the Photoactivity of Electron Donor-Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. 10.1021/jacs.0c01416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.-S.; Liu K.-T.; Su Y. O. Electrochemical Reduction of Substituted α,α,α-Trifluoroacetophenones. Linear Relationship between Cyclic Voltammetric Peak Potentials and Hammett Substituent Constants. J. Phys. Org. Chem. 1990, 3, 723–731. 10.1002/poc.610031105. [DOI] [Google Scholar]
- Seo H.; Jamison T. F. Catalytic Generation and Use of Ketyl Radical from Unactivated Aliphatic Carbonyl Compounds. Org. Lett. 2019, 21, 10159–10163. 10.1021/acs.orglett.9b04235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamy E.; Nadjo L.; Saveant J. M. Standard Potential and Kinetic Parameters of the Electrochemical Reduction of Carbon Dioxide in Dimethylformamide. J. Electroanal. Chem. Interfacial Electrochem. 1977, 78, 403–407. 10.1016/S0022-0728(77)80143-5. [DOI] [Google Scholar]
- Seo H.; Katcher M. H.; Jamison T. F. Photoredox Activation of Carbon Dioxide for Amino Acid Synthesis in Continuous Flow. Nat. Chem. 2017, 9, 453–456. 10.1038/nchem.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo H.; Liu A.; Jamison T. F. Direct β-Selective Hydrocarboxylation of Styrenes with CO2 Enabled by Continuous Flow Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 13969–13972. 10.1021/jacs.7b05942. [DOI] [PubMed] [Google Scholar]
- Cossy J.; Belotti D. Generation of Ketyl Radical Anions by Photoinduced Electron Transfer (PET) between Ketones and Amines. Synthetic Applications. Tetrahedron 2006, 62, 6459–6470. 10.1016/j.tet.2006.03.062. [DOI] [Google Scholar]
- Filardo G.; Gambino S.; Silvestri G.; Gennaro A.; Vianello E. Electrocarboxylation of Styrene through Homogeneous Redox Catalysis. J. Electroanal. Chem. Interfacial Electrochem. 1984, 177, 303–309. 10.1016/0022-0728(84)80232-6. [DOI] [Google Scholar]
- Phillips J. P.Rearrangements of Radical Anions Generated from Cyclopropyl Ketones. Ph.D. Thesis, Virginia Polytechnic Institute, 1998. [Google Scholar]
- Griller D.; Ingold K. U. Free-Radical Clocks. Acc. Chem. Res. 1980, 13 (9), 317–323. 10.1021/ar50153a004. [DOI] [Google Scholar]
- Nonhebel D. C. The Chemistry of Cyclopropylmethyl and Related Radicals. Chem. Soc. Rev. 1993, 22, 347–359. 10.1039/cs9932200347. [DOI] [Google Scholar]
- Tlahuext-Aca A.; Garza-Sanchez R. A.; Glorius F. Multicomponent Oxyalkylation of Styrenes Enabled by Hydrogen- Bond-Assisted Photoinduced Electron Transfer. Angew. Chem., Int. Ed. 2017, 56, 3708–3711. 10.1002/anie.201700049. [DOI] [PubMed] [Google Scholar]
- Reich D.; Trowbridge A.; Gaunt M. J. Rapid Syntheses of (−)-FR901483 and (+)-TAN1251C Enabled by Complexity-Generating Photocatalytic Olefin Hydroaminoalkylation. Angew. Chem., Int. Ed. 2020, 59, 2256–2261. 10.1002/anie.201912010. [DOI] [PubMed] [Google Scholar]
- Sakamoto K.; Tsujii E.; Abe F.; Nakanishi T.; Yamashita M.; Shigematsu N.; Izumi S.; Okuhara M. FR901483, a Novel Immunosuppressant Isolated from Cladobotryum Sp. No. 11231 Taxonomy of the Producing Organism, Fermentation, Isolation, Physico-Chemical Properties and Biological Activities. J. Antibiot. 1996, 49, 37–44. 10.7164/antibiotics.49.37. [DOI] [PubMed] [Google Scholar]
- Lefebvre Q.; Porta R.; Millet A.; Jia J.; Rueping M. One Amine-3 Tasks: Reductive Coupling of Imines with Olefins in Batch and Flow. Chem. - Eur. J. 2020, 26, 1363–1367. 10.1002/chem.201904483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van As D. J.; Connell T. U.; Brzozowski M.; Scully A. D.; Polyzos A. Photocatalytic and Chemoselective Transfer Hydrogenation of Diarylimines in Batch and Continuous Flow. Org. Lett. 2018, 20, 905–908. 10.1021/acs.orglett.7b03565. [DOI] [PubMed] [Google Scholar]
- Wang Z. S.; Chen Y. B.; Zhang H. W.; Sun Z.; Zhu C.; Ye L. W. Ynamide Smiles Rearrangement Triggered by Visible-Light-Mediated Regioselective Ketyl-Ynamide Coupling: Rapid Access to Functionalized Indoles and Isoquinolines. J. Am. Chem. Soc. 2020, 142, 3636–3644. 10.1021/jacs.9b13975. [DOI] [PubMed] [Google Scholar]
- Hossain A.; Pagire S. K.; Reiser O. Visible-Light-Mediated Synthesis of Pyrazines from Vinyl Azides Utilizing a Photocascade Process. Synlett 2017, 28, 1707–1714. 10.1055/s-0036-1590888. [DOI] [Google Scholar]
- Lu Z.; Yang Y. Q.; Li H. X. Photoinduced Aromatization of Dihydropyridines. Synthesis 2016, 48, 4221–4227. 10.1055/s-0035-1560562. [DOI] [Google Scholar]
- Deng Q. H.; Zou Y. Q.; Lu L. Q.; Tang Z. L.; Chen J. R.; Xiao W. J. De Novo Synthesis of Imidazoles by Visible-Light-Induced Photocatalytic Aerobic Oxidation/[3+2] Cycloaddition/Aromatization Cascade. Chem. - Asian J. 2014, 9, 2432–2435. 10.1002/asia.201402443. [DOI] [PubMed] [Google Scholar]
- Wang Z. S.; Chen Y. B.; Wang K.; Xu Z.; Ye L. W. One-Pot Synthesis of 2-Hydroxymethylindoles: Via Photoredox-Catalyzed Ketyl-Ynamide Coupling/1,3-Allylic Alcohol Transposition. Green Chem. 2020, 22, 4483–4488. 10.1039/D0GC01522A. [DOI] [Google Scholar]
- Robert-Banchereau E.; Lacombe S.; Ollivier J. Unsensitized Photooxidation of Sulfur Compounds with Molecular Oxygen in Solution. Tetrahedron 1997, 53, 2087–2102. 10.1016/S0040-4020(96)01158-1. [DOI] [Google Scholar]
- Machrouhi F.; Namy J. L. Samarium Diiodide/Nickel Diiodide an Efficient System for Homo and Heterocoupling Reactions of Imines. Tetrahedron Lett. 1999, 40, 1315–1318. 10.1016/S0040-4039(98)02673-2. [DOI] [Google Scholar]
- Takahashi M.; Micalizio G. C. Regio- and Stereoselective Cross-Coupling of Substituted Olefins and Imines. A Convergent Stereoselective Synthesis of Saturated 1,5-Aminoalcohols and Substituted Piperidines. J. Am. Chem. Soc. 2007, 129, 7514–7516. 10.1021/ja071974v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M.; Micalizio G. C. Concerning the Potential Reversibility of Carbometalation in Alkoxide-Directed Ti(Oi-Pr)4-Mediated Reductive Cross-Coupling of Homoallylic Alcohols with Aromatic Imines. Chem. Commun. 2010, 46, 3336–3338. 10.1039/b927430h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson G.; Py S.; Vallée Y. Samarium Diiodide-Induced Reductive Cross-Coupling of Nitrones with Aldehydes and Ketones. Angew. Chem., Int. Ed. 2002, 41, 1772–1775. . [DOI] [PubMed] [Google Scholar]
- Okamoto S.; Kojiyama K.; Tsujioka H.; Sudo A. Metal-Free Reductive Coupling of CO and CN Bonds Driven by Visible Light: Use of Perylene as a Simple Photoredox Catalyst. Chem. Commun. 2016, 52, 11339–11342. 10.1039/C6CC05867A. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Rouch W. D.; McCulla R. D. Conjugated Polymers as Photoredox Catalysts: Visible-Light-Driven Reduction of Aryl Aldehydes by Poly(p-Phenylene). Eur. J. Org. Chem. 2012, 2012 (31), 6187–6196. 10.1002/ejoc.201200437. [DOI] [Google Scholar]
- Miyake G. M.; Theriot J. C. Perylene as an Organic Photocatalyst for the Radical Polymerization of Functionalized Vinyl Monomers through Oxidative Quenching with Alkyl Bromides and Visible Light. Macromolecules 2014, 47, 8255–8261. 10.1021/ma502044f. [DOI] [Google Scholar]
- Okamoto S.; Ariki R.; Tsujioka H.; Sudo A. A Metal-Free Approach to 1,2-Diamines via Visible Light-Driven Reductive Coupling of Imines with Perylene as a Photoredox Catalyst. J. Org. Chem. 2017, 82, 9731–9736. 10.1021/acs.joc.7b01838. [DOI] [PubMed] [Google Scholar]
- Bartholomew R. F.; Brimage D. R. G.; Davidson R. S. The Photosensitised Oxidation of Amines. Part IV. The Use of Aromatic Hydrocarbons as Sensitisers. J. Chem. Soc. C 1971, (3482), 3482–3484. 10.1039/j39710003482. [DOI] [Google Scholar]
- Pischel U.; Zhang X.; Hellrung B.; Haselbach E.; Muller P. A.; Nau W. M. Fluorescence Quenching of n,π*-Excited Azoalkanes by Amines: What Is a Sterically Hindered Amine?. J. Am. Chem. Soc. 2000, 122, 2027–2034. 10.1021/ja992508b. [DOI] [Google Scholar]
- Parker V. D. Energetics of Electrode Reactions. II. The Relationship between Redox Potentials, Ionization Potentials, Electron Affinities, and Solvation Energies of Aromatic Hydrocarbons. J. Am. Chem. Soc. 1976, 98, 98–103. 10.1021/ja00417a017. [DOI] [Google Scholar]
- Zhu J.; Yuan Y.; Wang S.; Yao Z. J. Synthesis of 2,3-Dialkylated Tartaric Acid Esters via Visible Light Photoredox-Catalyzed Reductive Dimerization of α-Ketoesters. ACS Omega 2017, 2, 4665–4677. 10.1021/acsomega.7b00749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualandi A.; Rodeghiero G.; Della Rocca E.; Bertoni F.; Marchini M.; Perciaccante R.; Jansen T. P.; Ceroni P.; Cozzi P. G. Application of Coumarin Dyes for Organic Photoredox Catalysis. Chem. Commun. 2018, 54, 10044–10047. 10.1039/C8CC04048F. [DOI] [PubMed] [Google Scholar]
- Rong J.; Seeberger P. H.; Gilmore K. Chemoselective Photoredox Synthesis of Unprotected Primary Amines Using Ammonia. Org. Lett. 2018, 20, 4081–4085. 10.1021/acs.orglett.8b01637. [DOI] [PubMed] [Google Scholar]
- Caron A.; Morin E.; Collins S. K. Bifunctional Copper-Based Photocatalyst for Reductive Pinacol-Type Couplings. ACS Catal. 2019, 9, 9458–9464. 10.1021/acscatal.9b01718. [DOI] [Google Scholar]
- Christ P.; Lindsay A. G.; Vormittag S. S.; Neudörfl J. M.; Berkessel A.; O’Donoghue A. C. pKa Values of Chiral Brønsted Acid Catalysts: Phosphoric Acids/Amides, Sulfonyl/Sulfuryl Imides, and Perfluorinated TADDOLs (TEFDDOLs). Chem. - Eur. J. 2011, 17, 8524–8528. 10.1002/chem.201101157. [DOI] [PubMed] [Google Scholar]
- Alam M. J.; Alam O.; Alam P.; Naim M. J. A Review on Pyrazole Chemical Entity and Biological Activity. Int. J. Pharma Sci. Res. 2015, 6, 1433–1442. [Google Scholar]
- Vaǐpolin A. A.; Nikolaev Y. A.; Rud’ V. Y.; Rud’ Y. V.; Terukov E. I.; Fernelius N. Fabrication and Properties of Photosensitive Structures Based on ZnIn2S4 Single Crystals. Semiconductors 2003, 37, 178–182. 10.1134/1.1548661. [DOI] [Google Scholar]
- Ye L.; Li Z. ZnIn2S4: A Photocatalyst for the Selective Aerobic Oxidation of Amines to Imines under Visible Light. ChemCatChem 2014, 6, 2540–2543. 10.1002/cctc.201402360. [DOI] [Google Scholar]
- Chen Z.; Xu J.; Ren Z.; He Y.; Xiao G. High Efficient Photocatalytic Selective Oxidation of Benzyl Alcohol to Benzaldehyde by Solvothermal-Synthesized ZnIn2S4 Microspheres under Visible Light Irradiation. J. Solid State Chem. 2013, 205, 134–141. 10.1016/j.jssc.2013.07.015. [DOI] [Google Scholar]
- Han G.; Liu X.; Cao Z.; Sun Y. Photocatalytic Pinacol C-C Coupling and Jet Fuel Precursor Production on ZnIn2S4 Nanosheets. ACS Catal. 2020, 10, 9346–9355. 10.1021/acscatal.0c01715. [DOI] [Google Scholar]
- Mayeda E. A.; Miller L. L.; Wolf J. F. Electrooxidation of Benzylic Ethers, Esters, Alcohols, and Phenyl Epoxides. J. Am. Chem. Soc. 1972, 94, 6812–6816. 10.1021/ja00774a039. [DOI] [Google Scholar]
- Chalkley M. J.; Garrido-Barros P.; Peters J. C. A Molecular Mediator for Reductive Concerted Proton-Electron Transfers via Electrocatalysis. Science 2020, 369, 850–854. 10.1126/science.abc1607. [DOI] [PubMed] [Google Scholar]
- Chen M.; Zhao X.; Yang C.; Xia W. Visible-Light-Triggered Directly Reductive Arylation of Carbonyl/Iminyl Derivatives through Photocatalytic PCET. Org. Lett. 2017, 19, 3807–3810. 10.1021/acs.orglett.7b01677. [DOI] [PubMed] [Google Scholar]
- Lehnherr D.; Lam Y. H.; Nicastri M. C.; Liu J.; Newman J. A.; Regalado E. L.; Dirocco D. A.; Rovis T. Electrochemical Synthesis of Hindered Primary and Secondary Amines via Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2020, 142, 468–478. 10.1021/jacs.9b10870. [DOI] [PubMed] [Google Scholar]
- Dong J.; Wang Z.; Wang X.; Song H.; Liu Y.; Wang Q. Ketones and Aldehydes as Alkyl Radical Equivalents for Direct C-H Alkylation of Heteroarenes. Sci. Adv. 2019, 5, eaax9955. 10.1126/sciadv.aax9955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatgilialoglu C.; Crich D.; Komatsu M.; Ryu I. Chemistry of Acyl Radicals. Chem. Rev. 1999, 99, 1991–2070. 10.1021/cr9601425. [DOI] [PubMed] [Google Scholar]
- Kang Z.; Zhang D.; Hu W. Regio- and Diastereoselective Three-Component Reactions via Trapping of Ammonium Ylides with N-Alkylquinolinium Salts: Synthesis of Multisubstituted Tetra- and Dihydroquinoline Derivatives. Org. Lett. 2017, 19, 3783–3786. 10.1021/acs.orglett.7b01664. [DOI] [PubMed] [Google Scholar]
- Leitch J. A.; Rogova T.; Duarte F.; Dixon D. J. Dearomative Photocatalytic Construction of Bridged 1,3-Diazepanes. Angew. Chem., Int. Ed. 2020, 59, 4121–4130. 10.1002/anie.201914390. [DOI] [PubMed] [Google Scholar]
- Betori R. C.; Scheidt K. A. Reductive Arylation of Arylidene Malonates Using Photoredox Catalysis. ACS Catal. 2019, 9, 10350–10357. 10.1021/acscatal.9b03608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald B. R.; Scheidt K. A. Intermolecular Reductive Couplings of Arylidene Malonates via Lewis Acid/Photoredox Cooperative Catalysis. Org. Lett. 2018, 20, 6877–6881. 10.1021/acs.orglett.8b02893. [DOI] [PubMed] [Google Scholar]
- Betori R. C.; McDonald B. R.; Scheidt K. A. Reductive Annulations of Arylidene Malonates with Unsaturated Electrophiles Using Photoredox/Lewis Acid Cooperative Catalysis. Chem. Sci. 2019, 10, 3353–3359. 10.1039/C9SC00302A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P. P.; Srivastava V. Recent Advances in Using 4DPAIPN in Photocatalytic Transformations. Org. Biomol. Chem. 2021, 19, 313–321. 10.1039/D0OB01884H. [DOI] [PubMed] [Google Scholar]
- Mcdevitt P.; Vittimberga B. M. The Electron Transfer Reactions of Cyano Substituted Pyridines and Quinolines with Thermally Generated Diphenyl Ketyl. J. Heterocycl. Chem. 1990, 27, 1903–1908. 10.1002/jhet.5570270711. [DOI] [Google Scholar]
- Kolthoff I. M.; Chantooni M. K.; Bhowmik S. Dissociation Constants of Uncharged and Monovalent Cation Acids in Dimethyl Sulfoxide. J. Am. Chem. Soc. 1968, 90, 23–28. 10.1021/ja01003a005. [DOI] [Google Scholar]
- Leifert D.; Studer A. The Persistent Radical Effect in Organic Synthesis. Angew. Chem., Int. Ed. 2020, 59, 74–108. 10.1002/anie.201903726. [DOI] [PubMed] [Google Scholar]
- Jeffrey J. L.; Petronijević F. R.; Macmillan D. W. C. Selective Radical-Radical Cross-Couplings: Design of a Formal β-Mannich Reaction. J. Am. Chem. Soc. 2015, 137, 8404–8407. 10.1021/jacs.5b05376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Dong G. Catalytic β-Functionalization of Carbonyl Compounds Enabled by α,β-Desaturation. ACS Catal. 2020, 10, 6058–6070. 10.1021/acscatal.0c01519. [DOI] [Google Scholar]
- Schoeller W. W.; Niemann J.; Rademacher P. On the Electrochemical Oxidation of Enamines. J. Chem. Soc., Perkin Trans. 2 1988, (3), 369–373. 10.1039/p29880000369. [DOI] [Google Scholar]
- Pirnot M. T.; Rankic D. A.; Martin D. B. C.; MacMillan D. W. C. Photoredox Activation for the Direct β-Arylation of Ketones and Aldehydes. Science 2013, 339, 1593–1596. 10.1126/science.1232993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrett J. A.; Clift M. D.; MaCmillan D. W. C. Direct β-Alkylation of Aldehydes via Photoredox Organocatalysis. J. Am. Chem. Soc. 2014, 136, 6858–6861. 10.1021/ja502639e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronijević F. R.; Nappi M.; MacMillan D. W. C. Direct β-Functionalization of Cyclic Ketones with Aryl Ketones via the Merger of Photoredox and Organocatalysis. J. Am. Chem. Soc. 2013, 135, 18323–18326. 10.1021/ja410478a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHovitz J. S.; Loh Y. Y.; Kautzky J. A.; Nagao K.; Meichan A. J.; Yamauchi M.; MacMillan D. W. C.; Hyster T. K. Static to Inducibly Dynamic Stereocontrol: The Convergent Use of Racemic β-Substituted Ketones. Science 2020, 369, 1113–1118. 10.1126/science.abc9909. [DOI] [PubMed] [Google Scholar]
- Li W.; Duan Y.; Zhang M.; Cheng J.; Zhu C. A Photoredox Catalyzed Radical-Radical Coupling Reaction: Facile Access to Multi-Substituted Nitrogen Heterocycles. Chem. Commun. 2016, 52, 7596–7599. 10.1039/C6CC02027E. [DOI] [PubMed] [Google Scholar]
- Xia Q.; Tian H.; Dong J.; Qu Y.; Li L.; Song H.; Liu Y.; Wang Q. N-Arylamines Coupled with Aldehydes, Ketones, and Imines by Means of Photocatalytic Proton-Coupled Electron Transfer. Chem. - Eur. J. 2018, 24, 9269–9273. 10.1002/chem.201801886. [DOI] [PubMed] [Google Scholar]
- Willms J. A.; Gleich H.; Schrempp M.; Menche D.; Engeser M. Investigations of the Copper-Catalyzed Oxidative Cross-Coupling of Tetrahydroisoquinolines with Diethylzinc by a Combination of Mass Spectrometric and Electrochemical Methods. Chem. - Eur. J. 2018, 24, 2663–2668. 10.1002/chem.201704914. [DOI] [PubMed] [Google Scholar]
- Tambe S. D.; Min K. H.; Iqbal N.; Cho E. J. Distinctive Reactivity of N-Benzylidene-[1,1’-Biphenyl]-2-Amines under Photoredox Conditions. Beilstein J. Org. Chem. 2020, 16, 1335–1342. 10.3762/bjoc.16.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N. R.; Kelly C. B.; Siegenfeld A. P.; Molander G. A. Mild, Redox-Neutral Alkylation of Imines Enabled by an Organic Photocatalyst. ACS Catal. 2017, 7, 1766–1770. 10.1021/acscatal.6b03665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. H.; Yu S. Visible-Light-Induced Radical Acylation of Imines with α-Ketoacids Enabled by Electron-Donor-Acceptor Complexes. Org. Lett. 2019, 21, 3711–3715. 10.1021/acs.orglett.9b01169. [DOI] [PubMed] [Google Scholar]
- Yang S.; Zhu S.; Lu D.; Gong Y. Formylation of Fluoroalkyl Imines through Visible-Light-Enabled H-Atom Transfer Catalysis: Access to Fluorinated α-Amino Aldehydes. Org. Lett. 2019, 21, 2019–2024. 10.1021/acs.orglett.9b00128. [DOI] [PubMed] [Google Scholar]
- Uraguchi D.; Kinoshita N.; Kizu T.; Ooi T. Synergistic Catalysis of Ionic Brønsted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines. J. Am. Chem. Soc. 2015, 137, 13768–13771. 10.1021/jacs.5b09329. [DOI] [PubMed] [Google Scholar]
- Pan S.; Jiang M.; Hu J.; Xu R.; Zeng X.; Zhong G. Synthesis of 1,2-Amino Alcohols by Decarboxylative Coupling of Amino Acid Derived α-Amino Radicals to Carbonyl Compounds: Via Visible-Light Photocatalyst in Water. Green Chem. 2020, 22, 336–341. 10.1039/C9GC03470F. [DOI] [Google Scholar]
- Marangoni D. G.; Smith R. S.; Roscoe S. G. Surface Electrochemistry of the Oxidation of Glycine at Pt. Can. J. Chem. 1989, 67, 921–926. 10.1139/v89-141. [DOI] [Google Scholar]
- Zhang L.; Chu Y.; Ma P.; Zhao S.; Li Q.; Chen B.; Hong X.; Sun J. Visible-Light-Mediated Photocatalytic Cross-Coupling of Acetenyl Ketones with Benzyl Trifluoroborate. Org. Biomol. Chem. 2020, 18, 1073–1077. 10.1039/C9OB02624J. [DOI] [PubMed] [Google Scholar]
- Yasu Y.; Koike T.; Akita M. Visible Light-Induced Selective Generation of Radicals from Organoborates by Photoredox Catalysis. Adv. Synth. Catal. 2012, 354, 3414–3420. 10.1002/adsc.201200588. [DOI] [Google Scholar]
- Tellis J. C.; Primer D. N.; Molander G. A. Single-Electron Transmetalation in Organoboron Cross-Coupling by Photoredox/Nickel Dual Catalysis. Science 2014, 345, 433–436. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. Single-Electron Transmetalation: An Enabling Technology for Secondary Alkylboron Cross-Coupling. J. Am. Chem. Soc. 2015, 137, 2195–2198. 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui J. K.; Lang S. B.; Heitz D. R.; Molander G. A. Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development. ACS Catal. 2017, 7, 2563–2575. 10.1021/acscatal.7b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chciuk T. V.; Anderson W. R.; Flowers R. A. Proton-Coupled Electron Transfer in the Reduction of Carbonyls by Samarium Diiodide-Water Complexes. J. Am. Chem. Soc. 2016, 138, 8738–8741. 10.1021/jacs.6b05879. [DOI] [PubMed] [Google Scholar]
- Connell T. U.; Fraser C. L.; Czyz M. L.; Smith Z. M.; Hayne D. J.; Doeven E. H.; Agugiaro J.; Wilson D. J. D.; Adcock J. L.; Scully A. D.; et al. The Tandem Photoredox Catalysis Mechanism of [Ir(ppy)2(dtbbpy)]+ Enabling Access to Energy Demanding Organic Substrates. J. Am. Chem. Soc. 2019, 141, 17646–17658. 10.1021/jacs.9b07370. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Cornella J.; Juliá-Hernández F.; Martin R. Visible-Light-Promoted Atom Transfer Radical Cyclization of Unactivated Alkyl Iodides. ACS Catal. 2017, 7, 409–412. 10.1021/acscatal.6b03205. [DOI] [Google Scholar]
- Forni J. A.; Micic N.; Connell T. U.; Weragoda G.; Polyzos A. Tandem Photoredox Catalysis: Enabling Carbonylative Amidation of Aryl and Alkylhalides. Angew. Chem., Int. Ed. 2020, 59, 18646–18654. 10.1002/anie.202006720. [DOI] [PubMed] [Google Scholar]
- Czyz M. L.; Taylor M. S.; Horngren T. H.; Polyzos A. Reductive Activation and Hydrofunctionalization of Olefins by Multiphoton Tandem Photoredox Catalysis. ACS Catal. 2021, 11, 5472–5480. 10.1021/acscatal.1c01000. [DOI] [Google Scholar]
- Guo X.; Wenger O. S. Reductive Amination by Photoredox Catalysis and Polarity-Matched Hydrogen Atom Transfer. Angew. Chem., Int. Ed. 2018, 57, 2469–2473. 10.1002/anie.201711467. [DOI] [PubMed] [Google Scholar]
- Xi Z. W.; Yang L.; Wang D. Y.; Pu C. D.; Shen Y. M.; Wu C. De.; Peng X. G. Visible-Light Photocatalytic Synthesis of Amines from Imines via Transfer Hydrogenation Using Quantum Dots as Catalysts. J. Org. Chem. 2018, 83, 11886–11895. 10.1021/acs.joc.8b01651. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Zheng W.; Zhu D.; Chang H. Theoretical Modeling of pKa’s of Thiol Compounds in Aqueous Solution. New J. Chem. 2019, 43, 5239–5254. 10.1039/C8NJ06259E. [DOI] [Google Scholar]
- Koh K.; Sanyal U.; Lee M. S.; Cheng G.; Song M.; Glezakou V. A.; Liu Y.; Li D.; Rousseau R.; Gutiérrez O. Y.; et al. Electrochemically Tunable Proton-Coupled Electron Transfer in Pd-Catalyzed Benzaldehyde Hydrogenation. Angew. Chem., Int. Ed. 2020, 59, 1501–1505. 10.1002/anie.201912241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murarka S. N-(Acyloxy)Phthalimides as Redox-Active Esters in Cross-Coupling Reactions. Adv. Synth. Catal. 2018, 360, 1735–1753. 10.1002/adsc.201701615. [DOI] [Google Scholar]
- de Sarkar S.; Murarka S.; Parida S. K.; Mandal T.; Das S.; Hota S. K. Single Electron Transfer-Induced Redox Processes Involving N-(Acyloxy)Phthalimides. ACS Catal. 2021, 11, 1640–1683. 10.1021/acscatal.0c04756. [DOI] [Google Scholar]
- Okada K.; Okamoto K.; Oda M. A New and Practical Method of Decarboxylation: Photosensitized Decarboxylation of N-Acyloxyphthalimides via Electron-Transfer Mechanism. J. Am. Chem. Soc. 1988, 110, 8736–8738. 10.1021/ja00234a047. [DOI] [Google Scholar]
- Okada K.; Okubo K.; Morita N.; Oda M. Reductive Decarboxylation of N-(Acyloxy)Phthalimides via Redox-Initiated Radical Chain Mechanism. Tetrahedron Lett. 1992, 33, 7377–7380. 10.1016/S0040-4039(00)60192-2. [DOI] [Google Scholar]
- Okada K.; Okamoto K.; Oda M. Photochemical Chlorodecarboxylation via an Electron Transfer Mechanism. J. Chem. Soc., Chem. Commun. 1989, 20, 1636–1637. 10.1039/c39890001636. [DOI] [Google Scholar]
- Okada K.; Okamoto K.; Morita N.; Okubo K.; Oda M. Photosensitized Decarboxylative Michael Addition through N-(Acyloxy)Phthalimides via an Electron-Transfer Mechanism. J. Am. Chem. Soc. 1991, 113, 9401–9402. 10.1021/ja00024a074. [DOI] [Google Scholar]
- Okada K.; Okubo K.; Morita N.; Oda M. Redox-Mediated Decarboxylative Photo-Phenylselenenylation of N-Acyloxyphthalimides. Chem. Lett. 1993, 22, 2021–2024. 10.1246/cl.1993.2021. [DOI] [Google Scholar]
- Dedeian K.; Djurovich P. I.; Garces F. O.; Carlson G.; Watts R. J. A New Synthetic Route to the Preparation of a Series of Strong Photoreducing Agents: Fac Tris-Ortho-Metalated Complexes of Iridium(III) with Substituted 2-Phenylpyridines. Inorg. Chem. 1991, 30, 1685–1687. 10.1021/ic00008a003. [DOI] [Google Scholar]
- Marcus Y. The Effectiveness of Solvents as Hydrogen Bond Donors. J. Solution Chem. 1991, 20, 929–944. 10.1007/BF01074953. [DOI] [Google Scholar]
- Sha W.; Ni S.; Han J.; Pan Y. Access to Alkyl-Substituted Lactone via Photoredox-Catalyzed Alkylation/Lactonization of Unsaturated Carboxylic Acids. Org. Lett. 2017, 19, 5900–5903. 10.1021/acs.orglett.7b02899. [DOI] [PubMed] [Google Scholar]
- Kong W.; Yu C.; An H.; Song Q. Photoredox-Catalyzed Decarboxylative Alkylation of Silyl Enol Ethers to Synthesize Functionalized Aryl Alkyl Ketones. Org. Lett. 2018, 20, 349–352. 10.1021/acs.orglett.7b03587. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Chen J. R.; Xiao W. J. Visible-Light Photocatalytic Decarboxylative Alkyl Radical Addition Cascade for Synthesis of Benzazepine Derivatives. Org. Lett. 2018, 20, 224–227. 10.1021/acs.orglett.7b03588. [DOI] [PubMed] [Google Scholar]
- Balfour J. A.; Goa K. L. Benazepril: A Review of Its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Efficacy in Hypertension and Congestive Heart Failure. Drugs 1991, 42, 511–539. 10.2165/00003495-199142030-00008. [DOI] [PubMed] [Google Scholar]
- Kaski J. C.; Gloekler S.; Ferrari R.; Fox K.; Lévy B. I.; Komajda M.; Vardas P.; Camici P. G. Role of Ivabradine in Management of Stable Angina in Patients with Different Clinical Profiles. Open Hear. 2018, 5, e000725 10.1136/openhrt-2017-000725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur K.; Kaushal S.; Chopra S. C. Varenicline for Smoking Cessation: A Review of the Literature. Curr. Ther. Res. 2009, 70, 35–54. 10.1016/j.curtheres.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia J. T. M.; Piva da Silva G.; André E.; Paixão M. W. Photoredox Decarboxylative Alkylation/(2+2+1) Cycloaddition of 1,7-Enynes: A Cascade Approach Towards Polycyclic Heterocycles Using N-(Acyloxy)Phthalimides as Radical Source. Adv. Synth. Catal. 2019, 361, 5558–5564. 10.1002/adsc.201900657. [DOI] [Google Scholar]
- Curran D. P.; Shen W. Radical Translocation Reactions of Vinyl Radicals: Substituent Effects on 1,5-Hydrogen-Transfer Reactions. J. Am. Chem. Soc. 1993, 115, 6051–6059. 10.1021/ja00067a021. [DOI] [Google Scholar]
- Jiao M. J.; Liu D.; Hu X. Q.; Xu P. F. Photocatalytic Decarboxylative [2+2+1] Annulation of 1,6-Enynes with: N-Hydroxyphthalimide Esters for the Synthesis of Indene-Containing Polycyclic Compounds. Org. Chem. Front. 2019, 6, 3834–3838. 10.1039/C9QO01166H. [DOI] [Google Scholar]
- Duffy M. P.; Delaunay W.; Bouit P. A.; Hissler M. π-Conjugated Phospholes and Their Incorporation into Devices: Components with a Great Deal of Potential. Chem. Soc. Rev. 2016, 45, 5296–5310. 10.1039/C6CS00257A. [DOI] [PubMed] [Google Scholar]
- Liu L.; Dong J.; Yan Y.; Yin S. F.; Han L. B.; Zhou Y. Photoredox-Catalyzed Decarboxylative Alkylation/Cyclization of Alkynylphosphine Oxides: A Metal- and Oxidant-Free Method for Accessing Benzo[b]Phosphole Oxides. Chem. Commun. 2019, 55, 233–236. 10.1039/C8CC08689C. [DOI] [PubMed] [Google Scholar]
- Sherwood T. C.; Xiao H. Y.; Bhaskar R. G.; Simmons E. M.; Zaretsky S.; Rauch M. P.; Knowles R. R.; Dhar T. G. M. Decarboxylative Intramolecular Arene Alkylation Using N-(Acyloxy)Phthalimides, an Organic Photocatalyst, and Visible Light. J. Org. Chem. 2019, 84, 8360–8379. 10.1021/acs.joc.9b00432. [DOI] [PubMed] [Google Scholar]
- Kachkovskyi G.; Faderl C.; Reiser O. Visible Light-Mediated Synthesis of (Spiro)Anellated Furans. Adv. Synth. Catal. 2013, 355, 2240–2248. 10.1002/adsc.201300221. [DOI] [Google Scholar]
- Faderl C.; Budde S.; Kachkovskyi G.; Rackl D.; Reiser O. Visible Light-Mediated Decarboxylation Rearrangement Cascade of Aryl-N-(Acyloxy)Phthalimides. J. Org. Chem. 2018, 83, 12192–12206. 10.1021/acs.joc.8b01538. [DOI] [PubMed] [Google Scholar]
- Cheng W. M.; Shang R.; Fu Y. Photoredox/Brønsted Acid Co-Catalysis Enabling Decarboxylative Coupling of Amino Acid and Peptide Redox-Active Esters with N-Heteroarenes. ACS Catal. 2017, 7, 907–911. 10.1021/acscatal.6b03215. [DOI] [Google Scholar]
- Cheng W. M.; Shang R.; Fu M. C.; Fu Y. Photoredox-Catalysed Decarboxylative Alkylation of N-Heteroarenes with N-(Acyloxy)Phthalimides. Chem. - Eur. J. 2017, 23, 2537–2541. 10.1002/chem.201605640. [DOI] [PubMed] [Google Scholar]
- Proctor R. S. J.; Davis H. J.; Phipps R. J. Catalytic Enantioselective Minisci-Type Addition to Heteroarenes. Science 2018, 360, 419–422. 10.1126/science.aar6376. [DOI] [PubMed] [Google Scholar]
- Sherwood T. C.; Li N.; Yazdani A. N.; Dhar T. G. M. Organocatalyzed, Visible-Light Photoredox-Mediated, One-Pot Minisci Reaction Using Carboxylic Acids via N-(Acyloxy)Phthalimides. J. Org. Chem. 2018, 83, 3000–3012. 10.1021/acs.joc.8b00205. [DOI] [PubMed] [Google Scholar]
- Papaioannou N.; Fray M. J.; Rennhack A.; Sanderson T. J.; Stokes J. E. Regioselective Amidomethylation of 4-Chloro-3-Fluoropyridine by Metalation and Minisci-Type Reactions. J. Org. Chem. 2020, 85, 12067–12079. 10.1021/acs.joc.0c01168. [DOI] [PubMed] [Google Scholar]
- Jin C.; Yan Z.; Sun B.; Yang J. Visible-Light-Induced Regioselective Alkylation of Coumarins via Decarboxylative Coupling with N-Hydroxyphthalimide Esters. Org. Lett. 2019, 21, 2064–2068. 10.1021/acs.orglett.9b00327. [DOI] [PubMed] [Google Scholar]
- Yan Z.; Sun B.; Zhang X.; Zhuang X.; Yang J.; Su W.; Jin C. Construction of C(sp2)-C(sp3) Bond between Quinoxalin-2(1H)-Ones and N-Hydroxyphthalimide Esters via Photocatalytic Decarboxylative Coupling. Chem. - Asian J. 2019, 14, 3344–3349. 10.1002/asia.201900904. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Xu J.; Zhou M.; Zhao J.; Zhang P.; Li W. The Visible-Light-Triggered Regioselective Alkylation of Quinoxalin-2(1H)-Ones via Decarboxylation Coupling. Org. Biomol. Chem. 2019, 17, 10201–10208. 10.1039/C9OB02203A. [DOI] [PubMed] [Google Scholar]
- Ren L.; Cong H. Visible-Light-Driven Decarboxylative Alkylation of C-H Bond Catalyzed by Dye-Sensitized Semiconductor. Org. Lett. 2018, 20, 3225–3228. 10.1021/acs.orglett.8b01077. [DOI] [PubMed] [Google Scholar]
- Boyington A. J.; Riu M. L. Y.; Jui N. T. Anti-Markovnikov Hydroarylation of Unactivated Olefins via Pyridyl Radical Intermediates. J. Am. Chem. Soc. 2017, 139, 6582–6585. 10.1021/jacs.7b03262. [DOI] [PubMed] [Google Scholar]
- Zhou W. J.; Cao G. M.; Shen G.; Zhu X. Y.; Gui Y. Y.; Ye J. H.; Sun L.; Liao L. L.; Li J.; Yu D. G. Visible-Light-Driven Palladium-Catalyzed Radical Alkylation of C-H Bonds with Unactivated Alkyl Bromides. Angew. Chem., Int. Ed. 2017, 56, 15683–15687. 10.1002/anie.201704513. [DOI] [PubMed] [Google Scholar]
- Chan C. M.; Xing Q.; Chow Y. C.; Hung S. F.; Yu W. Y. Photoredox Decarboxylative C(sp3)-N Coupling of α-Diazoacetates with Alkyl N-Hydroxyphthalimide Esters for Diversified Synthesis of Functionalized N-Alkyl Hydrazones. Org. Lett. 2019, 21, 8037–8043. 10.1021/acs.orglett.9b03020. [DOI] [PubMed] [Google Scholar]
- He J.; Chen G.; Zhang B.; Li Y.; Chen J. R.; Xiao W. J.; Liu F.; Li C. Catalytic Decarboxylative Radical Sulfonylation. Chem. 2020, 6, 1149–1159. 10.1016/j.chempr.2020.02.003. [DOI] [Google Scholar]
- Shang T. Y.; Lu L. H.; Cao Z.; Liu Y.; He W. M.; Yu B. Recent Advances of 1,2,3,5-Tetrakis(Carbazol-9-yl)-4,6-Dicyanobenzene (4CzIPN) in Photocatalytic Transformations. Chem. Commun. 2019, 55, 5408–5419. 10.1039/C9CC01047E. [DOI] [PubMed] [Google Scholar]
- Smith J. M.; Dixon J. A.; Degruyter J. N.; Baran P. S. Alkyl Sulfinates: Radical Precursors Enabling Drug Discovery. J. Med. Chem. 2019, 62, 2256–2264. 10.1021/acs.jmedchem.8b01303. [DOI] [PubMed] [Google Scholar]
- Knauber T.; Chandrasekaran R.; Tucker J. W.; Chen J. M.; Reese M.; Rankic D. A.; Sach N.; Helal C. Ru/Ni Dual Catalytic Desulfinative Photoredox C(sp2)-C(sp3) Cross-Coupling of Alkyl Sulfinate Salts and Aryl Halides. Org. Lett. 2017, 19, 6566–6569. 10.1021/acs.orglett.7b03280. [DOI] [PubMed] [Google Scholar]
- Yan M.; Lo J. C.; Edwards J. T.; Baran P. S. Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc. 2016, 138, 12692–12714. 10.1021/jacs.6b08856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro E. J.; Kummerle A. E.; Fraga C. A. M. The Methylation Effect in Medicinal Chemistry. Chem. Rev. 2011, 111, 5215–5246. 10.1021/cr200060g. [DOI] [PubMed] [Google Scholar]
- Schönherr H.; Cernak T. Profound Methyl Effects in Drug Discovery and a Call for New C-H Methylation Reactions. Angew. Chem., Int. Ed. 2013, 52, 12256–12267. 10.1002/anie.201303207. [DOI] [PubMed] [Google Scholar]
- Cernak T.; Dykstra K. D.; Tyagarajan S.; Vachal P.; Krska S. W. The Medicinal Chemist’s Toolbox for Late Stage Functionalization of Drug-like Molecules. Chem. Soc. Rev. 2016, 45, 546–576. 10.1039/C5CS00628G. [DOI] [PubMed] [Google Scholar]
- Duncton M. A. J. Minisci Reactions: Versatile CH-Functionalizations for Medicinal Chemists. MedChemComm 2011, 2, 1135–1161. 10.1039/c1md00134e. [DOI] [Google Scholar]
- Krska S. W.; DiRocco D. A.; Dreher S. D.; Shevlin M. The Evolution of Chemical High-Throughput Experimentation to Address Challenging Problems in Pharmaceutical Synthesis. Acc. Chem. Res. 2017, 50, 2976–2985. 10.1021/acs.accounts.7b00428. [DOI] [PubMed] [Google Scholar]
- Collins K. D.; Gensch T.; Glorius F. Contemporary Screening Approaches to Reaction Discovery and Development. Nat. Chem. 2014, 6, 859–871. 10.1038/nchem.2062. [DOI] [PubMed] [Google Scholar]
- Mennen S. M.; Alhambra C.; Allen C. L.; Barberis M.; Berritt S.; Brandt T. A.; Campbell A. D.; Castañón J.; Cherney A. H.; Christensen M.; et al. The Evolution of High-Throughput Experimentation in Pharmaceutical Development and Perspectives on the Future. Org. Process Res. Dev. 2019, 23, 1213–1242. 10.1021/acs.oprd.9b00140. [DOI] [Google Scholar]
- DiRocco D. A.; Dykstra K.; Krska S.; Vachal P.; Conway D. V.; Tudge M. Late-Stage Functionalization of Biologically Active Heterocycles through Photoredox Catalysis. Angew. Chem., Int. Ed. 2014, 53, 4802–4806. 10.1002/anie.201402023. [DOI] [PubMed] [Google Scholar]
- Morlet-Savary F.; Wieder F.; Fouassier J. P. Sensitized Dissociation of Peroxides and Peresters in the Presence of Thiopyrylium Salts. J. Chem. Soc., Faraday Trans. 1997, 93, 3931–3937. 10.1039/a704951j. [DOI] [Google Scholar]
- Komai T.; Matsuyama K.; Matsushima M. Homolytic Decomposition of T-Alkyl 2,2-Dimethylperoxypropionates. Bull. Chem. Soc. Jpn. 1988, 61, 1641–1646. 10.1246/bcsj.61.1641. [DOI] [Google Scholar]
- Huff C. A.; Cohen R. D.; Dykstra K. D.; Streckfuss E.; DiRocco D. A.; Krska S. W. Photoredox-Catalyzed Hydroxymethylation of Heteroaromatic Bases. J. Org. Chem. 2016, 81, 6980–6987. 10.1021/acs.joc.6b00811. [DOI] [PubMed] [Google Scholar]
- Baron R.; Darchen A.; Hauchard D. Electrode Reaction Mechanisms for the Reduction of Tert-Butyl Peracetate, Lauryl Peroxide and Dibenzoyl Peroxide. Electrochim. Acta 2006, 51, 1336–1341. 10.1016/j.electacta.2005.06.022. [DOI] [Google Scholar]
- Yayla H. G.; Peng F.; Mangion I. K.; McLaughlin M.; Campeau L. C.; Davies I. W.; Dirocco D. A.; Knowles R. R. Discovery and Mechanistic Study of a Photocatalytic Indoline Dehydrogenation for the Synthesis of Elbasvir. Chem. Sci. 2016, 7, 2066–2073. 10.1039/C5SC03350K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Xia Q.; Lv X.; Yan C.; Song H.; Liu Y.; Wang Q. Photoredox-Mediated Direct Cross-Dehydrogenative Coupling of Heteroarenes and Amines. Org. Lett. 2018, 20, 5661–5665. 10.1021/acs.orglett.8b02389. [DOI] [PubMed] [Google Scholar]
- Jin J.; MacMillan D. W. C. Direct α-Arylation of Ethers through the Combination of Photoredox-Mediated C-H Functionalization and the Minisci Reaction. Angew. Chem., Int. Ed. 2015, 54, 1565–1569. 10.1002/anie.201410432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Dong J.; Hao Y.; Li Y.; Liu Y.; Song H.; Wang Q. Photoredox-Mediated Minisci C-H Alkylation Reactions between N-Heteroarenes and Alkyl Iodides with Peroxyacetate as a Radical Relay Initiator. J. Org. Chem. 2019, 84, 16245–16253. 10.1021/acs.joc.9b02848. [DOI] [PubMed] [Google Scholar]
- Hawari J. A.; Kanabus-Kaminska J. M.; Wayner D. D. M.; Griller D.. Substituent Effects on the Thermochemistry and Electrochemistry of Free Radicals. Substituent Effects in Radical Chemistry; Springer: Netherlands, 1986; pp 91–105. [Google Scholar]
- Klopman G.; Doddapaneni N. Electrochemical Behavior of Cis and Trans Azobenzenes. J. Phys. Chem. 1974, 78, 1825–1828. 10.1021/j100611a011. [DOI] [Google Scholar]
- White J. R.; Bard A. J. Electrochemical Investigation of Photocatalysis at CdS Suspensions in the Presence of Methylviologen. J. Phys. Chem. 1985, 89, 1947–1954. 10.1021/j100256a027. [DOI] [Google Scholar]
- Reinheimer A.; Van Eldik R.; Kisch H. On the Mechanism of Radical C-N Coupling in Type B Semiconductor Photocatalysis: A High-Pressure Study. J. Phys. Chem. B 2000, 104, 1014–1024. 10.1021/jp992702k. [DOI] [Google Scholar]
- Hopfner M.; Weiß H.; Meissner D.; Heinemann F. W.; Kisch H. Semiconductor Photocatalysis Type B: Synthesis of Unsaturated α-Amino Esters from Imines and Olefins Photocatalyzed by Silica-Supported Cadmium Sulfide. Photochem. Photobiol. Sci. 2002, 1, 696–703. 10.1039/B204569A. [DOI] [PubMed] [Google Scholar]
- Keck H.; Schindler W.; Knoch F.; Kisch H. Type B Semiconductor Photocatalysis: The Synthesis of Homoallyl Amines by Cadmium Sulfide-Catalyzed Linear Photoaddition of Olefins and Enol/Allyl Ethers to N-Phenylbenzophenone Imine. Chem. - Eur. J. 1997, 3, 1638–1645. 10.1002/chem.19970031013. [DOI] [Google Scholar]
- Schindler W.; Kisch H. Heterogeneous Photocatalysis XV. Mechanistic Aspects of Cadmium Sulfide-Catalyzed Photoaddition of Olefins to Schiff Bases. J. Photochem. Photobiol., A 1997, 103, 257–264. 10.1016/S1010-6030(96)04556-X. [DOI] [Google Scholar]
- Gärtner M.; Ballmann J.; Damm C.; Heinemann F. W.; Kisch H. Support-Controlled Chemoselective Olefin-Imine Addition Photocatalyzed by Cadmium Sulfide on a Zinc Sulfide Carrier. Photochem. Photobiol. Sci. 2007, 6, 159–164. 10.1039/B616070K. [DOI] [PubMed] [Google Scholar]
- Pehlivanugullari H. C.; Sumer E.; Kisch H. Semiconductor Photocatalysis Type B: Synthesis of Unsaturated α-Cyano-Homoallylamines from Imines and Olefins Photocatalysed by Silica- And Cellulose-Supported Cadmium Sulphide. Res. Chem. Intermed. 2007, 33, 297–309. 10.1163/156856707779238595. [DOI] [Google Scholar]
- Aldemir M.; Heinemann F. W.; Kisch H. Photochemical Synthesis of N-Adamantylhomoallylamines through Addition of Cyclic Olefins to Imines Catalyzed by Alumina Grafted Cadmium Sulfide. Photochem. Photobiol. Sci. 2012, 11, 908–913. 10.1039/C1PP05298E. [DOI] [PubMed] [Google Scholar]
- Su Y. L.; Liu G. X.; Liu J. W.; Tram L.; Qiu H.; Doyle M. P. Radical-Mediated Strategies for the Functionalization of Alkenes with Diazo Compounds. J. Am. Chem. Soc. 2020, 142, 13846–13855. 10.1021/jacs.0c05183. [DOI] [PubMed] [Google Scholar]
- Brookhart M.; Studabaker W. B. Cyclopropanes from Reactions of Transition-Metal-Carbene Complexes with Olefins. Chem. Rev. 1987, 87, 411–432. 10.1021/cr00078a008. [DOI] [Google Scholar]
- Doyle M. P.; Duffy R.; Ratnikov M.; Zhou L. Catalytic Carbene Insertion into C-H Bonds. Chem. Rev. 2010, 110, 704–724. 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]
- Kadam H. K.; Tilve S. G. Advancement in Methodologies for Reduction of Nitroarenes. RSC Adv. 2015, 5, 83391–83407. 10.1039/C5RA10076C. [DOI] [Google Scholar]
- Fukuzumi S.; Tokuda Y. Efficient Six-Electron Photoreduction of Nitrobenzene Derivatives by 10-Methyl-9,10-Dihydroacridine in the Presence of Perchloric Acid. Bull. Chem. Soc. Jpn. 1992, 65, 831–836. 10.1246/bcsj.65.831. [DOI] [Google Scholar]
- Fukuzumi S.; Chiba M.; Tanaka T. Acid-Catalysed Multi-Electron Reduction of Nitrobenzene Derivatives by a Dihydronicotinamide Adenine Dinucleotide (NADH) Model Compound, 9,10-Dihydro-10-Methylacridine. J. Chem. Soc., Chem. Commun. 1989, (14), 941–943. 10.1039/c39890000941. [DOI] [Google Scholar]
- Fukuzumi S.; Chiba M. Multi-Electron Reduction of Nitrobenzene Derivatives by an Acid-Stable NADH Analogue via Acid-Catalysed Electron Transfer Radical-Chain Reactions. J. Chem. Soc., Perkin Trans. 2 1991, (9), 1393–1398. 10.1039/p29910001393. [DOI] [Google Scholar]
- Todorov A. R.; Aikonen S.; Muuronen M.; Helaja J. Visible-Light-Photocatalyzed Reductions of N-Heterocyclic Nitroaryls to Anilines Utilizing Ascorbic Acid Reductant. Org. Lett. 2019, 21, 3764–3768. 10.1021/acs.orglett.9b01205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery E. L.; Chatterjee A. K.; Winzeler E. A. Antimalarial Drug Discovery — Approaches and Progress towards New Medicines. Nat. Rev. Microbiol. 2013, 11, 849–862. 10.1038/nrmicro3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. J.; Chen B.; Zheng L. Q.; Wu L. Z.; Tung C. H. Highly Efficient and Selective Photocatalytic Hydrogenation of Functionalized Nitrobenzenes. Green Chem. 2014, 16, 1082–1086. 10.1039/C3GC42042F. [DOI] [Google Scholar]
- Konev M. O.; Cardinale L.; Jacobi Von Wangelin A. Catalyst-Free N-Deoxygenation by Photoexcitation of Hantzsch Ester. Org. Lett. 2020, 22, 1316–1320. 10.1021/acs.orglett.9b04632. [DOI] [PubMed] [Google Scholar]
- Wu J.; Grant P. S.; Li X.; Noble A.; Aggarwal V. K. Catalyst-Free Deaminative Functionalizations of Primary Amines by Photoinduced Single-Electron Transfer. Angew. Chem., Int. Ed. 2019, 58, 5697–5701. 10.1002/anie.201814452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J. F.; Lauber M. B.; Muhr V.; Kratzer D.; Paradies J. Readily Available Hydrogen Bond Catalysts for the Asymmetric Transfer Hydrogenation of Nitroolefins. Org. Biomol. Chem. 2011, 9, 4323–4327. 10.1039/c1ob05059a. [DOI] [PubMed] [Google Scholar]
- Bernardi R.; Caronna T.; Morrocchi S.; Ursini M. Photochemically Induced Reactions of Selected Ketones with 4-Cyanopyridine in Neutral and Acidic Medium. J. Heterocycl. Chem. 1996, 33, 1137–1142. 10.1002/jhet.5570330422. [DOI] [Google Scholar]
- Vittimberga B. M.; Minisci F.; Morrocchi S. The Photoinitiated Electron Transfer-Substitution Reaction of Diphenyl Ketyl with Protonated 4-Cyanopyridine. J. Am. Chem. Soc. 1975, 97, 4397–4398. 10.1021/ja00848a044. [DOI] [Google Scholar]
- Caronna T.; Morrocchi S.; Vittimberga B. M. The Photoinitiated Substitution Reactions of 2-Pyridinecarbonitrile with Benzophenone in Aqueous 2-Propanol. J. Heterocycl. Chem. 1980, 17, 399–400. 10.1002/jhet.5570170239. [DOI] [Google Scholar]
- Carolina T.; Clerici A.; Coggiola D.; Morrocchi S. Photochemical Substitution Reactions of 4-Pyridinecarbonitrile with Aliphatic Alcohols in Neutral and Acidified Medium. J. Heterocycl. Chem. 1981, 18, 1421–1423. 10.1002/jhet.5570180729. [DOI] [Google Scholar]
- Furihata T.; Sugimori A. Competitive Photo-Ethoxylation and -Hydroxyethylation of Pyridine-2-Carbonitrile in Acidic Ethanolic Solutions. J. Chem. Soc., Chem. Commun. 1975, (7), 241–242. 10.1039/c3975000241b. [DOI] [Google Scholar]
- Bernardi R.; Caronna T.; dal Pio Luogo D.; Morrocchi S.; Poggi G.; Vittimberga B. M. A Study of Solvent Effect on Photochemically Induced Reations between Pyridinedicarbonitriles and Alkenes: An Easy Approach to the Synthesis of Cyclopenta[b or c]Pyridines. J. Chem. Soc., Perkin Trans. 1 1996, 13, 1593–1600. 10.1039/p19960001593. [DOI] [Google Scholar]
- Caronna T.; Morrocchi S.; Vittimberga B. M. Importance of Acidity on the Energetically Unfavorable Electron-Transfer Reaction. An Extension of the Rehm-Weller Equation. Photoreaction of Triplet 2,4-Pyridinedicarbonitrile with 2-Propanol. J. Am. Chem. Soc. 1986, 108, 2205–2208. 10.1021/ja00269a013. [DOI] [PubMed] [Google Scholar]
- McNally A.; Prier C. K.; MacMillan D. W. C. Discovery of an α-Amino C-H Arylation Reaction Using the Strategy of Accelerated Serendipity. Science 2011, 334, 1114–1117. 10.1126/science.1213920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Z.; MacMillan D. W. C. Decarboxylative Arylation of α-Amino Acids via Photoredox Catalysis: A One-Step Conversion of Biomass to Drug Pharmacophore. J. Am. Chem. Soc. 2014, 136, 5257–5260. 10.1021/ja501621q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshikawa T.; Inoue M. Photoinduced Direct 4-Pyridination of C(sp3)-H Bonds. Chem. Sci. 2013, 4, 3118–3123. 10.1039/c3sc51080h. [DOI] [Google Scholar]
- Kamijo S.; Hoshikawa T.; Inoue M. Photochemically Induced Radical Transformation of C(sp3)-H Bonds to C(sp3)-CN Bonds. Org. Lett. 2011, 13, 5928–5931. 10.1021/ol202659e. [DOI] [PubMed] [Google Scholar]
- Hoshikawa T.; Yoshioka S.; Kamijo S.; Inoue M. Photoinduced Direct Cyanation of C(sp3)-H Bonds. Synthesis 2013, 45, 874–887. 10.1055/s-0032-1318325. [DOI] [Google Scholar]
- Hoshikawa T.; Kamijo S.; Inoue M. Photochemically Induced Radical Alkynylation of C(sp3)-H Bonds. Org. Biomol. Chem. 2013, 11, 164–169. 10.1039/C2OB26785C. [DOI] [PubMed] [Google Scholar]
- Yoshioka S.; Nagatomo M.; Inoue M. Application of Two Direct C(sp3)-H Functionalizations for Total Synthesis of (+)-Lactacystin. Org. Lett. 2015, 17, 90–93. 10.1021/ol503291s. [DOI] [PubMed] [Google Scholar]
- Nicastri M. C.; Lehnherr D.; Lam Y. H.; Dirocco D. A.; Rovis T. Synthesis of Sterically Hindered Primary Amines by Concurrent Tandem Photoredox Catalysis. J. Am. Chem. Soc. 2020, 142, 987–998. 10.1021/jacs.9b10871. [DOI] [PubMed] [Google Scholar]
- Callier-Dublanchet A. C.; Quiclet-Sire B.; Zard S. Z. A New Source of Nitrogen Centered Radicals. Tetrahedron Lett. 1995, 36, 8791–8794. 10.1016/0040-4039(95)01878-L. [DOI] [Google Scholar]
- Holubek J.; Volke J. Polarography of Heterocyclic Aromatic Compounds. XIII. Polarographic Fission of Carbon-Halogen Bonds in Monohalogenopyridines. Collect. Czech. Chem. Commun. 1962, 27, 680–692. 10.1135/cccc19620680. [DOI] [Google Scholar]
- Andrieux C. P.; Blocman C.; Dumas-Bouchiat J. M.; Saveant J. M. Heterogeneous and Homogeneous Electron Transfers to Aromatic Halides. An Electrochemical Redox Catalysis Study in the Halobenzene and Halopyridine Series. J. Am. Chem. Soc. 1979, 101, 3431–3441. 10.1021/ja00507a002. [DOI] [Google Scholar]
- Enemærke R. J.; Christensen T. B.; Jensen H.; Daasbjerg K. Application of a New Kinetic Method in the Investigation of Cleavage Reactions of Haloaromatic Radical Anions. J. Chem. Soc. Perkin Trans. 2 2001, 1, 1620–1630. 10.1039/b102835a. [DOI] [Google Scholar]
- Ghosh I.; Marzo L.; Das A.; Shaikh R.; König B. Visible Light Mediated Photoredox Catalytic Arylation Reactions. Acc. Chem. Res. 2016, 49, 1566–1577. 10.1021/acs.accounts.6b00229. [DOI] [PubMed] [Google Scholar]
- Heinrich M. R. Intermolecular Olefin Functionalisation Involving Aryl Radicals Generated from Arenediazonium Salts. Chem. - Eur. J. 2009, 15, 820–833. 10.1002/chem.200801306. [DOI] [PubMed] [Google Scholar]
- Fehler S. K.; Heinrich M. R. How the Structural Elucidation of the Natural Product Stephanosporin Led to New Developments in Aryl Radical and Medicinal Chemistry. Synlett 2015, 26, 580–603. 10.1055/s-0034-1378945. [DOI] [Google Scholar]
- Kindt S.; Heinrich M. R. Recent Advances in Meerwein Arylation Chemistry. Synthesis 2016, 48, 1597–1606. 10.1055/s-0035-1561586. [DOI] [Google Scholar]
- Demir A. S.; Reis Ö.; Emrullahoglu M. Generation of Aryl Radicals from Arylboronic Acids by Manganese(III) Acetate: Synthesis of Biaryls and Heterobiaryls. J. Org. Chem. 2003, 68, 578–580. 10.1021/jo026466c. [DOI] [PubMed] [Google Scholar]
- Lima F.; Kabeshov M. A.; Tran D. N.; Battilocchio C.; Sedelmeier J.; Sedelmeier G.; Schenkel B.; Ley S. V. Visible Light Activation of Boronic Esters Enables Efficient Photoredox C(sp2)-C(sp3) Cross-Couplings in Flow. Angew. Chem., Int. Ed. 2016, 55, 14085–14089. 10.1002/anie.201605548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima F.; Sharma U. K.; Grunenberg L.; Saha D.; Johannsen S.; Sedelmeier J.; Van der Eycken E. V.; Ley S. V. A Lewis Base Catalysis Approach for the Photoredox Activation of Boronic Acids and Esters. Angew. Chem., Int. Ed. 2017, 56, 15136–15140. 10.1002/anie.201709690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan G.; Yang M.; Wu X. Synthetic Applications of Arylboronic Acid via an Aryl Radical Transfer Pathway. Org. Biomol. Chem. 2013, 11, 7999–8008. 10.1039/c3ob41851k. [DOI] [PubMed] [Google Scholar]
- Bunton C. A.; Minch M. J.; Wolfe B. B. Decomposition of Pyridine-2- and −4-Diazotates. J. Am. Chem. Soc. 1974, 96, 3267–3275. 10.1021/ja00817a040. [DOI] [Google Scholar]
- Cox P. A.; Reid M.; Leach A. G.; Campbell A. D.; King E. J.; Lloyd-Jones G. C. Base-Catalyzed Aryl-B(OH)2 Protodeboronation Revisited: From Concerted Proton Transfer to Liberation of a Transient Aryl Anion. J. Am. Chem. Soc. 2017, 139, 13156–13165. 10.1021/jacs.7b07444. [DOI] [PubMed] [Google Scholar]
- Garden S. J.; Avila D. V.; Beckwith A. L. J.; Bowry V. W.; Ingold K. U.; Lusztyk J. Absolute Rate Constant for the Reaction of Aryl Radicals with Tri-n-Butyltin Hydride. J. Org. Chem. 1996, 61, 805–809. 10.1021/jo951245a. [DOI] [PubMed] [Google Scholar]
- Arora A.; Teegardin K. A.; Weaver J. D. Reductive Alkylation of 2-Bromoazoles via Photoinduced Electron Transfer: A Versatile Strategy to Csp2-Csp3 Coupled Products. Org. Lett. 2015, 17, 3722–3725. 10.1021/acs.orglett.5b01711. [DOI] [PubMed] [Google Scholar]
- Singh A.; Arora A.; Weaver J. D. Photoredox-Mediated C-H Functionalization and Coupling of Tertiary Aliphatic Amines with 2-Chloroazoles. Org. Lett. 2013, 15, 5390–5393. 10.1021/ol402751j. [DOI] [PubMed] [Google Scholar]
- Ji C.; Peters D. G. Electrochemical Reduction of 2-Bromothiazole at Carbon Cathodes in Acetonitrile. J. Electroanal. Chem. 1998, 455, 147–152. 10.1016/S0022-0728(98)00158-2. [DOI] [Google Scholar]
- Stirling M. J.; Sweeney G.; Macrory K.; Blacker A. J.; Page M. I. The Kinetics and Mechanism of the Organo-Iridium-Catalysed Enantioselective Reduction of Imines. Org. Biomol. Chem. 2016, 14, 3614–3622. 10.1039/C6OB00245E. [DOI] [PubMed] [Google Scholar]
- Arora A.; Weaver J. D. Photocatalytic Generation of 2-Azolyl Radicals: Intermediates for the Azolylation of Arenes and Heteroarenes via C-H Functionalization. Org. Lett. 2016, 18, 3996–3999. 10.1021/acs.orglett.6b01718. [DOI] [PubMed] [Google Scholar]
- Seath C. P.; Jui N. T. Intermolecular Reactions of Pyridyl Radicals with Olefins via Photoredox Catalysis. Synlett 2019, 30, 1607–1614. 10.1055/s-0037-1611527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aycock R. A.; Wang H.; Jui N. T. A Mild Catalytic System for Radical Conjugate Addition of Nitrogen Heterocycles. Chem. Sci. 2017, 8, 3121–3125. 10.1039/C7SC00243B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aycock R. A.; Vogt D. B.; Jui N. T. A Practical and Scalable System for Heteroaryl Amino Acid Synthesis. Chem. Sci. 2017, 8, 7998–8003. 10.1039/C7SC03612D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inouye S.; Shomura T.; Tsuruoka T.; Ogawa Y.; Watanabe H.; Yoshida J.; Niida T. L-β-(5-Hydroxy-2-Pyridyl)-Alanine and L-β-(3-Hydroxyureido)-Alanine from Streptomyces. Chem. Pharm. Bull. 1975, 23, 2669–2677. 10.1248/cpb.23.2669. [DOI] [PubMed] [Google Scholar]
- Izawa M; Takayama S; Shindo-Okada N; Doi S; Kimura M; Katsuki M; Nishimura S Inhibition of Chemical Carcinogenesis in Vivo by Azatyrosine. Biomed. Pharmacother. 1992, 52, 1628–1630. 10.1016/0753-3322(92)90225-V. [DOI] [PubMed] [Google Scholar]
- Beckers T.; Bernd M.; Kutscher B.; Kühne R.; Hoffmann S.; Reissmann T. Structure-Function Studies of Linear and Cyclized Peptide Antagonists of the GnRH Receptor. Biochem. Biophys. Res. Commun. 2001, 289, 653–663. 10.1006/bbrc.2001.5939. [DOI] [PubMed] [Google Scholar]
- Ma X.; Dang H.; Rose J. A.; Rablen P.; Herzon S. B. Hydroheteroarylation of Unactivated Alkenes Using N-Methoxyheteroarenium Salts. J. Am. Chem. Soc. 2017, 139, 5998–6007. 10.1021/jacs.7b02388. [DOI] [PubMed] [Google Scholar]
- Green S. A.; Matos J. L. M.; Yagi A.; Shenvi R. A. Branch-Selective Hydroarylation: Iodoarene-Olefin Cross-Coupling. J. Am. Chem. Soc. 2016, 138, 12779–12782. 10.1021/jacs.6b08507. [DOI] [PubMed] [Google Scholar]
- Seath C. P.; Vogt D. B.; Xu Z.; Boyington A. J.; Jui N. T. Radical Hydroarylation of Functionalized Olefins and Mechanistic Investigation of Photocatalytic Pyridyl Radical Reactions. J. Am. Chem. Soc. 2018, 140, 15525–15534. 10.1021/jacs.8b10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorov A. R.; Wirtanen T.; Helaja J. Photoreductive Removal of O-Benzyl Groups from Oxyarene N-Heterocycles Assisted by O-Pyridine-Pyridone Tautomerism. J. Org. Chem. 2017, 82, 13756–13767. 10.1021/acs.joc.7b02775. [DOI] [PubMed] [Google Scholar]
- Collot A. G.; Courtney M.; Coyne D.; Eustace S. E.; More O’Ferrall R. A. Binding Catalysis and Inhibition by Metal Ions and Protons in the Enolization of Phenylacetylpyrazine. J. Org. Chem. 2009, 74, 3356–3369. 10.1021/jo802650s. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Mayer J. M. Surprisingly Long-Lived Ascorbyl Radicals in Acetonitrile: Concerted Proton-Electron Transfer Reactions and Thermochemistry. J. Am. Chem. Soc. 2008, 130, 7546–7547. 10.1021/ja802055t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumler W. D.; Daniels T. C. Titration Curves and Dissociation Constants of L-Ascorbic Acid (Vitamin C) and Diethyl Dihydroxymaleate. J. Am. Chem. Soc. 1935, 57, 1929–1930. 10.1021/ja01313a053. [DOI] [Google Scholar]
- Shevchenko G. A.; Oppelaar B.; List B. An Unexpected α-Oxidation of Cyclic Ketones with 1,4-Benzoquinone by Enol Catalysis. Angew. Chem., Int. Ed. 2018, 57, 10756–10759. 10.1002/anie.201804445. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Fu L.; Chen P.; Zou J.; Liu G. Proton-Coupled Electron Transfer Enables Tandem Radical Relay for Asymmetric Copper-Catalyzed Phosphinoylcyanation of Styrenes. Org. Lett. 2019, 21, 5015–5020. 10.1021/acs.orglett.9b01607. [DOI] [PubMed] [Google Scholar]
- Wang C.; Qin J.; Shen X.; Riedel R.; Harms K.; Meggers E. Asymmetric Radical-Radical Cross-Coupling through Visible-Light-Activated Iridium Catalysis. Angew. Chem., Int. Ed. 2016, 55, 685–688. 10.1002/anie.201509524. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Li Y.; Han B.; Gong L.; Meggers E. Enantioselective Catalytic β-Amination through Proton-Coupled Electron Transfer Followed by Stereocontrolled Radical-Radical Coupling. Chem. Sci. 2017, 8, 5757–5763. 10.1039/C7SC02031G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W.; Zhou Z.; Gong L.; Meggers E. Asymmetric Alkylation of Remote C(sp3)-H Bonds by Combining Proton-Coupled Electron Transfer with Chiral Lewis Acid Catalysis. Chem. Commun. 2017, 53, 8964–8967. 10.1039/C7CC04941B. [DOI] [PubMed] [Google Scholar]
- Gentry E. C.; Rono L. J.; Hale M. E.; Matsuura R.; Knowles R. R. Enantioselective Synthesis of Pyrroloindolines via Noncovalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products. J. Am. Chem. Soc. 2018, 140, 3394–3402. 10.1021/jacs.7b13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q.; Gentry E. C.; Knowles R. R. Catalytic Carbocation Generation Enabled by the Mesolytic Cleavage of Alkoxyamine Radical Cations. Angew. Chem., Int. Ed. 2016, 55, 9969–9973. 10.1002/anie.201604619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K.; Tong X.; Li T.; Shi B.; Wang H.; Yan P.; Xia C. Enantioselective Radical Cyclization of Tryptamines by Visible Light-Excited Nitroxides. J. Org. Chem. 2018, 83, 10948–10958. 10.1021/acs.joc.8b01597. [DOI] [PubMed] [Google Scholar]
- Yin Y.; Dai Y.; Jia H.; Li J.; Bu L.; Qiao B.; Zhao X.; Jiang Z. Conjugate Addition-Enantioselective Protonation of N-Aryl Glycines to α-Branched 2-Vinylazaarenes via Cooperative Photoredox and Asymmetric Catalysis. J. Am. Chem. Soc. 2018, 140, 6083–6087. 10.1021/jacs.8b01575. [DOI] [PubMed] [Google Scholar]
- Qiao B.; Li C.; Zhao X.; Yin Y.; Jiang Z. Enantioselective Reduction of Azaarene-Based Ketones: Via Visible Light-Driven Photoredox Asymmetric Catalysis. Chem. Commun. 2019, 55, 7534–7537. 10.1039/C9CC03661J. [DOI] [PubMed] [Google Scholar]
- Costentin C. Electrochemical Approach to the Mechanistic Study of Proton-Coupled Electron Transfer. Chem. Rev. 2008, 108, 2145–2179. 10.1021/cr068065t. [DOI] [PubMed] [Google Scholar]
- Yi H.; Zhang G.; Wang H.; Huang Z.; Wang J.; Singh A. K.; Lei A. Recent Advances in Radical C-H Activation/Radical Cross-Coupling. Chem. Rev. 2017, 117, 9016–9085. 10.1021/acs.chemrev.6b00620. [DOI] [PubMed] [Google Scholar]
- Silvi M.; Verrier C.; Rey Y. P.; Buzzetti L.; Melchiorre P. Visible-Light Excitation of Iminium Ions Enables the Enantioselective Catalytic β-Alkylation of Enals. Nat. Chem. 2017, 9, 868–873. 10.1038/nchem.2748. [DOI] [PubMed] [Google Scholar]
- Guo X.; Okamoto Y.; Schreier M. R.; Ward T. R.; Wenger O. S. Enantioselective Synthesis of Amines by Combining Photoredox and Enzymatic Catalysis in a Cyclic Reaction Network. Chem. Sci. 2018, 9, 5052–5056. 10.1039/C8SC01561A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegasiewicz K. F.; Cooper S. J.; Emmanuel M. A.; Miller D. C.; Hyster T. K. Catalytic Promiscuity Enabled by Photoredox Catalysis in Nicotinamide-Dependent Oxidoreductases. Nat. Chem. 2018, 10, 770–775. 10.1038/s41557-018-0059-y. [DOI] [PubMed] [Google Scholar]
- Martens F. M.; Verhoeven J. W.; Gase R. A.; Pandit U. K.; de Boer T. J. On the Question of One-Electron Transfer in the Mechanism of Reduction by Nadh-Models. Tetrahedron 1978, 34, 443–446. 10.1016/0040-4020(78)80030-1. [DOI] [Google Scholar]
- Sandoval B. A.; Kurtoic S. I.; Chung M. M.; Biegasiewicz K. F.; Hyster T. K. Photoenzymatic Catalysis Enables Radical-Mediated Ketone Reduction in Ene-Reductases. Angew. Chem., Int. Ed. 2019, 58, 8714–8718. 10.1002/anie.201902005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGilvray K. L.; Decan M. R.; Wang D.; Scaiano J. C. Facile Photochemical Synthesis of Unprotected Aqueous Gold Nanoparticles. J. Am. Chem. Soc. 2006, 128, 15980–15981. 10.1021/ja066522h. [DOI] [PubMed] [Google Scholar]
- Gachard E.; Remita H.; Khatouri J.; Keita B.; Nadjo L.; Belloni J. Radiation-Induced and Chemical Formation of Gold Clusters. New J. Chem. 1998, 22, 1257–1265. 10.1039/a804445g. [DOI] [Google Scholar]
- Scaiano J. C.; Stamplecoskie K. G.; Hallett-Tapley G. L. Photochemical Norrish Type I Reaction as a Tool for Metal Nanoparticle Synthesis: Importance of Proton Coupled Electron Transfer. Chem. Commun. 2012, 48, 4798–4808. 10.1039/c2cc30615h. [DOI] [PubMed] [Google Scholar]
- Shukla D.; Ahearn W. G.; Farid S. Enhancement of Chain Amplification in Photoreactions of N-Methoxypyridinium Salts with Alcohols. Photochem. Photobiol. 2006, 82, 146. 10.1562/2005-06-28-RA-594. [DOI] [PubMed] [Google Scholar]
- Stamplecoskie K. G.; Scaiano J. C. Light Emitting Diode Irradiation Can Control the Morphology and Optical Properties of Silver Nanoparticles. J. Am. Chem. Soc. 2010, 132, 1825–1827. 10.1021/ja910010b. [DOI] [PubMed] [Google Scholar]
- Pacioni N. L.; Pardoe A.; McGilvray K. L.; Chrétien M. N.; Scaiano J. C. Synthesis of Copper Nanoparticles Mediated by Photogenerated Free Radicals: Catalytic Role of Chloride Anions. Photochem. Photobiol. Sci. 2010, 9, 766–774. 10.1039/b9pp00200f. [DOI] [PubMed] [Google Scholar]
- Scaiano J. C.; Netto-Ferreira J. C.; Alarcon E.; Billone P.; Alejo C. J. B.; Crites C. O. L.; Decan M.; Fasciani C.; González-Béjar M.; Hallett-Tapley G. Tuning Plasmon Transitions and Their Applications in Organic Photochemistry. Pure Appl. Chem. 2011, 83, 913–930. 10.1351/PAC-CON-11-01-09. [DOI] [Google Scholar]
- Wee T. L.; Sherman B. D.; Gust D.; Moore A. L.; Moore T. A.; Liu Y.; Scaiano J. C. Photochemical Synthesis of a Water Oxidation Catalyst Based on Cobalt Nanostructures. J. Am. Chem. Soc. 2011, 133, 16742–16745. 10.1021/ja206280g. [DOI] [PubMed] [Google Scholar]
- Scaiano J. C. Photochemical and Free-Radical Processes in Benzil-Amine Systems. Electron-Donor Properties of α-Aminoalkyl Radicals. J. Phys. Chem. 1981, 85, 2851–2855. 10.1021/j150619a031. [DOI] [Google Scholar]
- Slaughter B. V.; Khurshid S. S.; Fisher O. Z.; Khademhosseini A.; Peppas N. A. Hydrogels in Regenerative Medicine. Adv. Mater. 2009, 21, 3307–3329. 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischakov R.; Adler-Abramovich L.; Buzhansky L.; Shekhter T.; Gazit E. Peptide-Based Hydrogel Nanoparticles as Effective Drug Delivery Agents. Bioorg. Med. Chem. 2013, 21, 3517–3522. 10.1016/j.bmc.2013.03.012. [DOI] [PubMed] [Google Scholar]
- Raeburn J.; Cardoso A. Z.; Adams D. J. The Importance of the Self-Assembly Process to Control Mechanical Properties of Low Molecular Weight Hydrogels. Chem. Soc. Rev. 2013, 42, 5143–5156. 10.1039/c3cs60030k. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Li Y.; Qin M.; Cao Y.; Wang W. Photo-Cross-Linking Approach to Engineering Small Tyrosine-Containing Peptide Hydrogels with Enhanced Mechanical Stability. Langmuir 2013, 29, 13299–13306. 10.1021/la4029639. [DOI] [PubMed] [Google Scholar]
- Fancy D. A.; Kodadek T. Chemistry for the Analysis of Protein-Protein Interactions: Rapid and Efficient Cross-Linking Triggered by Long Wavelength Light. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 6020–6024. 10.1073/pnas.96.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp C. P.; Peterson I. L.; Knoff D. S.; Melcher L. G.; Maxwell C. J.; Cohen A. T.; Wertheimer A. M.; Kim M. Non-Cytotoxic Dityrosine Photocrosslinked Polymeric Materials With Targeted Elastic Moduli. Front. Chem. 2020, 8, 173. 10.3389/fchem.2020.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I.; Bang W. Y.; Park W. H.; Han E. H.; Lee E. Photo-Crosslinkable Elastomeric Protein-Derived Supramolecular Peptide Hydrogel with Controlled Therapeutic CO-Release. Nanoscale 2019, 11, 17327–17333. 10.1039/C9NR06115K. [DOI] [PubMed] [Google Scholar]
- Donnelly P. E.; Chen T.; Finch A.; Brial C.; Maher S. A.; Torzilli P. A. Photocrosslinked Tyramine-Substituted Hyaluronate Hydrogels with Tunable Mechanical Properties Improve Immediate Tissue-Hydrogel Interfacial Strength in Articular Cartilage. J. Biomater. Sci., Polym. Ed. 2017, 28, 582–600. 10.1080/09205063.2017.1289035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork J. W.; Johnson S. L.; Tranquillo R. T. Ruthenium-Catalyzed Photo Cross-Linking of Fibrin-Based Engineered Tissue. Biomaterials 2011, 32, 2479–2488. 10.1016/j.biomaterials.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen J. D.; Matsuura B. S.; Stephenson C. R. J. A Photochemical Strategy for Lignin Degradation at Room Temperature. J. Am. Chem. Soc. 2014, 136, 1218–1221. 10.1021/ja4113462. [DOI] [PubMed] [Google Scholar]
- Nguyen S. T.; Murray P. R. D.; Knowles R. R. Light-Driven Depolymerization of Native Lignin Enabled by Proton-Coupled Electron Transfer. ACS Catal. 2020, 10, 800–805. 10.1021/acscatal.9b04813. [DOI] [Google Scholar]
- Wang Y.; Liu Y.; He J.; Zhang Y. Redox-Neutral Photocatalytic Strategy for Selective C-C Bond Cleavage of Lignin and Lignin Models via PCET Process. Sci. Bull. 2019, 64, 1658–1666. 10.1016/j.scib.2019.09.003. [DOI] [PubMed] [Google Scholar]
- Ishida M.; Kim S. J.; Preihs C.; Ohkubo K.; Lim J. M.; Lee B. S.; Park J. S.; Lynch V. M.; Roznyatovskiy V. V.; Sarma T.; et al. Protonation-Coupled Redox Reactions in Planar Antiaromatic Meso-Pentafluorophenyl-Substituted o-Phenylene-Bridged Annulated Rosarins. Nat. Chem. 2013, 5, 15–20. 10.1038/nchem.1501. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Ren S.; Guo X. Switching Effects in Molecular Electronic Devices. Top. Curr. Chem. 2017, 375, 56. 10.1007/s41061-017-0144-5. [DOI] [PubMed] [Google Scholar]
- Sarma T.; Kim G.; Sen S.; Cha W. Y.; Duan Z.; Moore M. D.; Lynch V. M.; Zhang Z.; Kim D.; Sessler J. L. Proton-Coupled Redox Switching in an Annulated π-Extended Core-Modified Octaphyrin. J. Am. Chem. Soc. 2018, 140, 12111–12119. 10.1021/jacs.8b06938. [DOI] [PubMed] [Google Scholar]
- Tanaka T.; Osuka A. Chemistry of Meso-Aryl-Substituted Expanded Porphyrins: Aromaticity and Molecular Twist. Chem. Rev. 2017, 117, 2584–2640. 10.1021/acs.chemrev.6b00371. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wang S.; Wang X.; Zhang W.; Zheng W.; Zhang Y. M.; Zhang S. X. A. A Multicolour Bistable Electronic Shelf Label Based on Intramolecular Proton-Coupled Electron Transfer. Nat. Mater. 2019, 18, 1335–1342. 10.1038/s41563-019-0471-8. [DOI] [PubMed] [Google Scholar]
- Shao Z.; Wu Q.; Han X.; Zhao Y.; Xie Q.; Wang H.; Hou H. Proton Coupled Electron Transfer: Novel Photochromic Performance in a Host-Guest Collaborative MOF. Chem. Commun. 2019, 55, 10948–10951. 10.1039/C9CC05498G. [DOI] [PubMed] [Google Scholar]
- Conway B. E.Electrochemical Supercapacitors, Scientific Fundamentals and Technological Applications; Springer: Berlin, 1999. [Google Scholar]
- Vivekchand S. R. C.; Rout C. S.; Subrahmanyam K. S.; Govindaraj A.; Rao C. N. R. Graphene-Based Electrochemical Supercapacitors. Proc. - Indian Acad. Sci., Chem. Sci. 2008, 120, 9–13. 10.1007/s12039-008-0002-7. [DOI] [Google Scholar]
- Roldán S.; Blanco C.; Granda M.; Menéndez R.; Santamaría R. Towards a Further Generation of High-Energy Carbon-Based Capacitors by Using Redox-Active Electrolytes. Angew. Chem., Int. Ed. 2011, 50, 1699–1701. 10.1002/anie.201006811. [DOI] [PubMed] [Google Scholar]
- Zheng J. P.; Cygan P. J.; Jow T. R. Hydrous Ruthenium Oxide as an Electrode Material for Electrochemical Capacitors. J. Electrochem. Soc. 1995, 142, 2699–2703. 10.1149/1.2050077. [DOI] [Google Scholar]
- Singh C.; Paul A. Physisorbed Hydroquinone on Activated Charcoal as a Supercapacitor: An Application of Proton-Coupled Electron Transfer. J. Phys. Chem. C 2015, 119, 11382–11390. 10.1021/acs.jpcc.5b01322. [DOI] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical Methods: Fundamentals and Applications; Wiley, 1980. [Google Scholar]
- Xie X.; Bakker E. Photoelectric Conversion Based on Proton-Coupled Electron Transfer Reactions. J. Am. Chem. Soc. 2014, 136, 7857–7860. 10.1021/ja503491k. [DOI] [PubMed] [Google Scholar]
- Motoyama D.; Yoshikawa K.; Ozawa H.; Tadokoro M.; Haga M.-a. Energy-Storage Applications for a pH Gradient between Two Benzimidazole-Ligated Ruthenium Complexes That Engage in Proton-Coupled Electron-Transfer Reactions in Solution. Inorg. Chem. 2017, 56, 6419–6428. 10.1021/acs.inorgchem.7b00518. [DOI] [PubMed] [Google Scholar]